
Abstract
Hepatocellular carcinoma (HCC) is a highly heterogeneous malignancy with high incidence, recurrence, and metastasis rates. The emergence of immunotherapy has improved the treatment of advanced HCC, but problems such as drug resistance and immune-related adverse events still exist in clinical practice. The immunosuppressive tumor microenvironment (TME) of HCC restricts the efficacy of immunotherapy and is essential for HCC progression and metastasis. Therefore, it is necessary to elucidate the mechanisms behind immunosuppressive TME to develop and apply immunotherapy. This review systematically summarizes the pathogenesis of HCC, the formation of the highly heterogeneous TME, and the mechanisms by which the immunosuppressive TME accelerates HCC progression and metastasis. We also review the status of HCC immunotherapy and further discuss the existing challenges and potential therapeutic strategies targeting immunosuppressive TME. We hope to inspire optimizing and innovating immunotherapeutic strategies by comprehensively understanding the structure and function of immunosuppressive TME in HCC.
Similar content being viewed by others

Introduction
As the third leading cause of cancer-caused death globally, the high incidence, recurrence, and metastasis rates of hepatocellular carcinoma (HCC) have caused a monumental burden on public health systems [1, 2]. Because the early symptoms of HCC are not obvious, most patients have progressed to unresectable advanced HCC at the time of diagnosis. Moreover, traditional treatments such as radiotherapy, chemotherapy, radiofrequency ablation, molecular targeted agents, and transarterial chemoembolization exhibit limited effects against advanced HCC [3]. As tumor immunology research advances, the emergence of immunotherapy, including immune checkpoint inhibitors (ICIs), provides optimism for HCC treatment. Over the past five years, the Food and Drug Administration (FDA) has approved a combination regimen of anti-programmed death ligand-1 (PD-L1) atezolizumab and anti-vascular endothelial growth factor (VEGF) bevacizumab as the first-line therapy for HCC [4, 5]. Additionally, anti-programmed cell death protein 1 (PD-1) and anti-PD-L1 durvalumab plus anti-cytotoxic T-lymphocyte antigen-4 (CTLA-4) tremelimumab have been approved as second-line therapies [6, 7]. However, drug resistance and immune-related adverse events (irAEs) are still challenges in clinical practice [8].
In order to enhance the efficacy of ICIs and explore novel immunotherapeutic strategies, it is imperative to acquire a more profound comprehension of the mechanisms underlying the immunosuppressive tumor microenvironment (TME). Immunosuppressive cells, such as regulatory T cells (Tregs), tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), tumor-associated neutrophils (TANs), and regulatory B cells (Bregs), act as main functional components of the immunosuppressive TME, undergo complex crosstalk with HCC cells via multiple molecules and exosomes to promote immune escape [9]. In addition, extracellular matrix (ECM) remodeling and metabolic reprogramming are closely related to immunosuppressive TME, promoting HCC progression and metastasis [10, 11].
This review comprehensively summarizes the pathogenesis of HCC, the formation of the highly heterogeneous TME, and the mechanisms through which the immunosuppressive TME promotes HCC progression and metastasis. We also review the current applications, existing challenges, and future development of immunotherapies in the clinical treatment of HCC. We aim to offer insights to optimize and innovate immunotherapeutic strategies by comprehensively understanding the formation of the immunosuppressive TME and its role in promoting HCC progression and metastasis.
Liver microenvironment: from normal to precancerous
Normal microenvironment
As an essential organ responsible for metabolism, immune response, and detoxification, the liver is constantly exposed to antigens from the intestine during its functioning. To maintain homeostasis and proper function, the liver establishes an immunosuppressive microenvironment comprising immune cells, stromal cells, and hepatocytes [12]. In the physiological state, dendritic cells (DCs), Kupffer cells (KCs), and natural killer (NK) cells present in the hepatic sinus are mostly tolerogenic phenotypes, which have a low response to antigen stimulation. These cells, along with resident Tregs and MDSCs in the hepatic sinus and hepatic stellate cells (HSCs) in the perisinusoidal space, secrete immunosuppressive factors to maintain immune system homeostasis [13].
Chronic inflammation and fibrosis
The immune tolerance balance is disrupted when hepatotoxic factors, such as viruses, bacteria, and alcohol metabolites, cause liver damage and cell death. Signaling pathways such as nuclear factor-kappa B (NF-κB) in damaged hepatocytes will be activated, inducing the release of pro-inflammatory cytokines and chemokines like interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and C–C motif chemokine ligand 2 (CCL2) [14]. In addition, the disruption of hepatocyte membranes leads to the release of damage-associated molecular patterns, which facilitate the mobilization and activation of macrophages, DCs, and neutrophil granulocytes [15]. As liver-resident macrophages, KCs sense liver damage signals through pattern recognition receptors and migrate from the hepatic sinusoids to the injury site. Subsequently, they generate various pro-inflammatory mediators, further recruiting and activating immune cells, resulting in inflammatory changes in the liver microenvironment [16, 17]. Chronic inflammation due to persistent liver damage can induce fibrosis by disrupting the normal healing response [18]. Studies have shown that high fibrosis index and liver stiffness are positively correlated with HCC risk, and about 80–90% of HCC cases have underlying fibrosis [19,20,21].
HSCs, as critical fibroblasts involved in liver fibrosis, are regulated by liver sinus endothelial cells (LESCs). Under physiological conditions, VEGF released by hepatocytes, cholangiocytes, and hematopoietic stem cells induces LSEC differentiation by stimulating nitric oxide (NO) production, thus inhibiting HSC activation and reversing the activated HSCs [22]. However, VEGF secreted by activated HSCs promotes angiogenesis during liver fibrosis and further activates HSCs to facilitate fibrosis formation [23]. When liver damage occurs, LSECs become capillarized, leading to a reduction in NO production and an increase in the release of signaling molecules, including transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF), thus promoting HSC activation [18]. In chronic inflammatory states, TGF-β secreted by LSECs and immune cells such as KCs is crucial in HSC activation. Inhibition of TGF-β has been proven effective in preventing and resolving liver fibrosis [24]. It activates HSCs via mitogen-activated protein kinase (MAPK) signaling pathways, including p38, extracellular signal-regulated kinase (ERK), and cjun Nterminal kinase (JNK) [25]. TGF-β1 also promotes HSC activation and differentiation into myofibroblasts expressing α-smooth muscle actin through the Notch pathway [26]. In HSCs, TGF-β stimulates the synthesis of ECM proteins such as type I and II collagen by activating SMAD2/3 and inhibiting their degradation, leading to collagen deposition [27]. Activated HSCs produce leptin, which promotes HSCs proliferation, migration, vasoconstriction, and secretion of ECM molecules and plays a crucial role in autocrine activation by upregulating TGF-β [28]. TGF-β also inhibits HSCs apoptosis induced by NK cells and affects the anti-fibrotic function of NK cells [29]. ECM and activated HSCs/CAFs, as crucial components in liver fibrosis, also contribute to HCC development within the TME. Their characteristics and functions in HCC progression will be detailed in subsequent Sects. (4.1.2. ECM and 4.1.3. CAFs). Targeting fibrosis represents a potential preventive and therapeutic strategy for HCC. The molecular mechanism by which fibrosis components initiate and promote HCC deserves further investigation.
Apart from liver fibrosis, the continuous cycle of destruction-regeneration process and oxidative stress in chronic inflammation causing DNA damage repair dysfunction and gene mutation in hepatocytes also promote HCC initiation [30]. In a healthy liver, controlled compensatory proliferation induced by hepatic progenitor cells restores injury-induced hepatocyte death [31]. However, as cells proliferate in an environment conducive to accumulating genetic mutations and the induction of oncogenic signaling pathways, proliferating cells are endowed with malignant potential [30, 32]. Continuous exposure to hepatotoxic factors transforms the liver microenvironment from a normal to a pro-inflammatory state and a subsequent precancerous microenvironment characterized by chronic inflammation and fibrosis, creating favorable conditions for HCC initiation. (Fig. 1).

Liver microenvironment from healthy to precancerous state. The persistent exposure to hepatotoxic factors induces changes in a the state of liver parenchymal cells, b phenotype and function of non-parenchymal cells, and c the structure of the ECM, resulting in a progression of the liver microenvironment from normal to an inflammatory and ultimately to a precancerous state distinguished by chronic inflammation and fibrosis
Heterogeneity of TME in HCC
With the continuous production and accumulation of HCC cells, the liver microenvironment is eventually transformed into TME composed of immune cells (T cells, B cells, neutrophils, macrophages, DCs, NK cells, and MDSCs), cytokines (IL, TNF, TGF, interferon (IFN), and chemokines) and non-immune components (HSCs, carcinoma-associated fibroblasts (CAFs), ECM, metabolite, exosomes, and vasculature) [33,34,35]. However, the TME of HCC exhibits significant heterogeneity among individuals and within tumors, resulting in different disease progression and prognosis that present challenges to HCC treatment [36]. An accurate understanding of TME heterogeneity in HCC is critical for developing more effective immunotherapeutic strategies.
Inter-patient heterogeneity: etiology
During HCC initiation, the liver microenvironment undergoes a dynamic progression from normal to precancerous, eventually leading to the TME. Nevertheless, the specific mechanisms of liver carcinogenesis induced by different etiologies vary, thereby influencing the composition of the TME [37] (Fig. 2). Further investigation of the TME characteristics in HCC arising from various etiologies via direct comparison is imperative for determining tailored treatment options for patients.

TME in HCC induced by various etiologies (HBV, HCV, NASH, and ASH)
Virus-related HCC
Currently, there are five recognized forms of viral hepatitis, with Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) showing the most vital connection to HCC development [38].
HBV is a potent and transient inducer of oxidative DNA damage in hepatocytes and rapidly activates double-stranded DNA repair mechanisms, leading to HBV DNA integration into host DNA [39]. The integrated HBV DNA drives HCC by inducing insertional alteration of HCC-associated genes, chromosomal instability, and expression of HBV proteins with oncogenic potential [40]. HBV proteins affect the immune microenvironment of the liver. HBsAg can suppress the activation of signal transducer and activator of transcription 3 (STAT3) in NK cells, leading to HBV clearance disorder and accelerating the progression from HBV hepatitis to HCC [41]. HBeAg recruits monocytic MDSCs (M-MDSCs) and triggers their expansion, thus inhibiting indoleamine 2,3-dioxygenase (IDO)-mediated CD8+ T cell responses in vitro [42]. NK cells are activated during acute HBV infection and are the primary immune cells responsible for combating viral infections. However, in the chronic phase of the disease, the function of NK cells is compromised. In patients with chronic hepatitis B (CHB), the expression of NK cell-activating receptor NKp30 was significantly decreased, accompanied by an increase in the expression of the inhibitory receptor NKG2A compared to normal controls [43]. T cells also demonstrate exhaustion in CHB patients, as evidenced by a substantial increase in the inhibitory receptor PD-1 expression in CD4+ and CD8+ T cells [43, 44]. Compared with non-virus-associated HCC, HBV-associated HCC exhibits selective enrichment of Treg and CD8+ resident memory T cells. These enriched cells express a higher level of PD-1 and their function is more suppressive and exhausted, associated with a poor prognosis [45]. Studies have shown that NK and T cell disorders in peripheral blood in CHB patients may be related to increased Bregs and decreased DCs [43]. Excessive levels of IL-10-expressing Bregs are also found within the TME of HBV-related HCC, which impairs the antitumor effect mediated by cytotoxic CD4+ T cells [46].
In contrast, HCV does not integrate into the host genome and can be eliminated with antiviral drugs, partially reducing the incidence of HCC. However, it is still a significant risk factor [47]. HCV indirectly promotes mutagenesis by eliciting oxidative stress and generating reactive oxygen species (ROS), which causes chromosomal and mitochondrial DNA damage and introduces mutations during DNA repair. Additionally, it promotes the accumulation of mutagenic factors by inducing endoplasmic reticulum (ER) stress [48]. Innate immune responses mediated by NK cells and adaptive immune responses mediated by virus-specific CD4+ T, CD8+ T, and B cells induced by antigen-presenting cells (APCs) in the early stages of HCV infection can lead to infection resolution in a small number of individuals. However, in most cases, the rapid diversification of the HCV genome and the suppression of immune response by HCV proteins mediate immune escape, thus promoting HCC initiation and progression [49]. The HCV core protein inhibits the initiation of antigen-specific CD4+ and CD8+ T cell responses by suppressing the expression of major histocompatibility complex (MHC) and costimulatory molecules on APCs [50, 51]. In chronic HCV-infected patients, T cells are exhausted and dysfunctional, characterized by low IFN-γ production and CD127 expression levels, lack of proliferation, and upregulation of PD-1 and T cell immunoglobulin domain and mucin domain-3 (TIM-3) [38]. HCV proteins also directly promote HCC by regulating oncogenes, promoting cell proliferation, and inhibiting apoptosis [52].
Non-viral HCC
Metabolic disorders in the liver lead to increased lipotoxicity, ER and oxidative stress, and immune system activation. These abnormalities drive a cycle of liver necro-inflammation and regeneration, resulting in nonalcoholic steatohepatitis (NASH) progression to HCC [53]. Platelets in the context of steatosis promote the initial inflammatory process by interacting with KCs and inflammatory monocytes, thereby driving the NASH-HCC transition. Prophylactic and therapeutic antiplatelet therapy reduces NASH-related HCC progression in mice and humans [54]. CD8+ T cells play an essential role in NASH-HCC progression. In preclinical models of NASH-induced HCC, CD8+ T cells accumulate in the liver with phenotypes combined tissue residency (CXCR6) with effector (granzyme) and exhaustion (PD-1). These CD8+ T cells auto-aggressively eliminate cells in an MHC-I-independent manner, which is associated with HCC progression in NASH patients driven by chronic liver injury [55]. Anti-PD-1 treatment increases the accumulation of aggressive CXCR6+ PD-1+ CD8+ T cells in the TME but fails to alleviate tumor burden [56]. Patients with NASH-related HCC who received anti-PD-1 or anti-PD-L1 treatment have a lower overall survival rate than other HCC etiologies due to impaired immune surveillance [56]. In addition, studies have shown that hepatocyte-derived extracellular vesicles in fatty liver induced by a high-fat diet can induce pre-metastatic niche and immunosuppressive TME, thereby promoting liver metastasis in colorectal cancer [57].
Alcohol-related liver disease (ALD) is the most common type of chronic liver disease worldwide. It accounts for approximately 30% of HCC cases and HCC-specific deaths [58]. Long-term alcohol consumption destroys the physiological structure and function of the liver by causing steatosis, steatohepatitis, and cirrhosis, thus inducing HCC [59]. In hepatocytes, alcohol is oxidized to acetaldehyde by alcohol dehydrogenase in the cytosol and oxidized by cytochrome P450 2E1 in microsomes, inducing ROS formation [60]. Acetaldehyde is a mutagenic compound that directly forms a variety of proteins and DNA adducts. These adducts contribute to DNA repair failure, lipid peroxidation, and mitochondrial damage, thus leading to HCC [61]. ROS accumulation and secondary oxidative stress also play a significant role in HCC initiation via DNA mutagenesis and lipid peroxidation [60]. Studies have found that granulocytic MDSCs (G-MDSCs) are recruited in the livers of ALD patients, inhibiting the function of NK cells and preventing the NK-induced HSCs apoptosis, thus accelerating hepatic cirrhosis and HCC progression. G-MDSCs also produce high levels of arginase 1(ARG1), TGF-β, and IL-10, suppressing T cell responses [62]. In the mouse alcohol-DEN-HCC model, ethanol can recruit macrophages and promote the transition to the M2 phenotype by increasing hepatic CCL2 expression [63]. In addition, alcohol induces microbiota dysbiosis and alters intestinal permeability, consequently facilitating the translocation of lipopolysaccharide (LPS) derived from bacteria into the liver [64]. In KCs, LPS interacts with toll-like receptor 4 (TLR4), triggering the production of pro-inflammatory cytokines and promotes HCC progression via the IL-6/STAT3 and TNF-α/NF-κB axes [58].
Intra-tumor heterogeneity: time and space
In addition to inter-patient heterogeneity, TMEs within the same tumor also showed heterogeneity at different times and spaces of progression. Intrinsic tumor events, such as genomic instability and epigenetic modifications, or extrinsic events, such as environmental perturbations and therapeutic stress, affect the structure of the TME [65,66,67].
Temporal heterogeneity
Tumors undergo dynamic immunoediting during progression, resulting in temporal heterogeneity in TME. Immunoediting refers to the process of the TME from limiting tumor growth (immune surveillance) to shaping tumor immunogenicity to create an environment conducive to tumor progression (immune tolerance), which can be divided into three stages: elimination, equilibrium, and escape [68, 69]. (Fig. 3).

Three stages of tumor immunoediting: elimination, equilibrium, and escape. a Elimination: The immune response induced by tissue damage due to tumor growth destroys tumor cells and further triggers specific recognition and killing of highly immunogenic subclones. b Equilibrium: The destruction and growth of tumor cells gradually reach a dynamic equilibrium, manifested as temporary tumor dormancy. c Escape: The immunogenicity of tumor cells gradually decreases, eventually obtaining an immune escape phenotype and inducing the formation of immunosuppressive TME
In the elimination stage, tumor growth destroys surrounding tissues and releases inflammatory signals, which recruit innate immune cells, including NK cells, γδ+ T cells, DCs, and macrophages. Upon activation, these immune cells can produce IFN-γ, which recruits more NK cells and macrophages through positive feedback to exert anti-tumor functions [70]. Additionally, IFN-γ directly induces tumor cell death through anti-proliferative mechanisms and impedes intra-tumoral angiogenesis by inducing chemokines, such as C-X-C motif chemokine ligand 9 (CXCL9), CXCL10, and CXCL11 [71, 72]. DCs can phagocytose dead tumor cells and migrate to the nearby lymph nodes, where they present tumor antigens on their surface and activate CD4+ and CD8+ T cells with highly immunogenic antigens. The activated lymphocytes can then access the circulation and home to the TME, attracted by chemokines produced by the tumor or its stromal cells. Once in the tumor, T cells can specifically recognize highly immunogenic tumor subclones through their T cell receptors (TCRs) and eliminate them via various effector mechanisms, such as cytotoxicity, cytokine secretion, and regulation of immune cells [68].
When the number of cancer cells reaches a dynamic equilibrium due to proliferation and elimination, the tumor subclones capable of surviving elimination enter an immune-mediated equilibrium phase, which can manifest as tumor mass dormancy [73, 74]. However, due to the constant pressure from the adaptive immune system and the genetic instability of tumor cells, tumor subclones with reduced immunogenicity have a survival advantage and gradually accumulate as they can evade immune recognition and destruction [75]. After completing immunogenic editing and developing the ability to hinder immune response, tumor cells can encourage immune cell depletion or differentiation into an immunosuppressive phenotype (Tregs, MDSCs, M2-TAMs, N2-TANs, CAFs) by releasing immune suppressive molecules (TGF-β, IL-10, VEGF, galectin, or IDO). The cells create an immunosuppressive TME, leading to immune escape [76].
Spatial heterogeneity
In different regions within the same tumor, there is significant heterogeneity in the genetic composition and antigen expression of tumor cells. They co-evolve with the local microenvironment, ultimately contributing to the spatial heterogeneity of TME [65, 77]. With the development of single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics technology, the spatial heterogeneity of TME is gradually revealed. A study in 2021 showed the first genome-wide spatial transcriptome map of HCC and analyzed the TME characteristics in normal, frontier, and tumor regions. The findings reveal that tumor samples with complete fibrous capsules consisting of fibroblasts and endothelial cells have higher spatial cluster continuity, lower transcriptome diversity and lower immune cell infiltration in tumor regions [78]. Another HCC spatial transcriptome study has shown that cancer clusters from the carcinoma sector exhibit significantly increased gene expression abundance and higher proliferative capacity compared to fiber cord and para-carcinoma sectors [79]. The researchers annotated the carcinoma sector at the end of the HCC progression trajectory as the core region through pseudo-time analysis. Within this core region, they observed a significant upregulation of CCL15, which promotes the immunosuppressive TME by recruiting macrophages and inducing M2 polarization. The high expression of CCL15 and M2 macrophage marker CD163 is associated with a poor prognosis for survival [79]. A study of the ecosystem around the tumor margin identified a 500 µm-wide zone centered on the tumor border in HCC patients, called “the invasive zone” [80]. Compared with other regions, the local TME in the invasion zone is highly immunosuppressive, characterized by increased expression of immune checkpoint genes, such as CTLA-4, CD96, and TIGIT, creating a favorable environment for tumor progression. HCC cells in this region exhibit metabolic reprogramming of fatty acid metabolism, making them more aggressive. Overexpression of serum amyloid A1 and A2 in hepatocytes severely damaged by direct invasion of tumor cells can recruit macrophages and induce M2 polarization, further promoting tumor immune evasion [80]. In addition, a study has analyzed the structural differences of TME among patients with varying responses to HCC immunotherapy and found a special spatial structure near the tumor boundary in non-responsive patients, called tumor immune barrier (TIB) [81]. TIB is formed by the interaction of SPP1+ macrophages and CAF, which limits the immune cell infiltration of tumor core. In mouse tumor models, blockade or macrophage-specific knockout of SPP1 can destroy the TIB structure and enhance the efficacy of anti-PD-1 therapy [81].
Immunosuppressive TME involved in HCC progression and metastasis
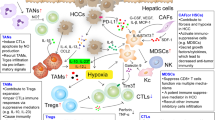
The TME of HCC exhibits significant spatiotemporal heterogeneity, which plays a dual role in tumor progression, thus leading to individual differences in prognosis. Immunosuppressive TME is undoubtedly the driver of HCC. (Fig. 4) Elucidating its composition and mechanisms by which it fosters tumor progression can provide valuable insights into identifying novel and effective targets for clinical treatment.

Components and features of immunosuppressive TME. The composition of the immunosuppressive TME is complex, mainly including HCC cells, immune cells, CAFs, ECM, metabolites, exosomes, and many functional molecules. Collectively, they interact to shape and enhance the features of the immunosuppressive TME that promote HCC progression and metastasis. a HCC cells exert robust proliferative ability by acquiring continuous proliferation signals, evasion of growth inhibition signals, resistance to cell death, and attainment of unlimited replication potential. b HCC cells unlock phenotypic plasticity through various mechanisms, including epithelial-mesenchymal transition (EMT) and transformation into cancer stem cells (CSCs), enhancing their aggressiveness. CAFs and TANs promote the conversion of HCC cells to CSCs, and TAMs participate in EMT. c HCC cells undergo metabolic reprogramming that affects the phenotype and function of other cells. Abnormal metabolites disrupt the function of CD4+ T, CD8+ T, and NK cells while promoting the recruitment of Tregs, TAMs, MDSCs, and CAFs, thereby contributing to immunosuppressive TME formation. (Table 1) d. Cytokines and exosomes released by tumors are significant in the complex crosstalk network of the immunosuppressive TME. In addition to promoting the recruitment and polarization of immunosuppressive cells, they also participate in angiogenesis, ECM remodeling, and EMT. (Tables 2 and 3) e HCC cells and CAFs promote ECM remodeling, thus facilitating HCC progression and metastasis. f The hypoxic environment due to ECM remodeling and the rapid proliferation of HCC cells promote angiogenesis and the recruitment of MDSCs and TAMs. g HCC cells and CAFs promote angiogenesis by secreting VEGF. h CD4+ T, CD8+ T, and NK cells acting as crucial components in inhibiting tumor growth, experience functional disruption within the immunosuppressive TME. i Immunosuppressive cells, acting as critical functional components within the immunosuppressive TME, can facilitate immune escape by directly influencing the phenotype of HCC cells or inhibiting the tumor-killing mechanisms of immune cells. They also mutually regulate each other through various functional molecules, synergistically exerting immunosuppressive functions. (Table 3) j In addition to remodeling the ECM, CAFs induce immune cells to transform into a suppressive phenotype
Non-Immune components
HCC cells
The malignant features of HCC cells are essential for shaping the immunosuppressive TME and directly promoting tumor progression and metastasis.
Firstly, HCC cells exhibit strong proliferation ability. The overexpression of hepatocyte growth factor (HGF) and abnormal activation of its tyrosine-protein kinase receptor c-MET induce auto-cellular signaling of self-growth within HCC cells [82]. Abnormal activation of growth factor signaling pathways, including HGF/c-MET and related genetic mutations such as RAS and PTEN, can activate RAS/RAF/MAPK and PI3K/AKT/mTOR pathway, which are essential for the regulation of cell metabolism, proliferation, differentiation, survival, and apoptosis [83]. ALKBH5, a demethylase, reduce PAQR4 expression, thus inhibiting the activation of the PI3K / AKT pathway. However, ALKBH5 is down-regulated in HCC and promotes HCC proliferation and invasion by affecting epigenetics [84, 85]. Cell division cycle associated 8 (CDCA8) has been identified as a novel oncogene in HCC, which enhances tumor cell viability and DNA synthesis, promoting HCC progression and metastasis [86].Mutations in tumor suppressor genes TP53 and RB drive the evasion of anti-proliferative signals during HCC progression by regulating the cell cycle [87]. Reactivation of telomerase in HCC cells prevents telomere shortening, enabling indefinite replication and enhancing cell aggressiveness via the accumulation of gene mutations [88].
In addition, HCC cells promote tumor heterogeneity by unlocking phenotypic plasticity during proliferation through dedifferentiation, differentiation inhibition, and transdifferentiation [89]. Several cytokines, including IL-1β, IL-6, IL-8, IL-11, CCL22, TNF-α, and TGF-β, can drive the dedifferentiation of HCC cells into CSCs via reverse transcription [90]. Due to the heterogeneity of CSCs, molecules such as CD44, CD90, and EpCAM are expressed differently in multiple subtypes, except for CD133 as a common molecular marker [91]. Numerous studies have demonstrated the association between high CD133 expression in HCC and increased tumor grade, advanced tumor stage, poor overall survival, high recurrence rate, and drug resistance [92].
EMT is also a crucial mechanism for the dedifferentiation of HCC cells, enhancing their invasive and metastatic abilities and allowing them to adapt to the rapid changes in the TME and, therefore, transform into more aggressive tumors [93]. The RHO GTPases upregulated in HCC control the movement of tumor cells, promoting migration, invasion, and metastasis, ultimately leading to EMT [94]. Disruption of E-cadherin/β-catenin complexes at cell boundaries also participates in EMT while blocking TGF-β can upregulate E-cadherin and reduce migration and invasion of HCC cells [95].
Furthermore, HCC cells are involved in immune evasion. They inhibit the activation of T/B cells through the downregulation of MHC and the immune co-stimulatory ligands CD80/86. The secretion of immunosuppressive molecules, such as TGF-β, IL-10, IDO, and ARG, also plays a complex role in the TME [96, 97]. The up-regulated circRanGAP1 in tumor cells induces TAM infiltration through the miR-27b-3p/NRAS/ERK axis to promote HCC progression [98]. Targeting the malignant features of HCC cells may inhibit tumor growth or improve immunosuppressive TME directly.
ECM
ECM in tumors undergoes various remodeling mechanisms, including deposition, modification, and degradation, to alter their physical and chemical properties, thus promoting tumor cell migration and invasion [99, 100] (Fig. 5). The subsequent formation of a distal premetastatic niche promotes metastatic colonization of tumor cells [99].

Mechanisms of ECM remodeling. a Tumor-derived factors facilitate the activation of CAFs, thus inducing the secretion of ECM molecules. b ECM-modifying enzymes enhance the density and rigidity of the ECM, thereby forming an immune barrier and hypoxic environment, which is conducive to the immune escape of HCC. c ECM-degrading enzymes and force-mediated physical remodeling facilitate HCC migration in ECM. d ECM remodeling occurs at potential metastatic sites to promote circulating tumor cell metastatic colonization
During the early stage of tumor progression, tumor-derived factors induce stromal cells to differentiate into CAFs, thereby promoting the production of ECM molecules and secreting them extracellularly after post-translational modifications, resulting in ECM deposition [101, 102].
ECM-modifying enzymes such as lysyl oxidase (LOX) and transglutaminase expressed by tumor cells further promote covalent cross-linking and linear arrangement of fibers [103, 104]. The high density and rigidity of ECM may serve as a physical barrier for T cells. Therefore, these cells cannot infiltrate into tumors, thus hindering their ability to recognize and kill cancer cells [105, 106]. ECM deposition can also exacerbate the restriction of oxygen and nutrient supply diffusion in tumor tissue. In hypoxic tumor cells, hypoxia-inducible factor-1 (HIF-1) expression exerts immunosuppressive function by inhibiting the activity of effector T cells and NK cells and recruiting immunosuppressive cells such as MDSCs and Tregs [107,108,109]. Hypoxia-induced adenosine also promotes immunosuppressive TME and inhibits the efficacy of ICIs, while HIF-1 promotes adenosine efflux in HCC [108, 110].
CAFs and tumor cells also secrete ECM-degrading enzymes, particularly matrix metalloproteinases (MMPs). In regions of dense collagen fibers, proteolysis is critical for tumor cell migration because it breaks down the migration barrier to reduce drag on migration trajectories [111]. The ECM-bound growth factors and cytokines, such as VEGF and TGF-β, can be released due to ECM degradation, leading to several signaling pathways that promote tumor growth and activation of angiogenesis [112, 113]. In addition to proteolysis, force-mediated physical remodeling plays a vital role in ECM disruption [114]. Increased ECM rigidity generates mechanical signals detected and transmitted by tumor cells and CAFs through transmembrane receptors, such as integrins [115]. Integrins bind to ECM molecules and link them to contractile structures in the cytoplasm, which respond to environmental stress signals. They form gaps in the stromal matrix, facilitating cell movement over longer distances by following the organized collagen fibers [116, 117]. Integrins also increase NF-κB or PI3K/AKT activity, reduce p53 activation, and increase the expression of pro-survival molecules BCL-2 and FLIP (also known as CFLAR) to enhance cell survival potential [118]. The release of antigens after tumor cell death, the first and critical step in initiating anti-cancer immunity, is weakened by the enhancement of cell survival potential [102]. In addition, integrins regulate regional activation of latent TGF-β contained in ECM and cell surface reservoirs. TGF-β is a pivotal immunosuppressive molecule that triggers the production of ECM-modifying enzymes [119, 120].
ECM remodeling promotes the migration of tumor cells into the bloodstream and facilitates metastatic colonization. Primary tumor-derived factors induce stromal cells at potential metastasis sites to activate CAF, leading to ECM remodeling and forming a pre-metastatic niche [121]. Circulating tumor cells evade the immune system by overexpressing ECM molecules, and the integrins on their surface can induce metastatic colonization at distantly remodeled ECM sites [99].
CAFs
CAFs in HCC originate from multiple cell types, including HSCs, HCC cells undergoing EMT, and mesenchymal stromal cells, leading to complex functional and phenotypic heterogeneity [107]. The currently recognized markers of CAF include α-SMA, FAP, FSP-1, Periostin, NG2, Tenascin-C, Podoplanin, and MFAP5 [122].
Apart from remodeling the ECM to promote the HCC progression, CAFs, as essential components in the immunosuppressive TME, are also involved in the interaction between immune cells to exert the immunosuppressive function. CAFs can produce Prostaglandin E2 (PGE2), IDO, and several co-signaling molecules of the B7 family to inhibit NK cell function and promote fibrosis and tumor cell growth. Combined use of PGE2 or IDO inhibitors and drugs targeting B7-H1 (PD-L1) can restore the anti-tumor effect of immune cells in TME [33]. CAFs in HCC can recruit DCs and secrete IL-6, which activates STAT3 in DCs. This activation transforms DCs into regulatory DCs, characterized by low expression of co-stimulatory molecules, high suppressive cytokine production, and enhanced immunosuppressiveness [123]. IL-6 secreted by CAFs also stimulates stem-cell-like properties by amplifying STAT3/Notch signaling in HCC cells [124]. Antigen-presenting CAF subtypes expressing MHC-II interact with and induce naïve CD4+ T cells into Tregs in an antigen-specific fashion [125]. CAFs can also enhance immunosuppression by inducing macrophages to polarize to the M2 phenotype and promoting the activity of MDSCs in the TME [107, 126].
Furthermore, CAFs also have a direct promoting effect on HCC cells. CAFs induce FOXQ1 expression, leading to the activation of the FOXQ1/NDRG1 axis in tumor cells to enhance HCC initiation. This activation also recruits more HSCs to TME as a complement for CAFs [127]. The positive feedback loop between CAFs and HCC cells promotes HCC initiation and progression, presenting a potential therapeutic target.
Metabolic reprogramming
Metabolic disorders, including glucose, lipids, and amino acids, are commonly found in the TME. HCC reprograms its metabolism to facilitate tumor proliferation and invasion.
According to the "Warburg effect", tumor cells mainly produce energy through glycolysis under aerobic conditions instead of oxidative phosphorylation [128, 129]. HCC increases adenosine triphosphate (ATP) production by augmenting glucose uptake and aerobic glycolysis [130]. Concurrently, the excessive production of lactic acid leads to acidification of the TME, thereby facilitating the infiltration and metastasis of HCC cells [131, 132]. Tumor cells also generate ATP for cell survival via the tricarboxylic acid cycle, which can accommodate limited quantities of metabolites originating from various pathways [133]. Reduced activity of succinate dehydrogenase and fumarate hydratase in HCC triggers the accumulation of succinate and fumaric acid, which in turn stabilizes HIF-1α, initiating glycolytic activation and angiogenesis while hindering the function of anti-tumor immunity [134, 135].
In addition to breaking down glucose to provide ATP, tumor cells utilize lipids, amino acids, and other substances to promote their proliferation [136]. Linoleic acid is one of the essential fatty acids in the human body, which has been found to suppress HCC cell proliferation and promote apoptosis. However, the level of linoleic acid in the portal vein blood of HCC patients is reduced [137]. On the contrary, the level of arachidonic acid synthesized by linoleic acid in the serum of HCC patients is elevated [138]. Arachidonic acid can be metabolized into various pro-inflammatory mediators, such as prostaglandins and leukotrienes, aggravating hepatic inflammation and facilitating HCC progression. Moreover, excessive cholesterol intake is widely regarded as a risk factor that independently promotes HCC progression [139]. HCC contains abundant cholesterol-rich membrane microdomains. These domains promote the proliferation and migration of HCC cells by inducing the upregulation of TLR7 expression [140]. HCC also displays abnormal levels of amino acids and metabolic enzymes. Tryptophan, an amino acid essential for the survival and proliferation of T cells, is depleted by the upregulation of the rate-limiting enzymes IDO1 and tryptophan-2, 3-dioxygenase 2 (TDO2) by HCC cells, thus suppressing immune surveillance [141]. In addition, an MYC-dependent metabolic switch from glutaminase 2 (GLS2) to GLS1 may occur during HCC progression, enabling metabolic rewiring by increasing glutamine uptake and decomposition rate in tumor cells [11]. The metabolic reprogramming of HCC cells can impact metabolites and enzymes in the TME, thereby facilitating the development of immunosuppressive TME [142] (Table 1).
Exosomes
The macromolecule profiles in exosomes released by HCC cells significantly differ from those of normal cells [143]. These exosomes can be taken up and internalized by other cells to promote angiogenesis, vascular permeability, EMT, ECM remodeling, and immunosuppression, exerting a driving force in the formation of immunosuppressive TME. (Table 2) Exosomes secreted by other cells recruited or activated during HCC progression also play a significant role as functional carriers. M2 exosomal miR-660-5p promotes the growth of HCC cells by regulating KLF3 [144]; miR-27a-3p increases stemness, proliferation, drug resistance, migration, invasion, and in vivo tumorigenicity of HCC cells by down-regulating TXNIP [145]. LncRNA TUG1 in exosomes derived from CAFs promotes the migration, invasion, and glycolysis of HCC cells via the miR-524-5p/SIX1 axis [146]. The loss of anti-tumoral miR-150-3p and miR-320a in CAFs-derived exosomes is also associated with HCC progression [147, 148].
Vascular populations
HCC has high levels of vascular transformations and angiogenesis during progression [149]. Vascular populations are involved in energy supply, signal crosstalk and tumor metastasis in TME, thereby promoting tumor progression [150]. With tumor growth and ECM deposition, increased hypoxia within the tumor promotes the secretion of pro-angiogenic factors, such as VEGF and PDGF. They stimulate the proliferation and migration of endothelial cells (ECs) in surrounding tissues to achieve angiogenesis [151]. Studies have shown differences in ECs between tumors and adjacent normal tissues. Plasmalemma vesicle-associated protein (PLVAP)+ ECs are enriched in tumor tissues and are identified as HCC-specific [152]. PLVAP can induce monocytes to differentiate into FOLR2+ TAM through NOTCH signaling, which promotes the formation of immunosuppressive TME and attenuates the response to immunotherapy [152, 153]. ECs also regulate tumor progression and metastasis by secreting angiocrine factors, such as IL-6, TGFβ, and VEGF [154]. Studies have shown that VEGF disrupts the tight junction of ECs and leads to increased vascular permeability, which promotes tumor metastasis [155]. The level of angiocrine factors secreted by ECs in metastatic tumors is higher than those in non-metastatic tumors [156]. In addition, some researchers have developed HCC-endothelial co-culture models to simulate angiocrine crosstalk in HCC. They found that angiocrine signaling produces an inflammatory microenvironment that affects the recruitment and status of immune cells [157].
Tumors are not entirely dependent on the host ECs to form vascular populations. Vasculogenic mimicry (VM) is a process in which tumor cells autonomously form vascular-like channels. These channels are connected with the host circulatory system and contain blood components such as red blood cells, platelets, and hemoglobin [158]. In HCC, VM is associated with high tumor grade, invasiveness, and poor prognosis [159, 160]. The mechanism of VM formation is very complex. TME components such as hypoxia, EMT, CSC, CAF, ECM, cytokines, and non-coding RNA are all associated with VM [161]. The specific formation and regulation mechanism of VM in HCC remains to be further studied. As an alternative perfusion route for tumors, VM may be an important mechanism leading to the non-response of current anti-angiogenesis therapy [162]. Inhibition of both angiogenesis and VM formation is a possible strategy to optimize the efficacy of HCC.
Immunosuppressive cells
Tregs: the hard core
Tregs, an immunosuppressive subtype of CD4+ T cells, constitutively express the CD25 and CTLA-4 on their surface and the forkhead box P3 (Foxp3) transcription factor in their nucleus [163]. Tregs originate from different sources and can be categorized into two groups: thymic Tregs, also known as natural Tregs, and peripheral Tregs, also called induced Tregs [164]. The former are derived from developing T cells in the thymus with intermediate TCR affinity for self-peptide/MHC ligands. The latter are derived from naïve CD4+ T cells in the periphery that are activated in the presence of immunosuppressive molecules such as TGF-β and IL-2 [164, 165].
Tregs express multiple chemokine receptors, enabling their recruitment to the TME through chemokine gradients, such as Chemokine receptor 4 (CCR4)-CCL17/22, CCR5-CCL5, CCR8-CCL1, and CCR10-CCL28 [164]. Both the proportion and absolute number of Tregs in the surrounding area of HCC were notably elevated [166]. The presence of Tregs in the peripheral blood of HCC patients also markedly surpasses that in healthy individuals. AS HCC progression, Treg frequency escalates, thus enhancing invasiveness and metastatic potential [167]. The prognosis of HCC patients is significantly negatively correlated with the high expression of Tregs [168].
Tumor-infiltrating Tregs (TI-Tregs), characterized by CD45RA− CD25hi Foxp3hi CTLA-4 hi phenotype, promote tumor evasion through various mechanisms [169]. Most expanded Tregs do not express CD45RA, which indicates that Tregs possess a memory phenotype. Memory Tregs exposed to tumor antigens are significantly amplified in HCC [170]. Foxp3 is a crucial regulatory transcription factor that generates immunosuppressive Tregs [171]. Foxp3 can suppress glycolysis, promote fatty acid-fueled oxidative phosphorylation, and enable the use of lactate as an energy source via monocarboxylic acid transporter 1, thereby providing Tregs in HCC the metabolic flexibility that is essential for their survival and efficient functional capacity in the nutrient-restricted and lactate-rich microenvironment brought about by tumor metabolic reprogramming [164, 172]. In addition, elevated levels of Foxp3 expression in TI-Tregs can promote CD25 (IL-2Rα) expression, greatly enhancing affinity for IL-2, allowing Treg to bind to IL-2 preferentially [173]. IL-2 sequestration by Tregs may be accompanied by a lack of co-stimulation, leading to an inability to effectively activate CD8+ T and NK cells [174, 175]. CTLA-4 belongs to the immunoglobulin superfamily, structurally similar to CD28, but has a higher affinity to CD80/CD86, thereby exerting inhibitory effects on T cell responses [176]. In naive T cells, CTLA-4 is initially situated intracellularly and translocates to the cell surface after receiving stimulation signals. However, CTLA-4 is constitutively expressed on Tregs and participates in their immunosuppressive functions [177]. The highly expressed TGF-β1 in the TME of HCC upregulates the expression of CTLA-4 and PD-1 on T cells, thereby attenuating the cytotoxicity of T cells against HCC cells [178]. In addition to the cell surface receptors directly involved in immunosuppression, Tregs present on their surface many other molecules that can participate indirectly in tumor immune evasion, such as CD39 and CD73, act as ectonucleotidases that convert ATP/ADP to AMP and AMP to adenosine, respectively. The co-expression of CD39 and CD73 by Tregs enables them to enhance the levels of cyclic adenosine monophosphate (cAMP) in anti-tumor T cells through the activation of adenosine receptors, which diminish TCR signaling and IFN-γ production [179, 180]. Downregulation of cAMP levels requires the involvement of TCR/CD28-mediated recruitment of phosphodiesterases to enhance T cell activation [181]. However, Tregs inhibit the CD28 co-stimulus signal, thus aggravating the suppression of T cell activation. In addition, Tregs secrete inhibitory cytokines like TGF-β, IL-10, and IL-35 to promote immunosuppressive TME [182].
Bregs: the evil side
B cells are the second most abundant tumor-infiltrating lymphocytes and include effector B cells and Breg cells, which play a dual role in tumor immunity [183]. Many studies have shown that Breg cells promote immune evasion and correlate with advanced disease and poorer prognosis. TIM-1+ and PD-1hi Breg subtypes can be identified in HCC, characterized as CD5high CD24− CD27−/+ CD38+/high and CD5hi CD24−/+ CD27hi/+ CD38dim [184, 185].
Myeloid cells in TME can induce TIM-1+Breg cells to produce IL-10 through TIM-1/TIM-4 signaling [184]. Bregs induced by increased TLR4-mediated BCL6 expression can increase the expression of PD-1, which suppresses specific T cell immune response, while also producing IL-10 [185]. IL-10 secreted by Bregs suppresses the activity of cytotoxic CD8+ T cells, Th1 and Th17, and induces the conversion of CD4+ T cells to Tregs, thereby promoting tumor immune evasion [186]. In addition, the classic Breg, characterized by the CD19+ CD24+ CD38+ phenotype, can promote HCC growth and invasiveness through direct interaction with HCC cells via the CD40/CD154 signaling pathway [187].
MDSCs: brothers in arms
The differentiation of myeloid cells frequently changes under carcinogenic conditions, leading to the accumulation of immature myeloid cells, termed MDSCs. These cells exhibit potent immunosuppressive activity and impaired antigen presentation capabilities [188]. MDSCs have two major types: M-MDSC and G-MDSC, also known as polymorphonuclear MDSC (PMN-MDSC), which have similar morphological features to monocytes and neutrophils, respectively [189]. In humans, M-MDSCs are characterized by the expression of surface markers CD11b+ CD33hi HLA-DR− CD14+ CD15−, while PMN-MDSCs are defined as CD11b+ CD33dim HLA-DR− CD14− CD15+ CD66b+. In mice, M-MDSCs are mainly described as CD11b+ Ly6Chi Ly6G− and PMN-MDSCs are defined as CD11b+ Ly6Clo Ly6G+ [190, 191]. M-MDSCs and TAMs can be differentiated in humans based on the expression of HLA-DR, an MHC-II molecule. In mice, they can be distinguished by the differential expression of Ly6C and S100A9, as well as the high expression of MCSF, F4/80, IRF8 and CSF1R in TAMs. However, the phenotypic differentiation between PMN-MDSCs and TANs is currently controversial. The effective identification markers remain elusive in mice. Gradient centrifugation can potentially segregate PMN-MDSC and neutrophils in human peripheral blood, with the former exhibiting a lower density [193]. Recent studies have identified lectin-type oxidized LDL receptor 1 (LOX-1) as a promising distinctive surface marker for distinguishing human PMN-MDSCs from neutrophils, warranting further investigation for validation [194]. Elevated LOX-1+ CD15+ PMN-MDSC levels can be detected in patients with HCC [195]. Generally, PMN-MDSCs are prevalent in the TME, while M-MDSCs amass in the peripheral blood and demonstrate stronger inhibitory activity [196].
During cancer progression, the MDSCs generation and activation process can be divided into two phases where the signal factors overlap significantly. The first phase is the proliferation of immature myeloid cells (IMCs) with inhibited differentiation function in the bone marrow, mainly mediated by tumor-derived cytokines, such as IL-6, IL-11, IL-17A, granulocyte–macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), and TNFα. It also involves the activation of signaling pathways including STAT3, Notch, NLRP3, RB1, IRF8, adenosine receptors A2b, and C/EBPβ [196, 197]. In the second phase, IMCs are converted into MDSCs in peripheral tissues mainly due to the action of pro-inflammatory cytokines from tumor-associated stromal cells and activated immune cells, such as TNF-α, multiple ILs, PGE2, COX2, and involves signaling pathways, including NF-κB, STAT1, STAT6, and ER stress pathways [196, 197]. Activated MDSCs and IMCs then migrate from BM or peripheral lymphoid tissue into the bloodstream, colonizing the tumor site [198].
Alterations in the expression of enzymes, including receptor-interacting protein kinase 3, apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3B, and cell cycle-related kinase, activate multiple signal transduction pathways in HCC cells, leading to the generation of IL-6, chemokines (CXCL1, CCL2, and CCL15), and growth factors, inducing the recruitment and proliferation of MDSCs [199,200,201,202]. PGE2 and COX2 derived from HSCs also aid in accumulating MDSCs in tumor sites [203]. In addition, MDSCs tend to infiltrate into hypoxic areas of HCC tissue. HIFs can activate the transcription of CCL26 in HCC cells, recruiting CX3CR1-expressing MDSCs into tumors [204]. HIF-1 also upregulates the expression of exonucleoside triphosphate diphosphate hydrolase 2, resulting in an increase in extracellular 5′-AMP levels, which prevents the differentiation of MDSCs into non-immune suppressive DCs [205].
In HCC, ARG1 and inducible nitric oxide synthase, highly expressed by MDSCs, compete to consume L-arginine [206]. L-arginine is a conditionally essential amino acid for T cells. Knockout of L-arginine blocks the cell cycle of tumor-infiltrating T cells in the G0-G1 phase to interfere with the T cells cycle [207]. Depletion of L-arginine also affects the assembly and stabilization of the TCR-CD3 complexes and impairs the formation of immune synapses between APCs and T cells [208, 209]. MDSC can also induce the production of high levels of ROS and reactive nitrogen species (RNS). ROS harms T cells by damaging proteins, nucleic acids, and lipids [210]. High levels of RNS induce the dissociation of the TCR-CD3 complex and reduce T cell migration and tumor infiltration induced by chemotactic factors through nitration reactions [211]. In addition to suppressing the immune response of T cells, MDSCs promote clonal expansion of antigen-specific natural Tregs and generate induced Tregs from naïve CD4+ T cells [212]. The inhibitory effect of MDSCs on other immune cells also plays a significant role in HCC progression and metastasis. MDSCs can decrease the expression of co-stimulatory molecules CD86 and MHC-II on KCs surface and upregulate the co-inhibitory molecule PD-L1, which ultimately suppresses the antigen presentation proficiency of KCs [213]. In HCC patients, MDSCs inhibit cytotoxicity and IFN-γ release through direct cell-to-cell contact via NKp30 receptors on NK cell surfaces [214]. MDSCs also inhibit TLR-ligand-induced IL-12 production by DCs through IL-10 production and suppress the T cell-stimulating activity of DCs in HCC [215]. In addition to immunosuppression, MDSCs are highly adaptable and respond to microenvironmental signals. In peripheral tissues, the presence of inflammatory factors derived from tumors can induce the differentiation of MDSCs into immunosuppressive macrophages and inhibit the functional maturation of DCs. Under hypoxic conditions within the TME, MDSCs can differentiate into TAMs [197].
TAMs: friends or foes?
Macrophages in the liver are composed of KCs and monocytes. KCs originate from yolk sac-derived precursors during embryogenesis and differentiate into non-migratory tissue-resident macrophages in the liver, essential for liver and systemic homeostasis [216]. After sensing danger signals, KCs control inflammation and recruit immune cells to the liver. In HCC, monocytes are recruited to TME by several factors, including VEGF, PDGF, TGF-β, CCL2, or macrophage colony-stimulating factor (M-CSF), and eventually differentiate into TAMs [217]. Macrophages are typically classified into two subtypes: M1, characterized by pro-inflammatory and anti-tumor activity, and M2, known for anti-inflammatory and pro-tumor characteristics [218]. M1 phenotype is stimulated by pro-inflammatory cytokines such as IFN-γ, TNF, and TLR ligands. They can present antigens and express several pro-inflammatory cytokines such as IL-1, IL-6, IL-12, IL-23, IFN, TNF-α, CXCL1-3, CXCL-5, and CXCL8-10 [219]. In contrast, M2 phenotype is triggered by cytokines like IL-4, IL-10, and IL-13. They express immunosuppressive molecules including IL-10, TGF-β, and PD-L1 and demonstrate limited capability for antigen presentation [220, 221].
During the elimination and equilibrium stages of tumor immunoediting, macrophages are polarized to M1 under the stimulation of immune cell-derived IFN-γ [70, 222]. M1 phenotype promotes Th1 antitumor immune response by producing cytokines [223]. However, with the progression of HCC, TAMs will undergo a phenotypic transition from M1 to M2 [222]. This is closely related to IL-4, IL-10, and TGF-β secreted by HCC cells [224]. In addition, the M2 phenotype can promote this phenotypic transformation by secreting CCL2 [225]. Defective NF-κB activation and IL-10 overexpression are the causes of M2 immunosuppression [226]. As tumor progression, the transition from M1 to M2 is paralleled with a gradual decline in NF-κB activity [227]. An extensive nuclear localization of p50 NF-κB inhibitory homodimers leads to the lack of M1-functions and tumor progression [228]. Intriguingly, in the pre-cancerous chronic inflammation stage, the full activation of NF-κB can exacerbate local M1 inflammation, also promoting tumorigenesis [229, 230]. This suggests that the M1 phenotype may have a dual role in tumor progression. Future investigations on TAMs should consider both their polarization state and the stage of HCC. Although TAMs are traditionally categorized into M1 and M2 polarized phenotypes, recent studies have shown that the distinction is not absolute [231]. A study utilizing scRNA-seq examined macrophages in HCC and identified a cell cluster that co-expressed genes characteristic of M1 and M2 polarization states [232]. In another study focusing on HBV/HCV-related HCC, macrophages with both M1 and M2 features were also detected [233]. Some researchers have proposed a functional adaptation model suggesting that macrophages can gradually alter their functional phenotypes in response to evolving signals within the TME [234].
Although the classification and polarization mechanisms are controversial, most HCC-infiltrated TAMs have markers of the M2 phenotype, including HLA-DR, CD68, CD163, and CD206, and exert immunosuppressive functions [235]. TAM invasion frequency is associated with a dismal survival prognosis and an increased risk of recurrence in HCC [235]. TAMs are preferentially attracted to hypoxic areas in the tumor [221]. Hypoxia-induced HIF-1α and necrotic debris of HCC cells promote the secretion of IL-1β secretion by TAM, which, in turn, upregulates the production of HIF-1α in HCC cells. The HIF-1α/IL-1β signaling loop between HCC cells and TAM promotes EMT and metastasis of HCC [236]. TAMs play a critical role in suppressing the T cell immune response. Elevated levels of IL-6 in patients with HCC activate the STAT3/C-Myc pathway to increase the transcript levels of miR-25-3p in TAMs, resulting in inhibition of phosphatase receptor type O. This inhibition increases the expression of PD-L1, which in turn causes T cell exhaustion [237]. IL-12 secreted by TAMs in HCC can activate T cells to release IFN-γ, which causes TAMs to secrete IDO, an immunomodulatory enzyme that inhibits T cell responses. The feedback inhibition mechanism of the IFN-γ-IDO-T cell dysfunction axis impairs T cell proliferation and effector cytokine production [238]. TAMs also express galactoeglutinin-9 (Gal-9), which binds to TIM-3, a co-inhibitory receptor with an elevated expression on T cells in TME. Blocking the TIM-3-Gal-9 signaling pathway reactivates T cell-mediated anti-tumor immunity [239]. In addition to immunosuppressive function, TAMs promote EMT and tumor stem cell differentiation in the Wnt/β-catenin pathway by secreting TNF-α [240]. Moreover, TAMs secrete several proteases, including MMPs, cysteine cathepsins, and serine proteases, which cleave ECM and basement membrane components and destroy cell adhesion junctions, promoting tumor cell invasion and metastasis [221].
TANs: accelerators or brakes?
Neutrophils play a crucial role in the human body's immune response, aiding in the resistance to infection and tissue damage. They also migrate into tumors and differentiate into two phenotypes: anti-tumorigenic N1 and pro-tumorigenic N2, depending on the various signal inductions present in the TME [241]. It has been suggested that the pathologically activated N2 phenotype with immunosuppressive activity looks the same as PMN-MDSC [242]. Currently, there is no established method to clearly distinguish TANs and PMN-MDSCs, especially in mice. However, due to technical limitations in obtaining TANs from human tumor tissues, most studies on TANs have been conducted in mouse models, potentially compromising the dependability of experimental findings.
Although N1/N2 classification is a convenient model to describe TAN, a simple binary representation is not enough to characterize the phenotype and function of neutrophils. In 2022, a study based on scRNA-seq first described significant neutrophil heterogeneity in HCC patients and preclinical models [36]. The researchers identified six transcriptionally distinct neutrophil clusters from tumor tissues in HCC and analyzed the potential function of each cluster in TME. IFIT1+, SPP1+ and CCL4+ TANs were found to be pro-tumor, linked to a worse prognosis and expressing a high level of PD-L1; CD74+ TANs were associated with a better prognosis, but their relatively high PD-L1 expression raises doubts about their role in tumor progression; MMP8+ and APOA2+ TANs exhibited lower PD-L1 levels, correlating with a better prognosis and suggesting a potential anti-tumor role in HCC [36]. However, this study was conducted on resected samples, which may not accurately reflect patients with advanced HCC unable to undergo resection. With new technology development, TAN at different stages of tumor progression may be further categorized into distinct subtypes. The current research on the pro-tumor mechanism of TAN still focuses on the N2 phenotype.
CAFs play a vital role in neutrophil accumulation and N2 polarization. They recruit peripheral blood neutrophils through the SDF1a/CXCR4 signaling pathway and induce them to differentiate into PDL1+ N2 phenotype with a capacity to suppress T cell immunity via the IL6-STAT3-PDL1 signaling cascade [243]. CAF-derived cardiotrophin-like cytokine factor 1 (CLCF-1) also induces TAN infiltration and N2 polarization. CLCF-1 stimulates HCC cells to secrete CXCL6 and TGF-β, which can activate the ERK1/2 signaling pathway in CAFs. This activation increases CLCF-1 production, forming a positive feedback loop between CSCs and TANs, thus regulating HCC progression [244]. In addition, TANs themselves are involved in the recruitment. They secrete bone morphogenetic protein 2 and TGF-β2 and regulate the gene expression in HCC cells, thus increasing their stem-like characteristics. These TAN-induced HCC stem-cell-like cells are highly active in the NF-κB signaling pathway, secreting higher levels of CXCL5, thereby attracting more TAN infiltration [245]. Significant expressions of GM-CSF, TNF-α, and TGF-β in the peritumor region of HCC promote a phenotypic transition from an N1 to N2 phenotype [246, 247].
High infiltration of TANs within the TME frequently correlates with an adverse prognosis. GM-CSF derived from HCC cells can promptly activate ERK1/2, p38, and NF-κB in TANs to stimulate HGF production. Activation of highly expressed c-MET in HCC cells promotes HCC progression and metastasis [248]. The expression of CCL2 and CCL17 in TANs is also associated with prognosis in HCC patients. CCL2 facilitates macrophage recruitment, while CCL17 facilitates Treg recruitment, promoting immune evasion [249]. The dual regulatory effect of TANs on the vasculature plays an important role in promoting tumor progression and metastasis. TANs facilitate tumor angiogenesis by secreting MMP9 [250]. However, in tumor regions with established vasculature, TANs can compromise vascular integrity by reducing tight junctions between endothelial cells, thereby promoting tumor invasiveness through the release of neutrophil extracellular traps (NETs) [251]. The formation of NETs can also act as a protective barrier around tumor cells, shielding them from CD8+ T cell and NK cell-mediated cytotoxicity. By obstructing physical contact between immune cells and tumor cells, the NETs can contribute to tumor progression and metastasis [252].
Immunosuppressive molecules
Multiple cells in TME synergistically facilitate tumor immune evasion through complex crosstalk, in which immunosuppressive molecules play a crucial role. Immune checkpoints, as inhibitory molecules on the surface of immune cells, have emerged as significant targets for immunotherapy. In addition to the constitutively expressed CTLA-4 on Tregs involved in immune evasion, PD-1, TIM-3, and lymphocyte activating gene 3 (LAG-3) also have significant associations in HCC progression. Antibodies directed against these immune checkpoints restore tumor antigen-induced T cell responses in HCC, and their combination exhibits an additive effect [253,254,255,256,257].
The PD-1/PD-Ls pathway regulates the induction and maintenance of immune tolerance in the TME [258, 259]. PD-1, a member of the immunoglobulin superfamily, serves as a transmembrane co-inhibitory receptor mainly found on the surfaces of activated T cells, B cells, and NK cells [260]. PD-L1 and PD-L2 are ligands for PD-1 and have different expression patterns. In general, PD-L1 is constitutively expressed at low levels on hepatocytes, HSCs, LSECs, and KCs. PD-L2 is expressed on DCs and macrophages after activation. PD-Ls are also highly expressed on the surface of tumor cells [260]. Pro-inflammatory cytokines, such as IFNs, TNF-α, and VEGF, produced in the immune response upregulate the expression of PD-Ls [261]. In HCC patients, CD8+ T cells that recognize tumor neoantigens and are activated express PD-1 and produce IFN-γ, inducing high expression of PD-Ls on APC and HCC cells [261, 262]. The combination of PD-1 and PD-Ls selectively inhibits the proliferation and immune activity of tumor-specific T cells, thus achieving tumor immune escape [263].
TIM-3 is expressed on T cells, DCs, macrophages, and NK cells. It has several ligands, including Gal-9, phosphatidylserine, carcinoembryonic antigen cell adhesion molecule 1, and high-mobility group protein 1 [239]. TIM-3 induces CD4+ and CD8+ T cell exhaustion and enhances the immunosuppressive function of Tregs via increased expression of IL-10 and a shift to a more glycolytic metabolic phenotype [264]. Activation of TIM-3 on resident DCs hinders the recruitment of nucleic acids into DC endosomes, thereby inhibiting the immune response against nucleic acids derived from tumors [265]. In addition, TIM-3 participates in the M2 polarization of macrophages and promotes tumor growth via the NF-κB/IL-6 axis [266]. TIM-3 is upregulated on NK cells in HCC, blocking cytokine secretion and cytotoxicity through a PI3K/Akt/mTOR-dependent way [267]. HCC cells also express TIM-3, which promotes HCC metastasis by increasing EMT [268].
LAG-3 is a transmembrane protein predominantly expressed in activated T cells that regulates T cell proliferation, activation, effector functions, and homeostasis [269]. It shares significant structural homology with CD4 and exhibits a greater affinity for MHC-II [270]. Monoclonal antibodies that block the interaction between LAG-3 and MHC-II have antitumor activity [271]. In addition, fibrinogen-like proteins, Gal-3, and lymph node sinusoidal EC C-type lectin, can also activate LAG-3 [268]. FGL1 is an emerging hepatic factor expressed in the liver under steady-state conditions to induce an immunosuppressive environment of physiological states. However, the expression of FGL1 is significantly increased in HCC, which is associated with poor prognosis and immunotherapy resistance [269, 272]. Increased LAG-3 expression of CD8+ T cells in advanced HCC inhibits the production of cytokine and granzyme in T cells and promotes differentiation into Tregs [273]. Recent studies have shown that LAG-3 may synergize with PD-1/PD-L1, and the combination of monoclonal antibodies targeting LAG-3 and PD-1 can ameliorate tumors resistant to PD-L1 inhibitors [274].
In addition to immune checkpoints, soluble molecules such as cytokines and enzymes also exert immunosuppressive functions and act as signaling molecules to influence the phenotype and function of other cells. (Table 3) The intricate compensation and feedback mechanisms of the signaling network, formed through the participation of immunosuppressive molecules, pose significant challenges to immunotherapy. Understanding the crosstalk mechanism among various components and further exploring the effects of targeting key molecules can offer valuable insights for optimizing therapeutic strategies.
Application of immunosuppressive TME in clinical treatment
The current immunotherapy landscape
Approved drugs
In traditional local therapies, the limited availability of treatment options for patients with advanced HCC is hindered by chemical resistance and the risk of radioactivity. The results from the 2007 SHARP trial demonstrated the superior efficacy of multi-kinase inhibitor (MKI) sorafenib therapy over placebo, marking the advent of systemic therapies based on immuno-oncology [275]. Up to now, FDA has approved three first-line therapies (sorafenib, lenvatinib, and atezolizumab plus bevacizumab) and six second-line therapies (regorafenib, nivolumab, cabozantinib, ramucirumab, pembrolizumab, and nivolumab plus ipilimumab), encompassing MKIs (targets VEGFR, PDGFR, FGFR, KIT, RET, MET, MAPK, etc.), monoclonal antibodies (anti-VEGFA), and ICIs (targeted PD-1/PD-L1 and CTLA-4) [4,5,6,7, 275,276,277,278,279,280,281] (Fig. 6a). The TME can be reshaped by MKIs and monoclonal antibodies, transforming a cold tumor into a hot tumor with infiltration of effector T cells [282] (Fig. 6b). The combination of them with ICIs has the potential to improve therapeutic efficacy. In 2020, the IMbrave150 trial demonstrated the superiority of the atezolizumab plus bevacizumab combination over sorafenib in all clinical endpoints, heralding a new era dominated by combination immunotherapy [4, 5]. Over the last seventeen years, the continuous innovation of immunotherapy has significantly improved patient survival outcomes, including overall survival, progression-free survival, and overall response rate. (Fig. 7).

Targets and mechanisms of FDA-approved immunotherapies. a. The direct targets of FDA-approved first-line (blue background) and second-line (yellow background) immunotherapies in the HCC microenvironment. b. MKIs and monoclonal antibodies affect the immunosuppressive TME after acting on the targets. Inhibiting the infiltration and function of immunosuppressive cells while promoting the infiltration and activation of immune cells can transform cold tumors into hot tumors

The development process of immunotherapy with increasing efficacy
Development of drug therapy
The immunosuppressive TME plays a crucial role in enabling immune evasion. Therefore, remodeling the TME is a key focus of numerous emerging immunotherapies [283,284,285]. Immunosuppressive cells, as essential components of intricate immune networks, offer three ideas for immunotherapy based on their distinct mechanisms of recruitment, polarization, and exertion of immunosuppressive functions: 1. Disrupt the pathways involved in the recruitment and polarization of immunosuppressive cells in the TME or induce their apoptosis to decrease the infiltration of these cells. 2. Destroy the regulatory mechanisms between immunosuppressive cells and tumor cells, thus preventing the immunosuppressive effect. 3. Remodel immunosuppressive cells or activate cellular immune function. Currently, the development of single-agent regimens for potential immunotherapies, such as inhibiting novel immune checkpoints, activating co-stimulatory molecules in immune cells, and targeting critical molecules involved in cellular crosstalk, basically follow the abovementioned ideas. (Table 4).
The diversity and crosstalk of cells and molecules in the TME provide many potential effective targets for drug development; however, a substantial amount of clinical evidence indicates that the efficacy of a single drug has a ceiling effect. In contrast, combination therapy can collaboratively reshape the immunosuppressive TME through various mechanisms, improving drug sensitivity and overcoming drug resistance [17, 286,287,288]. However, compared to single-drug therapy, combination therapy may lead to unexpected drug overlapping toxicity and specific toxicity, often accompanied by higher incidence and a broader spectrum of adverse events. Based on the incidence rate of grade 3 or 4 treatment-related adverse events, it appears that the combination of ICIs has the best safety profile in HCC, followed by the combination of ICIs with anti-angiogenesis antibodies and the combination of ICIs with MKIs [289]. Several ongoing clinical trials aim to confirm the efficacy and safety of emerging therapies and combination treatment regimens. In order to maximize the benefits of combination therapy, it is necessary to conduct further research and adjust dosage and intervention sequences based on the mechanisms of the drugs.
Emerging immunotheraputic strategies
In addition to drug-targeted therapy, the emergence of innovative approaches such as tumor vaccines and adoptive cell therapy (ACT) offers promising prospects for immunotherapy [290]. However, additional research and development are required to address concerns regarding their preparation, stability, and safety before gaining approval from the FDA for clinical applications.
Tumor vaccines developed based on tumor-associated antigens (TAAs) can enhance patient-specific immune responses. Since the development of DNA sequencing technology makes it possible to identify patient-specific TAAs, therapeutic vaccine-induced immune response is a promising strategy for HCC treatment [291].
Peptide-based or DC-based vaccines are two types of traditional HCC vaccines. HCC peptide vaccines are derived from TAAs, effectively instigating targeted anti-tumor responses. Currently, only vaccines based on alpha-fetoprotein (AFP), telomerase reverse transcriptase (TERT), and glypican-3 (GPC3) have been evaluated in clinical trials with limited results [292]. DC cells extracted from peripheral blood are induced by cytokines such as GM-CSF, IL-3, and IL-4 to mature and then are loaded with TAAs through appropriate strategies [293]. An in vitro study has demonstrated that whole tumor cell lysate-pulsed DCs activate the HCC cell-specific cytotoxic activity of T cells [294]. In the orthotopic murine model for HCC, the DC vaccine loaded with Hepa1-6 cell lysates can alter immunosuppressive TME by reducing the accumulation of Tregs and TGF-β, thus promoting tumor regression [295]. Recent clinical trials have also shown that DC vaccines prepared by different strategies have promising clinical application prospects [296].
Oncolytic virus is a new type of tumor vaccine. It selectively infects tumor cells and replicates within them, ultimately inducing tumor cell lysis while preserving normal cells. TAAs and progeny virus particles released after tumor cell lysis can further induce anti-tumor responses [297]. The results of a phase I trial revealed that the intratumoral injection of JX-594, a targeted oncolytic poxvirus, in refractory HCC patients demonstrates favorable anti-tumor efficacy [298]. Subsequent randomized clinical trials have also confirmed that it can significantly improve the overall survival of advanced HCC patients [299].
ACT is a highly personalized passive immunotherapy that combats tumors by extracting immune cells from patients or healthy donors, processing them in vitro, and then infusing them into the patients. The transferred cells can proliferate in vivo and maintain their anti-tumor efficacy [300]. The cells used in ACT mainly include cytokine-induced killer (CIK) cell, tumor-infiltrating lymphocyte (TIL), chimeric antigen receptor T cell (CAR-T), and TCR-engineered T cell (TCR-T).
CIK cells mainly include CD3+CD56+ cells, CD3+CD56− cytotoxic T cells, and CD3−CD56+ NK cells, which are rapidly expanded in vitro induced by cytokines such as anti-CD3 antibody, IFN-γ, and IL-2 [301, 302]. After CIK cells bind to tumor target cells through the adhesion molecule lymphocyte function-associated antigen-1, their tumor cell lysis activity is triggered in a non-MHC-restricted manner. Their surface receptor structure determines dual roles as CD8-specific effector T cells and NK-like cells, exerting anti-tumor functions via granule exocytosis, cytotoxicity, and cytokine secretion [303]. In a multi-center randomized phase III trial, researchers used IL-2 and anti-CD3 antibody co-stimulation to culture peripheral blood mononuclear cells from patients in vitro, generating CIK cells for subsequent reinfusion [304].
TILs are obtained from the patient's surgically excised tumor tissue. After selective culture, they can expand to plenty of pure lymphocytes and then be reinfused into the patient. They can recognize various TAAs in patients and exert personalized tumor inhibition [300]. A randomized trial demonstrated that HCC patients who received TIL therapy following radical resection can reduce the recurrence rate and prolong the recurrence-free survival [305].
CAR-T cells refer to specific T cells redirected using gene transfer technology. They are equipped with CAR, an engineering receptor consisting of an extracellular single-chain variable fragment that binds to tumor antigens, a transmembrane domain, and an intracellular costimulatory domain, such as CD28 and 4-1BB and CD3ζ chains [306, 307]. After infusion, CAR-T cells are transplanted and transported to the tumor site, where they identify and eliminate tumor cells in an MHC-independent manner. This process stimulates the proliferation of CAR-T cells and the release of tumor antigens, thus activating further anti-tumor responses [306]. The most concerned TAA for CAR-T therapy in HCC is GPC3. Anti-GPC3 CAR-T cells have demonstrated efficacy in inducing cytotoxicity against HCC cells in numerous studies [308, 309]. Using similar technology, NK cells can be genetically engineered to produce CAR-NK cells, which have higher safety than CAR-T cells for HCC patients and deserve further study [310, 311]. TCR-T cell therapy uses genetic engineering to introduce TCRαβ chains that recognize TAA into T cells to form a functional TCR-CD3 complex with endogenous CD3ζ chains. Therefore, TCR-T cells have a high affinity for the TAA-MHC complex and exert anti-tumor function [312]. Studies have shown that TCR-T cells targeting AFP exhibit specific cytotoxicity to HCC cells in vitro and in tumor-bearing mice [313].
Existing challenges and future directions
Although immunotherapy has brought new hope for HCC patients, it still faces numerous challenges. Apart from the quest for more effective therapeutic strategies, it is essential to address the existing problems during the clinical implementation of approved drugs.
First, addressing the prevention and management of adverse drug reactions in immunotherapy is necessary. ICIs, as pivotal drugs in tumor immunotherapy, can alleviate the immune system suppression of tumor tissue, thereby enhancing the activation and proliferation of lymphocytes to achieve anti-tumor immune therapy. However, they may also induce an imbalance in immune tolerance and trigger irAEs, which impact various organ systems, with the most frequent occurrence observed in the skin, digestive, endocrine, nervous, blood, and cardiovascular systems [314]. ICIs can enhance the activity of T cells against shared antigens (present in both tumor and healthy tissues) and self-antigens, increase the levels of pre-existing antibodies or inflammatory cytokines in the body, or enhance complement-mediated inflammatory responses through direct binding with antibodies against CTLA-4, which are all possible causes of irAEs [315]. Due to similarities in the underlying mechanisms of anti-tumor immunity and the induction of irAEs, patients with more pronounced irAEs may have better anti-tumor outcomes [316]. For ICIs that have complementary effects in tumor treatment, the mechanisms through which they trigger irAEs also vary, which may explain why combination therapy usually carries higher safety risks [316, 317]. If the prevention and treatment of irAEs induced by various medications are attainable, it will significantly enhance the clinical implementation of combination therapy.
Apart from adverse reactions induced by therapy, drug resistance as a significant cause of treatment failure in HCC patients receiving immunotherapy is also challenging in clinical practice. Immunotherapy resistance can be divided into two main categories: primary resistance, which refers to complete non-response to treatment, and acquired resistance, referring to initial response to treatment but disease progression occurs after some time [318]. The heterogeneity and dynamic changes of TME play pivotal roles in driving drug resistance and represent a breakthrough to improve the therapeutic effect.
Identifying biomarkers for response prediction and patient selection is crucial for primary drug resistance. Among the approved immunotherapeutic drugs, Ramucirumab is the only biomarker-guided therapy. It provides survival benefits over placebo only in patients with AFP levels ≥ 400 ng/ml, and the overall efficacy is affected by changes in AFP levels during treatment [280]. As a cancer biomarker of HCC, the predictive effect of AFP in other drugs deserves further investigation. Recent studies have demonstrated that a ≥ 75% reduction or ≤ 10% increase in AFP levels measured six weeks after starting atezolizumab plus bevacizumab therapy is associated with longer overall survival and progression-free survival [319]. This potential prognostic value is particularly evident in patients with HBV etiology [319], highlighting the importance of considering etiology in treatment selection. A recent meta-analysis has shown that patients with virus-induced HCC have better responses to immunotherapy than those with non-viral HCC [56]. NASH seems to be a predictor of adverse outcomes in HCC patients treated with ICIs [56]. There remains a lack of comparative studies on the TME characteristics and therapy response rates of HCC under different etiologies, which deserves our attention. Intestinal microbiota also has potential predictive value as an emerging role affecting TME. Some studies have focused on it and found the diversity and taxonomic signatures are different in patients with or without responses to immunotherapy [320, 321]. Further investigation of the dynamic variation of the intestinal microbiome may provide early prediction of the therapeutic efficacy. Currently, the most studied biomarkers are tumor mutational burden and PD-L1 expression. However, their prognostic value remains controversial and cannot be used as a binary marker for immunotherapy. Many factors in immunosuppressive TME promoting HCC progression affect the efficacy of immunotherapy and are potential predictive markers. However, due to the difficulty in determining the threshold of efficacy prediction, their application is limited. Finding effective early biomarkers or combining multiple markers to improve the predictive effect is essential to avoid primary resistance.
Research on the formation mechanisms of acquired resistance during treatment is scarce. The activation of alternative pathways in the TME to compensate for the inhibitory effects of the drugs on their intended target is a possible reason. Studies have shown that lenvatinib can serve as one of the immunotherapy mechanisms for HCC by inhibiting the FGF pathway. However, this inhibition can lead to feedback activation of the EGFR-PAK2-ERK5 signaling axis, resulting in drug resistance. EGFR inhibition can block this signaling axis and achieve resistance reversal [322]. Additionally, lenvatinib can promote neutrophil polarization towards the N2 phenotype and upregulate the expression of PD-L1, which also hinders its efficacy [323]. Combining anti-PD-L1 therapy may ameliorate this situation. These studies indicate that the development of combination therapy based on compensatory changes in TME is beneficial in improving acquired resistance. However, there is a paucity of studies on the molecular mechanisms underlying TME alterations after drug treatment. It is imperative to undertake fundamental investigations and clinical trials to elucidate the activation mechanism of alternative pathways to guide the optimization of combination therapy. Combining drugs with different mechanisms will not only improve acquired resistance but may also cover more patients with initial responses to improve primary resistance. However, this may lead to the superposition of adverse reactions, which need special attention and prevention.
Conclusion
As a highly heterogeneous malignant tumor, the mechanisms of HCC progression and metastasis are still unclear, which brings significant challenges to clinical treatment. With an increasing understanding of immunosuppressive TME, immunotherapy has become a first-line choice for advanced HCC. However, drug resistance and safety remain issues for existing immunotherapies. It is necessary to thoroughly study the specific mechanisms of complex interactions between various cells in the immunosuppressive TME. In addition, TME is a dynamically evolving environment in the context of tumor immune editing. During HCC progression, cellular composition, cytokines, extracellular matrix, and substance metabolism in TME undergo changes that significantly impact the function and anti-tumor response of immune cells. We can better regulate and enhance anti-tumor response against HCC by exploring the mechanisms underlying these changes. In conclusion, there is a need for further exploration of the complex mechanistic network in the immunosuppressive TME to provide an essential theoretical basis for the development of more effective immunotherapies in patients with HCC.
Availability of data and materials
Not applicable.
Abbreviations
- ATP:
-
Adenosine triphosphate
- ALD:
-
Alcohol-related liver disease
- AFP:
-
Alpha-fetoprotein
- APCs:
-
Antigen-presenting cells
- ARG1:
-
Arginase1
- CSCs:
-
Cancer stem cells
- CAFs:
-
Carcinoma-associated fibroblasts
- CLCF-1:
-
Cardiotrophin-like cytokine factor 1
- CCL2:
-
C-C motif chemokine ligand 2
- JNK:
-
Cjun Nterminal kinase
- M-CSF:
-
Colony-stimulating factor
- CXCL9:
-
C-X-C motif chemokine ligand 9
- cAMP:
-
Cyclic adenosine monophosphate
- CTLA-4:
-
Cytotoxic T-lymphocyte antigen-4
- DCs:
-
Dendritic cells
- ECs:
-
Endothelial cells
- EMT:
-
Epithelial-mesenchymal transition
- ECM:
-
Extracellular matrix
- ERK:
-
Extracellular signal-regulated kinase
- FDA:
-
Food and Drug Administration
- Foxp3:
-
Forkhead box p3
- Gal-9:
-
Galactoeglutinin-9
- GLS2:
-
Glutaminase 2
- G-CSF:
-
Granulocyte colony-stimulating factor
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- G-MDSCs:
-
Granulocytic MDSCs
- VEGF:
-
Growth factor
- HSCs:
-
Hepatic stellate cells
- HBV:
-
Hepatitis B Virus
- HCV:
-
Hepatitis C Virus
- HCC:
-
Hepatocellular carcinoma
- HGF:
-
Hepatocyte growth factor
- HIF-1:
-
Hypoxia-inducible factor-1
- IMCs:
-
Immature myeloid cells
- ICIs:
-
Immune checkpoint inhibitors
- irAEs:
-
Immune-related adverse events
- IFN:
-
Interferon
- IL-6:
-
Interleukin-6
- KCs:
-
Kuepfer cells
- LOX-1:
-
Lectin-type oxidative LDL receptor 1
- LPS:
-
Lipopolysaccharide
- LSECs:
-
Liver sinus endothelial cells
- LAG3:
-
Lymphocyte activating gene 3
- LOX:
-
Lysyl oxidase
- MHC:
-
Major histocompatibility complex
- MMPs:
-
Matrix metalloproteinases
- MAPK:
-
Mitogen-activated protein kinase
- M-MDSCs:
-
Monocytic MDSCs
- MKI:
-
Multi-kinase inhibitor
- MDSCs:
-
Myeloid-derived suppressor cells
- NK:
-
Natural killer
- NETs:
-
Neutrophil extracellular traps
- NO:
-
Nitric oxide
- NASH:
-
Nonalcoholic steatohepatitis
- NF-κB:
-
Nuclear factor-kappa B
- PDGF:
-
Platelet-derived growth factor
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed death ligand-1
- PGE2:
-
Prostaglandin E2
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- Bregs:
-
Regulatory B cells
- Tregs:
-
Regulatory T cells
- STAT3:
-
Signal transducer and activator of transcription 3
- scRNA-seq:
-
Single-cell RNA sequencing
- TIM-3:
-
T cell immunoglobulin domain and mucin domain-3
- TCRs:
-
T cell receptors
- TLR4:
-
Toll-like receptor 4
- TGF-β:
-
Transforming growth factor-β
- TDO2:
-
Tryptophan-2,3-dioxygenase 2
- TIB:
-
Tumor immune barrier
- TME:
-
Tumor microenvironment
- TNF-α:
-
Tumor necrosis factor-α
- TAMs:
-
Tumor-associated macrophages
- TAN:
-
Tumor-associated neutrophils
- TI-Tregs:
-
Tumor-infiltrating Tregs
- VM:
-
Vasculogenic mimicry
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–27.
Zhao M, Huang H, He F, Fu X. Current insights into the hepatic microenvironment and advances in immunotherapy for hepatocellular carcinoma. Front Immunol. 2023;14:1188277.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. New Engl J Med. 2020; https://www.nejm.org/doi/https://doi.org/10.1056/NEJMoa1915745
Cheng A-L, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol. 2022;76:862–73.
Finn RS, Ryoo B-Y, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind Phase III Trial. J Clin Oncol. 2020;38:193–202.
Yau T, Kang Y-K, Kim T-Y, El-Khoueiry AB, Santoro A, Sangro B, et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the checkmate 040 randomized clinical trial. JAMA Oncol. 2020;6: e204564.
Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. 2021;16:223–49.
Liu P, Kong L, Liu Y, Li G, Xie J, Lu X. A key driver to promote HCC: cellular crosstalk in tumor microenvironment. Front Oncol. 2023;13:1135122.
Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S, et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther. 2021;6:153.
Foglia B, Beltrà M, Sutti S, Cannito S. Metabolic reprogramming of HCC: a new microenvironment for immune responses. Int J Mol Sci. 2023;24:7463.
Yoon J-S, Lee C-W. Protein phosphatases regulate the liver microenvironment in the development of hepatocellular carcinoma. Exp Mol Med. 2022;54:1799–813.
Li X, Ramadori P, Pfister D, Seehawer M, Zender L, Heikenwalder M. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat Rev Cancer. 2021;21:541–57.
Ignat S-R, Dinescu S, Hermenean A, Costache M. Cellular interplay as a consequence of inflammatory signals leading to liver fibrosis development. Cells. 2020;9:461.
Mihm S. Danger-Associated Molecular Patterns (DAMPs): molecular triggers for sterile inflammation in the liver. Int J Mol Sci. 2018;19:3104.
Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. 2015;62:279–91.
Cao M, Wang Z, Lan W, Xiang B, Liao W, Zhou J, et al. The roles of tissue resident macrophages in health and cancer. Exp Hematol Oncol. 2024;13:3.
Pei Q, Yi Q, Tang L. Liver fibrosis resolution: from molecular mechanisms to therapeutic opportunities. Int J Mol Sci. 2023;24:9671.
Suh B, Park S, Shin DW, Yun JM, Yang H-K, Yu SJ, et al. High liver fibrosis index FIB-4 is highly predictive of hepatocellular carcinoma in chronic hepatitis B carriers. Hepatology. 2015;61:1261–8.
Kim MN, Kim SU, Kim BK, Park JY, Kim DY, Ahn SH, et al. Increased risk of hepatocellular carcinoma in chronic hepatitis B patients with transient elastography-defined subclinical cirrhosis. Hepatology. 2015;61:1851–9.
Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:S35-50.
Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, et al. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology. 2012;142:918-927.e6.
Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol. 2017;66:212–27.
Park H, Lee EJ, Moon D, Yun H, Cha A, Hwang I, et al. Discovery of 3,7-dimethoxyflavone that inhibits liver fibrosis based on dual mechanisms of antioxidant and inhibitor of activated hepatic stellate cell. Free Radic Biol Med. 2023;204:195–206.
Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411.
Aimaiti Y, Yusufukadier M, Li W, Tuerhongjiang T, Shadike A, Meiheriayi A, et al. TGF-β1 signaling activates hepatic stellate cells through Notch pathway. Cytotechnology. 2019;71:881–91.
Dooley S, ten Dijke P. TGF-β in progression of liver disease. Cell Tissue Res. 2012;347:245–56.
Saxena NK, Anania FA. Adipocytokines and hepatic fibrosis. Trends Endocrinol Metab. 2015;26:153–61.
Jeong W-I, Park O, Suh Y-G, Byun J-S, Park S-Y, Choi E, et al. Suppression of innate immunity (natural killer cell/interferon-γ) in the advanced stages of liver fibrosis in mice. Hepatology. 2011;53:1342–51.
Sakurai T, He G, Matsuzawa A, Yu G-Y, Maeda S, Hardiman G, et al. Hepatocyte necrosis induced by oxidative stress and IL-1α release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156.
Carpino G, Renzi A, Franchitto A, Cardinale V, Onori P, Reid L, et al. Stem/progenitor cell niches involved in hepatic and biliary regeneration. Stem Cells Int. 2016;2016:3658013.
Flecken T, Schmidt N, Hild S, Gostick E, Drognitz O, Zeiser R, et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology. 2014;59:1415–26.
The immunosuppressive tumor microenvironment in hepatocellular carcinoma-current situation and outlook. Mol Immunol. 2022;151:218–30.
Sas Z, Cendrowicz E, Weinhäuser I, Rygiel TP. Tumor microenvironment of hepatocellular carcinoma: challenges and opportunities for new treatment options. Int J Mol Sci. 2022;23:3778.
Belhabib I, Zaghdoudi S, Lac C, Bousquet C, Jean C. Extracellular matrices and cancer-associated fibroblasts: targets for cancer diagnosis and therapy? Cancers (Basel). 2021;13:3466.
Xue R, Zhang Q, Cao Q, Kong R, Xiang X, Liu H, et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature. 2022;612:141–7.
Llovet JM, Castet F, Heikenwalder M, Maini MK, Mazzaferro V, Pinato DJ, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19:151–72.
Datfar T, Doulberis M, Papaefthymiou A, Hines IN, Manzini G. Viral Hepatitis and Hepatocellular Carcinoma: State of the Art. Pathogens. 2021;10:1366.
Michalak TI. The Initial Hepatitis B virus-hepatocyte genomic integrations and their role in hepatocellular oncogenesis. Int J Mol Sci. 2023;24:14849.
Tu T, Budzinska MA, Shackel NA, Urban S. HBV DNA integration: molecular mechanisms and clinical implications. Viruses. 2017;9:75.
Zheng B, Yang Y, Han Q, Yin C, Pan Z, Zhang J. STAT3 directly regulates NKp46 transcription in NK cells of HBeAg-negative CHB patients. J Leukoc Biol. 2019;106:987–96.
F Y, X Y, C Z, R M, M Z, H Z, et al. Hepatitis B e antigen induces the expansion of monocytic myeloid-derived suppressor cells to dampen T-cell function in chronic hepatitis B virus infection. PLoS pathogens [Internet]. 2019 [cited 2023 Nov 7];15. Available from: https://pubmed.ncbi.nlm.nih.gov/30998767/
Jin X, Yan Z, Lu L, Lu S, Zhang G, Lin W. Peripheral immune cells exhaustion and functional impairment in patients with chronic hepatitis B. Front Med (Lausanne). 2021;8: 759292.
Baudi I, Kawashima K, Isogawa M. HBV-specific CD8+ T-cell tolerance in the liver. Front Immunol. 2021;12: 721975.
Lim CJ, Lee YH, Pan L, Lai L, Chua C, Wasser M, et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut. 2019;68:916–27.
Xue H, Lin F, Tan H, Zhu Z-Q, Zhang Z-Y, Zhao L. Overrepresentation of IL-10-Expressing B Cells Suppresses Cytotoxic CD4+ T Cell Activity in HBV-induced hepatocellular carcinoma. PLoS ONE. 2016;11: e0154815.
Carrat F, Fontaine H, Dorival C, Simony M, Diallo A, Hezode C, et al. Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: a prospective cohort study. Lancet. 2019;393:1453–64.
Hayes CN, Zhang P, Zhang Y, Chayama K. Molecular mechanisms of hepatocarcinogenesis following sustained virological response in patients with chronic hepatitis c virus infection. Viruses. 2018;10:531.
Chigbu DI, Loonawat R, Sehgal M, Patel D, Jain P. Hepatitis C virus infection: host-virus interaction and mechanisms of viral persistence. Cells. 2019;8:376.
Zimmermann M, Flechsig C, La Monica N, Tripodi M, Adler G, Dikopoulos N. Hepatitis C virus core protein impairs in vitro priming of specific T cell responses by dendritic cells and hepatocytes. J Hepatol. 2008;48:51–60.
Hirano J, Yoshio S, Sakai Y, Songling L, Suzuki T, Itoh Y, et al. Hepatitis C virus modulates signal peptide peptidase to alter host protein processing. Proc Natl Acad Sci USA. 2021;118: e2026184118.
Banerjee A, Ray RB, Ray R. Oncogenic potential of hepatitis C virus proteins. Viruses. 2010;2:2108–33.
Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908–22.
Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med. 2019;25:641–55.
Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M, et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature. 2021;592:444–9.
Pfister D, Núñez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592:450–6.
Wang Z, Kim SY, Tu W, Kim J, Xu A, Yang YM, et al. Extracellular vesicles in fatty liver promote a metastatic tumor microenvironment. Cell Metab. 2023;35:1209-1226.e13.
Ganne-Carrié N, Nahon P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J Hepatol. 2019;70:284–93.
Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, Lackner C, et al. Alcoholic liver disease. Nat Rev Dis Primers. 2018;4:16.
Huang DQ, Mathurin P, Cortez-Pinto H, Loomba R. Global epidemiology of alcohol-associated cirrhosis and HCC: trends, projections and risk factors. Nat Rev Gastroenterol Hepatol. 2023;20:37–49.
Rumgay H, Murphy N, Ferrari P, Soerjomataram I. Alcohol and cancer: epidemiology and biological mechanisms. Nutrients. 2021;13:3173.
Gao M, Huang A, Sun Z, Sun Y, Chang B, Zhang J, et al. Granulocytic myeloid-derived suppressor cell population increases with the severity of alcoholic liver disease. J Cell Mol Med. 2019;23:2032–41.
Yan G, Wang X, Sun C, Zheng X, Wei H, Tian Z, et al. Chronic Alcohol Consumption Promotes Diethylnitrosamine-Induced Hepatocarcinogenesis via Immune Disturbances. Scientific Reports [Internet]. 2017 [cited 2023 Nov 15];7. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5451469/
Bajaj JS, Kakiyama G, Zhao D, Takei H, Fagan A, Hylemon P, et al. Continued alcohol misuse in human cirrhosis is associated with an impaired gut-liver axis. Alcohol Clin Exp Res. 2017;41:1857–65.
Jia Q, Wang A, Yuan Y, Zhu B, Long H. Heterogeneity of the tumor immune microenvironment and its clinical relevance. Exp Hematol Oncol. 2022;11:24.
Wang B, Liu Y, Liao Z, Wu H, Zhang B, Zhang L. EZH2 in hepatocellular carcinoma: progression, immunity, and potential targeting therapies. Exp Hematol Oncol. 2023;12:52.
Dentro SC, Leshchiner I, Haase K, Tarabichi M, Wintersinger J, Deshwar AG, et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell. 2021;184:2239-2254.e39.
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8.
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural Innate and Adaptive Immunity to Cancer. Annu Rev Immunol. 2011;29:235–71.
Ikeda H, Old LJ, Schreiber RD. The roles of IFNγ in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13:95–109.
Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–48.
Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36:705–16.
Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50.
Endo H, Inoue M. Dormancy in cancer. Cancer Sci. 2019;110:474–80.
O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16:151–67.
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70.
Fu T, Dai L-J, Wu S-Y, Xiao Y, Ma D, Jiang Y-Z, et al. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J Hematol Oncol. 2021;14:98.
Wu R, Guo W, Qiu X, Wang S, Sui C, Lian Q, et al. Comprehensive analysis of spatial architecture in primary liver cancer. Sci Adv. 7:3750.
Wang Y, Yuan S, Jiang H, Li Z, Yin H, Tan J, et al. Spatial maps of hepatocellular carcinoma transcriptomes reveal spatial expression patterns in tumor immune microenvironment. Theranostics. 2022;12:4163–80.
Wu L, Yan J, Bai Y, Chen F, Zou X, Xu J, et al. An invasive zone in human liver cancer identified by Stereo-seq promotes hepatocyte–tumor cell crosstalk, local immunosuppression and tumor progression. Cell Res. 2023;33:585–603.
Liu Y, Xun Z, Ma K, Liang S, Li X, Zhou S, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023;78:770–82.
Lachenmayer A, Alsinet C, Chang CY, Llovet JM. Molecular approaches to treatment of HCC. Dig Liver Dis. 2010;42:S264–72.
Ma X-L, Nie Y-Y, Xie S-H, Zheng H, Tong Y, Wang Y-C, et al. ASAP2 interrupts c-MET-CIN85 interaction to sustain HGF/c-MET-induced malignant potentials in hepatocellular carcinoma. Exp Hematol Oncol. 2023;12:38.
Wang W, Huang Q, Liao Z, Zhang H, Liu Y, Liu F, et al. ALKBH5 prevents hepatocellular carcinoma progression by post-transcriptional inhibition of PAQR4 in an m6A dependent manner. Exp Hematol Oncol. 2023;12:1.
Fang Z, Mei W, Qu C, Lu J, Shang L, Cao F, et al. Role of m6A writers, erasers and readers in cancer. Exp Hematol Oncol. 2022;11:45.
Chen E, He Y, Jiang J, Yi J, Zou Z, Song Q, et al. CDCA8 induced by NF-YA promotes hepatocellular carcinoma progression by regulating the MEK/ERK pathway. Exp Hematol Oncol. 2023;12:9.
Nault J-C, Martin Y, Caruso S, Hirsch TZ, Bayard Q, Calderaro J, et al. Clinical Impact of Genomic Diversity From Early to Advanced Hepatocellular Carcinoma. Hepatology. 2020;71:164–82.
Ningarhari M, Caruso S, Hirsch TZ, Bayard Q, Franconi A, Védie A-L, et al. Telomere length is key to hepatocellular carcinoma diversity and telomerase addiction is an actionable therapeutic target. J Hepatol. 2021;74:1155–66.
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46.
Dubois-Pot-Schneider H, Fekir K, Coulouarn C, Glaise D, Aninat C, Jarnouen K, et al. Inflammatory cytokines promote the retrodifferentiation of tumor-derived hepatocyte-like cells to progenitor cells. Hepatology. 2014;60:2077–90.
Fang X, Yan Q, Liu S, Guan X-Y. Cancer stem cells in hepatocellular carcinoma: intrinsic and extrinsic molecular mechanisms in stemness regulation. Int J Mol Sci. 2022;23:12327.
Kim BH, Park J-W, Kim JS, Lee S-K, Hong EK. Stem cell markers predict the response to sorafenib in patients with hepatocellular carcinoma. Gut Liver. 2019;13:342–8.
Sengez B, Carr BI, Alotaibi H. EMT and inflammation: crossroads in HCC. J Gastrointest Cancer. 2023;54:204–12.
Wang T, Rao D, Yu C, Sheng J, Luo Y, Xia L, et al. RHO GTPase family in hepatocellular carcinoma. Exp Hematol Oncol. 2022;11:91.
Fransvea E, Angelotti U, Antonaci S, Giannelli G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology. 2008;47:1557–66.
Shi X, Yang J, Deng S, Xu H, Wu D, Zeng Q, et al. TGF-β signaling in the tumor metabolic microenvironment and targeted therapies. J Hematol Oncol. 2022;15:135.
Makarova-Rusher OV, Medina-Echeverz J, Duffy AG, Greten TF. The yin and yang of evasion and immune activation in HCC. J Hepatol. 2015;62:1420–9.
Lin X-H, Liu Z-Y, Zhang D-Y, Zhang S, Tang W-Q, Li D-P, et al. circRanGAP1/miR-27b-3p/NRAS Axis may promote the progression of hepatocellular Carcinoma. Exp Hematol Oncol. 2022;11:92.
Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11:5120.
Massagué J, Obenauf AC. Metastatic Colonization. Nature. 2016;529:298–306.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582–98.
Yuzhalin AE, Lim SY, Kutikhin AG, Gordon-Weeks AN. Dynamic matrisome: ECM remodeling factors licensing cancer progression and metastasis. Biochim Biophys Acta Rev Cancer. 2018;1870:207–28.
Mangala LS, Mehta K. Tissue transglutaminase (TG2) in cancer biology. Prog Exp Tumor Res. 2005;38:125–38.
Barker HE, Cox TR, Erler JT. The rationale for targeting the LOX family in cancer. Nat Rev Cancer. 2012;12:540–52.
O’Connor RS, Hao X, Shen K, Bashour K, Akimova T, Hancock WW, et al. Substrate rigidity regulates human T cell activation and proliferation. J Immunol. 2012;189:1330–9.
Moreau J-F, Pradeu T, Grignolio A, Nardini C, Castiglione F, Tieri P, et al. The emerging role of ECM crosslinking in T cell mobility as a hallmark of immunosenescence in humans. Ageing Res Rev. 2017;35:322–35.
Naik A, Leask A. Tumor-associated fibrosis impairs the response to immunotherapy. Matrix Biol. 2023;119:125–40.
Cheu JW-S, Chiu DK-C, Kwan KK-L, Yang C, Yuen VW-H, Goh CC, et al. Hypoxia-inducible factor orchestrates adenosine metabolism to promote liver cancer development. Sci Adv. 9:eade5111.
Chen Z-Q, Zuo X-L, Cai J, Zhang Y, Han G-Y, Zhang L, et al. Hypoxia-associated circPRDM4 promotes immune escape via HIF-1α regulation of PD-L1 in hepatocellular carcinoma. Exp Hematol Oncol. 2023;12:17.
Wu H, Gong Y, Ji P, Xie Y, Jiang Y-Z, Liu G. Targeting nucleotide metabolism: a promising approach to enhance cancer immunotherapy. J Hematol Oncol. 2022;15:45.
Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. 2007;9:893–904.
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–44.
Rodríguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2010;1803:39–54.
Wolf K, te Lindert M, Krause M, Alexander S, te Riet J, Willis AL, et al. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol. 2013;201:1069–84.
Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin signaling. Cell. 2009;139:891–906.
Attieh Y, Clark AG, Grass C, Richon S, Pocard M, Mariani P, et al. Cancer-associated fibroblasts lead tumor invasion through integrin-β3–dependent fibronectin assembly. J Cell Biol. 2017;216:3509.
Fraley SI, Wu P, He L, Feng Y, Krisnamurthy R, Longmore GD, et al. Three-dimensional matrix fiber alignment modulates cell migration and MT1-MMP utility by spatially and temporally directing protrusions. Sci Rep. 2015;5:14580.
Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22.
Postovit L-M, Abbott DE, Payne SL, Wheaton WW, Margaryan NV, Sullivan R, et al. Hypoxia/reoxygenation: a dynamic regulator of lysyl oxidase-facilitated breast cancer migration. J Cell Biochem. 2008;103:1369–78.
Nolte M, Margadant C. Controlling Immunity and Inflammation through Integrin-Dependent Regulation of TGF-β. Trends Cell Biol. 2020;30:49–59.
Liu Y, Cao X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell. 2016;30:668–81.
Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. 2016;99:186–96.
Jt C, Yn D, Hm Y, Gy W, Bs F, Wj C, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis [Internet]. 2016 [cited 2023 May 11];5. Available from: https://pubmed.ncbi.nlm.nih.gov/26900950/
Xiong S, Wang R, Chen Q, Luo J, Wang J, Zhao Z, et al. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am J Cancer Res. 2018;8:302–16.
Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell. 2022;40:656-673.e7.
Chen S, Morine Y, Tokuda K, Yamada S, Saito Y, Nishi M, et al. Cancer-associated fibroblast-induced M2-polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor-1 pathway. Int J Oncol. 2021;59:59.
Luo Q, Wang C-Q, Yang L-Y, Gao X-M, Sun H-T, Zhang Y, et al. FOXQ1/NDRG1 axis exacerbates hepatocellular carcinoma initiation via enhancing crosstalk between fibroblasts and tumor cells. Cancer Lett. 2018;417:21–34.
Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211–8.
Zhong X, He X, Wang Y, Hu Z, Huang H, Zhao S, et al. Warburg effect in colorectal cancer: the emerging roles in tumor microenvironment and therapeutic implications. J Hematol Oncol. 2022;15:160.
Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95.
Gatenby RA, Gawlinski ET. The glycolytic phenotype in carcinogenesis and tumor invasion: insights through mathematical models. Cancer Res. 2003;63:3847–54.
Bose S, Le A. Glucose Metabolism in Cancer. Adv Exp Med Biol. 2018;1063:3–12.
Huang D, Li T, Wang L, Zhang L, Yan R, Li K, et al. Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress. Cell Res. 2016;26:1112–30.
Cheng J, Yan J, Liu Y, Shi J, Wang H, Zhou H, et al. Cancer-cell-derived fumarate suppresses the anti-tumor capacity of CD8+ T cells in the tumor microenvironment. Cell Metabolism [Internet]. 2023 [cited 2023 May 15]; Available from: https://www.sciencedirect.com/science/article/pii/S1550413123001717
Satriano L, Lewinska M, Rodrigues PM, Banales JM, Andersen JB. Metabolic rearrangements in primary liver cancers: cause and consequences. Nat Rev Gastroenterol Hepatol. 2019;16:748–66.
Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:749.
Liu J, Geng W, Sun H, Liu C, Huang F, Cao J, et al. Integrative metabolomic characterisation identifies altered portal vein serum metabolome contributing to human hepatocellular carcinoma. Gut. 2022;71:1203–13.
Jee SH, Kim M, Kim M, Yoo HJ, Kim H, Jung KJ, et al. Metabolomics profiles of hepatocellular carcinoma in a korean prospective cohort: the korean cancer prevention study-II. Cancer Prev Res (Phila). 2018;11:303–12.
Liang JQ, Teoh N, Xu L, Pok S, Li X, Chu ESH, et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat Commun. 2018;9:4490.
Liu Y, Guo X, Wu L, Yang M, Li Z, Gao Y, et al. Lipid rafts promote liver cancer cell proliferation and migration by up-regulation of TLR7 expression. Oncotarget. 2016;7:63856–69.
Krishnamurthy S, Gilot D, Ahn SB, Lam V, Shin J-S, Guillemin GJ, et al. Involvement of kynurenine pathway in hepatocellular carcinoma. Cancers (Basel). 2021;13:5180.
Yan Y, Huang L, Liu Y, Yi M, Chu Q, Jiao D, et al. Metabolic profiles of regulatory T cells and their adaptations to the tumor microenvironment: implications for antitumor immunity. J Hematol Oncol. 2022;15:104.
Chen W, Mao Y, Liu C, Wu H, Chen S. Exosome in Hepatocellular Carcinoma: an update. J Cancer. 2021;12:2526–36.
Tian B, Zhou L, Wang J, Yang P. miR-660-5p-loaded M2 macrophages-derived exosomes augment hepatocellular carcinoma development through regulating KLF3. Int Immunopharmacol. 2021;101: 108157.
Li W, Xin X, Li X, Geng J, Sun Y. Exosomes secreted by M2 macrophages promote cancer stemness of hepatocellular carcinoma via the miR-27a-3p/TXNIP pathways. Int Immunopharmacol. 2021;101: 107585.
Lu L, Huang J, Mo J, Da X, Li Q, Fan M, et al. Exosomal lncRNA TUG1 from cancer-associated fibroblasts promotes liver cancer cell migration, invasion, and glycolysis by regulating the miR-524-5p/SIX1 axis. Cell Mol Biol Lett. 2022;27:17.
Yugawa K, Yoshizumi T, Mano Y, Itoh S, Harada N, Ikegami T, et al. Cancer-associated fibroblasts promote hepatocellular carcinoma progression through downregulation of exosomal miR-150-3p. Eur J Surg Oncol. 2021;47:384–93.
Zhang Z, Li X, Sun W, Yue S, Yang J, Li J, et al. Loss of exosomal miR-320a from cancer-associated fibroblasts contributes to HCC proliferation and metastasis. Cancer Lett. 2017;397:33–42.
Taskaeva I, Bgatova N. Microvasculature in hepatocellular carcinoma: an ultrastructural study. Microvasc Res. 2021;133: 104094.
Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–8.
Yao C, Wu S, Kong J, Sun Y, Bai Y, Zhu R, et al. Angiogenesis in hepatocellular carcinoma: mechanisms and anti-angiogenic therapies. Cancer Biol Med. 2023;20:25–43.
Sharma A, Seow JJW, Dutertre C-A, Pai R, Blériot C, Mishra A, et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. 2020;183:377-394.e21.
Li Z, Pai R, Gupta S, Currenti J, Guo W, Di Bartolomeo A, et al. Presence of onco-fetal neighborhoods in hepatocellular carcinoma is associated with relapse and response to immunotherapy. Nat Cancer. 2024;5:167–86.
Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer. 2010;10:138–46.
Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–34.
Ohga N, Ishikawa S, Maishi N, Akiyama K, Hida Y, Kawamoto T, et al. Heterogeneity of tumor endothelial cells: comparison between tumor endothelial cells isolated from high- and low-metastatic tumors. Am J Pathol. 2012;180:1294–307.
Lim JTC, Kwang LG, Ho NCW, Toh CCM, Too NSH, Hooi L, et al. Hepatocellular carcinoma organoid co-cultures mimic angiocrine crosstalk to generate inflammatory tumor microenvironment. Biomaterials. 2022;284: 121527.
Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Peer J, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739–52.
Liu W, Xu G, Jia W, Li J, Ma J, Chen K, et al. Prognostic significance and mechanisms of patterned matrix vasculogenic mimicry in hepatocellular carcinoma. Med Oncol. 2011;28(Suppl 1):S228-238.
Sun B, Zhang S, Zhang D, Du J, Guo H, Zhao X, et al. Vasculogenic mimicry is associated with high tumor grade, invasion and metastasis, and short survival in patients with hepatocellular carcinoma. Oncol Rep. 2006;16:693–8.
Zheng N, Zhang S, Wu W, Zhang N, Wang J. Regulatory mechanisms and therapeutic targeting of vasculogenic mimicry in hepatocellular carcinoma. Pharmacol Res. 2021;166: 105507.
Shi Y, Shang J, Li Y, Zhong D, Zhang Z, Yang Q, et al. ITGA5 and ITGB1 contribute to Sorafenib resistance by promoting vasculogenic mimicry formation in hepatocellular carcinoma. Cancer Med. 2023;12:3786–96.
Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87.
Koyama S, Nishikawa H. Mechanisms of regulatory T cell infiltration in tumors: implications for innovative immune precision therapies. J Immunother Cancer. 2021;9: e002591.
Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol. 2014;14:377–91.
Yang XH, Yamagiwa S, Ichida T, Matsuda Y, Sugahara S, Watanabe H, et al. Increase of CD4+ CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J Hepatol. 2006;45:254–62.
Lan Y-T, Fan X-P, Fan Y-C, Zhao J, Wang K. Change in the Treg/Th17 cell imbalance in hepatocellular carcinoma patients and its clinical value. Medicine [Internet]. 2017 [cited 2023 May 30];96. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556221/
Lee W-C, Wu T-J, Chou H-S, Yu M-C, Hsu P-Y, Hsu H-Y, et al. The impact of CD4+ CD25+ T cells in the tumor microenvironment of hepatocellular carcinoma. Surgery. 2012;151:213–22.
Tay C, Tanaka A, Sakaguchi S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell. 2023;41:450–65.
Yoshizawa K, Abe H, Kubo Y, Kitahara T, Aizawa R, Matsuoka M, et al. Expansion of CD4(+)CD25(+)FoxP3(+) regulatory T cells in hepatitis C virus-related chronic hepatitis, cirrhosis and hepatocellular carcinoma. Hepatol Res. 2010;40:179–87.
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61.
Bai Y, Chen D, Cheng C, Li Z, Chi H, Zhang Y, et al. Immunosuppressive landscape in hepatocellular carcinoma revealed by single-cell sequencing. Front Immunol. 2022;13: 950536.
Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, et al. Foxp3 Transcription-Factor-Dependent and -Independent Regulation of the Regulatory T Cell Transcriptional Signature. Immunity. 2007;27:786–800.
Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to Interleukin-2 elicit selective T cell subset potentiation through distinct conformational mechanisms. Immunity. 2015;42:815–25.
Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T Cells and Human Disease. Annu Rev Immunol. 2020;38:541–66.
Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001;19:565–94.
Chen X, Du Y, Hu Q, Huang Z. Tumor-derived CD4+CD25+regulatory T cells inhibit dendritic cells function by CTLA-4. Pathol Res Pract. 2017;213:245–9.
Bao S, Jiang X, Jin S, Tu P, Lu J. TGF-β1 Induces Immune Escape by Enhancing PD-1 and CTLA-4 Expression on T Lymphocytes in Hepatocellular Carcinoma. Front Oncol. 2021;11: 694145.
Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. 2017;276:121–44.
Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA. 2006;103:13132–7.
Abrahamsen H, Baillie G, Ngai J, Vang T, Nika K, Ruppelt A, et al. TCR- and CD28-mediated recruitment of phosphodiesterase 4 to lipid rafts potentiates TCR signaling. J Immunol. 2004;173:4847–58.
Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. 2019;20:724–35.
Milardi G, Lleo A. Tumor-Infiltrating B lymphocytes: promising immunotherapeutic targets for primary liver cancer treatment. Cancers (Basel). 2023;15:2182.
Ye L, Zhang Q, Cheng Y, Chen X, Wang G, Shi M, et al. Tumor-derived exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by promoting TIM-1+ regulatory B cell expansion. J Immunother Cancer. 2018;6:145.
Xiao X, Lao X-M, Chen M-M, Liu R-X, Wei Y, Ouyang F-Z, et al. PD-1hi Identifies a Novel Regulatory B-cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov. 2016;6:546–59.
Qin M, Wang D, Fang Y, Zheng Z, Liu X, Wu F, et al. Current Perspectives on B lymphocytes in the immunobiology of hepatocellular carcinoma. Front Oncol. 2021;11: 647854.
Shao Y, Lo CM, Ling CC, Liu XB, Ng KT-P, Chu ACY, et al. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett. 2014;355:264–72.
Wu Y, Yi M, Niu M, Mei Q, Wu K. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer. 2022;21:184.
Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget. 2016;8:3649–65.
Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150.
Joshi S, Sharabi A. Targeting myeloid-derived suppressor cells to enhance natural killer cell-based immunotherapy. Pharmacol Ther. 2022;235: 108114.
Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S, Totaro MG, et al. RORC1 Regulates Tumor-Promoting “Emergency” Granulo-Monocytopoiesis. Cancer Cell. 2015;28:253–69.
Moses K, Brandau S. Human neutrophils: their role in cancer and relation to myeloid-derived suppressor cells. Semin Immunol. 2016;28:187–96.
Condamine T, Dominguez GA, Youn J-I, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016;1:8943.
Nan J, Xing Y, Hu B, Tang J, Dong H, He Y, et al. Endoplasmic reticulum stress induced LOX-1+ CD15+ polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunology. 2018;154:144–55.
Wang Y, Zhang T, Sun M, Ji X, Xie M, Huang W, et al. Therapeutic values of myeloid-derived suppressor cells in hepatocellular carcinoma: facts and hopes. Cancers (Basel). 2021;13:5127.
Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther. 2021;6:362.
Karin N. The development and homing of myeloid-derived suppressor cells: from a two-stage model to a multistep narrative. Front Immunol. 2020;11: 557586.
Li Y-M, Liu Z-Y, Wang J-C, Yu J-M, Li Z-C, Yang H-J, et al. Receptor-Interacting Protein Kinase 3 Deficiency Recruits Myeloid-Derived Suppressor Cells to Hepatocellular Carcinoma Through the Chemokine (C-X-C Motif) Ligand 1-Chemokine (C-X-C Motif) Receptor 2 Axis. Hepatology. 2019;70:1564–81.
Sun H, Yang W, Tian Y, Zeng X, Zhou J, Mok MTS, et al. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat Commun. 2018;9:5214.
Liu L-Z, Zhang Z, Zheng B-H, Shi Y, Duan M, Ma L-J, et al. CCL15 recruits suppressive monocytes to facilitate immune escape and disease progression in hepatocellular carcinoma. Hepatology. 2019;69:143–59.
Lin Y, Yang X, Liu W, Li B, Yin W, Shi Y, et al. Chemerin has a protective role in hepatocellular carcinoma by inhibiting the expression of IL-6 and GM-CSF and MDSC accumulation. Oncogene. 2017;36:3599–608.
Li J, Li H, Yu Y, Liu Y, Liu Y, Ma Q, et al. Mannan-binding lectin suppresses growth of hepatocellular carcinoma by regulating hepatic stellate cell activation via the ERK/COX-2/PGE2 pathway. Oncoimmunology. 2018;8: e1527650.
Chiu DK-C, Xu IM-J, Lai RK-H, Tse AP-W, Wei LL, Koh H-Y, et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology. 2016;64:797–813.
Chiu DK-C, Tse AP-W, Xu IM-J, Di Cui J, Lai RK-H, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8:517.
Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–91.
Gabrilovich DI, Nagaraj S. Myeloid-derived-suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74.
Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–49.
Feldmeyer N, Wabnitz G, Leicht S, Luckner-Minden C, Schiller M, Franz T, et al. Arginine deficiency leads to impaired cofilin dephosphorylation in activated human T lymphocytes. Int Immunol. 2012;24:303–13.
Ohl K, Tenbrock K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front Immunol. 2018;9:2499.
Kasic T, Colombo P, Soldani C, Wang CM, Miranda E, Roncalli M, et al. Modulation of human T-cell functions by reactive nitrogen species. Eur J Immunol. 2011;41:1843–9.
Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135:234–43.
Lacotte S, Slits F, Orci LA, Meyer J, Oldani G, Delaune V, et al. Impact of myeloid-derived suppressor cell on Kupffer cells from mouse livers with hepatocellular carcinoma. Oncoimmunology. 2016;5: e1234565.
Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50:799–807.
Hu C-E, Gan J, Zhang R-D, Cheng Y-R, Huang G-J. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol. 2011;46:156–64.
Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306–21.
Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, et al. The Inflammatory Microenvironment in Hepatocellular Carcinoma: A Pivotal Role for Tumor-Associated Macrophages. Biomed Res Int. 2013;2013: 187204.
Zhu S, Yi M, Wu Y, Dong B, Wu K. Roles of tumor-associated macrophages in tumor progression: implications on therapeutic strategies. Exp Hematol Oncol. 2021;10:60.
Tian Z, Hou X, Liu W, Han Z, Wei L. Macrophages and hepatocellular carcinoma Cell Biosci. 2019;9:79.
Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83.
Wan S, Kuo N, Kryczek I, Zou W, Welling TH. Myeloid Cells in Hepatocellular Carcinoma. Hepatology. 2015;62:1304–12.
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–95.
Qin R, Ren W, Ya G, Wang B, He J, Ren S, et al. Role of chemokines in the crosstalk between tumor and tumor-associated macrophages. Clin Exp Med. 2023;23:1359–73.
Ge Z, Ding S. The Crosstalk Between Tumor-Associated Macrophages (TAMs) and Tumor Cells and the Corresponding Targeted Therapy. Front Oncol. 2020;10: 590941.
Sierra-Filardi E, Nieto C, Domínguez-Soto A, Barroso R, Sánchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. 2014;192:3858–67.
Sica A, Saccani A, Bottazzi B, Polentarutti N, Vecchi A, van Damme J, et al. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J Immunol. 2000;164:762–7.
Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18:349–55.
Saccani A, Schioppa T, Porta C, Biswas SK, Nebuloni M, Vago L, et al. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006;66:11432–40.
Nasrollahzadeh E, Razi S, Keshavarz-Fathi M, Mazzone M, Rezaei N. Pro-tumorigenic functions of macrophages at the primary, invasive and metastatic tumor site. Cancer Immunol Immunother. 2020;69:1673–97.
Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6.
Xiao N, Zhu X, Li K, Chen Y, Liu X, Xu B, et al. Blocking siglec-10hi tumor-associated macrophages improves anti-tumor immunity and enhances immunotherapy for hepatocellular carcinoma. Exp Hematol Oncol. 2021;10:36.
Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell. 2019;179:829-845.e20.
Song G, Shi Y, Zhang M, Goswami S, Afridi S, Meng L, et al. Global immune characterization of HBV/HCV-related hepatocellular carcinoma identifies macrophage and T-cell subsets associated with disease progression. Cell Discov. 2020;6:90.
Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005;175:342–9.
Dong P, Ma L, Liu L, Zhao G, Zhang S, Dong L, et al. CD86+/CD206+, Diametrically Polarized Tumor-Associated Macrophages, Predict Hepatocellular Carcinoma Patient Prognosis. Int J Mol Sci. 2016;17:320.
Zhang J, Zhang Q, Lou Y, Fu Q, Chen Q, Wei T, et al. Hypoxia-inducible factor-1α/interleukin-1β signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology. 2018;67:1872–89.
Zhang W, Liu Y, Yan Z, Yang H, Sun W, Yao Y, et al. IL-6 promotes PD-L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma. J Immunother Cancer. 2020;8: e000285.
Zhao Q, Kuang D-M, Wu Y, Xiao X, Li X-F, Li T-J, et al. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J Immunol. 2012;188:1117–24.
Ganjalikhani Hakemi M, Jafarinia M, Azizi M, Rezaeepoor M, Isayev O, Bazhin AV. The Role of TIM-3 in Hepatocellular Carcinoma: A Promising Target for Immunotherapy? Front Oncol. 2020;10: 601661.
Chen Y, Wen H, Zhou C, Su Q, Lin Y, Xie Y, et al. TNF-α derived from M2 tumor-associated macrophages promotes epithelial-mesenchymal transition and cancer stemness through the Wnt/β-catenin pathway in SMMC-7721 hepatocellular carcinoma cells. Exp Cell Res. 2019;378:41–50.
Sionov RV. Leveling Up the Controversial Role of Neutrophils in Cancer: When the Complexity Becomes Entangled. Cells. 2021;10:2486.
Zhang Y, Guoqiang L, Sun M, Lu X. Targeting and exploitation of tumor-associated neutrophils to enhance immunotherapy and drug delivery for cancer treatment. Cancer Biol Med. 2020;17:32–43.
Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018;9:422.
Song M, He J, Pan Q-Z, Yang J, Zhao J, Zhang Y-J, et al. Cancer-Associated Fibroblast-Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology. 2021;73:1717–35.
Zhou S-L, Yin D, Hu Z-Q, Luo C-B, Zhou Z-J, Xin H-Y, et al. A Positive Feedback Loop Between Cancer Stem-Like Cells and Tumor-Associated Neutrophils Controls Hepatocellular Carcinoma Progression. Hepatology. 2019;70:1214–30.
He G, Zhang H, Zhou J, Wang B, Chen Y, Kong Y, et al. Peritumoural neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signalling pathway in hepatocellular carcinoma. J Exp Clin Cancer Res. 2015;34:141.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil (TAN) Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–94.
He M, Peng A, Huang X-Z, Shi D-C, Wang J-C, Zhao Q, et al. Peritumoral stromal neutrophils are essential for c-Met-elicited metastasis in human hepatocellular carcinoma. Oncoimmunology. 2016;5: e1219828.
Zhou S-L, Zhou Z-J, Hu Z-Q, Huang X-W, Wang Z, Chen E-B, et al. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to sorafenib. Gastroenterology. 2016;150:1646-1658.e17.
Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2007;104:20262–7.
Jiang Z-Z, Peng Z-P, Liu X-C, Guo H-F, Zhou M-M, Jiang D, et al. Neutrophil extracellular traps induce tumor metastasis through dual effects on cancer and endothelial cells. Oncoimmunology. 11:2052418.
Teijeira Á, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52:856-871.e8.
Qin S, Dong B, Yi M, Chu Q, Wu K. Prognostic Values of TIM-3 Expression in Patients With Solid Tumors: A Meta-Analysis and Database Evaluation. Front Oncol. 2020;10:1288.
Wang R, He S, Long J, Wang Y, Jiang X, Chen M, et al. Emerging therapeutic frontiers in cancer: insights into posttranslational modifications of PD-1/PD-L1 and regulatory pathways. Exp Hematol Oncol. 2024;13:46.
Wang Y, Zhang H, Liu C, Wang Z, Wu W, Zhang N, et al. Immune checkpoint modulators in cancer immunotherapy: recent advances and emerging concepts. J Hematol Oncol. 2022;15:111.
Ma W, Xue R, Zhu Z, Farrukh H, Song W, Li T, et al. Increasing cure rates of solid tumors by immune checkpoint inhibitors. Exp Hematol Oncol. 2023;12:10.
Dai M, Liu M, Yang H, Küçük C, You H. New insights into epigenetic regulation of resistance to PD-1/PD-L1 blockade cancer immunotherapy: mechanisms and therapeutic opportunities. Exp Hematol Oncol. 2022;11:101.
Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD-L1 expression in the tumor microenvironment. J Hematol Oncol. 2021;14:10.
Liu Z, Yu X, Xu L, Li Y, Zeng C. Current insight into the regulation of PD-L1 in cancer. Exp Hematol Oncol. 2022;11:44.
Mocan T, Sparchez Z, Craciun R, Bora CN, Leucuta DC. Programmed cell death protein-1 (PD-1)/programmed death-ligand-1 (PD-L1) axis in hepatocellular carcinoma: prognostic and therapeutic perspectives. Clin Transl Oncol. 2019;21:702–12.
Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767–78.
Gao Q, Wang X-Y, Qiu S-J, Yamato I, Sho M, Nakajima Y, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15:971–9.
Iwai Y, Hamanishi J, Chamoto K, Honjo T. Cancer immunotherapies targeting the PD-1 signaling pathway. J Biomed Sci. 2017;24:26.
Banerjee H, Nieves-Rosado H, Kulkarni A, Murter B, McGrath KV, Chandran UR, et al. Expression of Tim-3 drives phenotypic and functional changes in Treg cells in secondary lymphoid organs and the tumor microenvironment. Cell Rep. 2021;36: 109699.
Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13:832–42.
Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, et al. Tim-3 fosters HCC development by enhancing TGF-β-mediated alternative activation of macrophages. Gut. 2015;64:1593–604.
Tan S, Xu Y, Wang Z, Wang T, Du X, Song X, et al. Tim-3 Hampers Tumor Surveillance of Liver-Resident and Conventional NK Cells by Disrupting PI3K Signaling. Cancer Res. 2020;80:1130–42.
Sharafi F, Hasani SA, Alesaeidi S, Kahrizi MS, Adili A, Ghoreishizadeh S, et al. A comprehensive review about the utilization of immune checkpoint inhibitors and combination therapy in hepatocellular carcinoma: an updated review. Cancer Cell Int. 2022;22:269.
Yang C, Qian Q, Zhao Y, Huang B, Chen R, Gong Q, et al. Fibrinogen-like protein 1 promotes liver-resident memory T-cell exhaustion in hepatocellular carcinoma. Front Immunol. 2023;14:1112672.
LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171:1393–405.
Williams JB, Horton BL, Zheng Y, Duan Y, Powell JD, Gajewski TF. The EGR2 targets LAG-3 and 4–1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J Exp Med. 2017;214:381–400.
Qian W, Zhao M, Wang R, Li H. Fibrinogen-like protein 1 (FGL1): the next immune checkpoint target. J Hematol Oncol. 2021;14:147.
Durham NM, Nirschl CJ, Jackson CM, Elias J, Kochel CM, Anders RA, et al. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE. 2014;9: e109080.
Guo M, Yuan F, Qi F, Sun J, Rao Q, Zhao Z, et al. Expression and clinical significance of LAG-3, FGL1, PD-L1 and CD8+T cells in hepatocellular carcinoma using multiplex quantitative analysis. J Transl Med. 2020;18:306.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90.
El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–502.
Kudo M, Finn RS, Qin S, Han K-H, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163–73.
Abou-Alfa GK, Meyer T, Cheng A-L, El-Khoueiry AB, Rimassa L, Ryoo B-Y, et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N Engl J Med. 2018;379:54–63.
Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19:940–52.
Zhu AX, Kang Y-K, Yen C-J, Finn RS, Galle PR, Llovet JM, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20:282–96.
Bruix J, Qin S, Merle P, Granito A, Huang Y-H, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma Nat Rev Dis Primers. 2021;7:1–28.
Tie Y, Tang F, Wei Y, Wei X. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. 2022;15:61.
Wang Y, Johnson KCC, Gatti-Mays ME, Li Z. Emerging strategies in targeting tumor-resident myeloid cells for cancer immunotherapy. J Hematol Oncol. 2022;15:118.
Cheng X, Wang H, Wang Z, Zhu B, Long H. Tumor-associated myeloid cells in cancer immunotherapy. Journal of Hematology & Oncology [Internet]. 2023 [cited 2024 Jul 9];16. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10324139/
Wu M, Huang Q, Xie Y, Wu X, Ma H, Zhang Y, et al. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J Hematol Oncol. 2022;15:24.
Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21:28.
Yi M, Niu M, Wu Y, Ge H, Jiao D, Zhu S, et al. Combination of oral STING agonist MSA-2 and anti-TGF-β/PD-L1 bispecific antibody YM101: a novel immune cocktail therapy for non-inflamed tumors. J Hematol Oncol. 2022;15:142.
Yang C, Zhang H, Zhang L, Zhu AX, Bernards R, Qin W, et al. Evolving therapeutic landscape of advanced hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2023;20:203–22.
Zhang P, Zhang G, Wan X. Challenges and new technologies in adoptive cell therapy. J Hematol Oncol. 2023;16:97.
Mandlik DS, Mandlik SK, Choudhary HB. Immunotherapy for hepatocellular carcinoma: Current status and future perspectives. World J Gastroenterol. 2023;29:1054–75.
Tagliamonte M, Petrizzo A, Mauriello A, Tornesello ML, Buonaguro FM, Buonaguro L. Potentiating cancer vaccine efficacy in liver cancer. Oncoimmunology. 2018;7: e1488564.
Shang N, Figini M, Shangguan J, Wang B, Sun C, Pan L, et al. Dendritic cells based immunotherapy. Am J Cancer Res. 2017;7:2091–102.
El-Ashmawy NE, El-Zamarany EA, Khedr EG, El-Bahrawy HA, El-Feky OA. Antigen-loaded dendritic cells triggers a specific cytotoxic T lymphocytes immune response against hepatocellular carcinoma: in vitro study. Clin Transl Oncol. 2019;21:636–45.
Wang Q, Luan W, Warren L, Kadri H, Kim KW, Goz V, et al. Autologous tumor cell lysate-loaded dendritic cell vaccine inhibited tumor progression in an orthotopic murine model for hepatocellular carcinoma. Ann Surg Oncol. 2016;23:574–82.
Jeng L-B, Liao L-Y, Shih F-Y, Teng C-F. Dendritic-cell-vaccine-based immunotherapy for hepatocellular carcinoma: clinical trials and recent preclinical studies. Cancers (Basel). 2022;14:4380.
Chen L, Zuo M, Zhou Q, Wang Y. Oncolytic virotherapy in cancer treatment: challenges and optimization prospects. Front Immunol. 2023;14:1308890.
Park B-H, Hwang T, Liu T-C, Sze DY, Kim J-S, Kwon H-C, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533–42.
Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–36.
Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–8.
Schmidt-Wolf IG, Lefterova P, Mehta BA, Fernandez LP, Huhn D, Blume KG, et al. Phenotypic characterization and identification of effector cells involved in tumor cell recognition of cytokine-induced killer cells. Exp Hematol. 1993;21:1673–9.
Schmidt-Wolf IG, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J Exp Med. 1991;174:139–49.
Pievani A, Borleri G, Pende D, Moretta L, Rambaldi A, Golay J, et al. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. 2011;118:3301–10.
Lee JH, Lee J-H, Lim Y-S, Yeon JE, Song T-J, Yu SJ, et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology. 2015;148:1383-1391.e6.
Takayama T, Sekine T, Makuuchi M, Yamasaki S, Kosuge T, Yamamoto J, et al. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: a randomised trial. Lancet. 2000;356:802–7.
June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med. 2018;379:64–73.
Yan T, Zhu L, Chen J. Current advances and challenges in CAR T-Cell therapy for solid tumors: tumor-associated antigens and the tumor microenvironment. Exp Hematol Oncol. 2023;12:14.
Jiang Z, Jiang X, Chen S, Lai Y, Wei X, Li B, et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front Immunol. 2016;7:690.
Dargel C, Bassani-Sternberg M, Hasreiter J, Zani F, Bockmann J-H, Thiele F, et al. T Cells Engineered to Express a T-cell receptor specific for glypican-3 to recognize and kill hepatoma cells in vitro and in mice. Gastroenterology. 2015;149:1042–52.
Kalathil SG, Thanavala Y. Natural Killer Cells and T cells in hepatocellular carcinoma and viral hepatitis: current status and perspectives for future immunotherapeutic approaches. Cells. 2021;10:1332.
Lamers-Kok N, Panella D, Georgoudaki A-M, Liu H, Özkazanc D, Kučerová L, et al. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J Hematol Oncol. 2022;15:164.
Watanabe K, Nishikawa H. Engineering strategies for broad application of TCR-T- and CAR-T-cell therapies. Int Immunol. 2021;33:551–62.
Sun L, Guo H, Jiang R, Lu L, Liu T, He X. Engineered cytotoxic T lymphocytes with AFP-specific TCR gene for adoptive immunotherapy in hepatocellular carcinoma. Tumour Biol. 2016;37:799–806.
Haanen J, Obeid M, Spain L, Carbonnel F, Wang Y, Robert C, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up☆. Ann Oncol. 2022;33:1217–38.
Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378:158–68.
Machairas N, Tsilimigras DI, Pawlik TM. Current landscape of immune checkpoint inhibitor therapy for hepatocellular carcinoma. Cancers (Basel). 2022;14:2018.
Zhang B, Wu Q, Zhou YL, Guo X, Ge J, Fu J. Immune-related adverse events from combination immunotherapy in cancer patients: a comprehensive meta-analysis of randomized controlled trials. Int Immunopharmacol. 2018;63:292–8.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–23.
Zhu AX, Dayyani F, Yen C-J, Ren Z, Bai Y, Meng Z, et al. Alpha-fetoprotein as a potential surrogate biomarker for atezolizumab + bevacizumab treatment of hepatocellular carcinoma. Clin Cancer Res. 2022;28:3537–45.
Mao J, Wang D, Long J, Yang X, Lin J, Song Y, et al. Gut microbiome is associated with the clinical response to anti-PD-1 based immunotherapy in hepatobiliary cancers. J Immunother Cancer. 2021;9: e003334.
Wu H, Zheng X, Pan T, Yang X, Chen X, Zhang B, et al. Dynamic microbiome and metabolome analyses reveal the interaction between gut microbiota and anti-PD-1 based immunotherapy in hepatocellular carcinoma. Int J Cancer. 2022;151:1321–34.
Jin H, Shi Y, Lv Y, Yuan S, Ramirez CFA, Lieftink C, et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature. 2021;595:730–4.
Deng H, Kan A, Lyu N, He M, Huang X, Qiao S, et al. Tumor-derived lactate inhibit the efficacy of lenvatinib through regulating PD-L1 expression on neutrophil in hepatocellular carcinoma. J Immunother Cancer. 2021;9: e002305.
Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20:28.
Liu N, Luo J, Kuang D, Xu S, Duan Y, Xia Y, et al. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2α–mediated tumor progression. J Clin Invest. 129:631–46.
Cho E-H. Succinate as a regulator of hepatic stellate cells in liver fibrosis. Front Endocrinol (Lausanne). 2018;9:455.
Westheim AJF, Stoffels LM, Dubois LJ, van Bergenhenegouwen J, van Helvoort A, Langen RCJ, et al. The modulatory effects of fatty acids on cancer progression. Biomedicines. 2023;11:280.
Perrone F, Minari R, Bersanelli M, Bordi P, Tiseo M, Favari E, et al. The prognostic role of high blood cholesterol in advanced cancer patients treated with immune checkpoint inhibitors. J Immunother. 2020;43:196–203.
Liu F, Tian T, Zhang Z, Xie S, Yang J, Zhu L, et al. Long non-coding RNA SNHG6 couples cholesterol sensing with mTORC1 activation in hepatocellular carcinoma. Nat Metab. 2022;4:1022–40.
Chen C, Guan J, Gu X, Chu Q, Zhu H. Prostaglandin E2 and receptors: insight into tumorigenesis, tumor progression, and treatment of hepatocellular carcinoma. Front Cell Dev Biol. 2022;10: 834859.
Luan B, Yoon Y-S, Le Lay J, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci U S A. 2015;112:15642–7.
Du D, Liu C, Qin M, Zhang X, Xi T, Yuan S, et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm Sin B. 2022;12:558–80.
Phillips MM, Sheaff MT, Szlosarek PW. Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges. Cancer Res Treat. 2013;45:251–62.
Lin X-J, Fang J-H, Yang X-J, Zhang C, Yuan Y, Zheng L, et al. Hepatocellular Carcinoma Cell-Secreted Exosomal MicroRNA-210 Promotes Angiogenesis In Vitro and In Vivo. Mol Ther Nucleic Acids. 2018;11:243–52.
Wang Q, Wang G, Niu L, Zhao S, Li J, Zhang Z, et al. Exosomal MiR-1290 promotes angiogenesis of hepatocellular carcinoma via targeting SMEK1. J Oncol. 2021;2021:6617700.
Dong S-S, Dong D-D, Yang Z-F, Zhu G-Q, Gao D-M, Chen J, et al. Exosomal mir-3682-3p Suppresses Angiogenesis by Targeting ANGPT1 via the RAS-MEK1/2-ERK1/2 Pathway in Hepatocellular Carcinoma. Front Cell Dev Biol. 2021;9: 633358.
Moh-Moh-Aung A, Fujisawa M, Ito S, Katayama H, Ohara T, Ota Y, et al. Decreased miR-200b-3p in cancer cells leads to angiogenesis in HCC by enhancing endothelial ERG expression. Sci Rep. 2020;10:10418.
Fang J-H, Zhang Z-J, Shang L-R, Luo Y-W, Lin Y-F, Yuan Y, et al. Hepatoma cell-secreted exosomal microRNA-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology. 2018;68:1459–75.
Yokota Y, Noda T, Okumura Y, Kobayashi S, Iwagami Y, Yamada D, et al. Serum exosomal miR-638 is a prognostic marker of HCC via downregulation of VE-cadherin and ZO-1 of endothelial cells. Cancer Sci. 2021;112:1275–88.
Yang B, Feng X, Liu H, Tong R, Wu J, Li C, et al. High-metastatic cancer cells derived exosomal miR92a-3p promotes epithelial-mesenchymal transition and metastasis of low-metastatic cancer cells by regulating PTEN/Akt pathway in hepatocellular carcinoma. Oncogene. 2020;39:6529–43.
Li D, Zhang Y, Zhang H, Zhan C, Li X, Ba T, et al. CADM2, as a new target of miR-10b, promotes tumor metastasis through FAK/AKT pathway in hepatocellular carcinoma. Journal of Experimental & Clinical Cancer Research : CR [Internet]. 2018 [cited 2023 May 24];37. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5836378/
Liu Y, Yang Y, Wang T, Wang L, Wang X, Li T, et al. Long non-coding RNA CCAL promotes hepatocellular carcinoma progression by regulating AP-2α and Wnt/β-catenin pathway. Int J Biol Macromol. 2018;109:424–34.
Wang R, Yu Z, Chen F, Xu H, Shen S, Chen W, et al. miR-300 regulates the epithelial-mesenchymal transition and invasion of hepatocellular carcinoma by targeting the FAK/PI3K/AKT signaling pathway. Biomed Pharmacother. 2018;103:1632–42.
Xu Q, Liu X, Liu Z, Zhou Z, Wang Y, Tu J, et al. MicroRNA-1296 inhibits metastasis and epithelial-mesenchymal transition of hepatocellular carcinoma by targeting SRPK1-mediated PI3K/AKT pathway. Mol Cancer. 2017;16:103.
He M, Qin H, Poon TCW, Sze S-C, Ding X, Co NN, et al. Hepatocellular carcinoma-derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis. 2015;36:1008–18.
Zhou Y, Ren H, Dai B, Li J, Shang L, Huang J, et al. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J Exp Clin Cancer Res. 2018;37:324.
Fang T, Lv H, Lv G, Li T, Wang C, Han Q, et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. 2018;9:191.
Yin C, Han Q, Xu D, Zheng B, Zhao X, Zhang J. SALL4-mediated upregulation of exosomal miR-146a-5p drives T-cell exhaustion by M2 tumor-associated macrophages in HCC. Oncoimmunology. 2019;8:1601479.
Xu Y, Luan G, Liu F, Zhang Y, Li Z, Liu Z, et al. Exosomal miR-200b-3p induce macrophage polarization by regulating transcriptional repressor ZEB1 in hepatocellular carcinoma. Hepatol Int. 2023;
Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, et al. Endoplasmic reticulum stress promotes liver cancer cells to release exosomal miR-23a-3p and up-regulate PD-L1 expression in macrophages. Hepatology. 2019;70:241–58.
Li X, Lei Y, Wu M, Li N. Regulation of macrophage activation and polarization by HCC-derived exosomal lncRNA TUC339. Int J Mol Sci. 2018;19:2958.
Zhang P-F, Gao C, Huang X-Y, Lu J-C, Guo X-J, Shi G-M, et al. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol Cancer. 2020;19:110.
Wang X, Shen H, Zhangyuan G, Huang R, Zhang W, He Q, et al. 14-3-3ζ delivered by hepatocellular carcinoma-derived exosomes impaired anti-tumor function of tumor-infiltrating T lymphocytes. Cell Death Dis. 2018;9:159.
Fan F, Chen K, Lu X, Li A, Liu C, Wu B. Dual targeting of PD-L1 and PD-L2 by PCED1B-AS1 via sponging hsa-miR-194-5p induces immunosuppression in hepatocellular carcinoma. Hepatol Int. 2021;15:444–58.
Numata Y, Akutsu N, Ishigami K, Koide H, Wagatsuma K, Motoya M, et al. Synergistic effect of IFN-γ and IL-1β on PD-L1 expression in hepatocellular carcinoma. Biochem Biophys Rep. 2022;30: 101270.
He Q, Liu M, Huang W, Chen X, Zhang B, Zhang T, et al. IL-1β-Induced Elevation of Solute Carrier Family 7 Member 11 Promotes Hepatocellular Carcinoma Metastasis Through Up-regulating Programmed Death Ligand 1 and Colony-Stimulating Factor 1. Hepatology. 2021;74:3174.
Dang Y, Chen J, Feng W, Qiao C, Han W, Nie Y, et al. Interleukin 1β-mediated HOXC10 Overexpression Promotes Hepatocellular Carcinoma Metastasis by Upregulating PDPK1 and VASP. Theranostics. 2020;10:3833–48.
Guo C, Ouyang Y, Cai J, Xiong L, Chen Y, Zeng X, et al. High expression of IL-4R enhances proliferation and invasion of hepatocellular carcinoma cells. Int J Biol Markers. 2017;32:e384–90.
Murray PJ. Macrophage Polarization. Annu Rev Physiol. 2017;79:541–66.
Xu J, Lin H, Wu G, Zhu M, Li M. IL-6/STAT3 Is a Promising Therapeutic Target for Hepatocellular Carcinoma. Front Oncol. 2021;11: 760971.
Xue J-C, Zhou H-Y, Yu L-S-Y, Wu Y-Y. Effect of IL-8 on hepatocellular carcinoma-associated metastasis by targeting MMP9 in mice. Transl Cancer Res. 2022;11:2050–60.
Sun F, Wang J, Sun Q, Li F, Gao H, Xu L, et al. Interleukin-8 promotes integrin β3 upregulation and cell invasion through PI3K/Akt pathway in hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38:449.
Peng S, Chen Y, Gong Y, Li Z, Xie R, Lin Y, et al. Predictive value of intratumour inflammatory cytokine mRNA levels of hepatocellular carcinoma patients and activation of two distinct pathways govern IL-8 induced epithelial-mesenchymal transition in human hepatic cancer cell lines. Cytokine. 2019;119:81–9.
Xiao P, Long X, Zhang L, Ye Y, Guo J, Liu P, et al. Neurotensin/IL-8 pathway orchestrates local inflammatory response and tumor invasion by inducing M2 polarization of Tumor-Associated macrophages and epithelial-mesenchymal transition of hepatocellular carcinoma cells. Oncoimmunology. 2018;7: e1440166.
Zhang S, Gan X, Qiu J, Ju Z, Gao J, Zhou J, et al. IL-10 derived from Hepatocarcinoma cells improves human induced regulatory T cells function via JAK1/STAT5 pathway in tumor microenvironment. Mol Immunol. 2021;133:163–72.
Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology. 2016;6: e1264562.
Schülke S. Induction of Interleukin-10 producing dendritic cells as a tool to suppress allergen-specific T Helper 2 Responses. Front Immunol. 2018;9:455.
He Y, Pei J, Li X, Chi G. IL-35 promotes EMT through STAT3 activation and induces MET by promoting M2 macrophage polarization in HCC. Biochem Biophys Res Commun. 2021;559:35–41.
Liu X, Ren H, Guo H, Wang W, Zhao N. Interleukin-35 has a tumor-promoting role in hepatocellular carcinoma. Clin Exp Immunol. 2021;203:219–29.
Malfettone A, Soukupova J, Bertran E, Crosas-Molist E, Lastra R, Fernando J, et al. Transforming growth factor-β-induced plasticity causes a migratory stemness phenotype in hepatocellular carcinoma. Cancer Lett. 2017;392:39–50.
Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-β1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–7.
Batlle E, Massagué J. Transforming Grown Factor-β Signaling in Immunity and Cancer. Immunity. 2019;50:924–40.
Papaspyridonos M, Matei I, Huang Y, do Rosario Andre M, Brazier-Mitouart H, Waite JC, et al. Id1 suppresses anti-tumour immune responses and promotes tumour progression by impairing myeloid cell maturation. Nat Commun. 2015;6:6840.
Ioannis K, Eralda S, Egal ESA, Beswick E. G-CSF in tumors: aggressiveness, tumor microenvironment and immune cell regulation. Cytokine. 2021;142: 155479.
Abrams SI, Waight JD. Identification of a G-CSF-Granulocytic MDSC axis that promotes tumor progression. Oncoimmunology. 2012;1:550–1.
Wang J, Wang Y, Chu Y, Li Z, Yu X, Huang Z, et al. Tumor-derived adenosine promotes macrophage proliferation in human hepatocellular carcinoma. J Hepatol. 2021;74:627–37.
Liu W-T, Jing Y-Y, Yu G, Chen H, Han Z, Yu D, et al. Hepatic stellate cell promoted hepatoma cell invasion via the HGF/c-Met signaling pathway regulated by p53. Cell Cycle. 2016;15:886–94.
Zhang T, Wang Y, Xie M, Ji X, Luo X, Chen X, et al. HGF-mediated elevation of ETV1 facilitates hepatocellular carcinoma metastasis through upregulating PTK2 and c-MET. J Exp Clin Cancer Res. 2022;41:275.
Deng H, Kan A, Lyu N, Mu L, Han Y, Liu L, et al. Dual vascular endothelial growth factor receptor and fibroblast growth factor receptor inhibition elicits antitumor immunity and enhances programmed cell death-1 checkpoint blockade in hepatocellular carcinoma. Liver Cancer. 2020;9:338–57.
Qi Y, Song Y, Cai M, Li J, Yu Z, Li Y, et al. Vascular endothelial growth factor A is a potential prognostic biomarker and correlates with immune cell infiltration in hepatocellular carcinoma. J Cell Mol Med. 2023;27:538–52.
Asghar K, Farooq A, Zulfiqar B, Rashid MU. Indoleamine 2,3-dioxygenase: As a potential prognostic marker and immunotherapeutic target for hepatocellular carcinoma. World J Gastroenterol. 2017;23:2286–93.
Cui N, Hu M, Khalil RA. Biochemical and biological attributes of matrix metalloproteinases. Prog Mol Biol Transl Sci. 2017;147:1–73.
Miyoshi A, Kitajima Y, Sumi K, Sato K, Hagiwara A, Koga Y, et al. Snail and SIP1 increase cancer invasion by upregulating MMP family in hepatocellular carcinoma cells. Br J Cancer. 2004;90:1265–73.
Xun X, Zhang C, Wang S, Hu S, Xiang X, Cheng Q, et al. Cyclooxygenase-2 expressed hepatocellular carcinoma induces cytotoxic T lymphocytes exhaustion through M2 macrophage polarization. Am J Transl Res. 2021;13:4360–75.
Wang Q, Lu D, Fan L, Li Y, Liu Y, Yu H, et al. COX-2 induces apoptosis-resistance in hepatocellular carcinoma cells via the HIF-1α/PKM2 pathway. Int J Mol Med. 2019;43:475–88.
Zhou Y, Fu C, Kong Y, Pan D, Wang Y, Huang S, et al. Antitumor and immunomodulatory effects of a novel multitarget inhibitor, CS2164, in mouse hepatocellular carcinoma models. Anticancer Drugs. 2019;30:909–16.
Wei C-Y, Zhu M-X, Zhang P-F, Huang X-Y, Wan J-K, Yao X-Z, et al. PKCα/ZFP64/CSF1 axis resets the tumor microenvironment and fuels anti-PD1 resistance in hepatocellular carcinoma. J Hepatol. 2022;77:163–76.
Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157–67.
Yao W, Ba Q, Li X, Li H, Zhang S, Yuan Y, et al. A Natural CCR2 antagonist relieves tumor-associated macrophage-mediated immunosuppression to produce a therapeutic effect for liver cancer. EBioMedicine. 2017;22:58–67.
Ochoa-Callejero L, Pérez-Martínez L, Rubio-Mediavilla S, Oteo JA, Martínez A, Blanco JR. Maraviroc, a CCR5 antagonist, prevents development of hepatocellular carcinoma in a mouse model. PLoS ONE. 2013;8: e53992.
Xiao Z, Chung H, Banan B, Manning PT, Ott KC, Lin S, et al. Antibody mediated therapy targeting CD47 inhibits tumor progression of hepatocellular carcinoma. Cancer Lett. 2015;360:302–9.
Lo J, Lau EYT, So FTY, Lu P, Chan VSF, Cheung VCH, et al. Anti-CD47 antibody suppresses tumour growth and augments the effect of chemotherapy treatment in hepatocellular carcinoma. Liver Int. 2016;36:737–45.
Li G, Zhao L. Sorafenib-loaded hydroxyethyl starch-TG100-115 micelles for the treatment of liver cancer based on synergistic treatment. Drug Deliv. 2019;26:756–64.
Wu L, Zhang X, Zheng L, Zhao H, Yan G, Zhang Q, et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res. 2020;8:710–21.
Lu S, Gao Y, Huang X, Wang X. Cantharidin exerts anti-hepatocellular carcinoma by Mir-214 modulating macrophage polarization. Int J Biol Sci. 2014;10:415–25.
Acknowledgements
All figures were generated by BioRender (https://biorender.com/).
Funding
This work was supported by grants from the National Natural Science Foundation of China No. 82273374 (K.W.), No. U23A20451 (L.X.), No. 82273310 (L.X.), the Natural Science Foundation of Hubei Province 2022CFA016 (L.X.), and the Basic Research Support Program of Huazhong University of Science and Technology 2023BR038 (L.X.).
Author information
Authors and Affiliations
Contributions
LX, KW, DF and YN offered directions on this manuscript. YY drafted the manuscript. YY, WF and JC made the figures and table. YY, WF and XC reviewed the literature. GW, SW, XX and LX reviewed the manuscript and gave comments. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yin, Y., Feng, W., Chen, J. et al. Immunosuppressive tumor microenvironment in the progression, metastasis, and therapy of hepatocellular carcinoma: from bench to bedside. Exp Hematol Oncol 13, 72 (2024). https://doi.org/10.1186/s40164-024-00539-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-024-00539-x