
Abstract
The purpose of this review is to report on the state-of-the-art on the interaction of moisture with natural materials and fabricated biomimetic functional materials, with an emphasis upon the hygro-responsive behaviour of wood. The primary objective is to examine how water sorption affects dimensional behaviour and how knowledge of this property in natural plant-based (mainly, but not exclusively wood) materials can be used to inform biomimetic design of moisture-responsive materials and devices. The study examines the literature on natural and bio-inspired materials, concentrating upon sorption kinetics, water migration and location of the sorbed water in the materials and their microstructure and mechanical response of the microstructure and how this affects molecular mobility of the sorbate translating to macrostructural changes. Also included within this review, it is an overview of the main experimental techniques which have been used to investigate the interaction of water with these materials at molecular length scales and how modern techniques can resolve the response of these materials at the cell wall level.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
This review explores the relationship between sorption behaviour and structure in composite materials that have a hygro-responsive matrix combined with non-responsive elements such as fibres, rods, plates or sheets. The particular focus is upon furthering the understanding of the sorption behaviour of wood in particular and natural fibres to a lesser extent, but biomimetic materials based upon these principles are also included. This type of material exhibits swelling behaviour in the presence of moisture, but because the ‘inert’ phase exhibits some form of structural heterogeneity, the observed swelling behaviour is consequently anisotropic. This is often a problem with wood-based materials but can be used to advantage when designing biomimetic structures.
The interaction of moisture with natural hygro-responsive materials (such as wood and textiles) has been a topic of immense scientific interest for over a century. Much initial research was directed at trying to better understand the water vapour sorption behaviour of wood to improve kiln-drying schedules and to reduce the negative properties associated with dimensional instability [1]. One of the key pieces of information for understanding the relationship between solids and water vapour is the sorption isotherm, which is the relationship between the relative humidity (RH—also referred to as water activity aw) of the surrounding atmosphere and the moisture content (MC) of the material at equilibrium (equilibrium moisture content, EMC) at a constant temperature. One critical aspect of making such measurements is to ensure that a true equilibrium state has been achieved, although wood in service conditions seldom achieves an equilibrium state. There are many sorption isotherm models that have been developed to describe the sorption behaviour of natural materials, and this topic has been very well explored elsewhere e.g. [2,3,4,5,6,7,8]. However, these models suffer from various limitations, and there is still a need to develop a theory that better explains the observed behaviour [2, 9], not just for wood, but other lignocellulosic/cellulosic materials as well as foodstuffs, textiles, etc.
Hygro-responsive materials change dimensions when they interact with water vapour. They comprise a dynamic polymeric structure which absorbs water vapour, but they may also have an essentially inert fibrillar or layered structure embedded within the hygro-responsive matrix. In the latter case, these materials exhibit anisotropic responses when they gain or lose water molecules. For lignocellulosic materials, such as wood, sorption models additionally need to include consideration of hygro-inert reinforcing crystalline cellulose microfibrils embedded in a hygro-responsive amorphous lignin/hemicellulose matrix. It is argued in this review that the properties of these materials are best described by consideration of the molecular dynamics in these materials, giving rise to the phenomena, such as swelling pressure, the sigmoidal sorption isotherm, sorption hysteresis, characteristic dynamic sorption behaviour and associated changes in static and dynamic mechanical properties (modulus, strength and viscoelastic behaviour).
Studies of moisture interactions with natural materials are very instructive from a biomimetic design perspective, and the review also briefly covers this subject from the perspective of how water sorption/desorption leads to dimensional changes. The intention of the review is not to analyse the existing and very comprehensive literature of biomimetic moisture-activated materials; but rather to approach the subject from the point of view of how location of sorbed water in hierarchical heterogenous natural materials (such as wood) affects the dimensional behaviour of these materials. Such studies may provide insights into refining the design of new biomimetic moisture-activated ‘smart’ materials.
The next section of the review considers some basic principles which may assist with a generic understanding of the sorption properties of the dynamic polymeric matrix of natural materials and how this relates to dimensional changes.
General considerations regarding polymer microstructure and moisture-induced behaviour
Water in rubbery and glassy polymers
There are materials which absorb water vapour because they contain pores of small dimensions allowing for capillary condensation. However, these materials are inert and do not swell to accommodate the absorbed water (an example in this category would be activated carbon [10]); materials of this type generally exhibit IUPAC Type V sorption isotherms [11], or Type I if they are hydrophilic [12] (Fig. 1).

Definitions of different sorption isotherms according to IUPAC. Lignocellulosic materials exhibit Type II isotherms, but also with hysteresis.
Hygro-responsive materials are, for the purposes of this review, defined as a class of polymeric materials that contain chemical moieties that interact with water molecules and exhibit molecular mobility. They can therefore change their structure to accommodate the sorbed water molecules and to allow for transportation of these penetrant molecules through the macromolecular network. This molecular mobility is facilitated by the presence of the sorbed water molecules which creates additional void volume within the structure (usually referred to as plasticization or softening) resulting in macroscopic viscoelastic behaviour [13, 14]. Transport therefore does not rely on an interconnected porous structure (which is necessary in inert materials), but rather mobility of the polymeric matrix molecules. Materials of this class exhibit isotherms which are classed as Type II, but also often exhibit hysteresis between the absorption and desorption branches of the isotherm. Before a discussion of the sorption behaviour of these systems, there is a brief introduction to how solvent systems interact with dynamic polymers.
The Flory–Huggins (F–H) model is a relatively simple theory that is often invoked to describe the solvent-induced swelling behaviour of polymers. The F–H approach uses a lattice-based model, which considers the entropy of mixing of a polymer chain in a solvent. The parameters used to describe the sorption behaviour are the volume fraction of the polymer, volume fraction of the solvent and a term to describe the solvation of the polymer. The sorption behaviour is expressed as follows:
where: p1 is the pressure of the solvent, p10 is the saturation pressure of the solvent, ϕ1 is the volume fraction of the sorbate and ϕ2 is the volume fraction of the polymer. The term χ is the Flory–Huggins interaction parameter, which is related to the difference in energy between the solvent molecule in the pure solvent (in this case, liquid water) and the molecule in the polymer matrix. The F−H interaction parameter is a measure of thermodynamic miscibility of a solvent with a polymer in binary systems and is treated as a constant in the F–H theory. The relationship between the sorption curve and the interaction parameter is shown in Fig. 2. The F–H polymer dissolution model produces an isotherm that exhibits a continuous upward curvature as the RH increases up to a maximum, but finite value at total saturation (IUPAC Type III), with the steepness of the curve controlled by the magnitude of the interaction parameter (χ). A strong interaction between the solvent and polymer yields a low value of χ (Fig. 2).

Sorption isotherm generated by the Flory–Huggins model, showing the influence of variation in the interaction parameter. A decrease in this parameter indicates a stronger solvent-polymer interaction.
Although a popular approach, the F–H model has been shown to have shortcomings. It is possible to determine χ independently using, e.g. scattering methods to determine polymer–solvent interactions [15], but this can be problematical and different measurement methods usually give different values for χ [16]. When fitting to sorption isotherm data, it is more common to use the interaction parameter as a fitting variable for experimental isotherm data, rather than using it as a fixed parameter obtained from an independent experiment, which is unsatisfactory for a fundamental approach to the problem. According to F–H theory, the interaction parameter should be independent of the concentration and molecular weight of the polymer, but in practice, this independence does not usually hold, especially for polar systems [17,18,19]. It has been shown that the interaction parameter can vary depending on the inhomogeneity of distribution of the solvent molecules [20] as well as exhibiting a concentration dependence [20, 21]. Furthermore, the underlying lattice model has been shown to have limitations when representing the behaviour of real polymers [22].
Polymer hydrogels (PHGs) are a sub-class of hygro-responsive polymers. Sorption isotherms of PHGs typically exhibit a shape that is characterized by IUPAC Type II or Type III curves. PHGs can exhibit extremely high levels of water uptake, a property that is at least partially determined by the crosslinking density of the polymer structure [23]. They are not soluble in water unless the crosslinking structure is not stable in the presence of water or can be disturbed by the application of mechanical energy. There also exist ‘dual network’ hydrogels which have chemical and physical (e.g. hydrogen bonding) crosslinking regions. Such materials exhibit mechanical behaviour that is dependent upon an applied strain rate [24].
In the context of the present review, the hydrophilic chemical entities associated with the polymers that are of most interest are hydroxyl (OH) groups. These groups are capable of hydrogen bonding to sorbed water molecules and are usually referred to as primary sorption sites for this reason. A higher concentration of OH groups in the polymer increases the hydrophilicity of the polymer and consequently affects the interaction parameter with water as a solvent [25].
It is thought that hydrogen bonding to OH groups attached to the polymer chains reduces the mobility of sorbed water molecules [26]. As the concentration of water in the expanding polymer network increases, these incoming molecules are associated with other water molecules rather than the essentially static polymeric OH groups and consequently have much higher mobility compared to those H-bonded to the sorption sites. However, the whole system is dynamic, with constant exchange of water molecules between these different environments [27]. This leads to the question of whether the different water environments can be readily identified and whether there are any measurable differences in terms of mobility, which is discussed later.
The water molecules in the polymer matrix also create space between molecules within the network, which results in increased mobility of the polymer chains. This phenomenon is referred to as plasticization. Before any further discussion, it is helpful at this stage to consider the different types of volume that exist within a polymeric matrix. According to the model of Duda and Zielinski, polymer volume is divided into three components [28]:
-
Occupied volume is the volume occupied by the polymeric components and is constant for all temperatures.
-
Interstitial free volume is the free volume that is not accessible by penetrant molecules and represents the free volume arising from vibrational motion of the polymer and increases slightly with temperature.
-
Hole free volume (herein referred to as free volume) is the volume arising from relaxation and plasticization of the polymer upon heating and cooling. This free volume is accessible by penetrant molecules and can be changed by the presence of these molecules. In rubbery polymers, the free volume is in equilibrium, but in glassy polymers, the molecular relaxations are slow and extra hole free volume may become trapped within the structure.
In some hydrogel systems at low MC, there is insufficient free volume for the polymer segments to change structure without cooperative motion of adjacent segments [29], which is characteristic of glassy polymers below the glass transition temperature (Tg) [30]. A glassy state in a polymer system occurs when the rate of cooling of that system is faster than the rate of relaxation of that system and free volume can be trapped within the matrix. Glassy polymers exhibit non-Fickian diffusion processes because the rate of diffusion is determined by the polymer relaxation processes, rather than a concentration gradient. The viscoelastic relaxation of amorphous polymers in the glass transition region is best described in terms of a relaxation spectrum, with the characteristic relaxation times spanning several decades [29, 31]. Solvents can increase the free volume in the polymer matrix, which results in an increase in molecular mobility through the matrix. Characteristics of the sorption behaviour of glassy polymers are the sigmoidal shape of the sorption isotherm, hysteresis between the adsorption and desorption loops, as well as non-Fickian sorption kinetics [30, 32].
A popular model describing the changes in free volume of glassy polymers due to penetration by solvent molecules has been developed by Vrentas and Vrentas [33, 34], which produces the observed sigmoidal isotherm. This model is an extension of the F–H model (Eq. 1), with the introduction of a new term, F, which takes account of the elastic energy stored in the polymer matrix, when sorption occurs below Tg. The model is represented as follows:
The magnitude of the F term can be calculated from first principles, given knowledge of the heat capacities of the polymer above and below Tg, the molecular weight of the penetrant, the Tg of the polymer–solvent combination and the mass fraction of the polymer [35]. At Tg, the term F becomes zero and the equation consequently reduces to the F–H expression.
The model of Vrentas and Vrentas was further developed to account for hysteresis between the absorption and desorption branches of the sorption isotherm in glassy polymeric materials, with the introduction of a new parameter (k), which is calculated based upon knowledge of the glass transition temperature of the pure polymer [34].
A key concept on which the model is based is that removal of penetrant molecules from the polymer-penetrant mixture results in the creation of a glassy structure and that this process can be considered mathematically equivalent to cooling the polymer-penetrant mixture. Since the terms F and k can both be calculated from first principles, this means that both the absorption and desorption branches of the isotherm can be predicted without the use of arbitrary fitting parameters (at least in principle) [36].
Both the F–H and the Vrentas–Vrentas (V–V) models were initially developed to explain the sorption behaviour of non-polar polymer–solvent systems. However, the use of the V–V model has been extended to describe polar polymer systems interacting with water, but with mixed results [36,37,38,39,40,41]. Although sorption and desorption in a glassy polymer are a non-equilibrium process [42], Argatov and Kocherbitov argue that models of the sorption and diffusion phenomena can still be based upon a classical thermodynamic approach [41]. They have developed a model that is based upon the F–H isotherm but reintroduces the idea of a solvent concentration dependence of the χ interaction parameter, which is related to the V–V F term in the following way:
They maintain that the use of a concentration dependent interaction parameter is a requirement for non-equilibrium systems in order to describe observed experimental behaviour of sorption with glassy polymers and note that this has also been used elsewhere [43]. Argatov and Kocherbitov have studied the variation in the interaction parameter using experimental data and used this information to determine the onset of the glass transition temperature [41]. There is much interest in this subject in the coatings field, and researchers have developed diffusion models for glassy polymers which are based upon irreversible rather than classical thermodynamics [44]. There exists a considerable body of literature on this topic which falls outside the boundaries of this review, but some references can be found in Arya et al. [30].
Much of the early work on water sorption in gel systems was conducted by Wilfred Barkas of the Forest Products Laboratory in Princes Risborough, UK. He explored the use of gel models to explain the sorption behaviour of wood and noted that in circumstances where the swelling of a gel was restricted in some way, that shear stresses are developed; resulting in sorption hysteresis being observed [45]. It has been shown that the water uptake rate and extent in sorption isotherms is reduced in gels which have a higher crosslink density [20, 46, 47]. In one study, it was shown that the extent of hysteresis decreased as the crosslink density increased, which is contrary to what would be predicted [47]. However, the hysteresis in this case was attributed to water that was somehow trapped within the polymer network during the desorption cycle, but this is not the origin of the hysteresis phenomenon discussed here. The intention when recording reliable sorption isotherms is to avoid water or solvents of any kind being trapped as inclusion compounds within the polymer matrix [48, 49]. It is interesting to note that the observed sorption isotherm and associated hysteresis loop of crosslinked hydrogels is affected by the previous history of the sample [50]. Sorption behaviour which is dependent of the previous sorption history of the sample is typically observed with glassy polymers [33, 51,52,53,54].
The essential point made in this review is that an understanding of sorption phenomena in many hygro-responsive natural materials can only be obtained through an approach that considers glassy polymers, the glass transition temperature, free volume and the related mobility of the matrix polymers as affected by the sorbate solvent molecules (in this case water). For solution-based models, the presence of OH groups affects the interaction between water and the polymer, but in such models, it is not necessary to consider the these groups as sorption sites.
Sorption sites, water clustering and nano/microporosity
It is usually assumed that at low levels of uptake, sorbed water molecules will diffuse in some manner through the polymer matrix until they reach a ‘sorption site’ (usually a hydroxyl group). In models of this type, OH groups have a very significant role to play in determining sorption behaviour but usually exclude other important phenomena, such as mechano-sorptive responses. The experimental determination of accessible OH groups in polymeric materials commonly uses hydrogen/deuterium exchange between deuterium oxide as a sorbate and the material of interest. Quantification of the thus-generated -OD groups can use gravimetric, IR or NMR methods [48, 55,56,57]. The distribution of sorbed water molecules in the polymer matrix will depend upon the concentration of OH groups that are available as sorption sites within a given volume (accessible OH groups) and a balance between the tendency of water molecules to cluster around these sorption sites or to distribute throughout the polymer matrix in the manner of a solvent.
A common method used to determine the extent of water clustering in polymer-water systems is that introduced by Zimm and Lundberg [32, 43, 58, 59]. The Zimm–Lundberg (Z–L) method has the advantage that clustering data can be determined directly from the equilibrium sorption isotherm but suffers from the disadvantage that it is not a direct measure of actual cluster size. In order to determine applicability to water-polymer systems, there have been studies where the predictions of the Z–L model have been independently tested using FTIR spectroscopy, which showed that the Z–L approach tended to underestimate cluster size. [60]. As already noted with the F–H model, the failure of the Z–L model to correctly describe water solubility and water clustering in glassy polymers is partially due to the equilibrium constraints on these models in contrast with the non-equilibrium conditions which apply below Tg [60, 61]. In addition (as with the F–H model), the Z–L model was originally developed to describe solvent solubility in weakly interacting non-polar systems, which means that extension to water-polymer systems may not be reliable. This has resulted in the development of improved models for the clustering of water molecules in hydrophilic polymers, e.g. [62, 63]. Other clustering models are also available, such as the Langmuir/Flory–Huggins model [64]. It should be noted that the clustering of water molecules within the polymer matrix can lead to marked deviations from Henry’s Law [28].
The water molecules which are involved in H-bonding with the polymeric OH groups are usually referred to as ‘bound’ water, whereas the water molecules which are not directly associated with sorption sites are called ‘free’, or sometimes ‘intermediate’ water [65]. These terms can lead to confusion, in that some papers refer to bound water as the water that is located within the polymer matrix and free water as existing in larger pores that may be located in the matrix. In wood, the bound water usually means the water located within the cell wall, whereas the free water is located in larger void spaces, such as the cell lumen. Water molecules that hydrogen bond to polymer sorption sites have less mobility compared to water molecules that are not so directly bonded. However, the residence times of water molecules in association with matrix OH groups may be very short, meaning that such molecules are in reality indistinguishable from the free water molecules, due to very rapid exchange [27]. Water mobility can be determined using nuclear magnetic resonance (NMR) relaxation times or dielectric relaxation spectroscopy (DRS), [65], as is discussed later. In NMR, the relaxation of protons is mainly dipolar and in water both translation and rotation relaxation mechanisms are important. In addition, the exchange of protons between the water molecules and polymer hydroxyl sites is also a significant relaxation mechanism [27, 66,67,68]. This makes the determination of water mobility in the matrix problematical. The water molecules in ‘bound’ and ‘unbound’ environments would be expected to exhibit different mobilities, although exchange between the different environments can be very rapid, so individual molecules are unlikely to exist in a specific state for long.
There exist nm-sized pores within the polymer matrix that are often termed micropores (IUPAC definition meaning pores with diameter less than 2 nm), but the more general term ‘nanopores’ is used in this review to mean nm-sized pores, with no strict definition. Water that is confined within the nanopores of the polymer structure exhibits a freezing point depression compared to unconfined water [69]. This freezing point depression is caused by a combination of osmotic and capillary effects. It is possible to determine the relationship between the freezing point depression and water amount using techniques, such as NMR [27, 70] or calorimetry [71]. The results are usually expressed as a pore size distribution (PSD), where the pore size is related to the freezing point depression by the Gibbs–Thomson equation. There is also a proportion of the water that is described as non-freezing, in that it does not exhibit a phase change at reduced temperatures and this population is assigned to water that is in a ‘bound’ state, i.e. closely associated with the polymer network [72]. However, it is not necessary to invoke a special state of water (‘bound’) to explain non-freezing water and it is difficult to provide unambiguous evidence for the existence of water that might be described as ‘bound’ [27]. Other experiments with non-polar solvents in swollen polymer networks have also cast doubt on the hypothesis that non-freezing water is necessarily attributable to hydrogen bonding with polar groups on the polymer [73]. It is clearly problematical to assign water populations unambiguously to ‘bound’ or ‘unbound’ states.
The pre-existing (residual) free volume within the polymer matrix consists of nm-sized pores, but the geometry of this nanoporosity can evolve as water molecules enter and exit the polymer matrix and this dynamic behaviour is referred to as transient porosity. The evolving properties of this nanoporosity can be investigated using positron annihilation lifetime spectroscopy (PALS) [74]. For example, PALS measurements in carbohydrate matrices in sorption experiments show that the nanopore size initially decreases but then increases as the MC is raised, due to the accompanying plasticization of the polymer chains [75,76,77]. The initial decrease is observed because the initially ‘empty’ voids associated with residual free volume begin to fill with water molecules. The existence of transient microporosity in polymers, gels and the cell wall of plant materials is an important factor which must be considered when explaining sorption and diffusion phenomena. This dynamic behaviour is quite different from an inert porous material, such as a zeolite or nanoporous carbon, in terms of hygroscopic properties and origin of hysteresis [78, 79].
Sorption kinetics and diffusion within the polymer matrix
When water molecules enter a polymeric matrix, the macromolecules will rearrange their structure to accommodate the sorbed water molecules at a rate that depends on the ambient temperature and the concentration of solvent, in addition to steric effects related to the chemical structure. The relative time scales of the diffusion of solvent molecules and polymer relaxation determine the nature of the transport process. This can be represented in terms of a diffusion Deborah number (De), which is the ratio of the characteristic relaxation time and the diffusion time [80]. For a value of De less than 1, the changes in polymer structure occur much faster than the rate of diffusion of the solvent molecules and classical Fickian diffusion is observed. This is typical for rubbery polymers above Tg. When De is greater than 1, the polymer chains rearrange much more slowly compared to the rate of diffusion of solvent molecules and the diffusion process is relaxation-limited rather than determined by a concentration gradient. This behaviour is characteristic of polymers below Tg, and diffusion of water through glassy polymers is non-Fickian in nature [60]. When De is close to 1, anomalous diffusion is observed, with a coupling of relaxation-limited and Fickian processes. Pseudo-Fickian models of this type include that developed by Berens and Hopfenberg, amongst others [81,82,83,84]. The relaxation term is actually an average of many relaxation processes, but for many polymer/penetrant systems, a single relaxation term is sufficient to represent the behaviour in macroscopic systems [85]. The sorption kinetics of glassy polymers is often interpreted using a model which incorporates two independent relaxation terms describing contributions from diffusion and relaxation processes [81, 82]. Another interpretation for a two-component kinetic model is the movement of a penetrant solvent front through the sample with associated swelling [86].
This two-component anomalous diffusion process is not to be confused with the dual-mode model which has been developed to describe sorption in glassy polymers. The dual-mode model assumes that there are two populations of water, one of which acts as a solvent for the polymer (called the Henry’s Law population) and is therefore intimately mixed within the structure. The other population is sorbed onto the surface of nanopores that are considered to exist within the structure (called the Langmuir population) as a consequence of excess free volume that is frozen into the glassy matrix below the glass transition temperature [61, 87, 88].
Although dual-mode sorption models can quite successfully model the sorption behaviour of glassy polymers, they are empirical; requiring temperature-specific parameters to be evaluated for sorption and different ones for desorption. Furthermore, in order to describe differences between absorption and desorption isotherms, it is necessary to assume that there is an exchange between the two water populations (a proportion of the dissolved population now becomes part of the Langmuir population). In addition, it must be assumed that this change must be instantaneous at the point of transition from absorption to desorption (which does not appear to be represented by a realistic physical phenomenon). The explanation for this behaviour assumes that the hole population increases with the concentration of sorbate molecules. For this to occur, there must be sufficient free volume for the polymer chains to move and allow for diffusion of the sorbate molecule. Furthermore, when the polymer relaxes back to the original configuration, this then prevents the penetrant molecule from diffusing back to its original position. The diffusing molecule is effectively ‘trapped’ in the new site that has been created, with a large energy barrier preventing further movement [89]. Based upon this modification of the dual-mode model, the Tg represents the point at which the last hole is created as the temperature is reduced until the structure is ‘frozen’ [90].
The ability of sorbed water molecules to act as a plasticiser for glassy polymer networks and to increase the free volume (thereby facilitating polymer reconfiguration) has been widely discussed [91,92,93,94,95,96]. In free volume models, diffusion of low molecular weight penetrants into amorphous polymers above Tg is considered to take place by the hopping of the penetrant molecules into free-volume nanopores which are formed by random thermal fluctuations of the matrix molecules [97, 98], as the MC increases, this ‘stop/go’ motion is facilitated [99]. Below Tg, rearrangement of the polymer networks requires cooperative relaxation processes [29, 100, 101] and as the temperature of the polymer is reduced, the size of the domain of cooperation increases, reducing the likelihood of reconfiguration events. However, the presence of water molecules acting as a plasticiser within the matrix creates free volume, which facilitates rearrangement of the polymers and consequently reduces the Tg of the system. The rate of diffusion of water in the nanopores of the gel is affected by the connectivity or tortuosity of the network [68, 102], depends on the MC [103] and decreases as free volume decreases [104]. However, this porosity network is dynamic, rather than fixed as it would be in an inert material with concepts such as tortuosity giving the misleading impression that interconnectivity between nanopores is persistent.
Transport of sorbate molecules through the dynamic nanoporous network within the matrix is therefore dependent upon molecular mobility of the matrix. Where the sorption behaviour is dominated by bulk diffusion, the principal model that has been used to describe sorption kinetics has been based upon Fickian diffusion. However, for materials that exhibit sorption-induced dimensional changes, it has been long realized that a simple Fickian diffusion model is inadequate to describe sorption kinetics; applying particularly to glassy polymer/penetrant systems, where deviations from Fickian behaviour are commonly noted [105]. This has led to the development of alternative models where the kinetics is defined as being relaxation limited and is dominated by the relatively slow viscous relaxation of the polymer matrix [106]. Models of this type consider the dimensions of the penetrant molecules, the interactions between penetrant and polymer (solvation) and the creation of free volume within the polymer matrix. Because of the dramatic change in behaviour observed with glassy polymers, the effect of penetrant molecules upon the Tg is of major importance [84, 107, 108].
Hydrogels
Hydrogels are three-dimensional crosslinked hydrophilic polymeric materials, which can absorb large amounts of water, with associated swelling of the structure, but without dissolution. Hydrogels consist of a region where crosslinking occurs between sections where there is high polymer chain mobility, plus chemical groups that interact with water molecules [109]. The sorption behaviour of non-ionic hydrogels is controlled by factors that control the swelling of the network structure, namely:
-
The hydrophilic properties of the polymer chain which affect the polymer-water mixing and the swelling of the network (represented by the F–H interaction parameter).
-
The elastic response of the crosslinked network (entropic in nature), which counteracts the swelling.
Although crosslinking can be achieved through the formation of non-covalently-bonded regions (known as physical hydrogels), these may not be stable when subjected to mechanical forces, and covalent linkages are usually required to ensure long-term stability, although metal ion-complexed crosslinks are usually hydrolytically-stable under neutral conditions [110]. A higher crosslink density results in an increase in mechanical strength and modulus, but a decrease in the degree of swelling and rate of diffusion of water molecules through the polymer network [20, 111,112,113,114,115]. The amount of water that hydrogels can hold is a function of the crosslink density and stability within the polymer network, as well as the nature of the hydrophilic groups on the polymer backbones [23].
The dipole–dipole interaction between the water molecules and the polymer network creates an osmotic pressure within the matrix that results in swelling when the hydrogel is exposed to water. When a hydrogel is exposed to moisture, it will exchange water molecules with its environment until it reaches an EMC; where there is a balance between the elastic strain stored in the polymer matrix and the osmotic pressure [116, 117]. The sorption process is viewed as not being one of molecules attaching to sorption sites on a surface (internal, or external), but rather analogous to a process of dissolution of the polymer chains.
Natural hydrogels are based upon polysaccharides (e.g. cellulose, starch, gels, carrageenan, alginates, dextran, pullulans, chitosan, chitin) [118], proteins [119,120,121,122,123,124] and polyphenols (e.g. lignin) [109, 125]. The most commonly used polymers for synthetic hydrogels are poly(vinyl alcohol), poly(lactic acid), poly(ethylene glycol|), poly(ethylene oxide), poly(acylic acid), poly(arylamide), poly(vinylpyrrolidone) and poly(caprolactone) [23]. This group of materials also includes nanocomposite hydrogels, which comprise inorganic and organic components, such as exfoliated clay platelets dispersed in a hydrogel matrix [126].
The swelling kinetics is either diffusion-limited or relaxation-limited. In the first case, the diffusion of water molecules through the polymer matrix occurs at a much faster rate than the relaxation of the polymer chains, and the rate of swelling is controlled by the concentration gradient. In the latter case, the rate of penetration of the water molecules is controlled by the relaxation of the polymer network.
A development of the F–H model is the Flory–Rehner (F–R) theory that describes the swelling behaviour of gel networks [116, 127]. The swelling of the polymer network is determined by the elastic energy of the polymer chains and a mixing function that is determined by the interaction between the water molecules and the polymer network (and hence the interaction parameter described earlier). The free energy of the hydrogel can therefore be expressed as:
where ΔGgel represents the contribution of the elastic strain energy and ΔGmix is the energy of mixing.
However, F–R theory does not completely describe the swelling behaviour of gels, and various modifications have therefore been proposed [128]. Furthermore, the F–H/F–R models are only able to describe the sorption behaviour of polymer solvent systems in the rubbery state (above the Tg). One characteristic of the sorption isotherm of glassy polymers is the presence of a pronounced ‘shoulder’ at low sorbate concentrations, which is not predicted by F–H theory. Attempts have been made to model this behaviour by making the interaction parameter (χ) adjustable, but such an approach is not really satisfactory, since it reduces χ to an arbitrary fitting parameter rather than a well-defined constant [38].
Many gels exhibit a sigmoidal isotherm curve (IUPAC Type II), for which a modification of the F–H/F–R model is required. One approach is to combine the F–H model with a polymer structural relaxation model [129]. Water molecules entering the polymer network can result in a plasticising effect, resulting in non-ideal volumetric changes. The result is that there is an additional free energy change beyond that predicted by the F–H model. Different models attempted to take account of this behaviour and explain the observed Type II isotherm by taking account of the free volume created by the sorbate molecules [33, 35, 130].
These free volume methods take account of the polymer being in a glassy state, which however is not an equilibrium state and, in principle, classical thermodynamic-based theories of sorption are not applicable [131]. However, it has been shown that free volume theories do have a physical basis, because the excess sorption found in the glassy state is related to the elastic energy stored in the polymer network [132]. During the process of absorption, the polymer network swells to accommodate the penetrant molecules resulting in changes in the configuration of the polymer network, which is stored as strain energy. This is an entropic property, since the configuration of the polymers has changed as a result of the presence of the penetrant molecules. The equilibrium point is reached when the energy of mixing and the elastic strain energy are balanced [133].
The properties of water absorbed in hydrogels are determined by the polymer–water interactions and by the geometry of the nanopores in the 3D structure. Different water states have been claimed to exist in hydrogels—‘bound’ water associated with sorption sites, ‘intermediate’ water and ‘free’ water with no specific association with the polymer chains [65, 134]. Molecular dynamic studies of hydrogels have shown that the structure of water is significantly modified in the region of the polymer chains [135]. Calorimetric measurements in a hydrogel of poly(2-hydroxyethylmethacrylate) indicate that water undergoes a glass to liquid transition in two stages, which was interpreted as water interacting with either the hydrophobic or hydrophilic segments of the polymer chains [136].
The interaction of moisture with wood and other lignocellulosic materials
The molecular components of plant cell wall lignocellulosic materials and their interaction with moisture
This subject area has been extensively covered in numerous reviews, e.g. [137,138,139,140,141], and only a brief introduction is included here, giving a generalized description. Lignification occurs in various plant cell types (tracheary elements, schlerenchyma cells, endodermal cells, seed coat cells and siliques cells) and can occur as a natural part of cell development or in response to biotic or abiotic stresses [142]. The discussion that follows and the main subject of the review is concerned with the tracheary elements, with an emphasis on wood products.
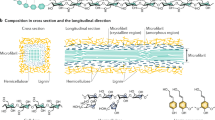
The properties of wood and other lignocellulosic materials are derived from the behaviour of the constituent molecules, and this behaviour is strongly influenced by the presence of water [143,144,145,146]. The influence that water has upon the molecular dynamics of the cell wall matrix molecules (lignin-hemicellulose) is particularly important. The cell wall of lignocellulosic materials comprises long thin microfibrils of crystalline cellulose embedded within a matrix of hemicelluloses in combination with lignin (Fig. 3). The structure and composition of the lignin and hemicelluloses vary within and between plant species with some plant fibres (e.g. cotton) being essentially lignin-free [147]. The majority of the OH content is associated with the polysaccharidic components of the cell wall. However, although the cellulose component has a high OH/C ratio, only a proportion of this OH content is accessible to water molecules, with the remainder being inaccessible because it is located in crystalline regions within the core of the microfibril [57, 148,149,150,151,152,153,154]. Because of the extended crystalline structure and extensive hydrogen bonding network within the microfibrils, they are very strong and stiff [155,156,157,158,159,160,161]. Since the interior of the microfibrils is not accessible to water molecules, they remain largely unaffected by the presence of moisture in the cell wall, although it has been shown that the crystalline lattice of cellulose is changed by the absorption and desorption of water [162, 163]. This observation has been attributed as being due to external pressure applied by the surrounding matrix material upon cell wall hydration [164]. However, Paajanen et al. [165] noted that that the crystalline lattice expands in the [200] direction (and becomes distorted) in the dry state due to interaction with the neighbouring fibrils and the matrix polysaccharides. In the swollen state, the cellulose crystals can adopt a higher degree of order with a smaller lattice spacing in the [200] direction. There therefore is no need to explain this phenomenon by invoking a swelling pressure and MD models produced the correct results without any restriction of swelling at high MC. It is difficult to treat each cell wall component in isolation, for example when determining properties, such as elastic modulus, although values for cellulose have been reported in the literature [166, 167].

A diagram showing the location of the cell wall within the wood structure and illustrating the different cell wall levels with a microscopic view of the microfibrils embedded within a matrix (composed of lignin and hemicelluloses) (a). In b, the effect of the presence of sorbed water molecules in the inter-microfibrillar matrix results in the expansion of the matrix (leaving the microfibrils unaffected). In the upper part of (b), the microfibrils are oriented in a longitudinal direction and this results in expansion in the transverse direction only.
By contrast with the cellulose, the hemicellulosic components have a lower crystalline content and consequently contain a much higher proportion of accessible OH content [168,169,170,171]. The lignin is an amorphous three-dimensional network polymer comprised of phenolic building blocks that has a much lower OH/C ratio compared with the polysaccharide content [172,173,174]. Simplistically, the structure of the cell wall can be viewed as similar to a fibre-reinforced composite, with the cellulose microfibrils being the reinforcing element, the lignin the matrix material, and the hemicelluloses acting as an interfacial coupling agent [139]. Reliably measuring the softening point of lignin and hemicellulose is extremely difficult, and although values of Tg for these components have been reported, these are expected to be different when determined in situ, rather than when determined for the isolated molecular species [166, 175,176,177,178,179]. There is not a single well-defined glass transition temperature of wood, due to its complex composition and structure, and this makes application of models invoking a relationship between sorption properties and Tg problematical [36]. A useful approach in this context is to study the same species but use genetic manipulation to change one of the molecular components and study the resulting change in properties. For example, a study of the effect of different lignin types on the thermal softening of transgenic aspen showed that a reduction in lignin content reduced the softening temperature, but that changing the syringyl/guaiacyl ratio did not affect the Tg [180].
The presence of water in the enveloping hemicellulose-lignin matrix has a significant influence upon molecular motion of the matrix molecules in a manner analogous to a water-gel polymeric network [181]. When dry wood absorbs moisture, this causes the structure to swell and if restraint is placed upon the wood sample, then a swelling pressure is generated. The absorption of water into the cell wall results in swelling of the cell wall matrix which creates additional void volume [182, 183], resulting in an increase in the spacing between adjacent microfibrils [184]. However, it must be noted that wood in its native (never-dried) state is already saturated with water and that macroscopic stresses arise when the wood is dried from this native state to a MC below the fibre saturation point [185]. These macroscopic strains can be annealed out by re-wetting the wood (often with the application of heat). These macroscopic stresses are not the same as the micromechanical stresses that arise in the macromolecular matrix when water molecules enter the structure giving rise to stored elastic strain energy which is released when water leaves the cell wall. The amount of water that is contained within the cell wall is determined by the thermodynamics of the system. When the bound water chemical potential is in equilibrium with the free water chemical potential, then a state of equilibrium is realized [186]. A recent paper has presented an alternative view of the interaction of water with wood and other semi-rigid swelling materials [187]. In this model, internal conformational strain is stored in the structure during the drying, with the swelling of the materials having an enthalpic origin. It is further pointed out in this paper that the relationship between the temperature-dependent isotherm, heat of swelling and swelling pressure/work of swelling is incompletely understood and needs further experimental investigation.
In terms of molecular mobility, the hemicelluloses are the component most affected by the presence of water in the cell wall; because the cellulose is constrained by extensive hydrogen bonding networks and the lignin by extensive crosslinking [139, 166, 188,189,190,191,192,193,194,195]. The interface between the surface of the cellulose microfibril and the hemicellulosic component has an important role to play when considering the properties of lignocellulosic materials under load in the presence of moisture [196, 197].
As a result of the presence of essentially hygro-inert microfibril elements embedded within a dynamic matrix, the orientation of the microfibrils gives rise to anisotropic behaviour. For example, the microfibril angle within the S-2 layer of the wood cell wall affects the swelling behaviour of the wood at a macroscopic level. In most types of wood, the microfibrillar winding angle is typically 10–30° relative to the longitudinal axis of the cell wall, which results in swelling in the transverse orientation being greater than in the longitudinal direction. In certain types of wood (such as compression wood in gymnosperms or juvenile wood), higher winding axes result in much greater transverse swelling [198, 199]. This is shown in Fig. 3.
However, usually the microfibrils are oriented at a finite angle to the longitudinal cell axis, which results in some expansion in the longitudinal as well as the transverse direction. This property is determined by the magnitude of the microfibril angle (Fig. 3).
An interesting approach to furthering understanding the interactions of moisture with lignocellulosic materials is to use molecular modelling [200]. For example, a study of the molecular dynamics of biopolymers considered the effect of moisture and heat upon the hygric swelling, thermal expansion and mechanical properties of lignin [201]. This study investigated an uncondensed lignin (a linear polymer) with a degree of polymerisation (DP) of 100 and consisting of five chains, as a model for the condensed (crosslinked) lignin found in the cell wall of plants. Water molecules were inserted into the model and effect on molecular dynamics investigated. A molecular modelling study which included all of the cell wall components found that the hygromechanical behaviour of the cell wall composite was not much affected by the lignin, which was found to be largely hydration independent [202]. The main problem with such an approach is the complexity of the molecular arrangements in the cell wall which requires simplification for the models to work. Furthermore, the exact molecular arrangements are not known, so when creating the models, it is necessary to base these on limited experimental evidence. The challenge is not to simplify to the point where the model no longer offers useful insights.
The sorption isotherm
The relationship of wood and other cellulosic materials with water vapour has been extensively studied for more than a century, yet remains incompletely understood. Before proceeding further, it is important to distinguish the difference between an isotherm obtained with previously-dried, compared to desorption of water-saturated lignocellulosic material (Fig. 4). It was originally thought that the boundary curve followed in the first desorption cycle from the ‘green’ state was unique and that subsequent desorption cycles would follow a different path. However, this has been shown to be erroneous and that subsequent desorption cycles will rejoin this boundary curve, although the exact behaviour depends strongly on the drying conditions employed after the first desorption cycle [203]. The isotherms discussed in this paper exclusively refer to the sorption behaviour of lignocellulosic material that has been previously-dried. The sorption isotherm of lignocellulosic materials is a characteristic sigmoidal IUPAC Type II curve exhibiting hysteresis between the absorption and desorption isotherm branches [204,205,206,207,208,209,210]. The properties of the desorption isotherm depend upon the point at which desorption is initiated and whether it is a scanning or boundary desorption isotherm curve [211, 212].

Sorption isotherm showing the first desorption from the water-saturated state and subsequent sorption cycles (lhs) and absolute hysteresis for these two cycles (adapted from [211]).
There are many sorption models that have been applied to experimental sorption isotherms of hygro-responsive natural materials. Models which treat the substrate as an inert porous material, with a defined internal surface are not appropriate [2, 204], but nonetheless have been extensively applied to sorption isotherms of wood and many other natural materials, foodstuffs, etc. [2, 3]. In models of this type, the internal surface area and monolayer versus multilayer coverage are parameters of interest but have no physical meaning when applied to dynamic systems. Many of these static material models either consider the substrate as a planar surface or an inert porous material, but take no account of the changes in dimension occurring when the material interacts with moisture. In the static pore models, hysteresis arises from consideration of mechanisms of pore filling and pore emptying, which is applicable to inert porous, but not hygro-responsive lignocellulosic or cellulosic materials. Although inert substrate models can apparently reproduce the experimental data, they do not provide any insights into the physical nature of sorption and desorption of natural materials (such as wood and plant fibres) that exhibit dimensional changes as a result. They certainly provide no information regarding how the mechanical properties are changed by the presence of moisture.
At a given temperature, a sample of wood will absorb or desorb water until it reaches a state of equilibrium, which can take a considerable amount of time, even for small (mg) samples [214, 215]. The equilibrium MC represents the point at which the MC at a particular RH remains constant and where the rates of absorption or desorption are the same [216]. Establishing these points experimentally can be very time-consuming and usually involves the setting of a weight change criterion to establish an approximate ‘equilibrium’ point. This usually involves a decision where the weight change over a period of time is no more than a small percentage of the ‘true equilibrium’ value [3]. This can be a particular problem with using dynamic sorption equipment if only one sample is tested per sorption run, since the weight change criteria necessary to establish an ‘equilibrium’ value can lead to extremely long time periods (months) being required to establish one isotherm. In addition, it is recommended that a minimum of two and preferably three isotherm runs be performed on the same sample consecutively in order to establish reproducibility of the data [217]. Furthermore, several samples of the same material should also be measured to establish reproducibility. This makes production of isotherm data logistically impractical with single sample devices. Glass et al. [214] have identified slow sorption processes with characteristic times of the order of 500–2000 min that can only be identified if hold times of the order of 24–50 h are used for each RH step. The question of whether the more logistically practical equilibrium criteria can be used in comparative studies remains open.
However, even when great care is taken to establish the EMC, this state is not considered a true thermodynamic equilibrium because there is hysteresis between the absorption and desorption branches. This does not mean that a ‘true’ equilibrium would be established if the experiment was run for a sufficient length of time, since the differences between the absorption and desorption branches of the isotherm are not related to the time of exposure in the experiment. Most sorption isotherms are recorded up to a RH of 95% since there is an increasing likelihood of capillary condensation of water within the macropore structure of the cell wall and difficulties in maintaining an accurate stable RH much above 95% RH [211]. There have been attempts to project the absorption isotherm curve up to a value of 100% RH and use this cross-over point to estimate a fibre saturation point (FSP). This point may, or may not, coincide with a region where there is a change in physical wood properties with cell wall MC [218,219,220]. Thybring et al. point out that FSP (determined by changes in wood properties with cell wall MC) should not be confused with maximum cell wall MC, for which values depend upon the measurement method employed [70].
Sorption hysteresis
Sorption hysteresis is the property where the sorption behaviour of the material is different, depending upon whether the MC is increasing or decreasing. For the purposes of this review, only hysteresis within the hygroscopic range (from 0% to approximately 98% RH) is considered here [211, 213]. Water sorption within the hygroscopic range occurs in the cell wall, and sorption behaviour is linked to the mechanical response of the cell wall (and hence macromolecular dynamics). Although universally observed in the sorption isotherms of lignocellulosic materials (as well as other hygro-responsive materials), there is still much discussion regarding the origins of hysteresis. Some general observations are here made about sorption hysteresis in lignocellulosic materials:
-
Sorption hysteresis is generally determined by subtracting the absorption EMC from the desorption EMC at the same RH value—this is referred to as absolute hysteresis (simply referred to as hysteresis in this review) (Fig. 4). An alternative approach is to divide the desorption EMC by the absorption EMC to provide the hysteresis ratio – not referred to any further in this paper.
-
Empirical observations have shown that the absolute hysteresis is dependent upon the lignin content of the sample—with materials having a larger lignin content displaying a higher hysteresis value [221].
-
Thermally modified wood shows a lower level of moisture uptake throughout the isotherm overall, but the hysteresis is greater than that observed with unmodified wood of the same species [139, 222]. However, this only applies to the first sorption cycle; in subsequent cycles, the sorption hysteresis is considerably reduced. This behaviour has been attributed to changes in cell wall mechanical properties.
-
The area bounded by the sorption hysteresis loop decreases as the isotherm temperature is raised [221, 223,224,225,226,227].
-
Sorption hysteresis is history dependent, with the previous sorption history of the sample affecting the behaviour [49, 204, 217, 228, 229].
Early attempts to explain sorption hysteresis were based upon models that assumed that the availability of sorption sites was different during absorption and desorption cycles [230] or by considering the sorption behaviour of porous materials [231]. Models of this type treat the cell wall substance as an essentially inert porous material with hysteresis explained by considering the physics of pore filling and emptying [221]. This model considers both film-forming in pores, which does not exhibit hysteretic behaviour and capillary condensation, which does. Independent domain theory has been applied to this underlying theory to model a series of scanning desorption curves, which can, in turn, be used to create a desorption boundary curve [232,233,234,235,236]. The modified Mualem model [237] is a simplification of the independent domain model in which no interaction between pores is considered and where the adsorption and desorption of the pore system are only determined by the pore necks and pore bodies. This model requires the use of an adjustable parameter which is determined from a selected calibration point on the desorption curve, with good fits to the desorption curve obtained using this process [237]. However, the use of models which treat the cell wall as a static porous network do not offer useful insights into the behaviour of dynamic swelling systems, even less so if the model relies on arbitrary fitting parameters. The independent domain model can be applied to populations of sorption sites [235], but the details of the interaction of water with these sites are not considered in the model. The model is concerned with the interaction of a material with a sorbate where the sorption steps are finite, with the sorption sites being in one of two possible states [238, 239]. An alternative approach is to model the number of excess absorption sites which are postulated to exist during the desorption process [240]. Why extra sites would be generated at the instant of desorption is hard to explain through any known physical process (similar to the problem with applying the dual-mode model). Invoking the state or number of sorption sites as an explanation of hysteresis also does not provide insights into the links with dynamic swelling behaviour and molecular motion within the cell wall. In an interesting development, the relationship between swelling behaviour and sorption hysteresis was explored, in particular by examining differences in hydrogen bond networks formed during absorption and desorption cycles of the isotherm [241]. This model was further refined to include the effect of cellulose nanocrystal elements embedded within an amorphous matrix, the presence of which limited the swelling of the matrix, leading to a reduction in sorption and hysteresis [242], a result that has been experimentally observed [243].
There is a considerable body of evidence indicating that sorption hysteresis in hygro-responsive materials has to be linked in some way to the molecular mobility behaviour of the cell wall polymer network and this approach has been used to explain hysteresis in various water–polymer systems [41, 221, 225, 243,244,245,246,247]. According to many such models, the phenomenon of hysteresis is observed in glassy polymers, where the response of the polymer matrix to the ingress or egress of sorbate molecules is not instantaneous, but limited, due to a lack of void volume resulting in a restriction in molecular motion. This results in the matrix being in different configurations during absorption and desorption. Plasticization of the matrix by sorbate molecules will lead to a reduction in hysteresis, as will an increase in isotherm temperature. According to such a model, a collapse of sorption hysteresis will be observed when the Tg of the matrix is exceeded. The idea that hysteresis is linked to plastic deformation of the cell wall, which can be considered an analogue of a gel-like polymer system, is certainly not new and was first proposed by Barkas as far back as the 1930s [45, 183, 248,249,250,251].
Noting that dry, native cellulose decomposes before reaching its Tg [252], Salmén and Larsson used a modified cellulose polymer which exhibited a moisture-induced softening within the temperature range of 20–65°C [225]. By studying this material using humidity scanning dynamic mechanical analysis (DMA) and conducting sorption experiments, at different temperatures, they were able to determine the relationship between Tg and hysteresis of the isothermal sorption loops. This work showed that there was such a relationship and, furthermore, that sorption hysteresis vanished above the glass transition temperature, in agreement with previous work reported for glucomannan [243] and more recently for other water-polymer systems [41, 245]. This explanation for hysteresis is able to explain the disappearance of hysteresis above Tg, or where the moisture levels are sufficient to change the state of the polymer network from glassy to rubbery, but there are examples of cellulosic systems where the hysteresis is mainly in the higher part of the hygroscopic range, but essentially absent at lower moisture contents [253, 254]. It is well known that the sorption behaviour of lignocellulosic materials is influenced by the previous sorption history of the material, and it is for this reason that the ‘gripped-box’ model was introduced to describe sorption hysteresis [228]; however, this model is empirical and does not relate to any of the physical properties of the material. It should be noted that at very high levels of relative humidity (above 95%), an additional hysteresis phenomenon is observed, which has been thoroughly discussed elsewhere [213, 255] and will not be considered further in this review.
Sorption sites
The primary sorption sites in wood are the OH groups associated with the cell wall polymers (cellulose, hemicellulose and lignin), plus some ionic groups [26]. The main method for determining the concentration of accessible OH groups in wood and cellulosic materials is to use deuterium (2H, or D) exchange with D2O; a subject that has been thoroughly reviewed [204, 256]. Dynamic vapour sorption (DVS) equipment of various types has proven to be very useful for these deuterium exchange experiments, but it is essential to use the correct protocols in order to obtain reliable results [48]. By the use of such studies in wood samples, it has been found that accessible OH content is not affected by the MC of the sample, but that there was a low concentration of OD groups persisting after deuterium exchange and subsequent re-protonation [257]. This latter observation is probably linked to the existence of both fast and slow exchange processes. The slower process is affected by many experimental variables, such as sample geometry, deuteration conditions, RH and temperature; but the crystalline core of the microfibrils remains resistant to these exchange processes [258]. In Fig. 5 below, a comparison of the number of water molecules per OH group (‘sorption site’) is shown, based upon the Z–L model, or based upon the assumption that there are 9 mmol of accessible OH groups per gram of dry wood material. Also shown is an isotherm from which these data were derived. It can be seen that the number of water molecules per accessible OH group or mean cluster size does not exceed 2 water molecules even at 95% RH.

A comparison of the sorption isotherm with mean cluster size (MCS, determined using the Zimm–Lundberg theory) and the ratio of sorbed water molecules divided by the accessible hydroxyl groups (H2O/OH) (assuming 9.0 mmol accessible OH groups per dry gram of wood).
Water in the cell wall of lignocellulosic materials is usually considered to either be directly hydrogen bonded to the primary sorption sites or hydrogen bonded to other water molecules and is thus considered to form two populations within the cell wall. This idea was supported by the observation in DSC scans that there were both freezing and non-freezing water populations. However, as was noted earlier in this review with previous work on polysaccharide gels [27], it has been shown that the presence of freezing and non-freezing water in the wood cell wall is not necessarily due to distinct water populations but may be an experimental artefact [204, 259]. Experimental techniques, such as NMR and inelastic neutron scattering, have been used as evidence for identifying different water populations, as is discussed later. There has been some analysis of water clustering in the wood cell wall using the Z–L model [260,261,262,263,264,265,266,267,268,269]; which could possibly be interpreted in terms of different water populations (i.e. directly H-bonded to polymeric OH groups and indirectly-bonded via an intermediary water molecule), but the applicability of the Z–L model for such purposes is questionable. The issue of whether water tends to form clusters around sorption sites or is more evenly distributed is an important one and continues to elicit interest [6, 260, 261, 263, 266, 270, 271]. Even if such separate populations do exist, it is possible that the dynamics of exchange between these populations is over such short time scales that they cannot be resolved.
Various estimates can be found for the total accessible OH content of wood samples, and it is essential to be aware that some experiments are compromised because of the application of inappropriate experimental protocols, but values in the region of 8.5–9.5 mmol g−1 appear to be representative [257, 272]. Various estimates can be made for the saturation level of water in the wood cell wall, which typically varies between 20–50% by mass, depending upon the method used for measurement (higher values have been reported) [70]. These data allow for a very crude estimation of the number of water molecules per polymeric OH site in wood (between 1.2 and 3.3 water molecules per OH group at cell wall saturation): Berthold et al. estimated this to be 1–2 water molecules per OH site at 92% RH [26]. This is an average value and assumes an even distribution of water molecules throughout the matrix in the manner of a solvent. Obviously, as the MC of the cell wall reduces, so does the ratio of water molecules to accessible OH groups, with the possibility of water molecules bridging adjacent OH sites at lower MCs.
Hygro-responsive behaviour
Dimensional changes occur in response to variation in the cell wall MC of the lignocellulosic material. With an increase in cell wall MC, swelling occurs in the lignin–hemicellulose matrix, resulting in an increase in the distance between the cellulose microfibrils [273, 274] and some disaggregation of supramolecular microfibrillar structures [275], leading to macroscopic dimensional changes which are controlled by microfibrillar orientation [167, 276, 277] and accompanied by an increase of the water diffusion coefficient in the surrounding matrix [165, 182, 274, 278]. These dimensional changes in the cell wall couple to produce visible anisotropic changes in the shape of macroscopic specimens in a complex way [1, 186, 279, 280] manifested as swelling/shrinking of the samples and the development of macroscopic stresses if the material is restrained [281,282,283]. The main cell wall layer that is associated with dimensional changes in wood is the S-2 layer, with the microfibrillar orientation of the S-2 layer having a major influence on the anisotropy of swelling [284], although other cell wall layers and the surrounding wood substance do influence this behaviour [285,286,287].
When attempting to model the interaction of moisture with swelling materials, such as wood, the molecular dynamics of the interfibrillar matrix in the presence of moisture must be considered [107]. If a gel model for the hygro-responsive behaviour of the cell wall is adopted, then the equilibrium established during absorption/desorption is linked to the elastic strain energy stored in the matrix and the relationship of this to the activity of the water at different RH levels [45]. However, the gel behaviour of the cell wall is, in reality, restricted to the hemicellulose/lignin matrix, and the global behaviour is better represented by consideration of an inert reinforcement (the microfibrils) embedded within the matrix [288] and models for such composite morphologies have been developed [289]. The presence of moisture-inert reinforcing elements (microfibrils) within the hygro-dynamic gel-like matrix gives rise to anisotropic dimensional changes in response to changing RH [284]. Many other materials in nature also employ a composite structure with fibres, rods or sheets embedded in a gel-like matrix to impart properties such as toughness or specific geometrical changes [200].
In lignocellulosic materials, the mobility of the molecular chains of the lignin–hemicellulose matrix is an important factor that must be considered when modelling sorption and diffusion behaviour [107, 246, 290] and, as noted, also applies to the phenomenon of hysteresis [221, 225, 243, 246, 291]. Although there is still much debate regarding the dynamic response of wood and other lignocellulosic materials to changes in RH of the surroundings, there does appear to be the development of a consensus that some sort of relaxation-limited model is the best approach [107].
It is very well known that the mechanical properties of wood change with variation in cell wall MC. There is a reduction in stiffness and strength, but an increase in toughness as the MC of the wood is raised [198, 292, 293]. These properties change up to a nominal FSP but remain approximately constant thereafter; although this correlation is not as simple as is sometimes assumed [70, 294]. Carrington noted that the mechanical properties of wood change at approximately 5% MC [295], an observation subsequently confirmed by others [296,297,298]. The reasons for this are not fully explained, but it is assumed that at lower cell wall MC, there is some disruption of H-bonding within the polymer network and that at higher cell wall MC plasticization of the polymer network occurs [299]. A recent review of the DMA literature has shown that there is distinct minimum in tan − δ at 5% MC in almost all studies for measurements in the longitudinal direction [300]. That review is comprehensive, and the reader is directed to this reference for further analysis and discussion regarding the use of DMA for investigating the interaction of moisture with wood and the effect on dynamic mechanical behaviour. Useful DMA experiments for studying the interaction of moisture with natural materials should vary the surrounding RH and keep the temperature constant (as with isotherms). Unfortunately, DMA studies that only vary the temperature are of little to no value for understanding the hygro-dynamic behaviour of wood.
Mechanosorption is a term that is used to define the mechanical properties of a material when it is simultaneously exposed to atmospheric moisture and is concerned with the time-dependent properties. It is also observed that the sorption behaviour of wood is affected by any external mechanical forces that may be applied [301, 302]. It is generally assumed that mechanosorptive behaviour has two components. One is an instantaneous phenomenon which is linked to the mechanical response of wood during drying or wetting when it is under load. The second is a delayed effect, which is referred to as mechanosorptive creep and which is accelerated under conditions of RH change [303,304,305,306,307]. It is known that wood and other lignocellulosic materials, such as natural fibres [308], will exhibit creep when subjected to constant load and that creep is magnified if the load is applied under conditions of changing RH [309, 310]. Partial creep recovery occurs if a wood sample is subject to a static load and then remoistened [311]. The mechanosorptive creep effect can result in premature failure of wood samples depending on the rate of loading [312]. It has been observed that the magnitude of deformation occurring due to mechanosorptive creep is dependent upon the rate at which the MC of the wood changes, with much larger deformations occurring in environments which exhibit corresponding rapid changes in RH [306]. Most studies of wood behaviour examine properties either under conditions of equilibrium or constant RH, but wood in service is subjected to fluctuating environmental conditions, meaning that dynamic experiments are of great interest [223]. When water molecules enter the cell wall of wood, some of the hydrogen bonds associated with the matrix or matrix-microfibril interface are broken and replaced by water-OH bonds, resulting in a decrease in stiffness of the cell wall [196]. This is linked with a decrease in the energy barrier associated with the breaking and reforming of hydrogen bonds and facilitates viscoelastic creep under an external load. Mechanosorptive creep results in the so called ‘hygro-lock’ effect which occurs when wood is dried under load and which locks in distortion of the material. This can be reversed during a subsequent moistening period. It has be postulated that this permanent deformation effect is related to the ‘stick–slip’ mechanism of breaking and reforming of hydrogen bonding networks within the polymer structure [276, 313,314,315,316,317,318,319,320,321,322,323]. This hydrogen bond breaking is facilitated by the presence of interstitial water between the surface of the microfibrils and the matrix polymers in the hydrated cell wall [140].
Cell wall nanoporosity
The cell wall of wood is essentially non-porous in the dry state [324,325,326,327,328], in common with other lignified materials [329,330,331,332], but porosity is generated as water molecules enter the cell wall. This transient porosity evolves as the MC of the cell wall changes. It is problematical to image or measure this porosity, since many potential techniques require the samples to be dry [324]. Much of the earlier work used solute exclusion to determine the accessible microporosity of the water-saturated cell wall [324]. More recently, dual-axis electron tomography has been used to probe the architecture of the S-2 layer of the wood cell wall of never-dried Norway spruce at nanometre scale [333]. However, this experiment required mild-delignification prior to heavy metal staining. This technique allowed for resolution of the cell wall polymers and the micropore geometry, the latter having diameters predominantly below 3 nm. It is not clear how representative this geometry is of a water-saturated lignified cell wall, but diameters of this order are often reported using other techniques [324]. Another physical method to resolve cell wall microporosity uses silicon nanocasts [334]. This technique involves the penetration of swollen wood samples with a silica sol–gel, followed by calcination in a furnace to remove all the organic material, producing a negative cast of the wood, which was then examined using scanning electron microscopy and nitrogen sorption. Techniques of this type can be used to prove the existence of cell wall nanopores but cannot give information about pore dynamics.
A variety of techniques have been applied to investigating the nanoporosity of the cell wall, including: atomic force microscopy [335], NMR [336, 337], neutron scattering [338], fluorescent probes [339], nitrogen sorption [340], krypton sorption [326] and solute exclusion [341]. Thermoporosimetry is based on the observation that water-saturated porous materials (e.g. wood, pulp fibres) contain both freezing and non-freezing water, with the latter thought to be associated with water located in the cell wall nanopores [68]. This has led to the development of isothermal measurement methods to determine the amounts of freezing and non-freezing water in cellulosic samples [342]. However, differential scanning thermoporosimetry is claimed to give a more accurate measurement of pore size distribution compared to isothermal methods [71]. Note earlier comments regarding the interpretation of freezing and non-freezing water.
Sorption kinetics
The rate at which lignocellulosic materials take up, or lose, moisture depends on a range of factors:
-
Sample size—it is self-evident that the surface of the sample being directly exposed to the atmosphere will respond much faster to changes in RH compared to the interior of the sample [343]. However, even with small samples, moisture transport must be considered [344, 345].
-
Orientation of the sample—in thin samples, the orientation of the sample is important and affects the sorption kinetics [344].
-
Temperature—increased temperature leads to a higher rate of penetration of the water molecules in the sample.
-
Concentration gradient—the rate at which the sample changes mass depends upon the difference in concentration between the penetrant in the sample and in the surrounding environment.
The issue with any sorption kinetic experiments of lignocellulosic materials has been obtaining sufficiently accurate data to allow for the fitting of robust sorption models. An example of the problems encountered is illustrated by the debate regarding the use of curve fitting to data obtained using DVS. Early work on the use of curve-fitting software in combination with DVS data for flax fibres showed that the experimental data were well described by using a model having two exponential relaxation terms (the parallel exponential kinetics, PEK model) [346]. Unfortunately, the data obtained from the DVS are compromised by the finite time that it takes the instrument to step change from one RH value to another and with potential stability problems of the balance over long experimental time periods [216]. Furthermore, the arbitrary setting of ‘equilibrium’ criteria has a profound effect upon the parameters obtained from the curve fits to the double exponential model [347]. Curve fitting is highly sensitive to these factors. Rather than attempting to fit the sorption data to a pre-conceived model, an alternative approach is to fit the experimental points using a model that comprises a spectrum of exponential functions [216, 347], as is done for low-field NMR relaxometry experiments [348]. In this way, no a priori assumptions are made regarding the number of decay functions present, and more robust fitting to the data is to be expected. Unfortunately, the data quality is severely compromised by the aforementioned finite time period taken to change from one RH state to another, unlike the experimental conditions pertaining in the low-field NMR experiments, where the magnetic field is essentially instantaneously removed, allowing for unambiguous recording of the decay events [349]. The reliability of the experimental data is not of sufficient quality to allow for unambiguous repeatable parameters to be obtained in any curve-fitting exercise. This will apply to any kinetic experiment involving a change in RH in a chamber containing a sample of finite size, since the change cannot be instantaneous and the results will in any case also be strongly influenced by sample size/geometry. Whether these issues are resolvable, it remains to be seen.
Biomimetic hygro-responsive materials
The purpose of a moisture-activated material is to convert the chemical potential energy in atmospheric water vapour into mechanical work [350]. This is an area which has received a tremendous amount of research attention over the past decade and been the subject of numerous reviews. Humidity-responsive behaviour is widely encountered in nature, with examples such a pine cone flakes and wheat awns, often quoted as examples [162, 277, 351,352,353,354,355,356]. Based upon these observations, there has been much interest in developing bespoke hygro-responsive materials using biomimetic principles, with the response fine-tuned by the combination of materials which have different moisture-responsive properties [354, 357,358,359,360,361,362,363,364,365]. There have also been moisture-activated actuators constructed using natural fibres [366, 367], cellulose [354, 357, 368, 369] and lignin [370].
Homogeneous hydrogel materials expand isotropically when exposed to moisture, whereas in bilayer hygro-responsive materials, one of the layers (the active layer) changes its volume reversibly or irreversibly when exposed to moisture, whereas the other layer (the passive layer) does not respond. On exposure to moisture, a strain is developed at the interface between the two layers (called the activation strain), which results in deformation of the material. A strong bond is required at the interface of the two materials to prevent delamination, and the passive layer needs to be flexible to allow for deformation to occur. Apart from a simple bilayer structure, it is also possible to produce patterned hydrogel materials which allow for programmable deformation [371]. One method to obtain patterned structures is to use photolithography to control crosslink density in one of the polymer layers. The extent of swelling in the presence of water is reduced as the crosslink density increases. In addition to using bilayer approaches to producing deformable materials, it is possible to use a single homogeneous gel in which the properties are tuned, for example by controlling the crosslink density, polymer composition or some other material property [372]. Controlled deformation can also be achieved by using molecular organization of the hydrogel material, by, for example, using liquid crystal structures/self-assembly and stimulus-responsive materials to created hygro-responsive materials [373, 374].
Many natural materials have complex 3D geometries which produce shape changing behaviour and a major challenge when constructing hygro-responsive materials with tuneable properties is to ensure that a desired aligned structure is achieved. One method for obtaining programmed deformation is to produce materials containing oriented rigid high aspect ratio particles (platelets or fibres). Anisotropy in swelling behaviour can be achieved by using shear alignment of microfibrils in materials, by using 3D printing [371, 375]. Rather than embed non-responsive materials in a matrix, controlled swelling behaviour in hydrogels can be achieved by producing variations in crosslink density [376]. Similar effects can be achieved by using different levels of substitution of the OH groups of cellulose to create a bilayer film [377]. The use of 3D printing technology to create structures to produce a desired shape transformation when exposed to moisture is a technique that is referred to as 4D printing [378,379,380,381,382,383,384,385,386,387]. Applications for such structures include biomedical [388, 389], energy [390], smart textiles [361], biomimetic building skins [391], moisture-activated motors [392, 393], activators [371, 394,395,396] sensors [397], shape memory and nanocomposite structures [398], self-folding/rolling materials [394], nacre-biomimetic structures [399, 400] and programmable deformation [371, 401] (Fig. 6).

Examples of design principles for hygro-responsive materials: a example of a bi-component material where the upper layer is hygro-responsive and the lower layer is non-responsive due to crosslinking between adjacent polymers, b the bi-component material is created by the chemical substitution of active chemical groups in the lower layer (e.g. acetylation of OH groups), and c programmed behaviour obtained by targeted crosslinking of the polymer matrix.
Apart from the design of such materials being inspired by natural moisture-activated structures, the design of these materials also allows for the possibility of providing insights into the moisture sorption behaviour of lignocellulosics. For example, controlling the crosslink density of a hydrogel will affect its Tg, and this would be expected to also influence the onset of hysteresis. It is possible to change the interaction between the surface of a microfibril and hydrogel matrix by chemical substitution and/or introducing chemical crosslinks, which would affect creep behaviour.
Instrumental methods to determine the interaction of water with natural materials at a molecular scale
The presence of water in natural materials can lead to various responses at different length scales. Water in the cell wall of wood can reduce the energy barriers to molecular relaxation processes, giving rise to viscoelastic behaviour such as creep. Despite the importance of moisture in determining the physical properties of many natural materials, our understanding of the interaction of moisture with these materials remains incomplete. A few instrumental techniques are available to study the interaction of moisture with natural materials at a molecular level, including vibration spectroscopy (infra-red (IR), Raman), nuclear magnetic resonance (NMR), neutron or X-ray scattering and diffraction and dielectric relaxation spectroscopy (DRS). This section reviews these techniques in detail for the purposes of improving our understanding of the interaction of water with wood at the cell wall scale. The use of these techniques to investigate the structure of wood at a molecular level has been reviewed elsewhere [138].
Vibrational spectroscopy
Vibrational spectroscopy probes molecular vibrations at different frequencies in order to understand chemical properties based upon characteristic frequencies. These characteristic frequencies depend upon bond strength and atomic masses. Vibrational spectra are obtained with two main techniques, infra-red (IR) and Raman spectroscopy, which are based on different physical mechanisms. In IR spectroscopy, the molecules within materials are exposed to a polychromatic light source and those photons that have energies that match the gap between the ground state and an excited vibrational state are absorbed. In contrast, Raman spectroscopy is based on the molecule’s inelastic scattering of monochromatic light, which shifts the frequency of the scattered light by the frequency of the molecular vibration. The vibrational transitions probed by Raman and IR spectroscopy are the same or different depending on the molecule, and the two techniques often provide complementary information [402]. Bonds with a strong dipole moment (such as OH bonds in water) are IR active, but the polarizability is usually low, which results in a weak Raman signal [403].
The molecular vibrations of water have been widely used to study the sorption of water in biomaterials [404,405,406,407,408,409]. Covalent bonds in liquid water, for example, result in absorbance bands at ca. 3400 cm−1 (OH stretching vibrations) and 1650 cm−1 (HOH bending vibrations) within the mid-IR spectrum, which correspond to overtone and combination bands in the near-IR spectrum at ca. 5200, 7000 and 8500 cm−1 [410, 411]. The molecular vibrations of water allow the quantification of MC in materials based on band area changes [412,413,414] or multivariate calibration models [415,416,417]. Vibrational spectroscopy is also sensitive to changes in the hydrogen-bonded structure of water [410, 411, 418], which has been used to differentiate between different states of water molecules in the wood [405]. A shift in the OH stretching vibrations can also be induced by the deuteration of materials. Deuteration exchanges the protium (1H) for deuterium (2H) in functional groups that form hydrogen bonds with deuterium oxide. The band shift that results from the mass increase of the vibrating hydrogen has been used to study the water accessibility of hydroxyls in wood and cellulose [55, 409, 419,420,421,422]. However, dissimilar molar absorptivities of 1HO and 2HO stretching vibrations [272, 423, 424] and a proportion of non-exchanging hydroxyls in cellulosic materials [149] may affect the quantification of accessible hydroxyls.
With the advancement of instrumentation, molecular vibrations can even be followed in spatial dimensions, which offers new insights into the moisture distribution within biomaterials. Hyperspectral images can be collected with different imaging setups and acquisition modes, but they have in common that each pixel contains a continuous spectrum [425]. The collected image data are arranged into a three-dimensional hypercube, with one spectral dimension and two in-plane spatial dimensions. The spatial resolution is ultimately determined by the diffraction limit, which is dependent on the wavelength.
Near-IR imaging offers a lower spatial resolution than mid-IR or Raman micro-spectroscopy. When collected with modern push broom (line-scanning) instruments, however, near-IR hyperspectral images are obtained fast and with minimal sample preparation [426]. Such instruments have been applied successfully to map the MC across wood surfaces. The prediction of the MC in individual pixels requires a calibration model, which is typically built from gravimetrically measured MCs and spectra averaged over the corresponding samples based on partial least square (PLS) regression. The approach has been applied successfully to map MC variations during air drying of water-soaked wood [427,428,429] or after conditioning of untreated, thermally modified or acetylated wood between 0–95% RH [430, 431]. This revealed the spatially resolved variation in MC, for example between outer edges and central parts of the sample or between early and latewood regions. In addition to quantitative variations, principal component analysis (PCA) applied to near-IR hyperspectral images classified different hydrogen-bonded structures of free and bound water across wood surfaces during drying [429]. However, near-IR hyperspectral imaging has a limited penetration depth and thus maps the MC only at the surface. Furthermore, the prediction of MC or other properties based on near-IR spectra measured in reflectance mode is influenced by surface roughness [432, 433].
Vibrational micro-spectroscopy based on mid-IR or Raman spectroscopy requires more sample preparation but allows a lateral resolution at the micron scale. These techniques are typically applied to thin wood or plant sections to follow the chemical composition in relation to the microstructure [403]. Vibrational micro-spectroscopy has been applied to study local variations of moisture-related spectral bands within the hygroscopic range for wood [413], cellulose nanofibers [434] or lignin [435]. In these studies, a sample cell controlled the relative humidity and enabled the acquisition of mid-IR or Raman spectra from selected areas. Mid-IR micro-spectroscopy allows a lateral resolution of ca. 5–20 µm [403]; hence, variation in water-related bands between different wood cell walls could be studied [413]. Confocal Raman micro-spectroscopy even achieves a sub-micron resolution [403], and this has offered insights into the impact of absorbed water in distinct regions within the cell walls and the compound middle lamella of wood [413]. Mid-IR imaging with micrometre spatial resolution has already been applied to quantify the moisture distribution in cellulose nanofiber films [436]. A localization of absorbed water at a sub-micron resolution was recently achieved by confocal Raman mapping of untreated and acetylated Norway spruce cell walls [437]. The study used deuterium oxide to condition the samples, and thus, absorbed water and water-accessible hydroxyls caused an increase of the 2HO stretching band at 2300–2685 cm−1 and not the 1HO stretching band at 3100–3700 cm−1. Thereby, the study could also distinguish between water-accessible and -inaccessible hydroxyl groups within the wood cell wall.
In addition to the linear spectroscopy methods, there are nonlinear vibrational spectroscopy methods that are less frequently used. Sum frequency generation (SFG) spectroscopy can distinguish the signal of crystalline cellulose from the other fibre wall components and has been used to detect irreversible changes caused by drying in plant primary cell walls [438]. It also provides information on the arrangements of cellulose crystals and their changes during drying and rehydration procedures [439]. Nonlinear vibrational spectroscopy can also be applied for hyperspectral imaging at a high spatial and temporal resolution [440]. This has been used for the time-resolved imaging of water diffusion into different materials based on coherent anti-Stokes Raman scattering (CARS) microscopy [441,442,443].
NMR spectroscopy
Nuclear magnetic resonance (NMR) spectroscopy covers multiple techniques, in which the alignment of nuclear spins in a constant magnetic field is perturbed by an oscillating field that resonates with the magnetic states of the nuclei at specific frequencies. The resonance frequencies depend on the strength of the static magnetic field, the isotope of observation and its chemical environment. The most common nuclei observed by NMR spectroscopy are 1H and 13C. Different NMR techniques yield different information on water in natural materials and the molecular structure of the materials themselves [444,445,446].
Proton NMR relaxometry, a low-field NMR technique in the time-domain, uses radio-frequency pulses to perturb the alignment of magnetic spins of 1H nuclei and measures their time-dependent relaxation. Such experiments can detect especially the signal from water, which is affected by the local environment of the molecules. By utilizing two-dimensional analysis with correlation mapping to resolve overlapping peaks, under some simplifying assumptions, the method can distinguish between different binding states of water in wood, from ‘free’ water to ‘bound’ water, based on the mobility of the molecules [447,448,449]. Also determining the FSP is possible [70]. A further extension of the method by cryoporometry can provide information on the size of nanopores in which the water resides [450, 451]. Furthermore, pulsed field gradient (PFG) NMR has been used to study the self-diffusion of water for instance in paper and wood, including its dependence on the MC [102, 452]. For diffusion experiments, pulsed field-gradient stimulated spin echo (PFG STE) NMR is used, but results obtained from such a technique can be influenced by cross relaxation between the water and macromolecular matrix [68].
Magnetic resonance imaging, a time-domain 1H NMR technique with additional gradient magnetic fields, can detect spatial information with a typical resolution of around 1 mm [446]. It has been used to study the spatial distribution of water in wood samples and its changes with water uptake and drying [453, 454]. An analysis based on relaxation times allows separation of water molecules with different mobility, leading for instance to observations of the drying kinetics of free and bound water separately [455].
The nuclear spin interactions depend on spatial direction, but this effect is cancelled out in liquid-state NMR by the random motion of the molecules. In solid samples, on the other hand, line broadening caused by the anisotropic interactions is reduced by using the magic-angle spinning (MAS) technique. Especially 13C cross-polarization (CP), MAS has been widely used to study the molecular conformations in cellulose crystals, allowing the characterization of different levels of crystalline order and the degree of cellulose crystallinity [444]. Moisture-related changes in these conformations and the proximity of different molecular groups to water have been observed in wood, pulp and various other cellulosic samples [140, 456, 457]. Spin diffusion experiments also yield information on the spatial proximity of plant cell wall polysaccharides and water [140, 458]. Utilizing the deuteration of hydroxyl groups, 2H MAS NMR was able to differentiate between two populations (mobile and immobile) of bound water in microcrystalline cellulose [459].
X-ray computed tomography
Although this and some of the other techniques presented in this part of the paper are not used to resolve the interaction of water with wood and other plant materials at a molecular level, they do nonetheless provide unique information regarding the behaviour of the cell wall when absorbing and desorbing moisture and are included for this reason. The aim of computed tomography (CT) is to obtain a three-dimensional representation of the internal structure of an object in a non-destructive manner. This three-dimensional representation is reconstructed from two-dimensional projections (radiographs) that are taken from different angles by rotating a sample over a defined axis. Ionizing electromagnetic radiation (X-rays or gamma-rays) has sufficient energy to penetrate the lignocellulosic cell wall material and to interact with its microscopic structures in different ways. In the standard X-ray tomography method and absorption tomography, the tomographic images produced with X-rays show variation in the attenuation coefficient, which is generally proportional to the mass density of the material. When assuming the oven-dry cell wall density to be constant, density variations shown in the tomographic images are due to the anatomic structure and the water content in cell walls and lumens [460, 461]. More advanced X-ray CT methods are also available especially at synchrotrons, including phase-contrast imaging and other techniques that take advantage of coherent X-ray beams and can provide better contrast for samples of biological origin [462].
X-ray CT scanners have been used to analyse the water distribution in tree trunks [463, 464] and timber boards during kiln-drying [465, 466] or capillary water uptake [467]. The determination of MC typically requires two CT images from the same sample, one image at the unknown moisture state and one at a known MC, i.e. in the oven-dry state. The local MC can then be calculated based on the density differences. However, wood volume changes due to shrinkage must be considered for a pixel-wise measurement of the MC, and different mathematical approaches exist to achieve this [468, 469].
The emergence of X-ray micro-CT scanners within the last decades allows the three-dimensional visualization of plant tissue at resolutions down to ca. 1 µm [470, 471]. Synchrotron beamlines offer full-field micro- and/or nano-tomographic imaging setups with even higher spatial resolution and shorter acquisition times (several minutes or only seconds) [472,473,474]. This has allowed time-resolved studies of the distribution of water in the cellular structure of wood and the determination of contact angles in the cell lumens during water uptake [475] and drying of wood [454]. When combined with a climate-controlled in situ sample cell, X-ray CT offers a three-dimensional characterization of materials during exposure to mechanical load, temperature and/or water vapour [476]. Thereby, moisture-induced dimensional changes have been analysed in micron-sized wood samples [182, 278, 285], individual wood fibres [145] and wood cell wall micropillars [286]. This has provided insights into local strain variations, hygroexpansion properties of the cell walls and other deformations at the cellular scale.
Neutron radiography
Neutron imaging or radiography follows similar principles as transmission measurements based on gamma- or X-rays by measuring the attenuation of the incoming radiation by an object within the beam path. In contrast to X-ray photons, neutrons have a high probability to interact with light elements such as hydrogen, while heavier elements are practically transparent for neutrons [460, 477, 478]. The high sensitivity to hydrogen (or deuterium) makes neutron radiography particularly suitable for the analysis of moisture distributions in materials at the micron scale. If the image signal is corrected for contributions of scattered neutrons, quantitative evaluations are possible [479, 480]. Neutron radiography has been used to visualize the liquid water transport in wood in spatial and temporal dimensions. This has provided insights into the preferred water pathways in the wood microstructure [481,482,483,484]. The method has also been applied to analyse the variation and transport of water vapour and bound water in wood [485,486,487,488,489] and cellulose materials [490]. However, for quantitative moisture measurements in the hygroscopic range, volume changes in swelling materials must be considered because they hinder the pixel-wise comparison between radiographs taken in a dry and moist state [491]. In addition to the two-dimensional neutron radiography, recent improvements in neutron instrumentation allow fast time-resolved neutron tomography, which has been successfully applied to follow the uptake of deuterated water by plant roots with a temporal resolution of 10 s [492].
X-ray and neutron scattering
Elastic scattering of X-rays or neutrons can be used to study the molecular level and nanoscale structure of natural materials at different moisture conditions and therefore learn about their water interactions [493, 494]. Although the theoretical background is essentially the same, the scattering techniques are often divided into small-angle scattering and wide-angle scattering (or diffraction) according to the scattering angle range and the length scale being studied.
Small-angle scattering techniques, i.e. small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS), are sensitive to spatial variation of scattering length density, and they provide information on structures at the nanometre scale [495]. The scattering length density is proportional to electron density in SAXS, whereas in SANS, it is specific to the element and isotope. In natural cellulosic fibres, these methods show contributions from the cellulose microfibrils and their aggregates, embedded in a matrix of lower density, as well as any pores or voids, provided that sufficient scattering length density contrast exists between the different phases [493, 494]. The data analysis usually requires model fitting in the reciprocal space, which can be done based on analytical models for simple geometrical shapes or by more approximate approaches supported by complementary techniques like microscopy. For instance, SAXS and SANS data from wood samples can be analysed using the WoodSAS model, which has been utilized to quantify moisture-dependent changes in the packing distance of microfibrils [496]. The basis for this model lies in the earlier interpretations that the shoulder feature or peak corresponding to about 4 nm in the wet state originates from the regular packing distance between the microfibrils [184, 273, 497]. Similar results have been reported for pulp samples or fibrillated celluloses based on a shift of a peak appearing in a Kratky plot, which is a simple way to present the data in a form that enhances the shoulder feature related to the microfibril packing distance [498,499,500]. Also, a contribution of the regular size of microfibril bundles (aggregates of individual microfibrils) in SANS data has been detected in wood samples, and it shows swelling with increasing MC [501]. Moreover, as the scattering is sensitive to the contrast created by water, the time-dependent penetration and diffusion of water at the different hierarchical levels of the fibre wall structure can be observed either by SAXS/SANS through changes in the amount of water during drying [274, 502] or by SANS through a change in the H2O/D2O ratio within the nanoscale structures [338].
Wide-angle X-ray scattering (WAXS) or X-ray diffraction (XRD) has been widely used to observe moisture-related changes in the lattice parameters of cellulose crystallites and the size of ordered domains [162,163,164, 503]. Although various explanations (e.g. compression of the cellulose crystallites noted earlier) for these changes have been proposed in the literature (summarized in [165]), the latest evidence obtained with the aid of molecular dynamics simulations points to the effect of lateral aggregation of the fibrils, which distorts the cellulose crystallites and increases the lattice spacing perpendicular to the hydrogen-bonded molecular sheets of cellulose close to the dry state [165]. This idea is supported by the widely reported broadening of the diffraction peaks of cellulose with drying, which indicates a lower degree of crystalline order [504]. In addition to its effects on the cellulose crystallites, water produces a broad isotropic scattering contribution in WAXS data, which allows for examining the total MC of the sample. In this way, it can be possible to correlate the changes in scattering data to changes in MC in situ without any additional experiments [502]. In neutron scattering, the special sensitivity to deuterium allows enhancing the scattering from OH (or OD) groups that are accessible to deuterated water. This has been utilized for instance in efforts to distinguish the specific contribution of microfibril surfaces in wide-angle neutron scattering (WANS) intensities [505].
In addition to their coverage over multiple levels of the structural hierarchy, a clear benefit of X-ray and neutron scattering methods is that they can be used to study the water interaction of natural fibres in situ, without damaging the sample or cell wall structure during the sample preparation or the experimental observations. They also allow time-resolved studies with a time resolution down to fractions of a second, for instance following a change in humidity or temperature, or in response to a mechanical stimulus such as stretching. Spatially-resolved data can be obtained by mapping the sample with an X-ray beam having its diameter in the micro or nanoscale or by using a CT method based on scattering or diffraction [462, 506]. On the downside, scattering data especially in the small-angle regime may be challenging to interpret and often require special expertise. With the development of established ways to interpret scattering data from materials like natural fibres, they may, however, provide highly efficient tools to follow the effects of water on the nanostructure of these materials. In such efforts, scattering data calculated from molecular models have proven particularly useful [165].
Besides the more commonly used elastic scattering methods, in which the energy of the scattering photon or neutron is conserved, also inelastic scattering (energy not conserved) has been demonstrated in this field. Especially quasi-elastic neutron scattering has been utilized to study the mobility of water in cellulosic materials, which has allowed the distinction between different populations of water based on their mobility [507, 508]. These populations were assigned to water which is closely associated with the surface of the cellulose microfibril (attributed to non-freezing water) and water that occupies the nanopore space between the microfibrils, based upon similar assignments being made for porous silica materials [507]. Other inelastic neutron scattering studies have drawn connections between the water-accessibility and degree of order in the polymers forming natural cellulosic fibres [509] or the effects of water on the hydrogen bonding network [510].
Dielectric relaxation spectroscopy
Dielectric relaxation spectroscopy (DRS) is a technique that is used to study the rotational dynamics of molecules having permanent dipole moments and has potential for studying the interaction of water with lignocellulosic materials and cellulose [181, 511,512,513]. Sudo et al. studied the molecular dynamics of water in wood along the fibre direction over a large frequency range (from 40 to 10 GHz) [514]. This showed evidence of water confined in the cell wall having restricted motion. A DRS study of the molecular dynamics of the cell wall components at different MCs was conducted by Nakao et al. [299] who pointed out that although the relaxation behaviour of a polymer with a simple structure can be analysed by linear viscoelastic theory using a distribution function, the same cannot be done for wood over a wide time region. This is because there is no valid time–temperature superposition principle for a multiple component system.
Conclusions
A realistic model describing the sorption isotherm and sorption hysteresis loop observed for water vapour interactions with lignocellulosic and cellulosic materials is still lacking in the scientific literature. This situation continues despite the phenomenon being studied for over a century. A satisfactory model would have to incorporate the following (inter-related) properties:
-
1.
A link to the dynamic behaviour of the constituent macromolecules explaining how the dynamic behaviour shifts with changing MC.
-
2.
Changing dimensions of the material when absorbing moisture and desorbing moisture.
-
3.
Behaviour which is dependent upon the previous sorption history of the material.
-
4.
An explanation for sorption hysteresis which is related to the physical behaviour of the materials, especially taking account of sorption properties of glassy materials.
-
5.
The model should explain mechanosorptive behaviour, predicting the effect of changing MC upon mechanical properties and also consider how the application of external loads changes the sorption behaviour. When explaining phenomena such as creep, attention must be given to the interaction of moisture with the cellulose microfibril-hemicellulose interface.
-
6.
The model should be predictive and not rely upon arbitrary adjustable parameters for fitting to the experimental data.
-
7.
Many materials of interest exhibit anisotropic behaviour and involve mixtures of essentially inert elements (such as fibrils or platelets) embedded within an amorphous matrix. A description of this anisotropic behaviour needs to be incorporated in the model.
In order to reach a comprehensive model, it is important to gain realistic information on the molecular level and nanostructure of the materials. This could be enabled by the new possibilities of various in situ characterization methods that can follow the water and the related (nano)structural changes under various moisture conditions. It is essential that the models developed have a physical basis which is informed by the physics of the interaction of moisture with the cell wall polymers of lignocellulosic materials. These instrumental techniques offer the potential for understanding these interactions at levels of detail that offers the possibility of gaining fundamental insights.
Data availability
Not applicable.
Code availability
Not applicable.
References
Hill C, Kymäläinen M, Rautkari L (2022) Review of the use of solid wood as an external cladding material in the built environment. J Mater Sci 57:9031–9076. https://doi.org/10.1007/s10853-022-07211-x
Thybring EE, Boardman CR, Zelinka SL, Glass SV (2021) Common sorption isotherm models are not physically valid for water in wood. Colloids Surf A 627:127214. https://doi.org/10.1016/j.colsurfa.2021.127214
Zelinka SL, Glass SV, Thybring EE (2020) Evaluation of previous measurements of water vapor sorption in wood at multiple temperatures. Wood Sci Technol 54:769–786. https://doi.org/10.1007/s00226-020-01195-0
Basu S, Shivhare US, Mujumdar AS (2006) Models for sorption isotherms for foods: a review. Drying Technol 24:917–930. https://doi.org/10.1080/07373930600775979
Chirife J, Iglesias HA (2007) Equations for fitting water sorption isotherms of foods: part 1—a review. Int J Food Sci Technol 13:159–174. https://doi.org/10.1111/j.1365-2621.1978.tb00792.x
Willems W (2015) A critical review of the multilayer sorption models and comparison with the sorption site occupancy (SSO) model for wood moisture sorption isotherm analysis. Holzforschung 69:67–75. https://doi.org/10.1515/hf-2014-0069
Peleg M (2020) Models of sigmoid equilibrium moisture sorption isotherms with and without the monolayer hypothesis. Food Eng Rev 12:1–13. https://doi.org/10.1007/s12393-019-09207-x
Parker ME, Bronlund JE, Mawson AJ (2006) Moisture sorption isotherms for paper and paperboard in food chain conditions. Packag Technol Sci 19:193–209. https://doi.org/10.1002/pts.719
Zelinka SL, Glass SV, Thybring EE (2018) Myth versus reality: Do parabolic sorption isotherm models reflect actual wood–water thermodynamics? Wood Sci Technol 52:1701–1706. https://doi.org/10.1007/s00226-018-1035-9
Liu L, Tan S, Horikawa T et al (2017) Water adsorption on carbon—A review. Adv Colloid Interface Sci 250:64–78. https://doi.org/10.1016/j.cis.2017.10.002
Thommes M, Kaneko K, Neimark AV et al (2015) Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl Chem 87:1051–1069. https://doi.org/10.1515/pac-2014-1117
Ng E-P, Mintova S (2008) Nanoporous materials with enhanced hydrophilicity and high water sorption capacity. Microporous Mesoporous Mater 114:1–26. https://doi.org/10.1016/j.micromeso.2007.12.022
Laftah WA, Hashim S, Ibrahim AN (2011) Polymer hydrogels: a review. Polym Plast Technol Eng 50:1475–1486. https://doi.org/10.1080/03602559.2011.593082
Ullah F, Othman MBH, Javed F et al (2015) Classification, processing and application of hydrogels: a review. Mater Sci Eng, C 57:414–433. https://doi.org/10.1016/j.msec.2015.07.053
Miquelard-Garnier G, Roland S (2016) Beware of the Flory parameter to characterize polymer-polymer interactions: a critical reexamination of the experimental literature. Eur Polymer J 84:111–124. https://doi.org/10.1016/j.eurpolymj.2016.09.009
Willis JD, Beardsley TM, Matsen MW (2020) Simple and accurate calibration of the flory-huggins interaction parameter. Macromolecules 53:9973–9982. https://doi.org/10.1021/acs.macromol.0c02115
Eliassi A, Modarress H, Mansoori GA (1999) Measurement of activity of water in aqueous poly(ethylene glycol) solutions (effect of excess volume on the Flory−Huggins χ-parameter). J Chem Eng Data 44:52–55. https://doi.org/10.1021/je980162z
Thakral S, Thakral NK (2013) Prediction of drug-polymer miscibility through the use of solubility parameter based flory-huggins interaction parameter and the experimental validation: peg as model polymer. J Pharm Sci 102:2254–2263. https://doi.org/10.1002/jps.23583
Eckelt J, Sugaya R, Wolf BA (2008) Pullulan and dextran: uncommon composition dependent Flory-Huggins interaction parameters of their aqueous solutions. Biomacromol 9:1691–1697. https://doi.org/10.1021/bm800217y
Shafee EE, Naguib HF (2003) Water sorption in cross-linked poly(vinyl alcohol) networks. Polymer 44:1647–1653. https://doi.org/10.1016/S0032-3861(02)00865-0
Ferrer GG, Pradas MM, Gómez Ribelles JL, Sánchez MS (2004) Thermodynamical analysis of the hydrogel state in poly(2-hydroxyethyl acrylate). Polymer 45:6207–6217. https://doi.org/10.1016/j.polymer.2004.06.039
Bawendi MG, Freed KF (1988) Systematic corrections to Flory-Huggins theory: polymer–solvent–void systems and binary blend–void systems. J Chem Phys 88:2741–2756. https://doi.org/10.1063/1.454005
Bashir S, Hina M, Iqbal J et al (2020) Fundamental concepts of hydrogels: synthesis, properties, and their applications. Polymers 12:2702. https://doi.org/10.3390/polym12112702
Hu X, Zhou J, Daniel WFM et al (2017) Dynamics of dual networks: strain rate and temperature effects in hydrogels with reversible H-bonds. Macromolecules 50:652–659. https://doi.org/10.1021/acs.macromol.6b02422
van der Sman RGM (2019) Scaling of Flory-Huggins interaction parameter for polyols with chain length and number of hydroxyl groups. Food Hydrocolloids 96:396–401. https://doi.org/10.1016/j.foodhyd.2019.05.042
Berthold J, Rinaudo M, Salmeń L (1996) Association of water to polar groups; estimations by an adsorption model for ligno-cellulosic materials. Colloids Surf A 112:117–129. https://doi.org/10.1016/0927-7757(95)03419-6
Belton PS (1997) NMR and the mobility of water in polysaccharide gels. Int J Biol Macromol 21:81–88. https://doi.org/10.1016/S0141-8130(97)00045-7
van der Wel GK, Adan OCG (1999) Moisture in organic coatings—a review. Prog Org Coat 37:1–14. https://doi.org/10.1016/S0300-9440(99)00058-2
Matsuoka S, Hale A (1997) Cooperative relaxation processes in polymers. J Appl Polym Sci 64:77–93. https://doi.org/10.1002/(SICI)1097-4628(19970404)64:1%3c77::AID-APP7%3e3.0.CO;2-O
Arya RK, Thapliyal D, Sharma J, Verros GD (2021) Glassy polymers—diffusion, sorption. Ageing Appl Coat 11:1049. https://doi.org/10.3390/coatings11091049
Xiao R, Li H (2021) Glass transition in gels. Phys Rev Materials 5:065604. https://doi.org/10.1103/PhysRevMaterials.5.065604
Weinmüller C, Langel C, Fornasiero F et al (2006) Sorption kinetics and equilibrium uptake for water vapor in soft-contact-lens hydrogels. J Biomed Mater Res 77A:230–241. https://doi.org/10.1002/jbm.a.30598
Vrentas JS, Vrentas CM (1991) Sorption in glassy polymers. Macromolecules 24:2404–2412. https://doi.org/10.1021/ma00009a043
Vrentas JS, Vrentas CM (1996) Hysteresis effects for sorption in glassy polymers. Macromolecules 29:4391–4396. https://doi.org/10.1021/ma950969l
Vrentas JS, Vrentas CM (1990) Predictions of volumetric behavior for glassy polymer-penetrant systems. J Polym Sci B Polym Phys 28:241–244. https://doi.org/10.1002/polb.1990.090280210
Hill C, Beck G (2017) On the applicability of the Flory-Huggins and Vrentas models for describing the sorption isotherms of wood. Int Wood Prod J 8:50–55. https://doi.org/10.1080/20426445.2016.1275094
Shamblin SL, Hancock BC, Zografi G (1998) Water vapor sorption by peptides, proteins and their formulations. Eur J Pharm Biopharm 45:239–247. https://doi.org/10.1016/S0939-6411(98)00006-X
Hancock BC, Zografi G (1993) The use of solution theories for predicting water vapor absorption by amorphous pharmaceutical solids: a test of the flory-huggins and vrentas models. Pharm Res 10:1262–1267. https://doi.org/10.1023/A:1018901325842
Pierlot AP (1999) Water in wool. Text Res J 69:97–103. https://doi.org/10.1177/004051759906900204
Ormondroyd GA, Curling SF, Mansour E, Hill CAS (2016) The water vapour sorption characteristics and kinetics of different wool types. J Text Inst. https://doi.org/10.1080/00405000.2016.1224442
Argatov I, Kocherbitov V (2021) An empirical model for sorption by glassy polymers: an assessment of thermodynamic parameters. Polym Test 99:107220. https://doi.org/10.1016/j.polymertesting.2021.107220
Doghieri F, Sarti GC (1996) Nonequilibrium lattice fluids: a predictive model for the solubility in glassy polymers. Macromolecules 29:7885–7896. https://doi.org/10.1021/ma951366c
Pandis C, Spanoudaki A, Kyritsis A et al (2011) Water sorption characteristics of poly(2-hydroxyethyl acrylate)/silica nanocomposite hydrogels: water sorption characteristics of PHEA/silica. J Polym Sci B Polym Phys 49:657–668. https://doi.org/10.1002/polb.22225
Verros GD (2015) Application of irreversible thermodynamics to the solvent diffusion in an amorphous glassy polymer: a comprehensive model for drying of toluene-poly(methyl methacrylate) coatings. Can J Chem Eng 93:2298–2306. https://doi.org/10.1002/cjce.22340
Barkas WW (1942) Wood water relationships—VII. swelling pressure and sorption hysteresis in gels. Trans Faraday Soc 38:194–209. https://doi.org/10.1039/TF9423800194
Kildeeva N, Chalykh A, Belokon M et al (2020) Influence of genipin crosslinking on the properties of chitosan-based films. Polymers 12:1086. https://doi.org/10.3390/polym12051086
Ruike M, Inoue T, Takada S et al (1999) Water sorption and drying behaviour of crosslinked dextrans. Biosci Biotechnol Biochem 63:271–275. https://doi.org/10.1271/bbb.63.271
Uimonen T, Hautamäki S, Altgen M et al (2020) Dynamic vapour sorption protocols for the quantification of accessible hydroxyl groups in wood. Holzforschung 74:412–419. https://doi.org/10.1515/hf-2019-0058
Hill CAS, Ramsay J, Gardiner B (2015) Variability in water vapour sorption isotherm in Japanese Larch (Larix kaempferi Lamb.)—earlywood and latewood influences. Int Wood Prod J 6:53–59. https://doi.org/10.1179/2042645314Y.0000000090
Passauer L, Struch M, Schuldt S et al (2012) Dynamic moisture sorption characteristics of xerogels from water-swellable oligo(oxyethylene) lignin derivatives. ACS Appl Mater Interfaces 4:5852–5862. https://doi.org/10.1021/am3015179
Wonders AG, Paul DR (1979) Effect of CO2 exposure history on sorption and transport in polycarbonate. J Membr Sci 5:63–75. https://doi.org/10.1016/S0376-7388(00)80438-X
Berens AR (1977) Effects of sample history, time, and temperature on the sorption of monomer vapor by PVC. J Macromole Sci Part B 14:483–498. https://doi.org/10.1080/00222347708212233
Lu Y, Pignatello JJ (2002) Demonstration of the “conditioning effect” in soil organic matter in support of a pore deformation mechanism for sorption hysteresis. Environ Sci Technol 36:4553–4561. https://doi.org/10.1021/es020554x
Lu Y, Pignatello JJ (2004) History-dependent sorption in humic acids and a lignite in the context of a polymer model for natural organic matter. Environ Sci Technol 38:5853–5862. https://doi.org/10.1021/es049774w
Taniguchi T, Harada H, Nakato K (1978) Determination of water adsorption sites in wood by a hydrogen-deuterium exchange. Nature 272:230–231. https://doi.org/10.1038/272230a0
Phuong LX, Takayama M, Shida S et al (2007) Determination of the accessible hydroxyl groups in heat-treated styrax tonkinensis (Pierre) Craib ex Hartwich wood by hydrogen-deuterium exchange and H-2 NMR spectroscopy. Holzforschung 61:488–491. https://doi.org/10.1515/hf2007.086
Lindh EL, Salmén L (2017) Surface accessibility of cellulose fibrils studied by hydrogen–deuterium exchange with water. Cellulose 24:21–33. https://doi.org/10.1007/s10570-016-1122-8
Zimm BH (1953) Simplified relation between thermodynamics and molecular distribution functions for a mixture. J Chem Phys 21:934–935. https://doi.org/10.1063/1.1699065
Zimm BH, Lundberg JL (1956) Sorption of vapors by high polymers. J Phys Chem 60:425–428. https://doi.org/10.1021/j150538a010
Davis EM, Elabd YA (2013) Water clustering in glassy polymers. J Phys Chem B 117:10629–10640. https://doi.org/10.1021/jp405388d
Davis EM, Elabd YA (2013) Prediction of water solubility in glassy polymers using nonequilibrium thermodynamics. Ind Eng Chem Res 52:12865–12875. https://doi.org/10.1021/ie401713h
Favre E, Clément R, Nguyen QT et al (1993) Sorption of organic solvents into dense silicone membranes. Part 2—development of a new approach based on a clustering hypothesis for associated solvents. J Chem Soc Faraday Trans 89:4347–4353. https://doi.org/10.1039/FT9938904347
Perrin L, Nguyen QT, Sacco D, Lochon P (1997) Experimental studies and modelling of sorption and diffusion of water and alcohols in cellulose acetate. Polym Int 42:9–16. https://doi.org/10.1002/(SICI)1097-0126(199701)42:1%3c9::AID-PI637%3e3.0.CO;2-A
Hakalahti M, Faustini M, Boissière C et al (2017) Interfacial mechanisms of water vapor sorption into cellulose nanofibril films as revealed by quantitative models. Biomacromol 18:2951–2958. https://doi.org/10.1021/acs.biomac.7b00890
Pissis P, Kyritsis A (2013) Hydration studies in polymer hydrogels. J Polym Sci B Polym Phys 51:159–175. https://doi.org/10.1002/polb.23220
McConville P, Pope JM (2001) 1H NMR T2 relaxation in contact lens hydrogels as a probe of water mobility. Polymer 42:3559–3568. https://doi.org/10.1016/S0032-3861(00)00714-X
Barbieri R, Quaglia M, Delfini M, Brosio E (1998) Investigation of water dynamic behaviour in poly(HEMA) and poly(HEMA-co-DHPMA) hydrogels by proton T2 relaxation time and self-diffusion coefficient n.m.r. measurements. Polymer 39:1059–1066. https://doi.org/10.1016/S0032-3861(97)00403-5
Topgaard D, Söderman O (2002) Self-diffusion of nonfreezing water in porous carbohydrate polymer systems studied with nuclear magnetic resonance. Biophys J 83:3596–3606. https://doi.org/10.1016/S0006-3495(02)75360-5
Hatakeyama H, Hatakeyama T (1998) Interaction between water and hydrophilic polymers. Thermochim Acta 308:3–22. https://doi.org/10.1016/S0040-6031(97)00325-0
Thybring EE, Digaitis R, Nord-Larsen T et al (2020) How much water can wood cell walls hold? A triangulation approach to determine the maximum cell wall moisture content. PLoS ONE 15:e0238319. https://doi.org/10.1371/journal.pone.0238319
Zhong X, Ma E (2022) A novel approach for characterizing pore size distribution of wood cell wall using differential scanning calorimetry thermoporosimetry. Thermochim Acta 718:179380. https://doi.org/10.1016/j.tca.2022.179380
Debta S, Bhutia SZ, Satapathy DK, Ghosh P (2022) Intrinsic-water desorption induced thermomechanical response of hydrogels. Soft Matter 18:8285–8294. https://doi.org/10.1039/D2SM01054B
Salmerón Sánchez M, Monleón Pradas M, Gómez Ribelles JL (2002) Thermal transitions of benzene in a poly(ethyl acrylate) network. J Non-Cryst Solids 307–310:750–757. https://doi.org/10.1016/S0022-3093(02)01557-0
Chen HM, Van Horn JD, Jean YC (2012) Applications of positron annihilation spectroscopy to life science. DDF 331:275–293. https://doi.org/10.4028/www.scientific.net/DDF.331.275
Roussenova M, Murith M, Alam A, Ubbink J (2010) Plasticization, antiplasticization, and molecular packing in amorphous carbohydrate-glycerol matrices. Biomacromol 11:3237–3247. https://doi.org/10.1021/bm1005068
Kilburn D, Claude J, Mezzenga R et al (2004) Water in glassy carbohydrates: opening it up at the nanolevel. J Phys Chem B 108:12436–12441. https://doi.org/10.1021/jp048774f
Kilburn D, Claude J, Schweizer T et al (2005) Carbohydrate polymers in amorphous states: an integrated thermodynamic and nanostructural investigation. Biomacromol 6:864–879. https://doi.org/10.1021/bm049355r
Factorovich MH, Gonzalez Solveyra E, Molinero V, Scherlis DA (2014) Sorption isotherms of water in nanopores: relationship between hydropohobicity, adsorption pressure, and hysteresis. J Phys Chem C 118:16290–16300. https://doi.org/10.1021/jp5000396
Nakamura M, Ohba T, Branton P et al (2010) Equilibration-time and pore-width dependent hysteresis of water adsorption isotherm on hydrophobic microporous carbons. Carbon 48:305–308. https://doi.org/10.1016/j.carbon.2009.09.008
Vrentas JS, Jarzebski CM, Duda JL (1975) A Deborah number for diffusion in polymer-solvent systems. AIChE J 21:894–901. https://doi.org/10.1002/aic.690210510
Berens AR, Hopfenberg HB (1978) Diffusion and relaxation in glassy polymer powders: 2 separation of diffusion and relaxation parameters. Polymer 19:489–496. https://doi.org/10.1016/0032-3861(78)90269-0
Berens AR, Hopfenberg HB (1979) Induction and measurement of glassy-state relaxations by vapor sorption techniques. J Polym Sci Polym Phys Ed 17:1757–1770. https://doi.org/10.1002/pol.1979.180171011
Wind MM, Lenderink HJW (1996) A capacitance study of pseudo-fickian diffusion in glassy polymer coatings. Prog Org Coat 28:239–250. https://doi.org/10.1016/0300-9440(95)00601-X
Frisch HL (1980) Sorption and transport in glassy polymers-a review. Polym Eng Sci 20:2–13. https://doi.org/10.1002/pen.760200103
Stanford JP, Pfromm PH, Rezac ME (2017) Effect of vapor phase ethylenediamine crosslinking of matrimid on alcohol vapor sorption and diffusion. J Appl Polym Sci 134:44771. https://doi.org/10.1002/app.44771
Firestone BA, Siegel RA (1991) Kinetics and mechanisms of water sorption in hydrophobic, ionizable copolymer gels. J Appl Polym Sci 43:901–914. https://doi.org/10.1002/app.1991.070430507
Wang L, Corriou J-P, Castel C, Favre E (2013) Transport of gases in glassy polymers under transient conditions: limit-behavior investigations of dual-mode sorption theory. Ind Eng Chem Res 52:1089–1101. https://doi.org/10.1021/ie2027102
Stannett V, Haider M, Koros WJ, Hopfenberg HB (1980) Sorption and transport of water vapor in glassy poly(acrylonitrile). Polym Eng Sci 20:300–304. https://doi.org/10.1002/pen.760200414
Guo J, Barbari TA (2009) Unified dual mode description of small molecule sorption and desorption kinetics in a glassy polymer. Macromolecules 42:5700–5708. https://doi.org/10.1021/ma9007576
Kamiya Y, Hirose T, Mizoguchi K, Naito Y (1986) Gravimetric study of high-pressure sorption of gases in polymers. J Polym Sci B Polym Phys 24:1525–1539. https://doi.org/10.1002/polb.1986.090240711
Kelley SS, Rials TG, Glasser WG (1987) Relaxation behaviour of the amorphous components of wood. J Mater Sci 22:617–624. https://doi.org/10.1007/BF01160778
Hishikawa Y, Togawa E, Kataoka Y, Kondo T (1999) Characterization of amorphous domains in cellulosic materials using a FTIR deuteration monitoring analysis. Polymer 40:7117–7124. https://doi.org/10.1016/S0032-3861(99)00120-2
Biliaderis C (1999) Glass transition and physical properties of polyol-plasticised pullulan–starch blends at low moisture. Carbohyd Polym 40:29–47. https://doi.org/10.1016/S0144-8617(99)00026-0
Paul DR (1999) Water vapor sorption and diffusion in glassy polymers. Macromol Symp 138:13–20. https://doi.org/10.1002/masy.19991380104
Liu Y, Bhandari B, Zhou W (2006) Glass transition and enthalpy relaxation of amorphous food saccharides: a review. J Agric Food Chem 54:5701–5717. https://doi.org/10.1021/jf060188r
Champion D, Le Meste M, Simatos D (2000) Towards an improved understanding of glass transition and relaxations in foods: molecular mobility in the glass transition range. Trends Food Sci Technol 11:41–55. https://doi.org/10.1016/S0924-2244(00)00047-9
Lützow N, Tihminlioglu A, Danner RP et al (1999) Diffusion of toluene and n-heptane in polyethylenes of different crystallinity. Polymer 40:2797–2803. https://doi.org/10.1016/S0032-3861(98)00473-X
Xi L, Shah M, Trout BL (2013) Hopping of water in a glassy polymer studied via transition path sampling and likelihood maximization. J Phys Chem B 117:3634–3647. https://doi.org/10.1021/jp3099973
Kulasinski K, Guyer R, Derome D, Carmeliet J (2015) Water diffusion in amorphous hydrophilic systems: a stop and go process. Langmuir 31:10843–10849. https://doi.org/10.1021/acs.langmuir.5b03122
Owen AJ, Bonart R (1985) Cooperative relaxation processes in polymers. Polymer 26:1034–1038. https://doi.org/10.1016/0032-3861(85)90225-3
Matsuoka S (1997) Entropy, free volume, and cooperative relaxation. J Res Natl Inst Stand Technol 102:213. https://doi.org/10.6028/jres.102.017
Topgaard D, Söderman O (2001) Diffusion of water absorbed in cellulose fibers studied with 1H-NMR. Langmuir 17:2694–2702. https://doi.org/10.1021/la000982l
Hoch G, Chauhan A, Radke CJ (2003) Permeability and diffusivity for water transport through hydrogel membranes. J Membr Sci 214:199–209. https://doi.org/10.1016/S0376-7388(02)00546-X
Thimmegowda MC, Sathyanarayana PM, Shariff G et al (2002) A free volume microprobe study of water sorption in a contact lens polymer. J Biomater Sci Polym Ed 13:1295–1311. https://doi.org/10.1163/15685620260449705
Crank J (1977) Diffusion in polymers. Print Acad Pr, London
Park GS (1986) Transport principles—solution, diffusion and permeation in polymer membranes. In: Bungay PM, Lonsdale HK, Pinho MN (eds) Synthetic membranes: science, engineering and applications. Springer, Netherlands, Dordrecht, pp 57–107
Jakes JE, Hunt CG, Zelinka SL et al (2019) Effects of moisture on diffusion in unmodified wood cell walls: a phenomenological polymer science approach. Forests 10:1084. https://doi.org/10.3390/f10121084
Ogieglo W, Wessling M, Benes NE (2014) Polymer relaxations in thin films in the vicinity of a penetrant- or temperature-induced glass transition. Macromolecules 47:3654–3660. https://doi.org/10.1021/ma5002707
Karoyo AH, Wilson LD (2021) A review on the design and hydration properties of natural polymer-based hydrogels. Materials 14:1095. https://doi.org/10.3390/ma14051095
Buwalda SJ, Boere KWM, Dijkstra PJ et al (2014) Hydrogels in a historical perspective: from simple networks to smart materials. J Control Release 190:254–273. https://doi.org/10.1016/j.jconrel.2014.03.052
Jeon O, Song SJ, Lee K-J et al (2007) Mechanical properties and degradation behaviors of hyaluronic acid hydrogels cross-linked at various cross-linking densities. Carbohyd Polym 70:251–257. https://doi.org/10.1016/j.carbpol.2007.04.002
Lee KY, Rowley JA, Eiselt P et al (2000) Controlling mechanical and swelling properties of alginate hydrogels independently by cross-linker type and cross-linking density. Macromolecules 33:4291–4294. https://doi.org/10.1021/ma9921347
Tanaka Y, Gong JP, Osada Y (2005) Novel hydrogels with excellent mechanical performance. Prog Polym Sci 30:1–9. https://doi.org/10.1016/j.progpolymsci.2004.11.003
Wu Y, Joseph S, Aluru NR (2009) Effect of cross-linking on the diffusion of water, ions, and small molecules in hydrogels. J Phys Chem B 113:3512–3520. https://doi.org/10.1021/jp808145x
Tighe BJ, Peppas N (1987) Hydrogels in medicine and pharmacy. CRC Press, Boca Raton
Buenger D, Topuz F, Groll J (2012) Hydrogels in sensing applications. Prog Polym Sci 37:1678–1719. https://doi.org/10.1016/j.progpolymsci.2012.09.001
Mathews MB, Decker L (1977) Comparative studies of water sorption of hyaline cartilage. Biochimica et Biophysica Acta BBA General Sub 497:151–159. https://doi.org/10.1016/0304-4165(77)90148-9
Sannino A, Demitri C, Madaghiele M (2009) Biodegradable cellulose-based hydrogels: design and applications. Materials 2:353–373. https://doi.org/10.3390/ma2020353
Li H, Kong N, Laver B, Liu J (2016) Hydrogels constructed from engineered proteins. Small 12:973–987. https://doi.org/10.1002/smll.201502429
Zhang Q, Liu Y, Yang G et al (2023) Recent advances in protein hydrogels: from design, structural and functional regulations to healthcare applications. Chem Eng J 451:138494. https://doi.org/10.1016/j.cej.2022.138494
Kim M, Tang S, Olsen BD (2013) Physics of engineered protein hydrogels. J Polym Sci B Polym Phys 51:587–601. https://doi.org/10.1002/polb.23270
Tang Y, Wang H, Liu S et al (2022) A review of protein hydrogels: Protein assembly mechanisms, properties, and biological applications. Colloids Surf B 220:112973. https://doi.org/10.1016/j.colsurfb.2022.112973
Zheng H, Zuo B (2021) Functional silk fibroin hydrogels: preparation, properties and applications. J Mater Chem B 9:1238–1258. https://doi.org/10.1039/D0TB02099K
Tang Y, Zhang X, Li X et al (2022) A review on recent advances of protein-polymer hydrogels. Eur Polymer J 162:110881. https://doi.org/10.1016/j.eurpolymj.2021.110881
Thakur VK, Thakur MK (2015) Recent advances in green hydrogels from lignin: a review. Int J Biol Macromol 72:834–847. https://doi.org/10.1016/j.ijbiomac.2014.09.044
Haraguchi K (2007) Nanocomposite hydrogels. Curr Opin Solid State Mater Sci 11:47–54. https://doi.org/10.1016/j.cossms.2008.05.001
Flory PJ, Rehner J (1943) Statistical mechanics of cross-linked polymer networks I. Rubberlike elasticity. J Chem Phys 11:512–520. https://doi.org/10.1063/1.1723791
Friesen S, Hannappel Y, Kakorin S, Hellweg T (2022) Comparison of different approaches to describe the thermotropic volume phase transition of smart microgels. Colloid Polym Sci 300:1235–1245. https://doi.org/10.1007/s00396-022-04950-w
Zhang J, Zografi G (2000) The Relationship between “BET” and “free volume”-derived parameters for water vapor absorption into amorphous solids. J Pharm Sci 89:1063–1072. https://doi.org/10.1002/1520-6017(200008)89:8%3c1063::AID-JPS11%3e3.0.CO;2-0
Rosenbaum S (2007) Solution of water in polymers: the keratin-water isotherm. J polym sci C Polym symp 31:45–55. https://doi.org/10.1002/polc.5070310107
Jin X, van der Sman RGM, van Maanen JFC et al (2014) Moisture sorption isotherms of broccoli interpreted with the Flory-Huggins free volume theory. Food Biophys 9:1–9. https://doi.org/10.1007/s11483-013-9311-6
Leibler L, Sekimoto K (1993) On the sorption of gases and liquids in glassy polymers. Macromolecules 26:6937–6939. https://doi.org/10.1021/ma00077a034
Ganji F, Vasheghani-farahani S, Vasheghani-farahani E (2010) Theoretical description of hydrogel swelling: a review. Iran Polym J 19:375–398
Jhon MS, Andrade JD (1973) Water and hydrogels. J Biomed Mater Res 7:509–522. https://doi.org/10.1002/jbm.820070604
Alegre-Requena JV, Saldías C, Inostroza-Rivera R, Díaz Díaz D (2019) Understanding hydrogelation processes through molecular dynamics. J Mater Chem B 7:1652–1673. https://doi.org/10.1039/C8TB03036G
Hofer K, Mayer E, Johari G (1990) Glass-liquid transition of water and ethylene glycol solution in poly(2-hydroxyethyl methacrylate) hydrogel. J Phys Chem 94:2689–2696. https://doi.org/10.1021/j100369a083
Plaza NZ (2019) On the Experimental assessment of the molecular-scale interactions between wood and water. Forests 10:616. https://doi.org/10.3390/f10080616
Toumpanaki E, Shah DU, Eichhorn SJ (2021) Beyond what meets the eye: imaging and imagining wood mechanical-structural properties. Adv Mater 33:2001613. https://doi.org/10.1002/adma.202001613
Hill C, Altgen M, Rautkari L (2021) Thermal modification of wood—a review: chemical changes and hygroscopicity. J Mater Sci 56:6581–6614. https://doi.org/10.1007/s10853-020-05722-z
Cresswell R, Dupree R, Brown SP et al (2021) Importance of water in maintaining softwood secondary cell wall nanostructure. Biomacromol 22:4669–4680. https://doi.org/10.1021/acs.biomac.1c00937
Emonet A, Hay A (2022) Development and diversity of lignin patterns. Plant Physiol 190:31–43. https://doi.org/10.1093/plphys/kiac261
Barros J, Serk H, Granlund I, Pesquet E (2015) The cell biology of lignification in higher plants. Ann Bot 115:1053–1074. https://doi.org/10.1093/aob/mcv046
Salmén L, Burgert I (2009) Cell wall features with regard to mechanical performance. A review COST Action E35 wood machining—micromechanics and fracture. Holzforschung 63:121–129. https://doi.org/10.1515/hf.2009.011
Gamstedt EK, Bader TK, De Borst K (2013) Mixed numerical–experimental methods in wood micromechanics. Wood Sci Technol 47:183–202. https://doi.org/10.1007/s00226-012-0519-2
Joffre T, Isaksson P, Dumont PJJ et al (2016) A Method to measure moisture induced swelling properties of a single wood cell. Exp Mech 56:723–733. https://doi.org/10.1007/s11340-015-0119-9
Eder M, Arnould O, Dunlop JWC et al (2013) Experimental micromechanical characterisation of wood cell walls. Wood Sci Technol 47:163–182. https://doi.org/10.1007/s00226-012-0515-6
Chokshi S, Parmar V, Gohil P, Chaudhary V (2022) Chemical composition and mechanical properties of natural fibers. Journal of Natural Fibers 19:3942–3953. https://doi.org/10.1080/15440478.2020.1848738
Wada M, Nishiyama Y, Chanzy H et al (2008) The structure of celluloses. Powder Diffr 23:92–95. https://doi.org/10.1154/1.2912442
Lindh EL, Bergenstråhle-Wohlert M, Terenzi C et al (2016) Non-exchanging hydroxyl groups on the surface of cellulose fibrils: the role of interaction with water. Carbohyd Res 434:136–142. https://doi.org/10.1016/j.carres.2016.09.006
Kondo T (1997) The assignment of IR absorption bands due to free hydroxyl groups in cellulose. Cellulose 4:281–292. https://doi.org/10.1023/A:1018448109214
Verlhac C, Dedier J, Chanzy H (1990) Availability of surface hydroxyl groups in valonia and bacterial cellulose. J Polym Sci A Polym Chem 28:1171–1177. https://doi.org/10.1002/pola.1990.080280517
Kargarzadeh H, Mariano M, Gopakumar D et al (2018) Advances in cellulose nanomaterials. Cellulose 25:2151–2189. https://doi.org/10.1007/s10570-018-1723-5
O’Sullivan AC (1997) Cellulose: the structure slowly unravels. Cellulose 4:173–207. https://doi.org/10.1023/A:1018431705579
Matthews JF, Skopec CE, Mason PE et al (2006) Computer simulation studies of microcrystalline cellulose Iβ. Carbohyd Res 341:138–152
Eichhorn SJ, Young RJ (2001) The Young’s modulus of a microcrystalline cellulose. Cellulose 8:197–207. https://doi.org/10.1023/A:1013181804540
Nishiyama Y (2009) Structure and properties of the cellulose microfibril. J Wood Sci 55:241–249. https://doi.org/10.1007/s10086-009-1029-1
Eichhorn SJ, Dufresne A, Aranguren M et al (2010) Review: current international research into cellulose nanofibres and nanocomposites. J Mater Sci 45:1–33. https://doi.org/10.1007/s10853-009-3874-0
Eichhorn SJ (2011) Cellulose nanowhiskers: promising materials for advanced applications. Soft Matter 7:303–315. https://doi.org/10.1039/C0SM00142B
Huber T, Müssig J, Curnow O et al (2012) A critical review of all-cellulose composites. J Mater Sci 47:1171–1186. https://doi.org/10.1007/s10853-011-5774-3
Benítez AJ, Walther A (2017) Cellulose nanofibril nanopapers and bioinspired nanocomposites: a review to understand the mechanical property space. J Mater Chem A 5:16003–16024. https://doi.org/10.1039/C7TA02006F
Mokhena TC, Sadiku ER, Mochane MJ et al (2021) Mechanical properties of cellulose nanofibril papers and their bionanocomposites: a review. Carbohyd Polym 273:118507. https://doi.org/10.1016/j.carbpol.2021.118507
Zabler S, Paris O, Burgert I, Fratzl P (2010) Moisture changes in the plant cell wall force cellulose crystallites to deform. J Struct Biol 171:133–141. https://doi.org/10.1016/j.jsb.2010.04.013
Salmén L, Stevanic JS, Holmqvist C, Yu S (2021) Moisture induced straining of the cellulosic microfibril. Cellulose 28:3347–3357. https://doi.org/10.1007/s10570-021-03712-1
Abe K, Yamamoto H (2005) Mechanical interaction between cellulose microfibril and matrix substance in wood cell wall determined by X-ray diffraction. J Wood Sci 51:334–338. https://doi.org/10.1007/s10086-004-0667-6
Paajanen A, Zitting A, Rautkari L et al (2022) Nanoscale mechanism of moisture-induced swelling in wood microfibril bundles. Nano Lett 22:5143–5150. https://doi.org/10.1021/acs.nanolett.2c00822
Salmén L (2004) Micromechanical understanding of the cell-wall structure. CR Biol 327:873–880. https://doi.org/10.1016/j.crvi.2004.03.010
Burgert I (2006) Exploring the micromechanical design of plant cell walls. Am J Bot 93:1391–1401
Gatenholm P, Tenkanen M (2003) Hemicelluloses: science and technology. American Chemical Society, Washington
Ibn Yaich A, Edlund U, Albertsson A-C (2017) Transfer of biomatrix/wood cell interactions to hemicellulose-based materials to control water interaction. Chem Rev 117:8177–8207. https://doi.org/10.1021/acs.chemrev.6b00841
Terrett OM, Dupree P (2019) Covalent interactions between lignin and hemicelluloses in plant secondary cell walls. Curr Opin Biotechnol 56:97–104. https://doi.org/10.1016/j.copbio.2018.10.010
Rao J, Lv Z, Chen G, Peng F (2023) Hemicellulose: structure, chemical modification, and application. Prog Polym Sci 140:101675. https://doi.org/10.1016/j.progpolymsci.2023.101675
Li M, Pu Y, Ragauskas AJ (2016) Current understanding of the correlation of lignin structure with biomass recalcitrance. Front Chem. https://doi.org/10.3389/fchem.2016.00045
Sun R (2020) Lignin source and structural characterization. Chemsuschem 13:4385–4393. https://doi.org/10.1002/cssc.202001324
Rico-García D, Ruiz-Rubio L, Pérez-Alvarez L et al (2020) Lignin-based hydrogels: synthesis and applications. Polymers 12:81. https://doi.org/10.3390/polym12010081
Placet V, Passard J, Perré P (2008) Viscoelastic properties of wood across the grain measured under water-saturated conditions up to 135 °C: evidence of thermal degradation. J Mater Sci 43:3210–3217. https://doi.org/10.1007/s10853-008-2546-9
Assor C, Placet V, Chabbert B et al (2009) Concomitant changes in viscoelastic properties and amorphous polymers during the hydrothermal treatment of hardwood and softwood. J Agric Food Chem 57:6830–6837. https://doi.org/10.1021/jf901373s
Salmén L (1984) Viscoelastic properties of in situ lignin under water-saturated conditions. J Mater Sci 19:3090–3096. https://doi.org/10.1007/BF01026988
Chowdhury S, Fabiyi J, Frazier CE (2010) Advancing the dynamic mechanical analysis of biomass: comparison of tensile-torsion and compressive-torsion wood DMA. Holzforschung 64:747–756. https://doi.org/10.1515/hf.2010.123
Zhan T, Jiang J, Lu J (2015) The viscoelastic properties of Chinese fir during water-loss process under hydrothermal conditions. Drying Technol 33:1739–1745
Horvath B, Peralta P, Frazier C, Peszlen IM (2011) Thermal softening of transgenic aspen. BioResources 6:2125–2134
Einfeldt J, Meißner D, Kwasniewski A (2004) Molecular interpretation of the main relaxations found in dielectric spectra of cellulose—experimental arguments. Cellulose 11:137–150. https://doi.org/10.1023/B:CELL.0000025404.61412.d6
Patera A, Derome D, Griffa M, Carmeliet J (2013) Hysteresis in swelling and in sorption of wood tissue. J Struct Biol 182:226–234. https://doi.org/10.1016/j.jsb.2013.03.003
Barkas WW (1949) The swelling of wood under stress. H.M. Stationary Office, London
Fernandes AN, Thomas LH, Altaner CM et al (2011) Nanostructure of cellulose microfibrils in spruce wood. Proc Natl Acad Sci 108:E1195–E1203. https://doi.org/10.1073/pnas.1108942108
Yin Q, Liu H-H (2021) Drying stress and strain of wood: a review. Appl Sci 11:5023. https://doi.org/10.3390/app11115023
Arzola-Villegas X, Lakes R, Plaza NZ, Jakes JE (2019) Wood moisture-induced swelling at the cellular scale—Ab intra. Forests 10:996. https://doi.org/10.3390/f10110996
Nishiyama Y (2023) Thermodynamics of the swelling work of wood and non-ionic polysaccharides: a revisit. Carbohyd Polym 320:121227. https://doi.org/10.1016/j.carbpol.2023.121227
Cousins WJ (1976) Elastic modulus of lignin as related to moisture content. Wood SciTechnol 10:9–17. https://doi.org/10.1007/BF00376380
Hess KM, Killgore JP, Srubar WV (2018) Nanoscale hygromechanical behavior of lignin. Cellulose 25:6345–6360. https://doi.org/10.1007/s10570-018-2045-3
Nissan AH (1977) The elastic modulus of lignin as related to moisture content. Wood Sci Technol 11:147–151. https://doi.org/10.1007/BF00350992
Cousins WJ (1978) Young’s modulus of hemicellulose as related to moisture content. Wood SciTechnol 12:161–167. https://doi.org/10.1007/BF00372862
Youssefian S, Jakes JE, Rahbar N (2017) Variation of nanostructures, molecular interactions, and anisotropic elastic moduli of lignocellulosic cell walls with moisture. Sci Rep 7:2054. https://doi.org/10.1038/s41598-017-02288-w
Kulasinski K, Salmén L, Derome D, Carmeliet J (2016) Moisture adsorption of glucomannan and xylan hemicelluloses. Cellulose 23:1629–1637. https://doi.org/10.1007/s10570-016-0944-8
Pan Y, Zhong Z (2016) Micromechanical modeling of the wood cell wall considering moisture absorption. Compos B Eng 91:27–35. https://doi.org/10.1016/j.compositesb.2015.12.038
Kojima Y, Yamamoto H (2004) Properties of the cell wall constituents in relation to the longitudinal elasticity of wood. Wood Sci Technol 37:427–434. https://doi.org/10.1007/s00226-003-0177-5
Engelund ET, Salmén L (2012) Tensile creep and recovery of Norway spruce influenced by temperature and moisture. Holzforschung 66:959–965. https://doi.org/10.1515/hf-2011-0172
Kulasinski K, Guyer R, Derome D, Carmeliet J (2015) Water adsorption in wood microfibril-hemicellulose system: role of the crystalline-amorphous interface. Biomacromol 16:2972–2978. https://doi.org/10.1021/acs.biomac.5b00878
Dinwoodie JM (2000) Timber: its nature and behaviour. CRC Press, Florida
Burgert I, Eder M, Gierlinger N, Fratzl P (2007) Tensile and compressive stresses in tracheids are induced by swelling based on geometrical constraints of the wood cell. Planta 226:981–987. https://doi.org/10.1007/s00425-007-0544-9
Zhou S, Jin K, Buehler MJ (2021) Understanding plant biomass via computational modeling. Adv Mater 33:2003206. https://doi.org/10.1002/adma.202003206
Zhang C, Shomali A, Guyer R et al (2020) Disentangling heat and moisture effects on biopolymer mechanics. Macromolecules 53:1527–1535. https://doi.org/10.1021/acs.macromol.9b01988
Zhang C, Chen M, Keten S et al (2021) Hygromechanical mechanisms of wood cell wall revealed by molecular modeling and mixture rule analysis. Sci Adv 7:eabi8919. https://doi.org/10.1126/sciadv.abi8919
Hoffmeyer P, Engelund Emil T, Thygesen Lisbeth G (2011) Equilibrium moisture content (EMC) in Norway spruce during the first and second desorptions. hfsg 65:875. https://doi.org/10.1515/hf.2011.112
Thybring EE, Fredriksson M, Zelinka SL, Glass SV (2022) Water in wood: a review of current understanding and knowledge gaps. Forests 13:2051. https://doi.org/10.3390/f13122051
Thybring EE, Kymäläinen M, Rautkari L (2018) Experimental techniques for characterising water in wood covering the range from dry to fully water-saturated. Wood Sci Technol 52:297–329. https://doi.org/10.1007/s00226-017-0977-7
Skaar C (1988) Wood-water relations. Springer, Berlin
Tsoumis GT (2009) Science and technology of wood: structure, properties, utilization. Verlag Kessel, Remagen-Oberwinter
Simpson W (1980) Sorption theories applied to wood. Wood and Fiber Science 3:183–195
Siau JF (2012) Transport processes in wood. Springer Science & Business Media, Berlin
Ananthakrishnaiyer V (1970) Sorption of aqueous and nonaqueous media by wood and cellulose. Chem Rev 70:619–637. https://doi.org/10.1021/cr60268a001
Fredriksson M, Thybring EE (2018) Scanning or desorption isotherms? Characterising sorption hysteresis of wood. Cellulose 25:4477–4485. https://doi.org/10.1007/s10570-018-1898-9
Patera A, Derluyn H, Derome D, Carmeliet J (2016) Influence of sorption hysteresis on moisture transport in wood. Wood Sci Technol 50:259–283. https://doi.org/10.1007/s00226-015-0786-9
Fredriksson M, Thybring EE (2019) On sorption hysteresis in wood: separating hysteresis in cell wall water and capillary water in the full moisture range. PLoS ONE 14:e0225111. https://doi.org/10.1371/journal.pone.0225111
Glass SV, Boardman CR, Zelinka SL (2017) Short hold times in dynamic vapour sorption measurements mischaracterize the equilibrium moisture content of wood. Wood Sci Technol 51:243–260
Glass SV, Boardman CR, Thybring EE, Zelinka SL (2018) Quantifying and reducing errors in equilibrium moisture content measurements with dynamic vapor sorption (DVS) experiments. Wood Sci Technol. https://doi.org/10.1007/s00226-018-1007-0
Thybring EE, Glass VS, Zelinka LS (2019) Kinetics of water vapor sorption in wood cell walls: state of the art and research needs. Forests. https://doi.org/10.3390/f10080704
Popescu C-M, Hill CAS (2013) The water vapour adsorption–desorption behaviour of naturally aged Tilia cordata Mill. wood. Polym Degrad Stab 98:1804–1813. https://doi.org/10.1016/j.polymdegradstab.2013.05.021
Barkas WW (1935) Fibre saturation point of wood. Nature 135:545–545. https://doi.org/10.1038/135545b0
Babiak M, Kudela J (1995) A contribution to the definition of the fiber saturation point. Wood SciTechnol. https://doi.org/10.1007/BF00204589
Hill CAS (2008) The reduction in the fibre saturation point of wood due to chemical modification using anhydride reagents: a reappraisal. Holzforschung 62:423–428. https://doi.org/10.1515/HF.2008.078
Hill CAS, Norton A, Newman G (2009) The water vapor sorption behavior of natural fibers. J Appl Polym Sci 112:1524–1537. https://doi.org/10.1002/app.29725
Hill C, Ramsay J, Keating B et al (2012) The water vapour sorption properties of thermally modified and densified wood. J Mater Sci 47:3191–3197. https://doi.org/10.1007/s10853-011-6154-8
Yang T, Wang J, Sheng N, Ma E (2018) Comparison of dynamic sorption and hygroexpansion of wood by different cyclic hygrothermal changing effects II. J Building Phys 41:360–376. https://doi.org/10.1177/1744259117708353
Kelsey K (1957) The sorption of water vapor by wood. Aust J Appl Sci 8:42–54
Salmén L, Larsson PA (2018) On the origin of sorption hysteresis in cellulosic materials. Carbohyd Polym 182:15–20. https://doi.org/10.1016/j.carbpol.2017.11.005
Djolani B (1972) Hystérèse et effets de second ordre de la sorption d’humidité dans le bois aux températures de 5°, 21°, 35°, 50° C. Annales des Sciences Forestières 29:465–474
Krupińska B, Strømmen I, Pakowski Z, Eikevik TM (2007) Modeling of sorption isotherms of various kinds of wood at different temperature conditions. Drying Technol 25:1463–1470. https://doi.org/10.1080/07373930701537062
Rémond R, Almeida G, Perré P (2018) The gripped-box model: a simple and robust formulation of sorption hysteresis for lignocellulosic materials. Constr Build Mater 170:716–724. https://doi.org/10.1016/j.conbuildmat.2018.02.116
Mmari W, Johannesson B (2020) Modeling transient and hysteretic hygrothermal processes in wood using the hybrid mixture theory. Int J Heat Mass Transf 163:120408. https://doi.org/10.1016/j.ijheatmasstransfer.2020.120408
Urquhart AR (1929) The mechanism of the adsorption of water by cotton. J Text Inst Trans 20:T125–T132. https://doi.org/10.1080/19447022908661485
Chen C-M, Wangaard FF (1968) Wettability and the hysteresis effect in the sorption of water vapor by wood. Wood Sci Technol 2:177–187. https://doi.org/10.1007/BF00350907
Peralta PN (1995) Modeling wood moisture sorption hysteresis using the independent-domain theory. Wood Fiber Sci 27:250–257
Peralta PN (1996) Moisture sorption hysteresis and the independent-domain theory: the moisture distribution function. Wood Fiber Sci 28:406–410
Peralta PN (1998) Modeling wood moisture sorption hysteresis based on similarity hypothesis. Part 1. Direct Approach Wood Fiber Sci 30:48–55
Lund Frandsen H, Svensson S, Damkilde L (2007) A hysteresis model suitable for numerical simulation of moisture content in wood. Holzforschung 61:175–181. https://doi.org/10.1515/HF.2007.031
Shi J, Avramidis S (2017) Water sorption hysteresis in wood: I review and experimental patterns—geometric characteristics of scannig curves. Holzforschung 71:307–316
Zhang X, Zillig W, Künzel HM et al (2015) Evaluation of moisture sorption models and modified Mualem model for prediction of desorption isotherm for wood materials. Build Environ 92:387–395. https://doi.org/10.1016/j.buildenv.2015.05.021
Enderby JA (1955) The domain model of hysteresis. Part 1.—independent domains. Trans Faraday Soc 51:835–848. https://doi.org/10.1039/TF9555100835
Everett DH (1954) A general approach to hysteresis. Part 3—A formal treatment of the independent domain model of hysteresis. Trans Faraday Soc 50:1077–1096. https://doi.org/10.1039/TF9545001077
Willems W (2014) The water vapor sorption mechanism and its hysteresis in wood: the water/void mixture postulate. Wood Sci Technol 48:499–518. https://doi.org/10.1007/s00226-014-0617-4
Chen M, Coasne B, Guyer R et al (2018) Role of hydrogen bonding in hysteresis observed in sorption-induced swelling of soft nanoporous polymers. Nat Commun 9:3507. https://doi.org/10.1038/s41467-018-05897-9
Chen M, Coasne B, Derome D, Carmeliet J (2020) Role of cellulose nanocrystals on hysteretic sorption and deformation of nanocomposites. Cellulose 27:6945–6960. https://doi.org/10.1007/s10570-020-03247-x
Keating BA, Hill CAS, Sun D et al (2013) The water vapor sorption behavior of a galactomannan cellulose nanocomposite film analyzed using parallel exponential kinetics and the Kelvin-Voigt viscoelastic model. J Appl Polym Sci 129:2352–2359. https://doi.org/10.1002/app.39132
van der Sman RGM (2023) Effects of viscoelasticity on moisture sorption of maltodextrins. Food Hydrocolloids 139:108481. https://doi.org/10.1016/j.foodhyd.2023.108481
Björklund S, Kocherbitov V (2019) Water vapor sorption-desorption hysteresis in glassy surface films of mucins investigated by humidity scanning QCM-D. J Colloid Interface Sci 545:289–300. https://doi.org/10.1016/j.jcis.2019.03.037
Hill CAS, Keating BA, Jalaludin Z, Mahrdt E (2012) A rheological description of the water vapour sorption kinetics behaviour of wood invoking a model using a canonical assembly of Kelvin-Voigt elements and a possible link with sorption hysteresis. Holzforschung. https://doi.org/10.1515/HF.2011.115
Bryan WP (1987) Thermodynamic models for water-protein sorption hysteresis. Biopolymers 26:1705–1716. https://doi.org/10.1002/bip.360261005
Morrison JL, Dzieciuch MA (1959) The thermodynamic properties of the system cellulose—water vapor. Can J Chem 37:1379–1390. https://doi.org/10.1139/v59-202
Barkas WW (1936) Wood-water relationships: (I) molecular sorption and capillary retention of water by Sitka spruce wood. Proc Phys Soc 48:1–17. https://doi.org/10.1088/0959-5309/48/1/302
Barkas WW (1937) Wood-water relationships, part III. Molecular sorption of water by sitka spruce wood. Proc Phys Soc 49:237
Smith SE (1947) The Sorption of Water Vapor by High Polymers. J Am Chem Soc 69:646–651. https://doi.org/10.1021/ja01195a053
Salmen N, Back E (1977) The influence of water on the glass transition temperature of cellulose. Tappi 60:137–140
Xie Y, Hill CAS, Jalaludin Z, Sun D (2011) The water vapour sorption behaviour of three celluloses: analysis using parallel exponential kinetics and interpretation using the Kelvin-Voigt viscoelastic model. Cellulose 18:517–530. https://doi.org/10.1007/s10570-011-9512-4
Mihranyan A, Llagostera AP, Karmhag R et al (2004) Moisture sorption by cellulose powders of varying crystallinity. Int J Pharm 269:433–442. https://doi.org/10.1016/j.ijpharm.2003.09.030
Almeida G, Hernández RE (2006) Changes in physical properties of tropical and temperate hardwoods below and above the fiber saturation point. Wood Sci Technol 40:599–613. https://doi.org/10.1007/s00226-006-0083-8
Tarmian A, Burgert I, Thybring EE (2017) Hydroxyl accessibility in wood by deuterium exchange and ATR-FTIR spectroscopy: methodological uncertainties. Wood Sci Technol 51:845–853. https://doi.org/10.1007/s00226-017-0922-9
Altgen M, Rautkari L (2021) Humidity-dependence of the hydroxyl accessibility in Norway spruce wood. Cellulose 28:45–58. https://doi.org/10.1007/s10570-020-03535-6
Väisänen S, Pönni R, Hämäläinen A, Vuorinen T (2018) Quantification of accessible hydroxyl groups in cellulosic pulps by dynamic vapor sorption with deuterium exchange. Cellulose 25:6923–6934. https://doi.org/10.1007/s10570-018-2064-0
Zelinka SL, Lambrecht MJ, Glass SV et al (2012) Examination of water phase transitions in Loblolly pine and cell wall components by differential scanning calorimetry. Thermochim Acta 533:39–45. https://doi.org/10.1016/j.tca.2012.01.015
Hartley ID, Kamke FA, Peemoeller H (1992) Cluster theory for water sorption in wood. Wood Sci Technol 26:83–99. https://doi.org/10.1007/BF00194465
Hartley ID, Avramidis S (1993) Analysis of the wood sorption isotherm using clustering theory. Holzforschung 47:163–167. https://doi.org/10.1515/hfsg.1993.47.2.163
Hartley ID, Kamke FA, Peemoeller H (1994) Absolute moisture content determination of aspen wood below the fiber saturation point using pulsed NMR. Holzforschung 48:474–479. https://doi.org/10.1515/hfsg.1994.48.6.474
Rawat SPS, Khali DP (1998) Clustering of water molecules during adsorption of water in wood. J Polym Sci B Polym Phys 36:665–671. https://doi.org/10.1002/(SICI)1099-0488(199803)36:4%3c665::AID-POLB12%3e3.0.CO;2-D
Khali DP, Rawat SPS (2000) Clustering of water molecules during adsorption of water in brown rot decayed and undecayed wood blocks of Pinus sylvestris. Holz als Roh- und Werkstoff 58:340–341. https://doi.org/10.1007/s001070050441
Shi J, Avramidis S (2021) Nanopore-level wood-water interactions—a molecular simulation study. Forests 12:356. https://doi.org/10.3390/f12030356
Kymäläinen M, Rautkari L, Hill CAS (2015) Sorption behaviour of torrefied wood and charcoal determined by dynamic vapour sorption. J Mater Sci 50:7673–7680. https://doi.org/10.1007/s10853-015-9332-2
Engelund ET, Thygesen LG, Svensson S, Hill CAS (2013) A critical discussion of the physics of wood–water interactions. Wood Sci Technol 47:141–161. https://doi.org/10.1007/s00226-012-0514-7
Himmel S, Mai C (2015) Effects of acetylation and formalization on the dynamic vapor sorption behavior of wood. Holzforschung 69:633–643. https://doi.org/10.1515/hf-2014-0161
Li J, Ma E (2021) Characterization of water in wood by time-domain nuclear magnetic resonance spectroscopy (TD-NMR): a review. Forests 12:886. https://doi.org/10.3390/f12070886
Willems W (2018) Hygroscopic wood moisture: single and dimerized water molecules at hydroxyl-pair sites? Wood Sci Technol 52:777–791. https://doi.org/10.1007/s00226-018-0998-x
Walker JCF (2006) Water in wood. Primary wood processing. Springer, Netherlands, pp 69–94
Thybring EE, Thygesen LG, Burgert I (2017) Hydroxyl accessibility in wood cell walls as affected by drying and re-wetting procedures. Cellulose 24:2375–2384. https://doi.org/10.1007/s10570-017-1278-x
Plaza NZ, Pingali SV, Qian S et al (2016) Informing the improvement of forest products durability using small angle neutron scattering. Cellulose 23:1593–1607. https://doi.org/10.1007/s10570-016-0933-y
Zitting A, Paajanen A, Rautkari L, Penttilä PA (2021) Deswelling of microfibril bundles in drying wood studied by small-angle neutron scattering and molecular dynamics. Cellulose 28:10765–10776. https://doi.org/10.1007/s10570-021-04204-y
Grunin YB, Grunin LYu, Schiraya VYu et al (2020) Cellulose–water system’s state analysis by proton nuclear magnetic resonance and sorption measurements. Bioresour Bioprocess 7:41. https://doi.org/10.1186/s40643-020-00332-8
Fratzl P, Burgert I, Keckes J (2022) Mechanical model for the deformation of the wood cell wall. Int J Mater Res 95:579–584. https://doi.org/10.1515/ijmr-2004-0112
Burgert I, Fratzl P (2009) Actuation systems in plants as prototypes for bioinspired devices. Phil Trans R Soc A 367:1541–1557. https://doi.org/10.1098/rsta.2009.0003
Derome D, Griffa M, Koebel M, Carmeliet J (2011) Hysteretic swelling of wood at cellular scale probed by phase-contrast X-ray tomography. J Struct Biol 173:180–190. https://doi.org/10.1016/j.jsb.2010.08.011
Perstorper M, Johansson M, Kliger R, Johansson G (2001) Distortion of Norway spruce timber. Holz als Roh- und Werkstoff 59:94–103. https://doi.org/10.1007/s001070050481
Ormarsson S, Dahlblom O, Johansson M (2009) Finite element study of growth stress formation in wood and related distortion of sawn timber. Wood Sci Technol 43:387–403. https://doi.org/10.1007/s00226-008-0209-2
Angst V, Malo KA (2010) Moisture induced stresses perpendicular to the grain in glulam: Review and evaluation of the relative importance of models and parameters. Holzforschung. https://doi.org/10.1515/hf.2010.089
Cheng W, Morooka T, Wu Q, Liu Y (2007) Characterization of tangential shrinkage stresses of wood during drying under superheated steam above 100°C. For Prod J 57:39–43
Garat W, Le Moigne N, Corn S et al (2020) Swelling of natural fibre bundles under hygro- and hydrothermal conditions: determination of hydric expansion coefficients by automated laser scanning. Compos A Appl Sci Manuf 131:105803. https://doi.org/10.1016/j.compositesa.2020.105803
Yamamoto H, Sassus F, Ninomiya M, Gril J (2001) A model of anisotropic swelling and shrinking process of wood. Wood Sci Technol 35:167–181. https://doi.org/10.1007/s002260000074
Patera A, Jefimovs K, Rafsanjani A et al (2014) Micro-scale restraint methodology for humidity induced swelling investigated by phase contrast X-ray tomography. Exp Mech 54:1215–1226. https://doi.org/10.1007/s11340-014-9894-y
Rafsanjani A, Stiefel M, Jefimovs K et al (2014) Hygroscopic swelling and shrinkage of latewood cell wall micropillars reveal ultrastructural anisotropy. J R Soc Interface 11:20140126. https://doi.org/10.1098/rsif.2014.0126
Chauhan SS, Aggarwal P (2004) Effect of moisture sorption state on transverse dimensional changes in wood. Holz Roh Werkst 62:50–55. https://doi.org/10.1007/s00107-003-0437-y
Hofer U, Pichler C, Maderebner R, Lackner R (2019) Lomnitz-type viscoelastic behavior of clear spruce wood as identified by creep and relaxation experiments: influence of moisture content and elevated temperatures up to 80 °C. Wood Sci Technol 53:765–783. https://doi.org/10.1007/s00226-019-01099-8
Derrien K, Gilormini P (2009) The effect of moisture-induced swelling on the absorption capacity of transversely isotropic elastic polymer–matrix composites. Int J Solids Struct 46:1547–1553. https://doi.org/10.1016/j.ijsolstr.2008.11.014
Dubois F, Husson J-M, Sauvat N, Manfoumbi N (2012) Modeling of the viscoelastic mechano-sorptive behavior in wood. Mech Time Depend Mater 16:439–460. https://doi.org/10.1007/s11043-012-9171-3
Hill CAS, Xie Y (2011) The dynamic water vapour sorption properties of natural fibres and viscoelastic behaviour of the cell wall: is there a link between sorption kinetics and hysteresis? J Mater Sci 46:3738–3748. https://doi.org/10.1007/s10853-011-5286-1
Dinwoodie JM (1989) Wood: nature’s cellular, polymeric fibre-composite. Institute of Metals, London
Desch HE, Dinwoodie JM (1996) Timber: structure, properties, conversion and use, 7th edn. Macmillan, Basingstoke
RogerE H (1993) Influence of moisture sorption history on the swelling of sugar maple wood and some tropical hardwoods. Wood SciTechnol 27:337–345. https://doi.org/10.1007/BF00192220
Carrington H (1922) The elastic constants of spruce as influenced by moisture. Aeronaut J 26:462–471. https://doi.org/10.1017/S2398187300139465
Kollmann F, Krech H (1960) Dynamische Messung der elastischen Holzeigenschaften und der Dämpfung Ein Beitrag zur zerstörungsfreien Werkstoffprüfung. Holz als Roh-und Werkstoff 18:41–54. https://doi.org/10.1007/BF02615616
Kaijita S, Yamada T, Suzuki M (1961) Studies on rheological properties of wood. I Mokuzai Gakkaishi 7:29–33
James W (1961) Internal friction and speed in Douglas fir. For Prod J 11:383–388
Nakao S, Nakano T (2011) Analysis of molecular dynamics of moist wood components by applying the stretched-exponential function. J Mater Sci 46:4748–4755. https://doi.org/10.1007/s10853-011-5385-z
Brémaud I, Gril J (2021) Moisture content dependence of anisotropic vibrational properties of wood at quasi equilibrium: analytical review and multi-trajectories experiments. Holzforschung 75:313–327. https://doi.org/10.1515/hf-2020-0028
Armstrong L, Kingston R (1960) Effect of moisture changes on creep in wood. Nature 185:862–863
Grossman PUA (1976) Requirements for a model that exhibits mechano-sorptive behaviour. Wood Sci Technol 10:163–168. https://doi.org/10.1007/BF00355737
Saifouni O, Destrebecq J-F, Froidevaux J, Navi P (2016) Experimental study of the mechanosorptive behaviour of softwood in relaxation. Wood Sci Technol 50:789–805. https://doi.org/10.1007/s00226-016-0816-2
Holzer SM, Loferski JR, Dillard DA (1989) A review of creep in wood: concepts relevant to develop long-term behavior predictions for wood structures. Wood Fiber Sci 4:376–392
Mohager S, Toratti T (1992) Long term bending creep of wood in cyclic relative humidity. Wood SciTechnol. https://doi.org/10.1007/BF00203409
Takahashi C, Ishimaru Y, Iida I, Furuta Y (2005) The creep of wood destabilized by change in moisture content. Part 2: the creep behaviors of wood during and immediately after adsorption. Holzforschung 59:46–53. https://doi.org/10.1515/HF.2005.008
Liu T (1993) Creep of wood under a large span of loads in constant and varying environments. Pt. 1: Experimental observations and analysis. Holz als Roh-und Werkstoff (Germany)
Hill C, Hughes M (2010) Natural fibre reinforced composites opportunities and challenges. J Biobased Mat Bioenergy 4:148–158. https://doi.org/10.1166/jbmb.2010.1079
Svensson S, Toratti T (2002) Mechanical response of wood perpendicular to grain when subjected to changes of humidity. Wood Sci Technol 36:145–156. https://doi.org/10.1007/s00226-001-0130-4
Houška M, Koc P (2000) Sorptive stress estimation: an important key to the mechano-sorptive effect in wood. Mech Time Depend Mater 4:81–98. https://doi.org/10.1023/A:1009897822196
Montero C, Gril J, Legeas C et al (2012) Influence of hygromechanical history on the longitudinal mechanosorptive creep of wood. Holzforschung 66:757–764. https://doi.org/10.1515/hf-2011-0174
Hassani MM, Wittel FK, Hering S, Herrmann HJ (2015) Rheological model for wood. Comput Methods Appl Mech Eng 283:1032–1060. https://doi.org/10.1016/j.cma.2014.10.031
Zhang C, Keten S, Derome D, Carmeliet J (2021) Hydrogen bonds dominated frictional stick-slip of cellulose nanocrystals. Carbohyd Polym 258:117682. https://doi.org/10.1016/j.carbpol.2021.117682
Sinko R, Keten S (2015) Traction–separation laws and stick–slip shear phenomenon of interfaces between cellulose nanocrystals. J Mech Phys Solids 78:526–539. https://doi.org/10.1016/j.jmps.2015.02.012
McKenzie WM, Karpovich H (1968) The frictional behaviour of wood. Wood SciTechnol 2:139–152. https://doi.org/10.1007/BF00394962
Adler DC, Buehler MJ (2013) Mesoscale mechanics of wood cell walls under axial strain. Soft Matter 9:7138. https://doi.org/10.1039/c3sm50183c
Keckes J, Burgert I, Frühmann K et al (2003) Cell-wall recovery after irreversible deformation of wood. Nature Mater 2:810–813. https://doi.org/10.1038/nmat1019
He Z, Wu H, Xia J et al (2023) How weak hydration interfaces simultaneously strengthen and toughen nanocellulose materials. Extreme Mech Lett 58:101947. https://doi.org/10.1016/j.eml.2022.101947
Zhang C, Chen M, Keten S et al (2021) Towards unraveling the moisture-induced shape memory effect of wood: the role of interface mechanics revealed by upscaling atomistic to composite modeling. NPG Asia Mater 13:74. https://doi.org/10.1038/s41427-021-00342-8
Altaner CM, Jarvis MC (2008) Modelling polymer interactions of the ‘molecular Velcro’ type in wood under mechanical stress. J Theor Biol 253:434–445. https://doi.org/10.1016/j.jtbi.2008.03.010
Hoffmeyer P, Davidson RW (1989) Mechano-sorptive creep mechanism of wood in compression and bending. Wood Sci Technol 23:215–227. https://doi.org/10.1007/BF00367735
Kretschmann D (2003) Velcro mechanics in wood. Nat Mater 2:775
Placet V, Cissé O, Lamine Boubakar M (2014) Nonlinear tensile behaviour of elementary hemp fibres. Part I: investigation of the possible origins using repeated progressive loading with in situ microscopic observations. Compos A Appl Sci Manuf 56:319–327. https://doi.org/10.1016/j.compositesa.2012.11.019
Hill CAS, Papadopoulos AN (2001) A review of methods used to determine the size of the cell wall microvoids of wood. J Inst Wood Sci 15:337–345
Papadopoulos AN, Hill CAS, Gkaraveli A (2003) Determination of surface area and pore volume of holocellulose and chemically modified wood flour using the nitrogen adsorption technique. Holz Roh Werkst 61:453–456. https://doi.org/10.1007/s00107-003-0430-5
Nopens M, Sazama U, König S et al (2020) Determination of mesopores in the wood cell wall at dry and wet state. Sci Rep 10:9543. https://doi.org/10.1038/s41598-020-65066-1
Kojiro K, Miki T, Sugimoto H et al (2010) Micropores and mesopores in the cell wall of dry wood. J Wood Sci 56:107–111. https://doi.org/10.1007/s10086-009-1063-z
Maloney T (1999) The formation of pores in the cell wall. J Pulp Pap Sci 25:430–436
Chesson A, Gardner PT, Wood TJ (1997) Cell wall porosity and available surface area of wheat straw and wheat grain fractions. J Sci Food Agric 75:289–295. https://doi.org/10.1002/(SICI)1097-0010(199711)75:3%3c289::AID-JSFA879%3e3.0.CO;2-R
Gardner PT, Wood TJ, Chesson A, Stuchbury T (1999) Effect of degradation on the porosity and surface area of forage cell walls of differing lignin content. J Sci Food Agric 79:11–18. https://doi.org/10.1002/(SICI)1097-0010(199901)79:1%3c11::AID-JSFA159%3e3.0.CO;2-6
Donaldson L (2007) Cellulose microfibril aggregates and their size variation with cell wall type. Wood Sci Technol 41:443–460. https://doi.org/10.1007/s00226-006-0121-6
Carmona C, Langan P, Smith JC, Petridis L (2015) Why genetic modification of lignin leads to low-recalcitrance biomass. Phys Chem Chem Phys 17:358–364. https://doi.org/10.1039/C4CP05004E
Fernando D, Kowalczyk M, Guindos P et al (2023) Electron tomography unravels new insights into fiber cell wall nanostructure; exploring 3D macromolecular biopolymeric nano-architecture of spruce fiber secondary walls. Sci Rep 13:2350. https://doi.org/10.1038/s41598-023-29113-x
Persson PV, Hafrén J, Fogden A et al (2004) Silica nanocasts of wood fibers: a study of cell-wall accessibility and structure. Biomacromol 5:1097–1101. https://doi.org/10.1021/bm034532u
Fahlén J, Salmen L (2005) Pore and matrix distribution in the fiber wall revealed by atomic force microscopy and image analysis. Biomacromol 6:433–438. https://doi.org/10.1021/bm040068x
Hinedi ZR, Chang AC, Anderson MA, Borchardt DB (1997) Quantification of microporosity by nuclear magnetic resonance relaxation of water imbibed in porous media. Water Resour Res 33:2697–2704. https://doi.org/10.1029/97WR02408
Li X, Zhao Z (2020) Time domain-NMR studies of average pore size of wood cell walls during drying and moisture adsorption. Wood Sci Technol 54:1241–1251. https://doi.org/10.1007/s00226-020-01209-x
Penttilä PA, Zitting A, Lourençon T et al (2021) Water-accessibility of interfibrillar spaces in spruce wood cell walls. Cellulose 28:11231–11245. https://doi.org/10.1007/s10570-021-04253-3
Donaldson LA, Kroese HW, Hill SJ, Franich RA (2015) Detection of wood cell wall porosity using small carbohydrate molecules and confocal fluorescence microscopy. J Microsc 259:228–236. https://doi.org/10.1111/jmi.12257
Liang R, Zhu Y-H, Wen L et al (2020) Exploration of effect of delignification on the mesopore structure in poplar cell wall by nitrogen absorption method. Cellulose 27:1921–1932. https://doi.org/10.1007/s10570-019-02921-z
Hill CAS, Forster SC, Farahani MRM et al (2005) An investigation of cell wall micropore blocking as a possible mechanism for the decay resistance of anhydride modified wood. Int Biodeterior Biodegradation 55:69–76. https://doi.org/10.1016/j.ibiod.2004.07.003
Park S, Venditti RA, Jameel H, Pawlak JJ (2006) Changes in pore size distribution during the drying of cellulose fibers as measured by differential scanning calorimetry. Carbohyd Polym 66:97–103. https://doi.org/10.1016/j.carbpol.2006.02.026
Murr A (2019) The relevance of water vapour transport for water vapour sorption experiments on small wooden samples. Transp Porous Med 128:385–404. https://doi.org/10.1007/s11242-019-01253-7
Murr A (2022) Water vapour sorption and moisture transport in and across fibre direction of wood. Cellulose 29:4135–4152. https://doi.org/10.1007/s10570-022-04520-x
Murr A, Lackner R (2018) Analysis on the influence of grain size and grain layer thickness on the sorption kinetics of grained wood at low relative humidity with the use of water vapour sorption experiments. Wood Sci Technol 52:753–776. https://doi.org/10.1007/s00226-018-1003-4
Kohler R, Alex R, Brielmann R, Ausperger B (2006) A new kinetic model for water sorption isotherms of cellulosic materials. Macromol Symp 244:89–96. https://doi.org/10.1002/masy.200651208
Thybring EE, Boardman CR, Glass SV, Zelinka SL (2018) The parallel exponential kinetics model is unfit to characterize moisture sorption kinetics in cellulosic materials. Cellulose. https://doi.org/10.1007/s10570-018-2134-3
Fredriksson M, Thygesen LG (2017) The states of water in Norway spruce ( Picea abies (L.) Karst.) studied by low-field nuclear magnetic resonance (LFNMR) relaxometry: assignment of free-water populations based on quantitative wood anatomy. Holzforschung 71:77–90. https://doi.org/10.1515/hf-2016-0044
Beck G, Thybring EE, Thygesen LG (2018) Brown-rot fungal degradation and de-acetylation of acetylated wood. Int Biodeterior Biodegradation 135:62–70. https://doi.org/10.1016/j.ibiod.2018.09.009
Zhang Y, Jiang H, Li F et al (2017) Graphene oxide based moisture-responsive biomimetic film actuators with nacre-like layered structures. J Mater Chem A 5:14604–14610. https://doi.org/10.1039/C7TA04208F
Fratzl P, Weinkamer R (2007) Nature’s hierarchical materials. Prog Mater Sci 52:1263–1334. https://doi.org/10.1016/j.pmatsci.2007.06.001
Fratzl P, Barth FG (2009) Biomaterial systems for mechanosensing and actuation. Nature 462:442–448. https://doi.org/10.1038/nature08603
Bertinetti L, Hangen UD, Eder M et al (2015) Characterizing moisture-dependent mechanical properties of organic materials: humidity-controlled static and dynamic nanoindentation of wood cell walls. Phil Mag 95:1992–1998. https://doi.org/10.1080/14786435.2014.920544
Solhi L, Guccini V, Heise K et al (2023) Understanding nanocellulose-water interactions: turning a detriment into an asset. Chem Rev. https://doi.org/10.1021/acs.chemrev.2c00611
Jin K, Qin Z, Buehler MJ (2015) Molecular deformation mechanisms of the wood cell wall material. J Mech Behav Biomed Mater 42:198–206. https://doi.org/10.1016/j.jmbbm.2014.11.010
Li S, Wang KW (2016) Plant-inspired adaptive structures and materials for morphing and actuation: a review. Bioinspir Biomim 12:011001. https://doi.org/10.1088/1748-3190/12/1/011001
Etale A, Onyianta AJ, Turner SR, Eichhorn SJ (2023) Cellulose: a review of water interactions, applications in composites, and water treatment. Chem Rev. https://doi.org/10.1021/acs.chemrev.2c00477
Behl M, Lendlein A (2007) Shape-memory polymers. Mater Today 10:20–28. https://doi.org/10.1016/S1369-7021(07)70047-0
Behl M, Zotzmann J, Lendlein A (2009) Shape-memory polymers and shape-changing polymers. In: Lendlein A (ed) Shape-memory polymers. Springer, Berlin, pp 1–40
Zhao Q, Behl M, Lendlein A (2013) Shape-memory polymers with multiple transitions: complex actively moving polymers. Soft Matter 9:1744–1755. https://doi.org/10.1039/C2SM27077C
Hu J, Meng H, Li G, Ibekwe SI (2012) A review of stimuli-responsive polymers for smart textile applications. Smart Mater Struct 21:053001. https://doi.org/10.1088/0964-1726/21/5/053001
Montero de Espinosa L, Meesorn W, Moatsou D, Weder C (2017) Bioinspired polymer systems with stimuli-responsive mechanical properties. Chem Rev 117:12851–12892. https://doi.org/10.1021/acs.chemrev.7b00168
Oliver K, Seddon A, Trask RS (2016) Morphing in nature and beyond: a review of natural and synthetic shape-changing materials and mechanisms. J Mater Sci 51:10663–10689. https://doi.org/10.1007/s10853-016-0295-8
Jakes JE, Plaza N, Zelinka SL et al (2014) Wood as inspiration for new stimuli-responsive structures and materials. In: Lakhtakia A (ed) San Diego. California, USA, p 90550K
Berglund LA, Burgert I (2018) Bioinspired wood nanotechnology for functional materials. Adv Mater 30:1704285. https://doi.org/10.1002/adma.201704285
Le Duigou A, Requile S, Beaugrand J et al (2017) Natural fibres actuators for smart bio-inspired hygromorph biocomposites. Smart Mater Struct 26:125009. https://doi.org/10.1088/1361-665X/aa9410
Mirvakili SM, Hunter IW (2018) Artificial muscles: mechanisms, applications, and challenges. Adv Mater 30:1704407. https://doi.org/10.1002/adma.201704407
Qiu X, Hu S (2013) “Smart” materials based on cellulose: a review of the preparations, properties, and applications. Materials 6:738–781. https://doi.org/10.3390/ma6030738
Kontturi E, Laaksonen P, Linder MB et al (2018) Advanced materials through assembly of nanocelluloses. Adv Mater 30:1703779. https://doi.org/10.1002/adma.201703779
Dallmeyer I, Chowdhury S, Kadla JF (2013) Preparation and characterization of kraft lignin-based moisture-responsive films with reversible shape-change capability. Biomacromol 14:2354–2363. https://doi.org/10.1021/bm400465p
Agarwal S, Jiang S, Chen Y (2019) Progress in the field of water- and/or temperature-triggered polymer actuators. Macromol Mater Eng 304:1800548. https://doi.org/10.1002/mame.201800548
Falahati M, Ahmadvand P, Safaee S et al (2020) Smart polymers and nanocomposites for 3D and 4D printing. Mater Today 40:215–245. https://doi.org/10.1016/j.mattod.2020.06.001
Joshi S, Rawat K et al (2020) 4D printing of materials for the future: opportunities and challenges. Appl Mater Today 18:100490. https://doi.org/10.1016/j.apmt.2019.100490
Qi J, Wu T, Wang W et al (2022) Solid-phase molecular self-assembly facilitated supramolecular films with alternative hydrophobic/hydrophilic domains for skin moisture detection. Aggregate. https://doi.org/10.1002/agt2.173
Wang Q, Sun J, Yao Q et al (2018) 3D printing with cellulose materials. Cellulose 25:4275–4301. https://doi.org/10.1007/s10570-018-1888-y
Guvendiren M, Yang S, Burdick JA (2009) Swelling-induced surface patterns in hydrogels with gradient crosslinking density. Adv Funct Mater 19:3038–3045. https://doi.org/10.1002/adfm.200900622
Zhang K, Geissler A, Standhardt M et al (2015) Moisture-responsive films of cellulose stearoyl esters showing reversible shape transitions. Sci Rep 5:11011. https://doi.org/10.1038/srep11011
Li Y-C, Zhang YS, Akpek A et al (2016) 4D bioprinting: the next-generation technology for biofabrication enabled by stimuli-responsive materials. Biofabrication 9:012001. https://doi.org/10.1088/1758-5090/9/1/012001
Akbar I, El Hadrouz M, El Mansori M, Lagoudas D (2022) Toward enabling manufacturing paradigm of 4D printing of shape memory materials: open literature review. Eur Polymer J 168:111106. https://doi.org/10.1016/j.eurpolymj.2022.111106
Ding H, Zhang X, Liu Y, Ramakrishna S (2019) Review of mechanisms and deformation behaviors in 4D printing. Int J Adv Manuf Technol 105:4633–4649. https://doi.org/10.1007/s00170-019-03871-3
Khalid MY, Arif ZU, Noroozi R et al (2022) 4D printing of shape memory polymer composites: a review on fabrication techniques, applications, and future perspectives. J Manuf Process 81:759–797. https://doi.org/10.1016/j.jmapro.2022.07.035
Le Duigou A, Correa D, Ueda M et al (2020) A review of 3D and 4D printing of natural fibre biocomposites. Mater Des 194:108911. https://doi.org/10.1016/j.matdes.2020.108911
Saritha D, Boyina D (2021) A concise review on 4D printing technology. Mater Today Proc 46:692–695. https://doi.org/10.1016/j.matpr.2020.12.016
Shie M-Y, Shen Y-F, Astuti SD et al (2019) Review of polymeric materials in 4d printing biomedical applications. Polymers 11:1864. https://doi.org/10.3390/polym11111864
Tamay DG, Dursun Usal T, Alagoz AS et al (2019) 3D and 4D printing of polymers for tissue engineering applications. Front Bioeng Biotechnol 7:164. https://doi.org/10.3389/fbioe.2019.00164
Zafar MQ, Zhao H (2020) 4D Printing: future insight in additive manufacturing. Met Mater Int 26:564–585. https://doi.org/10.1007/s12540-019-00441-w
Zhang Z, Demir KG, Gu GX (2019) Developments in 4D-printing: a review on current smart materials, technologies, and applications. Int J Smart Nano Mater 10:205–224. https://doi.org/10.1080/19475411.2019.1591541
Kirillova A, Ionov L (2019) Shape-changing polymers for biomedical applications. J Mater Chem B 7:1597–1624. https://doi.org/10.1039/C8TB02579G
Delaey J, Dubruel P, Van Vlierberghe S (2020) Shape-memory polymers for biomedical applications. Adv Funct Mater 30:1909047. https://doi.org/10.1002/adfm.201909047
Park Y, Chen X (2020) Water-responsive materials for sustainable energy applications. J Mater Chem A 8:15227–15244. https://doi.org/10.1039/D0TA02896G
Al-Obaidi KM, Azzam Ismail M, Hussein H, Abdul Rahman AM (2017) Biomimetic building skins: an adaptive approach. Renew Sustain Energy Rev 79:1472–1491. https://doi.org/10.1016/j.rser.2017.05.028
Cheng H, Hu Y, Zhao F et al (2014) Moisture-activated torsional graphene-fiber motor. Adv Mater 26:2909–2913. https://doi.org/10.1002/adma.201305708
Liu Y, Chen Z, Han D et al (2021) Bioinspired soft robots based on the moisture-responsive graphene oxide. Adv Sci 8:2002464. https://doi.org/10.1002/advs.202002464
Janbaz S, Hedayati R, Zadpoor AA (2016) Programming the shape-shifting of flat soft matter: from self-rolling/self-twisting materials to self-folding origami. Mater Horiz 3:536–547. https://doi.org/10.1039/C6MH00195E
Ilami M, Bagheri H, Ahmed R et al (2021) Materials, actuators, and sensors for soft bioinspired robots. Adv Mater 33:2003139. https://doi.org/10.1002/adma.202003139
Pu W, Wei F, Yao L, Xie S (2022) A review of humidity-driven actuator: toward high response speed and practical applications. J Mater Sci 57:12202–12235. https://doi.org/10.1007/s10853-022-07344-z
Han Y-G (2019) Relative humidity sensors based on microfiber knot resonators—a review. Sensors 19:5196. https://doi.org/10.3390/s19235196
Kumar Patel K, Purohit R (2018) Future prospects of shape memory polymer nano-composite and epoxy based shape memory polymer—a review. Mater Today Proc 5:20193–20200. https://doi.org/10.1016/j.matpr.2018.06.389
Liu A, Berglund LA (2012) Clay nanopaper composites of nacre-like structure based on montmorrilonite and cellulose nanofibers—Improvements due to chitosan addition. Carbohyd Polym 87:53–60. https://doi.org/10.1016/j.carbpol.2011.07.019
Das P, Malho J-M, Rahimi K et al (2015) Nacre-mimetics with synthetic nanoclays up to ultrahigh aspect ratios. Nat Commun 6:5967. https://doi.org/10.1038/ncomms6967
Ma J-N, Zhang Y-L, Han D-D et al (2020) Programmable deformation of patterned bimorph actuator swarm. Natl Sci Rev 7:775–785. https://doi.org/10.1093/nsr/nwz219
Siebert F, Hildebrandt P (2008) Vibrational spectroscopy in life science. Wiley-VCH Verlag GmbH & Co, Weinheim, Germany
Gierlinger N (2018) New insights into plant cell walls by vibrational microspectroscopy. Appl Spectrosc Rev 53:517–551. https://doi.org/10.1080/05704928.2017.1363052
Inagaki T, Yonenobu H, Tsuchikawa S (2008) Near-infrared spectroscopic monitoring of the water adsorption/desorption process in modern and archaeological wood. Appl Spectrosc 62:860–865. https://doi.org/10.1366/000370208785284312
Tsuchikawa S, Tsutsumi S (1998) Adsorptive and capillary condensed water in biological material. J Mater Sci Lett 17:661–663. https://doi.org/10.1023/A:1006672324163
Célino A, Gonçalves O, Jacquemin F, Fréour S (2014) Qualitative and quantitative assessment of water sorption in natural fibres using ATR-FTIR spectroscopy. Carbohyd Polym 101:163–170. https://doi.org/10.1016/j.carbpol.2013.09.023
Olsson A-M, Salmén L (2004) The association of water to cellulose and hemicellulose in paper examined by FTIR spectroscopy. Carbohyd Res 339:813–818. https://doi.org/10.1016/j.carres.2004.01.005
Laity PR, Hay JN (2000) Measurement of water diffusion through cellophane using attenuated total reflectance-fourier transform infrared spectroscopy. Cellulose 7:387–397. https://doi.org/10.1023/A:1009263118424
Hofstetter K, Hinterstoisser B, Salmén L (2006) Moisture uptake in native cellulose—the roles of different hydrogen bonds: a dynamic FT-IR study using Deuterium exchange. Cellulose 13:131–145. https://doi.org/10.1007/s10570-006-9055-2
Brubach J-B, Mermet A, Filabozzi A et al (2005) Signatures of the hydrogen bonding in the infrared bands of water. J Chem Phys 122:184509. https://doi.org/10.1063/1.1894929
Libnau FO, Kvalheim OM, Christy AA, Toft J (1994) Spectra of water in the near- and mid-infrared region. Vib Spectrosc 7:243–254. https://doi.org/10.1016/0924-2031(94)85014-3
Shou J-J, Wang F, Zeng G, Zhang Y-H (2011) Adsorption and desorption kinetics of water in lysozyme crystal investigated by confocal raman spectroscopy. J Phys Chem B 115:3708–3712. https://doi.org/10.1021/jp112404b
Guo Xin Wu, Yiqiang YN (2017) Characterizing spatial distribution of the adsorbed water in wood cell wall of Ginkgo biloba L. by μ-FTIR and confocal Raman spectroscopy. Holzforschung 71:415. https://doi.org/10.1515/hf-2016-0145
Schuttlefield J, Al-Hosney H, Zachariah A, Grassian VH (2007) Attenuated total reflection fourier transform infrared spectroscopy to investigate water uptake and phase transitions in atmospherically relevant particles. Appl Spectrosc 61:283–292. https://doi.org/10.1366/000370207780220868
Thygesen LG, Lundqvist S-O (2000) NIR measurement of moisture content in wood under unstable temperature conditions. Part 1. Thermal effects in near infrared spectra of wood. J Near Infrared Spectrosc 8:183–189. https://doi.org/10.1255/jnirs.277
Thygesen LG, Lundqvist S-O (2000) NIR measurement of moisture content in wood under unstable temperature conditions. Part 2. Handling temperature fluctuations. J Near Infrared Spectrosc 8:191–199. https://doi.org/10.1255/jnirs.278
Leblon B, Adedipe O, Hans G et al (2013) A review of near-infrared spectroscopy for monitoring moisture content and density of solid wood. For Chron 89:595–606. https://doi.org/10.5558/tfc2013-111
Fornés V, Chaussidon J (1978) An interpretation of the evolution with temperature of the ν2+ν3 combination band in water. J Chem Phys 68:4667–4671. https://doi.org/10.1063/1.435576
Suchy M, Virtanen J, Kontturi E, Vuorinen T (2010) Impact of drying on wood ultrastructure observed by deuterium exchange and photoacoustic FT-IR spectroscopy. Biomacromol 11:515–520. https://doi.org/10.1021/bm901268j
Altaner C, Apperley DC, Jarvis MC (2006) Spatial relationships between polymers in Sitka spruce: proton spin-diffusion studies. 60:665–673. https://doi.org/10.1515/HF.2006.112
Salmén L, Stevanic JS (2018) Effect of drying conditions on cellulose microfibril aggregation and “hornification.” Cellulose 25:6333–6344. https://doi.org/10.1007/s10570-018-2039-1
Mann J, Marrinan HJ (1956) The reaction between cellulose and heavy water. Part 1. A qualitative study by infra-red spectroscopy. Trans Faraday Soc 52:481–487. https://doi.org/10.1039/TF9565200481
Mann J, Marrinan HJ (1956) The reaction between cellulose and heavy water. Part 3—a quantitative study by infra-red spectroscopy. Trans Faraday Soc 52:492–497. https://doi.org/10.1039/TF9565200492
Venyaminov SYu, Prendergast FG (1997) Water (H2O and D2O) molar absorptivity in the 1000–4000 cm−1 range and quantitative infrared spectroscopy of aqueous solutions. Anal Biochem 248:234–245. https://doi.org/10.1006/abio.1997.2136
Amigo JM (2020) Hyperspectral imaging. Elsevier, Amsterdam
Boldrini B, Kessler W, Rebner K, Kessler RW (2012) Hyperspectral imaging: a review of best practice, performance and pitfalls for in-line and on-line applications. J Near Infrared Spectrosc 20:483–508. https://doi.org/10.1255/jnirs.1003
Kobori H, Gorretta N, Rabatel G et al (2013) Applicability of Vis-NIR hyperspectral imaging for monitoring wood moisture content (MC). Holzforschung 67:307–314. https://doi.org/10.1515/hf-2012-0054
Stefansson P, Thiis T, Gobakken LR, Burud I (2021) Hyperspectral NIR time series imaging used as a new method for estimating the moisture content dynamics of thermally modified Scots pine. Wood Mater Sci Eng 16:49–57. https://doi.org/10.1080/17480272.2020.1772366
Ma T, Morita G, Inagaki T, Tsuchikawa S (2022) Moisture transport dynamics in wood during drying studied by long-wave near-infrared hyperspectral imaging. Cellulose 29:133–145. https://doi.org/10.1007/s10570-021-04290-y
Awais M, Altgen M, Mäkelä M et al (2022) Quantitative prediction of moisture content distribution in acetylated wood using near-infrared hyperspectral imaging. J Mater Sci 57:3416–3429. https://doi.org/10.1007/s10853-021-06812-2
Ma T, Schimleck L, Inagaki T, Tsuchikawa S (2021) Rapid and nondestructive evaluation of hygroscopic behavior changes of thermally modified softwood and hardwood samples using near-infrared hyperspectral imaging (NIR-HIS). Holzforschung 75:345–357. https://doi.org/10.1515/hf-2019-0298
dos Santos LM, Amaral EA, Nieri EM et al (2021) Estimating wood moisture by near infrared spectroscopy: testing acquisition methods and wood surfaces qualities. Wood Mat Sci Eng 16:336–343. https://doi.org/10.1080/17480272.2020.1768143
Costa EVS, Rocha MFV, Hein PRG et al (2018) Influence of spectral acquisition technique and wood anisotropy on the statistics of predictive near infrared–based models for wood density. J Near Infrared Spectrosc 26:106–116. https://doi.org/10.1177/0967033518757070
Guo X, Liu L, Wu J et al (2018) Qualitatively and quantitatively characterizing water adsorption of a cellulose nanofiber film using micro-FTIR spectroscopy. RSC Adv 8:4214–4220. https://doi.org/10.1039/C7RA09894D
Xiao T, Yuan H, Ma Q et al (2019) An approach for in situ qualitative and quantitative analysis of moisture adsorption in nanogram-scaled lignin by using micro-FTIR spectroscopy and partial least squares regression. Int J Biol Macromol 132:1106–1111. https://doi.org/10.1016/j.ijbiomac.2019.04.043
Guo X, Wu Y (2018) IN SITU visualization of water adsorption in cellulose nanofiber film with micrometer spatial resolution using micro-FTIR imaging. J Wood Chem Technol 38:361–370. https://doi.org/10.1080/02773813.2018.1488869
Ponzecchi A, Thybring EE, Digaitis R, et al (2022) Raman micro-spectroscopy of two types of acetylated Norway spruce wood at controlled relative humidity. Frontiers in Plant Science 13:
Huang S, Makarem M, Kiemle SN et al (2018) Dehydration-induced physical strains of cellulose microfibrils in plant cell walls. Carbohyd Polym 197:337–348. https://doi.org/10.1016/j.carbpol.2018.05.091
Makarem M, Kim H, Emami P et al (2020) Impact of drying on meso- and nanoscale structures of citrus fiber: a study by SFG, ATR-IR, XRD, and DLS. Ind Eng Chem Res 59:2718–2724. https://doi.org/10.1021/acs.iecr.9b06194
Chung C-Y, Boik J, Potma EO (2013) Biomolecular imaging with coherent nonlinear vibrational microscopy. Annu Rev Phys Chem 64:77–99. https://doi.org/10.1146/annurev-physchem-040412-110103
Hansen D, Brewer JR, Eiler J et al (2020) Water diffusion in polymer composites probed by impedance spectroscopy and time-resolved chemical imaging. ACS Appl Polym Mater 2:837–845. https://doi.org/10.1021/acsapm.9b01107
Iachina I, Lomholt MA, Eriksen JH, Brewer JR (2022) Multilayer diffusion modeling and coherent anti-stokes raman scattering microscopy for spatially resolved water diffusion measurements in human skin. J Biophotonics 15:e202200110. https://doi.org/10.1002/jbio.202200110
Ibata K, Takimoto S, Morisaku T et al (2011) Analysis of aquaporin-mediated diffusional water permeability by coherent anti-stokes raman scattering microscopy. Biophys J 101:2277–2283. https://doi.org/10.1016/j.bpj.2011.08.045
Foston M (2014) Advances in solid-state NMR of cellulose. Curr Opin Biotechnol 27:176–184. https://doi.org/10.1016/j.copbio.2014.02.002
Rodin VV (2020) NMR techniques in studying water in biotechnological systems. Biophys Rev 12:683–701. https://doi.org/10.1007/s12551-020-00694-5
Li J, Ma E (2021) Characterization of water in wood by time-domain nuclear magnetic resonance spectroscopy (TD-NMR): a review. Forests. https://doi.org/10.3390/f12070886
Li J, Ma E (2022) 2D time-domain nuclear magnetic resonance (2D TD-NMR) characterization of cell wall water of Fagus sylvatica and Pinus taeda L. Cellulose 29:8491–8508. https://doi.org/10.1007/s10570-022-04789-y
Cox J, McDonald PJ, Gardiner BA (2010) A study of water exchange in wood by means of 2D NMR relaxation correlation and exchange. 64:259–266 https://doi.org/10.1515/hf.2010.036
Bonnet M, Courtier-Murias D, Faure P et al (2017) NMR determination of sorption isotherms in earlywood and latewood of Douglas fir. Identif bound water compon relat local environ 71:481–490. https://doi.org/10.1515/hf-2016-0152
Li J, Ma E, Yang T (2019) Differences between hygroscopicity limit and cell wall saturation investigated by LF-NMR on Southern pine (Pinus spp.). Holzforschung 73:911–921. https://doi.org/10.1515/hf-2018-0257
Gao X, Shouzeng Z, Jin J, Cao P (2015) Bound water content and pore size distribution in swollen cell walls determined by NMR technology. BioResources 10:8208–8224
Perkins EL, Batchelor WJ (2012) Water interaction in paper cellulose fibres as investigated by NMR pulsed field gradient. Carbohyd Polym 87:361–367. https://doi.org/10.1016/j.carbpol.2011.07.065
Zhou M, Caré S, Courtier-Murias D et al (2018) Magnetic resonance imaging evidences of the impact of water sorption on hardwood capillary imbibition dynamics. Wood Sci Technol 52:929–955. https://doi.org/10.1007/s00226-018-1017-y
Penvern H, Zhou M, Maillet B et al (2020) How bound water regulates wood drying. Phys Rev Applied 14:054051. https://doi.org/10.1103/PhysRevApplied.14.054051
Gezici-Koç Ö, Erich SJF, Huinink HP et al (2017) Bound and free water distribution in wood during water uptake and drying as measured by 1D magnetic resonance imaging. Cellulose 24:535–553. https://doi.org/10.1007/s10570-016-1173-x
Park S, Johnson DK, Ishizawa CI et al (2009) Measuring the crystallinity index of cellulose by solid state 13C nuclear magnetic resonance. Cellulose 16:641–647. https://doi.org/10.1007/s10570-009-9321-1
Garvey CJ, Simon GP, Whittaker AK, Parker IH (2019) Moisture-activated dynamics on crystallite surfaces in cellulose. Colloid Polym Sci 297:521–527. https://doi.org/10.1007/s00396-018-04464-4
White PB, Wang T, Park YB et al (2014) Water-polysaccharide interactions in the primary cell wall of arabidopsis thaliana from polarization transfer solid-state NMR. J Am Chem Soc 136:10399–10409. https://doi.org/10.1021/ja504108h
Lindh EL, Terenzi C, Salmén L, Furó I (2017) Water in cellulose: evidence and identification of immobile and mobile adsorbed phases by 2H MAS NMR. Phys Chem Chem Phys 19:4360–4369. https://doi.org/10.1039/C6CP08219J
Bucur V (2003) Ionizing radiation computed tomography. In: Bucur V (ed) Nondestructive characterization and imaging of wood. Springer, Berlin, pp 13–73
Bucur V (2003) Techniques for high resolution imaging of wood structure: a review. Meas Sci Technol 14:R91. https://doi.org/10.1088/0957-0233/14/12/R01
Hémonnot CYJ, Köster S (2017) Imaging of Biological materials and cells by x-ray scattering and diffraction. ACS Nano 11:8542–8559. https://doi.org/10.1021/acsnano.7b03447
Fromm JH, Sautter I, Matthies D et al (2001) Xylem water content and wood density in spruce and oak trees detected by high-resolution computed tomography. Plant Physiol 127:416–425. https://doi.org/10.1104/pp.010194
Tognetti R, Raschi A, Béres C et al (1996) Comparison of sap flow, cavitation and water status of Quercus petraea and Quercus cerris trees with special reference to computer tomography. Plant, Cell Environ 19:928–938. https://doi.org/10.1111/j.1365-3040.1996.tb00457.x
Pang S, Wiberg P (1998) Model predicted and CT scanned moisture distribution in aPinus radiata board during drying. Holz Roh Werkst 56:9–14. https://doi.org/10.1007/s001070050256
Alkan S, Zhang Y, Lam F (2007) Moisture distribution changes and wetwood behavior in subalpine fir wood during drying using high x-ray energy industrial CT scanner. Drying Technol 25:483–488. https://doi.org/10.1080/07373930601184023
Johansson J, Kifetew G (2010) CT-scanning and modelling of the capillary water uptake in aspen, oak and pine. Eur J Wood Wood Prod 68:77–85. https://doi.org/10.1007/s00107-009-0359-4
Couceiro J (2019) X-ray computed tomography to study moisture distribution in wood. PhD thesis, Luleå University of Technology
Watanabe K, Lazarescu C, Shida S, Avramidis S (2012) A novel method of measuring moisture content distribution in timber during drying using CT scanning and image processing techniques. Drying Technol 30:256–262. https://doi.org/10.1080/07373937.2011.634977
Brodersen CR (2013) Visualizing wood anatomy in three dimensions with high-resolution X-ray micro-tomography (μCT )—a review. IAWA J 34:408–424. https://doi.org/10.1163/22941932-00000033
Van den Bulcke J, Boone MA, Dhaene J et al (2019) Advanced X-ray CT scanning can boost tree ring research for earth system sciences. Ann Bot 124:837–847. https://doi.org/10.1093/aob/mcz126
Indore NS, Karunakaran C, Jayas DS (2022) Synchrotron tomography applications in agriculture and food sciences research: a review. Plant Methods 18:101. https://doi.org/10.1186/s13007-022-00932-9
Koddenberg T, Greving I, Hagemann J et al (2021) Three-dimensional imaging of xylem at cell wall level through near field nano holotomography. Sci Rep 11:4574. https://doi.org/10.1038/s41598-021-83885-8
Flenner S, Storm M, Kubec A et al (2020) Pushing the temporal resolution in absorption and Zernike phase contrast nanotomography: enabling fast ıt in situ experiments. J Synchrotron Radiat 27:1339–1346. https://doi.org/10.1107/S1600577520007407
Zhou M, Caré S, King A et al (2019) Wetting enhanced by water adsorption in hygroscopic plantlike materials. Phys Rev Res 1:033190. https://doi.org/10.1103/PhysRevResearch.1.033190
Vonk NH, Dekkers ECA, van Maris MPFHL, Hoefnagels JPM (2019) A Multi-loading, climate-controlled, stationary roi device for in-situ x-ray CT hygro-thermo-mechanical testing. Exp Mech 59:295–308. https://doi.org/10.1007/s11340-018-0427-y
Bucur V (2003) Neutron imaging. In: Bucur V (ed) Nondestructive characterization and imaging of wood. Springer, Berlin, pp 281–298
Mannes D, Josic L, Lehmann E, Niemz P (2009) Neutron attenuation coefficients for non-invasive quantification of wood properties. 63:472–478. https://doi.org/10.1515/HF.2009.081
Hassanein R, Lehmann E, Vontobel P (2005) Methods of scattering corrections for quantitative neutron radiography. Nucl Instrum Methods Phys Res Sect A 542:353–360. https://doi.org/10.1016/j.nima.2005.01.161
Raventos M, Harti RP, Lehmann E, Grünzweig C (2017) A method for neutron scattering quantification and correction applied to neutron imaging. Phys Procedia 88:275–281. https://doi.org/10.1016/j.phpro.2017.06.038
Sedighi-Gilani M, Vontobel P, Lehmann E et al (2014) Liquid uptake in Scots pine sapwood and hardwood visualized and quantified by neutron radiography. Mater Struct 47:1083–1096. https://doi.org/10.1617/s11527-013-0112-7
Sedighi-Gilani M, Griffa M, Mannes D et al (2012) Visualization and quantification of liquid water transport in softwood by means of neutron radiography. Int J Heat Mass Transf 55:6211–6221. https://doi.org/10.1016/j.ijheatmasstransfer.2012.06.045
Desmarais G, Gilani MS, Vontobel P et al (2016) Transport of polar and nonpolar liquids in softwood imaged by neutron radiography. Transp Porous Media 113:383–404. https://doi.org/10.1007/s11242-016-0700-4
Rosner S, Riegler M, Vontobel P et al (2012) Within-ring movement of free water in dehydrating Norway spruce sapwood visualized by neutron radiography. Holzforschung 66:751–756. https://doi.org/10.1515/hf-2011-0234
Sonderegger W, Hering S, Mannes D et al (2010) Quantitative determination of bound water diffusion in multilayer boards by means of neutron imaging. Eur J Wood Wood Prod 68:341–350. https://doi.org/10.1007/s00107-010-0463-5
Mannes D, Sanabria S, Funk M et al (2014) Water vapour diffusion through historically relevant glutin-based wood adhesives with sorption measurements and neutron radiography. Wood Sci Technol 48:591–609. https://doi.org/10.1007/s00226-014-0626-3
Lanvermann C, Sanabria SJ, Mannes D, Niemz P (2014) Combination of neutron imaging (NI) and digital image correlation (DIC) to determine intra-ring moisture variation in Norway spruce. Holzforschung 68:113–122. https://doi.org/10.1515/hf-2012-0171
Gilani MS, Vontobel P, Lehmann E et al (2014) Moisture migration in wood under heating measured by thermal neutron radiography. Exp Heat Transf 27:160–179. https://doi.org/10.1080/08916152.2012.757677
Zhou X, Desmarais G, Carl S et al (2022) Investigation of coupled vapor and heat transport in hygroscopic material during adsorption and desorption. Build Environ 214:108845. https://doi.org/10.1016/j.buildenv.2022.108845
Bridarolli A, Odlyha M, Burca G et al (2021) Controlled environment neutron radiography of moisture sorption/desorption in nanocellulose-treated cotton painting canvases. ACS Appl Polym Mater 3:777–788. https://doi.org/10.1021/acsapm.0c01073
Sanabria SJ, Lanvermann C, Michel F et al (2015) Adaptive neutron radiography correlation for simultaneous imaging of moisture transport and deformation in hygroscopic materials. Exp Mech 55:403–415. https://doi.org/10.1007/s11340-014-9955-2
Tötzke C, Kardjilov N, Manke I, Oswald SE (2017) Capturing 3D water flow in rooted soil by ultra-fast neutron tomography. Sci Rep 7:6192. https://doi.org/10.1038/s41598-017-06046-w
Penttilä PA, Paajanen A, Ketoja JA (2021) Combining scattering analysis and atomistic simulation of wood-water interactions. Carbohyd Polym 251:117064. https://doi.org/10.1016/j.carbpol.2020.117064
Martínez-Sanz M, Gidley MJ, Gilbert EP (2015) Application of X-ray and neutron small angle scattering techniques to study the hierarchical structure of plant cell walls: a review. Carbohyd Polym 125:120–134. https://doi.org/10.1016/j.carbpol.2015.02.010
Roe R-J (2000) Methods of x-ray and neutron scattering in polymer science. Oxford U Press, New York
Penttilä PA, Rautkari L, Österberg M, Schweins R (2019) Small-angle scattering model for efficient characterization of wood nanostructure and moisture behaviour. J Appl Crystallogr 52:369–377. https://doi.org/10.1107/S1600576719002012
Jakob HF, Tschegg SE, Fratzl P (1996) hydration dependence of the wood-cell wall structure in picea abies. Small Angle X-ray Scatter Study Macromol 29:8435–8440. https://doi.org/10.1021/ma9605661
Mao J, Heck B, Abushammala H et al (2019) A structural fibrillation parameter from small angle X-ray scattering to quantify pulp refining. Cellulose 26:4265–4277. https://doi.org/10.1007/s10570-019-02386-0
Virtanen T, Penttilä PA, Maloney TC et al (2015) Impact of mechanical and enzymatic pretreatments on softwood pulp fiber wall structure studied with NMR spectroscopy and X-ray scattering. Cellulose 22:1565–1576. https://doi.org/10.1007/s10570-015-0619-x
Penttilä PA, Várnai A, Fernández M et al (2013) Small-angle scattering study of structural changes in the microfibril network of nanocellulose during enzymatic hydrolysis. Cellulose 20:1031–1040. https://doi.org/10.1007/s10570-013-9899-1
Penttilä PA, Altgen M, Awais M et al (2020) Bundling of cellulose microfibrils in native and polyethylene glycol-containing wood cell walls revealed by small-angle neutron scattering. Sci Rep 10:20844. https://doi.org/10.1038/s41598-020-77755-y
Leppänen K, Bjurhager I, Peura M et al (2011) X-ray scattering and microtomography study on the structural changes of never-dried silver birch. Eur Aspen Hybrid Aspen Drying 65:865–873. https://doi.org/10.1515/HF.2011.108
Fang L, Catchmark JM (2014) Structure characterization of native cellulose during dehydration and rehydration. Cellulose 21:3951–3963. https://doi.org/10.1007/s10570-014-0435-8
Penttilä PA, Altgen M, Carl N et al (2020) Moisture-related changes in the nanostructure of woods studied with X-ray and neutron scattering. Cellulose 27:71–87
Thomas LH, Forsyth VT, Martel A et al (2014) Structure and spacing of cellulose microfibrils in woody cell walls of dicots. Cellulose 21:3887–3895. https://doi.org/10.1007/s10570-014-0431-z
Ahvenainen P, Dixon PG, Kallonen A et al (2017) Spatially-localized bench-top X-ray scattering reveals tissue-specific microfibril orientation in Moso bamboo. Plant Methods 13:5. https://doi.org/10.1186/s13007-016-0155-1
O’Neill H, Pingali SV, Petridis L et al (2017) Dynamics of water bound to crystalline cellulose. Sci Rep 7:11840. https://doi.org/10.1038/s41598-017-12035-w
Plaza NZ (2017) Neutron scattering studies of nano-scale wood-water interactions. PhD thesis, University of Wisconsin-Madison
Müller M, Czihak C, Schober H et al (2000) All disordered regions of native cellulose show common low-frequency dynamics. Macromolecules 33:1834–1840. https://doi.org/10.1021/ma991227l
Araujo C, Freire CSR, Nolasco MM et al (2018) Hydrogen bond dynamics of cellulose through inelastic neutron scattering spectroscopy. Biomacromol 19:1305–1313. https://doi.org/10.1021/acs.biomac.8b00110
Einfeldt J, Kwasniewski A (2002) Characterization of different types of cellulose by dielectric spectroscopy. Cellulose 9:225–238. https://doi.org/10.1023/A:1021184620045
Maeda H, Fukada E (1987) Effect of bound water on piezoelectric, dielectric, and elastic properties of wood. J Appl Polym Sci 33:1187–1198. https://doi.org/10.1002/app.1987.070330411
Roig F, Ramanantsizehena G, Lahatra Razafindramisa F et al (2017) Dielectric and mechanical properties of various species of Madagascan woods. Wood Sci Technol 51:1389–1404. https://doi.org/10.1007/s00226-017-0936-3
Sudo S, Suzuki Y, Abe F et al (2018) Investigation of the molecular dynamics of restricted water in wood by broadband dielectric measurements. J Mater Sci 53:4645–4654. https://doi.org/10.1007/s10853-017-1824-9
Funding
Open Access funding provided by Aalto University.
Author information
Authors and Affiliations
Contributions
LR contributed the original concept of the paper, CH wrote the first draft of the paper except for the section on instrumental methods which was written by MA and PP. LR, MA and PP contributed to the second draft of the paper. MA produced all figures apart from Figs. 3 and 6.
Corresponding author
Ethics declarations
Conflict of interest
No conflicts of interest or competing interests.
Ethical approval
Not applicable.
Additional information
Handling Editor: Stephen Eichhorn.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hill, C., Altgen, M., Penttilӓ, P. et al. Review: interaction of water vapour with wood and other hygro-responsive materials. J Mater Sci 59, 7595–7635 (2024). https://doi.org/10.1007/s10853-024-09636-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-024-09636-y