
Abstract
The hereditary spastic paraplegias (HSPs) are a group of genetic conditions in which spastic paralysis of the legs is the principal clinical feature. This is caused by a relatively selective distal axonal degeneration involving the longest axons of the corticospinal tracts. Consequently, these conditions provide an opportunity to identify genes, proteins and cellular pathways that are critical for axonal health. In this review, we will provide a brief overview of the classification, clinical features and genetics of HSP, highlighting selected HSP subtypes (i.e. those associated with thin corpus callosum or cerebellar ataxia) that are of particular clinical interest. We will then discuss appropriate investigation strategies for HSPs, suggesting how these might evolve with the introduction of next-generation sequencing technology. Finally, we will discuss the management of HSP, an area somewhat neglected by HSP research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The hereditary spastic paraplegias (HSPs) are a group of genetic conditions in which spastic paralysis of the legs is the principal clinical feature [1, 2]. In most subtypes of HSP, this is caused by a relatively selective distal axonal degeneration involving the longest axons of the corticospinal tracts [3]. As a group, these conditions are moderately rare, with prevalence estimates in different geographical regions ranging from 0.1 to 9.6 per 105 [4]. However, as they are chronic and sometimes severe conditions that frequently present in childhood and young adult life, they represent a significant disease burden. In addition, they are scientifically important, since they provide an opportunity to identify genes, proteins and cellular pathways that are critical for axonal health.
In this review, we will provide a brief overview of the classification, clinical features and genetics of HSP, highlighting selected HSP subtypes that are of particular clinical interest. We will then discuss appropriate investigation strategies for HSPs, suggesting how these might evolve with the introduction of next-generation sequencing technology. Finally, we will discuss the management of HSP, an area somewhat neglected by HSP research.
Overview of main HSP clinical categories
Traditionally, the hereditary spastic paraplegias are subcategorised into “pure” and “complex” (or “complicated”) subtypes [3]. In northern European populations, pure forms are the most frequent. The clinical features of pure HSP have been thoroughly described in previous research articles and reviews, and diagnostic criteria have been proposed [1, 5–9]. Briefly, the typical clinical picture is of a slowly progressive, predominantly symmetrical, spastic paraplegia. This is frequently accompanied by minor sensory abnormalities (such as absent vibration sensation) and neurological bladder involvement, but bowel involvement is rare. If seen at all, arm involvement is minimal and does not extend beyond hyper-reflexia and minor weakness (e.g. difficulty unscrewing a tight bottle top); more significant arm involvement should raise the possibility of alternative diagnoses such as primary lateral sclerosis or amyotrophic lateral sclerosis, while bulbar involvement is incompatible with a diagnosis of pure HSP. Age at onset varies from childhood to late adult life. Within many multiplex families it is highly variable, although mutations in certain genes, notably spastic paraplegia gene (SPG) 3A/atlastin1, are predominantly associated with a childhood age at onset.
While pure HSPs present a relatively homogenous clinical picture, complex HSPs are a disparate group of distinct disorders (Table 1) [1–3]. As most complex HSPs are inherited in an autosomal recessive pattern, they are the commonest forms of HSP in populations where consanguinity is frequent; for example, in a Tunisian series of 38 families, approximately 70 % of families had autosomal recessive complex HSP [10]. However, it is important to recognise that these complex subtypes of HSP are found worldwide, e.g. HSP with thin corpus callosum caused by SPG11 or SPG15 mutations has been described in many populations and is the most common type of complex HSP that we see in our own clinical practice in Cambridge, UK.
Genetics of HSPs
The HSPs show a remarkable degree of genetic heterogeneity. The chromosomal location of more than 70 SPGs is known, and presently more than 50 of the genes have been fully identified (Table 1). Moreover, additional syndromes such as Warburg Micro syndrome, previously considered distinct, have recently been incorporated under the rubric of HSP by reason of overlapping cellular pathology [11]. This new knowledge has led to an evolution of the classification of HSP, which, while still referring to phenotypic information, is increasingly focussed on the gene involved. In addition, gene identification has resulted in the distinction between pure and complex HSP becoming somewhat blurred as it has become apparent that mutations in the same gene may cause either a pure or complex phenotype. An example of this is the rare identification of mutations in SPG4/SPAST, SPG31/REEP1 or SPG3A/atlastin1, three genes typically associated with pure HSP, in patients who have spastic paraplegia accompanied by cerebellar ataxia or peripheral neuropathy [12–14].
Pure HSP phenotype
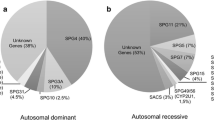
In northern European populations, autosomal dominant inheritance accounts for the majority of patients from families with pure HSP [15]. Autosomal recessive inheritance accounts for most of the remainder, while clear-cut X-linked inheritance is infrequent. Rarely, gonadal mosaicism for a dominant gene has been described and this can be mistaken for autosomal recessive inheritance [16]. In addition, some patients present without a family history. Potential genetic explanations for this are varied and include singleton autosomal recessive cases, new autosomal dominant mutations, or inherited autosomal dominant mutations with non-penetrance in a parent. The estimated relative frequency of mutations in selected genes associated with pure HSP is given in Table 2. From this table, it can be seen that testing for three genes, SPG4/SPAST, SPG3A/Atlastin1 and SPG31/REEP1, will identify the responsible mutation in approximately 50 % of families with autosomal dominant pure HSP. For SPG4/SPAST, this testing should include methodologies to pick up whole exon deletions, as these are a common mutational class in this subtype of HSP (they have also been described in other forms including SPG3A/Atlastin1 and SPG31/REEP1-HSP) [14, 17–19]. For families with an autosomal recessive inheritance pattern, screening SPG5 (CYP7B1) will pick up the causative mutations in approximately 5–10 % of cases. However, the yield from testing each individual additional gene beyond these commoner genes is low. Using traditional “one at a time” sequential gene testing to detect these rare mutations can be very expensive and may not be justifiable for publically funded health services. Next-generation sequencing approaches are currently revolutionising this situation (see below).
Complex HSP phenotypes
As mentioned above, the complex HSPs are a diverse set of conditions, each with distinctive clinical features and each caused by one or a small set of genes. Here, we will focus on two of the more common presentations, HSP with thin corpus callosum and HSP accompanied by cerebellar ataxia.
HSP with thin corpus callosum
A thin corpus callosum on MRI scanning is a characteristic feature of patients who have autosomal recessive SPG11 and SPG15 mutations. These patients have a variable combination of clinical features which, as well as thin corpus callosum, includes typically childhood or teenage onset spastic paraplegia, learning difficulties with progressive cognitive decline, cerebellar signs, ocular involvement, peripheral motor axonopathy and extrapyramidal features including parkinsonism. SPG11/15 mutations are associated with relatively severe gait involvement and patients often eventually require a wheelchair. It is important to note that a thin corpus callosum is not a universal finding in SPG11/15 HSP (e.g. in large series, it was found in 90 % of SPG11 cases and 60 % of SPG15 patients), and that learning difficulties may be present before the spastic gait develops [20–22].
Cell biological studies on the SPG11 and SPG15 proteins have revealed why mutations in these genes lead to a similar clinical phenotype. Both SPG11 and SPG15 proteins interact with a 4-protein complex termed AP5 (adapter protein 5), which localises to small intracellular membrane-bound vesicles called endosomes [23, 24]. Although AP5’s function is not fully understood, it probably acts to sort membrane cargoes away from the endosomal compartment [23, 24]. Intriguingly, recessive mutations in a component of the core AP5 complex (SPG48/AP5Z1) also cause a very similar clinical phenotype to SPG11/15 mutations, with cognitive defects, thin corpus callosum and spastic paraplegia [25]. Thus, HSP with thin corpus callosum is an excellent example of the principle that diseases with similar phenotypes are often caused by mutations in functionally related genes. At least four other subtypes of recessive HSP may also present with thin corpus callosum, SPG21/Maspardin, SPG35/FA2H and SPG46/GBA2 and SPG54/DDHD2 ([25] and see Table 1); it remains to be seen whether any of the encoded proteins are functionally related to the AP5 complex.
HSP with cerebellar ataxia
Patients who present with a mixed cerebellar ataxia and spastic paraplegia phenotype represent a particular diagnostic challenge, as the differential diagnosis is very broad. It can be helpful to first make a decision as to whether spastic paraplegia or cerebellar ataxia is the dominant feature. There are consensus diagnostic pathways for patients with predominant cerebellar ataxia and we will not review these further here [26]. In patients where spastic paraplegia is the predominant feature, it is useful to subdivide into pure and complex subtypes, analogous to the subdivision of HSP.
“Complex” spastic ataxia in which the cerebellar ataxia is a (sometimes variable) feature of a more complex syndromal picture. Such conditions include HSP with thin corpus callosum (SPG11/15 mutations), SPG35/FA2H-associated HSP, Troyer syndrome (SPG20) and Mast syndrome (SPG21), SPG26/B4GALNT1, SPG30/KIF1A, SPG39/PNPLA6 and SPG46/GBA2. This group also contains a variety of rare metabolic conditions (reviewed in [27, 28]), including cerebrotendinous xanthomatosis, triple H syndrome, cerebral folate deficiency, metachromatic leukodystrophy, Type III 3-methylglutaconic aciduria, as well as other conditions such as Alexander disease and vanishing white matter disease. Rare patients with Chediak–Higashi syndrome (caused by mutations in the lysosomal trafficking regulator LYST gene) may also present with spastic paraplegia, cerebellar ataxia and peripheral neuropathy, without the hypopigmentary or immune deficiency typically associated with this condition [29].
“Pure” spastic ataxia There are many patients in whom spastic paraplegia with cerebellar involvement is the exclusive clinical presentation. This can occur in three main scenarios:
-
(a)
As an unusual presentation of mutations in genes that are classically associated with “pure” hereditary spastic paraplegia, including autosomal dominant SPG4/spastin and SPG31/REEP1 and autosomal recessive SPG5/CYP7B1 [12, 14, 30].
-
(b)
As a presentation of genes that are classically associated with cerebellar ataxia. This would especially include Friedreich ataxia and SCA3, in which pyramidal signs may be the presenting feature [31, 32].
-
(c)
As a presentation of mutations in genes that typically cause a spastic paraplegia/cerebellar ataxia overlap syndrome. These genes are often classified under the spastic ataxia (SPAX) nomenclature. Well-recognised examples include the autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS; SPAX6) and SPG7/paraplegin [33, 34]. They also include VAMP1 (SPAX1), KIF1C gene (SPAX2/SPG58), MARS2 (SPAX3), MTPAP (SPAX4) and AFG3L2 (SPAX5/SCA28) (Table 1) [32, 35].
Investigation of a patient with suspected HSP
The traditional approach to investigating a patient with possible HSP is to define the phenotype and inheritance pattern, exclude alternative causes, then attempt to make a definitive molecular genetic diagnosis, where possible using clinical and family history information to focus genetic investigations appropriately.
Definition of phenotype, inheritance and exclusion of other causes
As specific investigations of a patient with suspected HSP depend on their clinical features and inheritance pattern, careful clinical phenotyping of index cases and potentially affected family members is crucial. A detailed medical history should include developmental milestones and a three generational family tree. Affected individuals and ideally, apparently unaffected family members should undergo a clinical neurological examination, to document specific neurological features and identify asymptomatic family members who may have subtle abnormal signs on examination. This is particularly important for apparently sporadic cases or isolated sibships, where positive examination findings in an asymptomatic parent will point towards the probability of autosomal dominant inheritance.
Even in individuals who have typical clinical features of HSP and a positive family history of spastic paraparesis, it is important to consider alternative causes and importantly exclude other treatable familial conditions. In his regard, MR imaging of the brain and spinal cord is crucial and should be undertaken in all sporadic cases and at least one family member of familial presentations. Imaging will not only identify many structural, inflammatory or metabolic abnormalities but is also useful to guide investigations into specific genetic causes (for example, by highlighting atrophy patterns). Note that imaging does have an appreciable false negative rate, for example, it can be normal in certain metabolic or inflammatory conditions, including adrenoleukodystrophy or primary progressive multiple sclerosis.
A baseline metabolic blood screen in the index case could include Vitamins B12 and E, creatine kinase, very long chain fatty acids, white cell enzymes, anti-nuclear antibody and copper levels. However, individuals with more complex clinical features or an unclear family history may require further baseline tests. For example, in presumed sporadic pure HSP, a set of more extensive investigations is appropriate, as an inherited basis for the disease has not been proven. This includes CSF examination (including oligoclonal bands, HTLV serology), Syphilis and HIV serology as well as vasculitis screen [36–38]. An oral short trial of Levodopa will identify those individuals with dopa-responsive dystonia and neurophysiology can be useful if there is an associated neuropathy or suspicion of amyotrophic lateral sclerosis [39]. Table 3 summarises a list of conditions to be considered in the differential diagnosis and investigation of sporadic pure HSP.
For those individuals in whom the above baseline investigations are unremarkable and/or where the family history and clinical features point towards a specific genetic diagnosis, it is then appropriate to test for the suspected mutation.
Genetic investigations
There is considerable clinical utility for HSP families in detecting their causative mutation. It gives diagnostic certainty, prevents further unnecessary investigations and opens the possibility of predictive and prenatal testing. However, until very recently, in many healthcare systems, economic factors necessitated that genetic investigations were restricted to analysis of HSP genes in which there was the highest chance of detecting a mutation, as the expense of analysing rarely-causative genes was not justified by the low mutation detection rate per additional gene analysed. However, this has changed with the introduction of next-generation sequencing, which allows cost-effective analysis of many genes together, using approaches including (1) sequencing of large panels of genes, (2) sequencing of all coding exons in the genome, or (3) sequencing of entire genomes. At present, a common approach uses next-generation sequencing to screen exons of panels of genes causally associated with specific conditions or groups of conditions. These new approaches hold the prospect of substantially increasing the mutational pick-up rate for HSP patients. Once fully integrated into molecular diagnostic services, it is possible to envisage that genetic testing could be introduced at an early stage in patient work-up, perhaps avoiding the need for other costly or painful investigations if a definitive molecular genetic diagnosis is made.
Although these developments are undeniably positive, there are pitfalls and caveats associated with next-generation sequencing approaches. Sequence variation is very common in the genome (for example, many millions of single nucleotide variants have been identified) [40], with the vast majority of these sequence changes having a neutral effect with no clinical consequence. In addition, many of these sequence changes are individually rare. Thus, next-generation approaches typically detect numerous rare sequence variants; the more genes that are analysed, the larger this problem becomes. A major problem lies in deciding which, if any, of the detected sequence changes are pathogenic. In most cases, this can be resolved using bioinformatics and other approaches (e.g. identifying previously reported and validated mutations, excluding known rare population polymorphisms, determining the likely effect on the amino acid sequence of the encoded protein and how deleterious this effect is likely to be, excluding mutations that do not co-segregate with disease) [41]. However, inevitably in some cases, a single conclusively pathogenic mutation cannot be identified and one or more candidate pathogenic mutations remain. This is a particular issue with mutations of the missense class (in which only a single amino acid of the encoded protein is altered), since it can be difficult to predict whether the resulting effect on the protein will have functional consequences. Such sequence changes are often reported as variants of uncertain significance. Defining approaches to determining which of these mutations are pathogenic will be a major future challenge, as will be determining ethical approaches to whether and how the uncertainty associated with such sequence changes is reported back to patients.
In addition, exon-sequencing approaches do not detect all mutational classes. A particular problem arises in detecting large-scale deletions or duplications involving whole exons, or in detecting promoter or deep intronic mutations. This is an issue for at least some HSP genes, e.g. whole exon deletions are a common cause of SPG4/SPAST HSP (see above). We anticipate that in the relatively near future this issue will be resolved by whole-genome sequencing, which has the capacity to identify large deletions/duplications, as well as promoter and deep intronic mutations. In a research setting, genome sequencing may also allow identification of causative mutations in genes that have not previously been associated with the disease under consideration. In the meantime, additional testing for deletions/duplications should be considered in selected genes for appropriate patients, prioritised based on clinical features, in whom mutations have not been identified in exon-sequencing panels.
High-throughput sequencing approaches also may present ethical issues if so-called “incidental findings” (clinically relevant changes in genes unrelated to the condition being tested) are detected. These are particularly important when whole-exome or whole-genome approaches are taken, where, given sufficient numbers of tested subjects, it is inevitable that other potentially clinically significant abnormalities, e.g. mutations in cancer predisposing genes or detection of carrier status for autosomal recessive disease, will be detected. Whether and how such findings are reported back to patients requires careful consideration of their clinical validity and utility, and the ethical issues surrounding this are currently a topic of much debate [42].
Finally, while next-generation sequencing approaches have the potential to increase rates of molecular genetic diagnosis in HSP, they do not remove the need for careful phenotyping––this is still important, as it can help to focus bioinformatics analysis onto the most relevant genes. Careful phenotyping may also better define the clinical spectrum associated with pathogenic mutations in particular genes.
Rehabilitation and therapy for HSP patients
People with HSP complain of muscle stiffness, pain, spasms and cramps, tripping over their toes due to weakness of ankle dorsiflexion and hip flexion, loss of balance, effortful walking and progressively more flexed standing posture. Eventually, walking becomes impossible for some patients due to a combination of (a) spasticity, (b) weakness, particularly of ankle dorsiflexion, leading to (c) loss of range of movement at ankle, knee and hip, making it impossible to stand straight, and (d) loss of motor control leading to delayed postural reflexes and loss of balance.
A home exercise programme supervised by a physiotherapist, concentrating on stretches to maintain range of movement and reduce spasticity, accompanied by balance exercises in patients with more advanced disease, is the cornerstone of management. This is usually supported by a trial of oral muscle relaxants such as Baclofen, Tizanidine, or Gabapentin/Pregabalin. Problematic spasticity in specific groups of muscles, most commonly in the ankle plantarflexors and hip adductors, may be treated by Botulinum toxin without the risk of sedation associated with oral muscle relaxants [43]. The role of Botulinum toxin is to support and facilitate stretching and splinting, rather than to replace it. Functional electrical stimulation (FES) is popular among patients and physiotherapists and is as effective as simple off the shelf ankle foot orthoses (AFOs) in the early stages before calf shortening has developed: after that, FES is less useful as it cannot provide support or compensation during stance phase [44–46].
Intrathecal Baclofen, delivered by an implanted pump, is the most effective method of reducing very high tone in the lower limbs, and can bring immediate relief of pain and improvement in sitting posture, along with a reduction in effort transferring from wheelchair to bed or car [47]. Effective control of muscle tone often improves quality of sleep for the patient and their partner, and permits stretching and splinting with the aim of preventing further deterioration of flexion contractors. The question of when to start intrathecal baclofen in this condition has not been addressed by published trials. While most often performed much later in the disease, a case can be made for implanting a pump relatively early to prevent the development of contractures, with a view to maintaining upright gait and retaining the option of supporting weak knee extensors and ankle dorsiflexors with orthoses [48].
Once ankle contractures have developed, and people can no longer stand with knees and hips straight while their heels are on the ground, heel wedging needs to be incorporated into shoes or AFOs to compensate. The lighter “off the shelf” AFOs, or FES of the peroneal nerve, are no longer appropriate in this situation as these only prevent passive plantarflexion during the swing phase of gait and are not designed to stabilise the foot and ankle during the stance phase of gait. The necessary custom moulded AFOs with compensatory heel wedging will often not fit in patients’ usual shoes; if larger trainers are impractical, expensive bespoke footwear may be required. Custom AFOs may also be designed to compensate for weak knee extensors using the ground reaction force to hold the lower leg in a more vertical position, but this is only possible if the knee still extends fully.
Conclusion
The last decade has seen astonishing progress in the identification of HSP genes. Coupled to the introduction of high-throughput sequencing approaches, we are quickly moving towards the ideal situation where every HSP patient will have a defined molecular diagnosis if they choose to have it. This will give important immediate benefits to HSP families, including diagnostic certainty, prevention of unnecessary additional investigations and accurate risk prediction for clinically unaffected family members. In the future, as HSP therapies emerge, it may also be a pre-requisite for the personalised selection of appropriate treatment. In the meantime, it is important that supportive therapy, which can make a real difference to patients’ lives, is not neglected.
References
Harding AE (1993) Hereditary spastic paraplegias. Semin Neurol 13:333–336
Reid E, Rugarli E (2010) Hereditary spastic paraplegias. In: Valle D et al (eds) The online metabolic and molecular bases of inherited diseases. McGraw Hill, New York
Harding AE (1984) The hereditary ataxias and related disorders. Churchill Livingston, Edinburgh
Ruano L, Melo C, Silva MC, Coutinho P (2014) The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 42:174–183
Harding AE (1981) Hereditary “pure” spastic paraplegia: a clinical and genetic study of 22 families. J Neurol Neurosurg Psychiatry 44:871–883
Reid E, Grayson C, Rogers MT, Rubinsztein DC (1999) Locus-phenotype correlations in autosomal dominant pure hereditary spastic paraplegia. A clinical and molecular genetic study of 28 United Kingdom families. Brain 122(Pt 9):1741–1755
Durr A et al (1994) The phenotype of “pure” autosomal dominant spastic paraplegia. Neurology 44:1274–1277
Reid E (1997) Pure hereditary spastic paraplegia. J Med Genet 34:499–503
Fink JK et al (1996) Hereditary spastic paraplegia: advances in genetic research. Hereditary Spastic Paraplegia Working group. Neurology 46:1507–1514
Boukhris A et al (2009) Tunisian hereditary spastic paraplegias: clinical variability supported by genetic heterogeneity. Clin Genet 75:527–536
Gerondopoulos A et al (2014) Rab18 and a Rab18 GEF complex are required for normal ER structure. J Cell Biol 205:707–720
Nielsen JE et al (2004) Hereditary spastic paraplegia with cerebellar ataxia: a complex phenotype associated with a new SPG4 gene mutation. Eur J Neurol 11:817–824
Ivanova N et al (2007) Hereditary spastic paraplegia 3A associated with axonal neuropathy. Arch Neurol 64:706–713
Goizet C et al (2011) REEP1 mutations in SPG31: frequency, mutational spectrum, and potential association with mitochondrial morpho-functional dysfunction. Hum Mutat 32:1118–1127
Erichsen AK, Koht J, Stray-Pedersen A, Abdelnoor M, Tallaksen CM (2009) Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study. Brain 132:1577–1588
Depienne C et al (2007) A de novo SPAST mutation leading to somatic mosaicism is associated with a later age at onset in HSP. Neurogenetics 8:231–233
Depienne C et al (2007) Exon deletions of SPG4 are a frequent cause of hereditary spastic paraplegia. J Med Genet 44:281–284
Beetz C et al (2006) High frequency of partial SPAST deletions in autosomal dominant hereditary spastic paraplegia. Neurology 67:1926–1930
Sulek A et al (2013) Screening for the hereditary spastic paraplaegias SPG4 and SPG3A with the multiplex ligation-dependent probe amplification technique in a large population of affected individuals. Neurol Sci 34:239–242
Stevanin G et al (2007) Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet 39:366–372
Stevanin G et al (2008) Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 131:772–784
Hanein S et al (2008) Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet 82:992–1002
Hirst J et al (2013) Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol Biol Cell 24:2558–2569
Hirst J et al (2011) The fifth adaptor protein complex. PLoS Biol 9:e1001170
Pensato V et al (2014) Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain 137:1907–1920
van de Warrenburg BPC et al (2014) EFNS/ENS Consensus on the diagnosis and management of chronic ataxias in adulthood. Eur J Neurol 21:552–562
Sedel F, Lyon-Caen O, Saudubray JM (2007) Therapy insight: inborn errors of metabolism in adult neurology––a clinical approach focused on treatable diseases. Nature clinical practice. Neurology 3:279–290
de Bot ST, van de Warrenburg BP, Kremer HP, Willemsen MA (2010) Child neurology: hereditary spastic paraplegia in children. Neurology 75:e75–e79
Shimazaki H et al (2014) Autosomal-recessive complicated spastic paraplegia with a novel lysosomal trafficking regulator gene mutation. J Neurol Neurosurg Psychiatry 85:1024–1028
Goizet C et al (2009) CYP7B1 mutations in pure and complex forms of hereditary spastic paraplegia type 5. Brain 132:1589–1600
Wang Y-G et al (2009) Six cases of SCA3/MJD patients that mimic hereditary spastic paraplegia in clinic. J Neurol Sci 285:121–124
de Bot ST, Willemsen MA, Vermeer S, Kremer HP, van de Warrenburg BP (2012) Reviewing the genetic causes of spastic-ataxias. Neurology 79:1507–1514
Casari G et al (1998) Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 93:973–983
Wilkinson PA et al (2004) A clinical, genetic and biochemical study of SPG7 mutations in hereditary spastic paraplegia. Brain 127:973–980
Bourassa Cynthia V et al (2012) VAMP1 mutation causes dominant hereditary spastic ataxia in newfoundland families. Am J Hum Genet 91:548–552
Ropper AH, Ayata C, Adelman L (2003) Vasculitis of the spinal cord. Arch Neurol 60:1791–1794
Araujo AQ, Silva MT (2006) The HTLV-1 neurological complex. Lancet Neurol 5:1068–1076
Price RW (1996) Neurological complications of HIV infection. Lancet 348:445–452
Furukawa Y, Graf WD, Wong H, Shimadzu M, Kish SJ (2001) Dopa-responsive dystonia simulating spastic paraplegia due to tyrosine hydroxylase (TH) gene mutations. Neurology 56:260–263
Liu X, Jian X, Boerwinkle E (2013) dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat 34:E2393–E2402
Pittman A, Hardy J (2013) Genetic analysis in neurology: the next 10 years. JAMA Neurol 70:696–702
Berg JS et al (2013) Processes and preliminary outputs for identification of actionable genes as incidental findings in genomic sequence data in the Clinical Sequencing Exploratory Research Consortium. Genet Med 15:860–867
Geva-Dayan K, Domenievitz D, Zahalka R, Fattal-Valevski A (2010) Botulinum toxin injections for pediatric patients with hereditary spastic paraparesis. J Child Neurol 25:969–975
Kluding PM et al (2013) Foot drop stimulation versus ankle foot orthosis after stroke: 30-week outcomes. Stroke 44:1660–1669
Karimi MT (2013) Functional walking ability of paraplegic patients: comparison of functional electrical stimulation versus mechanical orthoses. Eur J Orthop Surg Traumatol orthop traumatol 23:631–638
Bethoux F et al (2014) The effects of peroneal nerve functional electrical stimulation versus ankle-foot orthosis in patients with chronic stroke: a randomized controlled trial. Neurorehab Neural Repair 28:688–697
Meythaler JM et al (1992) Intrathecal baclofen in hereditary spastic paraparesis. Arch Phys Med Rehabil 73:794–797
Lambrecq V et al. (2007) Intrathecal baclofen in hereditary spastic paraparesis: benefits and limitations. Annales de readaptation et de medecine physique : revue scientifique de la Societe francaise de reeducation fonctionnelle de readaptation et de medecine physique 50:577–581
Fonknechten N et al (2000) Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum Mol Genet 9:637–644
Patrono C et al (2005) Autosomal dominant hereditary spastic paraplegia: DHPLC-based mutation analysis of SPG4 reveals eleven novel mutations. Hum Mutat 25:506
Sauter S et al (2002) Mutation analysis of the spastin gene (SPG4) in patients in Germany with autosomal dominant hereditary spastic paraplegia. Hum Mutat 20:127–132
Shoukier M et al (2009) Expansion of mutation spectrum, determination of mutation cluster regions and predictive structural classification of SPAST mutations in hereditary spastic paraplegia. Eur J Hum Genet 17:187–194
Meijer HA, Thomas AA (2002) Control of eukaryotic protein synthesis by upstream open reading frames in the 5′-untranslated region of an mRNA. Biochem J367:1–11
Park SY et al (2005) Mutation analysis of SPG4 and SPG3A genes and its implication in molecular diagnosis of Korean patients with hereditary spastic paraplegia. Arch Neurol 62:1118–1121
Depienne C et al (2006) Spastin mutations are frequent in sporadic spastic paraparesis and their spectrum is different from that observed in familial cases. J Med Genet 43:259–265
Crippa F et al (2006) Eight novel mutations in SPG4 in a large sample of patients with hereditary spastic paraplegia. Arch Neurol 63:750–755
Brugman F et al (2005) Spastin mutations in sporadic adult-onset upper motor neuron syndromes. Ann Neurol 58:865–869
Braschinsky M, Luus SM, Gross-Paju K, Haldre S (2009) The prevalence of hereditary spastic paraplegia and the occurrence of SPG4 mutations in Estonia. Neuroepidemiology 32:89–93
Abel A et al (2004) Early onset autosomal dominant spastic paraplegia caused by novel mutations in SPG3A. Neurogenetics 5:239–243
Durr A et al (2004) Atlastin1 mutations are frequent in young-onset autosomal dominant spastic paraplegia. Arch Neurol 61:1867–1872
Zhao X et al (2001) Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet 29:326–331
Schlang KJ, Arning L, Epplen JT, Stemmler S (2008) Autosomal dominant hereditary spastic paraplegia: novel mutations in the REEP1 gene (SPG31). BMC Med Genet 9:71
Beetz C et al (2008) REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain 131:1078–1086
Hewamadduma C et al (2009) New pedigrees and novel mutation expand the phenotype of REEP1-associated hereditary spastic paraplegia (HSP). Neurogenetics 10:105–110
Zuchner S et al (2006) Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am J Hum Genet 79:365–369
Schule R et al (2008) SPG10 is a rare cause of spastic paraplegia in European families. J Neurol Neurosurg Psychiatry 79:584–587
Tessa A et al (2008) A novel KIF5A/SPG10 mutation in spastic paraplegia associated with axonal neuropathy. J Neurol 255:1090–1092
Goizet C et al (2009) Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum Mutat 30:E376–E385
Valdmanis PN et al (2007) Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia. Am J Hum Genet 80:152–161
de Bot S et al (2013) Pure adult-onset Spastic Paraplegia caused by a novel mutation in the KIAA0196 (SPG8) gene. J Neurol 260:1765–1769
Montenegro G et al (2012) Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J Clin Investig 122:538–544
Beetz C et al (2008) Screening of hereditary spastic paraplegia patients for alterations at NIPA1 mutational hotspots. J Neurol Sci 268:131–135
Klebe S et al (2007) NIPA1 (SPG6) mutations are a rare cause of autosomal dominant spastic paraplegia in Europe. Neurogenetics 8:155–157
Brugman F et al (2008) Paraplegin mutations in sporadic adult-onset upper motor neuron syndromes. Neurology 71:1500–1505
Arnoldi A et al (2008) A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum Mutat 29:522–531
Elleuch N et al (2006) Mutation analysis of the paraplegin gene (SPG7) in patients with hereditary spastic paraplegia. Neurology 66:654–659
Caragine LP Jr, Halbach VV, Ng PP, Dowd CF (2002) Vascular myelopathies-vascular malformations of the spinal cord: presentation and endovascular surgical management. Semin Neurol 22:123–132
Barker RA, Revesz T, Thom M, Marsden CD, Brown P (1998) Review of 23 patients affected by the stiff man syndrome: clinical subdivision into stiff trunk (man) syndrome, stiff limb syndrome, and progressive encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry 65:633–640
Saleh S, Saw C, Marzouk K, Sharma O (2006) Sarcoidosis of the spinal cord: literature review and report of eight cases. J Natl Med Assoc 98:965–976
Pema PJ, Horak HA, Wyatt RH (1998) Myelopathy caused by nitrous oxide toxicity. AJNR Am J Neuroradiol 19:894–896
Shaw-Smith CJ, Lewis SJ, Reid E (2004) X-linked adrenoleukodystrophy presenting as autosomal dominant pure hereditary spastic paraparesis. J Neurol Neurosurg Psychiatry 75:686–688
Müller vom Hagen J, Karle KN, Schüle R, Krägeloh-Mann I, Schöls L (2014) Leukodystrophies underlying cryptic spastic paraparesis: frequency and phenotype in 76 patients. Eur J Neurol 21:983–988
Jaiser SR, Winston GP (2010) Copper deficiency myelopathy. J Neurol 257:869–881
Goldwein JW (1987) Radiation myelopathy: a review. Med Pediatr Oncol 15:89–95
Acknowledgments
We thank Rhys Roberts for reviewing the manuscript. This work was supported by grants from the UK Medical Research Council [MR/M00046X/1]; the Wellcome Trust [082381]; the Tom Wahlig Stiftung; and the UK HSP Support Group. The Cambridge Institute for Medical Research is supported by a Wellcome Trust Strategic Award [100140].
Conflicts of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Hensiek, A., Kirker, S. & Reid, E. Diagnosis, investigation and management of hereditary spastic paraplegias in the era of next-generation sequencing. J Neurol 262, 1601–1612 (2015). https://doi.org/10.1007/s00415-014-7598-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-014-7598-y