
Abstract
As integral components of the immune microenvironment, tissue resident macrophages (TRMs) represent a self-renewing and long-lived cell population that plays crucial roles in maintaining homeostasis, promoting tissue remodeling after damage, defending against inflammation and even orchestrating cancer progression. However, the exact functions and roles of TRMs in cancer are not yet well understood. TRMs exhibit either pro-tumorigenic or anti-tumorigenic effects by engaging in phagocytosis and secreting diverse cytokines, chemokines, and growth factors to modulate the adaptive immune system. The life-span, turnover kinetics and monocyte replenishment of TRMs vary among different organs, adding to the complexity and controversial findings in TRMs studies. Considering the complexity of tissue associated macrophage origin, macrophages targeting strategy of each ontogeny should be carefully evaluated. Consequently, acquiring a comprehensive understanding of TRMs' origin, function, homeostasis, characteristics, and their roles in cancer for each specific organ holds significant research value. In this review, we aim to provide an outline of homeostasis and characteristics of resident macrophages in the lung, liver, brain, skin and intestinal, as well as their roles in modulating primary and metastatic cancer, which may inform and serve the future design of targeted therapies.
Similar content being viewed by others

Introduction
Macrophages played pivotal roles in maintaining homeostasis through sensing various signals, phagocytosis and orchestrating subsequent immune response, accounting for only 1–5% in organs under steady state and the number can be as high as to 40% in cancer tissues [1,2,3]. The traditional view holds that tumor associated macrophages (TAMs) mainly arise from the differentiation of hematopoietic precursors [4, 5]. In the past decade, TAMs are considered to be derived from different origins with distinct phenotypes and divergent functionality, that embryonic-derived macrophages also contribute to the generation of TAMs in the pancreas, brain, lung and breast [5,6,7,8]. It is now accepted that TAMs can be either originated from long lived yolk-sac or fetal liver progenitors during organogenesis or recruited from bone marrow myeloid progenitors, namely tissue resident macrophages (TRMs) and bone marrow derived macrophages (BMDMs) respectively [1, 9,10,11]. Under steady-state conditions, resident macrophages, including brain microglia, liver Kupffer cells and epidermal Langerhans cells are locally self-maintained, independently of BMDMs across most organs in adult mice [12]. While TRMs of peritoneal and pancreatic stroma are both self-renewed and slightly replaced by BMDMs [1]. Intestinal resident macrophages are quite different in that they are constantly replaced by BMDMs [13].
Despite from their ontogeny differences, tissue resident macrophages varied from BMDMs in several aspects. Tissue resident macrophages are located at specific sub-tissular niches in each organ [1]. Alveolar macrophages are conjunct to alveolar epithelial cells, receiving GM-CSF from epithelial cells and clearing surface surfactants [14, 15]. Lymphatic vessel endothelial hyaluronan receptor (LYVE+) vascular associated macrophages and MHCII+ nerve associated macrophages has been identified due to their specified microanatomical niche across different organs [16]. Besides, unique tissue resident macrophages localization endowed with tissue specific functions that may not be easily replaced by bone marrow derived macrophages. For example, microglia are capable of mechanically prune neurons and remodel synaptic connectivity [17, 18]. Kupffer cells are equipped with machinery for uptake and recycling erythrocytes while recruited monocyte do not [19]. Osteoclasts are required for bone morphogenesis and hematopoietic niche maintenance [20]. Moreover, tissue resident macrophages are early witness and encounters during oncogenesis and micro metastasis formation, sensing cancer-related soluble products, while BMDMs can be recruited at relatively later stage of cancer progression [1, 3]. In a single-cell sequencing study, macrophages accumulated in cancer tissues demonstrate enhanced monocyte-like characteristics, apart from liver tumors, where the increased macrophage populations correspond to monocyte-derived macrophages, highlighting a cancer-type specific recruitment manner [21]. The time sequence of macrophages in tumor microenvironment from varied ontogeny may modulate tumor progression in different ways.
In this review, we outlined our current understanding for the roles of tissue resident macrophages of lung, liver, brain, skin and intestine in cancer. These five organs were selected for the extensive and in-depth cancer related research on particular tissue resident macrophages. Besides, the prevalence and mortality rate of malignancies in these five organs are among the top of public health concern worldwide.
Pulmonary tissue resident macrophages
Homeostasis of tissue resident macrophages in lung
Pulmonary macrophages play a crucial role in defending against pathogens and maintaining cellular debris clearance, ensuring lung homeostasis [22, 23]. The lung harbors three populations of tissue resident macrophages, namely alveolar macrophages (AMs), and interstitial macrophages (IMs) including vascular-associated interstitial macrophages, and nerve-associated interstitial macrophages (NAMs) [22, 24]. Among these, AMs represent the predominant population, accounting for 90–95% of pulmonary resident immune cells, and are mainly distributed in the lung's diffusing area [14, 25, 26]. Vascular-associated interstitial macrophages reside preferentially alongside blood vessels, while NAMs are located in proximity to nerves within the bronchial tree [22, 24]. Recent studies have indicated that both mouse and human AMs originate from embryonic precursors and occupy the alveolar niche before birth. Throughout adulthood, they undergo minimal proliferation in the absence of inflammation, relying on self-renewal mechanisms, but can be replenished by BM-derived cells under stressful conditions [11, 27,28,29,30,31]. On the other hand, IMs are believed to originate from the yolk sac and are subsequently replaced by fetal liver monocytes [32]. Depending on the specific subtype, IMs can either self-replenish without input from circulating monocytes or gradually undergo replacement by monocytes at varying rates and extents [33, 34]. Notably, a study conducted by Ural et al. during pregnancy demonstrated that NAMs are derived from the yolk sac during pregnancy and persist at least several months after birth [22]. Interestingly, NAMs in other organs, such as the skin and gut, exhibit self-renewing capacities and share common features with microglia [13, 35, 36].
Characteristics of pulmonary resident macrophages
Alveolar macrophages (AMs) rely on the activation of GM-CSF and TGF-βR signaling pathways, which in turn activate the transcription factor PPAR-γ, to facilitate their differentiation and survival [37]. Deficiencies in GM-CSF, either due to autoantibodies or gene deletions, can lead to the excessive accumulation of surfactant lipids and proteins produced by type 2 alveolar epithelial cells in the alveolar space, resulting in a condition known as pulmonary alveolar proteinosis [38,39,40]. Additionally, conditional deletions of TGF-βR at various time points in murine models have been found to halt the development and differentiation of AMs [41]. In contrast, the development and survival of IMs rely on the CSF1R signaling axis [22] (Fig. 1).

The identity of TRMs is determined by their specific niche of residence. TRMs in different tissues originate from the embryo and rely on distinct niche signals and transcription factors to facilitate their differentiation, survival and self-maintenance. Additionally, TRMs in the lung, liver, brain, skin, and intestine secrete various cytokines and exhibit distinct markers. These factors collectively define the specific identity of TRMs
Different types of pulmonary tissue-specific macrophages exhibit distinct phenotypic and transcriptional features. AMs reside on the luminal side of the alveolar niche [32]. In humans, AMs are often characterized by the co-expression of HLA-DR, CD43, CD169, CD206, CD11b, CD141, CD64, and low levels of CD14, while in mice, they are identified by CD11C, CD64, F4/80, CD45, CD36, CD206, MERTK, and SIGLECF [32, 42]. The pan-cancer analysis of single myeloid cells across 15 human cancer types unveiled significant findings, indicating that AMs express genes such as FABP4, MARCO, MRC1( also known as CD206), MSR1 and PPAR-γ, which are involved in self-replenishment through the secretion of (TGF-β [43,44,45]. In addition, M2 AMs secrete anti-inflammatory mediators, such as IL-10, PGE2, and TGF-β, while M1 AMs produce inflammatory mediators, such as INOS, IL-1β, IL-6, TNF-α, and MIP1α [46]. Vascular-associated interstitial macrophages are recognized as CD206+LYVE-1hiMHCIIloCX3CR1− cells, expressing an immunoregulatory signature, including TGF-β2, PLAUR and FCNA, while nerve-associated macrophages are characterized as CD206−LYVE-1loMHCIIhiCX3CR1+ cells, closely associated with nerve bundles or endings and highly express pro-inflammatory molecules such as IL-1β and CXCL12 with antigen presenting function. Nerve-associated macrophages secrete immunomodulatory IL-10 [22, 24, 37].
Lung resident macrophages in cancer
Like monocyte derived macrophages, tissue resident macrophages such as AMs also possess substantial diversity in response to tumor-microenvironmental cues and employ considerable plasticity in orchestrating immune responses to cancer. Besides, a recent cross-tissue single-cell sequencing study indicated that LYVE1+ macrophages that are often HES1+, with overlapped features of fetal liver macrophages, highlighting the possibility that tissue resident macrophages can be reverted to an embryonic state with potential diversity and plasticity applicable to different functions [21]. Recent studies involving treatment non-small cell lung cancer (NSCLC) patients and a mouse model using KrasG12D and P53 null (KP) epithelial cells have highlighted the significant role of tissue-resident AMs in driving lung tumorigenesis [47]. In line with these findings, a subset of P16 and CXCR1-expressing AMs with features resembling cellular senescence have been identified as suppressors of the antitumor cytotoxic T cell response during Kras-driven murine lung cancer development [48]. Importantly, the removal of these senescent AMs has been shown to substantially diminish tumorigenesis. Furthermore, similar senescent AMs have been detected in early-stage treatment human NSCLCs. Lineage tracing experiments have revealed that as NSCLC lesions develop in mice, tumor-associated macrophages accumulate near tumor cells, facilitating invasion, epithelial-mesenchymal transition, and increased dispersion of tumor cells over time. Moreover, these tumor-associated macrophages induce potent regulatory T cell responses that confer protection to tumor cells against adaptive immunity [8, 47, 49]. AMs may promote lung tumors through ROS/BACH1/PDLIM2/STAT3 signaling pathway with impaired phagocytosis [50]. In a murine lung cancer model, the expression of Inhibin beta A (INHBA) is upregulated in AMs during tumor-bearing conditions, resulting in the secretion of activin A. Interestingly, this activin A secretion has been found to inhibit the proliferation of lung cancer cells. Additionally, experimental models have shown that the postnatal deletion of INHBA/activin A can restrict tumor growth [51]. AMs-derived extracellular vesicles containing SOCS3 inhibited STAT3 activation as well as proliferation and survival of lung adenocarcinoma cells. Recombinant SOCS3 delivered in synthetic liposomes inhibits in vitro proliferation and survival of lung adenocarcinoma cells and suppresses the malignant transformation of normal endothelial cells, suggesting its potential as a lung cancer treatment strategy [52]. Consistent with this notion, it has been observed that BMDMs facilitated metastatic tumor spreading in the lung cancer models, whereas TRMs supported proliferation of cancer cell at the primary tumor site [8]. Other single-cell sequencing studies have also identified TRMs-TAMs as macrophages resembling normal TRMs but with high expression of embryonic precursor features [47, 53], and their application in lung cancer has shown elevated expression of MARCO, scavenger receptors, and FABP4, similar to AMs [54, 55]. TRMs-TAMs promote tumor invasiveness by inducing Epithelial-mesenchymal transition (EMT) and recruiting regulatory T cells in the KP lung cancer model [56].
In addition to primary pulmonary carcinoma, lung TRMs can play a crucial role in promoting metastatic lesions in the lungs. In a murine melanoma lung metastasis model, researchers have observed that resident AMs maintain the expression of a mixed pro-inflammatory/anti-tumor gene profile over time as the lesion grows, including upregulation of pro-inflammatory genes, such as IL-12β, IL-1α, and IL-1β, and also anti-inflammatory genes, including Smad3 of the TGF-β signaling pathway, and a considerable downregulation of the inhibitory Smad, Smad7 from the early to the late stage [57]. Conversely, IMs undergo a shift in their gene expression from an initial pro-inflammatory state, characterized by upregulation of genes such as SOCS1 and downregulation of CD38, IGF1, andCD206, to a later tumor-promoting profile, with substantial induction of Arginase-1. During metastatic growth, AMs within the macrophage pool gradually get replaced by infiltrative macrophages, which may contribute to metastatic progression [57]. Mechanistically, the WNT/β-catenin/TNF-α pro-metastatic axis in AMs has been implicated, revealing potential therapeutic implications for targeting tumors that are resistant to the anti-neoplastic effects of TNF-α [58]. Moreover, targeting the recruitment of Monocytic-myeloid-derived suppressor cells (MO-MDSC) through CXCL10/CXCR3 and TLR4/CCL12 axis in AMs holds promise as a therapeutic strategy to suppress lung metastasis [59] (Fig. 2, Table 1).

Dynamic crosstalk between pro- and antitumorigenic alveolar macrophages (AMs) and tumor cells in primary and metastatic cancers. In primary tumors, AMs expressing P16 and CXCR1 suppress CTL response and promoting the progression of lung tumor via the ROS, BACH1, PDLIM2 and STAT3 signaling pathway; Upregulation of INHBA expression in AMs leads to the secretion of activin A, which inhibits the proliferation of lung cancer cells. Additionally, AMs-derived extracellular vesicles containing SOCS3 suppresses STAT3 activation in cancer cells leading to inhibits the proliferation and survival of lung adenocarcinoma cells. Upregulation of pro-inflammatory genes, such as IL-12β, IL-1α, and IL-1β, and also anti-inflammatory genes, including Smad3 of the TGF-β signaling pathway, and downregulation of Smad7. During cancer metastasis, AMs are recruited from MO-MDSCs through the CXCL10-CXCR3 and TLR4-CCL12 axis, and contributing to the metastatic progression through the Wnt/β-catenin/TNF-α axis
Liver tissue resident macrophages
Homeostasis of tissue resident macrophages in liver
With the largest population of resident tissue macrophages in all solid organs, liver-resident macrophages perform various functions crucial for liver homeostasis, including the clearance of erythrocytes and debris, regulation of iron and lipid metabolism, maintenance of immune tolerance, and sensing tissue damage [60,61,62,63]. KCs are primarily of embryonic origin and self-maintain throughout life [64,65,66], reside within the lumen of the liver sinusoids and do not originate from circulating monocytes under steady state [67,68,69]. While they share common functions with tissue macrophages, such as responding to tissue damage and antigen presentation, KCs also have specialized roles in iron scavenging and uptake of digested particles from the portal blood [63]. Monocyte-derived macrophages, on the other hand, are mainly localized at the portal triad in the healthy liver and also contribute to iron and cholesterol metabolism [70, 71]. Kupffer cells are major producers of cytokines such as IL-1β, IL-6, IL-10 and TGF-β1, chemokines such as CXCL1, CXCL2, CXCL8, CXCL16, CCL2 and other bioactive molecules including YAP in response to appropriate stimulation, playing a crucial role in maintaining liver homeostasis [72,73,74,75]. KCs can be replaced by monocyte derived KCs in disease model, for example, only when the proliferation of embryo-derived KCs is impaired, KCs secrete chemokines like CCL2, which leads to the excessive infiltration of LY6Chi bone marrow monocytes and subsequent liver injury [76,77,78]. It is noteworthy that repopulation of monocytes appears to be relatively inefficient compared to proliferation of resident cells and they lack the necessary machinery for the uptake and recycling of erythrocytes [68, 79, 80].
Characteristics of liver resident macrophages
Kupffer cells depend on the activation of the macrophage colony-stimulating factor 1 receptor, which is stimulated by M-CSF and IL-34, as well as rely on the transcription factor ID3 and LXR-α to facilitate their differentiation, survival and self-maintenance [10, 37]. Inactivation of ID3 impairs the development of liver macrophages and leads to selective KCs deficiency in adults [31]. Additionally, the transcription factor ZEB2 maintains the expression of LXR-α, its loss results in change in KCs identity and their disappearance [81]. Recent study has shown that the induction and maintenance of KCs identity require a combination of interactions involving DLL4, TGF-β family ligands, and endogenous LXR ligands [19]. Moreover, the expression of DLL4 by liver sinusoidal endothelial cells is crucial for driving the differentiation of monocytes into KCs, as well as this process is facilitated by the rapid induction of LXR-α expression and an increased responsiveness to LXR-α-inducing signals produced by hepatic stellate cells [19, 79, 81].
Kupffer cells exhibit specific surface markers and transcriptional features that differ from other TRMs. The transcription factor CLEC4F has been identified as a specific marker for KCs, while TIM4 is expressed in various long-lived tissue-resident macrophage subsets, throughout development and postnatally [81,82,83,84]. In mouse, KCs are characterized by F4/80+CD11B+CD68+TIM4+CLEC4F+ surface phenotype [72, 85], as well as exhibit a diverse repertoire of immune receptors, including toll-like receptors (TLRs) like TLR4 and TLR9, scavenger receptors and complement receptors, which play a vital role in maintaining liver tolerance by orchestrating the induction of regulatory T cells [61, 86]. In contrast, the heterogeneity of human liver macrophages is less well-defined compared to mouse Kupffer cells. In humans, KCs have been defined as a CD163+MARCO+CD5L+TIM4+CLEC4F+ macrophage population in human liver based on single-cell sequencing studies conducted by three separate research groups [87,88,89]. In addition, to demonstrate the heterogeneity of KCs, the relevant study revealed the existence of two distinct KC populations in the steady-state murine liver: KC1 (CD206loESAM−), and KC2 (CD36hiCD206hiESAM+). Notably, the CD36hiCD206hiESAM+ KC2 subset is specifically involved in the regulation of liver fatty acid metabolism [90]. However, these markers may not be sufficient to distinguish KCs from recruited macrophages, highlighting the need for further research in this area.
Kupffer cells in cancer
Numerous studies have proven that significant role of Kupffer cells (KCs) in the development of Hepatocellular carcinoma (HCC). The latest research has shown that KCs, acting as incomplete APCs, can induce CD8+ T cell tolerance and revert T cell exhaustion in the context of HBV infection model [91, 92]. In addition, two clusters of FOLR2+ TAM1 populations with different origins were identified: monocyte derived while another appears to be embryonic-origin tissue-resident macrophages. FOLR2+ TAM1 displayed immunosuppressive interactions with Tregs, supporting the onco-fetal reprogramming of tumor microenvironment (TME) in HCC [93]. It can be said unequivocally that KCs inhibit the antitumor response by activating signaling pathways involving PD-L1/PD-1 and TIM-3/Galectin-9 in T cells [94,95,96]. In response to stimulation from cancer cells, the expression of the TREM-1 increases, promoting KCs activation and HCC progression, while TREM-1 deficiency has been found to decrease the release of IL-1β, IL-6, CCL2, and CXCL10 by KCs, resulting in suppressed HCC growth [97]. Others studies suggest that ROS and paracrine TNF produced by KCs contribute to biliary tract cell proliferation and may eventually causing intrahepatic cholangiocarcinoma [73].Overall, KCs play a vital role in the liver immune microenvironment by secreting an array of soluble proteins such as cytokines, chemokines, and growth factors.
Apart from HCC, KCs also play a significant role in liver metastasis of various cancers. In the gastric cancer liver metastasis, KCs have been suggested to undergo a transition between M1 polarization, characterized by adherence, tumor cell phagocytosis, and induction of cell apoptosis, and M2 polarization, characterized by secretion of IL-6, HGF, VEGF, and MMP-9. This transition allows KCs to exert bidirectional effects on tumor cells through different mechanisms [98]. In an orthotopic murine model of colorectal cancer liver metastasis, a study investigating KCs depletion revealed the dualistic nature of KCs in influencing tumor growth. The early depletion of KCs was observed to enhance tumor progression by modulating the expression of INOS and VEGF within the TME. Conversely, late-stage KCs depletion exhibited a contrasting effect, impeding tumor growth by augmenting the infiltration of CD3 infiltrating immune cells and promoting apoptosis [99]. Interestingly, in a WAG/Rij rat syngeneic model of CRC liver metastasis induced by staged hepatectomy, it was observed that KCs expressed COX-2, while BMDMs primarily expressed Arginase-1 [100]. By utilizing the CRISPR/CasΦ mechanism to edit tumor-associated genes in KCs, it was observed that specific clearance of KCs plays an important anti-tumor role in the early stage of liver metastasis, while dysfunction occurs in the later stage of liver metastasis. In this research, the authors overcome KC dysfunction and elicit remarkable curative effects against several types of metastatic liver cancer in mice by using constructed MAFB- and MAF-targeting dual sgRNA CRISPR/CasΦ Vector [101]. Therefore, finding proper ways to regulate KCs in anti-cancer treatment will pose a major challenge (Fig. 3).

Dynamic interaction between pro- and antitumorigenic Kupffer cells (KCs) and tumor cells in primary and metastatic cancer. In primary tumors, KCs inhibit the anti-tumor response by activating signaling pathways involving PD-L1/PD-1 and galectin-9/TIM-3 in T cells. Stimulation from cancer cells leads to an increase in TREM-1 expression in KCs, promoting the progression of hepatocellular carcinoma (HCC). TREM-1 deficiency results in the reduced release of IL-1β, IL-6, CCL2, and CXCL10 by KCs, thereby suppressing HCC growth. ROS and TNF produced by KCs contribute to tumor cell proliferation. Constructed MAFB- and MAF-targeting dual sgRNA CRISPR/CasΦ vector in KCs achieved therapeutic effects. During cancer metastasis, KCs was found to decrease tumor growth by altering the expression of INOS and VEGF in cancer cells. Conversely, KCs increased tumor growth through decreased infiltration of T cells and upregulation of COX-2 expression
Central nervous system (CNS) resident macrophages
Homeostasis of tissue resident microglia in CNS
As part of the CNS’s innate immune system, microglia are vital for preserving immune defense and resolution, neuronal support, tissue maintenance, and synaptic integrity [102,103,104,105]. Microglia, which are widely distributed throughout the brain parenchyma, comprising approximately 10–15% of all glial cells and are commonly recognized as the CNS’s tissue-resident macrophages [106]. Contrary to studies conducted in healthy adult mice, research on human microglia has revealed significant spatial heterogeneity in their transcriptional programs, particularly in relation to grey and white matter regions [107,108,109,110]. Consistent with other TRMs origin, microglia colonize the brain as embryonic microglia during the initial phases of development [106, 111]. These immature myeloid cells, characterized by their high proliferative potential and amoeboid morphology, migrate into the brain through various routes. In mice, they enter via the meninges and ventricles [112,113,114], Similarly, in humans, microglia traverse through the leptomeninges, choroid plexus, and ventricular zone [115,116,117]. The initial colonization of microglia primarily takes place in the white matter regions, including the internal capsule, external capsule, and cerebral peduncle. As microglial cells undergo proliferation and migration, they subsequently extend their colonization to the subplate and cortical plate regions in both radial and tangential manners [117]. Microglia release trophic factors that promote neuronal circuit formation and survival, including IGF-1, which supports the postnatal survival of layer V cortical neurons [113]. Microglia possess the ability to detect harmful stimuli and mount a response by producing inflammatory cytokines such as TNF-α, IL-6, IL-1β, IFN-γ, and several chemokines such as CCL2, CX3CL1, CXCL10 [118]. Additionally, microglia, acting as accessory cells, can induce programmed cell death and clear cellular debris. This has been observed in the developing chick eye, where microglia release nerve growth factor to trigger apoptosis of retinal nerve cells [119], and in the murine hippocampus, where microglia induce programmed cell death in neurons [120]. Microglia actively participate in neuronal pruning during development, respond to synaptic activity and plasticity, and play a crucial role in maintaining synaptic homeostasis. They achieve this by releasing trophic factors and synaptogenic signals, including DAP12 signaling [121] 0.4.2 Characteristics of CNS resident microglia.
Microglia sustain their population by means of their extended lifespan and limited self-renewal capabilities within their microenvironment, primarily through the activation of CSF1R signaling pathways [106, 122,123,124]. The transcription factor SALL1 and IRF-8, expressed exclusively by microglia, is involved in regulating microglial identity and physiological properties, distinct from other members of the mononuclear phagocyte system or other CNS-resident cells in the CNS [125, 126]. Simultaneously, the transcription factors IRF-8 and PU.1 have an impact on CNS microglia by regulating their abundance, activation state, and developmental processes [111, 127]. It plays a vital role in maintaining microglial homeostasis and may control their activation through mechanisms involving apoptosis-related genes [111, 128]. Mouse models with IRF-8 deficiency (IRF-8 KO mice) demonstrate a significant loss of microglial signature genes accompanied by severely altered microglial morphology [129].
In the latest study, TMEM119 and Purinergic receptor P2RY12 are gaining ground as presumedly more specific microglia markers [130]. In adult mouse, microglia have been extensively characterized as TMEM119+P2RY12+CX3CR1+MRC1− [108, 131], while TMEM119+P2RY12+CX3CR1+CD206− in adult human [110, 122]. Furthermore, distinguishing microglia residing in the CNS from peripheral macrophages in tissues is challenging due to their shared expression of several markers, including CD11B, F4/80, CX3CR1, CD45, and Ionized calcium-binding adapter molecule 1 (IBA-1) [132]. However, studies have reported that CD44 is exclusively expressed by infiltrating cells and not by resident microglia [133]. Another study comparing the gene transcription profiles of adult microglia and peripheral cells revealed the absence of CD169 in microglia [134]. In certain situations, quantitative discrimination of markers is possible, such as detecting subtle differences in CD45 protein levels between CD11B+CD45lo in microglia and CD11B+CD45hi in macrophages from the adult brain [135, 136]. However, caution must be exercised as CD45 expression in microglia increases during inflammation and aging [137, 138]. Transcription factors PU.1 and MYB have also been identified as differentiating factors for microglia (PU.1-dependent transcription) and peripheral macrophages (MYB-dependent transcription) [133, 139, 140]. Therefore, discerning the unique characteristics of microglia in comparison to other myeloid cells is of utmost importance for comprehending brain development and diseases.
CNS resident microglia in cancer
Microglia, as the largest population of TAMs in the CNS, have been recognized for their role in promoting tumor proliferation and invasion in the initiation and progression of brain tumors [141, 142]. In an vitro study, it was reported that murine glioma cells moved threefold more easily when exposed to microglial and migrated faster with a higher rate compared with tumor cells incubated without microglia [142]. Further investigations, have revealed that microglia can modulate tumor growth by secreting various cytokines. For instance, microglia were found to stimulate glioblastoma cell invasion by releasing EGF [143]. STI1, a ligand of the cellular prion protein, is synthesized and secreted by microglia to enhance the proliferation and migration of glioblastomas both in vitro and in vivo [144]. TGF-β released by microglia could promote glioma cells migration through strengthening the expression and function of integrin in co-culture systems [145]. Additionally, microglia could also be converted into a pro-tumorigenic phenotype by CSF1 released by tumor cells [146]. Human glioma cells induce microglia to secrete and release IL-6 through CCL2/CCR2 axis, thus promoting glioma invasion [147, 148]. In a mouse glioblastoma model primarily containing TRMs, combined inhibition of MERTK with radiotherapy demonstrated a significantly pronounced growth delay compared to radiotherapy alone [149]. In addition, inhibiting BMDMs could delay the recurrence of glioblastoma after radiotherapy due to the increased abundance of BMDMs post-irradiation [150]. However, some BMDMs probably undergo transformation into TRMs during growth, implying a functional switch that poses challenges to TAMs-based targeted therapies [151].
In addition to promoting tumor proliferation in primary intracranial tumors, microglia also exert a tumorigenic effect in brain metastasis. Various primary cancers, such as lung, melanoma, and breast cancer, can colonize the brain, with lung cancer showing the highest incidence of brain metastasis [152]. Microglia, when exposed to metastatic lung cancer in the brain, exhibit considerable activation and an increased number of cells labeled with the specific microglial marker [153]. Moreover, in the study of non-small cell lung cancer develop brain metastasis (NSCLC-BM), researchers found that brain-specific metastatic cells A549-F3 induce polarization of microglia towards an M2 phenotype characterized by high expression of CD206 and Arginase-1 via the IL-6/JAK2/STAT3 signaling pathway and microglia, in turn, promote NSCLC-BM development by affecting the colonization of metastatic cells [154]. On the contrary, the supernatant from LPS-activated microglia in vitro induces apoptosis in metastatic lung cancer cells in a dose- and time-dependent manner. Yet at lower concentrations, it demonstrates trophic effects, leading to some cancer cells becoming resistant to microglial cytotoxicity [153]. Microglia activation, also observed prior to brain metastasis in MMTV-Wnt1 mice models, is promoted by ANXA1 secreted from metastatic cancer cells, enhancing tumor cell migration. This process can be inhibited by silencing ANXA1 or using inhibitors, leading to reduced microglial migration and activation of STAT3 [155]. Concurrently, elevated expression of TGF-β in microglia fosters tolerance towards these metastatic melanoma cells by anti-tumor cytotoxic T cells [156]. Nonetheless, microglia can potentially be converted into tumor-unsupportive cells depending on the microenvironmental conditions and disease states. Recent research has shown that systemic administration of CPG-C, a toll-like receptor 9 agonist, combats brain metastases by activating microglia, which play a pivotal role in tumor suppression and phagocytosis upon direct tumor interaction. This is supported by elevated gene expression related to these processes both in vitro and in vivo [157]. Recent studies have highlighted the role of microglia as effector cells with a crucial role in anti-tumor responses. The CD47/SIRPα axis, a key innate immune checkpoint that suppresses phagocytic activity in myeloid cells [158]. Blocking this axis with anti-CD47 antibodies inhibits tumor growth and enhances survival in mice with human-derived glioblastoma multiforme cells by re-educating glioma-associated microglia [159]. Targeting the CD47/SIRPα axis shows promise in preventing various adult and pediatric brain tumors [160]. Of note, single-cell technology has unveiled that monocyte derived macrophages (MDMs) but not microglias play a crucial role in providing favorable signaling cues such as CD40 to Th cells and controlling the proliferation of metastatic cancer cells. Additionally, they contribute to the modulation of the TME in the brain by supporting the function of blood vessels, that perivascular M1-like MDMs exhibited higher CD40 expression than those further away from blood vessels, whereas perivascular M2-like MDMs expressed high levels of OX40L (also known as CD134L). Notably, a distinctive subset of macrophages expressing myeloperoxidase has been identified, which is associated with long-term survival [161]. The re-educating of microglia offers a more promising and positive therapeutic approach compared with conventional therapies that target the depletion of microglia to suppress their tumorigenesis properties. However, one of the major challenges in understanding the role of microglia in tumors is the lack of specific surface markers to distinguish microglia in the tumor microenvironment from bone marrow-derived macrophages [162]. The development of single-cell sequencing and transcriptome sequencing technology will help clarify the molecular mechanisms of cross-talk between microglia, the tumor microenvironment, and tumor cells. Due to their strong plasticity, microglia will likely become one of the most promising immunotherapeutic targets in the future (Fig. 4).

Dynamic interplay between pro- and antitumorigenic microglia and tumor cells in primary and metastatic cancers. In primary tumors, microglia enhance the proliferation and migration of cancer cells in both in vitro and in vivo settings by releasing TGF-β, EGF, and STI1. Cancer cells induce microglia to secrete and release IL-6 through the CCL2/CCR2 axis, thereby promoting glioma invasion。During cancer metastasis, tumor cells stimulate the JAK2/STAT3 signaling pathway in microglia by secreting IL-6 and promote tumor cell migration through the secretion of ANXA1, which reduces microglial migration and activates the STAT3. Elevated expression of TGF-β in microglia fosters tolerance towards cancer cells by anti-tumor CTL. CpG-C, inhibits brain metastasis by activating microglia, while the CD47-SIRPα axis acts as a crucial innate immune checkpoint suppressing phagocytic activity in myeloid cells
Skin tissue resident macrophages
Homeostasis of tissue resident macrophages in skin
Skin resident macrophages have diverse functions in maintaining skin homeostasis, inducing immunogenic, tolerant responses and involved in the development of skin diseases, such as psoriasis, atopic dermatitis, and psoriasis-like dermatitis [163,164,165]. They consist of two main cell types known as Langerhans cells (LCs) and dermal macrophages (DMs), serving as the frontline defense in the cutaneous immune system [37]. As the majority of skin tissue macrophages, LCs exhibit unique characteristics shared by both macrophages and DCs [166]. They are mainly located in the middle and upper part of the epidermis, residing between the epidermal spinous cells, and can also be found in the dermis, oral mucosa, vaginal epithelium and esophagus while DMs predominantly populate the dermis [36, 167]. Furthermore, LCs constitute approximately 3–5% of all nucleated cell in the adult epidermis [168]. LCs are thought to originate from hematopoietic precursors that colonize the skin during early embryonic development. The initial population of LCs is derived from yolk sac-derived CD16+ myeloid progenitor cells, which populate the skin prior to the onset of fetal liver hematopoiesis. During embryogenesis, these cells are largely replaced by CD14+ fetal hepatic mononuclear cells. Thus, the decline of adult LCs is mainly due to fetal hepatic mononuclear cells, with a smaller contribution from fetal yolk sac monocytes [169]. Lineage tracing studies have provided compelling evidence challenging the conventional notion that adult mouse LCs originate solely from bone marrow (BM)-derived DCs precursors. Instead, these studies have revealed that adult LCs are primarily derived from two distinct developmental sources: embryonic yolk sac-derived macrophages and fetal hepatic monocytes. This finding highlights the remarkable developmental plasticity of LCs and underscores the diverse origins contributing to their population in adult skin [170, 171]. However, recent breakthrough research has revealed that murine LCs present in mucosal epithelia are derived from circulating BM precursors and undergo continuous replenishment [172, 173]. Tongue LCs play a crucial role in antifungal immunity [174] and exhibit a protective role against cancer [175]. On the other hand, DMs primarily provide protection to newborn infants immediately at birth, safeguarding the outermost body surface from contact with bacteria colonizing the maternal genital tract and the skin [176]. Unlike LCs, DMs cannot migrate into the skin-draining lymph nodes and have limited antigen processing and presentation capabilities to T cells [177]. The single-cell transcriptomics, fate mapping, and imaging studies have revealed that DMs self-maintained with minimal postnatal input from hematopoietic stem cells but receive continuous contributions from circulating monocytes [36, 178]
Characteristics of skin resident macrophages
The murine model has shown that the development and differentiation of Langerhans cells (LCs) are dependent on several transcription factors associated with TGF-β1 signaling, including PU.1, BMPR1A/ALK3, RUNX3, and ID2, as well as the interaction between CSF1 and IL-34 [179,180,181,182]. Some studies have demonstrated that PU.1 regulates LCs differentiation by controlling the expression of the critical TGF-β-responsive transcription factor, RUNX3 [183]. However, it has been found that the TGF-β1/Smad3 signaling pathway does not significantly affect LCs homeostasis and maturation. Further investigations have shown that blocking Smad2 or Smad4 alone, or in combination, in LCs lineages does not have a significant impact on the maintenance, maturation, antigen uptake, and migration of LCs in steady-state conditions, both in vivo and in vitro. However, disruption of the Smad2 and Smad4 pathways in the myeloid system leads to notable inhibition of BM-derived LCs in the inflammatory state [184]. Additionally, during epidermal ontogeny, the spatial and temporal availability of TGF-β family members, along with Notch ligands, collaborate to promote LCs differentiation [185, 186]. LCs are considered to be macrophage with DCs functions, as they originate from a MAFB-expressing progenitor, indicating a macrophage origin, as well as express the transcription factor ZBTB46, which reinforces their DCs identity [187, 188]. LCs are self-renewing in homeostatic conditions and are long-lived cells that can migrate and mature into DCs [189]. Dermal macrophages rely on the activation of the CSF1R and the cytokines on IL-10 and IL-4 for self-sustainment proliferation for self-maintenance [178], while LCs secreted IL-1β, low levels of TNF-α and IL-8, but not IL-6 or IL-10 [190].
Langerhans cells (LCs) possess distinct markers that aid in their identification. Specifically, the c-type lectin receptor langerin (CD207), which is a novel interferon-stimulated gene, serves as a specific marker for distinguishing LCs from other DCs subsets [191,192,193]. Moreover, LCs are characterized by the presence of unique rod- or tennis racket-shaped Birbeck granule in electron microscopy [194]. By employing single-cell sequencing and mass cytometry analysis on human LCs derived from CD34+ hemopoietic stem cells obtained from the cord blood, researchers have successfully identified four distinct subgroups of human LCs: LC1 (CD207hi, CD1a, EPCAM), LC2 (CD207lo, CD1c, CD1b, HLA-DR), activated LCs (Alc) (CCR7lo, CD83, CD40), and migratory LCs (migLC) (CCR7hi, CXCR4) [195]. LCs play a crucial role in inducing tolerance to protein antigens in intact skin through the action of Langerin. Additionally, targeting LCs via Langerin may hold potential for regulating systemic immune responses [196]. The LC1 and LC2 subgroups can be discerned by their distinct expression patterns of the C-type lectin receptor “Langerin,” the CD1 family of non-classical antigen-presenting receptors (CD1a, CD1b, CD1c), and EPCAM. The LC1 subset demonstrates elevated levels of langerin, CD1a, and EPCAM, whereas the LC2 subset exhibits lower langerin expression and higher levels of CD1c and CD1b [197].In addition, Early growth response 1 (EGR1) and Notch pathways have been found to have a significant impact on the bifurcation of LC1 and LC2, where LC1 may function as “effector LCs” with classical LCs functions, and LC2 may act as “regulatory LCs” that primarily play an immunoregulatory role under inflammatory conditions. In most cases, LC1 and LC2 subsets work collaboratively to interact with the external environment and orchestrate immune responses, thereby contributing to the maintenance of skin homeostasis [195]. In contrast to human LCs, mouse LCs lack CD205 and CD207 expression, and instead express CD14, CD204, and low levels of MHCII and TLR4 molecules at birth. Intriguingly y, the CD204+ and CD14+ LCs disappear after four days, and MHCII, CD11c, and CD207 are acquired during the first week of life [198].
Langerhans cell in cancer
The important role of skin tissue resident macrophages in skin cancer has been highlighted in a recent study on human Squamous cell carcinoma (SCC). The study findings revealed that LCs, a subset of DCs, can induce proliferation of CD4+ and CD8+ T cells and enhance production of IFN-γ more efficiently compared to other DCs subsets. LCs were discovered to possess a heightened capacity for inducing type 1 T cell responses within the SCC microenvironment, subsequently exerting a suppressive effect on antitumor activity [199, 200]. The potency of LCs to enhance the CD8+ T cell response can be further augmented by activating LCs with the toll-like receptor 3 ligand polyinosine [201]. Additionally, LCs have been found to promotes epithelial DNA damage and squamous cell carcinoma by metabolizing carcinogenic agents [202]. It is predicted that melanoma fail to activate the migration of LCs to lymph nodes until the tumor reaches a critical size, which is determined by a positive TNF-α feedback loop within the melanoma in silico model [203]. In non-melanoma skin cancer, a higher presence of LCs was observed in Basal cell carcinoma (BCC) cases compared to SCC. The reduction of Langerhans cells in SCC may indicate their role as a barrier against metastasis, whereas the marked reduction of LCs in SCC compared to BCC suggests their potential implicated in the intricate processes of non-melanoma skin cancer development and progression [204, 205]. P21-activated kinase 1 (PAK1), previously reported to have oncogenic activity in various types of cancer, was found to regulates the number of epidermal stem cell by altering LCs properties and functions in skin carcinogenesis. This underscores the emerging significance of PAK1 in the regulation of LCs, as well as its potential holds great promise for the treatment of skin immune diseases and the management of carcinogenesis [206]. LCs and dermal DCs are the main cells that induce antigen-specific immunity in the skin, and the two mainstream strategies targeting LCs are either transcutaneous vaccination in situ or manipulating LCs ex vivo to facilitate efficient anti-tumoral response [207]. In the field of transcutaneous cancer vaccines, LCs have emerged as prime targets. Through an endocytic C-type lectin receptor called Langerin (CD207) and a glycomimetic Langerin ligand (liposomes 22), specific and efficient targeting of LCs in human skin has been achieved. These transcutaneous cancer vaccines induce protective T cell immunity, and their effectiveness relies on the efficient activation of LCs [208]. Recently Glyceryl monooleate (MO) was chosen as a skin permeation enhancer, and the MO-based reverse micellar carrier enabled the successful delivery of antigen to Langerhans cells and dermal dendritic cells [209]. (Fig. 5).

Langerhans cells (LCs) exhibit anti-tumor effects. LCs induce proliferation of T cells and enhance the production of IFN-γ. The potency of LCs in T cell response is further enhanced through activation with TLR-3. PAK1 expressed on LCs regulates the number of epidermal stem cells, thereby exerting inhibitory effects on tumor proliferation. CD207 and a glycomimetic Langerin ligand liposomes 22 can serve as transcutaneous cancer vaccine agents via inducing protective T cell immunity
Intestinal resident macrophages
Homeostasis of tissue resident macrophages in intestine
Intestinal resident macrophages, leveraging their robust phagocytic capacity, play a vital role in maintaining host defense and tissue homeostasis in the gastrointestinal tract [210]. However, unlike other murine tissue-resident macrophages, intestinal macrophages derived from embryos are replaced by circulating monocytes at 3 weeks of age and replenished from peripheral circulating monocytes through a “monocyte waterfall” process depending on CCL2/CCR2 axis [211, 212]. During embryonic development, embryo-derived macrophages turnover, and monocytes defined as Ly6ChiCD64−CX3CR1intMHCII− enter the gastrointestinal tract, transitioning through subsequent differentiation stages to become mature CD64+CX3CR1hiMHCIIhi macrophages in mice [211, 213], and CD64+CD11C+MHCIIhi in human [214]. Among them, the most characterized macrophage population resides in the lamina propria (LP) [210], macrophages in the intestine can be categorized based on their anatomical positioning, primarily distinguishing between lamina propria macrophages (LPMs) and muscularis macrophages (MMs) [13, 215,216,217]. For many years, it was widely believed that intestinal macrophages were an exception among tissue-resident macrophages, deriving exclusively from circulating monocytes. Recent findings from two research groups have spotlighted a distinct population of long-lived and self-maintain colonic macrophages that endure from early stages into adulthood, without being supplanted by circulating monocytes. These macrophages reside deep within the gut wall, closely associated with blood vessels and enteric neurons of both the submucous and myenteric plexus [13, 82]. This discovery challenges our previous understanding of tissue-resident macrophages in the colon. Recent studies in humans have confirmed the coexistence of long-lived macrophages and those rapidly replaced by incoming monocytes in the intestinal macrophage pool. Notably, the adult small intestine harbors both subsets, with the long-lived macrophages predominantly located in the villi and submucosa [216].
Characteristics of intestinal resident macrophages
As mentioned above, lamina propria macrophages (LPMs) primarily consist of monocyte-derived macrophages, with a relatively lower proportion of long-lived macrophages in comparison to the deeper intestinal layers. The intestinal lamina propria is characterized by an inflammatory microenvironment resulting from continuous exposure to luminal commensal bacteria and dietary antigens. Despite the presence of this reactive setting, the immune system is tightly controlled to prevent unintentional self-harm, and LPMs play a crucial role as key regulators in establishing a tolerogenic environment [218]. Among the factors involved in immune regulation, IL-10 is the most extensively studied tolerogenic factor in intestinal LPMs, and its complete absence results in the development of spontaneous enterocolitis [219]. In an immature state, LPMs exhibit a failure to upregulate IL-10 and excessive production of inflammatory cytokines such as IL-1β, TNF-α, IL-6, IL-12, and chemokines, that further promotes the influx of LY6Chi monocytes [212, 220]. LPMs also play a crucial role in maintaining the intestinal stem cell niche. Depletion of CSF1R-dependent LPMs impairs paneth cells differentiation and reduces LGR5+ stem cells, affecting the differentiation and replenishment of other intestinal epithelial cells [221]. These observations indicate that a subset of CSF1R-dependent LPMs is essential for maintaining intestinal crypt homeostasis. In recent findings, researchers identified that a predominant portion of self-sustaining macrophages in the lamina propria align closely with submucosal neurons and blood vessels [13]. Remarkably, their depletion led to the degeneration of these neurons, vessel disruption, and increased vascular leakage [222].
In contrast to LPMs, a significant portion of intestinal resident macrophages, known as muscularis macrophages (MMs), are derived from embryonic origins and mainly reside in the myenteron, forming a specialized “macrophage niche” within the intestinal environment [13, 82]. Within the myenteric plexus, MMs closely interact with enteric neurons, executing unique functions. Their activation is largely influenced by neuron-derived cues, given their expression of various neurotransmitter receptors that modulate their behavior [215, 223,224,225,226]. Moreover, the absence of muscularis macrophages in CSF1op/op mice is associated with a higher density of enteric neurons and a more disorganized structure of the myenteric plexus [217]. Muscularis neuron-associated macrophages within the myenteric plexus play a crucial role in maintaining intestinal homeostasis by synthesizing BMP2, which is essential for orchestrating peristaltic activity [215]. Animal studies have demonstrated that upon pathogen stimulation, MMs upregulate various transcription factors such as NF-Kb, STAT, and P38-MAPK, induce pro-inflammatory gene expression, as well as release chemokines and cytokines including IL-1β, MCP-1, IL-6, and TNF-α [45, 227, 228].
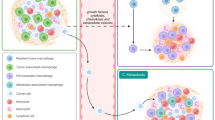
6.3 Intestinal resident macrophages in cancerTissue-resident macrophages in the colon have been identified as TAMs and play a crucial role in the fate of tumors, including their occurrence, development, and elimination [229]. In vitro experiments have demonstrated that TAMs derived from resident macrophages in colorectal cancer exhibit an M2-like phenotype characterized by express high levels of pattern recognition receptors such as MR and CD163, as well as secretion of chemokines including CCL17, CCL22, and CCL24, along with immune regulatory cytokines such as IL-10 and TGF-β. These TAMs play a role in promoting tumor occurrence and development [230]. The transcription factor C-MYC does not involved in macrophage proliferation and survival but is expressed in TAMs and regulates the expression of pro-tumoral genes, such as VEGF, HIF-1α and TGF-β [231]. In addition, it has been demonstrated that lactic acid, a byproduct of tumor cell glycolysis, acts as a potential signal triggering TAMs to adopt an M2 phenotype characterized by the expression of VEGF and Arginase-1, with HIF-1α facilitating this process [232]. In the realm of gastrointestinal neoplasms, tumors of the small intestine, predominantly of the neuroendocrine subtype, manifest infrequently, especially when compared to the higher incidence of colorectal carcinomas [233, 234]. While the direct relationship between these tumors and intestinal macrophages remains underexplored, macrophages play pivotal roles in various inflammatory and tumorigenic processes, suggesting potential involvement in small intestine tumor progression [235]. Consequently, in the subsequent sections, the focus of the review will primarily be on colorectal tumor.
Additionally, literature reports have indicated that colon cancer cells secrete IL-34, influencing the polarization of monocytes in the mucosal lamina propria towards M2 macrophages expressing CD206 and CD163, as well as secreting IL-10., thereby further enhancing tumor proliferation and metastasis [236]. Similarly, Irene Soncin et al. found a resident subsets CCR2−independent F4/80+MHCIIlo macrophage in the microenvironment of colorectal adenoma, which had self-renewal capacity in TME and played pro-tumorigenic roles in tumor progression. However, the process of self-renewal depended on isolated niche and CSF1 may be a key facilitator [237]. In addition, a portion of CD169+ macrophages (characterized by CD115+CD169+CD11B+F4/80loCD11Clo), reside in the colonic lamina propria, primarily surround the crypt and arise from both tissue macrophage self-renewal and blood stem cell input, with their development relying on vitamin A [238]. The presence of these cells in adjacent lymph nodes of colon cancer patients correlates with clinical stage, overall survival, and prognosis [239], with a higher concentration suggesting extended survival and a positive clinical outlook for individuals with tumors [239]. Colonic macrophages also play a significant role in metastatic tumors. Lim, S.Y. et al. elucidated that TAMs prompt the production of S100A8/A9 mRNA within the colon cancer TME through the ERK signaling pathway, subsequently enhancing tumor migration [240]. On a related note, Wei, C. et al. highlighted that TAMs, by secreting IL-6, instigate the EMT process in colorectal cancer, bolstering its migration and invasion via the JAK2/STAT3/ FOXQ1 axis [241]. Both intestinal resident macrophages replenished from peripheral circulating monocytes and the newly discovered embryos-derived and self-renewal macrophages have shown the characteristics of promoting tumors, although they show different functions in maintaining tissue homeostasis. Therefore, the concept of “intestinal resident macrophage” is still attractive candidates for therapeutic intervention. However, it is still unclear of the tumor promoting capacity of these two populations of resident macrophages. This also presents a major challenge of the potential therapeutic targets on exhausted macrophages and reprogram macrophage phenotypes [242, 243]. It is well believed that an in-depth understanding of its function and underlying mechanism will help us develop effective macrophage-based therapeutic approaches (Fig. 6).

Intestinal resident macrophages facilitate tumor cell invasion. The transcription factor C-MYC regulates the expression of pro-tumoral genes, such as VEGF, HIF-1α and TGF-β in Tumor-associated macrophages (TAMs), which are characterized by the expression of pattern recognition receptors CD163 and MP, as well as the secretion of cytokines IL-10 and TGF-β, as well as chemokines including CCL17, CCL22 and CCL24. Tumor cells produce lactic acid through the mediation of HIF-1α, which induces VEGF and arginase-1 expression in intestinal resident macrophages. Cancer cells secrete IL-34, promoting the polarization of monocyte towards M2 macrophages, thereby further enhancing tumor proliferation and metastasis. TAMs enhance tumor migration and invasion by the ERK signaling pathway, secreting IL-6, and this effect is mediated via the JAK2/STAT3/FoxQ1 axis
Conclusions
The advances and progress of fate-mapping, parabiosis models and single cell sequencing techniques facilitates the better understanding of macrophage function regarding their origin during past decade [67]. Although the maintain and expansion of TRMs are instructed by three main factors derived from stromal cells (CSF1, CSF2 and IL-34) via two receptors on macrophages (CSF1R and CSF2R) [244], niche specific signals also played central roles in governing TRMs such as TGF-β signaling controlling PPAR-γ in alveolar macrophages [41], and retinoic acid signaling regulating GATA6 expression in peritoneal cavity-resident macrophages [245]. The roles of TRMs in regulating cancer growth is organ specific in that the expansion of TRMs and flooding influx of bone marrow derived macrophage varied among organs [3]. The number of TRMs decreased while BM-derived monocytes expanded in murine breast cancer model [246, 247]. While in a pancreatic cancer model, TRMs gradually increased [248]. As a result, depletion of bone marrow derived circulation monocyte leads to tumor shrinkage in breast cancer model and ablation of TRMs significantly impaired tumor growth in pancreatic adenocarcinoma model [6, 246,247,248]. Apart from varied roles of TRMs in primary cancer, their functions in regulating metastatic cancer could be quite different. In a KCs-deficient mice, the number of colorectal carcinoma metastasis increased [249]. While depletion of alveolar macrophages reduced metastatic tumor spreading [250]. Additionally, in breast cancer primary tumors, FOLR2+ tissue-resident macrophages are positioned in the perivascular areas of the tumor stroma, where they interact with CD8+ T cells, and their density in tumors is positively associated with improved patient survival rates [251]. TIM-4+ cavity-resident macrophages have been associated with a reduction in the population of CD8+ T cells. Mechanistically, CD8+ T cells upregulate phosphatidylserine, rendering them susceptible to being sequestered away from tumor targets and suppressed by the proliferating TIM-4+ cavity-resident macrophages. However, blocking TIM-4 effectively prevents this sequestration and proliferation-induced suppression, leading to enhance effectiveness of anti-PD-1 therapy and adoptive T cell therapy in mouse models by enabling improved tumor targeting and activation of CD8+ T cells [252]. Specific depletion of CD163+ TIM-4+ macrophages prevent metastasis of ovarian cancer [253, 254]. The distinct macrophage lineage may constantly adjust themselves to environmental cues, adding the complexity of phenotypic study. Ultimately, the dynamics of TRMs and BM-derived monocytes at different stages of cancer progression and niche specific markers as well as the microenvironment profiles should all be investigated for a better understanding before tissue specific macrophages can be therapeutically targeted.
Data availability
Not applicable.
Abbreviations
- TRMs:
-
Tissue resident macrophages
- TAMs:
-
Tumor associated macrophages
- BMDMs:
-
Bone marrow derived macrophages
- BM:
-
Bone marrow
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- LYVE:
-
Lymphatic vessel endothelial hyaluronan receptor
- MHCII:
-
Major histocompatibility complex class II
- AMs:
-
Alveolar macrophages
- NAMs:
-
Nerve-associated interstitial macrophages
- IMs:
-
Interstitial macrophages
- TGF-βR:
-
Transforming growth factor-beta receptor
- PPAR-γ:
-
Peroxisome proliferator-activated receptor gamma
- CSF1R:
-
Colony-stimulating factor 1-receptor
- HLA-DR:
-
Human leukocyte antigen-DR
- MERTK:
-
Mer tyrosine kinase
- SIGLECF:
-
Sialic acid-binding immunoglobulin-like lectin F
- FABP4:
-
Fatty acid-binding protein 4
- MARCO:
-
Macrophage receptor with collagenous structure
- MRC1:
-
Mannose receptor c-type 1
- MSR1:
-
Macrophage scavenger receptor 1
- TGF-β:
-
Transforming growth factor beta
- IL-10:
-
Interleukin-10
- IL-12:
-
Interleukin-12
- IL-4:
-
Interleukin-4
- IL-6:
-
Interleukin-6
- IL-8:
-
Interleukin-8
- IL-1β:
-
Interleukin-1 beta
- PGE2:
-
Prostaglandin E2
- INOS:
-
Inducible nitric oxide synthase
- TNF-α:
-
Tumor necrosis factor alpha
- MIP1α:
-
Macrophage inflammatory protein-1 alpha
- CX3CR1:
-
C-X3-C motif chemokine receptor 1
- CCR2:
-
C–C chemokine receptor type2
- TGF-β2:
-
Transforming Growth Factor-beta 2
- PLAUR:
-
Plasminogen Activator Urokinase Receptor
- FCNA:
-
Fibronectin 1
- NSCLC:
-
Non-small cell lung cancer
- CXCR1:
-
C-X-C chemokine receptor 1
- CTL:
-
Cytotoxic T cell
- ROS:
-
Reactive oxygen species
- BACH1:
-
BTB and cnc homology 1
- PDLIM2:
-
PDZ and lim domain 2
- STAT3:
-
Signal transducer and activator of transcription 3
- SOCS1:
-
Suppressor of Cytokine Signaling 1
- INHBA:
-
Inhibin beta A
- SOCS3:
-
Suppressor of cytokine signaling 3
- EMT:
-
Epithelial-mesenchymal transition
- IL-12β:
-
Interleukin-12 beta
- IL-1α:
-
Interleukin-1 alpha
- MO-MDSC:
-
Monocytic-myeloid-derived suppressor cells
- CXCL10:
-
C-X-C motif chemokine ligand 10
- CXCR3:
-
C-X-C chemokine receptor 3
- TLR4:
-
Toll-like receptor 4
- CCL12:
-
C–C motif chemokine ligand 12
- WNT:
-
Wingless-related integration site
- KCs:
-
Kupffer cells
- TGF-β1:
-
Transforming growth factor beta 1
- CXCL1:
-
C-X-C motif chemokine ligand 1
- CXCL2:
-
C-X-C motif chemokine ligand 2
- CXCL8:
-
C-X-C motif chemokine ligand 8
- CXCL12:
-
C-X-C motif chemokine ligand 12
- CXCL16:
-
C-X-C motif chemokine ligand 16
- CCL2:
-
C–C motif chemokine ligand 2
- YAP:
-
Yes-associated protein
- M-CSF:
-
Macrophage-colony stimulating factor
- IL-34:
-
Interleukin-34
- ID3:
-
Inhibitor of DNA binding 3
- LXR-α:
-
Liver X receptor-alpha
- ZEB2:
-
Zinc finger e-box binding homeobox 2
- DLL4:
-
Delta-like ligand 4
- TIM-4:
-
T-cell immunoglobulin and mucin domain-containing protein 4
- CLEC4F:
-
C-type lectin domain family 4 member F
- TLRs:
-
Toll-like receptors
- TLR4:
-
Toll-like receptor 4
- TLR9:
-
Toll-like receptor 9
- PD-L1:
-
Programmed cell death-ligand 1
- PD-1:
-
Programmed death 1
- TIM-3:
-
T cell immunoglobulin domain and mucin domain-3
- ESAM:
-
Endothelial cell-selective adhesion molecule
- HCC:
-
Hepatocellular carcinoma
- APCs:
-
Antigen presenting cells
- FOLR2:
-
Folate receptor beta
- MT1G:
-
Metallothionein 1G
- TIGIT:
-
T cell immunoreceptor with Ig and ITIM domains
- CTLA4:
-
Cytotoxic T-lymphocyte-associated protein 4
- TREM-1:
-
Triggering receptor expressed on myeloid cells 1
- TNF:
-
Tumor necrosis factor
- VEGF:
-
Vascular endothelial growth factor
- COX-2:
-
Cyclooxygenase-2
- HGF:
-
Hepatocyte growth factor
- MMP-9:
-
Matrix metalloproteinase-9
- LCs:
-
Langerhans cells
- DMs:
-
Dermal macrophages
- DCs:
-
Dendritic cells
- PU.1:
-
Spi-1 proto-oncogene transcription factor
- BMPR1A:
-
Bone morphogenetic protein receptor type 1a
- ALK3:
-
Activin receptor-like kinase 3
- RUNX3:
-
Runt-related transcription factor
- ID2:
-
Inhibitor of DNA binding 2
- Smad2:
-
Mothers against decapentaplegic homolog 2
- Smad3:
-
Mothers against decapentaplegic homolog 3
- Smad4:
-
Mothers against decapentaplegic homolog 4
- Smad7:
-
Mothers against decapentaplegic homolog 7
- MAFB:
-
V-MAF avian musculoaponeurotic fibrosarcoma oncogene homolog B
- C-maf:
-
Cellular-musculoaponeurotic fibrosarcoma oncogene homolog
- ZBTB46:
-
Zinc finger and BTB domain-containing protein 46
- CD207:
-
C-type lectin receptor langerin
- EPCAM:
-
Epithelial cell adhesion molecule
- CCR7:
-
C–C chemokine receptor 7
- CXCR4:
-
C-X-C chemokine receptor 4
- EGR1:
-
Early growth response 1
- SCC:
-
Squamous cell carcinoma
- BCC:
-
Basal cell carcinoma
- IFN-γ:
-
Interferon-gamma
- PAK1:
-
P21-activated kinase 1
- CNS:
-
Central nervous system
- IGF-1:
-
Insulin-like growth factor-1
- SALL1:
-
Spalt-like transcription factor 1
- IRF-8:
-
Interferon-regulatory factor 8
- TMEM119:
-
Transmembrane protein 119
- P2RY12:
-
Purinergic receptor P2Y g-protein coupled 12
- CXCR3:
-
C-X-C chemokine receptor 3
- IBA-1:
-
Ionized calcium-binding adapter molecule 1
- MYB:
-
Myeloblastosis oncogene
- EGF:
-
Epidermal growth factor
- STI1:
-
Stress-inducible protein 1
- CCR2:
-
C–C chemokine receptor 2
- NSCLC-BM:
-
Non-small cell lung cancer develop brain metastasis
- JAK2:
-
Janus kinase 2
- STAT3:
-
Signal transducer and activator of transcription 3
- ANXA1:
-
Annexin A1
- CpG-C:
-
CPG oligodeoxynucleotide type c
- SIRPα:
-
Signal regulatory protein alpha
- TME:
-
Tumor microenvironment
- LPS :
-
Lipopolysaccharide
- MMTV-Wnt1:
-
Mouse mammary tumor virus- wingless-type MMTV integration site family, member 1
- LP:
-
Lamina propria
- LPMs:
-
Lamina propria macrophages
- MMs:
-
Muscularis macrophages
- MO:
-
Monooleate
- LGR5:
-
Leucine-rich repeat-containing g-protein coupled receptor 5
- BMP2:
-
Bone morphogenetic protein 2
- NF-KB:
-
Nuclear factor kappa B
- STAT:
-
Signal transducer and activator of transcription, Activator of transcription
- P38-MAPK:
-
P38 mitogen-activated protein kinase
- MCP-1:
-
Monocyte chemoattractant protein-1
- MR:
-
Mannose receptor
- CCL17:
-
C–C motif chemokine ligand 17
- CCL22:
-
C–C motif chemokine ligand 22
- CCL24:
-
C–C motif chemokine ligand 24
- C-MYC:
-
Cellular myelocytomatosis oncogene
- HIF-1α:
-
Hypoxia-inducible factor-1 alpha
- ERK:
-
Extracellular signal-regulated kinase
- MET:
-
Epithelial-Mesenchymal Transition
- FOXQ1:
-
Forkhead box q1
- CSF2:
-
Colony-stimulating factor 2
- CSF2R:
-
Colony-stimulating factor 2-receptor
- GATA6:
-
GATA-binding protein 6
- FOLR2:
-
Folate Receptor 2
References
Cox N, Pokrovskii M, Vicario R, et al. Origins, biology, and diseases of tissue macrophages. Annu Rev Immunol. 2021;39:313–44.
Wu SY, Watabe K. The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front Biosci (Landmark Ed). 2017;22(10):1805–29.
Christofides A, Strauss L, Yeo A, et al. The complex role of tumor-infiltrating macrophages. Nat Immunol. 2022;23(8):1148–56.
Cortez-Retamozo V, Etzrodt M, Newton A, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A. 2012;109(7):2491–6.
Franklin RA, Liao W, Sarkar A, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–5.
Zhu Y, Herndon JM, Sojka DK, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. 2017;47(2):323-38.e6.
Bowman RL, Klemm F, Akkari L, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. 2016;17(9):2445–59.
Loyher PL, Hamon P, Laviron M, et al. Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med. 2018;215(10):2536–53.
Hume DA, Irvine KM, Pridans C. The mononuclear phagocyte system: the relationship between monocytes and macrophages. Trends Immunol. 2019;40(2):98–112.
Perdiguero EG, Klapproth K, Schulz C, et al. The origin of tissue-resident macrophages: when an erythro-myeloid progenitor is an erythro-myeloid progenitor. Immunity. 2015;43(6):1023–4.
van de Laar L, Saelens W, de Prijck S, et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity. 2016;44(4):755–68.
Lazarov T, Juarez-carreño S, Cox N, et al. Physiology and diseases of tissue-resident macrophages. Nature. 2023;618(7966):698–707.
de Schepper S, Verheijden S, Aguilera-Lizarraga J, et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell. 2018;175(2):400-15.e13.
Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. 2014;14(2):81–93.
Trapnell BC, Whitsett JA. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol. 2002;64:775–802.
Mass E, Nimmerjahn F, Kierdorf K, et al. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat Rev Immunol. 2023;23(9):563–79.
Lalancette-Hébert M, Gowing G, Simard A, et al. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27(10):2596–605.
Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9(4):259–70.
Sakai M, Troutman TD, Seidman JS, et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain kupffer cell identity. Immunity. 2019;51(4):655-70.e8.
Jacome-Galarza CE, Percin GI, Muller JT, et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature. 2019;568(7753):541–5.
Mulder K, Patel AA, Kong WT, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. 2021;54(8):1883-900.e5.
Ural BB, Yeung ST, Damani-Yokota P, et al. Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci Immunol. 2020. https://doi.org/10.1126/sciimmunol.aax8756.
Barlow JL, McKenzie ANJ. Innate lymphoid cells of the lung. Annu Rev Physiol. 2019;81:429–52.
Chakarov S, Lim HY, Tan L, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. 2019;363(6432):0964.
Hume PS, Gibbings SL, Jakubzick CV, et al. Localization of macrophages in the human lung via design-based stereology. Am J Respir Crit Care Med. 2020;201(10):1209–17.
Mowat AM, Scott CL, Bain CC. Barrier-tissue macrophages: functional adaptation to environmental challenges. Nat Med. 2017;23(11):1258–70.
Yao Y, Liu Q, Adrianto I, et al. Histone deacetylase 3 controls lung alveolar macrophage development and homeostasis. Nat Commun. 2020;11(1):3822.
Xu-Vanpala S, Deerhake ME, Wheaton JD, et al. Functional heterogeneity of alveolar macrophage population based on expression of CXCL2. Sci Immunol. 2020. https://doi.org/10.1126/sciimmunol.aba7350.
Evren E, Ringqvist E, Doisne JM, et al. CD116+ fetal precursors migrate to the perinatal lung and give rise to human alveolar macrophages. J Exp Med. 2022. https://doi.org/10.1084/jem.20210987.
Gomez Perdiguero E, Klapproth K, Schulz C, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–51.
Mass E, Ballesteros I, Farlik M, et al. Specification of tissue-resident macrophages during organogenesis. Science. 2016;353(6304):aaf4238.
Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity. 2022;55(9):1564–80.
Misharin AV, Morales-Nebreda L, Mutlu GM, et al. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. 2013;49(4):503–10.
Branchett WJ, Lloyd CM. Regulatory cytokine function in the respiratory tract. Mucosal Immunol. 2019;12(3):589–600.
Wang PL, Yim AKY, Kim KW, et al. Peripheral nerve resident macrophages share tissue-specific programming and features of activated microglia. Nat Commun. 2020;11(1):2552.
Kolter J, Feuerstein R, Zeis P, et al. A subset of skin macrophages contributes to the surveillance and regeneration of local nerves. Immunity. 2019;50(6):1482-97.e7.
Nobs SP, Kopf M. Tissue-resident macrophages: guardians of organ homeostasis. Trends Immunol. 2021;42(6):495–507.
Stanley E, Lieschke GJ, Grail D, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A. 1994;91(12):5592–6.
Dranoff G, Crawford AD, Sadelain M, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. 1994;264(5159):713–6.
Uchida K, Beck DC, Yamamoto T, et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med. 2007;356(6):567–79.
Yu X, Buttgereit A, Lelios I, et al. The cytokine TGF-β promotes the development and homeostasis of alveolar macrophages. Immunity. 2017;47(5):903-12.e4.
Guilliams M, de Kleer I, Henri S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210(10):1977–92.
Serezani APM, Pascoalino BD, Bazzano JMR, et al. Multiplatform single-cell analysis identifies immune cell types enhanced in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2022;67(1):50–60.
Leach SM, Gibbings SL, Tewari AD, et al. Human and mouse transcriptome profiling identifies cross-species homology in pulmonary and lymph node mononuclear phagocytes. Cell Rep. 2020;33(5):108337.
Wehner S, Schwarz NT, Hundsdoerfer R, et al. Induction of IL-6 within the rodent intestinal muscularis after intestinal surgical stress [J]. Surgery. 2005;137(4):436–46.
Bissonnette EY, Lauzon-Joset JF, Debley JS, et al. Cross-talk between alveolar macrophages and lung epithelial cells is essential to maintain lung homeostasis. Front Immunol. 2020;11:583042.
Casanova-Acebes M, Dalla E, Leader AM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021;595(7868):578–84.
Prieto LI, Sturmlechner I, Graves SI, et al. Senescent alveolar macrophages promote early-stage lung tumorigenesis. Cancer Cell. 2023;41(7):1261-75.e6.
Tissue-resident macrophages help early-stage lung tumors develop. Cancer Discov. 2021;11(8):1873.
Li L, Sun F, Han L, et al. PDLIM2 repression by ROS in alveolar macrophages promotes lung tumorigenesis. JCI Insight. 2021. https://doi.org/10.1172/jci.insight.144394.
Taniguchi S, Matsui T, Kimura K, et al. In vivo induction of activin A-producing alveolar macrophages supports the progression of lung cell carcinoma. Nat Commun. 2023;14(1):143.
Speth JM, Penke LR, Bazzill JD, et al. Alveolar macrophage secretion of vesicular SOCS3 represents a platform for lung cancer therapeutics. JCI Insight. 2019. https://doi.org/10.1172/jci.insight.131340.
Cheng S, Li Z, Gao R, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184(3):792-809.e23.
Zilionis R, Engblom C, Pfirschke C, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. 2019;50(5):1317-34.e10.
Kim N, Kim HK, Lee K, et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun. 2020;11(1):2285.
Ma RY, Black A, Qian BZ. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 2022;43(7):546–63.
Tapmeier TT, Howell JH, Zhao L, et al. Evolving polarisation of infiltrating and alveolar macrophages in the lung during metastatic progression of melanoma suggests CCR1 as a therapeutic target. Oncogene. 2022;41(46):5032–45.
Kramer ED, Tzetzo SL, Colligan SH, et al. β-Catenin signaling in alveolar macrophages enhances lung metastasis through a TNF-dependent mechanism. JCI Insight. 2023. https://doi.org/10.1172/jci.insight.160978.
Shang C, Sun Y, Wang Y, et al. CXCL10 conditions alveolar macrophages within the premetastatic niche to promote metastasis. Cancer Lett. 2022;537:215667.
Scott CL, Guilliams M. The role of Kupffer cells in hepatic iron and lipid metabolism. J Hepatol. 2018;69(5):1197–9.
You Q, Cheng L, Kedl RM, et al. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology. 2008;48(3):978–90.
Ramachandran P, Matchett KP, Dobie R, et al. Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nat Rev Gastroenterol Hepatol. 2020;17(8):457–72.
Bennett H, Troutman TD, Sakai M, et al. Epigenetic regulation of kupffer cell function in health and disease. Front Immunol. 2020;11:609618.
Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. 2016;44(3):439–49.
Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14(6):392–404.
Schneider C, Kopf M. tEMPting Fate MaYBe the Solution. Immunity. 2015;42(4):597–9.
Yona S, Kim KW, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91.
Scott CL, Zheng F, de Baetselier P, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. 2016;7:10321.
Soysa R, Lampert S, Yuen S, et al. Fetal origin confers radioresistance on liver macrophages via p21(cip1/WAF1). J Hepatol. 2019;71(3):553–62.
Gammella E, Buratti P, Cairo G, et al. Macrophages: central regulators of iron balance. Metallomics. 2014;6(8):1336–45.
Theurl I, Hilgendorf I, Nairz M, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med. 2016;22(8):945–51.
Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17(5):306–21.
Yuan D, Huang S, Berger E, et al. Kupffer cell-derived tnf triggers cholangiocellular tumorigenesis through jnk due to chronic mitochondrial dysfunction and ROS. Cancer Cell. 2017;31(6):771-89.e6.
Ma HY, Yamamoto G, Xu J, et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J Hepatol. 2020;72(5):946–59.
Song K, Kwon H, Han C, et al. Yes-associated protein in kupffer cells enhances the production of proinflammatory cytokines and promotes the development of nonalcoholic steatohepatitis. Hepatology. 2020;72(1):72–87.
Dal-Secco D, Wang J, Zeng Z, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med. 2015;212(4):447–56.
Mossanen JC, Krenkel O, Ergen C, et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology. 2016;64(5):1667–82.
Wen Y, Lambrecht J, Ju C, et al. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. 2021;18(1):45–56.
Bonnardel J, T’Jonck W, Gaublomme D, et al. Stellate cells, hepatocytes, and endothelial cells imprint the kupffer cell identity on monocytes colonizing the liver macrophage niche. Immunity. 2019;51(4):638-54.e9.
Louwe PA, Badiola Gomez L, Webster H, et al. Recruited macrophages that colonize the post-inflammatory peritoneal niche convert into functionally divergent resident cells. Nat Commun. 2021;12(1):1770.
Scott CL, T’Jonck W, Martens L, et al. The transcription factor ZEB2 is required to maintain the tissue-specific identities of macrophages. Immunity. 2018;49(2):312-25.e5.
Shaw TN, Houston SA, Wemyss K, et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J Exp Med. 2018;215(6):1507–18.
Dick SA, Macklin JA, Nejat S, et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20(1):29–39.
Siwicki M, Gort-Freitas NA, Messemaker M, et al. Resident Kupffer cells and neutrophils drive liver toxicity in cancer immunotherapy. Sci Immunol. 2021. https://doi.org/10.1126/sciimmunol.abi7083.
Lefere S, Degroote H, van Vlierberghe H, et al. Unveiling the depletion of Kupffer cells in experimental hepatocarcinogenesis through liver macrophage subtype-specific markers. J Hepatol. 2019;71(3):631–3.
Heymann F, Peusquens J, Ludwig-Portugall I, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. 2015;62(1):279–91.
Macparland SA, Liu JC, Ma XZ, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. 2018;9(1):4383.
Aizarani N, Saviano A, Sagar N, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. 2019;572(7768):199–204.
Ramachandran P, Dobie R, Wilson-Kanamori JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575(7783):512–8.
Blériot C, Barreby E, Dunsmore G, et al. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity. 2021;54(9):2101-16.e6.
Dou L, Shi X, He X, et al. Macrophage phenotype and function in liver disorder. Front Immunol. 2019;10:3112.
de Simone G, Andreata F, Bleriot C, et al. Identification of a Kupffer cell subset capable of reverting the T cell dysfunction induced by hepatocellular priming. Immunity. 2021;54(9):2089-100.e8.
Sharma A, Seow JJW, Dutertre CA, et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. 2020;183(2):377-94.e21.
Li H, Wu K, Tao K, et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology. 2012;56(4):1342–51.
Wu K, Kryczek I, Chen L, et al. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7–H1/programmed death-1 interactions. Cancer Res. 2009;69(20):8067–75.
Sheng J, Zhang J, Wang L, et al. Topological analysis of hepatocellular carcinoma tumour microenvironment based on imaging mass cytometry reveals cellular neighbourhood regulated reversely by macrophages with different ontogeny. Gut. 2022;71(6):1176–91.
Wu J, Li J, Salcedo R, et al. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012;72(16):3977–86.
Li D, Zhang X, Jiang L. Molecular mechanism and potential therapeutic targets of liver metastasis from gastric cancer. Front Oncol. 2022;12:1000807.
Wen SW, Ager EI, Christophi C. Bimodal role of Kupffer cells during colorectal cancer liver metastasis. Cancer Biol Ther. 2013;14(7):606–13.
García-Pérez R, Ferrer Fábrega J, Varona-Bosque A, et al. Role of Kupffer cells in the progression of CRC liver metastases after the first stage of ALPPS. Sci Rep. 2018;8(1):8089.
Liu W, Zhou X, Yao Q, et al. In situ expansion and reprogramming of Kupffer cells elicit potent tumoricidal immunity against liver metastasis. J Clin Invest. 2023. https://doi.org/10.1172/JCI157937.
Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32:367–402.
Hohsfield LA, Najafi AR, Ghorbanian Y, et al. Subventricular zone/white matter microglia reconstitute the empty adult microglial niche in a dynamic wave. Elife. 2021. https://doi.org/10.7554/eLife.66738.
Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol. 2017;79:619–43.
Salter MW, Stevens B. Microglia emerge as central players in brain disease [J]. Nat Med. 2017;23(9):1018–27.
Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–5.
Li Q, Cheng Z, Zhou L, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron. 2019;101(2):207-23.e10.
Hammond TR, Dufort C, Dissing-Olesen L, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50(1):253-71.e6.
Masuda T, Sankowski R, Staszewski O, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566(7744):388–92.
Böttcher C, Schlickeiser S, Sneeboer MAM, et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat Neurosci. 2019;22(1):78–90.
Kierdorf K, Erny D, Goldmann T, et al. Microglia emerge from erythromyeloid precursors via Pu. 1- and Irf8-dependent pathways. Nat Neurosci. 2013;16(3):273–80.
Lelli A, Gervais A, Colin C, et al. The NADPH oxidase Nox2 regulates VEGFR1/CSF-1R-mediated microglial chemotaxis and promotes early postnatal infiltration of phagocytes in the subventricular zone of the mouse cerebral cortex. Glia. 2013;61(9):1542–55.
Ueno M, Fujita Y, Tanaka T, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16(5):543–51.
Swinnen N, Smolders S, Avila A, et al. Complex invasion pattern of the cerebral cortex bymicroglial cells during development of the mouse embryo. Glia. 2013;61(2):150–63.
Tay TL, Savage JC, Hui CW, et al. Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J Physiol. 2017;595(6):1929–45.
Ginhoux F, Lim S, Hoeffel G, et al. Origin and differentiation of microglia. Front Cell Neurosci. 2013;7:45.
Verney C, Monier A, Fallet-Bianco C, et al. Early microglial colonization of the human forebrain and possible involvement in periventricular white-matter injury of preterm infants. J Anat. 2010;217(4):436–48.
Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014;11:98.
Frade JM, Barde YA. Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron. 1998;20(1):35–41.
Wakselman S, Bechade C, Roumier A, et al. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci. 2008;28(32):8138–43.
Roumier A, Bechade C, Poncer JC, et al. Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J Neurosci. 2004;24(50):11421–8.
Sankowski R, Böttcher C, Masuda T, et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat Neurosci. 2019;22(12):2098–110.
Guldner IH, Wang Q, Yang L, et al. CNS-native myeloid cells drive immune suppression in the brain metastatic niche through Cxcl10. Cell. 2020;183(5):1234-48.e25.
Elmore MR, Najafi AR, Koike MA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–97.
Buttgereit A, Lelios I, Yu X, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol. 2016;17(12):1397–406.
Feinberg PA, Becker SC, Chung L, et al. Elevated TNF-α leads to neural circuit instability in the absence of interferon regulatory factor 8. J Neurosci. 2022;42(32):6171–85.
Zhou N, Liu K, Sun Y, et al. Transcriptional mechanism of IRF8 and PU.1 governs microglial activation in neurodegenerative condition. Protein Cell. 2019;10(2):87–103.
Huang Y, Xu Z, Xiong S, et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci. 2018;21(4):530–40.
Zhang P, Schlecht A, Wolf J, et al. The role of interferon regulatory factor 8 for retinal tissue homeostasis and development of choroidal neovascularisation. J Neuroinflammation. 2021;18(1):215.
Kenkhuis B, Somarakis A, Kleindouwel LRT, et al. Co-expression patterns of microglia markers Iba1, TMEM119 and P2RY12 in Alzheimer’s disease. Neurobiol Dis. 2022;167:105684.
Jordao MJC, Sankowski R, Brendecke SM, et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. 2019;363(6425):7554.
Amici SA, Dong J, Guerau-De-arellano M. Molecular mechanisms modulating the phenotype of macrophages and microglia. Front Immunol. 2017;8:1520.
Bennett ML, Bennett FC, Liddelow SA, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113(12):E1738–46.
Butovsky O, Siddiqui S, Gabriely G, et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest. 2012;122(9):3063–87.
Khan F, Pang L, Dunterman M, et al. Macrophages and microglia in glioblastoma: heterogeneity, plasticity, and therapy. J Clin Invest. 2023. https://doi.org/10.1172/JCI163446.
Jurga AM, Paleczna M, Kuter KZ. Overview of general and discriminating markers of differential microglia phenotypes. Front Cell Neurosci. 2020;14:198.
Benmamar-Badel A, Owens T, Wlodarczyk A. Protective microglial subset in development, aging, and disease: lessons from transcriptomic studies. Front Immunol. 2020;11:430.
Haage V, Semtner M, Vidal RO, et al. Correction to: Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathol Commun. 2020;8(1):1.
Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 2018;19(10):622–35.
Lavin Y, Winter D, Blecher-Gonen R, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159(6):1312–26.
Silvin A, Ginhoux F. Microglia heterogeneity along a spatio-temporal axis: more questions than answers. Glia. 2018;66(10):2045–57.
Bettinger I, Thanos S, Paulus W. Microglia promote glioma migration. Acta Neuropathol. 2002;103(4):351–5.
Coniglio SJ, Eugenin E, Dobrenis K, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012;18(1):519–27.
Carvalho Da Fonseca AC, Wang H, Fan H, et al. Increased expression of stress inducible protein 1 in glioma-associated microglia/macrophage. J Neuroimmunol. 2014;274(1–2):71–7.
Wesolowska A, Kwiatkowska A, Slomnicki L, et al. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion–an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene. 2008;27(7):918–30.
Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–72.
Zhang J, Sarkar S, Cua R, et al. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis. 2012;33(2):312–9.
Saederup N, Cardona AE, Croft K, et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE. 2010;5(10):e13693.
Wu J, Frady LN, Bash RE, et al. MerTK as a therapeutic target in glioblastoma. Neuro Oncol. 2018;20(1):92–102.
Akkari L, Bowman RL, Tessier J, et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci Transl Med. 2020. https://doi.org/10.1126/scitranslmed.aaw7843.
Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–21.
Fidler IJ. The biology of brain metastasis: challenges for therapy. Cancer J. 2015;21(4):284–93.
He BP, Wang JJ, Zhang X, et al. Differential reactions of microglia to brain metastasis of lung cancer. Mol Med. 2006;12(7–8):161–70.
Jin Y, Kang Y, Wang M, et al. Targeting polarized phenotype of microglia via IL6/JAK2/STAT3 signaling to reduce NSCLC brain metastasis. Signal Transduct Target Ther. 2022;7(1):52.
Foo SL, Sachaphibulkij K, Lee CLY, et al. Breast cancer metastasis to brain results in recruitment and activation of microglia through annexin-A1/formyl peptide receptor signaling. Breast Cancer Res. 2022;24(1):25.
Zhang C, Zhang F, Tsan R, et al. Transforming growth factor-beta2 is a molecular determinant for site-specific melanoma metastasis in the brain. Cancer Res. 2009;69(3):828–35.
Benbenishty A, Gadrich M, Cottarelli A, et al. Prophylactic TLR9 stimulation reduces brain metastasis through microglia activation. PLoS Biol. 2019;17(3):e2006859.
Veillette A, Chen J. SIRPalpha-CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. 2018;39(3):173–84.
Willingham SB, Volkmer JP, Gentles AJ, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109(17):6662–7.
Hutter G, Theruvath J, Graef CM, et al. Microglia are effector cells of CD47-SIRPalpha antiphagocytic axis disruption against glioblastoma. Proc Natl Acad Sci U S A. 2019;116(3):997–1006.
Karimi E, Yu MW, Maritan SM, et al. Single-cell spatial immune landscapes of primary and metastatic brain tumours. Nature. 2023;614(7948):555–63.
Soto MS, Sibson NR. The multifarious role of microglia in brain metastasis. Front Cell Neurosci. 2018;12:414.
Zhang X, Li X, Wang Y, et al. Abnormal lipid metabolism in epidermal Langerhans cells mediates psoriasis-like dermatitis. JCI Insight. 2022. https://doi.org/10.1172/jci.insight.150223.
Marschall P, Wei R, Segaud J, et al. Dual function of Langerhans cells in skin TSLP-promoted T(FH) differentiation in mouse atopic dermatitis. J Allergy Clin Immunol. 2021;147(5):1778–94.
Tanaka R, Ichimura Y, Kubota N, et al. The role of PD-L1 on langerhans cells in the regulation of psoriasis. J Invest Dermatol. 2022;142(12):3167-74.e9.
Atmatzidis DH, Lambert WC, Lambert MW. Langerhans cell: exciting developments in health and disease. J Eur Acad Dermatol Venereol. 2017;31(11):1817–24.
Capucha T, Mizraji G, Segev H, et al. Distinct murine mucosal langerhans cell subsets develop from pre-dendritic cells and monocytes. Immunity. 2015;43(2):369–81.
Chorro L, Sarde A, Li M, et al. Langerhans cell (LC) proliferation mediates neonatal development, homeostasis, and inflammation-associated expansion of the epidermal LC network. J Exp Med. 2009;206(13):3089–100.
Hoeffel G, Wang Y, Greter M, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med. 2012;209(6):1167–81.
Ginhoux F, Schultze JL, Murray PJ, et al. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. 2016;17(1):34–40.
Yao Y, Martin C, Yin C, et al. Micro RNAs are required for Langerhans cell, skin- and lung-resident macrophage ontogeny. J Allergy Clin Immunol. 2018;142(3):976-8.e2.
Hovav AH. Mucosal and skin langerhans cells—nurture calls. Trends Immunol. 2018;39(10):788–800.
Horev Y, Salameh R, Nassar M, et al. Niche rather than origin dysregulates mucosal Langerhans cells development in aged mice. Mucosal Immunol. 2020;13(5):767–76.
Sparber F, Dolowschiak T, Mertens S, et al. Langerin+ DCs regulate innate IL-17 production in the oral mucosa during Candida albicans-mediated infection. PLoS Pathog. 2018;14(5):e1007069.
Saba Y, Aizenbud I, Matanes D, et al. Early antitumor activity of oral Langerhans cells is compromised by a carcinogen. Proc Natl Acad Sci U S A. 2022. https://doi.org/10.1073/pnas.2118424119.
Feuerstein R, Kolter J, Henneke P. Dynamic interactions between dermal macrophages and Staphylococcus aureus. J Leukoc Biol. 2017;101(1):99–106.
Tamoutounour S, Guilliams M, Montanana Sanchis F, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity. 2013;39(5):925–38.
Lee SH, Charmoy M, Romano A, et al. Mannose receptor high, M2 dermal macrophages mediate nonhealing Leishmania major infection in a Th1 immune environment. J Exp Med. 2018;215(1):357–75.
Zhang X, Gu J, Yu FS, et al. TGF-β1-induced transcription factor networks in Langerhans cell development and maintenance. Allergy. 2016;71(6):758–64.
Lonardi S, Scutera S, Licini S, et al. CSF1R is required for differentiation and migration of langerhans cells and langerhans cell histiocytosis. Cancer Immunol Res. 2020;8(6):829–41.
Hochgerner M, Bauer T, Zyulina V, et al. BMPR1a Is required for the optimal TGFβ1-dependent CD207(+) langerhans cell differentiation and limits skin Inflammation through CD11c(+) Cells. J Invest Dermatol. 2022;142(9):2446-54.e3.
Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. 2014. https://doi.org/10.1101/cshperspect.a021857.
Chopin M, Seillet C, Chevrier S, et al. Langerhans cells are generated by two distinct PU1-dependent transcriptional networks. J Exp Med. 2013;210(13):2967–80.
Huang L, Li GH, Yu Q, et al. Smad2/4 signaling pathway is critical for epidermal langerhans cell repopulation under inflammatory condition but not required for their homeostasis at steady state. Front Immunol. 2020;11:912.
Strobl H, Krump C, Borek I. Micro-environmental signals directing human epidermal Langerhans cell differentiation. Semin Cell Dev Biol. 2019;86:36–43.
Bellmann L, Zelle-Rieser C, Milne P, et al. Notch-mediated generation of monocyte-derived langerhans cells: phenotype and function. J Invest Dermatol. 2021;141(1):84-94.e6.
Wu X, Briseño CG, Durai V, et al. Mafb lineage tracing to distinguish macrophages from other immune lineages reveals dual identity of Langerhans cells. J Exp Med. 2016;213(12):2553–65.
Satpathy AT, Kc W, Albring JC, et al. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med. 2012;209(6):1135–52.
Mielcarek M, Kirkorian AY, Hackman RC, et al. Langerhans cell homeostasis and turnover after nonmyeloablative and myeloablative allogeneic hematopoietic cell transplantation. Transplantation. 2014;98(5):563–8.
Rozis G, Benlahrech A, Duraisingham S, et al. Human Langerhans’ cells and dermal-type dendritic cells generated from CD34 stem cells express different toll-like receptors and secrete different cytokines in response to toll-like receptor ligands. Immunology. 2008;124(3):329–38.
Mizumoto N, Takashima A. CD1a and langerin: acting as more than Langerhans cell markers. J Clin Invest. 2004;113(5):658–60.
Zhou N, Ge Y, Fang K, et al. BRAF wild-type recurrent indeterminate dendritic cell tumour presenting with leonine facies. J Eur Acad Dermatol Venereol. 2020;34(5):e230–1.
Maarifi G, Czubala MA, Lagisquet J, et al. Langerin (CD207) represents a novel interferon-stimulated gene in Langerhans cells. Cell Mol Immunol. 2020;17(5):547–9.
Romani N, Clausen BE, Stoitzner P. Langerhans cells and more: langerin-expressing dendritic cell subsets in the skin. Immunol Rev. 2010;234(1):120–41.
Liu X, Zhu R, Luo Y, et al. Distinct human Langerhans cell subsets orchestrate reciprocal functions and require different developmental regulation. Immunity. 2021;54(10):2305-20.e11.
Luo Y, Wang S, Liu X, et al. Langerhans cells mediate the skin-induced tolerance to ovalbumin via Langerin in a murine model. Allergy. 2019;74(9):1738–47.
Collin M, Bigley V. Many Langerhans make light work of skin immunity. Immunity. 2021;54(10):2188–90.
Becerril-García MA, Yam-Puc JC, Maqueda-Alfaro R, et al. Langerhans cells from mice at birth express endocytic- and pattern recognition-receptors, migrate to draining lymph nodes ferrying antigen and activate neonatal T Cells in vivo. Front Immunol. 2020;11:744.
Fujita H, Suárez-Fariñas M, Mitsui H, et al. Langerhans cells from human cutaneous squamous cell carcinoma induce strong type 1 immunity. J Invest Dermatol. 2012;132(6):1645–55.
Saeidi V, Doudican N, Carucci JA. Understanding the squamous cell carcinoma immune microenvironment. Front Immunol. 2023;14:1084873.
Fehres CM, Duinkerken S, Bruijns SC, et al. Langerin-mediated internalization of a modified peptide routes antigens to early endosomes and enhances cross-presentation by human Langerhans cells. Cell Mol Immunol. 2017;14(4):360–70.
Modi BG, Neustadter J, Binda E, et al. Langerhans cells facilitate epithelial DNA damage and squamous cell carcinoma. Science. 2012;335(6064):104–8.
Howell R, Davies J, Clarke MA, et al. Localized immune surveillance of primary melanoma in the skin deciphered through executable modeling. Sci Adv. 2023;9(15):1992.
Maraee A, Farag AGA, Gadallah MM, et al. Tumour-infiltrating Langerhans cells in non-melanoma skin cancer, a clinical and immunohistochemical study. Ecancermedicalscience. 2020;14:1045.
Pogorzelska-Dyrbuś J, Szepietowski JC. Density of langerhans cells in nonmelanoma skin cancers: a systematic review. Mediators Inflamm. 2020;2020:8745863.
Okumura K, Saito M, Yoshizawa Y, et al. Pak1 maintains epidermal stem cells by regulating Langerhans cells and is required for skin carcinogenesis. Oncogene. 2020;39(24):4756–69.
Neagu M, Constantin C, Jugulete G, et al. Langerhans cells-revising their role in skin pathologies. J Pers Med. 2022. https://doi.org/10.3390/jpm12122072.
Wamhoff EC, Schulze J, Bellmann L, et al. A specific, glycomimetic langerin ligand for human langerhans cell targeting. ACS Cent Sci. 2019;5(5):808–20.
Kozaka S, Tahara Y, Wakabayashi R, et al. Transcutaneous cancer vaccine using a reverse micellar antigen carrier. Mol Pharm. 2020;17(2):645–55.
Viola MF, Boeckxstaens G. Niche-specific functional heterogeneity of intestinal resident macrophages. Gut. 2021;70(7):1383–95.
Bain CC, Bravo-Blas A, Scott CL, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15(10):929–37.
Bain CC, Scott CL, Uronen-Hansson H, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6(3):498–510.
Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Front Immunol. 2018;9:2733.
Muller PA, Matheis F, Mucida D. Gut macrophages: key players in intestinal immunity and tissue physiology. Curr Opin Immunol. 2020;62:54–61.
Gabanyi I, Muller PA, Feighery L, et al. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell. 2016;164(3):378–91.
Bujko A, Atlasy N, Landsverk OJB, et al. Transcriptional and functional profiling defines human small intestinal macrophage subsets. J Exp Med. 2018;215(2):441–58.
Muller PA, Koscso B, Rajani GM, et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell. 2014;158(2):300–13.
Hine AM, Loke P. Intestinal macrophages in resolving inflammation. J Immunol. 2019;203(3):593–9.
Kuhn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75(2):263–74.
Zigmond E, Jung S. Intestinal macrophages: well educated exceptions from the rule. Trends Immunol. 2013;34(4):162–8.
Sehgal A, Donaldson DS, Pridans C, et al. The role of CSF1R-dependent macrophages in control of the intestinal stem-cell niche. Nat Commun. 2018;9(1):1272.
Viola MF, Boeckxstaens G. Intestinal resident macrophages: multitaskers of the gut. Neurogastroenterol Motil. 2020;32(8):e13843.
Froh M, Thurman RG, Wheeler MD. Molecular evidence for a glycine-gated chloride channel in macrophages and leukocytes. Am J Physiol Gastrointest Liver Physiol. 2002;283(4):G856–63.
Ho WZ, Lai JP, Zhu XH, et al. Human monocytes and macrophages express substance P and neurokinin-1 receptor. J Immunol. 1997;159(11):5654–60.
Marques-Da-silva C, Burnstock G, Ojcius DM, et al. Purinergic receptor agonists modulate phagocytosis and clearance of apoptotic cells in macrophages. Immunobiology. 2011;216(1–2):1–11.
Nemethova A, Michel K, Gomez-Pinilla PJ, et al. Nicotine attenuates activation of tissue resident macrophages in the mouse stomach through the beta2 nicotinic acetylcholine receptor. PLoS ONE. 2013;8(11):e79264.
Farro G, Gomez-Pinilla PJ, di Giovangiulio M, et al. Smooth muscle and neural dysfunction contribute to different phases of murine postoperative ileus. Neurogastroenterol Motil. 2016;28(6):934–47.
Türler A, Schwarz NT, Türler E, et al. MCP-1 causes leukocyte recruitment and subsequently endotoxemic ileus in rat. Am J Physiol Gastrointest Liver Physiol. 2002;282(1):G145–55.
Sica A, Larghi P, Mancino A, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18(5):349–55.
Edin S, Wikberg ML, Rutegard J, et al. Phenotypic skewing of macrophages in vitro by secreted factors from colorectal cancer cells. PLoS ONE. 2013;8(9):e74982.
Pello OM, de Pizzol M, Mirolo M, et al. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood. 2012;119(2):411–21.
Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–63.
Kamei DJ, Shiguihara RS, Araujo FR. Neuroendocrine tumor of the small intestine: case report. Arq Bras Cir Dig. 2020;33(1):e1492.
Yan H, Yin L, Han H, et al. Relationship between primary tumor resection for metastatic small intestine neuroendocrine tumors and survival: a propensity score-matched analysis. J Invest Surg. 2022;35(6):1239–47.
Nakanishi R, Henmi S, Marusawa H. Small bowel bleeding caused by myeloid sarcoma. Intern Med. 2022;61(1):123–4.
Franze E, Laudisi F, di Grazia A, et al. Macrophages produce and functionally respond to interleukin-34 in colon cancer. Cell Death Discov. 2020;6(1):117.
Soncin I, Sheng J, Chen Q, et al. The tumour microenvironment creates a niche for the self-renewal of tumour-promoting macrophages in colon adenoma. Nat Commun. 2018;9(1):582.
Hiemstra IH, Beijer MR, Veninga H, et al. The identification and developmental requirements of colonic CD169(+) macrophages. Immunology. 2014;142(2):269–78.
Ohnishi K, Komohara Y, Saito Y, et al. CD169-positive macrophages in regional lymph nodes are associated with a favorable prognosis in patients with colorectal carcinoma. Cancer Sci. 2013;104(9):1237–44.
Lim SY, Yuzhalin AE, Gordon-Weeks AN, et al. Tumor-infiltrating monocytes/macrophages promote tumor invasion and migration by upregulating S100A8 and S100A9 expression in cancer cells. Oncogene. 2016;35(44):5735–45.
Wei C, Yang C, Wang S, et al. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol Cancer. 2019;18(1):64.
Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16(7):447–62.
Schultze JL. Reprogramming of macrophages–new opportunities for therapeutic targeting. Curr Opin Pharmacol. 2016;26:10–5.
Blériot C, Chakarov S, Ginhoux F. Determinants of resident tissue macrophage identity and function. Immunity. 2020;52(6):957–70.
Buechler MB, Kim KW, Onufer EJ, et al. A stromal niche defined by expression of the transcription factor WT1 mediates programming and homeostasis of cavity-resident macrophages. Immunity. 2019;51(1):119-30.e5.
Robinson A, Burgess M, Webb S, et al. Systemic influences of mammary cancer on monocytes in mice. Cancers (Basel). 2022. https://doi.org/10.3390/cancers14030833.
Ma RY, Zhang H, Li XF, et al. Monocyte-derived macrophages promote breast cancer bone metastasis outgrowth. J Exp Med. 2020. https://doi.org/10.1084/jem.20191820.
Baer JM, Zuo C, Kang LI, et al. Fibrosis induced by resident macrophages has divergent roles in pancreas inflammatory injury and PDAC. Nat Immunol. 2023;24(9):1443–57.
Matsumura H, Kondo T, Ogawa K, et al. Kupffer cells decrease metastasis of colon cancer cells to the liver in the early stage. Int J Oncol. 2014;45(6):2303–10.
Fritz JM, Tennis MA, Orlicky DJ, et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front Immunol. 2014;5:587.
Nalio Ramos R, Missolo-Koussou Y, Gerber-Ferder Y, et al. Cell. 2022;185(7):1189-207.e25.
Chow A, Schad S, Green MD, et al. Tim-4(+) cavity-resident macrophages impair anti-tumor CD8(+) T cell immunity. Cancer Cell. 2021;39(7):973-88.e9.
Etzerodt A, Moulin M, Doktor TK, et al. Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J Exp Med. 2020. https://doi.org/10.1084/jem.20191869.
Guo S, Chen X, Guo C, et al. Tumour-associated macrophages heterogeneity drives resistance to clinical therapy. Expert Rev Mol Med. 2022;24:e17.
Acknowledgements
The figures were created with BioRender.com.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81903135), Sichuan Science and technology Foundation Project, China (Grant No. 2023NSFSC0720, 2023NSFSC0703, 2023YFH0079), Open Project of Sichuan Provincial Key Laboratory for Clinical Immunology Translational Medicine (Grant No. LCMYZHYX-KFKT202306), Fundamental Research Funds for the Central Universities (Grant No. ZYGX2021YGCX008), Sichuan Provincial Cadre Health Research Project (Grant No. 2023–803) and Sichuan Special Funding for Postdoctoral Research Projects (Grant No. TB2022090).
Author information
Authors and Affiliations
Contributions
MC, ZW, WL, WL and BX collected the related paper and wrote the original manuscript and made figures. MC, ZW, WL, WL, BX, JZ, XL and YW participated in the design of this review. WL, JZ, YW and YZ obtained funding. SZ, SL and JL gave constructive guidance and made critical revisions. YZ participated in the revise of this review and gave constructive guidance. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cao, M., Wang, Z., Lan, W. et al. The roles of tissue resident macrophages in health and cancer. Exp Hematol Oncol 13, 3 (2024). https://doi.org/10.1186/s40164-023-00469-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-023-00469-0