
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by motor neuron loss. ALS is now associated with mutations in numerous genes, many of which cause disease in part through toxic gain-of-function mechanisms. Antisense oligonucleotides (ASOs) are small sequences of DNA that can reduce expression of a target gene at the post-transcriptional level, making them attractive for neutralizing mutant or toxic gene products. Advancements in the medicinal chemistries of ASOs have improved their pharmacodynamic profile to allow safe and effective delivery to the central nervous system. ASO therapies for ALS have rapidly developed over the last two decades, and ASOs that target SOD1, C9orf72, FUS, and ATXN2 are now in clinical trials for familial or sporadic forms of ALS. This review discusses the current state of ASO therapies for ALS, outlining their successes from preclinical development to early clinical trials.
Similar content being viewed by others


Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressing, devastating neurodegenerative disease that causes motor neuron loss and muscle atrophy. Most cases of ALS occur sporadically, whereas 10% are familial (fALS) and can usually be attributed to a single dominantly inherited genetic risk factor [1]. Patients with a monogenic cause of ALS establish a population that shares a common etiology and an apparent druggable target [1]. Since the identification of mutations in copper-zinc superoxide dismutase 1 (SOD1) as the first genetic cause of fALS [2], advancements in genome technologies have linked mutations in > 30 genes to ALS pathogenesis [3]. It is now recognized that most fALS cases can be attributed to a hexanucleotide expansion in the first intron of chromosome 9 open reading frame 72 (C9orf72) [4,5,6]. Decades of mechanistic studies have revealed that many monogenic causes of ALS occur in part through toxic gain of function (GOF) by the mutated gene [7]. For these cases, emerging therapeutic strategies that directly alter disease-causing genes and can neutralize mutant or toxic gene products carry immense promise.
Antisense oligonucleotides (ASOs) enable precise modulation of gene expression without altering host DNA, a limitation of current gene transfer approaches. ASOs modify protein expression by acting directly on target RNA either prior to or during translation [8]. Owing to their efficacy, ASOs have been powerful tools for mechanistic investigation for decades. Recent advancements in the medicinal chemistries of ASOs have elevated ASOs from investigational tools to safe, tolerable, and efficacious drugs with clinical utility [8]. ASOs are now approved for the treatment of > 10 conditions that include spinal muscular atrophy (SMA), a pediatric-onset motor neuron disease [9]. For diseases with a pathogenesis tied to single gene mutations or mechanisms, ASOs have become a top consideration, owing to straight-forward validation in disease model systems and to demonstrated successes in clinical translation.
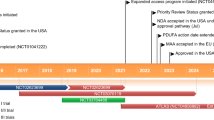
Cellular and animal model systems that phenotypically and pathologically resemble ALS have facilitated ASO drug advancement. Skin biopsies and peripheral blood cells from patients carrying disease mutations have allowed ASO testing in patient-derived cells or reprogrammed induced pluripotent stem cell (iPSC) lines. Remarkably, these models phenocopy many cellular features of disease and are now a mainstay in the first phase of ASO drug development. Candidate ASOs with convincing on-target action and efficacy have since advanced to clinical trials. Here, we will discuss advances and challenges in ASO technologies for SOD1, C9orf72, FUS, and ATXN2 (Fig. 1) and the pipelines that have elevated each from ASO discovery to human clinical trials for ALS.

Antisense oligonucleotide therapies under investigation in preclinical studies and human clinical trials for amyotrophic lateral sclerosis. ASO candidates targeting genes associated with ALS, including SOD1 [29,30,31, 45, 46], C9orf72 [68,69,70, 72,73,74], FUS [85], and ATXN2 [99], have demonstrated target efficacy, distribution throughout the central nervous system, and a strong safety profile. ASOs depicted target the human form of the gene and have been tested in preclinical and/or clinical trials (see references for more detail). ASO depictions do not represent exact binding locations. Note: the binding site of WVE-004 within the C9orf72 gene [71] is not included in this figure. Figure created with BioRender.com
Antisense Oligonucleotide Design and Mechanisms
ASOs are single-stranded DNA oligomers of 10–50 base pairs in length that bind to a target RNA through Watson-Crick-Franklin base pairing. Native, unmodified oligonucleotides have weak binding affinities and are highly susceptible to nucleases. However, modifications to the phosphate backbone or sugar rings confer favorable binding affinity and stability to ASOs and define their mechanisms of action [10].
ASO Chemistry and Modifications
One of the most common ASO chemistries involves modifications to the phosphodiester linkages forming the phosphate backbone [10]. Here, the substitution of the non-bridging phosphate oxygen atom for sulfur creates a phosphorothioate (PS) backbone that confers drug-like properties of many ASOs. First, the PS backbone promotes ASO stability and affords greater nuclease resistance, increasing the likelihood for target RNA engagement [10]. Second, the PS backbone can recruit the enzyme RNase H to cleave a target transcript, conferring PS-ASOs with degrading capability [10].
In addition to backbone modifications, ASOs can be modified at the 2′-position of the ribose moiety to further increase stability and binding [10]. Most used and represented in clinical trials are the 2′-O-methyl (2′-O-Me) and 2′-O-methoxyethyl (2′-MOE) modifications, which strengthen binding affinity to its target and promote nuclease resistance [9, 11]. Importantly, inclusion of 2′-O-Me or 2′-MOE bases reduces immune stimulation, which might occur due to PS use [12,13,14]. ASOs with “fully modified” sugar rings, in which all bases contain a 2′ modification, support non-degrading ASO mechanisms such as splicing manipulations but do not support RNase H enzymatic activity. However, a “gapmer” ASO design — in which 2′-O-Me or 2′-MOE-modified bases flank a central “gap” of unmodified PS bases — can degrade target RNA but maintain a low immunostimulatory profile [9, 11, 13, 15].
Recent innovations in ASO design include mixed backbone ASOs with varying combinations of base modifications. One interesting hypothesis is that creating ASOs with a predefined stereochemistry might improve potency by stabilizing 2′ modifications and enhancing RNase H recruitment, and some of these ASOs are currently in clinical trials [11, 16]. Other modifications to the backbone or ribose sugar, such as morpholinos or locked nucleic acid (LNA) formulations, have great potential and are also progressing through clinical trials [17, 18]. Morpholinos have high binding affinity but do not recruit RNase H [10]. LNAs are a variant on 2′-O-Me structures with superior binding affinity and nuclease resistance that have been limited due to potential toxicity profile, though new formulations might mitigate some concerns [10, 19,20,21]. Designing ASOs with diverse backbone types has become popular in recent years, but whether these modifications improve ASO function or attenuate toxicity over unmixed formulations is still not known.
Mechanisms of Action
The mechanism of action for an ASO depends on its sequence and design. Once an ASO enters a cell, it can readily translocate to the nucleus, where it binds complementarily to target RNA. The ASO sequence confers extremely high target specificity, and its presence in the nucleus allows target engagement at the earliest stages of processing [15]. Here, backbone and 2′ sugar modifications determine the activity of an ASO, which we divide into RNase-mediated degrading or non-degrading mechanisms [10].
ASOs working through RNase H-dependent cleavage mechanisms (Fig. 2) are most commonly used in preclinical testing and in clinical trials for ALS. RNase H responds to RNA–DNA heteroduplexes, cleaves the RNA strand, and releases intact DNA [22, 23]. The templating of an ASO to its target RNA mimics this RNA–DNA pairing, which prompts RNase H to cleave the RNA transcript while the intact ASO is released to bind new transcripts. Cleaved transcripts cannot be translated to protein, so repeated ASO binding events will exhaust any pools available for translation, resulting in sustained protein reduction. As described earlier, ASOs operating by this mechanism must have at least a portion containing bases unmodified at the 2′ position, such as with a gapmer design.

Antisense oligonucleotide-mediated degradation of a gene target. Once delivered to the cerebrospinal fluid, antisense oligonucleotides (ASOs) are taken up into cells and can enter the nucleus. ASOs then bind to their gene target in a complementary fashion, creating a RNA–DNA hybrid that recruits the enzyme RNase H to cleave the target RNA. This action results in degradation of the gene target but spares the ASO, allowing it to persist in the cell. Without the RNA template, the gene will not be translated into protein, and the protein product is reduced over time. ASO therapeutics under investigation for amyotrophic lateral sclerosis (ALS) commonly employ this RNA-degrading strategy. Figure created with BioRender.com
Fully modified ASOs bind to a target transcript with high affinity and act through non-degrading mechanisms by sterically hindering trans-acting elements at the targeted sequence. This property allows fully modified ASOs to influence mRNA processing steps, such as alternative splicing or translational control [10]. Splicing events occur at ~ 90% of mRNA transcripts in human cells, and each requires that a full complement of splice factors act in concert at stereotyped sequence motifs. Thus, ASOs that occlude recognition motifs for splice enhancers/repressors or that mask conventional intron/exon junctions can redirect mRNA splicing [24]. Non-degrading ASOs may also modify RNA translation, stability, or localization by targeting critical post-transcriptional regulatory hubs or trans-acting elements, such as microRNAs (miRNAs), that act on these hubs [25,26,27].
Therapeutic Uses for ASOs and Current Limitations
Current challenges for some ASO therapies, particularly those with central nervous system (CNS) targets, include immune stimulation and CNS delivery. Toll-like receptors recognize foreign DNA and respond to specific sequence motifs [12, 28]. For example, CpG oligodeoxynucleotides activate toll-like receptor 9 to induce a potent interferon response [12, 28]. At high concentrations, early ASOs with a PS backbone could stimulate an inflammatory response, but advancements in backbone and 2′ sugar modifications now can mitigate some undue immune stimulation [12, 13]. ASOs cannot readily cross the blood brain barrier. Thus, administration of ASOs to the CNS in preclinical and clinical testing requires intracerebroventricular (i.c.v.) and intrathecal routes, respectively, into the cerebrospinal fluid (CSF). Fortunately, direct injection into the CSF allows the ASO to distribute throughout the CNS, and initial trials using SOD1-targeted ASOs have demonstrated intrathecal administration as a well-tolerated, safe, and efficacious approach to ASO delivery [29,30,31]. New delivery approaches such as conjugating ASOs to lipid nanoparticles or cholesterols may soon enable peripheral administration or cell type targeting in the CNS [32,33,34].
Despite these challenges, the sequence and chemistry of ASOs make them powerful and precise preclinical investigational tools that can be designed with clinical translation in mind. ASOs that recruit RNase H are particularly useful in combating disease processes linked to toxicity of a single gene product (protein or RNA), a feature shared across many neurodegenerative diseases [10]. Non-degrading ASOs have also achieved remarkable success, most notably with the FDA approval of the splice-switching ASO nusinersen for the treatment of SMA [35, 36]. New uses for ASOs are under active investigation that may soon broaden their applicability to other disease mechanisms, such as genetic haploinsufficiency.
ASOs Targeting SOD1
Mutations in SOD1 account for about 2% of all ALS cases and 15–25% of familial cases [37]. Post-mortem tissue from patients with SOD1-ALS demonstrates SOD1 aggregation, motor neuron degeneration, and muscle denervation. These features also appear in rodent model systems that express mutant human SOD1 [38] but are absent in rodents lacking SOD1 [39], supporting a toxic GOF mechanism for mutant SOD1. Transgenic mice and rats carrying copies of human mutated SOD1G93A are a well-validated cornerstone of preclinical ALS research. Like SOD1-ALS, SOD1G93A rodents develop aggregates of SOD1 in the cytoplasm of motor neurons, exhibit progressive motor neuron loss and weakness, and die prematurely [40, 41]. Mutations in SOD1 also exacerbate disease processes including oxidative stress [42], mitochondrial dysfunction [43], and excitotoxicity [44]. Collectively, these studies support the prediction that reducing SOD1 might slow ALS progression.
Preclinical
The first ASO therapy to target and decrease SOD1 protein emerged from testing in human cells and mutant SOD1 animal models [45]. The first candidate SOD1-lowering ASOs achieved robust, dose-dependent responses in cell culture, and i.c.v. infusion into a non-transgenic rat afforded > 60% knockdown of SOD1 mRNA throughout the brain and spinal cord [45]. The widespread distribution of ASO throughout the brain and spinal cord as demonstrated in this first study was unexpected given that ASOs are relatively large molecules and highly charged [8]. This distribution remains incompletely understood but has been documented in multiple subsequent studies that are reviewed below. When tested in SOD1G93A rats, ASOs reduced SOD1 mRNA by 40–60% throughout the brainstem and spinal cord and extended survival by 37% after disease onset [45]. As discussed below, these studies along with toxicology studies led to a successful phase 1 study (NCT01041222) using candidate ASO 333611, which was subsequently redeveloped to incorporate more advanced ASO technologies. Next generation ASOs containing 2’-MOE and 2′-O-ethyl modifications were advanced through a development pipeline, demonstrating superior target engagement and stronger knockdown over ASO 333611 in in vitro and in vivo pharmacokinetic studies [46]. In addition, i.c.v. injection of these ASOs into transgenic SOD1G93A rodents before disease onset profoundly extended survival, slowed motor impairment, and dampened neuromuscular injury [46]. ASOs also decreased serum levels of phosphorylated neurofilament heavy chain (pNFH), which increases alongside neuronal injury and has emerged as an important biomarker for ALS [1]. When delivered after disease onset, ASOs suppressed further increases in neurofilament and remarkably restored neuromuscular activity to near baseline. When advanced to nonhuman primates (NHPs), intrathecally delivered ASOs achieved favorable target engagement and distribution [46]. ASOs lowered SOD1 in NHPs across the CNS in a dose-dependent manner, with the highest dose achieving ~ 50% knockdown throughout the spinal cord. The profound functional rescue observed in rodent models and favorable pharmacokinetics in NHPs motivated another phase 1 a/b clinical trial (NCT02623699) for the new ASO BIIB067, discussed below.
Tracking ASO target engagement and patient response to therapy would enable real-time feedback of ASO efficacy and help inform upon trial design. To achieve this goal, a reasonable approach might be to measure the targeted protein from the CSF as a surrogate measure of ASO activity. However, SOD1 has a long half-life in the CSF (>20 days) [47], so therapies that lower its expression would lag actual ASO activity. An alternate approach might be measuring changes in SOD1 synthesis, rather than absolute levels. This strategy using stable isotope labeling and mass spectrometry was demonstrated in SOD1 transgenic mice treated with SOD1-lowering ASO [48]. At 30 days post i.c.v. injection, SOD1 in the CSF was minimally lowered, but a > 50% reduction in newly synthesized SOD1 labeled with 13C6-leucine could be detected as early as 10 days after dosing [48]. Importantly, levels of labeled SOD1 matched SOD1 mRNA levels observed in tissue from the brain and spinal cord. For human clinical trials, this approach could provide an early measure of target engagement in CNS tissues and guide trial design for CSF draws and dosing.
Clinical
Despite common neuropathology, individuals with SOD1-ALS may display considerable heterogeneity in clinical presentation and history [49]. Of note, a natural history study on SOD1-ALS cases reported that the specific mutation in SOD1 is an important modifier for disease progression rate [50]. For example, the SOD1A4V mutation comprises most SOD1-ALS cases in the USA and results in a characteristically aggressive disease. SOD1A4V and several other mutations are now associated with “fast-progressing” disease, which has become an important measure to stratify clinical trial group design. Incorporating these and other modifiers for disease will be crucial for designing and interpreting clinical trials that are powered to detect treatment effects.
Convincing target engagement and disease rescue from initial animal studies combined with an acceptable profile in toxicology studies prompted a phase 1, double-blind, placebo-controlled clinical trial to study the safety and tolerability of intrathecally delivered ASO 333611 in SOD1-ALS patients [30]. This trial marked the first-in-man for a CNS-targeted ASO, predating trials of nusinersen or similar ASO. Given the first-in-man nature of the study, a single course (12 h slow intrathecal infusion) of a low dose (0.15 to 3 mg) was given. Results from this trial (NCT01041222) supported intrathecal infusion of ASOs as a favorable and well-tolerated route for administration to the CNS with no serious adverse effects. Although a reduction in SOD1 protein levels in the CSF was not detected, the trial tested conservatively low doses and a short time interval [30]. These studies [30, 47] were a landmark for ASO therapy, demonstrating for the first time that intrathecal infusion of ASOs is safe in humans and efficacious in animals and establishing a pipeline to rapidly advance ASOs from initial selection to clinical trials.
Following the initial SOD1 ASO trial, a phase 1–2, randomized, double-blind, placebo-controlled trial (VALOR; NCT02623699) was conducted starting in January 2016 to assess tolerability and pharmacokinetics of tofersen, the redeveloped SOD1-targeting ASO (BIIB067) in patients with SOD1-associated ALS [29]. Fifty participants were recruited and assigned to one of four dose cohorts (20, 40, 60, or 100 mg). Within a cohort, each participant was randomized to receive five doses of tofersen or placebo administered by five intrathecal injections over 12 weeks. Unlike the first SOD1 ASO trial where the ASO was delivered via intrathecal catheter and external pump, this trial (and subsequent ASO trials) used an intrathecal bolus.
Tofersen was overall well-tolerated and safe. The most common adverse events were minor and related to lumbar puncture. Consistent with predicted SOD1 pharmacokinetics in CSF, SOD1 protein levels in the CSF were unchanged through study day 30. At study day 85, SOD1 was reduced in the 40 mg (27%), 60 mg (21%), and 100 mg (36%) groups, demonstrating effective target engagement [29]. Forty-two percent of participants receiving tofersen had lymphocytic pleocytosis, compared with 8% in placebo groups, although the significance of this finding is unclear.
Exploratory clinical measures in this trial included functional rating, lung vital capacity, muscle strength by handheld dynamometry, and both phosphorylated heavy chain (pNFH) and neurofilament light chain (NfL) concentrations in the CSF and plasma. Subgroup post hoc analysis for exploratory measures was performed for participants with fast-progressing disease. Though not powered to detect an effect on clinical measures, the progression of disability was tracked with ALS Functional Rating Scale-Revised (ALSFRS-R), which is a well-established, investigator-obtained scale [1, 51]. There was a trend toward slowing of decline in ALSFRS-R measured in the 100 mg group, which was further magnified in the fast-progressor subgroup. This trend was also observed for predicted slow vital capacity and handheld dynamometry mega-score. Levels of plasma and CSF pNFH and NfL were reduced by day 85 in the 100 mg group compared to the day 1 baseline or placebo group. These promising results also supported segregating analysis on slow and fast progressors in subsequent trials.
The favorable safety profile and effective target engagement achieved in phases 1–2 motivated the advancement of tofersen to a 28-week phase 3, randomized, double-blind, placebo-controlled trial (VALOR; NCT02623699). This study evaluated the efficacy and safety of intrathecally administered tofersen at 100 mg (top dose from phases 1–2) in SOD1-ALS patients. A total of 108 participants with clinically manifest disease were enrolled and randomized to tofersen (n = 72) or placebo (n = 36) treatment arms. Consistent with analysis from the tofersen phase 1–2 trial [29] and to reduce the effects of the known heterogeneity of this genetic population, subgroups of fast and slow progressive patients were prespecified with the effect on progression in the fast-progressing group as the primary outcome. Sixty participants were enrolled for “fast-progressing” ALS. This trial continued through 2021, and results from this study were presented at the American Neurological Association Annual (ANA) Meeting in October 2021. A phase 3 long-term, open-label extension study (NCT03070119) continues to follow participants who received tofersen to assess long-term safety, tolerability, and efficacy.
Data slides from the ANA meeting were made publicly available and are the source of the results described here. The most common adverse events with tofersen at 100 mg were related to the lumbar puncture procedure [31]. Serious adverse events occurred at similar rates for tofersen (18.1%) and placebo (13.9%). No placebo-treated participants discontinued the study, compared with 5.6% of participants receiving tofersen. Of note, 4.8% of tofersen-receiving patients reported a severe neurologic event, including two cases of reversible transverse myelitis. There were increases in CSF protein in both placebo (8%) and tofersen (13%)-treated groups and a “shift to high leukocytes” in only tofersen-treated groups (10%). The significance of these CSF changes remains unclear. No data have yet been presented regarding the correlation of these changes with clinical outcomes or other measures, but this information will be important for understanding these data.
The study missed the primary outcome for efficacy, which was change from baseline across 28 weeks in ALSFRS-R [31]. However, multiple secondary and exploratory endpoints supported favorable clinical and biomarker trends for tofersen treatment. Within the fast-progressing group, tofersen decreased CSF SOD1 (38%) and plasma NfL (67%). Slow progressors showed similar but smaller reductions in SOD1 (26%) and NfL (48%). Among fast progressors, tofersen showed trends toward benefit in respiratory (slow vital capacity) and muscle strength (handheld dynamometry) with differences between placebo and tofersen most apparent in the open-label extension (OLE). Slow progressors initially treated with tofersen showed very little decline in ALSFRS-R, slow vital capacity, and muscle strength, and slow progressors on placebo showed some apparent disease stabilization after being treated with tofersen in OLE. Data from this study are anticipated to continue to emerge and will be important to understand the long-term trends in OLE as well as the effect on survival.
Given the favorable safety profile and clear evidence of SOD1 protein lowering, the SOD1 ASO is now being considered for treatment of asymptomatic gene carriers. A reasonable hypothesis is that treating individuals with SOD1-lowering drugs at the earliest evidence of biomarker changes, but prior to development of overt motor neuron disease, may slow or prevent their decline to clinically manifest disease. This prediction has motivated another phase 3 trial using tofersen (ATLAS; NCT04856982) to investigate its efficacy in presymptomatic adults with SOD1 mutations who have elevated neurofilament indicating imminent phenoconversion. This randomized, placebo-controlled trial will recruit ~150 participants and extend from May 2021 to August 2027. Presymptomatic participants without clinically manifest ALS will be followed longitudinally and will have frequent monitoring of serum NfL levels. If a participant exhibits high NfL, they will then be randomized to receive either 100 mg tofersen or placebo monthly for 2 years. An open-label extension period will allow participants who develop clinical ALS to receive tofersen in the unblinded arms of the study. The primary measure in this trial is the percentage of participants that clinically manifest ALS within 12 months of treatment. Important secondary measures include time to manifest ALS, ALSFRS-R scores, and concentrations of CSF SOD1 and plasma NfL.
ASOs Targeting C9orf72
Hexanucleotide expansion in intron 1 of C9orf72 is the most frequent genetic cause of frontotemporal dementia (FTD) and ALS, accounting for ~ 40% of fALS cases [4,5,6]. How C9orf72 expansion causes disease remains an open question, but post-mortem samples and model systems of disease have highlighted multiple mechanisms contributing to C9orf72 pathogenicity [52]. First, the mutant C9orf72 gene can be transcribed bi-directionally, resulting in expression of sense and antisense RNA strands. Toxicity may occur from the intranuclear deposition of sense and antisense C9orf72 into RNA foci and the folding of the GGGGCC repeats into a G-quadruplex, which itself can sequester important RNA-binding proteins and chromatin modifiers [53,54,55]. Second, both strands can produce strings of dipeptide repeats (DPRs), owing to a noncanonical repeat-associated non-ATG-mediated (RAN) translation mechanism. These dipeptides may be toxic, perhaps by disrupting proteostasis or nucleolar machinery [56,57,58,59]. Intracellular deposits of five DPRs appear in human post-mortem tissue, further supporting a toxic GOF mechanism [60,61,62]. Third, haploinsufficiency of C9orf72 limits its physiological functions, which include vesicle trafficking, autophagy, lysosomal processing, and immune response [63, 64], suggesting loss of normal C9orf72 can contribute to disease [65]. It is likely that both gain- and loss-of-function mechanisms synergize to cause disease [52, 66]. Altogether, the above findings largely support ASO reduction of expanded C9orf72 RNA, but not wild-type C9orf72, as a promising therapeutic strategy to reduce toxicity due to DPRs and repeat-containing RNA.
Preclinical
Cells derived from patients with C9orf72 expansions phenocopy key cellular features seen in post-mortem tissue, including intranuclear RNA foci and intracytoplasmic DPR deposits [67]. Because these features are likely toxic and an immediate consequence of mutant C9orf72 expression, these provide excellent read-outs for ASO efficacy in suppressing C9orf72 and resolving toxicity. Three reports, published almost simultaneously in 2013, were first to explore the therapeutic potential of ASO-mediated degradation of C9orf72 [68,69,70]. When delivered to patient-derived fibroblasts [68], iPSC neurons [70], or motor neurons [69, 70], C9orf72-targeting gapmer ASOs could potently reduce repeat-containing C9orf72 transcripts and clear intranuclear RNA foci. Since 2013, new ASO formulations have shown promise in cellular models. Two studies further demonstrated that ASOs with a stereopure backbone [71, 72] or mixed backbone containing LNAs [73] also could reduce expansion-containing transcripts and attenuate pathology using C9orf72-ALS patient-derived cells.
One unique opportunity for C9orf72 targeting comes from the multiple transcript variants produced. Splice variants 1 and 3 contain the intron that harbors the expansion, whereas variant 2 does not. Variant 1 leads to a short isoform protein; variants 2 and 3 produce the same long isoform proteins. By targeting intron 1, multiple groups [68,69,70,71,72,73] (referenced above) have achieved lowering of the expansion-containing transcripts while preserving variant 2 and thus not further suppressing C9orf72 protein levels. This quality is important because it is likely not desirable to degrade all C9orf72 transcripts, as C9orf72 protein has important physiological functions [52]. However, one remaining challenge for C9orf72 is that the expanded C9orf72 DNA is transcribed bi-directionally, producing both sense and antisense RNA strands that deposit in RNA foci. At present, all ASOs in development only bind and degrade the sense strand, while DPRs and RNA foci produced from antisense C9orf72 remain. In cellular studies, C9orf72-targeting ASOs did decrease RNA foci, but this reduction occurred predominantly through clearance of foci composed of sense strand RNA without affecting antisense foci [68]. This inability to neutralize simultaneously both sense and antisense strands remains an important limitation to current C9orf72 ASO therapies [70].
Transgenic mice carrying a bacterial artificial chromosome with the full human repeat-containing C9orf72 gene are commonly used rodent models for C9orf72-associated disease [74, 75]. These animals develop RNA foci and DPR proteins throughout the cortex and spinal cord but do not exhibit neurodegeneration or motor impairments. Some, however, develop progressive cognitive impairment and anxiety phenotypes reminiscent of FTD [74]. Across multiple studies [71,72,73,74], ASOs targeting C9orf72 were delivered by i.c.v. injection prior to phenotypic onset. ASOs decreased repeat-containing C9orf72 throughout the brain and spinal cord without affecting total C9orf72 RNA. Expectedly, ASOs decreased DPR levels and frequency of sense foci by > 50%, without appreciable effects on amounts of antisense foci [72,73,74]. Intron-targeting ASOs delivered after phenotypic onset lowered DPRs [72,73,74] and rescued behavioral assays for anxiety and cognitive function to near levels seen in non-transgenic mice at 12 or 15 months [74]. These encouraging data from cellular and animal models have since motivated testing in humans. Below, we discuss a phase 1 trial (NCT03626012) for the ASO BIIB078, a recently initiated phase 1b/2a trial (NCT04931862) for the stereopure ASO WVE-004 [71, 72], and early results from a single individual receiving the mixed backbone ASO afinersen under an Investigational New Drug (IND, “compassionate use”) application [73].
With clinical trial design in mind, measuring RAN proteins produced from expanded C9orf72 RNA could provide a way to track ASO target engagement and monitor response to therapy [76]. The RAN product poly(GP) is produced from RAN translation of both C9orf72 RNA strands and can be detected in the CSF of mutation carriers. Notably, CSF poly(GP) is present only in C9orf72 individuals and remains stable over time in expansion carriers despite disease progression [76, 77]. Upon knockdown of C9orf72 by ASOs in repeat-expressing transgenic mice, poly(GP) in the CSF sharply falls, which correlates robustly with reductions observed in tissue poly(GP) levels, RNA foci, and repeat-containing C9orf72 RNA. Altogether, the specificity of poly(GP) to mutation carriers, the stability across disease progression, and its strong correlation to cellular pathology post-treatment make CSF poly(GP) an excellent pharmacodynamic marker that could be used in emerging clinical trials [76].
Clinical
Understanding clinical measures in C9orf72-ALS is critical for designing powered studies that target C9orf72-ALS patients and measure clinical outcomes most relevant to this population. A natural history study tracking C9orf72-ALS disease identified that C9orf72 cases have more homogenous clinical presentation, later onset, and largely similar disease duration, compared to sporadic cases [78]. As a biomarker, CSF levels of poly(GP) are stable over disease course and can segregate C9orf72-ALS from sporadic cases. Importantly, CSF poly(GP) correlates strongly with poly(GP) concentrations in post-mortem cortex and cerebellum [78]. These findings should provide guidance in designing appropriately powered studies and support measuring poly(GP) in the CSF as a correlate to target engagement.
Based on favorable results using ASOs in patient-derived cells [68,69,70] and transgenic mice [74], a randomized, placebo-controlled phase 1 trial (NCT03626012) was conducted to examine the safety and tolerability of the C9orf72-lowering ASO BIIB078. In this first-in-man, ascending dose trial, 114 participants with C9orf72-ALS were recruited and administered BIIB078 or placebo by intrathecal infusion. Inclusion criteria also required a prestudy ALSFRS-R slope of less than 0.4 points per month, which excludes “fast progressors.” This trial continued from September 2018 to November 2021, and early results were announced in a press release in March 2022 [79]. BIIB078 was well-tolerated with only mild to moderate adverse effects. Secondary endpoints evaluated pharmacokinetics and clinical measures, such as change in ALSFRS-R, slow vital capacity, and muscle strength. None of these endpoints were met, and participants that received BIIB078 trended toward greater clinical decline. Further development on BIIB078 has been discontinued.
A randomized, placebo-controlled phase 1b/2a trial (NCT04931862) was also initiated for the stereopure C9orf72-lowering ASO WVE-004 [71, 72] in patients with ALS and/or FTD due to C9orf72 expansions. WVE-004 or placebo will be administered intrathecally to participants at four ascending doses to evaluate safety and tolerability. Secondary outcome measures will assess pharmacokinetics and pharmacodynamics, including changes in poly(GP) levels in the CSF. This trial began in July 2021 and is expected to continue through February 2023 with estimated enrollment of 42 participants. A press release in April 2022 announced early results for a portion of participants. Most adverse events related to WVE-004 were mild to moderate, and no elevations in CSF leukocytes or proteins were observed. A single dose of WVE-004 achieved durable reductions in poly(GP) in the CSF, indicating successful target engagement. No change in clinical outcome measures was observed in participants receiving WVE-004, and CSF NfL was slightly elevated.
An IND application (IND141673) was approved by the FDA for the testing of the mixed backbone ASO afinersen in a single participant, a 60-year-old male with a C9orf72 expansion [73]. Prior to this application, toxicology assessments in rodents, sheep, and monkeys supported safety of afinersen [73]. At treatment initiation, this individual had mild motor changes with elevated poly(GP) in the CSF. Eight escalating doses were administered intrathecally over 60 weeks, starting in August 2019. Afinersen distributed well throughout the CSF, and compared to baseline, CSF poly(GP) levels dropped by 80%, suggesting that afinersen engaged and decreased C9orf72. ALSFRS-R remained stable throughout treatment. In contrast with studies of tofersen for SOD1-ALS, afinersen treatment was associated with increased pNFH and NfL in the CSF [73]. At the time of this review, no clinical trial is yet registered for afinersen.
ASOs Targeting FUS
Mutations in fused in sarcoma (FUS) cause a rare and aggressive form of ALS with early and often juvenile onset [7]. FUS is an RNA-binding protein that is critical in DNA repair and RNA metabolism, including the splicing and translation of mRNA [80]. ALS-associated mutations in FUS may cause partial loss of function (LOF), but mechanistic studies have demonstrated GOF mechanisms are significant drivers of motor neuron loss in FUS-ALS [81]. While most forms of ALS commonly show TDP-43 pathology, postmortem tissue from FUS-ALS patients exhibits intracytoplasmic aggregation of FUS in the absence of TDP-43 pathology [82]. The role of FUS in RNA metabolism requires frequent translocation between the nucleus and cytoplasm [80], but mis-localization and sequestration of FUS in aggregates likely impairs normal functions, which argues for a LOF mechanism. However, this conclusion contrasts with findings from in vivo models in which loss of FUS in motor neurons induced no motor neuron loss or denervation [83], whereas transgenic mice overexpressing wild-type FUS develop aggressive motor neuron loss and intracellular inclusions composed of FUS and other RNA-binding proteins [84]. Furthermore, intracellular aggregates of FUS appear to sequester or decrease solubility of other RNA-binding proteins, such as hnRNPA1 or UPF1, that are critical for RNA metabolism. Collectively, these studies using FUS-ALS human tissue and rodent model systems support a toxic GOF mechanism contributing to FUS-ALS. Therefore, ASO therapies that decrease FUS expression may hold promise for individuals with FUS-ALS.
Preclinical
Knock-in mice that express wild-type and mutant FUSP525L from the FUS locus develop progressive loss of spinal motor neurons and intracellular aggregates of insoluble FUS but do not exhibit motor impairments or reduced survival [85]. Identified from in vitro screen, the ASO ION363 targeting the 6th intron of FUS decreases expression of both mutant and wild-type transcripts. I.c.v injection of ION363 in FUSP525L mice decreased both mutant and wild-type FUS by 20–50% in the brain and spinal cord by 1 month post-injection. ION363 reduced levels of insoluble FUS and insoluble RNA-binding proteins that associate with FUS aggregates, such as hnRNPA1. Moreover, ION363 prevented neurodegeneration of lumbar motor neurons and loss of neuromuscular junction innervation, which were sustained at 4 and 6 months [85]. Reversal of neurodegeneration and reductions in FUS in FUSP525L mice motivated an IND (“compassionate use”) application for testing in humans with FUS mutations [85].
Clinical
An IND application was approved by the FDA for the use of ION363 in a single ALS patient with a FUSP525L mutation (age 26). Under this application, no toxicology assessments in rodents or monkeys were needed. The participant received 12 injections between June 2019 and March 2020 before her dying from ALS in May 2020 [85]. She received monthly ascending doses intrathecally, starting from 20 mg up to maximum 120 mg. Treatment began at 6 months post-clinical onset, at which point her disease reached advanced stage. The participant had no serious adverse events. Since her death, more than a dozen individuals carrying FUS mutations with presymptomatic or symptomatic ALS have also received ION363 through Expanded Access Program IND applications.
Neuropathological examination was performed for the first participant, alongside a non-ALS control and an ALS patient with the FUSP525L mutation who did not receive therapy [85]. Relative to ALS control, the FUS ASO-treated participant had less total FUS and mutant FUS protein, including insoluble FUS, in the lumbar spinal cord. Levels of other insoluble RNA-binding proteins, such as hnRNPA1, were also lower. In the spinal cord and motor cortex, there was little nuclear FUS staining, and aggregates containing FUS in motor neurons were sparse compared to the untreated ALS-FUSP525L control in which FUS aggregates were abundant. These examinations suggest that ION363 treatment in this participant correlates with reduced FUS pathology.
A phase 3 clinical trial (FUSION; NCT04768972) to assess efficacy, safety, and pharmacology of ION363 (now jacifusen) was initiated for patients with FUS-ALS. Over April 2021 through March 2024, 64 patients will be enrolled and assigned to jacifusen or placebo at a 2:1 ratio, including any who had previously received jacifusen under IND applications. Jacifusen will be administered monthly or bimonthly for 29 weeks in part 1 of the study. Part 2 of the study will be a subsequent 72 week open-label extension period in which all participants will receive jacifusen. A unique component of this trial is the involvement of a “rescue.” Specifically, if a participant shows significant functional decline during part 1, they will be moved to the part 2/open-label extension of the study. The primary outcome is a joint rank analysis of ALSFRS-R, time to rescue (as described above), and ventilation assistance-free survival. Secondary outcomes include muscle and lung function, survival, and change in CSF FUS protein and neurofilaments.
ASOs Targeting ATXN2
Trinucleotide repeat expansions in ataxin-2 (ATXN2) are a cause of the neurodegenerative disease spinocerebellar ataxia type-2 (SCA2) and an identified risk factor for ALS. ATXN2 is an important regulator of mRNA polyadenylation and stress granule assembly that contains an unstable polyglutamine (polyQ) tract encoded by repeats of CAG [86]. Normal alleles usually have 22 or 23 repeats, while patients with SCA2 carry an allele with a longer expansion of >34 repeats [87]. Intermediate length expansions of 27–33 repeats in ATXN2 are now recognized as a significant heritable modifier of ALS risk [7]. The link between ATXN2 and ALS was first identified when a genetic screen in yeast found that upregulating Pbp1 (ATXN2 homolog) severely worsens cell survival due to TDP-43 overexpression, while lowering Pbp1 suppresses this toxicity [88]. This finding was important, as the nuclear depletion, cytoplasmic mislocalization, and pathologic aggregation of TDP-43 are hallmarks across > 95% of ALS cases. Furthermore, mutations in the gene encoding TDP-43, TARDBP, are a well-established cause of familial ALS. Thus, TDP-43 dysfunction likely plays a critical role in the vast majority of ALS cases and targeting ATXN2 may limit TDP-43 dysfunction. Neuropathological examination of post-mortem tissue from patients with ALS with and without intermediate length ATXN2 expansions also revealed that spinal motor neurons often contain large intracytoplasmic deposits of ATXN2 [88, 89]. In human population genetics studies, sequencing for polyQ repeat lengths from >900 patients with ALS and control cases revealed a 2.8-fold enrichment of intermediate expansions in the ALS cohort, detected in 4.8% of cases [88]. These results have since been validated in additional population-based cohorts, for example, showing 5.4- [90], 4.8- [91], or 3.0-fold [92] enrichments.
Mounting evidence supports ATXN2 and intermediate-length polyQ expansions in ATXN2 in altering RNA metabolism or stimulating the pathological transformation and deposition of TDP-43. First, intermediate length expansions can directly promote TDP-43 aggregation by activating caspases and downstream effectors that cleave and phosphorylate TDP-43, though these events do not occur with non-expanded ATXN2 [93]. Second, intermediate length expansions increase the stability of ATXN2 and allow for greater molecular association with TDP-43, which is especially apparent under stress conditions when stress granules are induced [88]. While other mechanisms related to RNA processing or receptor trafficking may be involved, it is proposed that non-expanded ATXN2 modifies disease through modulating stress granule dynamics. Stress granules are dynamic, membrane-less cytoplasmic condensates composed of RNA and proteins that assemble during cellular stress to temporarily stall translation [94]. If cellular stress does not resolve, such as in disease, stress granules persist, which may limit protein production, sequester critical proteins, and provide a crucible for RNA-binding proteins to interact [95]. As ATXN2 is a critical organizer of stress granules, the increased stability afforded to intermediate length expansions in ATXN2 might exacerbate disease by affecting their resolution or prolonging sequestration [86]. For example, TDP-43 often localizes within stress granules, and these condensates may promote TDP-43 self-association and mis-localization to the cytoplasm [88, 96]. As alluded to above, ATXN2 modifies cell toxicity or survival due to overexpressed TDP-43 in yeast [88], flies [88], and mice [97]. Because TDP-43-proteinopathy and disrupted RNA processing are hallmarks across most forms of ALS [98], the ability for ATXN2 to modify these processes makes ATXN2 an attractive target beyond ATXN2 expansion-related ALS.
Preclinical
ASO development and preclinical testing for ATXN2 knockdown were simultaneously performed for model systems of ATXN2-repeat expansion disease SCA2 and an ALS model system of TDP-43-proteinopathy [97, 99]. In the initial development of ASOs for SCA2, a promising ASO candidate was selected that targets exon 11 of human ATXN2 [99]. In two SCA2 models of ATXN2 expansion (ATXN2-Q127 and BAC-Q72), ASOs reduced human ATXN2 ~ 75% in the cerebellum, cortex, and spinal cord. This result was accompanied by striking rescue of disease-relevant gene expression and functional readouts, such as firing frequency of cerebellar Purkinje cells [99]. This study was a promising demonstration of ATXN2-lowering therapy in a model of ATXN2-expansion disease, supporting its translation to animal models of ALS.
ATXN2 lowering therapy for ALS was evaluated in a transgenic mouse model of TDP-43-proteinopathy that drives pan-neuronal expression of human wild-type TDP-43 [97]. This model is aggressive and results in rapid and robust motor dysfunction and death around postnatal day 24. Genetic ablation of ATXN2 dramatically extended lifespan by 80%, limited gait dysfunction, and slowed disease progression [97]. Consistent with a stress granule-mediated mechanism, loss of ATXN2 did not affect total TDP-43 levels but did reduce TDP-43 inclusions. When administered on postnatal day 1 to TDP-43-proteinopathy mice, an ATXN2-lowering ASO extended lifespan by 35% and improved gait performance, despite the aggressive decline in this model system [97]. These studies supported the initiation of a clinical trial for ATXN2-lowering ASOs in ATXN2-associated ALS and sporadic ALS.
Clinical
A randomized, placebo-controlled phase 1 trial (NCT04494256) was initiated in September 2020 to evaluate the safety and pharmacokinetics of an intrathecally delivered ASO designed to lower ATXN2 (BIIB105) in patients with ATXN2-associated ALS and sporadic ALS. The study expects enrollment of 70 participants with an end date of February 2023. Dosing arms will segregate individuals with sporadic ALS from ATXN2-associated ALS. Within each group, participants will be assigned to receive placebo or one of five sequentially escalating doses of the ATXN2 ASO.
Limitations and Future Directions
Limitations to Treating ALS with ASO Therapy
Limitations to treating ALS with ASOs fall into three general categories: challenges associated with the disease itself, ASO pharmacokinetic/pharmacodynamic properties, and potential on and off target toxicities of the ASO. Regarding ALS disease, there may be challenges in initiating any therapy after the disease has started. While there are good examples of reversals of function or pathology in animal models of neurodegeneration, it is not clear yet whether the neurodegenerative disease process may be halted in humans. Regarding ASO pharmacokinetic/pharmacodynamics, there are multiple favorable properties of ASOs including widespread distribution, long half-life, and proven target reduction in humans for multiple targets including SOD1. However, the details of drug delivery are not entirely clear in humans with limited data on correlations between CSF measurements and tissue levels. Whether an individual ASO has lowered the target sufficiently in the cell types of interest, for example, motor neurons, remains incompletely understood. Toxicities may also limit the effectiveness of the ASO. On-target toxicity associated with lowering levels of the target gene product may introduce some unknown side effects. While the toxicity of a dominantly inherited mutation likely outweighs the risk of lowering the gene product, this possibility will need further consideration. Off-target toxicity, particularly activation of the immune system, has been a longstanding potential concern with ASO use. The presence of immune cells, elevated protein composition, and a few cases of myelitis seen in human clinical trials may indicate some pro-inflammatory effects. While the full data from the recent SOD1 ASO trial are not yet available, there were no correlations observed yet between changes in CSF parameters and disease course or response to the ASO.
New Targets for ASO Therapy
Dozens of genes are now linked with ALS, and each represents a putative target for ASO therapy. Most cases of fALS can be linked to mutations in SOD1, C9orf72, FUS, or ATXN2, which has likely hastened ASO development for these genetic forms of disease. ASOs are now in development to target genes associated with other forms of fALS. These may be supported by new ASO strategies that are under active investigation, including approaches to increase gene expression. ASOs that block the action of translational repressors on a target transcript might increase protein translation. Gene upregulation would have implications for treating haploinsufficiency disorders, such as TBK1-associated ALS, and for targeting pathways associated with sporadic ALS.
ASOs may also be effective therapeutics for non-genetic forms of ALS, as may be the case for ATXN2. Stathmin-2 and UNC13A are two important splice targets of TDP-43 that are essential for neuron health and activity. Loss of nuclear TDP-43, a hallmark across ALS cases, induces mRNA splicing defects, and mis-splicing of stathmin-2 and UNC13A causes their reduced protein expression and function [100,101,102,103,104]. ASOs that correct splicing defects and restore stathmin-2 or UNC13A protein might ameliorate some downstream consequences of TDP-43-proteinopathy.
Conclusions Regarding ASO as Therapeutics
The successful progression of ASOs through the drug development pipeline has made them strong candidates for many diseases with single gene mutations or prominent toxic species. Beyond ALS, ASO trials have consistently demonstrated effective target engagement and action, with a marked change to biomarkers that reflect disease processes. The number of implicated genes for ALS grows with each year, and with the emergence of streamlined pipelines to clinical trial, ASO development for rare diseases becomes increasingly feasible. While complete understanding of ASO distribution and toxicities have lagged behind their clinical application, advancements in ASO technology will continue to expand their delivery, cell targeting properties, applicability in disease, and efficacy, enabling new promising therapeutics to keep pace with breakthroughs in ALS pathophysiology.
References
van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390(10107):2084–98.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62.
Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17(1):17–23.
Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 2012;11(3):232–40.
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56.
Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206.
Schoch KM, Miller TM. Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron. 2017;94(6):1056–70.
Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;1–22.
Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol. 2010;50(1):259–93.
Scoles DR, Minikel EV, Pulst SM. Antisense oligonucleotides. Neurology. Genetics. 2019;5(2):e323.
Agrawal S, Kandimalla ER. Antisense and siRNA as agonists of Toll-like receptors. Nat Biotechnol. 2004;22(12):1533–7.
Shen W, Hoyos CLD, Migawa MT, Vickers TA, Sun H, Low A, et al. Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index. Nat Biotechnol. 2019;37(6):640–50.
Robbins M, Judge A, Liang L, McClintock K, Yaworski E, MacLachlan I. 2′-O-methyl-modified RNAs act as TLR7 antagonists. Mol Ther. 2007;15(9):1663–9.
Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliver Rev. 2015;87:46–51.
Iwamoto N, Butler DCD, Svrzikapa N, Mohapatra S, Zlatev I, Sah DWY, et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat Biotechnol. 2017;35(9):845–51.
Janssen HLA, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV Infection by Targeting MicroRNA. New Engl J Medicine. 2013;368(18):1685–94.
Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. 2016;79(2):257–71.
Hagedorn PH, Persson R, Funder ED, Albæk N, Diemer SL, Hansen DJ, et al. Locked nucleic acid: modality, diversity, and drug discovery. Drug Discov Today. 2018;23(1):101–14.
Kasuya T, Hori SI, Watanabe A, Nakajima M, Gahara Y, Rokushima M, et al. Ribonuclease H1-dependent hepatotoxicity caused by locked nucleic acid-modified gapmer antisense oligonucleotides. Sci Rep-uk. 2016;6(1):30377.
Moazami MP, Rembetsy-Brown JM, Wang F, Krishnamurthy PM, Weiss A, Marosfoi M, et al. Quantifying and mitigating motor phenotypes induced by antisense oligonucleotides in the central nervous system. Biorxiv. 2021. https://doi.org/10.1101/2021.02.14.431096.
Wu H, Lima WF, Zhang H, Fan A, Sun H, Crooke ST. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs*. J Biol Chem. 2004;279(17):17181–9.
Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes in eukaryotes. Febs J. 2009;276(6):1494–505.
Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016;44(14):6549–63.
Jia L, Mao Y, Ji Q, Dersh D, Yewdell JW, Qian SB. Decoding mRNA translatability and stability from the 5′ UTR. Nat Struct Mol Biol. 2020;27(9):814–21.
Bray N. The power of 3′ UTRs. Nat Rev Neurosci. 2018;19(6):319–319.
Kuersten S, Goodwin EB. The power of the 3′ UTR: translational control and development. Nat Rev Genet. 2003;4(8):626–37.
Younis HS, Vickers T, Levin AA, Henry SP. CpG and non-CpG oligodeoxynucleotides induce differential proinflammatory gene expression profiles in liver and peripheral blood leukocytes in mice. J Immunotoxicol. 2008;3(2):57–68.
Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, et al. Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. New Engl J Med. 2020;383(2):109–19.
Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435–42.
Miller T, Cudkowicz M. Results from the phase 3 VALOR study and its open-label extension: evaluating the clinical efficacy and safety of tofersen in adults with ALS and confirmed SOD1 mutation. American Neurological Association; 2021.
Kulkarni JA, Cullis PR, van der Meel R. Lipid nanoparticles enabling gene therapies: from concepts to clinical utility. Nucleic Acid Ther. 2018;28(3):146–57.
Suhr OB, Coelho T, Buades J, Pouget J, Conceicao I, Berk J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis. 2015;10(1):109.
Nagata T, Dwyer CA, Yoshida-Tanaka K, Ihara K, Ohyagi M, Kaburagi H, et al. Cholesterol-functionalized DNA/RNA heteroduplexes cross the blood–brain barrier and knock down genes in the rodent CNS. Nat Biotechnol. 2021;1–8.
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. New Engl J Medicine. 2017;377(18):1723–32.
U.S. Food and Drug Administration. FDA approves first drug for spinal muscular atrophy [Internet]. 2016. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy. Accessed 27 Apr 2022.
Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurology Neurosurg Psychiatry. 2017;88(7):540.
Gurney ME, Pu H, Chiu AY, Canto MCD, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–5.
Reaume A, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DR, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13(1):43–7.
Dawson TM, Golde TE, Lagier-Tourenne C. Animal models of neurodegenerative diseases. Nat Neurosci. 2018;21(10):1370–9.
Philips T, Rothstein JD. Rodent models of amyotrophic lateral sclerosis. Curr Protoc Pharmacol. 2015;69(1):5–67.
Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118(2):659–70.
Liu J, Lillo C, Jonsson PA, Velde CV, Ward CM, Miller TM, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43(1):5–17.
Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc National Acad Sci. 2002;99(3):1604–9.
Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Investig. 2006;116(8):2290–6.
McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Investig. 2018;128(8):3558–67.
Crisp MJ, Mawuenyega KG, Patterson BW, Reddy NC, Chott R, Self WK, et al. In vivo kinetic approach reveals slow SOD1 turnover in the CNS. J Clin Invest. 2015;125(7):2772–80.
Self WK, Schoch KM, Alex J, Barthélemy N, Bollinger JG, Sato C, et al. Protein production is an early biomarker for RNA-targeted therapies. Ann Clin Transl Neur. 2018;5(12):1492–504.
Picher-Martel V, Valdmanis PN, Gould PV, Julien JP, Dupré N. From animal models to human disease: a genetic approach for personalized medicine in ALS. Acta Neuropathologica Commun. 2016;4(1):70.
Bali T, Self W, Liu J, Siddique T, Wang LH, Bird TD, et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J Neurology Neurosurg Psychiatry. 2017;88(2):99.
Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3(1):17071.
Gendron TF, Petrucelli L. Disease mechanisms of C9ORF72 repeat expansions. Csh Perspect Med. 2018;8(4):a024224.
Reddy K, Zamiri B, Stanley SYR, Macgregor RB, Pearson CE. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures*. J Biol Chem. 2013;288(14):9860–6.
Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Štalekar M, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5(5):1178–86.
Rossi S, Serrano A, Gerbino V, Giorgi A, Francesco LD, Nencini M, et al. Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J Cell Sci. 2015;128(9):1787–99.
May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014;128(4):485–503.
Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014;128(4):505–24.
Zhang YJ, Gendron TF, Ebbert MTW, O’Raw AD, Yue M, Jansen-West K, et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med. 2018;24(8):1136–42.
Schmitz A, Marques JP, Oertig I, Maharjan N, Saxena S. Emerging perspectives on dipeptide repeat proteins in C9ORF72 ALS/FTD. Front Cell Neurosci. 2021;15:637548.
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–8.
Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126(6):881–93.
Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PEA, Caulfield T, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126(6):829–44.
O’Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AKMG, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351(6279):1324–9.
Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016;8(347):347ra93-347ra93.
Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24(3):313–25.
Zhu Q, Jiang J, Gendron TF, McAlonis-Downes M, Jiang L, Taylor A, et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 2020;23(5):615–24.
Guo W, Fumagalli L, Prior R, Bosch LVD. Current advances and limitations in modeling ALS/FTD in a dish using induced pluripotent stem cells. Front Neurosci-switz. 2017;11:671.
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc National Acad Sci. 2013;110(47):E4530–9.
Sareen D, O’Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149-208ra149.
Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Heusler AR, Mistry NA, et al. RNA Toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–28.
Liu Y, Andreucci A, Iwamoto N, Yin Y, Yang H, Liu F, Patil S, Mohapatra S, Purcell-Estabrook E, Taborn K, Dale E, Vargeese C. WVE-004, an investigational stereopure antisense oligonucleotide for the treatment of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (2302). Neurology Apr 2021, 96 (15 Supplement) 2302.
Liu Y, Dodart JC, Tran H, Berkovitch S, Braun M, Byrne M, et al. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat Commun. 2021;12(1):847.
Tran H, Moazami MP, Yang H, McKenna-Yasek D, Douthwright CL, Pinto C, et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med. 2021;1–8.
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, et al. Gain of Toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90(3):535–50.
Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, et al. Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron. 2015;88(5):902–9.
Gendron TF, Chew J, Stankowski JN, Hayes LR, Zhang YJ, Prudencio M, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci Transl Med. 2017;9(383):7866.
Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 2014;83(5):1043–50.
Cammack AJ, Atassi N, Hyman T, van den Berg LH, Harms M, Baloh RH, et al. Prospective natural history study of C9orf72 ALS clinical characteristics and biomarkers. Neurology. 2019;93(17):e1605–17.
Biogen and Ionis announce topline phase 1 study results of investigational drug in C9orf72 amyotrophic lateral sclerosis | Biogen [Internet]. 2022. Available from: https://investors.biogen.com/news-releases/news-release-details/biogen-and-ionis-announce-topline-phase-1-study-results. Accessed 27 Apr 2022.
Ratti A, Buratti E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem. 2016;138(S1):95–111.
Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet. 2013;22(13):2676–88.
Vance C, Rogelj B, Hortobágyi T, Vos KJD, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–11.
Kino Y, Washizu C, Kurosawa M, Yamada M, Miyazaki H, Akagi T, et al. FUS/TLS deficiency causes behavioral and pathological abnormalities distinct from amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2015;3(1):24.
Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, et al. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun. 2016;7(1):10465.
Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P, Shneider NA. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med. 2022;1–13.
Ostrowski LA, Hall AC, Mekhail K. Ataxin-2: from RNA control to human health and disease. Genes-basel. 2017;8(6):157.
Paulson HL. The spinocerebellar ataxias. J Neuro-ophthalmol. 2009;29(3):227–37.
Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–75.
Watanabe R, Higashi S, Nonaka T, Kawakami I, Oshima K, Niizato K, et al. Intracellular dynamics of ataxin-2 in the human brains with normal and frontotemporal lobar degeneration with TDP-43 inclusions. Acta Neuropathol Commun. 2020;8(1):176.
Conforti FL, Spataro R, Sproviero W, Mazzei R, Cavalcanti F, Condino F, et al. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology. 2012;79(24):2315–20.
Chio A, Calvo A, Moglia C, Canosa A, Brunetti M, Barberis M, et al. ATXN2 polyQ intermediate repeats are a modifier of ALS survival. Neurology. 2015;84(3):251–8.
Sproviero W, Shatunov A, Stahl D, Shoai M, van Rheenen W, Jones AR, et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol Aging. 2017;51:178.e1-178.e9.
Hart MP, Gitler AD. ALS-associated ataxin 2 polyQ expansions enhance stress-induced caspase 3 activation and increase TDP-43 pathological modifications. J Neurosci. 2012;32(27):9133–42.
Dudman J, Qi X. Stress granule dysregulation in amyotrophic lateral sclerosis. Front Cell Neurosci. 2020;14:598517.
Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biology. 2013;201(3):361–72.
Nihei Y, Ito D, Suzuki N. Roles of ataxin-2 in pathological cascades mediated by TAR DNA-binding protein 43 (TDP-43) and fused in sarcoma (FUS)*. J Biol Chem. 2012;287(49):41310–23.
Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544(7650):367–71.
Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS Genetics, mechanisms, and therapeutics: where are we now? Front Neurosci-switz. 2019;13:1310.
Scoles DR, Meera P, Schneider M, Paul S, Dansithong W, Figueroa KP, et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 2017;544(7650):362–6.
Pottier C, Ren Y, Perkerson RB, Baker M, Jenkins GD, van Blitterswijk M, et al. Genome-wide analyses as part of the international FTLD-TDP whole-genome sequencing consortium reveals novel disease risk factors and increases support for immune dysfunction in FTLD. Acta Neuropathol. 2019;137(6):879–99.
Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22(2):180–90.
Prudencio M, Humphrey J, Pickles S, Brown AL, Hill SE, Kachergus JM, et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J Clin Invest. 2020;130(11):6080–92.
Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature. 2022;603(7899):131–7.
Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, et al. TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature. 2022;603(7899):124–30.
Funding
Support for this work was provided by the National Institute of Neurological Disorders and Stroke (R01NS078398) to T.M.M.
Author information
Authors and Affiliations
Contributions
Disclosure forms provided by the authors are available with the online version of this article.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Boros, B.D., Schoch, K.M., Kreple, C.J. et al. Antisense Oligonucleotides for the Study and Treatment of ALS. Neurotherapeutics 19, 1145–1158 (2022). https://doi.org/10.1007/s13311-022-01247-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-022-01247-2