
Abstract
Cell toxicity may result in organ dysfunction and cause severe health problem. Recent studies revealed many toxicants may induced the over production of Nitric oxide, reactive oxygen species and the subsequent oxidative stress, cause cell toxicity. Mitochondrion dysfunction maybe the subsequent consequence of oxidative stress and has been recognized as another contributing factor in cell toxicity. Besides, oxidative products induced by some toxicants may also produce the compounds that damage cell DNA, leading to toxicity. Especially, the significance of nanoparticle induced cell toxicity was disclosed recently and attract more concern. The mechanism mainly includes inflammation, oxidative stress and DNA damage. On the other side, some biomarkers of cell toxicity including autophagy, cytokines, miRNA has been identified. The understanding of these phenomenon may enable us to clarify the cell toxicity mechanism then contribute to cell toxicity protection, disease treatment and drug side effect prevention.
Similar content being viewed by others

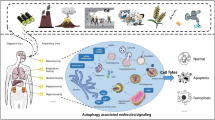

Introduction
Cell toxicity is caused by exogenous toxicant which can damage cells, especially when the toxicant can cause cell death and serious organ dysfunction [1]. The effects of a toxicant are usually dose-dependent and species–specific. Cell toxicant include chemical agent, environment pollutant, natural plants extract and pharmaceutical drugs [2,3,4]. The mechanism of cell toxicity has been investigated in several decades and still attract interest of today.
The mechanisms of cell toxicity are widely involved. It has long been proved that toxicant may induce overproduction of Nitric oxide (NO), reactive oxygen species (ROS) and the subsequent oxidative stress [5]. The high level of NO, ROS and the subsequent oxidative burst have been identified as one main mechanism of severe cell toxicity or even organ dysfunction. Besides, mitochondrion dysfunction was also noticed as a consequence of oxidative stress, especially in neuro cells toxicity [6,7,8]. Toxicity agents may also induce and release compounds that directly damage DNA, causing cell apoptosis and toxicity [9,10,11].
Oxide nanoparticles (NP) induced cell toxicity has been gradually revealed and attract significant attention as their great application in medicine. Nanoparticles are particles between 1 and 100 nm (nm) in size and may be absorbed by human cells and may increase the occupational and public exposure and yield extraordinary hazards for human health. NPs in human cells may cause a serials of cell physiological change, ultimately resulted in cell toxicity [12, 13]. Some studies showed exposure to NPs induced cell toxicity were inflammation involved, such as IL-8 [14, 15].
These recent discovered cell toxicants and their toxicity mechanism may enable us to alleviate the cell dysfunction and promote cell protection. Here we review these articles about cell toxicity, its toxic mechanism and the associated biomarkers, try to summarize the recent progress and hotspot in this area and found some potential therapy target in cell toxicity prevention.
NO overproduction
NO is believed to play duel role in cell function, either detrimental or protective. NO regulation plays a critical role in cell function, especially in endothelial cells. NO may regulate the normal vascular tone and the repression of NO may cause proatherogenic situation [16, 17]. However, NO overproduction may also cause cell dysfunction or even cell toxicity. The high level of NO and related oxidative burst have been identified as one main mechanism of several cell toxicity. For instance, doxorubicin may induce skeletal muscle dysfunction and cardiotoxicity through NO. The administration of doxorubicin may cause the increased intracellular and interstitial NO concentrations in the SM, leading to SM dysfunction and myocardial cell toxicity (Fig. 1) [18]. NO may also be involved in NPs included oxidative burst of granulocytes and impair phagocyte function. The high concentrations of carboxyl polystyrene particles may stimulate myeloperoxidase release of granulocytes and NO over production in macrophages, cause cell toxicity [19]. MgNPs also has cell toxicity in a normal biological system. MgNPs may impair the proliferation of human umbilical vein vascular endothelial cells. MgNPs exposure may increase the activity of endothelial NO synthase, cause the over production of NO and leading to cell toxicity (Fig. 2) [20].

The mechanism of cell toxicity mainly include: a NO or cytokines overproduction induced oxidative burst, b ROS and oxidative stress, c DNA damage, d Mitochondrial dysfunction. Toxicant were shown as green

Mechanism of Metal oxide NPs induced cell toxicity. Include inflammatory environment, membrane damage, excess NO and Mitochondria dysfunction
Several pathways may involve in NO overproduction induced cell toxicity. NO may suppress NF-κB pathway, potentiates TNF-α induced neurotoxicity [21]. NO may also activate p38 MAPK and p53 pathways, further increase DNA double strand breaks in microglia. However, the activation of both Akt and ERK cascades may alleviate the DNA damages induced by NO [22]. In diabetes and Alzheimer’s disease, the upregulates of NO synthase was also identified with the involved pathway of PPAR-γ, P38. These signaling changes are blocked by PPAR-γ small-interfering RNA transfection, and is also blocked by the NO inhibitor and p38 inhibitor [23].
ROS and oxidative stress
ROS was identified as a pivotal modulator factor in immune system, neural system, infection and cancer development [24,25,26,27]. ROS homeostasis is essential in the sustain of cells function. Toxicity agents induced the over production of ROS and the subsequent oxidative stress may cause severe cell toxicity.
Researches have revealed ROS was involved in cell toxicity induced by variety of exogenous toxicant. For instance, acetaminophen is a well-known liver toxicant, acetaminophen-treated liver cells showed the significant increased ROS production and glutathione depletion, cause hepatocytes toxicity and death [28]. Pentachlorophenol, a pesticides, may cause high toxicological impact in hepatocytes with the involvement of ROS overproduction and oxidative stress. Antioxidants such as ascorbic acid and quercetin may modulate the toxicity effects of Pentachlorophenol [29]. Metal pollutants such as Aluminum may generate intracellular ROS and triggers a metabolic shift towards lipogenesis in astrocytes and hepatocytes. ROS activation is associated with impaired mitochondrial activity, anaerobiosis and the channeling of α-ketoacids towards anti-oxidant defense. These process leads to a reduction in ATP synthesis, were potential cause of brain and liver disorders [30]. The ROS induced cell toxicity have been revealed by temporal imaging. Cytoplasmic sensor in neuroblastoma cells demonstrated the 6-hydroxydopamine may induced oxidative stress and glucose deprivation enhanced ROS, cause neuroblastoma cells toxicity (Fig. 1) [31].
Cell toxicity of particulates intake in human cells has attract concern recently. Especially, airborne particulate with a diameter less than 2.5 μm (PM2.5) in considered to be a main cancerogen for lung cancer. A study revealed an increase of intracellular ROS with a time-dependent manner when human type II alveolar epithelial A549 cells exposed to PM2.5 particles. PM2.5 induced ROS may activate Nrf2-mediated defenses, such as HO-1 expression, against oxidative stress through PI3K/AKT signaling pathway [32]. Another nanoparticle, silica, is commonly used in food products, also revealed cell toxicity problem. High concentration of silica may cause significant cytotoxic effects on human lung fibroblast cell. Silica may induces a dose-dependent cell toxicity and elevated ROS levels, and meanwhile trigger the protective gene expression levels of stress-responsive genes (CAT, GSTA4, TNF, CYP1A, POR, SOD1, GSTM3, GPX1, and GSR1), which may be the potential therapy medicine for lung fibrosis [33]. AgNPs used in medical supplies may cause cytotoxic effect by ROS with decreased activities of superoxide dismutase and glutathione peroxides and the increased level of LPO [34]. Iron oxide nanoparticles also have potential toxicity by disrupt the barrier function of epithelium. Human placental cell line BeWo b30 was grown as epithelia and subsequently been assessed for epithelial integrity when exposure to α-Fe2O3 nanoparticles. Transepithelial electrical resistance indicated that exposure to the α-Fe2O3 nanomaterial resulted in leakiness of the epithelium and increases in cell death and ROS. Genotoxicity as assessed by DNA microarray and confirmed by QPCR indicated that the large diameter particles (78 nm) induce apoptosis in these cells (Fig. 2) [35].
Mitochondrial dysfunction
Mitochondrion is critical in energy metabolism and playing key roles in biochemical synthesis, redox control and apoptosis. Alterations in mitochondrial function are increasingly being recognized as a contributing factor in many human diseases [36,37,38,39]. Recent understanding showed mitochondrion dysfunction and oxidative stress are usually co-existing in toxicant induced cell toxicity, and maybe a prevention method in cell toxicity [40].
Bromobenzene is a toxin may cause liver and kidney damage through oxidative stress. It may also induce mitochondrial dysfunction with the decreased activities of mitochondrial enzymes [41]. Formaldehyde induced toxicity is also with the involvement of oxidative stress and mitochondrial dysfunction. Formaldehyde may reduce mitochondrial membrane potential, inhibit mitochondrial respiratory enzymes such as NADH dehydrogenase (complex I), cytochrome c oxidase (complex IV), and promote cell apoptosis through initiator caspase-9 and apoptosis-effector caspase 3/7 [42]. Similarly, Quinocetone may triggers toxicity on HepG2 cells by oxidative stress and mitochondrial apoptotic. Quinocetone may reducing the activities of endogenous antioxidant enzymes and further promote apoptosis through c-MYC-dependent activation of the mitochondrial apoptotic pathway [43]. Toxicant cyclosporine A and cannabidiol may induce ROS in monocytic cell line. Impaired mitochondrial function was accompanied by elevated ROS and cell apoptosis level. Mitochondrial dysfunction may be also one mechanism of cytotoxicity in immune cells [44].
Some toxicant may directly induce mitochondrial dysfunction, result in cell apoptosis. For instance, FK506 is an important immunosuppressive medication and may provoke neurotoxicity and nephrotoxicity. FK506 may provokes an important decrease in oxygen consumption, reduction in the synthesis of mitochondria DNA-encoded proteins [45]. These mitochondrial dysfunction results are similar to those triggered by rapamycin immunosuppressive properties. Efavirenz induced mitochondrial depolarization may trigger mitochondrial morphology alteration and mitochondria mediated apoptosis [46]. It is also similar in the imatinib mesylate influences mitochondrial signaling leading to mitochondrial dysfunction and cardiotoxicity (Fig. 1) [47].
DNA damage
DNA damage is another common mechanism of cell toxicity. Toxicant may induce compounds that damage DNA and un-repaired DNA damages may cause cell death. Besides, these types of alteration can be replicated and passed on to subsequent cell generations. These cell toxicities may regulate gene expression and change gene function, possibly contribute to progression to cancer.
Chratoxin A may induce DNA damage as a mechanism of cell toxicity. Chratoxin A treated BME-UV1 and MDCK epithelial cells showed a significant increase of cell apoptosis with the increased level of DNA integrity impairment [48]. Mustard gas is an alkylating agent that increases cell toxicity and the incidence of cancer with the mechanism of induce DNA damage. Mustard gas analog may induce centrosome amplification and chromosome instability in cells, which may hasten the mutation rate necessary for tumorigenesis [49]. Naphthalimido compounds may induced cell toxicity by intercalating to DNA via the major groove and induce DNA damage. A lower concentration of Naphthalimido may also induced a significant decrease in repair of DNA damage [50].
Besides, DNA damage may also be synergetic with oxidative stress to induce cell toxicity. Hydroquinone exposure in air pollution may cause genotoxicity on human lung cells. Chromosomal aberration and associated DNA damage was observed in hydroquinone treated lung epithelium cell A549, meanwhile an increase oxidized glutathione and the reduced anti oxidization glutathione was also observed (Fig. 1) [51]. In cell toxicity of silver nanoparticles, DNA damage and ROS may also have synergetic effect. It is proved by the co-existence of decreased mitochondrial membrane potential, cell G2/M phase arrest and the increased of anti-peroxide effect in AgNP exposure cells (Fig. 2) [34].
Cell toxicity biomarker and potential therapy target
Autophagy is a phenomenon of cell toxicity, and may also be an effective marker in anti-cancer efficiency of cancer treatment. A study suggests that erlotinib combined with radiotherapy may remarkably induce apoptosis in lung cancer cells with provoked autophagy. The cell toxicity of lung cancer cells may be reversed by autophagy inhibitor chloroquine [52]. Besides, pinus radiata bark extract may induces cytotoxic effects in human breast cancer cells with increased accumulation of autophagic markers [53].
ROS is the marker and may be also the potential therapy target of cell toxicity. LPO, increase catalase activity and mitochondria metoclopramide groups are the markers of ROS and mitochondria associated cell toxicity. The extract of Sambucus ebulus L. fruit may protect neuro cells through relieve ROS in brain mitochondria. Increased plasma antioxidants or scavenging of free radicals were observed in the extract treated patients. The improvement of the two biomarkers may be therapy target of the restore of mitochondria function [54].
The increased cytokines may be the marker of cell toxicity in some inflammation associated diseases. For example, severe systemic inflammation may trigger lung cell toxicity Some cytokines group may be the marker for inflammation induced ARDS. These biomarkers including bone morphogenetic protein-15, CXCL16, CXCR3, IL-6, protein NOV homolog, glypican 3, IGFBP-4, IL-5, IL-5R alpha, IL-22 BP, leptin, MIP-1d, and orexin B. The overexpressed IL-6, CXCL16, or IGFBP-4 may also represent the severity of the disease [55]. Another example is renal allograft rejection. Some inflammation factors may provide diagnostic biomarkers for predicting cell toxicity induced rejection. Proteomic study found 33 inflammatory proteins and the protein–protein network analyzes suggest ICAM-1 is the main biomarker in chronic rejection [56].
MicroRNA profiling could be a useful tool for the discovery of cell toxicity biomarkers and target therapy genes. The neuronal toxicity of MeHgCl may characterized by the overexpression of a signature composed of five miRNAs (miR-302b, miR-367, miR-372, miR-196b and miR-141) that are known to be involved in the regulation of developmental processes and cellular stress response mechanisms [57]. Besides, miR-146a, the target gene of MCL1 which encoding an anti-apoptotic protein, is a marker of H2O2-induced PC12 cells toxicity. MCL1 may activate JAK/STAT signaling pathway and attenuated H2O2-induced cell toxicity [58].
Conclusion
Cell toxicity induced by environment toxicant and medicines may be resulted in severe organ dysfunction and disease. Recent studies has revealed the main cell toxicity mechanism includes NO overproduction, ROS induced oxidative stress, mitochondrial dysfunction, DNA damage. Especially, the toxicity of metal oxide NPs has been recognized in the recent years. All of which may enable us to clarify the cell toxicity mechanism. However, the toxicity prevention methods require further explicated to prevent toxicity and contribute to disease treatment.
References
Furue H (1995) Toxicity criteria. Gan To Kagaku Ryoho. 22(5):616–626
Das KK, Reddy RC, Bagoji IB, Das S, Bagali S, Mullur L et al (2018) Primary concept of nickel toxicity—an overview. J Basic Clin Physiol Pharmacol. https://doi.org/10.1515/jbcpp-2017-0171
Kharchoufa L, Merrouni IA, Yamani A, Elachouri M (2018) Profile on medicinal plants used by the people of North Eastern Morocco: toxicity concerns. Toxicon 154:90–113
Wang W, Zhou H, Liu L (2018) Side effects of methotrexate therapy for rheumatoid arthritis: a systematic review. Eur J Med Chem 158:502–516
Beckman JS, Koppenol WH (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271(5 Pt 1):C1424–C1437
Mondragón-Rodríguez S, Perry G, Zhu X, Moreira PI, Acevedo-Aquino MC, Williams S (2013) Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: implications for Alzheimer’s disease. Oxid Med Cell Longev 2013:940603
Fang D, Zhang Z, Li H, Yu Q, Douglas JT, Bratasz A et al (2016) Increased electron paramagnetic resonance signal correlates with mitochondrial dysfunction and oxidative stress in an Alzheimer’s disease mouse brain. J Alzheimer’s Dis. 51(2):571–580
Zhou Y, Wang ZF, Li W, Hong H, Chen J, Tian Y et al (2018) Protective effects of microRNA-330 on amyloid β-protein production, oxidative stress, and mitochondrial dysfunction in Alzheimer’s disease by targeting VAV1 via the MAPK signaling pathway. J Cell Biochem 119(7):5437–5448
Hu G, Mancl ME, Barnes BJ (2005) Signaling through IFN regulatory factor-5 sensitizes p53-deficient tumors to DNA damage-induced apoptosis and cell death. Cancer Res 65(16):7403–7412
Roos WP, Kaina B (2006) DNA damage-induced cell death by apoptosis. Trends Mol Med. 12(9):440–450
Roos WP, Kaina B (2013) DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 332(2):237–248
Kumar V, Kumari A, Guleria P, Yadav SK (2012) Evaluating the toxicity of selected types of nanochemicals. Rev Environ Contam Toxicol 215:39–121
Djurišić AB, Leung YH, Ng AM, Xu XY, Lee PK, Degger N et al (2015) Toxicity of metal oxide nanoparticles: mechanisms, characterization, and avoiding experimental artefacts. Small 11(1):26–44
Sun J, Wang S, Zhao D, Hun FH, Weng L, Liu H (2011) Cytotoxicity, permeability, and inflammation of metal oxide nanoparticles in human cardiac microvascular endothelial cells: cytotoxicity, permeability, and inflammation of metal oxide nanoparticles. Cell Biol Toxicol 27(5):333–342
Abbott CTE, Schwab KJ (2013) Toxicity of commercially available engineered nanoparticles to Caco-2 and SW480 human intestinal epithelial cells. Cell Biol Toxicol 29(2):101–116
Buie JJ, Renaud LL, Muise-Helmericks R, Oates JC (2017) IFN-α Negatively regulates the expression of endothelial nitric oxide synthase and nitric oxide production: implications for systemic lupus erythematosus. J Immunol 199(6):1979–1988
Miyata S, Noda A, Hara Y, Ueyama J, Kitaichi K, Kondo T et al (2017) Nitric oxide plasma level as a barometer of endothelial dysfunction in factory workers. Exp Clin Endocrinol Diabetes 125(10):684–689
Fabris S, MacLean DA (2016) Doxorubicin chemotherapy affects intracellular and interstitial nitric oxide concentrations in skeletal muscle: effect of doxorubicin on intracellular and interstitial NO in skeletal muscle. Cell Biol Toxicol 32(2):121–131
Prietl B, Meindl C, Roblegg E, Pieber TR, Lanzer G, Fröhlich E (2014) Nano-sized and micro-sized polystyrene particles affect phagocyte function. Cell Biol Toxicol 30(1):1–16
Meng N, Han L, Pan X, Su L, Jiang Z, Lin Z et al (2015) Nano-Mg(OH)2-induced proliferation inhibition and dysfunction of human umbilical vein vascular endothelial cells through caveolin-1-mediated endocytosis. Cell Biol Toxicol 31(1):15–27
Nakaizumi A, Horie T, Kida T, Kurimoto T, Sugiyama T, Ikeda T et al (2012) Nitric oxide potentiates TNF-α-induced neurotoxicity through suppression of NF-κB. Cell Mol Neurobiol 32(1):95–106
Wang JY, Lee CT, Wang JY (2014) Nitric oxide plays a dual role in the oxidative injury of cultured rat microglia but not astroglia. Neuroscience 281:164–177
Kuo TY, Huang CL, Yang JM, Huang WJ, Huang NK, Chen YW et al (2012) The role of ribosylated-BSA in regulating PC12 cell viability. Cell Biol Toxicol 28(4):255–267
Bittar A, Jun J, La JH, Wang J, Leem JW, Chung JM (2017) Reactive oxygen species affect spinal cell type-specific synaptic plasticity in a model of neuropathic pain. Pain 158(11):2137–2146
Kalashnikova I, Mazar J, Neal CJ, Rosado AL, Das S, Westmoreland TJ et al (2017) Nanoparticle delivery of curcumin induces cellular hypoxia and ROS-mediated apoptosis via modulation of Bcl-2/Bax in human neuroblastoma. Nanoscale. 9(29):10375–10387
Kim HO, Yeom M, Kim J, Kukreja A, Na W, Choi J et al (2017) Reactive oxygen species-regulating polymersome as an antiviral agent against influenza virus. Small 13(32):1700818
Yigit B, Wang N, Chen SS, Chiorazzi N, Terhorst C (2017) Inhibition of Reactive oxygen species (ROS) limits expansion of chronic lymphocytic leukemia cells. Leukemia 31(10):2273
Leclerc E, Hamon J, Claude I, Jellali R, Naudot M, Bois F (2015) Investigation of acetaminophen toxicity in HepG2/C3a microscale cultures using a system biology model of glutathione depletion. Cell Biol Toxicol 31(3):173–185
Pietsch C, Hollender J, Dorusch F, Burkhardt-Holm P (2014) Cytotoxic effects of pentachlorophenol (PCP) and its metabolite tetrachlorohydroquinone (TCHQ) on liver cells are modulated by antioxidants. Cell Biol Toxicol 30(4):233–252
Han S, Lemire J, Appanna VP, Auger C, Castonguay Z, Appanna VD (2013) How aluminum, an intracellular ROS generator promotes hepatic and neurological diseases: the metabolic tale. Cell Biol Toxicol 29(2):75–84
Dooley CT, Li L, Misler JA, Thompson JH (2012) Toxicity of 6-hydroxydopamine: live cell imaging of cytoplasmic redox flux. Cell Biol Toxicol 28(2):89–101
Deng X, Rui W, Zhang F, Ding W (2013) PM2.5 induces Nrf2-mediated defense mechanisms against oxidative stress by activating PIK3/AKT signaling pathway in human lung alveolar epithelial A549 cells. Cell Biol Toxicol 29(3):143–157
Athinarayanan J, Periasamy VS, Alsaif MA, Al-Warthan AA, Alshatwi AA (2014) Presence of nanosilica (E551) in commercial food products: TNF-mediated oxidative stress and altered cell cycle progression in human lung fibroblast cells. Cell Biol Toxicol 30(2):89–100
Song XL, Li B, Xu K, Liu J, Ju W, Wang J et al (2012) Cytotoxicity of water-soluble mPEG-SH-coated silver nanoparticles in HL-7702 cells. Cell Biol Toxicol 28(4):225–237
Faust JJ, Zhang W, Chen Y, Capco DG (2014) Alpha-Fe(2)O(3) elicits diameter-dependent effects during exposure to an in vitro model of the human placenta. Cell Biol Toxicol 30(1):31–53
Mossoba ME, Flynn TJ, Vohra S, Wiesenfeld PL, Sprando RL (2015) Human kidney proximal tubule cells are vulnerable to the effects of Rauwolfia serpentina. Cell Biol Toxicol 31(6):285–293
Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW, Kitsis RN et al (2016) Mitochondrial function, biology, and role in disease: a scientific statement from the american heart association. Circ Res 118(12):1960–1991
Chiang S, Kalinowski DS, Jansson PJ, Richardson DR, Huang ML (2017) Mitochondrial dysfunction in the neuro-degenerative and cardio-degenerative disease, Friedreich’s ataxia. Neurochem Int 117:35–48
van den Berg M, Hooijman PE, Beishuizen A, de Waard MC, Paul MA, Hartemink KJ et al (2017) Diaphragm atrophy and weakness in the absence of mitochondrial dysfunction in the critically Ill. Am J Respir Crit Care Med 196(12):1544–1558
Wu D, Wang X, Sun H (2018) The role of mitochondria in cellular toxicity as a potential drug target. Cell Biol Toxicol 34(2):87–91
Vedi M, Sabina EP (2016) Assessment of hepatoprotective and nephroprotective potential of withaferin A on bromobenzene-induced injury in Swiss albino mice: possible involvement of mitochondrial dysfunction and inflammation. Cell Biol Toxicol 32(5):373–390
Zerin T, Kim JS, Gil HW, Song HY, Hong SY (2015) Effects of formaldehyde on mitochondrial dysfunction and apoptosis in SK-N-SH neuroblastoma cells. Cell Biol Toxicol 31(6):261–272
Zhang K, Zheng W, Zheng H, Wang C, Wang M, Li T et al (2014) Identification of oxidative stress and responsive genes of HepG2 cells exposed to quinocetone, and compared with its metabolites. Cell Biol Toxicol 30(6):313–329
Schultze N, Wanka H, Zwicker P, Lindequist U, Haertel B (2017) Mitochondrial functions of THP-1 monocytes following the exposure to selected natural compounds. Toxicology 377:57–63
Palacín M, Coto E, Llobet L, Pacheu-Grau D, Montoya J, Ruiz-Pesini E (2013) FK506 affects mitochondrial protein synthesis and oxygen consumption in human cells. Cell Biol Toxicol 29(6):407–414
Ganta KK, Mandal A, Chaubey B (2016) Depolarization of mitochondrial membrane potential is the initial event in non-nucleoside reverse transcriptase inhibitor efavirenz induced cytotoxicity. Cell Biol Toxicol 33(1):69–82
Chambers TP, Santiesteban L, Gomez D, Chambers JW (2017) Sab mediates mitochondrial dysfunction involved in imatinib mesylate-induced cardiotoxicity. Toxicology 382:24–35
Giromini C, Rebucci R, Fusi E, Rossi L, Saccone F, Baldi A (2016) Cytotoxicity, apoptosis, DNA damage and methylation in mammary and kidney epithelial cell lines exposed to ochratoxin A. Cell Biol Toxicol 32(3):249–258
Bennett RA, Behrens E, Zinn A, Duncheon C, Lamkin TJ (2014) Mustard gas surrogate, 2-chloroethyl ethylsulfide (2-CEES), induces centrosome amplification and aneuploidy in human and mouse cells: 2-CEES induces centrosome amplification and chromosome instability. Cell Biol Toxicol 30(4):195–205
Bestwick CS, Ralton LD, Milne L, Kong TLP, Duthie SJ (2011) The influence of bisnaphthalimidopropyl polyamines on DNA instability and repair in Caco-2 colon epithelial cells. Cell Biol Toxicol 27(6):455–463
Peng C, Arthur D, Liu F, Lee J, Xia Q, Lavin MF et al (2013) Genotoxicity of hydroquinone in A549 cells. Cell Biol Toxicol 29(4):213–227
Keta O, Bulat T, Golić I, Incerti S, Korać A, Petrović I et al (2016) The impact of autophagy on cell death modalities in CRL-5876 lung adenocarcinoma cells after their exposure to γ-rays and/or erlotinib. Cell Biol Toxicol 32(2):83–101
Venkatesan T, Choi YW, Mun SP, Kim YK (2016) Pinus radiata bark extract induces caspase-independent apoptosis-like cell death in MCF-7 human breast cancer cells. Cell Biol Toxicol 32(5):451–464
Fathi H, Ebrahimzadeh MA, Ziar A, Mohammadi H (2015) Oxidative damage induced by retching; antiemetic and neuroprotective role of Sambucus ebulus L. Cell Biol Toxicol 31(4–5):231–239
Chen C, Shi L, Li Y, Wang X, Yang S (2016) Disease-specific dynamic biomarkers selected by integrating inflammatory mediators with clinical informatics in ARDS patients with severe pneumonia. Cell Biol Toxicol 32(3):169–184
Zhu D, Liu Z, Pan Z, Qian M, Wang L, Zhu T et al (2016) A new method for classifying different phenotypes of kidney transplantation. Cell Biol Toxicol 32(4):323–332
Pallocca G, Fabbri M, Sacco MG, Gribaldo L, Pamies D, Laurenza I et al (2013) miRNA expression profiling in a human stem cell-based model as a tool for developmental neurotoxicity testing. Cell Biol Toxicol 29(4):239–257
Yang X, Mao X, Ding X, Guan F, Jia Y, Luo L et al (2018) miR-146a down-regulation alleviates H2O2-induced cytotoxicity of PC12 cells by regulating MCL1/JAK/STAT pathway: miR-146a down-regulation relieves H2O2-induced PC12 cells cytotoxicity by MCL1/JAK/STAT. Cell Biol Toxicol. https://doi.org/10.1007/s10565-018-9424-2
Authors’ contributions
YZ contribute fully to this article. The author read and approved the final manuscript.
Acknowledgements
The author is grateful for the collaboration of Zhongshan Hospital, Fudan University, and their stuff.
Competing interests
The author declare that he have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
The author is consent for the publication of this article.
Ethics approval and consent to participate
Not applicable.
Funding
The work was supported by Shanghai Leading Academic Discipline Project (B115), The National Nature Science Foundation of China (31400713).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, Y. Cell toxicity mechanism and biomarker. Clin Trans Med 7, 34 (2018). https://doi.org/10.1186/s40169-018-0212-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40169-018-0212-7