
Abstract
Rice production needs to be sustained in the coming decades, with changing climatic conditions becoming more conducive to the prevalence of disease outbreaks. Major rice diseases collectively cause enormous economic damage and yield instability. Breeding for disease-resistant rice varieties could be one of the best options to counter these disease outbreaks. Disease-screening protocols and newer technologies are essential for effective phenotyping and would aid in gene discovery and function. Understanding the genetics of disease mechanisms and stacking of broad-spectrum disease-resistance genes could lead to faster development of rice varieties with multiple disease resistance. New molecular breeding approaches are discussed for the development of these varieties. The molecular biology of disease resistance is now better understood and could be well manipulated for improved resilience. Transgenic approaches for disease resistance are discussed. Genome-editing tools for the development of disease-resistant rice varieties are thoroughly discussed. The use of bioinformatics tools to speed up the process and to obtain a better understanding of molecular genetics mechanisms of disease resistance is explained.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Rice (Oryza sativa L.) is a staple and the most crucial food security crop in the world. It plays a vital role in the human diet and feeds more than 50% of the world’s population (Rathna Priya et al. 2019). By 2050, global demand for rice is projected to rise more than 40% to feed the rapidly growing world population (Milovanovic and Smutka 2017). Despite impressive global increases in production from 289 million tons in 1968 to 782 million tons in 2018, this quantum jump still has to keep pace with demand for rice from the rising population (FAOSTAT 2020). At present, rice cultivation throughout South Asia and in ASEAN countries is facing significant threats because of a few major biotic stresses (Yugander et al. 2017). Approximately 52% of the global productivity of rice grain yield is severely damaged by biotic factors, of which nearly 31% is due to various diseases such as bacterial blight (caused by Xanthomonas oryzae), blast (caused by Magnaporthe grisea), sheath blight (caused by Rhizoctonia solani), and tungro disease (tungro bacilliform virus and tungro spherical virus) (Park et al. 2008). Detailed information about the symptoms caused by these major diseases, along with the favorable conditions required by these pathogens and yield losses incurred, is presented in Table 1. The severity of biotic stresses in rice production is increasing at a startling pace of late because of rapid changes in climate (Jamaloddin et al. 2020). Changing climatic conditions are contributing to the emergence of new virulent races and the occurrence of diseases in new localities. Many diseases considered as minor thus far have become economically significant in many rice-cultivating areas and are exacerbating their impact (Anderson et al. 2004). According to Zhang et al. (2009), rice crops are affected by around 70 pathogens, especially viruses, bacteria, fungi, and nematodes. Estimated yield loss because of pathogens globally and as per hotspot range for rice is 30% (24.6–40.9%) (Savary et al. 2019). Over the past 150 years, many rice diseases have caused outbreaks and spread rapidly in different parts of the world. Rice diseases were observed for the first time in different locations, such as bacterial blight and sheath blight in Japan during 1884–1885 and 1910, respectively; false smut in the United States during 1906; blast in Africa during 1922; rice tungro in the Philippines during 1940; rice brown spot in India during 1942; bacterial leaf streak in India during 1963; and rice yellow mottle disease in Kenya during 1966. These diseases, along with a few newly emerging epidemics, are becoming a significant threat to rice production.
Despite so many alternatives for crop protection, plant pathogens still pose a challenge to agriculture. Several management practices have been adopted to decrease their impact, such as chemical control, biological control, optimum fertilizer application, appropriate planting dates, and disease forecasting. However, not all of these methods are environment-friendly and alone are not enough to control the diseases completely. The present situation thus requires environment-friendly and cost-effective modern technologies such as the development and cultivation of disease-resistant cultivars. The development of these varieties using only conventional breeding methods consumes a lot of time, land, and labor. In this context, molecular markers come to the rescue of plant breeders by helping them decrease the time between breeding and achieving the desired product. The discovery of DNA markers led to a new tool in plant breeding called marker-assisted selection (MAS), which is one of the widely used components of a discipline called molecular breeding. The application of DNA markers to plant breeding significantly increased its efficiency and precision. MAS is now one of the most advanced methodologies on hand for the transfer of one or more desired genes/genomic regions into elite rice varieties in more durable combinations. Deploying a single R-gene often leads to resistance breakdown in a short period as the pathogen evolves and makes itself resistant to the action of the gene. Therefore, pyramiding of multiple R-genes imparting resistance against different races of a pathogen through MAS is an efficient way to attain long-term and broad-spectrum resistance. Although this is an advanced method, it has some disadvantages. The main drawback of this approach is that one parent, or even both parents, used in the breeding program may carry quantitative trait loci (QTL) alleles that are either similar or exact to the ones present in the elite germplasm accessions used in other breeding programs. In such a situation, the QTL being introgressed may contribute only partially to the trait improvement. In other cases, the impact of a QTL may differ based on the genetic background as a result of interactions with other loci or epistasis (Holland 2001). Moreover, there are many more important traits for which no genes have been reported so far. In such situations in which a gene is not available in the gene pool, researchers are forced to look outside the gene pool toward other genera or sometimes toward another kingdom to find the desired gene.
Genetic modification (GM) technology has been developed to make changes to an organism’s genes to give it new traits that would not occur in nature or to eliminate undesirable characteristics. GM technology using recombinant DNA technology is useful for developing disease-resistant varieties but still has not reached farmers because of a lack of public acceptance and political issues in many countries. Under these situations, researchers are left with an option to create mutations in the gene pool with an expectation to generate variation for a trait not naturally present in the gene pool.
Mutation breeding is helpful in creating novel mutants with genetic variations for plant breeding and functional genomics. It could be used for rice crop improvement programs. Mutation induction can be of advantage to produce cultivars with desired characteristics within defined germplasm pools. Normally, gamma-rays (γ-rays) and ethyl methane sulfonate (EMS) have been used extensively to develop rice mutants. In rice, there have been reports of some important mutant collections developed to carry out functional studies and Hirochika et al. (2004) made available a list of the mutant libraries. Madamba et al. (2009) found a gamma-ray-induced IR64 mutant, G978, that gave enhanced resistance to blast and bacterial blight. The resistance was found to be quantitative and nonrace-specific against bacterial and fungal pathogens. The mutation was shown to be inherited as a single recessive gene, Bsdr1, and it caused a shorter stature relative to IR64 and was mapped as a QTL to a 3.8-Mb region on chromosome 12. Comparison of the gene expression profiles of the mutant and wild type showed the candidate gene to encode a U-box domain-containing protein. The disrupted gene exhibited a loss of expression in the mutant and cosegregated with the mutant phenotype (Madamba et al. 2009). These techniques of causing mutations have a problem of creating more undesirable than desirable phenotypes. In other words, these techniques result in random mutations in the genome. The frequency of variations can be controlled but not in the genomic region where they are desired to occur. To achieve a desirable outcome from these experiments, a large population of mutants has to be screened, and this requires a lot of time, space, and resources.
Ultimately, the new era of genome engineering technologies offers vast potential for crop improvement as they allow site-specific modifications of DNA sequences to be executed under laboratory conditions. The accessibility to vast genomic resources and an easy-to-handle genome size make rice more amenable for GM technologies. Advances in genomics and the development of various genome-editing technologies using engineered site-specific nucleases (SSNs) have made the application of genome engineering to crops easy. Among various SSNs, the CRISPR/Cas9 system is commonly applied because of its simplicity, robustness, and high efficiency (Wang et al. 2018). In comparison with other genome-editing tools such as zinc-finger nucleases and transcription activator-like effector nucleases (TALENs), this technique is versatile and simple (Ma et al. 2015b). This technology has been applied to agricultural crop plants with the aim of crop improvement. Oliva et al. (2019) used the CRISPR-Cas9 system to introduce mutations in three SWEET gene promoters to make robust and broad-spectrum bacterial blight-resistant lines. There is still much scope for its use and application.
In the future, the challenge for scientists is not only to develop rice varieties for specific diseases but also to select for horizontal resistance without altering other desirable traits of elite rice varieties. A systematically designed experiment involving highly efficient molecular tools would make it possible to achieve this outcome. Hence, the current chapter amalgamates details on the present status of the key diseases that affect rice production, various molecular strategies for attaining disease resistance, and prospects of molecular breeding for disease resistance in rice.
2 Phenotypic Screening Techniques for Major Diseases of Rice: Pathogen Inoculum, Plant Infection Assays, and Disease Scoring
2.1 Bacterial Blight
Bacterial blight (BB) , caused by Xanthomonas oryzae pv. oryzae (Xoo), targets the seedling stage of rice, resulting in leaves turning grayish green and rolling up. Usually, BB inoculation can be done in two ways, either in the field or in the greenhouse. Many techniques are available to infect the plant with inoculum such as clipping, needle prick, paint-brush, and spray methods. But the most preferable, efficient, and feasible for inoculation is the clipping method (Jabeen et al. 2011). Individually, collected Xoo strains are multiplied and stored on modified Wakimoto’sagar (Sundaram et al. 2009) and the selected rice plants at 45-days-old stage are clip-inoculated with a freshly prepared bacterial suspension (~108–9 cfu/mL) by the following method given by Kauffman et al. (1973). In this method, 1–2-cm tips of five leaves are clipped with scissors, after they were already dipped in bacterial suspension culture, and disease score is recorded 2 weeks post-inoculation both by visual scoring and by measuring the lesion length (LL) as per the Standard Evaluation System (SES) scale of the International Rice Research Institute (IRRI 1996) (0–3 = resistant, 3–5 = moderately resistant (MR), 5–7 = moderately susceptible (MS), and 7–9 = susceptible).
2.2 Blast Disease
The causal organism for blast disease is a fungus, Magnaporthe oryzae (Mo) . Symptoms of blast can appear during any developmental stage and on all parts of the rice plant, including leaves, leaf collars, necks, panicles, pedicels, and seeds. Standard screening protocols of rice varieties for susceptibility to rice blast are usually carried out by spraying the plant with conidial suspensions under greenhouse and field conditions using local isolates of the pathogen (Takahashi et al. 2009). However, for screening against exotic strains, quarantine restrictions are frequently applied to control any escape of the pathogen into the surrounding environment (Jia et al. 2003). In field conditions, artificial leaf blast disease screening usually takes place in a Uniform Blast Nursery (UBN) (Jamaloddin et al. 2020). Applying an excess rate of nitrogen fertilizer (150 kg N/ha) makes rice more vulnerable to spreading blast infection. Artificial inoculation is done with a highly virulent blast race (fungal conidial suspension at a concentration of 1 × 105 spores/mL) by spraying on UBN beds 25–30 days after sowing (DAS). Later, the beds are covered with polythene sheets during the night to create humid conditions for disease development. The disease score is collected 10–15 days after infection, depending on the severity of the infection on the susceptible check using the SES (IRRI 1996). In in vitro conditions, spot inoculation and filter paper inoculation methods are used for inoculation at the vegetative and reproductive stages of rice plants (Jia et al. 2003; Takahashi et al. 2009).
2.3 Sheath Blight
Sheath blight (ShB) disease is caused by a fungus, Rhizoctonia solani . The fungus attacks the rice plant from tillering to heading stage. The early symptoms of sheath blight involve oval circles on leaves just above the waterline. Various screening methods have been developed to screen for ShB in greenhouse and field conditions. Eizenga et al. (2002) delineated a growth-chamber screening technique for sheath blight on Oryza spp. Later, Jia et al. (2003) developed the detached-leaf method. For screening ShB under greenhouse conditions, three inoculation methods have been described: liquid-cultured mycelia ball, mycelia suspension, and agar block. Out of these, the liquid culture mycelia ball is a more efficient and better method for successful inoculation (Park et al. 2008). Field screening at the reproductive stage is the most commonly used method. But field trials require a lot of labor and a large amount of seed material, inoculum, and high-humidity conditions for up to 3–5 months to complete the evaluations (Jia et al. 2007). Normally, screening of selected material for ShB tolerance/susceptibility is done using a highly virulent isolate of Rhizoctonia solani. Initially, ShB isolate is maintained on a potato dextrose agar (PDA, extract from 200 g/L of potato, 20 g/L of dextrose, and 20 g/L of agar) plate and incubated at 28 °C in darkness. For plant inoculations, Typha stem pieces (3–4 cm) are cut and autoclaved in plastic covers. This sterile Typha is inoculated with a 5-mm mycelial plug of R. solani from a 3-day-old PDA plate and incubated in the dark for 10 days at 28 °C. The colonized Typha pieces will be used for inoculating the rice plants at a rate of three to four pieces per hill.
Disease phenotype will be scored 2 weeks after inoculation by measuring the relative lesion height (RLH) as per the following formula:
The IRRI (1996) phenotype scale is used to classify the plants based on their disease severity index from 0 to 9.
2.4 Sheath Rot of Rice
Sheath rot (ShR) is a symptom that is observed in rice plants when infected by any of the following pathogens: Sarocladium oryzae, Fusarium sp., Pseudomonas sp., and Cochliobolus lunatus. Other pathogens have been reported to cause similar symptoms in rice (Bigirimana et al. 2015). Multiple screening techniques that are being used for sheath rot disease resistance in rice include the mealybug inoculation method, rice grain/hull inoculum, leaf piece inoculum, cotton swabbing of conidial spores, spraying or injecting conidial suspension on the sheath, and detached tiller–based assays (Mahadevaiah et al. 2015; Samiyappan et al. 2003). The established screening methods differ depending on the causal agent of ShR as well as the growth stage of the plant. The pathogen is cultured on PDA plates for up to 14 days at 28 °C (Panda and Mishra 2019). A study by Mahadevaiah et al. (2015) compared multiple inoculation methods for Sarocladium oryzae during different growth stages and observed that seed inoculation is a suitable screening method for screening for disease resistance in young plants or early infection. In this method, the seeds are soaked overnight in conidial suspension (105 spores/mL) and then germinated. The number of germinated plants and lesion lengths 14 days after inoculation are scored. For screening plants in peak vegetative to booting stage, foliar inoculation methods were able to provide reliable and conclusive results 15 days after infection. For a faster in vitro screening, the detached-tiller assay is recommended, in which the tillers are cut and placed on moist paper and inoculated with mycelial mats. Visible lesions are observed as early as 3 days postinoculation (Samiyappan et al. 2003; Mahadevaiah et al. 2017). Disease severity is estimated by measuring the area of the sheath and/or leaf affected (Mahadevaiah et al. 2017). Pseudomonas fuscovaginae also causes ShR in rice (Bigirimana et al. 2015). Adorada et al. (2013) reported and recommended multiple screening techniques for screening ShR caused by P. fuscovaginae. The bacteria are cultured using King’s medium B initially for about 24 h. For plant inoculations, the following methods were found to be effective: (1) pin-pricking the upper leaf sheath using a needle dipped in bacterial suspension (107 cfu/mL) and measuring disease severity 14 days postinoculation in plants at the booting stage; (2) spraying the inoculum was found effective and is recommended for mass screening for ShR resistance in plants at the booting stage; (3) for early-stage resistance, soaking seeds in bacterial inoculum before germination is recommended, followed by measuring the decrease in seedling height 10 days later (Adorada et al. 2013).
2.5 False Smut
The fungus Ustilaginoidea virens causes false smut of rice. This fungus attacks the developing panicles and leads to the formation of smutted balls (cottony flakes around the grains). The fungus is generally cultured on potato sucrose agar plates or potato sucrose broth for mass production of conidial suspension (Panguluri and Kumar 2013). Screening for false smut is done during the booting stage of the plants through the following methods. Spraying conidial suspension (5 × 104 spores/mL) at the booting stage is one of the recommended ways for screening for false smut (Kaur and Singh 2017). Another method involves injecting the conidial suspension into the boot (Panguluri and Kumar 2013; Kaur 2014). It has been observed that spraying spores has produced a higher disease incidence and this is suitable for screening for resistant varieties (Kaur 2014). Disease severity is scored by calculating the percentages of infected tillers and infected grains per panicle and a score is assigned as recommended by Rice SES (IRRI 2013; Chaudhari et al. 2019).
2.6 Tungro Disease of Rice
Rice tungro is caused by two viruses, RTBV (rice tungro bacilliform virus) and RTSV (rice tungro spherical virus) , and is transmitted by green leafhopper (GLH: Nephotettix virescens (Dist.)). The viral infection is manifested by the stunted growth of rice plants and yellowing of leaves (Anjaneyulu et al. 1982; Panguluri and Kumar 2013). Nursery screening for tungro resistance in rice is carried out by letting three to five viruliferous GLH per plant (20–30 days old) feed in a closed environment and scoring the disease symptoms 14 days later as recommended by Rice SES (Anjaneyulu et al. 1982; Sebastian et al. 1996).
2.7 Bacterial Leaf Streak
Bacterial leaf streak (BLS) of rice is caused by the bacterium Xanthomonas oryzae pv. oryzicola (Xoc). BLS is manifested as water-soaked lesions on the leaf surface, which can result in decreased photosynthesis and hence diminished yield (He et al. 2012). Screening for BLS resistance is mainly performed using either of two methods. For screening seedlings, bacteria are initially grown in peptone sucrose broth, and a bacterial suspension from 108 to 109 cfu/mL is used for infiltrating the expanded leaves using a needleless syringe. The disease symptoms are scored 14 days postinoculation (Ju et al. 2017). For field screening or screening older plants, matured leaves are pin-pricked with needles that are dipped in bacterial inoculum on either side of the leaves. The lesions caused are measured 20 days postinoculation (Tang et al. 2000; Chen et al. 2006a, b; He et al. 2012). Disease severity is scored as per Rice SES.
3 Genetics of Disease Resistance
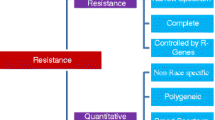
Deployment of genes conferring host-plant resistance provides an economical, durable, effective, and environmentally safe approach to combat plant diseases and decrease yield losses (Fig. 1). Major resistance genes from different resistance donors have been reported for various rice diseases. So far, more than 44 resistance genes have been identified against bacterial blight (Kim and Reinke 2019). More than 100 distinctive blast-resistance genes have been reported on different rice chromosomes and, out of these, 21 genes have been cloned (Devi et al. 2020). Two major sheath blight QTLs (qShB9-2 and qSBR11-1) have been reported (Channamallikarjuna et al. 2010). But, thus far, genetic diversity for high resistance to/tolerance of ShB has not been reported in either cultivated rice or its wild relatives; thus, cloning of genes for ShB resistance is straggling (Bonman 1992). For bacterial leaf streak (BLS), no major resistance genes (R-genes) have been identified and only a few QTLs have been mapped. Out of these, qXO-2-1, qXO-4-1, and qXO-11-2 were showing resistance to more than nine Xoc and Xoo strains (Bossa-Castro et al. 2018). In the case of tungro disease, a resistance QTL was found in Indian landrace ARC 11554 and was localized on chromosome 4 (Wang et al. 2016). False smut resistance in several rice cultivars has been identified as a quantitative trait controlled by multiple genes (Andargie et al. 2018; Han et al. 2020). But, to date, no rice variety has been identified to show complete resistance to false smut, whereas many cultivars exhibit considerable differences in quantitative field resistance to the pathogen (Huang et al. 2019). According to previous studies, the genetics of sheath rot disease resistance was dissected by studying the segregating pattern in an F2 population (Rajashekara et al. 2014; Mvuyekure et al. 2017) and recombinant inbred lines (Graichen et al. 2010; Mahadevaiah et al. 2017). Some of these R-genes or loci have been extensively used in MAS breeding programs, and some of them have been fine-mapped and are undergoing cloning efforts. Detailed information on resistance genes/QTLs for economically important rice diseases (i.e., bacterial blight, blast, and sheath blight) appears in Tables 2, 3, and 4, respectively.

(a) An illustration of the different biotic diseases in rice and (b) a funnel diagram representing the sources of valuable traits (tolerance of biotic diseases, yield components, and superior grain quality) that exist in traditional and wild rice germplasm. Using various phenotypic screening techniques and genome sequencing technologies can enable us to understand the molecular genetics and physiological mechanisms of stress tolerance. The identified genomic regions of QTLs and genes associated with the key traits play a vital role in understanding the interactions and further improving disease tolerance and superior grain quality traits with the help of marker-assisted selection and genomic selection approaches for crop improvement
4 Breeding for Disease Resistance
Rice breeders have come up with many disease-resistant cultivars adapted to different rice-growing regions worldwide by applying conventional breeding approaches. Because of the dominance and epistasis effects of genes conferring resistance to a few diseases, gene pyramiding through conventional breeding methods becomes a challenge. Also, genes having similar responses to two or more races of a pathogen are difficult to recognize and transfer by conventional approaches (Joseph et al. 2004; Sundaram et al. 2009; Rajpurohit et al. 2011). The exercise of breeding for disease resistance could never obtain a break because of the emergence of new pathotypes, which could overcome the resistance. Advances in rice genomics provided tools such as molecular markers for plant breeders to effectively develop cultivars with resistance against various diseases, which is an environment-friendly alternative vis-à-vis the use of agrochemicals (Miah et al. 2013). Molecular markers can be used to map and introgress one or more desired genes for biotic and abiotic stress resistance from diverse gene pools (Suh et al. 2009). Marker-assisted selection for pyramiding desired genes without altering other quality characteristics of a rice cultivar is crucial in rice improvement (Sundaram et al. 2008; Suh et al. 2009; Shanti et al. 2010). As an added advantage, the availability of gene-linked molecular markers for the resistance genes eases the identification of plants harboring two or more R-genes at any growth stage without a bioassay (Sundaram et al. 2008; Shanti et al. 2010; Bainsla and Meena 2016).
Three bacterial blight-resistance genes (xa5, xa13, and Xa21) were pyramided into susceptible cultivar PR106 using MAS. The introgression lines were tested against 17 Xoo isolates under both glasshouse and field conditions. The trials suggested that the combination of genes provided broad-spectrum resistance against the pathogen races predominant in the region (Singh et al. 2001). Recent advances in DNA sequencing have made fine-mapping and characterization of the mapped genes easier, thus contributing significantly to the use of MAS for the development of resistant cultivars. The complete list of cloned genes was collected from Q-TARO (http://qtaro.abr.affrc.go.jp/) and OryGenesDB (https://orygenesdb.cirad.fr/data.html) (Fig. 2). These genes were mainly associated with bacterial blight and blast resistance in rice. Interestingly, the regions on chromosomes 1, 4, and 5 were associated with multiple resistance genes, and these genes were colocalized in the same regions. These genomic regions play a major role in the resistance/tolerance mechanisms for diseases. To date, there are 46 BB R-genes mapped from different sources, out of which 29 are dominant, 12 are recessive, nine cloned, and nine fine-mapped (Chen et al. 2020). More than 100 R-genes (Pi) have been reported, and around 500 QTLs were associated with blast resistance. However, only 25 Pi genes were cloned and characterized (Sharma et al. 2012; Ashkani et al. 2015). Several R QTLs were reported against bacterial leaf streak, but their study was limited to inheritance analysis (He et al. 2012).

Physical position of major biotic disease-resistance genes in rice. The chromosome left side indicates the location of genes and the right side shows the names of genes collected from the Q-TARO and Oryza base databases. The color indicates the genes related to various diseases such as bacterial leaf blight (red color) and blast (blue color), and green color indicates insect resistance in rice
4.1 MAS/MABB Foreground/Background Selection
To address the limitations of conventional breeding, molecular breeding through MAS is among the most precise tools used to introgress multiple resistance genes into an elite varietal background at one time. Plant breeders were already successful in using this tool in developing resistant rice cultivars by deploying broad-spectrum multiple-R-genes with the help of MAS (Huang et al. 1997; Sanchez et al. 2000; Sundaram et al. 2008; Hari et al. 2013; Hajira et al. 2016; Balachiranjeevi et al. 2018; Swathi et al. 2019; Jamaloddin et al. 2020). In marker-assisted backcross breeding (MABB), a combination of foreground selection and background selection followed by continuous backcrossing can recover up to 99% of the recurrent parent genome (RPG) (Tanksley et al. 1989). In foreground selection, gene-linked markers, or functional markers (SSRs, InDels, and SNPs), are applied to detect the associated R-genes in the target population at any stage of plant growth. In contrast, background selection applied by using polymorphic information (SSRs, SNPs) between the donor and recurrent parents can estimate RPG recovery in each backcross generation at any plant growth stage (Singh et al. 2001). Recently, the Green Super Rice (GSR) breeding strategy proved that one backcross followed by selfing could recover more than 90% of the recurrent parent genome (Balachiranjeevi et al. 2019).
4.2 Pyramiding Disease-Resistance Genes
Pyramiding of various biotic disease-resistance genes into a rice cultivar makes it a good candidate for breeders to introgress the resistance into locally adapted varieties that produce higher yield but are susceptible to diseases. The process of gene pyramiding through conventional breeding alone becomes difficult because the linkage between some undesirable traits is difficult to break even after repeated backcrossing (Tanksley et al. 1989). Pyramiding of two or more resistance genes renders the phenotypic assessment of rice genotypes ineffective as distinguishing the effect of each individual gene precisely becomes difficult since each gene imparts resistance to more than one race of the pathogen. Moreover, when a dominant and a recessive allele are present, the effect of the recessive gene is concealed. The availability of tightly linked markers for each of the resistance genes thus eases the recognition of plants with multiple genes. Initially, in rice, Huang et al. (1997) successfully introgressed four major BB resistance genes (Xa4, Xa5, Xa13, and Xa21) and developed breeding lines with combinations of two, three, and four genes. In an extension of this work, several research institutions in India and other countries have studied the effectiveness of the pyramided genes against BB disease, to which most of the popular varieties were susceptible. This research has opened the gates in India to address the susceptibility of popular rice varieties such as PR106 (Singh et al. 2001), Pusa Basmati-1 (Joseph et al. 2004), and Samba Mahsuri (Sundaram et al. 2008) by pyramiding the BB R-genes (xa5, xa13, and Xa21) in the initial phase of improvement. Later, the improvement of popular rice varieties and parental lines continued mainly against BB (Xa21, Xa23, xa5, xa13, Xa4, Xa7, Xa33, and Xa38) and blast disease (Pi genes Pi2, Pi9, Pi40, Pi54, Piz, and Pi1) separately or by combining R-genes for both diseases (Gopalakrishnan et al. 2008; Sundaram et al. 2009; Hari et al. 2013; Balachiranjeevi et al. 2015; Yugander et al. 2018; Rekha et al. 2018; Swathi et al. 2019; Jamaloddin et al. 2020). The possibility of recombination between the gene of interest and the linked marker has led to the selection of false-positive rice genotypes in the marker-assisted selection process, which could be overcome by using gene-specific functional markers (Ingvardsen et al. 2008). Many genetic markers, also called functional markers, have been identified for different disease-resistance genes in rice, such as BB-resistance genes xa5 (Iyer-Pascuzzi and McCouch 2007), xa13 (Chu et al. 2006), and Xa21 (Song et al. 1995). The gene-pyramided lines enable the conducting of quantitative analysis to assess the effect of each gene and interactions between them and, most importantly, enhancing the performance, stability, and longevity of genetic resistance.
4.3 Varieties Improved and Developed
Highly accepted varieties and parental lines were improved against multiple diseases through MAS. For the first time, Huang et al. (1997) developed lines pyramided with two, three, and four genes through MAS and tested their resistance against BB. The resistance levels of introgressed lines showed an elevated resistance compared with lines containing a single gene. Later, Singh et al. (2001) improved Indian rice cultivar PR106 against BB through MAS by pyramiding xa5, xa13, and Xa21 genes, followed by Joseph et al. (2004), who improved popular basmati variety Pusa Basmati-1, and Sundaram et al. (2008) improved popular variety Samba Mahsuri for BB and reported more than 95% RPG recovery through MABB. Through MAS, three blast genes (Pi1, Pi2, and Pi33) were introduced in the background of popular Russian rice variety Kuboyar. The improved lines of Kuboyar were used to develop blast-resistant hybrids by using them as hybrid parental lines. Similarly, Hari et al. (2011) improved restorer line KMR3R for resistance against BB by transferring the Xa21 gene along with Rf3 and Rf4 (restorer of fertility) genes through MABB. Balachiranjeevi et al. (2015) imparted resistance to a maintainer line (DRR17B) by introgressing Xa21 and Pi54 genes against BB and blast disease, respectively.
4.4 Multiple Disease-Resistance Breeding Strategies
In breeding for disease resistance, multiple methodologies such as pedigree, modified bulk, single seed descent (SSD), doubled-haploid (DH), and MAB have been used to develop resistant rice varieties (Mackill et al. 1996; Khush 2005; Collard et al. 2013). In addition to these strategies, the GSR breeding program was one of the successful strategies that involved vigorous phenotypic screening at early backcross stages (BC1F2 to BC1F4) combined with three successive rounds of stringent selection for the best plant type to come up with climate-resilient rice varieties. This strategy could develop homozygous inbred cultivars within a short span of 4–5 years vis-à-vis 9–10 years with a conventional breeding program (Yu et al. 2020). The GSR breeding strategy is carried out in three steps. The first is to develop early backcross BC1F2 populations by crossing a widely adapted recipient variety with a diverse set of donors. Second is to simultaneously do phenotypic screening of early backcross-derived lines of BC1F2, BC1F3, and BC1F4 generations under the different abiotic and biotic stress conditions in a rigorous manner to identify and select introgression lines (ILs) tolerant of different stresses as compared to the tolerant and susceptible checks. The third step is the mapping of genomic regions influenced by particular climate fluctuations and their characterization to decode the molecular and physiological basis of the identified genomic regions (Ali et al. 2017). Three rounds of screening of populations from BC1F2 to BC1F4 for different diseases simultaneously could help in the development of varieties with tolerance of multiple biotic stresses. The GSR breeding strategy led to successful mapping of the Xa39 gene and deploying it in the background of Huang-Hua-Zhang (Zhang et al. 2015). Further, through the designed QTL pyramiding approach, one could combine selective ILs carrying different biotic and abiotic stress-tolerance genes/QTLs derived from different donors but having a common recipient parent. Similar to the GSR breeding program, breeders have simultaneously pyramided multiple disease-resistance genes (BB + blast) with different combinations such as Xa21 + Pi54, Xa21 + Pi54 + Pi2, and xa5 + xa13 + Xa21 + Pi54 + Pi2 into the background of an elite cultivar by employing MAS and MABB (Jiang et al. 2015; Jamaloddin et al. 2020). Recently, one of the successful breeding strategies (the GSR breeding program) revealed lots of hidden genetic diversity for disease resistance through MAS and also proved that RPG recovery could surpass 90% with one backcross followed by selfing (Balachiranjeevi et al. 2019). Furthermore, Feng et al. (2018) reported that pyramiding the detected QTLs effectively broadened the genetic base. Research is being extended to dissect the detected QTLs in order to identify candidate genes through functional validation using a map-based cloning approach.
5 Molecular Mechanisms of Disease Resistance
A wide variety of pathogens , including bacteria, fungi, and viruses, attacks crop plants. Either a pathogen can successfully invade, leading to the development of disease, or the plant can resist the pathogen using an active or passive form of resistance. Different strategies have been developed by various pathogens to enter, infect, and reproduce in plants. Pathogens are mainly classified as necrotrophs and biotrophs based on the method they use to invade, infect, and attack a plant (Oliver and Ipcho 2004). Necrotrophic pathogens kill the host-plant tissue soon after they establish infection and then develop and feed on the dead tissue. Unlike these, biotrophic pathogens require a live-host tissue for their growth and reproduction.
Specific defense mechanisms work effectively against biotrophs through a hypersensitive response developed by rapid local cell death surrounding infection, and this serves to hinder the growth and invasion of pathogens into other plant parts. This mechanism arises when the first level of the defense mechanism is breached by the pathogen (Zipfel and Felix 2005). Usually, most pathogens that infect plants, such as fungi, harbor secretory proteins, which disrupt these barriers (Serrano et al. 2014). After the entry of the pathogen into the host cell, it is recognized by special molecules called microbe-/pathogen-associated molecular patterns (MAMPs or PAMPs) , which include ergosterol, peptidoglycan, lipopolysaccharide, and bacterial flagella in proteins. The innate immune system recognizes these proteins with the help of host plasma membrane-bound receptors called pattern recognition receptors (PRRs) to further obstruct the growth of infection, providing MAMP-triggered immunity (MTI). PRRs also detect molecules that become released in the host when the pathogens cause damage (damage-associated molecular patterns, DAMPs). The binding of these components also triggers pattern-triggered immunity (PTI) and downstream defense responses (Tena et al. 2011). Overall, the recognition of PAMP/MAMP or DAMP results in the activation of PTI, triggering the production of different reactive oxygen species (ROS), initiation of mitogen-activated protein (MAP) kinase activity, and various transcription factor activation, thus limiting the spread of pathogens completely (Nürnberger and Kemmerling 2009).
The widely accepted model of plant disease resistance is explained by a two-level innate immune system. The two levels include PTI, which is usually a weak, basal, and generic immune response, and the other is effector-triggered immunity (ETI), which is a potent response and is specific to the pathogen in question (Jones and Dangl 2006). PTI is mediated by the PRRs that recognize molecular patterns associated with the pathogens or the resulting damage products (PAMPs or DAMPs). On the other hand, ETI includes recognition of a pathogen-specific factor and results in a severe and rapid form of immune response leading to localized cell death (also known as a hypersensitive response or HR) to hinder the pathogen from spreading any further. ETI is achieved by a gene-to-gene interaction and is thus specific to the race of the pathogen. While PRRs mediate PTI, ETI is mediated by specific genes that belong to the nucleotide-binding-leucine-rich repeat (NB-LRR) domain-containing proteins, otherwise called resistance (R) genes. The recognition of cognate ligands results in activation of signaling events that in turn results in the generation of different forms of immune response such as callose and lignin deposition, production of antimicrobial compounds, induction of cell death, changes in primary and secondary metabolic flux, and synthesis of secondary metabolites depending on the type of elicitors. Other classifications of genes involved in disease resistance include major resistance (MR) genes and defense-related genes (DR), whose roles cannot be explained by the definition of PTI- and ETI-associated genes (Ke et al. 2017). PTI is considered to be quantitative in nature, that is, multiple genes function together to achieve immunity, also known as a QTL. ETI against a pathogen strain is controlled by a single gene and is specific only to those strains that contain the cognate avirulence (Avr) protein that the R-gene recognizes, thus leading to a qualitative resistance. Studies in the past few decades established a framework of how the resistance mechanisms act using model pathosystems. Along this line, rice resistance to its major pathogens such as Xanthomonas oryzae ssp., Magnaporthe oryzae, and Rhizoctonia solani has been studied to a reasonable extent. However, rice resistance to other pathogens still needs more investigation to come to a consensus. This section summarizes the established mechanisms of disease resistance in rice.
Plant resistance is dictated by the type of resistance genes and a network of signaling pathways (Chisholm et al. 2006). Broadly, the plant defense system can be categorized into two classes: basal defense and specific defense. The basal defense system is much more effective against necrotrophic pathogens (Singh et al. 2018). Elicitors are molecules that induce a plant defense response at very low concentrations (Thakur and Sohal 2013). The role of the basal defense system is to check the entry of pathogens and provide immunity at the starting stage of infection. This defense response involves membrane permeability, activating ion fluxes (Ca2+, K+, H+), generating ROS, producing nitric oxide (NO), and phosphorylation/dephosphorylation of proteins by protein kinases and phosphatases. It also includes the production of signaling molecules such as jasmonic acid (JA), salycilic acid (SA), and ethylene (ET). These proteins are characteristic players in the regulation of defense signal transduction cascades. These steps further trigger an array of signaling that leads to the regulation of the expression of defense-related genes and the stimulation of defense responses. These responses include cell-wall strengthening (callose and lignin deposition), phytoalexin synthesis, and activation of kinase cascades escorted by a hypersensitive response (Jones and Dangl 2006).
5.1 Resistance to Bacterial Blight
To date, 46 resistance genes have been identified to confer resistance to Xoo in rice. Among them, 11 genes were cloned and functionally characterized. Some of the resistance genes are quantitative in nature, whereas others confer qualitative resistance (Ke et al. 2017; Jiang et al. 2020; Chen et al. 2020). The 11 cloned genes fall under different classes of resistance genes: LRR-RLKs (leucine-rich repeat receptor-like kinases), NB-LRR, a wall-associated kinase, executor R proteins, SWEET (sugars will eventually be exported transporters) genes, and a transcription factor gamma subunit protein. Three of the cloned resistance genes, Xa3/Xa26, Xa4, and Xa21, code for kinases. Xa4 is a wall-associated kinase (Ke et al. 2017; Jiang et al. 2020) that provides resistance to certain races of Xoo through cell-wall reinforcement. Xa3/Xa26 and Xa21 are LRR-RLKs that recognize Xoo-associated molecules AvrXa3 and sulphated RaxX, respectively. Xa21- and Xa3/Xa26-mediated resistance has been found to be positively regulated by OsSERK2 (rice somatic embryogenesis receptor kinase 2). Nine genes were found to be regulating Xa21-mediated resistance positively or negatively. Xa4-mediated resistance leads to the accumulation of phytoalexins. Xa1, an NB-LRR, recognizes intact transcription activation-like effectors (TALEs) from Xoo and thus leads to resistance. SWEET genes code for sugar transporters and were identified to be targets of different Xoo TALEs, thereby acting as susceptibility factors. Natural polymorphisms were identified in the promoters of three SWEET genes, OsSWEET11/Os8N3/xa13, OsSWEET13/xa25, and OsSWEET14/Os11N3/xa41, which promote their induction by cognate TALEs, thus providing recessive resistance. Genes, including Xa10, Xa23, and Xa27, are classified as executor R-genes as the expression of the respective resistance alleles is induced by Xoo TALEs. These genes are characterized by the presence of multiple potential transmembrane domains whose expression induction results in HR and thus resistance to Xoo. Another recessive resistance gene, xa5, codes for transcription factor IIA gamma subunit 5 (TFIIAγ5) with valine to glutamine mutation in the 39th position. The susceptible allele, Xa5, is hijacked by the TALEs to induce the expression of other host susceptibility genes. The mutation disrupts the ability of TALEs to bind to TFIIAγ5, thus leading to resistance (Ke et al. 2017; Jiang et al. 2020).
5.2 Resistance to Bacterial Leaf Streak
To date, no major BLS-resistance genes have been identified. However, the xa5 gene was mapped to be a major resistance QTL for Xoc resistance. It was previously observed that TALEs from Xoc also hijack TFIIAγ5 for inducing host susceptibility genes. In another study, Xo1, a resistance locus in an American rice variety, was identified to be responsible for resistance to African Xoc strains but not to Asian strains. Xa21 was identified to provide weak resistance to Xoc through the recognition of Ax21, a quorum-sensing molecule produced by Xoc (Jiang et al. 2020). Three major broad-spectrum resistance QTLs, qXO-2-1, qXO-4-1, and qXO-11-2, were identified to confer resistance to Xoo and Xoc (Bossa-Castro et al. 2018).
5.3 Resistance to Rice Blast
More than 100 resistance genes and 500 QTLs are known to be associated with blast resistance in rice. However, to date, only 25 genes have been cloned (Li et al. 2019b). These 25 cloned R-genes are called Pi genes. Of the 25 Pi genes, 22 encode NB-LRR family proteins. A majority of these R-genes trigger ETI, thus leading to qualitative or race-specific resistance. So far, seven R-genes have been identified to confer broad-spectrum resistance to blast: Pi7, Pi9, Pi21, Pi50, Pi57, Pigm, and Ptr. Apart from canonical R-genes, so far, five defense-related genes were also shown to confer resistance to blast: bsr-d1, bsr-k1, spl11, spl33, and OsBBI1, Pi9, Pi50, Pigm, Ptr, and OsBBI1 are dominant resistance genes or positive regulators of blast resistance, whereas the rest of them are recessive resistance genes, in other words, their wild-type alleles negatively regulate blast resistance (Li et al. 2019b).
5.4 Resistance to Sheath Blight
Information on the mechanisms that govern ShB resistance in rice is just being uncovered. There are no reports on a single resistance gene that confers resistance to ShB. However, many QTLs have been identified to be associated with ShB resistance. Most of the QTLs were reported to provide a minor contribution to the resistance phenotype, whereas two QTLs (qShB9-2 and qShB11-1) were found to contribute more than 10% to ShB resistance. Sequence analyses revealed the presence of various defense-associated genes in these QTLs. qShB9-2 was identified in many rice varieties that exhibit resistance to ShB. It was observed that qShB9-2 contains a β-1,3-glucanase, OsWAK91, and 12 other possible candidate genes. On the other hand, the qShB11-1 interval was shown to have receptor-like kinases, a lipase, and a tandem array of 11 chitinase genes. Tens of minor QTLs were found to be associated with ShB resistance. Nevertheless, no information is available on the gene(s) responsible for the resistance. Studies using resistant cultivars shed light on the possible mechanisms by which rice fights Rhizoctonia solani. Various studies showed changes in metabolic pathways, including primary and secondary metabolites. Intermediates of glycolysis and tricarboxylic acid cycle were found to be accumulated in rice post R.solani infection, indicating the possible involvement of primary metabolism in response to the pathogen. Also, the accumulation of secondary metabolites such as phytoalexins, chlorogenic acid, polyphenols, and flavonoids was reported to be higher in the tolerant varieties than in the susceptible varieties postchitin treatment (Molla et al. 2020). ROS deregulation has been observed to delay pathogen colonization in resistant cultivars (Oreiro et al. 2019).
5.5 Broad-Spectrum Resistance Genes
From a breeder’s point of view, a single locus/gene is more preferred as it would permit easier introgression. Many defense-related genes have been identified to provide broad-spectrum resistance to either multiple races of a pathogen (vertical resistance) or multiple pathogens altogether (horizontal resistance). Such responses are quantitative in nature and hence can be highly durable and practical to keep infectious diseases at bay. Several previous studies have been reported that the expression of defense-response genes (DR genes) such as rice germin-like proteins (OsGLP) or a class of DR genes present in a QTL along with R genes is also most probably associated with rice resistance, as knockdown of these genes escalated the susceptibility against two major rice fungal diseases, blast and sheath blight (Manosalva et al. 2009). OsPAL4 is reported to impart broad-spectrum resistance to rice (Tonnessen et al. 2015). A LysM receptor-like kinase (RLS), OsCERK1, regulates cytoplasmic OsRLCK176 and OsRLCK185 recognizes chitin and peptidoglycans activating immune signaling pathways in rice against blast and bacterial blight diseases. OsSERK1, OsWAK25, OsWRKY45-1, OsWRKY45-2, OsWRKY13, OsDR8, OsMPK6, OsPAL4, OsNH1, OsLYP4, OsBSR1, and OSK35 have all been shown to regulate resistance to bacterial blight and rice blast positively . OsPAD4 and OsPAL4 positively regulate resistance to ShB, whereas OsWAK25 negatively regulates ShB resistance (Ke et al. 2017). These genes, although identified in different studies, play a highly connected role in helping rice fight the invading pathogens. More comprehensive studies are needed to link the dots to construct a complete map of rice resistance to diseases.
6 Impact of Major Nutrient Fertilizers on Biotic Disease Resistance in Rice
The rapidly increasing world population requires a sustainable nutritional global food supply, which is a significant concern for crop production. Changing climatic scenarios and decreasing natural resources suggest that there is a need to intensify agricultural production using an efficient agronomic nutrient management (ANM) system. Following efficient ANM technologies can enable us to understand and mitigate the adverse impacts of stress, inadequate soil fertility status, pathogens, and pests (Dordas 2008).
Several efficient screening technologies exist, such as smart water irrigation systems, integrated fertilizer applications, and disease biocontrol strategies, that have been developed and adopted in different ecosystems to control various diseases in rice (Bargaz et al. 2018). Among these, the rate of fertilizer used, judicious and timely applications of nutrients, and availability of these nutrients play a crucial role in plant growth and also in developing defense mechanisms against various pests and diseases (Fageria et al. 2008; Sun et al. 2020). The management of nutrient statuses in the soil, especially nitrogen (N), phosphorus (P), and potassium (K), is an eco-friendly strategy to control different biotic stresses instead of frequent application of pesticides. Globally, the efficiency of fertilizer use by the crop and the correct rates of fertilizer applications are poorly studied. Earlier studies have indicated that only 30–35% of N, 10–25% of P, and 35–50% of K are taken up by plants. Particularly in China, the amount of fertilizer used has increased drastically from 270 to 350 kg/ha, which is more than 75% of the global average of fertilizer application. This excessive amount of N fertilizer leads to leaching, which is a significant cause of groundwater pollution and degradation of soil quality (Teng et al. 2016).
Developing sustainable agriculture is one of the major strategies to increase global rice production. Application of nutrient fertilizer at the right rate and stage and also microorganisms are the key factors in disease control. The essential nutrient elements can decrease disease severity but also increase the severity of disease incidence (Dordas 2008). Nitrogen is one of the key elements for plant growth and development, which are involved in the major physiological and metabolic pathways related to N assimilation (Bolton and Thomma 2008; Mur et al. 2017). Plenty of research has been conducted on the role of N and its interaction with disease resistance and the results are inconsistent, with a poor understanding of the resistance mechanisms in physiological and metabolic pathways. These differences may be due to various stress signaling mechanisms caused by the different forms of N (NH4+ and NO3−), the type of pathogen specificity, and the stage of N application (Dordas 2008). However, several researchers have suggested that the correct time application of fertilizers has been significantly increasing disease resistance and decreasing the use of fungicides (Anderson 2002; Hervieux et al. 2002; Bhat et al. 2013). Recently, Sun et al. (2020) reviewed N applications and their critical role in the defense mechanisms in various diseases such as blast, downy mildew, stem rot, powdery mildew, leaf rust, stem rust, and rice blast diseases in plants. Balancing of these nutrients is imperative to understand the cellular structure and composition, which mainly affect plant defense mechanisms. For instance, high rates of N application lead to a significant impact on susceptibility by decreasing the thickness of cell-wall components (cellulose and lignin), whereas decreasing N applications lead to an increase in lodging resistance by changes in stem lignification and secondary cell-wall synthesis (Zhang et al. 2017b; Sun et al. 2018). Also, decreasing N fertilizer significantly increases the incidence of major insect pests, including brown planthopper, leaffolder, and stem borer, the key insect pests in the major rice-growing areas in Asian countries (Lu et al. 2007). Some reports have suggested that N applications significantly influence the size of leaf blast lesions (Matsuyama 1973; Kaur et al. 1979). Sime et al. (2017) studied the different rates of nutrient fertilizer application and their relation to blast disease. The combination of NPK (20-10-10) at a rate of 200 kg/ha has a remarkable impact on decreasing blast disease in all phases of plant growth. Similarly, Reddy et al. (1979) reported an optimal rate of N application (76 kg/ha) to maximize grain yield and also minimize disease. One of the major diseases is bacterial leaf blight of rice, caused by Xanthomonas oryzae , which increased significantly when a higher amount of N fertilizer (>100 kg/ha) was applied, and yield decreased. Begum et al. (2011) reported that a balanced application of nutrient fertilizers, including K, significantly decreased the percentage of BLB. The application of K fertilizer has dramatically decreased the intensity of various infectious diseases such as BLB, sheath blight, and stem rot in rice, and also in other cereal crops (Sharma et al. 2005a). Decreasing BLB severity by applying K topdressing is a viable approach just before disease-occurring stages and this makes it possible to maximize grain yield and have lesser disease development.
Using slag-based silicon (Si) fertilizer in rice fields is an alternative approach to control the major disease brown spot, which is caused by the fungus Bipolaris oryzae . This disease causes significant yield losses, mainly in tropical and subtropical areas, where the frequent occurrences of heavy rainfall and high temperature are the main factors in decreasing the Si content in highly weathered soils (Raven 2003). The major role of these Si applications is to mediate resistance mechanisms through the physiological and metabolic pathways that can lead to creating more pronounced cell silicification in rice leaves, and the strong leaf epidermal surface might increase the resistance to fungal penetration (Hayasaka et al. 2008; Sun et al. 2010; Ning et al. 2014). These Si fertilizers provided clear evidence showing the importance of increasing the thickness of the silicon layer in the epidermal cell walls that are supposed to be the main site for conferring resistance to brown spot disease in rice. Interestingly, Wu et al. (2017) experimented with the transcriptional responses in two different nutrient fertilizers, Si and N concentrations, and their relation to BPH infestation. These two elements had a trade-off mechanism in terms of resistance. The interaction of these two elements clearly showed decreases in the expression of Si transporters such as OsLsi1 and OsLsi2 under high rates of N application, whereas, in the N transporters OsNRT1:1, OsGS2, OsFd-GOGAT, OsNADH-GOGAT2, and OsGDH2, expression increased under a high rate of Si fertilizer. This demonstrated that N and Si had antagonistic interactions in rice (Wu et al. 2017). Similarly, Robichaux (2001) identified a significant decrease in the major disease sheath blight, caused by the fungal pathogen Rhizoctonia solani, by adding calcium silicate in greenhouse and field conditions. Rice grain yield is increased by almost 13% from the use of a calcium silicate application rate of 3.3 mg/ha and also a significant decrease in ShB in different soil types. These results have proven that Si fertilizer can diminish fungal disease severity by increasing the Si concentration in rice leaves and boosting grain yield.
7 Genome-Editing Tools for Improving Disease Resistance
Diseases cause a considerable yield loss annually (Heinrichs and Muniappan 2017; Mushtaq et al. 2019). Breeding for disease resistance has been pursued for a long time. The traditional practice is to introgress disease resistance into elite cultivars through breeding techniques. Although a successful method, it has its downside (Zafar et al. 2020). The traditional way is time-, labor-, and resource-consuming (Romero and Gatica-Arias 2019). With the arrival of the genomics era, identifying disease-resistance genes has become highly efficient, and resistance alleles can be identified at a single base resolution. With such a massive potential in hand and constant improvement in various genome-editing (GE) tools such as site-specific mutagenesis (SSM), meganucleases (MNs), zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein (CRISPR/Cas), this has opened a novel path to achieve the improvement of disease resistance in various crops (Zhang et al. 2017a; Mishra et al. 2018; Zafar et al. 2020). Recently, several researchers have reviewed the various GE tool applications and their limitations in target gene specificity and accuracy (Abdallah et al. 2015; Mishra et al. 2018; Zafar et al. 2020). As compared with SSM, MNs, and ZFNs, the most widely used GE tools such as TALENs and the CRISPR/Cas system have a versatile, fast, and relatively efficient GE method. Over the past several years, these two methods have transformed the field of genome engineering, and they can easily edit and also recognize specific genomic regions (Gaj et al. 2016). These methods have a significant impact on the genomic revolution that has accelerated the discovery of novel sequence variations and breakthroughs in the scientific knowledge to demonstrate the power of these GE tools in establishing resistance to pathogens in various diseases. The rapid progress in the CRISPR-Cas9 system makes it a highly accurate and efficient method that can edit in multiple genes at multiple locations using a single molecular construct (Cong et al. 2013).
7.1 Site-Specific Mutagenesis: The Path So Far
Site-specific mutagenesis is achieved by deploying a class of enzymes called “nucleases” fused with DNA-binding motifs to target specific sequences in the genome. The activity of nuclease results in double-strand breaks at the target site, which are then repaired by the host DNA repair mechanisms via nonhomologous end joining (NHEJ) or homologous recombination (Feng et al. 2013). During this process, small insertions or deletions occur in the genome, thus disrupting the gene sequence (Mishra et al. 2018). SSM is an evolving area of research with newer tools often emerging with improved precision and efficiency.
7.1.1 Meganucleases
Meganucleases (MNs) are endonucleases (enzymes that cut within a strand) that occur naturally and possess sequence-specific DNA-binding and nuclease activities. The application of MNs for site-targeted mutagenesis began in the 1980s. Owing to the recognition of long DNA sequences (18–40 bp), MNs were a good choice. On the other hand, the number of naturally occurring MNs was limited, thus diminishing their wider application. Moreover, custom modification of MNs is a viable but expensive option (Abdallah et al. 2015).
7.1.2 Zinc-Finger Nucleases and TALENs
Zinc-finger nucleases (ZFNs) kick-started the wider application of site-specific mutagenesis in 1996 (Kim et al. 1996). The zinc-finger motif is one of the most copious DNA-binding motifs present in eukaryotes (Klug and Schwabe 1995). Each ZF motif recognizes a specific 3-bp sequence in the major groove of DNA. Thus, tandemly placing multiple ZF motifs of different base specificity and fusing them to a nuclease can result in the generation of a molecular scissor that can precisely cut the target site. Modular assembly-based methods enabled the construction of ZFNs that can virtually target any DNA sequence (Gaj et al. 2013). However, limitations of using ZFNs exist. The modular assembly is a complex and expensive process that requires many optimizations. Off-target cleavage is another challenge that many SSM techniques face (Ramirez et al. 2008; Gupta and Musunuru 2014).
Transcription activator-like effectors (TALEs) are proteins that naturally occur in the genus Xanthomonas , which predominantly consists of phytopathogenic bacteria (Boch et al. 2009; Moscou and Bogdanove 2009). TALEs are employed by Xanthomonas oryzae pv. Oryzae (Xoo) to target and activate the expression of specific host genes to increase the susceptibility of the host. The specific binding to DNA is achieved by a 33–35-amino-acid-long tandem repeat domain, each of which targets a specific base. The base specificity is conferred by the amino acids that are located in the 12th and 13th positions of the series. These positions are called repeat variable di-residues (RVDs) (Boch et al. 2009; Moscou and Bogdanove 2009; Gaj et al. 2013; Abdallah et al. 2015). Exploiting this brought in a revolution in the field of genome editing called TALENs. TALENs are TALE nucleases wherein the DNA-binding motif of a TALE is fused with a catalytic domain of a nuclease, thus allowing the domain to target and cleave a specific sequence in the genome. By modifying the RVDs, one could define the target site and thus assemble a custom TALEN to target any region of interest in the genome (Christian et al. 2010; Boch 2011). The design and delivery of TALENs, however, pose a setback for the technique owing to their large size (Abdallah et al. 2015).
7.1.3 CRISPR-Based Genome Editing
Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas), collectively called CRISPR-Cas, is a microbial adaptive immune strategy that works based on an RNA-guided nuclease complex to cleave foreign genetic elements. A CRISPR-Cas locus is a cluster of Cas genes, noncoding RNA, and an array of repetitive elements. The repeated elements are interspaced with protospacers (short variable repeats that are derived from foreign DNA targets). Together, the noncoding RNA and protospacers constitute the CRISPR RNA (crRNA). Each protospacer is associated with a protospacer adjacent motif (PAM) that differs between the types of CRISPR systems. Depending on the organization and composition of the nuclease genes, the CRISPR-Cas system is classified into Class I and Class II. Each class has three types of CRISPR-Cas system each. The Class I CRISPR system is less used owing to its limited knowledge and associated complexities. The Class II system, on the other hand, is a well-characterized and highly used genome-editing system. Class II is further subclassified into three types (types II, V, and VI) based on the specificities for nucleotide substrates, PAM, and the Cas genes that affect the substrate cleavage (Koonin and Makarova 2019; Moon et al. 2019). A brief overview is discussed below.
CRISPR-Cas9 from Streptomyces pyogenes is the founding system for CRISPR-based genome editing, which is economical, easier, and more efficient. Cas9 is an RNA-guided nuclease that causes double-strand breaks in the genomic region that is complementary to the crRNA provided that the 3′ of the DNA sequence is 5′-NGG-3′ (G-rich PAM). In addition, the CRISPR-Cas9 system needs a trans-acting crRNA (tracrRNA) to be functional (Deltcheva et al. 2011; Jinek et al. 2012). Cas9 proteins of different bacterial origins have different PAM specificities, spacer lengths, and sizes. Improvements in the techniques made it possible to multiplex genome editing with the use of polycistronic tRNA-gRNA (PTG), wherein the tRNA processing system is used to construct a tandem array of tRNA and gRNA (attached to the spacer), which would be transcribed as a full primary transcript that is later processed and cleaved to release individual single-guide RNAs (sgRNAs), each of which targets a unique region in the genome (Zafar et al. 2020).
Cpf1, also known as Cas12a, belongs to the type V CRISPR-Cas system. Unlike Cas9, Cas12a does not need tracrRNA for the complex to be functional and it recognizes a 5′ T-rich PAM sequence (5′-TTTN-3′ or 5′-TTN-3′) (Zetsche et al. 2015). Also, Cpf1 exhibits RNase activity that can cleave pre-crRNAs to mature crRNA, thus enabling the possibility of including multiple crRNAs in a single cassette. Cpf1 allows the use of longer gRNAs of up to 100 nt (Zetsche et al. 2015; Mishra et al. 2018). This system is gaining usage because of its higher specificity and enhanced efficiency. Advances are made in terms of increasing the range of targets by engineering the complex to target other PAMs.
Base editing can be used to modify single bases in the genome, thus opening an avenue to increase the allelic diversity of the genes and also to create specific mutations to alter a gene function. The use of the CRISPR-Cas system achieves this in conjunction with base-modifying enzymes such as cytidine deaminase (to induce C:G to A:T mutations) or adenine deaminase (to induce A:T to G:C mutations). Base-editing techniques eliminate the need for double-strand breaks and thus the activation of DNA repair pathways (Lu and Zhu 2017; Hao et al. 2019).
Gene knock-in thus far required double-strand breaks and activation of homology-directed repair (HDR), which uses a donor template (carrying the gene copy to be knocked-in) to incorporate the new copy in the genome. This technique is extremely limited because of the less frequent and cell cycle stage-dependent nature of HDR. Also, the effective delivery of the donor template has posed a serious challenge. To overcome this, an elegant method was devised, called prime editing. Prime editing depends on a two-component system that includes (1) Cas9 nickase fused with a reverse transcriptase and (2) a prime editing guide RNA (pegRNA) that carries the desired edit(s) to be incorporated into the genome. Once delivered, the complex is guided to the target site by pegRNA and a nick is created in the genome. The nicked DNA serves as a primer that the reverse transcriptase uses to reverse transcribe the pegRNA, thus incorporating the edit into the genome (Anzalone et al. 2019; Lin et al. 2020). All the above-discussed techniques were successfully applied in rice and other plant species to edit various genes. The following few paragraphs will provide a glimpse of the application of genome editing in rice, focusing mainly on disease resistance.
7.2 Application of Genome Editing in Biotic Stress Tolerance in Rice
Conventional breeding has been successfully employed to date to develop disease-resistant rice varieties by introgressing resistance genes from wild rice varieties or landraces into elite cultivars. Although successful, it is a time-consuming procedure, and also the wild germplasm does not contain genes/loci for all the economically important traits that are of concern for breeders. With the advent of genome editing, several research groups started testing the possibility of using genome editing in rice and were by and large successful.
7.2.1 Resistance to Biotic Stress Factors
Genome editing has been successfully employed to generate rice plants resistant to various biotic stress factors, including bacterial blight, bacterial leaf streak, blast, and tungro virus. The application of genome-editing techniques for BB resistance began with modifying the promoter of a BB susceptibility gene, OsSWEET14 (Os11N3), using TALENs (Li et al. 2012). SWEET genes are sugar transporters, and the expression of certain SWEET genes is induced upon Xoo infection by the action of TALEs. So far, the SWEET genes, including OsSWEET11, OsSWEET12, OsSWEET13, OsSWEET14, and OsSWEET15, have been shown to be induced by Xoo and could act as susceptibility factors (Streubel et al. 2013). TAL effectors bind to the effector-binding elements (EBEs) in the target promoter and activate the expression of the downstream gene, which tends to be a susceptibility factor in many cases. Different TAL effectors induce many such susceptibility genes, and their cognate EBEs were also deciphered. Li et al. (2012) have successfully employed TALENs to modify the EBEs of OsSWEET14. This study established the possibility of using genome-editing techniques to generate disease-resistant varieties as well as to understand the targets of different TAL effectors. Jiang et al. (2020) have conducted a proof-of-concept study to confirm the applicability of the CRISPR-Cas9 system in rice by editing the promoters of OsSWEET11 and OsSWEET14 genes (Jiang et al. 2013). In a study using CRISPR-Cas9, Zhou et al. (2015) created a null mutant of OsSWEET13 to show that PthXo2 (an Xoo TAL effector)-dependent disease occurrence needs intact OsSWEET13. Xu et al. (2019) used CRISPR-Cas9 to edit OsSWEET11 and OsSWEET14 to engineer broad-spectrum resistance to BB in rice variety Kitaake. In addition to that, they identified new EBEs in the promoter of OsSWEET13 and successfully used CRISPR-Cas9 to disrupt the EBE, thus generating a rice line that was resistant to all the tested Xoo isolates (n = 133) (Xu et al. 2019). Oliva et al. (2019) generated five mutations in the promoters of OsSWEET11, OsSWEET13, and OsSWEET14 in three rice lines, Kitaake, IR64, and Ciherang-Sub1. All the lines were reported to show robust and broad-spectrum resistance in the paddy trials (Oliva et al. 2019). Zhou et al. (2018) used CRISPR-Cas9 to create a knockout of a susceptibility gene called BsrK-1 (broad-spectrum resistance Kitaake-1), which resulted in resistance to BB as well as blast. BsrK-1 is a tetratricopeptide domain-containing protein that was shown to bind to the mRNAs of multiple OsPAL (phenylalanine ammonia-lyase) genes and promote their turnover. In BsrK-1 knockouts, the accumulation of OsPAL mRNA was observed along with increased resistance to diseases (Zhou et al. 2018). The feasibility of the transgene-free method of genome editing was tested by mutating Os8N3/OsSWEET11 (Kim et al. 2019). Cai et al. (2017) demonstrated that a TAL effector (Tal7) from X. oryzae pv. oryzicola RS105 targets the promoter of rice Cyclin-D4-1 and induces its expression. They have successfully applied TALEN-based genome editing to disrupt the EBE in the promoter of Cyclin-D4-1, which leads to resistance to RS105 infection.
The applicability of CRISPR-Cas9 for generating blast-resistant rice lines has been demonstrated by performing both single-site and multisite-targeted mutagenesis of OsERF922, a negative regulator of blast resistance, to produce knockouts. All the mutants showed blast resistance while not having any adverse effect on other agronomic traits (Wang et al. 2016). Rice tungro disease (RTD) is a disease caused by rice tungrospherical virus (RTSV) and rice tungrobacilliform virus (RTBV) and is transmitted by green leafhoppers. RTD results in yellowing of leaves, decreased tiller numbers, and stunted growth (Azzam and Chancellor 2002; Lee et al. 2010). Macovei et al. (2018) generated RTSV-resistant lines in the background of IR64 using the CRISPR-Cas9 system. In this study, the eIF4G gene was successfully mutated independently, using three different gRNAs, and the mutant plants showed heritable resistance to RTSV (Macovei et al. 2018).
7.2.2 Summary of Nonbiotic Stress-Related Phenotypes
The application of genome editing in rice is rising with time. Other than for biotic stress tolerance, genome editing has been successfully applied to edit several genes having various roles, including nutritional value, yield, and abiotic stress tolerance (Shan et al. 2015; Li et al. 2016; Sun et al. 2016; Shen et al. 2017; Tang et al. 2017; Abe et al. 2018; Endo et al. 2019; Romero and Gatica-Arias 2019).
7.3 Improvements in the Techniques
Currently, transgene-free methods are being tested and employed for genome editing wherein the mutant plants do not contain any of the CRISPR-Cas9 components. This is achieved in several ways, including using Cas9-gRNA ribonucleoproteins (RNPs). This RNP complex is directly delivered into plant cells by transfection or particle bombardment. The RNP complex can perform the editing and will be degraded by the cellular types of machinery. Another approach is to transiently express CRISPR-Cas9 from DNA or RNA in plants from regenerated calli. Both methods suffer from the possibility of component degradation, which might lead to less-efficient editing. To eliminate this disadvantage, He et al. (2018) came up with the suicide gene-based method of a transgene-free CRISPR-Cas9 approach in rice. In this method, a pair of suicidal genes, encoding toxic proteins that kill plant cells, are incorporated into the CRISPR-Cas9 cassette. Therefore, no plant with a CRISPR-Cas9 construct will survive, thus eliminating the plants containing the transgenes. Among other surviving plants, the true mutants can be screened and identified using appropriate techniques. In addition to protein-coding genes, miRNAs are being targeted for editing owing to their involvement in various growth, development, and stress-response pathways. The use of the CRISPR-Cas system to edit miRNAs has been functionally validated in rice (Zhou et al. 2017; Mangrauthia et al. 2017).
8 Bioinformatics Tools for Disease Resistance and Management
Bioinformatics is an interdisciplinary field that uses computational tools to capture and interpret the function of various genes. The advent of genomics has revolutionized every aspect of life science. The availability of a large amount of data has necessitated better ways to analyze, interpret, and organize the results for the scientific community (Bayat 2002; Vassilev et al. 2005). Thousands of databases and repositories are available for various datasets such as for the genome, gene and protein sequences, expression and coexpression of genes, and genomic variations such as SNPs and InDels, to name a few (Garg and Jaiswal 2016). With time, sequencing platforms have seen an astounding revolution and are becoming more efficient and affordable day by day. Since the first report on the whole-genome sequence of rice in 2005, many varieties were further sequenced as a part of the 3000 Rice Genomes Project (Matsumoto et al. 2005; Li et al. 2014). The data obtained from the project resulted in establishing rice variation databases and these data have provided invaluable insights into rice evolution and domestication (Chen et al. 2019). Moreover, the readily available data can guide breeders to wisely choose varieties and markers for breeding various traits from one cultivar to another. Procedures to score the expression of genes have also undergone an overwhelming transformation from methods such as serial analysis of gene expression (SAGE) to microarrays to RNA-sequencing (Perez-de-Castro et al. 2012). As a result, other than genome databases, gene and protein expression databases play an important role in elucidating the various mechanisms that control a given trait, such as days to flowering, growth and development, abiotic stress tolerance, and disease resistance, among others. Multiple other tools and databases are available to study and acquire information on different aspects, including phylogenomics, protein-protein interaction, promoter analysis, gene and QTL information, marker-trait association, and metabolite profiles (Garg and Jaiswal 2016). This section aims to provide an overview of the application of bioinformatics in breeding for disease resistance in rice.
8.1 The Role of Bioinformatics in Mapping Genomic Loci
8.1.1 Mapping QTLs and Genes Associated with Disease Resistance
Mapping the loci responsible for a desired trait has been successfully carried out for years using a conventional method such as simple sequence repeat (SSR)-based genotyping of a mapping population. With the arrival of affordable sequencing techniques, QTLs and genes can now be mapped at a gene-level resolution. Methods such as MutMap and QTLseq have made it possible not only to identify the genomic locus responsible for a trait but also to pinpoint the causal variation within a gene that led to the phenotype (Abe et al. 2012; Takagi et al. 2013b). Thus, SNP markers that are highly associated with a trait can be identified and employed in breeding programmed for efficient introgression of the trait. In a proof-of-concept study, Takagi et al. (2013a) had identified a QTL conferring partial resistance to blast disease of rice. Following this attempt, multiple studies have successfully used this procedure to map QTLs for various traits in rice and other species. A rice blast resistance gene called Pii was mapped by another method called MutMap-Gap (Takagi et al. 2013b).
Sequencing data have been successfully used to compare the genomes of different cultivars and obtain the resistance alleles of cloned rice blast resistance genes (Mahesh et al. 2016). Genes with highly repetitive sequences pose a challenge in accurately characterizing them in context with short-read sequences. A recent study by Read et al. (2020) addressed this challenge by using a long-read sequencing approach called nanopore sequencing in combination with Illumina sequencing to assemble the genome of American rice variety Carolina Gold Select and identify 529 complete or partial NB-LRR domain-containing protein genes that are highly repetitive in nature. The study identified a major disease resistance locus called Xo1 that confers resistance to Xanthomonas oryzae pv. oryzae (the causal agent of bacterial blight of rice) and X. oryzae pv. oryzicola (the causal agent of bacterial leaf streak of rice). Also, a blast resistance gene called Pi63 at the Xo1 locus was identified (Read et al. 2020). Another study compared the genomes of 13 domesticated and wild rice relatives and shed light on the complex phylogeny of the Oryza genus and identified many haplotypes of disease-resistance genes that can be of potential use for breeding (Stein et al. 2018). Using the genome sequence of rice variety Tetep, an extensive set of molecular markers was designed for breeding novel resistant varieties (Wang et al. 2019).
8.1.2 Genome-Wide Association Studies
Genome-wide association studies (GWAS) exploit the natural variation among different cultivars to identify trait-associated genes (Hu et al. 2018). This is one of the preferred methods for the identification of gene targets for breeding. The availability of genome sequences and phenotype data, along with powerful statistical and bioinformatics tools, have made it possible to analyze hundreds or thousands of genomes in one go and identify genes and haplotypes that are associated with given traits. A GWAS on 373 indica rice sequences identified SNPs associated with 14 different agronomic characteristics (Huang et al. 2010). A GWAS with a panel of 584 rice accessions led to the identification of a gene called PiPR1 that confers partial resistance to blast disease of rice (Liu et al. 2020). Other GWAS have identified tens of QTLs and new alleles of known blast-resistance genes (Li et al. 2019a).
8.1.3 Speeding Up Breeding
The main setback with conventional breeding methods is the time taken for developing a new variety. Also, for durable disease resistance and other complex traits, it is essential to efficiently identify minor-effect QTLs and use the associated markers in breeding programs. The existing methods, including QTL mapping from biparental crossing and GWAS, are not up to the mark to efficiently identify such minor-effect QTLs (Bhat et al. 2016). To address both of these concerns, genomic selection (GS) was proposed (Meuwissen et al. 2001). Unlike MAS, GS does not necessarily need QTL information before selection. GS uses reference population data containing phenotype and high-density marker data to predict breeding values for all the markers. Based on the predicted values, the breeding population data will be analyzed to select the individual that possesses the desirable phenotype (Perez-de-Castro et al. 2012; Hu et al. 2018). In this way, it is possible to introgress even minor-effect QTLs efficiently, as there are no biased marker effects, unlike with MAS. Studies on other plant species have shown the higher prediction accuracy of GS in genetic gains and a significant decrease in the time taken for breeding (Hu et al. 2018). Although proposed two decades ago, the implementation of GS in crop breeding has just begun, mainly because of the advent of high-throughput and affordable genotyping methods that produce dense marker information such as genotyping-by-sequencing and automated phenotyping (Bhat et al. 2016). Given the importance of disease-resistance breeding in rice, the application of GS could be of tremendous benefit.
8.1.4 Using Machine Learning and Artificial Intelligence
The field of computational biology is advancing at an unprecedented rate with the arrival of machine learning (ML) and artificial intelligence (AI) . In ML, the machine gains experience by identifying patterns in given datasets and using that experience to interpret the data in question. ML has applications in various aspects of plant sciences, including phenotyping and increasing the accuracy of sequence analysis pipelines, such as differentiating true SNPs from spurious SNPs (Hu et al. 2018). ML was successfully used to phenotype and categorize foliar stresses in soybean with high accuracy (Ghosal et al. 2018). Various parameters such as yield, developmental stage, weed status, crop quality, water and soil management, and disease occurrence were successfully predicted using ML (Liakos et al. 2018). In rice, bakanae disease (caused by Fusarium fujikuroi) was predicted with an accuracy of 87.9% using support vector machine classifiers, a popular ML tool that is often used to overcome classification and regression problems (Chung et al. 2016). Although few in number, these studies have put forth the applicability of advanced computational strategies to improve agriculture.
9 Future Prospects and Conclusions
The rice crop plays an essential role in ensuring global food security and providing nutritional security for the rapidly growing world population. Increasing grain yield is a significant target for plant breeders apart from identifying resistance to/tolerance of biotic and abiotic stresses. Enhancing genetic gain is also a primary concern to meet the food demand of the ever-increasing world population, especially with global climate change. In recent years, the innovations in rice breeding programs and advanced genomics technologies such as next-generation sequencing and high-throughput genotyping have been fully exploited to understand trait interactions and select promising rice genotypes for use in breeding programs. The genetic improvements in yield component traits and increasing yield significantly under biotic and abiotic stresses have not been achieved to a great extent due to the complex nature of these stresses. The knowledge of integrated genomics and high-throughput phenomics technologies has laid the foundation to understand these complex traits and also associated molecular genetics and physiological mechanisms that can enable breeders to find better rice genotypes and to move forward as knowledge-based rice breeding is the most acceptable approach in developing climate-smart stress-tolerant and high-yielding rice genotypes. This approach has advanced at a fast pace with low-cost, efficiency, and high resolution of genetic mapping for QTLs and genes and also haplotype blocks to find allelic variations for the target trait of interest. The current advances in CRISPR/Cas9 genome-editing tools have led to significant targeted changes in specific trait-associated genes and changes in single base levels that promise to accelerate crop improvement. These genomic-assisted breeding tools are breeder-friendly, and smart decisions in breeding programs can enhance the efficiency of the selection of rice genotypes in a short period.
References
Abdallah NA, Prakash CS, McHughen AG (2015) Genome editing for crop improvement: challenges and opportunities. GM Crops Food 6:183–205
Abe A, Kosugi S, Yoshida K et al (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30:174–178. https://doi.org/10.1038/nbt.2095
Abe K, Araki E, Suzuki Y et al (2018) Production of high oleic/low linoleic rice by genome editing. Plant Physiol Biochem 131:58–62. https://doi.org/10.1016/j.plaphy.2018.04.033
Adorada DL, Stodart BJ, Cruz CV et al (2013) Standardizing resistance screening to Pseudomonas fuscovaginae and evaluation of rice germplasm at seedling and adult plant growth stages. Euphytica 192:1–16. https://doi.org/10.1007/s10681-012-0804-z
Ahn S, Kim Y, Han S et al (1996) Molecular mapping of a gene for resistance to a Korean isolate of rice blast. Rice Genet Newsl 13:74–76
Ahn SN, Kim YK, Hong HC et al (1997) Mapping of genes conferring resistance to Korean isolates of rice blast fungus using DNA markers. Korean J Breed 29:416–423
Ali J, Xu JL, Gao YM et al (2017) Harnessing the hidden genetic diversity for improving multiple abiotic stress tolerance in rice (Oryza sativa L.). PLoS One 12:e0172515
Andargie M, Li L, Feng A et al (2018) Mapping of the quantitative trait locus (QTL) conferring resistance to rice false smut disease. Curr Plant Biol 15:38–43. https://doi.org/10.1016/j.cpb.2018.11.003
Anderson S (2002) The relationship between nutrients and other elements to plant diseases. Spectrum Analytic Inc, Washington CH, OH, pp 26–32
Anderson PK, Cunningham AA, Patel NG et al (2004) Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends Ecol Evol 19:535–544
Anjaneyulu A, Singh SK, Shenoi MM (1982) Evaluation of rice varieties for tungro resistance by field screening techniques. Trop Pest Manag 28:147–155. https://doi.org/10.1080/09670878209370692
Anzalone AV, Randolph PB, Davis JR et al (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576:149–157. https://doi.org/10.1038/s41586-019-1711-4
Ashikawa I, Hayashi N, Yamane H et al (2008) Two adjacent nucleotide-binding site-leucine-rich repeat class genes are required to confer Pikm-specific rice blast resistance. Genetics 180:2267–2276. https://doi.org/10.1534/genetics.108.095034
Ashkani S, Rafii MY, Shabanimofrad M et al (2015) Molecular breeding strategy and challenges towards improvement of blast disease resistance in rice crop. Front Plant Sci 6:886
Azzam O, Chancellor TCB (2002) The biology, epidemiology, and management of rice tungro disease in Asia. Plant Dis 86:88–100
Bainsla NK, Meena HP (2016) Breeding for resistance to biotic stresses in plants. In: Yadav P, Kumar S, Jain V (eds) Recent advances in plant stress physiology. Daya Publishing House, New Delhi
Balachiranjeevi C, Bhaskar NS, Abhilash V et al (2015) Marker-assisted introgression of bacterial blight and blast resistance into DRR17B, an elite, fine-grain type maintainer line of rice. Mol Breed 35:151. https://doi.org/10.1007/s11032-015-0348-8
Balachiranjeevi CH, Bhaskar Naik S, Abhilash Kumar V et al (2018) Marker-assisted pyramiding of two major, broad-spectrum bacterial blight-resistance genes, Xa21 and Xa33 into an elite maintainer line of rice, DRR17B. PLoS One 13:e0201271. https://doi.org/10.1371/journal.pone.0201271
Balachiranjeevi CH, Prahalada GD, Mahender A et al (2019) Identification of a novel locus, BPH38 (t), conferring resistance to brown planthopper (Nilaparvata lugens Stal.) using early backcross population in rice (Oryza sativa L.). Euphytica 215:185
Bargaz A, Lyamlouli K, Chtouki M et al (2018) Soil microbial resources for improving fertilizers efficiency in an integrated plant nutrient management system. Front Microbiol 9:1606
Barman SR, Gowda M, Venu RC, Chattoo BB (2004) Identification of a major blast resistance gene in the rice cultivar “Tetep”. Plant Breed 123:300–302. https://doi.org/10.1111/j.1439-0523.2004.00982.x
Bayat A (2002) Bioinformatics. Br Med J 324:1018–1022
Begum N, Rahman MM, Bashar MA et al (2011) Effect of potassium fertilizer on development of bacterial blight of rice. Bangladesh J Sci Ind Res 46:69–76
Berruyer R, Adreit H, Milazzo J et al (2003) Identification and fine-mapping of Pi33, the rice resistance gene corresponding to the Magnaporthe grisea avirulence gene ACE1. Theor Appl Genet 107:1139–1147. https://doi.org/10.1007/s00122-003-1349-2
Bhasin H, Bhatia D, Raghuvanshi S et al (2012) New PCR-based sequence-tagged site marker for bacterial blight-resistance gene Xa38 of rice. Mol Breed 30:607–611. https://doi.org/10.1007/s11032-011-9646-y
Bhat ZA, Ahangar MA, Sanghera GS, Mubarak T (2013) Effect of cultivar, fungicide spray and nitrogen fertilization on management of rice blast under temperate ecosystem. Int J Sci Environ Technol 2:410–415
Bhat JA, Ali S, Salgotra RK et al (2016) Genomic selection in the era of next-generation sequencing for complex traits in plant breeding. Front Genet 7:221
Bigirimana VP, Hua GKH, Nyamangyoku OI, Hòfte M (2015) Rice sheath rot: an emerging ubiquitous destructive disease complex. Front Plant Sci 6:1066
Boch J (2011) TALEs of genome targeting. Nat Biotechnol 29:135–136
Boch J, Scholze H, Schornack S et al (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512. https://doi.org/10.1126/science.1178811
Bolton MD, Thomma BPHJ (2008) The complexity of nitrogen metabolism and nitrogen-regulated gene expression in plant pathogenic fungi. Physiol Mol Plant Pathol 72:104–110
Bonman JM (1992) Durable resistance to rice blast disease-environmental influences. Euphytica 63:115–123. https://doi.org/10.1007/BF00023917
Bossa-Castro AM, Tekete C, Raghavan C et al (2018) Allelic variation for broad-spectrum resistance and susceptibility to bacterial pathogens identified in a rice MAGIC population. Plant Biotechnol J 16:1559–1568. https://doi.org/10.1111/pbi.12895
Bunawan H, Dusik L, Bunawan SN, Amin NM (2014) Rice tungro disease: from identification to disease control. World Appl Sci J 31:1221–1226. https://doi.org/10.5829/idosi.wasj.2014.31.06.610
Busungu C, Taura S, Sakagami JI, Ichitani K (2016) Identification and linkage analysis of a new rice bacterial blight-resistance gene from XM14, a mutant line from IR24. Breed Sci 66:636–645. https://doi.org/10.1270/jsbbs.16062
Cai L, Cao Y, Xu Z et al (2017) A transcription activator-like effector Tal7 of Xanthomonas oryzae pv. oryzicola activates rice gene Os09g29100 to suppress rice immunity. Sci Rep 7:1–13. https://doi.org/10.1038/s41598-017-04800-8
Causse MA, Fulton TM, Cho YG et al (1994) Saturated molecular map of the rice genome based on an interspecific backcross population. Genetics 138:1251–1274
Cesari S, Thilliez G, Ribot C et al (2013) The rice resistance protein pair RGA4/RGA5 recognizes the Magnaporthe oryzae effectors AVR-Pia and AVR1-CO39 by direct binding. Plant Cell 25:1463–1481. https://doi.org/10.1105/tpc.112.107201
Channamallikarjuna V, Sonah H, Prasad M et al (2010) Identification of major quantitative trait loci qSBR11-1 for sheath blight resistance in rice. Mol Breed 25:155–166. https://doi.org/10.1007/s11032-009-9316-5
Chaudhari AK, Rakholiya KB, Baria TT (2019) Screening of rice cultivars against false smut [Ustilaginoidea virens (Cooke) Takahashi] of rice. Int J Curr Microbiol App Sci 8:2786–2793. https://doi.org/10.20546/ijcmas.2019.806.336
Cheema KK, Grewal NK, Vikal Y et al (2008) A novel bacterial blight-resistance gene from Oryza nivara mapped to 38 kb region on chromosome 4L and transferred to Oryza sativa L. Genet Res (Camb) 90:397–407. https://doi.org/10.1017/S0016672308009786
Chen DH, Dela Viña M, Inukai T et al (1999) Molecular mapping of the blast resistance gene, Pi44(t), in a line derived from a durably resistant rice cultivar. Theor Appl Genet 98:1046–1053. https://doi.org/10.1007/s001220051166
Chen XW, Li SG, Xu JC et al (2004) Identification of two blast resistance genes in a rice variety, Digu. J Phytopathol 152:77–85. https://doi.org/10.1046/j.1439-0434.2003.00803.x
Chen C, Zheng W, Huang X et al (2006a) Major QTL conferring resistance to rice bacterial leaf streak. Agric Sci China 5:216–220. https://doi.org/10.1016/S1671-2927(06)60041-2
Chen X, Shang J, Chen D et al (2006b) A B-lectin receptor kinase gene conferring rice blast resistance. Plant J 46:794–804. https://doi.org/10.1111/j.1365-313X.2006.02739.x
Chen S, Huang Z, Zeng L et al (2008) High-resolution mapping and gene prediction of Xanthomonas oryzae pv. oryzae resistance gene Xa7. Mol Breed 22:433–441. https://doi.org/10.1007/s11032-008-9187-1
Chen J, Shi Y, Liu W et al (2011a) A Pid3 allele from rice cultivar Gumei2 confers resistance to Magnaporthe oryzae. J Genet Genom 38:209–216. https://doi.org/10.1016/j.jgg.2011.03.010
Chen S, Liu X, Zeng L et al (2011b) Genetic analysis and molecular mapping of a novel recessive gene xa34(t) for resistance against Xanthomonas oryzae pv. oryzae. Theor Appl Genet 122:1331–1338. https://doi.org/10.1007/s00122-011-1534-7
Chen J, Peng P, Tian J et al (2015) Pike, a rice blast resistance allele consisting of two adjacent NBS–LRR genes, was identified as a novel allele at the Pik locus. Mol Breed 35:1–15. https://doi.org/10.1007/s11032-015-0305-6
Chen E, Huang X, Tian Z et al (2019) The genomics of Oryza species provides insights into rice domestication and heterosis. Annu Rev Plant Biol 70:639–665. https://doi.org/10.1146/annurev-arplant-050718-100320
Chen S, Wang C, Yang J et al (2020) Identification of the novel bacterial blight-resistance gene Xa46(t) by mapping and expression analysis of the rice mutant H120. Sci Rep 10:12642. https://doi.org/10.1038/s41598-020-69639-y
Chisholm ST, Coaker G, Day B et al (2006) Host-microbe interactions: shaping the evolution of the plant immune response. Cell 124:803–814
Christian M, Cermak T, Doyle EL et al (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186:756–761. https://doi.org/10.1534/genetics.110.120717
Chu Z, Fu B, Yang H et al (2006) Targeting xa13, a recessive gene for bacterial blight-resistance in rice. Theor Appl Genet 112:455–461. https://doi.org/10.1007/s00122-005-0145-6
Chung CL, Huang KJ, Chen SY et al (2016) Detecting Bakanae disease in rice seedlings by machine vision. Comput Electron Agric 121:404–411. https://doi.org/10.1016/j.compag.2016.01.008
Collard BC, Ismail AM, Hardy B (eds) (2013) EIRLSBN: twenty years of achievements in rice breeding. International Rice Research Institute, Los Baños. 145 p
Cong L, Ran FA, Cox D et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Deltcheva E, Chylinski K, Sharma CM et al (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. https://doi.org/10.1038/nature09886
Deng Y, Zhai K, Xie Z et al (2017) Epigenetic regulation of antagonistic receptors confers rice blast resistance with yield balance. Science 355:962–965. https://doi.org/10.1126/science.aai8898
Devi SJSR, Singh K, Umakanth B et al (2020) Identification and characterization of a large effect QTL from Oryza glumaepatula revealed Pi68(t) as putative candidate gene for rice blast resistance. Rice 13:17. https://doi.org/10.1186/s12284-020-00378-4
Dordas C (2008) Role of nutrients in controlling plant diseases in sustainable agriculture. A review. Agron Sustain Dev 28:33–46
Eizenga GC, Lee FN, Rutger JN (2002) Screening Oryza species plants for rice sheath blight resistance. Plant Dis 86:808–812
Eizenga GC, Prasad B, Jackson AK, Jia MH (2013) Identification of rice sheath blight and blast quantitative trait loci in two different O. sativa/O. nivara advanced backcross populations. Mol Breed 31:889–907. https://doi.org/10.1007/s11032-013-9843-y
Endo M, Mikami M, Endo A et al (2019) Genome editing in plants by engineered CRISPR–Cas9 recognizing NG PAM. Nat Plants 5:14–17. https://doi.org/10.1038/s41477-018-0321-8
Fageria NK, Baligar VC, Li YC (2008) The role of nutrient efficient plants in improving crop yields in the twenty first century. J Plant Nutr 31:1121–1157
FAOSTAT (2020) Statistics data. Food and Agriculture Organization of the United Nations, Rome. http://www.fao.org/faostat/en/#dat
Feng Z, Zhang B, Ding W et al (2013) Efficient genome editing in plants using a CRISPR/Cas system. Cell Res 23:1229–1232
Feng B, Chen K, Cui Y et al (2018) Genetic dissection and simultaneous improvement of drought and low nitrogen tolerances by designed QTL pyramiding in rice. Front Plant Sci 9:306. https://doi.org/10.3389/fpls.2018.00306
Fjellstrom R, Conaway-Bormans CA, McClung AM et al (2004) Development of DNA markers suitable for marker-assisted selection of three Pi genes conferring resistance to multiple Pyricularia grisea pathotypes. Crop Sci 44:1790–1798. https://doi.org/10.2135/cropsci2004.1790
Fu D, Chen L, Yu G et al (2011) QTL mapping of sheath blight resistance in a deep-water rice cultivar. Euphytica 180:209–218. https://doi.org/10.1007/s10681-011-0366-5
Fujii K, Hayano-Saito Y, Shumiya A et al (1995) Genetical mapping based on the RFLP analysis for the panicle blast resistance derived from a rice parental line St. No. 1. Breed Sci 45:209
Fukuoka S, Saka N, Koga H et al (2009) Loss of function of a proline-containing protein confers durable disease resistance in rice. Science 325:998–1001. https://doi.org/10.1126/science.1175550
Gaj T, Gersbach CA, Barbas CF (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31:397–405
Gaj T, Sirk SJ, Shui S, Liu J (2016) Genome-editing technologies: principles and applications. Cold Spring Harb Perspect Biol 8:a023754
Garg P, Jaiswal P (2016) Databases and bioinformatics tools for rice research. Curr Plant Biol 7–8:39–52. https://doi.org/10.1016/j.cpb.2016.12.006
Ghosal S, Blystone D, Singh AK et al (2018) An explainable deep machine vision framework for plant stress phenotyping. Proc Natl Acad Sci U S A 115:4613–4618. https://doi.org/10.1073/pnas.1716999115
Goad DM, Jia Y, Gibbons A et al (2020) Identification of novel QTL conferring sheath blight resistance in two weedy rice mapping populations. Rice 13:21. https://doi.org/10.1186/s12284-020-00381-9
Gopalakrishnan S, Sharma RK, Anand Rajkumar K et al (2008) Integrating marker-assisted background analysis with foreground selection for identification of superior bacterial blight-resistant recombinants in Basmati rice. Plant Breed 127:131–139. https://doi.org/10.1111/j.1439-0523.2007.01458.x
Goto I (1970) Genetic studies on the resistance of rice plant to the blast fungus. Jpn J Phytopathol 36:304–312. https://doi.org/10.3186/jjphytopath.36.304
Goto I (1988) Genetic studies on resistance of rice plant to blast fungus (VII): blast resistance genes of Kuroka. Ann Phytopathol Soc Jpn 54:460–465
Gowda M, Roy-Barman S, Chattoo BB (2006) Molecular mapping of a novel blast resistance gene Pi38 in rice using SSLP and AFLP markers. Plant Breed 125:596–599. https://doi.org/10.1111/j.1439-0523.2006.01248.x
Graichen FAS, Martinelli JA, Federizzi LC et al (2010) Inheritance of resistance to oat crown rust in recombinant inbred lines. Sci Agric 67:435–440. https://doi.org/10.1590/s0103-90162010000400010
Gu K, Yang B, Tian D et al (2005) R-gene expression induced by a type-III effector triggers disease resistance in rice. Nature 435:1122–1125. https://doi.org/10.1038/nature03630
Guo SB, Zhang DP, Lin XH (2010) Identification and mapping of a novel bacterial blight-resistance gene Xa35(t) originated from Oryza minuta. Scientia Agricultura Sinica 43:2611–2618
Gupta RM, Musunuru K (2014) Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J Clin Invest 124:4154–4161
Hajira SK, Sundaram RM, Laha GS et al (2016) A single-tube, functional marker-based multiplex PCR assay for simultaneous detection of major bacterial blight-resistance genes Xa21, xa13 and xa5 in rice. Rice Sci 23:144. https://doi.org/10.1016/j.rsci.2015.11.004
Han Y, Li D, Yang J et al (2020) Mapping quantitative trait loci for disease resistance to false smut of rice. Phytopathol Res 2:20. https://doi.org/10.1186/s42483-020-00059-6
Hao L, Ruiying Q, Xiaoshuang L et al (2019) CRISPR/Cas9-mediated adenine base editing in rice genome. Rice Sci 26:125–128
Hari Y, Srinivasarao K, Viraktamath BC et al (2011) Marker-assisted improvement of a stable restorer line, KMR-3R and its derived hybrid KRH2 for bacterial blight resistance and grain quality. Plant Breed 130:608–616. https://doi.org/10.1111/j.1439-0523.2011.01881.x
Hari Y, Srinivasarao K, Viraktamath BC et al (2013) Marker-assisted introgression of bacterial blight and blast resistance into IR 58025B, an elite maintainer line of rice. Plant Breed 132:586–594. https://doi.org/10.1111/pbr.12056
Hayasaka T, Fujii H, Ishiguro K (2008) The role of silicon in preventing appressorial penetration by the rice blast fungus. Phytopathology 98:1038–1044
Hayashi K, Yoshida H (2009) Refunctionalization of the ancient rice blast disease resistance gene Pit by the recruitment of a retrotransposon as a promoter. Plant J 57:413–425. https://doi.org/10.1111/j.1365-313X.2008.03694.x
Hayashi K, Yoshida H, Ashikawa I (2006) Development of PCR-based allele-specific and InDel marker sets for nine rice blast-resistance genes. Theor Appl Genet 113:251–260. https://doi.org/10.1007/s00122-006-0290-6
Hayashi N, Inoue H, Kato T et al (2010) Durable panicle blast-resistance gene Pb1 encodes an atypical CC-NBS-LRR protein and was generated by acquiring a promoter through local genome duplication. Plant J 64:498–510. https://doi.org/10.1111/j.1365-313X.2010.04348.x
He W, Huang D, Li R et al (2012) Identification of a resistance gene bls1 to bacterial leaf streak in wild rice Oryza rufipogon Griff. J Integr Agric 11:962–969. https://doi.org/10.1016/S2095-3119(12)60087-2
He Y, Zhu M, Wang L et al (2018) Programmed self-elimination of the CRISPR/Cas9 construct greatly accelerates the isolation of edited and transgene-free rice plants. Mol Plant 11:1210–1213
Heinrichs EA, Muniappan R (2017) IPM for tropical crops: rice. CAB Rev 12:1
Hervieux V, Yaganza ES, Arul J, Tweddell RJ (2002) Effect of organic and inorganic salts on the development of Helminthosporium solani, the causal agent of potato silver scurf. Plant Dis 86:1014–1018
Hirochika H, Guiderdoni E, An G et al (2004) Rice mutant resources for gene discovery. Plant Mol Biol 54:325–334. https://doi.org/10.1023/B:PLAN.0000036368.74758.66
Holland JB (2001) Epistasis and pant breeding. Plant Breed Rev 21:27–92
Hu H, Scheben A, Edwards D (2018) Advances in integrating genomics and bioinformatics in the plant breeding pipeline. Agriculture 8:75
Hua L, Wu J, Chen C et al (2012) The isolation of Pi1, an allele at the Pik locus which confers broad spectrum resistance to rice blast. Theor Appl Genet 125:1047–1055. https://doi.org/10.1007/s00122-012-1894-7
Huang N, Angeles ER, Domingo J et al (1997) Pyramiding of bacterial blight-resistance genes in rice: marker-assisted selection using RFLP and PCR. Theor Appl Genet 95:313–320. https://doi.org/10.1007/s001220050565
Huang X, Wei X, Sang T et al (2010) Genome-wide asociation studies of 14 agronomic traits in rice landraces. Nat Genet 42:961–967. https://doi.org/10.1038/ng.695
Huang S, Liu L, Wang L, Hou Y (2019) Research on advance of rice false smut Ustilaginoidea virens (Cooke) Takah worldwide: Part I. Research status of rice false smut. J Agric Sci 11:240. https://doi.org/10.5539/jas.v11n15p240
Hutin M, Sabot F, Ghesquière A et al (2015) A knowledge-based molecular screen uncovers a broad-spectrum OsSWEET14 resistance allele to bacterial blight from wild rice. Plant J 84:694–703. https://doi.org/10.1111/tpj.13042
Ingvardsen CR, Schejbel B, Lübberstedt T (2008) Functional markers in resistance breeding. Springer, Berlin, pp 61–87
Inukai T, Nelson RJ, Zeigler RS et al (1996) Genetic analysis of blast resistance in tropical rice cultivars using near-isogenic lines. In: Khush GS (ed) Rice genetics III. International Rice Research Institute, Los Baños, pp 447–455
Inukai T, Nagashima S, Kato M (2019) Pid3-I1 is a race-specific partial-resistance allele at the Pid3 blast resistance locus in rice. Theor Appl Genet 132:395–404. https://doi.org/10.1007/s00122-018-3227-y
IRRI (1996) Standard evaluation system for rice, 4th edn. International Rice Research Institute, Los Baños
IRRI (2013) Standard evaluation system for rice, 5th edn. International Rice Research Institute, Los Baños
Iwata N (1996) Registration of new gene symbols. Rice Genet 13:12–18
Iyer-Pascuzzi AS, McCouch SR (2007) Functional markers for xa5-mediated resistance in rice (Oryza sativa, L.). Mol Breed 19:291–296. https://doi.org/10.1007/s11032-006-9055-9
Jabeen R, Rahman SU, Rais A (2011) Evaluating BLB resistance/aggressiveness in rice through best inoculum concentration, inoculation and application methods. Pak J Bot 43:2635–2638
Jamaloddin M, Durga Rani CV, Swathi G et al (2020) Marker assisted gene pyramiding (MAGP) for bacterial blight and blast resistance into mega rice variety “Tellahamsa”. PLoS One 15:e0234088. https://doi.org/10.1371/journal.pone.0234088
Jeung JU, Kim BR, Cho YC et al (2007) A novel gene, Pi40(t), linked to the DNA markers derived from NBS-LRR motifs confers broad spectrum of blast resistance in rice. Theor Appl Genet 115:1163–1177. https://doi.org/10.1007/s00122-007-0642-x
Jia Y, Valent B, Lee FN (2003) Determination of host responses to Magnaporthe grisea on detached rice leaves using a spot inoculation method. Plant Dis 87:129–133. https://doi.org/10.1094/PDIS.2003.87.2.129
Jia Y, Correa-Victoria F, McClung A et al (2007) Rapid determination of rice cultivar responses to the sheath blight pathogen Rhizoctonia solani using a micro-chamber screening method. Plant Dis 91:485–489. https://doi.org/10.1094/PDIS-91-5-0485
Jia L, Yan W, Zhu C et al (2012) Allelic analysis of sheath blight resistance with association mapping in rice. PLoS One 7:e32703. https://doi.org/10.1371/journal.pone.0032703
Jiang W, Zhou H, Bi H et al (2013) Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res 41:e188–e188
Jiang J, Yang D, Ali J, Mou T (2015) Molecular marker-assisted pyramiding of broad-spectrum disease resistance genes, Pi2 and Xa23, into GZ63-4S, an elite thermo-sensitive genic male-sterile line in rice. Mol Breed 35:1–12. https://doi.org/10.1007/s11032-015-0282-9
Jiang N, Yan J, Liang Y et al (2020) Resistance genes and their interactions with bacterial blight/leaf streak pathogens (Xanthomonas oryzae) in rice (Oryza sativa L.)—an updated review. Rice 13:3. https://doi.org/10.1186/s12284-019-0358-y
Jinek M, Chylinski K, Fonfara I et al (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. https://doi.org/10.1126/science.1225829
Jones JDG, Dangl JL (2006) The plant immune system. Nature 444:323–329
Joseph M, Gopalakrishnan S, Sharma RK et al (2004) Combining bacterial blight resistance and Basmati quality characteristics by phenotypic and molecular marker-assisted selection in rice. Mol Breed 13:377–387. https://doi.org/10.1023/B:MOLB.0000034093.63593.4c
Ju Y, Tian H, Zhang R et al (2017) Overexpression of OsHSP18.0-CI enhances resistance to bacterial leaf streak in rice. Rice 10:12. https://doi.org/10.1186/s12284-017-0153-6
Kauffman HE, Reddy APK, Hsieh SPY, Merca SD (1973) An improved technique for evaluating resistance of rice varieties to Xanthomonas oryzae. Plant Dis Rep 57:537–541
Kaur Y (2014) Development of screening technique for false smut of rice. Dissertation. Punjab Agricultural University, Ludhiana
Kaur T, Singh I (2017) Standardization of screening technique for false smut of rice. Progress Agric 17:10. https://doi.org/10.5958/0976-4615.2017.00007.2
Kaur S, Padmanabhan SY, Kaur P (1979) Effect of nitrogen on the intracellular spread of Pyricularia oryzae. Indian Phytopathol 32:285–286
Ke Y, Deng H, Wang S (2017) Advances in understanding broad-spectrum resistance to pathogens in rice. Plant J 90:738–748. https://doi.org/10.1111/tpj.13438
Khush GS (2005) What it will take to feed 5.0 billion rice consumers in 2030. Plant Mol Biol 59:1–6
Khush GS, Angeles ER (1999) A new gene for resistance to race 6 of bacterial blight in rice, Oryza sativa. Rice Genet Newsl 16:92–93
Kim SM (2018) Identification of novel recessive gene xa44(t) conferring resistance to bacterial blight races in rice by QTL linkage analysis using an SNP chip. Theor Appl Genet 131:2733–2743. https://doi.org/10.1007/s00122-018-3187-2
Kim SM, Reinke RF (2019) A novel resistance gene for bacterial blight in rice, Xa43(t) identified by GWAS, confirmed by QTL mapping using a bi-parental population. PLoS One 14:e0211775. https://doi.org/10.1371/journal.pone.0211775
Kim YG, Cha J, Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A 93:1156–1160. https://doi.org/10.1073/pnas.93.3.1156
Kim SM, Suh JP, Qin Y et al (2015) Identification and fine-mapping of a new resistance gene, Xa40, conferring resistance to bacterial blight races in rice (Oryza sativa L.). Theor Appl Genet 128:1933–1943. https://doi.org/10.1007/s00122-015-2557-2
Kim YA, Moon H, Park CJ (2019) CRISPR/Cas9-targeted mutagenesis of Os8N3 in rice to confer resistance to Xanthomonas oryzae pv. oryzae. Rice 12:67. https://doi.org/10.1186/s12284-019-0325-7
Kinoshita T, Kiyosawa S (1997) Some considerations on linkage relationships between Pii and Piz in the blast resistance of rice. Rice Genet Newsl 14:57–59
Klug A, Schwabe JWR (1995) Zinc fingers. FASEB J 9:597–604. https://doi.org/10.1096/fasebj.9.8.7768350
Koonin EV, Makarova KS (2019) Origins and evolution of CRISPR-Cas systems. Philos Trans R Soc B Biol Sci 374:20180087
Korinsak S, Sriprakhon S, Sirithanya P et al (2009) Identification of microsatellite markers (SSR) linked to a new bacterial blight-resistance gene xa33(t) in rice cultivar “Ba7”. Maejo Int J Sci Technol 3:235–247
Kou Y, Wang S (2013) Bacterial blight resistance in rice. In: Translational genomics for crop breeding. John Wiley & Sons Ltd., Chichester, pp 11–30
Kurata N, Yamazaki Y (2006) Oryzabase:an integrated biological and genome information database for rice. Plant Physiol 140:12–17. https://doi.org/10.1104/pp.105.063008
Lee KS, Rasabandith S, Angeles ER, Khush GS (2003) Inheritance of resistance to bacterial blight in 21 cultivars of rice. Phytopathology 93:147–152. https://doi.org/10.1094/PHYTO.2003.93.2.147
Lee SK, Song MY, Seo YS et al (2009) Rice Pi5-mediated resistance to Magnaporthe oryzae requires the presence of two coiled-coil-nucleotide-binding-leucine-rich repeat genes. Genetics 181:1627–1638. https://doi.org/10.1534/genetics.108.099226
Lee JH, Muhsin M, Atienza GA et al (2010) Single nucleotide polymorphisms in a gene for translation initiation factor (elF4G) of rice (Oryza sativa) associated with resistance to rice tungro spherical virus. Mol Plant-Microbe Interact 23:29–38. https://doi.org/10.1094/MPMI-23-1-0029
Lei C, Huang D, Li W et al (2005) Molecular mapping of a blast resistance gene in an indica rice cultivar Yanxian No. 1. Rice Genet Newsl 22:76–77
Li T, Liu B, Spalding MH et al (2012) High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat Biotechnol 30:390
Li Z, Fu BY, Gao YM et al (2014) The 3,000 rice genomes project. Giga Sci 3:7. https://doi.org/10.1186/2047-217X-3-7
Li M, Li X, Zhou Z et al (2016) Reassessment of the four yield-related genes Gn1a, DEP1, GS3, and IPA1 in rice using a CRISPR/Cas9 system. Front Plant Sci 7:377
Li C, Wang D, Peng S et al (2019a) Genome-wide association mapping of resistance against rice blast strains in South China and identification of a new Pik allele. Rice 12:47. https://doi.org/10.1186/s12284-019-0309-7
Li W, Chern M, Yin J et al (2019b) Recent advances in broad-spectrum resistance to the rice blast disease. Curr Opin Plant Biol 50:114–120
Liakos KG, Busato P, Moshou D et al (2018) Machine learning in agriculture: a review. Sensors 18:2674
Lin F, Chen S, Que Z et al (2007) The blast resistance gene Pi37 encodes a nucleotide binding site-leucine-rich repeat protein and is a member of a resistance gene cluster on rice chromosome 1. Genetics 177:1871–1880. https://doi.org/10.1534/genetics.107.080648
Lin Q, Zong Y, Xue C et al (2020) Prime genome editing in rice and wheat. Nat Biotechnol 38:582–585. https://doi.org/10.1038/s41587-020-0455-x
Liu XQ, Wang L, Chen S et al (2005) Genetic and physical mapping of Pi36(t), a novel rice blast resistance gene located on rice chromosome 8. Mol Gen Genomics 274:394–401. https://doi.org/10.1007/s00438-005-0032-5
Liu X, Yang Q, Lin F et al (2007) Identification and fine-mapping of Pi39(t), a major gene conferring the broad-spectrum resistance to Magnaporthe oryzae. Mol Gen Genomics 278:403–410. https://doi.org/10.1007/s00438-007-0258-5
Liu W, Jin S, Zhu X et al (2008) Improving blast resistance of a thermo-sensitive genic male sterile rice line GD-8S by molecular marker-assisted selection. Rice Sci 15:179–185. https://doi.org/10.1016/S1672-6308(08)60040-2
Liu Q, Yuan M, Zhou Y et al (2011) A paralog of the MtN3/saliva family recessively confers race-specific resistance to Xanthomonas oryzae in rice. Plant Cell Environ 34:1958–1969. https://doi.org/10.1111/j.1365-3040.2011.02391.x
Liu G, Jia Y, McClung A et al (2013) Confirming QTLs and finding additional loci responsible for resistance to rice sheath blight disease. Plant Dis 97:113–117. https://doi.org/10.1094/PDIS-05-12-0466-RE
Liu W, Liu J, Triplett L et al (2014) Novel insights into rice innate immunity against bacterial and fungal pathogens. Annu Rev Phytopathol 52:213–241. https://doi.org/10.1146/annurev-phyto-102313-045926
Liu MH, Kang H, Xu Y et al (2020) Genome-wide association study identifies an NLR gene that confers partial resistance to Magnaporthe oryzae in rice. Plant Biotechnol J 18:1376–1383. https://doi.org/10.1111/pbi.13300
Lu Y, Zhu JK (2017) Precise editing of a target base in the rice genome using a modified CRISPR/Cas9 system. Mol Plant 10:523–525
Lu Z, Yu X, Heong K, Cui HU (2007) Effect of nitrogen fertilizer on herbivores and its stimulation to major insect pests in rice. Rice Sci 14:56–66
Ma J, Lei C, Xu X et al (2015a) Pi64, encoding a novel CC-NBS-LRR protein, confers resistance to leaf and neck blast in rice. Mol Plant-Microbe Interact 28:558–568. https://doi.org/10.1094/MPMI-11-14-0367-R
Ma X, Zhang Q, Zhu Q et al (2015b) A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8:1274–1284. https://doi.org/10.1016/j.molp.2015.04.007
Mackill DJ, Coffman WR, Garrity DP (1996) Rainfed lowland rice improvement. International Rice Research Institute, Los Baños. 242 p
Macovei A, Sevilla NR, Cantos C et al (2018) Novel alleles of rice eIF4G generated by CRISPR/Cas9-targeted mutagenesis confer resistance to Rice tungro spherical virus. Plant Biotechnol J 16:1918–1927. https://doi.org/10.1111/pbi.12927
Madamba MRS, Sugiyama N, Bordeos A et al (2009) A recessive mutation in rice conferring non-race-specific resistance to bacterial blight and blast. Rice 2:104–114. https://doi.org/10.1007/s12284-009-9027-x
Mahadevaiah C, Hittalmani S, Uday G, Kumar MKP (2015) Standardization of disease screening protocol for sheath rot disease in rice. Int J Agric Sci Res 5:129–137
Mahadevaiah C, Kumar MP, Hittalmani S (2017) Dissecting parameters associated with sheath rot (Sarocladium oryzae [(Sawada) W. Gams &D. Hawksw.]) disease in rice (Oryza sativa L.). Curr Sci 112:151–155. https://doi.org/10.18520/cs/v112/i01/151-155
Mahesh HB, Shirke MD, Singh S et al (2016) Indica rice genome assembly, annotation and mining of blast disease-resistance genes. BMC Genomics 17:242. https://doi.org/10.1186/s12864-016-2523-7
Mangrauthia SK, Maliha A, Prathi NB, Marathi B (2017) MicroRNAs: potential target for genome editing in plants for traits improvement. Indian J Plant Physiol 22:530–548
Manosalva PM, Davidson RM, Liu B et al (2009) A germin-like protein gene family functions as a complex quantitative trait locus conferring broad-spectrum disease resistance in rice. Plant Physiol 149:286–296. https://doi.org/10.1104/pp.108.128348
Matsumoto T, Wu J, Kanamori H et al (2005) The map-based sequence of the rice genome. Nature 436:793–800. https://doi.org/10.1038/nature03895
Matsuyama N (1973) Effect of nitrogenous fertilizer on biochemical processes that could affect lesion size of rice blast. Phytopathology 63:1202–1203
McCouch S, Nelson R, Tohme J, Zeigler R (1994) Mapping of blast resistance genes in rice. In: Zeigler RS, Leong SA, Teng PS (eds) Rice blast disease. CAB International, Wallingford, UK, pp 167–186
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Miah G, Rafii M, Ismail M et al (2013) A review of microsatellite markers and their applications in rice breeding programs to improve blast disease resistance. Int J Mol Sci 14:22499–22528
Miao LL, Wang CL, Zheng CK et al (2010) Molecular mapping of a new gene for resistance to rice bacterial blight. Sci Agric Sin 43:2611
Milovanovic V, Smutka L (2017) Asian countries in the global rice market. Acta Univ Agric Silvic Mendel Brun 65:679–688
Mishra R, Joshi RK, Zhao K (2018) Genome editing in rice: recent advances, challenges, and future implications. Front Plant Sci 9:1361
Molla KA, Karmakar S, Molla J et al (2020) Understanding sheath blight resistance in rice: the road behind and the road ahead. Plant Biotechnol J 18:895–915. https://doi.org/10.1111/pbi.13312
Moon SB, Kim DY, Ko JH, Kim YS (2019) Recent advances in the CRISPR genome editing tool set. Exp Mol Med 51:1–11
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326:1501. https://doi.org/10.1126/science.1178817
Mur LAJ, Simpson C, Kumari A et al (2017) Moving nitrogen to the centre of plant defense against pathogens. Ann Bot 119:703–709
Mushtaq M, Sakina A, Wani SH et al (2019) Harnessing genome editing techniques to engineer disease resistance in plants. Front Plant Sci 10:550
Mvuyekure SM, Sibiya J, Derera J et al (2017) Genetic analysis of mechanisms associated with inheritance of resistance to sheath rot of rice. Plant Breed 136:509–515. https://doi.org/10.1111/pbr.12492
Nakamura S, Asakawa S, Ohmido N et al (1997) Construction of an 800-kb contig in the near-centromeric region of the rice blast resistance gene Pi-ta2 using a highly representative rice BAC library. Mol Gen Genet 254:611–620. https://doi.org/10.1007/s004380050459
Naqvi NI, Bonman JM, Mackill DJ et al (1995) Identification of RAPD markers linked to a major blast resistance gene in rice. Mol Breed 1:341–348. https://doi.org/10.1007/BF01248411
Neelam K, Mahajan R, Gupta V et al (2020) High-resolution genetic mapping of a novel bacterial blight- resistance gene xa-45(t) identified from Oryza glaberrima and transferred to Oryza sativa. Theor Appl Genet 133:689–705. https://doi.org/10.1007/s00122-019-03501-2
Nelson JC, Oard JH, Groth D et al (2012) Sheath-blight resistance QTLs in japonica rice germplasm. Euphytica 184:23–34. https://doi.org/10.1007/s10681-011-0475-1
Ning D, Song A, Fan F et al (2014) Effects of slag-based silicon fertilizer on rice growth and brown-spot resistance. PLoS One 9:e102681
Nürnberger T, Kemmerling B (2009) PAMP-triggered basal immunity in plants. Adv Bot Res 51:1–38
Ogawa T (1988) Near-isogenic lines as international differentials for resistance to bacterial blight of rice. Rice Genet Newsl 5:106
Ogawa T (2008) Monitoring race distribution and identification of genes for resistance to bacterial leaf blight. Rice Genet III 3:456–459
Ogawa T, Lin L, Tabien RE et al (1987) A new recessive gene for resistance to bacterial blight of rice. Rice Genet Newsl 4:98
Okuyama Y, Kanzaki H, Abe A et al (2011) A multifaceted genomics approach allows the isolation of the rice Pia-blast resistance gene consisting of two adjacent NBS-LRR protein genes. Plant J 66:467–479. https://doi.org/10.1111/j.1365-313X.2011.04502.x
Oliva R, Ji C, Atienza-Grande G et al (2019) Broad-spectrum resistance to bacterial blight in rice using genome editing. Nat Biotechnol 37:1344–1350. https://doi.org/10.1038/s41587-019-0267-z
Oliver RP, Ipcho SVS (2004) Arabidopsis pathology breathes new life into the necrotrophs-vs.-biotrophs classification of fungal pathogens. Mol Plant Pathol 5:347–352
Oreiro EG, Grimares EK, Atienza-Grande G et al (2019) Genome-wide associations and transcriptional profiling reveal ROS regulation as one underlying mechanism of sheath blight resistance in rice. Mol Plant-Microbe Interact 33:212–222. https://doi.org/10.1094/MPMI-05-19-0141-R
Pan Q, Tanisaka T (1997) Studies on the genetics and breeding of blast resistance in rice. VII. Gene analysis for the blast resistance of Indian native cultivar, Aus 373. Breed Sci 47:35
Pan Q, Wang L, Ikehashi H, Tanisaka T (1996) Identification of a new blast resistance gene in the indica rice cultivar Kasalath using Japanese differential cultivars and isozyme markers. Phytopathology 86:1071–1075
Panda KK, Mishra MK (2019) Studies on physiological characteristics of Sarocladium oryzae causing sheath rot of rice. Int J Curr Microbiol App Sci 8:1767–1774. https://doi.org/10.20546/ijcmas.2019.808.209
Panguluri SK, Kumar AA (2013) Phenotyping for plant breeding: applications of phenotyping methods for crop improvement. Springer, New York
Park DS, Sayler RJ, Hong YG et al (2008) A method for inoculation and evaluation of rice sheath blight disease. Plant Dis 92:25–29. https://doi.org/10.1094/PDIS-92-1-0025
Peeters KJ, Haeck A, Harinck L et al (2020) Morphological, pathogenic and toxigenic variability in the rice sheath rot pathogen Sarocladium oryzae. Toxins (Basel) 12:109. https://doi.org/10.3390/toxins12020109
Perez-de-Castro A, Vilanova S, Canizares J et al (2012) Application of genomic tools in plant breeding. Curr Genom 13:179–195. https://doi.org/10.2174/138920212800543084
Petpisit V, Khush GS, Kauffman HE (1977) Inheritance of resistance to bacterial blight in rice. 1. Crop Sci 17:551–554. https://doi.org/10.2135/cropsci1977.0011183x001700040018x
Qu S, Liu G, Zhou B et al (2006) The broad-spectrum blast resistance gene Pi9 encodes a nucleotide-binding site-leucine-rich repeat protein and is a member of a multigene family in rice. Genetics 172:1901–1914. https://doi.org/10.1534/genetics.105.044891
Rajashekara H, Ellur RK, Khanna A et al (2014) Inheritance of blast resistance and its allelic relationship with five major R-genes in a rice landrace “Vanasurya”. Indian Phytopathol 67:365–369
Rajpurohit D, Kumar R, Kumar M et al (2011) Pyramiding of two bacterial blight resistance and a semidwarfing gene in Type 3 Basmati using marker-assisted selection. Euphytica 178:111–126. https://doi.org/10.1007/s10681-010-0279-8
Ramirez CL, Foley JE, Wright DA et al (2008) Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods 5:374–375
Rathna Priya TS, Eliazer Nelson ARL, Ravichandran K, Antony U (2019) Nutritional and functional properties of coloured rice varieties of South India: a review. J Ethn Foods 6:11
Raven JA (2003) Cycling silicon: the role of accumulation in plants. New Phytol 158:419–421
Read AC, Moscou MJ, Zimin AV et al (2020) Genome assembly and characterization of a complex zfBED-NLR gene-containing disease resistance locus in Carolina Gold Select rice with Nanopore sequencing. PLoS Genet 16:e1008571. https://doi.org/10.1371/journal.pgen.1008571
Reddy APK, Katyal JC, Rouse DI, MacKenzie DR (1979) Relationship between nitrogen fertilization, bacterial leaf blight severity and yield of rice. Phytopathology 69:970–973
Rekha G, Abhilash Kumar V, Viraktamath BC et al (2018) Improvement of blast resistance of the popular high-yielding, medium slender-grain type, bacterial blight-resistant rice variety, Improved Samba Mahsuri by marker-assisted breeding. J Plant Biochem Biotechnol 27:463–472. https://doi.org/10.1007/s13562-018-0455-9
Robichaux CR (2001) The effect of calcium silicate on rice yield and sheath blight disease. Fitopatol Bras 30:457–469
Romero FM, Gatica-Arias A (2019) CRISPR/Cas9: development and application in rice breeding. Rice Sci 26:265–281
Sallaud C, Lorieux M, Roumen E et al (2003) Identification of five new blast resistance genes in the highly blast-resistant rice variety IR64 using a QTL mapping strategy. Theor Appl Genet 106:794–803. https://doi.org/10.1007/s00122-002-1088-9
Samiyappan R, Amutha G, Kandan A et al (2003) Purification and partial characterization of a phytotoxin produced by Sarocladium oryzae, the rice sheath rot pathogen (Reinigung und partielle charakterisierung eines vom reisblattscheidenfäule-pathogen sarocladium oryzae erzeugten phytoto). Arch Phytopathol Plant Protect 36:247–256. https://doi.org/10.1080/03235400310001617879
Sanchez AC, Brar DS, Huang N et al (2000) Sequence tagged site marker-assisted selection for three bacterial blight-resistance genes in rice. Crop Sci 40:792–797. https://doi.org/10.2135/cropsci2000.403792x
Savary S, Willocquet L, Pethybridge SJ et al (2019) The global burden of pathogens and pests on major food crops. Nat Ecol Evol 3:430–439. https://doi.org/10.1038/s41559-018-0793-y
Sebastian LS, Ikeda R, Huang N et al (1996) Molecular mapping of resistance to rice tungro spherical virus and green leafhopper. Phytopathology 86:25–30. https://doi.org/10.1094/Phyto-86-25
Serrano M, Coluccia F, Torres M et al (2014) The cuticle and plant defense to pathogens. Front Plant Sci 5:274. https://doi.org/10.3389/fpls.2014.00274
Shan Q, Zhang Y, Chen K et al (2015) Creation of fragrant rice by targeted knockout of the OsBADH2 gene using TALEN technology. Plant Biotechnol J 13:791–800. https://doi.org/10.1111/pbi.12312
Shanti ML, Kumar Varm CM, Premalatha P et al (2010) Understanding the bacterial blight pathogen-combining pathotyping and molecular marker studies. Int J Plant Pathol 1:58–68. https://doi.org/10.3923/ijpp.2010.58.68
Sharma S, Duveiller E, Basnet R et al (2005a) Effect of potash fertilization on Helminthosporium leaf blight severity in wheat, and associated increases in grain yield and kernel weight. Field Crop Res 93:142–150
Sharma TR, Madhav MS, Singh BK et al (2005b) High-resolution mapping, cloning and molecular characterization of the Pi-kh gene of rice, which confers resistance to Magnaporthe grisea. Mol Gen Genomics 274:569–578. https://doi.org/10.1007/s00438-005-0035-2
Sharma TR, Rai AK, Gupta SK et al (2012) Rice blast management through host-plant resistance: retrospect and prospects. Agric Res 1:37–52
Shen C, Que Z, Xia Y et al (2017) Knock out of the annexin gene OsAnn3 via CRISPR/Cas9-mediated genome editing decreased cold tolerance in rice. J Plant Biol 60:539–547
Shinoda H, Toriyama K, Yunoki T et al (1971) Studies on the varietal resistance of rice to blast. 6. Linkage relationship of blast resistance genes. Jpn Chugoku Nogyo Shikengo Fukuyama Bull Ser A 20:1
Sidhu GS, Khush GS, Mew TW (1978) Genetic analysis of bacterial blight-resistance in seventy-four cultivars of rice, Oryza sativa L. Theor Appl Genet 53:105–111. https://doi.org/10.1007/BF00272687
Sime HD, Mbong GA, Malla DK, Suh C (2017) Effect of different doses of NPK fertilizer on the infection coefficient of rice (Orysa sativa L.) blast in Ndop, North West of Cameroon. Agron Afr 29:245–255
Singh S, Sidhu JS, Huang N et al (2001) Pyramiding three bacterial blight-resistance genes (xa5, xa13 and Xa21) using marker-assisted selection into indica rice cultivar PR106. Theor Appl Genet 102:1011–1015. https://doi.org/10.1007/s001220000495
Singh PK, Nag A, Arya P et al (2018) Prospects of understanding the molecular biology of disease resistance in rice. Int J Mol Sci 19:1141
Song WY, Wang GL, Chen LL et al (1995) A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 270:1804. https://doi.org/10.1126/science.270.5243.1804
Stein JC, Yu Y, Copetti D et al (2018) Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat Genet 50:285–296. https://doi.org/10.1038/s41588-018-0040-0
Streubel J, Pesce C, Hutin M et al (2013) Five phylogenetically close rice SWEET genes confer TAL effector-mediated susceptibility to Xanthomonas oryzae pv. oryzae. New Phytol 200:808–819. https://doi.org/10.1111/nph.12411
Su J, Wang W, Han J et al (2015) Functional divergence of duplicated genes results in a novel blast resistance gene Pi50 at the Pi2/9 locus. Theor Appl Genet 128:2213–2225
Suh J-P, Noh T-H, Kim K-Y et al (2009) Expression levels of three bacterial blight-resistance genes against K3a race of Korea by molecular and phenotype analysis in japonica rice (O. sativa L.). J Crop Sci Biotechnol 12:103–108. https://doi.org/10.1007/s12892-009-0103-y
Sun W, Zhang J, Fan Q et al (2010) Silicon-enhanced resistance to rice blast is attributed to silicon-mediated defense resistance and its role as physical barrier. Eur J Plant Pathol 128:39–49
Sun Y, Zhang X, Wu C et al (2016) Engineering herbicide-resistant rice plants through CRISPR/Cas9-mediated homologous recombination of acetolactate synthase. Mol Plant 9:628–631
Sun Q, Liu X, Yang J et al (2018) MicroRNA528 affects lodging resistance of maize by regulating lignin biosynthesis under nitrogen-luxury conditions. Mol Plant 11:806–814
Sun Y, Wang M, Mur LAJ et al (2020) Unravelling the roles of nitrogen nutrition in plant disease defenses. Int J Mol Sci 21:572
Sundaram RM, Vishnupriya MR, Biradar SK et al (2008) Marker-assisted introgression of bacterial blight resistance in Samba Mahsuri, an elite indica rice variety. Euphytica 160:411–422. https://doi.org/10.1007/s10681-007-9564-6
Sundaram RM, Vishnupriya MR, Laha GS et al (2009) Introduction of bacterial blight resistance into Triguna, a high-yielding, mid-early duration rice variety. Biotechnol J 4:400–407. https://doi.org/10.1002/biot.200800310
Swathi G, Durga Rani CV, Md J et al (2019) Marker-assisted introgression of the major bacterial blight- resistance genes, Xa21 and xa13, and blast resistance gene, Pi54, into the popular rice variety, JGL1798. Mol Breed 39:1–12. https://doi.org/10.1007/s11032-019-0950-2
Tabien RE, Li Z, Paterson AH et al (2002) Mapping QTLs for field resistance to the rice blast pathogen and evaluating their individual and combined utility in improved varieties. Theor Appl Genet 105:313–324. https://doi.org/10.1007/s00122-002-0940-2
Taguchi-Shiobara F, Ozaki H, Sato H et al (2013) Mapping and validation of QTLs for rice sheath blight resistance. Breed Sci 63:301–308. https://doi.org/10.1270/jsbbs.63.301
Takagi H, Abe A, Yoshida K et al (2013a) QTL-seq: rapid mapping of quantitative trait loci in rice by whole- genome resequencing of DNA from two bulked populations. Plant J 74:174–183
Takagi H, Uemura A, Yaegashi H et al (2013b) MutMap-Gap: whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast-resistance gene Pii. New Phytol 200:276–283. https://doi.org/10.1111/nph.12369
Takahashi W, Miura Y, Sasaki T (2009) A novel inoculation method for evaluation of grey leaf spot resistance in Italian ryegrass. J Plant Pathol 91:171–176. https://doi.org/10.4454/jpp.v91i1.638
Tan GX, Ren X, Weng QM et al (2004) Mapping of a new resistance gene to bacterial blight in rice line introgressed from Oryza officinalis. Acta Genet Sin 31:724–729
Tang D, Wu W, Li W et al (2000) Mapping of QTLs conferring resistrance to bacterial leaf streak in rice. Theor Appl Genet 101:286–291. https://doi.org/10.1007/s001220051481
Tang L, Mao B, Li Y et al (2017) Knockout of OsNramp5 using the CRISPR/Cas9 system produces low Cd-accumulating indica rice without compromising yield. Sci Rep 7:14438. https://doi.org/10.1038/s41598-017-14832-9
Tanksley SD, Young ND, Paterson AH, Bonierbale MW (1989) RFLP mapping in plant breeding: new tools for an old science. Bio/Technology 7:257–264. https://doi.org/10.1038/nbt0389-257
Tanweer FA, Rafii MY, Sijam K et al (2015) Current advance methods for the identification of blast resistance genes in rice. C R Biol 338:321–334. https://doi.org/10.1016/j.crvi.2015.03.001
Tena G, Boudsocq M, Sheen J (2011) Protein kinase signaling networks in plant innate immunity. Curr Opin Plant Biol 14:519–529
Teng Q, Hu X-F, Chang Y-Y et al (2016) Effects of different fertilisers on rice resistance to pests and diseases. Soil Res 54:242–253
Thakur M, Sohal BS (2013) Role of elicitors in inducing resistance in plants against pathogen infection: a review. Int Sch Res Not 2013:762412
Tonnessen BW, Manosalva P, Lang JM et al (2015) Rice phenylalanine ammonia-lyase gene OsPAL4 is associated with broad spectrum disease resistance. Plant Mol Biol 87:273–286. https://doi.org/10.1007/s11103-014-0275-9
Vassilev D, Leunissen J, Atanassov A et al (2005) Application of bioinformatics in plant breeding. Biotechnol Biotechnol Equip 19:139. https://doi.org/10.1080/13102818.2005.10817293
Vikal Y, Chawla H, Sharma R et al (2014) Mapping of bacterial blight-resistance gene xa8 in rice (Oryza sativa L.). Indian J Genet Plant Breed 74:589. https://doi.org/10.5958/0975-6906.2014.00895.5
Wang GL, Mackill DJ, Bonman JM et al (1994) RFLP mapping of genes conferring complete and partial resistance to blast in a durably resistant rice cultivar. Genetics 136:1421–1434
Wang ZX, Yano M, Yamanouchi U et al (1999) The Pib gene for rice blast resistance belongs to the nucleotide binding and leucine-rich repeat class of plant disease resistance genes. Plant J 19:55–64. https://doi.org/10.1046/j.1365-313X.1999.00498.x
Wang W, Zhai W, Luo M et al (2001) Chromosome landing at the bacterial blight-resistance gene Xa4 locus using a deep coverage rice BAC library. Mol Gen Genet 265:118–125. https://doi.org/10.1007/s004380000382
Wang C, Wen G, Lin X et al (2009) Identification and fine-mapping of the new bacterial blight-resistance gene, Xa31(t), in rice. Eur J Plant Pathol 123:235–240. https://doi.org/10.1007/s10658-008-9356-4
Wang F, Wang C, Liu P et al (2016) Enhanced rice blast resistance by CRISPR/Cas9-targeted mutagenesis of the ERF transcription factor gene OsERF922. PLoS One 11:e0154027
Wang M, Wang S, Liang Z et al (2018) From genetic stock to genome editing: gene exploitation in wheat. Trends Biotechnol 36:160–172
Wang L, Zhao L, Zhang X et al (2019) Large-scale identification and functional analysis of NLR genes in blast resistance in the Tetep rice genome sequence. Proc Natl Acad Sci U S A 116:8479–18487. https://doi.org/10.1073/pnas.1910229116
Wen ZH, Zeng YX, Ji ZJ, Yang CD (2015) Mapping quantitative trait loci for sheath blight disease resistance in Yangdao 4 rice. Genet Mol Res 14:1636–1649. https://doi.org/10.4238/2015.March.6.10
Wu KS, Tanksley SD (1993) Abundance, polymorphism and genetic mapping of microsatellites in rice. Mol Gen Genet 241:225–235. https://doi.org/10.1007/BF00280220
Wu JL, Fan YY, Li DB et al (2005) Genetic control of rice blast resistance in the durably resistant cultivar Gumei 2 against multiple isolates. Theor Appl Genet 111:50–56. https://doi.org/10.1007/s00122-005-1971-2
Wu KS, Martinez C, Lentini Z et al (2008) Cloning a blast resistance gene by chromosome walking. Rice Genet III:669. https://doi.org/10.1142/97898128142789_0082
Wu X, Yu Y, Baerson SR et al (2017) Interactions between nitrogen and silicon in rice and their effects on resistance toward the brown planthopper Nilaparvata lugens. Front Plant Sci 8:28
Xiang Y, Cao Y, Xu C et al (2006) Xa3, conferring resistance for rice bacterial blight and encoding a receptor kinase-like protein, is the same as Xa26. Theor Appl Genet 113:1347–1355. https://doi.org/10.1007/s00122-006-0388-x
Xu Q, Yuan X, Yu H et al (2011) Mapping quantitative trait loci for sheath blight resistance in rice using double haploid population. Plant Breed 130:404–406. https://doi.org/10.1111/j.1439-0523.2010.01806.x
Xu X, Hayashi N, Wang CT et al (2014) Rice blast resistance gene Pikahei-1(t), a member of a resistance gene cluster on chromosome 4, encodes a nucleotide-binding site and leucine-rich repeat protein. Mol Breed 34:691–700. https://doi.org/10.1007/s11032-014-0067-6
Xu Z, Xu X, Gong Q et al (2019) Engineering broad-spectrum bacterial blight resistance by simultaneously disrupting variable TALE-binding elements of multiple susceptibility genes in rice. Mol Plant 12:1434–1446. https://doi.org/10.1016/j.molp.2019.08.006
Yadav S, Anuradha G, Kumar RR et al (2015) Identification of QTLs and possible candidate genes conferring sheath blight resistance in rice (Oryza sativa L.). Springerplus 4:175. https://doi.org/10.1186/s40064-015-0954-2
Yoshimura S, Yamanouchi U, Katayose Y et al (1998) Expression of Xa1, a bacterial blight-resistance gene in rice, is induced by bacterial inoculation. Proc Natl Acad Sci U S A 95:1663–1668. https://doi.org/10.1073/pnas.95.4.1663
Yu S, Ali J, Zhang C et al (2020) Correction to: Genomic breeding of green super rice varieties and their deployment in Asia and Africa. Theor Appl Genet 133:1337
Yuan B, Zhai C, Wang W et al (2011) The Pik-p resistance to Magnaporthe oryzae in rice is mediated by a pair of closely linked CC-NBS-LRR genes. Theor Appl Genet 122:1017–1028. https://doi.org/10.1007/s00122-010-1506-3
Yuan C, Yuxiang Z, Zhijuan J et al (2019) Identification of stable quantitative trait loci for sheath blight resistance using recombinant inbred line. Rice Sci 26:331–338. https://doi.org/10.1016/j.rsci.2019.08.007
Yugander A, Sundaram RM, Ladhalakshmi D et al (2017) Virulence profiling of Xanthomonas oryzae pv. oryzae isolates, causing bacterial blight of rice in India. Eur J Plant Pathol 149:171–191. https://doi.org/10.1007/s10658-017-1176-y
Yugander A, Sundaram RM, Singh K et al (2018) Incorporation of the novel bacterial blight-resistance gene Xa38 into the genetic background of elite rice variety Improved Samba Mahsuri. PLoS One 13:e0198260. https://doi.org/10.1371/journal.pone.0198260
Zafar K, Sedeek KEM, Rao GS et al (2020) Genome editing technologies for rice improvement: progress, prospects, and safety concerns. Front Genome Ed 2:5. https://doi.org/10.3389/fgeed.2020.00005
Zenbayashi K, Ashizawa T, Tani T, Koizumi S (2002) Mapping of the QTL (quantitative trait locus) conferring partial resistance to leaf blast in rice cultivar Chubu 32. Theor Appl Genet 104:547–552. https://doi.org/10.1007/s00122-001-0779-y
Zeng YX, Xia LZ, Wen ZH et al (2015) Mapping resistant QTLs for rice sheath blight disease with a doubled haploid population. J Integr Agric 14:801–810. https://doi.org/10.1016/S2095-3119(14)60909-6
Zetsche B, Gootenberg JS, Abudayyeh OO et al (2015) Cpf1 is a single RNA-guided endonuclease of a Class 2 CRISPR-Cas system. Cell 163:759–771. https://doi.org/10.1016/j.cell.2015.09.038
Zhai C, Lin F, Dong Z et al (2011) The isolation and characterization of Pik, a rice blast-resistance gene which emerged after rice domestication. New Phytol 189:321–334. https://doi.org/10.1111/j.1469-8137.2010.03462.x
Zhang Q, Wanf CL, Zhao KJ et al (2001) The effectiveness of advanced rice lines with new resistance gene Xa23 to rice bacterial blight. Rice Genet Newsl 18:71–72
Zhang H, Li G, Li W, Song F (2009) Transgenic strategies for improving rice disease resistance. Afr J Biotechnol 8:1750–1757
Zhang F, Zhuo DL, Zhang F et al (2015) Xa39, a novel-dominant gene conferring broad-spectrum resistance to Xanthomonas oryzae pv. oryzae in rice. Plant Pathol 64:568–575. https://doi.org/10.1111/ppa.12283
Zhang H, Zhang J, Lang Z et al (2017a) Genome editing: principles and applications for functional genomics research and crop improvement. CRC Crit Rev Plant Sci 36:291–309
Zhang W, Wu L, Ding Y et al (2017b) Nitrogen fertilizer application affects lodging resistance by altering secondary cell wall synthesis in japonica rice (Oryza sativa). J Plant Res 130:859–871
Zhao H, Wang X, Jia Y et al (2018) The rice blast-resistance gene Ptr encodes an atypical protein required for broad-spectrum disease resistance. Nat Commun 9:1–12. https://doi.org/10.1038/s41467-018-04369-4
Zheng CK, Wang CL, Yu YJ et al (2009) Identification and molecular mapping of Xa32(t), a novel resistance gene for bacterial blight (Xanthomonas oryzae pv. oryzae) in rice. Acta Agron Sin 35:1173–1180. https://doi.org/10.1016/S1875-2780(08)60089-9
Zhou JH, Wang JL, Xu JC et al (2004) Identification and mapping of a rice blast-resistance gene Pi-g(t) in the cultivar Guangchangzhan. Plant Pathol 53:191–196. https://doi.org/10.1111/j.0032-0862.2004.00986.x
Zhou B, Qu S, Liu G et al (2006) The eight amino-acid differences within three leucine-rich repeats between Pi2 and Piz-t resistance proteins determine the resistance specificity to Magnaporthe grisea. Mol Plant-Microbe Interact 19:1216–1228. https://doi.org/10.1094/MPMI-19-1216
Zhou J, Peng Z, Long J et al (2015) Gene targeting by the TAL effector PthXo2 reveals cryptic resistance gene for bacterial blight of rice. Plant J 82:632–643
Zhou J, Deng K, Cheng Y et al (2017) CRISPR-Cas9 based genome editing reveals new insights into microRNA function and regulation in rice. Front Plant Sci 8:1598. https://doi.org/10.3389/fpls.2017.01598
Zhou X, Liao H, Chern M et al (2018) Loss of function of a rice TPR-domain RNA-binding protein confers broad-spectrum disease resistance. Proc Natl Acad Sci U S A 115:3174–3179. https://doi.org/10.1073/pnas.1705927115
Zhuang JY, Ma WB, Wu JL et al (2002) Mapping of leaf and neck blast-resistance genes with resistance gene analog, RAPD and RFLP in rice. Euphytica 128:363–370. https://doi.org/10.1023/A:1021272710294
Zipfel C, Felix G (2005) Plants and animals: a different taste for microbes? Curr Opin Plant Biol 8:353–360
Zuo S, Yin Y, Pan C et al (2013) Fine-mapping of qSB-11LE, the QTL that confers partial resistance to rice sheath blight. Theor Appl Genet 126:1257–1272. https://doi.org/10.1007/s00122-013-2051-7
Zuo S, Zhang Y, Yin Y et al (2014) Fine-mapping of qSB-9 TQ, a gene conferring major quantitative resistance to rice sheath blight. Mol Breed 34:2191–2203. https://doi.org/10.1007/s11032-014-0173-5
Acknowledgments
The authors would like to thank and acknowledge the Bill & Melinda Gates Foundation for providing a research grant to ZL for the Green Super Rice project under ID OPP1130530. We would like to thank the Department of Agriculture, Philippines, for providing funds to JA under the Next-Gen project and also thank and acknowledge IRRI Communications for English-language editing and the anonymous internal reviewer’s valuable suggestions and constructive comments that helped improve this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Jamaloddin, M. et al. (2021). Molecular Approaches for Disease Resistance in Rice. In: Ali, J., Wani, S.H. (eds) Rice Improvement. Springer, Cham. https://doi.org/10.1007/978-3-030-66530-2_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-66530-2_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-66529-6
Online ISBN: 978-3-030-66530-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)