
Abstract
RHO GTPases are a subfamily of the RAS superfamily of proteins, which are highly conserved in eukaryotic species and have important biological functions, including actin cytoskeleton reorganization, cell proliferation, cell polarity, and vesicular transport. Recent studies indicate that RHO GTPases participate in the proliferation, migration, invasion and metastasis of cancer, playing an essential role in the tumorigenesis and progression of hepatocellular carcinoma (HCC). This review first introduces the classification, structure, regulators and functions of RHO GTPases, then dissects its role in HCC, especially in migration and metastasis. Finally, we summarize inhibitors targeting RHO GTPases and highlight the issues that should be addressed to improve the potency of these inhibitors.
Similar content being viewed by others

Introduction
The RHO GTPases form a subfamily of the RAS superfamily of GTP-binding proteins with a size of 21 to 25 kDa which are found in all eukaryotic cells. The first RHO GTPase protein was discovered in the abdominal ganglia of Aplysia in 1985 [1]. RHO GTPases have a conserved primary structure with 50–55% sequence similarity to each other [2]. However, it was observations reported years later that put forward the perception of the cellular function of RHO GTPases, mainly linked to the actin cytoskeleton [3,4,5], and further studies demonstrated that they regulate many other signal transduction pathways. They are involved in the modulation of cell proliferation control, cell polarity, development, vesicular transport pathways and other aspects of cell biology [6]. Conversely, the dysregulation of RHO GTPases is associated with various diseases such as inactivating mutation in T cell lymphoma [7], overexpression in hypertension [8], and abnormal activation in arthritis [9].
Hepatocellular carcinoma (HCC) is the leading cause of cancer-related death worldwide, especially the overall survival of patients with advanced HCC should be improved [10]. Exploring the pathogenesis of HCC may help us out of this woods. Strikingly, aberrant expression of RHO GTPase was found to contribute to HCC progression [11], especially migration and metastasis. Furthermore, RHO GTPases are regarded as potential diagnostic biomarkers or therapeutic targets for cancer [12, 13]. However, the underlying mechanisms through which RHO GTPases contribute to HCC initiation and progression remain poorly understood.
In this review, we first introduce the classification, structure, regulators and functions of RHO GTPases, then dissect their role in HCC initiation and development, especially in migration. Finally, we discuss inhibitors targeting the RHO GTPases and highlight the issues that should be addressed to improve the potency of these inhibitors.
RHO GTPase family in biology
According to their structural similarity, sequence, and intracellular functions, the RAS superfamily of GTPases is divided into five main groups: RAS, RHO, RAB, ARF and RAN. Among them, RHO family GTPases have recently been expanded to over 20 members [14], and some of them have been extensively studied, including RHOA, RAC1 and CDC42. What’s more, most of them are highly conserved among eukaryotic species [15]. Activated by extracellular factors such as soluble molecules, adhesion interactions, and mechanical stress, RHO GTPases can initiate signaling cascades that span a wide range of targets or effectors, including kinases and scaffold/adaptor-like proteins [16]. The activation and inactivation of RHO GTPases is mainly regulated by three upstream factors: the guanine nucleotide dissociation inhibitors (GDIs), guanine nucleotide exchange factors (GEFs), and GTPase-activating proteins (GAPs).
RHO Family GTPases
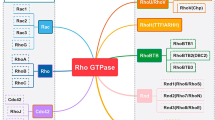
RHO GTPases can be classified into eight subfamilies based on their sequence homology, including RHO (RHOA–RHOC), RAC (RAC1–RAC3 and RHOG), CDC42 (CDC42, RHOJ/TCL and RHOQ/TC10), RHODF (RHOD and RHOF/RIF), RHOUV (RHOU/WRCH1 and RHOV/CHP), RND (RND1, RND2, RND3/RHOE), RHOH, and RHOBTB (RHOBTB1 and RHOBTB2) (Fig. 1). RHO GTPases typically contain a G domain and a C-terminal hypervariable region (Fig. 2). The G domain consists of five conserved sequence motifs, G1 to G5, which constitute a conserved GDP/GTP-binding domain that participates in nucleotide binding and hydrolysis. The C-terminal hypervariable region ends with a common sequence, which is known as CAAX (C: cysteine, A: any aliphatic amino acid, and X: any amino acid) [17] and modification at this sequence are crucial for the subcellular localization of RHO GTPases [18]. Switch I/II regions of RHO GTPase are the common binding sites for GEFs, GDIs, GAPs, or effectors, and change their conformation during the nucleotide exchange and hydrolysis cycle (Fig. 2) [17]. RHO GTPases are generally expressed ubiquitously, but there are exceptions such as RHOH, RAC2, RHOBTB and RND3 which express in the specific cell [15].

RHO GTPase family. The unrooted phylogenetic tree of the RHO GTPase family was based on the Clustal Omega program alignment of the amino-acid sequences of the 20 RHO GTPase proteins. RHO GTPases can be classified into eight subfamilies: RHO, RAC, CDC42, RHODF, RHOUV, RND, RHOH and RHOBTB. These subfamilies are highlighted with circles and labeled on the right side. The classical RHO GTPases include four subfamilies: RHO, RAC, CDC42 and RHODF, which are regulated by the GDP/GTP cycle. The atypical RHO GTPases comprise the RHOUV, RND, RHOH and RHOBTB subfamilies. These GTPases are regulated by gene expression, localization, phosphorylation, and/or protein stability and not by GEF and GAP

Domains of RHO GTPases. RHO GTPases typically contain a G domain, which is a conserved GDP/GTP-binding domain that participates in nucleotide binding and hydrolysis. Switch I/II regions of RHO GTPase are the common binding sites for GEFs, GDIs, GAPs, or effectors, and undergo conformational changes during the nucleotide exchange and hydrolysis cycle. The C-terminal hypervariable region ends with a common sequence, which is known as CAAX, and modification at this sequence is crucial for subcellular localization of RHO GTPases
RHO GTPases can also be classified into a classical subfamily and an atypical subfamily bases on their structure, kinetic properties and other features. The former includes the four subfamilies RHO, RAC, CDC42 and RHODF, which are regulated by GDP/GTP exchange and exert the main functions of the RHO GTPase family [19]. By contrast, some RHO GTPase members do not follow this classical GTPase cycle, and are consequently considered atypical. Some of them have a remarkably enhanced intrinsic GDP/GTP activity, while some fail to hydrolyze GTP, and were respectively named the fast-cycling RHO GTPases (RHOUV), and hydrolysis-deficient RHO GTPases (RND, RHOH and RHOBTB) [20]. In addition to control by GEFs and GAPs, these GTPases are regulated at the level of gene expression, localization, phosphorylation, and/or protein stability, and their functions involve additional domains that are not present in typical RHO proteins [21].
Upstream regulators of RHO GTPases
Similar to other members of the RAS superfamily, the activity of RHO GTPases is determined by the ratio of their GTP/GDP-bound forms inside the cell. Although the RHO switch itself that transmits extracellular cues to intracellular signaling pathways is straightforward, it is intricately regulated by three main classes regulatory proteins (Fig. 3): GEFs, GDIs, and GAPs. Among them, GEFs positively regulate RHO GTPases, while GAPs and GDIs exert negative regulation. In humans, there are 85 GEFs, 66 GAPs, and 3 GDIs, which mediate the precise regulation of RHO GTPases [17].

The classical RHO GTPase cycle. RHOGEFs bind to the GDP-bound form RHO GTPase and induce the exchange of GDP for GTP to activate RHO. Subsequently, the conformation of RHO GTPases changes, which allows them to interact with their specific effectors and triggers biological effects such as cell migration, polarity and adhesion. RHOGAPs bind to the GTP-bound form of RHO GTPases to promote their intrinsic GTP hydrolysis activity, thereby acting as an inhibitor of RHO GTPases. RHOGDIs can extract geranylgeranylated RHO GTPases from the membrane and inhibit nucleotide exchange and hydrolysis to restrict GDP/GTP cycling. RHOGDIs maintain inactive pools of RHO GTPases in the cytosol and protect the prenyl group of the RHO GTPase in the hydrophobic pocket, preventing RHO GTPases from inappropriate activation, misfolding, or degradation
GEFs
RHO GTPases are generally activated by GEFs, which can bind to the GDP-bound form, destabilize the GDP-GTPase complex and stabilize a nucleotide-free reaction intermediate by deforming the phosphate-binding site of RHO GTPase at the same time, instead of directly stimulating the binding of the RHO GTPase to GDP/GTP [22]. Then, the high intracellular concentration of the nucleotide becomes the only determinant of the binding of RHO GTPase to GTP. As the free nucleotide-binding site of RHO GTPase has a similar affinity for GTP and GDP, while the GTP concentration is tenfold higher than that of GDP, RHO GTPase will bind to GTP and be activated [22]. Subsequently, the bound GTP replaces the GEF and the conformation of RHO GTPase changes again. This process enables RHO GTPases to interact with their specific effectors, thereby facilitating downstream signal transduction.
According to their structure, GEFs are classified into two families, the dedicator of cytokinesis (DOCK) homology region domain family and the DBL-homology (DH) domain family [23]. The majority of RHO GEFs belong to the DBL family, while the DOCK family functions as GEFs for CDC42 and/or RAC, but not RHOA [24]. In addition, it is believed that the action of GEFs is specific not only to their substrates, i.e. RHO GTPases, but also to the cell types and upstream signals [25]. For example, GEFs act immediately upstream of RHO GTPases, providing a direct link between the activation of RHO and various cell surface receptors for growth factors, adhesion molecules, cytokines, and G protein-coupled receptors [23].In addition, RHO GEFs play an essential scaffolding function and coordinate downstream signaling in response to upstream cell stimuli by interacting with a definite set of targets and bringing these effector proteins into the vicinity of the RHO GTPases which they activate [24].
There are four times as many RHO GEFs as RHO GTPases, allowing a more rigorous spatiotemporal control of various activities. Several GEFs have high specificity for a single GTPase (such as FGD1, which activates CDC42), while others may not be as specific (such as α-PIX, which activates both CDC42 and RAC) [23]. However, it remains challenging to predict the substrate specificity of most GEFs.
GAPs
Members of the RHOGAP family possess a conserved 150-residue RHOGAP domain, which mediates the binding to the GTP-bound form of RHO proteins and accelerates their intrinsic GTP hydrolysis activity [26], thereby converting RHO GTPases to their inactive, GDP-bound form. GAPs therefore act as an inhibitor of RHO GTPases, and are often considered signal terminators. There are 66 members of the RHOGAP family in humans, which far outnumbers the RHO GTPases, indicating that several RHOGAPs can impart specific functions to individual RHO GTPase [17]. It is necessary to tightly control the activity of each RHOGAP to ensure an appropriate balance between the GDP- and GTP-bound states of RHO proteins [14]. In addition to activating GTP hydrolysis, GAPs may act as RHO GTPase effectors to mediate other downstream target functions [11].
GDIs
In mammals, there are three RHOGDI proteins, which bind to distinct RHO GTPases [14]. Among them, RHOGDI1 is the best-characterized and most abundant. It is extensively expressed and interacts with various RHO GTPases, including RHOA, RHOC, CDC42, RAC1, and RAC2 [27]. RHOGDI3 is usually expressed at low levels and is likely to interact with RHOB and RHOG [28]. RHOGDIs consist of a C-terminal domain, including a geranylgeranyl-binding pocket that is indispensable for extracting geranylgeranylated RHO GTPases from the membrane, as well as an N-terminal domain which interacts with the switch I and switch II domains of RHO GTPases, inhibiting exchange and hydrolysis to restrict GDP/GTP cycling [27, 29]. Thus, RHOGDI can form an inactive complex with RHO GTPases, and thereby sequester them to maintain inactive pools of RHO GTPases in the cytosol [14]. This complex also allows inactive RHO GTPases to quickly be translocated to any membrane in the cell to rapidly respond to specific signals [27]. In addition, they can serve as a chaperone to carry RHO GTPases between membranes, protecting the prenyl group of the RHO GTPase in the hydrophobic pocket. RHOGDI may therefore also contribute to the activation of RHO GTPases. Taken together, RHOGDIs prevent the inappropriate activation of RHO GTPases from and protect them from misfolding and degradation [30]. The classical model of the RHO GTPase cycle assumes that GDIs only extract with a GTPase when it has been inactivated by GAP [27]. However, recent studies indicate that RHOGDIs can not only promote the passive shuttling of inactive RHO GTPases (GDP-bound) in the cytoplasm, but also extract active RHO GTPases (GTP-bound) form membranes [31], which indicates that RHOGDIs effectively contribute to the spatiotemporal control of RHO GTPases.
Another aspect of the complex RHO regulation is the fact that GEFs and GAPs are multidomain proteins subject to complex regulation, which is modulated by posttranslational modifications, protein interactions, and binding of second messengers, which in turn regulate their localization, specificity, and activity [22]. In addition to regulation by GEFs, GAPs and GDIs, RHO GTPases are also controlled by post-translational modifications, including lipid modifications, ubiquitination, phosphorylation, and SUMOylation, which can significantly affect their function [16], especially in atypical RHO GTPases. Likewise, the expression of RHO GTPases can be regulated at the transcriptional or post-transcriptional level. For example, micro-RNAs (miRNAs) can regulate the posttranscriptional processing of RHO GTPase-encoding mRNAs [32]. These factors create a complex network of interactions that determine the precise spatiotemporal activation of RHO GTPases, and thereby determine their final effects on the cell.
Cellular functions of RHO GTPases
Under stimulation by diverse upstream signals, RHO GTPases play various roles in cell motility, cell cycle, phagocytosis, membrane trafficking and other aspects (Fig. 4).

RHO GTPase function. Under stimulation by various upstream signals, including integrin, GPCRs and tyrosine kinase receptors, RHO GEFs are activated. Subsequently, activated RHOA, RAC1 and CDC42 bind to and specifically activate their downstream effectors, including protein kinases (e.g., PKN, ROCK, and Citron) and scaffolding proteins (e.g., WASP, IRSp53 and mDia). These target proteins activate distinct signaling pathways with multiple roles in cell motility, cell cycle, phagocytosis and membrane trafficking
Cell motility
Cell motility is a multistep process (Fig. 5), including (1). Protrusion of the leading edge, (2). Local formation of new adhesions, (3). Cell body contraction, and (4). Detachment of the trailing edge [33].

RHO GTPase-driven cell migration modes. As a response to various signals, migrating cells enter the cell motility cycle. At the leading edge, RAC1 induces the formation of actin-rich lamellipodia. CDC42 determines the formation of filopodia and the direction of motion. New protrusions adhere to the ECM through the formation of focal complexes, which are controlled mainly by RAC1 and RHOA. Then the cell body contracts depending on the formation of stress fibers and the contraction of actin-myosin fibers, which is mediated by RHOA and ROCKs. Finally, the rear adhesions are dissolved, the cell tail retracts, and the cell moves forward
The initial step of cell migration is the extension of cytoplasm, which is induced by actin polymerization at the leading edge and determines the movement direction. Membrane protrusion involves new actin polymerization and requires actin nucleators, such as Ena/VASP protein and the actin-related protein-2/3 (Arp2/3) complex. The structure of membrane protrusions can be divided into finger-like structures called filopodia and sheet-like structures named lamellipodia, which are respectively induced by CDC42 and RAC1. CDC42 is activated in response to various external stimuli such as chemo-attractants, integrins, or receptors for soluble ligands. By releasing intramolecular interactions, CDC42-GTP can relieve the autoinhibitory mechanism of Wiskott–Aldrich syndrome protein (WASP) and Neuronal-Wiskott–Aldrich syndrome protein (N-WASP) to activate them. When activating these effectors, CDC42 contributes to the indirect activation of the Arp2/3 complex [34] to initiate peripheral actin polymerization [6], leading to filopodia formation. CDC42 can also induce cytoskeletal polarization through the polarity protein partitioning defective-6 (PAR6) pathway with PAR3 and/or isoforms of atypical protein kinase C (aPKC) [35]. Furthermore, CDC42 also plays important roles in cytoskeleton regulation and cell motility through many targets including the formin-family protein mammalian diaphanous (mDia), IRSp53 and myotonic dystrophy-related CDC42-binding protein kinases (MRCK) α/β [19, 36,37,38]. RAC activates the WASP-family verprolin-homologous protein (WAVE) complex, in turn which activates Arp2/3 to stimulate the formation of a “dendritic” actin network together with WAVE, contributing to lamellipodium extension [39, 40]. IRSp53 is another RAC target that contributes to this process by binding to RAC and WAVE [41]. However, RAC can also control the termination of Arp2/3 activation through Arpin [42]. RAC possibly also activates mDia2, which nucleates unbranched actin filaments [19]. By directly regulating the interaction between Lamellipodin (Lpd) and the WAVE complex, RAC activates Ena/VASP, which regulates the length of actin filaments at the cell front by temporarily safeguarding actin filament ends against capping protein while also recruiting polymerization-competent profilin-bound G-actin [43]. The binding of active RAC/CDC42 disturbs the autoinhibitory conformation of PAK and then activates its catalytic domain through phosphorylation [36]. RAC/CDC42 phosphorylates LIM motif-containing protein kinase (LIMK) to activate it via PAK1, which in turn efficiently induces the phosphorylation and inactivation of cofilin, leading to decreased depolymerization of F-actin [44]. As mentioned above, many effectors of CDC42 are also downstream of RAC, and CDC42 is consequently considered a potential regulator that drives the activity of RAC-dependent lamellipodia [45]. Strikingly, RHOA was found to be activated at the leading edge to promote the polymerization of actin [45]. Consequently, the combined activity of RAC, CDC42 and RHOA induces the formation of a protrusion at the leading edge to facilitate cell migration. Next, RAC and RHOA induce the formation of focal adhesions [46], in which actin-myosin fibers are connected to some discrete points on the inner plasma membrane, where dynamic protein complexes that are conducive to cell adhesion to the extracellular matrix (ECM) are localized [47].
Subsequent cell contraction is mainly mediated by stress fibers, a contractile device consisting of bundles of F-actin and myosin II [47]. The formation of stress fibers is mainly induced by RHOA/ROCK, which targets various cytoskeletal regulatory proteins. These proteins include the myosin light chain (MLC), MLC phosphatase, and LIMK [6]. ROCK phosphorylates and activates LIMK, which can further phosphorylate cofilin to inactivate it and inhibit its actin-depolymerization activity, resulting in the stabilization of actin filaments [48]. In addition, ROCK promotes the phosphorylation of MLC by directly phosphorylating it or inactivating myosin phosphatase, which finally causes myosin II activation and actomyosin-driven contractility [49]. mDia, another significant effector of RHOA, can cooperate with ROCK to assemble actomyosin bundles [50, 51]. Furthermore, ezrin/radixin/moesin (ERM), eukaryotic elongation factor 1-α1 (eEF1a1), and adducin are downstream effectors of ROCK that engage in actin cytoskeleton assembly [52]. Thus, RHOA not only strengthens actin polymerization, but also reduces its depolymerization to drive cell body contraction. Finally, RHOA/ROCK mediates tail retraction at the rear of the cell [46].
An epithelial-mesenchymal transition (EMT) is a biological process, which allows a polarized epithelial cell to assume a mesenchymal cell phenotype, playing important roles in embryo implantation, embryogenesis, organ development, tissue regeneration, organ fibrosis well as cancer progression and metastasis [53]. There are two migration fashions of invasive cells—a mesenchymal or an amoeboid mode of migration [54], and the core of the former is RAC and the latter is RHOA. RAC1 is closely related to the mesenchymal migration mode of invasive cells and is characterized by the elongated morphology of cells and a leading edge with active membrane ruffles [54]. Furthermore, the activity of RAC1 is central to modulating the switch between amoeboid and mesenchymal migration fashion. RAC1 activates WAVE2 to promote mesenchymal movement by actin assembly and to inhibit actomyosin contractility and amoeboid movement by suppressing ROCK activity [55]. In contrast, during amoeboid movement, ROCK inhibits RAC1 via stimulating ArhGAP22, a RAC GAP [55]. Amoeboid migration is characterized by round cells which have weak adhesion with surrounding matrices [56]. Increased RHOA induces amoeboid motility via stimulating membrane blebbing through ROCK-dependent phosphorylation of myosin II and consequent actomyosin contractility [56]. This migration approach displays high levels of actomyosin contractility and can squeeze through the matrix via deforming the cell body, in a proteolysis-independent way [57]. Furthermore, RHOA/ROCK can also promote EMT by upregulating EMT-related genes via AP-1 [58].
Cell cycle
The cell cycle includes four phases, termed G1, S, G2, and M. In order to avoid abnormal proliferation or apoptosis caused by abnormal passage through the cell cycle, there are many checkpoints that can arrest the cell cycle through proteins such as cyclins and cyclin-dependent kinases [59]. RHOA/ROCK induces the phosphorylation of glycogen synthase kinase-3β (GSK-3β) and accumulation of β-catenin, which contributes to increased expression of c-Myc and cyclin D1, leading to cell proliferation and migration [60]. RHOA is also involved in the cell cycle by Citron to regulate cytokinesis [61]. RAC1 is important for cellular transformation through the regulation of antiapoptotic signals and cell cycle machinery. RAC1 promotes the progression of the G1 phase by promoting the biosynthesis of cyclin D1 [62]. RAC1 can in turn also be activated by cyclin-dependent kinase 1 (CDK1) to promote mitosis through PAK [63]. In addition, RAC1 influences transformation by regulating nuclear factor-κB (NF-kB) [64]. By activating NF-κB in the nucleus, RAC1 leads to an inflammatory response that induces the nuclear translocation of nuclear factor erythroid 2-related factor 2 (NRF2) and increases its activity, which can block RAC1-dependent NF-κB activation through the NRF2/ARE pathway [65]. Thus, RAC1 has a unique nuclear function. In addition, RAC1 was also reported to control cell proliferation and transcription through p70 ribosomal S6 kinase (p70S6k) [66, 67].
Phagocytosis and membrane trafficking
Phagocytosis plays multiple roles in the organism, including tissue homeostasis, remodeling, and immune defense. RHO GTPases, especially RAC1, are involved in phagocytosis. RAC1/2 modulate Arp2/3 recruitment and actin polymerization at the phagosome to regulate phagocytosis mediated by integrins and Fc-gamma receptors (FcγRs) [68]. In phagocytic cells, RAC1 was found to regulate the activity of NADPH oxidase through p67 to promote phagocytosis [69]. Recent studies demonstrated that RAC1 and RHOA can induce phagocytosis of apoptotic bodies in hepatic stellate cells (HSCs) [70]. Moreover, RHO GTPases participate in membrane trafficking. RHOA is involved in endosomal trafficking by protein kinase N (PKN) [71]. Furthermore, CDC42/RAC1 is also involved in microtubule dynamics by inhibiting microtubule plus end disassembly through phosphorylation of Op18/stathmin to inactive it via PAK [6].
Other functions
RHO GTPases also have many other features. For instance, RHOU activates PAK1 and JNK1 to induce filopodia and regulate intercellular tight junctions [72]. RND proteins have been demonstrated to modulate the organization of the actin cytoskeleton in some tissues, participate in neurite extension, and regulate contractility of smooth muscles [21]. RHO GTPases play essential roles in the nervous system, for example, RHOA inhibits axon outgrowth by mediating the effects of myelin-associated axon growth inhibitors such as myelin-associated glycoprotein (MAG) through ROCK [73]. There are also findings implicating RHO GTPases in the regulation of metabolism. RAC1, which enhances the translocation of glucose transporter 4 (GLUT4) through a RalA-dependent downstream pathway, was shown to have the ability to regulate glucose uptake [74].
However, there are still many issues in the research of RHO GTPases, especially concerning atypical RHO GTPases such as RHOBTB, RND, RHOH, RHOV and RHOU. Further questions remain surrounding their regulatory mechanisms, the nature of their downstream signals, and also their functions in the cell and whole organism.
RHO GTPase family in HCC
Hepatocellular carcinoma (HCC) is the fifth most common cancer in the world, as well as the third leading cause of cancer-related deaths [75, 76]. Moreover, the annual incidence of HCC is expected to surpass 1 million by 2025 [77]. HCC commonly occurs in the context of chronic liver disease due to risk factors such as hepatitis B or C, alcohol abuse or nonalcoholic hepatic steatosis and diabetes [78]. While novel diagnostic approaches and therapeutic strategies have led to substantial improvement, the long-term prognosis of patients with HCC remains unsatisfactory, with median 5 year survival of 18% [78], and metastasis as the main potential reason for high mortality [79]. Moreover, the median survival of patients with advanced HCC is only 6–8 months, and the main molecularly targeted drug, sorafenib, can only extend the median overall survival to 10.7–14.7 months [80, 81].
Strictly controlled cell migration is indispensable for the development of multicellular organisms, and its deregulation is a hallmark of metastatic cancer [82]. Recent studies have shown frequent dysregulation of RHO GTPases in a variety of human cancers, which is mainly caused by the dysregulation of their upstream regulators, GEFs, GAPs and GDIs. RHO GTPases have a well-recognized role in the acquisition of malignant features by cancer cells, and contribute to the modulation of aggressive biological behaviors of tumor cells by affecting the cytoskeleton [12]. Given that RHO GTPases participate in various cellular functions, there is no doubt that they are connected with almost every stage of cancer development and progression, such as the dysregulation of cell proliferation, angiogenesis, resistance to apoptosis, tissue invasion, and metastasis [14]. Therefore, exploring the roles of the RHO GTPase family in HCC initiation and progression may contribute to finding a cure for this aggressive malignancy (Fig. 6).

RHO GTPase in HCC.RHO GTPases promote HCC progression via various approaches and play an essential role in HCC A migration and invasion, B proliferation, C tumor microenvironment and D apoptosis
RHO GTPases in HCC cell migration and invasion
The ability of cancer cells to spread to other parts of the body via metastasis is an important feature of cancer and a key factor determining the prognosis [83]. It is well known that RHO GTPases modulate cell motility and hence play an essential role in the migration, invasion, and metastasis of cancer cells [36, 84].
Many studies show that upregulation of RHOA expression promotes migration and invasion of HCC cells. For example, RHOA/ROCK can be activated by hypoxia-induced upregulation of supervillin to activate the extracellular regulated protein kinase (ERK)/p38 pathway, leading to the promotion of HCC cell migration and invasion [85]. Recent studies found that RHOA is frequently overexpressed in HCC, which is strongly correlated with satellite lesions, venous invasion and advanced TNM stage [86, 87]. Additionally, RHOA/ROCK2 was reported to promote the ubiquitination and degradation of dual-specificity phosphatase-1 (DUSP1/MKP1) to downregulate the protein expression of MKP1, resulting in the promotion of HCC invasion [88]. RHOA can also block the ubiquitination and degradation of MMP2 via ROCK2 to facilitate invasion of HCC [89]. Another study demonstrated that RHOA promotes HCC cell migration and invasion by activating ROCK1/MLC signaling, and facilitates HCC metastasis by enhancing the activity of the FAK/SRC pathway [90]. Moreover, RHOA activity can also be inhibited by Snail to recruit SRC-phosphorylated p190 RHOGAP, thereby promoting collective migration and the formation of circulating tumor cell clusters [91]. When activated by transcription factor HOXD10, RHOC promotes HCC metastasis via the urokinase-type plasminogen activator receptor (uPAR)/MMP pathway [92]. Another study demonstrated that RHOA can be activated by matricellular protein SPON2-α4β1 integrin signaling, and inactivated by SPON2-α5β1 integrin signaling or integrin α9 to control HCC cell migration [93, 94].
In contrast to primary HCC cell lines, an elevated active RAC1 level was found in a metastatic HCC cell line [95]. RAC1-WAVE2 signaling can be activated by SRC, which inhibits RHOA-ROCK signaling at the same time, leading to mesenchymal-type movement of HCC cells [96]. Following activation by RAB23, RAC1 increases the expression of TGF-β, resulting in the promotion of EMT and migration of HCC cells [97]. The activity of RAC1 can be suppressed by integrin α9 to decrease the phosphorylation of FAK and SRC, thereby inhibiting the migration and invasion of HCC cells [94]. In addition, elevated expression of RAC1B, a RAC splice variant with more repaid GDP/GTP exchange, was found in many cancers[98,99,100], including liver cancer [101]. The expression of RAC1B can be enhanced by ARHGAP11A to promote invasion, migration and EMT of HCC cells by decreasing N-cadherin and Snail expression while increasing E-cadherin expression [101].
However, CDC42 has contradictory effects in HCC. Although CDC42 was found to be upregulated in a variety of tumors [16], the loss of CDC42 in the liver leads to tumorigenesis and progressive development of HCC [102], suggesting a potential tumor suppressor role of CDC42 [102]. Strikingly, CDC42 was found to enhance the ability of HCC cells to invade surrounding tissues by inducing filopodia formation [103]. The expression of CDC42 in HCC can be increased by decreasing the expression of epithelial growth factor receptor (EGFR) to induce Myosin II activation, thus promoting HCC migration and invasion [104]. The transcriptional activity of the CDC42 promoter can be increased by RAB5a to upregulate CDC42 expression and thereby facilitate HCC progression [105]. Conversely, miR-185 was found to suppress this process by directly downregulating CDC42 expression [106]. In addition, RAC1 and CDC42 are correlated with cancer metastasis, upregulating the PI3K signaling pathway to promote the migration and invasion of various types of cancer, including HCC [107]. Furthermore, RAC1/CDC42 induces the phosphorylation of Ser215 of p53 by PAK4, further eliminating the inhibitory effect of p53 on the invasion and migration of HCC cells [108]. RAC1/CDC42 also promotes HCC EMT and metastasis via PAK1 [109, 110], which induces cancer metastasis via the phosphorylation of paxillin and activation of JNK [111].
Several other atypical RHO GTPases also play important roles in HCC metastasis, such as RHOF, which interacts with AMP-activated protein kinase (AMPK) and enhances its phosphorylation. This in turn increases RAB3d expression, amplifying the Warburg effect to promote the migration and invasion of HCC cells [112]. It was also reported that RND1 is downregulated in HCC, enhancing the activity of RHOA and leading to EMT-mediated migration and metastasis of HCC cells via the RAF/MEK/ERK signaling pathway [113].
Consequently, RHO GTPases are considered a key factor in regulating the migration, invasion, EMT and metastasis of HCC cells.
RHO GTPase family in HCC cell proliferation
In the early stages of HCC development, uncontrolled cell proliferation is critical, and RHO GTPase family is involved in the dysregulation of proliferation. For example, RHOA activates the ERM pathway and EMT via ROCK1 to facilitate HCC cell proliferation [114]. Furthermore, RHOA/ Rhotekin (RTKN) promotes HCC cell proliferation by activating NF-κB signaling [115]. RHOA can also promote HCC cell proliferation and cell cycle progression by upregulating cell cycle-associated proteins, CDK1 levels and proliferating cell nuclear antigen through RTKN2 [116]. Another study demonstrated that RHOA can also be activated by epithelial cell transforming sequence 2 (ECT2) to facilitate the proliferation of HCC cells via the RHOA/F-actin/Hippo-YAP signaling axis [117]. Similarly, RHOA/actin promotes HCC cell proliferation via the transcriptional regulator serum response factor (SRF) [118]. Other studies showed that RHOA facilitate HCC cell proliferation not only via ROCK2 [119], but also ERK [120]. Of note, RHOC can promote cell cycle progression by upregulating cyclin D1 and CDK4 as well as downregulating cyclin-dependent kinase inhibitors, p16 and p21, thus promoting HCC cell proliferation [121]. RAC1 can also be activated by RAB23 to increase the expression of TGF-β or activate PI3K/AKT signaling, leading to the growth and proliferation of HCC cells [97, 122]. Conversely, RAC1 can be inhibited by miR-365 and miR-194, leading to the suppression of HCC dedifferentiation and cancer stem cell proliferation [123, 124]. In addition, RAC and CDC42 are able to inhibit cell cycle arrest in HCC via PAK5 and thereby facilitate tumor growth cells [125]. Strikingly, RHOE enables HCC cell to bypass senescence and promotes their proliferation [126].
Thus, the crucial roles of RHO GTPases in cell proliferation are becoming increasingly clear, providing a theoretical basis for a deeper understanding of HCC pathogenesis.
RHO GTPases in HCC microenvironment
RHO GTPase family has important regulatory roles in inflammation and angiogenesis in the tumor microenvironment of HCC. Tumor capillary endothelial cells (ECs) show abnormally high levels of RHOA, resulting in aberrant mechanosensing and excessive angiogenesis [127]. RHOA/ROCK can also remodel the ECM in the tumor microenvironment to facilitate HCC cell invasion [128, 129]. RHOC/ROCK2 promotes vasculogenic mimicry (VM) in HCC through ERK/MMPs, which significantly improves the tumor blood supply [130]. Furthermore, knockdown of RHOC in HCC cells reduced VEGF expression as well as the migration and organization of ECs to decrease HCC-induced angiogenesis [131]. In addition, RHOB was also found to promote angiogenesis by enhancing VEGFA-VEGFR2 signaling to contribute to HCC malignancy [132]. The degradation of RHOB is blocked by TNFAIP1 downregulation, thereby activating the p38/JNK MAPK pathway to induce the expression of IL-6 and IL-8 in TNF-α-stimulated HCC cells [133]. These pro-inflammatory cytokines can modulate the tumor inflammatory microenvironment to facilitate cancer development and progression.
Conversely, the expression and activity of RHO GTPases can also be modulated by the tumor microenvironment. For instance, a recent study found that the expression of RHOA/ROCK and RAC1/PAK can be increased by hypoxia, inducing VM through the stabilization of HIF-1α and p-Vimentin-activated EMT, which ultimately promotes the invasion and metastasis of HCC [134].
RHO GTPase in hcc cell apoptosis
Escape from apoptosis is a hallmark of cancer [135], and the RHO GTPase family is also involved in the process of apoptosis. For example, RHOA inhibits HCC cell apoptosis via RTKN [116], which can activate NF-κB signaling [115]. Moreover, RHOA can also block apoptosis of HCC cells through ROCK2 [119, 136]. RHOA also inhibits HCC cell apoptosis when activated by ECT2 [137]. Similar to RHOA, RHOC plays an antiapoptotic role in HCC by upregulating the antiapoptotic gene BCL2 [138], and downregulating the proapoptotic gene BAX [121]. Another study unambiguously showed that CDC42/RAC1 mediates cisplatin resistance by inhibiting cell cycle arrest and apoptosis in HCC cells through PAK5 [125]. However, the exact mechanism requires further studies.
Treatment strategies
While novel diagnostic approaches and therapeutic strategies have substantially improved, the cure rates and long-term survival of patients with HCC remain unsatisfactory [76]. Hence, there is an urgent need to explore the molecular mechanisms underlying tumorigenesis, metastasis and chemoresistance in HCC to identify new therapeutic targets.
As mentioned above, the dysregulation of RHO GTPases is involved in the various aspects of malignancy, which makes RHO GTPase family an appealing target for cancer therapy [12, 13]. However, because of the sub-nanomolar binding affinity of RHO GTPases for their substrates GDP or GTP, as well as the high concentration of GTP in the cells [139], and the lack of any suitable binding sites in RHO GTPases, they are generally presumed to be undruggable clinical targets. Therefore, drug development for targeting RHO family proteins is limited. So far, the major pharmacological interventions targeting RHO GTPases are the lipid modification on their carboxy-terminal region, the interface of RHO with RHOGEF [139], and downstream effectors [13] (Fig. 7, Table. 1). Nevertheless, because of many problems such as high toxicity, low selectivity and insufficient efficacy of existing inhibitors, there are no clinically efficacious drugs targeting RHO GTPases for cancer treatment available [140].

Targeting the RHO GTPase signaling. There are many approaches to target RHO GTPase signaling, including disrupting the interactions between RHO and GEF, inhibiting RHO proteins directly and inhibiting the effect of RHO effectors
Inhibitors targeting the RHO GTPase
There are two approaches to inhibiting RHO GTPase: inhibiting RHO-nucleotide interactions or regulating the localization of RHO. For instance, EHT 1864, a nucleotide-binding inhibitor of RAC, which suppresses both guanine nucleotide association and Rac1-Tiam1 complex formation, keeping Rac in an inactive state [141]. R-ketorolac, a specific inhibitor of RAC and CDC42, can inhibit cancer cell migration and invasion in vivo [142]. Otherwise, the specific CDC42 inhibitor ML141 (CID2950007) and its analog CID44216842 can inhibit GTP binding to CDC42 to block CDC42-driven cancer cell migration [143]. Because of the absence of stable binding pockets on the surface of RHOA, the generation of RHOA-specific inhibitors presents several challenges [144]. Furthermore, membrane association is a prerequisite of RHO GTPase activation, whereas their ability to anchor at the membranes depends on the presence of the prenyl group. Splice variants of SmgGDS are major regulators of the prenylation of RHO family members [145]. However, statins can increase SmgGDS expression via targeting HMG CoA reductase pathway and thus diminishing lipid modifications needed by Rho GTPases to suppress their activation [146, 147].
Inhibitors targeting the interaction of RHO and their effectors
A more common strategy to suppress the role of RHO GTPases in facilitating malignancy is targeting its downstream effector proteins. Most kinase inhibitors target the kinase ATP binding site in the active state of kinase, reversibly competing with ATP [148]. As mentioned above, the RHOA effector ROCK plays a promoting role in various stages of cancer. Even though considerable efforts have been made to develop ROCK inhibitors, most are still at the preclinical evaluation stage. For example, studies of ATS907 and AR-12286 for glaucoma were discontinued due to their adverse effects [148]. Y-27632 consistently suppresses RHOA-induced, ROCK-mediated formation of focal adhesions and stress fibers [149, 150], and inhibits intrahepatic metastasis of human HCC [151]. In Japan and China, the ROCK inhibitor fasudil is approved for the acute clinical treatment of cerebral vasospasm [152] and shows therapeutic potential in HCC [153]. However, its pharmacokinetic characteristics make it unsuitable for use in chemotherapy [154]. The high similarity of the homologous ATP-binding regions of ROCK and several other protein kinases, such as PKA and PKC, restricts the development of highly selective ROCK inhibitors [52, 148]. Taken together, ROCK inhibitors may have significant potential for treating cancer and other diseases, with clinical trials for human cancers currently under way [148]. Furthermore, the RAC1/CDC42 inhibitor MBQ-167 may be a promising anticancer drug that specifically suppresses downstream PAK signaling and STAT3 activity to inhibit the growth, migration and EMT of cancer cells [155]. Another study found that BDP9066 is a selective small-molecule inhibitor of the Cdc42-binding MRCK kinases [156]. Although rationally designed small molecule inhibitors have shown promising preclinical results, there are no clinically effective drugs approved for cancer treatment, and there are also no related clinical trials at present.
Inhibitors targeting the interaction of RHO and RHOGEF
Another approach to design inhibitors of the RHO GTPase family is to target the interaction with their upstream regulators. NSC23766 can impair the interaction of RAC1 with TIAM1 and Trio GEFs, but cannot disturb the activation of RHOA and CDC42 [157]. Therefore, this inhibitor can suppress the CAMSAP2-dependent RAC1/JNK pathway, or the cysteine-rich domains-1-RAC1 pathway to inhibit the invasion and migration of human HCC [158, 159]. However, the potency of this compound is relatively low, limiting its further development as a clinical candidate. EHop-016 is another RAC1 inhibitor that binds more tightly to the effector domain, moreover, it is a derivative of NSC23766 but is 100 times more potent than NSC23766 [160]. However, EHop-016 is no longer specific to RAC1 at higher concentrations but also suppresses CDC42 activity without influencing RHOA. The specific RAC1 inhibitor 1D-142 can modulate the RAC1-related transcriptional programme in HCC via regulating the interaction of RAC and RACGEF to significantly inhibit tumor growth and intrahepatic metastasis [161].These inhibitors can be further developed as pharmacological inhibitors of RAC in metastatic cancer cells. Rhosin, the first developed RHOA-specific inhibitor, impedes the docking between RHOA and various GEFs to suppress cancer progression [162]. DC-Rhoin, a novel inhibitor of RHO GTPase, can covalently modify RHO protein at Cys107 to disrupt the interaction of RHO with GEF and GDI [140]. Because of the inactivation of RHO GTPase by RHOGAP [163], enhancing RHOGAP activity is a promising treatment strategy. However, several RHOGAPs overexpressed in HCC exert a negative effect on HCC progression, therefore confounding the development of RHOGAP activators as anticancer agents. Preventing the release of RHO GTPases from RHOGDIs may also be a potential inhibitory strategy.
Although inhibitors of RHO GTPase family have not been used clinically for cancer treatment to date, rational targeting to the RHO GTPase family still carries significant potential in the discovery of new anticancer drugs, particularly for future combinatorial therapies. We should further evaluate the benefits and risks of the inhibitors as well as develop inhibitors with higher potency and less toxicity.
Conclusions
There is accumulating evidence for the crucial roles of the RHO GTPase family in the development of HCC. RHO GTPases directly control the movement of cancer cell, promoting migration, invasion and metastasis, ultimately leading to the EMT and a switch of cancer cell migration between mesenchymal and amoeboid modes. In addition, the RHO GTPase regulators GEFs, GAPs and GDIs are also usually dysregulated in HCC. Therefore, this subfamily is considered a promising therapeutic target for HCC. However, the development of inhibitors for RHO GTPases is limited due to several issues such as their intrinsic structure, which lacks stable binding pockets. We need to further clarify the regulatory mechanism of the RHO GTPase family to open up novel directions for the design of additional therapeutic interventions and pay more attention to the design of inhibitors targeting RHO effectors and regulators. Furthermore, the majority of studies on the roles of RHO GTPase and its regulators in cancer progression have been performed in vitro. To understand whether they also contribute to the migration and invasion of cancer cells in vivo, future studies should establish pre-clinical in vivo models. Finally, we mainly focus on RHOA, RAC1 and CDC42, with few studies investigating the other members. Therefore, further studies are needed to understand the roles of these less-characterized RHO GTPases in the development of HCC.
Availability of data and materials
Not applicable.
Abbreviations
- GDIs:
-
Guanine nucleotide dissociation inhibitors
- GEFs:
-
Guanine nucleotide exchange factors
- GAPs:
-
GTPase-activating proteins
- DOCK:
-
Dedicator of cytokinesis
- DH:
-
DBL-homology
- miRNAs:
-
Micro-RNAs
- WASP:
-
Wiskott–Aldrich syndrome protein
- N-WASP:
-
Neuronal-Wiskott–Aldrich syndrome protein
- Arp2/3:
-
Actin-related protein-2/3
- PAR6:
-
Partitioning defective-6
- aPKC:
-
Atypical protein kinase C
- mDia:
-
Mammalian diaphanous
- MRCK:
-
Myotonic dystrophy-related CDC42-binding protein kinases
- WAVE:
-
WASP-family verprolin-homologous protein
- Lpd:
-
Lamellipodin
- ECM:
-
Extracellular matrix
- MLC:
-
Myosin light chain
- LIMK:
-
LIM motif-containing protein kinase
- ERM:
-
Ezrin/radixin/moesin
- eEF1a1:
-
Eukaryotic elongation factor 1-α1
- GSK-3β:
-
Glycogen synthase kinase-3β
- CDK1:
-
Cyclin-dependent kinase 1
- NF-kB:
-
Nuclear factor-κB
- NRF2:
-
Nuclear factor erythroid 2-related factor 2
- p70S6k:
-
P70 ribosomal S6 kinase
- FcγRs:
-
Fc-gamma receptors
- HSCs:
-
Hepatic stellate cells
- PKN:
-
Protein kinase N
- MAG:
-
Myelin-associated glycoprotein
- RTKN:
-
Rhotekin
- GLUT4:
-
Glucose transporter 4
- HCC:
-
Hepatocellular carcinoma
- ERK:
-
Extracellular regulated protein kinase
- DUSP1:
-
Dual-specificity phosphatase-1
- uPAR:
-
Urokinase-type plasminogen activator receptor
- EGFR:
-
Epithelial growth factor receptor
- AMPK:
-
AMP-activated protein kinase
- ECT2:
-
Epithelial cell transforming sequence 2
- SRF:
-
Serum response factor
- ECs:
-
Endothelial cells
- VM:
-
Vasculogenic mimicry
- MDR:
-
Multidrug resistance
- YAP:
-
YES-associated protein
- mTOR:
-
Mammalian target of rapamycin
- EMT:
-
Epithelial-mesenchymal transition
- SSO:
-
Splice-switching oligonucleotides
References
Madaule P, Axel R. A novel ras-related gene family. Cell. 1985;41(1):31–40. https://doi.org/10.1016/0092-8674(85)90058-3.
Aelst LV, Souza-Schorey CD. Rho GTPases and signaling networks. Genes Dev. 1997;11(18):2295–322. https://doi.org/10.1101/gad.11.18.2295.
Ridley AJ, Paterson HF, Johnston CL, et al. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70(3):401–10. https://doi.org/10.1016/0092-8674(92)90164-8.
Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70(3):389–99. https://doi.org/10.1016/0092-8674(92)90163-7.
Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81(1):53–62. https://doi.org/10.1016/0092-8674(95)90370-4.
Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–69. https://doi.org/10.1146/annurev.cellbio.21.020604.150721.
Yoo HY, Sung MK, Lee SH, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46(4):371–5. https://doi.org/10.1038/ng.2916.
Nguyen H, Chiasson VL, Chatterjee P, et al. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97(4):696–704. https://doi.org/10.1093/cvr/cvs422.
Malmhall-Bah E, Andersson KME, Erlandsson MC, et al. Rho-GTPase dependent leukocyte interaction generates pro-inflammatory thymic Tregs and causes arthritis. J Autoimmun. 2022;130:102843. https://doi.org/10.1016/j.jaut.2022.102843.
Medavaram S, Zhang Y. Emerging therapies in advanced hepatocellular carcinoma. Exp Hematol Oncol. 2018;7:17. https://doi.org/10.1186/s40164-018-0109-6.
Grise F, Bidaud A, Moreau V. Rho GTPases in hepatocellular carcinoma. Biochim Biophys Acta. 2009;1795(2):137–51. https://doi.org/10.1016/j.bbcan.2008.12.003.
Jung H, Yoon SR, Lim J, et al. Dysregulation of Rho GTPases in Human Cancers. Cancers. 2020. https://doi.org/10.3390/cancers12051179.
Clayton NS, Ridley AJ. Targeting Rho GTPase signaling networks in cancer. Front Cell Dev Biol. 2020;8:222. https://doi.org/10.3389/fcell.2020.00222.
Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol. 2016;17(8):496–510. https://doi.org/10.1038/nrm.2016.67.
Boureux A, Vignal E, Faure S, et al. Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol Biol Evol. 2007;24(1):203–16. https://doi.org/10.1093/molbev/msl145.
Haga RB, Ridley AJ. Rho GTPases: regulation and roles in cancer cell biology. Small GTPases. 2016;7(4):207–21. https://doi.org/10.1080/21541248.2016.1232583.
Mosaddeghzadeh N, Ahmadian MR. The RHO Family GTPases: Mechanisms of Regulation and Signaling. Cells. 2021. https://doi.org/10.3390/cells10071831.
Roberts PJ, Mitin N, Keller PJ, et al. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J Biol Chem. 2008;283(37):25150–63. https://doi.org/10.1074/jbc.M800882200.
Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690–701. https://doi.org/10.1038/nrm2476.
Aspenstrom P. Fast-cycling Rho GTPases. Small GTPases. 2020;11(4):248–55. https://doi.org/10.1080/21541248.2017.1391365.
Chardin P. Function and regulation of Rnd proteins. Nat Rev Mol Cell Biol. 2006;7(1):54–62. https://doi.org/10.1038/nrm1788.
Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129(5):865–77. https://doi.org/10.1016/j.cell.2007.05.018.
Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6(2):167–80. https://doi.org/10.1038/nrm1587.
Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene. 2014;33(31):4021–35. https://doi.org/10.1038/onc.2013.362.
Satoh T. Diverse physiological functions and regulatory mechanisms for signal-transducing small GTPases. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21197291.
Tsai MH, Hall A, Stacey DW. Inhibition by phospholipids of the interaction between R-ras, rho, and their GTPase-activating proteins. Mol Cell Biol. 1989;9(11):5260–4. https://doi.org/10.1128/mcb.9.11.5260-5264.1989.
Garcia-Mata R, Boulter E, Burridge K. The “invisible hand”: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12(8):493–504. https://doi.org/10.1038/nrm3153.
de Leon-Bautista MP, Cardenas-Aguayo MD, Casique-Aguirre D, et al. Immunological and functional characterization of RhoGDI3 and its molecular targets RhoG and RhoB in human pancreatic cancerous and normal cells. PLoS ONE. 2016;11(11):e0166370. https://doi.org/10.1371/journal.pone.0166370.
Keep NH, Barnes M, Barsukov I, et al. A modulator of rho family G proteins, rhoGDI, binds these G proteins via an immunoglobulin-like domain and a flexible N-terminal arm. Structure. 1997;5(5):623–33. https://doi.org/10.1016/s0969-2126(97)00218-9.
Boulter E, Garcia-Mata R, Guilluy C, et al. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol. 2010;12(5):477–83. https://doi.org/10.1038/ncb2049.
Golding AE, Visco I, Bieling P, et al. Extraction of active RhoGTPases by RhoGDI regulates spatiotemporal patterning of RhoGTPases. Elife. 2019;12(5):864.
Liu M, Bi F, Zhou X, et al. Rho GTPase regulation by miRNAs and covalent modifications. Trends Cell Biol. 2012;22(7):365–73. https://doi.org/10.1016/j.tcb.2012.04.004.
Svitkina T. The actin cytoskeleton and actin-based motility. Cold Spring Harb Perspect Biol. 2018. https://doi.org/10.1101/cshperspect.a018267.
Rohatgi R, Ma L, Miki H, et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97(2):221–31. https://doi.org/10.1016/s0092-8674(00)80732-1.
Hurd TW, Gao L, Roh MH, et al. Direct interaction of two polarity complexes implicated in epithelial tight junction assembly. Nat Cell Biol. 2003;5(2):137–42. https://doi.org/10.1038/ncb923.
Hanna S, El-Sibai M. Signaling networks of Rho GTPases in cell motility. Cell Signal. 2013;25(10):1955–61. https://doi.org/10.1016/j.cellsig.2013.04.009.
Kage F, Winterhoff M, Dimchev V, et al. FMNL formins boost lamellipodial force generation. Nat Commun. 2017;8:14832. https://doi.org/10.1038/ncomms14832.
Zihni C, Vlassaks E, Terry S, et al. An apical MRCK-driven morphogenetic pathway controls epithelial polarity. Nat Cell Biol. 2017;19(9):1049–60. https://doi.org/10.1038/ncb3592.
Whitelaw JA, Swaminathan K, Kage F, et al. The WAVE regulatory complex is required to balance protrusion and adhesion in migration. Cells. 2020. https://doi.org/10.3390/cells9071635.
Laurin M, Cote JF. Insights into the biological functions of dock family guanine nucleotide exchange factors. Genes Dev. 2014;28(6):533–47. https://doi.org/10.1101/gad.236349.113.
Thomas A, Mariani-Floderer C, Lopez-Huertas MR, et al. Involvement of the Rac1-IRSp53-Wave2-Arp2/3 signaling pathway in HIV-1 Gag particle release in CD4 T cells. J Virol. 2015;89(16):8162–81. https://doi.org/10.1128/JVI.00469-15.
Fregoso FE, van Eeuwen T, Simanov G, et al. Molecular mechanism of Arp2/3 complex inhibition by Arpin. Nat Commun. 2022;13(1):628. https://doi.org/10.1038/s41467-022-28112-2.
Law AL, Vehlow A, Kotini M, et al. Lamellipodin and the Scar/WAVE complex cooperate to promote cell migration in vivo. J Cell Biol. 2013;203(4):673–89. https://doi.org/10.1083/jcb.201304051.
Edwards DC, Sanders LC, Bokoch GM, et al. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1(5):253–9. https://doi.org/10.1038/12963.
Machacek M, Hodgson L, Welch C, et al. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461(7260):99–103. https://doi.org/10.1038/nature08242.
Chauhan BK, Lou M, Zheng Y, et al. Balanced Rac1 and RhoA activities regulate cell shape and drive invagination morphogenesis in epithelia. Proc Natl Acad Sci USA. 2011;108(45):18289–94. https://doi.org/10.1073/pnas.1108993108.
Julian L, Olson MF. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases. 2014;5:e29846. https://doi.org/10.4161/sgtp.29846.
Geneste O, Copeland JW, Treisman R. LIM kinase and diaphanous cooperate to regulate serum response factor and actin dynamics. J Cell Biol. 2002;157(5):831–8. https://doi.org/10.1083/jcb.200203126.
Kimura K, Ito M, Amano M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 1996;273(5272):245–8. https://doi.org/10.1126/science.273.5272.245.
Watanabe N, Kato T, Fujita A, et al. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1(3):136–43. https://doi.org/10.1038/11056.
Watanabe N, Madaule P, Reid T, et al. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997;16(11):3044–56. https://doi.org/10.1093/emboj/16.11.3044.
Liu J, Wada Y, Katsura M, et al. Rho-associated coiled-coil kinase (ROCK) in molecular regulation of angiogenesis. Theranostics. 2018;8(21):6053–69. https://doi.org/10.7150/thno.30305.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8. https://doi.org/10.1172/JCI39104.
Yamada KM, Sixt M. Mechanisms of 3D cell migration. Nat Rev Mol Cell Biol. 2019;20(12):738–52. https://doi.org/10.1038/s41580-019-0172-9.
Sanz-Moreno V, Gadea G, Ahn J, et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135(3):510–23. https://doi.org/10.1016/j.cell.2008.09.043.
Saito K, Ozawa Y, Hibino K, et al. FilGAP, a Rho/Rho-associated protein kinase-regulated GTPase-activating protein for Rac, controls tumor cell migration. Mol Biol Cell. 2012;23(24):4739–50. https://doi.org/10.1091/mbc.E12-04-0310.
Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5(8):711–9. https://doi.org/10.1038/ncb1019.
Wang Q, Yang X, Ying Xu, et al. RhoA/Rho-kinase triggers epithelial-mesenchymal transition in mesothelial cells and contributes to the pathogenesis of dialysis-related peritoneal fibrosis. Oncotarget. 2018;9(18):14397–412.
Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17(2):93–115. https://doi.org/10.1038/nrc.2016.138.
Kim JG, Kim MJ, Choi WJ, et al. Wnt3A induces GSK-3beta Phosphorylation and beta-catenin accumulation through RhoA/ROCK. J Cell Physiol. 2017;232(5):1104–13. https://doi.org/10.1002/jcp.25572.
Bassi ZI, Verbrugghe KJ, Capalbo L, et al. Sticky/Citron kinase maintains proper RhoA localization at the cleavage site during cytokinesis. J Cell Biol. 2011;195(4):595–603. https://doi.org/10.1083/jcb.201105136.
Mettouchi A, Klein S, Guo W, et al. Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol Cell. 2001;8(1):115–27. https://doi.org/10.1016/s1097-2765(01)00285-4.
Whalley HJ, Porter AP, Diamantopoulou Z, et al. Cdk1 phosphorylates the Rac activator Tiam1 to activate centrosomal Pak and promote mitotic spindle formation. Nat Commun. 2015;6:7437. https://doi.org/10.1038/ncomms8437.
Perona R, Montaner S, Saniger L, et al. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11(4):463–75. https://doi.org/10.1101/gad.11.4.463.
Cuadrado A, Martin-Moldes Z, Ye J, et al. Transcription factors NRF2 and NF-kappaB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J Biol Chem. 2014;289(22):15244–58. https://doi.org/10.1074/jbc.M113.540633.
Chou MM, Blenis J. The 70 kDa S6 kinase complexes with and is activated by the Rho family G proteins Cdc42 and Rac1. Cell. 1996;85(4):573–83. https://doi.org/10.1016/s0092-8674(00)81257-x.
Campa CC, Ciraolo E, Ghigo A, et al. 2015 Crossroads of PI3K and Rac pathways. Small GTPases. 2014. https://doi.org/10.4161/21541248989789.
Moradinasab S, Pourbagheri-Sigaroodi A, Ghaffari SH, et al. Targeting macrophage-mediated tumor cell phagocytosis: an overview of phagocytosis checkpoints blockade, nanomedicine intervention, and engineered CAR-macrophage therapy. Int Immunopharmacol. 2022;103:108499. https://doi.org/10.1016/j.intimp.2021.108499.
Diekmann D, Abo A, Johnston C, et al. Interaction of Rac with p67phox and regulation of phagocytic NADPH oxidase activity. Science. 1994;265(5171):531–3. https://doi.org/10.1126/science.8036496.
Jiang JX, Mikami K, Shah VH, et al. Leptin induces phagocytosis of apoptotic bodies by hepatic stellate cells via a Rho guanosine triphosphatase-dependent mechanism. Hepatology. 2008;48(5):1497–505. https://doi.org/10.1002/hep.22515.
Thumkeo D, Watanabe S, Narumiya S. Physiological roles of Rho and Rho effectors in mammals. Eur J Cell Biol. 2013;92(10–11):303–15. https://doi.org/10.1016/j.ejcb.2013.09.002.
Tao W, Pennica D, Xu L, et al. Wrch-1, a novel member of the Rho gene family that is regulated by Wnt-1. Genes Dev. 2001;15(14):1796–807. https://doi.org/10.1101/gad.894301.
Stern S, Hilton BJ, Burnside ER, et al. RhoA drives actin compaction to restrict axon regeneration and astrocyte reactivity after CNS injury. Neuron. 2021;109(21):3436-3455.e9. https://doi.org/10.1016/j.neuron.2021.08.014.
Takenaka N, Nakao M, Matsui S, et al. A crucial role for the small GTPase Rac1 downstream of the protein kinase Akt2 in insulin signaling that regulates glucose uptake in mouse adipocytes. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20215443.
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. https://doi.org/10.3322/caac.21492.
Galle PR, Forner A, Llovet JM, et al. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236. https://doi.org/10.1016/j.jhep.2018.03.019.
Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. https://doi.org/10.1038/s41572-020-00240-3.
Craig AJ, von Felden J, Garcia-Lezana T, et al. Tumour evolution in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2020;17(3):139–52. https://doi.org/10.1038/s41575-019-0229-4.
Yang JD, Hainaut P, Gores GJ, et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604. https://doi.org/10.1038/s41575-019-0186-y.
Qin S, Bi F, Shanzhi G, et al. Donafenib versus sorafenib in first-line treatment of unresectable or metastatic hepatocellular carcinoma: a randomized, open-label, parallel-controlled phase II-III trial. J Clin Oncol. 2021;39(27):3002–11. https://doi.org/10.1200/JCO.21.00163.
Zhu XD, Sun HC. Emerging agents and regimens for hepatocellular carcinoma. J Hematol Oncol. 2019;12(1):110. https://doi.org/10.1186/s13045-019-0794-6.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3(5):362–74. https://doi.org/10.1038/nrc1075.
Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 2015;36:103–12. https://doi.org/10.1016/j.ceb.2015.08.005.
Chen X, Zhang S, Wang Z, et al. Supervillin promotes epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma in hypoxia via activation of the RhoA/ROCK-ERK/p38 pathway. J Exp Clin Cancer Res. 2018;37(1):128. https://doi.org/10.1186/s13046-018-0787-2.
Bai Yi, Xie F, Miao F, et al. The diagnostic and prognostic role of RhoA in hepatocellular carcinoma. Aging. 2019;11(14):5158–72. https://doi.org/10.18632/aging.102110.
Wang D, Dou K, Xiang H, et al. Involvement of RhoA in progression of human hepatocellular carcinoma. J Gastroenterol Hepatol. 2007;22(11):1916–20. https://doi.org/10.1111/j.1440-1746.2006.04534.x.
Du Y, Shan Lu, Ge J, et al. ROCK2 disturbs MKP1 expression to promote invasion and metastasis in hepatocellular carcinoma. Am J Cancer Res. 2020;10(3):884–96.
Huang D, Du X, Yuan R, et al. Rock2 promotes the invasion and metastasis of hepatocellular carcinoma by modifying MMP2 ubiquitination and degradation. Biochem Biophys Res Commun. 2014;453(1):49–56. https://doi.org/10.1016/j.bbrc.2014.09.061.
Zhan Y, Zheng NX, Teng F, et al. MiR-199a/b-5p inhibits hepatocellular carcinoma progression by post-transcriptionally suppressing ROCK1. Oncotarget. 2017;8(40):67169–80. https://doi.org/10.18632/oncotarget.18052.
Li CF, Chen JY, Ho YH, et al. Snail-induced claudin-11 prompts collective migration for tumour progression. Nat Cell Biol. 2019;21(2):251–62. https://doi.org/10.1038/s41556-018-0268-z.
Liao CG, Kong LM, Zhou P, et al. miR-10b is overexpressed in hepatocellular carcinoma and promotes cell proliferation, migration and invasion through RhoC, uPAR and MMPs. J Transl Med. 2014;12:234. https://doi.org/10.1186/s12967-014-0234-x.
Zhang YL, Li Q, Yang XM, et al. SPON2 promotes M1-like macrophage recruitment and inhibits hepatocellular carcinoma metastasis by distinct integrin-Rho GTPase-Hippo pathways. Cancer Res. 2018;78(9):2305–17. https://doi.org/10.1158/0008-5472.CAN-17-2867.
Zhang YL, Xing X, Cai LB, et al. Integrin alpha9 suppresses hepatocellular carcinoma metastasis by Rho GTPase signaling. J Immunol Res. 2018;2018:4602570. https://doi.org/10.1155/2018/4602570.
Lee TK, Man K, Ho JW, et al. Significance of the Rac signaling pathway in HCC cell motility: implications for a new therapeutic target. Carcinogenesis. 2005;26(3):681–7. https://doi.org/10.1093/carcin/bgi002.
Wang SJ, Cui H-Y, Liu Y-M, et al. promotes Src-dependent activation of Rac1 signaling through during the motility of hepatocellular carcinoma cells. Oncotarget. 2015. https://doi.org/10.18632/oncotarget.2801.
Zhang L, Zhang B, You W, et al. Rab23 promotes hepatocellular carcinoma cell migration Via Rac1/TGF-beta signaling. Pathol Oncol Res. 2020;26(1):301–6. https://doi.org/10.1007/s12253-018-0463-z.
Gudino V, Pohl SO, Billard CV, et al. RAC1B modulates intestinal tumourigenesis via modulation of WNT and EGFR signalling pathways. Nat Commun. 2021;12(1):2335. https://doi.org/10.1038/s41467-021-22531-3.
Gudino V, Cammareri P, Billard CV, et al. Negative regulation of TGFbeta-induced apoptosis by RAC1B enhances intestinal tumourigenesis. Cell Death Dis. 2021;12(10):873. https://doi.org/10.1038/s41419-021-04177-7.
Ungefroren H, Kumarasinghe A, Musfeldt M, et al. RAC1B Induces SMAD7 via USP26 to suppress TGFbeta 1-dependent cell migration in mesenchymal-subtype carcinoma cells. Cancers. 2020. https://doi.org/10.3390/cancers12061545.
Dai B, Zhang X, Shang R, et al. Blockade of ARHGAP11A reverses malignant progress via inactivating Rac1B in hepatocellular carcinoma. Cell Commun Signal. 2018;16(1):99. https://doi.org/10.1186/s12964-018-0312-4.
van Hengel J, D’Hooge P, Hooghe B, et al. Continuous cell injury promotes hepatic tumorigenesis in cdc42-deficient mouse liver. Gastroenterology. 2008;134(3):781–92. https://doi.org/10.1053/j.gastro.2008.01.002.
Zhang J, Yang C, Gong L, et al. RICH2, a potential tumor suppressor in hepatocellular carcinoma. Front Biosci. 2019;24(8):1363–76. https://doi.org/10.2741/4784.
Lopez-Luque J, Bertran E, Crosas-Molist E, et al. Downregulation of Epidermal growth factor receptor in hepatocellular carcinoma facilitates transforming growth factor-beta-induced epithelial to amoeboid transition. Cancer Lett. 2019;464:15–24. https://doi.org/10.1016/j.canlet.2019.08.011.
Yang X, Liu Z, Li Y, et al. Rab5a promotes the migration and invasion of hepatocellular carcinoma by up-regulating Cdc42. Int J Clin Exp Pathol. 2018;11(1):224–31.
Zhang Q, Chen Y, Liu K. miR-185 inhibits cell migration and invasion of hepatocellular carcinoma through CDC42. Oncol Lett. 2018;16(3):3101–7. https://doi.org/10.3892/ol.2018.8971.
Rane CK, Minden A. P21 activated kinase signaling in cancer. Semin Cancer Biol. 2019;54:40–9. https://doi.org/10.1016/j.semcancer.2018.01.006.
Xu HT, Lai W-L, Liu H-F, et al. PAK4 Phosphorylates p53 at Serine 215 to promote liver cancer metastasis. Cancer Res. 2016. https://doi.org/10.1158/0008-5472.CAN-15-3373.
Zhou Y, Fan RG, Qin CL, et al. LncRNA-H19 activates CDC42/PAK1 pathway to promote cell proliferation, migration and invasion by targeting miR-15b in hepatocellular carcinoma. Genomics. 2019;111(6):1862–72. https://doi.org/10.1016/j.ygeno.2018.12.009.
Ji X, Chen X, Zhang B, et al. T-box transcription factor 19 promotes hepatocellular carcinoma metastasis through upregulating EGFR and RAC1. Oncogene. 2022. https://doi.org/10.1038/s41388-022-02249-2.
Lopez-Colome AM, Lee-Rivera I, Benavides-Hidalgo R, et al. Paxillin: a crossroad in pathological cell migration. J Hematol Oncol. 2017;10(1):50. https://doi.org/10.1186/s13045-017-0418-y.
Li S, Liu Y, Bai Y, et al. Ras Homolog family member F, Filopodia associated promotes hepatocellular carcinoma metastasis by altering the metabolic status of cancer cells through RAB3D. Hepatology. 2021;73(6):2361–79. https://doi.org/10.1002/hep.31641.
Qin CD, Ma DN, Zhang SZ, et al. The Rho GTPase Rnd1 inhibits epithelial-mesenchymal transition in hepatocellular carcinoma and is a favorable anti-metastasis target. Cell Death Dis. 2018;9(5):486. https://doi.org/10.1038/s41419-018-0517-x.
Tang J, Liu C, Xu B, et al. ARHGEF10L contributes to liver tumorigenesis through RhoA-ROCK1 signaling and the epithelial-mesenchymal transition. Exp Cell Res. 2019;374(1):46–68. https://doi.org/10.1016/j.yexcr.2018.11.007.
Zhou J, Zhang Y, Qi Y, et al. MicroRNA-152 inhibits tumor cell growth by directly targeting RTKN in hepatocellular carcinoma. Oncol Rep. 2017;37(2):1227–34. https://doi.org/10.3892/or.2016.5290.
Wei W, Chen H, Liu S. Knockdown of Rhotekin 2 expression suppresses proliferation and invasion and induces apoptosis in hepatocellular carcinoma cells. Mol Med Rep. 2016;13(6):4865–71. https://doi.org/10.3892/mmr.2016.5113.
Yang XM, Cao XY, He P, et al. Overexpression of Rac GTPase activating protein 1 contributes to proliferation of cancer cells by reducing hippo signaling to promote cytokinesis. Gastroenterology. 2018;155(4):1233–49. https://doi.org/10.1053/j.gastro.2018.07.010.
Ohrnberger S, Thavamani A, Braeuning A, et al. Dysregulated serum response factor triggers formation of hepatocellular carcinoma. Hepatology. 2015;61(3):979–89. https://doi.org/10.1002/hep.27539.
Zhang L, Zhou H, Wei G. miR-506 regulates cell proliferation and apoptosis by affecting RhoA/ROCK signaling pathway in hepatocellular carcinoma cells. Int J Clin Exp Pathol. 2019;12(4):1163.
Wang MY, Chen DP, Qi B, et al. Pseudogene RACGAP1P activates RACGAP1/Rho/ERK signalling axis as a competing endogenous RNA to promote hepatocellular carcinoma early recurrence. Cell Death Dis. 2019;10(6):426. https://doi.org/10.1038/s41419-019-1666-2.
Xie S, Zhu M, Lv G, et al. The role of RhoC in the proliferation and apoptosis of hepatocellular carcinoma cells. Med Oncol. 2012;29(3):1802–9. https://doi.org/10.1007/s12032-011-0003-0.
Yue X, Wu F, Li Y, et al. Gain of function mutant p53 protein activates AKT through the Rac1 signaling to promote tumorigenesis. Cell Cycle. 2020;19(11):1338–51. https://doi.org/10.1080/15384101.2020.1749790.
Jiang ZB, Ma BQ, Liu SG, et al. miR-365 regulates liver cancer stem cells via RAC1 pathway. Mol Carcinog. 2019;58(1):55–65. https://doi.org/10.1002/mc.22906.
Ran RZ, Chen J, Cui LJ, et al. miR-194 inhibits liver cancer stem cell expansion by regulating RAC1 pathway. Exp Cell Res. 2019;378(1):66–75. https://doi.org/10.1016/j.yexcr.2019.03.007.
Zhang DG, Zhang J, Mao LL, et al. p21-Activated kinase 5 affects cisplatin-induced apoptosis and proliferation in hepatocellular carcinoma cells. Tumour Biol. 2015;36(5):3685–91. https://doi.org/10.1007/s13277-014-3007-5.
Basbous S, Paysan L, Sena S, et al. Silencing of RND3/RHOE inhibits the growth of human hepatocellular carcinoma and is associated with reversible senescence. Cancer Gene Ther. 2022. https://doi.org/10.1038/s41417-022-00445-6.
Ghosh K, Thodeti CK, Dudley AC, et al. Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc Natl Acad Sci USA. 2008;105(32):11305–10. https://doi.org/10.1073/pnas.0800835105.
Sun C, Hu A, Wang S, et al. ADAM17-regulated CX3CL1 expression produced by bone marrow endothelial cells promotes spinal metastasis from hepatocellular carcinoma. Int J Oncol. 2020;57(1):249–63. https://doi.org/10.3892/ijo.2020.5045.
Narumiya S, Thumkeo D. Rho signaling research: history, current status and future directions. FEBS Lett. 2018;592(11):1763–76. https://doi.org/10.1002/1873-3468.13087.
Zhang JG, Zhang DD, Liu Y, et al. RhoC/ROCK2 promotes vasculogenic mimicry formation primarily through ERK/MMPs in hepatocellular carcinoma. Biochim Biophys Acta Mol Basis Dis. 2019;1865(6):1113–25. https://doi.org/10.1016/j.bbadis.2018.12.007.
Wang W, Wu F, Fang F, et al. RhoC is essential for angiogenesis induced by hepatocellular carcinoma cells via regulation of endothelial cell organization. Cancer Sci. 2008;99(10):2012–8. https://doi.org/10.1111/j.1349-7006.2008.00902.x.
Jin L, Liu W-R, Tian M-n, et al. CCL24 contributes to HCC malignancy via angiogenesis pathway and indicates poor prognosis. Oncotarget. 2017;8(3):5135–514. https://doi.org/10.18632/oncotarget.14095.
Liu Y, Zhang W, Wang S, et al. Cullin3-TNFAIP1 E3 ligase controls inflammatory response in hepatocellular carcinoma cells via ubiquitination of RhoB. Front Cell Dev Biol. 2021;9:617134. https://doi.org/10.3389/fcell.2021.617134.
Zhang JG, Zhou HM, Zhang X, et al. Hypoxic induction of vasculogenic mimicry in hepatocellular carcinoma: role of HIF-1 alpha, RhoA/ROCK and Rac1/PAK signaling. BMC Cancer. 2020;20(1):32. https://doi.org/10.1186/s12885-019-6501-8.
Diepstraten ST, Anderson MA, Czabotar PE, et al. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer. 2022;22(1):45–64. https://doi.org/10.1038/s41568-021-00407-4.
Ma W, Sze KM, Chan LK, et al. RhoE/ROCK2 regulates chemoresistance through NF-κB/IL-6/ STAT3 signaling in hepatocellular carcinoma. Oncotarget. 2016;7(27):41445–59. https://doi.org/10.18632/oncotarget.9441.
Chen J, Xia H, Zhang X, et al. ECT2 regulates the Rho/ERK signalling axis to promote early recurrence in human hepatocellular carcinoma. J Hepatol. 2015;62(6):1287–95. https://doi.org/10.1016/j.jhep.2015.01.014.
Perini GF, Ribeiro GN, Pinto Neto JV, et al. BCL-2 as therapeutic target for hematological malignancies. J Hematol Oncol. 2018;11(1):65. https://doi.org/10.1186/s13045-018-0608-2.
Gray JL, von Delft F, Brennan PE. Targeting the small GTPase superfamily through their regulatory proteins. Angew Chem Int Ed Engl. 2020;59(16):6342–66. https://doi.org/10.1002/anie.201900585.
Sun Z, Zhang H, Zhang Y, et al. covalent inhibitors allosterically block the activation of rho family proteins and suppress cancer cell invasion. Adv Sci (Weinh). 2020;7(14):2000098. https://doi.org/10.1002/advs.202000098.
Shutes A, Onesto C, Picard V, et al. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007;282(49):35666–78. https://doi.org/10.1074/jbc.M703571200.
Peretti AS, Dominguez D, Grimes MM, et al. The R-enantiomer of ketorolac delays mammary tumor development in mouse mammary tumor virus-polyoma middle T Antigen (MMTV-PyMT) mice. Am J Pathol. 2018;188(2):515–24. https://doi.org/10.1016/j.ajpath.2017.10.018.
Hong L, Kenney SR, Phillips GK, et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J Biol Chem. 2013;288(12):8531–43. https://doi.org/10.1074/jbc.M112.435941.
Lin Y, Zheng Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin Drug Discov. 2015;10(9):991–1010. https://doi.org/10.1517/17460441.2015.1058775.
Brandt AC, Koehn OJ, Williams CL. SmgGDS: An emerging master regulator of prenylation and trafficking by small GTPases in the Ras and Rho families. Front Mol Biosci. 2021;8: 685135. https://doi.org/10.3389/fmolb.2021.685135.
Tanaka S, Fukumoto Y, Nochioka K, et al. Statins exert the pleiotropic effects through small GTP-binding protein dissociation stimulator upregulation with a resultant Rac1 degradation. Arterioscler Thromb Vasc Biol. 2013;33(7):1591–600. https://doi.org/10.1161/ATVBAHA.112.300922.
Al-Haidari AA, Syk I, Thorlacius H. HMG-CoA reductase regulates CCL17-induced colon cancer cell migration via geranylgeranylation and RhoA activation. Biochem Biophys Res Commun. 2014;446(1):68–72. https://doi.org/10.1016/j.bbrc.2014.02.078.
Feng Y, LoGrasso PV, Defert O, et al. Rho kinase (ROCK) inhibitors and their therapeutic potential. J Med Chem. 2016;59(6):2269–300. https://doi.org/10.1021/acs.jmedchem.5b00683.
Maekawa M, Ishizaki T, Boku S, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285(5429):895–8. https://doi.org/10.1126/science.285.5429.895.
Uehata M, Ishizaki T, Satoh H, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389(6654):990–4. https://doi.org/10.1038/40187.
Takamura M, Sakamoto M, Genda T, et al. Inhibition of intrahepatic metastasis of human hepatocellular carcinoma by Rho-associated protein kinase inhibitor Y-27632. Hepatology. 2001;33(3):577–81. https://doi.org/10.1053/jhep.2001.22652.
Hartmann DA, Berthiaume AA, Grant RI, et al. Brain capillary pericytes exert a substantial but slow influence on blood flow. Nat Neurosci. 2021;24(5):633–45. https://doi.org/10.1038/s41593-020-00793-2.
Zhao Y, Zhang Y, Vazirinejad Mehdiabad M, et al. Enhanced anti-tumor effect of liposomal Fasudil on hepatocellular carcinoma in vitro and in vivo. PLoS ONE. 2019;14(10):e0223232. https://doi.org/10.1371/journal.pone.0223232.
Rath N, Munro J, Cutiongco MF, et al. Rho kinase inhibition by AT13148 blocks pancreatic ductal adenocarcinoma invasion and tumor growth. Cancer Res. 2018;78(12):3321–36. https://doi.org/10.1158/0008-5472.CAN-17-1339.
Humphries-Bickley T, Castillo-Pichardo L, Hernandez-O’Farrill E, et al. Characterization of a dual Rac/Cdc42 inhibitor MBQ-167 in metastatic cancer. Mol Cancer Ther. 2017;16(5):805–18. https://doi.org/10.1158/1535-7163.MCT-16-0442.
Unbekandt M, Belshaw S, Bower J, et al. Discovery of potent and selective mrck inhibitors with therapeutic effect on skin cancer. Cancer Res. 2018;78(8):2096–114. https://doi.org/10.1158/0008-5472.CAN-17-2870.
Gao Y, Bradley Dickerson J, Guo F, et al. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA. 2004;101(2):7618–23. https://doi.org/10.1073/pnas.0307512101.
Li D, Ding X, Xie M, et al. CAMSAP2-mediated noncentrosomal microtubule acetylation drives hepatocellular carcinoma metastasis. Theranostics. 2020;10(8):3749–66. https://doi.org/10.7150/thno.42596.
Chang CY, Lin SC, Su WH, et al. Somatic LMCD1 mutations promoted cell migration and tumor metastasis in hepatocellular carcinoma. Oncogene. 2012;31(21):2640–52. https://doi.org/10.1038/onc.2011.440.
Montalvo-Ortiz BL, Castillo-Pichardo L, Hernandez E, et al. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J Biol Chem. 2012;287(16):13228–38. https://doi.org/10.1074/jbc.M111.334524.
Bayo J, Fiore EJ, Dominguez LM, et al. Bioinformatic analysis of RHO family of GTPases identifies RAC1 pharmacological inhibition as a new therapeutic strategy for hepatocellular carcinoma. Gut. 2021;70(7):1362–74. https://doi.org/10.1136/gutjnl-2020-321454.
Shang X, Marchioni F, Sipes N, et al. Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem Biol. 2012;19(6):699–710. https://doi.org/10.1016/j.chembiol.2012.05.009.
He H, Huang J, Wu S, et al. The roles of GTPase-activating proteins in regulated cell death and tumor immunity. J Hematol Oncol. 2021. https://doi.org/10.1186/s13045-021-01184-1.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 82173313 and No. 81871191). T.W. conceptualized and designed the review, researched data for the articles and wrote the first draft. T.W., D.R., C.Y., J.S., Y.L., M.X. and W.H. reviewed/edited the manuscript before its submission. All the authors contributed to manuscript revision and approved the submitted final version. T.W. contributed to this work as the first author.
Funding
This work was supported by National Natural Science Foundation of China, No. 82173313 and No. 81871191.
Author information
Authors and Affiliations
Contributions
TW conceptualized and designed the review, researched the data for the article and wrote the first draft. TW, DR, CY, JS, YL, MX and WH reviewed/edited the manuscript before its submission. All the authors contributed to manuscript revision and approved the submitted final version. TW contributed to this work as first author. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no relevant financial or nonfinancial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, T., Rao, D., Yu, C. et al. RHO GTPase family in hepatocellular carcinoma. Exp Hematol Oncol 11, 91 (2022). https://doi.org/10.1186/s40164-022-00344-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-022-00344-4