
Abstract
Programmed cell death protein 1(PD-1) is a type of immune-inhibitory checkpoint protein, which delivers inhibitory signals to cytotoxic T cells by binding to the programmed death ligand-1 (PD-L1) displayed on the surface of cancer cells. Antibodies blocking PD-1/PD-L1 interaction have been extensively used in treatment of human malignancies and have achieved promising outcomes in recent years. However, gradual development of resistance to PD-1/PD-L1 blockade has decreased the effectiveness of this immunotherapy in cancer patients. The underlying epigenetic mechanisms need to be elucidated for application of novel strategies overcoming this immunotherapy resistance. Epigenetic aberrations contribute to cancerogenesis by promoting different hallmarks of cancer. Moreover, these alterations may lead to therapy resistance, thereby leading to poor prognosis. Recently, the epigenetic regulatory drugs have been shown to decrease the resistance to PD-1/PD-L1 inhibitors in certain cancer patients. Inhibitors of the non-coding RNAs, DNA methyltransferases, and histone deacetylases combined with PD-1/PD-L1 inhibitors have shown considerable therapeutic efficacy against carcinomas as well as blood cancers. Importantly, DNA methylation-mediated epigenetic silencing can inhibit antigen processing and presentation, which promotes cancerogenesis and aggravates resistance to PD-1/PD-L1 blockade immunotherapy. These observations altogether suggest that the combination of the epigenetic regulatory drugs with PD-1/PD-L1 inhibitors may present potential solution to the resistance caused by monotherapy with PD-1/PD-L1 inhibitors.
Similar content being viewed by others
Introduction
Over the past few years, cancer immunotherapy has been at the forefront of research. A breakthrough in the treatment of advanced stage cancers has been the targeting of immunological checkpoints, particularly the interplay between programmed death 1 (PD-1) and programmed death ligand 1 (PD-L1). PD-1 inhibits T cell activation in the tumor microenvironment by binding to two ligands: PD-L1 and PD-L2 expressed on the surface of tumor cells [1,2,3,4]. PD-1/PD-L1 inhibitors can boost T cell effector activity against tumor cells by interfering with their interactions. Multiple solid tumors, including non-small cell lung cancer (NSCLC) [5], melanoma [6], triple negative breast cancer (TNBC] [7], liver cancer [8], and cervical cancer [9], as well as blood cancers like Hodgkin’s lymphoma (HL) [10] and NK/T-cell lymphoma [11], have been treated with PD-1/PD-L1 inhibitory drugs. However, drug resistance has gradually developed as a result of the widespread use of PD-1/PD-L1 inhibitors in cancer treatments. A majority of patients developed resistance to PD-1/PD-L1 inhibitors even if they initially showed good response to PD-1/PD-L1 therapy [12].
Among NSCLC patients treated with PD-1 inhibitors, almost half of the patients developed drug resistance [13]. The development of NSCLC was shown to be associated with genomic instability promoted by alterations in DNA methylation patterns [14, 15]. Among melanoma patients with resistance to PD-1 antibodies, the therapeutic outcomes were reported to be associated with the expression levels of certain non-coding RNAs [16]. In NSCLC, for instance, it was demonstrated that p53 down-regulates PD-L1 via miR-34, which could serve as a predictive biomarker for PD-1 inhibitor immunotherapy [17, 18]. Histone deacetylase 6 (HDAC6), which has the potential to be a predictive biomarker for melanoma and ovarian cancer[19, 20], also regulates epigenetic resistance to PD-1 immunotherapy in melanoma patients. Numerous studies on the epigenetic modulation of the PD-1/PD-L1 immune checkpoints have shown that a variety of epigenetic inheritance mechanisms play a significant role in the interaction between epigenetic and immune modulation [21, 22]. Here, we summarized the potential clinical benefits of epigenetic regulation on reversing resistance to PD-1/PD-L1 blockade for patients with cancers.
Epigenetic therapies in cancer
The term ‘epigenetics’ defines all meiotically and mitotically heritable changes in gene expression that are not coded in the DNA sequence itself. Three epigenetic mechanisms, that is, DNA methylations, histone modifications, and non-coding RNAs, were shown to initiate and sustain epigenetic silencing [23]. Recently, numerous studies have suggested that epigenetic alterations play critical roles in a wide range of cancer types [24, 25]. Cancer-associated epigenetic alterations in tumors reveal potentially reversible targets for existing drugs, and for an increasing repertoire of new drugs [26, 27].
The types of epigenetic aberrations in cancer
Methylation of the C5 position of cytosine bases in the context of CpG dinucleotides in DNA has long been recognized as an epigenetic silencing mechanism of fundamental importance [28]. The methylation of CpG sites within the human genome is generated or maintained by a number of DNA methyltransferases (DNMTs) that have multifaceted roles, including silencing of transposable elements, defense against viral sequences, and transcriptional repression of certain genes [29]. Aberrant methylation of CpG islands is a hallmark of human cancers and is observed early during carcinogenesis [30]. The aberrant silencing of genes, including tumor-suppressor genes, are linked to focal increases in methylation in promoter-associated CpG islands [31].
Post-translational histone modifications have also been defined as epigenetic processes playing critical roles in cancer development [32]. In general, hyperacetylation of histones activates transcription of genes, whereas hypoacetylation of them inactivates transcription [33]. Histone deacetylases (HDACs) regulate biological processes, which include autophagy, DNA damage repair, metabolism, apoptosis, senescence, and cell cycle control [34, 35, 36]. This transcriptional modulation regulates the expression of tumor suppressor, antigen-processing and presentation machinery, and tumor antigen genes that were silenced during tumorigenesis in cancer cells [37].
The role of non-coding RNAs in post-transcriptional silencing has attracted much interest. However, long noncoding RNAs (lncRNAs), in particular antisense transcripts, can also lead to mitotically heritable transcriptional silencing by the formation of heterochromatin [38, 39]. Therefore, lncRNAs might be a key trigger to direct histone modifications and DNA methylations to specific genomic loci, thereby leading to heritable and stable silencing of genes [40, 41]. The therapeutic activation of abnormally silenced genes thus requires drugs that can target the multifaceted changes. Besides, except for lncRNAs and miRNAs, circular RNAs (circRNAs) have also been identified to regulate the PD-1/PD-L1 pathway and thus participate in immune response and immunotherapy.
Epigenetic drugs used for cancer treatment
Epigenetic alterations have fundamental roles in cancer progression characterized by reversibility and susceptibility to external factors. They are emerging as promising targets for cancer therapies. Typically, transcriptional repression is linked to the actions of DNMTs and HDACs [42]. Thus, drugs targeting these proteins can augment expression of involved genes, with many consequences for pathways downstream of this gene activation. When incorporated into DNA, DNMT inhibitors (DNMTi) act as cytidine analogues that block the catalytic actions of DNMTs, thereby triggering DNA demethylation [43, 44]. Cytosine methylation at CpG dinucleotides in DNA varies significantly in almost all cancers [45, 46]. Tumor suppressor genes were reactivated by DNMTis, which induced expression of genes that were silenced by promoter DNA methylation [47]. Multiple types of tumor cells were temporarily exposed to low doses of DNMTis, which resulted in marginal changes in apoptosis or cell cycle, and diminished cancer stem cell functions [48]. Due to promising clinical efficacy of DNMTis, such as 5-azacytidine and 5-aza-2-deoxycytidine (decitabine) for treating hematologic neoplasms, these DNMTi drugs were approved by FDA for treatment of myelodysplastic syndrome, a benign neoplasm, precursor of leukemia and acute myeloid leukemia [49].
For the treatment of cutaneous T-cell lymphoma (CTCL) and peripheral T-cell lymphoma, HDAC inhibitors (HDACi) have been approved [50, 51, 52]. HDACis have pleiotropic effects, often dose and compound dependent. Some of them affect the acetylation status of non-histone and/or non-nuclear proteins or cause off-target effects [53], while others clearly cause epigenetic alterations and affect histone acetylation [54, 55]. Given the importance of epigenetic regulation in different cancer types, it is not surprising that the epigenetic targeting is becoming an attractive treatment strategy in cancer therapy. Epigenetic treatment may therefore benefit cancer patients as monotherapy or combination [56].
Resistance to PD-1/PD-L1 immunotherapy is epigenetically regulated
PD-1 is expressed not only in different kinds of tumor-infiltrating lymphocytes but also in some cancer tissues [57]. PD-1/PD-L1 interaction inhibits immune-surveillance against cancer cells through elimination of specific or non-specific immune responses that eliminate tumor cells [58]. Globally, dozens of PD-1 or PD-L1 inhibitors were approved for marketing as they yielded good clinical responses [59,60,61,62]. However, a number of recent studies have shown that anti-PD-1/PD-L1 resistance is linked to poor drug responses in some cancer patients [63, 64]. These findings underscore the need for new approaches to combating anti-PD-1/PD-L1 resistance as well as a deeper comprehension of the underlying mechanisms. The total response rate of anti-PD-1/PD-L1 treatment for cancers is less than 20% [65].
Through inducing IFN-γ activity triggered by tumor infiltrating lymphocytes (TILs), cancer cells alter PD-L1 expression [66]. DNA methylation and enhancer of zeste homolog 2 (EZH2) activity were found to induce PD-1 resistance in melanoma by inhibiting IFN-γ transcription and the RAS and PI3K pathways’ subsequent functions. The IFN- response was triggered by hypermetylation of PD-L1-regulating genes, limiting the activities of PD-1/PD-L1 blockade. On the other hand, cytotoxic T cell exhaustion is largely caused by DNA methylation. These methylation changes do have specific mechanisms which are still unclear [70].
Epigenetic processes other than DNA methylation were also reported to be associated with the resistance to PD-1 inhibitors. Modification by N6-methyladenosine (m6A) of RNA is a kind of reversible regulation. The m6A was found in most mRNA of eukaryotic cells [71]. For the melanoma cells, the fat mass and obesity-associated protein (FTO) is the first m6A demethylase identified [73]. YT521-B homology (YTH) domain family (YTHDF) proteins tend to accelerate the metabolism of mRNAs modified by m6A. The increasing FTO level decreases m6A modification of PD-1 and degradation of RNA induced by YTHDF so that the tumorigenic cells grow faster. Knocking-out FTO in the melanoma cells through in vitro experiments sensitized to IFN-γ, and increased the immune response of PD-1 inhibitor in mice melanomas. These data showed that the regulation of m6A is a potential way to eliminate the resistance to anti-PD-1 therapy [73].
The influence of epigenetic regulatory drugs on PD-1/PD-L1 immunotherapy resistance
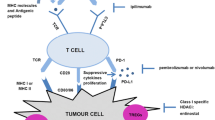
Given that there is an association between epigenetic regulation and cancer development, drugs targeting epigenetic alterations are widely used in clinical trials in combination with PD-1/PD-L1 inhibitors. The following is a list of the most frequently used epigenetic regulatory drugs: BET inhibitors, histone methyltransferase inhibitors, DNA methyltransferase inhibitors, IDH2 or TET2 inhibitors, and histone acetyltransferase inhibitors. Figure 1 demonstrates the effects of epigenetic regulatory drugs on PD-1/PD-L1 immunotherapy.

Epigenetic regulatory drugs promote anti-tumor immune signaling to synergize with PD1/PD-L1 immunotherapy
DNA methyltransferase inhibitors
DNA methylation in genomes of mammalian cells is usually generated through addition of methyl group of S-adenosylmethionine (SAM) to the upstream of promoter or cytosine residues within CpG (5’-C-phosphate-G-3’) islands at the 5’ end by DNA methyltransferases (DNMT) [74]. Genome-wide alterations in methylation levels are frequently observed in many malignant tumors with hypermethylations, especially in promoter-associated CpG islands and hypomethylations in rest of the genome[75,76,77].
DNA methyltransferase inhibitors (i.e., azacitidine and decitabine [5-aza-2’-deoxycytidine]) are used in the treatment of certain blood cancers [78, 79, 80]. Of note, decitabine was reported to increase the response rate to paclitaxel/carboplatin chemotherapy even when it is used in low-doses (7 mg/m2/day), which has more than 70% of disease control rate (DCR) in 55 patients with platinum-resistance. Indeed, the combination of cytokine-induced killer (CIK) cell immunotherapy with decitabine almost completely reversed resistance (DCR is 100%), indicating that the efficacy of immunotherapy in cancer treatment may be significantly enhanced by decitabine [81].
The combination of decitabine and PD-1 inhibitor was also evaluated in a Phase 2 trial for classical Hodgkin lymphoma (cHL) [84]. Generally, the objective response rate (ORR) for anti-PD-1 monotherapy of cancer is no more than 50%, and the complete rate (CR) is less than 10% [83]. Although patients with cHL had a high ORR of 80–90% in response to PD-1 inhibitor, CR was still no more than 20% [84]. In a clinical trial with 120 refractory classical Hodgkin lymphoma patients, addition of low-dose decitabine (10 mg/d) to anti–PD-1 antibody camrelizumab yielded an ORR of 95%, and a CR of 71% which was twice superior compared to PD-1 inhibitor monotherapy. Even in patients with anti-PD-1 monotherapy resistance, ORR and CR were estimated to be 62% and CR 28%, respectively, with decitabine plus camrelizumab. The reasons for such improvements observed in therapeutic efficacy were interpreted as the ability of decitabine in increasing sensitivity to PD-1 inhibitors [84, 85].
Histone deacetylase inhibitors
Post-translational acetylation of histones is highly dynamic and regulated by the activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs) [87]. Currently, four HDAC inhibitors have been approved by FDA: vorinostat, romidepsin, belinostat, and panobinostat. HDAC inhibitors alter RNA polymerase II-driven transcription through acute hyperacetylation of histones and epigenetic regulators at the chromatin interface [33]. This transcriptional modulation includes the re-expression of genes that were silenced during tumorigenesis in cancer cells, including tumor antigens, antigen-processing and presentation machinery genes, and tumor suppressor genes [38]. These HDAC inhibitors were approved by FDA for treatment of different types of lymphoid malignancies: (1) vorinostat and romidepsin for cutaneous T-cell lymphomas [88]; (2) romidepsin and belinostat for peripheral T-cell lymphomas [89]; and (3) panobinostat in combination with dexamethasone for the treatment of multiple myeloma [90].
The anti-PD-1 immunotherapy was considered as a potential way to improve clinical outcomes of Hodgkin lymphoma and multiple myeloma cases because PD-L1 was shown to be highly expressed in tumor cells of these patients [91, 92]. Clinical trials have also been conducted for multiple myeloma and certain other B cell malignancies in some countries [93, 94]; however, the therapeutic efficacy of monotherapy was not satisfactory [95]. In vitro experiments have revealed that HDACs might have an adjuvant function in modulation of sensitivity of the PD-1/PD-L1 signaling [98]. Hence, it is logical to use these two different types of drugs for more effective cancer treatment. Clinical trials involving a combination of PD-1/PD-L1 and HDAC inhibitors were carried out in some cancer types such as Merkel cell carcinoma (MCC). In 2019, four MCC patients with no response to anti-PD-1 monotherapy received a combination of panobinostat and anti-PD-1 therapy, and the results showed that HDACi may increase the effectiveness of anti-PD-1/PD-L1 therapy [99]. Currently, multiple clinical trials including combination of these drugs are carried out in the USA for different cancer types (Table 1).
Histone methyltransferase inhibitors
Histone methyltransferase (HMT) enzymes methylate residues on specific lysine residues of histones, which activate or repress transcription in a very residue and methyl group number-specific manner. Enhancer of zeste homologue 2 (EZH2), SET domain bifurcated 1 (SETDB1), and disruptor of telomeric silencing 1-like (DOT1L), all of which produce silencing marks, have been utilized as therapeutic targets. As a result, similar to the effects of DNMTis and HDACis, inhibiting these HMTs opens up chromatin structure and activates gene expression. Only small molecule inhibitors of EZH2 have currently been approved for clinical interventions [100].
Melanoma suffers from decreased immunogenicity and loss of antigen presentation when the expression of EZH2 is increased. Addition of an EZH2 inhibitor to anti-CTLA-4 or IL-2 treatment reversed many of above immunosuppressive effects and significantly improved immune therapy in preclinical models [101]. Zhou et al. identified EZH2 as a therapeutic target for enhancing tumor cell antigen presentation and subsequently decreasing resistance to anti-PD-1 therapy in HNSCC [102].
A histone H3 lysine 79 (H3K79) methyltransferase called DOT1L is recruited by aberrant fusion proteins that are part of the mixed-lineage leukemia (MLL) HMT. This creates a permissive chromatin state that makes it easier for HATs and BRD4 to promote leukemia through driving transcription in an abnormal way. Pharmacological inhibition or genetic depletion of DOT1L can alleviate H3K79 methylation at the promoters of pro-inflammatory cytokines such as IL-6 and IFN-β, which is reported to be mediated by DOT1L [103]. Emilie et al. found that the combination of histone deacetylase inhibitor (SAHA) with DOT1L inhibitors (EPZ5676 or SGC0946) or BET bromodomain inhibitor (PFI-1) were efficient to partially reverse TGF-β1 effects by decreasing PD-L1 expression, suggesting that combination of epigenetic compounds might enhance clinical responses to PD-L1 [104].
BET inhibitors
Functionally associated with transcriptional co-activators, bromodomain and extra-terminal (BET) proteins positively regulate RNA Pol II-dependent transcription at active enhancer and promoter regions. In a wide range of genetically diverse solid and hematological cancers, BET inhibitors have demonstrated broad efficacy. Suppressing Pol II-driven oncogenic transcription, particularly MYC and MYC-dependent transcriptional programs in hematological malignancies, is where BET inhibitors exert their anti-tumor effects [105].
In genetically diverse tumor models, a number of researchers have demonstrated that promoter- and enhancer-bound BET proteins are necessary for the transcription of immune checkpoint ligands PD-L1 and PD-L2. In addition, ectopic expression of PD-L1 in lymphoma was sufficient to reduce the efficacy of a BET inhibitor (i.e., JQ1) in vivo. Importantly, IRF1-driven PD-L1 expression induced by IFN-γ was also suppressed by BET inhibition, which is known to be an adaptive immune evasion mechanism. Changes in MYC expression, which may regulate PD-L1 in particular cellular contexts, had no effect on BET inhibition’s ability to suppress PD-L1 [106].
BET inhibitors have been shown to improve the efficacy of cancer immunotherapies in recent preclinical studies. In the context of MYC-driven B cell lymphoma, BET inhibitors exhibited improved activity when treated with anti-PD-1 and agonistic anti-CD137 (4-1BB) [107]. JQ1 increased the effectiveness of anti-PD-1 therapy in KRAS-driven NSCLC, which was linked to less CD4 + FOXP3 + regulatory T (Treg) cell infiltration [108]. These studies, taken together, suggest that BET bromodomain inhibitors activate the immune system of the host, which could be used to boost immune responses against tumors.
Non-coding RNAs
Recently, non-coding RNAs have been observed to play important roles in tumor immunity [109, 110]. Non-coding RNAs are a special class of functional RNAs including microRNAs (miRNA) and long non-coding RNAs (lncRNA), which play critical roles in initiation and development of tumors [111] miRNAs secreted by tumor cells results in immunosuppression during cancer development [112]. miR-21, miR-146, and miR-195 were all reported to promote immunity through toll-like receptor (TLR) signaling [113]. Some miRNAs such as miR-21 are regulated by the CSF1-ETS2 (V-ets erythroblastosis virus E26 oncogene homolog 2) pathway, which promotes tumorigenesis and angiogenesis due to M2 reprogramming of the tumor infiltrating myeloid cells [114].
Cancer cells have multiple immune escape mechanisms to evade T-cell responses, with PD-1 pathway being a classical example. Bioinformatics analyses showed that several key lncRNAs contribute to immune escape. An important lncRNA, EPIC1, was shown to enhance methylation of the 27th amino acid in histone H3 (H3K27) by binding the enhancer of zeste homolog 2 (EZH2) protein, leading to low transcript level of IFN-γ and inhibition of IFN-JAK-STAT1 signal pathway. Cancer cells with EPIC1 overexpression have a strong resistance to PD-1 blocking treatment [115]. According to the tight relationship between non-coding RNAs and resistance to PD-1 blockade, miR/PD-1 and lncRNA/PD-1 signaling pathways have been investigated as targets for potential cancer immunotherapy drugs. The compounds downregulating the miRNAs and lncRNAs targeting PD-1/PD-L1 axis may be candidates for new inhibitors [116].
Circular RNAs (circRNAs) were firstly found as single-stranded covalently closed circular RNA molecules structures of viroids [117]. The major functions of circRNAs include regulation of gene transcription, protein binding, serving as templates for protein translation, or miRNA sponge through which binding sites for miRNAs are supplied [118]. The expression of circRNAs was observed in different diseases and dysregulated expression may affect tumor occurrence and development [119, 120]. It has been shown that circRNAs play an important role in cancer immunology by increasing surface PD-L1 expression [121]. For example, in melanoma cells circ-0020710 binds more competitively to miR-370-3p than chemokine (C-X-C motif) ligand 12 (CXCL12) as miRNA sponge. Accumulation of CXCL12 can then recruit more immune suppressor cells which results in cytotoxic lymphocyte (CTL) exhaustion, and introduce the formation of immunosuppressive microenvironment which contributes to the resistance of PD-1/PD-L1 pathway blockade. Therefore, inhibition of circ-0020710 or CXCL12 increases efficacy of cancer treatment when combined with PD-1 inhibitors [122]. Furthermore, the expression of PD-1 in T cells and PD-L1 in cancer cells are found to be affected by multiple circRNAs, which also work on the development of resistance to PD-1/PD-L1 blockade. For PD-1 expression level in the T cells, circRNAs mostly act through miRNA sponging and influence downstream reaction of related mRNAs [123,124,125]. On the other hand, mainly through miRNA sponge function, alteration of circRNAs would affect the PD-L1 expression in cancer cells [126,127,128,129,130,131]. Figure 2 summarizes the relationship between non-coding RNAs and PD-1/PD-L1 axis in cancer cells.

The regulation of PD-1/PD-L1 axis via non-coding RNAs
Future directions and conclusions
PD-1/PD-L1 immunotherapy has proven to be an exciting and productive area of research for treatment of malignancies; however, this type of therapy only shows long-term efficacy in a minority of patients. The technologies and drugs related to epigenetic regulation have great potential for improving the clinical efficacy of PD-1/PD-L1 immunotherapy. DNMTi, HDACi, and inhibitors of non-coding RNAs altogether are promising therapeutic agents to reverse resistance to PD-1/PD-L1 blockade in a wide variety of cancers. We eagerly await results of ongoing clinical trials summarized in Table 1, which will provide information on the safety, efficacy, and potential biomarkers for these combinatory treatments. Utilizing epigenetic therapies to eliminate PD-1/PD-L1 inhibitor resistance may prove to be a safe and effective way for treating multiple types of cancer. Although combination with epigenetic drugs and inhibitors of PD-1/PD-L1 showed promising results on cancer treatment, the mechanism for such increased efficacy remains elusive, and further clinical studies are needed for elucidation of the optimal treatment conditions for the patients. The resistance induced by PD-L1 expression was regulated by two different mechanisms in cancer cells: primary resistance (intrinsic resistance) and acquired resistance. In current studies, epigenetic regulatory drugs including DNA methyltransferase inhibitors, histone deacetylase inhibitors, histone methyltransferase inhibitors, BET inhibitors, and non-coding RNAs can affect the oncogenic signaling in the cancer cells, and the resistance induced by those belongs to the primary resistance [80, 88, 104, 107]. On the other hand, some histone methyltransferase drugs, BET inhibitors and non-coding RNAs can also trigger TIL changes regulated by IFN-γ that is responsible for PD-L1 expression on immune system cells and cancer cells which reflects the mechanisms of acquired resistance [107, 116].
Data availability
Not applicable.
Change history
19 December 2022
A Correction to this paper has been published: https://doi.org/10.1186/s40164-022-00359-x
Abbreviations
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed death ligand-1
- NSCLC:
-
Non-small cell lung cancer
- TNBC:
-
Triple negative breast cancer
- HL:
-
Hodgkin’s lymphoma
- HDAC6:
-
Histone deacetylase 6
- DNMTs:
-
DNA methyltransferases
- HDACs:
-
Histone deacetylases
- lncRNAs:
-
Long noncoding RNAs
- DNMTi:
-
DNA methyltransferases inhibitors
- CTCL:
-
Cutaneous T-cell lymphoma
- TILs:
-
Tumor infiltrating lymphocytes
- EZH2:
-
Enhancer of zeste homolog 2
- SAM:
-
S-adenosylmethionine
- CIK:
-
Cytokine-induced killer
- DCR:
-
Disease control rate
- ORR:
-
Objective response rate
- CR:
-
complete rate
- HATs:
-
Histone acetyltransferases
- MCC:
-
Merkel cell carcinoma
- HMT:
-
Histone methyltransferase
- SETDB1:
-
SET domain bifurcated 1
- MLL:
-
Mixed-lineage leukemia
- BET:
-
Bromodomain and extra-terminal
- miRNA:
-
microRNAs
- TLR:
-
Toll-like receptor
- TAM:
-
Tumor-associated macrophage
- CTA:
-
Cancer testis antigens
- dsRNA:
-
Double-stranded RNA
- ERVs:
-
Endogenous retroviruses
- IFN-γ:
-
Interferon gamma
- HDACi:
-
Histone deacetylase inhibitors
- HATi:
-
Histone acetyltransferase inhibitors
- BETi:
-
BET inhibitors
References
Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224(1):166–82.
Brahmer JR. Harnessing the immune system for the treatment of non-small-cell lung cancer. J Clin Oncol. 2013;31(8):1021–8.
Miller JF, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell. 2015;27(4):439–49.
Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018;18(3):153–67.
Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, Engl. et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer.N. J Med. 2018;378(21):1976–86.
Menzies AM, Johnson DB, Ramanujam S, Atkinson VG, Wong ANM, Park JJ, et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann Oncol. 2017;28(2):368–76.
Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 StudyJ Clin Oncol. 2016, 34(21):2460–2467.
Yau T, Hsu C, Kim TY, Choo SP, Kang YK, Hou MM, et al. Nivolumab in advanced hepatocellular carcinoma: Sorafenib-experienced Asian cohort analysis. J Hepatol. 2019;71(3):543–52.
Rischin D, Gil-Martin M, González-Martin A, Braña I, Hou JY, Cho D, Falchook GS, et al. PD-1 blockade in recurrent or metastatic cervical cancer: Data from cemiplimab phase I expansion cohorts and characterization of PD-L1 expression in cervical cancer.Gynecol Oncol. 2020, 159(2): 322–328.
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–9.
Li X, Cheng Y, Zhang M, Yan J, Li L, Fu X, Zhang X, Chang Y, Sun Z, Yu H, Zhang L. Activity of pembrolizumab in relapsed/refractory NK/T-cell lymphoma. J Hematol Onco. 2018;11(1):1–8.
Syn, NLTeng, MWL.Mok, TSKA, Soo R. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017;18:e731–41.
Walsh RJ, Soo RA. Resistance to immune checkpoint inhibitors in non-small cell lung cancer: biomarkers and therapeutic strategies. Ther Adv Med Oncol. 2020;3(12):1758835920937902.
Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4(9):707–17.
Cai L, Bai H, Duan J, Wang Z, Gao S, Wang D, et al. Epigenetic alterations are associated with tumor mutation burden in non-small cell lung cancer. J immunotherapy cancer. 2019;7(1):198.
Veronica Huber V, Vallacchi V, Fleming X, Cova HA, Dugo M, et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J Clin Invest. 2018;128(12):5505–16.
Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst. 2015;108(1):303.
Ding L, Lu S, Li Y. Regulation of PD-1/PD-L1 Pathway in Cancer by Noncoding RNAs. Pathol Oncol Res. 2020;6(2):651–63.
Zhicheng Hu, Rong Y, Li S, Qu S. Shaobin Huang. Upregulated Histone Deacetylase 6 Associates with Malignant Progression of Melanoma and Predicts the Prognosis of Patients. Cancer Manage Res. 2020;12:12993–3001.
Bitler BG, Wu S, Park PH, Hai Y, Aird KM, Wang Y, et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat Cell Biol. 2017;19(8):962–73.
Yan Y, Gao R, Trinh TLP, Grant MB. Immunodeficiency in Pancreatic Adenocarcinoma with Diabetes Revealed by Comparative Genomics. Clin Cancer Res. 2017;23(20):6363–73.
Jinyang Li, Salina Yuan, Robert J Norgard, Fangxue Yan, Yu H Sun, Il-Kyu Kim, et al. Epigenetic and Transcriptional Control of the Epidermal Growth Factor Receptor Regulates the Tumor Immune Microenvironment in Pancreatic Cancer. Cancer Discov. 2021, 11(3):736-753.
Allis C, Jenuwein T, Reinberg D, Caparros M. Epigenetics. Cold Spring Harbor Lab. Press, NY, USA, 2015.
Heninger E, Krueger TE, Lang JM. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol. 2015;4(6):29.
Ali MA, Matboli M, Tarek M, Reda M, Kamal KM, Nouh M, et al. Epigenetic regulation of immune checkpoints: another target for cancer immunotherapy? Immunotherapy. 2017, 9(1): 99–108.
Jones PA, Ohtani H, Chakravarthy A, De Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer. 2019;19(3):151–61.
Gerhard Jung, Eva Hernández-Illán, Leticia Moreira, Francesc Balaguer, Ajay Goel. Epigenetics of colorectal cancer: biomarker and therapeutic potentialNat Rev Gastroenterol Hepatol. 2020 , 17(2):111-130.
Zhou Z, Li HQ, Liu F. DNA Methyltransferase Inhibitors and their Therapeutic Potential. Curr Top Med Chem. 2018, 18(28): 2448–2457.
Pan Y, Liu G, Zhou F, Su B, Li Y. DNA methylation profiles in cancer diagnosis and therapeutics. Clin Exp Med. 2018, 18(1): 1–14.
Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl Acad. Sci. USA 2002, 99(6): 3740–3745.
Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28.
Margarita E Neganova, Sergey G Klochkov, Yulia R Aleksandrova, Gjumrakch Aliev. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin Cancer Biol. 2022, 83:452-471.
Martin, B.J.E., Brind’Amour, J., Kuzmin, A. et al. Transcription shapes genome-wide histone acetylation patterns. Nat Commun. 2021;12:210. https://doi.org/10.1038/s41467-020-20543-z
Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, Foiani M. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471(7336):74-79.
King J, Patel M, Chandrasekaran S, Metabolism, HDACs, Inhibitors HDACA Systems Biology Perspective. Metabolites. 2021;11(11):792.
Zhang J, Zhong Q. Histone deacetylase inhibitors and cell death. Cell Mol Life Sci. 2014;71(20)3885–3901.
Yoon S, Eom GH. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med J. 2016;52(1):1–11.
Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci. 2017 Jul 1;18(7): 1414.
Richard I Joh, Christina M Palmieri, Ian T Hill, Mo Motamedi. Regulation of histone methylation by noncoding RNAs. Biochim Biophys Acta. 2014, 1839(12):1385-94.
Xunlei Zhang, Xingsong Zhang, Lili Shen, Li Song, Jindong Wu, Guangxin Cao, et al. Comprehensive analysis of differentially expressed lncRNAs as diagnostic and prognostic markers for colorectal cancer. Exp Ther Med. 2019, 18(6):4481-4489.
Merry CR, Forrest ME, Sabers JN, Beard L, Gao XH, Hatzoglou M, Jackson MW, Wang Z, Markowitz SD, Khalil AM. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum Mol Genet. 2015;24(21):6240–53.
Miao-Chih Tsai, Ohad Manor, Yue Wan, Nima Mosammaparast, Jordon K Wang, Fei Lan, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329(5992):689-93.
Stresemann C, Brueckner B, Musch T, Stopper H, Lyko F. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res. 2006;66:2794–800.
Hervouet E, Peixoto P, Delage-Mourroux R, Boyer-Guittaut M, Cartron PF. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin Epigenetics. 2018;10:17.
Angelique Bruyer, Ken Maes, Laurie Herviou, Alboukadel Kassambara, Anja Seckinger, Guillaume Cartron, et al. DNMTi/HDACi combined epigenetic targeted treatment induces reprogramming of myeloma cells in the direction of normal plasma cells. Br J Cancer. 2018, 118(8):1062-1073.
Das PM, Singal R. DNA methylation and cancer. J Clin Oncol. 2004 Nov 15;22(22):4632–42.
Clements EG, Mohammad HP, Leadem BR, Easwaran H, Cai Y, Van Neste L, et al. DNMT1 modulates gene expression without its catalytic activity partially through its interactions with histone-modifying enzymes. Nucleic Acids Res. 2012;40:4334–46.
Baylin SB, Jones PA. Epigenetic Determinants of Cancer. In: Allis CD, Carparros M-L, Jenuwein T, Reinberg D, editors. Epigenetics, 2nd edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratories, 2015.
Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–46.
Jeannine Diesch, Anabel Zwick, Anne-Kathrin Garz, Anna Palau, Marcus Buschbeck, Katharina S Götze. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin Epigenetics. 2016, 21;8:71.
Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109:31–9.
Zhang Q, Wang S, Chen J, Yu Z Histone Deacetylases (HDACs) Guided Novel Therapies for T-cell lymphomas.Int J Med Sci. 2019 Jan 29;16(3):424–442.
Clawson GA. Histone deacetylase inhibitors as cancer therapeutics. Ann Transl Med. 2016;4(15):287.
VanderMolen KM, McCulloch W, Pearce CJ, Oberlies NH. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. J Antibiot (Tokyo). 2011 Aug;64(8):525–31.
Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–91.
Xiaopeng Peng, Zhiqiang Sun, Peihua Kuang, Jianjun Chen. Recent progress on HDAC inhibitors with dual targeting capabilities for cancer treatmentEur J Med Chem. 202, 208:112831.
Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–7.
Salmaninejad A, Valilou SF, Shabgah AG, Aslani S, Alimardani M, et al. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J Cell Physiol. 2019;234(10):16824–37.
Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, et al. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer. 2015;112(9):1501–9.
Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. 2007 Jul;19(7):813–24.
Constantinidou A, Alifieris C, Trafalis DT. Targeting Programmed Cell Death – 1 (PD-1) and Ligand (PD-L1): A new era in cancer active immunotherapy. Pharmacol Ther. 2019, 194: 84–106.
Hayashi H, Nakagawa K. Combination therapy with PD-1 or PD-L1 inhibitors for cancer. Int. J Clin Oncol. 2020;25(5):818–30.
Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, De Macedo MP, Austin-Breneman JL, Jiang H, Chang Q, Reddy SM, Chen WS, Tetzlaff MT, Broaddus RJ, Davies MA, Gershenwald JE, Haydu L, Lazar AJ, Patel SP, Hwu P, Hwu WJ, Diab A, Glitza IC, Woodman SE, Vence LM, Wistuba II, Amaria RN, Kwong LN, Prieto V, Davis RE, Ma W, Overwijk WW, Sharpe AH, Hu J, Futreal PA, Blando J, Sharma P, Allison JP, Chin L, Wargo JA. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016;6(8):827–37.
Armand, P., Shipp, M. A., Ribrag, V., Michot, J.-M., Zinzani, P, et al. Programmed death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after brentuximab vedotin failure. Journal of Clinical Oncology. 2016, 34(31): 3733-3739.
Maria de Miguel, Emiliano Calvo.Clinical Challenges of Immune Checkpoint Inhibitors Cancer Cell. 2020, 38(3):326-333.
Asano T, Meguri Y, Yoshioka T, Kishi Y, Iwamoto M, Nakamura M, et al. PD-1 modulates regulatory T-cell homeostasis during low-dose interleukin-2 therapy. Blood. 129(15): 2186–2197.
Wakabayashi G, Lee YC, Luh F, Kuo CN, Chang WC, Yen Y. Development and clinical applications of cancer immunotherapy against PD-1 signaling pathway. J Biomed Sci. 2019, 26(1): 96.
Yuan Y, Adam A, Zhao C, Chen H. Recent Advancements in the Mechanisms Underlying Resistance toPD-1/PD-L1 Blockade Immunotherapy. Cancers (Basel). 2021, 13(4): 663.
Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016, 22(2): 128–134.
Wu X, Gu Z, Chen Y, Chen B, Chen W, Weng L, et al. Application of PD-1 Blockade in Cancer Immunotherapy. Comput Struct Biotechnol J. 2019;17:661–74.
Emran AA, Chatterjee A, Rodger EJ, et al. Targeting DNA methylation and EZH2 activity to overcome melanoma resistance to immunotherapy. Trends Immunol. 2019;40:328–44.
Stephanie Oerum, Vincent Meynier, Marjorie Catala, Carine Tisné. A comprehensive review of m6A/m6Am RNA methyltransferase structures. Nucleic Acids Res. 2021, 49(13):7239-7255.
Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10(1):2782.
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013, 38(1): 23–38.
Jin B, Robertson KD. DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol. 2013;754:3–29.
Wong KY, Chim CS. DNA methylation of tumor suppressor protein-coding and non-coding genes in multiple myeloma. Epigenomics. 2015;7(6):985–1001.
Giri AK, Aittokallio T. DNMT Inhibitors Increase Methylation in the Cancer Genome. Front Pharmacol. 2019;10:385.
Küçük C, Hu X, Jiang B, Klinkebiel D, Geng H, Gong Q, et al. Global promoter methylation analysis reveals novel candidate tumor suppressor genes in natural killer cell lymphoma. Clin Cancer Res. 2015;21(7):1699–711.
Kaminskas E, Farrell AT, Wang YC, et al. FDA drug approval summary: azacitidine (5-azacytidine, VidazaTM) for injectable suspension. Oncologist. 2005;10(3):176–82.
Gore SD, Jones C, Kirkpatrick P. Decitabine[J]. Nat Rev Drug Discov. 2006;5(11):891–3.
Zhang Y, Mei Q, Liu Y, Li X, Brock MV, Chen M, et al. The safety efficacy, and treatment outcomes of a combination of low-dose decitabine treatment in patients with recurrent ovarian cancer. Oncoimmunology. 2017;6(9):e1323619.
Wu X, Gu Z, Chen Y, Chen B, Chen W, Weng L, et al. Application of PD-1 Blockade in Cancer Immunotherapy. Comput Struct Biotechnol J. 2019;23:17: 661–74.
Chunmeng Wang, Yang Liu, Liang Dong, Xiang Li, Qingming Yang, Malcolm V , et al. BrockEfficacy of Decitabine plus Anti-PD-1 Camrelizumab in Patients with Hodgkin Lymphoma Who Progressed or Relapsed after PD-1 Blockade Monotherapy. Clin Cancer Res. 2021, 27(10):2782-2791.
Nie J, Wang C, Liu Y, Yang Q, Mei Q, Dong L, et al. Addition of Low-Dose Decitabine to Anti–PD-1 Antibody Camrelizumab in Relapsed/Refractory Classical Hodgkin Lymphoma. J Clin Oncol. 2019;37(17):1479–89.
Gourd E. New treatment for relapsed or refractory Hodgkin’s lymphoma. Lancet Oncol. 2019, 20(6): e298.
M Janaki Ramaiah, Anjana Devi Tangutur, Rajasekhar Reddy Manyam. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 277:119504.
Lopez AT, Bates S, Geskin L. Current Status of HDAC Inhibitors in Cutaneous T-cell Lymphoma. Am J Clin Dermatol. 2018;19(6):805–19.
Mulvey E, Ruan J. Biomarker-driven management strategies for peripheral T cell lymphoma. J Hematol Oncol. 2020, 13(1): 59.
Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011;29(3):255–65.
Maouche N, Kishore B, Bhatti Z, Basu S, Karim F, Sundararaman S, Collings F, Tseu B, Leary H, Ryman N, Reddy U, Vallance GD, Kothari J, Ramasamy K Panobinostat in combination with bortezomib and dexamethasone in multiply relapsed and refractory myeloma; UK routine care cohort. PLoS One. 2022;17(7):e0270854.
Lee B-H, Park Y, Kim JH, Kang K-W, Lee SJ, Kim SJ, et al. PD-L1 expression in bone marrow plasma cells as a biomarker to predict multiple myeloma prognosis: developing a nomogram-based prognostic model. Sci Rep. 2020 Jul 28;10(1):12641.
Tiemann M, Samoilova V, Atiakshin D, Buchwalow I. Immunophenotyping of the PD-L1-positive cells in angioimmunoblastic T cell lymphoma and Hodgkin disease. BMC Res Notes. 2020 Mar 7;13(1):139.
Jelinek T, Paiva B, Hajek R. Update on PD-1/PD-L1 Inhibitors in Multiple Myeloma. Front Immunol. Nov. 2018;16:9:2431.
Tomassetti S, Chen R, Dandapani S.The role of pembrolizumab in relapsed/refractory primary mediastinal large B-cell lymphoma.Ther Adv Hematol. 2019, 22;10: 2040620719841591.
Oliva S, Troia R, D’Agostino M, Boccadoro M, Gay F. Promises and Pitfalls in the Use of PD-1/PD-L1 Inhibitors in Multiple Myeloma. Front Immunol. 2018 Nov;27:9:2749.
Xinyi Tu B, Qin Y, Zhang C, Zhang M, Kahila S, Nowsheen, et al. PD-L1 (B7-H1) Competes with the RNA Exosome to Regulate the DNA Damage Response and Can Be Targeted to Sensitize to Radiation or Chemotherapy. Mol Cell. 2019 Jun 20;74(6):1215–1226.
Yang Gao NT, Nihira X, Bu C, Chu J, Zhang A, Kolodziejczyk, et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol. 2020;22(9):1064–75.
Selma Ugurel I, Spassova J, Wohlfarth C, Drusio A, Cherouny A, Melior, et al. MHC class-I downregulation in PD-1/PD-L1 inhibitor refractory Merkel cell carcinoma and its potential reversal by histone deacetylase inhibition: a case series. Cancer. Immunol Immunother. 2019;68(6):983–90.
Yang C, Zhang J, Ma Y, Wu C, Cui W, Wang L. Histone methyltransferase and drug resistance in cancers. J Exp Clin Cancer Res. 2020;39(1):173.
Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, et al. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017;20(4):854–67.
Liye Zhou T, Mudianto X, Ma R, Riley R, Uppaluri. Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents Anti-PD-1 Resistance in Head and Neck Cancer. Clin Cancer Res. 2020;26(1):290–300.
Chen X, Liu X, Zhang Y, Huai W, Zhou Q, Xu S, Chen X, Li N, Cao X. Methyltransferase Dot1l preferentially promotes innate IL-6 and IFN-β production by mediating H3K79me2/3 methylation in macrophages. Cell Mol Immunol. 2020;17(1):76–84.
Emilie Evanno J, Godet N, Piccirilli J, Guilhot S, Milin JM, Gomber, et al. Tri-methylation of H3K79 is decreased in TGF-β1-induced epithelial-to-mesenchymal transition in lung cancer. Clin Epigenetics. 2017, 9: 80.
Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13(5):337–56.
Ebine K, Kumar K, Pham TN, Shields MA, Collier KA, Shang M, DeCant BT, Urrutia R, Hwang RF, Grimaldo S, Principe DR, Grippo PJ, Bentrem DJ, Munshi HG. Interplay between interferon regulatory factor 1 and BRD4 in the regulation of PD-L1 in pancreatic stellate cells. Sci Rep. 2018;8(1):13225.
Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, Beavis PA, Darcy PK, Martin BP, Spencer A, Traunbauer AK, Sadovnik I, Bauer K, Valent P, Bradner JE, Zuber J, Shortt J, Johnstone RW. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017;18(9):2162–2174.
Abruzzese MP, et al. Inhibition of bromodomain and extra-terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: role of cMYC-IRF4-miR-125b interplay. J Hematol Oncol. 2016;9(1):134.
Abruzzese MP, Bilotta MT, Fionda C, Zingoni A, Soriani A, Vulpis E, et al. BET bromodomain inhibition cooperates with PD-1 blockade to facilitate antitumor response in kras-mutant non-small cell lung cancer. Cancer Immunol. Res.2018, 6, 1234–1245.
Adeegbe DO, Liu S, Hattersley MM, Bowden M, Zhou CW, Li S, Vlahos R, Grondine M, et al. BET bromodomain inhibition cooperates with PD-1 blockade to facilitate antitumor response in kras-mutant non-small cell lung cancer. Cancer Immunol. Res. 2018;6:1234–45.
Zhou Y, Kong Y, Fan W, Tao T, Xiao Q, Li N, et al. Principles of RNA methylation and their implications for biology and medicine. Biomed Pharmacother. 2020 Nov;131:110731.
Jiang MC, Ni JJ, Cui WY, Wang BY, Zhuo W, et al. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am J Cancer Res. 2019;9(7):1354–66.
Bayraktar R, Bertilaccio MT, Calin GA. The interaction between two worlds: microRNAs and Toll-like receptors. Frontiers in immunology. 2019;10:1053.
Mathsyaraja H, Thies K, Taffany DA, Deighan C, Liu T, Yu L, Fernandez SA, Shapiro C, Otero J, Timmers C, Lustberg MB, Chalmers J, Leone G, Ostrowski MC. CSF1-ETS2-induced microRNA in myeloid cells promote metastatic tumor growth. Oncogene. 2015;34(28):3651–61.
Guo W, Wang Y, Yang M, Wang Z, Wang Y, Chaurasia S, Wu Z, Zhang M, Yadav GS, Rathod S, Concha-Benavente F, Fernandez C, Li S, Xie W, Ferris RL, Kammula US, Lu B, Yang D. LincRNA-immunity landscape analysis identifies EPIC1 as a regulator of tumor immune evasion and immunotherapy resistance. Sci Adv. 2021;7(7):eabb3555.
Huber V, Vallacchi V, Fleming V, et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in mela?noma. J Clin Invest. 2018;128:5505–16.
Ashrafizadeh M, Zarrabi A, Hushmandi K, Zarrin V, Moghadam ER, Zabolian A, et al. PD-1/PD-L1 axis regulation in cancer therapy: The role of long non-coding RNAs and microRNAs. Life Sci. 2020;256:117899.
Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A. 1976 Nov;73(11):3852–6.
Huang A, Zheng H, Wu Z, et al. Circular RNA-protein interactions: functions, mechanisms, and identification. Theranostics. 2020;10(8):3503–17.
Kristensen LS, Andersen MS, Stagsted LVW, et al. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019 Nov;20(11):675–91.
Xu X, Zhang J, Tian Y, et al. CircRNA inhibits DNA damage repair by interacting with host gene. Mol Cancer. 2020 Aug 24;19(1):128.
Tanaka E, Miyakawa Y, Kishikawa T, et al. Expression of circular RNA CDR1-AS in colon cancer cells increases cell surface PD-L1 protein levels. Oncol Rep. 2019;42(4):1459–66.
Wei CY, Zhu MX, Lu NH, et al. Circular RNA circ_0020710 drives tumor progression and immune evasion by regulating the miR-370-3p/CXCL12 axis in melanoma. Mol Cancer. 2020;19(1):84.
Wang J, Zhao X, Wang Y, et al. circRNA-002178 act as a ceRNA to promote PDL1/PD1 expression in lung adenocarcinoma. Cell Death Dis. 2020;11(1):32.
Wang X, Yao Y, Jin M. Circ-0001068 is a novel biomarker for ovarian cancer and inducer of PD1 expression in T cells. Aging. 2020;12(19):19095–106.
Zhang PF, Pei X, Li KS, et al. Circular RNA circFGFR1 promotes progression and anti-PD-1 resistance by sponging miR-381-3p in non-small cell lung cancer cells. Mol Cancer. 2019;18(1):179.
Hong W, Xue M, Jiang J, et al. Circular RNA circ-CPA4/ let-7 miRNA/PD-L1 axis regulates cell growth, stemness, drug resistance and immune evasion in non-small cell lung cancer (NSCLC). J Exp Clin Cancer Res. 2020;39(1):149.
Li L, Zhang Q, Lian K. Circular RNA circ_0000284 plays an oncogenic role in the progression of non-small cell lung cancer through the miR-377-3p-mediated PD-L1 promotion. Cancer Cell Int. 2020;20:247.
Yang J, Jia Y, Wang B, et al. Circular RNA CHST15 sponges miR-155-5p and miR-194-5p to promote the immune escape of lung cancer cells mediated by PD-L1. Front Oncol. 2021;11:595609.
Yang Z, Chen W, Wang Y, et al. CircKRT1 drives tumor progression and immune evasion in oral squamous cell carcinoma by sponging miR-495-3p to regulate PDL1 expression. Cell Biol Int. 2021;45(7):1423–35.
Jiang Z, Hou Z, Liu W, et al. Circ-Keratin 6c promotes malignant progression and immune evasion of colorectal cancer through microRNA-485-3p/programmed cell death receptor ligand 1 axis. J Pharmacol Exp Ther. 2021;377(3):358–67.
Xu G, Zhang P, Liang H, et al. Circular RNA hsa_circ_0003288 induces EMT and invasion by regulating hsa_circ_0003288/miR-145/PD-L1 axis in hepatocellular carcinoma. Cancer Cell Int. 2021;21(1):212.
Acknowledgements
We thank the current and former members of our laboratories and collaborators for their contributions to the publications cited in this review article, and we apologize for not being able to cite all the recent publications, due to space limitations.
Funding
The work in the lab was supported by the National Natural Science Foundation of China (81911530169) and the University Special Innovative Research Program of the Department of Education of Guangdong Province (2020KQNCX073). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Mengyuan Dai, Miao Liu, Can Küçük, Hua Yang and Hua You conceived the hypothesis, did the literature search, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Incorrect version of Figure 1 replaced and missing citation and references are added in the article.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dai, M., Liu, M., Yang, H. et al. New insights into epigenetic regulation of resistance to PD-1/PD-L1 blockade cancer immunotherapy: mechanisms and therapeutic opportunities. Exp Hematol Oncol 11, 101 (2022). https://doi.org/10.1186/s40164-022-00356-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-022-00356-0