
Abstract
Immune checkpoint inhibitors targeting programmed cell death protein 1, programmed death-ligand 1, and cytotoxic T-lymphocyte-associated protein 4 provide deep and durable treatment responses which have revolutionized oncology. However, despite over 40% of cancer patients being eligible to receive immunotherapy, only 12% of patients gain benefit. A key to understanding what differentiates treatment response from non-response is better defining the role of the innate immune system in anti-tumor immunity and immune tolerance. Teleologically, myeloid cells, including macrophages, dendritic cells, monocytes, and neutrophils, initiate a response to invading pathogens and tissue repair after pathogen clearance is successfully accomplished. However, in the tumor microenvironment (TME), these innate cells are hijacked by the tumor cells and are imprinted to furthering tumor propagation and dissemination. Major advancements have been made in the field, especially related to the heterogeneity of myeloid cells and their function in the TME at the single cell level, a topic that has been highlighted by several recent international meetings including the 2021 China Cancer Immunotherapy workshop in Beijing. Here, we provide an up-to-date summary of the mechanisms by which major myeloid cells in the TME facilitate immunosuppression, enable tumor growth, foster tumor plasticity, and confer therapeutic resistance. We discuss ongoing strategies targeting the myeloid compartment in the preclinical and clinical settings which include: (1) altering myeloid cell composition within the TME; (2) functional blockade of immune-suppressive myeloid cells; (3) reprogramming myeloid cells to acquire pro-inflammatory properties; (4) modulating myeloid cells via cytokines; (5) myeloid cell therapies; and (6) emerging targets such as Siglec-15, TREM2, MARCO, LILRB2, and CLEVER-1. There is a significant promise that myeloid cell-based immunotherapy will help advance immuno-oncology in years to come.
Similar content being viewed by others


Introduction
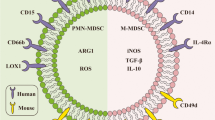
Tumors are often described as “wounds that do not heal” [1]. This is likely due in part to the inhibition of myeloid cells within the tumor microenvironment (TME). Myeloid cells are innate immune cells that function as the front line in host defense against pathogens and play important roles in tissue repair after clearance of pathogens [2]. Myeloid cells are important in all stages of tumor development and orchestrate innate and adaptive immune responses [3,4,5]. In early stages of tumorigenesis, innate immune cells, including macrophages and dendritic cells (DCs), trigger an inflammatory response to induce myelopoiesis and recruitment of other immune cells to eliminate tumor cells [3, 6,7,8,9]. However, failure of cytotoxic immune cells to clear the tumor cells due to somatic mutations results in unresolved, persistent inflammation, which continuously recruits immune cell infiltration and gradually reprograms them to support tumorigenesis [10,11,12,13]. Myeloid cells, including macrophages, DCs, neutrophils, monocytes, and myeloid-derived suppressor cells (MDSCs) imprinted by the TME, display distinct yet overlapping functions (Fig. 1). Given the development of multi-omics technologies, myeloid cells are now known to have high heterogeneity and complexity, which both create challenges and have implications for the development of myeloid cell-targeting immunotherapies [11, 14,15,16,17,18].

Myeloid cells in the TME: friend or foe? A. Myeloid cells can be molded by the TME or therapeutic strategies to exert either pro-tumor or anti-tumor functions. TAMs, tolerogenic DCs, neutrophils, and MDSCs mainly foster cancer progression through supporting tumor cell transition and proliferation, promoting metastasis through enhanced vascularization and preparation of metastatic niche, as well as mediating immunosuppression, through the secretion of soluble factor, extracellular vesicles or direct ligand–receptor interaction. TAMs, DCs, and neutrophils can be programmed toward an anti-tumor phenotype. cDC1s and cDC2s are major APCs that present tumor-associated antigens to T cells and prime T cell responses. TAMs can be reprogrammed to serve as APCs. Both TAMs and DCs, once properly activated, express cytokines such as type I IFN, CXCL9, and CXCL10 to recruit T cells into the TME. Neutrophils can perform direct cancer killing through the generation of ROS or indirect killing induced by death signals such as TRAIL and TNF. B. Summary of the cross talk between myeloid cells and tumor cells in the TME. Tumor cells secreted a variety of soluble factors including chemo attractants that recruit myeloid cells. The recruited myeloid cells further amplify these signals and in turn fuel tumor growth and metastasis by producing factors that remodulate surrounding tissue structure, growth factors, and immunosuppressive molecules. Red: anti-tumor effects; Black: pro-tumor effects
Targeting myeloid cells with immunotherapy was discussed at length during the 2021 China Cancer Immunotherapy workshop in Beijing, the sixth annual conference organized by the Chinese American Hematologist and Oncologist Network (CAHON), China Center for Food and Drug International Exchange (CCFDIE), China National Medical Product Administration (NMPA), and Tsinghua University. Researchers from both the USA and China discussed their most recent work on myeloid cells in immuno-oncology, ranging from the deconvolution of the myeloid compartment in the TME to discovering novel targets for manipulating myeloid cells for improved immunotherapy. In this review, we discuss the mechanisms of myeloid cell-mediated tumor immunity and evasion. We will highlight selected approaches for modulation of myeloid cells and include data presented at the 2021 China Cancer Immunotherapy Workshop. Finally, we provide a comprehensive review of the ongoing clinical trials involving novel agents that target myeloid cells for the purpose of cancer treatment.
Major myeloid cell populations in the tumor microenvironment and their mechanism of immunosuppression
Tumor-associated macrophages
Tumor-associated macrophages (TAMs) are the most abundant immune cells within the TME. High infiltration of TAMs or enrichment of the TAM gene signature is associated with poor prognosis in solid tumors including breast cancer, bladder cancer, and cervical cancer [11, 19,20,21]. Macrophages have different origins dependent on tissue types and thus are not always categorically “myeloid” cells. They can be yolk sac- or fetal liver-derived tissue-resident macrophages (TRMs) in addition to those differentiated from hematopoietic stem cells (HSCs) [22,23,24]. TAMs include both TRM and monocyte-derived macrophages [17, 25,26,27,28,29]. Despite differential origins, TAMs are programmed by the surrounding environment to primarily suppress anti-tumor immunity, while the anti-tumor functions of TAMs in response to certain treatments, such as low-dose irradiation and histone deacetylase (HDAC) inhibitors, have also been described [30, 31].
Macrophages can be polarized in vitro into pro-inflammatory M1 phenotype by IFNγ and lipopolysaccharide (LPS) treatment, or anti-inflammatory M2 phenotype when treated with interleukin-4 (IL-4) [32,33,34]. M1 macrophages produce pro-inflammatory cytokines such as tumor necrosis factor α (TNFα), IL-1β, IL-12, and IL-18 and upregulate major histocompatibility complex (MHC) class II (MHC-II) as well as co-stimulatory molecules including CD80 and CD86. M2 macrophages are characterized as immunosuppressive through the expression of immune inhibitory molecules including transforming growth factor β (TGFβ), IL-10, arginase 1 (Arg1), and CD206. The M1/M2 macrophages retain plasticity after polarization, which can be reversed depending on the culture condition [35]. However, the M1/M2 classification is oversimplified for TAMs because TAMs consist of a heterogenous population and express both M1 and M2 signatures phenotypically [36].
The TME is a hostile environment due to rapid tumor growth and high metabolic demand and is characterized by hypoxia, restricted nutrition availability, acidosis, and other factors. The interplay between a tumor cell and the surrounding cells pressures the infiltrating immune cells to shift their phenotypes to adapt to the TME [37]. Macrophages are initially recruited to the tumor sites through macrophage colony-stimulating factor 1 (CSF-1) signaling [38], and amplified through a variety of cytokines including C–C motif chemokine ligand 2 (CCL2), TNFα, vascular endothelial growth factor (VEGF), C-X-C motif chemokine ligand 12 (CXCL12), and TGFβ [26, 39,40,41,42]. Hypoxia and lactic acid polarized TAMs produce a wide range of soluble factors including VEGF, platelet-derived growth factor (PDGF), hepatocyte growth factor (HGF), and basic fibroblast growth factors (bFGF) as well as inflammatory cytokines including TGFβ, IL-1β, TNFα, and CCL18 to promote tumor growth, angiogenesis, tumor plasticity, and metastasis [3, 43, 44].
In addition to soluble factors, tumor cells may also skew myeloid cell differentiation and phenotype through extracellular vesicles (EV) which transfer DNA, RNA, proteins, lipids, metabolites, or miRNAs [45, 46]. Tumor-derived exosomes (TDEs) are a subclass of EV which are taken up by macrophages, induce PD-L1 expression on macrophages and enhance their immunosuppressive capacity [47]. Although the underlying mechanism is not well elucidated, a recent study suggests that TDEs metabolically reprogram macrophages by engaging with toll-like receptors 2 (TLR2) and triggering MyD88 and NF-κB signaling, leading to increased glycolytic activity, elevated lactate production and polarizing macrophages into an immunosuppressive phenotype [47]. TDEs also engage with other TLRs such as TLR4 and TLR7 on other phagocytes including monocytes and neutrophils in which miRNA, noncoding RNA, and high-mobility group box 1 (HMGB1) transferred by TDEs are implicated in driving the pro-tumor phenotype of these myeloid cells [48,49,50].
TAMs play a central role in mediating immunosuppression, inhibiting tumor cell clearance by cytotoxic T cells via direct contact or secretion of soluble factors. PD-L1 expression is upregulated in TAMs in mouse models and in human cancers including hepatocellular carcinoma (HCC), melanoma, breast, and ovarian cancer [51,52,53]. PD-L1 expression on TAMs or other myeloid cells contributes to CD8+ T cell suppression and resistance to immune checkpoint inhibitor (ICI) therapy [52, 53]. Another co-inhibitory molecule B7-H4 expressed on TAMs mediates T cell dysfunction in HCC and ovarian cancer [54, 55]. L-Arginine is essential for T cell metabolic fitness and survival as well as the generation of memory T cells [56]. Arginase produced by TAMs depletes L-Arginine in the TME and represses T cell receptor (TCR) expression on activated T cells, resulting in impaired anti-tumor T cell responses [57]. TAMs also act through intermediate cells to suppress T cell activity. For example, IL-10 secreted by TAMs promotes regulatory T cell (Treg) function and inhibits IL-12 production by CD103+ DCs, leading to T cell suppression and diminished T cell activation [58]. TAMs confer therapeutic resistance to chemotherapy, immunotherapy, and radiation [59]. In triple-negative breast cancer (TNBC), chemotherapy-induced reactive oxygen species (ROS) which upregulates PD-L1 on TAMs, leading to reduced efficacy of paclitaxel [60]. In a recent study, monocyte-derived macrophages in liver metastasis eliminate anti-tumor CD8+ T cells through induction of Fas-dependent apoptosis, thus mediating resistance to ICI therapies and may explain the immunosuppressive TME within the liver [61].
Because of the high abundance, durability, and adaptability of macrophages, TAMs reeducated by the TME play pivotal roles in fueling tumor progression. However, the versatility of TAMs also provides opportunities for manipulation for therapeutic purposes. Although TAMs do not fit nicely into M1/M2 classification, a high M1 signature over M2 signature predicts better survival in ovarian cancer, strengthening the rationale of targeting TAMs for cancer treatment [62]. Strategies ranging from TAM depletion, repolarization, metabolic reprogramming, and even engineered macrophages are being developed. Given the heterogeneity of TAM populations, targeting a specific TAM subpopulation may enhance the likelihood of effective tumor suppression. In the 2021 China Cancer Immunotherapy Workshop, Edgar G. Engleman (Stanford University) noted that a C-type lectin receptor Dectin-2 is most highly expressed in TAMs and dictates a highly immunosuppressive phenotype. Accumulation of Dectin-2+ TAMs promotes tumor growth in mouse tumor models. However, intratumoral administration of Dectin-2 ligand reprograms TAMs into an immune-activating phenotype and contributes to enhanced anti-tumor immunity. Emerging TAM targets will be discussed later in this review.
Dendritic cells
Dendritic cells (DCs) are key antigen-presenting cells (APCs) that prime and activate T cells. Despite their low abundance in the TME, DCs play a vital role in bridging innate immunity with adaptive immunity and orchestrating anti-tumor responses by T cells (Fig. 1). Conventional DCs (cDCs) and plasmacytoid DCs (pDCs) are two major types of DCs. cDCs are differentiated from common DC precursors (CDP) and are divided into cDC1s and cDC2s [63]. The cDC1 population, which are recognized as CD103+ DCs in mice and CD141+ DCs in humans, can cross-present tumor antigens to CD8+ T cells through MHC class I (MHC-I) in addition to stimulating Th1 polarization of CD4+ T cells [64,65,66]. The cross-presentation capacity by cDC1s is critical for priming CD8+ T-cell-mediated anti-tumor immunity both in situ and in the lymph nodes [66,67,68,69]. cDC1 infiltration correlates with improved clinical outcomes to immunotherapy [66,67,68,69]. cDC2s are defined as CD11b+ DCs in mouse models and CD1c+ in humans. They induce CD4+ T cell responses through MHC-II presentation and contribute to immune surveillance in the TME [70, 71]. A recent study found a subset of cDC2s expressing interferon-stimulated genes (ISGs) also has antigen cross-presentation ability and fosters CD8+ T cell-dependent anti-tumor immunity [72]. In addition, cDC1s are more effective in MHC-II presentation than cDC2s [73, 74]. cDC1s activate CD4+ T cells and are licensed by CD40 signaling via CD4+ T cells, for optimal CD8+ T cell priming [75].
In addition to the antigen-specific signal by MHC molecules, mature DCs express co-stimulatory molecules such as CD86, CD80, CD40, OX40L, GITRL, and 4-1BBL, which are essential for optimal T cell activation and survival [5]. Type I interferon (IFN) plays an important role in host anti-tumor immunity [76,77,78]. Activation of the cytosolic DNA sensing pathway mediated by cyclic GMP-AMP synthase (cGAS) and Stimulator of interferon genes (STING) in DCs promote DC maturation and type I IFN production, which augments T cell cytotoxicity to eradicate tumors [79,80,81]. Tumor-resident DCs stimulated by type I IFN produce CXCL9 and CXCL10 which promotes T cells trafficking to the TME [67]. IL-12 produced by cDC1s is required for anti-tumor immunity by T cells and response to anti-PD-1 therapy [82].
DCs are modulated by the TME to drive immune tolerance. Co-inhibitory molecules such as PD-L1, PD-L2, V-domain immunoglobulin suppressor of T cell activation (VISTA), and CD31 are induced on DCs to restrain T cell function [5, 83,84,85]. Inflammasomes are cytosolic multiprotein complex triggered by pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs), which initiate a pyroptotic inflammatory response [86]. A subset of TIM-3+ DCs with reduced DNA uptake capacity suppresses anti-tumor immunity through inflammasome activation, which can be reversed by TIM-3 blockade [87,88,89]. CTLA-4 ligation with CD80 and CD86 induces indoleamine 2,3-dioxygenase 1 (IDO1) by DCs, which converts the essential amino acid tryptophan to kynurenine, inhibiting T cell proliferation and favoring Treg cell differentiation [90].
During the 2021 China Cancer Immunotherapy Workshop, Miriam Merad (Mount Sinai) discussed the identification of a new DC cluster, “mature DCs enriched in immunoregulatory molecules” (mregDCs), which were present in non-small cell lung cancer (NSCLC), HCC, and colorectal cancer (CRC) using single-cell RNA sequencing and cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) [91]. These mregDCs co-express immune regulatory genes such as CD274, Pdcd1lg2, and CD200 and maturation genes (CD40, Ccr7, and Il12rb). Both cDC1s and cDC2s can acquire mregDC signature upon sensing or uptake of tumor-associated antigen, partially driven by IL-4 signaling and AXL signaling, whereas IFNγ is required for IL-12 production by mregDCs. IL-4 blockade enhances IL-12 production in mregDCs and promotes T cell effector function [91].
pDCs are differentiated from CDPs or lymphoid progenitors [92]. They are potent type I IFN producers when encountering pathogens but poor antigen presenters [93]. pDCs in the TME have impaired type I IFN production (mediated in part by TGFβ) and increased induction of Treg differentiation, hence supporting tumor growth in breast and ovarian cancer [94,95,96,97,98,99]. However, the full role of pDCs in the TME is not yet clear.
Neutrophils
Neutrophils are the most abundant myeloid population that developed from granulocyte–monocyte progenitors (GMPs) in the bone marrow. The high neutrophil-to-lymphocyte ratio (NLR) in the peripheral blood of cancer patients is associated with poor prognosis in many cancers, including NSCLC, CRC, HCC, and prostate cancer [100,101,102,103,104,105,106]. A higher baseline NLR is associated with worse survival and decreased clinical response with ICI therapy across many cancer types including advanced melanoma and NSCLC [107,108,109,110]. Neutrophilic infiltration is seen in the majority of solid tumors; however, the prognostic relevance remains controversial and inconsistent [21]. For example, in HCC, a high density of tumor-associated neutrophils is associated with poor prognosis while in CRC, mixed conclusions are reported [111,112,113,114,115].
Circulating neutrophils are divided into two populations based on density: high-density neutrophils (HDNs), and low-density neutrophils (LDNs) that are found within the mononuclear cell fraction after density gradient centrifugation of blood, indicating an immature phenotype [116]. LDNs are pleiotropic and can be either immunosuppressive or pro-inflammatory, depending on the disease context [116]. Tumor-associated neutrophils (TANs) are classified into anti-tumor N1 and pro-tumor N2, which mimics the nomenclature of M1/M2 polarized macrophages [116,117,118]. The diversity of the neutrophile composition within tissues and variations between disease states likely contributes to these inconsistent implications.
Neutrophil recruitment to the tissues is mainly dependent on CXCL8-CXCR1/CXCR2 axis [119]. Cytokines produced by tumor and surrounding cells such as GM-CSF, G-CSF, and IL-6 stimulate granulopoiesis in the bone marrow and recruit neutrophils to the tumor site [3, 117, 120, 121]. IL-1β and G-CSF prolong neutrophil survival in the TME [122]. Other molecules such as IL-17 produced by γδ T cells are also involved in neutrophil recruitment in the TME [123]. Neutrophils promote cancer progression through both unique and shared mechanisms as TAMs. They can modulate the extracellular matrix (ECM) by producing matrix metallopeptidase (MMP) 8 and 9 along with neutrophil elastase (NE), inducing VEGF production to promote metastasis [124, 125]. They also release ROS and reactive nitrogen species (RNS) to induce DNA damage in epithelial cells to facilitate carcinogenesis [118]. Neutrophils express a wide repertoire of cytokines and inhibitory ligands that mediate immunosuppression via crosstalk with other immune cells. For example, Arg1, PD-L1, and VISTA expressed by neutrophils dampen T cell function in the TME [117].
Despite the pro-tumoral activities discussed above, neutrophils also play anti-tumoral roles and prevent metastasis in the TME. Neutrophils can eliminate cancer cells through ROS-dependent killing, which induces lethal Ca2+ influx in target cells, dependent on transient receptor potential melastatin 2 (TRPM2) that is highly expressed in cancer cells [126, 127]. Neutrophils can also elicit tumor-killing functions by the expression of NO, TRAIL, and TNF [128, 129]. In addition to direct killing, neutrophils are shown to express immune stimulatory molecules such as CD86, OX40L, and 4-1BBL to enhance T cell function [130].
Neutrophils are known to form neutrophil extracellular traps (NETs) to confine pathogens from dissemination and exert immune modulatory functions. Like neutrophils themselves, NETs possibly play multifaceted roles in tumor immunity. They potentially facilitate tumor progression by the release of NE, cathepsin G, and MMP9, as well as tumor metastasis [131,132,133,134]. NETs may also shield tumor cells and protect them from CD8+ T cell and NK cell cytotoxicity [135]. By contrast, there is evidence that NETs degrade pro-inflammatory cytokines and reduce inflammation in chronic inflammation [136], which may have implications for the positive role of NETs in tumor. More studies are required to elucidate the role of NETs in the TME.
Monocytes
Monocytes are classified into three major populations distinguished by differential expression of CD14 and CD16 in human, and in mouse Ly6c and TREML4: classical (CD14++CD16− in human and Ly6c++TREML4− in mouse), intermediate (CD14++CD16+ in human and Ly6cint in mouse) and non-classical (CD14+CD16++ in human and Ly6c−TREML4++ in mouse) monocytes [4]. Non-classical monocytes, also known as “patrolling” monocytes, play an important role in maintaining vessel integrity by clearing dying endothelial cells and preventing tumor metastasis [137]. Classical monocytes are more abundant than non-classical and are recruited to tissue via CCL2–CCR2 axis. VEGF-A and CSF-1 play redundant roles in monocyte recruitment [42, 138]. Upon encountering tumor-derived signals, monocytes sequentially differentiate into TAMs, promoting cancer progression, metastasis as well as mediating immunosuppression [4]. Genetic ablation of CSF-1 reduced TAMs infiltration and delayed tumor progression in mouse mammary tumor models [138].
TDEs are reported to modulate monocyte function in different directions depending on the source of TDEs [139,140,141]. TDEs secreted by highly metastatic melanoma recruit pro-tumor monocytes to the pre-metastatic niche, while TDEs from non-metastatic tumors induce the expansion of anti-tumor “patrolling” monocytes and prevent lung metastasis by clearing tumor cells at the pre-metastatic niche [140].
Myeloid-derived suppressor cells
During myelopoiesis, immature myeloid cells are found in circulation and tumor sites. They are similar to monocytes and neutrophils but exhibit potent immunosuppressive activity, and are termed MDSCs [142, 143]. There are two types of MDSCs: monocytic MDSC (M-MDSC) and granulocytic/polymorphonuclear MDSC (PMN-MDSC). The distinction of MDSCs from neutrophils and monocytes has long been challenging. In the TME, it is likely that monocytic cells undergo sequential differentiation stages, from monocytes to M-MDSCs, and eventually become TAMs [144]. MHC class II is widely used for distinguishing M-MDSCs (CD14+CD15−HLD-DRlo/− in human) from monocytes (CD14+CD15−HLD-DRhi in human) while this may not be sufficient [145]. Although TAMs can be phenotypically distinguished from M-MDSCs in mice through increased expression of F4/80, CD115, and IRF8 (CD68 and CD163 in human) and lower expression of Ly6c and S100A9, a specific marker for M-MDSC is needed to better address the difference in monocytic cells [144]. It is even more difficult to identify PMN-MDSCs from neutrophils because they share the same phenotypical markers and they have overlapped functions. Both LDNs and N2 TANs refer to PMN-MDSCs in cancer-related studies. The lack of a uniform nomenclature for granulocytic cells creates confusion in studying their roles, especially between pro-tumor N2 TANs and PMN-MDSCs [144]. Now more markers such as LOX1 which distinguishes PMN-MDSCs from neutrophils in humans have emerged to help better identify these myeloid cell subpopulations [146].
With more in-depth study of MDSCs using transcriptomic and proteomic technologies, there is an updated view that MDSCs are pathologically activated neutrophils and monocytes during persistent myelopoiesis [145]. Tumor-derived factors such as GM-CSF, CSF-1, and G-CSF signal through signal transducer and activator of transcription 3 (STAT3), CCAAT/enhancer-binding protein β (C/EBPβ) and IRF8 to promote myelopoiesis [147, 148]. Downregulation of IRF8 in myeloid progenitors prevents terminal differentiation, therefore leading to the accumulation of immature myeloid cells [147, 149, 150]. The secondary signals from tumor- and tumor stroma-derived factors including HMGB1, TLRs, TGFβ, and endoplasmic reticulum (ER) stress then pathologically activate MDSCs through STAT6, STAT1, and NF-κb signaling pathways [142, 147, 151].
Like TAMs, MDSCs remodel the TME by producing VEGF, bFGF, and MMP9 to facilitate cancer progression and metastasis [147, 152, 153]. MDSCs also exert immunosuppression by suppressing T cell function through direct ligand–receptor engagement, release of soluble inhibitory cytokines and sequestration of amino acids essential for T cells. In most cancer types, PMN-MDSCs are the major population (~ 80%) of MDSCs [147]. STAT3 phosphorylation is increased in MDSCs and results in elevated nicotinamide adenine dinucleotide phosphate (NADPH) level, leading to ROS accumulation [154]. ROS and ROS-mediated peroxynitrite (PNT) accumulation nitrates TCR and block TCR binding with MHC molecules, impairing T cell responsiveness to antigens [155]. Hyperproduction of PNT also inhibits T cell infiltration by nitrating the chemokines that are responsible for attracting T cells [155,156,157,158]. M-MDSCs are rapidly converted to TAMs in tumor hypoxia regions to enhance immunosuppression [159, 160]. Both of PMN-MDSCs and M-MDSCs produce Arg1, iNOS, and IDO1 to suppress T cell function [142, 147, 161]. MDSCs also impair other immune cell functions including DCs, B cells, and NK cells but promote Tregs by producing IL-10 and TGFβ [13, 162,163,164].
In summary, the major myeloid cell populations in the TME, including TAMs, DCs, neutrophils, and MDSCs, are “aberrantly programmed” by the TME. Once activated, these cells exert effects on the TME which promote tumor growth. Strategies have been developed to recalibrate these myeloid cells and harness their power to restore anti-tumor immunity (Fig. 2 and Table 1). We will discuss preclinical data and clinical data as it relates to each target. We will also discuss some emerging targets for myeloid cell manipulation.

Myeloid-specific targets as immune adjuncts for the management of solid malignancies. Targets were categorized based on their functional role: proliferation, differentiation, recruitment, polarization, functional blockade, cytokine signaling, epigenetic reprogramming, and metabolic reprograming. For cellular therapies, myeloid-specific approaches include dendritic cell vaccines and chimeric antigen receptor macrophages
Targeting strategies against myeloid cells for cancer immunotherapy
In this section, we discuss ongoing strategies targeting the myeloid compartment in the preclinical and clinical settings which include: (1) altering myeloid cell composition within the TME through enhanced differentiation, proliferation, and recruitment of myeloid cells; (2) functional blockade of immune-suppressive myeloid cells; (3) reprogramming via either polarization, metabolic, or epigenetic modification of myeloid cells to acquire pro-inflammatory properties; (4) modulating myeloid cells via cytokines; (5) myeloid cell therapies; and (6) emerging targets such as Siglec-15, triggering receptor expressed on myeloid cells 2 (TREM2), macrophage receptor with collagenous structure (MARCO), leukocyte immunoglobulin-like receptor B2 (LILRB2), and common lymphatic endothelial and vascular endothelial receptor 1 (CLEVER-1) (Table 1).
Strategies to alter myeloid cell differentiation, proliferation, and recruitment with the tumor microenvironment
In response to tumor-derived factors, immunosuppressive myeloid cells are consistently recruited, expanded, or differentiated to fuel tumor progression. One of the most straightforward strategies of targeting myeloid cells for cancer treatment is to alter the myeloid population composition, reducing the pro-tumor myeloid cell infiltration and increasing the abundance of anti-tumor immune cells. Strategies ranging from chemoattractant blockade to myeloid growth factors have been studied extensively in both preclinical animal models and clinical trials.
CCL2–CCR2 axis
The CCL2–CCR2 plays an integral role in the recruitment of myeloid cells including inflammatory monocytes, TAMs, and MDSCs. In metastatic CRC models, liver metastases which contain TAMs with high CCR2 expression are linked to a worse prognosis [165]. Inhibition of the CCL2–CCR2 axis suppresses tumor metastasis through reduced angiogenesis in preclinical models, in both direct manner, since CCL2 itself exerts an angiogenic effect, and indirect manner, which is through reduced chemoattraction of monocytes and macrophages [166,167,168]. A variety of inhibitors have been studied in the clinical setting to assess tumor response, which are summarized below.
Carlumab (CNTO 888) is a human monoclonal anti-CCL2 antibody with primarily negative clinical results. Carlumab was ineffective as monotherapy, as seen in a phase II study (NCT00992186) involving second-line therapy for metastatic castrate-resistant prostate cancer, where the objective response rate (ORR) was 0% and the median progression-free survival (mPFS) was only 2.7 months [135]. However, carlumab in combination with conventional chemotherapy (docetaxel, paclitaxel, carboplatin, gemcitabine, and PEGylated liposomal doxorubicin) for advanced solid tumors demonstrated improved clinical responses, including an ORR of 37.5% and a duration of response (DOR) of 6.3 months [169, 170]. Unfortunately, the effects of carlumab may be short-lived based on median CCL2 serum concentrations collected throughout the study period. While there was an initial reduction in total levels at the two-hour mark following initiation, there was a subsequent threefold to fivefold increase with further doses compared to baseline, regardless of the chemotherapy backbone, suggesting chemotherapy alone may have resulted in tumor response. Based on safety data, carlumab is well tolerated with the chemotherapy, with the most common grade 3 treatment-related adverse events (TRAEs) being cytopenias, fatigue, and stomatitis.
PF-04136309 is a small-molecule oral CCR2 inhibitor. In two small phase I trials (NCT01413022 NCT02732938), PF-04136309 was added to chemotherapy (FOLFIRINOX, nab-paclitaxel, and gemcitabine) in the management of advanced pancreatic cancer and produced response rates ranging from 23.8 to 48.5% [171, 172]. Pulmonary toxicity was reported in 24% when PF-04136309 was combined with nab-paclitaxel and gemcitabine. In the exploratory analysis, almost all recipients of PF-04136309 were found to have a decrease in peripheral blood CD14+ CCR2+ monocytes, though CCR2+ TAMs remained present in the majority of biopsy samples.
BMS-813160 is a small-molecule inhibitor that antagonizes both CCR2 and CCR5 and is currently under investigation in combination with nivolumab for the treatment of a variety of tumor types (NCT03496662, NCT03767582, NCT03184870, NCT04123379, and NCT02996110). Neither carlumab nor PF-04136309 has ongoing trials at this time.
CSF-1R
CSF-1 is a major lineage regulator and chemoattractant for TAMs. Preclinical data have demonstrated that inhibition of CSF-1R signaling repolarizes TAMs from M2-like to M1-like anti-tumor phenotype rather than simply depleting TAMs [173, 174]. One issue encountered with CSF-1R blockade has been compensatory upregulation of PD-L1 and CTLA-4 to maintain tolerogenic abilities, so clinical models have focused on a dual inhibitory approach involving CSF-1R blockade and ICIs to overcome this effect [175, 176].
While antagonists like sunitinib grossly block class III receptor tyrosine kinases (c-KIT, FLT3, CSF-1R, and PDGFR), dedicated CSF1R inhibitors have been developed, including small-molecule agents (pexidartinib, ARRY-382, BLZ945, and vimseltinib) and monoclonal antibodies (emactuzumab, cabiralizumab, IMC-CS4, AMG820, lacnotuzumab, PD-0360324, and axatilimab) [177, 178]. However, few have been able to demonstrate meaningful clinical activity. Two phase I trials involving LY3022855 monotherapy (NCT02265536, NCT01346358) and one phase I trial involving AMG 820 (NCT01444404) in the management of advanced solid tumors reported zero objective responses (0/86 and 0/25, respectively), though decreases in TAMs were noted in addition to elevations in circulation CSF-1 levels, indicating that proper target engagement occurred [179,180,181]. When LY302285 is used in combination with ICIs including tremelimumab (anti-CTLA-4) or durvalumab (anti-PD-L1), ORR approaches 4.2% (3/72) [182]. Similarly, for AMG820, when combined with pembrolizumab (anti-PD-1) for advanced solid tumors, ORR has been documented at 2.6% (3/116), well below expected response rates seen with pembrolizumab monotherapy [183].
One area of promise for CSF-1 inhibitors is in the management of tenosynovial giant cell tumors (TGCTs) and pigmented villonodular synovitis (PVNS) which are both rare, nonmalignant tumors that originate from the synovium of musculoskeletal joints and occur because of CSF-1 overexpression due to CSF-1/COL6A3 translocations [184]. Pexidartinib received FDA approval in 2019 following the results of the phase III trial (ENLIVEN) which randomized patients with unresectable TGCTs to receive pexidartinib vs. placebo. Following a 25-week follow-up period, the ORR was 38% (vs. 0% placebo, p < 0.0001) with a complete response (CR) rate of 15% [185]. Interestingly, ORR rates were similar between placebo crossovers and the initial pexidartinib arm, with crossover participants experiencing less hepatotoxicity, so the FDA did not include a loading dose in the approval [186]. Unique adverse events reported in ENLIVEN included changes to hair color (67%), transaminitis (39%), and nausea (38%), and both periorbital (13%) and peripheral (13%) edema among others.
CXCR1/2
The release of IL-8 by malignant cells and its subsequent binding to CXCR1 and CXCR2 on circulating myeloid cells and surrounding endothelial cells leads to the recruitment of MDSCs to the TME and the promotion of angiogenesis [187]. Ibuprofen inhibits IL-8 signaling, both through cyclooxygenase-2 (COX2) and non-COX2 pathways, and has been used as a base model for the development of novel CXCR1/2 inhibitors, including reparixin and ladarixin [188]. Other backbones have been explored as well, including nicotinamide antagonists (SX-682) and thiazolopyrimidine derivatives (AZD 5069).
Reparixin showed promising single-arm phase I trial data when combined with weekly paclitaxel in metastatic HER2-negative breast cancer (ORR 30%) [189]. However, subsequent randomized, two-arm data from the phase II fRIDA trial failed to detect a difference in the primary endpoint of mPFS when comparing the combination therapy to paclitaxel alone (5.5 vs. 5.6 months, respectively) [190]. Ladarixin is a second-generation dual inhibitor with stronger affinity for CXCR2, slowed melanoma progression in preclinical models, but clinical trials remain absent at this time [191]. Ongoing trials involving allosteric, reversible, small-molecule inhibitors SX-682 and navarixin as monotherapies and in combination with PD-1/PD-L1 agents are currently underway (NCT04245397, NCT03161431, NCT04599140, NCT04477343, NCT04574583, and NCT03473925).
Indirect methods of CXCR1/2 inhibition are also emerging, including the development of monoclonal antibodies which bind and sequester IL-8, such as HuMax-IL8 (BMS-986253) which has been shown in preclinical models to reduce PMN-MDSCs and prevent the mesenchymalization of TNBC [192]. Following a phase I study, HuMax-IL8 was found to provide no objective response as monotherapy, but multiple follow-up trials are ongoing involving its use in combination with immunotherapy agents (NCT04848116, NCT03689699, NCT02451982, NCT04050462, NCT03400332, NCT04572451, and NCT04123379) and chemotherapy (NCT05148234) [193].
FLT3L
FMS-like tyrosine kinase 3 receptor ligand (FLT3L) plays an active role in the maturation of macrophage-dendritic progenitors (MDPs) into pDCs and cDCs [194]. Preclinical studies have suggested that recombinant human FLT3 ligand (rhuFLT3L) agonism can lead to an enhancement in immunologic therapies, including PD-L1 inhibition [68]. Additionally, rhuFLT3L use has been shown to aid in the abscopal effect of radiation therapy by promoting immunogenic cell death [195, 196]. A similar abscopal effect has been noted when rhuFLT3L is combined with DC vaccine therapies [197]. Finally, in PD-L1 resistant mouse models, a combination approach involving FLT3L, radiotherapy, and TLR3/CD40 stimulation promotes CD8+ T cell influx, PD-L1 responsiveness, and tumor regression both locally and in distant untreated lesions, leading researchers to focus on this combination approach for clinical trials [198].
CDX-301 is a soluble rhuFLT3L developed using Chinese hamster ovary cells, and it has been shown to be a viable, well-tolerated option for combination trials [199]. Though it provides no clinical response on its own and public-domain clinical data remain scarce, preliminary phase II data (NCT0283925) involving CDX-301 in combination with single lesion SBRT resulted in 31% of analyzed subjects (9/29) recorded partial response (PR) involving distant lesions on PET imaging 2 months following therapy, further highlighting its abscopal potential [200].
STAT3
STAT3 has been implemented in immune escape and the promotion of tumor proliferation. The immunosuppressive potential of MDSCs occurs partially due to hindrances in myeloid progenitor differentiation as activated STAT3 inhibits the expression of protein kinase C βII (PKCβII) signaling [201]. Within the tumors themselves, constitutively activated STAT3 results in increased expression of PD-L1 along with the release of immunosuppressive cytokines (IL-6, IL-10, etc.) and growth factors such as CSF-1 and VEGF [202].
Considering STAT3 contributes to both tumor growth and the promotion of tolerogenic immune cells, it is an ideal target for cancer therapy development [203]. STAT3 activation occurs following phosphorylation by Janus kinases (JAKs) and subsequent homodimerization, leading it to translocate to the nucleus and perform its transcription functions. STAT3 and JAKs are then deactivated through Src homology domain-containing tyrosine phosphatases (SHP-1/2). While certain compounds have been found to impact STAT3 phosphorylation through drug repositioning studies (celecoxib, niclosamide, and pyrimethamine) or through known JAK inhibitors (ruxolitinib and pacritinib), more selective STAT3 inhibitors have since been developed including small-molecule inhibitors (napabucasin, TTI-101, OPB-51602, OPB-31121, OPB-111077, BP-1-102, and S3I-201) and oligonucleotides (danvatirsen and STAT3 DECOY) [202, 204, 205].
While the majority of trials (NCT02753127, NCT02993731, NCT01839604, NCT00955812, NCT00657176, NCT01406574, NCT01344876, NCT01711034, NCT02178956, NCT02315534, and NCT02279719) have failed to document meaningful clinical efficacy, as monotherapy or in combination (FOLFIRI, gemcitabine, paclitaxel, sorafenib, and temozolomide), several agents that have off-target effects that lead to lower STAT3 activity are currently being explored, including SHP-1/2 agonists like SC-43 [NCT04733521] and IL-6R inhibitors like tocilizumab (NCT02767557, NCT04940299, and NCT04691817) and siltuximab (NCT04191421) [202, 206,207,208].
Strategies to functionally block immune-suppressive myeloid cells
CD47-SIRP⍺
CD47 is ubiquitously expressed on the surface of normal tissue in order to allow for immune self-recognition. This occurs when CD47 binds to SIRP⍺ which is found on macrophages and DCs [209]. Tumor cells take advantage of this system via overexpression of CD47, providing a unique immune escape mechanism that has garnered considerable interest. Within TAMs, SIRPα expression also remains high and binding to CD47 within the TME further assists TAMs in maintaining their immunosuppressive phenotype through SHP-1/2 signaling [210]. Preclinical studies have found that antagonizing CD47/SIRPα signaling results not only in augmented phagocytosis, but also in DC activation, CD8+ T cell priming, and a decrease in myeloid-driven immunosuppression through macrophage polarization and an increased M1 to M2 ratio [211, 212]. Unique inhibitors of the CD47-SIRP⍺ axis include monoclonal antibodies against CD47 (magrolimab also known as Hu5F9-G4, evorpacept, CC-90002, SRF231, letaplimab, lemzoparlimab, AO-176, TJ011133, SHR-1603, and ZL-1201), monoclonal antibodies against SIRP⍺ (BI765063, GS-0189, CC-95251), and recombinant SIRP⍺-Fc fusion proteins (TTI-621, TTI-622, and evorpacept) [213, 214]. Bispecific antibodies are also emerging with secondary targets including CD19 (TG-1801), CD20 (IMM0306), CD40L (SL-172154), PD-1 (HX009), and PD-L1 (IBI322) [215].
Developing a monoclonal antibody toward SIRPα can be challenging considering that various SIRP homologs exist alongside various SIRPα alleles, so agents require pan-allele sensitivity while avoiding SIRP homolog activity [216]. Advantages, however, include the fact that SIRP is not ubiquitously expressed, allowing for anti-SIRPα therapies to avoid the destruction of bystanders such as red blood cells, as seen with anti-CD47 agents. This also allows them to be given at lower doses while theoretically maintaining efficacy due to decreased antigen sink. Many of the monoclonal anti-CD47 agents currently developed target different epitopes and as a result, a specific subset has been found to only weakly bind to red blood cell CD47 (lemzoparlimab, magrolimab, and AO-176), allowing them to spare these cells and prevent the development of anemia [217]. Additionally, newer anti-CD47 agents have been developed with inert Fc regions (evorpacept) to further avoid this effector function, though as a result these therapies become reliant on combination therapies involving a tumor-opsonizing antibody [218]. The SIRPα-Fc fusion products are made up of IgG Fc fused to the extracellular domain of SIRPα and this structure allows for SIRPα to bind to CD47 for a longer duration by slowing clearance through the presence of the Fc domain [219]. Though affinity for native SIRPα may be lower compared to anti-CD47 mAbs, SIRPα variants have been designed to overcome this deficiency. The small molecular weights seen with these fusion proteins may also assist with their ability to penetrate TME more readily. Bispecific antibodies aim to provide dual-signaling and improve immune cell proximity, though whether this correlates to improved efficacy remains to be seen.
The most promising clinical data involve the use of magrolimab in combination with rituximab ± chemotherapy (gemcitabine and oxaliplatin) for the treatment of relapsed/refractory B cell non-Hodgkin lymphoma (NHL), where researchers noted an ORR of 50% (11/22) with CR noted in 36% (8/36) of participants (NCT02953509) [220]. Contrast this to the phase I results involving magrolimab monotherapy in the treatment of advanced solid tumors where the ORR approached 5% (NCT02216409, NCT30811285) [221]. Similarly, evorpacept in combination with pembrolizumab ± trastuzumab for advanced solid tumors (ASPEN-01 and NCT03013218) resulted in an ORR of 0% (0/15) and a disease control rate (DCR) of 26.7% (4/15) [222]. Biopsies obtained from participants post-treatment showed increases in TAM populations on immunohistochemistry staining, and no increase in CD8+ tumor-infiltrating lymphocytes (TILs) was noted in either treatment arm.
CD24-Siglec-10
CD24 suppresses inflammatory responses through binding to sialic acid-binding immunoglobulin-type lectin-10 (Siglec-10) found on the surface of macrophages [223]. However, CD24 has also recently been found to provide a unique immune escape mechanism utilized by a variety of cancer cells [224]. Though CD24 is primarily expressed on immune progenitor cells and lymphoid tissue, certain tumor types have been found to express CD24 at high magnitudes [224, 225]. To elicit an effect, CD24 binds to TAMs via surface-bound Siglec-10, resulting in immune escape through SHP-1 and SHP-2 signaling, similar to CD47. To put this theory of immune escape to the test, researchers removed the CD24 protein gene from human breast cancer cell lines, then intermixed these CD24-deficient cells with wild-type cancer cells. They confirmed that macrophages cleared out the CD24-deficient populations more rapidly [224]. These cells were also significantly more sensitive to anti-CD47 therapies, suggesting a plausible synergistic role with some of the CD47-targeting agents mentioned previously. Finally, Siglec-10 knockout macrophages were also created, resulting in improved phagocytosis abilities compared to controls.
CD24 also plays a potential role in cancer migration in various cancer types along with prognostication [226, 227]. As a result, many preclinical studies now closely evaluate targeting this signaling pathway as a way of combating both malignancies and the TME. Initial models involved unconjugated monoclonal antibodies targeting the leucine–alanine–proline (LAP) epitope of CD24 (SWA11) which led to antibody-dependent cellular cytotoxicity (ADCC) in lung, ovary, bladder, myeloma, and lymphoma models, all while notably altering the cytokine milieu and hindering metastatic potential [228]. Bispecific antibodies involving MHC-I (cG7-MICA) and CD30 have also been examined with similar results reported. Success has also been noted with antibody–drug conjugates involving various payloads including nitric oxide, pseudomonas exotoxin, and even ricin A-chain immunotoxin [228,229,230,231]. More recently, anti-CD24 chimeric antigen receptor (CAR) T cells and NK products have been investigated in pancreatic and ovarian cancer models with the use of CARs derived from SWA11, with dual targeting seeming to help reduce the incidence of off-target events [228, 232, 233].
A humanized, affinity-matured version of anti-CD24 has already been developed (ONC-781) and this monoclonal antibody has been used to construct an antibody–drug conjugate (ONC-784), a bispecific antibody to CD3 (ONC-783), and a CAR-T therapy (ONC-782) for potential clinical trials [234]. Little remains publicly available regarding clinical trial prospects, but it seems fair to say that dual inhibition of immune escape mechanisms (PD-L1, CD47, and CD28) will likely be on the horizon.
Strategies to reprogram myeloid cells to acquire pro-inflammatory properties
TLR agonists
Sensing of DAMPs and PAMPs through TLRs expressed by APCs results in their activation and subsequent T cell priming [235]. TLR agonists are studied as adjunct therapies to tumor vaccines and immunotherapy agents to amplify treatment response. However, modifications of TLR agonists are required for clinical use to adjust for their short half-life, poor localization, and limited immunogenicity [236]. For the purpose of this review, we will be discussing TLR9 agonists which have been the most extensively studied TLR agonists within the clinical trial setting.
TLR9 is constitutively expressed within the endosomes of B Cells and pDCs, though additional myeloid subtypes have been found to express TLR9 when activated by immune triggers including infection [235]. TLR9 recognizes unmethylated cytosine-phosphate guanine (CpG) oligodeoxynucleotides (ODNs) found on modified or foreign DNA, resulting in robust activation of innate and adaptive immune cells through MyD88 signaling [237]. This discovery has led researchers to engineer TLR9 agonists based on CpG ODNs. Given these agonists are physiologic derivatives, they naturally carry shorter half-lives, but with modifications including a nuclease-resistant phosphonothioate backbone (CPG 7909, ISS 1018, CpG-28, IMO-2055, tilsotolimod, SD-101, GNKG168, and S-540956), the half-life of these agents has been increased from minutes to days [237]. Other modifications include the creation of double stem-loop immunomodulators (dSLIMs) which are CpG DNA molecules that have been covalently closed, forming a dumbbell-like shape that is resistant to DNase degradation (lefitolimod and EnanDIM) [238]. Additionally, various delivery vehicles have been explored to improve localization and bioavailability including nanoparticles (cavrotolimod) and viral-like particles (CMP-001 and NZ-TLR9) [238]. TLR9 agonists are also being investigated as conjugate payloads as part of antibody–drug conjugates for monoclonal antibodies, including anti-SIRPα (ALTA-002) and anti-CD22 (TAC-001). These have been collectively termed as “Toll-like receptor agonist antibody conjugates” (TRAAC) [239].
Preclinical data involving modified CpG ODNs in murine models have demonstrated that intratumoral injections result in tumor regression along with tumor-specific T cell responses and upregulation of immune checkpoint genes including PD-L1, OX40, and CTLA4 [240]. This has been a key justification for combining checkpoint inhibitors with CpG ODNs. Additionally, CpG ODNs are radiosensitizers in early lung cancer models with a sensitivity enhancement ratio (SER) of 1.28, further justifying a multi-therapy approach [241]. In the clinical setting, single-arm phase II results involving intratumoral injections of a CpG agonist (PF-3512676) plus local radiation in low-grade B cell lymphoma noted an ORR of 23.3% (7/30) with a DCR of 86.6% (26/30) [242]. Ongoing phase I studies are examining the use of CpG ODNs in combination with local radiation and immunotherapy agents for the management of refractory lymphomas (NCT03410901).
PF-3512676 (CPG 7909) is the most extensively studied clinical CpG ODN, particularly in combination with conventional chemotherapy (paclitaxel, carboplatin) for the treatment of NSCLC. Initial phase II trials appeared promising with improvements in OS compared to chemotherapy alone but following the release of interim results from two phase III trials, both trials were terminated due to high rates of sepsis-related events and minimal evidence of improved clinical efficacy [243]. CpG ODNs continue to be studied as adjuncts, particularly in the realm of cancer vaccine therapies given their immunostimulatory properties.
CD40 agonists
CD40 is readily expressed on antigen-presenting cells and is essential to their activation. Additionally, its ligand CD40L is found on a variety of immune and non-immune cells, including CD4+ T cells. CD40L helps with the cross-priming of CD4+ cells to non-self-antigens by providing a co-stimulatory effect [244]. Activation of CD40 on DCs leads to upregulation of MHC molecules, increase in IL-12 secretion, and the promotion of cytotoxic T cell activation [245]. Preclinical mouse models and pilot human studies involving CD40 agonist antibodies in combination with gemcitabine in the treatment of pancreatic cancer have shown that CD40 activation helps reverse immunosuppression with modest tumor response rates [246].
Recently developed CD40 agonists include fully human IgG monoclonal antibodies (selicrelumab, mitazalimab, CDX-1140, and 2141-V11), humanized IgG monoclonal antibodies (sotigalimab also known as APX005M, SEA-CD40, and dacetuzumab), chimeric IgG antibodies (ChiLob7/4), recombinant CD40L fusion proteins (MEDI5083), and vaccine-delivered transgenes (LOAd703 and NG-350A) [247]. One feature that separates the monoclonal antibodies apart is their antibody isotype, with most IgG1 models needing FcγR cross-linking to produce a signal (sotigalimab, ChiLon7/4, ADC-1013, and SEA-CD40) whereas IgG2 antibodies mimic CD40L signaling independent of FcγR cross-linking [245, 248]. Additionally, newer IgG1-based monoclonal antibodies have modified (non-fucosylated) Fc regions which help increase their affinity to FcγR in an attempt to improve ADCC (SEA-CD40 and APX005M). Another separating feature for antibodies is epitope binding, with studies showing that agonistic activity decreases for a given antibody as its epitope target draws closer to the cellular membrane, often leading to the development of antagonistic properties [249].
Overall, tumor response rates with single-agent CD40 monoclonal antibodies have been low to date. Single dose selicrelumab was able to produce an ORR of 27% (4/15) in melanoma participants, but in a separate trial involving weekly selicreulmab for advanced melanoma, the ORR was 0% (0/11) with evidence of T cell depletion observed in the exploratory analysis [250, 251]. In combination with tremelimumab for treatment-naive metastatic melanoma, selicreulmab provided an ORR of 27.3% (6/22) with a CR rate of 9.1% (2/22) and evidence of increased T cell infiltration and activation [252, 253]. This is an improvement when compared to separate tremelimumab monotherapy phase III trials where treatment-naïve patients with metastatic or unresectable melanoma achieved an ORR of only 10.7% (36/328) and a CR of 3% (11/328) [253].
Similarly, a phase I study of sotigalimab (APX005M) combined with nab-paclitaxel, gemcitabine, and PD-1 blockade (nivolumab) in metastatic pancreatic adenocarcinoma produced a promising ORR of 58% (14/24), though there were two treatment-related deaths attributed to sepsis (8.3%) [254]. Without sotigalimab, a separate phase I trial involving nab-paclitaxel, gemcitabine, and nivolumab in treatment-naïve stage IV pancreatic adenocarcinoma noted an ORR of only 18% [255].
Contrast these results to a phase 1b solid tumor trial involving selicreulmab in combination with atezolizumab which found an ORR of only 10% (8/80), though CD8+ T cell activation expansion was documented, and all responses were linked to subcutaneous dosing over IV dosing [256]. Finally, the use of dual TAM polarizing agents (sotigalimab and cabiralizumab) with or without nivolumab in NSCLC in the phase I setting resulted in no responses but did increase pro-inflammatory cytokine levels along with CD40 expression [257]. Though initial clinical data are underwhelming, further optimization of dosing frequencies and sequencing may help improve efficacy in subsequent studies.
PI3Kγ inhibitors
Phosphatidylinositol 3-kinase gamma (PI3Kγ) activation aids in the polarization of TAMs into the M2-like phenotype. The use of a PI3Kγ inhibitor reverses this partially due to the upregulation of IFNγ which signals TAMs to revert back to an M1 phenotype, thereby promoting anti-tumor immunity [258]. This has been documented in PI3Kγ−/− pancreatic murine models where blockade of PI3Kγ leads to TAM reprogramming and improved cytotoxic T cell mobilization into the TME [259]. PI3Kγ inhibitors also provide synergy when combined with anti-PD-L1 therapy in the realm of HNSCC which tends to be immunologically inert [260]. PI3K inhibitors vary based on their affinity to the four main class I PI3K isoforms: alpha, beta, delta, and gamma. While preclinical hematologic models have suggested that pan-PI3K inhibitors may provide modest improvements to cytotoxic potential compared to dual inhibitors, PI3K inhibition is often plagued with toxicities that limit their clinical utility, making selective inhibitors a desirable option in hopes of improving treatment tolerability [261]. While hyperglycemia has been linked more so to PI3Kα inhibition, whereas rates of severe colitis and pneumonitis are higher with PI3K-δ and PI3Kγ dual inhibitors (idelalisib and duvelisib) [262, 263].
Selective PI3Kγ inhibition is relatively new following the emergence of eganelisib, though others are currently in development with promising PI3Kγ affinity (AZD3458) [264]. Preclinical murine studies primarily focused on TNBC, melanoma, CRC, and lung models have shown that eganelisib reverts TAMs back to a M1 phenotype with increased IL-12 and iNOS levels. Additionally, combining eganelisib with both anti-CTLA4 and anti-PD-1 therapy results in CR rates of 30% in breast and 80% in melanoma models (B16-GM-CSF) and provided immunity to tumor re-implantation, whereas dual checkpoint inhibition alone did not result in any complete responses. This ultimately led to a phase I trial (MARIO-1) which involved eganelisib as monotherapy and in combination with nivolumab (anti-PD-1) for the treatment of advanced solid tumors [265]. Data from the melanoma and HNSCC expansion cohorts were later presented with combination therapy providing an ORR of 7.7% (3/39) and 10.0% (2/20), respectively, with a favorable safety profile and translational data demonstrating decreases in measured MDSC levels [266, 267]. In MARIO-3, eganelisib was combined with atezolizumab and nab-paclitaxel as first line therapy for TNBC with interim results including an ORR of 56.1% (23/41) in the intention-to-treat, an ORR of 48.1% (13/27) in PD-L1-negative participants, and a DCR of 81.4% (22/27) [268]. Researchers then compared survival outcomes to that of IMpassion130 as a historical control, with PD-L1 patients having an mPFS of 11.0 months (vs. 7.5 months), while PD-L1-negative patients carried an mPFS of 7.3 months (vs. 5.6 months). Only 14% of patients (n = 7) discontinued treatment due to adverse events, including hepatotoxicity, peripheral neuropathy, and rash. Finally, translational biopsy data documented increased PD-L1 expression at 2 months post-treatment, resulting in 5 out of 8 sampled PD-L1-negative tumors and surrounding immune cells converting to a PD-L1 positive status. Additional eganelisib trials remain underway, including MARIO-275, a phase II trial comparing nivolumab monotherapy to combination therapy in urothelial cancer, with initial data reporting an ORR of 30.3% (10/33) in the experimental arm versus 25% (4/16) with nivolumab alone and no notable difference in mPFS between arms at this time (9.1 vs. 8.0 months, HR 0.79, 95% CI 0.39–1.60) [269].
CD39/CD73/A2AR/A2BR
Apoptotic and hypoxic cells often release high amounts of adenosine triphosphate (ATP) into the extracellular domain, which in turn can signal a cascade of inflammatory responses. This occurs as a result of ATP binding to purinergic receptors P2X and P2Y, triggering inflammasome activation and neutrophil chemotaxis, respectively [270]. In order to counteract this inflammatory response, ATP is enzymatically broken down by enzymes CD39 and CD73 which are highly expressed on the surface of MDSCs and tumor cells [271]. CD39 converts ATP to adenosine monophosphate (AMP) whereas CD73 converts AMP into adenosine. Adenosine then acts as a powerful immunosuppressive metabolite through binding to adenosine receptors found on immune cells such as A2AR and A2BR, further inducing immunodormant states among TAMs, neutrophils, DCs, and MDSCs alike while also promoting Treg differentiation [272,273,274]. Given the malignant and tolerogenic nature of CD73, CD39, A2AR, and A2BR, numerous inhibitors have been developed for each target, including dual inhibitors of the targets above [275,276,277].
Regarding CD73, small-molecule inhibitors (quemliclustat and LY3475070), monoclonal antibodies (oleclumab, mupadolimab, Sym024, IBI325, JAB-BX102, INCA00186, NZV930, BMS-986179, HLX23, AK119, and uliledlimab), and bispecific antibodies (TGFβ: GS-1423) currently crowd the pipeline [275, 276]. Among the monoclonal antibodies, oleclumab has been the most widely investigated. This humanized IgG1 non-Fc-binding anti-CD73 antibody appears to act as an allosteric inhibitor and has been shown to have a picomolar affinity to CD73, but it comes with a few drawbacks. Initial phase I trial data involving oleclumab monotherapy remain unpublished, but in combination with durvalumab (anti-PD-L1), several trials were withdrawn due to reportedly low ORRs and one phase II trial involving ovarian cancer participants reported a DCR of only 27% [278]. Regarding newer iterations, interim data released from a phase I trial involving uliledlimab in combination with atezolizumab (anti-PD-L1) in the management of advanced solid tumors reported an ORR of 23% (3/13) with a DCR of 46% (6/13), and a significant trend toward increased CD73 expression among treatment responders compared to non-responders (78% vs. 23%) [279].
For CD39 inhibitors, the selection is less robust, with the majority of available agents being monoclonal antibodies that remain in early-stage clinical trials (TTX-030, SRF617, IPH5201), though others are currently under development (ES002) [275, 277]. The anti-tumor activity with IPH5201 has been shown in animal models involving human CD39 knock-in mice injected with melanoma cell lines (B16F10) with researchers able to link the blocking of ATP hydrolysis through inhibition of both membrane and soluble CD39 to the subsequent activation of TAMs and DCs [280]. Additionally, in human CD39 knock-in models, IPH5201 attenuated the anti-tumor activity of chemotherapy agents like oxaliplatin which cause effluxes of ATP from tumor cells. Antisense oligonucleotides are also under development using a locked nucleic acid methodology that allows for blockage of CD39 mRNA. To date, preclinical data suggest that following a dose-dependent suppression of CD39 mRNA expression in tumor-bearing mice, CD8+ T cell expansion shortly follows along with increases in PD-1 positive TIL expression and drops in Tregs, TAMs, and CD39 protein levels [281]. Dual inhibition with anti-PD-1 antibodies has been shown to further inhibit tumor within these murine models.
Finally, attention has been placed on inhibiting adenosine signaling through antagonism toward receptors found on immune cells, including A2AR on T cells, MDSCs, TAMs, DCs, and A2BR on NK cells, MDSCs, TAMs, and DCs. These agents have already been shown to be quite tolerable among vulnerable patient populations, including those with Parkinson's disease, as they have been linked to regulating dopamine signaling [282]. Among these agents include selective A2AR inhibitors (taminadenant, ciforadenant, AZD4635, inupadenant, and preladenant), selective A2BR inhibitors (PBF-1129), and dual inhibitors (etrumadenant) [283]. The A2AR antagonist AZD4635 has been studied as monotherapy in CRC murine models CT26 and MC38 and has been shown to slow tumor growth by 44% and 73%, respectively, with improvements to 73% and 91% with the addition of anti-PD-1 therapy [284]. AZD4365 also increases the presence of intratumoral CD103+ DCs and OVA antigen-specific CD8+ T cells. Clinical trial data are limited, but a similar inhibitor, ciforadenant, has been shown in phase I studies to provide an ORR of 8% (2/25) and a DCR of 60.0% (15/25) cumulatively in patients with renal cell carcinoma (RCC) and prostate cancer [285]. As for A2BR inhibitors, there is an ongoing phase I trial evaluating PBF-1129 in lung cancer (NCT03274479) but results are yet to be released. Finally, for dual inhibitors, early results from phase I studies involving etrumadenant (AB928) combined with anti-PD-1 therapy have shown linear pharmacokinetics, moderate tolerability (1 DLT, grade 2 rash), and an ORR of 8.3% (1/12) with a DCR of 33.3% (4/12) [286].
IDO1 inhibitors
As discussed above, myeloid cells including TAMs, DCs, and MSDCs express high levels of the enzyme IDO1, which is important for the degradation of L-tryptophan into kynurenine [287]. The subsequent depletion of L-tryptophan from the TME has been linked to the arrest of cytotoxic T cells [288]. Tumor draining lymph nodes tend to be the areas of highest IDO1 expression, particularly on the surface of APCs, with research suggesting this expression contributes to a tumor’s ability to form locoregional metastases, as seen in breast models [287, 289].
IDO inhibitors are primarily composed of small-molecule inhibitors such as epacadostat, navoximod, BMS-986205, EOS200271, KHK2455, LY3381916, and MK-7162. The first IDO inhibitor to advance through early-phase trials was epacadostat, eventually ending up in a phase III randomized, international, placebo-controlled trial as a therapy for unresectable stage III & IV melanoma in combination with pembrolizumab to assess whether it improved immune checkpoint efficacy [290]. Researchers reported no meaningful difference in terms of mPFS (4.7 vs. 4.9 months with placebo, HR 1.00), ORR (34 vs. 32%), CR (4% vs. 4%), DCR (51% vs. 51%), or treatment-related adverse events (10% vs. 9%), with 72–73% of participants having a positive PD-L1 status and 62–66% having a positive IDO1 status. While some developers have pivoted toward IDO1 inhibitor modifications to improve efficacy, others have set their sights on additional tryptophan metabolism pathways. Similar to IDO1, tryptophan-2,3-dioxygenase (TDO) serves the same function of degrading the L-tryptophan, into N-kynurenine, but what separates TDO from IDO1 is its expression patterns, with a higher predominance seen within the liver, bone marrow, brain, immune system, genitourinary tract, and gastrointestinal tract [291]. Like IDO1, TDO has been linked to immune resistance, including in mouse models where, in the presence of TDO inhibition, immune sensitivity was restored in those injected with TDO-expressing cancers. Within this same study, researchers also demonstrated that across multiple human cancer types, 32% expressed IDO1 alone, 35% expressed TDO alone, and 51% expressed both markers, making a case for dual IDO1/TDO inhibition. Currently developed IDO1/TDO inhibitors include HTI-1090, DN1406131, RG70099, and EPL-1410. Another avenue of active research includes the targeting of downstream signaling proteins, including aryl hydrocarbon receptor (AHR) which has been linked to Treg activation through kynurenine, along with other kynurenine metabolism enzymes such as KATI/II/III, KYNU, and KMO [292].
HDAC inhibitors
While certain DNA sequences may remain preserved within cancer cells, their expressional patterns can vary considerably depending on the presence of epigenetic modifications, including noncoding RNAs, DNA methylation, and histone modifications [293]. These changes have been linked to the metastatic potential of cancer cells as they continue to evolve, making these posttranslational modifications a key hallmark of malignancy [294]. Cellular metabolism plays a key role in the activity of certain HDAC that have been linked to immunosuppressive functions [295]. While certain HDACs (class III) require nicotinamide adenine dinucleotide (NAD+) as a cofactor, which is a byproduct of anaerobic glycolysis, histone acetyltransferases (HATs) require acetyl-CoA, the end product of aerobic glycolysis, in order to reverse these effects. This skewed ratio of NAD+ to acetyl-CoA within the TME further aids in the transformation of tumor cells and immune cells alike. HDAC activity within TAMs has been linked to decreased MHC-II expression, as seen in murine cancer models [296]. Additionally, HDAC has been shown to lessen MHC-I expression within cancerous cells to prevent them from presenting tumor-associated antigens to immune cells [297]. Both HDAC effects have been shown to be reversible with the introduction of an HDAC inhibitor (HDACi), resulting in tumor cell destruction [296,297,298]. Finally, HDAC inhibitors have been shown to deplete MDSCs within in vitro tumor models, further justifying their use as an immunotherapy adjunct [299].
Classical human HDAC enzymes tend to be zinc-dependent and come in various classes, with class I being ubiquitously expressed (HDAC1, HDAC2, HDAC3, and HDAC8), whereas class II (HDAC-4, HDAC-5, HDAC-6, HDAC-7, HDAC-9, and HDAC-10) and class IV (HDAC-11) have expression limited to cells of the central nervous system and muscular cells [300]. When it comes to HDAC inhibitors, most tend to have activity against class 1 HDACs (entinostat, romidepsin, mocetinostat, domatinostat, valproic acid, and phenylbutyric acid), though pan-inhibitors exist (panobinostat, abexinostat, givinostat, resminostat, quisinostat, pracinostat, belinostat, and vorinostat) as do more selective inhibitors (ricolinostat—HDAC6) [301, 302]. Vorinostat was the first dedicated HDACi to get propelled into clinical trials, particularly in the management of acute myeloid leukemia (AML). However, a phase II study of vorinostat as monotherapy in the management of high-risk AML published a CR of 2.7% (1/37), vastly underperforming the standard 40% CR rate seen with conventional therapies at the time [303]. Similarly, in a phase II randomized trial comparing azacitidine monotherapy to azacitidine plus vorinostat in AML found no difference in ORR (41 vs. 42%), CR (22% vs. 26%), or OS (9.6 vs. 11.0 months, p = 0.32) between the control and experimental arms, respectively [304]. Despite initial discouraging results, vorinostat rebounded as a potential lymphoma therapy based on small-scale phase I data. Eventually, two simultaneous phase II trials investigating vorinostat monotherapy in those with refractory cutaneous T cell lymphoma (CTCL) led to its FDA approval after investigators reported a cumulative ORR of 28.0% (30/107) with a median time to progression of 148 and 212 days for each study [305]. This was followed by FDA approval of romidepsin and belinostat for similar findings of durable treatment response in multicenter phase II trials.
For the majority of solid tumor types, however, results have been underwhelming. Single-agent HDACi therapy has failed to induce a partial or complete response in the vast majority of phase I and II trials involving HNSCC, breast cancer, thyroid cancer, ovarian cancer, and glioblastoma multiforme [306]. One phase II trial explored vorinostat in relapsed or refractory solid tumors including breast, CRC, and NSCLC with no reported responses and 68.8% (11/16) of participants discontinuing therapy due to adverse events including diarrhea, nausea, thrombocytopenia, fatigue, and anorexia [307]. One area of hope for HDACi therapy involves their use as adjuvant agents, particularly in combination with immunotherapy, as in vivo models of immune-resistant breast and pancreatic cancer have shown that the use of an HDACi-like entinostat can weaken MDSC-suppressive functions and improve CD8+ effector T cell activity compared to checkpoint therapy alone [308].
Strategies to modulate myeloid cells via cytokines
Type I interferons: IFNα and IFNβ
One of the first cytokines to be directly linked to anticancer activity is IFNα, a type I IFN produced many cells but most abundantly by the pDCs. IFNα is constitutively expressed in most cells and its production becomes pronounced when cells detect aberrant intracellular DNA or RNA, such as that seen in tumors or viral infection [81]. Activation of the cytosolic nucleic acid sensing pathways or TLRs leads to type I IFN and pro-inflammatory production, such as IL-12, TNFα, CXCL9, and CXCL10, that are important for T cell trafficking and function [309].
Using recombinant DNA technology, researchers were able to create the first FDA-approved immunotherapy against cancer, recombinant IFNα2 (rIFNα2), which received its approval in 1986 based on a single-arm, multicenter trial involving refractory hairy cell leukemia patients [310]. In the decades following, recombinant IFNα2 faded off into obscurity due to the development of more efficacious alternatives. Additionally, the tolerance of IFNα and IFNβ analogs has historically been poor, both due to flu-like symptoms and inconvenient dosing schedules. This led to the development of STING agonists, which mimic the physiologic cyclic dinucleotide (CDN) molecules cyclic 2’,3’-cGAMP, a product of cGAS [311]. Momentum for STING agonists grew after preclinical studies involving STING-deficient tumor-bearing mice were demonstrated as having fewer IFNγ-producing CD8+ T cells with increased Tregs and MDSCs [312]. Furthermore, cGAS deficient mice fail to mount a response to PD-L1 therapy compared to wild-type counterparts in a murine melanoma model [313]. Taken a step further, STING agonists like IACS-8803 have been shown to reverse these effects by repolarizing suppressive myeloid subsets in both human and mouse pancreatic cancer models, leading to increased sensitivity of orthotopic cancer cells to checkpoint inhibitor therapy [314].
One of the earlier STING agonists to reach large-scale clinical trials was vadimezan. A phase III trial investigating carboplatin and paclitaxel with or without vadimezan in advanced NSCLC observed the same median PFS (5.5 vs. 5.5 months) and ORR (25 vs. 25%) in both arms, but with a higher rate of grade 4 neutropenia (27% vs. 19% with control, respectively) and infusion site reactions (10% vs. 0.5%) [315]. Newer versions of STING agonists have been modified to improve STING affinity, cell permeability, and resist hydrolysis through ENPP1 (ectonucleotide pyrophosphatase/phosphodiesterase family member 1) [311]. Current STING agonists under clinical investigation include small-molecule agonists like MIW815, BMS-986301, E7766, ulevostinag, MK-2118, GSK3745417, TAK-676, SB-11285, and IMSA-101 [316]. In addition, small-molecule inhibitors of ENPP1 (MAVU-104, MV-626) have been developed as an off-target method of activating STING, with clinical trials already up and running [311, 316]. Finally, newer delivery methods for STING agonists are being explored, including STING agonist antibody–drug conjugates (CRD5500), exosomes (exoSTING), liposomal nanoparticles (STING-NPs), and vaccines (ONM-500 nanovaccine) [317].
Type II interferons: IFNγ
IFNγ is a type II interferon which is paramount to enhancing adaptive and innate cytotoxic activity. IFNγ exerts this effect of macrophages through JAK/STAT signaling, leading to activation of ISGs which control the production of inflammatory cytokines while also increasing MHC molecule and phagocytic receptor expression on macrophages, thereby activating them [318]. Within the TME, these actions by IFNγ promote the conversion of M2-like TAMs into M1 phenotypes, causing them to incite anti-tumor activity. However, IFNγ production and secretion from T cells and NK cells often requires interactions with M1 macrophages, hence why IFNγ remains low within the TME given the lack of a catalyst. Additionally, with extended periods of inflammation, IFNγ can activate negative feedback signals, leading to the increased PD-L1 expression and IDO1 expression on TAMs, which in turn inhibit NK cell activity [319, 320].
Using IFNγ as a therapeutic agent comes with unique challenges, including concerns for systemic cytotoxicity. This was best illustrated in a phase III trial involving advanced stage ovarian cancer patients where participants were randomized to receive chemotherapy with or without recurring subcutaneous IFNγ 1b injections [321]. The trial was stopped early due to a significantly higher mortality rate in the IFNγ 1b arm compared to placebo (39.7% vs. 30.4%, respectively) which appeared to be driven by higher rates of serious cytopenias (34.5% vs. 22.7%, respectively) and fever (20.6% vs. 5.0%). Despite these findings, given the link between IFNγ expression and its link to PD-L1 expression, a resurgence of interest has taken place, with multiple clinical trials involving recombinant IFNγ alongside anti-PD-1/PD-L1 agents (NCT02614456 and NCT03063632). Drug development has also geared toward incorporating IFNγ as an adjunct to vaccine therapies [322].
IL-12
APCs are the main source of IL-12, a vital pro-inflammatory cytokine that boosts IFNγ and TNFα production from cytotoxic cells and promotes effector cell activation. In the setting of malignancy, IL-12 can also play an important role in reprogramming MDSCs and TAMs [323, 324]. Through effector cell promotion, IL-12 therapy has been shown to guide anti-tumor activity in a wide variety of preclinical models, including lymphoma, renal cell, breast, ovarian, lung, melanoma, and sarcoma [325]. Despite these encouraging results, clinical studies have fallen short, both in terms of efficacy and safety. The most infamous case was a phase II trial in which 17 advanced RCC patients received daily recombinant IL-12 (rhIL-12) for a planned 5 consecutive days every 3 weeks [326]. However, despite reassuring safety data from phase I trials, 12 of the 17 patients required hospitalization due to severe side effects with two treatment-related deaths documented. No patients continued onto cycle 2 due to early termination of the trial. Reported grade 3/4 TRAEs included stomatitis, cytopenias, elevated liver enzymes, and gastrointestinal hemorrhage. Early phase I trials demonstrated that part of the limited efficacy seen in clinical trials relates back to negative feedback signaling through increasing IL-10 level with repeated doses of rhIL-12 [327]. Even when used in combined modality studies, it appeared that IL-12 provided little to no added clinical benefit while simultaneously worsening treatment tolerance [328].
Antibody-IL-12 fusion proteins have also been explored (M9241 also known as NHS-IL12) and although phase I/II trials remain active (NCT04633252, NCT04235777, NCT04756505, and NCT04708470), others have been terminated or suspended in part due to limited clinical efficacy (NCT02994953, NCT04327986). Based on the above issues, research has shifted toward remodeling IL-12 into an adjunct localized therapy as opposed to a non-specific systemic option. Gene therapy with IL-12 soon emerged, initially involving GEN-1, a human plasmid IL-12 (phIL-12) and a DNA delivery system within a lipopolymer which allows for local injections of IL-12-encoding DNA [328]. GEN-1 was first investigated as an intraperitoneal injection and gained traction after a phase I study demonstrated that platinum-sensitive ovarian cancer patients derived an ORR of 50.0% (6/12) with a DCR of 91.7% (11/12) and good tolerability [329]. However, multiple phase I and II trials shortly followed involving platinum-resistant cohorts which failed to replicate these results [330]. Given the above, the focus has since shifted to applying GEN-1 as an adjunct to neoadjuvant chemotherapy for curative intent in ovarian cancer patients, as seen in the phase I OVATION-I trial which found an ORR of 85.7% (12/14) with CR in 14.3% (2/14) of patients and subsequent surgical resection of all macroscopic disease (R0) achieved in 64.3% (9/13) of participants [331]. Translational data also showed reductions in Foxp3, IDO1, PD-1, and PD-L1 in 67–83% of post-treatment biopsies. A randomized phase I/II trial (OVATION-2 and NCT03393884) is currently ongoing to help confirm these encouraging results.
Alternative delivery systems were discussed by H. Kim Lyerly (Duke) at the 2021 China Cancer Immunotherapy workshop include viral (adenovirus and HSV-1) and cellular (CAR-T) vessels, but of the clinical data available regarding viral delivery, little efficacy has been seen [332]. Newer gene therapy agents also include intratumoral IL-12 mRNA (MEDI1191 and SAR441000). In preclinical models, these agents promote Th1 transformation and CD8+ T-cell-mediated tumor regression, leading to their advancement into early-phase clinical trials in combination with checkpoint inhibitors for the treatment of advanced solid tumors (NCT03946800 and NCT03871348) [333].
TNFα
TNFα is primarily a pro-inflammatory cytokine that assists in the extravasation and activation of effector cells while also triggering apoptosis in aberrant cells [334]. This occurs through the activation of TNF receptor TNFR1, though alternative receptors exist, including TNFR2. TNFR1 is ubiquitously expressed whereas TNFR2 is primarily limited to the CNS, endothelium, and on regulatory immune cells [335]. On abnormal cells, TNFR1 recruits the adaptor proteins TNFR1-associated death domain (TRADD) and Fas-associated death domain (FADD), leading to downstream apoptotic signaling through the caspase cascade [336]. On immune and endothelial cells, TNFR1 drives a pro-inflammatory cascade that leads to the recruitment and expansions of effector cells, which themselves produce TNFα, creating a positive feedback loop [337]. Conversely, TNFR2 signaling provides negative feedback through the activation and expansion of both Tregs and MDSCs [338, 339]. Furthermore, TNFR2 is expressed on a variety of tumor cells, with activation leading to proliferative (NF-κB), angiogenic, and anti-apoptotic effects [340]. TNFR2 expression in cancer subtypes has also been linked to a worsened prognosis [341].
Researchers have engineered ways to create selective TNFR2 inhibitors given the conflicting immune functions of TNFR1 and TNFR2. In culture-based ovarian cancer models (OVCAR3), TNFR2 inhibitors generate anti-tumor activity and reduce the presence of tumor-infiltrating Treg populations [342]. In addition, CRC and lung cancer models involving TNFR2 knockout mice demonstrated reduced metastatic potential and a measurable reduction in suppressive MDSC subsets, further linking TNFR2 to MDSC activity [343]. TNFR2 inhibitors include monoclonal antibodies such as APX601, HFB200301, BI-1808, BITR2101, and SIM0235. Clinical data remain absent, but trials involving HFB200301 (NCT05238883) and BI-1808 (NCT NCT04752826) have already begun accruing participants.
In parallel to immunostimulatory cytokines, some cytokines directly mediate immunosuppression by myeloid cells or exert their inhibitory effect on myeloid cells to promote cancer. Their functions are extremely context dependent. Here we call them immunomodulating cytokines for myeloid cells and described below some important examples.
IL-1β
Often a central player in tumor invasion and spread, IL-1β is also integral to the creation of an immunosuppressive network involving TAMs, Tregs, and MDSCs. This has been shown in preclinical HNSCC models where disruption of IL-1β production results a reduction in these tolerogenic cell types and an increase in CD8+ T cell presence [344]. TAMs often produce high concentrations of IL-1β as a result of their inflammasomes, further driving recruitment and expansion of MDSCs [345].
Ways of inhibiting IL-1β signaling include the use of recombinant IL-1R antagonists (anakinra), IL-1R accessory protein antagonists (CAN04), IL-1β sequestrants (rilonacept, canakinumab, and gevokizumab), and off-target inhibitors that impact components of either the inflammasome NLRP3 complex or downstream caspase-1 signaling [346, 347].
Oddly enough, interest in IL-1β inhibition as a potential cancer therapy grew considerably following a post hoc analysis from the CANTOS study, a randomized, double-blind, placebo-controlled trial involving the use of canakinumab in 10,061 patients with atherosclerosis and coronary artery disease [348]. Researchers noted that after a median follow-up of 3.7 years, canakinumab recipients had lower rates of cancer mortality (HR 0.49, p = 0.0009), lung cancer incidence (HR 0.61, p = 0.034), lung cancer mortality (HR 0.23, p = 0.0002) compared to those on placebo, along with reductions in C-reactive protein (CRP) and IL-6 levels. However, all-cause mortality was similar between treatment arms (HR 0.94, p = 0.31), a finding likely due to sepsis given higher rates of fatal infections seen in the canakinumab arms compared to placebo. This led Novartis to launch four separate large-scale trials (CANOPY-A, CANOPY-N, CANOPY-1, and CANOPY-2) which assessed canakinumab efficacy in the neoadjuvant, adjuvant, and metastatic setting for NSCLC. CANOPY 1 and 2 both failed to meet their primary endpoints of PFS and OS when adding canakinumab to the treatment of metastatic NSCLC, though prespecified subgroup analyses from CANOPY-1 currently suggest that clinically meaningful improvements to PFS and OS were seen in those with increased inflammatory biomarkers [349, 350]. However, in CANOPY-2, rates of fatal infection were elevated with canakinumab therapy (6.7% vs. 1.8%) as previously noted in the CANTOS analysis. The remaining phase II neoadjuvant trial (CANOPY-N, NCT03968419) and phase III adjuvant trial (CANOPY-A, NCT03447769) are still active at this time.
IL-6
As mentioned earlier, myeloid precursors are recruited to the bone marrow through cytokines like IL-6. Once differentiated into TAMs, these M2-like cells release IL-6 along with effector cells and tumor cells, thereby promoting tumor plasticity, a term referring to the ability of epithelial cells to transition to mesenchymal phenotypes [351]. This transition has been linked to the aggressiveness and metastatic potential of various cell lines and can also contribute to treatment resistance. One way to combat tumor plasticity while also limiting myeloid recruitment and M2 differentiation is using IL-6 inhibitors. These include IL-6R inhibitors (tocilizumab and sarilumab) and IL-6 sequestrants (siltuximab, sirukumab, olokizumab, and clazakizumab) [352]. The most recognized inhibitor is tocilizumab, a humanized antibody that has received numerous FDA approvals since 2010 in the management of rheumatologic disorders ranging from rheumatoid arthritis to Castleman’s disease. Additionally, tocilizumab is well known for its use in the management of cytokine release syndrome (CRS) during CAR-T therapy.
The first major study to investigate IL-6 sequestrant use in the realm of cancer management was a phase II randomized trial involving the addition of siltuximab to bortezomib, melphalan, and prednisone (VMP) for newly diagnosed, transplant-ineligible multiple myeloma patients [353]. Investigators found no evidence of meaningful clinical improvement in terms of CR rates (27% siltuximab + VMP vs. 22% VMP), ORR (88% siltuximab + VMP vs. 80% VMP), mPFS (17 vs. 17 months) and 1-year OS (88% vs. 88%). This was followed by a separate randomized phase II trial involving relapsed/refractory multiple myeloma patients who received siltuximab or placebo in addition to bortezomib with similar results reported regarding ORR, CR, PFS, and OS [354].
For IL-6R inhibitors, less is known regarding clinical impact. A phase I trial involving tocilzumab, PEGylated IFNα, carboplatin, and doxorubicin in the treatment of epithelial ovarian cancer revealed promising data with an ORR 52.3% (11/21), CR 14.3% (3/21), and DCR of 81.0% (17/21) [355]. The exploratory analysis also found that high-dose tocilizumab (8 mg/kg) resulted in an increase in M1 macrophage presence and secreted IFNγ levels. Regarding adverse events, unique post-market issues have arisen, including increased rates of pancreatitis and gastric perforation, though these events tend to occur in those with predisposing risk factors [356]. More research is necessary to validate the safety and efficacy of this treatment class.
IL-10
MDSCs have been shown in murine cancer models to be the primary source of IL-10 within the tumor microenvironment [357]. IL-10 provides autocrine signaling for MDSCs in order to promote their immunosuppressive phenotype. In addition, IL-10 has been shown to lead to a cascade of immunosuppressive effects within the TME, including increased Treg cell activity and inhibition of IL-12 secretion from Th1 helper cells through activation of STAT3 [358]. However, it also carries anti-tumor properties, as illustrated in murine knockout models and in human IL-10R deficiency analyses which found an increased propensity toward the development of malignancies including colon cancer and B cell lymphomas in the absence of IL-10/IL-10R signaling [359,360,361].
Given these conflicting roles, drug development has remained limited in this area. Recombinant versions of IL-10 have been developed, most promising being pegilodecakin (AM001), a PEGylated human IL-10 with a prolonged half-life. Preclinical data involving pegilodecakin in IL-10−/− mice with chemically induced skin cancers showed that a single dose of systemic pegilodecakin led to increased IFNγ level and cytotoxic T cells, tumor regression, and durable immune memory when mice were rechallenged with tumor cells up to 8 months later [362]. One of the larger clinical trials involving pegilodecakin was the SEQUOIA trial, a phase III study comparing FOLFOX ± pegilodecakin in gemcitabine-refractory pancreatic adenocarcinoma patients [363]. After a median follow-up of 15 months, no difference in median OS (5.8 vs. 6.3 months, p = 0.66), median PFS (2.1 vs. 2.1 months, p = 0.81), or ORR (4.6% [13/283] vs. 5.6% [16/268]) were reported between the experimental arm and control arm, respectively. Exploratory analyses did show an increase in Granzyme B, IFNγ, and IL-18 levels from baseline, which was more appreciable in the experimental arm, but overall the results were quite underwhelming.
TGFβ
TGFβ plays a crucial role in tumor proliferation while also providing immunosuppressive effects on surrounding immune cells, including promotion of M2 phenotypes for TAMs and preventing DC antigen presentation through downregulation of MHC-II [364]. Additionally, TGFβ released from MDSCs can inhibit NK cell activity through interference of IFNγ production while also promoting Treg cell recruitment and expansion. Through the years, multiple TGFβ and TGFβ receptor (TGFβR) inhibitors have emerged to help diminish signaling, including small-molecule TGFβR inhibitors (galunisertib, vactosertib, BMS-986260, LY3200882, LY2157299, PF-06952229, A83-01, SB-431542, RepSox, SM16, and AVID200), TGFβ sequestering monoclonal antibodies (ABBV-151, fresolimumab, SAR439459, NIS793), TGFβR monoclonal antibodies (XPA-42–089), bispecific TGFβR antibodies (BCA101: EGFR; Binstrafusp alfa: PD-L1; a-CTLA4-TGFβRIIecd: CTLA-4), and TGFβ antisense targeting agents (trabedersen, ISTH0036, TASO-001, gemogenovatucel-T, belagenpumatucel-L) [365, 366].
So far for antisense vaccine approaches, successful preclinical models have failed to translate into promising clinical trial data [367,368,369]. Part of the reason for this may tie into the complex nature of TGFβ signaling as it has been known to promote anti-tumor signaling in early malignancy settings before later evolving into pro-tumor signals as tumors progress. Additionally, inhibition of TGFβ may lead to compensatory immunosuppressive signaling, preventing tumor regression. Bispecific antibodies such as bintrafusp alfa may help counter this. This past year, pooled data from phase I (NCT02517398) and phase II (NCT03427411) trials involving binstrafusp alfa in advanced, pretreated, checkpoint-naive HPV-associated malignancies were presented at the European Society of Medical Oncology (ESMO) 2021 conference [370]. With a total of 75 patients, the reported ORR was 30.5% (23/75) with a CR rate of 6.7% and a median duration of response was 17.3 months. So similar to immune checkpoint inhibitors alone, a durable response was noted, but the rate of objective responses surpasses that of historical rates seen with PD-1 monotherapy, which is an encouraging sign.
Strategies to directly target myeloid cells
Vaccines
Cancer vaccines were first developed as a way to utilize the body’s immune system to combat malignancies. Initial formulations involved intratumoral whole cell vaccines with limited success due to antigen tolerance; therefore, adjuncts were often required to promote immune response, such as IL-2, GM-CSF, ODNs, and detoxified LPS [371]. Genetic engineering can be used to achieve this as well, as seen with talimogene laherparepvec (T-VEC), an FDA-approved therapy used in the management of stage 3 unresectable melanoma [372]. T-VEC is an oncolytic HSV-1 virus that has been modified with restricted replication in tumor cells and transgene GM-CSF expression [373]. This engineered virotherapy has also been applied to autologous tumor vaccines including GVAX (GM-CSF), FVAX (FLT3L), and TEGVAX (GM-CSF, TLR4 agonist, and TLR7/8 agonist) [374].
Specific targeting was later made possible due to the discovery of tumor-associated antigens (TAAs). These are categorized as either unique antigens (β-catenin-m, HSP70-2/m, Myosin/m, etc.) or shared antigens. Shared antigens include those overexpressed by cancerous cells (HER2, p53, survivn, and livin), antigens differentially expressed by certain tissue types (CEA, PSA, Mammoglobin-A, Tyrosinase, Gp100, MART-1, Melan-A globo-H, Muc1, sTn, and GM2), and antigens unique to germ cells (MAGE, NY-ESO-1, SSX, BAGE, and GAGE) [375]. Despite the discovery of these various antigens, eliciting an immune response to specific vaccines remained difficult without the process of immunostimulatory agents including TLR agonists, so combination therapies are favored. FDA-approved TLR agonists currently used in conjunction with viral vaccination therapies include monophosphoryl lipid A (TLR4 agonist) with hepatitis B and human papilloma virus (HPV), Imiquimod (TLR7 agonist) for anogenital HPV strains, and both Flagellin (TLR5 agonist) and CpG (TLR9 agonist) derivatives for influenza vaccines. While cancer vaccine studies have attempted to extrapolate these immunostimulants into clinical studies, TLR3 agonists have also been repurposed and remain as a leading area of investigation. These agonists typically involve double-stranded RNA complexes like poly-IC or synthetic derivatives for improved stability (rintatolimod) and reduced toxicity (poly-ICLC) which provide a robust innate and adaptive response [376,377,378]. Data from a phase I trial examining the efficacy and safety of an ovarian cancer peptide vaccine found that NY-ESO-1-specific antibody and CD8+ T cell presence improved from 46% (6/13) and 62% (8/13) of participants with vaccine alone to 91% (10/11) and 91% (10/11) when poly-ICLC was added [378, 379].
Another way to improve antigen immunogenicity is by saturating autologous DCs in antigen, whether it be in vivo or ex vivo, in hopes they will then present this antigen to adaptive immune cells. Peripheral blood monocytes can be used to derive dendritic cells ex vivo and antigen loading is possible through use of TAAs or whole tumor cells [380]. A classic example includes sipuleucel-T which is a cellular therapy derived from autologous peripheral monocytes which are activated ex vivo using a recombinant fusion protein (PA2024) consisting of PSA, prostatic acid phosphatase, and GM-CSF [381]. Sipuleucel-T was found to provide a survival benefit across 3 separate double-blind, placebo-controlled, multicenter trials in patients with metastatic castrate-resistant prostate cancer and is now FDA-approved [367, 369]. The majority of DC cancer vaccines that have followed have been lackluster in phase II/III trials, but recent preclinical and phase I data surrounding the identification and use of personalized neoantigens during ex vivo dendritic cell loading appear promising [382,383,384,385].
Additional dendritic options include the use of DC-derived exosomes (DCexos) which are inert vesicles expressing MHC-I and II that are unphased by the immunosuppressive state of the tumor microenvironment and provide improved stability and bioavailability [386]. Murine models have shown them to be a viable option for tumor eradication with T cell immunity; however, three early-phase clinical trials involving peptide-loaded DCexos extracted from autologous peripheral monocytes failed to demonstrate meaningful antigen-specific T-cell responses, though NK effector functions were reported [387,388,389,390]. The use of protein-loaded DCexos, however, has been documented to induce antigen-specific cytotoxic T cell responses in murine models [391, 392].
CAR-M
CAR-T therapy has revolutionized the way clinicians care for patients with B cell malignancies through targeting of CD19 or B cell maturation antigen (BCMA) [393]. However, unique challenges arise when this therapy is applied to solid tumors, including issues surrounding localization, persistence, exhaustion, tumor heterogeneity, and balancing toxicities [394]. Similar to vaccines, these effects are in part due to the presence of MDSCs, so a variety of chimeric co-receptors have been engineered to target key myeloid pathways including CD24 (ONC-782), TR2 (CAR.MUC1/TR2.41BB), and FLT3L among others [395, 396].
Given that pathologic recruitment of monocytes occurs within tumor microenvironments in order to create TAMs, researchers also began work on developing genetically engineered CAR macrophages (CAR-Ms) based on CD19 CAR-T models [397]. Early hypothesis testing involved transducing a human monocytic cell line THP-1 from acute myeloid leukemia with a first generation CD19 CAR-T encoding the intracellular domain of CD3ζ in order to signal antibody-dependent phagocytosis given its structural similarity to FcεRIγ [398]. These CAR-M macrophages successfully engulfed tumor cells in an antigen-specific fashion. Researchers were able to test out further CAR-M iterations by using an adenoviral vector (Ad5F35) to transduce macrophages with CARs targeting solid tumor antigens including mesothelin and HER2, which led to similar success. This was followed by two in vivo ovarian cancer (SKOV3) murine models in which a single infusion of CAR-M therapy led to a significant shrinkage in tumor burden and a considerable prolongation of overall survival, though progressions did eventually occur. Additionally in vivo studies involving biofluorescence also noted CAR-M trafficking and persistence in tumor tissue along with the liver, spleen, and lungs in explanted samples taken 5 days following a single infusion. Overall, CAR-M appears to provide a window of therapeutic opportunity that is currently still in its infancy but will hopefully evolve in years to come. Currently, clinical data are absent with one phase I trial (NCT03608618) involving mesothelin-targeting CAR-M therapy (MCY-M11) currently terminated reportedly as a result of sponsor interests and another phase I trial (NCT04660929) currently underway involving anti-HER2 CAR-M therapy (CT-0508) in HER2-expressing malignancies. In the anti-HER2 trial, one group will receive 3 separate IV infusions over a 5-day period to deliver 5 billion CAR-M cells, whereas another group will receive the 5 billion cells over a single infusion to assess safety, with assessments continuing over a 14-month follow-up period.
Already, separate research has led to the emergence of a new family of CAR-Ms termed CAR-iMacs, which are CAR-M therapies derived from induced pluripotent stem cells (iPSCs) transduced using lentiviral CAR delivery [399]. This process of using iPSCs allows manufacturers to yield high amounts of CAR-M cells from a single collected specimen, further easing production logistics. These CAR-iMacs have already been studied in both in vitro and in vivo solid tumor models with researchers noting antibody-dependent cell phagocytosis in vitro along with in vivo CAR-iMac expansion lasting 2–3 days, tumor burden shrinkage, and CAR-iMac persistence lasting 20–30 days. No word from developers regarding plans for CAR-iMac clinical trials in the near future as further adjustments to improve efficacy and persistence are currently planned.
Finally, non-viral CAR delivery techniques have been discovered, allowing researchers to create in vivo CAR-Ms through the use of available TAMs [400]. Given that macrophages highly overexpress mannose receptors, nanocomplexes such as mannose-conjugated polyethylenimine (MPEI) have been used to target TAMs within the TME and deliver DNA plasmids containing CARs along with IFN-γ in order to polarize them. Preclinical in vivo data involving anti-ALK (anaplastic lymphoma kinase) CAR for the targeting of neuroblastoma models appear to be promising in terms of immunomodulation and tumor shrinkage. If further validated, this new strategy could provide clinicians an off-the-shelf, readily available product that avoids the need for costly ex vivo manufacturing.
Emerging targets for therapeutic manipulation of myeloid cells
Siglec-15
Siglec-15 is a sialic acid recognition protein primarily expressed by select myeloid populations that has previously been linked to osteoclast differentiation and bone remodeling, making it a potential target for the management of osteoporosis [401, 402]. However, given its role in macrophage differentiation, it has recently been investigated as a potential tool for activating dormant myeloid cells.
What makes Siglec-15 unique is that unlike other Siglec family members like Siglec-10 which perform intracellular signaling through SHP-1/2 to initiate immunosuppressive actions, Siglec-15 utilizes the adapter protein DAP12 along with a tyrosine kinase called SYK in order to achieve this [403]. Therefore, a unique signaling pathway is available for targeting TAM polarization that avoids redundant antagonism. Siglec-15 ligands include sialic acid-containing glycans such as those with a sialyl-Tn (sTn) structure, a common ligand that is highly associated with a variety of malignancies [404, 405]. When activated in vitro and in vivo using murine models, Siglec-15 has been shown to suppress both T cell proliferation and activation [406]. Furthermore, inhibition of Siglec-15 gives rise to elevated IL-2 and TNFα levels, therefore promoting an inflammatory TME. Mouse models have demonstrated that dual inhibition of Siglec-15 and PD-1 lead to improved tumor responses, including CR, compared to monotherapy alone in either arm [406]. When reviewing expression data from The Cancer Genome Atlas (TCGA), Siglec-15 expression has been shown to be upregulated in a wide variety of malignancies, including CRC, thyroid, endometrial, lung, hepatic, renal, and bladder cancers [406].
Given these findings above, Siglec-15 inhibitors are under development. Currently, NC318, a humanized monoclonal antibody, remains as the sole inhibitor under clinical investigation. A phase I/II trial of NC318 therapy for advanced solid tumors noted no DLTs across dose levels during phase I studies [407]. Adverse events reported included diarrhea (16%), elevated pancreatic enzymes (6–8%), pruritis (6%), and immune-related adverse events that included vitiligo, uveitis, and pneumonitis. For phase II efficacy results, NC318 monotherapy provided an ORR of 4.8% (4/83) with a CR of 1.2% (1/83) and disease control rate (DCR) of 38% (32/83) regardless of PD-L1 or Siglec-15 expression status [408]. Median response was not reported, but 2 patients (CR and PR) remained on therapy beyond 2 years without signs of progression. With NC318 therapy came a dose-dependent increase in soluble Siglec-15, making it a helpful marker for monitoring NC318 activity. Researchers also performed a post hoc analysis using a Siglec-15 immunohistochemistry assay and found that based on screening biopsy samples, Siglec-15 expression on cancer cell membranes was predictive of PFS and duration on therapy [408]. Given the majority of responders to Siglec-15 monotherapy had NSCLC, a phase II trial (NCT04699123) is currently underway comparing NC318 with or without pembrolizumab in patients with advanced NSCLC.
TREM2
Similar to Siglec-15, TREM2 plays an important role in osteoclast differentiation [409, 410]. Furthermore, both use the adaptor protein DAP12 to transmit intracellular signaling via activation of the tyrosine kinase, Syk [410]. TREM2 expression is restricted within the majority of normal tissue, whereas approximately 75% of cancer types have been shown to express TREM2, making it a suitable target given its wide therapeutic window [411]. Preclinical experiments involving TREM2+ DCs and macrophages derived from bone marrow and lung cancer-bearing mice have shown that these innate immune cells inhibit T cell proliferation, secrete higher levels of IL-10, secrete lower levels of IL-12, and phagocytose OVA at reduced capacity [412]. Additionally, the injection of TREM2+ DCs into these cancer-bearing mice led to accelerated tumor progression and worsened survival. Individually, the use of IL-10 sequestrants, Syk inhibitors, and TREM-2 antagonists have been demonstrated to independently reverse these effects to varying degrees. Finally, the level of TREM2 presence within TAMs has been positively correlated with tumor staging, including the degree of nodal metastases. Similar inverse trends of TREM2 expression and overall survival have been documented with gastric, hepatic, colorectal, ovarian and breast cancers [411, 413, 414]. Other studies utilizing TREM2−/− mice and TREM2 inhibitors for sarcoma, colorectal, and breast cancer models have demonstrated that TREM2 deficiency leads to improved antigen presentation from TREM2−/− macrophages compared to wild type, along with improved CD8+ TIL presence and PD-1 expression, suggesting that TREM2 inhibition may be synergistic with ICI therapy [411].
Based on these findings, development of novel TREM2-targeting agents has been underway. PY314, a humanized monoclonal antibody against TREM2, was among the first to reach clinical trials, with a phase I study (NCT04691375) currently underway comparing PY314 therapy with or without pembrolizumab in patients with advanced solid tumors. While clinical data are currently ongoing, preclinical studies involving PY314 have demonstrated that this anti-TREM2 therapy can provide anti-tumor activity in certain breast cancer models (EMT6) while improving the immune landscape of the TME through increasing the presence of CD8+ TILs, NK cells, and MHC-II-expressing TAMs [413]. When combined with anti-PD-L1, PY314 further amplifies these immune cell changes, as seen via flow cytometry and IHC staining. Altogether, TREM2 pathway targeting provides researchers another potential tool in the management of MDSCs.
MARCO
MARCO represents a pattern recognition scavenger receptor who is expressed constitutively on M2-like subsets of macrophages and whose role has initially been linked to anti-inflammatory changes through cholesterol sequestration in the setting of cardiovascular disease [415]. Analysis of TCGA data shows that MARCO expression is most notable in malignancies of the pancreas, skin, cervix, testicles, thyroid, kidneys, and central nervous system (CNS) [416]. Other cancer types either showed similar or decreased MARCO expression compared to controls. Despite this, MARCO expression has not only been linked to worsened prognosis in glioblastoma and pancreatic cancer, but also gastroesophageal and lung malignancies [417,418,419]. Moreover, several preclinical models including melanoma, breast and CRC have demonstrated that anti-MARCO monoclonal antibodies not only reduce tumor volumes, but also appear to convert MARCO-expressing TAMs from an M2 to an M1 phenotype while also reducing Treg levels [420]. Separate models (melanoma) have found that the injection of anti-MARCO antibodies into tumor-bearing mice leads to an influx of NK cells and CD8+ T cells, and this effect is augmented by the addition of anti-PD-L1 antibodies [421]. Finally, MARCO expression in TAMs is positively correlated with phosphorylation levels of Syk and PI3K [422].
All things considered, MARCO appears to be a promising new immunotherapy target. Given MARCO ligands primarily include acetylated-LDL and negatively charged molecules, sulfatides (negatively charged glycolipids) became one of the first low molecular weight inhibitors to spark interest [423]. While the development of inhibitors against class A scavenger receptors like MARCO remains in its infancy, inhibitors against other scavenger receptor classes such as SR-B1, SR-B2, and LOX-1 are also available for evaluation [424].
LILRB2
A member of the leukocyte immunoglobulin-like receptor (LILR) family, LILRB2 is found primarily on myeloid immune cells and plays an integral role in providing negative feedback during inflammatory responses through binding to MHC-1 and HLA-G, a non-classical class I molecule [425]. LILRB2 has also been found on hematopoietic stem cells and binds to angiopoietin-like protein 2 (ANGPTL2), leading to the activation of the SHP2 signaling pathway and subsequent cell proliferation [426]. Within malignancies, enrichment of LILRB2 is often noted, including in AML, chronic lymphocytic leukemia (CLL), esophageal cancer, pancreatic cancer, NSCLC, and lobular breast cancers [427]. In vitro, LILRB2 inhibition involving NSCLC cancer cell line A549 leads to significant decrease in cancer migration and proliferation potential [428]. This has been replicated in pancreatic cancer models where silencing ANGPTL2 expression reduced migratory potential through reversion of tumor plasticity [429].
Development of LILRB2 inhibitors is ongoing, including MK-4830, a fully human IgG4 monoclonal antibody specific to LILRB2. Recently published phase I data involving MK-4830 with or without pembrolizumab in advanced solid tumors (NCT03564691) revealed that no DLTs were observed in either arm, with the most common side effects including fatigue (40%), nausea (28%), decreased appetite (22%), and diarrhea (20%) [430]. Regarding efficacy, MK-4830 monotherapy provided an ORR of 2.0% (1/50) and DCR of 24.0% (22/50), whereas combination therapy with pembrolizumab (not including cross overs) led to an ORR of 23.5% (8/34), CR of 2.9% (1/34) and DCR of 50.0% (17/34). At 6 months, DCR was 12% (6/50) and 41.2% (14/34), respectively. Improvements in both cytotoxic T cell levels and PD-L1 positivity were noted in responders, though two responders had a combined positive score (CPS) of 0 following combination therapy. Follow-up trials are in progress, including investigations of MK-4830 in combination with immunotherapy for the treatment of small cell lung cancer (SCC), NSCLC, RCC, CRC, and melanoma. Other LILRB2 inhibitors have also entered early-phase clinical trials, including monoclonal antibodies IO-108 (NCT05054348) and JTX 8064 (NCT04669899).
CLEVER-1
CLEVER-1 or stabilin 1 has been historically linked to cancer proliferation and spread as noted in Stab1 knockout mice [431, 432]. However, recent studies also highlight its immunologic impact in TAM polarization, with CLEVER-1 inhibition resulting in TAM conversion to an M1-like phenotype and subsequent T cell activation [433]. Additionally, high concentrations of CLEVER-1 expressing TAMs have been linked to worsened survival outcomes [434]. Given this newer discovery, clinical data in this area have been limited to date. The leading antagonist is bexmarilimab (FP-1305), a humanized IgG4 monoclonal antibody created from Chinese hamster ovary cells that is currently in phase I/II clinical trials involving solid tumors. Preliminary results released at ESMO 2021 suggest good tolerability (most common were fatigue [31%], abdominal pain [23%] and anemia [21%]) but underwhelming response rates with a DCR of 17.2% (19/110) and an ORR of 0% [435].
Conclusion
The majority of malignant tumors respond poorly to modern ICIs and other immunotherapy agents. Some tumors have innate resistance to immunotherapy, while others acquire resistance over time with many resistance mechanisms traced back to the TME and the lack of myeloid cells. With the advancement of single-cell multi-omics approaches, there is an increased appreciation of the heterogeneity and complexity of myeloid cell composition as described above. However, with a greater understanding of the various myeloid components and the impact of these myeloid subgroups on the TME as well as the tumor response to various therapies, there is a greater potential for manipulation of the myeloid compartment. It is our opinion that combination regimens are most likely to have the greatest impact as illustrated above with the various pleiotropic immune cells and immunocytokines. Employing several agents which target different components of the myeloid component in combination with ICIs or cytotoxic chemotherapy is most likely to have the greatest impact, although with the addition of more therapeutic agents comes the potential for great toxicity.
There has been an eruption of newly developed myeloid-targeted therapies with the majority of these agents still in the clinical trial phase, making it difficult for clinicians to navigate through the available literature and determine which agents appear most promising. In this review, we discussed past and current preclinical and clinical data to provide readers a detailed summary of where each potential target stands and where the future is headed. While T cells have long been the focus of immunotherapy, we believe that the future of immunotherapy will involve targeting myeloid cells.
Key areas of continued research include further investigation into the cross talk among cancer cells, myeloid cells, adaptive immune cells, and surrounding cells like epithelial cells and fibroblasts, to create tolerogenic environments, with the help of single-cell multi-omics technologies. Finally, as our understanding of non-T-cell-based immunotherapy continues to evolve, we are optimistic that the benefit of immunotherapy will be extended to more patients and ultimately save more lives.
Availability of data and materials
Not applicable.
Abbreviations
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- AHR:
-
Aryl hydrocarbon receptor
- AML:
-
Acute myeloid leukemia
- AMP:
-
Adenosine monophosphate
- ANGPTL2:
-
Angiopoietin-like protein 2
- APC:
-
Antigen-presenting cell
- Arg1:
-
Arginase 1
- ATP:
-
Adenosine triphosphate
- BCMA:
-
B cell maturation antigen
- bFGF:
-
Basic fibroblast growth factor
- C/EBPβ:
-
CCAAT/enhancer-binding protein β
- CAHON:
-
Chinese American Hematologist and Oncologist Network
- CAR-Ms:
-
CAR macrophages
- CAR:
-
Chimeric antibody receptor
- CCFDIE:
-
China Center for Food and Drug International Exchange
- CCL:
-
C–C motif chemokine ligand
- cDC:
-
Conventional DC
- CDN:
-
Cyclic dinucleotide
- CDP:
-
Common dendritic cell precursors
- cGAS:
-
Cyclic GMP-AMP synthase
- CITE-seq:
-
Cellular indexing of transcriptomes and epitopes by sequencing
- CLEVER-1:
-
Common lymphatic endothelial and vascular endothelial receptor 1
- CLL:
-
Chronic lymphocytic leukemia
- CNS:
-
Central nervous system
- COX-2:
-
Cyclooxygenase-2
- CpG:
-
Cytosine–phosphate–guanine
- CPS:
-
Combined positive score
- CR:
-
Complete response
- CRC:
-
Colorectal cancer
- CRS:
-
Cytokine release syndrome
- CSF-1:
-
Colony-stimulating factor 1
- CTCL:
-
Cutaneous T cell lymphoma
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CXCL:
-
C-X-C motif chemokine ligand
- DAMP:
-
Danger-associated molecular patterns
- DC:
-
Dendritic cell
- DCexos:
-
DC-derived exosomes
- DCR:
-
Disease control rate
- DNMT:
-
DNA methyltransferase
- DOR:
-
Duration of response
- dSLIMs:
-
Double stem-loop immunomodulators
- ECM:
-
Extracellular matrix
- ER:
-
Endoplasmic reticulum
- EV:
-
Extracellular vesicle
- FADD:
-
Fas-associated death domain
- FLT3L:
-
FMS-like tyrosine kinase 3 receptor ligand
- GMP:
-
Granulocyte–monocyte progenitor
- HATs:
-
Histone acetyltransferases
- HCC:
-
Hepatocellular carcinoma
- HDAC:
-
Histone deacetylase
- HDN:
-
High-density neutrophils
- HGF:
-
Hepatocyte growth factor
- HMGB1:
-
High-mobility group box 1
- HPV:
-
Human papilloma virus
- HSC:
-
Hematopoietic stem cell
- ICI:
-
Immune checkpoint inhibitor
- IDO1:
-
Indoleamine 2,3-dioxygenase 1
- IFN:
-
Interferon
- iPSC:
-
Induced pluripotent stem cell
- ISG:
-
Interferon-stimulated genes
- JAKs:
-
Janus kinases
- LAP:
-
Leucine–alanine–proline
- LDL:
-
Low-density lipoprotein
- LDN:
-
Low-density neutrophils
- LILR:
-
Leukocyte immunoglobulin-like receptor
- LILRB2:
-
Leukocyte immunoglobulin-like receptor B2
- LPS:
-
Lipopolysaccharide
- M-MDSC:
-
Monocytic MDSC
- MARCO:
-
Macrophage receptor with collagenous structure
- MDP:
-
Macrophage-dendritic progenitors
- MDSCs:
-
Myeloid-derived suppressor cells
- MHC:
-
Major histocompatibility complex
- MMP:
-
Matrix metallopeptidase
- MPEI:
-
Mannose-conjugated polyethyleneimine
- mPFS:
-
Median progression-free survival
- mregDC:
-
Mature DCs enriched in immunoregulatory molecules
- NAD+ :
-
Nicotinamide adenine dinucleotide
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NE:
-
Neutrophil elastase
- NETs:
-
Neutrophil extracellular traps
- NHL:
-
Non-Hodgkin lymphoma
- NLR:
-
Neutrophil-to-lymphocyte ratio
- NMPA:
-
China National Medical Product Administration
- NSCLC:
-
Non-small cell lung cancer
- ODN:
-
Oligodeoxynucleotide
- ORR:
-
Objective response rate
- PAMP:
-
Pathogen-associated molecular patterns
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed death-ligand 1
- pDC:
-
Plasmacytoid DC
- PDGF:
-
Platelet-derived growth factor
- PI3Kγ:
-
Phosphatidylinositol 3-kinase gamma
- PKCβII:
-
Protein kinase C βII
- PMN-MDSC:
-
Granulocytic/polymorphonuclear MDSC
- PNT:
-
Peroxynitrite
- PVNS:
-
Pigmented villonodular synovitis
- RCC:
-
Renal cell carcinoma
- rhuFLT3L:
-
Recombinant human FLT3 ligand
- rIFNα2:
-
Recombinant IFNα2
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- SCC:
-
Small cell lung cancer
- SHP-1/2:
-
Src homology domain-containing tyrosine phosphatases 1/2
- Siglec-10:
-
Sialic acid-binding immunoglobulin-type lectin-10
- STAT3:
-
Signal transducer and activator of transcription
- STING:
-
Stimulator of interferon genes
- TAAs:
-
Tumor-associated antigens
- TAM:
-
Tumor-associated macrophage
- TAN:
-
Tumor-associated neutrophils
- TCGA:
-
The Cancer Genome Atlas
- TCR:
-
T cell receptor
- TDEs:
-
Tumor-derived exosomes
- TDO:
-
Tryptophan-2,3-dioxygenase
- TGCT:
-
Tenosynovial giant cell tumor
- TGFβ:
-
Transforming growth factor β
- TIL:
-
Tumor-infiltrating lymphocytes
- TLR:
-
Toll-like receptors
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple-negative breast cancer
- TNFα:
-
Tumor necrosis factor α
- TRAAC:
-
Toll-like receptor agonist antibody conjugates
- TRADD:
-
TNFR1-associated death domain
- TRAEs:
-
Treatment-related adverse events
- Treg:
-
Regulatory T cell
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
- TRM:
-
Tissue-resident macrophage
- TRPM2:
-
Transient receptor potential melastatin 2
- T-VEC:
-
Talimogene laherparepvec
- VEGF:
-
Vascular endothelial growth factor
- VISTA:
-
V-domain immunoglobulin suppressor of T cell activation
References
Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–9.
Bassler K, et al. The myeloid cell compartment-cell by cell. Annu Rev Immunol. 2019;37:269–93.
Guc E, Pollard JW. Redefining macrophage and neutrophil biology in the metastatic cascade. Immunity. 2021;54(5):885–902.
Robinson A, et al. Monocyte regulation in homeostasis and malignancy. Trends Immunol. 2021;42(2):104–19.
Wculek SK, et al. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7–24.
Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. 1970;13:1–27.
Dunn GP, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–8.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–7.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–45.
Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16(7):447–62.
Cassetta L, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35(4):588.
Broz ML, Krummel MF. The emerging understanding of myeloid cells as partners and targets in tumor rejection. Cancer Immunol Res. 2015;3(4):313–9.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68.
Cheng S, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184(3):792-809 e23.
Lavin Y, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169(4):750-765 e17.
Zhang Q, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179(4):829-845 e20.
Klemm F, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell. 2020;181(7):1643-1660 e17.
Zilionis R, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. 2019;50(5):1317-1334 e10.
Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196(3):254–65.
Zhang QW, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE. 2012;7(12): e50946.
Gentles AJ, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–45.
Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–5.
Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336(6077):86–90.
Lavin Y, et al. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol. 2015;15(12):731–44.
Bowman RL, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. 2016;17(9):2445–59.
Chen Z, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 2017;77(9):2266–78.
Loyher PL, et al. Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med. 2018;215(10):2536–53.
Zhu Y, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. 2017;47(2):323-338 e6.
Franklin RA, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–5.
Klug F, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24(5):589–602.
Guerriero JL, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543(7645):428–32.
Nathan CF, et al. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 1983;158(3):670–89.
Stein M, et al. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176(1):287–92.
Mills CD, et al. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–73.
Davis MJ, et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio. 2013;4(3):e00264-e313.
DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19(6):369–82.
Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 2021;21(2):71–88.
Cecchini MG, et al. Role of colony stimulating factor-1 in the establishment and regulation of tissue macrophages during postnatal development of the mouse. Development. 1994;120(6):1357–72.
Kitamura T, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212(7):1043–59.
Sidibe A, et al. Angiogenic factor-driven inflammation promotes extravasation of human proangiogenic monocytes to tumours. Nat Commun. 2018;9(1):355.
Arwert EN, et al. A unidirectional transition from migratory to perivascular macrophage is required for tumor cell intravasation. Cell Rep. 2018;23(5):1239–48.
Lin EY, et al. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol Oncol. 2007;1(3):288–302.
Colegio OR, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–63.
Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66(2):605–12.
Ruivo CF, et al. The biology of cancer exosomes: insights and new perspectives. Cancer Res. 2017;77(23):6480–8.
Arkhypov I, et al. Myeloid cell modulation by tumor-derived extracellular vesicles. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21176319.
Morrissey SM, et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. 2021;33(10):2040-2058 e10.
Haderk F, et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci Immunol. 2017. https://doi.org/10.1126/sciimmunol.aah5509.
Hwang WL, et al. Tumor stem-like cell-derived exosomal RNAs prime neutrophils for facilitating tumorigenesis of colon cancer. J Hematol Oncol. 2019;12(1):10.
Zhang X, et al. Tumor-derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration. Mol Cancer. 2018;17(1):146.
Kuang DM, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206(6):1327–37.
Lin H, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest. 2018;128(2):805–15.
Petty AJ, et al. Hedgehog-induced PD-L1 on tumor-associated macrophages is critical for suppression of tumor-infiltrating CD8+ T cell function. JCI Insight. 2021. https://doi.org/10.1172/jci.insight.146707.
Kryczek I, et al. B7–H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203(4):871–81.
Li J, et al. Co-inhibitory molecule B7 superfamily member 1 expressed by tumor-infiltrating myeloid cells induces dysfunction of anti-tumor CD8(+) T cells. Immunity. 2018;48(4):773–7865.
Geiger R, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167(3):829-8421 e3.
Rodriguez PC, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64(16):5839–49.
Ruffell B, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623–37.
Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27(4):462–72.
Roux C, et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc Natl Acad Sci U S A. 2019;116(10):4326–35.
Yu J, et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med. 2021;27(1):152–64.
Zhang M, et al. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res. 2014;7:19.
Merad M, et al. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604.
Hildner K, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–100.
Jongbloed SL, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010;207(6):1247–60.
Broz ML, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26(5):638–52.
Spranger S, et al. Tumor-residing batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31(5):711-723 e4.
Salmon H, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. 2016;44(4):924–38.
Roberts EW, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. 2016;30(2):324–36.
Binnewies M, et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4(+) T cell immunity. Cell. 2019;177(3):556-5711 e6.
Alspach E, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574(7780):696–701.
Duong E, et al. Type I interferon activates MHC class I-dressed CD11b(+) conventional dendritic cells to promote protective anti-tumor CD8(+) T cell immunity. Immunity. 2022;55(2):308-323 e9.
Valdez Y, et al. Major histocompatibility complex class II presentation of cell-associated antigen is mediated by CD8alpha+ dendritic cells in vivo. J Exp Med. 2002;195(6):683–94.
Theisen DJ, et al. WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science. 2018;362(6415):694–9.
Ferris ST, et al. cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature. 2020;584(7822):624–9.
Diamond MS, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003.
Dunn GP, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6(7):722–9.
Fuertes MB, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208(10):2005–16.
Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142–9.
Deng L, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843–52.
Woo SR, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–42.
Garris CS, et al. Successful anti-PD-1 cancer immunotherapy requires T Cell-dendritic cell crosstalk involving the cytokines IFN-gamma and IL-12. Immunity. 2018;49(6):1148-1161 e7.
Chemnitz JM, et al. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173(2):945–54.
Clement M, et al. CD31 is a key coinhibitory receptor in the development of immunogenic dendritic cells. Proc Natl Acad Sci U S A. 2014;111(12):E1101–10.
Flies DB, et al. Coinhibitory receptor PD-1H preferentially suppresses CD4(+) T cell-mediated immunity. J Clin Invest. 2014;124(5):1966–75.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–20.
de Mingo Pulido A, et al. TIM-3 regulates CD103(+) dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell. 2018;33(1):60-74 e6.
de Mingo Pulido A, et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity. 2021;54(6):1154-1167 e7.
Dixon KO, et al. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature. 2021;595(7865):101–6.
Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37(3):193–207.
Maier B, et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature. 2020;580(7802):257–62.
Rodrigues PF, et al. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat Immunol. 2018;19(7):711–22.
Reizis B. Plasmacytoid dendritic cells: development, regulation, and function. Immunity. 2019;50(1):37–50.
Conrad C, et al. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer Res. 2012;72(20):5240–9.
Labidi-Galy SI, et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. 2011;71(16):5423–34.
Le Mercier I, et al. Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res. 2013;73(15):4629–40.
Sisirak V, et al. Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012;72(20):5188–97.
Sisirak V, et al. Breast cancer-derived transforming growth factor-beta and tumor necrosis factor-alpha compromise interferon-alpha production by tumor-associated plasmacytoid dendritic cells. Int J Cancer. 2013;133(3):771–8.
Terra M, et al. Tumor-derived TGFbeta alters the ability of plasmacytoid dendritic cells to respond to innate immune signaling. Cancer Res. 2018;78(11):3014–26.
Grenader T, et al. Derived neutrophil lymphocyte ratio is predictive of survival from intermittent therapy in advanced colorectal cancer: a post hoc analysis of the MRC COIN study. Br J Cancer. 2016;114(6):612–5.
Gu X, et al. Prognostic significance of neutrophil-to-lymphocyte ratio in prostate cancer: evidence from 16,266 patients. Sci Rep. 2016;6:22089.
Guthrie GJ, et al. The systemic inflammation-based neutrophil-lymphocyte ratio: experience in patients with cancer. Crit Rev Oncol Hematol. 2013;88(1):218–30.
Peng B, et al. Prognostic significance of the neutrophil to lymphocyte ratio in patients with non-small cell lung cancer: a systemic review and meta-analysis. Int J Clin Exp Med. 2015;8(3):3098–106.
Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. 2019;16(10):601–20.
Templeton AJ, et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J Natl Cancer Inst. 2014;106(6):dju124.
Terashima T, et al. Blood neutrophil to lymphocyte ratio as a predictor in patients with advanced hepatocellular carcinoma treated with hepatic arterial infusion chemotherapy. Hepatol Res. 2015;45(9):949–59.
Valero C, et al. Pretreatment neutrophil-to-lymphocyte ratio and mutational burden as biomarkers of tumor response to immune checkpoint inhibitors. Nat Commun. 2021;12(1):729.
Bagley SJ, et al. Pretreatment neutrophil-to-lymphocyte ratio as a marker of outcomes in nivolumab-treated patients with advanced non-small-cell lung cancer. Lung Cancer. 2017;106:1–7.
Cassidy MR, et al. Neutrophil to lymphocyte ratio is associated with outcome during ipilimumab treatment. EBioMedicine. 2017;18:56–61.
Zaragoza J, et al. High neutrophil to lymphocyte ratio measured before starting ipilimumab treatment is associated with reduced overall survival in patients with melanoma. Br J Dermatol. 2016;174(1):146–51.
Rao HL, et al. Increased intratumoral neutrophil in colorectal carcinomas correlates closely with malignant phenotype and predicts patients’ adverse prognosis. PLoS ONE. 2012;7(1): e30806.
Droeser RA, et al. High myeloperoxidase positive cell infiltration in colorectal cancer is an independent favorable prognostic factor. PLoS ONE. 2013;8(5): e64814.
Galdiero MR, et al. Occurrence and significance of tumor-associated neutrophils in patients with colorectal cancer. Int J Cancer. 2016;139(2):446–56.
Berry RS, et al. High levels of tumor-associated neutrophils are associated with improved overall survival in patients with stage II colorectal cancer. PLoS ONE. 2017;12(12): e0188799.
Wikberg ML, et al. Neutrophil infiltration is a favorable prognostic factor in early stages of colon cancer. Hum Pathol. 2017;68:193–202.
Silvestre-Roig C, et al. Neutrophil diversity in health and disease. Trends Immunol. 2019;40(7):565–83.
Jaillon S, et al. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020;20(9):485–503.
Powell DR, Huttenlocher A. Neutrophils in the tumor microenvironment. Trends Immunol. 2016;37(1):41–52.
Keeley EC, Mehrad B, Strieter RM. Chemokines as mediators of tumor angiogenesis and neovascularization. Exp Cell Res. 2011;317(5):685–90.
Casbon AJ, et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A. 2015;112(6):E566–75.
Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016;16(7):431–46.
Colotta F, et al. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80(8):2012–20.
Coffelt SB, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–8.
Dumitru CA, Lang S, Brandau S. Modulation of neutrophil granulocytes in the tumor microenvironment: mechanisms and consequences for tumor progression. Semin Cancer Biol. 2013;23(3):141–8.
Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103(33):12493–8.
Patel S, et al. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol. 2018;19(11):1236–47.
Gershkovitz M, et al. TRPM2 mediates neutrophil killing of disseminated tumor cells. Cancer Res. 2018;78(10):2680–90.
Finisguerra V, et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature. 2015;522(7556):349–53.
Koga Y, et al. Neutrophil-derived TNF-related apoptosis-inducing ligand (TRAIL): a novel mechanism of antitumor effect by neutrophils. Cancer Res. 2004;64(3):1037–43.
Eruslanov EB, et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest. 2014;124(12):5466–80.
Albrengues J, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018. https://doi.org/10.1126/science.aao4227.
Yang L, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583(7814):133–8.
Xiao Y, et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 2021;39(3):423-437 e7.
Cools-Lartigue J, et al. Neutrophil extracellular traps in cancer progression. Cell Mol Life Sci. 2014;71(21):4179–94.
Teijeira A, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52(5):856-871 e8.
Schauer C, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014;20(5):511–7.
Hanna RN, et al. Patrolling monocytes control tumor metastasis to the lung. Science. 2015;350(6263):985–90.
Lin EY, et al. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193(6):727–40.
Javeed N, et al. Immunosuppressive CD14(+)HLA-DR(lo/neg) monocytes are elevated in pancreatic cancer and “primed” by tumor-derived exosomes. Oncoimmunology. 2017;6(1): e1252013.
Plebanek MP, et al. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat Commun. 2017;8(1):1319.
Song X, et al. Cancer cell-derived exosomes induce mitogen-activated protein kinase-dependent monocyte survival by transport of functional receptor tyrosine kinases. J Biol Chem. 2016;291(16):8453–64.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74.
Gabrilovich DI, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67(1):425.
Bronte V, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150.
Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21(8):485–98.
Condamine T, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016. https://doi.org/10.1126/sciimmunol.aaf8943.
Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5(1):3–8.
Hegde S, Leader AM, Merad M. MDSC: Markers, development, states, and unaddressed complexity. Immunity. 2021;54(5):875–84.
Waight JD, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. 2013;123(10):4464–78.
Netherby CS, et al. The granulocyte progenitor stage is a key target of IRF8-mediated regulation of myeloid-derived suppressor cell production. J Immunol. 2017;198(10):4129–39.
Mohamed E, et al. The unfolded protein response mediator PERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity. 2020;52(4):668-682 e7.
Tartour E, et al. Angiogenesis and immunity: a bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer Metastasis Rev. 2011;30(1):83–95.
Shojaei F, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106(16):6742–7.
Corzo CA, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693–701.
Nagaraj S, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13(7):828–35.
Bronte V, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med. 2005;201(8):1257–68.
Lu T, et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. 2011;121(10):4015–29.
Molon B, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011;208(10):1949–62.
Kumar V, et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity. 2016;44(2):303–15.
Corzo CA, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207(11):2439–53.
Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–91.
Huang B, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66(2):1123–31.
Pan PY, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70(1):99–108.
Hoechst B, et al. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood. 2011;117(24):6532–41.
Grossman JG, et al. Recruitment of CCR2(+) tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. Oncoimmunology. 2018;7(9): e1470729.
Bonapace L, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515(7525):130–3.
Salcedo R, et al. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood. 2000;96(1):34–40.
Stamatovic SM, et al. CCL2 regulates angiogenesis via activation of Ets-1 transcription factor. J Immunol. 2006;177(4):2651–61.
ClinicalTrials.gov., An Open-Label, Multicenter, Phase 2 Study of Single-Agent CNTO 888 (an Anti-CCL2 Monoclonal Antibody) for the Treatment of Subjects With Metastatic Castrate-Resistant Prostate Cancer. , in Identifier NCT00992186.
Brana I, et al. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol. 2015;10(1):111–23.
Nywening TM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651–62.
Noel M, et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. 2020;38(3):800–11.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61.
Pyonteck SM, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–72.
Magkouta SF, et al. CSF1/CSF1R axis blockade limits mesothelioma and enhances efficiency of Anti-PDL1 immunotherapy. Cancers (Basel). 2021. https://doi.org/10.3390/cancers13112546.
Zhu Y, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–69.
Murray LJ, et al. SU11248 inhibits tumor growth and CSF-1R-dependent osteolysis in an experimental breast cancer bone metastasis model. Clin Exp Metastasis. 2003;20(8):757–66.
Cannarile MA, et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5(1):53.
Autio KA, et al. Immunomodulatory activity of a colony-stimulating factor-1 receptor inhibitor in patients with advanced refractory breast or prostate cancer: a phase I study. Clin Cancer Res. 2020;26(21):5609–20.
Dowlati A, et al. LY3022855, an anti-colony stimulating factor-1 receptor (CSF-1R) monoclonal antibody, in patients with advanced solid tumors refractory to standard therapy: phase 1 dose-escalation trial. Invest New Drugs. 2021;39(4):1057–71.
Papadopoulos KP, et al. First-in-human study of AMG 820, a monoclonal anti-colony-stimulating factor 1 receptor antibody, in patients with advanced solid tumors. Clin Cancer Res. 2017;23(19):5703–10.
Falchook GS, et al. A phase 1a/1b trial of CSF-1R inhibitor LY3022855 in combination with durvalumab or tremelimumab in patients with advanced solid tumors. Invest New Drugs. 2021;39(5):1284–97.
Razak AR, et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J Immunother Cancer. 2020. https://doi.org/10.1136/jitc-2020-001006.
West RB, et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc Natl Acad Sci U S A. 2006;103(3):690–5.
Tap WD, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet. 2019;394(10197):478–87.
Benner B, et al. Pexidartinib, a novel small molecule CSF-1R inhibitor in use for tenosynovial giant cell tumor: a systematic review of pre-clinical and clinical development. Drug Des Devel Ther. 2020;14:1693–704.
David JM, et al. The IL-8/IL-8R axis: a double agent in tumor immune resistance. Vaccines (Basel). 2016. https://doi.org/10.3390/vaccines4030022.
Che J, et al. Targeting CXCR1/2: The medicinal potential as cancer immunotherapy agents, antagonists research highlights and challenges ahead. Eur J Med Chem. 2020;185: 111853.
Schott AF, et al. Phase Ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2-negative metastatic breast cancer. Clin Cancer Res. 2017;23(18):5358–65.
Goldstein LJ, et al. A randomized, placebo-controlled phase 2 study of paclitaxel in combination with reparixin compared to paclitaxel alone as front-line therapy for metastatic triple-negative breast cancer (fRida). Breast Cancer Res Treat. 2021;190(2):265–75.
Kemp DM, et al. Ladarixin, a dual CXCR1/2 inhibitor, attenuates experimental melanomas harboring different molecular defects by affecting malignant cells and tumor microenvironment. Oncotarget. 2017;8(9):14428–42.
Dominguez C, et al. Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI Insight. 2017. https://doi.org/10.1172/jci.insight.94296.
Bilusic M, et al. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J Immunother Cancer. 2019;7(1):240.
Cueto FJ, Sancho D. The Flt3L/Flt3 axis in dendritic cell biology and cancer immunotherapy. Cancers (Basel). 2021. https://doi.org/10.3390/cancers13071525.
Habets TH, et al. Fractionated radiotherapy with 3 x 8 Gy induces systemic anti-tumour responses and abscopal tumour inhibition without modulating the humoral anti-tumour response. PLoS ONE. 2016;11(7): e0159515.
Chakravarty PK, et al. Flt3-ligand administration after radiation therapy prolongs survival in a murine model of metastatic lung cancer. Cancer Res. 1999;59(24):6028–32.
Fong L, et al. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci U S A. 2001;98(15):8809–14.
Oba T, et al. Overcoming primary and acquired resistance to anti-PD-L1 therapy by induction and activation of tumor-residing cDC1s. Nat Commun. 2020;11(1):5415.
Freedman RS, et al. Pilot study of Flt3 ligand comparing intraperitoneal with subcutaneous routes on hematologic and immunologic responses in patients with peritoneal carcinomatosis and mesotheliomas. Clin Cancer Res. 2003;9(14):5228–37.
Ohri N, et al. FLT3 ligand (CDX-301) and stereotactic radiotherapy for advanced non-small cell lung cancer. J Clin Oncol. 2020;38(15_suppl):9618–9618.
Su YL, et al. STAT3 in tumor-associated myeloid cells: multitasking to disrupt immunity. Int J Mol Sci. 2018. https://doi.org/10.3390/ijms19061803.
Zou S, et al. Targeting STAT3 in cancer immunotherapy. Mol Cancer. 2020;19(1):145.
Qin J-J, et al. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res. 2019;38(1):195.
Li H, et al. Fragment-based drug design and drug repositioning using multiple ligand simultaneous docking (MLSD): identifying celecoxib and template compounds as novel inhibitors of signal transducer and activator of transcription 3 (STAT3). J Med Chem. 2011;54(15):5592–6.
Verstovsek S, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–27.
Jonker DJ, et al. Napabucasin versus placebo in refractory advanced colorectal cancer: a randomised phase 3 trial. Lancet Gastroenterol Hepatol. 2018;3(4):263–70.
Huang TT, et al. Alteration of SHP-1/p-STAT3 signaling: a potential target for anticancer therapy. Int J Mol Sci. 2017. https://doi.org/10.3390/ijms18061234.
Bekaii-Saab T, et al. 1466P Napabucasin+ nab-paclitaxel with gemcitabine in patients (pts) with metastatic pancreatic adenocarcinoma (mPDAC): results from the phase III CanStem111P study. Ann Oncol. 2021;32:S1084–5.
Matozaki T, et al. Functions and molecular mechanisms of the CD47-SIRPalpha signalling pathway. Trends Cell Biol. 2009;19(2):72–80.
Pengam S, et al. SIRPalpha/CD47 axis controls the maintenance of transplant tolerance sustained by myeloid-derived suppressor cells. Am J Transplant. 2019;19(12):3263–75.
Liu X, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015;21(10):1209–15.
Sim J, et al. Discovery of high affinity, pan-allelic, and pan-mammalian reactive antibodies against the myeloid checkpoint receptor SIRPalpha. MAbs. 2019;11(6):1036–52.
Chao MP, et al. Therapeutic targeting of the macrophage immune checkpoint CD47 in myeloid malignancies. Front Oncol. 2019;9:1380.
Zhang W, et al. Advances in anti-tumor treatments targeting the CD47/SIRPalpha axis. Front Immunol. 2020;11:18.
Yang Y, Yang Z, Yang Y. Potential role of CD47-directed bispecific antibodies in cancer immunotherapy. Front Immunol. 2021;12: 686031.
Andrejeva G, et al. Novel SIRPalpha antibodies that induce single-agent phagocytosis of tumor cells while preserving T cells. J Immunol. 2021;206(4):712–21.
Chen YC, et al. Progress of CD47 immune checkpoint blockade agents in anticancer therapy: a hematotoxic perspective. J Cancer Res Clin Oncol. 2022;148(1):1–14.
Kauder SE, et al. ALX148 blocks CD47 and enhances innate and adaptive antitumor immunity with a favorable safety profile. PLoS ONE. 2018;13(8): e0201832.
Weiskopf K, et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341(6141):88–91.
Advani R, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-hodgkin’s lymphoma. N Engl J Med. 2018;379(18):1711–21.
Sikic BI, et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J Clin Oncol. 2019;37(12):946–53.
Lakhani NJ, et al. Evorpacept alone and in combination with pembrolizumab or trastuzumab in patients with advanced solid tumours (ASPEN-01): a first-in-human, open-label, multicentre, phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2021;22(12):1740–51.
Chen GY, et al. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science. 2009;323(5922):1722–5.
Barkal AA, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572(7769):392–6.
Fang X, et al. CD24: from A to Z. Cell Mol Immunol. 2010;7(2):100–3.
Kristiansen G, et al. CD24 expression is a new prognostic marker in breast cancer. Clin Cancer Res. 2003;9(13):4906–13.
Overdevest JB, et al. CD24 offers a therapeutic target for control of bladder cancer metastasis based on a requirement for lung colonization. Cancer Res. 2011;71(11):3802–11.
Altevogt P, et al. Novel insights into the function of CD24: A driving force in cancer. Int J Cancer. 2021;148(3):546–59.
Arber N, et al. A Novel Anti-CD24 Monoclonal Antibody: Unarmed, Conjugated and Bi-specific, for Targeting Human Malignancies: 204. Official journal of the American College of Gastroenterology | ACG, 2017. 112: p. S106.
Han Y, et al. CD24 targeting bi-specific antibody that simultaneously stimulates NKG2D enhances the efficacy of cancer immunotherapy. J Cancer Res Clin Oncol. 2019;145(5):1179–90.
Sun F, et al. Anti-CD24 antibody-nitric oxide conjugate selectively and potently suppresses hepatic carcinoma. Cancer Res. 2019;79(13):3395–405.
Maliar A, et al. Redirected T cells that target pancreatic adenocarcinoma antigens eliminate tumors and metastases in mice. Gastroenterology. 2012;143(5):1375-1384 e5.
Klapdor R, et al. Characterization of a novel third-generation anti-CD24-CAR against ovarian cancer. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20030660.
OncoC4 pharmaceutical company website pipeline. https://www.oncoc4.com/index.php/product-development/onc-781
Li TT, Ogino S, Qian ZR. Toll-like receptor signaling in colorectal cancer: carcinogenesis to cancer therapy. World J Gastroenterol. 2014;20(47):17699–708.
Zhang W, et al. Targeting CpG adjuvant to lymph node via dextran conjugate enhances antitumor immunotherapy. Bioconjug Chem. 2017;28(7):1993–2000.
Krieg AM. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene. 2008;27(2):161–7.
Karapetyan L, Luke JJ, Davar D. Toll-like receptor 9 agonists in cancer. Onco Targets Ther. 2020;13:10039–60.
Kuo TC, et al. Abstract 1721: TAC-001, a toll-like receptor 9 (TLR9) agonist antibody conjugate targeting B cells, promotes anti-tumor immunity and favorable safety profile following systemic administration in preclinical models. Cancer Res. 2021;81(13_supplement):1721–1721.
Wang D, et al. Modulation of the tumor microenvironment by intratumoral administration of IMO-2125, a novel TLR9 agonist, for cancer immunotherapy. Int J Oncol. 2018;53(3):1193–203.
Yuan S, et al. Toll-like receptor 9 activation by CpG oligodeoxynucleotide 7909 enhances the radiosensitivity of A549 lung cancer cells via the p53 signaling pathway. Oncol Lett. 2018;15(4):5271–9.
Kohrt HE, et al. Dose-escalated, intratumoral TLR9 agonist and low-dose radiation induce abscopal effects in follicular lymphoma. Blood. 2014;124(21):3092–3092.
Hirsh V, et al. Randomized phase III trial of paclitaxel/carboplatin with or without PF-3512676 (Toll-like receptor 9 agonist) as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol. 2011;29(19):2667–74.
Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–35.
Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19(5):1035–43.
Beatty GL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6.
Djureinovic D, Wang M, Kluger HM. Agonistic CD40 antibodies in cancer treatment. Cancers (Basel). 2021. https://doi.org/10.3390/cancers13061302.
Vonderheide RH. CD40 agonist antibodies in cancer immunotherapy. Annu Rev Med. 2020;71:47–58.
Yu X, et al. Complex interplay between epitope specificity and isotype dictates the biological activity of anti-human CD40 antibodies. Cancer Cell. 2018;33(4):664-675.e4.
Vonderheide RH, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25(7):876–83.
Rüter J, et al. Immune modulation with weekly dosing of an agonist CD40 antibody in a phase I study of patients with advanced solid tumors. Cancer Biol Ther. 2010;10(10):983–93.
Bajor DL, et al. Long-term outcomes of a phase I study of agonist CD40 antibody and CTLA-4 blockade in patients with metastatic melanoma. Oncoimmunology. 2018;7(10): e1468956.
Ribas A, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31(5):616–22.
O’Hara MH, et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol. 2021;22(1):118–31.
Wainberg ZA, et al. Open-label, phase I study of nivolumab combined with nab-paclitaxel plus gemcitabine in advanced pancreatic cancer. Clin Cancer Res. 2020;26(18):4814–22.
Barlesi F, et al. 291 Phase Ib study of selicrelumab (CD40 agonist) in combination with atezolizumab (anti-PD-L1) in patients with advanced solid tumors. J Immunother Cancer. 2020;8(Suppl 3):A178–A178.
Weiss SA, et al. A phase I study of APX005M and cabiralizumab with or without nivolumab in patients with melanoma, kidney cancer, or non-small cell lung cancer resistant to anti-PD-1/PD-L1. Clin Cancer Res. 2021;27(17):4757–67.
De Henau O, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539(7629):443–7.
Kaneda MM, et al. Macrophage PI3Kγ drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 2016;6(8):870–85.
Davis RJ, et al. Anti-PD-L1 efficacy can be enhanced by inhibition of myeloid-derived suppressor cells with a selective inhibitor of PI3Kδ/γ. Cancer Res. 2017;77(10):2607–19.
Lonetti A, et al. PI3K pan-inhibition impairs more efficiently proliferation and survival of T-cell acute lymphoblastic leukemia cell lines when compared to isoform-selective PI3K inhibitors. Oncotarget. 2015;6(12):10399–414.
Chandrasekaran S, et al. Strategies to overcome failures in T-cell immunotherapies by targeting PI3K-δ and -γ. Front Immunol. 2021;12: 718621.
Nunnery SE, Mayer IA. Management of toxicity to isoform α-specific PI3K inhibitors. Ann Oncol. 2019;30(10):x21–6.
Vanhaesebroeck B, et al. PI3K inhibitors are finally coming of age. Nat Rev Drug Discov. 2021;20(10):741–69.
Sullivan RJ, et al. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J Clin Oncol. 2018;36(15_suppl):3013–3013.
Cohen E, et al. 352 Updated clinical data from the squamous cell carcinoma of the head and neck (SCCHN) expansion cohort of an ongoing Ph1/1b Study of eganelisib (formerly IPI-549) in combination with nivolumab. J Immunother Cancer. 2020;8(Suppl 3):A214–5.
Postow M, et al. 434 Updated clinical data from the melanoma expansion cohort of an ongoing Ph1/1b study of eganelisib (formerly IPI-549) in combination with nivolumab. J Immunother Cancer. 2020;8(Suppl 3):A264–5.
Hatem S, et al. Abstract P5-16-02: Updated efficacy, safety and translational data from MARIO-3, a phase II open-label study evaluating a novel triplet combination of eganelisib (IPI-549), atezolizumab (atezo), and nab-paclitaxel (nab-pac) as first-line (1L) therapy for locally advanced or metastatic triple-negative breast cancer (TNBC). Cancer Res. 2022;82(4):P5-16-02-P5-16-02.
Tomczak P, et al. Preliminary analysis of a phase II, multicenter, randomized, active-control study to evaluate the efficacy and safety of eganelisib (IPI 549) in combination with nivolumab compared to nivolumab monotherapy in patients with advanced urothelial carcinoma. J Clinic Oncol. 2021;39(6_suppl):436–436.
Allard D, Turcotte M, Stagg J. Targeting A2 adenosine receptors in cancer. Immunol Cell Biol. 2017;95(4):333–9.
Antonioli L, et al. Switching off CD73: a way to boost the activity of conventional and targeted antineoplastic therapies. Drug Discov Today. 2017;22(11):1686–96.
Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414(6866):916–20.
Cekic C, et al. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014;74(24):7250–9.
Ghalamfarsa G, et al. CD73 as a potential opportunity for cancer immunotherapy. Expert Opin Ther Targets. 2019;23(2):127–42.
Jeffrey JL, Lawson KV, Powers JP. Targeting metabolism of extracellular nucleotides via inhibition of ectonucleotidases CD73 and CD39. J Med Chem. 2020;63(22):13444–65.
Harvey JB, et al. CD73’s potential as an immunotherapy target in gastrointestinal cancers. Front Immunol. 2020;11:508.
Moesta AK, Li XY, Smyth MJ. Targeting CD39 in cancer. Nat Rev Immunol. 2020;20(12):739–55.
Mirza M, et al. 1195 Results of NSGO-OV-UMB1/ENGOT-OV30 study: a phase II study of durvalumab and oleclumab in patients with relapsed ovarian cancer (OC). Int J Gynecol Cancer. 2021;31(Suppl 3):A376–A376.
Robert F, et al. Preliminary safety, pharmacokinetics (PK), pharmacodynamics (PD) and clinical efficacy of uliledlimab (TJ004309), a differentiated CD73 antibody, in combination with atezolizumab in patients with advanced cancer. J Clinic Oncol. 2021;39(15_suppl):2511–2511.
Perrot I, et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 2019;27(8):2411-2425.e9.
Kashyap AS, et al. Antisense oligonucleotide targeting CD39 improves anti-tumor T cell immunity. J Immunother Cancer. 2019;7(1):67.
Hauser RA, et al. Preladenant as an adjunctive therapy with levodopa in parkinson disease: two randomized clinical trials and lessons learned. JAMA Neurol. 2015;72(12):1491–500.
Churov A, Zhulai G. Targeting adenosine and regulatory T cells in cancer immunotherapy. Hum Immunol. 2021;82(4):270–8.
Chandra D, et al. Abstract A87: The A2AR antagonist AZD4635 prevents adenosine-mediated immunosuppression in tumor microenvironment and enhances antitumor immunity partly by enhancing CD103+ dendritic cells. Cancer Immunol Res. 2020;8(3_supplement):A87–A87.
Fong L, et al. Safety and clinical activity of adenosine A2a receptor (A2aR) antagonist, CPI-444, in anti-PD1/PDL1 treatment-refractory renal cell (RCC) and non-small cell lung cancer (NSCLC) patients. J Clin Oncol. 2017;35(15_suppl):3004–3004.
Powderly J, et al. 1206P-Phase I evaluation of AB928, a novel dual adenosine receptor antagonist, combined with chemotherapy or AB122 (anti-PD-1) in patients (pts) with advanced malignancies. Ann Oncol. 2019;30: v493.
Yu J, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190(7):3783–97.
Nakamura T, et al. Expression of indoleamine 2, 3-dioxygenase and the recruitment of Foxp3-expressing regulatory T cells in the development and progression of uterine cervical cancer. Cancer Sci. 2007;98(6):874–81.
Munn DH, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114(2):280–90.
Long GV, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20(8):1083–97.
Pilotte L, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109(7):2497–502.
Platten M, et al. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. 2019;18(5):379–401.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27.
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31–46.
Baardman J, et al. Metabolic-epigenetic crosstalk in macrophage activation. Epigenomics. 2015;7(7):1155–64.
Chang YC, et al. Epigenetic control of MHC class II expression in tumor-associated macrophages by decoy receptor 3. Blood. 2008;111(10):5054–63.
Skov S, et al. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005;65(23):11136–45.
Liu M, et al. Understanding the epigenetic regulation of tumours and their microenvironments: opportunities and problems for epigenetic therapy. J Pathol. 2017;241(1):10–24.
Wang HF, et al. Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors in vivo and in vitro. Cancer Immunol Immunother. 2017;66(3):355–66.
Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5(10):981–9.
Chen X, et al. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B. 2020;10(5):723–33.
San José-Enériz E et al. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers (Basel), 2019. 11(11).
Schaefer EW, et al. A phase 2 study of vorinostat in acute myeloid leukemia. Haematologica. 2009;94(10):1375–82.
Craddock CF, et al. Outcome of azacitidine therapy in acute myeloid leukemia is not improved by concurrent vorinostat therapy but is predicted by a diagnostic molecular signature. Clin Cancer Res. 2017;23(21):6430–40.
Mann BS, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12(10):1247–52.
Juo YY, et al. Epigenetic therapy for solid tumors: from bench science to clinical trials. Epigenomics. 2015;7(2):215–35.
Vansteenkiste J, et al. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest New Drugs. 2008;26(5):483–8.
Christmas BJ, et al. Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res. 2018;6(12):1561–77.
Borden EC. Interferons α and β in cancer: therapeutic opportunities from new insights. Nat Rev Drug Discov. 2019;18(3):219–34.
Golomb HM, et al. Alpha-2 interferon therapy of hairy-cell leukemia: a multicenter study of 64 patients. J Clin Oncol. 1986;4(6):900–5.
Flood BA, et al. STING pathway agonism as a cancer therapeutic. Immunol Rev. 2019;290(1):24–38.
Ohkuri T, et al. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res. 2014;2(12):1199–208.
Wang H, et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A. 2017;114(7):1637–42.
Ager CR, et al. High potency STING agonists engage unique myeloid pathways to reverse pancreatic cancer immune privilege. J Immunother Cancer. 2021. https://doi.org/10.1136/jitc-2021-003246.
Lara PN Jr, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29(22):2965–71.
Ding C, et al. Small molecules targeting the innate immune cGAS-STING-TBK1 signaling pathway. Acta Pharm Sin B. 2020;10(12):2272–98.
Amouzegar A et al. STING Agonists as Cancer Therapeutics. Cancers (Basel), 2021. 13(11).
Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18(9):545–58.
Zhang X, et al. PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol. 2017;22(6):1026–33.
Zhou J, Zhang S, Guo C. Crosstalk between macrophages and natural killer cells in the tumor microenvironment. Int Immunopharmacol. 2021;101(Pt B): 108374.
Alberts DS, et al. Randomized phase 3 trial of interferon gamma-1b plus standard carboplatin/paclitaxel versus carboplatin/paclitaxel alone for first-line treatment of advanced ovarian and primary peritoneal carcinomas: results from a prospectively designed analysis of progression-free survival. Gynecol Oncol. 2008;109(2):174–81.
Bourgeois-Daigneault MC, et al. Oncolytic vesicular stomatitis virus expressing interferon-γ has enhanced therapeutic activity. Mol Ther Oncolytics. 2016;3:16001.
Kerkar SP, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. 2011;121(12):4746–57.
Watkins SK, et al. IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. J Immunol. 2007;178(3):1357–62.
Brunda MJ, et al. Antitumor activity of interleukin 12 in preclinical models. Cancer Chemother Pharmacol. 1996;38(Suppl):S16-21.
Leonard JP, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90(7):2541–8.
Portielje JE, et al. Repeated administrations of interleukin (IL)-12 are associated with persistently elevated plasma levels of IL-10 and declining IFN-gamma, tumor necrosis factor-alpha, IL-6, and IL-8 responses. Clin Cancer Res. 2003;9(1):76–83.
Lasek W, Zagożdżon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother. 2014;63(5):419–35.
Anwer K, et al. Phase I trial of a formulated IL-12 plasmid in combination with carboplatin and docetaxel chemotherapy in the treatment of platinum-sensitive recurrent ovarian cancer. Gynecol Oncol. 2013;131(1):169–73.
Thaker PH, et al. GEN-1 immunotherapy for the treatment of ovarian cancer. Future Oncol. 2019;15(4):421–38.
Thaker PH, et al. GEN-1 in combination with neoadjuvant chemotherapy for patients with advanced epithelial ovarian cancer: a phase i dose-escalation study. Clin Cancer Res. 2021;27(20):5536–45.
Barton KN, et al. Phase I trial of oncolytic adenovirus-mediated cytotoxic and interleukin-12 gene therapy for the treatment of metastatic pancreatic cancer. Mol Ther Oncolytics. 2021;20:94–104.
Hewitt SL, et al. Intratumoral IL12 mRNA therapy promotes TH1 transformation of the tumor microenvironment. Clin Cancer Res. 2020;26(23):6284–98.
Old LJ. Tumor necrosis factor (TNF). Science. 1985;230(4726):630–2.
Ham B, et al. The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 2016;11(1):1–27.
Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10(1):45–65.
Cai X, et al. Inflammatory factor TNF-α promotes the growth of breast cancer via the positive feedback loop of TNFR1/NF-κB (and/or p38)/p-STAT3/HBXIP/TNFR1. Oncotarget. 2017;8(35):58338–52.
Chen X, Oppenheim JJ. Targeting TNFR2, an immune checkpoint stimulator and oncoprotein, is a promising treatment for cancer. Sci Signal. 2017. https://doi.org/10.1126/scisignal.aal2328.
Zhao X, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. 2012;122(11):4094–104.
Yang Y, et al. TNFR2: role in cancer immunology and immunotherapy. Immunotargets Ther. 2021;10:103–22.
Zhang YW, et al. Expression of tumor necrosis factor receptor 2 in human non-small cell lung cancer and its role as a potential prognostic biomarker. Thorac Cancer. 2019;10(3):437–44.
Torrey H, et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci Signal. 2017. https://doi.org/10.1126/scisignal.aaf8608.
Ham B, et al. TNF receptor-2 facilitates an immunosuppressive microenvironment in the liver to promote the colonization and growth of hepatic metastases. Cancer Res. 2015;75(24):5235–47.
Chen L, et al. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell Mol Life Sci. 2018;75(11):2045–58.
Tu S, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14(5):408–19.
Bettiol A, et al. Unveiling the efficacy, safety, and tolerability of anti-interleukin-1 treatment in monogenic and multifactorial autoinflammatory diseases. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20081898.
Lee C, et al. Inflammasome as a promising therapeutic target for cancer. Life Sci. 2019;231: 116593.
Ridker PM, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10105):1833–42.
Novartis top-line results for CANOPY-1 phase III study support further evaluation of canakinumab in lung cancer. News release. Novartis. [cited 2021 October 25]; https://bit.ly/3pyfhvt
Paz-Ares L, et al. 1194MO Canakinumab (CAN)+ docetaxel (DTX) for the second-or third-line (2/3L) treatment of advanced non-small cell lung cancer (NSCLC): CANOPY-2 phase III results. Ann Oncol. 2021;32:S953–4.
Liu Q, et al. Targeting interlukin-6 to relieve immunosuppression in tumor microenvironment. Tumour Biol. 2017;39(6):1010428317712445.
Choy EH, et al. Translating IL-6 biology into effective treatments. Nat Rev Rheumatol. 2020;16(6):335–45.
San-Miguel J, et al. Phase 2 randomized study of bortezomib-melphalan-prednisone with or without siltuximab (anti-IL-6) in multiple myeloma. Blood. 2014;123(26):4136–42.
Orlowski RZ, et al. A phase 2, randomized, double-blind, placebo-controlled study of siltuximab (anti-IL-6 mAb) and bortezomib versus bortezomib alone in patients with relapsed or refractory multiple myeloma. Am J Hematol. 2015;90(1):42–9.
Dijkgraaf EM, et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann Oncol. 2015;26(10):2141–9.
Genentech. Tocilizumab package insert in the USA. [cited 2022 Feb 5]; https://www.gene.com/download/pdf/actemra_prescribing.pdf.
Hart KM, et al. IL-10 immunomodulation of myeloid cells regulates a murine model of ovarian cancer. Front Immunol. 2011;2:29.
Oft M. IL-10: master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol Res. 2014;2(3):194–9.
Neven B, et al. A Mendelian predisposition to B-cell lymphoma caused by IL-10R deficiency. Blood. 2013;122(23):3713–22.
Berg DJ, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98(4):1010–20.
Glocker EO, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361(21):2033–45.
Emmerich J, et al. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res. 2012;72(14):3570–81.
Hecht JR, et al. Randomized phase III study of FOLFOX alone or with pegilodecakin as second-line therapy in patients with metastatic pancreatic cancer that progressed after gemcitabine (SEQUOIA). J Clin Oncol. 2021;39(10):1108–18.
Xue VW, et al. Transforming growth factor-β: a multifunctional regulator of cancer immunity. Cancers (Basel). 2020. https://doi.org/10.3390/cancers12113099.
Huang CY, et al. Recent progress in TGF-β inhibitors for cancer therapy. Biomed Pharmacother. 2021;134: 111046.
Zhu S, et al. Combination strategies to maximize the benefits of cancer immunotherapy. J Hematol Oncol. 2021;14(1):156.
Rocconi RP, et al. Gemogenovatucel-T (Vigil) immunotherapy as maintenance in frontline stage III/IV ovarian cancer (VITAL): a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Oncol. 2020;21(12):1661–72.
Teixeira AF, Ten Dijke P, Zhu HJ. On-target anti-TGF-β therapies are not succeeding in clinical cancer treatments: what are remaining challenges? Front Cell Dev Biol. 2020;8:605.
Giaccone G, et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur J Cancer. 2015;51(16):2321–9.
Strauss J, et al. 957O Long-term follow-up of patients (pts) with human papillomavirus (HPV)–associated malignancies treated with bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1. Ann Oncol. 2021;32:S829.
Dranoff G. GM-CSF-based cancer vaccines. Immunol Rev. 2002;188:147–54.
Liu BL, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303.
Goldsmith K, et al. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med. 1998;187(3):341–8.
Vafaei S, et al. Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier. Cancer Cell Int. 2022;22(1):2.
Buonaguro L, et al. Translating tumor antigens into cancer vaccines. Clin Vaccine Immunol. 2011;18(1):23–34.
Tagliamonte M, et al. Antigen-specific vaccines for cancer treatment. Hum Vaccin Immunother. 2014;10(11):3332–46.
Glavan TM, Pavelic J. The exploitation of Toll-like receptor 3 signaling in cancer therapy. Curr Pharm Des. 2014;20(42):6555–64.
Kyi C, et al. Therapeutic immune modulation against solid cancers with intratumoral poly-ICLC: a pilot trial. Clin Cancer Res. 2018;24(20):4937–48.
Sabbatini P, et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res. 2012;18(23):6497–508.
Schuler-Thurner B, et al. Rapid induction of tumor-specific type 1 T helper cells in metastatic melanoma patients by vaccination with mature, cryopreserved, peptide-loaded monocyte-derived dendritic cells. J Exp Med. 2002;195(10):1279–88.
Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22.
Higano CS, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115(16):3670–9.
Keskin DB, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–9.
Ott PA, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–21.
Schadendorf D, et al. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Ann Oncol. 2006;17(4):563–70.
Fu C, et al. DC-based vaccines for cancer immunotherapy. Vaccines (Basel). 2020. https://doi.org/10.3390/vaccines8040706.
Besse B, et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology. 2016;5(4): e1071008.
Morse MA, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. 2005;3(1):9.
Escudier B, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J Transl Med. 2005;3(1):10.
Zitvogel L, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998;4(5):594–600.
Lu Z, et al. Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. J Hepatol. 2017;67(4):739–48.
Hiltbrunner S, et al. Exosomal cancer immunotherapy is independent of MHC molecules on exosomes. Oncotarget. 2016;7(25):38707–17.
Liu H, et al. Novel strategies for immuno-oncology breakthroughs with cell therapy. Biomark Res. 2021;9(1):62.
Tumino N, et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J Hematol Oncol. 2021;14(1):191.
Nalawade SA, et al. Selectively targeting myeloid-derived suppressor cells through TRAIL receptor 2 to enhance the efficacy of CAR T cell therapy for treatment of breast cancer. J Immunother Cancer. 2021. https://doi.org/10.1136/jitc-2021-003237.
Lai J, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol. 2020;21(8):914–26.
Klichinsky M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38(8):947–53.
Sloas C, Gill S, Klichinsky M. Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Front Immunol. 2021;12: 783305.
Zhang L, et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J Hematol Oncol. 2020;13(1):153.
Kang M, et al. Nanocomplex-mediated in vivo programming to chimeric antigen receptor-M1 macrophages for cancer therapy. Adv Mater. 2021;33(43): e2103258.
Hiruma Y, et al. Impaired osteoclast differentiation and function and mild osteopetrosis development in Siglec-15-deficient mice. Bone. 2013;53(1):87–93.
Kameda Y, et al. Siglec-15 is a potential therapeutic target for postmenopausal osteoporosis. Bone. 2015;71:217–26.
Angata T, et al. Siglec-15: an immune system Siglec conserved throughout vertebrate evolution. Glycobiology. 2007;17(8):838–46.
Takamiya R, et al. The interaction between Siglec-15 and tumor-associated sialyl-Tn antigen enhances TGF-β secretion from monocytes/macrophages through the DAP12-Syk pathway. Glycobiology. 2013;23(2):178–87.
Munkley J. The role of Sialyl-Tn in cancer. Int J Mol Sci. 2016;17(3):275.
Wang J, et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med. 2019;25(4):656–66.
Sun J, et al. Siglec-15 as an emerging target for next-generation cancer immunotherapy. Clin Cancer Res. 2021;27(3):680–8.
Shum E, et al. 490 clinical benefit through siglec-15 targeting with NC318 antibody in subjects with Siglec-15 positive advanced solid tumors. J Immunother Cancer. 2021;9(Suppl 2):A520–1.
Cella M, et al. Impaired differentiation of osteoclasts in TREM-2-deficient individuals. J Exp Med. 2003;198(4):645–51.
Paloneva J, et al. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J Exp Med. 2003;198(4):669–75.
Molgora M, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing Anti-PD-1 immunotherapy. Cell. 2020;182(4):886-900.e17.
Yao Y, et al. TREM-2 serves as a negative immune regulator through Syk pathway in an IL-10 dependent manner in lung cancer. Oncotarget. 2016;7(20):29620–34.
Jahchan NS, et al. Abstract LB071: Tuning the tumor myeloid microenvironment (TME) by targeting TREM2+ tumor-associated macrophages to overcome resistance to immune checkpoint inhibitors. Cancer Res. 2021;81(13_Supplement):LB071–LB071.
Zhang X, et al. High TREM2 expression correlates with poor prognosis in gastric cancer. Hum Pathol. 2018;72:91–9.
Suzuki H, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386(6622):292–6.
Shi B, et al. The scavenger receptor MARCO expressed by tumor-associated macrophages are highly associated with poor pancreatic cancer prognosis. Front Oncol. 2021;11: 771488.
Chen AX, et al. Single-cell characterization of macrophages in glioblastoma reveals MARCO as a mesenchymal pro-tumor marker. Genome Med. 2021;13(1):88.
Jeremiasen M, et al. Tumor-associated CD68(+), CD163(+), and MARCO(+) macrophages as prognostic biomarkers in patients with treatment-naïve gastroesophageal adenocarcinoma. Front Oncol. 2020;10: 534761.
La Fleur L, et al. Expression of scavenger receptor MARCO defines a targetable tumor-associated macrophage subset in non-small cell lung cancer. Int J Cancer. 2018;143(7):1741–52.
Georgoudaki AM, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016;15(9):2000–11.
Eisinger S, et al. Targeting a scavenger receptor on tumor-associated macrophages activates tumor cell killing by natural killer cells. Proc Natl Acad Sci U S A. 2020;117(50):32005–16.
Xing Q, et al. Scavenger receptor MARCO contributes to macrophage phagocytosis and clearance of tumor cells. Exp Cell Res. 2021;408(2): 112862.
Yoshiizumi K, et al. Studies on scavenger receptor inhibitors. Part 1: synthesis and structure-activity relationships of novel derivatives of sulfatides. Bioorg Med Chem. 2002;10(8):2445–60.
Pandey M, et al. Inhibition of scavenger receptor class B type 1 (SR-B1) expression and activity as a potential novel target to disrupt cholesterol availability in castration-resistant prostate cancer. Pharmaceutics. 2021. https://doi.org/10.3390/pharmaceutics13091509.
Colonna M, et al. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J Immunol. 1998;160(7):3096–100.
Zheng J, et al. Inhibitory receptors bind ANGPTLs and support blood stem cells and leukaemia development. Nature. 2012;485(7400):656–60.
Zhang Y, Zheng J. Functions of immune checkpoint molecules beyond immune evasion. Adv Exp Med Biol. 2020;1248:201–26.
Chen HM, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest. 2018;128(12):5647–62.
Carbone C, et al. An angiopoietin-like protein 2 autocrine signaling promotes EMT during pancreatic ductal carcinogenesis. Oncotarget. 2015;6(15):13822–34.
Siu LL, et al. First-in-class anti-immunoglobulin-like transcript 4 myeloid-specific antibody MK-4830 abrogates a PD-1 resistance mechanism in patients with advanced solid tumors. Clin Cancer Res. 2022;28(1):57–70.
Hollmén M, Figueiredo CR, Jalkanen S. New tools to prevent cancer growth and spread: a “Clever” approach. Br J Cancer. 2020;123(4):501–9.
Karikoski M, et al. Clever-1/stabilin-1 controls cancer growth and metastasis. Clin Cancer Res. 2014;20(24):6452–64.
Virtakoivu R, et al. Systemic blockade of clever-1 elicits lymphocyte activation alongside checkpoint molecule downregulation in patients with solid tumors: results from a phase I/II clinical trial. Clin Cancer Res. 2021;27(15):4205–20.
Wang B, et al. Microlocalization and clinical significance of stabilin-1(+) macrophages in treatment-naïve patients with urothelial carcinoma of the bladder. World J Urol. 2020;38(3):709–16.
Bono P, et al. LBA38 Bexmarilimab, a novel macrophage re-programmer shows promising anti-tumour activity in phase I/II trial in several last line solid tumour types. Ann Oncol. 2021;32:S1313.
Acknowledgements
Not applicable.
Funding
ZL is supported by grants from the National Institutes of Health and Pelotonia.
Author information
Authors and Affiliations
Contributions
KCCJ and YW contributed equally to this paper and share first authorship. ZL and MEGM designed the project. All authors participated in literature review and the synthesis, drafting, and final approval of this manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests related to the words discussed.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Y., Johnson, K.C.C., Gatti-Mays, M.E. et al. Emerging strategies in targeting tumor-resident myeloid cells for cancer immunotherapy. J Hematol Oncol 15, 118 (2022). https://doi.org/10.1186/s13045-022-01335-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01335-y