
Abstract
Hepatocellular carcinoma (HCC) ranks the most common primary liver malignancy and the third leading cause of tumor-related mortality worldwide. Unfortunately, despite advances in HCC treatment, less than 40% of HCC patients are eligible for potentially curative therapies. Recently, cancer immunotherapy has emerged as one of the most promising approaches for cancer treatment. It has been proven therapeutically effective in many types of solid tumors, such as non-small cell lung cancer and melanoma. As an inflammation-associated tumor, it’s well-evidenced that the immunosuppressive microenvironment of HCC can promote immune tolerance and evasion by various mechanisms. Triggering more vigorous HCC-specific immune response represents a novel strategy for its management. Pre-clinical and clinical investigations have revealed that various immunotherapies might extend current options for needed HCC treatment. In this review, we provide the recent progress on HCC immunology from both basic and clinical perspectives, and discuss potential advances and challenges of immunotherapy in HCC.
Similar content being viewed by others


Background
Hepatocellular carcinoma (HCC) represents the most common type of primary liver cancer, with a global incidence of 500,000 new cases per year [1]. HCC is closely associated with chronic liver inflammation and some well-known risk factors, including chronic HBV and HCV infections, alcohol consumption, diabetes mellitus and several metabolic diseases [2]. The current therapeutic options available for HCC, such as transarterial chemoembolization (TACE), radiofrequency ablation, surgical resection and transplantation, are only curative for some patients in early stages. Other more effective approaches emerged in the past few years, such as tyrosine kinase inhibitors (TKIs) targeting angiogenesis (e.g. Sorafenib, lenvatinib, regorafenib) [3], clinically tested selective Cyclin dependent kinase 5 and 4/6 (Cdk5, Cdk4/6) inhibitors (Dinaciclib & Palbociclib) [4,5,6], and highly selective fibroblast growth factor receptor 4 (FGFR4) inhibitor H3B-6527 [7, 8], which pre-clinically and clinically show encouraging efficacy and have been rigorously pursued for advanced HCC.
The liver is a ‘tolerogenic’ organ that can arouse its immune responses to prevent undesirable pathogen attack and tumor initiation. However, as a typical inflammation-linked tumorigenesis, immune evasion is one of the features occurring during the initiation and evolution of HCC [9]. A number of immune suppressor mechanisms, including intratumoral accumulation of immunosuppressive cell populations, defective antigen presentation and activation of multiple inhibitory receptor-ligand pathways, favor tolerance over immunity, and promote progression of HCC [10, 11]. The magnitude of immune suppression in the tumor microenvironment (TME) is closely correlated with poor prognosis in HCC patients. Hence, for better arousing anti-tumor immunity, more details about suppressed immune landscape of HCC urgently needs to be elucidated.
The intricate immune network in TME of HCC
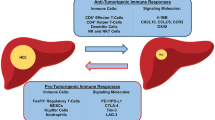
The HCC tumor microenvironment (TME) is a dynamic system, which comprises cancer cells, the intricate cytokine environment, extracellular matrix, immune cell subsets and other components [12]. It’s well established that the immune landscape of HCC has a strong suppressor feature. In this complex network, the pro-tumorigenic immune response, mediated by diverse immunosuppressive cell subsets, secretions and signaling, plays a pivotal role in driving immune evasion [13] (Fig. 1.). Moreover, ‘fatigue’ of anti-tumor immunity also contributes to tumor tolerance and progression. Here, we discuss new advances in the immunosuppressive picture of HCC.

The landscape of immunosuppressive tumor microenvironment of HCC. Diverse suppressive immune cell subsets infiltration, regulatory secretions and some inhibitory signaling mediate HCC immune evasion. (Notes: Tregs: regulatory T cells; TAMs: tumor-associated macrophages; TANs: tumor associated neutrophils; CTLs:cytotoxic T lymphocytes; CAF: cancer associated fibroblast; MDSCs: myeloid- derived suppressor cells; HSCs: hepatic stellate cells; NK: natural killer cell; KC: Kupffer cell)
Representative immunosuppressive components in TME of HCC
Myeloid-derived suppressor cells (MDSCs)
MDSCs is a heterogeneous population of immature myeloid cells (IMCs), which are expanded in pathological conditions and up-regulate expression of immune suppressive factors, such as arginase and inducible nitric oxide synthase (iNOS or NOS2) [14]. Various tumor originated cytokines, such as G-CSF, GM-CSF, VEGF, MCP-1 and IL-1β, have been demonstrated to induce MDSCs infiltration [15]. Cell cycle related kinase (CCRK) represents a novel signaling target for cancer immunotherapy [16]. Emerging evidence also indicates the hepatoma-intrinsic CCRK upregulates interlukin-6 (IL-6) production through EZH2/NF-κB signaling, which consequently induce MDSCs accumulation in TME [17]. Hepatic carcinoma related tumor-associated fibroblasts (TAFs), a stromal part in HCC, can induce peripheral blood monocyte migration and differentiation into CD14 + HLA-DR −/low MDSCs by SDF-1α. TAFs mediate the generation of MDSCs via IL-6/STAT3 signaling [18]. In addition, local hypoxia has been identified as a key regulator that can promote MDSCs accumulation through the Chemokine C-C motif Ligand 26 (CCL26)/CX3CR1 pathway [19]. Hypoxia-inducible factor 1α (HIF-1α) mediated ENTPD2 over-expression in HCC cells, has been proven to increase the extracellular level of 5′-AMP, which subsequently recruit MDSCs into TME [20].
As a powerful inhibitory immune modulator, infiltrated MDSCs in HCC damage effector T cells, expand immune checkpoink signaling, decrease NK cell cytotoxicity and cytokine production by diverse mechanisms [21, 22]. The MDSCs in fibrotic HCC tissue are notably correlated with reduced tumor infiltrating lymphocytes (TILs) and elevated tumorigenicity, aggressive phenotype, moreover, whose activation and infiltration contribute greatly to worse survival rate both in mouse model and HCC patients [23]. CD14+ HLA-DR −/low MDSCs can blunt HCC immunity through inducing activation of CD4+ CD25+ Foxp3+ regulatory T cells, inhibiting proliferation and IFN-γ secretion of CD3/CD28-stimulated autologous peripheral blood mononuclear cells (PBMCs) [24]. T cell function is impaired due to competition for energy resources (e.g. arginine and cysteine) with MDSCs [25], as well as involvement of several inhibitory receptor-ligand pathways in MDSCs-mediated immune evasion. Tumor-derived TGF-β triggers recruitment of MDSCs in a CXCL1/2/5- and CXCR2-dependent manner. The infiltrated MDSCs selectively suppress IFN-γ production deriving from NKT cells [26]. MDSCs also can express galectin-9 that binds to TIM-3 on T cells, inducing T-cell apoptosis [27]. In addition, it’s suggested that MDSCs in advanced HCC patients may interact with Kuppfer cells to induce PD-L1 expression, and mediate inhibited cytotoxicity and cytokine release of NK cells through the NKp30 receptor [28]. Taken together, MDSCs exert versatile immunosuppressive effects in HCC. Combined treatment with anti-PD-1/PD-L1 and concomitant targeting MDSCs (such as CCRK inhibition or p38 MAPK inhibitor) may synergistically enhance efficacy to eradicate HCC [17, 23]. In addition, recent evidence suggests radiation and IL-12 combination therapy (RT/IL-12) may elevate anti-tumor immunity in HCC by reducing MDSCs accumulation and the production of reactive oxygen species (ROS) [29]. Hence, MDSCs may serve as a potential target for resetting immunotorelant state in HCC tumors.
Tumor-associated macrophages (TAMs)
Macrophages arise from bone marrow-derived circulating monocytes, and then reside in normal tissues. The two polarizing phenotypes M1 and M2, are highly plastic in response to complex stimuli. Substantial clinical data and experimental research confirmed that alternatively activated status macrophages, the M2 phenotype, stimulate tumor initiation, progression and malignant metastasis by various mechanisms [30, 31]. In HCC, some specific populations of the immunosuppressive tumor-associated macrophages (TAMs) have emerged as a research hotspot recently. The well-identified HCC-derived cytokines, such as IL-4, IL-13, CSF-1, CCL2, CXCL12, connective tissue growth factor (CTGF) [32,33,34], induce TAMs differentiation from CCR2+ inflammatory monocytes, alternatively activated macrophages (AAMs) infiltration, then reduce innate or adaptive immunity [34, 35]. Osteopontin (OPN) expressed by HCC cells not only has a positive association with PD-L1 expression in HCC, moreover, it facilitates alternative activation and chemotactic migration of TAMs via CSF1-CSF1R pathway in TAMs [36]. HCC cells originated HIF-1α has been recently suggested to enhance IL-1β release by TAMs via TLR4/TRIF/NF-κB signaling pathway, which fosters EMT and immune evasion of HCC [37]. Crosstalk between MDSCs and TAMs results in decreased production of IL-6, IL-12, down-expression of MHCII, and elevated production of IL-10, a strong inhibitory mediator that impairs downstream CD8+ T cell and NK cell cytotoxicity [38]. TAMs-derived IL-10 also increases intratumoral Foxp3+ Tregs frequency, which then suppresses CD4+CD25− T cells activation [38, 39]. TAMs in the peritumoral stroma of HCC have been shown to secrete multiple key proinflammatory cytokines (e.g. IL-1β, IL-6, IL-23, and TNF-α) and contribute to the expansion of interleukin-17-producing CD4+ T helper 17 cells (Th17), which suppress anti-tumor immunity by over-expressing several activation markers, such as PD-1, CTLA-4, and GITR [40]. In addition, TAMs are highly involved in other immune inhibitory regulations [41, 42]. TGF-β in the TME of HCC promotes the Tim-3 expression on TAMs, subsequently enabling the activated TAMs to facilitate tumor growth and immune tolerance via NF-κB signaling and downstream IL-6 production [43]. TAMs derived IL-6/STAT3 signaling also has been validated to sustain HCC carcinogenesis by promoting its carcinoma stem cells (CSCs)-liked characteristics [44].
Notably, recent evidence indicates that PD-1− TAMs can capture anti-PD-1 monoclonal antibodies (aPD-1 mAbs) through Fcγ receptors (FcγRs) expressed on the surface binding to drug’s Fc domain glycan [45]. This novel investigation indicates that blockade of FcγRs before aPD-1 mAbs administration may substantially improve checkpoint blockade therapy.
Tumor associated neutrophils (TANs)
Heterogeneity of TANs is a fundamental property that allows TANs to perform corresponding functions for adaptations to changing microenvironment. Similar to macrophages, neutrophils differently affect tumor biological behaviors depending on their polarization, either anti-tumoral (N1) and pro-tumoral (N2) phenotypes [46]. In some solid tumor models, such as lung cancer, metastatic renal cell cancer (mRCC) and melanoma, it was previously reported that TANs infiltration or neutrophil-lymphocyte ratio (NLR) closely correlate with tumor progression, which can serve as a significant predictor for monitoring patients with advanced tumor receiving anti-PD-1/PD-L1 immunotherapy [47, 48]. TANs activation is modulated by cytokines, such as Type I interferons (IFNs) and TGF-β [49]. TANs mainly suppress anti-tumor immunity via interacting with CD8+ T cells, inducing CD8+ T cells apoptosis through nitric oxide (NO) production mediated by tumor necrosis factor-α (TNF-α) [50].
The facilitator role of TANs in pathological progression of HCC has become a topic of growing interest in recent years. Clinically, TANs play a key role in driving progression and poor prognosis of HCC, and NLR is an independent predictor of survival after hepatectomy in patients with HCC [51, 52]. The newest discovery shows that loss of hypoxia associated factor, HAF (encoded by SART1) results in inappropriate HIF-1 activation, and overproduction of downstream HIF-1 dependent chemokine, RANTES. HIF-1/RANTES upregulation accumulates TANs infiltration, which is associated with non-alcoholic steatohepatitis (NASH) driven HCC initiation and progression [53]. Moreover, recent studies suggested that TANs mediate the intratumoral infiltration of TAMs and regulatory T cells by overproducing some chemokines, such as CCL2 and CCL17, which then contributes to HCC progression, metastasis and resistance to sorafenib treatment [54]. A newly identified positive feedback loop implies that TANs induce HCC stem cell like characteristics via upregulating expression of miR-301b-3p in cancer cells, and maintain hyper-action in NF-kB signaling, lead to higher secretion level of C-X-C motif chemokine5 (CXCL5) and in turn recruit more TANs infiltration [55]. In general, TANs are strongly connected with immunosuppression in HCC, but direct interactions between TANs and other components in HCC tissue and the exact underlying mechanisms behind this regulation in HCC are not yet clear.
Tumor-infiltrating lymphocytes (TILs)
A high density of tumor-infiltrating lymphocytes (TILs) was once thought to be the host’s immune reaction against cancer. Some early clinical data suggested postoperative HCC patients with high level lymphocytes infiltration, especially T cells, had reduced recurrence and better survival [56]. However, accumulating evidence suggests that the overall degree of TILs in HCC is not capable of mounting effective anti-tumor immunity to control tumor progression [57]. Intrahepatic immune response involves diverse lymphocyte populations, which contribute differently to HCC immune surveillance. The intratumoral balance of regulatory and cytotoxic T cells plays a key role in evaluating the immune state and progression of HCC [57, 58].
Regulatory T cell (Treg)
Regulatory T cells (Tregs) can be derived from peripheral blood T lymphocytes, resident T cells and other cellular sources. Its recruitment has been found to be induced by the CCR6 (CC chemokine receptor type 6)–CCL20 (CC motif chemokine 20) axis. Tregs activation is induced by T cell receptor (TCR) engagement concurrent with IL-10 and TGF-β signaling [59]. Apart from activation via pro-inflammatory signals, recent investigations elucidate that long noncoding RNAs (LncRNAs) may play pivotal roles in driving Tregs differentiation and implications during HCC progression [60]. Overexpressed Lnc-epidermal growth factor receptor (Lnc-EGFR) in Tregs binds to EGFR and prevents its ubiquitination by c-CBL, augmenting activation of its downstream AP-1/NF-AT1 axis in Tregs thus to promote immunosuppression in HCC [60]. Moreover, Amphiregulin (AREG), a multifunctional player, may enhance Tregs suppressive function via the EGFR motivation as well [61].
The frequencies of Tregs are associated with HCC invasiveness and have a crucial role in hampering the development of effective anti-tumor responses in HCC [57, 62]. Recent evidence indicates that CD4+CD25+ Tregs in HCC patients can trigger a compromised immune response through various mechanisms [63]. A typical subset, CD4+ CD25+ Foxp3+ Tregs, may impair CD8+ T cells killing capacity via inhibiting the release and production of granzyme A, B (GrA, B), and perforin [64], concurrently, they also selectively suppress certain molecules (such as TNF-α, IFN-γ) involved in CD8+ T cell activation [64, 65]. Additionally, high expression of IL-35 in HCC tissue has been implicated positively to correlate with another newly identified subtype, CD39+ Foxp3+ Tregs infiltration [66], which serves as a better independent predictive indicator for recurrence in HCC patients after curative resection.
CD8+ cytotoxic T lymphocytes (CTLs)
The presence of CD8+ Cytotoxic T lymphocytes (CTLs) in HCC tissue is associated with improved survival. However, the efficacy of CTLs-mediated anti-tumor immune response is functionally limited through diverse mechanisms. Physical conditions (e.g. overload of lactic acid, low pH, hypoxia) [67], severe “metabolic competition” with tumor cells, a lack of CD4+ T cells help (moreover, interact with Tregs and other suppressor cells) [64, 68], and high expression of a large amount of immunoregulatory molecules in T cells or HCC cells (e.g. IL-10, Fas/FasL, CXCL17, VEGF, indoleamine-2,3-dioxygenase and so on) [67, 69,70,71], may be responsible for restricted tumor-associated antigens (TAAs)-specific CD8+ T cell responses and poor IFN-γ production of CTLs [72, 73]. Apart from the classic immunosuppressive cells in TME, other components critically manipulate the functions of CTLs as well. Liver fibrosis, a prominent characteristic of HCC, impairs platelet-derived CD44 recognition by CD8+ T cells, reducing effector CD8+ T cells infiltration, and adhering to liver sinusoids to perform immunosurveillance [74]. Expression of Fas/FasL in CD8+ T cells positively correlates with HCC anti-tumor immunity [69]. Recent evidence indicates that tumor-derived vascular endothelial growth factor A (VEGF-A) and prostaglandin E2 (PGE2) cooperatively induce FasL expression in endothelial cells, which leads to excessive turnover of CD8 + T cells and reduce anti-tumor immune responses [71]. CD14+ dendritic cells (CD14+ DCs), a newly discovered immune regulator of HCC, has been suggested to suppress CTLs via IL-10 and indoleamine-2,3-dioxygenase (IDO) production, and the two cytokines play central roles in various physiological and pathological immune responses and inflammatory processes [75].
Notably, immune checkpoint signaling, which involves enhancement of numerous inhibitory co-stimulatory molecules (e.g. PD-1, LAG-3, CTLA-4, Tim-3, 2B4), has been demonstrated to dramatically induce CTLs exhaustion [58, 76, 77]. More details will be discussed in the section “immune checkpoint pathways and related therapeutics”.
Innate immune players and stromal components
Natural killer (NK) cells
Natural killer (NK) cells constitute a large proportion of the innate immune system in the liver. As the first line of host defense against viral infections (e.g. HBV, HCV) and carcinogenesis, NK cells play a key role in maintaining the balance between immune defense and tolerance. Increasing evidence suggests that hypoxic stress in HCC tissue, switch of activating/inhibitory NK receptors (NKRs) and influences by immune regulatory components in TME, largely contribute to NK cells dysfunction, which significantly correlates with fatigue anti-tumor immunity and poor prognosis [78, 79].
α-Fetoprotein (AFP) overexpressed by HCC cells was demonstrated to indirectly impair interlukine-12 (IL-12) production from dendritic cells (DCs), which results in attenuated cytotoxic effector molecules release, decreased expression of natural killer group 2, member D (NKG2D), an activating receptor on NK cells, and subsequently inhibiting activation and ability of NK cells [80, 81]. A recent study also indicates AFP may exert dual effects on NK cells functions in a direct manner. Short-term exposure to AFP induces IL-2 hyperresponsive phenotype NK cells, accompanied with elevated secretion of IL-1β, IL-6 and TNF-α [82]. These pro-inflammatory cytokines were associated with a low recurrence rate and a prolonged overall survival (OS) of HBV-related HCC patients [83]. In contrast, extended effect of AFP negatively affects long-term NK cell viability [82].
Other modulators in TME of HCC also exert multiple effects on NK activities (Fig. 2.). As mentioned above, MDSCs and TAMs infiltration inhibit autologous NK cell cytotoxicity and cytokine production, and the suppression is mainly dependent on NKp30 on NK cells [28]. Tregs compete with NK cells for IL-2 availability and impair NK responses via cytokines release, such as IL-8, TGF-ß1 and IL-10, which then down-regulates expression of NKR ligands on hepatic stellate cells (HSCs) and inhibits their recognition by NKG2D [84]. Hepatocellular carcinoma-associated fibroblasts (CAFs or TAFs), has been shown to induce MDSCs generation through the IL-6/STAT3 axis and stromal cell-derived factor (SDF)-1α secretion [18]. In addition to its direct influence on immunosuppressive TME, CAFs-derived IDO and PGE2 attenuate NK cells-mediated TNF-α and IFN-γ production, which may be associated with persistent fibrosis in HCC and tumor cell immune evasion [85, 86].

Modulator role of NK cells in regulating HCC immune responses. NK cells exert multiple immune regulatory functions in HCC. Apart from the direct influences on tumor cells, interactions between NK cells and other immune cells or tumor stromal components have been demonstrated to mediate HCC immune evasion
Kupffer cells (KCs)
KCs have previously been demonstrated to constitute an important part in maintaining liver immune homeostasis. Some studies reported that IL-10-mediated suppression of KC-derived inflammatory TNF-α and NO production contribute to attenuation of hepatitis [87]. Although KCs were once regarded as a powerful line of defense against tumors in the liver, recently, KCs have commonly been explored as pro-carcinogenic stakeholders in the context of HCC, more underlying mechanisms about their immune regulator roles, and KCs-related innate or adaptive immune response have been gradually uncovered. Current investigations indicate that altered functions of KCs are mainly influenced by pro-inflammatory signals and other suppressive cells (e.g. MDSCs) [88]. Triggering receptors expressed on myeloid cells-1 (TREM-1) expressed by KCs, is a crucial factor in HCC initiation. New studies suggest that the potential ligand for TREM-1, high mobility group Box 1 (HMGB1) released by necrotic hepatocytes, is likely involved in activating KCs pro-inflammatory signaling and promoting HCC progression [89]. Autophagy-defective KCs, a novel non-parenchymal liver cellular degradation deficiency, has been shown to promote liver fibrosis, inflammation and hepatocarcinogenesis during the pre-neoplastic stage via enhancing the mitochondrial ROS/NF-κB/IL-1 pathway [90]. In addition, KCs-derived galectin-9, the natural ligand for the T cell immunoglobulin domain and mucin domain protein 3 (Tim-3), leads to expansion of CD4+ CD25+ FoxP3+ Tregs, contraction of CD4+ effector T cells, and apoptosis of CTLs in HCC [91]. Galectin-9 not only mediates T-cell senescence in HBV-associated HCC, significantly contributes to the inflammatory reactions and HCC immune escape [92], but notably also represents a potential biomarker of liver fibrosis and may emerge as a novel immunotherapeutic target for treating HCC and liver viral infections [92, 93].
Dendritic cells (DCs) and DC-based vaccines
Similar to NK cells, as another major player of innate immunity, DCs serve as professional antigen-presenting cells that are able to prime T-cells against tumor associated antigens (TAAs) involved in HCC progression. Recently, DCs have been an area of high interest as novel vaccines based on DCs have been developed and widely used in treating solid tumors including prostate cancer, melanoma, renal cancer and HCC [94]. DCs engineered with tumor-associated antigens (TAAs), which have been clarified by numerous in vitro and in vivo studies, are regarded as promising vaccines in HCC immunotherapy. In addition, autologous DCs pulsed ex vivo with the lysate of autologous tumor cells, HepG2 cells and telomerase peptides, have been evaluated in human clinical trials.
Recently, the Dendritic cell (DC)-derived exosomes (DEXs) and tumor cell–derived exosomes (TEXs), which elicit tumor regression in autochthonous HCC mouse models, form a new class of cell-free vaccines and extend options for HCC immunotherapeutic vaccines [95, 96] (Table 1.).
Representative immune inhibitory factors and modulators
The abundance of pro-inflammatory chemokines, cytokines and immunosuppressive molecules, which orchestrates a strongly immunosuppressive tumor milieu, play critical roles in reshaping TME, mediating intercellular crosstalk, and exerting immune evasion-promoting effects of HCC. Some of their specific functions have been mentioned while discussing immune cells of HCC, here, we summarize the representative players that current studies mainly highlight (Table 2.).
Current immunotherapeutic strategies for HCC
As an inflammation-associated cancer, HCC represents a promising target for immune based therapeutics. Clinically, the success of immune oncology in many types of cancer has encouraged implementation of immunotherapeutics in HCC. Recent studies have suggested that tumor antigen-specific immunotherapy and other approaches modulating immunogenicity have become attractive strategies for HCC treatment. Generally, these immunotherapeutic approaches for HCC could be mainly categorized into immune-checkpoint blockade (ICB), cell-based (mainly refers to DCs) /non-cell based vaccines, adoptive cell transfer (ACT), cytokine/antibody based immune regimens, and combination of immunotherapeutic agents with other drugs (Fig. 3.). Here, we collect some representative data on preclinical and clinical trials on immune based strategies of HCC, and discuss our current knowledge on their action mechanisms, rationale and application prospects for HCC treatment in the foreseeable future.

Current immunotherapeutic options for HCC. Immunotherapeutic approaches for HCC mainly include immune-checkpoint blockade (ICB), cell-based (mainly refers to DCs) /non-cell based vaccines, adoptive cell transfer (ACT), cytokine/antibody based immune regimens and oncolytic virus
Immune checkpoint inhibitors
Immune checkpoints are a specific sub-type of membrane-bound molecules that act as pivotal regulators of immune escape in cancers. The most studied immune checkpoints in HCC includes cytotoxic T lymphocyte protein 4 (CTLA-4), programmed cell death protein-1 and its ligand (PD-1, PD-L1), lymphocyte activation gene 3 protein (LAG-3) and mucin domain-containing molecule-3 (Tim-3).
Programmed cell death protein-1 and its ligand (PD-1, PD-L1)
PD-1, a regulator immunoglobulin expressed on activated CD4+, CD8+ T cells, B cells and NK cells, plays an important role in maintaining immune tolerance and repressing cytotoxicity of T lymphocytes [123]. Co-inhibitory signals in lymphocytes are mediated by binding of PD-1 to its ligands PD-L1 (B7-H1) and PD-L2 (B7-DC) [124]. In HCC, it’s clear that an increase in the number of both circulating and intratumoral PD-1+ CD8+ T cells predict high postoperative recurrences and poorer prognosis. It is also known that up-regulation of PD-L1 on HCC cells, which is induced by various cytokines, particularly IFN-γ, in turn contributes to impairing anti-tumor immunity and promotes CD8+ T cells apoptosis [110]. New in vitro and in vivo discoveries indicate that PD-1 overexpressed myeloid cells, such as DCs, suppress T cell responses in HCC. CD8+ T cells can be more potently activated to secrete IL-2 and IFN-γ via adoptive transfer of PD-1-deficient DCs [111].
Clinically, a representative phase 1/2 dose escalation and expansion trial on PD-1 immune checkpoint inhibitors Nivolumab (CheckMate 040 study) showed a promising role for immunotherapy in the treatment of advanced HCC, and relevant results were presented at the 2017 ASCO annual meeting [125]. In dose-escalation phase (enrolled number = 48), the objective response rate (ORR) was 15%, the disease control rate (DCR) was 58% and the median time to progression was 3.4 months. In dose-expansion phase (total number = 214; in 4 cohorts), generally, the ORR was reported as 20%, the DCR was 64%, the median time to progression was 4.1 months, and the 6-month and 9-month progression-free survival rates were 83 and 74% respectively. A subsequent CheckMate-040 based analysis compared the ORR and survival between intent-to-treat (ITT) overall population and Asian cohort. It suggested that Nivolumab showed similar mOS and manageable safety profile both in ITT population and Asian patients [126].
The efficacy of another anti-PD-1 monoclonal antibody, Pembrolizumab, was assessed in a phase II, open-label trial (KEYNOTE-224). In this study, Pembrolizumab was proven to be effective and well-tolerated in Sorafenib-experienced patients with advanced HCC, and PD-L1 expression level may act as a useful predictive bio-marker in selecting interested HCC patients. A total of 104 enrolled patients in this study represented 8-month median duration of response (mDOR), with median time to response of 2 months [127].
In addition, another phase 3 randomised clinical trial of nivolumab mono-therapy compared with sorafenib in the first-line setting is ongoing (NCT02576509). Moreover, combination therapies of anti-PD-L1 antibody (Duvalumab) with anti-CTL4–4 antibody (Tremelimumab) for unresectable HCC are under study as well (NCT02519348).
Other reported combination immunotherapy studies are encouraging and really open new avenues for HCC treatment [128,129,130], however, additional strategies are needed to uncover more sensitive predictive biomarkers besides PD-1/PD-L1 axis, optimize treatment selection, and improve HCC patients immune response. (More data of terminated or ongoing clinical trials are available in Table 3.).
Cytotoxic T lymphocyte protein 4 (CTLA-4)
Cytotoxic T lymphocyte protein 4 (CTLA-4), an inhibitory co-receptor that is expressed by activated T cells and is constitutively present on Tregs, has great affinity for competing with CD28 by binding to its ligands, CD80 and CD86, on antigen presenting cells (APCs). CTLA-4 plays a critical part in controlling CD4+ T cells function. In HCC and many other types of cancer, CTLA-4 suppresses the proliferation of T cells that have undergone TAA recognition and differentiation [131]. Additionally, inside HCC tissues, CTLA-4 further mediates immunosuppression by inducing Tregs activity and IDO and IL-10 productions in DCs [75].
Many clinical trials of antibodies targeting CTLA-4 are ongoing. A pilot clinical investigation testing anti-tumor and anti-viral effects of Tremelimumab in patients with HCC and HCV infection showed strong signs of anti-tumour efficacy (NCT01008358). The treatment presents a reliable safety profile, as no immune-related adverse events occurred. The median time to progression (TTP) was 6.48 months, and median overall survival (OS) was 8.2 months. The partial response rate was observed as 17.6%, and had a remarkable disease control rate of 76.4%. Moreover, 36% of the patients with AFP levels > 100 ng/ml showed more than 50% drop after Tremelimumab therapies [132]. In another phase 1 clinical study that tests the safety and effectiveness of Tremelimumab with radiofrequency (RFA) (NCT01853618), the median TTP and median OS were respectively 7.4 months (95% CI 4.7 to 19.4 months) and 12.3 months (95% CI 9.3 to 15.4 months). The 6-week HCC biopsies showed a clear increase in CD8+ T cells infiltration demonstrating that the combination of Tremelimumab with RFA in advanced HCC is feasible and results in stronger anti-tumor immunity [133].
Mucin domain-containing molecule-3 (Tim-3) and lymphocyte activation gene 3 protein (LAG-3)
Mucin domain-containing molecule-3 (Tim-3) is a transmembrane protein that is expressed on IFN-γ-secreting Th1 cells, NK cells and CTLs [113]. Tim-3 interacts with its soluble ligand galectin-9, and then negatively regulates T cell responses [91]. The expression of Tim-3 is increased in T cells infiltrating in chronic HBV infection [134], and the Tim-3/galectin-9 pathway consistently predicts poor prognosis in patients with HBV-associated HCC [76].
Lymphocyte activation gene 3 protein (LAG-3), a member of the immunoglobulin super-family proteins, which often binds MHC class II molecules with high affinity, represses the co-stimulatory functions of T cells [135]. Clinically, dual blockade of LAG-3 with anti-PD-1 therapy is being tested in a Phase I trial (NCT01968109) (Table 3.).
The immunosuppressive roles of both Tim-3 and LAG-3 in chronic viral hepatitis and HCC have been uncovered recently. However, their clinical values need to be further elucidated.
Adoptive cell transfer (ACT) based therapy in HCC
Besides the immune-checkpoint blockade (ICB), other effective immunotherapeutic options for HCC are urgently needed. In recent years, the exploration and development of cell-based immunotherapies in treating solid tumors have received considerable attentions. Adoptive cell transfer (ACT) offers robust and more durable anti-tumor immunity in cancer treatment. Recent translational research and clinical cases reported the success of engineered autologous HBV-specific T cell receptor (TCR) redirected therapeutics in treating HBV-associated HCC [136, 137], which broadens the immunotherapeutic approaches and might be used to treat a wider population of patients [138]. Based on the cell types, ACT used in HCC pre-clinical/clinical researches can be mainly classified as: (1) cytokine-induced killer (CIK) cells treatment, and (2) genetically modified NK cells or T cells (CAR-T).
CIK cells are a mixture of T lymphocytes, which are ex vivo expanded in the presence of cytokines (such as IL-1, IL-2, IFN-γ), comprising activated NKT cells, CD3−/CD56+ NK cells, and CD3+ /CD56− cytotoxic T cells [139]. CIK cells can be obtained in great numbers from peripheral blood mononuclear cells (PBMCs) and are very easily cultured. More importantly, it has been clarified that the absence of MHC restrictions favors CIK cells’ more potent anti-tumor efficacy compared with traditional CTLs [58, 139]. Previous studies suggested that CIK cells prevent HCC from progression, and effectively kill cancer stem cells (CSCs) mainly through NKG2d-ligands recognition [140]. A retrospective study demonstrated a significant correlation between high number of PD-1+ TILs and favorable outcome in CIK cells treated HCC group, which suggested that PD-1+ TILs may be utilized to predict the efficacy of CIK treatment in post-operative HCC patients [141]. A randomized, phase 3 clinical trial of the efficacy and safety of CIK cells treatment in 230 patients with HCC (NCT00699816) indicates that for post-curative treatment in HCC patients, adjuvant immunotherapy with CIK cells can prolong recurrence-free survival and OS (median RFS:44.0 months in treatment group, 30.0 months in the control group). Additionally, the proportion of patients with serious adverse events did not differ significantly between the treatment and control groups [142]. Several trials of CIK cells treatment in combination with other therapies, such as RFA, arterial chemoembolization and epitope-pulsed DCs, have been reported [143,144,145]. The evidence obtained from a growing body of literature confirms that CIK cells is a very promising adoptive immunotherapy that can be exploited for treatment and prevention of recurrence in HCC. However, a small fraction of patients undergoing standard therapies suffer from ‘immune fatigue’ status, and lack adequate leukocytes [139], a major obstacle for CIK cells treatment that needs to be overcome urgently. (Ongoing CIK-based clinical trials are available in Table 4.)
Another adoptive cell immunotherapy, which uses chimeric antigen receptor-modified T cells (CAR-T) or genetically modified NK cells, has been shown to be a promising strategy for the treatment of HCC. CAR-T can specifically recognize tumor-associated antigens and effectively eliminate tumor cells in a non-MHC restricted manner. Furthermore, additional genes could be used to enable resistance to immune suppression [146]. Successful use of CAR-T cells in treating haematological malignances includes ACT using CD19-specific CAR-T cells and the third-generation of CD20-specific CAR with CD28 and 4-1BB co-stimulatory domains. CAR-T cell treatment is well tolerated and can induce great remission in B-cell lymphoma [147, 148] (NCT00924326, NCT00621452), however, despite this, the achievements of CAR-T cells therapies against solid tumors are still modest. In HCC, recent studies revealed that glypican-3 (GPC-3), an oncofetal proteoglycan anchored to HCC cell membrane that functions to promote HCC progression and is associated with poor prognosis, provides a novel prognostic molecule therapeutic target in HCC immunotherapy [149]. Previous in vitro and orthotopic xenograft models of human HCC experiments both indicated that cytotoxic activities of T cells redirected to GPC-3 seemed to be positively correlated with GPC-3 expression levels in the targeting cell. This suggests that GPC-3-targeted CAR-T cells may offer a promising immune therapeutic intervention for GPC-3+ HCC [150, 151]. Moreover, a series of clinical studies conducted to test the safety and efficacy of CAR-T cells redirected to GPC-3 in HCC treatment are underway (Table 4.). Similarly, CAR-modified NKs represents a newly emerging immunotherapeutic modality for HCC therapy. Potent anti-tumor responses of GPC-3-specific NKs based therapeutics were observed in HCC xenografts with both high and low GPC-3 expression, which extends treatment options for patients with GPC-3+ HCC [152]. Another gene-modified NKs candidate, human interleukin-15 (hIL-15) gene-modified NKL cells (NKL-IL15), has been demonstrated to express high levels of cytolysis-related molecules (TNF-α, IFN-γ, NKp80 and so forth), which induces higher NKG2D ligand expression on target cells and results in increased susceptibility of HCC to NKs-mediated cytolysis [153].
Collectively, genetically modified NK cells and CAR-T cells based treatment provide new avenues for immunotherapies against HCC. Nevertheless, before being widely used as therapeutics in the clinic, their clinical efficacy and on-target-off tumor toxicity still require further assessments in more randomized trials.
Non-cell based vaccines and oncolytic viruses based immunotherapy in HCC
With the identification of a growing number of tumor-associated antigens (TAAs), and as a result, vaccines targeting HCC TAAs have been investigated and developed. A number of tumor antigens, such as human alpha-fetoprotein (AFP), GPC-3 and telomerase-reverse transcriptase (hTERT) have been identified as vaccine-based immunotherapeutic targets for HCC [154]. (Table 5.). Although increased efforts are being made to advance TAAs-based vaccines, the early clinical trials witnessed a mixed history of success and failure [155,156,157, 160]. The first HCC AFP-vaccine clinical trial was completed with only transient immunological responses detected, partially due to the limited number of antigens used or deficient CD4+ helper T cell support [157, 160, 161]. A vaccine with a single 16 amino acids sequence, hTERT-derived peptide (GV1001), and binds multiple HLA class II molecules, results in little clinical activity and no detected absolute antigen-specific CTLs responses [155]. On the other hand, partial clinical data on GPC-3 based vaccines demonstrated that the vaccine could induce measurable anti-tumor responses and are associated with prolonged OS of HCC patients [156, 162].
Apart from these classical TAAs, another attractive target is cancer-testis antigens (CTAs), which are considered to be novel targets for HCC immunotherapy due to the restricted expression patterns in a variety of tumors and normal tissues [163]. NY-ESO-1, also known as CTAG1, is one of the most immunogenic CTAs. A number of previous studies reported that NY-ESO-1 is highly expressed in many types of solid tumors, and a number of vaccine strategies targeting NY-ESO-1 are being developed [164,165,166]. In vitro investigations suggested that NY-ESO-1 expression is associated with poor tumor outcomes, and DCs loaded with NY-ESO-1 peptide can stimulate specific T cell responses against HCC cells [98, 167]. This implies that NY-ESO-1 has potential to be a valuable target molecule for immunotherapy against HCC. Clinically, vaccines targeting NY-ESO-1 expressing solid tumors (including HCC) are ongoing (NCT01522820).
Current studies demonstrate that DNA encoding epitope-optimized murine AFP and lentivector-mediated genetic immunization could induce potent AFP-specific CD8+ responses to generate a significant anti-tumor effect in autochthonous HCC model [168]. This may provide additional technology and new perspectives to further maximize the vaccines used in HCC therapy.
Oncolytic viruses (OVs) selectively replicate in tumor cells, damaging them, and subsequently spreading the virus in tumor tissue, while not harming normal cells. This characteristic endows OVs an effective platform for cancer immunotherapy [169]. Pre-clinical and clinical research highlights natural and genetically modified viruses, which are armed with immunomodulatory transgenes, that not only induce potent in situ anti-tumor immunity through mediating immunogenic cell death (ICD) and the inflammatory cascade, but also serve as vectors expressing therapeutic genes to improve cancer treatment [170, 171].
As for OVs used in HCC immunotherapy, previous in vitro and xenograft mice model studies indicated that a broad variety of therapeutic genes recombinant oncolytic adeno-associated viruses (AAVs) can exert a strong cytopathic effect on HCC cells (Fig. 4.). A tumor-selective replicating adenovirus expressing IFN-β, and ZD55-IFN-β, shows an elevated level of IFN-β expression, and 100-fold higher anti-tumor cytotoxicity than replicative adenovirus ONYX-015 [172]. The application of another recombinant AAVs model, AAV vectors containing human telomerase reverse transcriptase (hTERT) and tumor necrosis factor alpha related apoptosis inducing ligand (TRAIL) gene, namely AAV-hTERT-TRAIL, targets telomerase activity in HCC cells, and exhibits specific cytotoxicity and apoptosis to suppress the growth of HCC xenograft tumors [173]. An oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor (GM-CSF), Ad5-D24-GMCSF, induces tumor-specific and virus-specific immunity both in the syngeneic hamster model and patients. This suggests that oncolytic virus–mediated antitumor immunity may be a promising immunotherapeutic candidate for further clinical testing in HCC treatment [174].

Oncolytic viruses based immunotherapy in HCC. Oncolytic viruses (OVs) selectively replicate in and damage tumor cells, subsequently spread in tumor tissue
In early clinical studies, oncolytic poxvirus JX-594 (also known as PexaVec), showed robust virus replication-dependent oncolysis, well-tolerated character, anti-vascular effects and anti-tumor immunity in HCC and other solid tumors [158, 175, 176]. (NCT00629759) (Table 5.). Another randomized trial in advanced HCC demonstrated oncolytic and immunotherapy mechanisms of action (MOA), tumor responses and dose-related survival (high-dose JX-594 was associated with longer OS) in individuals with HCC [159] (NCT00554372).
In general, OVs is a potent therapeutic agent for cancer treatment, and it’s promising to extend immunotherapeutic options for HCC. Importantly, dosing regimens of OVs must be better defined for its clinical use, and in this regard, further results from clinical trials are awaited.
Conclusions
HCC is characterized by immune tolerance and comprises numerous infiltrated immune cells, a great number of suppressive molecules, complex pro-inflammatory/immunoregulatory signaling and intricate interactions between different components. The picture of immune microenvironment in HCC plays a key role in HCC progression and recurrence. Apparently, interactions of HCC tumor cells and various immune components in TME are really complicated and multifaceted, finally determining the plasticity and heterogeneity of its both innate and adaptive immune responses. Transcriptional and epigenetical alterations [177], metabolic reprogramming [178] and lack of co-stimulatory signals partially contribute to exhausted phenotype of TILs. Moreover, importantly, the benefit of current predictive biomarkers (e.g. PD-L1 expression level and tumor mutation burden (TMB)) in HCC patients receiving ICBs are still limited. Despite some impressive basic and translational discoveries, more details about underlying cellular or molecular mechanisms of immune evasion in HCC need to be further clarified. It’s clear that a better understanding of HCC immune landscape and will provide new breakthroughs in its clinical treatment.
Recently, immunotherapy brings great promises and new opportunities for HCC therapeutics. Its success has been evidenced by extensive studies. However, a subset of patients with HCC has little positive clinical responses to this treatment. In addition to the current combination regimens of ICBs with TKIs, or individualized cell therapeutic approaches, more effective ways to reinvigorate anti-tumor responses are urgently warranted. In this regard, combination of PD-1/PD-L1 monoclonal antibody and targeting co-stimulatory receptors (such as 4-1BB, OX40, CD27) with agonistic antibodies seems to be a potential therapeutic option for HCC, which may enhance and reverse functions of exhausted CD8+ TILs [179]. Additionally, strategies to target altered metabolic characteristics (e.g. the Warburg effect, abnormal glutamine metabolism, and urea cycle deficiency (UCD)) or interfere with the “key point molecules” (e.g. Arginase and indoleamine 2,3-dioxygenase (IDO)) that both influence metabolic reprogramming and T cell exhaustion may be a promising exploration clinically [180]. Moreover, some emerging pre-clinical investigations indicate the developing novel therapeutic approaches like epigenetic therapy using histone deacetylase inhibitors (HDACi) combined with CAR-T treatment are helpful for identification of more precise biomarkers and opening new avenues of HCC immunotherapy.
In general, immunotherapy is becoming one of the most promising approaches for HCC treatment, and it is likely to be more powerful in the foreseeable future.
Availability of data and materials
Not applicable.
Abbreviations
- ACT:
-
Adoptive cell transfer
- AFP:
-
A-fetoprotein
- APCs:
-
Antigen presenting cells
- CAF:
-
Cancer associated fibroblast
- CAR-T:
-
Chimeric antigen receptor-modified T cells
- CCL2:
-
Chemokine (C-C motif) ligand 2
- CIK:
-
Cytokine-induced killer
- CSCs:
-
Cancer stem cells
- CTAs:
-
Cancer-testis antigens
- CTLA-4:
-
Cytotoxic T lymphocyte protein 4
- CTLs:
-
Cytotoxic T lymphocytes
- CXCL17:
-
Chemokine (C-X-C motif) ligand 17
- CXCR4:
-
Chemokine (C-X-C motif) receptor 4
- DCs:
-
Dendritic cells
- DEXs:
-
Dendritic cell-derived exosomes
- GPC-3:
-
Glypican-3
- HCC:
-
Hepatocellular carcinoma
- HGF:
-
Hepatocyte growth factor
- HSCs:
-
Hepatic stellate cells
- hTERT:
-
human telomerase-reverse transcriptase
- ICB:
-
Immune-checkpoint blockade
- IDO:
-
Indoleamine-2,3-dioxygenase
- IFN-γ:
-
Interferon-γ
- IL-:
-
Interleukin-
- KC:
-
Kupffer cell
- LAG-3:
-
Lymphocyte activation gene 3
- MAGE-1:
-
Melanoma-associated antigen 1
- MCP-1:
-
Monocyte chemotactic protein 1
- MDSCs:
-
Myeloid- derived suppressor cells
- MTD:
-
Maximum-tolerated dose
- NK cell:
-
Natural killer cell
- OS:
-
Overall survival
- OVs:
-
Oncolytic viruses
- P:
-
Primary endpoint
- PD-1:
-
Programmed cell death protein-1
- PDGF:
-
Platelet-derived growth factor
- PD-L1:
-
Programmed cell death protein ligand-1
- PR:
-
Partial response
- RECIST:
-
Response evaluation criteria in solid tumors
- RR:
-
Response rate
- S:
-
Secondary endpoint
- SD:
-
Stable disease
- SDF-1α:
-
Stromal cell derived factor 1α
- TAA:
-
Tumor-associated antigens
- TAMs:
-
Tumor-associated macrophages
- TANs:
-
Tumor associated neutrophils
- TEXs:
-
Tumor cell–derived exosomes
- TGF-β:
-
Transforming growth factor-β
- Tim-3:
-
Mucin domain-containing molecule-3
- TME:
-
Tumor microenvironment
- Tregs:
-
Regulatory T cells
- TTP:
-
Time to progression
- TTSP:
-
Time to symptomatic progression
- VEGF:
-
Vascular endothelial growth factor
References
Thimme R, et al. Comprehensive analysis of the alpha-fetoprotein-specific CD8+ T cell responses in patients with hepatocellular carcinoma. Hepatology. 2008;48(6):1821–33.
McGlynn KA, Petrick JL, London WT. Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis. 2015;19(2):223–38.
Rimassa L, et al. Management of adverse events associated with tyrosine kinase inhibitors: improving outcomes for patients with hepatocellular carcinoma. Cancer Treat Rev. 2019;77:20–8.
Ardelt MA, et al. Inhibition of cyclin-dependent kinase 5: a strategy to improve Sorafenib response in hepatocellular carcinoma therapy. Hepatology. 2019;69(1):376–93.
Bollard J, et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut. 2017;66(7):1286–96.
Ehrlich SM, et al. Targeting cyclin dependent kinase 5 in hepatocellular carcinoma--a novel therapeutic approach. J Hepatol. 2015;63(1):102–13.
Joshi JJ, et al. H3B-6527 is a potent and selective inhibitor of FGFR4 in FGF19-driven hepatocellular carcinoma. Cancer Res. 2017;77(24):6999–7013.
Lang L,Teng Y. Fibroblast growth factor receptor 4 targeting in cancer: new insights into mechanisms and therapeutic strategies. Cells. 2019;8(1).Published online 2019 Jan 9. https://doi.org/10.3390/cells8010031.
Shlomai A, de Jong YP, Rice CM. Virus associated malignancies: the role of viral hepatitis in hepatocellular carcinoma. Semin Cancer Biol. 2014;26:78–88.
Hato T, et al. Immune checkpoint blockade in hepatocellular carcinoma: current progress and future directions. Hepatology. 2014;60(5):1776–82.
Zhang HH, et al. Regulatory T cells in chronic hepatitis B patients affect the immunopathogenesis of hepatocellular carcinoma by suppressing the anti-tumour immune responses. J Viral Hepat. 2010;17(Suppl 1):34–43.
Chew V, et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc Natl Acad Sci U S A. 2017;114(29):E5900–9.
Wu Y, et al. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology. 2013;57(3):1107–16.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74.
Kapanadze T, et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J Hepatol. 2013;59(5):1007–13.
Mok MT, et al. CCRK is a novel signalling hub exploitable in cancer immunotherapy. Pharmacol Ther. 2018;186:138–51.
Zhou J, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut. 2018;67(5):931-44. https://doi.org/10.1136/gutjnl-2017-314032.
Deng Y, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene. 2017;36(8):1090–101.
Chiu DK, et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology. 2016;64(3):797–813.
Chiu DK, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8(1):517.
Nan J, et al. Endoplasmic reticulum stress induced LOX-1(+) CD15(+) polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunology. 2018;154(1):144-55. https://doi.org/10.1111/imm.12876.
Kondo Y, Shimosegawa T. Significant roles of regulatory T cells and myeloid derived suppressor cells in hepatitis B virus persistent infection and hepatitis B virus-related HCCs. Int J Mol Sci. 2015;16(2):3307–22.
Liu M, et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut. 2019. https://doi.org/10.1136/gutjnl-2018-317257.
Hoechst B, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135(1):234–43.
Srivastava MK, et al. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70(1):68–77.
Zhang H, et al. Critical role of myeloid-derived suppressor cells in tumor-induced liver immune suppression through inhibition of NKT cell function. Front Immunol. 2017;8:129.
Dardalhon V, et al. Tim-3/galectin-9 pathway: regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J Immunol. 2010;185(3):1383–92.
Hoechst B, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799–807.
Wu CJ, et al. Combination of radiation and interleukin 12 eradicates large orthotopic hepatocellular carcinoma through immunomodulation of tumor microenvironment. Oncoimmunology. 2018;7(9):e1477459.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51.
Movahedi K, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70(14):5728–39.
Cai H, et al. Colony-stimulating factor-1-induced AIF1 expression in tumor-associated macrophages enhances the progression of hepatocellular carcinoma. Oncoimmunology. 2017;6(9):e1333213.
Huang W, et al. Interleukin-8 Induces Expression of FOXC1 to Promote Transactivation of CXCR1 and CCL2 in Hepatocellular Carcinoma Cell Lines and Formation of Metastases in Mice. Gastroenterology. 2015;149(4):1053–67 e14.
Wang TT, et al. CTGF secreted by mesenchymal-like hepatocellular carcinoma cells plays a role in the polarization of macrophages in hepatocellular carcinoma progression. Biomed Pharmacother. 2017;95:111–9.
Capece D, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int. 2013;2013:187204.
Zhu Y, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68(9):1653-66. https://doi.org/10.1136/gutjnl-2019-318419.
Zhang J, et al. HIF-1alpha/IL-1beta signaling enhances hepatoma epithelial-mesenchymal transition via macrophages in a hypoxic-inflammatory microenvironment. Hepatology. 2018;67(5):1872-89. https://doi.org/10.1002/hep.29681.
Zhou J, et al. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int J Cancer. 2009;125(7):1640–8.
Sharma S, et al. CD4+CD25+CD127(low) regulatory T cells play predominant anti-tumor suppressive role in hepatitis B virus-associated hepatocellular carcinoma. Front Immunol. 2015;6:49.
Kuang DM, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology. 2010;51(1):154–64.
Rani B, et al. Role of the tissue microenvironment as a therapeutic target in hepatocellular carcinoma. World J Gastroenterol. 2014;20(15):4128–40.
Lanaya H, et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat Cell Biol. 2014;16(10):972–7.
Yan W, et al. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut. 2015;64(10):1593–604.
Wan S, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147(6):1393–404.
Arlauckas SP, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med. 2017;9(389):eaal3604. https://doi.org/10.1126/scitranslmed.aal3604.
Nicolas-Avila JA, Adrover JM, Hidalgo A. Neutrophils in homeostasis, immunity, and Cancer. Immunity. 2017;46(1):15–28.
Ameratunga M, et al. Neutrophil-lymphocyte ratio kinetics in patients with advanced solid tumours on phase I trials of PD-1/PD-L1 inhibitors. Eur J Cancer. 2018;89:56–63.
Jeyakumar G, et al. Neutrophil lymphocyte ratio and duration of prior anti-angiogenic therapy as biomarkers in metastatic RCC receiving immune checkpoint inhibitor therapy. J Immunother Cancer. 2017;5(1):82.
Andzinski L, et al. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer. 2016;138(8):1982–93.
Michaeli J, et al. Tumor-associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFalpha and NO-dependent mechanism, promoting a tumor-supportive environment. Oncoimmunology. 2017;6(11):e1356965.
Margetts J, et al. Neutrophils: driving progression and poor prognosis in hepatocellular carcinoma? Br J Cancer. 2018;118(2):248–57.
Mano Y, et al. Preoperative neutrophil-to-lymphocyte ratio is a predictor of survival after hepatectomy for hepatocellular carcinoma: a retrospective analysis. Ann Surg. 2013;258(2):301–5.
Koh MY, et al. A new HIF-1alpha/RANTES-driven pathway to hepatocellular carcinoma mediated by germline haploinsufficiency of SART1/HAF in mice. Hepatology. 2016;63(5):1576–91.
Zhou SL, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to Sorafenib. Gastroenterology. 2016;150(7):1646–1658.e17.
Zhou SL, et al. A positive feedback loop between Cancer stem-like cells and tumor-associated neutrophils controls hepatocellular carcinoma progression. Hepatology. 2019. https://doi.org/10.1002/hep.30630.
Unitt E, et al. Tumour lymphocytic infiltrate and recurrence of hepatocellular carcinoma following liver transplantation. J Hepatol. 2006;45(2):246–53.
Gao Q, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007;25(18):2586–93.
Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12(12):681–700.
Chen KJ, et al. Selective recruitment of regulatory T cell through CCR6-CCL20 in hepatocellular carcinoma fosters tumor progression and predicts poor prognosis. PLoS One. 2011;6(9):e24671.
Jiang R, et al. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun. 2017;8:15129.
Zaiss DM, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity. 2013;38(2):275–84.
Kalathil S, et al. Higher frequencies of GARP(+)CTLA-4(+)Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res. 2013;73(8):2435–44.
Yuan CH, et al. Amphiregulin activates regulatory T lymphocytes and suppresses CD8+ T cell-mediated anti-tumor response in hepatocellular carcinoma cells. Oncotarget. 2015;6(31):32138–53.
Fu J, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132(7):2328–39.
Huang Y, et al. Tumor-infiltrating FoxP3+ Tregs and CD8+ T cells affect the prognosis of hepatocellular carcinoma patients. Digestion. 2012;86(4):329–37.
Fu YP, et al. Overexpression of interleukin-35 associates with hepatocellular carcinoma aggressiveness and recurrence after curative resection. Br J Cancer. 2016;114(7):767–76.
Ye LY, et al. Hypoxia-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res. 2016;76(4):818–30.
Kuang DM, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206(6):1327–37.
Guo CL, et al. Expression of Fas/FasL in CD8+ T and CD3+ Foxp3+ Treg cells--relationship with apoptosis of circulating CD8+ T cells in hepatocellular carcinoma patients. Asian Pac J Cancer Prev. 2014;15(6):2613–8.
Li L, et al. CXCL17 expression predicts poor prognosis and correlates with adverse immune infiltration in hepatocellular carcinoma. PLoS One. 2014;9(10):e110064.
Motz GT, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20(6):607–15.
Moreno-Cubero E, Larrubia JR. Specific CD8(+) T cell response immunotherapy for hepatocellular carcinoma and viral hepatitis. World J Gastroenterol. 2016;22(28):6469–83.
Flecken T, et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology. 2014;59(4):1415–26.
Guidotti LG, et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell. 2015;161(3):486–500.
Han Y, et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology. 2014;59(2):567–79.
Li H, et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology. 2012;56(4):1342–51.
Woo SR, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–27.
Kamimura H, et al. Reduced NKG2D ligand expression in hepatocellular carcinoma correlates with early recurrence. J Hepatol. 2012;56(2):381–8.
Hasmim M, et al. Critical role of tumor microenvironment in shaping NK cell functions: implication of hypoxic stress. Front Immunol. 2015;6:482.
Yamamoto M, et al. alpha-Fetoprotein impairs activation of natural killer cells by inhibiting the function of dendritic cells. Clin Exp Immunol. 2011;165(2):211–9.
Zhang C, et al. Interleukin-12 improves cytotoxicity of natural killer cells via upregulated expression of NKG2D. Hum Immunol. 2008;69(8):490–500.
Vujanovic L, et al. Tumor-derived alpha-fetoprotein directly drives human natural killer-cell activation and subsequent cell death. Cancer Immunol Res. 2017;5(6):493–502.
Sun C, et al. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol. 2015;12(3):292–302.
Langhans B, et al. Regulatory CD4+ T cells modulate the interaction between NK cells and hepatic stellate cells by acting on either cell type. J Hepatol. 2015;62(2):398–404.
Li T, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012;318(2):154–61.
Muhanna N, et al. Amelioration of hepatic fibrosis by NK cell activation. Gut. 2011;60(1):90–8.
Zhang M, et al. Apoptotic cells attenuate fulminant hepatitis by priming Kupffer cells to produce interleukin-10 through membrane-bound TGF-beta. Hepatology. 2011;53(1):306–16.
Lacotte S, et al. Impact of myeloid-derived suppressor cell on Kupffer cells from mouse livers with hepatocellular carcinoma. Oncoimmunology. 2016;5(11):e1234565.
Wu J, et al. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012;72(16):3977–86.
Sun K, et al. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-kappaB-IL1alpha/beta-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett. 2017;388:198–207.
Mengshol JA, et al. A crucial role for Kupffer cell-derived galectin-9 in regulation of T cell immunity in hepatitis C infection. PLoS One. 2010;5(3):e9504.
Kim HY, Park JW. Current immunotherapeutic strategies in hepatocellular carcinoma: recent advances and future directions. Ther Adv Gastroenterol. 2017;10(10):805–14.
Fujita K, et al. Correlation between serum galectin-9 levels and liver fibrosis. J Gastroenterol Hepatol. 2018;33(2):492–9.
Radford KJ, Tullett KM, Lahoud MH. Dendritic cells and cancer immunotherapy. Curr Opin Immunol. 2014;27:26–32.
Rao Q, et al. Tumor-derived exosomes elicit tumor suppression in murine hepatocellular carcinoma models and humans in vitro. Hepatology. 2016;64(2):456–72.
Lu Z, et al. Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. J Hepatol. 2017;67(4):739–48.
Cao DY, et al. alpha-fetoprotein and interleukin-18 gene-modified dendritic cells effectively stimulate specific type-1 CD4- and CD8-mediated T-Cell response from hepatocellular carcinoma patients in Vitro. Hum Immunol. 2007;68(5):334–41.
Chen Y, et al. Potential therapeutic value of dendritic cells loaded with NYESO1 protein for the immunotherapy of advanced hepatocellular carcinoma. Int J Mol Med. 2013;32(6):1366–72.
Vogt A, et al. Improving immunotherapy of hepatocellular carcinoma (HCC) using dendritic cells (DC) engineered to express IL-12 in vivo. Liver Int. 2014;34(3):447–61.
Gonzalez-Carmona MA, et al. CD40ligand-expressing dendritic cells induce regression of hepatocellular carcinoma by activating innate and acquired immunity in vivo. Hepatology. 2008;48(1):157–68.
Tada F, et al. Phase I/II study of immunotherapy using tumor antigen-pulsed dendritic cells in patients with hepatocellular carcinoma. Int J Oncol. 2012;41(5):1601–9.
Lee JH, et al. A phase I/IIa study of adjuvant immunotherapy with tumour antigen-pulsed dendritic cells in patients with hepatocellular carcinoma. Br J Cancer. 2015;113(12):1666–76.
Palmer DH, et al. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology. 2009;49(1):124–32.
El AM, et al. Immunotherapy by autologous dendritic cell vaccine in patients with advanced HCC. J Cancer Res Clin Oncol. 2013;139(1):39–48.
Butterfield LH, et al. A phase I/II trial testing immunization of hepatocellular carcinoma patients with dendritic cells pulsed with four alpha-fetoprotein peptides. Clin Cancer Res. 2006;12(9):2817–25.
Langhans B, et al. Intrahepatic IL-8 producing Foxp3(+)CD4(+) regulatory T cells and fibrogenesis in chronic hepatitis C. J Hepatol. 2013;59(2):229–35.
Ouyang FZ, et al. Dendritic cell-elicited B-cell activation fosters immune privilege via IL-10 signals in hepatocellular carcinoma. Nat Commun. 2016;7:13453.
Ambade A, et al. Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1alphainduced IL-10. Oncoimmunology. 2016;5(10):e1221557.
Cheng JT, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. 2016;5:e198.
Shi F, et al. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer. 2011;128(4):887–96.
Lim TS, et al. PD-1 expression on dendritic cells suppresses CD8(+) T cell function and antitumor immunity. Oncoimmunology. 2016;5(3):e1085146.
Zhou G, et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology. 2017;153(4):1107–1119.e10.
Rong YH, et al. Tim-3 expression on peripheral monocytes and CD3+CD16/CD56+natural killer-like T cells in patients with chronic hepatitis B. Tissue Antigens. 2014;83(2):76–81.
Pan K, et al. Expression and prognosis role of indoleamine 2,3-dioxygenase in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134(11):1247–53.
Chen Y, et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014;59(4):1435–47.
Sung YC, et al. Combined delivery of sorafenib and a MEK inhibitor using CXCR4-targeted nanoparticles reduces hepatic fibrosis and prevents tumor development. Theranostics. 2018;8(4):894–905.
Li X, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66(1):157–67.
Baeck C, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61(3):416–26.
Ren L, et al. Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget. 2016;7(46):75763–73.
Facciabene A, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T (reg) cells. Nature. 2011;475(7355):226–30.
Li YM, et al. RIP3 deficiency recruits myeloid-derived suppressor cells to hepatocellular carcinoma through the CXCL1-CXCR2 axis. 2019. https://doi.org/10.1002/hep.30676.
Liu LZ, et al. CCL15 recruits suppressive monocytes to facilitate immune escape and disease progression in hepatocellular carcinoma. Hepatology. 2019;69(1):143–59.
Wei SC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell. 2017;170(6):1120–33 e17.
Latchman Y, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2(3):261–8.
El-Khoueiry AB, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–502.
Yau T, et al. Nivolumab in advanced hepatocellular carcinoma: Sorafenib-experienced Asian cohort analysis. 2019;71(3):543-52. https://doi.org/10.1016/j.jhep.2019.05.014.
Zhu AX, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19(7):940–52.
Chowdhury PS, Chamoto K, Honjo T. Combination therapy strategies for improving PD-1 blockade efficacy: a new era in cancer immunotherapy. J Intern Med. 2018;283(2):110–20.
Mocan T, et al. Programmed cell death protein-1 (PD-1)/programmed death-ligand-1 (PD-L1) axis in hepatocellular carcinoma: prognostic and therapeutic perspectives. Clin Transl Oncol. 2019;21(6):702–12.
Harding JJ. Immune checkpoint blockade in advanced hepatocellular carcinoma: an update and critical review of ongoing clinical trials. Future Oncol. 2018;14(22):2293–302.
Kudo M. Immune checkpoint inhibition in hepatocellular carcinoma: basics and ongoing clinical trials. Oncology. 2017;92(Suppl 1):50–62.
Sangro B, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81–8.
Duffy AG, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol. 2017;66(3):545–51.
Nebbia G, et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS One. 2012;7(10):e47648.
Sharma P, et al. Primary, adaptive, and acquired resistance to Cancer immunotherapy. Cell. 2017;168(4):707–23.
Tan AT, et al. Use of expression profiles of HBV-DNA integrated into genomes of hepatocellular carcinoma cells to select T cells for immunotherapy. Gastroenterology. 2019;156(6):1862–1876.e9.
Qasim W, et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J Hepatol. 2015;62(2):486–91.
Zhang R, et al. Adoptive cell transfer therapy for hepatocellular carcinoma. Front Med. 2019;13(1):3–11.
Schmidt TL, Negrin RS, Contag CH. A killer choice for cancer immunotherapy. Immunol Res. 2014;58(2–3):300–6.
Rong XX, et al. Recognition and killing of cancer stem-like cell population in hepatocellular carcinoma cells by cytokine-induced killer cells via NKG2d-ligands recognition. Oncoimmunology. 2016;5(3):e1086060.
Chang B, et al. High number of PD-1 positive intratumoural lymphocytes predicts survival benefit of cytokine-induced killer cells for hepatocellular carcinoma patients. Liver Int. 2018;38(8):1449–58.
Lee JH, et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology. 2015;148(7):1383–91.e6.
Huang ZM, et al. Cytokine-induced killer cells in combination with transcatheter arterial chemoembolization and radiofrequency ablation for hepatocellular carcinoma patients. J Immunother. 2013;36(5):287–93.
Qiu Y, et al. Hepatocellular carcinoma-specific immunotherapy with synthesized alpha1,3- galactosyl epitope-pulsed dendritic cells and cytokine-induced killer cells. World J Gastroenterol. 2011;17(48):5260–6.
Pan K, et al. The efficacy of cytokine-induced killer cell infusion as an adjuvant therapy for postoperative hepatocellular carcinoma patients. Ann Surg Oncol. 2013;20(13):4305–11.
Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. 2013;13(8):525–41.
Kochenderfer JN, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–20.
Till BG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119(17):3940–50.
Szoor A, et al. T cell-activating mesenchymal stem cells as a biotherapeutic for HCC. Mol Ther Oncolytics. 2017;6:69–79.
Gao H, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. 2014;20(24):6418–28.
Jiang Z, et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front Immunol. 2016;7:690.
Yu M, et al. Development of GPC3-specific chimeric antigen receptor-engineered natural killer cells for the treatment of hepatocellular carcinoma. Mol Ther. 2018;26(2):366–78.
Jiang W, et al. hIL-15 gene-modified human natural killer cells (NKL-IL15) augments the anti-human hepatocellular carcinoma effect in vivo. Immunobiology. 2014;219(7):547–53.
Greten TF, Manns MP, Korangy F. Immunotherapy of HCC. Rev Recent Clin Trials. 2008;3(1):31–9.
Greten TF, et al. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer. 2010;10:209.
Sawada Y, et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res. 2012;18(13):3686–96.
Butterfield LH, et al. T-cell responses to HLA-A*0201 immunodominant peptides derived from alpha-fetoprotein in patients with hepatocellular cancer. Clin Cancer Res. 2003;9(16 Pt 1):5902–8.
Park BH, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9(6):533–42.
Heo J, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19(3):329–36.
Makarova-Rusher OV, et al. The yin and yang of evasion and immune activation in HCC. J Hepatol. 2015;62(6):1420–9.
Alisa A, et al. Analysis of CD4+ T-cell responses to a novel alpha-fetoprotein-derived epitope in hepatocellular carcinoma patients. Clin Cancer Res. 2005;11(18):6686–94.
Tada Y, et al. Analysis of cytotoxic T lymphocytes from a patient with hepatocellular carcinoma who showed a clinical response to vaccination with a glypican3derived peptide. Int J Oncol. 2013;43(4):1019–26.
Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009;100(11):2014–21.
Jager E, et al. Recombinant vaccinia/fowlpox NY-ESO-1 vaccines induce both humoral and cellular NY-ESO-1-specific immune responses in cancer patients. Proc Natl Acad Sci U S A. 2006;103(39):14453–8.
Chen JL, et al. NY-ESO-1 specific antibody and cellular responses in melanoma patients primed with NY-ESO-1 protein in ISCOMATRIX and boosted with recombinant NY-ESO-1 fowlpox virus. Int J Cancer. 2015;136(6):E590–601.
Kim SH, et al. Expression of cancer-testis antigens MAGE-A3/6 and NY-ESO-1 in non-small-cell lung carcinomas and their relationship with immune cell infiltration. Lung. 2009;187(6):401–11.
Nakamura S, et al. Expression and immunogenicity of NY-ESO-1 in hepatocellular carcinoma. J Gastroenterol Hepatol. 2006;21(8):1281–5.
Hong Y, et al. Epitope-optimized alpha-fetoprotein genetic vaccines prevent carcinogen-induced murine autochthonous hepatocellular carcinoma. Hepatology. 2014;59(4):1448–58.
Liu TC, Kirn D. Systemic efficacy with oncolytic virus therapeutics: clinical proof-of-concept and future directions. Cancer Res. 2007;67(2):429–32.
Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2(4):295–300.
Jiang H, et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous Cancer vaccination. Cancer Res. 2017;77(14):3894–907.
He LF, et al. Significant antitumor activity of oncolytic adenovirus expressing human interferon-beta for hepatocellular carcinoma. J Gene Med. 2008;10(9):983–92.
Wang Y, et al. Potent antitumor effect of TRAIL mediated by a novel adeno-associated viral vector targeting to telomerase activity for human hepatocellular carcinoma. J Gene Med. 2008;10(5):518–26.
Cerullo V, et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res. 2010;70(11):4297–309.
Heo J, et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol Ther. 2011;19(6):1170–9.
Hwang TH, et al. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol Ther. 2011;19(10):1913–22.
Khan O, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571(7764):211–8.
Scharping NE, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive Intratumoral T cell metabolic insufficiency and dysfunction. Immunity. 2016;45(2):374–88.
Kim HD, et al. 4-1BB delineates distinct activation status of exhausted tumor-infiltrating CD8(+) T cells in hepatocellular carcinoma. Hepatology. 2019. https://doi.org/10.1002/hep.30881.
Lee JS, et al. Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell. 2018;174(6):1559–70 e22.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 81172470, 81070362, 81372629 & 81772627), and two key projects from the Nature Science Foundation of Hunan Province (No. 2015JC3021 & No. 2016JC2037).
Author information
Authors and Affiliations
Contributions
YF, SL, SZ and HS designed and drafted the manuscript; YF, SZ and HS wrote figure legends and revised the article; SL drew the figures. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fu, Y., Liu, S., Zeng, S. et al. From bench to bed: the tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J Exp Clin Cancer Res 38, 396 (2019). https://doi.org/10.1186/s13046-019-1396-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-019-1396-4