
Abstract
In the last few years, our understanding of disease molecular mechanisms underpinning ALS has advanced greatly, allowing the first steps in translating into clinical practice novel research findings, including gene therapy approaches. Similarly, the recent advent of assistive technologies has greatly improved the possibility of a more personalized approach to supportive and symptomatic care, in the context of an increasingly complex multidisciplinary line of actions, which remains the cornerstone of ALS management. Against this rapidly growing background, here we provide an comprehensive update on the most recent studies that have contributed towards our understanding of ALS pathogenesis, the latest results from clinical trials as well as the future directions for improving the clinical management of ALS patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Motor neuron disease (MND) covers a heterogeneous group of neurological disorders defined and characterized by degeneration of motor neurons. With an incidence of approximately 2.16 per 100.000 person-years and a median survival time of 3 to 5 years, amyotrophic lateral sclerosis (ALS) is the most common and severe form, involving both lower motor neurons (LMN) and upper motor neurons (UMN), underpinned by a complex interplay between genetic predisposition and environmental factors [1, 2]. Starting from our previous update [3], this review summarizes the recent highlights in ALS research published in the Journal of Neurology and other relevant scientific Journals during the last 60 months.
Epidemiology
Recent studies confirmed that ALS incidence in European populations is the highest worldwide, probably due to genetic background and high life expectancy in developed regions [2, 4,5,6]. Moreover, the latest studies reported an increasing incidence of ALS year by year in Ireland, Scotland and some regions of Italy [2, 7,8,9,10,11]. The rising incidence of ALS in these countries may be related to multiple factors, such as the increased ascertainment, a reduced rate of emigration or increasing longevity [2, 9,10,11]. Conversely, in Asia, the overall incidence of ALS is lower than Europe and North America but, interestingly, the total prevalence is only second to that of West Europe [6], which may be in part linked to genetic differences, such as the lower prevalence of the C9orf72 hexanucleotide repeat expansion in Asian countries [2, 12, 13]. To increase the knowledge of the ALS genetic puzzle, it will also be crucial to conduct studies on admixed populations or communities with a non-European background [2, 14]. In this respect, preliminary results from the Latin American Epidemiologic study of ALS (LAENALS) corroborated the different genetic predisposition of these populations, supported by a low incidence and prevalence of ALS in these regions [15]. Previous studies initially reported an increased incidence of ALS among males, reporting a male-to-female ratio up to 2:1 [16]. However, more recent studies have refined the estimate of sex ratio and a recent meta-analysis described a male-to-female ratio of approximately 1.3:1, confirming that ALS is slightly more frequent in man. This difference may be due to sex hormones, unequal environmental exposure or sex chromosomes [17, 18]. Several factors have been addressed to explain this variability in sex ratio, such as potential differences in ascertainment strategy, increased life expectancy among females or the type of study (population vs non-population-based studies) [17]. Importantly, a recent study highlights new findings on the relationship between sex and age at onset, revealing a high incidence of males for both youngest and oldest onset and changing ratios at increasing age [17]. This latter observation may be related to the high association between the female gender and the pure UMN phenotype, which is correlated with a long survival [19,20,21]. Sexual dimorphism indeed entails relevant prognostic implication since man showed an higher weight loss and respiratory function deterioration than women, resulting in a worse prognosis [22]. However, the role of sex is more complex and controversial since it is also recognized that the flail arm phenotype, which is associated with a longer survival, is more common in man [18], and might also implicate differing structural and functional differences in the brain, as highlighted by recent neuroimaging studies [23, 24]. More studies are needed to clarify the interaction between sex, gene and time, which may confer susceptibility to the development of the disease [13, 17, 21].
Proposed disease mechanisms
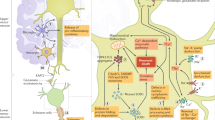
In the last few years, further advances have been made in our understanding of MND pathogenesis, a complex process involving a variety of genes and multiple pathways, such as an imbalance of protein homeostasis in neurons, alteration of RNA metabolism, mitochondrial dysfunction and the accumulation of protein aggregates in the cytoplasm of neurons with a toxic gain-of-function [3, 25,26,27,28].
Since the first ALS-causing gene Cu–Zn superoxide dismutase 1 (SOD1) was discovered almost 30 years ago [29], more than 40 genes have been associated with the inheritance or pathogenesis of ALS. To date, four major genes are known to cover up to 60% of familial forms of ALS and 10–13% of sporadic ALS cases, i.e., SOD1, the chromosome 9 open reading frame 72 (C9orf72), TAR DNA-binding protein (TARDBP) [30, 31], and fused in sarcoma (FUS). The C9orf72 hexanucleotide repeat expansion, discovered in 2011 [32, 33], is the most common genetic cause in European familial ALS (fALS) cases (more than 30%), but also accounts for approx. 7% cases of sporadic ALS (sALS). A great portion of familial FTD cases (approx. 25%) also carried the C9orf72 pathological expansion, explaining the genetic overlapping of ALS and FTD diseases.15, [34, 35]
Most of SOD1 disease-causing variants are missense mutations and account for 15–30% of fALS cases and less than 2% of sALS cases [36]. Mutations in TARDBP are found in approx. 4% of European fALS cases, whereas FUS is more common in Asian fALS cases (6.4% vs 2.8% of European fALS) [37]. Variants in other genes are found in less than 1% of patients [38]. A significant proportion of fALS patients still remain without a genetic diagnosis, suggesting a more complex disease pathogenesis and that other ALS genes may be involved and remain to be discovered [38].
An acceleration in ALS-related gene discovery came in the 2010s with the advent of next generation sequencing (NGS) and more recently, of whole-genome and exome sequencing (WGS/WES, respectively), technologies that facilitated the discovery of numerous genetic variants and enabled large-scale studies to identify ALS risk genes [39]. TANK-binding kinase 1 (TBK1) [40, 41] and NIMA-related kinase 1 (NEK1) [42] are two examples of genes discovered thanks to these large-scale studies, and subsequently confirmed as ALS-related genes [43, 44]. Broadly speaking, the advent of WGS/WES allows today the detection of more than 250 new genetic association each year, which includes a number of disease relevant variants [45], as well as many variants of uncertain significance (VUS). Interpreting the disease relevance of VUS represents a major challenge as many variants initially reported to be potentially pathogenic, from in silico prediction tools, are later reclassified as benign after additional experimental evidence is accumulated [39]. Loss-of-function (LoF) mutations of the TBK1 gene are nowadays accepted as the cause of a dominant form of ALS and FTD, potentially explaining up to 4% of fALS cases, including TBK1 in the ALS-major genes group. These mutations result in an impaired interaction of TBK1 with optineurin and p62, both already implicated in ALS pathogenesis, through a common pathway of autophagy regulation [41, 43, 46,47,48]. In the last few years, different studies have highlighted a significant enrichment of NEK1 LoF variants in ALS, and an additional role for the p.Arg261His missense variant in ALS susceptibility. A study conducted in an Italian ALS cohort suggested also a correlation between the presence of NEK1 variants and the flail arm-ALS phenotype [42, 44, 49].
A loss-of-function mechanism has also been associated with the DnaJ heat shock protein family member C7 (DNAJC7), which has been identified as a genetic risk factor for ALS according to a large-scale WES study [50]. Indeed, a functional study has recently shown reduced protein levels in ALS patients carrying a p.R156X variant, suggesting a role of DNAJC7 in the regulation of protein misfolding, which is a major molecular pathology in ALS, through its interaction with HSP90 and HSP70 [51].
Haploinsufficiency is a disease mechanism shared by different ALS-related genes, such as TBK1 and DNAJC7.[43, 51] A recent study reported a variant leading to small ubiquitin-like modifier (SUMO4) haploinsufficiency as a novel potential genetic risk factor for ALS. Recent evidence does point towards a role of the SUMO system in protecting from ALS pathology, by regulating both the assembly and dissolution of the stress granules, possibly acting in a synergistic manner with environmental oxidative stress-related factors [52].
Single-nucleotide variants in the gene encoding Kinesin Family Member 5A (KIF5A), a neuronal motor protein involved in anterograde transport along microtubules, have been also recently associated with ALS [53, 54]. Aberrant splicing has been identified as the mechanism by which KIF5A mutations cause ALS through a toxic gain-of-function, leading to the aggregation of KIF5A in distal axons and thus to neuronal toxicity. Numerous KIF5A mutations found in ALS patients are, indeed, clustered near the splice-site junctions of exon 27 and are predicted to alter the cargo-binding domain of KIF5A [55].
More recently, chloride channel CLIC-like 1 (CLCC1) has also been reported as a novel ALS-related gene. The loss of CLCC1 by a functional mutation was found to cause an ALS-like phenotype in mice [56]. CLCC1 is a transmembrane protein localized on the ER, which maintains ionic homeostasis between the ER and the cytoplasm. Another recent study reported different CLCC1 variants, all located at the C-terminus of the protein, which are predicted to affect protein stability and present with a similar ALS phenotype, namely an earlier age at onset with rapid progression and cognitive deficits. Thus, CLCC1 exon 10 might be a potential hotspot for ALS [57]. Interestingly, a study conducted in 2021 proposed the association of variants in serine palmitoyltransferase, long-chain base subunit 1 (SPTLC1) with juvenile ALS (namely ALS4), but not with adult-onset ALS, and implicated sphingolipid metabolism as a pathway in motor neuron disease [58]. This study also presented evidence, supported by other studies, that mutations in different genes might influence aspects of the ALS phenotype, as well as the disease progression rate and patient survival. The C9orf72 repeated expansion, for example, is associated with a faster progression rate while SOD1 mutations are associated with a slower course [59]. Moreover, the size of the C9orf72 expansion might represent a modifier of the cognitive phenotype. Indeed, a recent paper highlighted that C9-ALS patients with larger expansions had a more severe cognitive impairment, thereby supporting the emerging hypothesis that C9orf72 hexanucleotide repeats size might be a modifier of phenotype along the FTLD clinical spectrum [60]. Another recent study suggested an association between the C9orf72 and an accelerated decline in respiratory function. In addition, this association seemed to be more distinct in spinal-onset male patients [61]. Interestingly, polyglutamine (polyQ) intermediate expansions (24–34 CAG repeats) in the neuronal stress protein Ataxin 1 (ATXN-1) and Ataxin 2 (ATXN-2) genes have been recently reported to be independently associated with an increased risk for ALS [62, 63]. The risk of ALS increases exponentially with allele repeat size until the range known to be associated with spinocerebellar ataxia (SCA1 and 2) risk, even though the age of onset does not seem to be affected [63]. In addition, a study based on an Italian ALS cohort showed that the ATXN-2 intermediate-length polyQ repeat ALS risk correlated with a spinal phenotype and is associated with shorter survival [64].
A trend towards a lower male-to-female ratio in SOD1 ALS carriers has emerged in a recently published meta-analysis conducted on ALS patients harboring SOD1 mutations, leading the authors to suggest that differences in sex hormones may have less influence on ALS pathogenesis in the presence of a genetic mutation [13]. Lastly, the presence of variants in more than one ALS-related gene, with a potential contribution of hereditary peripheral neuropathy genes, might also influence the patient's phenotype, supporting an oligogenic model of ALS [65]. Recent reports also hypothesized that GRN gene variants could be ALS phenotypic modifiers [66] and that senataxin (SETX), the genetic cause of ALS type 4 (ALS4), could be a modifier of C9orf72 pathology. SETX mutations in ALS4, a rare juvenile-onset familial form of ALS, presumably cause a gain-of-function, as loss-of-function mutations in the SETX gene are responsible for autosomal recessive Ataxia with Oculomotor Apraxia type 2 (AOA2), and AOA2 carriers do not develop motor neuron disease [67, 68].
Environmental factors
Various environmental factors have been proposed to be associated with ALS, such as smoking, antioxidants intake, alcohol consumption, body mass index, physical exercise, head trauma, metabolic and inflammatory factors, cancer, and occupational or environmental exposures to electromagnetic fields, metals or pesticides [25, 26, 69,70,71,72,73]. However, to date the only well-established risk factors are represented by age, male gender, and family history [1]. Ref. [74] Previous studies have suggested that certain groups of athletes (especially soccer or American football players) may have an increased risk of ALS. Over the years, several studies tried to associate physical activity with the risk of developing ALS, leading to the hypothesis that the increased risk of ALS may be linked to factors related to sport rather than physical activity alone [75,76,77]. Indeed, recent studies evidenced that head impact exposure may increase the risk of neurodegenerative disease, in line with the observation that sports like soccer and football are those more linked to the development of ALS [78, 79]. However, other recent studies showed that physical activity in general and other sports such as skiing may be associated with the risk of developing ALS [80,81,82]. Several putative mechanism have been hypothesized, ranging from oxidative stress to exercise-induced changes in neuron morphology [80]. Nevertheless, most of these studies were retrospective, while prospective or population-based studies found no evidence for an association with the risk of ALS and on the contrary demonstrated that physical activity reduced the risk of ALS mortality [80]. Importantly, these controversies may lead one to hypothesize that head trauma and physical activity may represent a risk factor only in individuals with a specific genetic predisposing background, but to date, there is still a lack of studies investigating the interaction between these factors [83]. Preliminary results in C9orf72 carriers support this theory, since physical activity may increase the risk of ALS development and may be associated with an earlier age at onset of disease, but more studies are needed to confirm this hypothesis [12, 84, 85]. There are weaker or conflicting data for other putative environmental risk factors [1]. Nevertheless, a recent study on the geographical distribution of presymptomatic ALS patients revealed higher-incidence cluster areas, supporting the idea that exogenous factors may be involved in the ALS pathogenesis [86]. This hypothesis is in line with the results of another recent study focused on ALS incidence among immigrants in Sweden, which found that the risk of ALS was the same as native Swedish only in second-generation individuals, while it was lower in first-generation people [87]. Occupational risk factors have also been considered and a recent study reinforced the notion that veterans may have an increased risk of ALS, especially for Air Force personnel [88]. It remains unclear which factors may increase this risk, given the difficulties in identifying the exact environmental agents, but is supposed that it may be related to specific toxins such as pollutants, heavy metals or infectious agents [89,90,91,92].
ALS phenotype heterogeneity
The ALS clinical spectrum includes extremely heterogeneous and complex phenotypes marked by a varying involvement of upper and lower motor neurons, site of onset and rate of progression [93, 94]. Recognized MND phenotypes include classic, bulbar, flail arm, flail leg, pyramidal and respiratory ALS, primary lateral sclerosis (PLS), characterized predominantly by pure/predominant UMN degeneration and progressive muscular atrophy (PMA), characterized by pure/predominant LMN degeneration [95, 96]. The motor phenotype is so heterogeneous that it may overlap with other diseases of the MND spectrum, such as hereditary spastic paraparesis (HSP) or distal motor neuropathy (dSMA). Noteworthy, recent studies have demonstrated that ALS causative genes are not uniquely associated with a single clinical form but may be responsible for different phenotypes within the MND spectrum, ranging from predominant lower to upper motor neuron involvement. For instance, recent reports have highlighted how such genes as DCTN1 (dynactin 1) [97,98,99], GDAP1 (ganglioside-induced differentiation-associated protein 1) [97, 100, 101], DYNC1H1 (Dynein, cytoplasmic 1, heavy chain 1) [102, 103], KIF5A (Kinesin Heavy Chain Isoform 5A) [102, 104], and NEFH (neurofilament heavy chain gene) [105, 106] have been associated with a wide range of phenotypes, ranging from ALS to HSP [107], dSMA or even classic Charcot–Marie–Tooth disease [97, 102, 103, 108,109,110].
Bulbar ALS (bALS) is more homogeneous both in terms of progression [111] and neuropathological features [112, 113] and is characterized by rapid decline with short survival times (< 2 years post-diagnosis), and a significantly reduced quality of life [114, 115]. Epidemiologically, bALS represents 30% of all ALS presentation [95], and has recently been reported to be more prevalent among German than Chinese patients (35.9 vs. 22.8%) [14]. Pseudobulbar palsy is a variant primarily characterized by impairment of the cortico-bulbar tract that slowly spreads to the limbs: women are reported to be more affected than males, survival is longer and patients are particularly bothered by pseudobulbar affect [116, 117]. Although pseudobulbar palsy may be considered as an ALS variant, it is important to note that a varying degree of pseudobulbar affect (i.e., pathological crying and laughing or emotional lability) may be observed in up to 50% of ALS and PLS patients and has been related to cortico–pontine–cerebellar networks. Pseudobulbar affect may have a considerable impact on quality of life and should be promptly recognized in order to set up appropriate pharmacological interventions [118]. On the other hand, progressive bulbar palsy (PBP) electively involves lower motor neurons of the medulla oblongata nuclei, subsequently spreading to limbs [117]. Although disease progression and spreading is attended, median survival can be longer compared with classic bulbar-onset ALS [119]. Within this context, anti-IgLON5 disease is a proteoform neuro-immunological disorder syndrome, mimicking ALS, characterized by bulbar sign and symptoms, cognitive alteration, gait disturbance and sleep disorders [120]. Interestingly, four IgLON5 IgG seropositive bALS patients with vocal cord paresis at onset have recently been reported [121, 122]. Vocal cord paresis is rarely described as a presenting symptom in bALS, but if present is often found in association with the D90A mutation [123]. Another atypical and rare bALS variant is represented by Facial Onset Sensory Motor Neuropathy (FOSMN), whose hallmark is sensory trigeminal involvement at presentation [124], followed by weakness of the buccal muscles, swallowing disorders and subsequent spread to involve the limbs with cognitive behavioral impairment of a frontotemporal type. The balance of evidence suggests that FOSMN is most likely to be a TDP-43 proteinopathy [125].
Although accounting for a relatively small proportion al ALS patients (3–5%), respiratory onset ALS is a clinical phenotype that needs special mentioning since these patients may be misdiagnosed and referred to neurological attention for a formal diagnosis after the development of respiratory failure or even after being intubated [95, 126]. Notably, a recent study showed that this phenotype is more frequent in older men with predominant lower motor neuron involvement, weight loss, and shorted survival, needing prompt non-invasive ventilation (NIV) adaptation [126, 127].
Notably, a recent study have demonstrated how the cognitive profile in PLS resembles ALS–FTD, without prominent behavioral disturbances [128], in line with a report of a kindred with an association between hereditary PLS and progressive non-fluent aphasia [129]. In addition, an advanced MRI study showed that in PLS the disease burden in the motor cortex is more medial than in ALS, consistent with its lower limb predominance, while the extra-motor profile includes marked insular, inferior frontal, left pars opercularis and cerebellar white and grey matter degeneration, with the postcentral gyrus being relatively well preserved compared with ALS [130,131,132]. While suggesting a specific profile, these findings still support the notion that PLS lies on the ALS–FTD spectrum. However, the mechanisms underlying disease spread and slow disease progression are likely to be distinct in PLS [20, 128]. Similarly, a recent paper highlighted that in flail arm syndrome, a restricted MND phenotype characterized by progressive, predominantly proximal weakness and atrophy of the upper limbs, clinically covert involvement of the pyramidal tract is similar to classic ALS [18, 133], while another recent paper proposed diagnostic criteria for Mills’ syndrome, a rare MND variant characterized by a slowly progressive, spastic hemiparesis [134].
Cognitive and behavioral changes in ALS
ALS is a heterogeneous neurodegenerative disorder that can no longer be considered a disease limited to the motor system [25, 26, 135]. While physical deterioration account for the primary symptoms, cognitive and/or behavioral changes are experienced in up to half of patients with ALS and 5–15% of ALS cases meet the criteria for frontotemporal dementia (ALS-FTD) [136,137,138] with the behavioral variant (bvFTD) mostly represented within ALS-FTD patients [139,140,141]. However, the neuropsychological deficits in ALS are extremely heterogeneous [135, 138]. Originally construed as a disorder of behavior and executive impairment [142], the neuropsychological profile of ALS is now known also to be associated with alterations in language, social cognition (Table 1) and memory [143], albeit inconsistently [135, 144]. The revised consensus criteria for the diagnosis of cognitive and behavioral dysfunction in ALS has led to re-conceptualization that neuropsychological deficits fall along a spectrum: from no impairment to mild cognitive impairment (ALSci), mild behavioral impairment (ALSbi), both (ALScbi) or ALS-FTD; thus embracing the concept of a frontotemporal spectrum disorder [135]. Specifically, according to guidelines, ALSci is attributed to frontotemporal dysfunction since it is diagnosed when executive and/or language deficits and/or impairment of social cognition occur in non-demented ALS patients, while the ALSbi diagnosis requires the identification of apathy or the presence of at least two non-overlapping, supportive diagnostic features for bvFTD [135, 145]. However, most ALS-FTD patients present with heterogeneous combinations of deficits in behavior, language, or ‘frontal’ cognitive functions [146]. Overall, when present, cognitive deficits and behavioral changes significantly and adversely impact patient survival [147], psychological well-behind [148], consent capacity [149] and caregiver-burden [150]. Interestingly, the presence of ALSci seems to have limited relevance for the patients' and caregivers’ everyday life in comparison to the impact of behavioral alterations, namely apathy and disinhibition [150,151,152,153].
The extent to which cognitive and behavioral impairment occurs and whether or not it precedes the onset of motor symptoms is still unknown. In ALS-FTD, most patients have FTD features before motor onset, but others developed them at the same time or even after (even if less frequently) motor impairment. In ALS, a presymptomatic or prodromal period of varying duration, during which cognitive and behavioral involvement gradually unfolds, has been postulated in ALSci and/or ALSbi cases [172]. Studies from presymptomatic genetic carriers support this view: for instance, C9orf72 carriers may manifest dysexecutive and verbal fluency deficits before symptomatic disease onset [173, 174]. Despite two large cohort studies showing a higher frequency of cognitive and behavioral impairment in the later ALS stages [136, 175], it is still debated whether the frequency of neuropsychological changes is different across disease stages. It has been suggested that motor and cognitive components might worsen in parallel [136], and that cognitive worsening is more pronounced in patients with bulbar involvement [175]. But additional factors possibly modifying the clinical expression have been identified, namely C9orf72 gene mutations [60, 176], cognitive reserve [169] and apolipoproteinE genotypes [177].
From an anatomical perspective, the susceptibility to ALS is not only confined to motor neurons but it also involves extra-motor pathways [26, 178, 179], whose extension and severity seems to be related to the presence and degree of cognitive and behavioral involvement [180, 181]. Recent studies suggest that ALS pathological processes begin in motor neurons and propagate to other regions of the brain in a fashion that is predictable, based on proximity and connection by a corticofugal axonal spread [182,183,184]. The trajectory of the spreading pattern seems to be consistent with the inclusion and dissemination of phosphorylated 43-kDa transactive response DNA-binding protein (pTDP-43) [183, 185]. This suggests that ALS has a focal onset with subsequent spread along brain connections [186] in the prefrontal neocortex eventually developing “frontal type” cognitive deficits (i.e., executive and social), depending on disease duration and rapidity of propagation [183]. However, the timing of the manifestation of cognitive syndromes does not necessarily follow such sequential staging [187, 188]. A specific pathological course distinct from that of pure-motor (classic) ALS has been found and it is in keeping with the view that cognitive/behavioral involvement might represent a phenotypic marker for distinct ALS subtypes [147, 187].
Another central theme is the recognition of neuropsychiatric symptoms in ALS [189, 190]. Schizophrenia, depression, anxiety, dependence disorders, and psychosis have all been reported in both ALS and ALS-FTD in the years prior to or following diagnosis [191]. A predisposition to neuropsychiatric symptoms is more frequently described in patients with C9orf72 expansions and presymptomatic C9orf72 carriers [189]: ALS patients with this expansion exhibit high rates of delusions and hallucinations and may experience psychotic symptoms for years before the onset of motor or cognitive features. It has also been established that ALS patients with a family history of mood disorders or premorbid depressive symptoms have high risk of developing ALSbi, namely apathetic behavior, dysexecutive profile or ALS-FTD [189]. However, it is still unclear if neuropsychiatric symptoms contribute to the spectrum of frontotemporal disorders in ALS as there is considerable variation in the reporting of mood disorders in ALS, ranging from moderate depression to an absence of symptoms [192]. Nevertheless, ALS patients may exhibit symptoms of depression and anxiety (reported in about 30% of patients) at different stages which may reflect a reactions to their condition or alternatively an integral part of the disorders, as a result of frontotemporal and subcortical involvement [191, 193].
Despite the last decades having witnessed a substantial increase in the identification and understanding of frontotemporal dysfunction in ALS, measures designed to evaluate the non-motor features of ALS [194,195,196] have not been frequently employed in clinical care, research or trial design for ALS [197, 198]. Understanding the diverse clinical presentation of frontotemporal syndromes, their pathological and genetic substrates is crucial for improved early diagnosis, clinical management and the development of therapies tailored for specific ALS sub-groups.
Extra-motor involvement
Although ALS was initially regarded as selectively affecting the motor system, increasing evidence highlights extra motor involvement, indicating that ALS should be recognized as a multi-system disorder [25, 26, 199]. Ref. [94, 197, 200] In addition to the cognitive involvement, recent studies highlight that ALS may affect other CNS structures, such as the extrapyramidal system. Indeed, a recent prospective study showed that up to 30% of ALS patients may have extrapyramidal features meeting the diagnostic criteria for parkinsonism [201, 202]. These results are in line with another study which found that extrapyramidal deficits with impaired gait initiation in a subset of ALS patients [203]. Moreover, a recent report evidenced that the applause sign, which was initially reported as a specific sign of progressive supranuclear palsy, may also be present in about ~ 10% of ALS/FTLD patients [204]. It is unclear if these manifestations are related to basal ganglia involvement or affection of other brain circuitries and require further investigation [201, 203]. The involvement of cerebellar circuits is another finding that has been consistently—though only recently—highlighted in patients with ALS, especially in those carrying a C9orf72 gene expansion [205,206,207], although its role is still debated. A recent study has indicated focal damage to the anterior cerebellar regions in sporadic ALS patients, in contrast with a more posterior damage in C9orf72 expansion carriers, suggesting that cerebellar pathology might modulate different motor (i.e., dysarthria, coordination and gait deficits) and neurocognitive features (i.e., behavioral dysfunction and deficits in social cognition) according to the genetic background [208]. Intriguingly, focal cerebellar pathology has already been described also in PLS patients, involving especially the middle cerebellar peduncle, where it may be related to the development of pseudobulbar affect [132, 209]. The link between ALS and frontotemporal lobar degeneration (FTLD) is actually well acknowledged, especially in C9orf72 carriers [60]. A recent study confirmed the early prefrontal involvement in C9orf72 carriers, firstly suggesting an executive eye movement dysfunction, even in the presymptomatic phase [173]. The increasing interest on retinal involvement in neurodegenerative diseases in recent years also includes ALS with evidence that the retinal nerve fiber layer (RNFL) may be reduced, with an asymmetric involvement and correlating with disease progression [210]. However, these results need to be confirmed, since other reports while confirming the presence of retinal involvement, have not found that they correlate with disease duration or severity [211,212,213]. More recent studies have focused also on the autonomic nervous system and skin nerve fibers in ALS patients. One such study performed on a small cohort of ALS patients confirmed the high frequency of skin biopsy abnormalities [214]. These results are in line with previous reports, disclosing an association between ALS patients and small-fiber neuropathy, especially among those with spinal-onset or sensory symptoms [200, 215,216,217,218]. Moreover, in this study, the authors reported the finding of phosphorylated TAR DNA-binding protein 43 (pTDP-43) deposition in Meissner’s corpuscles in ~ 30% ALS patients, highlighting a potential value of skin biopsy as an ALS biomarker [214]. In addition to small-fiber nerve pathology, recent research found that ALS patients may have autonomic dysfunction. In this work, performed on 102 patients and 41 healthy controls, the authors found an increased rate of autonomic symptoms among ALS patients (~ 70%), especially among those with a bulbar-onset [219]. These results are in line with some reports disclosing a co-occurrence of a rare cardiomyopathy (i.e., Tako-Tsubo syndrome), whose complex pathophysiology may be in part related to sympathetic hyperfunction, especially in bulbar-ALS patients [220]. Further studies are needed to better characterize the extra-motor involvement in ALS patients, which may reflect a specific disease mechanism [221].
Prognosis
ALS phenotypic heterogeneity is reflected in the variability of ALS prognosis: although approx. one-half of patients die within 30 months of symptom onset, approx. 20% of patients survive between 5 and 10 years [2, 3, 25, 26, 222, 223].
A major determinant of ALS prognosis is disease progression, which is in clinical practice monitored by using the multidomain ALS functional rating score-revised (ALSFRS-R), whose limits have extensively been previously highlighted, but still remains the gold standard for primary efficacy outcomes in clinical trials [25, 224,225,226,227]. To overcome these limitations, staging paradigms have been recently proposed such as the King’s [228], and ALS Milano-Torino Staging (ALS-MiToS) [229]. The King’s, based on El Escorial criteria, is more sensitive early in the disease course, while the ALS-MiToS, is based on loss-of-functionality in ALSFRS-R domains and so is better for more advanced disease states [230, 231].
Given the broad distribution of survival, the identification of the key prognostic factors is, therefore, important for appropriate timing of medical interventions and stratification in clinical trials.
Of note, a recent paper pointed out that prognostic factors, which are known to modify the course of disease in Caucasians, apply to Chinese patients as well, despite the apparent differences regarding genotype and clinical phenotype. Specifically, younger age of onset, spinal onset, high BMI and low progression rate were positive prognostic factors in China as well as in Germany [232]. Conversely, long survival has been linked to younger age at onset, increased prevalence of PLS and longer diagnostic delay, absence of the C9orf72 repeat expansion and increase frequencies of gene variants in FIG4, hnRNPA2B1, SETX, SQSTM1, TAF15, VAPB and the SOD1 p.(Ile114Thr) [233].
Recent data have confirmed the relevance of BMI, metabolism and nutritional status as independent predictors on survival in ALS [234,235,236,237,238]. Within this context, a recent paper has pointed out that body weight changes after diagnosis strongly predicts survival in ALS, and weight gain after diagnosis may improve survival prognosis [239]. In line with this, a hypometabolic or hypermetabolic state have been, respectively, associated with better or worse prognosis [240, 241].
Given that respiratory failure is the main cause of mortality in ALS, measurements of respiratory functionality are integrated not only in clinical practice, but also in ALS prognostic models [12, 242]. However, more recent studies have highlighted that not only slow (SVC) or forced vital capacity (FCV) [243] may be a useful prognostic marker but that other parameters such as arterial blood gas parameters and base excess may independently predict shorter survival in ALS [244, 245]. Within this context, a recent neurophysiological study pointed out that Diaphragmatic CMAP amplitude from phrenic nerve stimulation may also be able to predict functional decline in ALS [246].
In the last few years, in order to overcome the limitations linked to ALS complexity, several prognostic models have been proposed, combining multiple predictors [222, 247,248,249,250,251,252,253,254,255]. Most models present, however, different methodological limitations, as recently pointed out in a systematic review focused on this topic [247]. More recently, artificial intelligence (AI) and machine learning methods have been adopted in order to develop reliable prediction models, even if their translation into clinical practice is currently still limited [256, 257].
Diagnostic challenges
Clinical examination remains the cornerstone of ALS diagnosis [258]. The diagnosis of ALS has been traditionally categorized by the El Escorial criteria into various levels of certainty depending on the presence and progressive spread of UMN and LMN signs [259]. Subsequently, the Awaji criteria proposed fasciculations as a clinical and electrophysiologic sign of LMN involvement, aiming at increasing overall diagnostic sensitivity [260, 261]. However, El Escorial and Awaji criteria are hampered by ALS heterogeneity and do not capture the full disease spectrum [261, 262]. In addition, a significant proportion of ALS patients may never attain the criteria for clinically definite ALS [263]. In order to overcome these limitations, the Gold Coast criteria have been recently proposed classifying patients as having or not having ALS, streamlining diagnostic certainty and eliminating confusion from the El Escorial terminology [264]. However, cognitive and behavioral impairment [265] are not included in these criteria [261, 262, 264].
Although an ALS diagnosis is generally fairly simple [261], the false-positive rate has been estimated to be as high as eight to 10% [266], while the false-negative rate approaches 45% [267, 268]. Several disorders, often referred to as ALS mimics, may primarily affect the motor system, simulating ALS. These include Sandhoff disease, adult polyglucosan body disease, Hirayama disease, spinal dural defects associated with CSF leaks and neurogenic calf amyotrophy with CK elevation by entrapment radiculopathy [269,270,271,272,273]. In selected cases, motor nerve biopsy may be useful for an early differential diagnosis of patients presenting with an atypical LMN syndrome [274,275,276], while motor‑evoked potentials may prove essential in order to detect subclinical UMN involvement in LMN phenotype [277]. Most importantly, differentiation between MND, in particular progressive muscular atrophy (PMA), and multifocal motor neuropathy (MMN) may be extremely challenging. Although MMN has distinctive neurophysiological features and is often associated with anti-GM1 antibodies [278, 279], a recent large case–control study showed that both PMA and MMN, but not ALS and PLS, are associated with an IgM monoclonal gammopathy (8% and 7%, respectively), suggesting that a subset of patients presenting with PMA share pathogenic mechanisms with MMN.[280]
Biomarkers
The lack of specific biomarkers is recognized as a major limitation in ALS management and clinical research including diagnosis, prognosis and patient stratification in clinical trials. In addition, biomarkers may play a crucial role in assessing the efficacy of potential new treatments and so extensive efforts have been recently made in order to develop potential biomarkers with the aim that they will be specific, repeatable over time and minimally invasive for patients.
In the last years, neurofilament light chain (NfL) and phosphorylated neurofilament heavy chain (pNFH) have exhibited promising diagnostic and prognostic capabilities, making them well suited for integration into clinical workup [281,282,283,284,285,286]. Moreover, the development of ultrasensitive digital immunoassays (e.g., Simoa™ and ELLA™) able to measure accurately picogram levels have enabled the identification of cutoff values for serum NfL in clinical settings, thus encouraging their routine clinical use as biomarkers [287]. A recent genetic trial involving the antisense oligonucleotide tofersen in SOD1-related ALS further confirms the role of NfL as a prognostic biomarker since early reductions of NfL were able to predict the subsequent slowing of ALSFRS-R decline after Tofersen treatment [288]. Other studies have importantly demonstrated that NfL blood levels increase prior to the clinical manifestation of ALS [289,290,291], suggesting that NfL are valuable biomarkers especially in the earliest phases of the disease and to monitor phenoconversion. All these studies have positioned NfL and pNFH as the most promising biomarkers for ALS, placing them at the forefront of potential integration into clinical practice.
Consistent with the involvement of inflammatory processes in ALS, many studies have explored the role of inflammatory biomarkers in cerebrospinal fluid (CSF) revealing alterations in the concentrations of cytokines, chemokines, and complement proteins (CIT). In particular, recent studies have revealed that chitotriosidase (CHIT1), chitinase-3-like protein 1 (YKL-40, or CHI3L1), and chitinase-3-like protein 2 (YKL-39, or CHI3L2) demonstrate a significant increase in their concentrations in the CSF of ALS patients compared with mimicking conditions [292, 293]. The levels of urinary neopterin, a marker of cell-mediated inflammation, have been found to be higher in ALS patients compared with both patients with other neurodegenerative diseases and ALS-mimics, supporting a role for neopterin as a diagnostic biomarker [294, 295]. Urinary neopterin concentration reflects the severity of ALS, underscoring neopterin’s potential as a prognostic biomarker in ALS.207.
Given the recent advances in our understandings of the impact of metabolism in ALS disease progression [296, 297], recent studies have explored the potential role of metabolic biomarkers. A recent case–control study suggested that high triglycerides may be associated with longer survival, whereas higher levels of cholesterol were associated with a higher mortality [298], while another study explored the role of irisin, a peptide hormone released by muscle and involved in metabolism, showing higher levels of irisin in ALS patients with impaired metabolic status compared to normo-metabolic ALS patients and healthy subjects [299].
Recently, circular RNAs (circRNAs) are emerging as a novel candidate biomarkers in ALS patients. In a recent study exploring a microarray expression profile of circRNAs in leukocyte, samples from ALS patients have suggested that specific circRNAs candidates have value as potential novel blood-based biomarkers for ALS, displaying AUC values exceeding 0.95 in ROC curve analysis [300]. Further studies regarding circRNAs and their association with ALS should be done in order to validate these results.
Despite numerous studies aimed at validating different potential markers in ALS disease, none is currently adopted in the clinical settings. In a future perspective, a panel of different biomarkers reflecting various aspects of the disease, would be more suitable for routine clinical practice and would allow one to glean more information on the different mechanisms involved in ALS.
Advances in neuroimaging
Over the last decade, neuroimaging techniques have seen dramatic improvements, opening up exciting opportunities to investigate new aspects of CNS structure and function in ALS patients [117, 301, 302]. In fact, neuroimaging provides an ideal tool to explore quantitative, reproducible measures of UMN impairment and is sensitive to extra-motor brain involvement that frequently accompanies the development of the cognitive and behavioral impairments seen in ALS [303]. These techniques have proven their utility for phenotypic characterization, disease monitoring and patient stratification in relation to clinical outcomes, as well as providing new insights into pathogenesis.
Conventional brain MRI protocols provide only limited accuracy for supporting an MND diagnosis by the demonstration of T2-weighted hyperintensity within the corticospinal tract (CST) [304]. A large consecutive series of ALS patients reported that susceptibility-weighted imaging showed the presence of a hypointense signal in the cortical band along the precentral gyrus (i.e., the “motor band sign”) in approximately 80% of ALS patients [305], although the specificity of this radiological sign is still to be determined. Advanced computational neuroimaging approaches have been used to describe in greater detail patterns of grey (GM) and white matter (WM) structural damage characterizing specific groups of ALS patients. For example, CST metrics have been used to discriminate aggressive forms of ALS from more slowly progressing cases [306,307,308]. Patients with faster clinical progression showed more severe cortical thinning of the left precentral gyrus and fractional anisotropy reduction of the CST relative to those with slower rate of progression [307]. Recently, cluster analysis based on both GM and WM measures was used to identify, through a data-driven approach, two ALS subtypes, characterized by different clinical profiles and degree of frontotemporal cortical involvement, with clear prognostic implications [205]. Another recent study evaluating morphometric modifications and diffusion tensor (DT) MRI alterations showed distinctive patterns of GM and WM involvement in ALS and PLS, with greater damage to extra-motor cortical and cerebellar structures in PLS patients [130]. In MND patients with predominant LMN involvement, DT MRI metrics were also able to distinguish fast- from slow progressors, based on the greater involvement of the CST and extra-motor WM tracts in patients with a more rapid disease progression [309,310,311]. On the other hand, another study has suggested that the flail arm syndrome, a phenotypical variant of ALS, may show a similar degree of CST involvement as to that seem in patients with a ‘classical’ ALS presentation [133]. Subtle structural brain alterations have been shown to occur even in the earliest phases of the disease, as presymptomatic carriers of a C9orf72 expansion demonstrated significant, selective thalamic and focal hippocampal volume reductions [207, 312].
Structural neuroimaging has also shown its utility to track ALS progression. A study investigating cortical thickness changes over time in different ALS phenotypes found a significant decline of cortical thickness in frontal, temporal, and parietal regions over time [313]. Effects were independent of the clinical subtype, with the exception of the precentral gyrus: the LMN ALS variants demonstrated the highest rates of cortical thinning in the precentral gyrus, the UMN-dominant subjects exhibited intermediate rates of atrophy, and the classical ALS patients exhibited no real change as the atrophy of the precentral gyrus seen in classical ALS is at floor on the first assessment, resulting in a lack of further atrophy over time [313]. A probabilistic fiber tractography study estimated structural connectivity changes after three months in patients with ALS [314]. While CST damage did not worsen over time, DT MRI changes were observed in the occipito-temporal pathways, again suggesting an early involvement of motor networks with subsequent extra-motor damage [314]. Recently, a tract-based staging scheme has been proposed to assess cerebral progression of ALS pathology in vivo, supported by the fact that the progression of WM alterations across tracts using DT MRI was associated with clinical disease severity in patients with ALS [315].
In addition to brain alterations, spinal MRI has also been shown to have role in the prognostic stratification of ALS patients. A recent multicenter study demonstrated that multiple stepwise linear regression models based on clinical variables and a combination of cervical spinal cord cross-sectional areas, diameters and DT MRI parameters can provide an accurate prediction of motor capacity as early as at 3 months from diagnosis [316]. Another recent study exploited the combined acquisition of both brain and spinal cord MRI sets to test the competing hypotheses of anterograde (i.e., dying-forward) vs retrograde (i.e., dying-back) neurodegeneration in ALS [317]. The analysis of volumetric and diffusion data in a common domain identified step-changes in tissue damage measures between the cranial descending corticospinal tract and C1 spinal cord level, and between C5 and C6 cord levels, with an apparent cranio-caudal gradient [317], although further validation of these data is needed to draw definitive conclusions about this issue.
In addition to MRI, positron emission tomography (PET) imaging has also drawn interest as a tool to visualize disease-related metabolic and molecular changes occurring in the brain of individuals with ALS with different clinical phenotypes. For example, 18F-FDG PET has demonstrated a pattern of increasingly severe frontal hypometabolism associated with the degree of cognitive and behavioral impairment in a large cohort of patients with ALS, with additional cerebellar damage in those with comorbid FTD [318], whereas a metabolic signature of relative damage to the lateral areas of the precentral gyri was found in patients with a pure bulbar involvement, relative to those with a pure spinal involvement [319]. The severity of frontotemporal hypometabolic patterns also showed a prognostic significance, being associated with greater functional impairment and shorter survival in patients with ALS and PLS [320]. Moreover, genetic background was found to influence 18F-FDG PET findings in ALS, as hypometabolism in frontotemporal regions, basal ganglia, and thalami could be detected in patients carrying a C9orf72 gene expansion since the presymptomatic stages [321]. By utilizing radiotracers targeting various molecular pathways implicated in ALS pathology, PET imaging also enables the non-invasive assessment of disease-related alterations in vivo. Among the most intensively studied molecular targets in the last years, the translocator protein (TSPO) of the outer mitochondrial membrane has been mostly considered a marker of activated microglia. Increased local uptake of TSPO tracers (such as 11C-PK11195 and 11C-PBR28) was consistently detected in the motor cortex, prefrontal cortex, pons, and thalami of sALS patients, colocalizing with structural abnormalities of both grey and white matter and showing significant correlations with clinical measures of UMN burden [322, 323].Similar findings were shown in ALS patients with pathogenic genetic variations in the SOD1 gene, even in the presymptomatic phases [324].
All these findings suggest that an advanced multiparametric neuroimaging approach has the potential to improve our understanding of the neural substrates underlying the clinical impairment seen during the ALS disease course.
Advances in pharmacological treatments
Despite considerable efforts and numerous well-designed clinical trials, Riluzole remains the only drug with any evidence for a demonstrated ability to increase survival in ALS patients, even if the mechanisms of action are not yet fully understood [325,326,327]. Recent studies also confirmed that Riluzole is well tolerated with a suggestion that a prolonged duration of riluzole intake correlates with longer median survival [328, 329].
Intravenous edaravone, a free radical scavenger, is another approved drug in Japan, U.S., Canada, South Korea and Switzerland, as it showed a positive effect on disability progression in a subgroup of patients [330]. However, a prospective evaluation conducted by the Italian EDARAVALS study group showed that in the Italian cohort, Edaravone, although overall well tolerated, had no significant effect in slowing disease progression or respiratory function decline [331]. These data are in line with a longitudinal MRI study showing no significant changes in fractional anisotropy (FA) values of corticospinal tracts and cerebral cortex thickness (CT) in ALS patients treated with Edaravone [332].
Different disease mechanisms have been targeted during these years, often adopting the strategy of repurposing compounds already used in clinical practice for the management of other conditions, based on data from ALS preclinical models [333,334,335]. Acetyl‑l‑carnitine has recently been restudied in a retrospective cohort after a previous phase 2 trial showing efficacy on self-sufficiency, ALSFRS-R total score and forced vital capacity (FVC) [336]. Compared with the non-treated patients, the group receiving the 1.5 mg dose had a higher number of subjects still alive at 24 months after baseline, while no differences were detected with the 3 mg dose group. A confirmatory phase 2/3 trial is planned in 2024 (NCT06126315). Tauro-urso-deoxycholic acid (TUDCA), a small molecule with preclinical evidence of anti-apoptotic effects, was shown in two independent phase 2 trials, alone and in combination with sodium phenylbutyrate (PB), to be able to modify disease course in ALS patients, as further confirmed by a retrospective analysis of real-life patients [337, 338]. Based on the results of these phase 2 trials, the FDA has already approved Relyvrio® (PB and taurursodiol), event if preliminary announcement of topline results has reported that both the Phoenix trial and the TUDCA-ALS study did not meet the prespecified primary endpoints, even if the data from these two independent phase 3 trials on the effects of TUDCA and PB in ALS are sill pending [339, 340].
A phase two trial evaluated the effect of Perampanel (an anti-epileptic drug) in reducing disability and increasing survival. However, this study showed no effect of the treatment on the different endpoints, with a high number of adverse events and a more rapid decline of ALSFRS-R in the treatment group [341].
Several trials have targeted neuroinflammation. A pilot trial on 7 patients treated with leukapheresis followed by intravenously Treg infusions and IL-2 injections or placebo provided insights on a possible efficacy of this well tolerated treatment, especially on ALS patients with a neuroinflammatory profile [342]. Another therapeutic attempt in this field has been recently done with a small cohort of ALS patients who underwent autologous hematopoietic stem cell transplantation, but further studies should be carefully evaluated based on the low benefit/risk balance [343]. Rapamycin has been investigated in a multicenter, randomized, double-blind trial, in 63 ALS patients at the dose of 2 mg/m2/day, 1 mg/m2/day and placebo (EUDRACT 2016–002399-28; NCT03359538). Despite the primary outcome not been achieved, rapamycin was able to decrease mRNA relative expression and plasmatic protein levels of the pro-inflammatory cytokine IL-18, increasing the percentage of classical monocytes and memory switched B cells [344].
Two phase 3 trials testing drugs that increase muscle contraction, Levosimendan [345] and Reldesemtiv [346], failed to demonstrate efficacy on disease outcomes, despite a good safety profile.
One of the most promising therapeutic innovations in the fields of neuromuscular disorders and ALS is represented by the use of antisense oligonucleotides (ASOs) [334]: a phase 3 trial of Tofersen, an ASO targeting SOD1 mutations, showed important effects on disease biomarkers (SOD1 and neurofilaments), despite mild clinical efficacy [288]. The open-label extension is still ongoing, even if the FDA has already approved its use. Other phase I/III studies are evaluating the efficacy of the ASO Jacifusen (also known as ION363), directed at patients with mutations in the FUS (fused in sarcoma) gene, which is responsible for certain forms of very rapid, early-onset disease [347]. Also within the target therapy represented by ASO technology, it is worth mentioning the currently ongoing phase I study aimed at evaluating the efficacy of ASO BIIB105 in silencing the Ataxin 2 (ATXN-2) gene, a known disease modulator associated with an accelerated phenotype, in patients with mutations in both ATXN-2 and sALS [348]. Despite these encouraging results, an attempt to exploit the ASO technology (Tadnersen) in patients with the pathogenic repeat expansions in the C9orf72 gene did not show the hoped-for efficacy in a preliminary phase I trial [349].
Among the non-pharmacological interventions that have been tried, a trial of high calorie therapy showed efficacy in increasing weight and BMI, though other disease outcomes were not reached [350], while another study assessed the impact of physical therapy on ALS, comparing aerobic, flexibility and muscle-strengthening exercises vs muscle-strengthening exercises alone [351].
Clinical management
In the absence of a cure, the cornerstones of the management of patients with ALS focuses on symptoms control [352]. These treatments may not only alleviate symptoms but also maintain quality of life and improve survival, with greater benefit for patients managed in specialized ALS clinics by a multidisciplinary team, able to provide integrated palliative care strategies [242, 353,354,355,356,357]. Symptomatic care in ALS includes a wide range of patient-tailored interventions addressing pain, emotional lability, anxiety and depression, sleep disturbances, constipation and including physiotherapy and adaptive aids. Prevalence of muscle cramps has been estimated to be as high as 44–55% in ALS patients. As recently reviewed, therapeutic options include quinine sulfate, tetrahydrocannabinol, vitamin B-complex, diltiazem, natridrofuryl oxalate, mexiletine, carbamazepine and leveteracitam as well as non-prescription approaches such as muscle stretching [358, 359]. Interestingly, a small case series recently demonstrated that radiotherapy treatment of sialorrhea, which affects up to 25% of patients [360], may be feasible, efficient and safe, even in patients requiring non-invasive ventilation (NIV) [361].
Dysphagia and respiratory failure, which is the main cause of mortality for ALS patient, represent two crucial areas of symptomatic interventions for ALS patients [352]. Malnutrition is a negative prognostic factor [362] and aspiration pneumonia one of most feared causes of morbidity and mortality in ALS, and so current practice guidelines recommend gastrostomy feeding for patients with severe dysphagia [242, 363]. The two main methods of gastrostomy insertion are percutaneous endoscopic gastrostomy (PEG) and radiologically inserted gastrostomy (RIG), even when per-oral image-guided gastrostomy may be possible. Even though it has been suggested that RIG may be safer than PEG, in particular in patients with respiratory failure [364, 365], there is to date little evidence to indicate which is better and what is the optimum timing for the procedure. Indeed, a recent prospective observational study showed that PEG was a safe procedure even in patients with low FVC [366], an observation that was confirmed by another study proposing a modified approach in order to carefully select high-risk patients [367]. These results were further confirmed by another large prospective cohort study showing that the three methods seemed to be equally safe in relation to survival and procedural complications [368]. However, both studies agree that PEG might be less beneficial when delayed. Further studies, and preferably randomized controlled trials are needed in order to gain better evidence for the nutritional management and use of gastrostomy in ALS patients.
Respiratory failure is not only the main mortality cause in ALS, but may also be the presenting feature, in this instance requiring careful differential diagnosis to exclude other neuromuscular and non-neuromuscular disorders is needed.[369] Recommended pulmonary tests are spirometry (including Supine FVC), polysomnography, arterial gas analysis and sniff nasal inspiratory pressure (SNIP), which has recently been shown to be a potential prognostic factor of tracheostomy or death during the early phase of disease [370]. While a recent open-label, randomized controlled trial has shown that addition of diaphragm pacing to standard care was associated with decreased survival in patients with ALS [371], NIV has been shown to improve quality of life and survival in patients with ALS [372], even if there is still no consensus on the exact timing of NIV initiation or cough augmentation techniques [373, 374]. In patients with substantial bulbar impairment, the efficacy of NIV may be more limited; however, a recent study has demonstrated objective sleep and nocturnal respiratory outcomes in both non-bulbar and bulbar patients [375]. Moreover, NIV has also been reported to significantly increase survival by 19 months in patients with ALS-bulbar onset [376]. In more advanced disease states, invasive ventilation via tracheostomy is an option for prolongment of survival [377]. In the more advanced stages, palliative strategies, including respite care, have been demonstrated not only to provide a positive effect not only to meet the needs of people with ALS but also for their care partners [378].
Emerging assistive technologies for the management of amyotrophic lateral sclerosis
People with ALS face numerous challenges due to the progressive nature of the disease, leading to a rapid physical decline, which restricts mobility and impairs all activities of daily living. This includes speaking, eating, dressing, walking, and writing [379]. The relentless decline in these functions can lead to feelings of sadness, hopelessness, worthlessness, anxiety, and guilt. Despite the physical limitations, patients are aware of their limitations. This disease of losses raises a deep concern among caregivers and patients themselves in preserving autonomy, self-control, and decision-making for as long as possible. Emerging technologies for the management of ALS patients range from multidisciplinary teleconsults (for monitoring the dysphagia, respiratory function, and nutritional status), to robotics. The COVID-19 pandemic created a need to accelerate the development and implementation of such technologies in clinical practice, to improve the daily lives of both ALS patients and caregivers [380,381,382].
Assistive technologies (ATs) can support patients in preserving autonomy and control along with slowing disease progression. They are of great value to ALS patients, since their use may help to overcome severe functional limitations [383]. Autonomy and self-determination play a crucial role in the lives of ALS patients and can partly be maintained by the implementation of ATs and devices. There are many technology options available to support persons with neurodegenerative conditions, either mainstream or specifically designed products [384].
One of the key areas where ATs provide support is communication, also known as Augmentative and Alternative Communication (AAC). As speech intelligibility declines, support in communication becomes important in ALS management. Everyday digital technologies of the last decade, such as smartphones, tablet devices, and the Internet, may be used to assist persons with neurodegenerative conditions allowing them to perform daily tasks; such approaches include using voice-activated commands to control the environment and text-to-speech to communicate verbally [385]. AAC ATs also include brain–computer interfaces and eye tracking systems. For people with ALS, having access to an ecosystem that integrates multi-professional assistance and ATs, particularly AAC resources, has been shown to be essential in preserving communication and interaction skills and enhancing quality of life and survival as the disease advances [386]. ATs and devices can support ALS patients in their mobility, such as through powered wheelchairs and helping them control their domestic environment, and thus foster social participation and autonomy [383, 384].
Telehealth ATs may have a significant role in managing ALS. Potential applications include:
1. Telehealth technologies for remote monitoring of dysphagia, respiratory function, and nutritional status, which can increase the effectiveness of ALS management [380, 383, 387, 388].
2. Tele-rehabilitation for delivering physical therapy and other rehabilitative services to patients in their own homes. Therapy sessions can be conducted virtually via video calls, allowing patients to receive therapy from the comfort of their own homes [389].
3. Digital personal assistants that can be used to assist persons with neurodegenerative conditions in performing daily tasks, such as using voice-activated commands to control the environment [383, 384].
3D printing, also known as additive manufacturing, is a technology that also has the potential to significantly improve the management of ALS, addressing the challenges associated with the disease. 3D printing can be used to create customized assistive devices tailored to the specific needs of each ALS patient, ranging from modified utensils for eating to custom-fit braces and mobility aids. It can be used to create prosthetics and orthotics that are lightweight, comfortable, and tailored to fit the patient perfectly. This can help improve mobility and independence for ALS patients. Finally, 3D printing can also be used to create specialized medical equipment that can help in the management of ALS, such as modified respiratory equipment or specialized seating systems that improve comfort and mobility [390]. Despite its potential, there are several challenges associated with the use of 3D printing in ALS management: 1. limited material selection; 2. cost of pre- and post-processing; 3. cost of system equipment; 4. acceptance of technology among patients and carers that depends on various factors such as perceived skills and competencies in using the device, expectations, trust, and reliability; 5. regulation and standardization as 3D-printed medical devices must meet the same safety and efficacy standards as traditionally manufactured devices [391]. However, these challenges are not insurmountable. With continued research and development, as well as collaboration between healthcare providers, engineers, and patients, 3D printing has the potential to play a pivotal role in transforming the management of ALS.
Wearable devices, such as smart-tracker or smartwatch, and smartphone can track ALS disease progression and may serve as novel clinical trial outcome measures [392,393,394]. Several wearable devices collecting data daily on physical activity measures have demonstrated statistically significant changes over time with correlations to the ALS functional rating scale-revised (ALSFRS-RSE) and the Rasch Overall ALS Disability Scale (ROADS) [395]. Interestingly, an international survey on patient perspectives on digital healthcare technology in care and clinical trials for ALS showed that most patients had a positive attitude toward the use of digital technology in both care and clinical trial settings [396].
Assistive robotics can help ALS patients to perform daily tasks, thereby enhancing their autonomy and quality of life [380]. For instance, a soft robotic wearable developed by researchers from the Harvard John A. Paulson School of Engineering and Applied Sciences (SEAS) and Massachusetts General Hospital (MGH) was able to provide significant upper arm and shoulder support and improved mobility for those with ALS [397]. Some assistive robots are equipped with communication aids and can be customized to fit the specific needs of each ALS patient, which can make them more effective and comfortable to use [379].
Neural interfaces are being explored for their potential to restore lost functions in ALS patients. Brain–computer interfaces (BCIs) use neural signals for computer control and may allow people with late-stage ALS to communicate even when conventional technology falls short. BCIs are designed to restore voluntary motor control to paralyzed people by converting intent-to-move nerve impulses from the motor cortex into a digital signal. These technologies commonly use machine learning to interpret the nerve impulses, collected from tiny electrodes implanted in the brain, and transform them into specific digital actions [398].
In recent years, we have seen great progress in the development and validation of implanted BCIs, which place neural signal recording electrodes in or on the cortex [379]. The Wyss Center for Bio and Neuroengineering is planning to launch a trial of its wireless brain–computer interface (BCI), called ABILITY, in people to enable them to communicate using only their thoughts [399]. In a case study, a NeuroKey-based BCI is being tested in a man with a fast-progressing form of ALS, who advanced to a locked-in state and could no longer use assistive devices to communicate [400].
ATs can, therefore, help ALS patients overcome severe functional limitations, allowing patients to retain autonomy and control as their disease progresses1.[379] However, despite these benefits, there are challenges and limitations associated with the use of ATs.
1. Physical and environmental barriers: ALS patients may face physical and environmental barriers that limit the effectiveness of ATs.
2. Financial and resource constraints: the cost of ATs can be prohibitive for some patients, limiting their access to these technologies.
3. Poor policy implementation: in some cases, policies related to the provision and use of ATs may not be effectively implemented.
4. Societal ignorance: lack of awareness and understanding about ATs among society at large can also pose challenges.
5. Technical challenges: acceptance of technology among patients and carers depends on various factors such as perceived skills and competencies in using the device, expectations, trust, and reliability.
While ATs have a significant positive impact on ALS management, it is important to address these challenges to make these technologies more accessible and effective for all patients. Moreover, there is a pressing need for further research in this field to address the remaining challenges and so allow us to continue advancing these promising technologies.
Ethical issues in motor neuron disease
Being a relentlessly fatal disease with no curative therapy currently available, there are many ethical issues in ALS care, from the diagnosis, disease course to the latest stages of disease, as recently reviewed [401]. Despite the fact that many healthcare providers and physicians may find it difficult and stressful [402, 403], ALS patients generally welcome the opportunity to discuss end-of-life issues with their physician [404, 405]. PEG and NIV decision-making processes are complex and individual, comprising patient-centric factors and external factors, including the roles played by healthcare professionals, family information provided and concepts of timing of interventions [406]. Although there is no consensus on timing, it is suggested that options for respiratory support and end-of-life issues be discussed when the patient displays symptoms of hypoventilation [407]. However, end-of-life discussions, including gastrostomy, NIV and tracheostomy invasive mechanical ventilation (TIV) are often delayed for a number but not necessarily justifiable reasons, potentially leading to poor patient management and unplanned crisis interventions [408]. Recent studies demonstrated that caregivers and the general public significantly underestimate the QoL of ALS patients and overestimate the patients’ rate of depression, while the desire to shorten life was significantly lower in ALS patients compared to what healthy subjects thought the patient would wish. Moreover, of those with undecided or negative attitudes, 10% changed their attitude towards life-sustaining treatments during the following year, emphasizing the need for unbiased patient perspectives in order to secure patient-centered decision-making [409, 410], even if the potential executive and behavioral impairments linked to ALS raises further inevitable questions [411].
Many socio-cultural, ethical, legal and financial issues may influence the use of TIV in ALS patients, as reflected by the varied percentage of use between countries, ranging from 0% of patients from the UK, 1–14% in the USA, 11% in northern Italy, to 27–45% in Japan [412, 413]. The ethical implications of TIV have recently been analyzed accordingly to the four core principles of healthcare ethics: beneficence, nonmaleficence, respect for patient autonomy, and justice [414]. However, there are many specific and challenging aspects to be considered within the context of ALS. The choice of TIV is indeed a critical milestones as it may imply survival in a locked-in state [414, 415]. Hence, TIV in ALS affects not only the patient but also the next of kin and the professional caregivers, rendering highly relevant to discuss potential positive and negative consequences also for these other stakeholders [414, 415]. Respect for the patient’s right to accept or refuse medical interventions is the mainstays of modern healthcare. Prerequisites are competence and being informed [149]. However, cognitive and behavioral impairments seen in ALS make it questionable whether these requirements are in place in all circumstances, which is highly relevant to patient autonomy and decision-making [265, 414, 416]. Indeed, the recent awareness of potential executive and behavioral impairment in ALS patients represents a further ethical question, emphasizing the need to develop new methods of neuropsychological assessment suitable to the more advanced stages of disease [149, 409, 417].
Conclusions
ALS remains a relentlessly progressive and fatal disease and the only approved disease-modifying drug has a modest effect in slowing disease progression. Symptomatic and palliative care, provided in a multidisciplinary context, still remains the cornerstone of ALS management [326]. However, significant progress has been accomplished in the identification of new genetic factors and pathomechanisms underlying this disease, giving new hope for the development of gene therapy and novel mechanistic therapeutic approaches.
References
Al-Chalabi A, Hardiman O (2013) The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 9:617–628
Xu L, Liu T, Liu L, Yao X, Chen L, Fan D, Zhan S, Wang S (2020) Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol 267:944–953
Riva N, Agosta F, Lunetta C, Filippi M, Quattrini A (2016) Recent advances in amyotrophic lateral sclerosis. J Neurol 263:1241–1254
Lloyd-Sherlock P (2000) Population ageing in developed and developing regions: implications for health policy. Soc Sci Med 51:887–895
Marin B, Boumédiene F, Logroscino G, Couratier P, Babron M-C, Leutenegger AL, Copetti M, Preux P-M, Beghi E (2017) Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta-analysis. Int J Epidemiol 46:57–74
Wolfson C, Gauvin DE, Ishola F, Oskoui M (2023) Global prevalence and incidence of amyotrophic lateral sclerosis: a systematic review. Neurology 101:e613–e623
McFarlane R, Peelo C, Galvin M, Heverin M, Hardiman O (2023) Epidemiologic trends of amyotrophic lateral sclerosis in Ireland, 1996–2021. Neurology 101:e1905–e1912
Hanhisuanto M, Solje E, Jokela M, Sipilä JOT (2023) Amyotrophic Lateral Sclerosis in Southwestern and Eastern Finland. Neuroepidemiology 57:238–245
Leighton DJ, Newton J, Stephenson LJ, Colville S, Davenport R, Gorrie G, Morrison I, Swingler R, Chandran S, Pal S, Consortium C-M (2019) Changing epidemiology of motor neurone disease in Scotland. J Neurol 266:817–825
Gianferrari G, Martinelli I, Zucchi E, Simonini C, Fini N, Vinceti M, Ferro S, Gessani A, Canali E, Valzania F, Sette E, Pugliatti M, Tugnoli V, Zinno L, Stano S, Santangelo M, De Pasqua S, Terlizzi E, Guidetti D, Medici D, Salvi F, Liguori R, Vacchiano V, Casmiro M, Querzani P, Currò Dossi M, Patuelli A, Morresi S, Longoni M, De Massis P, Rinaldi R, Borghi A, Amedei A, Mandrioli J (2022) Epidemiological, clinical and genetic features of als in the last decade: a prospective population-based study in the Emilia Romagna Region of Italy. Biomedicines 10:819
Chiò A, Mora G, Moglia C, Manera U, Canosa A, Cammarosano S, Ilardi A, Bertuzzo D, Bersano E, Cugnasco P, Grassano M, Pisano F, Mazzini L, Calvo A, Piemonte VD, Register for ALS (2017) Secular trends of amyotrophic lateral sclerosis: the piemonte and Valle d’Aosta Register. JAMA Neurol 74:1097–1104
Westeneng H-J, Debray TPA, Visser AE, van Eijk RPA, Rooney JPK, Calvo A, Martin S, McDermott CJ, Thompson AG, Pinto S, Kobeleva X, Rosenbohm A, Stubendorff B, Sommer H, Middelkoop BM, Dekker AM, van Vugt JJFA, van Rheenen W, Vajda A, Heverin M, Kazoka M, Hollinger H, Gromicho M, Körner S, Ringer TM, Rödiger A, Gunkel A, Shaw CE, Bredenoord AL, van Es MA, Corcia P, Couratier P, Weber M, Grosskreutz J, Ludolph AC, Petri S, de Carvalho M, Van Damme P, Talbot K, Turner MR, Shaw PJ, Al-Chalabi A, Chiò A, Hardiman O, Moons KGM, Veldink JH, van den Berg LH (2018) Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol 17:423–433
Domi T, Schito P, Sferruzza G, Russo T, Pozzi L, Agosta F, Carrera P, Riva N, Filippi M, Quattrini A, Falzone YM (2023) Unveiling the SOD1-mediated ALS phenotype: insights from a comprehensive meta-analysis. J Neurol
Rosenbohm A, Liu M, Nagel G, Peter RS, Cui B, Li X, Kassubek J, Rothenbacher D, Lule D, Cui L, Ludolph AC, Group ALSRSS (2018) Phenotypic differences of amyotrophic lateral sclerosis (ALS) in China and Germany. J Neurol 265:774–782
Velez-Go MB, Perna A, Vazquez C, Ketzoian C, Lillo P, Godoy-Reyes G, Saez D, Zaldivar Vaillant T, Gutierrez Gil JV, Lara-Fernandez GE, Povedano M, Heverin M, McFarlane R, Logroscino G, Hardiman O (2023) LAENALS: epidemiological and clinical features of amyotrophic lateral sclerosis in Latin America. Amyotrophic Lateral Sclerosis Frontotemporal Degener:1–9
Logroscino G, Traynor BJ, Hardiman O, Chio A, Couratier P, Mitchell JD, Swingler RJ, Beghi E, Eurals F (2008) Descriptive epidemiology of amyotrophic lateral sclerosis: new evidence and unsolved issues. J Neurol Neurosurg Psychiatry 79:6–11
Fontana A, Marin B, Luna J, Beghi E, Logroscino G, Boumediene F, Preux PM, Couratier P, Copetti M (2021) Time-trend evolution and determinants of sex ratio in amyotrophic lateral sclerosis: a dose-response meta-analysis. J Neurol 268:2973–2984
Schito P, Ceccardi G, Calvo A, Falzone YM, Moglia C, Lunetta C, Marinou K, Ticozzi N, Scialo C, Sorarù G, Trojsi F, Conte A, Tortelli R, Russo M, Zucchi E, Pozzi L, Domi T, Carrera P, Agosta F, Quattrini A, Fazio R, Chiò A, Sansone VA, Mora G, Silani V, Volanti P, Caponnetto C, Querin G, Tedeschi G, Sabatelli M, Logroscino G, Messina S, Mandrioli J, Riva N, Filippi M (2020) Clinical features and outcomes of the flail arm and flail leg and pure lower motor neuron MND variants: a multicentre Italian study. J Neurol Neurosurg Psychiatry 91:1001–1003
Schito P, Russo T, Domi T, Mandelli A, Pozzi L, Carro UD, Carrera P, Agosta F, Quattrini A, Furlan R, Filippi M, Riva N (2023) Clinical features and biomarkers to differentiate primary and amyotrophic lateral sclerosis in patients with an upper motor neuron syndrome. Neurology
Schito P, Spinelli EG, Malvaso A, Russo T, Falzone YM, Agosta F, Quattrini A, Filippi M, Riva N (2022) Primary lateral sclerosis presenting with focal onset spreading through contiguous neuroanatomic regions. Neurology 98:503–504
Chiò A, Moglia C, Canosa A, Manera U, D’Ovidio F, Vasta R, Grassano M, Brunetti M, Barberis M, Corrado L, D’Alfonso S, Iazzolino B, Peotta L, Sarnelli MF, Solara V, Zucchetti JP, De Marchi F, Mazzini L, Mora G, Calvo A (2020) ALS phenotype is influenced by age, sex, and genetics: a population-based study. Neurology 94:e802–e810
Grassano M, Moglia C, Palumbo F, Koumantakis E, Cugnasco P, Callegaro S, Canosa A, Manera U, Vasta R, De Mattei F, Matteoni E, Fuda G, Salamone P, Marchese G, Casale F, De Marchi F, Mazzini L, Mora G, Calvo A, Chio A (2024) Sex differences in amyotrophic lateral sclerosis survival and progression: a multidimensional analysis. Ann Neurol
Bede P, Elamin M, Byrne S, Hardiman O (2014) Sexual dimorphism in ALS: exploring gender-specific neuroimaging signatures. Amyotrophic Lateral Sclerosis Frontotemporal Degener 15:235–243
Trojsi F, Di Nardo F, Caiazzo G, Siciliano M, D’Alvano G, Passaniti C, Russo A, Bonavita S, Cirillo M, Esposito F, Tedeschi G (2021) Between-sex variability of resting state functional brain networks in amyotrophic lateral sclerosis (ALS). J Neural Transm 128:1881–1897
Feldman EL, Goutman SA, Petri S, Mazzini L, Savelieff MG, Shaw PJ, Sobue G (2022) Amyotrophic lateral sclerosis. Lancet 400:1363–1380
van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, van den Berg LH (2017) Amyotrophic lateral sclerosis. Lancet 390:2084–2098
Zhou W, Xu R (2023) Current insights in the molecular genetic pathogenesis of amyotrophic lateral sclerosis. Front Neurosci 17:1189470
Muzio L, Sirtori R, Gornati D, Eleuteri S, Fossaghi A, Brancaccio D, Manzoni L, Ottoboni L, Feo L, Quattrini A, Mastrangelo E, Sorrentino L, Scalone E, Comi G, Marinelli L, Riva N, Milani M, Seneci P, Martino G (2020) Retromer stabilization results in neuroprotection in a model of Amyotrophic Lateral Sclerosis. Nat Commun 11:3848
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Chen Y, Li S, Su L, Sheng J, Lv W, Chen G, Xu Z (2015) Association of progranulin polymorphism rs5848 with neurodegenerative diseases: a meta-analysis. J Neurol 262:814–822
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Consortium I, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257-268
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, Chromosome ALSFTDC, French research network on FFA, Consortium I, Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11:323-330
Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, Wray S, Sidle K, Fratta P, Orrell RW, Hardy J, Holton J, Revesz T, Rossor MN, Warren JD (2015) C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol 14:291–301
Silverman JM, Fernando SM, Grad LI, Hill AF, Turner BJ, Yerbury JJ, Cashman NR (2016) Disease mechanisms in ALS: misfolded SOD1 transferred through exosome-dependent and exosome-independent pathways. Cell Mol Neurobiol 36:377–381
Bagyinszky E, Hulme J, An SSA (2023) Studies of Genetic and Proteomic Risk Factors of Amyotrophic Lateral Sclerosis Inspire Biomarker Development and Gene Therapy. Cells 12
Bartoletti-Stella A, Vacchiano V, De Pasqua S, Mengozzi G, De Biase D, Bartolomei I, Avoni P, Rizzo G, Parchi P, Donadio V, Chio A, Pession A, Oppi F, Salvi F, Liguori R, Capellari S, BoReAls (2021) Targeted sequencing panels in Italian ALS patients support different etiologies in the ALS/FTD continuum. J Neurol 268:3766–3776
McCann EP, Grima N, Fifita JA, Chan Moi Fat S, Lehnert K, Henden L, Blair IP, Williams KL (2023) Characterising the genetic landscape of amyotrophic lateral sclerosis: a catalogue and assessment of over 1,000 published genetic variants. J Neuromuscular Dis 10:1127–1141
Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, Ren Z, Keebler J, Han Y, Levy SE, Boone BE, Wimbish JR, Waite LL, Jones AL, Carulli JP, Day-Williams AG, Staropoli JF, Xin WW, Chesi A, Raphael AR, McKenna-Yasek D, Cady J, Vianney de Jong JM, Kenna KP, Smith BN, Topp S, Miller J, Gkazi A, Consortium FS, Al-Chalabi A, van den Berg LH, Veldink J, Silani V, Ticozzi N, Shaw CE, Baloh RH, Appel S, Simpson E, Lagier-Tourenne C, Pulst SM, Gibson S, Trojanowski JQ, Elman L, McCluskey L, Grossman M, Shneider NA, Chung WK, Ravits JM, Glass JD, Sims KB, Van Deerlin VM, Maniatis T, Hayes SD, Ordureau A, Swarup S, Landers J, Baas F, Allen AS, Bedlack RS, Harper JW, Gitler AD, Rouleau GA, Brown R, Harms MB, Cooper GM, Harris T, Myers RM, Goldstein DB (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347:1436-1441
Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, Marroquin N, Nordin F, Hubers A, Weydt P, Pinto S, Press R, Millecamps S, Molko N, Bernard E, Desnuelle C, Soriani MH, Dorst J, Graf E, Nordstrom U, Feiler MS, Putz S, Boeckers TM, Meyer T, Winkler AS, Winkelman J, de Carvalho M, Thal DR, Otto M, Brannstrom T, Volk AE, Kursula P, Danzer KM, Lichtner P, Dikic I, Meitinger T, Ludolph AC, Strom TM, Andersen PM, Weishaupt JH (2015) Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 18:631–636
Brenner D, Muller K, Wieland T, Weydt P, Bohm S, Lule D, Hubers A, Neuwirth C, Weber M, Borck G, Wahlqvist M, Danzer KM, Volk AE, Meitinger T, Strom TM, Otto M, Kassubek J, Ludolph AC, Andersen PM, Weishaupt JH (2016) NEK1 mutations in familial amyotrophic lateral sclerosis. Brain J Neurol 139:e28
Pozzi L, Valenza F, Mosca L, Dal Mas A, Domi T, Romano A, Tarlarini C, Falzone YM, Tremolizzo L, Soraru G, Cerri F, Ferraro PM, Basaia S, Agosta F, Fazio R, Comola M, Comi G, Ferrari M, Quattrini A, Lunetta C, Penco S, Bonanomi D, Carrera P, Riva N (2017) TBK1 mutations in Italian patients with amyotrophic lateral sclerosis: genetic and functional characterisation. J Neurol Neurosurg Psychiatry 88:869–875
Riva N, Pozzi L, Russo T, Pipitone GB, Schito P, Domi T, Agosta F, Quattrini A, Carrera P, Filippi M (2022) NEK1 variants in a cohort of italian patients with amyotrophic lateral sclerosis. Front Neurosci 16:833051
Fellner A, Goldberg Y, Basel-Salmon L (2023) Ordering genetic testing by neurologists: points to consider. J Neurol 270:3714–3722
Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, Brown P, Ravenscroft T, van Blitterswijk M, Nicholson AM, DeTure M, Knopman DS, Josephs KA, Parisi JE, Petersen RC, Boylan KB, Boeve BF, Graff-Radford NR, Veltman JA, Gilissen C, Murray ME, Dickson DW, Rademakers R (2015) Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol 130:77–92
Lattante S, Doronzio PN, Marangi G, Conte A, Bisogni G, Bernardo D, Russo T, Lamberti D, Patrizi S, Apollo FP, Lunetta C, Scarlino S, Pozzi L, Zollino M, Riva N, Sabatelli M (2019) Coexistence of variants in TBK1 and in other ALS-related genes elucidates an oligogenic model of pathogenesis in sporadic ALS. Neurobiol Aging 84:239 e239–239 e214
Borghero G, Pugliatti M, Marrosu F, Marrosu MG, Murru MR, Floris G, Cannas A, Occhineri P, Cau TB, Loi D, Ticca A, Traccis S, Manera U, Canosa A, Moglia C, Calvo A, Barberis M, Brunetti M, Gibbs JR, Renton AE, Errichiello E, Zoledziewska M, Mulas A, Qian Y, Din J, Pliner HA, Traynor BJ, Chio A, Italsgen CS (2016) TBK1 is associated with ALS and ALS-FTD in Sardinian patients. Neurobiol Aging 43(180):e181-185
Kenna KP, van Doormaal PT, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, van Rheenen W, van Eijk KR, Jones AR, Keagle P, Shatunov A, Sproviero W, Smith BN, van Es MA, Topp SD, Kenna A, Miller JW, Fallini C, Tiloca C, McLaughlin RL, Vance C, Troakes C, Colombrita C, Mora G, Calvo A, Verde F, Al-Sarraj S, King A, Calini D, de Belleroche J, Baas F, van der Kooi AJ, de Visser M, Ten Asbroek AL, Sapp PC, McKenna-Yasek D, Polak M, Asress S, Munoz-Blanco JL, Strom TM, Meitinger T, Morrison KE, Consortium S, Lauria G, Williams KL, Leigh PN, Nicholson GA, Blair IP, Leblond CS, Dion PA, Rouleau GA, Pall H, Shaw PJ, Turner MR, Talbot K, Taroni F, Boylan KB, Van Blitterswijk M, Rademakers R, Esteban-Perez J, Garcia-Redondo A, Van Damme P, Robberecht W, Chio A, Gellera C, Drepper C, Sendtner M, Ratti A, Glass JD, Mora JS, Basak NA, Hardiman O, Ludolph AC, Andersen PM, Weishaupt JH, Brown RH, Jr., Al-Chalabi A, Silani V, Shaw CE, van den Berg LH, Veldink JH, Landers JE (2016) NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nature genetics 48:1037-1042
Farhan SMK, Howrigan DP, Abbott LE, Klim JR, Topp SD, Byrnes AE, Churchhouse C, Phatnani H, Smith BN, Rampersaud E, Wu G, Wuu J, Shatunov A, Iacoangeli A, Al Khleifat A, Mordes DA, Ghosh S, Consortium A, Consortium F, Project Min EC, Eggan K, Rademakers R, McCauley JL, Schule R, Zuchner S, Benatar M, Taylor JP, Nalls M, Gotkine M, Shaw PJ, Morrison KE, Al-Chalabi A, Traynor B, Shaw CE, Goldstein DB, Harms MB, Daly MJ, Neale BM (2019) Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nature neuroscience 22:1966-1974
Wang S, Zheng X, Wei Q, Lin J, Yang T, Xiao Y, Jiang Q, Li C, Shang H (2023) Rare DNAJC7 variants may play a minor role in chinese patients with ALS. Mol Neurobiol
Osmanovic A, Forster A, Widjaja M, Auber B, Das AM, Christians A, Brand F, Petri S, Weber RG (2022) A SUMO4 initiator codon variant in amyotrophic lateral sclerosis reduces SUMO4 expression and alters stress granule dynamics. J Neurol 269:4863–4871
Brenner D, Yilmaz R, Muller K, Grehl T, Petri S, Meyer T, Grosskreutz J, Weydt P, Ruf W, Neuwirth C, Weber M, Pinto S, Claeys KG, Schrank B, Jordan B, Knehr A, Gunther K, Hubers A, Zeller D, Kubisch C, Jablonka S, Sendtner M, Klopstock T, de Carvalho M, Sperfeld A, Borck G, Volk AE, Dorst J, Weis J, Otto M, Schuster J, Del Tredici K, Braak H, Danzer KM, Freischmidt A, Meitinger T, Strom TM, Ludolph AC, Andersen PM, Weishaupt JH, German ALSnMNDNET (2018) Hot-spot KIF5A mutations cause familial ALS. Brain J Neurol 141:688–697
Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, Dominov JA, Kenna BJ, Nalls MA, Keagle P, Rivera AM, van Rheenen W, Murphy NA, van Vugt J, Geiger JT, Van der Spek RA, Pliner HA, Shankaracharya, Smith BN, Marangi G, Topp SD, Abramzon Y, Gkazi AS, Eicher JD, Kenna A, Consortium I, Mora G, Calvo A, Mazzini L, Riva N, Mandrioli J, Caponnetto C, Battistini S, Volanti P, La Bella V, Conforti FL, Borghero G, Messina S, Simone IL, Trojsi F, Salvi F, Logullo FO, D'Alfonso S, Corrado L, Capasso M, Ferrucci L, Genomic Translation for ALSCC, Moreno CAM, Kamalakaran S, Goldstein DB, Consortium ALSS, Gitler AD, Harris T, Myers RM, Consortium NA, Phatnani H, Musunuri RL, Evani US, Abhyankar A, Zody MC, Answer ALSF, Kaye J, Finkbeiner S, Wyman SK, LeNail A, Lima L, Fraenkel E, Svendsen CN, Thompson LM, Van Eyk JE, Berry JD, Miller TM, Kolb SJ, Cudkowicz M, Baxi E, Clinical Research in ALS, Related Disorders for Therapeutic Development C, Benatar M, Taylor JP, Rampersaud E, Wu G, Wuu J, Consortium S, Lauria G, Verde F, Fogh I, Tiloca C, Comi GP, Soraru G, Cereda C, French ALSC, Corcia P, Laaksovirta H, Myllykangas L, Jansson L, Valori M, Ealing J, Hamdalla H, Rollinson S, Pickering-Brown S, Orrell RW, Sidle KC, Malaspina A, Hardy J, Singleton AB, Johnson JO, Arepalli S, Sapp PC, McKenna-Yasek D, Polak M, Asress S, Al-Sarraj S, King A, Troakes C, Vance C, de Belleroche J, Baas F, Ten Asbroek A, Munoz-Blanco JL, Hernandez DG, Ding J, Gibbs JR, Scholz SW, Floeter MK, Campbell RH, Landi F, Bowser R, Pulst SM, Ravits JM, MacGowan DJL, Kirby J, Pioro EP, Pamphlett R, Broach J, Gerhard G, Dunckley TL, Brady CB, Kowall NW, Troncoso JC, Le Ber I, Mouzat K, Lumbroso S, Heiman-Patterson TD, Kamel F, Van Den Bosch L, Baloh RH, Strom TM, Meitinger T, Shatunov A, Van Eijk KR, de Carvalho M, Kooyman M, Middelkoop B, Moisse M, McLaughlin RL, Van Es MA, Weber M, Boylan KB, Van Blitterswijk M, Rademakers R, Morrison KE, Basak AN, Mora JS, Drory VE, Shaw PJ, Turner MR, Talbot K, Hardiman O, Williams KL, Fifita JA, Nicholson GA, Blair IP, Rouleau GA, Esteban-Perez J, Garcia-Redondo A, Al-Chalabi A, Project Min EALSSC, Rogaeva E, Zinman L, Ostrow LW, Maragakis NJ, Rothstein JD, Simmons Z, Cooper-Knock J, Brice A, Goutman SA, Feldman EL, Gibson SB, Taroni F, Ratti A, Gellera C, Van Damme P, Robberecht W, Fratta P, Sabatelli M, Lunetta C, Ludolph AC, Andersen PM, Weishaupt JH, Camu W, Trojanowski JQ, Van Deerlin VM, Brown RH, Jr., van den Berg LH, Veldink JH, Harms MB, Glass JD, Stone DJ, Tienari P, Silani V, Chio A, Shaw CE, Traynor BJ, Landers JE (2018) Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 97:1268-1283 e1266
Saez-Atienzar S, Dalgard CL, Ding J, Chio A, Alba C, Hupalo DN, Wilkerson MD, Bowser R, Pioro EP, Bedlack R, Traynor BJ (2020) Identification of a pathogenic intronic KIF5A mutation in an ALS-FTD kindred. Neurology 95:1015–1018
Guo L, Mao Q, He J, Liu X, Piao X, Luo L, Hao X, Yu H, Song Q, Xiao B, Fan D, Gao Z, Jia Y (2023) Disruption of ER ion homeostasis maintained by an ER anion channel CLCC1 contributes to ALS-like pathologies. Cell Res 33:497–515
Tang L, Tang X, Zhao Q, Li Y, Bu Y, Liu Z, Li J, Guo J, Shen L, Jiang H, Tang B, Xu R, Cao W, Yuan Y, Wang J (2023) Mutation and clinical analysis of the CLCC1 gene in amyotrophic lateral sclerosis patients from Central South China. Ann Clini Transl Neurol. https://doi.org/10.1002/acn3.51934
Johnson JO, Chia R, Miller DE, Li R, Kumaran R, Abramzon Y, Alahmady N, Renton AE, Topp SD, Gibbs JR, Cookson MR, Sabir MS, Dalgard CL, Troakes C, Jones AR, Shatunov A, Iacoangeli A, Al Khleifat A, Ticozzi N, Silani V, Gellera C, Blair IP, Dobson-Stone C, Kwok JB, Bonkowski ES, Palvadeau R, Tienari PJ, Morrison KE, Shaw PJ, Al-Chalabi A, Brown RH Jr, Calvo A, Mora G, Al-Saif H, Gotkine M, Leigh F, Chang IJ, Perlman SJ, Glass I, Scott AI, Shaw CE, Basak AN, Landers JE, Chio A, Crawford TO, Smith BN, Traynor BJ, Consortium FS, American Genome C, International ALSGC, Consortium I, Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp SD, Scotter EL, Kenna KP, Keagle P, Tiloca C, Vance C, Troakes C, Colombrita C, King A, Pensato V, Castellotti B, Baas F, Ten Asbroek A, McKenna-Yasek D, McLaughlin RL, Polak M, Asress S, Esteban-Perez J, Stevic Z, D’Alfonso S, Mazzini L, Comi GP, Del Bo R, Ceroni M, Gagliardi S, Querin G, Bertolin C, van Rheenen W, Rademakers R, van Blitterswijk M, Lauria G, Duga S, Corti S, Cereda C, Corrado L, Soraru G, Williams KL, Nicholson GA, Blair IP, Leblond-Manry C, Rouleau GA, Hardiman O, Morrison KE, Veldink JH, van den Berg LH, Al-Chalabi A, Pall H, Shaw PJ, Turner MR, Talbot K, Taroni F, Garcia-Redondo A, Wu Z, Glass JD, Gellera C, Ratti A, Brown RH Jr, Silani V, Shaw CE, Landers JE, Dalgard CL, Adeleye A, Soltis AR, Alba C, Viollet C, Bacikova D, Hupalo DN, Sukumar G, Pollard HB, Wilkerson MD, Martinez EM, Abramzon Y, Ahmed S, Arepalli S, Baloh RH, Bowser R, Brady CB, Brice A, Broach J, Campbell RH, Camu W, Chia R, Cooper-Knock J, Ding J, Drepper C, Drory VE, Dunckley TL, Eicher JD, England BK, Faghri F, Feldman E, Floeter MK, Fratta P, Geiger JT, Gerhard G, Gibbs JR, Gibson SB, Glass JD, Hardy J, Harms MB, Heiman-Patterson TD, Hernandez DG, Jansson L, Kirby J, Kowall NW, Laaksovirta H, Landeck N, Landi F, Le Ber I, Lumbroso S, MacGowan DJL, Maragakis NJ, Mora G, Mouzat K, Murphy NA, Myllykangas L, Nalls MA, Orrell RW, Ostrow LW, Pamphlett R, Pickering-Brown S, Pioro EP, Pletnikova O, Pliner HA, Pulst SM, Ravits JM, Renton AE, Rivera A, Robberecht W, Rogaeva E, Rollinson S, Rothstein JD, Scholz SW, Sendtner M, Shaw PJ, Sidle KC, Simmons Z, Singleton AB, Smith N, Stone DJ, Tienari PJ, Troncoso JC, Valori M, Van Damme P, Van Deerlin VM, Van Den Bosch L, Zinman L, Landers JE, Chio A, Traynor BJ, Angelocola SM, Ausiello FP, Barberis M, Bartolomei I, Battistini S, Bersano E, Bisogni G, Borghero G, Brunetti M, Cabona C, Calvo A, Canale F, Canosa A, Cantisani TA, Capasso M, Caponnetto C, Cardinali P, Carrera P, Casale F, Chio A, Colletti T, Conforti FL, Conte A, Conti E, Corbo M, Cuccu S, Dalla Bella E, D’Errico E, DeMarco G, Dubbioso R, Ferrarese C, Ferraro PM, Filippi M, Fini N, Floris G, Fuda G, Gallone S, Gianferrari G, Giannini F, Grassano M, Greco L, Iazzolino B, Introna A, La Bella V, Lattante S, Lauria G, Liguori R, Logroscino G, Logullo FO, Lunetta C, Mandich P, Mandrioli J, Manera U, Manganelli F, Marangi G, Marinou K, Marrosu MG, Martinelli I, Messina S, Moglia C, Mora G, Mosca L, Murru MR, Origone P, Passaniti C, Petrelli C, Petrucci A, Pozzi S, Pugliatti M, Quattrini A, Ricci C, Riolo G, Riva N, Russo M, Sabatelli M, Salamone P, Salivetto M, Salvi F, Santarelli M, Sbaiz L, Sideri R, Simone I, Simonini C, Spataro R, Tanel R, Tedeschi G, Ticca A, Torriello A, Tranquilli S, Tremolizzo L, Trojsi F, Vasta R, Vacchiano V, Vita G, Volanti P, Zollino M, Zucchi E (2021) Association of Variants in the SPTLC1 Gene With Juvenile Amyotrophic Lateral Sclerosis. JAMA Neurol 78:1236–1248
Borghero G, Pili F, Muroni A, Ercoli T, Pateri MI, Pilotto S, Maccabeo A, Defazio G (2023) Disease survival and progression in TARDBP ALS patients from Sardinia, Italy. J Neurol
Colombo E, Poletti B, Maranzano A, Peverelli S, Solca F, Colombrita C, Torre S, Tiloca C, Verde F, Bonetti R, Carelli L, Morelli C, Ratti A, Silani V, Ticozzi N (2023) Motor, cognitive and behavioural profiles of C9orf72 expansion-related amyotrophic lateral sclerosis. J Neurol 270:898–908
Miltenberger-Miltenyi G, Conceicao VA, Gromicho M, Pronto-Laborinho AC, Pinto S, Andersen PM, de Carvalho M (2019) C9orf72 expansion is associated with accelerated decline of respiratory function and decreased survival in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 90:118–120
Conforti FL, Spataro R, Sproviero W, Mazzei R, Cavalcanti F, Condino F, Simone IL, Logroscino G, Patitucci A, Magariello A, Muglia M, Rodolico C, Valentino P, Bono F, Colletti T, Monsurro MR, Gambardella A, La Bella V (2012) Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology 79:2315–2320
Sproviero W, Shatunov A, Stahl D, Shoai M, van Rheenen W, Jones AR, Al-Sarraj S, Andersen PM, Bonini NM, Conforti FL, Van Damme P, Daoud H, Del Mar AM, Fogh I, Forzan M, Gaastra B, Gellera C, Gitler AD, Hardy J, Fratta P, La Bella V, Le Ber I, Van Langenhove T, Lattante S, Lee YC, Malaspina A, Meininger V, Millecamps S, Orrell R, Rademakers R, Robberecht W, Rouleau G, Ross OA, Salachas F, Sidle K, Smith BN, Soong BW, Soraru G, Stevanin G, Kabashi E, Troakes C, van Broeckhoven C, Veldink JH, van den Berg LH, Shaw CE, Powell JF, Al-Chalabi A (2017) ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol Aging 51:178171–178179
Chio A, Calvo A, Moglia C, Canosa A, Brunetti M, Barberis M, Restagno G, Conte A, Bisogni G, Marangi G, Moncada A, Lattante S, Zollino M, Sabatelli M, Bagarotti A, Corrado L, Mora G, Bersano E, Mazzini L, D’Alfonso Parals S (2015) ATXN2 polyQ intermediate repeats are a modifier of ALS survival. Neurology 84:251–258
Scarlino S, Domi T, Pozzi L, Romano A, Pipitone GB, Falzone YM, Mosca L, Penco S, Lunetta C, Sansone V, Tremolizzo L, Fazio R, Agosta F, Filippi M, Carrera P, Riva N, Quattrini A (2020) Burden of Rare Variants in ALS and axonal hereditary neuropathy genes influence survival in ALS: insights from a next generation sequencing study of an Italian ALS Cohort. Int J Mol Sci. 21
Ghirelli A, Spinelli EG, Canu E, Domi T, Basaia S, Castelnovo V, Pozzi L, Magnani G, Caso F, Caroppo P, Prioni S, Villa C, Riva N, Quattrini A, Carrera P, Filippi M, Agosta F (2023) Case report: coexistence of C9orf72 expansion and progranulin mutation in a case of genetic frontotemporal dementia-clinical features and neuroimaging correlates. J Neurol 270:5102–5109
Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, Tranchant C, Aubourg P, Tazir M, Schols L, Pandolfo M, Schulz JB, Pouget J, Calvas P, Shizuka-Ikeda M, Shoji M, Tanaka M, Izatt L, Shaw CE, M’Zahem A, Dunne E, Bomont P, Benhassine T, Bouslam N, Stevanin G, Brice A, Guimaraes J, Mendonca P, Barbot C, Coutinho P, Sequeiros J, Durr A, Warter JM, Koenig M (2004) Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet 36:225–227
Bennett CL, Dastidar S, Arnold FJ, McKinstry SU, Stockford C, Freibaum BD, Sopher BL, Wu M, Seidner G, Joiner W, Taylor JP, West RJH, La Spada AR (2023) Senataxin helicase, the causal gene defect in ALS4, is a significant modifier of C9orf72 ALS G4C2 and arginine-containing dipeptide repeat toxicity. Acta Neuropathol Commun 11:164
Pupillo E, Messina P, Giussani G, Logroscino G, Zoccolella S, Chio A, Calvo A, Corbo M, Lunetta C, Marin B, Mitchell D, Hardiman O, Rooney J, Stevic Z, Bandettini di Poggio M, Filosto M, Cotelli MS, Perini M, Riva N, Tremolizzo L, Vitelli E, Damiani D, Beghi E, Consortium E (2014) Physical activity and amyotrophic lateral sclerosis: a European population-based case-control study. Ann Neurol 75:708–716
D’Ovidio F, Rooney JPK, Visser AE, Manera U, Beghi E, Logroscino G, Vermeulen RCH, Veldink JH, van den Berg LH, Hardiman O, Chio A, Mc E (2019) Association between alcohol exposure and the risk of amyotrophic lateral sclerosis in the Euro-MOTOR study. J Neurol Neurosurg Psychiatry 90:11–19
Visser AE, Rooney JPK, D’Ovidio F, Westeneng HJ, Vermeulen RCH, Beghi E, Chio A, Logroscino G, Hardiman O, Veldink JH, van den Berg LH, Mc E (2018) Multicentre, cross-cultural, population-based, case-control study of physical activity as risk factor for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 89:797–803
Pupillo E, Messina P, Logroscino G, Zoccolella S, Chio A, Calvo A, Corbo M, Lunetta C, Micheli A, Millul A, Vitelli E, Beghi E, Consortium E (2012) Trauma and amyotrophic lateral sclerosis: a case-control study from a population-based registry. Eur J Neurol Off J Eur Fed Neurol Soc 19:1509–1517
Pupillo E, Poloni M, Bianchi E, Giussani G, Logroscino G, Zoccolella S, Chio A, Calvo A, Corbo M, Lunetta C, Marin B, Mitchell D, Hardiman O, Rooney J, Stevic Z, Bandettini di Poggio M, Filosto M, Cotelli MS, Perini M, Riva N, Tremolizzo L, Vitelli E, Damiani D, Beghi E, Consortiumdagger E (2018) Trauma and amyotrophic lateral sclerosis: a european population-based case-control study from the EURALS consortium. Amyotrophic Lateral Sclerosis Frontotemporal Degener 19:118–125
Ingre C, Roos PM, Piehl F, Kamel F, Fang F (2015) Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol 7:181–193
Hamidou B, Couratier P, Besançon C, Nicol M, Preux PM, Marin B (2014) Epidemiological evidence that physical activity is not a risk factor for ALS. Eur J Epidemiol 29:459–475
Zhu Q, Zhou J, Zhang Y, Huang H, Han J, Cao B, Xu D, Zhao Y, Chen G (2023) Risk factors associated with amyotrophic lateral sclerosis based on the observational study: a systematic review and meta-analysis. Front Neurosci 17:1196722
Åberg M, Nyberg J, Robertson J, Kuhn G, Schiöler L, Nissbrandt H, Waern M, Torén K (2018) Risk factors in Swedish young men for amyotrophic lateral sclerosis in adulthood. J Neurol 265:460–470
Russell ER, Mackay DF, Stewart K, MacLean JA, Pell JP, Stewart W (2021) Association of field position and career length with risk of neurodegenerative disease in male former professional soccer players. JAMA Neurol 78:1057–1063
Chio A, Calvo A, Dossena M, Ghiglione P, Mutani R, Mora G (2009) ALS in Italian professional soccer players: the risk is still present and could be soccer-specific. Amyotrophic Lateral Sclerosis Off Publ World Fed Neurol Res Group Motor Neuron Dis 10:205–209
M H, Np R, (2020) Physical activity as a risk factor for amyotrophic lateral sclerosis-findings from three large European cohorts. J Neurol 267:2173–2175
Zheng X, Wang S, Huang J, Lin J, Yang T, Xiao Y, Jiang Q, Huang R, Li C, Shang H (2023) Physical activity as risk factor in amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol 270:2438–2450
Diekmann K, Kuzma-Kozakiewicz M, Piotrkiewicz M, Gromicho M, Grosskreutz J, Andersen PM, Carvalho M, Uysal H, Osmanovic A, Schreiber-Katz O, Petri S, Körner S (2020) Impact of comorbidities and co-medication on disease onset and progression in a large German ALS patient group. J Neurol 267:2130–2141
Chapman L, Cooper-Knock J, Shaw PJ (2023) Physical activity as an exogenous risk factor for amyotrophic lateral sclerosis: a review of the evidence. Brain J Neurol 146:1745–1757
Julian TH, Glascow N, Barry ADF, Moll T, Harvey C, Klimentidis YC, Newell M, Zhang S, Snyder MP, Cooper-Knock J, Shaw PJ (2021) Physical exercise is a risk factor for amyotrophic lateral sclerosis: convergent evidence from Mendelian randomisation, transcriptomics and risk genotypes. EBioMedicine 68:103397
Westeneng H-J, Veenhuijzen K, Spek RA, Peters S, Visser AE, Rheenen W, Veldink JH, Berg LH (2021) Associations between lifestyle and amyotrophic lateral sclerosis stratified by C9orf72 genotype: a longitudinal, population-based, case-control study. Lancet Neurol 20:373–384
Vasta R, Callegaro S, Sgambetterra S, Cabras S, Di Pede F, De Mattei F, Matteoni E, Grassano M, Bombaci A, De Marco G, Fuda G, Marchese G, Palumbo F, Canosa A, Mazzini L, De Marchi F, Moglia C, Manera U, Chiò A, Calvo A (2023) Presymptomatic geographical distribution of ALS patients suggests the involvement of environmental factors in the disease pathogenesis. J Neurol 270:5475–5482
Wändell P, Fredrikson S, Carlsson AC, Li X, Sundquist J, Sundquist K (2022) Amyotrophic lateral sclerosis (ALS) among immigrant groups and Swedish-born individuals: a cohort study of all adults 18 years of age and older in Sweden. J Neurol 269:1989–1995
Sagiraju HKR, Živković S, VanCott AC, Patwa H, Ruiz G, de Porras D, Amuan ME, Pugh MJV (2020) Amyotrophic lateral sclerosis among veterans deployed in support of post-9/11 U.S. Confl Mil Med 185:e501–e509
Re DB, Yan B, Calderón-Garcidueñas L, Andrew AS, Tischbein M, Stommel EW (2022) A perspective on persistent toxicants in veterans and amyotrophic lateral sclerosis: identifying exposures determining higher ALS risk. J Neurol 269:2359–2377
Goutman SA, Boss J, Jang D-G, Mukherjee B, Richardson RJ, Batterman S, Feldman EL (2023) Environmental risk scores of persistent organic pollutants associate with higher ALS risk and shorter survival in a new Michigan case/control cohort. J Neurol Neurosurg Psychiatry:jnnp-2023–332121
Gresham LS, Molgaard CA, Golbeck AL, Smith R (1986) Amyotrophic lateral sclerosis and occupational heavy metal exposure: a case-control study. Neuroepidemiology 5:29–38
Kamalian A, Foroughmand I, Koski L, Darvish M, Saghazadeh A, Kamalian A, Razavi SZE, Abdi S, Dehgolan SR, Fotouhi A, Roos PM (2023) Metal concentrations in cerebrospinal fluid, blood, serum, plasma, hair, and nails in amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Trace Elem Med Biol 78:127165
Ravits J (2014) Focality, stochasticity and neuroanatomic propagation in ALS pathogenesis. Exper Neurol 262 Pt B:121–126
Bendotti C, Bonetto V, Pupillo E, Logroscino G, Al-Chalabi A, Lunetta C, Riva N, Mora G, Lauria G, Weishaupt JH, Agosta F, Malaspina A, Basso M, Greensmith L, Van Den Bosch L, Ratti A, Corbo M, Hardiman O, Chio A, Silani V, Beghi E (2020) Focus on the heterogeneity of amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis Frontotemporal Degener 21:485–495
Chio A, Calvo A, Moglia C, Mazzini L, Mora G, group Ps (2011) Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry 82:740–746
Al-Chalabi A, Hardiman O, Kiernan MC, Chio A, Rix-Brooks B, van den Berg LH (2016) Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol 15:1182–1194
Schabhuttl M, Wieland T, Senderek J, Baets J, Timmerman V, De Jonghe P, Reilly MM, Stieglbauer K, Laich E, Windhager R, Erwa W, Trajanoski S, Strom TM, Auer-Grumbach M (2014) Whole-exome sequencing in patients with inherited neuropathies: outcome and challenges. J Neurol 261:970–982
Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann CO, Stumm G, Ludolph AC (2004) Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63:724–726
Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH Jr, Ludlow CL, Fischbeck KH (2003) Mutant dynactin in motor neuron disease. Nat Genet 33:455–456
Sevilla T, Cuesta A, Chumillas MJ, Mayordomo F, Pedrola L, Palau F, Vilchez JJ (2003) Clinical, electrophysiological and morphological findings of Charcot-Marie-Tooth neuropathy with vocal cord palsy and mutations in the GDAP1 gene. Brain J Neurol 126:2023–2033
Zimon M, Baets J, Fabrizi GM, Jaakkola E, Kabzinska D, Pilch J, Schindler AB, Cornblath DR, Fischbeck KH, Auer-Grumbach M, Guelly C, Huber N, De Vriendt E, Timmerman V, Suter U, Hausmanowa-Petrusewicz I, Niemann A, Kochanski A, De Jonghe P, Jordanova A (2011) Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 77:540–548
Strickland AV, Schabhuttl M, Offenbacher H, Synofzik M, Hauser NS, Brunner-Krainz M, Gruber-Sedlmayr U, Moore SA, Windhager R, Bender B, Harms M, Klebe S, Young P, Kennerson M, Garcia AS, Gonzalez MA, Zuchner S, Schule R, Shy ME, Auer-Grumbach M (2015) Mutation screen reveals novel variants and expands the phenotypes associated with DYNC1H1. J Neurol. https://doi.org/10.1007/s00415-015-7727-2
Weedon MN, Hastings R, Caswell R, Xie W, Paszkiewicz K, Antoniadi T, Williams M, King C, Greenhalgh L, Newbury-Ecob R, Ellard S (2011) Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal charcot-marie-tooth disease. Am J Hum Genet 89:308–312
Liu YT, Laura M, Hersheson J, Horga A, Jaunmuktane Z, Brandner S, Pittman A, Hughes D, Polke JM, Sweeney MG, Proukakis C, Janssen JC, Auer-Grumbach M, Zuchner S, Shields KG, Reilly MM, Houlden H (2014) Extended phenotypic spectrum of KIF5A mutations: from spastic paraplegia to axonal neuropathy. Neurology 83:612–619
Daud D, Griffin H, Douroudis K, Kleinle S, Eglon G, Pyle A, Chinnery PF, Horvath R (2015) Whole exome sequencing and the clinician: we need clinical skills and functional validation in variant filtering. J Neurol 262:1673–1677
Al-Chalabi A, Andersen PM, Nilsson P, Chioza B, Andersson JL, Russ C, Shaw CE, Powell JF, Leigh PN (1999) Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum Mol Genet 8:157–164
Hensiek A, Kirker S, Reid E (2015) Diagnosis, investigation and management of hereditary spastic paraplegias in the era of next-generation sequencing. J Neurol 262:1601–1612
Gentile F, Scarlino S, Falzone YM, Lunetta C, Tremolizzo L, Quattrini A, Riva N (2019) The peripheral nervous system in amyotrophic lateral sclerosis: opportunities for translational research. Front Neurosci 13:601
Scarlino S, Domi T, Pozzi L, Romano A, Pipitone GB, Falzone YM, Mosca L, Penco S, Lunetta C, Sansone V, Tremolizzo L, Fazio R, Agosta F, Filippi M, Carrera P, Riva N, Quattrini A (2020) Burden of rare variants in ALS and axonal hereditary neuropathy genes influence survival in ALS: insights from a next generation sequencing study of an Italian ALS cohort. Int J Mol Sci 21:3346
Previtali SC, Zhao E, Lazarevic D, Pipitone GB, Fabrizi GM, Manganelli F, Mazzeo A, Pareyson D, Schenone A, Taroni F, Vita G, Bellone E, Ferrarini M, Garibaldi M, Magri S, Padua L, Pennisi E, Pisciotta C, Riva N, Scaioli V, Scarlato M, Tozza S, Geroldi A, Jordanova A, Ferrari M, Molineris I, Reilly MM, Comi G, Carrera P, Devoto M, Bolino A (2019) Expanding the spectrum of genes responsible for hereditary motor neuropathies. J Neurol Neurosurg Psychiatry 90:1171–1179
Fang T, Al Khleifat A, Meurgey JH, Jones A, Leigh PN, Bensimon G, Al-Chalabi A (2018) Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose-ranging study. Lancet Neurol 17:416–422
Shellikeri S, Karthikeyan V, Martino R, Black SE, Zinman L, Keith J, Yunusova Y (2017) The neuropathological signature of bulbar-onset ALS: a systematic review. Neurosci Biobehav Rev 75:378–392
Shellikeri S, Keith J, Black SE, Zinman L, Yunusova Y (2020) Neuropathology of speech network distinguishes bulbar from nonbulbar amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 79:284–295
Mitsumoto H, Del Bene M (2000) Improving the quality of life for people with ALS: the challenge ahead. Amyotrophic Lateral Sclerosis Other Motor Neuron Disord Off Publ World Fed Neurol Res Group Motor Neur Dis 1:329–336
Goldstein LH, Atkins L, Leigh PN (2002) Correlates of Quality of Life in people with motor neuron disease (MND). Amyotrophic Lateral Sclerosis Other Motor Neuron Disord Off Publ World Fed Neurol Res Group Motor Neur Dis 3:123–129
Saleem F, Munakomi S (2024) Pseudobulbar Palsy. In: StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Sunil Munakomi declares no relevant financial relationships with ineligible companies.
Goutman SA, Hardiman O, Al-Chalabi A, Chio A, Savelieff MG, Kiernan MC, Feldman EL (2022) Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis. Lancet Neurol 21:480–493
Finegan E, Chipika RH, Li Hi Shing S, Hardiman O, Bede P (2019) Pathological crying and laughing in motor neuron disease: pathobiology, screening. Interven Front Neurol 10:260
Karam C, Scelsa SN, Macgowan DJ (2010) The clinical course of progressive bulbar palsy. Amyotrophic Lateral Sclerosis Off Publ World Fed Neurol Res Group Motor Neur Dis 11:364–368
Gaig C, Graus F, Compta Y, Hogl B, Bataller L, Bruggemann N, Giordana C, Heidbreder A, Kotschet K, Lewerenz J, Macher S, Marti MJ, Montojo T, Perez-Perez J, Puertas I, Seitz C, Simabukuro M, Tellez N, Wandinger KP, Iranzo A, Ercilla G, Sabater L, Santamaria J, Dalmau J (2017) Clinical manifestations of the anti-IgLON5 disease. Neurology 88:1736–1743
Sista SR, Crum B, Aboseif A, Devine MF, Zekeridou A, Hammami MB, Rezk MM, Truffert A, Lalive PH, Kunchok A, McKeon A, Dubey D (2022) Motor-neuron-disease-like phenotype associated with IgLON5 disease. J Neurol 269:6139–6144
Origone P, Caponnetto C, Mantero V, Cichero E, Fossa P, Geroldi A, Verdiani S, Bellone E, Mancardi G, Mandich P (2012) Fast course ALS presenting with vocal cord paralysis: clinical features, bioinformatic and modelling analysis of the novel SOD1 Gly147Ser mutation. Amyotrophic Lateral Sclerosis Off Publ World Fed Neurol Res Group Motor Neuron Dis 13:144–148
Capece G, Ceroni M, Alfonsi E, Palmieri I, Cereda C, Diamanti L (2021) Case Report: Laryngospasm as Initial Manifestation of Amyotrophic Lateral Sclerosis in a Long-Survival Patient With Heterozygous p.D90A - SOD1 Mutation. Front Neurol 12:708885
Dalla Bella E, Rigamonti A, Mantero V, Morbin M, Saccucci S, Gellera C, Mora G, Lauria G (2014) Heterozygous D90A-SOD1 mutation in a patient with facial onset sensory motor neuronopathy (FOSMN) syndrome: a bridge to amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 85:1009–1011
de Boer EMJ, Barritt AW, Elamin M, Anderson SJ, Broad R, Nisbet A, Goedee HS, Vazquez Costa JF, Prudlo J, Vedeler CA, Fernandez JP, Panades MP, Alberti Aguilo MA, Bella ED, Lauria G, Pinto W, de Souza PVS, Oliveira ASB, Toro C, van Iersel J, Parson M, Harschnitz O, van den Berg LH, Veldink JH, Al-Chalabi A, Leigh PN, van Es MA (2021) Facial onset sensory and motor neuronopathy: new cases, cognitive changes, and pathophysiology. Neurol Clin Pract 11:147–157
Pinto S, Gromicho M, Oliveira Santos MO, Swash M, De Carvalho M (2023) Respiratory onset in amyotrophic lateral sclerosis: clinical features and spreading pattern. Amyotrophic Lateral Sclerosis Frontotemporal Degener 24:40–44
Shoesmith CL, Findlater K, Rowe A, Strong MJ (2007) Prognosis of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry 78:629–631
Agarwal S, Highton-Williamson E, Caga J, Matamala JM, Dharmadasa T, Howells J, Zoing MC, Shibuya K, Geevasinga N, Vucic S, Hodges JR, Ahmed RM, Kiernan MC (2018) Primary lateral sclerosis and the amyotrophic lateral sclerosis-frontotemporal dementia spectrum. J Neurol 265:1819–1828
Gazulla J, Ferrer I, Izquierdo-Alvarez S, Alvarez S, Sanchez-Alcudia R, Bestue-Cardiel M, Seral M, Benavente I, Sierra-Martinez E, Berciano J (2019) Hereditary primary lateral sclerosis and progressive nonfluent aphasia. J Neurol 266:1079–1090
Finegan E, Chipika RH, Li Hi Shing S, Doherty MA, Hengeveld JC, Vajda A, Donaghy C, McLaughlin RL, Pender N, Hardiman O, Bede P (2019) The clinical and radiological profile of primary lateral sclerosis: a population-based study. J Neurol 266:2718–2733
Canu E, Agosta F, Galantucci S, Chio A, Riva N, Silani V, Falini A, Comi G, Filippi M (2013) Extramotor damage is associated with cognition in primary lateral sclerosis patients. PLoS ONE 8:e82017
Kleinerova J, Tahedl M, Tan EL, Delaney S, Hengeveld JC, Doherty MA, McLaughlin RL, Hardiman O, Chang KM, Finegan E, Bede P (2024) Supra- and infra-tentorial degeneration patterns in primary lateral sclerosis: a multimodal longitudinal neuroradiology study. J Neurol
Rosenbohm A, Del Tredici K, Braak H, Huppertz HJ, Ludolph AC, Muller HP, Kassubek J (2022) Involvement of cortico-efferent tracts in flail arm syndrome: a tract-of-interest-based DTI study. J Neurol 269:2619–2626
Jaiser SR, Mitra D, Williams TL, Baker MR (2019) Mills’ syndrome revisited. J Neurol 266:667–679
Strong MJ, Abrahams S, Goldstein LH, Woolley S, McLaughlin P, Snowden J, Mioshi E, Roberts-South A, Benatar M, HortobaGyi T, Rosenfeld J, Silani V, Ince PG, Turner MR (2017) Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotrophic Lateral Sclerosis Frontotemporal Degener 18:153–174
Crockford C, Newton J, Lonergan K, Chiwera T, Booth T, Chandran S, Colville S, Heverin M, Mays I, Pal S, Pender N, Pinto-Grau M, Radakovic R, Shaw CE, Stephenson L, Swingler R, Vajda A, Al-Chalabi A, Hardiman O, Abrahams S (2018) ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology 91:e1370–e1380
Beeldman E, Raaphorst J, Klein Twennaar M, de Visser M, Schmand BA, de Haan RJ (2016) The cognitive profile of ALS: a systematic review and meta-analysis update. J Neurol Neurosurg Psychiatry 87:611–619
Watanabe Y, Raaphorst J, Izumi Y, Yoshino H, Ito S, Adachi T, Takigawa H, Masuda M, Atsuta N, Adachi Y, Isose S, Arai K, Yokota O, Oda M, Ogino M, Ichikawa H, Hasegawa K, Kimura H, Shimizu T, Aiba I, Yabe H, Kanba M, Kusumi K, Aoki T, Hiroe Y, Watanabe H, Nishiyama K, Nomoto M, Sobue G, Beeldman E, Hanajima R, Nakashima K, group A-F-Q-Jr (2020) Cognitive and behavioral status in Japanese ALS patients: a multicenter study. J Neurol 267:1321–1330
Strong MJ (2017) Revisiting the concept of amyotrophic lateral sclerosis as a multisystems disorder of limited phenotypic expression. Curr Opin Neurol 30:599–607
Phukan J, Elamin M, Bede P, Jordan N, Gallagher L, Byrne S, Lynch C, Pender N, Hardiman O (2012) The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry 83:102–108
Vinceti G, Olney N, Mandelli ML, Spina S, Hubbard HI, Santos-Santos MA, Watson C, Miller ZA, Lomen-Hoerth C, Nichelli P, Miller BL, Grinberg LT, Seeley WW, Gorno-Tempini ML (2019) Primary progressive aphasia and the FTD-MND spectrum disorders: clinical, pathological, and neuroimaging correlates. Amyotroph Lateral Scler Frontotemporal Degener 20:146–158
Strong MJ, Grace GM, Freedman M, Lomen-Hoerth C, Woolley S, Goldstein LH, Murphy J, Shoesmith C, Rosenfeld J, Leigh PN, Bruijn L, Ince P, Figlewicz D (2009) Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 10:131–146
Consonni M, Catricalà E, Dalla Bella E, Gessa VC, Lauria G, Cappa SF (2016) Beyond the consensus criteria: multiple cognitive profiles in amyotrophic lateral sclerosis? Cortex 81:162–167
Maier PM, Iggena D, Meyer T, Finke C, Ploner CJ (2023) Memory-guided navigation in amyotrophic lateral sclerosis. J Neurol 270:4031–4040
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gorno-Tempini ML, Rosen H, Prioleau-Latham CE, Lee A, Kipps CM, Lillo P, Piguet O, Rohrer JD, Rossor MN, Warren JD, Fox NC, Galasko D, Salmon DP, Black SE, Mesulam M, Weintraub S, Dickerson BC, Diehl-Schmid J, Pasquier F, Deramecourt V, Lebert F, Pijnenburg Y, Chow TW, Manes F, Grafman J, Cappa SF, Freedman M, Grossman M, Miller BL (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134:2456–2477
Long Z, Irish M, Foxe D, Hodges JR, Piguet O, Burrell JR (2021) Heterogeneity of behavioural and language deficits in FTD-MND. J Neurol 268:2876–2889
Elamin M, Bede P, Byrne S, Jordan N, Gallagher L, Wynne B, O’Brien C, Phukan J, Lynch C, Pender N, Hardiman O (2013) Cognitive changes predict functional decline in ALS: a population-based longitudinal study. Neurology 80:1590–1597
Schrempf T, Finsel J, Uttner I, Ludolph AC, Lulé D (2022) Neuropsychological deficits have only limited impact on psychological well-being in amyotrophic lateral sclerosis. J Neurol 269:1369–1374
Spataro R, La Bella V (2021) The capacity to consent to treatment in amyotrophic lateral sclerosis: a preliminary report. J Neurol 268:219–226
Burke T, Hardiman O, Pinto-Grau M, Lonergan K, Heverin M, Tobin K, Staines A, Galvin M, Pender N (2018) Longitudinal predictors of caregiver burden in amyotrophic lateral sclerosis: a population-based cohort of patient-caregiver dyads. J Neurol 265:793–808
Burke T, Elamin M, Galvin M, Hardiman O, Pender N (2015) Caregiver burden in amyotrophic lateral sclerosis: a cross-sectional investigation of predictors. J Neurol 262:1526–1532
Consonni M, Telesca A, Dalla Bella E, Bersano E, Lauria G (2020) Amyotrophic lateral sclerosis patients’ and caregivers’ distress and loneliness during COVID-19 lockdown. J Neurol. https://doi.org/10.1007/s00415-020-10080-6
Caga J, Hsieh S, Highton-Williamson E, Zoing MC, Ramsey E, Devenney E, Ahmed RM, Kiernan MC (2018) Apathy and its impact on patient outcome in amyotrophic lateral sclerosis. J Neurol 265:187–193
Frith CD, Frith U (2007) Social cognition in humans. Curr Biol 17:R724-732
Arioli M, Crespi C, Canessa N (2018) Social cognition through the lens of cognitive and clinical neuroscience. Biomed Res Int 2018:4283427
Carelli L, Solca F, Tagini S, Torre S, Verde F, Ticozzi N, Consonni M, Ferrucci R, Pravettoni G, Poletti B, Silani V (2021) Emotional processing and experience in amyotrophic lateral sclerosis: a systematic and critical review. Brain Sci. https://doi.org/10.3390/brainsci11101356
Bora E (2017) Meta-analysis of social cognition in amyotrophic lateral sclerosis. Cortex 88:1–7
Girardi A, MacPherson SE, Abrahams S (2011) Deficits in emotional and social cognition in amyotrophic lateral sclerosis. Neuropsychology 25:53–65
Crespi C, Cerami C, Dodich A, Canessa N, Arpone M, Iannaccone S, Corbo M, Lunetta C, Scola E, Falini A, Cappa SF (2014) Microstructural white matter correlates of emotion recognition impairment in Amyotrophic Lateral Sclerosis. Cortex 53:1–8
van der Hulst EJ, Bak TH, Abrahams S (2015) Impaired affective and cognitive theory of mind and behavioural change in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 86:1208–1215
Cerami C, Dodich A, Canessa N, Crespi C, Iannaccone S, Corbo M, Lunetta C, Consonni M, Scola E, Falini A, Cappa SF (2014) Emotional empathy in amyotrophic lateral sclerosis: a behavioural and voxel-based morphometry study. Amyotrophic Lateral Sclerosis Frontotemporal Degener 15:21–29
Cavallo M, Adenzato M, Macpherson SE, Karwig G, Enrici I, Abrahams S (2011) Evidence of social understanding impairment in patients with amyotrophic lateral sclerosis. PLoS ONE 6:e25948
Stern Y (2006) Cognitive reserve and Alzheimer disease. Alzheimer Dis Assoc Disord 20:S69-74
Stern Y (2009) Cognitive reserve. Neuropsychologia 47:2015–2028
Stern Y, Arenaza-Urquijo EM, Bartrés-Faz D, Belleville S, Cantilon M, Chetelat G, Ewers M, Franzmeier N, Kempermann G, Kremen WS, Okonkwo O, Scarmeas N, Soldan A, Udeh-Momoh C, Valenzuela M, Vemuri P, Vuoksimaa E, the Reserve RsaPFPEDaCFW (2020) Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimers Dement 16:1305–1311
Rhodes E, Alfa S, Jin HA, Massimo L, Elman L, Amado D, Baer M, Quinn C, McMillan CT (2024) Cognitive reserve in ALS: the role of occupational skills and requirements. Amyotrophic Lateral Sclerosis Frontotemporal Degener. https://doi.org/10.1080/21678421.2024.2336113
Canosa A, Palumbo F, Iazzolino B, Peotta L, Di Pede F, Manera U, Vasta R, Grassano M, Solero L, Arena V, Moglia C, Calvo A, Chiò A, Pagani M (2021) The interplay among education, brain metabolism, and cognitive impairment suggests a role of cognitive reserve in Amyotrophic Lateral Sclerosis. Neurobiol Aging 98:205–213
Temp AGM, Prudlo J, Vielhaber S, Machts J, Hermann A, Teipel SJ, Kasper E (2021) Cognitive reserve and regional brain volume in amyotrophic lateral sclerosis. Cortex 139:240–248
Consonni M, Dalla Bella E, Bersano E, Telesca A, Lauria G (2021) Cognitive reserve is associated with altered clinical expression in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis Frontotemporal Degener 22:237–247
Costello E, Rooney J, Pinto-Grau M, Burke T, Elamin M, Bede P, McMackin R, Dukic S, Vajda A, Heverin M, Hardiman O, Pender N (2021) Cognitive reserve in amyotrophic lateral sclerosis (ALS): a population-based longitudinal study. J Neurol Neurosurg Psychiatry 92:460–465
Temp AGM, Kasper E, Machts J, Vielhaber S, Teipel S, Hermann A, Prudlo J (2022) Cognitive reserve protects ALS-typical cognitive domains: a longitudinal study. Ann Clin Transl Neurol 9:1212–1223
Benatar M, Wuu J, McHutchison C, Postuma RB, Boeve BF, Petersen R, Ross CA, Rosen H, Arias JJ, Fradette S, McDermott MP, Shefner J, Stanislaw C, Abrahams S, Cosentino S, Andersen PM, Finkel RS, Granit V, Grignon AL, Rohrer JD, McMillan CT, Grossman M, Al-Chalabi A, Turner MR, Workshop FIP-SA (2022) Preventing amyotrophic lateral sclerosis: insights from pre-symptomatic neurodegenerative diseases. Brain 145:27–44
Behler A, Knehr A, Finsel J, Kunz MS, Lang C, Müller K, Müller HP, Pinkhardt EH, Ludolph AC, Lulé D, Kassubek J (2021) Eye movement alterations in presymptomatic C9orf72 expansion gene carriers. J Neurol 268:3390–3399
Lulé DE, Müller HP, Finsel J, Weydt P, Knehr A, Winroth I, Andersen P, Weishaupt J, Uttner I, Kassubek J, Ludolph AC (2020) Deficits in verbal fluency in presymptomatic. J Neurol Neurosurg Psychiatry 91:1195–1200
Chiò A, Moglia C, Canosa A, Manera U, Vasta R, Brunetti M, Barberis M, Corrado L, D'Alfonso S, Bersano E, Sarnelli MF, Solara V, Zucchetti JP, Peotta L, Iazzolino B, Mazzini L, Mora G, Calvo A (2019) Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology
Iazzolino B, Peotta L, Zucchetti JP, Canosa A, Manera U, Vasta R, Grassano M, Palumbo F, Brunetti M, Barberis M, Sbaiz L, Moglia C, Calvo A, Chiò A (2021) Differential neuropsychological profile of patients with amyotrophic lateral sclerosis with and without. Neurology 96:e141–e152
Canosa A, Pagani M, Brunetti M, Barberis M, Iazzolino B, Ilardi A, Cammarosano S, Manera U, Moglia C, Calvo A, Cistaro A, Chiò A (2019) Correlation between Apolipoprotein E genotype and brain metabolism in amyotrophic lateral sclerosis. Eur J Neurol 26:306–312
Piao YS, Wakabayashi K, Kakita A, Yamada M, Hayashi S, Morita T, Ikuta F, Oyanagi K, Takahashi H (2003) Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol 13:10–22
Cividini C, Basaia S, Spinelli EG, Canu E, Castelnovo V, Riva N, Cecchetti G, Caso F, Magnani G, Falini A, Filippi M, Agosta F (2022) Amyotrophic lateral sclerosis-frontotemporal dementia: shared and divergent neural correlates across the clinical spectrum. Neurology 98:e402–e415
Consonni M, Contarino VE, Catricalà E, Dalla Bella E, Pensato V, Gellera C, Lauria G, Cappa SF (2018) Cortical markers of cognitive syndromes in amyotrophic lateral sclerosis. Neuroimage Clin 19:675–682
Consonni M, Cappa SF, Dalla Bella E, Contarino VE, Lauria G (2019) Cortical correlates of behavioural change in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 90:380–386
Braak H, Brettschneider J, Ludolph AC, Lee VM, Trojanowski JQ, Del Tredici K (2013) Amyotrophic lateral sclerosis–a model of corticofugal axonal spread. Nat Rev Neurol 9:708–714
Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, Suh E, Van Deerlin VM, Wood EM, Baek Y, Kwong L, Lee EB, Elman L, McCluskey L, Fang L, Feldengut S, Ludolph AC, Lee VM, Braak H, Trojanowski JQ (2013) Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38
Castelnovo V, Canu E, Calderaro D, Riva N, Poletti B, Basaia S, Solca F, Silani V, Filippi M, Agosta F (2020) Progression of brain functional connectivity and frontal cognitive dysfunction in ALS. NeuroImage Clinical 28:102509
Spinelli EG, Ghirelli A, Riva N, Canu E, Castelnovo V, Domi T, Pozzi L, Carrera P, Silani V, Chio A, Filippi M, Agosta F (2022) Profiling morphologic MRI features of motor neuron disease caused by TARDBP mutations. Front Neurol 13:931006
Lulé D, Böhm S, Müller HP, Aho-Özhan H, Keller J, Gorges M, Loose M, Weishaupt JH, Uttner I, Pinkhardt E, Kassubek J, Del Tredici K, Braak H, Abrahams S, Ludolph AC (2018) Cognitive phenotypes of sequential staging in amyotrophic lateral sclerosis. Cortex 101:163–171
Consonni M, Dalla Bella E, Contarino VE, Bersano E, Lauria G (2020) Cortical thinning trajectories across disease stages and cognitive impairment in amyotrophic lateral sclerosis. Cortex
Takeda T, Kokubun S, Saito Y, Tsuneyama A, Ishikawa A, Isose S, Ito K, Arai K, Koreki A, Sugiyama A, Kuwabara S, Honda K (2022) Progressive medial temporal degeneration with TDP-43 pathology is associated with upper limb and bulbar onset types of amyotrophic lateral sclerosis. J Neurol 269:5497–5509
McHutchison CA, Leighton DJ, McIntosh A, Cleary E, Warner J, Porteous M, Chandran S, Pal S, Abrahams S (2020) Relationship between neuropsychiatric disorders and cognitive and behavioural change in MND. J Neurol Neurosurg Psychiatry 91:245–253
Mioshi E, Caga J, Lillo P, Hsieh S, Ramsey E, Devenney E, Hornberger M, Hodges JR, Kiernan MC (2014) Neuropsychiatric changes precede classic motor symptoms in ALS and do not affect survival. Neurology 82:149–155
Zucchi E, Ticozzi N, Mandrioli J (2019) Psychiatric symptoms in amyotrophic lateral sclerosis: beyond a motor neuron disorder. Front Neurosci 13:175
Carvalho TL, de Almeida LM, Lorega CM, Barata MF, Ferreira ML, de Brito-Marques PR, CaC C (2016) Depression and anxiety in individuals with amyotrophic lateral sclerosis: a systematic review. Trends Psychiatry Psychother 38:1–5
Devenney EM, Tu S, Caga J, Ahmed RM, Ramsey E, Zoing M, Kwok J, Halliday GM, Piguet O, Hodges JR, Kiernan MC (2021) Neural mechanisms of psychosis vulnerability and perceptual abnormalities in the ALS-FTD spectrum. Ann Clin Transl Neurol 8:1576–1591
Abrahams S, Newton J, Niven E, Foley J, Bak TH (2014) Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener 15:9–14
Greco LC, Lizio A, Casiraghi J, Sansone VA, Tremolizzo L, Riva N, Solca F, Torre S, Ticozzi N, Filippi M, Silani V, Poletti B, Lunetta C (2022) A preliminary comparison between ECAS and ALS-CBS in classifying cognitive-behavioural phenotypes in a cohort of non-demented amyotrophic lateral sclerosis patients. J Neurol 269:1899–1904
Beeldman E, Govaarts R, de Visser M, van Es MA, Pijnenburg YAL, Schmand BA, Raaphorst J (2021) Screening for cognition in amyotrophic lateral sclerosis: test characteristics of a new screen. J Neurol 268:2533–2540
Beswick E, Forbes D, Hassan Z, Wong C, Newton J, Carson A, Abrahams S, Chandran S, Pal S (2022) A systematic review of non-motor symptom evaluation in clinical trials for amyotrophic lateral sclerosis. J Neurol 269:411–426
Beswick E, Park E, Wong C, Mehta AR, Dakin R, Chandran S, Newton J, Carson A, Abrahams S, Pal S (2021) A systematic review of neuropsychiatric and cognitive assessments used in clinical trials for amyotrophic lateral sclerosis. J Neurol 268:4510–4521
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Ferrari G, Grisan E, Scarpa F, Fazio R, Comola M, Quattrini A, Comi G, Rama P, Riva N (2014) Corneal confocal microscopy reveals trigeminal small sensory fiber neuropathy in amyotrophic lateral sclerosis. Front Aging Neurosci 6:278
Calvo A, Chiò A, Pagani M, Cammarosano S, Dematteis F, Moglia C, Solero L, Manera U, Martone T, Brunetti M, Balma M, Castellano G, Barberis M, Cistaro A, Artusi CA, Vasta R, Montanaro E, Romagnolo A, Iazzolino B, Canosa A, Carrara G, Valentini C, Li T-Q, Nobili F, Lopiano L, Rizzone MG (2019) Parkinsonian traits in amyotrophic lateral sclerosis (ALS): a prospective population-based study. J Neurol 266:1633–1642
Pupillo E, Bianchi E, Messina P, Chiveri L, Lunetta C, Corbo M, Filosto M, Lorusso L, Marin B, Mandrioli J, Riva N, Sasanelli F, Tremolizzo L, Beghi E, Eurals C (2015) Extrapyramidal and cognitive signs in amyotrophic lateral sclerosis: a population based cross-sectional study. Amyotrophic Lateral Sclerosis Frontotemporal Degener 16:324–330
Feron M, Couillandre A, Mseddi E, Termoz N, Abidi M, Bardinet E, Delgadillo D, Lenglet T, Querin G, Welter M-L, Le Forestier N, Salachas F, Bruneteau G, Del Mar AM, Debs R, Lacomblez L, Meininger V, Pélégrini-Issac M, Bede P, Pradat P-F, Marco G (2018) Extrapyramidal deficits in ALS: a combined biomechanical and neuroimaging study. J Neurol 265:2125–2136
Schönecker S, Hell F, Bötzel K, Wlasich E, Ackl N, Süßmair C, German FC, Otto M, Anderl-Straub S, Ludolph A, Kassubek J, Huppertz H-J, Diehl-Schmid J, Riedl L, Roßmeier C, Fassbender K, Lyros E, Kornhuber J, Oberstein TJ, Fliessbach K, Schneider A, Schroeter ML, Prudlo J, Lauer M, Jahn H, Levin J, Danek A (2019) The applause sign in frontotemporal lobar degeneration and related conditions. J Neurol 266:330–338
Bede P, Murad A, Lope J, Hardiman O, Chang KM (2022) Clusters of anatomical disease-burden patterns in ALS: a data-driven approach confirms radiological subtypes. J Neurol 269:4404–4413
Westeneng HJ, Walhout R, Straathof M, Schmidt R, Hendrikse J, Veldink JH, van den Heuvel MP, van den Berg LH (2016) Widespread structural brain involvement in ALS is not limited to the C9orf72 repeat expansion. J Neurol Neurosurg Psychiatry 87:1354–1360
Spinelli EG, Ghirelli A, Basaia S, Cividini C, Riva N, Canu E, Castelnovo V, Domi T, Magnani G, Caso F, Caroppo P, Prioni S, Rossi G, Tremolizzo L, Appollonio I, Silani V, Carrera P, Filippi M, Agosta F (2021) Structural MRI signatures in genetic presentations of the frontotemporal dementia/motor neuron disease spectrum. Neurology 97:e1594–e1607
Bede P, Chipika RH, Christidi F, Hengeveld JC, Karavasilis E, Argyropoulos GD, Lope J, Li Hi Shing S, Velonakis G, Dupuis L, Doherty MA, Vajda A, McLaughlin RL, Hardiman O (2021) Genotype-associated cerebellar profiles in ALS: focal cerebellar pathology and cerebro-cerebellar connectivity alterations. J Neurol Neurosurg Psychiatry 92:1197–1205
Finegan E, Siah WF, Li Hi Shing S, Chipika RH, Hardiman O, Bede P (2022) Cerebellar degeneration in primary lateral sclerosis: an under-recognized facet of PLS. Amyotrophic Lateral Sclerosis Frontotemporal Degener 23:542–553
Rohani M, Meysamie A, Zamani B, Sowlat MM, Akhoundi FH (2018) Reduced retinal nerve fiber layer (RNFL) thickness in ALS patients: a window to disease progression. J Neurol 265:1557–1562
Miscioscia A, Puthenparampil M, Blasi L, Rinaldi F, Perini P, Sorarù G, Gallo P (2023) Neurodegeneration in the retina of motoneuron diseases: a longitudinal study in amyotrophic lateral sclerosis and Kennedy’s disease. J Neurol 270:4478–4486
Cerveró A, Casado A, Riancho J (2021) Retinal changes in amyotrophic lateral sclerosis: looking at the disease through a new window. J Neurol 268:2083–2089
Abdelhak A, Hübers A, Böhm K, Ludolph AC, Kassubek J, Pinkhardt EH (2018) In vivo assessment of retinal vessel pathology in amyotrophic lateral sclerosis. J Neurol 265:949–953
Ren Y, Liu W, Li Y, Sun B, Li Y, Yang F, Wang H, Li M, Cui F, Huang X (2018) Cutaneous somatic and autonomic nerve TDP-43 deposition in amyotrophic lateral sclerosis. J Neurol 265:1753–1763
Truini A, Biasiotta A, Onesti E, Di Stefano G, Ceccanti M, La Cesa S, Pepe A, Giordano C, Cruccu G, Inghilleri M (2015) Small-fibre neuropathy related to bulbar and spinal-onset in patients with ALS. J Neurol 262:1014–1018
Taga A, Schito P, Trapasso MC, Zinno L, Pavesi G (2019) Pain at the onset of amyotrophic lateral sclerosis: a cross-sectional study. Clin Neurol Neurosurg 186:105540
Weis J, Katona I, Muller-Newen G, Sommer C, Necula G, Hendrich C, Ludolph AC, Sperfeld AD (2011) Small-fiber neuropathy in patients with ALS. Neurology 76:2024–2029
Gregory R, Mills K, Donaghy M (1993) Progressive sensory nerve dysfunction in amyotrophic lateral sclerosis: a prospective clinical and neurophysiological study. J Neurol 240:309–314
Dubbioso R, Provitera V, Pacella D, Santoro L, Manganelli F, Nolano M (2023) Autonomic dysfunction is associated with disease progression and survival in amyotrophic lateral sclerosis: a prospective longitudinal cohort study. J Neurol 270:4968–4977
Napoli G, Rubin M, Cutillo G, Schito P, Russo T, Quattrini A, Filippi M, Riva N (2023) Tako-tsubo syndrome in amyotrophic lateral sclerosis: single-center case series and brief literature review. Int J Mol Sci 24:12096
De Marchi F, Franjkic T, Schito P, Russo T, Nimac J, Chami AA, Mele A, Vidatic L, Kriz J, Julien J-P, Apic G, Russell RB, Rogelj B, Cannon JR, Baralle M, Agosta F, Hecimovic S, Mazzini L, Buratti E, Munitic I (2023) Emerging trends in the field of inflammation and proteinopathy in ALS/FTD spectrum disorder. Biomedicines 11:1599
Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, Traynor BG, Eurals C (2009) Prognostic factors in ALS: a critical review. Amyotrophic Lateral Sclerosis Off Publ World Fed Neurol Res Group Motor Neuron Dis 10:310–323
Calvo A, Moglia C, Lunetta C, Marinou K, Ticozzi N, Ferrante GD, Scialo C, Soraru G, Trojsi F, Conte A, Falzone YM, Tortelli R, Russo M, Chio A, Sansone VA, Mora G, Silani V, Volanti P, Caponnetto C, Querin G, Monsurro MR, Sabatelli M, Riva N, Logroscino G, Messina S, Fini N, Mandrioli J (2017) Factors predicting survival in ALS: a multicenter Italian study. J Neurol 264:54–63
van Eijk RPA, de Jongh AD, Nikolakopoulos S, McDermott CJ, Eijkemans MJC, Roes KCB, van den Berg LH (2021) An old friend who has overstayed their welcome: the ALSFRS-R total score as primary endpoint for ALS clinical trials. Amyotrophic Lateral Sclerosis Frontotemporal Degener 22:300–307
Fournier CN, Bedlack R, Quinn C, Russell J, Beckwith D, Kaminski KH, Tyor W, Hertzberg V, James V, Polak M, Glass JD (2020) Development and validation of the rasch-built overall amyotrophic lateral sclerosis disability scale (ROADS). JAMA Neurol 77:480–488
Creemers H, Grupstra H, Nollet F, van den Berg LH, Beelen A (2015) Prognostic factors for the course of functional status of patients with ALS: a systematic review. J Neurol 262:1407–1423
de Jongh AD, van Eijk RPA, Bakker LA, Bunte TM, Beelen A, van der Meijden C, van Es MA, Visser-Meily JMA, Kruitwagen ET, Veldink JH, van den Berg LH (2023) Development of a rasch-built amyotrophic lateral sclerosis impairment multidomain scale to measure disease progression in ALS. Neurology 101:e602–e612
Roche JC, Rojas-Garcia R, Scott KM, Scotton W, Ellis CE, Burman R, Wijesekera L, Turner MR, Leigh PN, Shaw CE, Al-Chalabi A (2012) A proposed staging system for amyotrophic lateral sclerosis. Brain J Neurol 135:847–852
Chio A, Hammond ER, Mora G, Bonito V, Filippini G (2015) Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 86:38–44
Luna J, Couratier P, Lahmadi S, Lautrette G, Fontana A, Tortelli R, Logroscino G, Preux PM, Copetti M, Benoit M (2021) Comparison of the ability of the King’s and MiToS staging systems to predict disease progression and survival in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis Frontotemporal Degener 22:478–485
Fang T, Al Khleifat A, Stahl DR, Lazo La Torre C, Murphy C, Uk-Mnd Lical S, Young C, Shaw PJ, Leigh PN, Al-Chalabi A (2017) Comparison of the King’s and MiToS staging systems for ALS. Amyotrophic Lateral Sclerosis Frontotemporal Degener 18:227–232
Dorst J, Chen L, Rosenbohm A, Dreyhaupt J, Hubers A, Schuster J, Weishaupt JH, Kassubek J, Gess B, Meyer T, Weyen U, Hermann A, Winkler J, Grehl T, Hagenacker T, Lingor P, Koch JC, Sperfeld A, Petri S, Grosskreutz J, Metelmann M, Wolf J, Winkler AS, Klopstock T, Boentert M, Johannesen S, Storch A, Schrank B, Zeller D, Liu XL, Tang L, Fan DS, Ludolph AC (2019) Prognostic factors in ALS: a comparison between Germany and China. J Neurol 266:1516–1525
Leighton DJ, Ansari M, Newton J, Parry D, Cleary E, Colville S, Stephenson L, Larraz J, Johnson M, Beswick E, Wong M, Gregory J, Carod Artal J, Davenport R, Duncan C, Morrison I, Smith C, Swingler R, Deary IJ, Porteous M, Aitman TJ, Chandran S, Gorrie GH, Pal S, Lothian Birth Cohorts G, the C-MNDC, (2023) Genotype-phenotype characterisation of long survivors with motor neuron disease in Scotland. J Neurol 270:1702–1712
Desport JC, Preux PM, Truong CT, Courat L, Vallat JM, Couratier P (2000) Nutritional assessment and survival in ALS patients. Amyotrophic Lateral Sclerosis Other Motor Neur Disord Off Publ Fed Neurol Res Group Motor Neur Dis 1:91–96
Kasarskis EJ, Berryman S, Vanderleest JG, Schneider AR, McClain CJ (1996) Nutritional status of patients with amyotrophic lateral sclerosis: relation to the proximity of death. Am J Clin Nutr 63:130–137
Jawaid A, Murthy SB, Wilson AM, Qureshi SU, Amro MJ, Wheaton M, Simpson E, Harati Y, Strutt AM, York MK, Schulz PE (2010) A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotrophic Lateral Sclerosis Off Publ World Fed Neurol Res Group Motor Neur Dis 11:542–548
Reich-Slotky R, Andrews J, Cheng B, Buchsbaum R, Levy D, Kaufmann P, Thompson JL (2013) Body mass index (BMI) as predictor of ALSFRS-R score decline in ALS patients. Amyotrophic Lateral Sclerosis Frontotemporal Degener 14:212–216
Ahmed RM, Mioshi E, Caga J, Shibata M, Zoing M, Bartley L, Piguet O, Hodges JR, Kiernan MC (2014) Body mass index delineates ALS from FTD: implications for metabolic health. J Neurol 261:1774–1780
Shimizu T, Nakayama Y, Matsuda C, Haraguchi M, Bokuda K, Ishikawa-Takata K, Kawata A, Isozaki E (2019) Prognostic significance of body weight variation after diagnosis in ALS: a single-centre prospective cohort study. J Neurol 266:1412–1420
Cattaneo M, Jesus P, Lizio A, Fayemendy P, Guanziroli N, Corradi E, Sansone V, Leocani L, Filippi M, Riva N, Corcia P, Couratier P, Lunetta C (2022) The hypometabolic state: a good predictor of a better prognosis in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 93:41–47
He J, Fu J, Zhao W, Ren C, Liu P, Chen L, Li D, Tang L, Zhou L, Zhang Y, Ma X, Zhang G, Li N, Fan D (2022) Hypermetabolism associated with worse prognosis of amyotrophic lateral sclerosis. J Neurol 269:1447–1455
Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, Kalra S, Katz JS, Mitsumoto H, Rosenfeld J, Shoesmith C, Strong MJ, Woolley SC (2009) Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 73:1227–1233
Calvo A, Vasta R, Moglia C, Matteoni E, Canosa A, Mattei A, La Mancusa C, Focaraccio L, Mazzini L, Chio A, D’Ovidio F, Manera U (2020) Prognostic role of slow vital capacity in amyotrophic lateral sclerosis. J Neurol 267:1615–1621
Engel M, Glatz C, Helmle C, Young P, Drager B, Boentert M (2021) Respiratory parameters on diagnostic sleep studies predict survival in patients with amyotrophic lateral sclerosis. J Neurol 268:4321–4331
Alarcan H, Cotet C, Lepine N, Morel J, Vourc’h P, Andres C, Corcia P, Blasco H (2023) Evaluation of arterial blood gas parameters as prognostic markers in amyotrophic lateral sclerosis. Eur J Neurol Off J Eur Fed Neurol Soc 30:1611–1618
Miranda B, Gromicho M, Pereira M, Pinto S, Swash M, de Carvalho M (2020) Diaphragmatic CMAP amplitude from phrenic nerve stimulation predicts functional decline in ALS. J Neurol 267:2123–2129
Xu L, He B, Zhang Y, Chen L, Fan D, Zhan S, Wang S (2021) Prognostic models for amyotrophic lateral sclerosis: a systematic review. J Neurol 268:3361–3370
Elamin M, Bede P, Montuschi A, Pender N, Chio A, Hardiman O (2015) Predicting prognosis in amyotrophic lateral sclerosis: a simple algorithm. J Neurol 262:1447–1454
Kimura F, Fujimura C, Ishida S, Nakajima H, Furutama D, Uehara H, Shinoda K, Sugino M, Hanafusa T (2006) Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 66:265–267
Czaplinski A, Yen AA, Simpson EP, Appel SH (2006) Predictability of disease progression in amyotrophic lateral sclerosis. Muscle Nerve 34:702–708
del Aguila MA, Longstreth WT Jr, McGuire V, Koepsell TD, van Belle G (2003) Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology 60:813–819
Elamin M, Phukan J, Bede P, Jordan N, Byrne S, Pender N, Hardiman O (2011) Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology 76:1263–1269
Montuschi A, Iazzolino B, Calvo A, Moglia C, Lopiano L, Restagno G, Brunetti M, Ossola I, Lo Presti A, Cammarosano S, Canosa A, Chio A (2015) Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J Neurol Neurosurg Psychiatry 86:168–173
Testa D, Lovati R, Ferrarini M, Salmoiraghi F, Filippini G (2004) Survival of 793 patients with amyotrophic lateral sclerosis diagnosed over a 28-year period. Amyotrophic Lateral Sclerosis Other Motor Neuron Disorders Off Publ World Fed Neurol Res Group Motor Neur Dis 5:208–212
Chio A, Mora G, Leone M, Mazzini L, Cocito D, Giordana MT, Bottacchi E, Mutani R, Piemonte VD, Register for ALS (2002) Early symptom progression rate is related to ALS outcome: a prospective population-based study. Neurology 59:99–103
Grollemund V, Le Chat G, Secchi-Buhour MS, Delbot F, Pradat-Peyre JF, Bede P, Pradat PF (2021) Manifold learning for amyotrophic lateral sclerosis functional loss assessment : development and validation of a prognosis model. J Neurol 268:825–850
Tavazzi E, Daberdaku S, Zandona A, Vasta R, Nefussy B, Lunetta C, Mora G, Mandrioli J, Grisan E, Tarlarini C, Calvo A, Moglia C, Drory V, Gotkine M, Chio A, Di Camillo B, VdARfALSftERRfALS P (2022) Predicting functional impairment trajectories in amyotrophic lateral sclerosis: a probabilistic, multifactorial model of disease progression. J Neurol 269:3858–3878
Lehn AC, Dionisio S, Airey CA, Brown H, Blum S, Henderson R (2014) The tibialis anterior response revisited. J Neurol 261:1340–1343
Brooks BR, Miller RG, Swash M, Munsat TL (2000) El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis Other Motor Neuron Disorders Off Publ World Fed Neurol Res Group Motor Neur Dis 1:293–299
Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved?
de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J, Mills K, Mitsumoto H, Nodera H, Shefner J, Swash M (2008) Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol Off J Int Fed Clin Neurophysiol 119:497–503
Brooks BR, Miller RG, Swash M, Munsat TL (2000) El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1:293–299
Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman OM (2000) Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: a population-based study. Arch Neurol 57:1171–1176
Shefner JM, Al-Chalabi A, Baker MR, Cui LY, de Carvalho M, Eisen A, Grosskreutz J, Hardiman O, Henderson R, Matamala JM, Mitsumoto H, Paulus W, Simon N, Swash M, Talbot K, Turner MR, Ugawa Y, van den Berg LH, Verdugo R, Vucic S, Kaji R, Burke D, Kiernan MC (2020) A proposal for new diagnostic criteria for ALS. Clin Neurophysiol Off J Int Fed Clin Neurophysiol 131:1975–1978
Strong MJ, Abrahams S, Goldstein LH, Woolley S, McLaughlin P, Snowden J, Mioshi E, Roberts-South A, Benatar M, HortobáGyi T, Rosenfeld J, Silani V, Ince PG, Turner MR (2017) Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotrophic Lateral Sclerosis Frontotemporal Degener 18:153–174
Davenport RJ, Swingler RJ, Chancellor AM, Warlow CP (1996) Avoiding false positive diagnoses of motor neuron disease: lessons from the Scottish Motor Neuron Disease Register. J Neurol Neurosurg Psychiatry 60:147–151
Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O (2000) Amyotrophic lateral sclerosis mimic syndromes: a population-based study. Arch Neurol 57:109–113
Belsh JM, Schiffman PL (1996) The amyotrophic lateral sclerosis (ALS) patient perspective on misdiagnosis and its repercussions. J Neurol Sci 139(Suppl):110–116
Boelmans K, Kaufmann J, Schmelzer S, Vielhaber S, Kornhuber M, Munchau A, Zierz S, Gaul C (2013) Hirayama disease is a pure spinal motor neuron disorder–a combined DTI and transcranial magnetic stimulation study. J Neurol 260:540–548
Hellmann MA, Kakhlon O, Landau EH, Sadeh M, Giladi N, Schlesinger I, Kidron D, Abramsky O, Reches A, Argov Z, Rabey JM, Chapman J, Rosenmann H, Gal A, Moshe Gomori J, Meiner V, Lossos A (2015) Frequent misdiagnosis of adult polyglucosan body disease. J Neurol. https://doi.org/10.1007/s00415-015-7859-4
Grunseich C, Schindler AB, Chen KL, Bakar D, Mankodi A, Traslavina R, Ray-Chaudhury A, Lehky TJ, Baker EH, Maragakis NJ, Tifft CJ, Fischbeck KH (2015) Peripheral neuropathy in a family with Sandhoff disease and SH3TC2 deficiency. J Neurol 262:1066–1068
Shibata M, Kasahara H, Makioka K, Ikeda M, Nagashima K, Fujita Y, Ikeda Y (2020) Neurogenic calf amyotrophy with CK elevation by entrapment radiculopathy; clinical, radiological, and pathological analyses of 18 cases. J Neurol 267:3528–3540
Morishima R, Takai K, Ando T, Nakata Y, Shimizu T, Taniguchi M (2019) Brachial multisegmental amyotrophy caused by cervical anterior horn cell disorder associated with a spinal CSF leak: a report of five cases. J Neurol 266:2679–2684
Riva N, Gallia F, Iannaccone S, Corbo M, Terenghi F, Lazzerini A, Cerri F, Comi G, Quattrini A, Nobile-Orazio E (2011) Chronic motor axonal neuropathy. J Periph Nerv Syst JPNS 16:341–346
Riva N, Iannaccone S, Corbo M, Casellato C, Sferrazza B, Lazzerini A, Scarlato M, Cerri F, Previtali SC, Nobile-Orazio E, Comi G, Quattrini A (2011) Motor nerve biopsy: clinical usefulness and histopathological criteria. Ann Neurol 69:197–201
Riva N, Gentile F, Cerri F, Gallia F, Podini P, Dina G, Falzone YM, Fazio R, Lunetta C, Calvo A, Logroscino G, Lauria G, Corbo M, Iannaccone S, Chio A, Lazzerini A, Nobile-Orazio E, Filippi M, Quattrini A (2022) Phosphorylated TDP-43 aggregates in peripheral motor nerves of patients with amyotrophic lateral sclerosis. Brain J Neurol 145:276–284
Zoccolella S, Mastronardi A, Scarafino A, Iliceto G, D’Errico E, Fraddosio A, Tempesta I, Morea A, Scaglione G, Introna A, Simone IL (2020) Motor-evoked potentials in amyotrophic lateral sclerosis: potential implications in detecting subclinical UMN involvement in lower motor neuron phenotype. J Neurol 267:3689–3695
Pestronk A, Cornblath DR, Ilyas AA, Baba H, Quarles RH, Griffin JW, Alderson K, Adams RN (1988) A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol 24:73–78
Cats EA, Jacobs BC, Yuki N, Tio-Gillen AP, Piepers S, Franssen H, van Asseldonk JT, van den Berg LH, van der Pol WL (2010) Multifocal motor neuropathy: association of anti-GM1 IgM antibodies with clinical features. Neurology 75:1961–1967
Vlam L, Piepers S, Sutedja NA, Jacobs BC, Tio-Gillen AP, Stam M, Franssen H, Veldink JH, Cats EA, Notermans NC, Bloem AC, Wadman RI, van der Pol WL, van den Berg LH (2015) Association of IgM monoclonal gammopathy with progressive muscular atrophy and multifocal motor neuropathy: a case-control study. J Neurol 262:666–673
Rossi D, Volanti P, Brambilla L, Colletti T, Spataro R, La Bella V (2018) CSF neurofilament proteins as diagnostic and prognostic biomarkers for amyotrophic lateral sclerosis. J Neurol 265:510–521
Schreiber S, Spotorno N, Schreiber F, Acosta-Cabronero J, Kaufmann J, Machts J, Debska-Vielhaber G, Garz C, Bittner D, Hensiek N, Dengler R, Petri S, Nestor PJ, Vielhaber S (2018) Significance of CSF NfL and tau in ALS. J Neurol 265:2633–2645
Abu-Rumeileh S, Vacchiano V, Zenesini C, Polischi B, de Pasqua S, Fileccia E, Mammana A, Di Stasi V, Capellari S, Salvi F, Liguori R, Parchi P, BoReAls, (2020) Diagnostic-prognostic value and electrophysiological correlates of CSF biomarkers of neurodegeneration and neuroinflammation in amyotrophic lateral sclerosis. J Neurol 267:1699–1708
Falzone YM, Domi T, Agosta F, Pozzi L, Schito P, Fazio R, Del Carro U, Barbieri A, Comola M, Leocani L, Comi G, Carrera P, Filippi M, Quattrini A, Riva N (2020) Serum phosphorylated neurofilament heavy-chain levels reflect phenotypic heterogeneity and are an independent predictor of survival in motor neuron disease. J Neurol 267:2272–2280
Falzone YM, Domi T, Mandelli A, Pozzi L, Schito P, Russo T, Barbieri A, Fazio R, Volonte MA, Magnani G, Del Carro U, Carrera P, Malaspina A, Agosta F, Quattrini A, Furlan R, Filippi M, Riva N (2022) Integrated evaluation of a panel of neurochemical biomarkers to optimize diagnosis and prognosis in amyotrophic lateral sclerosis. Eur J Neurol Off J Eur Fed Neurol Soc 29:1930–1939
Peng L, Wan L, Liu M, Long Z, Chen D, Yuan X, Tang Z, Fu Y, Zhu S, Lei L, Wang C, Peng H, Shi Y, He L, Yuan H, Wan N, Hou X, Xia K, Li J, Chen C, Qiu R, Tang B, Chen Z, Jiang H (2023) Diagnostic and prognostic performance of plasma neurofilament light chain in multiple system atrophy: a cross-sectional and longitudinal study. J Neurol 270:4248–4261
Brousse M, Delaby C, De La Cruz E, Kuhle J, Benkert P, Mondesert E, Ginestet N, Hirtz C, Camu W, Lehmann S, Esselin F (2023) Serum neurofilament light chain cut-off definition for clinical diagnosis and prognosis of amyotrophic lateral sclerosis. Eu J Neurol Off J Eur Fed Neurol Soc 30:1919–1927
Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, Chio A, Van Damme P, Ludolph AC, Glass JD, Andrews JA, Babu S, Benatar M, McDermott CJ, Cochrane T, Chary S, Chew S, Zhu H, Wu F, Nestorov I, Graham D, Sun P, McNeill M, Fanning L, Ferguson TA, Fradette Valor S, Group OLEW (2022) Trial of Antisense oligonucleotide tofersen for SOD1 ALS. New Engl J Med 387:1099–1110
Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A (2018) Neurofilament light: a candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol 84:130–139
De Schaepdryver M, Goossens J, De Meyer S, Jeromin A, Masrori P, Brix B, Claeys KG, Schaeverbeke J, Adamczuk K, Vandenberghe R, Van Damme P, Poesen K (2019) Serum neurofilament heavy chains as early marker of motor neuron degeneration. Ann Clin Transl Neurol 6:1971–1979
Bjornevik K, O’Reilly EJ, Molsberry S, Kolonel LN, Le Marchand L, Paganoni S, Schwarzschild MA, Benkert P, Kuhle J, Ascherio A (2021) Prediagnostic neurofilament light chain levels in amyotrophic lateral sclerosis. Neurology 97:e1466-1474
Gille B, De Schaepdryver M, Dedeene L, Goossens J, Claeys KG, Van Den Bosch L, Tournoy J, Van Damme P, Poesen K (2019) Inflammatory markers in cerebrospinal fluid: independent prognostic biomarkers in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry 90:1338–1346
Thompson AG, Gray E, Bampton A, Raciborska D, Talbot K, Turner MR (2019) CSF chitinase proteins in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 90:1215–1220
Shepheard SR, Karnaros V, Benyamin B, Schultz DW, Dubowsky M, Wuu J, Chataway T, Malaspina A, Benatar M, Rogers ML (2022) Urinary neopterin: A novel biomarker of disease progression in amyotrophic lateral sclerosis. Eur J Neurol Off J Eur Fed Neurol Soc 29:990–999
Lunetta C, Lizio A, Gerardi F, Tarlarini C, Filippi M, Riva N, Tremolizzo L, Diamanti S, Dellanoce CC, Mosca L, Sansone VA, Campolo J (2020) Urinary neopterin, a new marker of the neuroinflammatory status in amyotrophic lateral sclerosis. J Neurol 267:3609–3616
Ludolph A, Dupuis L, Kasarskis E, Steyn F, Ngo S, McDermott C (2023) Nutritional and metabolic factors in amyotrophic lateral sclerosis. Nat Rev Neurol 19:511–524
Ludolph AC, Dorst J, Dreyhaupt J, Weishaupt JH, Kassubek J, Weiland U, Meyer T, Petri S, Hermann A, Emmer A, Grosskreutz J, Grehl T, Zeller D, Boentert M, Schrank B, Prudlo J, Winkler AS, Gorbulev S, Roselli F, Schuster J, Dupuis L, Group L-AS (2020) Effect of high-caloric nutrition on survival in amyotrophic lateral sclerosis. Ann Neurol 87:206–216
Michels S, Kurz D, Rosenbohm A, Peter RS, Just S, Bazner H, Bortlein A, Dettmers C, Gold HJ, Kohler A, Naumann M, Ratzka P, Ludolph AC, Rothenbacher D, Nagel G, Dorst J, Group ALSRSS (2023) Association of blood lipids with onset and prognosis of amyotrophic lateral sclerosis: results from the ALS Swabia registry. J Neurol 270:3082–3090
Lunetta C, Lizio A, Tremolizzo L, Ruscica M, Macchi C, Riva N, Weydt P, Corradi E, Magni P, Sansone V (2018) Serum irisin is upregulated in patients affected by amyotrophic lateral sclerosis and correlates with functional and metabolic status. J Neurol 265:3001–3008
Dolinar A, Koritnik B, Glavac D, Ravnik-Glavac M (2019) Circular RNAs as potential blood biomarkers in amyotrophic lateral sclerosis. Mol Neurobiol 56:8052–8062
Filippi M, Agosta F, Grosskreutz J, Benatar M, Kassubek J, Verstraete E, Turner MR, Neuroimaging Society in ALS (2015) Progress towards a neuroimaging biomarker for amyotrophic lateral sclerosis. Lancet Neurol 14:786–788
Agosta F, Spinelli EG, Filippi M (2018) Neuroimaging in amyotrophic lateral sclerosis: current and emerging uses. Expert Rev Neurother 18:395–406
Spinelli EG, Riva N, Rancoita PMV, Schito P, Doretti A, Poletti B, Di Serio C, Silani V, Filippi M, Agosta F (2020) Structural MRI outcomes and predictors of disease progression in amyotrophic lateral sclerosis. NeuroImage Clin 27:102315
Fabes J, Matthews L, Filippini N, Talbot K, Jenkinson M, Turner MR (2017) Quantitative FLAIR MRI in amyotrophic lateral sclerosis. Acad Radiol 24:1187–1194
Roeben B, Wilke C, Bender B, Ziemann U, Synofzik M (2019) The motor band sign in ALS: presentations and frequencies in a consecutive series of ALS patients. J Neurol Sci 406:116440
Cardenas-Blanco A, Machts J, Acosta-Cabronero J, Kaufmann J, Abdulla S, Kollewe K, Petri S, Heinze HJ, Dengler R, Vielhaber S, Nestor PJ (2014) Central white matter degeneration in bulbar- and limb-onset amyotrophic lateral sclerosis. J Neurol 261:1961–1967
Zhang J, Yin X, Zhao L, Evans AC, Song L, Xie B, Li H, Luo C, Wang J (2014) Regional alterations in cortical thickness and white matter integrity in amyotrophic lateral sclerosis. J Neurol 261:412–421
Caiazzo G, Corbo D, Trojsi F, Piccirillo G, Cirillo M, Monsurro MR, Esposito F, Tedeschi G (2014) Distributed corpus callosum involvement in amyotrophic lateral sclerosis: a deterministic tractography study using q-ball imaging. J Neurol 261:27–36
Spinelli EG, Agosta F, Ferraro PM, Querin G, Riva N, Bertolin C, Martinelli I, Lunetta C, Fontana A, Soraru G, Filippi M (2019) Brain MRI shows white matter sparing in Kennedy’s disease and slow-progressing lower motor neuron disease. Hum Brain Mapp 40:3102–3112
Rosenbohm A, Muller HP, Hubers A, Ludolph AC, Kassubek J (2016) Corticoefferent pathways in pure lower motor neuron disease: a diffusion tensor imaging study. J Neurol 263:2430–2437
Agosta F, Ferraro PM, Riva N, Spinelli EG, Chio A, Canu E, Valsasina P, Lunetta C, Iannaccone S, Copetti M, Prudente E, Comi G, Falini A, Filippi M (2016) Structural brain correlates of cognitive and behavioral impairment in MND. Hum Brain Mapp 37:1614–1626
Bede P, Lule D, Muller HP, Tan EL, Dorst J, Ludolph AC, Kassubek J (2023) Presymptomatic grey matter alterations in ALS kindreds: a computational neuroimaging study of asymptomatic C9orf72 and SOD1 mutation carriers. J Neurol 270:4235–4247
Schuster C, Kasper E, Machts J, Bittner D, Kaufmann J, Benecke R, Teipel S, Vielhaber S, Prudlo J (2014) Longitudinal course of cortical thickness decline in amyotrophic lateral sclerosis. J Neurol 261:1871–1880
Steinbach R, Loewe K, Kaufmann J, Machts J, Kollewe K, Petri S, Dengler R, Heinze HJ, Vielhaber S, Schoenfeld MA, Stoppel CM (2015) Structural hallmarks of amyotrophic lateral sclerosis progression revealed by probabilistic fiber tractography. J Neurol. https://doi.org/10.1007/s00415-015-7841-1
Muller HP, Behler A, Munch M, Dorst J, Ludolph AC, Kassubek J (2023) Sequential alterations in diffusion metrics as correlates of disease severity in amyotrophic lateral sclerosis. J Neurol 270:2308–2313
Khamaysa M, Lefort M, Pelegrini-Issac M, Lackmy-Vallee A, Preuilh A, Devos D, Rolland AS, Desnuelle C, Chupin M, Marchand-Pauvert V, Querin G, Pradat PF, Pulse study g, (2023) Comparison of spinal magnetic resonance imaging and classical clinical factors in predicting motor capacity in amyotrophic lateral sclerosis. J Neurol 270:3885–3895
Toh C, Keslake A, Payne T, Onwuegbuzie A, Harding J, Baster K, Hoggard N, Shaw PJ, Wilkinson ID, Jenkins TM (2023) Analysis of brain and spinal MRI measures in a common domain to investigate directional neurodegeneration in motor neuron disease. J Neurol 270:1682–1690
Canosa A, Moglia C, Manera U, Vasta R, Torrieri MC, Arena V, D'Ovidio F, Palumbo F, Zucchetti JP, Iazzolino B, Peotta L, Calvo A, Pagani M, Chio A (2020) Metabolic brain changes across different levels of cognitive impairment in ALS: a (18)F-FDG-PET study. J Neurol Neurosurg Psychiatry
Canosa A, Martino A, Giuliani A, Moglia C, Vasta R, Grassano M, Palumbo F, Cabras S, Di Pede F, De Mattei F, Matteoni E, Polverari G, Manera U, Calvo A, Pagani M, Chio A (2023) Brain metabolic differences between pure bulbar and pure spinal ALS: a 2-[(18)F]FDG-PET study. J Neurol 270:953–959
Van Laere K, Vanhee A, Verschueren J, De Coster L, Driesen A, Dupont P, Robberecht W, Van Damme P (2014) Value of 18fluorodeoxyglucose-positron-emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol 71:553–561
De Vocht J, Blommaert J, Devrome M, Radwan A, Van Weehaeghe D, De Schaepdryver M, Ceccarini J, Rezaei A, Schramm G, van Aalst J, Chio A, Pagani M, Stam D, Van Esch H, Lamaire N, Verhaegen M, Mertens N, Poesen K, van den Berg LH, van Es MA, Vandenberghe R, Vandenbulcke M, Van den Stock J, Koole M, Dupont P, Van Laere K, Van Damme P (2020) Use of multimodal imaging and clinical biomarkers in presymptomatic carriers of C9orf72 repeat expansion. JAMA Neurol 77:1008–1017
Zurcher NR, Loggia ML, Lawson R, Chonde DB, Izquierdo-Garcia D, Yasek JE, Akeju O, Catana C, Rosen BR, Cudkowicz ME, Hooker JM, Atassi N (2015) Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: assessed with [(11)C]-PBR28. NeuroImage Clin 7:409–414
Alshikho MJ, Zurcher NR, Loggia ML, Cernasov P, Reynolds B, Pijanowski O, Chonde DB, Izquierdo Garcia D, Mainero C, Catana C, Chan J, Babu S, Paganoni S, Hooker JM, Atassi N (2018) Integrated magnetic resonance imaging and [(11) C]-PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann Neurol 83:1186–1197
Tondo G, Iaccarino L, Cerami C, Vanoli GE, Presotto L, Masiello V, Coliva A, Salvi F, Bartolomei I, Mosca L, Lunetta C, Perani D (2020) (11) C-PK11195 PET-based molecular study of microglia activation in SOD1 amyotrophic lateral sclerosis. Ann Clin Transl Neurol 7:1513–1523
Mitsumoto H, Brooks BR, Silani V (2014) Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol 13:1127–1138
Bensimon G, Lacomblez L, Meininger V (1994) A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 330:585–591
Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V (1996) Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 347:1425–1431
Mandrioli J, Malerba SA, Beghi E, Fini N, Fasano A, Zucchi E, De Pasqua S, Guidi C, Terlizzi E, Sette E, Ravasio A, Casmiro M, Salvi F, Liguori R, Zinno L, Handouk Y, Rizzi R, Borghi A, Rinaldi R, Medici D, Santangelo M, Granieri E, Mussuto V, Aiello M, Ferro S, Vinceti M, Group E (2018) Riluzole and other prognostic factors in ALS: a population-based registry study in Italy. J Neurol 265:817–827
Jayaprakash K, Glasmacher SA, Pang B, Beswick E, Mehta AR, Dakin R, Newton J, Chandran S, Pal S, Consortium C-M (2020) Riluzole prescribing, uptake and treatment discontinuation in people with amyotrophic lateral sclerosis in Scotland. J Neurol 267:2459–2461
Writing G, Edaravone ALSSG (2017) Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 16:505–512
Lunetta C, Moglia C, Lizio A, Caponnetto C, Dubbioso R, Giannini F, Mata S, Mazzini L, Sabatelli M, Siciliano G, Simone IL, Soraru G, Toriello A, Trojsi F, Vedovello M, D’Ovidio F, Filippi M, Calvo A, ESG, (2020) The Italian multicenter experience with edaravone in amyotrophic lateral sclerosis. J Neurol 267:3258–3267
Distaso E, Milella G, Mezzapesa DM, Introna A, D’Errico E, Fraddosio A, Zoccolella S, Dicuonzo F, Simone IL (2021) Magnetic resonance metrics to evaluate the effect of therapy in amyotrophic lateral sclerosis: the experience with edaravone. J Neurol 268:3307–3315
Massey TH, Robertson NP (2018) Repurposing drugs to treat neurological diseases. J Neurol 265:446–448
Chio A, Mazzini L, Mora G (2020) Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology 167:107986
Dalla Bella E, Bersano E, Antonini G, Borghero G, Capasso M, Caponnetto C, Chio A, Corbo M, Filosto M, Giannini F, Spataro R, Lunetta C, Mandrioli J, Messina S, Monsurro MR, Mora G, Riva N, Rizzi R, Siciliano G, Silani V, Simone I, Soraru G, Tugnoli V, Verriello L, Volanti P, Furlan R, Nolan JM, Abgueguen E, Tramacere I, Lauria G (2021) The unfolded protein response in amyotrophic later sclerosis: results of a phase 2 trial. Brain J Neurol 144:2635–2647
Sassi S, Bianchi E, Diamanti L, Tornabene D, Sette E, Medici D, Mata S, Leccese D, Sperti M, Martinelli I, Ghezzi A, Mandrioli J, Iuzzolino VV, Dubbioso R, Trojsi F, Passaniti C, D’Alvano G, Filosto M, Padovani A, Mazzini L, De Marchi F, Zinno L, Nuredini A, Bongioanni P, Dolciotti C, Canali E, Toschi G, Petrucci A, Perna A, Riso V, Inghilleri M, Libonati L, Cambieri C, Pupillo E (2023) Retrospective observational study on the use of acetyl-L-carnitine in ALS. J Neurol 270:5344–5357
Lo Giudice M, Cocco A, Reggiardo G, Lalli S, Albanese A (2023) Tauro-Urso-deoxycholic acid trials in amyotrophic lateral sclerosis: what is achieved and what to expect. Clin Drug Investig 43:893–903
Paganoni S, Macklin EA, Hendrix S, Berry JD, Elliott MA, Maiser S, Karam C, Caress JB, Owegi MA, Quick A, Wymer J, Goutman SA, Heitzman D, Heiman-Patterson T, Jackson CE, Quinn C, Rothstein JD, Kasarskis EJ, Katz J, Jenkins L, Ladha S, Miller TM, Scelsa SN, Vu TH, Fournier CN, Glass JD, Johnson KM, Swenson A, Goyal NA, Pattee GL, Andres PL, Babu S, Chase M, Dagostino D, Dickson SP, Ellison N, Hall M, Hendrix K, Kittle G, McGovern M, Ostrow J, Pothier L, Randall R, Shefner JM, Sherman AV, Tustison E, Vigneswaran P, Walker J, Yu H, Chan J, Wittes J, Cohen J, Klee J, Leslie K, Tanzi RE, Gilbert W, Yeramian PD, Schoenfeld D, Cudkowicz ME (2020) Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N Engl J Med 383:919–930
Amylyx Pharma (2024) Amylyx Pharmaceuticals announces topline results from global phase 3 PHOENIX Trial of AMX0035 in ALS. In: Amylyx to Host Investor Conference Call Today, March 8, at 8:00 a.m. ET.
TUDCA-ALS consortium (2024) The TUDCA-ALS consortium announces top-line results from the European phase 3 clinical trial of TUDCA in patients with amyotrophic lateral sclerosis (ALS).
Aizawa H, Kato H, Oba K, Kawahara T, Okubo Y, Saito T, Naito M, Urushitani M, Tamaoka A, Nakamagoe K, Ishii K, Kanda T, Katsuno M, Atsuta N, Maeda Y, Nagai M, Nishiyama K, Ishiura H, Toda T, Kawata A, Abe K, Yabe I, Takahashi-Iwata I, Sasaki H, Warita H, Aoki M, Sobue G, Mizusawa H, Matsuyama Y, Haga T, Kwak S (2022) Randomized phase 2 study of perampanel for sporadic amyotrophic lateral sclerosis. J Neurol 269:885–896
Thonhoff JR, Berry JD, Macklin EA, Beers DR, Mendoza PA, Zhao W, Thome AD, Triolo F, Moon JJ, Paganoni S, Cudkowicz M, Appel SH (2022) Combined regulatory T-Lymphocyte and IL-2 treatment is safe, tolerable, and biologically active for 1 year in persons with amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm. 9
Lunetta C, Lizio A, Cabona C, Gerardi F, Sansone VA, Corbo M, Scialo C, Angelucci E, Gualandi F, Marenco P, Grillo G, Cairoli R, Cesana C, Saccardi R, Melazzini MG, Mancardi G, Caponnetto C (2022) A phase I/IIa clinical trial of autologous hematopoietic stem cell transplantation in amyotrophic lateral sclerosis. J Neurol 269:5337–5346
Mandrioli J, D’Amico R, Zucchi E, De Biasi S, Banchelli F, Martinelli I, Simonini C, Lo Tartaro D, Vicini R, Fini N, Gianferrari G, Pinti M, Lunetta C, Gerardi F, Tarlarini C, Mazzini L, De Marchi F, Scognamiglio A, Soraru G, Fortuna A, Lauria G, Bella ED, Caponnetto C, Meo G, Chio A, Calvo A, Cossarizza A (2023) Randomized, double-blind, placebo-controlled trial of rapamycin in amyotrophic lateral sclerosis. Nat Commun 14:4970
Cudkowicz M, Genge A, Maragakis N, Petri S, van den Berg L, Aho VV, Sarapohja T, Kuoppamaki M, Garratt C, Al-Chalabi A, investigators R, (2021) Safety and efficacy of oral levosimendan in people with amyotrophic lateral sclerosis (the REFALS study): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Neurol 20:821–831
Shefner JM, Al-Chalabi A, Andrews JA, Chio A, De Carvalho M, Cockroft BM, Corcia P, Couratier P, Cudkowicz ME, Genge A, Hardiman O, Heiman-Patterson T, Henderson RD, Ingre C, Jackson CE, Johnston W, Lechtzin N, Ludolph A, Maragakis NJ, Miller TM, Mora Pardina JS, Petri S, Simmons Z, Van Den Berg LH, Zinman L, Kupfer S, Malik FI, Meng L, Simkins TJ, Wei J, Wolff AA, Rudnicki SA (2023) COURAGE-ALS: a randomized, double-blind phase 3 study designed to improve participant experience and increase the probability of success. Amyotrophic Lateral Sclerosis Frontotemporal Degener 24:523–534
Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P, Shneider NA (2022) Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med 28:104–116
Boros BD, Schoch KM, Kreple CJ, Miller TM (2022) Antisense oligonucleotides for the study and treatment of ALS. Neurotherapeut J Am Soc Exper NeuroTherapeut 19:1145–1158
Khamaysa M, Pradat PF (2022) Status of ALS treatment, insights into therapeutic challenges and dilemmas. J Personal Med 12:1601
Zhu Q, Xu D, Huang H, Li D, Yang D, Zhou J, Zhao Y (2023) The safety and effectiveness of high-calorie therapy for treating amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol 270:4729–4743
Kalron A, Mahameed I, Weiss I, Rosengarten D, Balmor GR, Heching M, Kramer MR (2021) Effects of a 12-week combined aerobic and strength training program in ambulatory patients with amyotrophic lateral sclerosis: a randomized controlled trial. J Neurol 268:1857–1866
Talbot K, Feneberg E, Scaber J, Thompson AG, Turner MR (2018) Amyotrophic lateral sclerosis: the complex path to precision medicine. J Neurol 265:2454–2462
Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O (2003) An outcome study of riluzole in amyotrophic lateral sclerosis–a population-based study in Ireland, 1996–2000. J Neurol 250:473–479
Van den Berg JP, Kalmijn S, Lindeman E, Veldink JH, de Visser M, Van der Graaff MM, Wokke JH, Van den Berg LH (2005) Multidisciplinary ALS care improves quality of life in patients with ALS. Neurology 65:1264–1267
Rooney J, Byrne S, Heverin M, Tobin K, Dick A, Donaghy C, Hardiman O (2015) A multidisciplinary clinic approach improves survival in ALS: a comparative study of ALS in Ireland and Northern Ireland. J Neurol Neurosurg Psychiatry 86:496–501
Ng L, Khan F, Mathers S (2009) Multidisciplinary care for adults with amyotrophic lateral sclerosis or motor neuron disease. Cochr Datab System Rev:CD007425
Hrastelj J, Robertson NP (2015) Improving survival in amyotrophic lateral sclerosis: future treatments in a modern service. J Neurol 262:1791–1793
Katzberg HD (2015) Neurogenic muscle cramps. J Neurol 262:1814–1821
Riva N, Mora G, Soraru G, Lunetta C, Ferraro OE, Falzone Y, Leocani L, Fazio R, Comola M, Comi G, Group CS (2019) Safety and efficacy of nabiximols on spasticity symptoms in patients with motor neuron disease (CANALS): a multicentre, double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol 18:155–164
Young CA, Ellis C, Johnson J, Sathasivam S, Pih N (2011) Treatment for sialorrhea (excessive saliva) in people with motor neuron disease/amyotrophic lateral sclerosis. Cochr Database System Rev:CD006981
Amador Mdel M, Assouline A, Gonzalez-Bermejo J, Pradat PF (2015) Radiotherapy treatment of sialorrhea in patients with amyotrophic lateral sclerosis requiring non-invasive ventilation. J Neurol 262:1981–1983
Desport JC, Torny F, Lacoste M, Preux PM, Couratier P (2005) Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegener Dis 2:202–207
Wada A, Kawakami M, Liu M, Otaka E, Nishimura A, Liu F, Otsuka T (2015) Development of a new scale for dysphagia in patients with progressive neuromuscular diseases: the Neuromuscular Disease Swallowing Status Scale (NdSSS). J Neurol
Allen JA, Chen R, Ajroud-Driss S, Sufit RL, Heller S, Siddique T, Wolfe L (2013) Gastrostomy tube placement by endoscopy versus radiologic methods in patients with ALS: a retrospective study of complications and outcome. Amyotrophic Lateral Sclerosis Frontotemporal Degener 14:308–314
Blondet A, Lebigot J, Nicolas G, Boursier J, Person B, Laccoureye L, Aube C (2010) Radiologic versus endoscopic placement of percutaneous gastrostomy in amyotrophic lateral sclerosis: multivariate analysis of tolerance, efficacy, and survival. J Vasc Interven Radiolo JVIR 21:527–533
Dorst J, Dupuis L, Petri S, Kollewe K, Abdulla S, Wolf J, Weber M, Czell D, Burkhardt C, Hanisch F, Vielhaber S, Meyer T, Frisch G, Kettemann D, Grehl T, Schrank B, Ludolph AC (2015) Percutaneous endoscopic gastrostomy in amyotrophic lateral sclerosis: a prospective observational study. J Neurol 262:849–858
Thompson AG, Blackwell V, Marsden R, Millard E, Lawson C, Nickol AH, East JE, Talbot K, Allan PJ, Turner MR (2017) A risk stratifying tool to facilitate safe late-stage percutaneous endoscopic gastrostomy in ALS. Amyotrophic Lateral Sclerosis Frontotemporal Degener 18:243–248
ProGas Study G (2015) Gastrostomy in patients with amyotrophic lateral sclerosis (ProGas): a prospective cohort study. Lancet Neurol 14:702–709
Pfeffer G, Povitz M, Gibson GJ, Chinnery PF (2015) Diagnosis of muscle diseases presenting with early respiratory failure. J Neurol 262:1101–1114
Capozzo R, Quaranta VN, Pellegrini F, Fontana A, Copetti M, Carratu P, Panza F, Cassano A, Falcone VA, Tortelli R, Cortese R, Simone IL, Resta O, Logroscino G (2015) Sniff nasal inspiratory pressure as a prognostic factor of tracheostomy or death in amyotrophic lateral sclerosis. J Neurol 262:593–603
Di PWC, Di PSGC (2015) Safety and efficacy of diaphragm pacing in patients with respiratory insufficiency due to amyotrophic lateral sclerosis (DiPALS): a multicentre, open-label, randomised controlled trial. Lancet Neurol 14:883–892
Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ (2006) Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol 5:140–147
Jacobs TL, Brown DL, Baek J, Migda EM, Funckes T, Gruis KL (2016) Trial of early noninvasive ventilation for ALS: a pilot placebo-controlled study. Neurology 87:1878–1883
Rafiq MK, Bradburn M, Proctor AR, Billings CG, Bianchi S, McDermott CJ, Shaw PJ (2015) A preliminary randomized trial of the mechanical insufflator-exsufflator versus breath-stacking technique in patients with amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis Frontotemporal Degener 16:448–455
Boentert M, Brenscheidt I, Glatz C, Young P (2015) Effects of non-invasive ventilation on objective sleep and nocturnal respiration in patients with amyotrophic lateral sclerosis. J Neurol. https://doi.org/10.1007/s00415-015-7822-4
Berlowitz DJ, Howard ME, Fiore JF Jr, Vander Hoorn S, O’Donoghue FJ, Westlake J, Smith A, Beer F, Mathers S, Talman P (2015) Identifying who will benefit from non-invasive ventilation in amyotrophic lateral sclerosis/motor neurone disease in a clinical cohort. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2014-310055
Andersen PM, Borasio GD, Dengler R, Hardiman O, Kollewe K, Leigh PN, Pradat PF, Silani V, Tomik B, Group EW (2007) Good practice in the management of amyotrophic lateral sclerosis: clinical guidelines. An evidence-based review with good practice points. EALSC Working Group. Amyotrophic Lateral Sclerosis : Off Publ World Fed Neurol Res Group Motor Neuron Dis 8:195–213
Wu JM, Tam MT, Buch K, Khairati F, Wilson L, Bannerman E, Guerrero A, Eisen A, Toyer W, Stevenson T, Robillard JM (2022) The impact of respite care from the perspectives and experiences of people with amyotrophic lateral sclerosis and their care partners: a qualitative study. BMC Palliat Care 21:26
Oberstadt MCF, Esser P, Classen J, Mehnert A (2018) Alleviation of psychological distress and the improvement of quality of life in patients with amyotrophic lateral sclerosis: adaptation of a short-term psychotherapeutic intervention. Front Neurol 9:231
Pugliese R, Sala R, Regondi S, Beltrami B, Lunetta C (2022) Emerging technologies for management of patients with amyotrophic lateral sclerosis: from telehealth to assistive robotics and neural interfaces. J Neurol 269:2910–2921
Glasmacher SA, Larraz J, Mehta AR, Kearns PKA, Wong M, Newton J, Davenport R, Gorrie G, Morrison I, Carod Artal J, Chandran S, Pal S, Consortium C-M (2021) The immediate impact of the COVID-19 pandemic on motor neuron disease services and mortality in Scotland. J Neurol 268:2038–2040
Costamagna G, Abati E, Bresolin N, Comi GP, Corti S (2021) Management of patients with neuromuscular disorders at the time of the SARS-CoV-2 pandemic. J Neurol 268:1580–1591
Londral A (2022) Assistive technologies for communication empower patients with ALS to generate and self-report health data. Front Neurol 13:867567
Eicher C, Kiselev J, Brukamp K, Kiemel D, Spittel S, Maier A, Oleimeulen U, Greuèl M (2019) Expectations and Concerns Emerging from Experiences with Assistive Technology for ALS Patients. Universal Access in Human-Computer Interaction. Springer International Publishing, Cham, pp 57–68
Linse K, Aust E, Joos M, Hermann A (2018) Communication matters-pitfalls and promise of hightech communication devices in palliative care of severely physically disabled patients with amyotrophic lateral sclerosis. Front Neurol 9:603
Fernandes F, Barbalho I, Bispo Junior A, Alves L, Nagem D, Lins H, Arrais Junior E, Coutinho KD, Morais AHF, Santos JPQ, Machado GM, Henriques J, Teixeira C, Dourado Junior MET, Lindquist ARR, Valentim RAM (2023) Digital alternative communication for individuals with amyotrophic lateral sclerosis: what we have. J Clin Med. https://doi.org/10.3390/jcm12165235
Garibaldi M, Siciliano G, Antonini G (2021) Telemedicine for neuromuscular disorders during the COVID-19 outbreak. J Neurol 268:1–4
Helleman J, Bakers JNE, Pirard E, van den Berg LH, Visser-Meily JMA, Beelen A (2022) Home-monitoring of vital capacity in people with a motor neuron disease. J Neurol 269:3713–3722
Kudritzki V, Howard IM (2023) Telehealth-based exercise in amyotrophic lateral sclerosis. Front Neurol 14:1238916
Diment LE, Thompson MS, Bergmann JHM (2017) Clinical efficacy and effectiveness of 3D printing: a systematic review. BMJ Open 7:e016891
Jabil (2024) Top 3D Printing Challenges (And How to Overcome Them)
Holdom CJ, van Unnik JWJ, van Eijk RPA, van den Berg LH, Henderson RD, Ngo ST, Steyn FJ (2023) Use of hip- versus wrist-based actigraphy for assessing functional decline and disease progression in patients with motor neuron disease. J Neurol 270:2597–2605
Beswick E, Fawcett T, Hassan Z, Forbes D, Dakin R, Newton J, Abrahams S, Carson A, Chandran S, Perry D, Pal S (2022) A systematic review of digital technology to evaluate motor function and disease progression in motor neuron disease. J Neurol 269:6254–6268
Godkin FE, Turner E, Demnati Y, Vert A, Roberts A, Swartz RH, McLaughlin PM, Weber KS, Thai V, Beyer KB, Cornish B, Abrahao A, Black SE, Masellis M, Zinman L, Beaton D, Binns MA, Chau V, Kwan D, Lim A, Munoz DP, Strother SC, Sunderland KM, Tan B, McIlroy WE, Van Ooteghem K (2022) Feasibility of a continuous, multi-sensor remote health monitoring approach in persons living with neurodegenerative disease. J Neurol 269:2673–2686
Johnson SA, Karas M, Burke KM, Straczkiewicz M, Scheier ZA, Clark AP, Iwasaki S, Lahav A, Iyer AS, Onnela JP, Berry JD (2023) Wearable device and smartphone data quantify ALS progression and may provide novel outcome measures. NPJ Digit Med 6:34
Helleman J, Johnson B, Holdom C, Hobson E, Murray D, Steyn FJ, Ngo ST, Henders A, Lokeshappa MB, Visser-Meily JMA, van den Berg LH, Hardiman O, Beelen A, McDermott C, van Eijk RPA (2022) Patient perspectives on digital healthcare technology in care and clinical trials for motor neuron disease: an international survey. J Neurol 269:6003–6013
Proietti T, O’Neill C, Gerez L, Cole T, Mendelowitz S, Nuckols K, Hohimer C, Lin D, Paganoni S, Walsh C (2023) Restoring arm function with a soft robotic wearable for individuals with amyotrophic lateral sclerosis. Sci Transl Med 15:eadd1504
Vansteensel MJ, Klein E, van Thiel G, Gaytant M, Simmons Z, Wolpaw JR, Vaughan TM (2023) Towards clinical application of implantable brain-computer interfaces for people with late-stage ALS: medical and ethical considerations. J Neurol 270:1323–1336
WYSS CENTER (2024) ABILITY - An implantable brain-computer interface (BCI) to restore movement and communication
CENTER W (2024) NeuroKey™ - Real-time neural signal processing platform
Seitzer F, Kahrass H, Neitzke G, Strech D (2015) The full spectrum of ethical issues in the care of patients with ALS: a systematic qualitative review. J Neurol. https://doi.org/10.1007/s00415-015-7867-4
Shaw J, Brown R, Heinrich P, Dunn S (2013) Doctors’ experience of stress during simulated bad news consultations. Patient Educ Couns 93:203–208
Shaw JM, Brown RF, Dunn SM (2013) A qualitative study of stress and coping responses in doctors breaking bad news. Patient Educ Couns 91:243–248
Benditt JO, Smith TS, Tonelli MR (2001) Empowering the individual with ALS at the end-of-life: disease-specific advance care planning. Muscle Nerve 24:1706–1709
Connolly S, Galvin M, Hardiman O (2015) End-of-life management in patients with amyotrophic lateral sclerosis. Lancet Neurol 14:435–442
Greenaway LP, Martin NH, Lawrence V, Janssen A, Al-Chalabi A, Leigh PN, Goldstein LH (2015) Accepting or declining non-invasive ventilation or gastrostomy in amyotrophic lateral sclerosis: patients’ perspectives. J Neurol 262:1002–1013
Diagnosis ETFo, Management of Amyotrophic Lateral S, Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, Van Damme P, Hardiman O, Kollewe K, Morrison KE, Petri S, Pradat PF, Silani V, Tomik B, Wasner M, Weber M (2012) EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)–revised report of an EFNS task force. Eur J Neurol Off J Eur Fed Neurol Soc 19:360–375
Munroe CA, Sirdofsky MD, Kuru T, Anderson ED (2007) End-of-life decision making in 42 patients with amyotrophic lateral sclerosis. Respir Care 52:996–999
Lule D, Ehlich B, Lang D, Sorg S, Heimrath J, Kubler A, Birbaumer N, Ludolph AC (2013) Quality of life in fatal disease: the flawed judgement of the social environment. J Neurol 260:2836–2843
Maessen M, Veldink JH, Onwuteaka-Philipsen BD, Hendricks HT, Schelhaas HJ, Grupstra HF, van der Wal G, van den Berg LH (2014) Euthanasia and physician-assisted suicide in amyotrophic lateral sclerosis: a prospective study. J Neurol 261:1894–1901
Lule D, Nonnenmacher S, Sorg S, Heimrath J, Hautzinger M, Meyer T, Kubler A, Birbaumer N, Ludolph AC (2014) Live and let die: existential decision processes in a fatal disease. J Neurol 261:518–525
Weber C, Fijalkowska B, Ciecwierska K, Lindblad A, Badura-Lotter G, Andersen PM, Kuzma-Kozakiewicz M, Ludolph AC, Lule D, Pasierski T, Lynoe N (2017) Existential decision-making in a fatal progressive disease: how much do legal and medical frameworks matter? BMC Palliat Care 16:80
Dybwik K, Tollali T, Nielsen EW, Brinchmann BS (2010) Why does the provision of home mechanical ventilation vary so widely? Chron Respir Dis 7:67–73
Magelssen M, Holmoy T, Horn MA, Fondenaes OA, Dybwik K, Forde R (2018) Ethical challenges in tracheostomy-assisted ventilation in amyotrophic lateral sclerosis. J Neurol 265:2730–2736
Dybwik K, Tollali T, Nielsen EW, Brinchmann BS (2011) “Fighting the system”: families caring for ventilator-dependent children and adults with complex health care needs at home. BMC Health Serv Res 11:156
Moglia C, Palumbo F, Veronese S, Group MNDIS, Calvo A (2023) Withdrawal of mechanical ventilation in amyotrophic lateral sclerosis patients: a multicenter Italian survey. Neurol Sci Off J Italian Neurol Soc Italian Soc Clin Neurophysiol 44:4349–4357
Keller J, Gorges M, Horn HT, Aho-Ozhan HE, Pinkhardt EH, Uttner I, Kassubek J, Ludolph AC, Lule D (2015) Eye-tracking controlled cognitive function tests in patients with amyotrophic lateral sclerosis: a controlled proof-of-principle study. J Neurol 262:1918–1926
Acknowledgements
This work was supported by AriSLA, Italian Research Foundation for ALS (Ideals Grant) to NR and AQ and by the Giovanni Marazzina Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all the authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Riva, N., Domi, T., Pozzi, L. et al. Update on recent advances in amyotrophic lateral sclerosis. J Neurol (2024). https://doi.org/10.1007/s00415-024-12435-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00415-024-12435-9