
Abstract
Key message
A bread wheat panel reveals rich genetic diversity in Turkish, Pakistani and Iranian landraces and novel resistance loci to diverse powdery mildew isolates via subsetting approaches in association studies.
Abstract
Wheat breeding for disease resistance relies on the availability and use of diverse genetic resources. More than 800,000 wheat accessions are globally conserved in gene banks, but they are mostly uncharacterized for the presence of resistance genes and their potential for agriculture. Based on the selective reduction of previously assembled collections for allele mining for disease resistance, we assembled a trait-customized panel of 755 geographically diverse bread wheat accessions with a focus on landraces, called the LandracePLUS panel. Population structure analysis of this panel based on the TaBW35K SNP array revealed an increased genetic diversity compared to 632 landraces genotyped in an earlier study and 17 high-quality sequenced wheat accessions. The additional genetic diversity found here mostly originated from Turkish, Iranian and Pakistani landraces. We characterized the LandracePLUS panel for resistance to ten diverse isolates of the fungal pathogen powdery mildew. Performing genome-wide association studies and dividing the panel further by a targeted subsetting approach for accessions of distinct geographical origin, we detected several known and already cloned genes, including the Pm2a gene. In addition, we identified 22 putatively novel powdery mildew resistance loci that represent useful sources for resistance breeding and for research on the mildew-wheat pathosystem. Our study shows the value of assembling trait-customized collections and utilizing a diverse range of pathogen races to detect novel loci. It further highlights the importance of integrating landraces of different geographical origins into future diversity studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bread wheat (Triticum aestivum L.) provides more calories and protein per person than any other crops on Earth (FAO 2020). This allohexaploid (2n = 6x = 42, AABBDD) crop species originated from successive hybridization events, where the latest polyploidization is thought to have occurred ~ 8,000 years ago in the Fertile Crescent (Glémin et al. 2019; Haas et al. 2019). After domestication, wheat was disseminated to Europe and Asia (Bonjean and Angus 2001), where accessions were selected based on the needs of individual farmers, thus becoming locally adapted traditional accessions, so-called landraces (Zeven 1998; Villa et al. 2005). With the Green Revolution in the 1960s, landraces were systematically replaced by advanced cultivars (Evenson and Gollin 2003) at the cost of a narrower genetic diversity due to the bottleneck effect of breeding (Tanksley and McCouch 1997; Reif et al. 2005). Consequently, modern cultivars are likely to lack a substantial proportion of the genetic diversity present in the wheat gene pool to combat abiotic and biotic stresses, including infection by pathogens. Plant diseases are a big threat to agriculture, with fungal pathogens playing a major role, causing an estimated annual yield loss of 18% in wheat (Savary et al. 2019). Among those, wheat powdery mildew, caused by the obligate biotrophic ascomycete Blumeria graminis f.sp. tritici (Bgt), is a major source of yield loss worldwide (Savary et al. 2019). Chemical control via pesticides is expensive and can negatively impact the surrounding ecosystem (Dormann et al. 2007; Bourguet and Guillemaud 2016). Besides, the European Commission proposed binding rules to reduce EU pesticide usage by 50% until 2030 (EU 2020). Therefore, alternative strategies are needed to control wheat mildew, especially in the context of climate change, as the geographical dispersal of pathogens and the severity of their infections are expected to increase rapidly (Singh et al. 2023).
In the farm-to-fork strategy, the deployment of disease-resistant wheat accessions is proposed as a sustainable and effective way to combat pathogens (EU 2020). Such resistance can be conferred by resistance (R) genes, which typically encode intracellular nucleotide-binding leucine-rich repeat receptors (NLRs), albeit not always, that recognize pathogen avirulence effector proteins (AVRs) (Dodds and Rathjen 2010; Sánchez-Martín and Keller 2021; Athiyannan et al. 2022b). R gene resistance acts in a race-specific manner when an R protein recognizes the corresponding AVR (Flor 1971; Dodds and Rathjen 2010). The over 90 genetically characterized R genes against powdery mildew (Pm genes) represent a cornerstone of wheat breeding (McIntosh et al. 2013). However, race-specific resistance can be rapidly overcome by pathogens by evolving AVRs to evade recognition (McDonald and Linde 2002; Mundt 2014; Brown 2015). Due to this constant arms race and the low efficacy of the currently cloned Pm genes (Dracatos et al. 2023), the identification of new resistance genes is needed for wheat breeding programs. More than 7000 distinct NLR-encoding genes are estimated to be present in the wheat gene pool (Walkowiak et al. 2020). Based on this genomic analysis, there are possibly hundreds of potentially active but unknown Pm genes in the wheat germplasm. The untapped genetic diversity of wheat landraces and their adaptation to individual environments with high disease pressure of locally adapted pathogens makes them promising candidates for containing such genes (Tanksley and McCouch 1997; Zeven 1998; Müller et al. 2018).
Together with cultivars and wild relatives, landraces are conserved in gene banks, where, to date, more than 800,000 Triticum accessions are stored (CGIAR 2023). However, these remain mostly uncharacterized for their potential in agriculture, and their adaptive mechanisms are poorly understood, limiting their use in breeding (Tanksley and McCouch 1997; Müller et al. 2018). Attempts to unlock this hidden potential have been made using different approaches. For example, Balfourier et al. (2019) focused on maximizing the representation of different breeding statuses, dates of registration and geographical origin to reduce the collection size of the INRAe bread wheat collection from about 12,000 to 4506 accessions. Genotyping these accessions improved understanding of wheat phylogeography and genetic diversity over time. Another study characterized the genetic diversity of 80,000 accessions, which represented a large part of the CIMMYT and ICARDA germplasm banks, covering not only domesticated hexaploid wheat but also tetraploids and crop wild relatives (Sansaloni et al. 2020). This revealed unexplored diversity in landraces and wheat selection footprints. In a third recent example, Schulthess et al. (2022) genotyped the IPK winter wheat collection of 7651 accessions and a reference panel of 325 European elite cultivars. Later, this collection was phenotyped with a single powdery mildew isolate, detecting 11 previously undescribed resistance loci (Hinterberger et al. 2022).
Here, we used a panel based on former bread wheat collections assembled for allele mining (Bhullar et al. 2009, 2010b). These former collections included a main panel selected using a focused identification of germplasm strategy (FIGS) approach, revealing accessions with potentially high selection pressure for powdery mildew resistance (Mackay and Street 2004; Bhullar et al. 2009). We have now reduced these collections, focusing on landraces and maximizing the diversity of geographical origin. The reduced size of the panel allowed us to phenotype it with a diverse set of ten Bgt isolates, constituting a trait-customized panel that is ideal for searching for powdery mildew-resistant accessions and the underlying genes (Mascher et al. 2019). Using genome-wide association studies (GWASs) and a targeted subset approach for accessions of shared geographical origin and isolate-specific resistance patterns, we detected 22 most likely novel genetic regions associated with powdery mildew resistance.
Materials and methods
Plant material and growth conditions
As starting material from which we selected our working panel, we utilized a formerly assembled bread wheat collection of 1320 landraces that had been selected based on FIGS of accessions with potentially high selection pressure for powdery mildew resistance (Bhullar et al. 2009). Later, this collection was complemented with 733 accessions of diverse geographical origins (Bhullar et al. 2010b). Assessment of accession type, i.e., landrace, cultivar, breeding/research material or unknown, was based on passport data at https://www.genesys-pgr.org. When no GPS data were available for collection sites of accessions, we projected GPS using https://maps.google.com from the most detailed description available, i.e., given villages up to countries. Accessions from the two combined wheat collections were phenotyped for powdery mildew resistance at seedling stage with the six Bgt isolates: CHE_94202, CHE_96224, CHE_97223, CHE_97266, CHE_98230 and GBR_JIW2. The infection phenotype was used to create a reduced panel of wheat accessions, consisting of approximately 50% that showed complete resistance (0% visible infection) to one or more isolates or resistance with a threshold of 20% to at least two isolates. The remaining 50% were susceptible to all six isolates and had the same ratio of spring wheat to winter wheat as the resistant part of the panel. Additionally, we used wheat accession origin as a proxy for relatedness, choosing a geographically close susceptible counterpart for each resistant accession. The resulting diverse LandracePLUS panel of 755 bread wheat accessions (Table S1), with a focus on the fertile crescent, was infected with four additional Bgt isolates at seedling stage, i.e., CHE_19004, CHN_46_30, ISR_106 and ISR_94.
Differential lines used to assess virulence patterns for 27 different Pm genes are shown in Table S2, including near-isogenic lines (NILs) and accessions containing the designated gene. NILs had been backcrossed multiple times with susceptible accessions “Federation” or “Chancellor”, depicted by /x*Accession, where x is the number of backcrosses to the designated accession (McIntosh et al. 2013). Other differential lines were used as original seeds from the USDA ARS (https://npgsweb.ars-grin.gov/gringlobal/search) or propagated using isolation bags per single spikes.
Seeds used for infection tests were obtained by propagating accessions in the field using single rows per genotype without isolation. Seedlings for infection tests were grown in 40-well plastic trays in a growth chamber cycled at 20 °C/16 °C, 16/8 h photoperiod with 80% relative humidity.
Powdery mildew isolates and infections
We used previously sampled and sequenced Bgt isolates CHE_94202, CHE_96224, CHE_97223, CHE_97266, CHE_98230, CHN_46_30, GBR_JIW2, ISR_94 and ISR_106, which are described by Sotiropolous (Sotiropoulos et al. 2022) (Table S3). We sampled chasmothecia of one additional isolate, CHE_19004 (Table S3), in 2019 from a wheat field at Reckenholz, Affoltern, Switzerland, which was revived and sequenced as previously described (Sotiropoulos et al. 2022).
Powdery mildew infection tests of the differential lines and the LandracePLUS panel accessions were carried out on the primary leaves of 10 to 15-day-old seedlings grown under the abovementioned conditions. Leaf segments were placed with their adaxial side up in Petri dishes filled with 0.5% Phyto agar containing 30 ppm benzimidazole. Fresh conidiospores were dispersed using 5-ml Pasteur glass pipettes in a settling tower (Lutz et al. 1992). Petri dishes with detached leaf segments were incubated for 7 to 9 days at 20 °C, 80% relative humidity with a 16 h light/8 h dark cycle and 50 μmol m−2 s−1 photon flux density. Infections were done in batches, with replicates of leaf segments from at least three independent seedlings per wheat accession on the same petri dish, infected at the same time. We used mildew susceptible accession Kanzler as a control for a proper mildew infection for all infection tests. Accessions Chancellor and Federation were used as additional susceptible controls for tests on differential lines. These susceptible controls were grown together with the tested accessions of each batch and distributed throughout the layout petri dish. If controls were not well infected and, in addition, the overall infection was low, the full infection test was repeated rather than using controls as a means to correct phenotypic values.
Disease levels were assessed 7 to 9 days after inoculation, depending on fungal growth in the batch, using a discrete quantitative scale with a score from 0 to 100 for the percentage of leaf area covered by sporulating mildew colonies, as described earlier (Kaur et al. 2008). Disease levels of differential lines were directly scored as resistant (< = 20) and susceptible (> 20).
DNA extraction and genotyping
DNA extraction of plant material was performed as previously described (Stein et al. 2001). DNA quality was assessed via agarose gels and genotyped using the TaBW35K single-nucleotide polymorphism (SNP) array (Paux et al. 2022). The SNP call dataset included marker positions and flanking sequences based on RefSeq v1.0 of Chinese Spring (IWGSC 2018).
Pm gene screening via polymerase chain reactions (PCRs) and sequencing
PCR analysis was performed using Pm4 (Sánchez-Martín et al. 2021) and Pm2 haplotype-specific markers (Manser et al. 2021). Four random landraces that were identified to carry Pm2 were then used for long-range and a following nested PCR to amplify the gene for Sanger sequencing as previously described (Sánchez-Martín et al. 2016).
General data analysis and visualization
Unless indicated otherwise, analyses were done using R version 3.6.3 (R Core Team 2022), including data handling with Tidyverse (Wickham et al. 2023) and visualizations with R package ggplot2 version 3.3.6 (Wickham 2016).
Kinship matrices for hierarchical clustering and visualization were done with GAPIT version 3 (Wang and Zhang 2021). Dendrogram formation and hierarchical clustering were performed using the stats R package version 3.6.3 (R Core Team 2022) functions hclust (method = ward.D2) and dist (method = euclidean). Defining clusters of genotypes was done using the package dendextend version 1.16.0 (Galili 2015).
Wheat Pm gene sequence assembly and alignment was done with CLC Genomics Workbench version 20.0.4 (Qiagen Bioinformatics, https://digitalinsights.qiagen.com/). Pathogen Avr gene sequence alignment was done using IGV version 2.15.4 (Robinson et al. 2011).
SNP filtering and file format
We filtered for “PolyHighResolution” or off-target variants (OTVs) and markers with known chromosomal positions in the Chinese Spring RefSeq v1.0 reference genome. Thresholds of 25% heterozygosity, 25% missing data per wheat accession and 5% missing data per marker were applied, and markers with duplicated positions were removed. Absent haplotypes of an OTV were translated to “NA” to facilitate their inclusion in downstream analyses. Taken together, this resulted in 29,965 polymorphic markers. These were brought into a Hapmap format with R and then transformed into a variant call format (VCF) file using the software TASSEL version 5.0 (Bradbury et al. 2007). This file was used as an input to generate plink files using vcftools version 0.1.16 (Danecek et al. 2011), which were then transformed together with phenotyping data into bed, bim and fam files using PLINK v1.07 (Purcell et al. 2007) for Admixture and association analyses.
Phenotypic data analysis
The raw median of the biological replicates was taken as the final phenotype for seedling resistance assessment. Inconclusive phenotypes, e.g., 50% resistant and 50% susceptible against the same isolate due to possible seed contamination or heterozygosity, were excluded from further analyses. Phenotypes with less than three replicates were also excluded. These values were transformed into two categories, where 0–20% = resistant and > 20% = susceptible for GWAS and Mantel tests. For Pearson’s correlation, phenotypic values of the differential lines were transformed to 0 and 1, respectively.
Pearson’s correlation between the isolate phenotypes was calculated using the stats package and visualized with the package corrplot version 0.92 (Wei and Simko 2021). Heritability was calculated for each pathogen isolate using R package lme4 version 1.1-34 (Bates et al. 2015) and a nested linear mixed model approach, where fixed variance is defined as the wheat genotype, and random variance as the infection test batch (“Round”) nested within the specific petri dish (“Plate”). P-values are based on ANOVA tests between the full and null models.
In silico genotyping of TaBW35K array SNP variants in high-quality sequenced wheat genomes
For the comparison of genetic diversity, Fielder (Sato et al. 2021), Renan (Aury et al. 2022) and the 10 + wheat reference genomes ArinaLrFor, Chinese Spring, Claire, Cadenza, Jagger, Julius, Landmark, Lancer, Mace, Paragon, Norin61, Robigus, Stanley, SYMattis and Weebill (IWGSC 2018; Walkowiak et al. 2020) were added. Flanking sequences of SNP array markers were queried using BLASTN searches against the publicly available wheat genomes. Blast results were filtered for hits with at least 60 bp alignment and 96% shared identity, allowing no more than three mismatched nucleotides. Positions of the SNPs were then extracted from BLASTN alignments on the respective genomes through in-house scripts. Merging the resulting dataset with the 29,965 previously used markers resulted in an overlap of 27,337 SNPs used for principal component analysis (PCA) and hierarchical cluster analysis.
Diversity Analysis via PCA and hierarchical clustering
PCAs for the LandracePLUS panel and high-quality sequenced genomes were done based on the 27,337 SNP set using the R package SNPRelate version 1.18.1 and gdsfmt version 1.20.0 (Zheng et al. 2012). For the PCA for the comparison to the 632 landraces from the INRAe study (Balfourier et al. 2019), we first filtered the genotyping data of the combined datasets provided by INRAe for the same cleaned 29,965 SNPs of the LandracePLUS panel. Because no creation of a Hapmap or VCF file was necessary for further downstream analysis, this dataset could be directly used for PCA using the prcomp function.
Hierarchical clustering analysis was performed using the kinship matrix of the 27,337 SNP set, including high-quality sequenced genomes. We visualized the dendrogram using dendextend version 1.16.0 (Galili 2015).
Admixture kinship analysis and comparative visualization
We used the.bed file of 29,965 SNPs (not including the chromosome-assembled genomes) as an input to assess population structure using ADMIXTURE version 1.3.0 (Alexander et al. 2009), where the most likely number of founder populations K can be estimated via running the model over a series of values of K and then choosing K around the lowest occurring cross-validation (CV) error. We ran the model for K = 2 to K = 20. However, the CV error steadily dropped with increasing K (Fig. S1). We, therefore, regarded Ks after the largest CV error drops of 33% in total as appropriate estimations for population structure, i.e., K = 4 to K = 6. A bootstrap of 500 and CV of 10 was used for this analysis.
A kinship matrix for the visualization was made with the same 29,965 SNP set using GAPIT version 3 (Wang and Zhang 2021). The kinship matrix was used as an input for hierarchical clustering for the comparative dendrogram. This dendrogram was visualized using the R package ggdendro version 0.1.23 (de Vries and Ripley 2022). Finally, these two plots and the admixture barplot were merged using the R package patchwork version 1.1.1 (Pedersen 2020).
Mantel test
To calculate the Mantel test, we first transformed the VCF file of 29,965 SNPs into genlight format using the R package vcfR version 1.14.0 (Knaus and Grünwald 2017). Then, we used this as an input for producing a Bray–Curtis genetic distance matrix with the vegdist function from R package vegan 2.6-4 (Oksanen et al. 2022). This genetic matrix was then correlated to the Euclidean phenotypic distance matrices using the vegan package mantel function with the Spearman method and 999 permutations to obtain Mantel r values.
Genome-wide association analysis (GWAS)
For GWAS, missing data from the 29,965 SNP set were imputed using general Beagle version 5.4 (Browning et al. 2018). GWAS and estimates of effect size—beta—were calculated using the GEMMA (Zhou and Stephens 2012) univariate linear mixed model. For the full LandracePLUS panel and each subset, bed, bim and fam files were created as described above, fam files were used as input for creating the kinship matrix in GEMMA using the options -gk 1 and -miss 1. MAF was set to 1% for the full LandracePLUS panel and all subsets above 300 accessions in size, while for the other runs, the MAF was set to 5%. This matrix was then integrated into the univariate linear-mixed model with option -miss 1 (Zhou and Stephens 2012). Phenotypic data for the association studies were added to the fam in R. P-values were based on the likelihood ratio test, and -log10 transformed for Manhattan plots. We used two thresholds to account for multiple testing: the false discovery rate (FDR) and the more conservative Bonferroni correction (BC). However, we regarded SNPs that passed the FDR test as significant.
Due to the design of the TaBW35K SNP array based on linkage disequilibrium (LD), meaningful LD decay analysis was not possible. We, therefore, decided to define GWAS peak intervals based on the LD decay results of a recent study on bread wheat using over 40 million SNPs (Liu et al. 2023). There, LD decay was found to be 6.0353, 2.3851 and 3.0278 Mb for subgenomes A, B and D, respectively. Hence, we defined peaks in a subgenome-dependent manner, as regions where at least two SNPs were significantly associated with the powdery mildew phenotype within the range of the above LD decay bp distances. We further considered significant single SNP associations as peaks if they uniquely mapped to one chromosome (weak homology on homoeologs, i.e., at least six additional SNPs/gaps), were surrounded by no or few SNPs, and (1) they occurred for more than one isolate or (2) their significance passed the more stringent BC threshold. The most significant SNP—the peak SNP—plus/minus the subgenome-specific LD decay distance was used to define each peak interval. Alternatively, if several peak SNPs occurred, both were used for the interval calculation.
To test if the peak on chromosome 5D was derived from Pm2 and accounted for its presence, we included a covariate (option –c) for binary information on the presence of Pm2 based on haplotype-specific PCR screening.
Random subsets were produced using R package tibble 3.1.8 (Müller and Wickham 2022). Subsets of geographical origin were based on countries of origin.
The physical position of previously described Pm genes was either taken from the corresponding publication when available or estimated by blasting the flanking markers using BLASTN against Chinese Spring RefSeq v1.0 as a reference.
Accessions that possibly carry the causal genes of a resistance-associated region were determined by 1) having at least 50% of the resistance-associated SNPs within an associated region and 2) being resistant to the respective isolate (< = 20% leaf coverage).
Results
We assembled a geographically diverse panel of 755 bread wheat accessions (Fig. 1a–c) based on the selective reduction of former collections used for allele mining of the powdery mildew resistance gene Pm3 (Bhullar et al. 2009, 2010b). In the selection process, we focused on landraces and combined data on geographical origin with phenotypes of powdery mildew seedling stage resistance to six Bgt isolates. The resulting panel, hereafter LandracePLUS panel, contains 521 winter wheat and 234 spring wheat accessions, including 576 landraces, 162 older cultivars (acquisition date from 1946 to 2003, with the main part from 60s to 70s), seven research or breeding lines and 11 unknown accessions (Table S1). We used the LandracePLUS panel, which covers a broad geographical distribution, with a focus on accessions originating from the Middle East (Fig. 1a, b), to detect genetic loci associated with resistance to the powdery mildew pathogen (Fig. 1d).

LandracePLUS panel diversity and powdery mildew symptoms: a World map with semitransparent purple dots representing the origin of each of the 744 of the 755 wheat accessions of the LandracePLUS panel with known origin, b number of accessions from the LandracePLUS panel per country of origin, c selection of diverse wheat spikes from the LandracePLUS panel. Scale bar, 5 cm, and d leaf of wheat cultivar “Kanzler” with powdery mildew symptoms. Scale bar, 1 cm (Color figure online)
Diversity analysis of the LandracePLUS panel reveals unexplored genetic diversity
A total of 29,965 high-quality, polymorphic SNPs derived from genotyping with the TaBW35K SNP array were used for diversity analysis. All these SNPs had known chromosomal positions based on the Chinese Spring reference genome assembly RefSeq v1.0 (IWGSC 2018), including 18,610 regular SNPs and 11,355 OTVs, i.e., markers that detect both presence–absence polymorphisms and nucleotide polymorphisms. The 29,965 markers were distributed across the wheat genome, similar to earlier findings (Liu et al. 2017; Alemu et al. 2021; Govta et al. 2022), with lower coverage of genome D compared to the A and B genomes: 11,684 (39.0%) markers on the A-genome, 13,589 (45.3%) on the B-genome and 4692 (15.7%) on the D-genome. On average, 1,427 markers were assigned per chromosome, resulting in an average density of one marker each 483 Kbp. The least markers were assigned to chromosome 1D and most to chromosome 2B, with 497 and 2,465 markers, respectively.
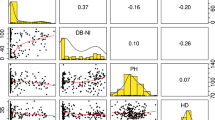
For a better interpretation of the observed genetic diversity within the LandracePLUS panel, wheat accessions with high-quality genome sequences, namely the 10 + wheat reference genomes (IWGSC 2018; Walkowiak et al. 2020), Fielder (Sato et al. 2021) and Renan (Aury et al. 2022) were included in the analysis with 27,337 out of the 29,965 SNPs that mapped unambiguously to chromosomes of these genomes. A PCA (Fig. 2a) and a dendrogram based on hierarchical clustering analysis (Fig. 2b) revealed four genetic clusters correlated with geographical origin overall. Group 1 was dominated by accessions from Iran and Pakistan, while group 2 was composed of accessions of mixed origin (Ethiopia, South Africa, USA, Canada, Mexico, Argentina, France, Switzerland, Kazakhstan, Kirgizstan, Tajikistan, Uzbekistan, Armenia and Russia). Group 3 predominantly contained accessions from Turkey, and group 4 was dominated by South, Southwest and East Asia (Iraq, Azerbaijan, India, Nepal, China and Japan). Most accessions with reference genomes available clustered in group 2 of the LandracePLUS panel, while Norin61 and Chinese Spring were on the edge of group 4 (Fig. 2a).

Genetic diversity and kinship analysis of the LandracePLUS panel: a PCA from 27,337 SNPs including high-quality sequenced wheat accessions labeled in the figure. PC1 = 8.4%, PC2 = 5.1%. Colors refer to the geographical origin of accessions and are indicated in b. b Dendrogram of a hierarchical clustering analysis from 27,337 SNPs including high-quality sequenced accessions indicated with stars. Colors represent the geographical origin of wheat accessions. Circled numbers on nodes refer to groups 1 to 4 when dividing into four clusters. c Alignment of Admixture plot, kinship matrix and dendrogram for the 755 wheat accessions based on 29,965 SNPs. The Admixture plot shows K = 5, where colors represent ancestral populations. In the kinship matrix, more saturated shades of blue indicate stronger relatedness. Dashed lines separate the four groups based on hierarchical clustering (Color figure online)
When highlighting the different types of accessions in the PCA, landraces covered almost the entire genetic diversity range of the LandracePLUS panel. In contrast, the 162 cultivars clustered mainly with group 2 and partially with group 4 (Fig. S2a). Therefore, landraces were genetically more diverse than cultivars and clustered apart from them. However, this was not always the case, as shown by the example of Ethiopian landraces, which clustered with cultivars in group 2 (Fig. S2b, Fig. 2a), suggesting that Ethiopian landraces have substantially contributed to breeding programs. The close clustering of accessions from all over the world described above (Fig. 2a, b) is likely driven by this separation between landraces and cultivars. As most of these geographically diverse accessions clustering together are cultivars, their genetic similarity is not derived from their origin but their breeding status as cultivars.
We assessed population structure performing hierarchical clustering on the 29,965 SNP set (excluding the high-quality genome sequences) and compared it to a kinship matrix and Admixture analysis (Fig. 2c). The found clusters reflected the four groups described earlier, except for a shift of several accessions of diverse origin from group 3 to group 2. Estimated ancestral populations K = 5 revealed a good fit for the LandracePLUS panel’s diversity into four groups (Fig. S1). Yet, the kinship matrix revealed additional subdivisions within Groups 3 and 4.
To assess whether the LandracePLUS panel represents a similar genetic diversity compared to former studies on wheat germplasm, we compared our data with a collection of 632 landraces that were genotyped with the TaBW280K SNP array (Rimbert et al. 2018), which contains all markers that were used for the LandracePLUS panel (Balfourier et al. 2019). A PCA with 29,965 filtered SNPs (Fig. S3) showed that the LandracePLUS panel covers the genetic diversity of this collection and further revealed unexplored genetic diversity absent in the study of Balfourier and colleagues (Balfourier et al. 2019). This additional diversity was mainly in groups 1 and 3, comprising Turkish, Pakistani and Iranian landraces.
Taken together, the LandracePLUS panel is a diverse selection of wheat accessions with a pronounced diversity of landraces compared to cultivars and high-quality sequenced genomes. Furthermore, the panel covers earlier found genetic diversity and additionally expands it, mainly with Turkish, Pakistani and Iranian landraces.
Differential lines reveal diverse virulence in a set of ten powdery mildew isolates
To maximize the chances of finding novel resistance genes, we first tested ten random isolates on a global collection of 27 differential lines, including NILs and donors of cloned or genetically described Pm genes (Fig. 3). On average, isolates were avirulent on ten out of 27 differential lines. While isolates CHE_96224, CHE_97266 and CHE_98230 were avirulent on 16, 18 and 16 resistant differential lines, respectively, isolates CHE_19004 and ISR_94 were more virulent, with five and two resistant lines, respectively. Each isolate had a distinct virulence pattern, reflecting that the ten chosen mildew isolates represent broad diversity in virulence, confirmed by haplotype analysis of molecularly cloned Avrs (Table S4).

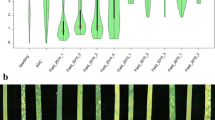
Virulence patterns of powdery mildew isolates and resistance distribution in the LandracePLUS panel. Virulence pattern of the ten powdery mildew isolates on differential lines with known Pm genes are shown as a heatmap on the top. On the bottom, the virulence pattern of the ten powdery mildew isolates on the LandracePLUS panel is depicted via violinplots and boxplots, highlighting median, 25 and 75 percentiles. The defined resistance threshold of 20 is indicated by a dashed line. Differential lines for the 27 Pm genes are listed in Table S2
Taken together, the resistance patterns of the differential lines reveal a phenotypically diverse set of ten powdery mildew isolates.
The LandracePLUS panel shows varying resistance to ten wheat powdery mildew isolates
The response of the 755 wheat accessions to the ten powdery mildew isolates revealed an overall susceptibility, with an average of 56 resistant accessions (rating < = 20) per isolate (Fig. 3). Considering the response to individual isolates, only 35, 36, 40 and 17 wheat accessions were resistant to CHE_19004, CHE_97223, GBR_JIW2 and ISR_94, respectively. This reflected the broad virulence of CHE_19004 and ISR_94 already found in the differential lines, while CHE_97223 and GBR_JIW2 seemed to be more virulent on the LandracePLUS panel compared to the set of differential lines. On the other side, 91 and 89 wheat accessions were resistant to CHE_96224 and CHE_98230, respectively, which were the most avirulent isolates on the differential lines along with CHE_97266.
Phenotypic Pearson’s correlation between the isolates based on the set of differential lines corroborated the diversity of virulence (Fig. S4a), with an average correlation coefficient of 0.31. The most similar isolates were CHE_96224 and CHE_97266, as well as CHE_98230 and GBR_JIW2, with a correlation of 0.69, while the two most diverging, though not significantly, were ISR_106 and ISR_94 with a negative correlation of 0.18. Pearson’s correlation between the isolates based on the LandracePLUS panel showed patterns resembling the differential line phenotypes (Fig. S4b). While the overall relation between isolates was similar, several correlation coefficients differed notably, mostly involving ISR_106, GBR_JIW2 and CHE_98230. This deviation could be caused by different contents of Pm genes in the LandracePLUS panel compared to the differential set. With a coefficient of 0.36, however, the average correlation was similar. The diversity in resistance reactions observed within the LandracePLUS panel against these Bgt isolates suggests highly diverse effector content, highlighting the chances of finding novel resistance loci in the LandracePLUS panel.
No wheat accession showed resistance to all ten isolates, while nine wheat accessions were resistant to nine of the isolates. On average, excluding accessions susceptible to all tested isolates, wheat accessions were resistant to three isolates.
We predicted the presence of known Pm genes in accessions of the LandracePLUS panel that matched the pattern of the corresponding Pm-containing differential line. For example, 23 accessions of the LandracePLUS panel had the same resistance pattern as the Pm2a differential line and, hence, are good candidates for containing Pm2a. Indeed, 15 of these 23 accessions contained Pm2 according to haplotype-specific markers (Manser et al. 2021) (Table S1). However, the potential presence of several Pm genes in the same accession can mask the resistance pattern of a specific Pm gene. This limits the use of differential lines to analyze overlapping resistance patterns and to predict specific genes. Nevertheless, the information on resistance patterns can be a useful tool to narrow down candidate accessions for the presence of a Pm gene of interest.
The LandracePLUS panel shows isolate-dependent heritability of mildew response phenotypes and low correlation between phenotype and genetic relatedness
We calculated the phenotypic heritability using a linear mixed model approach. The effect of the wheat genotype on mildew resistance was isolate-dependent, between 0.38 for ISR_94 and 0.79 for CHE_96224, and with a batch effect of 0.48 and 0.12, respectively (Table S5). This suggests that the observed response to CHE_96224 is highly reproducible, while the batch seemed to have a strong influence on resistance reaction for ISR_94. Half of the isolates (CHE_94202, CHE_96224, CHE_97223, CHE_97266 and GBR_JIW2) had a heritability of 0.7 or higher, while ISR_94 was the only mildew isolate with a value below 0.5, possibly indicating a mixture of powdery mildew races. Accordingly, we removed isolate ISR_94 from all subsequent analyses. To account for this batch effect for the remaining isolates, and since the described nature of R genes can be considered a binary one—resistant or susceptible—we decided to transform the phenotypic scoring values of 0 to 100 to these two categories, with a threshold of 20 or higher for susceptibility. Thus, differences between batches are weighted less, providing more reliable phenotypes and GWAS results.
Using these categorized phenotypes, we further tested the correlation between the genetic relatedness and the phenotype using a Mantel test (Mantel and Valand 1970). There were only slight differences between the ten different isolates, and the correlations were close to zero, with Mantel r values ranging from − 0.001 to 0.029, although not significant for most isolates (Table S6). This suggests that the applied approach of assembling the LandracePLUS panel minimized the effect of population structure on trait variation. The LandracePLUS panel should, therefore, provide improved power when conducting GWAS (Myles et al. 2009).
Association studies for seedling resistance to wheat powdery mildew in the LandracePLUS panel reveal previously cloned Pm genes as well as possibly novel genes
We conducted GWAS with the phenotyping data obtained for each of the nine powdery mildew isolates on the LandracePLUS panel with a MAF of 1%. We first tested for a good fit of the univariate linear mixed model based on QQ plots, which was confirmed for all isolates except CHE_97223 (Fig. S5). GWAS of the other eight isolates revealed five genomic regions associated with wheat mildew resistance on chromosomes 1A, 2B, 5D, 7A and 7D (Table 1, Fig. S6). To account for LD, we defined a peak region by adding the average LD decay distance per subgenome from a recent study in wheat (Liu et al. 2023) to either side of the peak SNP. The most significant peak was located on chromosome 5D for the AvrPm2-containing Bgt isolates CHE_94202, CHE_96224, CHE_97266, CHN_46_30 and ISR_106. When focusing on one representative AvrPm2-containing isolate, ISR_106, the mildew resistance association spanned the region from 40,919,172 to 46,974,772 bp of the short arm of chromosome 5D of Chinese Spring (Fig. 4a–c) and included the Pm2 resistance gene locus (Sánchez-Martín et al. 2016). To test if the presence of Pm2 was responsible for the significant association, we screened the LandracePLUS panel with a Pm2 haplotype-specific marker (Manser et al. 2021) and found that out of 66 wheat accessions resistant to ISR_106, 31 contained Pm2. In the whole panel, 39 accessions contained Pm2, of which 34 were landraces (Table S1). Most of these 34 accessions were from Turkey, with three landraces from Russia, Pakistan and Tajikistan (Fig. 4f). GWAS with a covariate for Pm2 presence resulted in the loss of the significant peak (Fig. 4d), corroborating that the peak was indeed caused by Pm2. Sequencing of the amplified Pm2 locus in four randomly selected landraces that were positive for the haplotype marker uniformly revealed the presence of the known allele Pm2a (Sánchez-Martín et al. 2016).

Analysis of the region associated with powdery mildew resistance on chromosome 5D. Manhattan plot for GWAS with all wheat accessions infected with isolate ISR_106 showing a all 21 chromosomes, b chromosome 5D and c the region of 40 to 50 Mb on chromosome 5D. The locus of the Chinese Spring version of Pm2 with a partially deleted gene (Sánchez-Martín et al. 2016) is indicated with a line. d Manhattan plot for GWAS with ISR_106 when adding a covariate for Pm2 presence in wheat accessions. e Manhattan plot for the Pm2 virulent isolate CHE_98230. Solid lines represent the threshold for false discovery rate, and dashed lines for Bonferroni correction. f Map with semitransparent brown dots depicting the origin of landraces that contain Pm2. Countries of origin are abbreviated with the three-letter country code of ISO 3166
In addition to Pm2, we detected a peak on chromosome 1AS for CHE_98230, which spanned the genomic region from 306,708 to 12,377,308 bp (Table 1, Fig. S6) and contains the locus of the cloned gene Pm3. Earlier studies have shown that out of 89 resistant accessions in the LandracePLUS panel, 21 contained functional Pm3 alleles, with 12 and five accessions containing Pm3c and Pm3b, respectively (Table S1) (Bhullar et al. 2009, 2010a, b). Therefore, we propose that the peak is caused by these functional Pm3 alleles.
A third, very large region on chromosome 2BL significantly showed CHE_96224 resistance-associated SNPs from 707,987,645 to 722,628,991 bp (Table 1, Fig. S6). Based on our criteria, this region was defined as three independent peaks. Upon closer inspection, the peaks are, however, only separated by small intervals showing no association. We therefore decided to consider them a single mildew-associated region. Indeed, it was shown that an introgression on chromosome 2BL is present in the wheat gene pool, most likely derived from the diploid wild relative Triticum timopheevii (Walkowiak et al. 2020; Keilwagen et al. 2022). This introgression is potentially present in the LandracePLUS panel. Many candidate Pm genes have already been described in this genomic interval, including Pm51 (Zhan et al. 2014), Pm63 (Tan et al. 2019), PmKN0816 (Wang et al. 2021), PmLS5082 (Wu et al. 2022), PmQ (Li et al. 2020b), pmYN99102 (Mu et al. 2022) and PmCG15-009 (Zhang et al. 2023).
We further detected a peak on chromosome 7AL for CHE_94202, CHE_97266, CHE_98230 and GBR_JIW2 that spanned from 717,179,300 to 733,267,996 bp (Table 1, Fig. S6). Possible candidate genes in this genomic interval include Pm59 (Tan et al. 2018), MlIw72 (Ji et al. 2008), Mlm2033 (Yao et al. 2007), Mlm80 (Yao et al. 2007), MlUM15 (Worthington et al. 2014), MlIw172s (Ouyang et al. 2014), PmG16 (Ben-David et al. 2010) and mlRd30 (Singrün et al. 2004). Alleles of Pm1 have also been mapped to chromosome 7AL (McIntosh et al. 2013). However, we could not confirm an overlap with our resistance-associated region because the Pm1 locus is absent in Chinese Spring (IWGSC 2018), and based on blast search using the genomic sequence of Pm1a as query, this locus is also absent in all other chromosome-scale-assembled wheat varieties (Walkowiak et al. 2020; Sato et al. 2021; Aury et al. 2022; Athiyannan et al. 2022a).
Finally, we detected a peak on chromosome 7DL for GBR_JIW2 that spanned from 631,068,212 to 637,123,812 bp (Table 1, Fig. S6, Fig. 5a), where no Pm gene has been described previously.

Manhattan plots of novel resistance-associated regions: a all phenotyped LandracePLUS panel accessions, b all phenotyped LandracePLUS panel accessions using a Pm2 covariate, c subset of accessions from Pakistan and Iran, and d subset of accessions from Turkey with CHE_97266 representing the peak occurring for multiple isolates. Isolates and chromosomes are depicted in the top-left corner. Solid lines represent the threshold for false discovery rate and dashed lines for Bonferroni correction
Association studies with a Pm2 covariate reveal five additional and novel resistance-associated loci
We used the information on the presence of Pm2 in LandracePLUS panel accessions to detect further resistance associations masked by the Pm2 gene. Incorporating the covariate in GWAS for isolates with the Pm2 peak, we detected five additional associations on chromosomes 1B, 3B, 5B and 7B (Table 1, Fig. S7, Fig. 5b). Two of these peaks appeared for CHE_96224, on chromosome 1BL, spanning from 610,940,301 to 615,710,501 bp and on chromosome 7BS from 68,921,370 to 73,691,570 bp. CHE_97266 showed an association on chromosome 3BL from 701,911,458 to 706,681,658 bp, and the last two associations belonged to CHN_46_30, with a peak on chromosome 3BL from 667,372,099 to 672,142,299 bp and another on chromosome 5BL from 647,102,735 to 651,872,935 bp. These associations occur in regions where no Pm gene has been reported earlier, highlighting the utility of using a known Pm gene as a covariate to discover new resistance loci in GWAS.
Pm4 alleles are widely present in the LandracePLUS panel but are not revealed by association studies
Based on the resistance of differential lines carrying genes Pm4a or Pm4b to five and four of the nine isolates, respectively (Sánchez-Martín et al. 2021), we expected to detect the cloned gene Pm4 in the LandracePLUS panel. Haplotype-specific markers detected the presence of the Pm4 haplotype in 62 accessions. However, no peak was produced at the Pm4 locus on chromosome 2AL for any isolate. To explain this missing association despite the broad presence of Pm4, we sequenced the locus in all 62 accessions, of which 51 and four accessions contained the non-functional alleles Pm4f and Pm4g, respectively (Sánchez-Martín et al. 2021), and four accessions with an undescribed allele, hereafter Pm4_42460, while the functional alleles Pm4b and Pm4d only occurred once and twice, respectively. The undescribed allele resembled Pm4f, except for one amino acid change in position 421 of splicing variant V1 (L421P). While allele Pm4g was present in accessions of diverse geographical origin, 42 out of 51 accessions carrying Pm4f and all four accessions containing Pm4_42460 were Turkish landraces. Our findings suggest that the non-functional alleles Pm4_42460 and Pm4f originated in Turkey.
Taken together, we find the presence of known Pm genes with GWAS of the LandracePLUS panel, giving insights into their geographic distribution and potential origin. In addition, we discovered six genomic regions where no Pm gene had been described earlier.
Utilizing subsets of the LandracePLUS panel discovers novel loci that are associated with powdery mildew resistance of distinct geographical origin
Some resistance genes are likely present only in certain groups of the LandracePLUS panel, e.g., accessions with similar geographical origins like for the Pm2 resistance gene. Accordingly, we first tested if subsets of the panel based on such groups were suitable to detect additional genomic regions associated with powdery mildew resistance. To assess the sensitivity of this approach, we used a re-sampling approach by creating 100 random subsets of 300, 200, 150 and 90 accessions from the LandracePLUS panel as input for GWAS with the Pm2 avirulent isolate ISR_106. Since the MAF threshold of 1% used above would not filter out SNPs that occur only twice for a subset of 200 to 299 accessions, we adjusted the MAF threshold to 5% for all subsets below a sample size of 300. We found the Pm2-associated peak in 100%, 87%, 78% and 52% of cases for the four subset sizes, respectively. Therefore, we considered subsets of a minimum of 150 to 200 accessions suitable for detecting loci associated with a major resistance gene in the LandracePLUS panel.
In our subsetting approach, we created sets of accessions based on their geographical origins. A subset of landraces from Pakistan and Iran showed a good fit to the GWAS model only for the mildew isolate CHE_98230 (Fig. S8), revealing five additional peaks compared to the full LandracePLUS panel, located on chromosomes 2B, 5A, 5B, 5D and 6A (Table 2, Fig. S9, Fig. 5c). While the peak on chromosome 2BL was located in a genomic region that has been described to contain the resistance gene Pm51 (Zhan et al. 2014) and the peak on chromosome 5BL has been implicated earlier with the resistance genes Pm53 (Petersen et al. 2015) and Ml3D232 (Zhang et al. 2010), the associated regions on chromosomes 5AL from 679,959,567 to 692,030,167 bp and on 5DS from 26,487,899 to 32,543,499 bp have not been associated with powdery mildew resistance before to our knowledge (Table 2). The last resistance-associated region, on chromosome 6AS, spanned the region from 41,508,045 to 53,578,645 (Fig. 5c). On chromosome arm 6AS, only Pm21 and Pm56 have been described. Pm21 originated and was cloned from the diploid grass Dasypyrum villosum (He et al. 2018; Xing et al. 2018). The gene was introduced in the hexaploid gene pool in China through a translocation line T6AL.6VS in the late 1980s (Chen et al. 1995). However, the translocated arm from D. villosum does not recombine with wheat homeologs (He et al. 2017). Hence, we expect to see a broad association covering the short arm of chromosome 6A in the case of Pm21 detection. Further, Pm21 was described to confer broad-spectrum resistance (He et al. 2017), whereas we observed the association for only one out of ten isolates. Therefore, we assume that the causal gene in our resistance-associated region differs from Pm21. Similarly, Pm56 was only recently introduced as a translocation line 6AL.6RS from rye (Hao et al. 2018) several years after we obtained and utilized the seeds of the initial collection. We conclude that this is a resistance-associated region on chromosome 6AS not previously described.
Finally, we used the same geographical approach to investigate only Turkish landraces, revealing a good fit to the model for all isolates except CHE_19004 (Fig. S10). This subsetting resulted in the discovery of 15 additional peaks on chromosomes 1A, 1B, 1D, 2A, 3A, 4B, 5A, 5B, 6A, 7A, 7B and 7D (Table 2, Fig. S11). In earlier studies, two of the 15 regions have been described with powdery mildew seedling resistance. The associated region on chromosome 2AL from 762,574,656 to 774,645,256 bp covers three known Pm genes, while the peak on chromosome 7BL from 701,729,427 to 706,653,524 bp overlaps with eight previously described Pm genes (Table 2). The association on chromosome 1BL from 664,434,720 to 669,204,920 bp includes the Pm39 locus. However, Pm39, also known as Lr46 (Lillemo et al. 2008), is an adult plant resistance gene and not active at seedling stage, and we conclude that the detected association is caused by an unknown, novel gene. The remaining 12 loci do not overlap with previously described Pm genes and therefore depict good candidates for novel powdery mildew resistance loci (Table 2).
Taken together, subsets based on geographical origin revealed 16 genomic regions where no Pm genes have been described previously, suggesting that these genes arose in the respective countries Pakistan, India and Turkey.
Candidate genes of five novel resistance loci include putative NLRs, serine/threonine kinases, a C2H2-type zinc finger and F-box-like proteins with leucine-rich repeat (LRR) domains
We investigated five of the 22 novel resistance loci more closely to get an insight into possible candidate genes (Fig. 5, Table S7). We chose the most significant peak with at least two significantly associated SNPs from GWAS of: 1) the full LandracePLUS panel, 2) the panel using the Pm2 covariate and 3) the Pakistan/Iran subset, on chromosomes 7DL, 7BS and 6AS, respectively. From the subset of Turkish landraces, we chose the highly significant single SNP association on chromosome 1DL and the peak on chromosome 1BL that occurred for five isolates, suggesting a more broad-spectrum resistance. For the associated region on chromosome 7DL, candidates annotated on Chinese Spring were ten putative NLRs, while the associated region on chromosome 7BS contained an annotation for a C2H2-type zinc finger and one F-box-like protein with an LRR domain. All associated regions except for 1BL had annotations for one serine/threonine kinase, the only candidate for the peak on chromosome 1DL. Both peaks on chromosome 6AS and 1BL contained annotations for one putative NLR, and the peak on chromosome 6AS included four additional F-box-like proteins with LRR domains (Fig. 5, Table S7). Further validation studies are necessary to confirm the resistant nature of these candidate genes. Due to the possibility of missing genes in the associated regions in the reference genome Chinese Spring, we also provide a list of candidate genes in all chromosome-scale-assembled wheat varieties (Supplementary_file4) (White et al. 2024). While Chinese Spring has 298 predicted genes in the five associated regions, the other 12 varieties ranged between 324, for Renan, and 471, for Lancer.
Novel resistance loci for breeding programs
We analyzed the gene pool of the 20 most modern cultivars of the LandracePLUS panel (registered between 1990 and 2003) to evaluate whether the 22 potentially novel powdery mildew genes are present in elite material or potentially novel in this gene pool (Supplementary_file3). For genomic regions on chromosomes 5BL (derived from the LandracePLUS panel using a Pm2 covariate) and 6AS (from the Pakistan/Iran subset), the most modern cultivars contained less than 50% of the resistance-associated SNPs, suggesting that the underlying resistance genes are likely not present in these cultivars. For six of the 22 associations, including the peaks on chromosomes 7DL and 1BL (described above in detail), some of the cultivars contained at least 50% of the resistance-associated SNPs, but none showed resistance toward the respective mildew isolate. We conclude that the underlying resistance genes are not present in this germplasm, at least not as active, resistance-conferring alleles. These findings suggest that the resistance loci have not been transferred to the modern gene pool from landraces. However, investigation of more recent cultivars would be needed to confirm these results.
Resistance-associated SNPs of the remaining 14 associated regions, including the peaks on chromosomes 7BS and 1DL discussed extensively, were present in the most modern germplasm. However, few accessions that harbored the alleles were resistant. Thus, the underlying resistance-conferring alleles seem to be partially present, meaning that breeders could integrate the corresponding cultivars we highlighted (Supplementary_file3) directly in their programs, avoiding possible yield penalties due to linkage drag. However, the combination of resistance and presence of resistance-associated SNPs occurred only for the three cultivars TRI 17181, TRI 17284 and TRI 16947. While these cultivars are attractive resistance-breeding candidates, it is difficult to dissect which detected regions are causing the observed resistance and, therefore, actually contain causative alleles.
Discussion
The LandracePLUS panel harbors untapped genetic diversity originating mainly from Turkish, Pakistani and Iranian landraces
We assembled a diverse panel of 755 bread wheat accessions with a focus on landraces. A FIGS approach laid the foundation for a trait-customized collection of accessions with potentially high selection pressure for powdery mildew resistance. A second step of selective reduction based on geographical origin resulted in the LandracePLUS panel. Mantel tests revealed a small impact of the population structure on powdery mildew resistance variation, reflecting the successful outcome of our targeted panel assembly. The resulting improved power of GWAS (Myles et al. 2009) facilitated the detection of novel Pm genes in the LandracePLUS panel.
The LandracePLUS panel revealed untapped genetic diversity in Turkish, Iranian and Pakistani landraces. It showed four main genetic clusters that correlate with the geographical origin, similar to a study which showed that wheat accessions from the Caucasus region, as well as Central, South and East Asian, are more diverse compared to other regions in the world (Balfourier et al. 2019). Landraces covered almost the full diversity of the LandracePLUS panel, whereas cultivars were limited to one cluster. This cluster also included the bread wheat accessions with high-quality sequenced genomes, except for the two landraces Chinese Spring and Norin61 (IWGSC 2018; Walkowiak et al. 2020; Sato et al. 2021; Aury et al. 2022). This observation highlights the need to include landraces in breeding programs to enlarge the genetic base of elite wheat varieties (Lopes et al. 2015; Marone et al. 2021). Future diversity studies of wheat should focus on landraces, in particular on those from regions with untapped genetic diversity, such as Turkey, Pakistan and Iran.
Pathogen virulence characterization guides Pm gene discovery
To discover novel R genes against powdery mildew in the LandracePLUS panel, we used a set of ten Bgt isolates that showed highly diverse virulence patterns when tested on differential lines with single, known Pm genes. As we observed avirulence for many differential lines, using these isolates should reveal the presence of most Pm genes for which differential lines are available.
The tested wheat accessions were rarely resistant to more than three isolates, in line with characteristic R gene-based race-specific resistance (Flor 1971). This implies that the observed single resistance genes would be of limited agricultural use, depending on the isolates present in the corresponding wheat-growing area. To broaden the resistance spectrum and prevent fast evolution of pathogen virulence, such genes should be deployed in a suitable manner, e.g., by gene stacking, gene pyramiding or transgenic overexpression (Mundt 2018; Koller et al. 2019). On the other hand, the accessions of the LandracePLUS panel susceptible to all tested powdery mildew isolates can be assumed to lack any major resistance gene active at the seedling stage. Thus, observed resistance in adult plants in the field would likely be durable, making such accessions attractive donors of adult plant resistance.
Currently, for the 27 genes represented in the differential set of Pm lines used in this study, only eight of the corresponding avirulence genes are molecularly known (Bourras et al. 2015, 2019; Praz et al. 2017; Hewitt et al. 2021; Müller et al. 2022; Kloppe et al. 2023; Kunz et al. 2023). Avirulence gene sequence comparison in our ten powdery mildew isolates revealed the presence of recognized haplotypes of seven of the cloned Avrs. This knowledge guided us to determine whether a resistance-associated region in the LandracePLUS panel was derived from a known Pm gene, as we showed for the cloned gene Pm2a. Further, the information on resistance to Bgt isolates of distinct geographical origin can guide an informed deployment of accessions in the respective agricultural areas (Vleeshouwers and Oliver 2014; Müller et al. 2022). For example, our findings suggest further work on landraces resistant to CHN_46_30 for potential deployment in China because they must contain effective R genes against this isolate of Chinese origin.
GWAS of the LandracePLUS panel detects known Pm genes and reveals six undescribed powdery mildew resistance-associated regions on chromosomes 1BL, 3BL, 5BL, 7BS and 7DL
GWAS for eight powdery mildew isolates on the LandracePLUS panel without subsetting revealed ten resistance-associated regions. Four of these were in genomic regions with previously described resistance genes. Of these, the peaks on chromosomes 2BL and 7AL overlapped with regions of genetically described, but not molecularly known Pm genes, while the peaks on chromosomes 1AS and 5DS were likely caused by the cloned genes Pm3 and Pm2, respectively. While Pm3 was known to be present in several LandracePLUS panel accessions (Bhullar et al. 2009, 2010a, b), we did not expect to find Pm2 widely in the gene pool of landraces. Pm2 was introduced into the breeding gene pool via the Russian cultivar Ulka (Pugsley and Carter 1953). It originated from the diploid wheat wild relative Ae. tauschii and has eight known haplotypes, of which only Pm2a was detected in hexaploid wheat (Manser et al. 2021). While Pm2a has been identified previously in six wheat landraces (Chen et al. 2019; Manser et al. 2021), we found its presence in 34, mostly Turkish landraces, suggesting Turkey as the geographical origin of the Pm2a resistance gene. While GWAS did not detect the cloned gene Pm4 in the LandracePLUS panel, we discovered a high frequency of two non-functional Pm4 alleles in Turkish landraces. Our findings suggest that these alleles originated in Turkey, fitting the origin of Pm4 from tetraploid wheat, which also arose in Turkey (Özkan et al. 2002; Sánchez-Martín et al. 2021). Despite the narrow representation of Pm4b and Pm4d in landraces, a wide presence of these functional alleles compared to Pm4f and Pm4g has been described in elite germplasm (Sánchez-Martín et al. 2021). This suggests that the breeding process for Pm4 mildew resistance was very effective.
We detected one region on chromosome 7DL not previously described as associated with mildew resistance. This region contained annotations for ten putative NLRs and one serine/threonine kinase. While NLRs are to date still the most common candidates for Pm genes, serine/threonine kinases have been described to play a role in defense response, including Pto, which confers resistance to bacterial speck disease in tomato (Martin et al. 1993; Loh and Martin 1995).
Using a Pm2 covariate, we detected five additional undescribed resistance associations in the LandracePLUS panel. One of them was located on chromosome 7BS, containing candidate genes putatively encoding a C2H2-type zinc finger, a serine/threonine kinase and an F-box-like protein with LRR domains. The latter have been described to facilitate hypersensitive cell death response in tobacco and tomato (Burg et al. 2008) and shown to be involved in defense response to stripe rust in wheat (Yin et al. 2018). On the other hand, zinc fingers of the C2H2-type were linked to plant defense response (Kim et al. 2004; Tian et al. 2010; Yin et al. 2020; Sharma et al. 2021), where some cases have shown that the transcriptional repression activity of the zinc finger was the mechanism behind this association (Weigel et al. 2005; Uehara et al. 2005).
Targeted GWAS subsets reveal 16 potentially novel resistance-associated loci
Utilizing targeted subsets, we discovered 16 most likely novel peaks on ten chromosomes (Table 2). GWAS is expected to be more powerful when conducted on large datasets where individuals are drawn randomly from the population (Uffelmann et al. 2021). However, important SNPs at a small regional scale might yet be diluted in species-wide panels and not detected by GWAS, which typically lacks the power to detect associations with rare alleles (Marees et al. 2018). Pending sufficient phenotypic and genetic variation, GWAS in local panels has proven very effective in Arabidopsis thaliana (Gloss et al. 2022). For this reason, resistance genes which may have been selected at small regional scales might be more efficiently detected in subsets of accessions from the same geographical origin.
Therefore, we focused on accessions originating from distinct countries that harbored novel genetic diversity, namely Pakistan, Iran and Turkey. This resulted in the detection of 16 additional loci where no Pm genes have been described. With a subset of accessions exclusively from Pakistan or Iran, we discovered a region on chromosome 6AS associated with mildew resistance. The region contains genes putatively encoding an NLR, a serine/threonine kinase and F-box-like proteins with LRR domains.
While we present novel regions and gene candidates, the molecular nature of the observed resistance must be confirmed in future studies, especially when considering that the reference genome Chinese Spring might lack (susceptible) alleles of the causal resistance genes. One approach would be the application of recent sequencing technologies, such as circular consensus sequencing (CCS) (Wenger et al. 2019). Such novel approaches have increased the feasibility of sequencing single donor accessions to assist in the cloning a gene of interest. For example, assembling the Kariega genome with CCS has demonstrated the usefulness of this approach in wheat and has led to the cloning of Yr27 (Athiyannan et al. 2022a). Another option to identify a gene of interest in a specific genotype depends on the availability of a pangenome, which ideally would capture the entire gene repertoire of a species (Tettelin et al. 2005). High-quality sequencing efforts have recently resulted in 19 wheat genomes, of which 14 have reference genome quality (IWGSC 2018; Walkowiak et al. 2020; Sato et al. 2021; Aury et al. 2022; Athiyannan et al. 2022a; Kale et al. 2022). Genome analysis has revealed structural rearrangements, introgressions and differences in gene content (Walkowiak et al. 2020). A successful example of using this resource is the cloning of Lr14a, which was based on the reference genome ArinaLrFor (Kolodziej et al. 2021). However, despite the recent advances in pangenome projects, the close clustering of the high-quality sequenced genomes compared to the LandracePLUS panel suggests that the currently available pangenome includes only a fraction of the diversity present in landraces, including resistance loci. Thus, it is essential to include more diverse wheat accessions, specifically landraces, in future work to increase the extent of the pangenome, particularly for NLR loci, which are rarely present across wheat genotypes. We propose to assemble high-quality genomes of several landraces from Turkey, Pakistan and Iran to achieve such a goal. Furthermore, contrasting phenotypes for traits of interest should be included when setting up pangenome consortia. This diversified selection could provide a resource that guides various trait-genotype associations. Finally, we suggest choosing a donor accession with confirmed resistance for each peak and sequencing the resistance-associated genome using CCS. This will reveal which gene candidates are present in the specific accessions and guide their molecular cloning and validation, e.g., via virus-induced gene silencing (Cakir et al. 2010).
The subset-based association analysis done in this work of the genetically diverse LandracePLUS panel challenged with ten Bgt isolates unraveled 22 potentially novel powdery mildew resistance genes. Therefore, this study can be used as an example for future work on similar collections in search of other traits of interest. Once a diversity panel is assembled, instead of focusing on single pathogen races, phenotyping with diverse isolates from geographically defined agricultural regions followed by subset-based analyses for the origin of accessions would reveal more resistance loci compared to studies done with single pathogen isolates on entire collections.
Data availability
The mildew isolate CHE_19004 whole-genome sequence data are available at NCBI’s Short Read Archive (SRA) under the accession code BioProject PRJNA945619. The nine isolate sequences from earlier work (Sotiropoulos et al. 2022) are available at NCBI under PRJNA625429 and SRP062198. The genotyping data of landraces from the study of Balfourier and colleagues (Balfourier et al. 2019) can be accessed at https://urgi.versailles.inra.fr/download/wheat/genotyping/Balfourier_et_al_Wheat_Phylogeography_DataS2.zip. All other data needed to evaluate the conclusions in this study are included in the article and its supplementary information files. All Blumeria graminis f.sp. tritici isolates used in this study are kept alive in the Department of Plant and Microbial Biology of the University of Zurich and are available upon request.
References
Alemu SK, Huluka AB, Tesfaye K et al (2021) Genome-wide association mapping identifies yellow rust resistance loci in Ethiopian durum wheat germplasm. PLoS ONE 16:e0243675. https://doi.org/10.1371/journal.pone.0243675
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664. https://doi.org/10.1101/gr.094052.109
Athiyannan N, Abrouk M, Boshoff WHP et al (2022a) Long-read genome sequencing of bread wheat facilitates disease resistance gene cloning. Nat Genet 54:227–231. https://doi.org/10.1038/s41588-022-01022-1
Athiyannan N, Aouini L, Wang Y, Krattinger SG (2022b) Unconventional R proteins in the botanical tribe Triticeae. Essays Biochem 66:561–569. https://doi.org/10.1042/EBC20210081
Aury JM, Engelen S, Istace B et al (2022) Long-read and chromosome-scale assembly of the hexaploid wheat genome achieves high resolution for research and breeding. Gigascience 11:1–18. https://doi.org/10.1093/gigascience/giac034
Balfourier F, Bouchet S, Robert S et al (2019) Worldwide phylogeography and history of wheat genetic div0065rsity. Sci Adv. https://doi.org/10.1126/sciadv.aav0536
Bates D, Mächler M, Bolker BM, Walker SC (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67:1–48. https://doi.org/10.18637/jss.v067.i01
Ben-David R, Xie W, Peleg Z et al (2010) Identification and mapping of PmG16, a powdery mildew resistance gene derived from wild emmer wheat. Theor Appl Genet 121:499–510. https://doi.org/10.1007/s00122-010-1326-5
Bhullar NK, Street K, Mackay M et al (2009) Unlocking wheat genetic resources for the molecular identification of previously undescribed functional alleles at the Pm3 resistance locus. Proc Natl Acad Sci U S A 106:9519–9524. https://doi.org/10.1073/pnas.0904152106
Bhullar NK, Mackay M, Keller B (2010a) Genetic diversity of the Pm3 powdery mildew resistance alleles in wheat gene bank accessions as assessed by molecular markers. Diversity (basel) 2:768–786. https://doi.org/10.3390/d2050768
Bhullar NK, Zhang Z, Wicker T, Keller B (2010b) Wheat gene bank accessions as a source of new alleles of the powdery mildew resistance gene Pm3: a large scale allele mining project. BMC Plant Biol 10:88. https://doi.org/10.1186/1471-2229-10-88
Blanco A, Gadaleta A, Cenci A et al (2008) Molecular mapping of the novel powdery mildew resistance gene Pm36 introgressed from Triticum turgidum var. dicoccoides in durum wheat. Theor Appl Genet 117:135–142. https://doi.org/10.1007/s00122-008-0760-0
Bonjean AP, Angus WJ (2001) The world wheat book: a history of wheat breeding. Lavoisier Publishing Inc.
Bourguet D, Guillemaud T (2016) The hidden and external costs of pesticide use. Springer, Cham, pp 35–120
Bourras S, McNally KE, Ben-David R et al (2015) Multiple avirulence loci and allele-specific effector recognition control the Pm3 race-specific resistance of wheat to powdery mildew. Plant Cell. https://doi.org/10.1105/tpc.15.00171
Bourras S, Kunz L, Xue M et al (2019) The AvrPm3-Pm3 effector-NLR interactions control both race-specific resistance and host-specificity of cereal mildews on wheat. Nat Commun 10:1–16. https://doi.org/10.1038/s41467-019-10274-1
Bradbury PJ, Zhang Z, Kroon DE et al (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635. https://doi.org/10.1093/bioinformatics/btm308
Brown JKM (2015) Durable resistance of crops to disease: a darwinian perspective. Annu Rev Phytopathol 53:513–539. https://doi.org/10.1146/annurev-phyto-102313-045914
Browning BL, Zhou Y, Browning SR (2018) A one-penny imputed genome from next-generation reference panels. Am J Hum Genet 103:338–348. https://doi.org/10.1016/j.ajhg.2018.07.015
Cakir C, Gillespie ME, Scofield SR (2010) Rapid determination of gene function by virus-induced gene silencing in wheat and barley. Crop Sci. https://doi.org/10.2135/cropsci2009.10.0567
CGIAR (2023) Genebank platform. https://www.genebanks.org/resources/crops/wheat/. Accessed
Chao K, Su W, Wu L et al (2019) Molecular mapping of a recessive powdery mildew resistance gene in wheat cultivar Tian Xuan 45 using bulked segregant analysis with polymorphic single nucleotide polymorphism relative ratio distribution. Phytopathology 109:828–838. https://doi.org/10.1094/PHYTO-03-18-0092-R
Chen PD, Qi LL, Zhou B et al (1995) Development and molecular cytogenetic analysis of wheat-haynaldia villosa 6VS/6AL translocation lines specifying resistance to powdery mildew. Theor Appl Genet 91:1125–1128. https://doi.org/10.1007/BF00223930
Chen F, Jia H, Zhang X et al (2019) Positional cloning of PmCH1357 reveals the origin and allelic variation of the Pm2 gene for powdery mildew resistance in wheat. Crop Journal 7:771–783. https://doi.org/10.1016/j.cj.2019.08.004
Chhuneja P, Yadav B, Stirnweis D et al (2015) Fine mapping of powdery mildew resistance genes PmTb7A.1 and PmTb7A.2 in Triticum boeoticum (Boiss.) using the shotgun sequence assembly of chromosome 7AL. Theor Appl Genet 128:2099–2111. https://doi.org/10.1007/s00122-015-2570-5
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Dodds PN, Rathjen JP (2010) Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet 11:539–548. https://doi.org/10.1038/nrg2812
Dong Z, Tian X, Ma C et al (2020) Physical mapping of Pm57, a powdery mildew resistance gene derived from Aegilops searsii. Int J Mol Sci 21:322. https://doi.org/10.3390/ijms21010322
Dormann CF, Schweiger O, Augenstein I et al (2007) Effects of landscape structure and land-use intensity on similarity of plant and animal communities. Glob Ecol Biogeogr 16:774–787. https://doi.org/10.1111/j.1466-8238.2007.00344.x
Dracatos PM, Lu J, Sánchez-Martín J, Wulff BBH (2023) Resistance that stacks up: engineering rust and mildew disease control in the cereal crops wheat and barley. Plant Biotechnol J 21:1938–1951. https://doi.org/10.1111/pbi.14106
Driscoll CJ, Bielig LM (1968) Mapping of the transec wheat-rye translocation. Can J Genet Cytol 10:421–425. https://doi.org/10.1139/g68-056
EU 2020 Farm to fork. https://ec.europa.eu/commission/presscorner/detail/en/qanda_22_3694. Accessed
Evenson RE, Gollin D (2003) Assessing the impact of the green revolution, 1960 to 2000. Science 300:758–762. https://doi.org/10.1126/science.1078710
FAO (2020) Food and Agriculture Organization of the United Nations, FAOSTAT statistics database, Food balances. https://www.fao.org/faostat/en/#data/FBS. Accessed
Flor HH (1971) Current status of the gene-for-gene concept. Annu Rev Phytopathol 9:275–296
Fu B, Chen Y, Li N et al (2013) pmX: a recessive powdery mildew resistance gene at the Pm4 locus identified in wheat landrace Xiaohongpi. Theor Appl Genet 126:913–921. https://doi.org/10.1007/s00122-012-2025-1
Galili T (2015) Dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 31:3718–3720. https://doi.org/10.1093/bioinformatics/btv428
Gao H, Zhu F, Jiang Y et al (2012) Genetic analysis and molecular mapping of a new powdery mildew resistant gene Pm46 in common wheat. Theor Appl Genet 125:967–973. https://doi.org/10.1007/s00122-012-1886-7
Glémin S, Scornavacca C, Dainat J et al (2019) Pervasive hybridizations in the history of wheat relatives. Sci Adv. https://doi.org/10.1126/sciadv.aav9188
Gloss AD, Vergnol A, Morton TC et al (2022) Genome-wide association mapping within a local Arabidopsis thaliana population more fully reveals the genetic architecture for defensive metabolite diversity. Philos Trans Royal Soc B: Biol Sci. https://doi.org/10.1098/rstb.2020.0512
Govta N, Polda I, Sela H et al (2022) Genome-wide association study in bread wheat identifies genomic regions associated with grain yield and quality under contrasting water availability. Int J Mol Sci. https://doi.org/10.3390/ijms231810575
Haas M, Schreiber M, Mascher M (2019) Domestication and crop evolution of wheat and barley: genes, genomics, and future directions. J Integr Plant Biol 61:204–225. https://doi.org/10.1111/jipb.12737
Han J, Zhang L-S, Li G-Q et al (2009) Molecular mapping of powdery mildew resistance gene MlWE18 in wheat originated from wild emmer (Triticum turgidum var. dicoccoides). Acta Agron Sin 35:1791–1797. https://doi.org/10.3724/sp.j.1006.2009.01791
Hao M, Liu M, Luo J et al (2018) Introgression of powdery mildew resistance gene pm56 on rye chromosome arm 6rs into wheat. Front Plant Sci 9:1040. https://doi.org/10.3389/fpls.2018.01040
He H, Ji Y, Zhu S et al (2017) Genetic, physical and comparative mapping of the powdery mildew resistance gene Pm21 originating from Dasypyrum villosum. Front Plant Sci 8:1914. https://doi.org/10.3389/fpls.2017.01914
He H, Zhu S, Zhao R et al (2018) Pm21, encoding a typical CC-NBS-LRR protein, confers broad-spectrum resistance to wheat powdery mildew disease. Mol Plant 11:879–882. https://doi.org/10.1016/j.molp.2018.03.004
Hewitt T, Müller MC, Molnár I et al (2021) A highly differentiated region of wheat chromosome 7AL encodes a Pm1a immune receptor that recognizes its corresponding AvrPm1a effector from Blumeria graminis. New Phytol 229:2812–2826. https://doi.org/10.1111/nph.17075
Hinterberger V, Douchkov D, Lück S et al (2022) Mining for new sources of resistance to powdery mildew in genetic resources of winter wheat. Front Plant Sci 13:366. https://doi.org/10.3389/fpls.2022.836723
Hsam SLK, Huang XQ, Zeller FJ (2001) Chromosomal location of genes for resistance to powdery mildew in common wheat (Triticum aestivum L. em Thell.) 6. Alleles at the Pm5 locus. Theor Appl Genet 102:127–133. https://doi.org/10.1007/s001220051627
Hu TZ, Li HJ, Liu ZJ et al (2008) Identification and molecular mapping of the powdery mildew resistance gene in wheat cultivar Yumai 66. Acta Agron Sin 34:545–550. https://doi.org/10.3724/sp.j.1006.2008.00545
Huang XQ, Hsam SLK, Zeller FJ (2002) Chromosomal location of genes for resistance to powdery mildew in Chinese wheat lines Jieyan 94-1-1 and Siyan 94-1-2. Hereditas 136:212–218. https://doi.org/10.1034/j.1601-5223.2002.t01-1-1360306.x
Iwgsc C, Appels R, Eversole K et al (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. https://doi.org/10.1126/science.aar7191
Ji X, Xie C, Ni Z et al (2008) Identification and genetic mapping of a powdery mildew resistance gene in wild emmer (Triticum dicoccoides) accession IW72 from Israel. Euphytica 159:385–390. https://doi.org/10.1007/s10681-007-9540-1
Jin Y, Xue F, Zhou Y et al (2020) Fine-mapping of the powdery mildew resistance gene mlxbd in the common wheat landrace Xiaobaidong. Plant Dis 104:1231–1238. https://doi.org/10.1094/PDIS-07-19-1347-RE
Kale SM, Schulthess AW, Padmarasu S et al (2022) A catalogue of resistance gene homologs and a chromosome-scale reference sequence support resistance gene mapping in winter wheat. Plant Biotechnol J 20:1730–1742. https://doi.org/10.1111/pbi.13843
Kaur N, Street K, Mackay M, et al (2008) Molecular approaches for characterization and use of natural disease resistance in wheat. In: European journal of plant pathology. Springer, pp 387–397
Keilwagen J, Lehnert H, Berner T et al (2022) Detecting major introgressions in wheat and their putative origins using coverage analysis. Sci Rep 12:1908. https://doi.org/10.1038/s41598-022-05865-w
Kim SH, Hong JK, Lee SC et al (2004) CAZFP1, Cys2/His2-type zinc-finger transcription factor gene functions as a pathogen-induced early-defense gene in Capsicum annuum. Plant Mol Biol 55:883–904. https://doi.org/10.1007/s11103-004-2151-5
Kloppe T, Whetten RB, Kim S et al (2023) Two pathogen loci determine Blumeria graminis f. sp. tritici virulence to wheat resistance gene Pm1a. New Phytologist. https://doi.org/10.1111/nph.18809
Knaus BJ, Grünwald NJ (2017) VCFR: a package to manipulate and visualize variant call format data in R. Mol Ecol Res 17(1):44–53
Koller T, Brunner S, Herren G et al (2019) Field grown transgenic Pm3e wheat lines show powdery mildew resistance and no fitness costs associated with high transgene expression. Transgenic Res 28:9–20. https://doi.org/10.1007/s11248-018-0099-5
Kolodziej MC, Singla J, Sánchez-Martín J et al (2021) A membrane-bound ankyrin repeat protein confers race-specific leaf rust disease resistance in wheat. Nat Commun 12:1–12. https://doi.org/10.1038/s41467-020-20777-x
Komáromi J, Jankovics T, Fábián A et al (2016) Powdery mildew resistance in wheat cultivar Mv Hombár is conferred by a new gene, PmHo. Phytopathology 106:1326–1334. https://doi.org/10.1094/PHYTO-03-16-0152-R
Kunz L, Sotiropoulos AG, Graf J et al (2023) The broad use of the Pm8 resistance gene in wheat resulted in hypermutation of the AvrPm8 gene in the powdery mildew pathogen. BMC Biol 21:29. https://doi.org/10.1186/s12915-023-01513-5
Li G, Cowger C, Wang X et al (2019) Characterization of Pm65, a new powdery mildew resistance gene on chromosome 2AL of a facultative wheat cultivar. Theor Appl Genet 132:2625–2632. https://doi.org/10.1007/s00122-019-03377-2
Li M, Dong L, Li B et al (2020a) A CNL protein in wild emmer wheat confers powdery mildew resistance. New Phytol 228:1027–1037. https://doi.org/10.1111/nph.16761
Li Y, Shi X, Hu J et al (2020b) Identification of a recessive gene PMQ conferring resistance to powdery mildew in wheat landrace Qingxinmai using BSR-seq analysis. Plant Dis 104:743–751. https://doi.org/10.1094/PDIS-08-19-1745-RE
Lillemo M, Asalf B, Singh RP et al (2008) The adult plant rust resistance loci Lr34/Yr18 and Lr46/Yr29 are important determinants of partial resistance to powdery mildew in bread wheat line Saar. Theor Appl Genet 116:1155–1166. https://doi.org/10.1007/s00122-008-0743-1
Liu J, He Z, Rasheed A et al (2017) Genome-wide association mapping of black point reaction in common wheat (Triticum aestivum L.). BMC Plant Biol 17:220. https://doi.org/10.1186/s12870-017-1167-3
Liu Y, Chen J, Yin C et al (2023) A high-resolution genotype–phenotype map identifies the TaSPL17 controlling grain number and size in wheat. Genome Biol 24:1–28. https://doi.org/10.1186/s13059-023-03044-2
Loh YT, Martin GB (1995) The Pto bacterial resistance gene and the Fen insecticide sensitivity gene encode functional protein kinases with serine/threonine specificity. Plant Physiol 108:1735–1739. https://doi.org/10.1104/pp.108.4.1735
Lopes MS, El-Basyoni I, Baenziger PS et al (2015) Exploiting genetic diversity from landraces in wheat breeding for adaptation to climate change. J Exp Bot 66:3477–3486. https://doi.org/10.1093/jxb/erv122
Luo PG, Luo HY, Chang ZJ et al (2009) Characterization and chromosomal location of Pm40 in common wheat: a new gene for resistance to powdery mildew derived from Elytrigia intermedium. Theor Appl Genet 118:1059–1064. https://doi.org/10.1007/s00122-009-0962-0
Lutz J, Limpert E, Bartos P, Zeller FJ (1992) Identification of powdery mildew resistance genes in common wheat (Triticum aestivum L.). I Czechoslovakian Cultivars Plant Breed 108:33–39. https://doi.org/10.1111/j.1439-0523.1992.tb00097.x
Lutz J, Hsam SLK, Limpert E, Zeller FJ (1995) Chromosomal location of powdery mildew resistance genes in triticum aestivum L. (common wheat). 2. genes pm2 and pm19 from Aegilops squarrosa L. Heredity (edinb) 74:152–156. https://doi.org/10.1038/hdy.1995.22
Mackay M, Street K (2004) Focused identification of germplasm strategy—FIGS. In: Proceedings of the 54th Australian cereal chemistry conference and the 11th wheat breeders’ assembly. cereal chemestry division, royal australian chemical institute (RACI), Melbourne, Victoria, Australia. pp 138–141
Manser B, Koller T, Praz CR et al (2021) Identification of specificity-defining amino acids of the wheat immune receptor Pm2 and powdery mildew effector AvrPm2. Plant J 106:993–1007. https://doi.org/10.1111/tpj.15214
Mantel N, Valand RS (1970) A technique of nonparametric multivariate analysis. Biometrics 26:547. https://doi.org/10.2307/2529108
Marees AT, de Kluiver H, Stringer S et al (2018) A tutorial on conducting genome-wide association studies: quality control and statistical analysis. Int J Methods Psychiatr Res. https://doi.org/10.1002/mpr.1608
Marone D, Russo MA, Mores A et al (2021) Importance of landraces in cereal breeding for stress tolerance. Plants 10:1267. https://doi.org/10.3390/plants10071267
Martin GB, Brommonschenkel SH, Chunwongse J et al (1993) Map-based cloning of a protein kinase gene conferring disease resistance in tomato. Science 262:1432–1436. https://doi.org/10.1126/science.7902614
Mascher M, Schreiber M, Scholz U et al (2019) Genebank genomics bridges the gap between the conservation of crop diversity and plant breeding. Nat Genet 51:1076–1081. https://doi.org/10.1038/s41588-019-0443-6
Maxwell JJ, Lyerly JH, Srnic G et al (2010) MlAB10: a Triticum turgidum Subsp. dicoccoides derived powdery mildew resistance gene identified in common wheat. Crop Sci 50:2261–2267. https://doi.org/10.2135/cropsci2010.04.0195
McDonald BA, Linde C (2002) Pathogen population genetics, evolutionary potential, and durable resistance. Annu Rev Phytopathol 40:349–379. https://doi.org/10.1146/annurev.phyto.40.120501.101443
McIntosh RA, Yamazaki Y, Dubcovsky J, et al (2013) Catalogue of gene symbols for wheat. In: Komugi wheat genetic resources database. https://wheat.pw.usda.gov/GG3/wgc. Accessed
Mohler V, Zeller FJ, Wenzel G, Hsam SLK (2005) Chromosomal location of genes for resistance to powdery mildew in common wheat (Triticum aestivum L. em Thell.). 9. Gene MlZec1 from the Triticum dicoccoides-derived wheat line Zecoi-1. Euphytica 142:161–167. https://doi.org/10.1007/s10681-005-1251-x
Mohler V, Bauer C, Schweizer G et al (2013) Pm50: a new powdery mildew resistance gene in common wheat derived from cultivated emmer. J Appl Genet 54:259–263. https://doi.org/10.1007/s13353-013-0158-9
Mu Y, Gong W, Qie Y et al (2022) Identification of the powdery mildew resistance gene in wheat breeding line Yannong 99102–06188 via bulked segregant exome capture sequencing. Front Plant Sci 13:3277. https://doi.org/10.3389/fpls.2022.1005627
Müller T, Schierscher-Viret B, Fossati D et al (2018) Unlocking the diversity of genebanks: whole-genome marker analysis of Swiss bread wheat and spelt. Theor Appl Genet 131:407–416. https://doi.org/10.1007/s00122-017-3010-5
Müller MC, Kunz L, Schudel S et al (2022) Ancient variation of the AvrPm17 gene in powdery mildew limits the effectiveness of the introgressed rye Pm17 resistance gene in wheat. Proc Natl Acad Sci U S A 119:e2108808119. https://doi.org/10.1073/pnas.2108808119
Müller K, Wickham H (2022) Tibble: simple data frames. https://CRAN.R-project.org/package=tibble
Mundt CC (2014) Durable resistance: a key to sustainable management of pathogens and pests. Infect Genet Evol 27:446–455. https://doi.org/10.1016/j.meegid.2014.01.011
Mundt CC (2018) Pyramiding for resistance durability: theory and practice. Phytopathology 108:792–802. https://doi.org/10.1094/PHYTO-12-17-0426-RVW
Myles S, Peiffer J, Brown PJ et al (2009) Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21:2194–2202. https://doi.org/10.1105/tpc.109.068437
Niu JS, Wang BQ, Wang YH et al (2008) Chromosome location and microsatellite markers linked to a powdery mildew resistance gene in wheat line Lankao 90(6). Plant Breeding 127:346–349. https://doi.org/10.1111/j.1439-0523.2007.01480.x
Oksanen J, Simpson Gavin L., Blanchet F. G., et al (2022) Vegan: community ecology package. https://CRAN.R-project.org/package=vegan. Accessed
Ouyang S, Zhang D, Han J et al (2014) Fine physical and genetic mapping of powdery mildew resistance gene MlIW172 originating from wild emmer (Triticum dicoccoides). PLoS ONE 9:e100160. https://doi.org/10.1371/journal.pone.0100160
Özkan H, Brandolini A, Schäfer-Pregl R, Salamini F (2002) AFLP analysis of a collection of tetraploid wheats indicates the origin of emmer and hard wheat domestication in Southeast Turkey. Mol Biol Evol 19:1797–1801. https://doi.org/10.1093/oxfordjournals.molbev.a004002
Paux E, Lafarge S, Balfourier F et al (2022) Breeding for economically and environmentally sustainable wheat varieties: an integrated approach from genomics to selection. Biology (basel) 11:149. https://doi.org/10.3390/biology11010149
Pedersen TL (2020) Patchwork: the composer of plots. https://CRAN.R-project.org/package=patchwork
Perugini LD, Murphy JP, Marshall D, Brown-Guedira G (2008) Pm37, a new broadly effective powdery mildew resistance gene from Triticum timopheevii. Theor Appl Genet 116:417–425. https://doi.org/10.1007/s00122-007-0679-x
Petersen S, Lyerly JH, Worthington ML et al (2015) Mapping of powdery mildew resistance gene Pm53 introgressed from Aegilops speltoides into soft red winter wheat. Theor Appl Genet 128:303–312. https://doi.org/10.1007/s00122-014-2430-8
Peusha H, Enno T, Priilinn O (2004) Chromosomal location of powdery mildew resistance genes and cytogenetic analysis of meiosis in common wheat cultivar Meri. Hereditas 132:29–34. https://doi.org/10.1111/j.1601-5223.2000.00029.x
Praz CR, Bourras S, Zeng F et al (2017) AvrPm2 encodes an RNase-like avirulence effector which is conserved in the two different specialized forms of wheat and rye powdery mildew fungus. New Phytol 213:1301–1314. https://doi.org/10.1111/nph.14372
Pugsley AT, Carter MV (1953) The resistance of twelve varieties of triticum vulgare to Erysiphe graminis tritici. Aust J Biol Sci 6:335–346. https://doi.org/10.1071/BI9530335
Purcell S, Neale B, Todd-Brown K et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. https://doi.org/10.1086/519795
Qie Y, Sheng Y, Xu H et al (2019) Identification of a new powdery mildew resistance gene pmDHT at or closely linked to the Pm5 locus in the Chinese wheat landrace dahongtou. Plant Dis 103:2645–2651. https://doi.org/10.1094/PDIS-02-19-0401-RE
Qiu YC, Zhou RH, Kong XY et al (2005) Microsatellite mapping of a Triticum urartu Tum. derived powdery mildew resistance gene transferred to common wheat (Triticum aestivum L.). Theor Appl Genet 111:1524–1531. https://doi.org/10.1007/s00122-005-0081-5
R Core Team R (2022) A language and environment for statistical computing. https://www.r-project.org/
Reif JC, Zhang P, Dreisigacker S et al (2005) Wheat genetic diversity trends during domestication and breeding. Theor Appl Genet 110:859–864. https://doi.org/10.1007/s00122-004-1881-8
Rimbert H, Darrier B, Navarro J et al (2018) High throughput SNP discovery and genotyping in hexaploid wheat. PLoS One. https://doi.org/10.1371/journal.pone.0186329
Robinson JT, Thorvaldsdóttir H, Winckler W et al (2011) Integrative genomics viewer. Nat Biotechnol 29:24–26. https://doi.org/10.1038/nbt.1754
Sánchez-Martín J, Keller B (2021) NLR immune receptors and diverse types of non-NLR proteins control race-specific resistance in Triticeae. Curr Opin Plant Biol 62:102053. https://doi.org/10.1016/j.pbi.2021.102053
Sánchez-Martín J, Steuernagel B, Ghosh S et al (2016) Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol 17:1–7. https://doi.org/10.1186/s13059-016-1082-1
Sánchez-Martín J, Widrig V, Herren G et al (2021) Wheat Pm4 resistance to powdery mildew is controlled by alternative splice variants encoding chimeric proteins. Nat Plants. https://doi.org/10.1038/s41477-021-00869-2
Sansaloni C, Franco J, Santos B et al (2020) Diversity analysis of 80,000 wheat accessions reveals consequences and opportunities of selection footprints. Nat Commun 11:4572. https://doi.org/10.1038/s41467-020-18404-w
Sato K, Abe F, Mascher M et al (2021) Chromosome-scale genome assembly of the transformation-amenable common wheat cultivar’ fielder. DNA Res. https://doi.org/10.1093/dnares/dsab008
Savary S, Willocquet L, Pethybridge SJ et al (2019) The global burden of pathogens and pests on major food crops. Nat Ecol Evol 3:430–439. https://doi.org/10.1038/s41559-018-0793-y
Schulthess AW, Kale SM, Liu F et al (2022) Genomics-informed prebreeding unlocks the diversity in genebanks for wheat improvement. Nat Genet 54:1544–1552. https://doi.org/10.1038/s41588-022-01189-7
Sharma R, Mahanty B, Mishra R, Joshi RK (2021) Genome wide identification and expression analysis of pepper C2H2 zinc finger transcription factors in response to anthracnose pathogen Colletotrichum truncatum. 3 Biotech. 11:1–18. https://doi.org/10.1007/s13205-020-02601-x
Shi AN, Leath S, Murphy JP (1998) A major gene for powdery mildew resistance transferred to common wheat from wild Einkorn wheat. Phytopathology 88:144–147. https://doi.org/10.1094/PHYTO.1998.88.2.144
Singh SP, Hurni S, Ruinelli M et al (2018) Evolutionary divergence of the rye Pm17 and Pm8 resistance genes reveals ancient diversity. Plant Mol Biol 98:249–260. https://doi.org/10.1007/s11103-018-0780-3
Singh BK, Delgado-Baquerizo M, Egidi E et al (2023) Climate change impacts on plant pathogens, food security and paths forward. Nat Rev Microbiol 21:640–656
Singrün C, Hsam SLK, Zeller FJ et al (2004) Localization of a novel recessive powdery mildew resistance gene from common wheat line RD30 in the terminal region of chromosome 7AL. Theor Appl Genet 109:210–214. https://doi.org/10.1007/s00122-004-1619-7
Sotiropoulos AG, Arango-Isaza E, Ban T et al (2022) Global genomic analyses of wheat powdery mildew reveal association of pathogen spread with historical human migration and trade. Nat Commun 13:16. https://doi.org/10.1038/s41467-022-31975-0
Stein N, Herren G, Keller B (2001) A new DNA extraction method for high-throughput marker analysis in a large-genome species such as Triticum aestivum. Plant Breeding 120:354–356. https://doi.org/10.1046/j.1439-0523.2001.00615.x
Sun M, Liu Q, Han Y et al (2022) PmSN15218: a potential new powdery mildew resistance gene on wheat chromosome 2AL. Front Plant Sci. https://doi.org/10.3389/fpls.2022.931778
Tan C, Li G, Cowger C et al (2018) Characterization of Pm59, a novel powdery mildew resistance gene in Afghanistan wheat landrace PI 181356. Theor Appl Genet 131:1145–1152. https://doi.org/10.1007/s00122-018-3067-9
Tan C, Li G, Cowger C et al (2019) Characterization of Pm63, a powdery mildew resistance gene in Iranian landrace PI 628024. Theor Appl Genet 132:1137–1144. https://doi.org/10.1007/s00122-018-3265-5
Tanksley SD, McCouch SR (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277:1063–1066. https://doi.org/10.1126/science.277.5329.1063
Tettelin H, Masignani V, Cieslewicz MJ et al (2005) Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc Natl Acad Sci U S A 102:13950–13955. https://doi.org/10.1073/pnas.0506758102
Tian Z-D, Zhang Y, Liu J, Xie C-H (2010) Novel potato C2H2-type zinc finger protein gene, StZFP1, which responds to biotic and abiotic stress, plays a role in salt tolerance. Plant Biol 12:689–697. https://doi.org/10.1111/j.1438-8677.2009.00276.x
Tosa Y, Tsujimoto H, Ogura H (1987) A gene involved in the resistance of wheat to wheatgrass powdery mildew fungus. Genome 29:850–852. https://doi.org/10.1139/g87-145
Uehara Y, Takahashi Y, Berberich T et al (2005) Tobacco ZFT1, a transcriptional repressor with a Cys2/His 2 type zinc finger motif that functions in spermine-signaling pathway. Plant Mol Biol 59:435–448. https://doi.org/10.1007/s11103-005-0272-0
Uffelmann E, Huang QQ, Munung NS et al (2021) Genome-wide association studies. Nat Rev Methods Primers 1:1–21. https://doi.org/10.1038/s43586-021-00056-9
Van Den Burg HA, Tsitsigiannis DI, Rowland O et al (2008) The F-box protein ACRE189/ACIF1 regulates cell death and defense responses activated during pathogen recognition in tobacco and tomato. Plant Cell 20:697–719. https://doi.org/10.1105/tpc.107.056978
Villa TCC, Maxted N, Scholten M, Ford-Lloyd B (2005) Defining and identifying crop landraces. Plant Genet Resour 3:373–384. https://doi.org/10.1079/pgr200591
Vleeshouwers VGAA, Oliver RP (2014) Effectors as tools in disease resistance breeding against biotrophic, hemibiotrophic, and necrotrophic plant pathogens. Mol Plant Microbe Interact 27:196–206. https://doi.org/10.1094/MPMI-10-13-0313-IA
de Vries A, Ripley BD (2022) Create dendrograms and tree diagrams using “ggplot2”
Walkowiak S, Gao L, Monat C et al (2020) Multiple wheat genomes reveal global variation in modern breeding. Nature 588:277–283. https://doi.org/10.1038/s41586-020-2961-x
Wan W, Xiao J, Li M et al (2020) Fine mapping of wheat powdery mildew resistance gene Pm6 using 2B/2G homoeologous recombinants induced by the ph1b mutant. Theor Appl Genet 133:1265–1275. https://doi.org/10.1007/s00122-020-03546-8
Wang J, Zhang Z (2021) GAPIT version 3: boosting power and accuracy for genomic association and prediction. Genom Proteom Bioinform 19:629–640. https://doi.org/10.1016/j.gpb.2021.08.005
Wang W, He H, Gao H et al (2021) Characterization of the powdery mildew resistance gene in wheat breeding line KN0816 and its evaluation in marker-assisted selection. Plant Dis 105:4042–4050. https://doi.org/10.1094/PDIS-05-21-0896-RE
Wang J, Li Y, Xu F et al (2022) Candidate powdery mildew resistance gene in wheat landrace cultivar Hongyoumai discovered using SLAF and BSR-seq. BMC Plant Biol 22:83. https://doi.org/10.1186/s12870-022-03448-5
Wei T, Simko V (2021) R package “corrplot”: visualization of a correlation matrix. Version 0.84. https://github.com/taiyun/corrplot
Weigel RR, Pfitzner UM, Gatz C (2005) Interaction of NIMIN1 with NPR1 modulates PR gene expression in Arabidopsis. Plant Cell 17:1279–1291. https://doi.org/10.1105/tpc.104.027441
Wenger AM, Peluso P, Rowell WJ et al (2019) Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol 37:1155–1162. https://doi.org/10.1038/s41587-019-0217-9
White B, Lux T, Rusholme-Pilcher RL, et al. (2024) De novo annotation of the wheat pan-genome reveals complexity and diversity within the hexaploid wheat pan-transcriptome. bioRxiv. https://doi.org/10.1101/2024.01.09.574802
Wickham H (2016) ggplot2: elegant graphics for data analysis. Springer-Verlag, New York
Wickham H, François R, Henry L, et al (2023) dplyr: a grammar of data manipulation. R package version 0.4. https://CRAN.R-project.org/paclage=dplyr
Worthington M, Lyerly J, Petersen S et al (2014) MlUM15: an Aegilops neglecta -derived powdery mildew resistance gene in common wheat. Crop Sci 54:1397–1406. https://doi.org/10.2135/cropsci2013.09.0634
Wu P, Hu J, Zou J et al (2019) Fine mapping of the wheat powdery mildew resistance gene Pm52 using comparative genomics analysis and the Chinese spring reference genomic sequence. Theor Appl Genet 132:1451–1461. https://doi.org/10.1007/s00122-019-03291-7
Wu L, Zhu T, He H et al (2022) Genetic dissection of the powdery mildew resistance in wheat breeding line LS5082 using BSR-Seq. Crop J 10:1120–1130. https://doi.org/10.1016/j.cj.2021.12.008
Xiao M, Song F, Jiao J et al (2013) Identification of the gene Pm47 on chromosome 7BS conferring resistance to powdery mildew in the Chinese wheat landrace Hongyanglazi. Theor Appl Genet 126:1397–1403. https://doi.org/10.1007/s00122-013-2060-6
Xie JZ, Wang L, li, Wang Y, et al (2017) Fine mapping of powdery mildew resistance gene PmTm4 in wheat using comparative genomics. J Integr Agric 16:540–550. https://doi.org/10.1016/S2095-3119(16)61377-1
Xing L, Hu P, Liu J et al (2018) Pm21 from Haynaldia villosa encodes a CC-NBS-LRR protein conferring powdery mildew resistance in wheat. Mol Plant 11:874–878. https://doi.org/10.1016/j.molp.2018.02.013
Xu H, Yao G, Xiong L et al (2008) Identification and mapping of pm2026: a recessive powdery mildew resistance gene in an einkorn (Triticum monococcum L.) accession. Theor Appl Genet 117:471–477. https://doi.org/10.1007/s00122-008-0791-6
Xu WG, Li CX, Hu L et al (2010) Molecular mapping of powdery mildew resistance gene PmHNK in winter wheat (Triticum aestivum L.) cultivar Zhoumai 22. Mol Breeding 26:31–38. https://doi.org/10.1007/s11032-009-9374-8
Xu W, Li C, Hu L et al (2011) Identification and molecular mapping of PmHNK54: a novel powdery mildew resistance gene in common wheat. Plant Breeding 130:603–607. https://doi.org/10.1111/j.1439-0523.2011.01882.x
Xu X, Li Q, Ma Z et al (2018) Molecular mapping of powdery mildew resistance gene PmSGD in Chinese wheat landrace Shangeda using RNA-seq with bulk segregant analysis. Mol Breeding 38:1–12. https://doi.org/10.1007/s11032-018-0783-4
Xu X, Liu W, Liu Z et al (2020) Mapping powdery mildew resistance gene pmYBL on chromosome 7B of Chinese wheat (Triticum aestivum L.) landrace Youbailan. Plant Dis 104:2411–2417. https://doi.org/10.1094/PDIS-01-20-0118-RE
Xue F, Ji W, Wang C et al (2012) High-density mapping and marker development for the powdery mildew resistance gene PmAS846 derived from wild emmer wheat (Triticum turgidum var. dicoccoides). Theor Appl Genet 124:1549–1560. https://doi.org/10.1007/s00122-012-1809-7
Yahiaoui N, Srichumpa P, Dudler R, Keller B (2004) Genome analysis at different ploidy levels allows cloning of the powdery mildew resistance gene Pm3b from hexaploid wheat. Plant J 37:528–538. https://doi.org/10.1046/j.1365-313X.2003.01977.x
Yao G, Zhang J, Yang L et al (2007) Genetic mapping of two powdery mildew resistance genes in einkorn (Triticum monococcum L.) accessions. Theor Appl Genet 114:351–358. https://doi.org/10.1007/s00122-006-0438-4
Yin JL, Fang ZW, Sun C et al (2018) Rapid identification of a stripe rust resistant gene in a space-induced wheat mutant using specific locus amplified fragment (SLAF) sequencing. Sci Rep 8:1–9. https://doi.org/10.1038/s41598-018-21489-5
Yin J, Wang L, Zhao J et al (2020) Genome-wide characterization of the C2H2 zinc-finger genes in Cucumis sativus and functional analyses of four CsZFPs in response to stresses. BMC Plant Biol 20:359. https://doi.org/10.1186/s12870-020-02575-1
Yu X, Ren S, Zhao L et al (2018) Molecular mapping of a novel wheat powdery mildew resistance gene Ml92145E8-9 and its application in wheat breeding by marker-assisted selection. Crop J 6:621–627. https://doi.org/10.1016/j.cj.2018.04.004
Zeller FJ, Kong L, Hartl L et al (2002) Chromosomal location of genes for resistance to powdery mildew in common wheat (Triticum aestivum L. em Thell.) 7. gene Pm29 in line Pova. Euphytica 123:187–194. https://doi.org/10.1023/A:1014944619304
Zeven AC (1998) Landraces: a review of definitions and classifications. Euphytica 104:127–139. https://doi.org/10.1023/A:1018683119237
Zhan H, Li G, Zhang X et al (2014) Chromosomal location and comparative genomics analysis of powdery mildew resistance gene Pm51 in a putative wheat-Thinopyrum ponticum introgression line. PLoS ONE 9:e113455. https://doi.org/10.1371/journal.pone.0113455
Zhang H, Guan H, Li J et al (2010) Genetic and comparative genomics mapping reveals that a powdery mildew resistance gene Ml3D232 originating from wild emmer co-segregates with an NBS-LRR analog in common wheat (Triticum aestivum L.). Theor Appl Genet 121:1613–1621. https://doi.org/10.1007/s00122-010-1414-6
Zhang R, Sun B, Chen J et al (2016) Pm55, a developmental-stage and tissue-specific powdery mildew resistance gene introgressed from Dasypyrum villosum into common wheat. Theor Appl Genet 129:1975–1984. https://doi.org/10.1007/s00122-016-2753-8
Zhang R, Fan Y, Kong L et al (2018) Pm62, an adult-plant powdery mildew resistance gene introgressed from Dasypyrum villosum chromosome arm 2VL into wheat. Theor Appl Genet 131:2613–2620. https://doi.org/10.1007/s00122-018-3176-5
Zhang D, Zhu K, Dong L et al (2019) Wheat powdery mildew resistance gene Pm64 derived from wild emmer (Triticum turgidum var. dicoccoides) is tightly linked in repulsion with stripe rust resistance gene Yr5. Crop Journal 7:761–770. https://doi.org/10.1016/j.cj.2019.03.003
Zhang W, Yu Z, Wang D et al (2023) Characterization and identification of the powdery mildew resistance gene in wheat breeding line ShiCG15–009. BMC Plant Biol 23:113. https://doi.org/10.1186/s12870-023-04132-y
Zheng X, Levine D, Shen J et al (2012) A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28:3326–3328. https://doi.org/10.1093/bioinformatics/bts606
Zhou X, Stephens M (2012) Genome-wide efficient mixed-model analysis for association studies. Nat Genet 44:821–824. https://doi.org/10.1038/ng.2310
Zhu Z, Zhou R, Kong X et al (2005) Microsatellite markers linked to 2 powdery mildew resistance genes introgressed from Triticum carthlicum accession PS5 into common wheat. Genome 48:585–590. https://doi.org/10.1139/G05-016
Zhu Z, Zhou R, Kong X et al (2006) Microsatellite marker identification of a Triticum aestivum-Aegilops umbellulata substitution line with powdery mildew resistance. Euphytica 150:149–153. https://doi.org/10.1007/s10681-006-9103-x
Zou S, Wang H, Li Y et al (2018) The NB-LRR gene Pm60 confers powdery mildew resistance in wheat. New Phytol 218:298–309. https://doi.org/10.1111/nph.14964
Acknowledgements
We would like to thank Nikolaos Minadakis, Benjamin Jaegle, Zoe Bernasconi, Alexandros Sotiropoulos, Thomas Wicker, Bruno Studer and Anne Roulin for their input and guidance in acquiring necessary techniques for this study. We would further like to thank Sarah Furrler, Garp Linder and Pietro Sassi for their contributions to this project in the laboratory. This project was financially supported by the University of Zurich, Swiss National Science Foundation grants 310030_204165 and 310030B_182833 to BK. JSM is recipient of the grants “Ramon y Cajal” Fellowship RYC2021-032699-I, funded by MCIN/AEI/https://doi.org/10.13039/501100011033 and by the “European Union NextGenerationEU/PRTR” and PID2022-142651OA-I00, funded by MCIN/AEI/https://doi.org/10.13039/501100011033 and “European Union NextGenerationEU/PRTR”.
Funding
Open access funding provided by University of Zurich. This study was funded by grants from the Swiss National Science Foundation (310030_204165 and 310030B_182833) and the Spanish Ministry of Science, Innovation and Universities (RYC2021-032699-I and PID2022-142651OA-I00).
Author information
Authors and Affiliations
Contributions
JSM and BK conceived the project. EJ and RL performed seed propagation. RL, VW and JSM performed infection tests. RL performed DNA extraction, PCR screens and allele detection. EP performed SNP array genotyping. RL and MH performed bioinformatics analysis. RL, JSM and BK analyzed the data and wrote the manuscript. All authors revised the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
Beat Keller is a member of the editorial board of TAG. The authors declare no other competing interests.
Additional information
Communicated by Jochen Reif.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file2 (XLSX 785 KB)
Table S1. Information on accessions of the LandracePlus panel. Alleles with asterisks are non-functional. Groups are based on the hierarchical clustering of the LandracePLUS panel, including high-quality sequenced genomes.
Table S2. Differential lines used for determining powdery mildew isolate virulence spectra and the respective Pm genes they contain. NILs had been backcrossed multiple times with susceptible accessions “Federation” or “Chancellor”, depicted by /x*Accession, where x is the number of backcrosses to the designated accession (McIntosh et al. 2013).
Table S3. Sampling information of the ten Bgt isolates.
Table S4. Molecularly known Avr and Svr haplotypes of the ten powdery mildew isolates used for phenotyping. H0 = published avirulent haplotype, H1 = published virulent haplotype, H2 = unpublished haplotype. Based on the phenotype of the differential lines containing cognate Pm genes, we propose that H2 of AvrPm1a and AvrPm3a/f are virulent, while H2 of AvrPm3d is proposed to be avirulent. H2 of AvrPm17 is likely virulent due to the resemblance of H1, with just one additional amino acid change. The resistant phenotype of the differential line containing Pm17 is suggested to be due to an additional Pm gene in Amigo. Amino acid (AA) changes are given based on H0 and depicted with original AA, position, new AA.
Table S5. Heritability of phenotypes for all ten powdery mildew isolates based on a linear mixed model.
Table S6. Correlation of genetic relatedness and phenotypes for the ten Bgt isolates.
Table S7. Gene candidates and annotations in Chinese Spring for regions associated to powdery mildew resistance. Highlighted genes are the best candidates based on their annotation.
Table S8. List of all significantly associated SNPs, their position, MAF, effect size beta and p-values for the LandracePLUS panel excluding subsets.
Table S9. List of all significantly associated SNPs, their position, MAF, effect size beta and p-values for the subsets of the LandracePLUS panel
Table S10. Raw phenotype data of the LandracePLUS panel infected with ten powdery mildew isolates. Plates refer to the individual petri dish the replicate was in and Rounds to a batch that was infected at the same time.
122_2024_4582_MOESM3_ESM.xlsx
Supplementary file3 (XLSX 303 KB) Alleles in the 20 most modern cultivars of the LandracePLUS panel for each potentially novel resistance-associated region. Resistance-associated alleles and accessions with more than 50% of these alleles and a resistance phenotype toward the peak-producing isolate are highlighted in bold. In addition, an overview of SNPs for all detected resistance-associated loci and the 755 wheat accessions with passport data and disease reaction is provided.
122_2024_4582_MOESM4_ESM.xlsx
Supplementary file4 (XLSX 282) Candidate genes in chromosome-scale-assembled wheat varieties ArinaLrFor, Fielder, Jagger, Julius, Kariega, Lancer, Landmark, Mace, SY Mattis, Norin61, Renan and Stanley based on syntenic analysis to regions associated to powdery mildew resistance in CS.
122_2024_4582_MOESM5_ESM.zip
Supplementary file Data S1. Raw genotyping data of the LandracePLUS panel with the TaBW35K SNP array. Empty cells are labeled “vide”, and wheat accession IDs are extended by “_cell identifier”.
122_2024_4582_MOESM6_ESM.txt
Supplementary file Data S2. Sample statistics of the raw genotyping data of the LandracePLUS panel. Total numbers are labeled “nb” and percentage “pc”.
122_2024_4582_MOESM7_ESM.txt
Supplementary file Data S3. Marker statistics of the raw genotyping data of the LandracePLUS panel. Total numbers are labeled “nb” and percentage “pc”.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leber, R., Heuberger, M., Widrig, V. et al. A diverse panel of 755 bread wheat accessions harbors untapped genetic diversity in landraces and reveals novel genetic regions conferring powdery mildew resistance. Theor Appl Genet 137, 88 (2024). https://doi.org/10.1007/s00122-024-04582-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-024-04582-4