
Abstract
Immunotherapies such as immune checkpoint blockade (ICB) and adoptive cell therapy (ACT) have revolutionized cancer treatment, especially in patients whose disease was otherwise considered incurable. However, primary and secondary resistance to single agent immunotherapy often results in treatment failure, and only a minority of patients experience long-term benefits. This review article will discuss the relationship between cancer immune response and mechanisms of resistance to immunotherapy. It will also provide a comprehensive review on the latest clinical status of combination therapies (e.g., immunotherapy with chemotherapy, radiation therapy and targeted therapy), and discuss combination therapies approved by the US Food and Drug Administration. It will provide an overview of therapies targeting cytokines and other soluble immunoregulatory factors, ACT, virotherapy, innate immune modifiers and cancer vaccines, as well as combination therapies that exploit alternative immune targets and other therapeutic modalities. Finally, this review will include the stimulating insights from the 2020 China Immuno-Oncology Workshop co-organized by the Chinese American Hematologist and Oncologist Network (CAHON), the China National Medical Product Administration (NMPA) and Tsinghua University School of Medicine.
Similar content being viewed by others


Introduction
Recent major breakthroughs in cancer immunotherapy lie in the identification of immune checkpoints that cancer cells hijack to suppress anti-cancer immunity. With the approval of immune checkpoint blockers (ICBs) across cancer types, immunotherapy has revolutionized cancer treatment, especially with metastatic cancers where some patients, previously considered to be incurable, can enjoy long-term remission and survival. So far, the US Food and Drug Administration (FDA) approved ICBs include antibodies targeting programmed cell death 1 (PD1), PD1 ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4).
With FDA approvals of multiple ICBs across cancer types, new applications and approvals of cancer immunotherapy have stagnated. More recently, adoptive cell therapy (ACT), such as chimeric antigen receptor-engineered T (CAR-T) cells, has emerged as an effective therapy in hematological malignancies. While ICBs restore suppressed pre-existing anti-cancer immunity, CAR-T cells bypass antigen presentation, T cell priming and activation, thus directly attacking cancer cells. After administration, ACT is still governed by the downstream resistance mechanisms, especially those at the tumor microenvironment (TME). In addition to ICBs and ACT, novel strategies of immunotherapy are being explored to further improve the treatment efficacy and/or decrease immune-mediated toxicities.
Even though ACT is, in general, associated with high response rates, many patients eventually develop secondary resistance. On the other hand, the response rate of ICB monotherapies is usually around the 20% range across solid tumors. One strategy to improve cancer immunotherapy is to develop biomarkers, such as PD-L1, that can be used to select potential responders and/or exclude potential non-responders. Another strategy is to combine agents with different mechanisms of action and target multiple resistant mechanisms. So far, several combination therapies have already been approved by the FDA across different cancer types (Table 1 and Fig. 1). This review article will review emerging combination therapies, some of which were updated at the 2020 China Immuno-Oncology (IO) Workshop co-organized by the Chinese American Hematologist and Oncologist Network (CAHON), the China National Medical Product Administration (NMPA) and Tsinghua University [1, 2].

Timeline of the FDA approvals of combination therapy
Cancer-immunity cycle
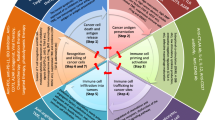
In 2013, Chen and Mellman (2013) used the concept of “the Cancer-Immunity Cycle,” which dissects the anti-cancer immune response process similar to the way the body mounts response toward any foreign antigens (Table 2 and Fig. 2) [3]. The cycle starts with cross-presentation of cancer-associated antigens from cancer cells to the major histocompatibility complex (MHC) molecules on the antigen presenting cells (APCs). Cancer antigens encompass cancer neoantigens from genomic alterations (mutations, translocations, readthrough and frame shifts), cancer associated proteins normally expressed at immune privileged sites, viral proteins and others (Step 1). APCs, upon capturing of cancer antigens, migrate to secondary lymphoid organs (Step 2). These APCs prime and activate naïve T cells via MHC-antigen-T cell receptor (TCR) interaction, along with a hierarchy of costimulatory signals, such as the CD28/B7-1/2-mediated signaling (Step 3). Activated immune cells then enter the circulation system (step 4), infiltrate into the tumor microenvironment (Step 5), recognize tumor cells through the interaction of the TCR and its cognate antigen presented on MHC of tumor cells (Step 6) and kill their target cancer cells (Step 7). After killing the targeted cancer cells, release of more tumor antigens further fuels the anti-cancer immunity cycle.

The cancer-immunity cycle, resistant mechanisms and potential solutions
Resistant mechanisms along the cancer-immunity cycle
Cancer cells have been found to have intrinsic mechanisms bypassing every possible step along the cancer-immunity cycle to evade anti-cancer immunity (Table 2 and Fig. 2). At the initiation of the anti-cancer immune response, some cancers with low tumor mutation burden or low immune cell infiltration (such as in prostate cancer) may not elicit sufficient immune responses. Loss of MHC expression, loss or mutation of β2-microglobulin and mutations within the TCR binding domain of MHC have all been associated with escape from anti-cancer immunity [4,5,6,7].
CTLA4 is the first target of ICBs approved by the FDA [8]. In addition to CTLA4, several other negative regulators such as T-cell immunoglobulin, mucin domain-3 protein (TIM-3), lymphocyte-activation gene 3 (LAG-3), T-cell immunoreceptor tyrosine-based inhibition motif domain (TIGIT) and V-domain immunoglobulin-containing suppressor of T-cell activation (VISTA) [9,10,11,12,13], have been identified and are currently being tested in clinical trials to determine their potential as targets for cancer immunotherapy. Other than negative regulators, suboptimal co-stimulation molecule expression, inefficient cytokine production and heightened infiltration of immunosuppressive immune cells have all been found to contribute to weakened anti-cancer immunity.
After immune cell priming and activation, any defects affecting immune cell trafficking, migration and infiltration into the tumor microenvironment can invalidate anti-cancer immunity. Vascular endothelial growth factor (VEGF) plays important roles in angiogenesis as well as multiple facets of anti-cancer immunity. It decreases trafficking and extravasation of cytotoxic T cells, promotes infiltration of Treg cells into the tumor bed [14] and enhances the expression of PD-1 and other inhibitory checkpoints involved in CD8+T cell exhaustion [15]. In mouse models, VEGF also impedes the commitment and progression of lymphoid progenitors to the T-cell lineage [16].
Cytokines within the TME not only affect immune cell migration and recruitment to the tumor site, but also modulate immune cell activities. Some cytokines, such as Chemokine (C-X-C motif) ligand 9 (CXCL9), CXCL10 and CXCL11, elicit chemotactic function and attract cytotoxic T cells while other cytokines, as seen with CCL5, CCL17, CCL22 and CXCL8, attract myeloid-derived suppressor cells (MDSCs) and Treg cells contributing to the immunosuppressive TME [17,18,19]. In addition to cytokines, transforming growth factor beta (TGF-β) is a multipotent growth factor that affects cell growth and differentiation, apoptosis and immunosuppression. It is present in high concentrations in the TME because of production by cancer, stromal and immune cells. In general, it inhibits anti-cancer immunity through inhibiting the function of effector immune cells and promoting suppressive cells [20]. Both cytokines and TGF-β have already been experimentally targeted for cancer immunotherapy.
Once immune cells enter the TME, numerous mechanisms have been identified to elicit resistance to anti-cancer immunity, including cancer cell intrinsic factors, immune cells and the immunosuppressive milieu. As discussed above, through immunoediting and selection pressure from anti-cancer immunity, cancer cells with loss or decrease of antigen presentation can survive anti-cancer immunity and proliferate to become resistant cancers. Upregulation of immunosuppressive signaling pathways, such as PD-1, PD-L1, LAG-3 and TIM-3, infiltration of immunosuppressive cells, such as Treg cells, MDSC, M2 macrophages, a hypoxic and acidic environment, or metabolic alterations in the tumor microenvironment, have all been found to negatively contribute to anti-cancer immunity.
Currently, the FDA-approved ICBs target the immune cell priming and activation (anti-CTLA4 antibody) or the final negative regulation of T effector cells (anti PD-1 and anti-PD-L1 antibodies). As these inhibitors only affect one to two steps of the anti-cancer immunity pathway, it is not surprising that only a minority (around 20%) of patients achieve cancer response with single agents. Slightly higher response rates have been observed with anti-CTLA4 and anti-PD1/PD-L1 combination treatments, at the cost of higher immune-mediated toxicities. Combination therapies are currently being extensively explored to target multiple defects along the immunity cycle and cancer intrinsic alterations and improve the anti-cancer efficacy, which will be covered in the following sections.
Combinations of chemotherapy and immunotherapy
Most chemotherapeutic agents were developed through its direct cytotoxic effects without consideration of the effects on immune system. The interplay between chemotherapy and immunotherapy has been demonstrated in mouse models where mice with intact immune systems had significantly improved tumor responses to anthracyclines [21]. To date, multiple studies have demonstrated the contribution of cytotoxic chemotherapy to anti-cancer immunity, leading to several FDA-approved combination therapies with immunotherapy (Table 2) [22].
Mechanisms of action
Debulking of tumors
One of the major benefits achieved by cytotoxic chemotherapy is tumor debulking. Tumor cells are the major contributor to immunosuppressive TME. Hence, reduction of cancer cell mass decreases production of immunosuppressive factors. Furthermore, reduction of tumor cell mass decreases the volume of cancer cells needed to be eliminated by immune cells. This can have dramatic consequences, especially in those tumors with limited immune cell infiltration at TME.
Immunogenic cell death (ICD)
ICD is a form of regulated cell death that is amenable to activating the adaptive immune response in immunocompetent hosts [23]. Numerous studies have shown that cytotoxic chemotherapy induces ICD and potentiates immunotherapy [24]. Insult of cancer cells by cytotoxic chemotherapy leads to release and relocation of damage-associated molecular patterns (DAMPs) that increase the adjuvanticity of cancer cells [25]. Release of intracellular molecules, such as ATP, enhances the recruitment of APCs; cytoplasmic annexin A1 released from cancer cells interacts with formyl peptide receptor 1 to promote interaction of dendritic cells and damaged cancer cells; exposure of endoplasmic reticulum chaperone proteins, such as heat shock protein 70 (HSP70), HSP90 and calreticulin, promotes the phagocytosis of stressed cancer cells by dendritic cells; cytosolic DNA and RNA stimulate the secretion of type I interferon and other proinflammatory cytokines through the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway, toll-like receptor 3 (TLR3) and TLR9; Type I interferon and other molecules released by stressed cancer cells, such as high mobility group box 1 (HMGB1), promote dendritic cell maturation and antigen presentation to T cells; and C–C motif chemokine ligand 2 (CCL2), C-X-C motif chemokine ligand 1 (CXCL1) and CXCL10 facilitate T-cell recruitment.
Increase in antigenicity of cancer cells
While ample evidence exists that chemotherapy increases the adjuvanticity of cancer cells through ICD, less is known about enhancement of antigenicity by chemotherapy. Many of the commonly used cytotoxic agents, such as anthracyclines, cyclophosphamide, platinum and taxanes, target cell cycle progression in proliferating cells and induce apoptosis. After tumor cell death, antigen-presenting cells engulf dying tumor cells and present tumor neoantigens to immune cells.
In addition, several other studies show that cytotoxic agents upregulate antigen-presenting machinery. Gemcitabine can significantly upregulate the expression of human leukocyte antigen (HLA)-A, B and C through increased expression of β2-microglobulin and alter the peptide antigen repertoire expressed on HLA class I [26]. A similar phenomenon is also observed with topotecan which upregulates HLA class I expression through activation of NF-κB/Interferon-β/MHC-I signaling axis [27]. As discussed above, ICD and stimulation of the cGAS/STING pathway induces type I interferon production which can upregulate HLA class I molecule expression and antigen presentation.
Depletion of immunosuppressive cells
Several subpopulations of immune cells are known to suppress anti-cancer immunity. Cytotoxic chemotherapy, such as platinum, cyclophosphamide, gemcitabine and 5-fluorouracil, can clearly reduce MDSCs in both humans and mice [28,29,30,31]. Trabectedin selectively depletes monocytes/macrophages through activation of caspase-8-dependent apoptosis [32]. Human Treg cells lack the expression of cyclophosphamide-excreting transporter ABCB1 and are more sensitive to cyclophosphamide treatment than other immune cells [33]. Furthermore, chemotherapy alters the TME and favors the differentiation of immune cells supporting anti-cancer immunity. For example, cyclophosphamide and doxorubicin favor the M1 differentiation of tumor-associated macrophages [34].
Modulation of gene expression
In addition to the cytotoxic chemotherapy, another major class of small molecular drugs are epigenetic modulators. Epigenetic modulation, such as DNA methylation, histone modification, chromatin remodeling and the readout of these modifications, has tremendous impact during oncogenesis and is a critical event in some cancers, such as loss of tumor suppressor genes from DNA methylation. Hence, epigenetic modulators constitute an ever-expanding class of anti-neoplasm agents.
In addition to direct induction of ICD and stimulation of antitumor immunity, as seen with histone deacetylase (HDAC) inhibitors vorinostat and panobinostat [35], another major contributing mechanism to the synergy between epigenetic modulators and immunotherapy is through gene expression modification. Both HDAC and DNA methyltransferase (DNMT) inhibitors have been shown to upregulate the antigen processing and presentation machinery. Both HLA class molecules [36, 37] and tumor-associated antigens [38] have been found to be upregulated by epigenetic modulators. Epigenetic modulators also have direct impacts on the immune system to potentiate anti-cancer immunity. They can upregulate co-stimulatory molecules, such as CD80, CD86 and ICAM-1, and immune checkpoints CTLA4, PD1 and PD-L1 [39]. Furthermore, cytokines can also be induced, and response to immunotherapy can be augmented by epigenetic modulators [40]. The innate immune system can be modified by epigenetic modulators as well. Activating receptor NKG2D on the surface of NK cells and stressing-inducing ligand MICA and MICB on tumor cells can all be induced by HDAC inhibitors to increase NK cell killing of tumor cells [41, 42].
Potentiation and restoration of sensitivity to chemotherapy
Several studies showed that potentiation of immunotherapy and cytotoxic chemotherapy is reciprocal. Some patients with chemoresistant tumors responded to chemotherapy re-challenge upon disease progression on anti-PD1 therapy. In both Hodgkin’s lymphoma and non-small cell lung cancer, increased response to salvage chemotherapy was observed after disease progression on immune checkpoint blockade [43, 44].
Detrimental effects of chemotherapy on immunotherapy
One of the major detrimental effects of chemotherapy to the immune system is lymphodepletion which can be immunosuppressive. In fact, some of the immunosuppressive drugs used in clinic to treat autoimmune diseases are cytotoxic chemotherapy used for cancer treatment, but with different doses and schedules. It is still controversial whether lymphodepletion induced by chemotherapy is suppressive for anti-cancer immunity. Lymphodepletion associated with cancer chemotherapy is usually associated with rebound of lymphocyte counts and an immune system “reset.” One study showed the uneven recovery of different immune cell subpopulations tilting to anti-cancer immunity [45].
Chemotherapy can also affect tertiary lymphoid structures (TLS) [46, 47]. TLS are ectopic lymphoid organizations developed in non-lymphoid tissues, including cancer, and display similar organization as secondary lymphoid organs, such as lymph nodes. Extensive data suggest that TLS function similarly to lymph nodes in recruiting lymphocytes into tumors, and mounting local and systemic immune response against cancers. Overall, the presence and high densities of TLS in tumors favorably correlate with prognosis in multiple cancer types, and sometimes independent of the pathological TNM (tumor-lymph node-metastasis) staging [46,47,48]. The lymphodepleting effect of chemotherapy can also affect TLS either by the direct cytotoxic effect of chemotherapeutic drugs or associated therapies, such as corticosteroids [49].
FDA-approved chemoimmunotherapy combinations
Many clinical trials with combinations of chemoimmunotherapy have been conducted in almost all major cancers with several FDA approvals (Fig. 1, Tables 1 and 3). The poster child of the combinations can be found in lung cancer. In the Keynote-189 trial with 616 lung adenocarcinoma patients, pembrolizumab with platinum-based doublet chemotherapy significantly improved the overall survival (OS) when compared to chemotherapy alone (HR 0.49, 95% CI 0.38–0.64, p < 0.001) [50]. While the benefit was greatest in patients whose tumors had PD-L1 > 50% expression, OS was improved across all patient subsets regardless of the PD-L1 status. In the Impower 130 clinical trial, anti-PD-L1 antibody atezolizumab was combined with carboplatin and nab-paclitaxel as it does not require corticosteroid. The combination group was associated with prolonged OS of 18.6 versus 13.9 months (HR 0.79, 95% CI 0·64–0·98, p = 0·033) [51]. Similar survival benefits were observed in metastatic lung squamous cell carcinoma, where pembrolizumab combined with carboplatin-doublet chemotherapy significantly improved OS (15.9 versus 11.3 months; HR 0.64, 95% CI 0.49–0.85, p < 0.001) [52].
In small cell lung cancer (SCLC), two immune checkpoint inhibitors, atezolizumab and durvalumab, were approved with the combination of standard of care platinum-based chemotherapy [53, 54]. The addition of atezolizumab improved the OS from 10.3 months to 12.3 months (HR 0.70, P = 0.007), while the addition of durvalumab improved the OS from 10.3 months to 13.0 months (HR 0.73, P = 0.0047).
In addition to lung cancers, the combination of chemotherapy and immunotherapy has also been approved in several other cancers. In breast cancer, atezolizumab plus nab-paclitaxel improved the OS of the intended population from 17.6 months of nab-paclitaxel alone to 21.3 months (HR 0.84; 95% CI, 0.69–1.02; p = 0.08) [55]. Furthermore, the addition of pembrolizumab to chemotherapy improved median progression free survival (PFS) from 5.6 months to 9.7 months in the population with PD-L1 expression at a combined positive score of 10 or higher (HR 0·65, 95% CI 0·49–0·86; one-sided p = 0·0012) [56]. In head and neck cancer, the addition of pembrolizumab to cisplatin/carboplatin + 5-fluouracil significantly improved OS when compared to the addition of cetuximab to chemotherapy in the group with the PD-L1 combined positive score of 1 or higher: median OS 13·6 versus 10·4 months (HR 0·65, 95% CI 0·53–0·80, p < 0·0001) [57].
The OS benefit has also been observed when immunotherapy was used as a maintenance therapy after completion of chemotherapy, as in bladder cancer. In the JAVELIN Bladder 100 trial, significantly improved OS was observed in patients with metastatic urothelial carcinoma who completed platinum-based chemotherapy without disease progression was subsequently treated with avelumab maintenance therapy: median OS 21.4 versus 14.3 months (HR 0.69, 95% CI 0.56–0.86, p = 0.001) [58].
However, chemo-immunotherapy combinations have not been a panacea in all solid tumors. In squamous NSCLC, even though the combination of pembrolizumab and chemotherapy improves OS, the addition of atezolizumab to chemotherapy did not (14.2 and 13.5 months, HR 0.88, 95% CI 0.73–1.05, p = 0.16) [59]. In metastatic urothelial cancer, chemo-immunotherapy combinations have been disappointing with minimal improvements over chemotherapy alone, in contrast to the Javelin Bladder 100 trial, where avelumab maintenance therapy significantly improved treatment outcomes. In part, this is likely due to patient selection from patients initially doing well after chemotherapy selected for the Javelin Bladder 100 trial and not delaying treatment until progression. More studies are needed to determine the optimal combination, sequence, drug choice and underlying mechanisms of different response.
Combination of radiation therapy with immunotherapy
The stimulation of anti-cancer immunity by radiotherapy (RT) was first suggested in case reports with regression of distant untreated tumors after local RT [60]. While this RT-induced abscopal phenomenon is rare and elusive, its effects on the induction of anti-cancer immune response are intriguing and have aroused tremendous interest with the advent of immune checkpoint blockade.
Potentiation of anti-cancer immunity by radiation
Both antigenicity and adjuvanticity are critical for immune response. RT can augment both antigenicity and adjuvanticity in addition to alteration of the local TME.
RT increases tumor antigenicity through multiple pathways. First, similar to chemotherapy as discussed above, radiation can induce MHC-I expression and enhance tumor antigen presentation [61]. Second, radiation induces ICD. During ICD, annexin A1 guides antigen-presenting cells to dying cancer cells while HSP70, HSP90, HMGB1 and other molecules promote uptake and cancer antigen presentation to T cells. It has been shown that radiation induces translocation of calreticulin to the plasma membrane [62], and release of HMGB1 [63]. Third, radiation downregulates CD47 expression on the cell surface and enhances the cancer cells’ uptake and antigen presentation [64]. CD47 presents as a “do not eat me” signal to APCs and is overexpressed in many cancer cells [65]. Fourth, reactive oxygen species (ROS) generated during ionizing radiation can modify macromolecules, such as proteins and DNA, and increase antigenicity. In addition to direct DNA damage, the presence of oxygen and generation of ROS are critical for radiation induced tissue injury [66].
Another important contribution of radiation to anti-cancer immunity is increased adjuvanticity. Radiation-induced DNA damage and cytoplasmic leakage of DNA from micronuclei activate the innate and adaptive immune response via cGAS/STING pathway and upregulate the expression of type I interferon pathway. This pathway is critical for radiation induced anti-cancer immunity. Silencing of cGAS in bone marrow-derived dendritic cells impairs T cell priming [67]. In addition to nuclear DNA, mitochondrial DNA breaks also have a role in activating a type I interferon response and synergizing with nuclear DNA breaks [68].
In addition to the cGAS-STING pathway, ICD, release of DAMPs and cytokines can enhance adjuvanticity, elicit migration of pro-anti-cancer immune subpopulation, decrease immunosuppressive cells, alter TME and tilt immune response to cancer cell killing. Overall, radiation converts cancer cells as an in situ vaccine to elicit anti-cancer immunity.
Inhibition of anti-cancer immunity by radiation
In contrast to what is discussed above, ample evidence also exists that radiation induces an immunosuppressive TME. In addition to cancer cells, radiation can kill normal cells, including immune cells, especially when broad field radiation is considered. Furthermore, radiation can alter the TME and, instead of tilting to anti-cancer immunity, induce an immunosuppressive milieu. Several studies showed that radiation induces infiltration and aggregation of MDSCs [69, 70], which contributes to the immunosuppressive TME through multiple pathways. The same STING pathway that contributes to the cancer adjuvanticity at least partially contributes to the aggregation of MDSCs in tumor tissues [71]. In addition, radiation can promote the expression of TGF-β and TGF-β family activin A, thus promoting the recruitment of Treg cells and reducing the infiltration of CD8+T cells [72]. TGF-β is upregulated upon radiation [73]. In a preclinical study, TGF-β neutralization and radiation increase T cell priming and decrease tumor growth and metastasis [74].
Other mechanisms of the immunosuppressive effects of radiation include the dysregulation of tumor blood vessels [75], hypoxia [76], stroma [77], tumor-associated macrophages (TAMs) [78], cancer-associated fibroblasts (CAFs) [79], cytokines [80, 81] and so on. Moreover, the abnormal expression of these components is also related to radiation resistance [82]. In conclusion, the formation of an immunosuppressive TME by radiation is a complicated process and targeting these immunosuppressive elements provides a new direction for enhancing RT-induced anti-tumor immunity.
Clinical consideration of radiation and immunotherapy combination
The first report showing the benefits of radiation and immunotherapy came from a patient with melanoma who had disease progression while on a clinical trial with ipilimumab, but subsequently had abscopal tumor shrinkage after radiation therapy [83]. A secondary analysis of the KEYNOTE-001 trial also showed that prior radiotherapy is associated with significant improvement of PFS and OS of patients with NSCLC treated with pembrolizumab [84]. Since then, there has been an eruption of clinical trials with radiotherapy and immunotherapy. Currently, over 800 active clinical trials are registered at clinicaltrials.gov, when using radiation and immunotherapy as the search key words.
So far, several clinical studies showed improved clinical outcomes when radiation is added to ICBs. In a meta-analysis including 20 clinical trials and 2,027 NSCLC patients, the combination of anti-PD1/PD-L1 inhibitors with radiotherapy was associated with significantly improved objective response rate (odds ratio [OR] 2.76, 95% CI 1.06–7.19, p = 0.038) and OS (2-year survival HR 1.77, 95% CI 1.35–2.33, p = 0.000) [85]. Currently, durvalumab has been approved as a maintenance therapy after platinum-based chemoradiation therapy for stage III NSCLC patients based on a Phase III PACIFIC trial [86, 87]. Addition of durvalumab significantly increased the median PFS (17.2 vs. 5.6 months; HR 0.51, 95% CI 0.41–0.63, p < 0.001) and OS (HR for death 0.68, 95% CI 0.54–0.86, p = 0.0025).
In addition to anti-PD1/PD-L1 antibodies, radiotherapy is already being studied with the combination of other immunotherapeutic agents such as cytokines, cell therapy, vaccines and other immune checkpoint modulators [88]. While most of these studies are still ongoing, some early reports show that these combinations are feasible and can potentially achieve synergistic effects. In a small Phase II trial, radiotherapy combined with CAR-T cell therapy improved the overall RR of diffuse large B-cell lymphoma (p = 0.033) [89].
Even though promising results were observed, other studies showed no improvement with the radioimmunotherapy combination. Several approaches are currently being explored to improve treatment outcomes. Selection of the right patients (biomarker development) and optimization of radiation techniques, including dose, schedule and timing, are both under intense investigation. Preclinical and clinical data suggest that dose and fraction, irradiated area, volume and sequence of administration can each have major impact in systemic anti-cancer immunity [90, 91]. Because radiation not only kills cancer cells, but also affects many aspects of immune response, such as cancer antigenicity, presentation, TIME, immune response at local drainage lymph nodes and in the whole system, it is not surprising that contradictory findings were observed regarding anticancer immunity with different dose and fractionation schedules. Lymphocytes have little DNA repair capacity and are highly sensitive to radiation even at the conventional dose of 1.8–2 Gy [92]. One study showed post-radiation immune cell re-population differs with lymphoid response observed more with hypofractionation while conventional dose/schedule induces more myeloid response, such as MDSCs and TAMs [93]. Several preclinical studies revealed that high-dose hypofractionation radiation stimulates more anticancer immune response than conventional fractionation radiation. High-dose radiation increases expression of MHC and death receptors critical for T cell-mediated cell killing [61], induces more T cell infiltration into tumors [94], triggers more robust abscopal effects [95] and synergizes more with anti-PD-L1 and anti-TIGIT therapies [93]. A US national database analysis also revealed that hypofractionated radiation therapy and immunotherapy achieved much higher three-year overall survival in metastatic melanoma patients than conventionally fractionated radiation plus immunotherapy (37.3% vs. 17.6%, p < 0.0001) [96]. However, less favorable results with high-dose hypofractionation were also observed in other preclinical studies. In a breast cancer model, the abscopal effect was only observed when anti-CTLA-4 therapy was combined with fractionated radiotherapy, but not with single high-dose therapy [91]. High-dose radiation induces DNA exonuclease Trex1 and dampens the cGAS-STING pathway activation [97]. Hence, well-designed prospective clinical trials are needed to determine the optimal radiation dose, schedule and fractionation to potentiate immunotherapy.
Combination of targeted therapy with immunotherapy
All cancers harbor genomic alterations that drive oncogenesis. Targeting these genomic alterations can have direct antitumor activities and can induce more responses than cytotoxic chemotherapy [98, 99]. For example, in patients with NSCLC, while the response rate of platinum-based doublet is less than 30% [100], a response rate of 80% is observed in patients with an epidermal growth factor receptor (EGFR) driver mutation treated with erlotinib [101]. In addition, many of the molecular drivers affect multiple steps along the cancer-immunity cycle.
Potential mechanisms
Direct antitumor activity and ICD
Elimination of cancer cells can not only decrease the number of cells for immune cells to target and destroy, but can also eliminate immunosuppressive factors and increase the efficacy of immunotherapy. The KEYNOTE-001 trial showed that smaller tumor sizes are an independent factor in predicting treatment outcomes [102]. An important factor to consider is ICD induced by targeted therapy. As discussed above in the sections of chemotherapy and radiation therapy, ICD induced by targeted therapy enhances cancer cell uptake and antigen presentation by antigen-presenting cells, prime and activate immune response, attract immune cells to tumor sites and potentiate anti-cancer immunity.
Antigen presentation
Many of the oncogenic pathways are directly involved in the regulation of the expression of antigen presentation machinery. The cyclin-dependent kinase 4 and 6 (CDK4/6) pathway is commonly activated in many cancers [103, 104]. Inhibition of the CDK4/6 pathway upregulates MHC expression [103]. Similar findings are also observed with the PI3K pathway. PI3K inhibitors have been approved in breast cancer and follicular lymphoma. These drugs have the potential to be effective in other cancers, such as bladder cancer [98, 99]. Activation of the PI3K pathway attenuates the expression of MHC class I and II expression, while inhibition of this pathway reverses the suppression of antigen presentation machinery via interferon γ [105].
Direct effect on immune cells
Many of the aberrant signaling activities have profound impacts on immune cells. The VEGF-VEGFR pathway plays critical roles in almost every subpopulation of immune cells. VEGFRs are expressed on activated and memory T cells [106]. Engagement of VEGF-VEGFR leads to activation of the downstream signaling pathways in T cells [106], inhibits TCR (T cell receptor)-dependent activation in T cells [107] and suppresses the cytotoxic activity of T cells [108]. In Treg cells, VEGFR2 is selectively expressed in FOXP3high Treg cells. Besides Treg cells, VEGF can activate JAK2 and STAT3 and induce accumulation of Gr1 + CD11b + MDSCs [109]. In dendritic cells, production of VEGF by human tumors inhibits dendritic cell maturation through the NF-kappa B pathway [110, 111]. Increased plasma VEGF levels are associated with increased number of immature dendritic cells, and surgical removal of tumors partially reverses these effects [112]. In applying these preclinical findings to clinical trials, the combination of angiogenesis inhibitors and ICB significantly improved the treatment outcomes in metastatic renal cell carcinoma and has gained several FDA approvals.
Similar direct effects on immune cells are also seen with many other targeted agents already approved by the FDA or in development. For example, ibrutinib is FDA approved for chronic lymphocytic leukemia/lymphoma (CLL), mantle cell lymphoma, marginal zone lymphoma and Waldenström’s macroglobulinemia. It modulates T cells by inhibiting Bruton's tyrosine kinase (BTK) and IL-2-inducible T cell kinase (ITK), and drives a Th1-selective pressure in T lymphocytes and a preferential inhibition of Th2 response [113]. In patients with CLL, it markedly increases CD4 + and CD8 + T cell numbers, decreases Treg/CD4 + T cell ratio, downregulates immunosuppressive CD200 and CD272 expression and decreases the production of immunosuppressive IL-10 production. Currently, seven clinical trials are ongoing to combine ibrutinib with immune checkpoint inhibitors for treatment of cancer.
Effects on tumor microenvironment
In addition to the direct antitumor activity, many of the signaling pathways have versatile functions on the tumor immune microenvironment (TIME) that can affect anti-cancer immunity. EGFR activation mutations occur in about 10–15% of all NSCLC and can upregulate PD-1 and PD-L1 expression which mediates immune escape [114]. Similarly, activation of the PI3K/AKT pathway, including the PTEN deletion, leads to constitutive expression of PD-L1 expression and resistance to immunotherapy [115].
Other than immune cells, no other cells play such a versatile array of functions in TIME as CAF [116, 117]. CAFs can secrete immunosuppressive cytokines, attract suppressive immune cell subpopulations, remodel tumor matrix and facilitate migration, invasion and metastasis of cancer cells. CAFs can alter local milieu and indirectly suppress anti-cancer immunity, and facilitate tumor cell growth [118]. Cross-communication between cancer cells and CAFs contributes to development of resistance to chemotherapy and immunotherapy. Many of the altered signaling transduction pathways, such as receptor tyrosine kinase receptors and their cognate ligands, Wnt signaling pathway, TGF-β pathway and others, contribute to activation of fibroblasts to CAFs [118, 119].
Many of the genomic alterations and oncogenic drivers alter metabolism and other constitutive components of the tumor microenvironment and negatively affect anti-cancer immunity. Oncogenic transformation leads to uncontrollable cancer cell proliferation, creates hypoxic and acidic tumor microenvironments and inhibits T cell function. Indolamine-2,3-dioxygenase (IDO) is a heme-containing enzyme that catalyzes the first and rate-limiting step of tryptophan catabolism. Depletion of the essential amino acid tryptophan and accumulation of the metabolic products, such as kynurenine, are highly immunosuppressive and tolerogenic. They can suppress effector T cell and NK cell function, stimulate Treg cells, promote expansion of MDSCs and tilt polarization of macrophages to more tolerogenic M2 phenotype [120]. It has been shown that multiple oncogenic pathways, such as the PI3K/AKT/mTOR, Ras/Raf/MEK/ERK and protein kinase C pathways, are all involved in the upregulation of IDO expression [121].
Clinical consideration of the targeted therapy and immunotherapy combination
Since many of the targeted therapeutic agents can directly or indirectly modulate immune cell functions, a plethora of clinical trials are currently ongoing to determine the efficacy and toxicity of combined targeted therapy and immunotherapy, mainly ICBs, in cancer [122,123,124]. As discussed above, anti-angiogenesis agents probably have the most versatile immune-modulative functions that affect almost all immune cell subpopulations [125]. Hence, it is not surprising that five out of six FDA-approved targeted and immunotherapy combinations target angiogenesis (Table 4): axitinib targets VEGFR 1–3 in addition to platelet-derived growth factor receptor (PDGFR) and c-Kit; cabozantinib inhibits VEGFR2 in addition to c-Met and Axl; lenvatinib targets VEGFR1-3 in addition to fibroblast growth factor receptors, PDGFR and RET; and bevacizumab is a monoclonal antibody against VEGF-A. The only targeted therapy combination that does not directly target angiogenesis is the combination of a BRAF inhibitor vemurafenib and a mitogen-activated extracellular kinase (MEK) inhibitor cobinetinib in combination with atezolizumab in advanced melanoma with BRAF V600 activation mutation. Three of the six combinations are indicated for advanced kidney cancer: pembrolizumab plus axitinib, avelumab plus axitinib and nivolumab plus cabozantinib.
In addition to anti-angiogenesis, almost every targeted therapy that has been shown to modulate the immune response is currently being combined and tested with immunotherapy, mainly ICBs. For example, PI3K inhibitors have been approved for the treatment of breast cancer and lymphoma. In addition to direct anti-cancer activity, it alters tumor local metabolism, downregulates antigen presentation machinery and has direct effects on immune cells as PI3K-δ is expressed in immune cells [126]. Over 10 clinical trials are currently ongoing that combine immunotherapy with agents targeting the PI3K/AKT/mTOR pathway [127].
In addition to ICBs, targeted therapy is also being combined with other immunotherapeutic agents. The BTK/ITK inhibitor, ibrutinib and acalabrutinib have been approved for the treatment of non-Hodgkin lymphoma and are known to increase T cell number and function. Currently, seven clinical trials have been designed to combine the BTK/ITK inhibitors with CAR-T cell therapy and one clinical trial combining ibrutinib with personalized multi-peptide cancer vaccine.
Cytokines and other soluble factors
Cytokines are small proteins or glycoproteins (< 30 KDa) that interact with cell surface receptors and exert critical roles in regulating humoral and cellular immune response through affecting cell trafficking, maturation, growth and responsiveness of target cells. Cytokines include chemokines, interleukins, interferons and tumor necrosis factors. Interleukin-2 (IL-2) and interferon α (IFN-α) are the first two cytokines approved for the treatment of cancers.
Chemokines
Chemokines are the largest subfamily of cytokines that play important roles in guiding immune cell trafficking and development, and can be classified into four main classes depending on the location of the first two cysteine (C) residues in their protein sequence: namely, the CC-chemokines, the CXC-chemokines, C-chemokines and CX3C-chemokines [128].
Different immune cell subpopulations respond to different chemokines, traffic into TME and affect anti-cancer immunity. For example, effector immune cells, such as CD8 + Teff cells, IFN-γ-expressing T helper 1 (TH1) cells and natural killer (NK) cells, can be attracted to the tumor microenvironment by CXC-chemokine ligand 9 (CXCL9), CXCL10 and CXCL11, and exert potent antitumor effects [129, 130]. Treg cells are immunosuppressive cells that can inhibit the functions of other immune cells through interaction of inhibitory cell surface receptor, such as: CTLA4-CD28 interaction [131], CTLA4-CD80/86 and LAG3/MHCII pairs; secretion of inhibitory cytokines such as TGF-β, IL-10 and IL-35; secretion of granzyme B and lysis of Teff cells; and metabolic disruption such as the adenosine pathway [132]. Treg cells express CCR4 and CCR10, and migrate into the tumor microenvironment in response to CCL22 and CCL28 [133, 134]. Dendritic cells can be attracted by CCL5, CCL20 and CXCL12 [135], while macrophages can be attracted by the CCL2-CCR2 signaling [136]. Sometimes the same chemokines can recruit different immune cells with different and even opposing immune functions. For example, CCL21 and CCL19 recruit CCR7 + dendritic cells and Treg cells, while CCL17 and CCL22 can directly recruit Treg and Th2 lymphocytes [137,138,139,140].
In addition to regulating immune cell trafficking and development, chemokines have direct effects on cancer cells. Cancer cells can express chemokine receptors and be stimulated by chemokines to promote cancer cell growth and proliferation [141,142,143]. Furthermore, chemokines can also facilitate cancer metastasis. The CXCL12/CXCR4 and CCL27/CCR10 pathways have both been found to be involved in cancer cell adhesion, migration and metastasis [144,145,146].
Interleukins and interferons
While the major function of chemokines is to regulate immune cell trafficking, interleukins and interferons have diverse functions in regulating the immune response. IL-2 and IFN-α were the first cytokines approved for cancer therapy. Since the first approval for the treatment of hairy cell leukemia [147], IFN-α has also been approved for the treatment of melanoma, follicular non-Hodgkin’s lymphoma, AIDS-related Kaposi’s sarcoma and renal cell carcinoma, while IL-2 was approved for the treatment of renal cell carcinoma and melanoma[148].
Pro-inflammatory interleukins and IFN-α act upon every step of the cancer immunity cycle and promote anti-cancer immunity, while immunosuppressive cytokines promote many aspects of oncogenesis and inhibit anti-cancer immunity. IL-2 plays key roles in the expansion of T lymphocytes and NK cells. Treg cells have high expression of IL-2 receptor alpha (IL-2Rα), which is a component of high-affinity IL-2 receptor. Hence, IL-2 can skew the expansion of T lymphocytes to Treg cells [149, 150]. To generate more favorable immune cell stimulation and decrease Treg cell proliferation, several strategies have been used to modify IL-2. One strategy is to conjugate recombinant IL-2 with polyethylene glycol (PEG), as in NKTR-214 (or bempegaldesleukin), that decreases the affinity to the high-affinity IL-2Rα receptor. Another strategy is to introduce a mutation to IL-2 to decrease its binding to IL-2Rα, known also as CD25. Both versions are currently in clinical development [151, 152].
Transforming Growth Factor-β (TGF-β)
TGF-β is a pleiotropic cytokine that plays key roles in embryogenesis and tissue homeostasis. It regulates cell proliferation, differentiation, adhesion, migration, metabolism and apoptosis in many normal cells. At early stage of oncogenesis, TGF-β can inhibit cancer growth, induce apoptosis and work more like a tumor suppressor. Once cancer develops, it is involved in promoting tumor fibrosis, epithelial–mesenchymal transition (EMT), tumor angiogenesis and suppression of immune response [153, 154]. Tumor fibrosis can prevent drugs and immune cells from accessing cancer cells, while EMT can lead to metastasis and resistance to therapy. Furthermore, TGF-β regulates many immune cell subtypes and is intensively involved in the immunosuppressive TME. It can suppress the expression of IL-2 which is critical for T cell proliferation [155], inhibit the differentiation of naïve T cells into Th1 cells [156] and mitigate the cytotoxic effects of CD8 + Teff cells through inhibiting the expression of five cytolytic gene products—namely, perforin, granzyme A, granzyme B, Fas ligand and interferon γ [157]. For Treg cells, TGF-β triggers the expression of FOXP3 which serves as the master transcription regulator for the Treg cell differentiation. Furthermore, Treg cells can carry latent TGF-β1, as well as a cell surface docking receptor GARP for latent TGF-β, to suppress anti-cancer immunity [158,159,160,161,162,163].
In addition to T cells, TGF-β has immunosuppressive effects on many other immune cells. In dendritic cells, TGF-β suppresses expression of MHC-II genes and inhibits antigen presentation [164, 165]. For natural killer cells, TGF-β blocks NK functions by decreasing the expression of NK cell surface receptors NKG2D and NKp30 [166], and inhibits Th1 response through suppressing IFN-γ and TBET expression [167, 168]. For macrophages, TGF-β induces macrophage differentiation to the M2 phenotype that is immunosuppressive in the TME [153, 169]. Furthermore, TGF-β plays important roles in tumor development and metastasis via MDSCs. Depletion of MDSCs abolishes the therapeutic effects of an anti-TGF-β antibody, at least in preclinical studies [170].
In addition to direct effects on immune cells, TGF-β plays major roles in the immunosuppressive TME. TGF-β produced by CAFs excludes CD4 + and CD8 + T cells from entering the tumor [171] and an anti-TGF-β antibody could reverse tumor T cell exclusion and sensitize tumors to PD-L1 treatment [172]. TGF-β produced in the TME can induce the expression of indoleamine 2,3-dioxygenase (IDO) and arginase which can suppress many effector immune cells [173].
Because of the pluripotent regulation of anti-cancer immunity functions by TGF-β, multiple therapeutic agents targeting TGF-β have been developed and are currently in clinical development (Table 5). Given the importance of TGF-β and its effects in the TME, Dr. James Gulley at the National Cancer Institute discussed the pathway and highlighted bintrafusp alfa at the 2020 China IO Meeting. Bintrafusp alfa is a bifunctional chimeric protein composed of the extracellular domain of the TGF-β receptor II (a TGF-β “trap”) fused to anti-PD-L1 human IgG. It is hypothesized that bintrafusp alfa carries the TGF-β trap to the cancer sites where PD-L1 is expressed, blocks both TGF-β and PD-L1, and enhances anti-cancer immunity. Preclinical studies showed significant anti-tumor effect, TME modification and reduction of EMT [174, 175]. A Phase I trial with bintrafusp alfa was conducted which showed promising anti-tumor effects with controllable toxicities [176]. So far, 30 clinical trials have been registered at clinicaltrials.gov with bintrafusp alfa in various cancers.
Strategies for clinical use and combination therapy
At the 2020 China IO meeting, Dr. Charles Drake from Columbia University and Dr. James Gulley from the National Institute of Health discussed strategies to target cytokines and other combinations for cancer immunotherapy. Dr. Drake first discussed the serendipitous findings from a clinical trial with an anti-IL-1β monoclonal antibody, canakinumab, in preventing cardiovascular diseases. People treated with canakinumab at 300 mg every 3 months had a relative risk of overall cancer incidence of 0.49 and fatal lung cancer of 0.23 when compared to the placebo cohort [177], suggesting that canakinumab has a protective effect. His group then confirmed that an anti-IL-1β antibody, especially in combination with anti-PD1 antibody, dramatically increased M1 macrophage and the M1/M2 macrophage ratio in the TME [178]. Subsequently, a pilot clinical trial was initiated to determine the efficacy and molecular correlative studies in kidney cancer (ClinicalTrials.gov Identifier: NCT04028245). He also discussed that cytokines can be significantly affected by androgen deprivation therapy (ADT) in prostate cancer that can possibly be targeted for cancer therapy. In mice, ADT significantly increases the expression of CXCL15, which is the mouse equivalent of human IL-8. This cytokine pathway is involved in infiltration of neutrophils and polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC) into the immunosuppressive TME. Based on those findings, a clinical trial was initiated with the anti-PD1 antibody nivolumab in combination with an anti-IL-8 antibody to synergize ADT in prostate cancer (Clinicaltrials.gov identifier No: NCCT03689699).
As discussed above, IL-2 and IFN-α are rarely used in clinic due to their systemic pro-inflammatory side effects. One future development strategy to use cytokines for cancer immunotherapy is to confine cytokines to the site of action, such as intratumoral injection of the cytokines or using gene therapy or other vehicles to express cytokines into the cancer sites. Intratumoral injection of IL-2 and IFN is one of the earliest formats of targeted delivery of cytokines to the site of action to minimize systemic pro-inflammatory reaction. Talimogene laherparepvec (TVEC) is a genetically engineered oncolytic herpes virus expressing human granulocyte–macrophage colony-stimulating factor (GM-CSF) that has been approved for intratumoral injection of melanoma [179].
Since intratumoral injection may not be practical in patients with multiple metastatic lesions or deep locations of cancer, another strategy is to modify cytokines and change their binding specificity. NKTR-214, a therapeutic where IL-2 is conjugated to polyethylene glycol (PEG), has decreased affinity for the high-affinity IL-2 receptor α and therefore lower associated toxicities compared to IL-2 [180]. Consistent with the findings in preclinical models, NKTR-214 significantly promotes cytotoxic immune cell infiltration and upregulates gene expression associated with effective cells with limited increase of Treg cells in tumors in a Phase I clinical trial [181], with further clinical development in urothelial and renal cancers.
More recently, cytokine-based bifunctional molecules have generated great interest in which the cytokine in the molecule exerts its immunoregulatory functions while the other part of the molecule acts as a carrier to deliver the cytokine to the site of action as seen in RO6874281, or as a carrier and functional domain as seen in bintrafusp alfa discussed above. RO6874281 contains a variant form of interleukin-2 (IL-2v) that completely lacks binding to the high-affinity IL-2 receptor α, but retains IL-2Rβγ binding. IL-2v is conjugated to a human monoclonal antibody directed against fibroblast activation protein-alpha (FAP) on CAF [182]. Both RO6874281 and bintrafusp alfa have shown clinical activities in addition to their reduced toxicity [182, 183]. In a Phase I trial with bintrafusp alfa as a second-line treatment for NSCLC, an overall response rate of 21.3% (17 of 80) was observed in the whole study population. It was 25.0% (10 of 40) at the recommended Phase 2 dose of 1200 mg every 2 weeks and 36.0% (10 of 27) in those with PD-L1-positive tumors [183].
Because cytokines can regulate every step along the anti-cancer immunity cycle, many clinical trials are ongoing to combine cytokines with other agents along the immunity cycle to determine whether the anti-cancer efficacy can be further improved. For immunostimulatory cytokines, such as IL-2, IL-10, IL-12 and IL-15, their native forms and genetically engineered cytokines have been combined with ICBs. For example, the combination of pegylated long-acting IL-10 and anti-PD1 antibody pembrolizumab or nivolumab had manageable toxicity profiles and showed preliminary antitumor activity [184]. For immunosuppressive cytokines, such as TGF-β, CCL2 and IL-8, their neutralizing antibodies or small molecule inhibitors have been tested in clinic with the combination of ICBs and chemotherapy.
Adoptive cell therapy
Brief history
Adoptive cell therapy (ACT) in cancer is the transfer of immune cells, either autologous or allogeneic, into patients with cancer to mount an anti-cancer immune response. In both cases, immune cells are isolated from patients themselves (autologous) or a donor (allogeneic), manipulated and expanded in vitro, and infused into patients for cancer therapy. The first ACT was performed in patients with metastatic melanoma using autologous tumor-infiltrating lymphocytes (TILs) [185]. TILs are available in only a minority of patients with selected tumors, usually melanoma, and associated with inconsistent response rates. With the development of chimeric antigen receptor (CAR) technology [186,187,188], melanoma tumors were shown to clinically regress after infusion of normal lymphocytes expressing an engineered T cell receptor targeting the MART-1 tumor antigen [189]. The research and clinical applications accelerated after 2010 with the demonstration of tumor regression in B cell lymphoma after administration of lymphocytes expressing CAR against the B cell antigen CD19 [190].
Because NK cells mirror the functions of CD8 + cytotoxic T cells [191], NK cells have also been engineered to express CARs for cancer immunotherapy [192]. The cytotoxic function of NK cells is upregulated via engagement of activating receptors, such as NKG2D. Hence, CAR NK cells usually use one of these activating receptors such as CAR NK cells expressing NKG2D-containing CARs [193, 194]. So far, several clinical trials with CAR NK cells targeting hematological malignancies (CD7, CD19, CD22, CD33, BCMA) and solid tumors (Robo1 and MUC1) are ongoing (www.clinicaltrials.gov).
Resistant mechanisms
Even with great success and FDA approvals of ACT, especially in hematological malignancies, 10–20% patients fail to achieve remission after receiving anti-CD19 CAR-T cell therapy, and 30–50% who achieve initial remission develop disease relapse [195, 196]. Some of the treatment failure can be secondary to logistic issues, such as manufacturing failure and delay, insufficient numbers of CAR-T cells, delay in insurance approval and disease progression to an irreversible end stage. More commonly, the same mechanisms of resistance to anti-cancer immunity, especially those at TME, are responsible for resistance to ACT.
CAR-T cells bypass the first three steps of the cancer immunity cycle: antigen release and presentation, immune cell priming and immune cell activation. However, like any other effector immune cells, ACT is still governed by the regulation of immune response and resistant mechanisms along the anti-cancer immunity cycle described above [3]. Since CAR-T cells are engineered T cells, the intrinsic T cell function status can affect the treatment outcomes. After infusion, CAR-T cells have 3 main characteristics to achieve long-lasting remission: expansion, persistence and tumor cytotoxicity. Hence, defects of the original T cells that affect T cell expansion, cytotoxic function and development of memory cells can also affect the efficacy. For example, the efficacy of ACT is inferior in chronic lymphocytic leukemia (CLL) than that in B-cell acute lymphoblastic leukemia (B-ALL), which may be related to the intrinsic T cell defects in CLL patients. Hence, generation of universal CAR-T cells from healthy donors or third-party donors is being explored [197, 198].
CAR-T cells contain a T cell receptor stimulatory domain and a co-stimulatory domain, both of which are required for T cell priming, activation and replication. Preclinical and observation studies showed that the co-stimulatory domain can significantly affect the persistence and cell function after infusion [199]. Optimization of CAR design to enhance CAR-T cell activation, replication and conversion to memory cells is ongoing. Compared to the second-generation CAR which contains a single costimulatory domain (CD28, 4-1BB or OX-40), the third-generation CAR contains two or more costimulatory domains which can exhibit strong short-term anti-tumor activity associated with CD28 and long-term persistence with 4-1BB [200, 201].
After infusion, CAR-T cells still need to go through cell trafficking, infiltration into the cancer sites and then recognition and killing of cancer cells. Dysregulation of cytokine and cytokine receptors, and an immunosuppressive TME can adversely affect CAR-T cell function. It has been shown that β-catenin- over-expressing tumors have an altered CXCR3-CXCL9/10 chemokine axis to attract effector T cells into tumors after adoptive transfer [202]. Delivery of CXCL11 to tumor sites significantly increases CAR-T cell infiltration and enhances anti-tumor activity [203].
Once inside tumors, suppressive signals produced in the TME, such as TGF-β, IDO1, IL-10 and adenosine, contribute to exhaustion of CAR-T cells. Tumor cells can produce suppressive signals, such as PD-L1, that suppress CAR-T cells. CAFs, MDSCs and TAMs can all contribute to the suppression of CAR cell function in the TME. Combination of ACT with other therapies to prevent exhaustion and enhance CAR-T cell functions is being explored. Blockade of adenosine 2A receptor significantly improves the efficacy of CAR-T cells [204]. PD1 is another negative regulator of Teff cells at the end cytotoxic stage and was found to be upregulated after CAR-T cell infusion [205].
In addition to the mechanisms along the cancer immunity cycle, alterations in malignant cells also contribute to primary and secondary resistance to ACT. Loss or modification of the target antigen has not only been identified in ACT, but also in other immunotherapy modalities [206, 207]. Loss of the target molecules could be secondary to alternative slicing [208] or interruption of antigen presentation to the cell surface [209]. Furthermore, a diminishment of target molecule density on the cell surface can lead to evasion of CAR-T cell therapy [210]. Low or loss of expression of target molecules on tumor cells at relapse can be secondary to pre-existing malignant cell heterogeneity [211] or lineage switching [212].
Novel construction and combination strategies to improve ACT efficacy
Novel design of CAR-T cells
Development of universal CAR-T cells is one approach to address the intrinsic defects of T cells from patients with hematological malignancies. To improve the proliferation of CAR-T cells, inclusion of a stronger co-stimulatory domain, such as 4-1BB instead of CD28, or incorporation of both 4-1BB and CD28 (the third-generation CAR-T cells), can improve the persistence of CAR-T cells [213, 214]. To alter the immunosuppressive TME, a fourth generation of CAR-T cells called TRUCKs (“T cells redirected for antigen‐unrestricted cytokine‐initiated killing”) has been designed and has entered early phase clinical trials. TRUCKs have a transgene, usually immunostimulatory cytokines, under the control of the NFAT‐responsive/IL‐2 minimal promoter. CAR engagement and activation leads to NFAT phosphorylation and transgene expression that acts in an autocrine fashion to stimulate CAR-T cells, or paracrine to modulate the immune cell environment [215]. Other than optimizing the CAR-T cell design, an alternative strategy is to combine ACT with another therapy and maximize the antitumor activity (Table 6).
Combination of ACT with immune checkpoint inhibitors
PD1 is upregulated after CAR-T cell infusion which can down-regulate the CD28 co-stimulatory signaling and induce the CAR-T cell dysfunction [205]. Both preclinical and several clinical trials suggest that the PD1/PD-L1 blockade and CAR-T combination therapies can achieve synergistic anti-tumor activity [216,217,218]. To eliminate the negative effects of PD-1/PD-L1 axis on the function of CAR-T cells, CAR-T cells have been modified with the knockdown of the PD1-encoding gene PDCD1 [219]. These PD1-deficient CAR-T cells possess increased antitumor activity similar to the combination of CAR-T cells and anti-PD1 antibodies without the systemic toxicity. In addition to PD1/PD-L1, inhibition of other immunosuppressive pathways has also been explored in ADT. For example, as discussed above, TGF-β is a major immunosuppressive regulator affecting multiple immune cells in the TME. Knockout of the TGF- β signaling in CAR-T cells enhances CAR-T cell proliferation and augments antitumor activity [220].
Combination with lymphodepletion
After CAR-T therapy, relapse with malignancies carrying the target antigen represents a potential opportunity to re-treat with the same CAR-T cell therapy. However, in many cases, a re-challenge with the same CAR-T cells frequently fails to induce a response [206]. One mechanism of resistance is that patients have developed an immune response to the single-chain variable fragment of CAR that can eliminate re-infused CAR-T cells. To prevent the development of immune response to the CAR, intensified lymphodepletion before CAR-T therapy has been used with improved clinical activities [221, 222]. Furthermore, lymphodepletion can minimize the effects of regulatory T cells, reduce other immune cells that compete for homeostatic cytokines and enhance APC activation.
Combination of two different CAR-T cell therapies
Instead of using lymphodepletion to prevent immune response and elimination of CAR-T cells, an alternative strategy is to combine two types of CAR-T cells with different structures of CARs targeting the same target molecules. In this case, the second type of CAR-T cells can survive and kill tumor cells even after the recipient patients develop immune response to the first CAR-T cells. It has been shown that humanized CD19 CAR-T therapy can overcome immune-mediated rejection of murine-derived anti-CD19 CAR-T therapy [223].
Another ACT combination strategy is to use CAR-T cells targeting two different antigens on tumor cells. One mechanism of ACT failure is tumor cell heterogeneity wherein some tumor cells do not express the target molecule. Targeting two different molecules on the same malignant cell can maximize tumor cell killing and decrease recurrence. This can be achieved through a tandem construct of a single CAR vector that targets two different antigens [224] or a combination of two different types of CAR-T cells, each targeting different antigens [210].
Combination with immune modulators
In order to create an immunostimulatory milieu, CAR-T cells can be combined with other immunostimulatory molecules, such as immunostimulatory cytokines, cytokine receptors and co-stimulatory molecules, or even modified to directly express these molecules as seen in the fourth generation of CAR-T cells [225]. IL-12 is one of the cytokines that has been extensively studied in preclinical models. It can enhance the cytotoxic activity of CD8 + T cells and NK cells, stimulate the Th1 helper T cell response and counteract the immunosuppression by Treg and MDSCs. A clinical trial with the fourth generation of CAR-T TRUCK cells expressing IL-12 has been initiated [226].
Combination with small molecule inhibitors
In addition to its direct antitumor activities, ibrutinib, a BTK inhibitor, has several other immunomodulatory effects. It can downregulate PD1 expression in CD4+ and CD8 + T cells, PD-L1 expression on CLL-affected B-cells and IL-10 production [227]. Furthermore, ibrutinib is an irreversible ITK inhibitor which leads to a preferential inhibition of Th2 in favor of the Th1 differentiation [113], and conversion of MDSCs to dendritic cells [228]. Currently, several clinical trials with the combination of ibrutinib and CAR-T cell therapy are ongoing. In addition to ibrutinib, several other tyrosine kinase inhibitors, such as EGFR inhibitors, are being explored in combination regimens.
Combination with oncolytic virotherapy (OV)
Several preclinical studies on the antitumor activity of oncolytic virotherapy with ACT have already been reported [229]. With this combination, oncolytic viruses can target and kill cancer cells, stimulate an innate immune response and create a stimulatory immune milieu to potentiate ACT. Because OVs are usually cancer-specific, they can express transgenes on the surface of cancer cells which can then be recognized by CAR-T cells. Furthermore, armed OVs can express cytokines that can attract CAR-T cells to the tumor sites and enhance the cytotoxicity of CAR-T cells [230]. So far, this combination has yet to be translated into clinical applications.
Virotherapy and innate immune modifiers
Innate immune system
Overview of the innate immune system
The innate immune system is the first line of defense against infections and foreign substances. In addition to anatomical barriers, it consists of different cells involved in broad-spectrum pattern-based recognition and response to foreign substances. It is not only an obligate prerequisite for the induction of adaptive immune response, but also a prerequisite of effective ways of clearing foreign substance, including killing tumor cells independent of T cells. Like adaptive immune response, the innate immune system is also tightly regulated through built-in stimulatory and inhibitory feedback networks which are being exploited for cancer immunotherapy. Considering the overall disappointing efficacy of ICBs, innate immune cells and their regulatory molecules represent attractive alternatives for improving and/or complementing ICBs for cancer therapy (Table 7). Natural killer cells and phagocytes, including macrophages and dendritic cells, are commonly studied for cancer immunotherapy [165, 169, 192, 231].
Enhancing the innate immune system for cancer immunotherapy
NK cells can be stimulated through the activating receptors, such as NKP46, NKP30, NKP44, NKG2D, CD16 and 2B4, and via cytokines, such as IL-2 and IL-15 [232]. Activation of NK cells can directly contribute to anti-cancer immunity through their direct cytotoxic activities, attraction of dendritic cells and cytokine production, such as IFN-γ. IL-2 can not only stimulate cytotoxic T cells, but also expand NK cells. ALT-803 includes an IL-15 mutant (IL-15N72D) and a dimeric IL-15 receptor α sushi domain-IgG1 Fc fusion protein. It acts as an IL-15 super-agonist [233]. Because of the critical roles that NK cells also play in adaptive immune response, clinical trials are currently ongoing not only for ALT-803 alone, but also in combination with ICBs (clinicaltrials.gov ID: NCT02523469, NCT03228667 and NCT03853317).
Dendritic cells play critical roles in antigen presentation, cross-priming and activation of cytotoxic T cells, and produce CXCL9 and CXCL10 to recruit tumor-specific T cells to the tumor sites. During immune response, NK cells release CC-chemokine ligand 5 (CCL5), XC-chemokine ligand 1 (XCL1), XCL2 and FLT3LG, which recruit dendritic cells to stimulate the immune response [234]. Dendritic cells are strongly activated by type I interferons which are induced upon activation of specific pattern-recognition receptors, such as toll-like receptor (TLR) 3, TLR 7 and TLR 9. Multiple drugs targeting TLRs have already reached the clinical trial stage in combination with standard of care or ICBs [235]. In one study, a TLR3 agonist poly-ICLC was combined with Fms-like tyrosine kinase 3 ligand (Flt3L) and localized radiotherapy in patients with indolent non-Hodgkin’s B cell lymphoma. FLT3L promotes the commitment of hematopoietic progenitor to the dendritic cell lineage, recruits intratumoral dendritic cells and enhances dendritic cell survival and proliferation. As discussed above, local radiotherapy converts cancer cells into an in situ vaccine while poly-ICLC activates dendritic cells. This combination increases dendritic cell number and activation at tumor sites and induces abscopal effects [236].
Type I IFN expression can be induced by the cGAS-STING pathway which plays critical roles in the innate immune system [237,238,239]. Upon binding to cytosolic DNA, cGAS triggers the reaction of GTP and ATP to form cyclic GMP-AMP which binds to STING. In addition to induction of Type I IFN expression, activation of the cGAS-STING pathway upregulates the expression of NK cell receptor NKG2D ligands that can lead to the recognition and destruction of cancer cells by NK cells and T cells. Because of the important roles of the cGAS-STING pathway in stimulating the anti-cancer immunity, several STING agonists have entered into clinical trials with the combination of ICBs to improve their clinical efficacy [240].
Macrophages are professional phagocytes that can internalize large particles, such as debris, apoptotic cells and pathogens, to maintain homeostasis in the human body. According to their functions, TAM can be classified into two major subsets: classically activated inflammatory macrophages (M1, CD80 + and CD86 +) that promote anti-cancer immunity and alternatively activated anti‐inflammatory macrophages (M2, CD 68 +, CD163 + and CD206 +) that are immunosuppressive. Given the key roles played by macrophages, several cytokines (such as IL-12), immunoagonists and inhibitors of TAMs are being explored to polarize and tilt macrophages to the M1 phenotype. For example, CpG oligonucleotide, which is also a TLR9 agonist, can not only induce macrophage polarization to the M1 phenotype [241], but also induce memory T cells and abscopal anti-tumor activity [242].
Suppression of the negative regulators of the innate immune system
Like the adaptive immune response, the innate immune system has many negative regulators that can be targeted to boost anti-cancer immunity. The immunoreceptor tyrosine-based inhibition motif (ITIM) is a conserved amino acid sequence found in the cytoplasmic tails of many inhibitory receptors on immune cells [243]. Some of the ITIM-containing receptors, such as PD1, has already been targeted for cancer therapy. The killer-cell immunoglobulin-like receptors (KIRs) are a family of ITIM-containing receptors identified from the seminal discoveries in 1990s by Alessandro Moretta et al. [244]. They are expressed on the plasma membrane of NK cells and some T cells. Most KIRs are inhibitory in that their recognition of MHC molecules suppresses the cytotoxic activity of NK cells [245]. Under normal conditions, these KIRs recognize autologous cells and prevent auto-reactive cytotoxicity which can also dampen the cytotoxicity of NK cells against HLA-expressing cancer cells. An anti-pan-KIR2D agent, lirilumab, has already reached clinical trials. The combination of lirilumab and nivolumab is well tolerated and showed promising clinical activities with an overall response of 76% (16/21) in relapsed/refractory classical Hodgkin lymphoma [246]. Other ITIM-containing receptors are being extensively studied [247]. For example, LIR-1 (binding to HLA-G and other low affinity HLA ligands) is expressed on approximately one-third of NK cells. An antibody blocking LIR-1 significantly potentiated the tumoricidal activity of NK cells in vitro and in vivo [248].
In addition to KIRs, other MHC-recognizing receptors are also important in regulating NK cell functions and tumor eradication. NKG2A (binding to HLA-E) is constitutively expressed in approximately 50% NK cells and can be induced in T cells upon cytokine stimulation or antigen-induced activation [249]. Preclinical studies showed that an anti-NKG2A antibody had remarkable antitumor effects and synergized with the anti-PD1 antibody durvalumab in unleashing NK and CD8 + T cell function [250]. Promising results were shown in a Phase II trial of the humanized anti-NKG2A antibody monalizumab in combination with cetuximab with a 31% objective response rate in patients with previously treated head and neck cancers [250].
Since most of the TAMs are M2 macrophages, several approaches have been explored to inhibit or deplete TAMs for cancer immunotherapy. Several cytokines, such as CCL5 and CCL2/CCR2, are involved in recruiting macrophages to the tumor sites and promoting cancer growth. Several preclinical studies showed that tumor growth inhibition could be achieved through targeting these cytokines or with depletion of TAMs. However, they have yet to be translated into clinical applications as these cytokines have other functions in addition to recruiting macrophages [251].
CD47 is a transmembrane protein expressed in all types of cells, serves as a self‐marker and interacts with signal regulatory protein α (SIRPα) to inhibit phagocytosis by immune cells [252]. SIRPα is a transmembrane protein expressed on macrophages, granulocytes, monocytes, dendritic cells and neurons. The recognition of CD47 by SIRPα generates a “don't eat me” signal and has been used by cancer cells to evade anti-cancer immunity. Many inhibitors have been developed to block the CD47‐SIRPα pathway and enhance tumor immunotherapy, including anti‐CD47 therapy and anti‐SIRPα therapy [253]. An early clinical trial showed that the combination of an anti-CD47 blocking antibody Hu5F9-G4 and the CD20 antibody rituximab had promising clinical activities with an objective response rate of 40% with relapsed or refractory diffuse large B-cell lymphoma and 71% with follicular lymphoma [254].
Virotherapy
Overview
Virotherapy uses viruses to target and kill cancer cells, induce innate and adaptive immune response for cancer treatment. Adenoviruses, herpes viruses, measles viruses, coxsackie viruses, polioviruses, reoviruses, poxviruses and Newcastle disease viruses, among others, are some of the oncolytic viruses (OVs) under preclinical and clinical development for cancer therapy [255]. Initially wild type viruses were used. Because those viruses are potentially associated with adverse events caused by viral replication in normal cells, almost all OVs under current development are modified with strong cancer tropism and usually armed with transgenes to enhance the efficacy and/or potentiate combination therapy. So far, three OVs have been approved and used at clinic: Rigvir (an oncolytic picornavirus) [256], H101 or Oncorine (an adenovirus) [257] and talimogene laherparepvec, also known as T-vec (a herpes simplex-1 virus encoding for GM-CSF) [179].
Mechanisms of action
Several mechanisms have been proposed for the action of OVs. First, OVs can directly target, lyse and kill cancer cells. Cancer tropism can occur naturally with OVs as oncogenic signaling pathways are more active in cancer cells which can facilitate viral replication. More recently, genetic engineering has been used to make cancer-specific OVs. For example, mutations/deletions can be introduced in genes that are required for replication in normal but not in cancer cells; critical viral genes are under the control of cell-specific promoters active in cancer cells, but not in normal cells; and expression of cancer-targeting viral receptors can guide cancer-specific tropism [258]. Second, OVs can activate the innate immune response. Viral replication and destruction of tumor cells and release of viral DNA can induce innate immune response locally to kill more cancer cells. Third, OVs can induce adaptive immune response. Viral replication and destruction of tumor cells alter TME, trigger chemotaxis and accumulation of cytotoxic lymphocytes to the site of infection, and can convert immune “cold” to “hot” tumors; viral replication induces ICD, and releases DAMPs, such as calreticulin, high-mobility group protein B1 (HMGB1) and ATP, along with tumor-associated antigens. Fourth, OVs can be engineered to express transgenes to stimulate immune response. For example, talimogene laherparepvec expresses GM-CSF and was FDA approved for treatment of recurrent melanoma. Several other immunoregulatory genes are currently being explored for their potentiation to stimulate immunotherapy with OVs, such as IL-2, IL-12, IFN-α/β, 4-1BB and CD40L [259].
OVs armed with transgenes
Even though oncolytic virotherapy showed promising preclinical activities, clinical activities are still moderate. In cutaneous melanoma, intratumoral injection of talimogene laherparepvec is associated with an overall response rate of 26.4% [179]. One advantage of OVs is that they can be armed with transgenes and combined with several therapeutic interventions. So far transgenes targeting every step along the anti-cancer immunity cycle have been studied at least in preclinical models (Table 7) [260]. For example, oncolytic adenovirus armed with bi-specific T cell engager (BiTE) has been demonstrated to induce both oncolysis by OV and engagement and activation of cytotoxic T cells which led to immune-mediated destruction of cancer cells both in vivo with tumor implants and in primary ex vivo patient specimens [261, 262].
Combination therapies with OVs in clinical development
While most OVs armed with the transgenes listed on Table 8 are still at the preclinical stages, several clinical trials combining OVs with another therapeutic agent have been initiated [263]. The most common combination therapy is OVs and ICBs. These two agents have complementary mechanisms of anti-cancer immunity in that ICBs release the suppression of anti-cancer immune response while OVs stimulate the immune response. The combination was first tested in preclinical models with an oncolytic Newcastle disease virus (NDV) in combination with systemic CTLA-4 blockade [264]. Local virotherapy induced abscopal effects and immune cell infiltration at distant tumors.
Since then, many trials combining OVs with anti-CTLA and anti-PD1/PD-L1 antibodies either have already been completed or are in progress to determine the efficacy and toxicity of the combinations. In one study with unresectable stage IIIB–IV melanoma, the combination of ipilimumab and talimogene laherparepvec significantly improved the objective response rate to 39% from 18% with ipilimumab alone (OR 2.9, 95% CI 1.5–5.5, P = 0.002), and induced abscopal effects on visceral lesions [265]. In addition to talimogene laherparepvec, several other major OVs are being combined with ICBs in clinical trials.
OVs have also been tested in clinical trials with chemotherapy. In a Phase I/II trial of carboplatin/paclitaxel plus reovirus, the combination therapy was well tolerated with significant clinical activity and minimal viral toxicity [266]. Another Phase I trial determined the toxicity of the combination of gemcitabine with replication-competent adenovirus Ad5-yCD/mutTK(SR39)rep-ADP (Ad5-DS) in locally advanced pancreatic cancer. This adenovirus expressed double-suicide genes: yeast cytosine deaminase (yCD) and herpes simplex virus 1 thymidine kinase (HSV-1 TK) [267]. This combination was well tolerated in early clinical trials.
Therapeutic cancer vaccine
Cancer vaccine is a targeted cancer immunotherapy that uses putative cancer antigen(s) or antigenic epitope(s) presented in protein, RNA, DNA, viral or bacterial vectors, cells or other means to stimulate anti-cancer immunity. In contrast to vaccination for infectious diseases in general, the efficacy of early cancer vaccines is disappointing, with objective response rates less than 5% [268]. More recently, with the understanding of anti-cancer immunity and development of vaccine technology, deep sequencing and bioinformatics, personalized cancer vaccines showed promising clinical activities. However, many patients do not respond. The reason for low efficacy for cancer vaccines is likely to be multifactorial. Cancer vaccines are used in patients whose immune system has already tolerated cancer and cancers can develop or have already developed an immunosuppressive TME that prevents anti-cancer immunity. In contrast, vaccines for infectious diseases are exogenous antigens that hosts have not developed resistance.
Cancer vaccine antigens
There are two major types of cancer vaccines: shared tumor-associated antigens (TAA) and unique tumor antigens (Table 9). Shared TAAs include cancer/testis antigens, cell differentiation antigens and overexpressed cell antigens. Cancer/testis antigens are a group of proteins, such as cancer/testis antigen 1 (CTAG1B, often referred to New York esophageal squamous cell carcinoma 1 or NY-ESO-1), melanoma-associated antigen 1 (MAGE-A1), MAGE-A3, etc., that are normally expressed in immune privileged germline cells, but upregulated in some cancer cells. Cell differentiation antigens are a group of antigens expressed in differentiated tissues from which some cancers develop and share those antigens, such as glycoprotein 100 (gp100), prostatic acid phosphate, and prostate-specific antigen (PSA). Overexpressed cell antigens are expressed in normal cells, but significantly upregulated in some cancer cells, such as mucin 1 (MUC1) or epithelial membrane antigen, mesothelin and HER2. There are two major issues associated with shared TAAs. First, the immune system has already developed tolerance to these antigens. Hence, even with strong adjuvants, co-stimulators or repeated vaccinations, anti-cancer immunity may develop, but is not sufficient to eliminate cancers. Second, these antigens are also expressed in some normal cells. Hence, untoward damage may develop against those normal cells/tissues expressing the target antigens.
Unique tumor antigens include oncogenic viral antigens associated with viral infection and tumor neoantigens associated with cancer genomic alterations, such as mutations, frame shift and gene fusion. The three most commonly studied viruses associated with cancers and cancer vaccines are hepatitis B virus, human papillomavirus (HPV) and Epstein–Barr virus (EBV). Because these unique tumor antigens appear after the immune system has already developed, the central tolerance to these antigens and cross-reactivity to normal tissues are usually low and the likelihood of induction of immune response to them is high.
Vaccine delivery vehicles
Several approaches have been used in clinic to deliver cancer antigens in order to elicit anti-cancer immunity (Table 9). Cell-based vaccines use autologous cancer cells, antigen-presenting cells or allogeneic cells to deliver cancer vaccine antigens. When cancer cells are used, cells are treated in vitro, usually with radiation, to prevent further cell division before administration. The advantage of cell-based vaccine is that specific target antigens do not need to be prospectively identified, and dendritic cells can present tumor antigens in the context of the MHC that can be further potentiated with a stimulatory cytokine. Sipuleucel-T is FDA approved for treatment of metastatic prostate cancer and is comprised of autologous dendritic cells activated ex vivo with expression of a chimeric protein of immune stimulatory cytokine, GM-CSF, fused to a cell differentiation tumor antigen PAP. In a Phase III clinical trial, the median overall survival (OS) of the sipuleucel-T group was about 4 months longer than the control cohort treated with placebo [269]. However, the vaccine treatment was not associated with any biomarker (PSA) or radiological response. In fact, subgroup analyses showed there was no OS difference between the therapeutic and control groups of young patients (< 65 years) who were supposed to have more robust immune response [270]. Several other cell-based vaccines are also at clinical trials. Both gemogenovatucel-T and GVAX are tumor cell vaccines expressing GM-CSF to promote antigen presentation, activation and survival of dendritic cells. Some other cellular vaccines are manipulated to down-regulate the expression of immunosuppressive factors. For example, belagenpumatucel-L is a mixture of four irradiated human NSCLC cell lines, transfected with a TGF-β2 antisense gene. A higher response rate was observed in patients with NSCLC treated with higher doses of Belagenpumatucel-L [271]. However, a Phase III trial failed to demonstrate the OS benefit when it was used as a maintenance therapy after platinum-based chemotherapy in Stage III/IV non-small cell lung cancer [272]. Gemogenovatucel-T expresses a bi-functional short hairpin RNA to knock down the expression of the enzyme furin which converts immunosuppressive TGF-β1 and TGF-β2 into active isoforms. Even though those cell-based vaccines showed promising activities in preclinical models and stimulated immune response in patients, their clinical efficacy has yet to be proven [273].
In addition to human cells, microorganisms such as bacteria and yeasts are also being explored for cancer vaccine therapy. Heated-inactivated bacteria and Bacillus Calmette-Guérin have been used in clinic to treat cancers for decades. But strictly, they are not cancer vaccines as they do not carry tumor antigens. Other bacteria, such as Listeria, can deliver DNA- and RNA-encoded tumor antigens directly into mammalian cells, including APCs, and are being tested as cancer vaccine vehicles [274].
For microorganisms, viral vectors have been studied as a vehicle to deliver cancer vaccines. For viral vectors, in addition to its ability to induce innate and adaptive immune response, exogenous genes can be incorporated and expressed, including cytokines and tumor antigens. One potential disadvantage of using a viral vector to deliver a cancer vaccine is that the immune response induced by previous vaccinations or infections of the same virus produces a neutralizing antibody and clears subsequent vaccinations. One approach is to administer the vaccine through intratumoral injection as talimogene laherparepvec. Another approach is to use a heterologous prime/boost strategy which cancer antigens are delivered by two different viral vectors at the primary and boost vaccinations (Table 10).
Peptide vaccines have also been used in vaccine studies as T cells recognize tumor antigens presented by MHC. When short peptides are used, they can directly bind to MHC molecules on any nucleated cells. As T cell activation requires the engagement of T cell receptors by antigens presented with MHC as well as a second signal from a co-stimulatory molecule, presentation of short peptide vaccines by nucleated cells other than APCs often induces T cell anergy and immune tolerance since other nucleated cells lack co-stimulatory signals. To overcome the immune tolerance associated with short peptides, synthetic long peptides (SLPs) have been used. SLPs are preferentially taken up and processed by dendritic cells which also provide co-stimulatory molecules to prime and activate T cell immunity. To further improve the efficacy, SLPs are often formulated with inflammatory adjuvants. For example, synthetic cancer neoantigen long peptide vaccines formulated with a toll-like receptor 3 (TLR3) ligand poly-ICLC (polyinosinic and polycytidylic acid) induced strong anti-cancer immunity and clinical response [275]. In this study, vaccination stimulated polyclonal neoantigen-specific CD4 + and CD8 + T cell responses and immune cell migration into intracranial glioblastoma tumors. Systemic immune response was abolished in those patients who received the immunosuppressive steroid dexamethasone during vaccine priming.
Peptides and proteins have also been formulated in nanoparticles to enhance its efficacy. Nanoparticles can protect peptides/proteins from degradation, prolong circulation time, control antigen release, confer targeted delivery through decorating cancer targeting peptides on the surface[276,277,278], and formulate with adjuvant agents to enhance immune response [279, 280]. Furthermore, by optimizing the structure, surface charge and cell penetrating peptides, the efficacy of nanoparticles can possibly be further enhanced [281, 282]. So far, various nanomaterials, such as polymeric materials, liposomes, micelles, silica nanoparticles, gold nanoparticles (AuNPs) and virus nanoparticles, have been studied in preclinical models with a few translated into clinical trials, but the efficacy is still yet to be validated [279].
Instead of using peptides and proteins, DNA and RNA vaccines have also been translated into clinical trials [283]. After administration, DNA and RNA can be taken up by antigen presenting cells that then translate into peptide/proteins and present to immune cells. In addition to serving as vaccines, exogenous DNA and RNA can serve as immune stimulators, trigger nucleic acid sensors and activate dendritic cells through certain TLR and STING pathways. One major disadvantage of naked DNA and RNA vaccine is their low delivery efficiency. To overcome this obstacle, various delivery methods have been developed, such as viral vectors and nanoparticles as discussed above, gene gun, electroporation and so on [284]. Two coronavirus (COVID-19) mRNA vaccines approved in the US are both RNA vaccines formulated in lipid nanoparticles [285, 286]. For cancer vaccines, RNA lipid nanoparticles, either alone or in combination with ICBs, induced durable clinical responses, associated with strong CD4 + and CD8 + T cell immunity against the vaccine antigens [287].
Combination therapy with cancer vaccines
Cancer vaccines are designed to bypass and/or stimulate the first three steps of the anti-cancer immunity cycle: cancer antigen release and presentation, immune cell priming and activation of immune cells. Once immune cells are activated, they still need to go through the remaining four steps along the cycle: mobilize in the periphery, infiltrate into cancer sites, recognize cancer cells and elicit cytotoxicity toward cancer cells. Hence, resistance mechanisms governing the anti-cancer immunity, especially those in the TME, can still dampen the efficiency of cancer vaccines and are being explored to potentiate cancer vaccines.
The most critical function of cancer vaccines is to present cancer antigens to prime and activate T cells and induce anti-cancer immunity. In many cancers, little or no anti-cancer immunity exists in patients that manifests as cold tumors with little immune cell infiltration at the tumor sites. These cold tumors usually do not respond to ICBs, as there is no ammunition to fire at cancer cells upon removal of the brake by ICBs. To improve their efficacy, cancer vaccines have been extensively studied to be combined with adjuvant agents, such as a TLR-3 agonist poly-ICLC, to stimulate immune response. Immune response to the cancer/testis antigen NY-ESO-1 is higher when NY-ESO-1 vaccine is combined with poly-ICLC [288].
Many trials are currently ongoing to determine the efficacy and toxicity of cancer vaccines in combination with cytokines. For example, IL-2 plays critical roles in key functions of immune response. Compared to IL-2 alone, the combination of IL-2 with a tumor-associated antigen gp100 significantly improved the overall response rate (ORR: 16% vs. 6%, p = 0.03) and progression-free survival (PFS: 2.2 versus 1.6 months, p = 0.008) [289]. In addition to IL-2, several other immunostimulatory cytokines are being explored. IL-12 is a multipotent cytokine that stimulates T and NK cells, regulates other cytokines and multiple aspects of immune response. Several clinical trials are currently ongoing to determine the efficacy and toxicity of IL-12 and cancer vaccine combinations.
Extensive preclinical studies as well as many clinical trials are currently ongoing to combine cancer vaccines with ICBs. GX-188E (tirvalimogene teraplasmid) is a therapeutic HPV DNA vaccine that encodes HPV-16 and HPV-18 E6 and E7 [290]. When GX-188E was combined with pembrolizumab in HPV-16/18-positive advanced cervical cancer, 11 of 26 patients (42%; 95% CI 23–63%) achieved a response; four patients (15%) had a CR at 24 weeks. This combination was well tolerated [291]. Similar results were observed in another Phase II trial with a synthetic long-peptide HPV-16 vaccine ISA101 combined with nivolumab in HPV-16-positive solid tumors; this combination showed a 33% ORR and 17.5-month median OS [292]. Other than concurrent use, ICB has also been shown to have activity as a salvage therapy after cancer vaccine failure. Ott et al. showed that pembrolizumab induced complete responses in melanoma patients after failure of treatment with synthetic long peptide vaccine of tumor neoantigens with poly-ICLC adjuvant [293]. With a follow-up of almost four years, long-term persistence of neoantigen-specific T cells was still observed with the development of memory T cell phenotype, tumor infiltration and epitope spreading [294].
In addition to multiple studies focusing on anti-PD1/PD-L1 and anti-CTLA4 antibodies, other immune co-inhibitory and co-stimulatory agents are being actively studied to enhance the efficacy of cancer vaccines. One Phase I clinical trial has already been reported with a melanoma-associated antigen recognized by T cells 1 (MART-1) peptide vaccine plus IMP321, a fusion protein consisting of four LAG-3 extracellular Ig-like domains fused to the Fc fraction of a human IgG1 (LAG-3Ig). The combination group is associated with significant increase of MART-1-specific CD8 T cells (p < 0.02), higher proportion of CCR7− CD45RA+/− CD8 T effector cells (p < 0.02) and reduced expansion of regulatory T cells (p < 0.04) [295].
Conclusion remarks
A few combination therapies have been approved by the FDA to improve clinical efficacy of ICIs. With increasing research in identifying action-driven reliable biomarkers in guiding clinical immuno-oncology decisions, IO combinations among ACT, novel ICIs, cancer vaccines and small molecule inhibitors are expected. In this regard, the future of cancer immunotherapy awaits for a truly patient-oriented, individualized approach.
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Abbreviations
- ACT:
-
adoptive cell therapy
- ADT:
-
androgen deprivation therapy
- APC:
-
antigen presenting cell
- ATP:
-
adenosine triphosphate
- B-ALL:
-
B-cell acute lymphoblastic leukemia
- BiTE:
-
bi-specific T cell engager
- BTK:
-
Bruton's tyrosine kinase
- CAF:
-
cancer-associated fibroblasts
- CAHON:
-
Chinese American Hematologist and Oncologist Network
- CAR:
-
chimeric antigen receptor
- CCL:
-
C–C motif chemokine ligand
- CD:
-
cluster of differentiation
- CDK:
-
cyclin-dependent kinase
- cGAS:
-
cyclic GMP-AMP synthase
- CLL:
-
chronic lymphocytic leukemia/lymphoma
- CTLA4:
-
cytotoxic T-lymphocyte-associated protein 4
- CXCL:
-
C-X-C motif chemokine ligand
- DAMP:
-
damage-associated molecular pattern
- dMMR:
-
defective DNA mismatch repair system
- DNA:
-
deoxyribonucleic acid
- DNMT:
-
DNA methyltransferase
- EBV:
-
Epstein–Barr virus
- EGFR:
-
epidermal growth factor receptor
- EMT:
-
epithelial–mesenchymal transition
- FDA:
-
Food and Drug Administration
- GM-CSF:
-
granulocyte–macrophage colony-stimulating factor
- HDAC:
-
histone deacetylase
- HLA:
-
human leukocyte antigen
- HMGB1:
-
high-mobility group protein B1
- HPV:
-
human papilloma virus
- HSP:
-
heat shock protein
- ICB:
-
immune checkpoint blocker
- ICD:
-
immunogenic cell death
- IDO:
-
indolamine-2,3-dioxygenase
- IFN:
-
interferon
- IL-2:
-
interleukin-2
- IO:
-
immuno-oncology
- ITIM:
-
immunoreceptor tyrosine-based inhibition motif
- ITK:
-
IL-2-inducible T cell kinase
- KIR:
-
killer-cell immunoglobulin-like receptor
- LAG-3:
-
lymphocyte-activation gene 3
- MAGE-A1:
-
melanoma-associated antigen 1
- MART-1:
-
melanoma-associated antigen recognized by T cells 1
- MDSC:
-
myeloid-derived suppressor cell
- MEK:
-
mitogen-activated extracellular kinase
- MHC:
-
major histocompatibility complex
- MSI-H:
-
microsatellite instability high
- NDV:
-
Newcastle disease virus
- NK:
-
natural killer
- NMPA:
-
China National Medical Product Administration
- NSCLC:
-
non-small cell lung cancer
- NY-ESO-1:
-
New York esophageal squamous cell carcinoma 1
- ORR:
-
overall response rate
- OS:
-
overall survival
- OV:
-
oncolytic virotherapy
- PD1:
-
programmed cell death 1
- PDGFR:
-
platelet-derived growth factor receptor
- PD-L1:
-
programmed cell death ligand 1
- PEG:
-
polyethylene glycol
- PFS:
-
progression-free survival
- PMN-MDSC:
-
polymorphonuclear myeloid-derived suppressor cell
- poly-ICLC:
-
polyinosinic and polycytidylic acid
- PSA:
-
prostate-specific antigen
- ROS:
-
reactive oxygen species
- RT:
-
radiotherapy
- SCLC:
-
small cell lung cancer
- SIRPα:
-
signal regulatory protein α
- STING:
-
stimulator of interferon genes
- TAM:
-
tumor-associated macrophage
- TCR:
-
antigen-T cell receptor
- TGF-β:
-
transforming growth factor beta
- TIGIT:
-
T-cell immunoreceptor tyrosine-based inhibition motif domain
- TIL:
-
tumor-infiltrating lymphocyte
- TIM-3:
-
T-cell immunoglobulin, mucin domain-3 protein
- TLR3:
-
toll-like receptor 3
- TME:
-
tumor microenvironment
- TRUCK:
-
T cells redirected for antigen‐unrestricted cytokine‐initiated killing
- TVEC:
-
talimogene laherparepvec
- VEGF:
-
vascular endothelial growth factor
- VISTA:
-
V-domain immunoglobulin-containing suppressor of T-cell activation
- XCL1:
-
XC-chemokine ligand 1
- yCD:
-
yeast cytosine deaminase
References
Li Z, Song W, Rubinstein M, Liu D. Recent updates in cancer immunotherapy: a comprehensive review and perspective of the 2018 China Cancer Immunotherapy Workshop in Beijing. J Hematol Oncol. 2018;11(1):142.
Pan C, Liu H, Robins E, Song W, Liu D, Li Z, Zheng L. Next-generation immuno-oncology agents: current momentum shifts in cancer immunotherapy. J Hematol Oncol. 2020;13(1):29.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10.
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61.
Pereira C, Gimenez-Xavier P, Pros E, Pajares MJ, Moro M, Gomez A, Navarro A, Condom E, Moran S, Gomez-Lopez G, et al. Genomic profiling of patient-derived xenografts for lung cancer identifies B2M inactivation impairing immunorecognition. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23(12):3203–13.
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–29.
Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;15(4):857–65.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology. 2017;6(1):e1261779.
Huang RY, Francois A, McGray AR, Miliotto A, Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology. 2017;6(1):e1249561.
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501.
Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, Zhao H, Chen J, Chen H, Efstathiou E, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. 2017;23(5):551–5.
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–61.
Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20(6):607–15.
Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212(2):139–48.
Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, Carbone DP. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101(12):4878–86.
Gorbachev AV, Kobayashi H, Kudo D, Tannenbaum CS, Finke JH, Shu S, Farber JM, Fairchild RL. CXC chemokine ligand 9/monokine induced by IFN-gamma production by tumor cells is critical for T cell-mediated suppression of cutaneous tumors. J Immunol. 2007;178(4):2278–86.
Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168(7):3195–204.
Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochem Biophys Acta. 2014;1843(11):2563–82.
Ungefroren H. Blockade of TGF-beta signaling: a potential target for cancer immunotherapy? Expert Opin Ther Targets. 2019;23(8):679–93.
Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202(12):1691–701.
Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28(6):690–714.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541.
Patel SA, Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. 2018;48(3):417–33.
Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, Chan TA, Coukos G, Demaria S, Deutsch E, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. 2020;8(1):e000337.
Gravett AM, Trautwein N, Stevanovic S, Dalgleish AG, Copier J. Gemcitabine alters the proteasome composition and immunopeptidome of tumour cells. Oncoimmunology. 2018;7(6):e1438107.
Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS ONE. 2012;7(3):e32542.
Welters MJ, van der Sluis TC, van Meir H, Loof NM, van Ham VJ, van Duikeren S, Santegoets SJ, Arens R, de Kam ML, Cohen AF, et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci Transl Med. 2016;8(334):334ra52.
Melief CJM, Welters MJP, Vergote I, Kroep JR, Kenter GG, Ottevanger PB, Tjalma WAA, Denys H, van Poelgeest MIE, Nijman HW, et al. Strong vaccine responses during chemotherapy are associated with prolonged cancer survival. Sci Transl Med. 2020;12(535):eaaz8235.
Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res Off J Am Assoc Cancer Res. 2005;11(18):6713–21.
Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rebe C, Ghiringhelli F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–61.
Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–62.
Dimeloe S, Frick C, Fischer M, Gubser PM, Razik L, Bantug GR, Ravon M, Langenkamp A, Hess C. Human regulatory T cells lack the cyclophosphamide-extruding transporter ABCB1 and are more susceptible to cyclophosphamide-induced apoptosis. Eur J Immunol. 2014;44(12):3614–20.
Buhtoiarov IN, Sondel PM, Wigginton JM, Buhtoiarova TN, Yanke EM, Mahvi DA, Rakhmilevich AL. Anti-tumour synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumour-associated macrophages. Immunology. 2011;132(2):226–39.
West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, Johnstone RW. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res. 2013;73(24):7265–76.
Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, Keiser N, Santaniello F, Tomasi TB. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165(12):7017–24.
Fonsatti E, Nicolay HJ, Sigalotti L, Calabro L, Pezzani L, Colizzi F, Altomonte M, Guidoboni M, Marincola FM, Maio M. Functional up-regulation of human leukocyte antigen class I antigens expression by 5-aza-2’-deoxycytidine in cutaneous melanoma: immunotherapeutic implications. Clin Cancer Res Off J Am Assoc Cancer Res. 2007;13(11):3333–8.
Coral S, Sigalotti L, Altomonte M, Engelsberg A, Colizzi F, Cattarossi I, Maraskovsky E, Jager E, Seliger B, Maio M. 5-aza-2’-deoxycytidine-induced expression of functional cancer testis antigens in human renal cell carcinoma: immunotherapeutic implications. Clin Cancer Res Off J Am Assoc Cancer Res. 2002;8(8):2690–5.
Dunn J, Rao S. Epigenetics and immunotherapy: the current state of play. Mol Immunol. 2017;87:227–39.
Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, Massengill C, Noyes DR, Martinez GV, Afzal R, et al. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2016;22(16):4119–32.
Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC, Gottschalk SM, Hughes DP, Kleinerman ES, Lee DA. The narrow-spectrum HDAC inhibitor entinostat enhances NKG2D expression without NK cell toxicity, leading to enhanced recognition of cancer cells. Pharm Res. 2015;32(3):779–92.
Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005;65(14):6321–9.
Rossi C, Gilhodes J, Maerevoet M, Herbaux C, Morschhauser F, Brice P, Garciaz S, Borel C, Ysebaert L, Oberic L, et al. Efficacy of chemotherapy or chemo-anti-PD-1 combination after failed anti-PD-1 therapy for relapsed and refractory Hodgkin lymphoma: a series from Lysa centers. Am J Hematol. 2018;93:1042–9.
Park SE, Lee SH, Ahn JS, Ahn MJ, Park K, Sun JM. Increased response rates to salvage chemotherapy administered after PD-1/PD-L1 inhibitors in patients with non-small cell lung cancer. J Thorac Oncol. 2018;13(1):106–11.
Moschella F, Valentini M, Arico E, Macchia I, Sestili P, D’Urso MT, Alessandri C, Belardelli F, Proietti E. Unraveling cancer chemoimmunotherapy mechanisms by gene and protein expression profiling of responses to cyclophosphamide. Cancer Res. 2011;71(10):3528–39.
Sautes-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, Dieu-Nosjean MC. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol. 2016;7:407.
Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19(6):307–25.
Lin L, Hu X, Zhang H, Hu H. Tertiary lymphoid organs in cancer immunology: mechanisms and the new strategy for immunotherapy. Front Immunol. 2019;10:1398.
Silina K, Soltermann A, Attar FM, Casanova R, Uckeley ZM, Thut H, Wandres M, Isajevs S, Cheng P, Curioni-Fontecedro A, et al. Germinal Centers Determine The Prognostic Relevance Of Tertiary Lymphoid Structures And Are Impaired By Corticosteroids In Lung Squamous Cell Carcinoma. Cancer Res. 2018;78(5):1308–20.
Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378(22):2078–92.
West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter HJ, Kopp HG, Daniel D, McCune S, Mekhail T, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(7):924–37.
Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, Hermes B, Cay Senler F, Csoszi T, Fulop A, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379(21):2040–51.
Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, Huemer F, Losonczy G, Johnson ML, Nishio M, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379(23):2220–9.
Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, Statsenko G, Hochmair MJ, Ozguroglu M, Ji JH, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet. 2019;394(10212):1929–39.
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–21.
Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, Gallardo C, Lipatov O, Barrios CH, Holgado E, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. 2020;396(10265):1817–28.
Burtness B, Harrington KJ, Greil R, Soulieres D, Tahara M, de Castro G, Psyrri A, Baste N, Neupane P, Bratland A, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394(10212):1915–28.
Powles T, Park SH, Voog E, Caserta C, Valderrama BP, Gurney H, Kalofonos H, Radulovic S, Demey W, Ullen A, et al. Avelumab maintenance therapy for advanced or metastatic urothelial carcinoma. N Engl J Med. 2020;383(13):1218–30.
Jotte R, Cappuzzo F, Vynnychenko I, Stroyakovskiy D, Rodriguez-Abreu D, Hussein M, Soo R, Conter HJ, Kozuki T, Huang KC, et al. Atezolizumab in combination with carboplatin and nab-paclitaxel in advanced squamous NSCLC (IMpower131): results from a randomized phase III trial. J Thorac Oncol. 2020;15(8):1351–60.
Abuodeh Y, Venkat P, Kim S. Systematic review of case reports on the abscopal effect. Curr Probl Cancer. 2016;40(1):25–37.
Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, Camphausen K, Luiten RM, de Ru AH, Neijssen J, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–71.
Gameiro SR, Jammeh ML, Wattenberg MM, Tsang KY, Ferrone S, Hodge JW. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget. 2014;5(2):403–16.
Yoshimoto Y, Oike T, Okonogi N, Suzuki Y, Ando K, Sato H, Noda SE, Isono M, Mimura K, Kono K, et al. Carbon-ion beams induce production of an immune mediator protein, high mobility group box 1, at levels comparable with X-ray irradiation. J Radiat Res. 2015;56(3):509–14.
Vermeer DW, Spanos WC, Vermeer PD, Bruns AM, Lee KM, Lee JH. Radiation-induced loss of cell surface CD47 enhances immune-mediated clearance of human papillomavirus-positive cancer. Int J Cancer. 2013;133(1):120–9.
Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24(2):225–32.
Spitz DR, Azzam EI, Li JJ, Gius D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23(3–4):311–22.
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843–52.
Tigano M, Vargas DC, Tremblay-Belzile S, Fu Y, Sfeir A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature. 2021;591(7850):477–81.
Kho VM, Mekers VE, Span PN, Bussink J, Adema GJ. Radiotherapy and cGAS/STING signaling: impact on MDSCs in the tumor microenvironment. Cell Immunol. 2021;362:104298.
Zhang MdJ, Zhang MdL, Yang MdY, Liu MdQ, Ma MdH, Huang MdA, Zhao MdY, Xia MdZ, Liu MdT, Wu MdG. Polymorphonuclear-MDSCs facilitate tumor regrowth after radiation by suppressing CD8(+) T cells. Int J Radiat Oncol Biol Phys. 2021;109(5):1533–46.
Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, Zheng W, Mauceri H, Mack M, Xu M, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. 2017;8(1):1736.
De Martino M, Daviaud C, Diamond JM, Kraynak J, Alard A, Formenti SC, Miller LD, Demaria S, Vanpouille-Box C. Activin A promotes regulatory T-cell-mediated immunosuppression in irradiated breast cancer. Cancer Immunol Res. 2021;9(1):89–102.
Klopp AH, Spaeth EL, Dembinski JL, Woodward WA, Munshi A, Meyn RE, Cox JD, Andreeff M, Marini FC. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007;67(24):11687–95.
Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, Barcellos-Hoff MH, Demaria S. TGFbeta is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 2015;75(11):2232–42.
Grabham P, Sharma P. The effects of radiation on angiogenesis. Vasc Cell. 2013;5(1):19.
Li Y, Patel SP, Roszik J, Qin Y. Hypoxia-driven immunosuppressive metabolites in the tumor microenvironment: new approaches for combinational immunotherapy. Front Immunol. 2018;9:1591.
Menon H, Ramapriyan R, Cushman TR, Verma V, Kim HH, Schoenhals JE, Atalar C, Selek U, Chun SG, Chang JY, et al. Role of radiation therapy in modulation of the tumor stroma and microenvironment. Front Immunol. 2019;10:193.
Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125(9):3365–76.
Zhang Z, Yang S, Wang Q. Impact of MET alterations on targeted therapy with EGFR-tyrosine kinase inhibitors for EGFR-mutant lung cancer. Biomark Res. 2019;7(1):27.
Zhang F, Manna S, Pop LM, Chen ZJ, Fu YX, Hannan R. Type I interferon response in radiation-induced anti-tumor immunity. Semin Radiat Oncol. 2020;30(2):129–38.
Wu CT, Chen MF, Chen WC, Hsieh CC. The role of IL-6 in the radiation response of prostate cancer. Radiat Oncol. 2013;8:159.
Mortezaee K, Najafi M. Immune system in cancer radiotherapy: Resistance mechanisms and therapy perspectives. Crit Rev Oncol Hematol. 2021;157:103180.
Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, Mu Z, Rasalan T, Adamow M, Ritter E, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366(10):925–31.
Shaverdian N, Lisberg AE, Bornazyan K, Veruttipong D, Goldman JW, Formenti SC, Garon EB, Lee P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017;18(7):895–903.
Geng Y, Zhang Q, Feng S, Li C, Wang L, Zhao X, Yang Z, Li Z, Luo H, Liu R, et al. Safety and efficacy of PD-1/PD-L1 inhibitors combined with radiotherapy in patients with non-small-cell lung cancer: a systematic review and meta-analysis. Cancer Med. 2021;10(4):1222–39.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, Yokoi T, Chiappori A, Lee KH, de Wit M, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377(20):1919–29.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, Kurata T, Chiappori A, Lee KH, de Wit M, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379(24):2342–50.
Jagodinsky JC, Harari PM, Morris ZS. The promise of combining radiation therapy with immunotherapy. Int J Radiat Oncol Biol Phys. 2020;108(1):6–16.
Qian Y, Gong Y, Fan Z, Luo G, Huang Q, Deng S, Cheng H, Jin K, Ni Q, Yu X, et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J Hematol Oncol. 2020;13(1):130.
Vanpouille-Box C, Formenti SC, Demaria S. Toward precision radiotherapy for use with immune checkpoint blockers. Clin Cancer Res Off J Am Assoc Cancer Res. 2018;24(2):259–65.
Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15(17):5379–88.
Kaur P, Asea A. Radiation-induced effects and the immune system in cancer. Front Oncol. 2012;2:191.
Grapin M, Richard C, Limagne E, Boidot R, Morgand V, Bertaut A, Derangere V, Laurent PA, Thibaudin M, Fumet JD, et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: a promising new combination. J Immunother Cancer. 2019;7(1):160.
Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174(12):7516–23.
Camphausen K, Moses MA, Menard C, Sproull M, Beecken WD, Folkman J, O’Reilly MS. Radiation abscopal antitumor effect is mediated through p53. Cancer Res. 2003;63(8):1990–3.
Wang SJ, Jhawar SR, Rivera-Nunez Z, Silk AW, Byun J, Miller E, Blakaj D, Parikh RR, Weiner J, Goyal S. The association of radiation dose-fractionation and immunotherapy use with overall survival in metastatic melanoma patients. Cureus. 2020;12(6):e8767.
Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618.
Pan CX, Zhang H, Tepper CG, Lin TY, Davis RR, Keck J, Ghosh PM, Gill P, Airhart S, Bult C, et al. Development and characterization of bladder cancer patient-derived xenografts for molecularly guided targeted therapy. PLoS ONE. 2015;10(8):e0134346.
Zeng SX, Zhu Y, Ma AH, Yu W, Zhang H, Lin TY, Shi W, Tepper CG, Henderson PT, Airhart S, et al. The phosphatidylinositol 3-kinase pathway as a potential therapeutic target in bladder cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23(21):6580–91.
Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH. Eastern cooperative oncology G: comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346(2):92–8.
Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–42.
Robert C, Ribas A, Hamid O, Daud A, Wolchok JD, Joshua AM, Hwu WJ, Weber JS, Gangadhar TC, Joseph RW, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol. 2018;36(17):1668–74.
Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, Khan N, Ubellacker JM, Xie S, Metzger-Filho O, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548(7668):471–5.
Long Q, Ma AH, Zhang H, Cao Z, Xia R, Lin TY, Sonpavde GP, de Vere WR, Guo J, Pan CX. Combination of cyclin-dependent kinase and immune checkpoint inhibitors for the treatment of bladder cancer. Cancer Immunol Immunother. 2020;69(11):2305–17.
Chandrasekaran S, Sasaki M, Scharer CD, Kissick HT, Patterson DG, Magliocca KR, Seykora JT, Sapkota B, Gutman DA, Cooper LA, et al. Phosphoinositide 3-kinase signaling can modulate MHC class I and II expression. Mol Cancer Res. 2019;17(12):2395–409.
Basu A, Hoerning A, Datta D, Edelbauer M, Stack MP, Calzadilla K, Pal S, Briscoe DM. Cutting edge: vascular endothelial growth factor-mediated signaling in human CD45RO+ CD4+ T cells promotes Akt and ERK activation and costimulates IFN-gamma production. J Immunol. 2010;184(2):545–9.
Kaur S, Chang T, Singh SP, Lim L, Mannan P, Garfield SH, Pendrak ML, Soto-Pantoja DR, Rosenberg AZ, Jin S, et al. CD47 signaling regulates the immunosuppressive activity of VEGF in T cells. J Immunol. 2014;193(8):3914–24.
Ziogas AC, Gavalas NG, Tsiatas M, Tsitsilonis O, Politi E, Terpos E, Rodolakis A, Vlahos G, Thomakos N, Haidopoulos D, et al. VEGF directly suppresses activation of T cells from ovarian cancer patients and healthy individuals via VEGF receptor type 2. Int J Cancer. 2012;130(4):857–64.
Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, Jove R, Gabrilovich D. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. 2004;172(1):464–74.
Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2(10):1096–103.
Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, Gabrilovich DI. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol. 1998;160(3):1224–32.
Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED, Carbone DP, Gabrilovich DI. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2000;6(5):1755–66.
Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, Hessler JD, Liu TM, Chang BY, Larkin KM, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122(15):2539–49.
Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3(12):1355–63.
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et al. Loss of tumor suppressor PTEN function increases B7–H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–8.
Han C, Liu T, Yin R. Biomarkers for cancer-associated fibroblasts. Biomark Res. 2020;8(1):64.
Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, Yin R. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12(1):86.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–86.
Yoshida GJ. Regulation of heterogeneous cancer-associated fibroblasts: the molecular pathology of activated signaling pathways. J Exp Clin Cancer Res. 2020;39(1):112.
Hornyák L, Dobos N, Koncz G, Karányi Z, Páll D, Szabó Z, Halmos G, Székvölgyi L. The role of indoleamine-2,3-dioxygenase in cancer development, diagnostics, and therapy. Front Immunol. 2018;9:151.
Hennequart M, Pilotte L, Cane S, Hoffmann D, Stroobant V, Plaen E, Van den Eynde BJ. Constitutive IDO1 expression in human tumors is driven by cyclooxygenase-2 and mediates intrinsic immune resistance. Cancer Immunol Res. 2017;5(8):695–709.
Huang L, Jiang S, Shi Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J Hematol Oncol. 2020;13(1):143.
Wu K, Yi M, Qin S, Chu Q, Zheng X, Wu K. The efficacy and safety of combination of PD-1 and CTLA-4 inhibitors: a meta-analysis. Exp Hematol Oncol. 2019;8:26.
Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer. 2019;18(1):155.
Qin S, Li A, Yi M, Yu S, Zhang M, Wu K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J Hematol Oncol. 2019;12(1):27.
Carnevalli LS, Sinclair C, Taylor MA, Gutierrez PM, Langdon S, Coenen-Stass AML, Mooney L, Hughes A, Jarvis L, Staniszewska A, et al. PI3Kalpha/delta inhibition promotes anti-tumor immunity through direct enhancement of effector CD8(+) T-cell activity. J Immunother Cancer. 2018;6(1):158.
Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. 2019;12(1):71.
Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702.
Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–72.
Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of IFN-γ in tumor progression and regression: a review. Biomarker Research. 2020;8(1):49.
Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11(12):852–63.
Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8(7):523–32.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9.
Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P, Zhang L, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475(7355):226–30.
Scarpino S, Stoppacciaro A, Ballerini F, Marchesi M, Prat M, Stella MC, Sozzani S, Allavena P, Mantovani A, Ruco LP. Papillary carcinoma of the thyroid: hepatocyte growth factor (HGF) stimulates tumor cells to release chemokines active in recruiting dendritic cells. Am J Pathol. 2000;156(3):831–7.
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–5.
Correale P, Rotundo MS, Botta C, Del Vecchio MT, Tassone P, Tagliaferri P. Tumor infiltration by chemokine receptor 7 (CCR7)(+) T-lymphocytes is a favorable prognostic factor in metastatic colorectal cancer. Oncoimmunology. 2012;1(4):531–2.
Mburu YK, Wang J, Wood MA, Walker WH, Ferris RL. CCR7 mediates inflammation-associated tumor progression. Immunol Res. 2006;36(1–3):61–72.
Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, Biota C, Doffin AC, Durand I, Olive D, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009;69(5):2000–9.
Cuesta-Mateos C, Fuentes P, Schrader A, Juárez-Sánchez R, Loscertales J, Mateu-Albero T, Vega-Piris L, Espartero-Santos M, Marcos-Jimenez A, Sánchez-López BA, et al. CCR7 as a novel therapeutic target in t-cell PROLYMPHOCYTIC leukemia. Biomark Res. 2020;8(1):54.
Smith MC, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, Luker GD. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64(23):8604–12.
Liang K, Liu Y, Eer D, Liu J, Yang F, Hu K. High CXC chemokine ligand 16 (CXCL16) expression promotes proliferation and metastasis of lung cancer via regulating the NF-kappaB pathway. Med Sci Monit. 2018;24:405–11.
Wang D, Yu W, Lian J, Wu Q, Liu S, Yang L, Li F, Huang L, Chen X, Zhang Z, et al. Th17 cells inhibit CD8+ T cell migration by systematically downregulating CXCR3 expression via IL-17A/STAT3 in advanced-stage colorectal cancer patients. J Hematol Oncol. 2020;13(1):68.
Darash-Yahana M, Pikarsky E, Abramovitch R, Zeira E, Pal B, Karplus R, Beider K, Avniel S, Kasem S, Galun E, et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004;18(11):1240–2.
Yang XL, Liu KY, Lin FJ, Shi HM, Ou ZL. CCL28 promotes breast cancer growth and metastasis through MAPK-mediated cellular anti-apoptosis and pro-metastasis. Oncol Rep. 2017;38(3):1393–401.
Mollica Poeta V, Massara M, Capucetti A, Bonecchi R. Chemokines and chemokine receptors: new targets for cancer immunotherapy. Front Immunol. 2019;10:379.
Solal-Celigny P, Lepage E, Brousse N, Reyes F, Haioun C, Leporrier M, Peuchmaur M, Bosly A, Parlier Y, Brice P, et al. Recombinant interferon alfa-2b combined with a regimen containing doxorubicin in patients with advanced follicular lymphoma. Groupe d’Etude des Lymphomes de l’Adulte. N Engl J Med. 1993;329(22):1608–14.
Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Perez-Gracia JL, Rodriguez-Ruiz ME, Ponz-Sarvise M, Castanon E, Melero I. Cytokines in clinical cancer immunotherapy. Br J Cancer. 2019;120(1):6–15.
Papiernik M, de Moraes ML, Pontoux C, Vasseur F, Penit C. Regulatory CD4 T cells: expression of IL-2R alpha chain, resistance to clonal deletion and IL-2 dependency. Int Immunol. 1998;10(4):371–8.
Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. 2002;169(9):4850–60.
Charych D, Khalili S, Dixit V, Kirk P, Chang T, Langowski J, Rubas W, Doberstein SK, Eldon M, Hoch U, et al. Modeling the receptor pharmacology, pharmacokinetics, and pharmacodynamics of NKTR-214, a kinetically-controlled interleukin-2 (IL2) receptor agonist for cancer immunotherapy. PLoS ONE. 2017;12(7):e0179431.
Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology. 2017;6(3):e1277306.
Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity. 2019;50(4):924–40.
Ciardiello D, Elez E, Tabernero J, Seoane J. Clinical development of therapies targeting TGFbeta: current knowledge and future perspectives. Ann Oncol. 2020;31(10):1336–49.
Brabletz T, Pfeuffer I, Schorr E, Siebelt F, Wirth T, Serfling E. Transforming growth factor beta and cyclosporin A inhibit the inducible activity of the interleukin-2 gene in T cells through a noncanonical octamer-binding site. Mol Cell Biol. 1993;13(2):1155–62.
Sad S, Mosmann TR. Single IL-2-secreting precursor CD4 T cell can develop into either Th1 or Th2 cytokine secretion phenotype. J Immunol. 1994;153(8):3514–22.
Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8(5):369–80.
Chen ML, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, Khazaie K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci U S A. 2005;102(2):419–24.
Budhu S, Schaer DA, Li Y, Toledo-Crow R, Panageas K, Yang X, Zhong H, Houghton AN, Silverstein SC, Merghoub T, et al. Blockade of surface-bound TGF-beta on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci Signal. 2017;10(494):eaak9702.
Metelli A, Wu BX, Riesenberg B, Guglietta S, Huck JD, Mills C, Li A, Rachidi S, Krieg C, Rubinstein MP, et al. Thrombin contributes to cancer immune evasion via proteolysis of platelet-bound GARP to activate LTGF-beta. Sci Transl Med. 2020;12(525):eaay4860.
Salem M, Wallace C, Velegraki M, Li A, Ansa-Addo E, Metelli A, Kwon H, Riesenberg B, Wu B, Zhang Y, et al. GARP dampens cancer immunity by sustaining function and accumulation of regulatory T cells in the colon. Cancer Res. 2019;79(6):1178–90.
Metelli A, Wu BX, Fugle CW, Rachidi S, Sun S, Zhang Y, Wu J, Tomlinson S, Howe PH, Yang Y, et al. Surface expression of TGFbeta docking receptor GARP promotes oncogenesis and immune tolerance in breast cancer. Cancer Res. 2016;76(24):7106–17.
de Streel G, Bertrand C, Chalon N, Lienart S, Bricard O, Lecomte S, Devreux J, Gaignage M, De Boeck G, Marien L, et al. Selective inhibition of TGF-beta1 produced by GARP-expressing Tregs overcomes resistance to PD-1/PD-L1 blockade in cancer. Nat Commun. 2020;11(1):4545.
Nandan D, Reiner NE. TGF-beta attenuates the class II transactivator and reveals an accessory pathway of IFN-gamma action. J Immunol. 1997;158(3):1095–101.
Wang Y, Xiang Y, Xin VW, Wang X-W, Peng X-C, Liu X-Q, Wang D, Li N, Cheng J-T, Lyv Y-N, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol. 2020;13(1):107.
Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, Biassoni R, Bottino C, Moretta L, Moretta A. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A. 2003;100(7):4120–5.
Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol. 2005;6(6):600–7.
Yu J, Wei M, Becknell B, Trotta R, Liu S, Boyd Z, Jaung MS, Blaser BW, Sun J, Benson DM Jr, et al. Pro- and antiinflammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity. 2006;24(5):575–90.
Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol. 2019;12(1):76.
Li Z, Pang Y, Gara SK, Achyut BR, Heger C, Goldsmith PK, Lonning S, Yang L. Gr-1+CD11b+ cells are responsible for tumor promoting effect of TGF-beta in breast cancer progression. Int J Cancer. 2012;131(11):2584–95.
Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Canellas A, Hernando-Momblona X, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–43.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–8.
Belladonna ML, Volpi C, Bianchi R, Vacca C, Orabona C, Pallotta MT, Boon L, Gizzi S, Fioretti MC, Grohmann U, et al. Cutting edge: autocrine TGF-beta sustains default tolerogenesis by IDO-competent dendritic cells. J Immunol. 2008;181(8):5194–8.
Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, Yu H, Qin G, Sircar A, Hernandez VM, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci Transl Med. 2018;10(424):eaan5488.
Knudson KM, Hicks KC, Luo X, Chen JQ, Schlom J, Gameiro SR. M7824, a novel bifunctional anti-PD-L1/TGFbeta Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology. 2018;7(5):e1426519.
Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, Lamping E, Marte JL, Donahue RN, Grenga I, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFbeta, in advanced solid tumors. Clin Cancer Res Off J Am Assoc Cancer Res. 2018;24(6):1287–95.
Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10105):1833–42.
Aggen DH, Ager CR, Obradovic AZ, Chowdhury N, Ghasemzadeh A, Mao W, Chaimowitz MG, Lopez-Bujanda ZA, Spina CS, Hawley JE, et al. Blocking interleukin-1 beta promotes tumor regression and remodeling of the myeloid compartment in a renal cell carcinoma model: multi-dimensional analyses. Clin Cancer Res Off J Am Assoc Cancer Res. 2021;27:608–21.
Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8.
Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, Sheng D, Liu X, Sims PW, VanderVeen LA, et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin Cancer Res Off J Am Assoc Cancer Res. 2016;22(3):680–90.
Bentebibel SE, Hurwitz ME, Bernatchez C, Haymaker C, Hudgens CW, Kluger HM, Tetzlaff MT, Tagliaferri MA, Zalevsky J, Hoch U, et al. A first-in-human study and biomarker analysis of NKTR-214, a novel IL2Rβγ-biased cytokine, in patients with advanced or metastatic solid tumors. Cancer Discov. 2019;9(6):711–21.
Soerensen MM, Ros W, Rodriguez-Ruiz ME, Robbrecht D, Rohrberg KS, Martin-Liberal J, Lassen UN, Bermejo IM, Lolkema MP, Tabernero J, et al. Safety, PK/PD, and anti-tumor activity of RO6874281, an engineered variant of interleukin-2 (IL-2v) targeted to tumor-associated fibroblasts via binding to fibroblast activation protein (FAP). J Clin Oncol. 2018;36(15_suppl):e15155.
Paz-Ares L, Kim TM, Vicente D, Felip E, Lee DH, Lee KH, Lin CC, Flor MJ, Di Nicola M, Alvarez RM, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in second-line treatment of patients with NSCLC: results from an expansion cohort of a phase 1 trial. J Thorac Oncol. 2020;15(7):1210–22.
Naing A, Wong DJ, Infante JR, Korn WM, Aljumaily R, Papadopoulos KP, Autio KA, Pant S, Bauer TM, Drakaki A, et al. Pegilodecakin combined with pembrolizumab or nivolumab for patients with advanced solid tumours (IVY): a multicentre, multicohort, open-label, phase 1b trial. Lancet Oncol. 2019;20(11):1544–55.
Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–80.
Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–8.
Zhang H, Zhao P, Huang H. Engineering better chimeric antigen receptor T cells. Exp Hematol Oncol. 2020;9(1):34.
Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, Gao L, Wen Q, Zhong JF, Zhang C, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. 2020;13(1):86.
Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–9.
Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–102.
Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331(6013):44–9.
Xu J, Niu T. Natural killer cell-based immunotherapy for acute myeloid leukemia. J Hematol Oncol. 2020;13(1):167.
Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73(6):1777–86.
Basher F, Dhar P, Wang X, Wainwright DA, Zhang B, Sosman J, Ji Z, Wu JD. Antibody targeting tumor-derived soluble NKG2D ligand sMIC reprograms NK cell homeostatic survival and function and enhances melanoma response to PDL1 blockade therapy. J Hematol Oncol. 2020;13(1):74.
Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16(6):372–85.
Nie Y, Lu W, Chen D, Tu H, Guo Z, Zhou X, Li M, Tu S, Li Y. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. Biomark Res. 2020;8(1):18.
Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, Potrel P, Bas C, Lemaire L, Galetto R, et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75(18):3853–64.
Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, Hakim FT, Halverson DC, Fowler DH, Hardy NM, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122(25):4129–39.
Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–90.
Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer. 2019;18(1):125.
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–5.
Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer cell. 2017;31(5):711–23.
Moon EK, Wang LS, Bekdache K, Lynn RC, Lo A, Thorne SH, Albelda SM. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. 2018;7(3):e1395997.
Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, Davenport AJ, John LB, Mardiana S, Slaney CY, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. 2017;127(3):929–41.
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–9.
Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, Bleakley M, Brown C, Mgebroff S, Kelly-Spratt KS, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–31.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17.
Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282–95.
Braig F, Brandt A, Goebeler M, Tony H-P, Kurze A-K, Nollau P, Bumm T, Böttcher S, Bargou RC, Binder M. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood. 2017;129(1):100–4.
Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, Wolters P, Martin S, Delbrook C, Yates B, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–8.
Piccaluga PP, Arpinati M, Candoni A, Laterza C, Paolini S, Gazzola A, Sabattini E, Visani G, Pileri SA. Surface antigens analysis reveals significant expression of candidate targets for immunotherapy in adult acute lymphoid leukemia. Leuk Lymphoma. 2011;52(2):325–7.
Jacoby E, Nguyen SM, Fountaine TJ, Welp K, Gryder B, Qin H, Yang Y, Chien CD, Seif AE, Lei H, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. 2016;7(1):12320.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra225.
Chmielewski M, Abken H. TRUCKS, the fourth-generation CAR T cells: current developments and clinical translation. Adv Cell Gene Ther. 2020;3(3):e84.
Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130–44.
Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, Lewis ID, Brenner MK, Brown MP. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther. 2016;24(6):1135–49.
Li AM, Hucks GE, Dinofia AM, Seif AE, Teachey DT, Baniewicz D, Callahan C, Fasano C, McBride B, Gonzalez V, et al. Checkpoint inhibitors augment CD19-directed chimeric antigen receptor (CAR) T cell therapy in relapsed B-cell acute lymphoblastic leukemia. Blood. 2018;132(Supplement 1):556–556.
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23(9):2255–66.
Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, Maus MV, Fraietta JA, Zhao Y, June CH. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26(7):1855–66.
Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, Hawkins R, Chaney C, Cherian S, Chen X, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8(355):355ra116.
Shalabi H, Shah NN, Fry TJ, Yates B, Delbrook C, Yuan C, Stetler-Stevenson M, Ramakrishna S. Intensification of lymphodepletion optimizes CAR re-treatment efficacy. Blood. 2017;130(Supplement 1):3889–3889.
Maude SL, Barrett DM, Rheingold SR, Aplenc R, Teachey DT, Callahan C, Baniewicz D, White C, Talekar MK, Shaw PA, et al. Efficacy of humanized CD19-targeted chimeric antigen receptor (CAR)-modified T cells in children and young adults with relapsed/refractory acute lymphoblastic leukemia. Blood. 2016;128(22):217–217.
Schneider D, Xiong Y, Wu D, Nӧlle V, Schmitz S, Haso W, Kaiser A, Dropulic B, Orentas RJ. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer. 2017;5:42.
Jaspers JE, Brentjens RJ. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacol Ther. 2017;178:83–91.
Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med. 2015;13:102.
Kondo K, Shaim H, Thompson PA, Burger JA, Keating M, Estrov Z, Harris D, Kim E, Ferrajoli A, Daher M, et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia. 2018;32(4):960–70.
Varikuti S, Singh B, Volpedo G, Ahirwar DK, Jha BK, Saljoughian N, Viana AG, Verma C, Hamza O, Halsey G, et al. Ibrutinib treatment inhibits breast cancer progression and metastasis by inducing conversion of myeloid-derived suppressor cells to dendritic cells. Br J Cancer. 2020;122(7):1005–13.
Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, Bouchier-Hayes L, Savoldo B, Dotti G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014;74(18):5195–205.
Tanoue K, Rosewell Shaw A, Watanabe N, Porter C, Rana B, Gottschalk S, Brenner M, Suzuki M. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 2017;77(8):2040–51.
Yilmaz A, Cui H, Caligiuri MA, Yu J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol. 2020;13(1):168.
Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18(11):671–88.
Liu B, Kong L, Han K, Hong H, Marcus WD, Chen X, Jeng EK, Alter S, Zhu X, Rubinstein MP, et al. A novel fusion of ALT-803 (interleukin (IL)-15 superagonist) with an antibody demonstrates antigen-specific antitumor responses. J Biol Chem. 2016;291(46):23869–81.
Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, Reis ESC. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172(5):1022–37.
Rothlin CV, Ghosh S. Lifting the innate immune barriers to antitumor immunity. J Immunother Cancer. 2020;8(1):e000695.
Hammerich L, Marron TU, Upadhyay R, Svensson-Arvelund J, Dhainaut M, Hussein S, Zhan Y, Ostrowski D, Yellin M, Marsh H, et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med. 2019;25(5):814–24.
Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–91.
Jiang M, Chen P, Wang L, Li W, Chen B, Liu Y, Wang H, Zhao S, Ye L, He Y, et al. cGAS-STING, an important pathway in cancer immunotherapy. J Hematol Oncol. 2020;13(1):81.
Li A, Yi M, Qin S, Song Y, Chu Q, Wu K. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J Hematol Oncol. 2019;12(1):35.
Harrington KJ, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, Tse A, Khilnani A, Marabelle A, Golan T. LBA15: preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann Oncol. 2018;29:viii712.
Huang Z, Zhang Z, Jiang Y, Zhang D, Chen J, Dong L, Zhang J. Targeted delivery of oligonucleotides into tumor-associated macrophages for cancer immunotherapy. J Control Release. 2012;158(2):286–92.
Brody JD, Ai WZ, Czerwinski DK, Torchia JA, Levy M, Advani RH, Kim YH, Hoppe RT, Knox SJ, Shin LK, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol. 2010;28(28):4324–32.
Veillette A, Latour S, Davidson D. Negative regulation of immunoreceptor signaling. Annu Rev Immunol. 2002;20:669–707.
Moretta L, Mantovani A. Alessandro Moretta 1953–2018. Nat Immunol. 2018;19(4):315–315.
Raulet DH, Vance RE, McMahon CW. Regulation of the natural killer cell receptor repertoire. Annu Rev Immunol. 2001;19:291–330.
Armand P, Lesokhin A, Borrello I, Timmerman J, Gutierrez M, Zhu L, Popa McKiver M, Ansell SM. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leukemia. 2021;35:777–86.
Kang X, Kim J, Deng M, John S, Chen H, Wu G, Phan H, Zhang CC. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle. 2016;15(1):25–40.
Chen H, Chen Y, Deng M, John S, Gui X, Kansagra A, Chen W, Kim J, Lewis C, Wu G, et al. Antagonistic anti-LILRB1 monoclonal antibody regulates antitumor functions of natural killer cells. J ImmunoTher Cancer. 2020;8(2):e000515.
Mingari MC, Ponte M, Bertone S, Schiavetti F, Vitale C, Bellomo R, Moretta A, Moretta L. HLA class I-specific inhibitory receptors in human T lymphocytes: interleukin 15-induced expression of CD94/NKG2A in superantigen- or alloantigen-activated CD8+ T cells. Proc Natl Acad Sci U S A. 1998;95(3):1172–7.
André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, Bléry M, Bonnafous C, Gauthier L, Morel A, et al. Anti-NKG2A mAb Is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175(7):1731-1743.e1713.
Xia Y, Rao L, Yao H, Wang Z, Ning P, Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater. 2020;32(40):2002054.
Eladl E, Tremblay-LeMay R, Rastgoo N, Musani R, Chen W, Liu A, Chang H. Role of CD47 in hematological malignancies. J Hematol Oncol. 2020;13(1):96.
Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. 2019;19(10):568–86.
Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, Kline J, Roschewski M, LaCasce A, Collins GP, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N Engl J Med. 2018;379(18):1711–21.
Hemminki O, dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol. 2020;13(1):84.
Alberts P, Tilgase A, Rasa A, Bandere K, Venskus D. The advent of oncolytic virotherapy in oncology: the Rigvir(R) story. Eur J Pharmacol. 2018;837:117–26.
Liang M. Oncorine, the world first oncolytic virus medicine and its update in China. Curr Cancer Drug Targets. 2018;18(2):171–6.
Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov. 2019;18(9):689–706.
Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2(4):295–300.
Ajina A, Maher J. Prospects for combined use of oncolytic viruses and CAR T-cells. J Immunother Cancer. 2017;5(1):90.
Fajardo CA, Guedan S, Rojas LA, Moreno R, Arias-Badia M, de Sostoa J, June CH, Alemany R. Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer Res. 2017;77(8):2052–63.
Freedman JD, Hagel J, Scott EM, Psallidas I, Gupta A, Spiers L, Miller P, Kanellakis N, Ashfield R, Fisher KD, et al. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol Med. 2017;9(8):1067–87.
Zhang B, Cheng P. Improving antitumor efficacy via combinatorial regimens of oncolytic virotherapy. Mol Cancer. 2020;19(1):158.
Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6(226):226ra232.
Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, Hamid O, Ross M, Friedlander P, Garbe C, et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36(17):1658–67.
Karapanagiotou EM, Roulstone V, Twigger K, Ball M, Tanay M, Nutting C, Newbold K, Gore ME, Larkin J, Syrigos KN, et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012;18(7):2080–9.
Lee JC, Shin DW, Park H, Kim J, Youn Y, Kim JH, Kim J, Hwang JH. Tolerability and safety of EUS-injected adenovirus-mediated double-suicide gene therapy with chemotherapy in locally advanced pancreatic cancer: a phase 1 trial. Gastrointest Endosc. 2020;92(5):1044-1052.e1041.
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22.
Huber ML, Haynes L, Parker C, Iversen P. Interdisciplinary critique of sipuleucel-T as immunotherapy in castration-resistant prostate cancer. J Natl Cancer Inst. 2012;104(4):273–9.
Nemunaitis J, Dillman RO, Schwarzenberger PO, Senzer N, Cunningham C, Cutler J, Tong A, Kumar P, Pappen B, Hamilton C, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J Clin Oncol. 2006;24(29):4721–30.
Giaccone G, Bazhenova LA, Nemunaitis J, Tan M, Juhász E, Ramlau R, van den Heuvel MM, Lal R, Kloecker GH, Eaton KD, et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur J Cancer. 2015;51(16):2321–9.
Rocconi RP, Grosen EA, Ghamande SA, Chan JK, Barve MA, Oh J, Tewari D, Morris PC, Stevens EE, Bottsford-Miller JN, et al. Gemogenovatucel-T (Vigil) immunotherapy as maintenance in frontline stage III/IV ovarian cancer (VITAL): a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Oncol. 2020;21(12):1661–72.
Bolhassani A, Naderi N, Soleymani S. Prospects and progress of Listeria-based cancer vaccines. Expert Opin Biol Ther. 2017;17(11):1389–400.
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–9.
Zhang H, Li Y, Lin TY, Xiao K, Haddad AS, Henderson PT, Jonas BA, Chen M, Xiao W, Liu R, et al. Nanomicelle formulation modifies the pharmacokinetic profiles and cardiac toxicity of daunorubicin. Nanomedicine (London). 2014;9(12):1807–20.
Lin TY, Li Y, Liu Q, Chen JL, Zhang H, Lac D, Zhang H, Ferrara KW, Wachsmann-Hogiu S, Li T, et al. Novel theranostic nanoporphyrins for photodynamic diagnosis and trimodal therapy for bladder cancer. Biomaterials. 2016;104:339–51.
Long Q, Lin TY, Huang Y, Li X, Ma AH, Zhang H, Carney R, Airhart S, Lam KS, deVere White RW, et al. Image-guided photo-therapeutic nanoporphyrin synergized HSP90 inhibitor in patient-derived xenograft bladder cancer model. Nanomedicine. 2018;14(3):789–99.
Tornesello AL, Tagliamonte M, Tornesello ML, Buonaguro FM, Buonaguro L. Nanoparticles to improve the efficacy of peptide-based cancer vaccines. Cancers (Basel). 2020;12(4):1049.
Lin TY, Li YP, Zhang H, Luo J, Goodwin N, Gao T, White Rde V, Lam KS, Pan CX. Tumor-targeting multifunctional micelles for imaging and chemotherapy of advanced bladder cancer. Nanomedicine (London). 2013;8(8):1239–51.
Koshy ST, Mooney DJ. Biomaterials for enhancing anti-cancer immunity. Curr Opin Biotechnol. 2016;40:1–8.
Fujita Y, Taguchi H. Current status of multiple antigen-presenting peptide vaccine systems: application of organic and inorganic nanoparticles. Chem Cent J. 2011;5(1):48.
Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–6.
Jorritsma SHT, Gowans EJ, Grubor-Bauk B, Wijesundara DK. Delivery methods to increase cellular uptake and immunogenicity of DNA vaccines. Vaccine. 2016;34(46):5488–94.
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C, et al. Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. N Engl J Med. 2020;383(27):2603–15.
Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med. 2021;384:403–16.
Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, Maurus D, Schwarck-Kokarakis D, Kuhn AN, Omokoko T, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585(7823):107–12.
Pavlick A, Blazquez AB, Meseck M, Lattanzi M, Ott PA, Marron TU, Holman RM, Mandeli J, Salazar AM, McClain CB, et al. Combined vaccination with NY-ESO-1 protein, poly-ICLC, and montanide improves humoral and cellular immune responses in patients with high-risk melanoma. Cancer Immunol Res. 2020;8(1):70–80.
Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K, Pockaj B, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–27.
Seo SH, Jin HT, Park SH, Youn JI, Sung YC. Optimal induction of HPV DNA vaccine-induced CD8+ T cell responses and therapeutic antitumor effect by antigen engineering and electroporation. Vaccine. 2009;27(42):5906–12.
Youn JW, Hur SY, Woo JW, Kim YM, Lim MC, Park SY, Seo SS, No JH, Kim BG, Lee JK, et al. Pembrolizumab plus GX-188E therapeutic DNA vaccine in patients with HPV-16-positive or HPV-18-positive advanced cervical cancer: interim results of a single-arm, phase 2 trial. Lancet Oncol. 2020;21(12):1653–60.
Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, Feng L, Lee JJ, Tran H, Kim YU, et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5(1):67–73.
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–21.
Hu Z, Leet DE, Allesøe RL, Oliveira G, Li S, Luoma AM, Liu J, Forman J, Huang T, Iorgulescu JB, et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat Med. 2021;27:515–25.
Romano E, Michielin O, Voelter V, Laurent J, Bichat H, Stravodimou A, Romero P, Speiser DE, Triebel F, Leyvraz S, et al. MART-1 peptide vaccination plus IMP321 (LAG-3Ig fusion protein) in patients receiving autologous PBMCs after lymphodepletion: results of a phase I trial. J Transl Med. 2014;12(1):97.
Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, Gentzler RD, Martins RG, Stevenson JP, Jalal SI, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016;17(11):1497–508.
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, Rodriguez-Abreu D, Moro-Sibilot D, Thomas CA, Barlesi F, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–301.
Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, Park K, Alexandru A, Lupinacci L, de la Mora JE, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. 2019;381(21):2020–31.
Paz-Ares L, Ciuleanu TE, Cobo M, Schenker M, Zurawski B, Menezes J, Richardet E, Bennouna J, Felip E, Juan-Vidal O, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(2):198–211.
Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthelemy P, Porta C, George S, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378(14):1277–90.
Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, Pouliot F, Alekseev B, Soulieres D, Melichar B, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1116–27.
Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, Venugopal B, Kollmannsberger C, Negrier S, Uemura M, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1103–15.
Choueiri TK, Powles T, Burotto M, Escudier B, Bourlon MT, Zurawski B, Oyervides Juarez VM, Hsieh JJ, Basso U, Shah AY, et al. Nivolumab plus cabozantinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2021;384(9):829–41.
FDA Approves Merck’s Keytruda (pembrolizumab) combined with trastuzumab and chemotherapy as first-line treatment in locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction adenocarcinoma. https://www.drugs.com/newdrugs/fda-approves-merck-s-keytruda-pembrolizumab-combined-trastuzumab-chemotherapy-first-line-locally-5511.html. Accessed 24 Sept 2021.
FDA Approves Merck’s Keytruda (pembrolizumab) plus platinum- and fluoropyrimidine-based chemotherapy for treatment of certain patients with locally advanced or metastatic esophageal or gastroesophageal junction (GEJ) carcinoma. https://www.drugs.com/newdrugs/fda-approves-merck-s-keytruda-pembrolizumab-plus-platinum-fluoropyrimidine-based-chemotherapy-5468.html. Accessed 24 Sept 2021.
Makker V, Taylor MH, Aghajanian C, Oaknin A, Mier J, Cohn AL, Romeo M, Bratos R, Brose MS, DiSimone C, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer. J Clin Oncol. 2020;38(26):2981–92.
Baas P, Scherpereel A, Nowak AK, Fujimoto N, Peters S, Tsao AS, Mansfield AS, Popat S, Jahan T, Antonia S, et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial. Lancet. 2021;397(10272):375–86.
Yau T, Kang YK, Kim TY, El-Khoueiry AB, Santoro A, Sangro B, Melero I, Kudo M, Hou MM, Matilla A, et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the checkmate 040 randomized clinical trial. JAMA Oncol. 2020;6(11):e204564.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–905.
Opdivo (nivolumab) + low-dose yervoy (ipilimumab) combination approved for previously treated MSI-H/dMMR metastatic colorectal cancer. https://www.drugs.com/newdrugs/opdivo-nivolumab-low-yervoy-ipilimumab-combination-approved-previously-treated-msi-h-dmmr-4779.html. Accessed 24 Sept 2021.
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–17.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34.
Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, Pereira RP, Eigentler T, Rutkowski P, Demidov L, et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;395(10240):1835–44.
Acknowledgements
The authors are indebted to all organizing committee members of 2020 China Cancer Immunotherapy Workshop, including those from CAHON (Ke Liu), China NMPA (Feng Zhu, Chenyan Gao, Zhimin Yang) and Tsinghua University School of Medicine (Chen Dong, Xin Lin), as well as all the invited speakers for their contributions to the success of the meeting. The authors apologize to colleagues whose work could not be fully detailed due to space limitations.
Funding
There is no funding for this article.
Author information
Authors and Affiliations
Contributions
SZ, TZ, DL, ZL and CP drafted the manuscript and finalized it with input from HL, LZ and WS. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
TZ: Research funding (to Duke) from Pfizer, Janssen, Acerta, Abbvie, Novartis, Merrimack, OmniSeq, PGDx, Merck, Mirati, Astellas and Regeneron; consulting/speaking with Genomic Health and Sanofi Aventis; and consulting/advisory board with AstraZeneca, Bayer, Pfizer, Foundation Medicine, Janssen, Amgen, BMS, Calithera, Dendreon and MJH Associates; Stock ownership/employment (spouse) from Capio Biosciences, Archimmune Therapeutics and Nanorobotics. LZ: grant support from Bristol-Meyer Squibb, Merck, AstraZeneca, iTeos, Amgen, NovaRock, Inxmed and Halozyme; paid consultant/Advisory Board Member at Biosion, Alphamab, NovaRock, Ambrx, Akrevia/Xilio, Datarevive, QED, Xilio, Natera, Novagenesis, Snow Lake Captials and Mingruizhiyao; and shareholder at Alphamab and Mingruzhiyao. HL: research support from Karyopharm and Bristol Myers Squibb, served at advisory board meetings of Agios and a consultant for Beigene, NGM Biopharm. WS: an employee of Kira Pharmaceuticals. DL attended advisory board meetings of AstraZeneca, Beigene, Rigel; serves on the Speaker bureaus of AstraZeneca, Beigene, Incyte, Janssen, Pharmacyclics and Rigel; and has research fundings from Acerta, Celgene, Denovo and Pfizer. ZL: Scientific advisory board member for Heat Biologics, Alphamab (chair), Hengenix and Ioknisys. CXP is a co-founder and shareholder of LP Therapeutics.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, S., Zhang, T., Zheng, L. et al. Combination strategies to maximize the benefits of cancer immunotherapy. J Hematol Oncol 14, 156 (2021). https://doi.org/10.1186/s13045-021-01164-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-021-01164-5