
Abstract
Accumulating evidence indicates that intratumoral heterogeneity contributes to the development of resistance to anticancer therapeutics. Fibroblasts, which are components of the paraneoplastic stroma, play a crucial role in the wound-healing process. Activated fibroblasts accumulate in the wound and are involved in many aspects of the tissue remodeling cascade that initiates the repair process and prevents further tissue damage. The pathophysiological roles of cancer-associated fibroblasts (CAFs) in the heterogeneous tumor microenvironment have attracted increasing interest. CAFs play crucial roles in tumor progression and the response to chemotherapy. Several cytokines and chemokines are involved in the conversion of normal fibroblasts into CAFs, and some of these form a feedback loop between cancer cells and CAFs. In addition, the physical force between tumor cells and CAFs promotes cooperative invasion or co-migration of both types of cells. Pro-inflammatory cytokines, such as leukemia inhibitory factor (LIF) and interleukin-6 (IL-6), are secreted by both cancer cells and CAFs, and mediate the epigenetic modification of CAFs. This enhances the pro-tumorigenic function of CAFs mediated by promoting actomyosin contractility and extracellular matrix remodeling to form the tracks used for collective cancer cell migration. The concept of intra-tumoral CAF heterogeneity refers to the presence of inflammatory CAFs with low levels of α-smooth muscle actin (α-SMA) and high levels of IL-6 expression, which are in striking contrast to transforming growth factor-β (TGF-β)-dependent myofibroblastic CAFs with high α-SMA expression levels. CAF populations that suppress tumor growth and progression through stroma-specific Hedgehog (Hh) activation have been detected in different murine tumor models including those of the bladder, colon, and pancreas. A new therapeutic strategy targeting CAFs is the “stromal switch,” in which tumor-promoting CAFs are changed into tumor-retarding CAFs with attenuated stromal stiffness. Several molecular mechanisms that can be exploited to design personalized anticancer therapies targeting CAFs remain to be elucidated. Strategies aimed at targeting the tumor stroma as well as tumor cells themselves have attracted academic attention for their application in precision medicine. This novel review discusses the role of the activation of EGFR, Wnt/β-catenin, Hippo, TGF-β, and JAK/STAT cascades in CAFs in relation to the chemoresistance and invasive/metastatic behavior of cancer cells. For instance, although activated EGFR signaling contributes to collective cell migration in cooperation with CAFs, an activated Hippo pathway is responsible for stromal stiffness resulting in the collapse of neoplastic blood vessels. Therefore, identifying the signaling pathways that are activated under specific conditions is crucial for precision medicine.
Similar content being viewed by others

Background
Fibroblasts are spindle-shaped cells that secrete collagen and have a cytoplasm with a predominant rough endoplasmic reticulum. Fibroblasts synthesize the extracellular matrix (ECM) of the connective tissue and play a crucial role in maintaining the structural integrity of most tissues including the skin [1,2,3]. The mammalian dermis represents an archetypal mesenchymal tissue that is largely composed of ECM elements, including type I and type III collagens, as well as proteoglycans and elastin [3]. Fibroblast heterogeneity depends on developmental stage and the tissue microenvironment [4, 5].
Fibroblasts exhibit distinct cellular phenotypes according to the surrounding microenvironment. Activated fibroblasts in tumor tissues are defined as cancer-associated fibroblasts (CAFs) [6,7,8,9]. In recent years, extensive research demonstrated that CAFs are the major cellular components of the tumor microenvironment in both primary and metastatic tumors; CAFs contribute to the regulation of a series of steps critical for malignant progression, including cancer initiation, proliferation, invasion, and metastasis, by producing various types of cytokines, chemokines, growth factors, and matrix-degrading enzymes [6, 8, 9]. CAFs are distinguished from their normal counterparts by the differential expression of markers such as α-smooth muscle actin (α-SMA), fibroblast activation protein (FAP), fibroblast-specific protein 1 (FSP1), and platelet-derived growth factor receptor (PDGFR) [7,8,9]. In addition to these markers, three proteins including collagen 11-α1, microfibrillar-associated protein 5, and asporin tend to be exclusively expressed in CAFs [9, 10]. The recent identification of proteins whose expression is restricted to CAFs may improve the reliable identification of CAFs and increase their value as candidate biomarkers and therapeutic targets.
CAFs and activated fibroblasts play similar roles in wound healing and fibrosis [11,12,13,14]. Fibroblasts are essential for tissue repair after damage and are involved in wound contraction, deposition of granulation tissue, and production and remodeling of the ECM in parallel with recruitment of platelets, neutrophils, and macrophages [14, 15] (Fig. 1). Fibroblasts in granulation tissue acquire a myofibroblastic phenotype characterized by α-SMA expression. On the other hand, CAFs alter the microenvironment by directly interacting with cancer cells and regulating paracrine signaling via inflammatory cytokines, control the immune response to neoplasia, deposit diverse ECM components, stimulate angiogenesis, and provide a scaffold for tumor metastasis and invasion [8].

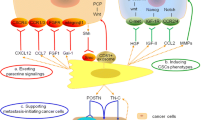
Activated fibroblasts in granulation tissue and carcinoma-associated fibroblasts (CAFs) closely resemble each other regarding their association with the microenvironment. a Hemostasis occurs at the first stage of wound healing, followed by the formation of clots by platelets at the injury site, which changes into fibrin (left). Neutrophils migrate to the granulation tissue, and cytokines are secreted, whereas activated fibroblasts are recruited to the wound-healing tissue. Endothelial progenitor cells are recruited in a manner dependent on the CXCL12-CXCR4 axis, which contributes to angiogenesis (right). During the proliferation stage of wound healing, macrophages infiltrate and initiate phagocytosis for the deposition of new extracellular matrix (ECM) [14]. b Robust neoangiogenesis occurs in the hypoxic tumor microenvironment. Unlike normal fibroblasts, CAFs stimulate tumor progression by secreting CXCL12. CXCL12 derived from CAFs promotes the recruitment of CXCR4-positive endothelial progenitor cells, which are essential for angiogenesis. Invasive metastasis to the bone marrow with high CXCL12 expression triggers CXCR4 activation in circulating tumor cells, which “hijack” the CXCL12-CXCR4 axis for homing to microenvironments that are normally restricted to hematopoietic progenitor cells (HPCs) [16]
CAFs can be recruited to the tumor from a distant source such as the bone marrow [7, 17]. The trans-differentiation of epithelial cells and pericytes can also give rise to CAF-like populations in response to epithelial-mesenchymal transition (EMT) and endothelial-mesenchymal transition (EndoMT), respectively [9, 18, 19]. To define and identify the origin of CAFs, it is important to consider that CAFs are ‘activated fibroblasts’, which, by striking contrast to non-activated (quiescent) tissue-resident fibroblasts, are an expanding population of cells that either proliferates in situ or is recruited to the tumor [7, 20]. The key features of CAFs that distinguish them from quiescent fibroblasts include metabolic adaptations that support their need for enhanced proliferation and biosynthetic activities, such as the production of ECM components and cytokines, growth factors, and enzymes to remodel the stroma [7, 9, 21]. However, the cellular origin of CAFs and the mechanisms underlying the reprogramming of normal fibroblasts into CAFs remain largely unknown.
The heterogeneity and mutual exclusivity of CAF marker expression patterns may be associated with unique functions in different types of malignancy. In breast cancer, CAFs positive for both FAP and podoplanin are immunosuppressive through a nitric oxide (NO)-dependent mechanism [22]. In prostate cancer, CAFs expressing high levels of CD90 play a pivotal role in promoting tumor progression through the upregulation of angiogenic factors, activation of the Hedgehog (Hh) signal, and decreased androgen receptor signaling [23]. In pancreatic ductal adenocarcinoma (PDAC), a specific subpopulation of CAFs was identified that is distinct from myofibroblastic CAFs strongly expressing α-SMA. These inflammatory CAFs express pro-inflammatory cytokines such as interleukin-6 (IL-6) and IL-11, thereby activating the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling pathway [24].
Therefore, this novel review article focuses on critical signal pathways for CAFs to regulate the malignant phenotype, given the crosstalk between tumor cells and CAFs as well as the heterogeneity of CAF population.
Heterogeneity of CAFs in the tumor-associated stroma
Increasing evidence strongly suggests that CAFs have diverse functions, implying that tumor-promoting CAF and tumor-suppressing CAFs coexist in the tumor stroma [7, 25, 26]. Current cancer immunotherapy strategies primarily target programmed cell death-1 ligand-1 (PD-L1), chimeric antigen receptors, and cytotoxic T lymphocyte-associated antigen 4 [27, 28]; however, the effects of CAFs on tumor immunosuppression remain relatively unexplored. FAP-positive CAF populations drive immunosuppression and promote resistance to anti-PD-L1 immunotherapy [29, 30]. Targeting the C-X-C motif chemokine 12 (CXCL12)–C-X-C chemokine receptor type 4 (CXCR-4) axis with AMD3100 (Plerixafor) reverses FAP-positive CAF-mediated immunosuppression and synergizes with anti-PD-L1 immunotherapy in PDAC [29]. Biffi et al. recently identified IL-1 and transforming growth factor-β (TGF-β) as tumor-secreted cytokines that promote CAF heterogeneity [31]. TGF-β signaling inhibits IL-1 receptor 1 (IL-1R1) expression, antagonizes IL-1α responses, and promotes the differentiation of paraneoplastic fibroblasts into myofibroblastic CAFs. By contrast, an IL-1-induced signaling cascade that activates JAK/STAT promotes the generation of inflammatory CAFs [31]. Therefore, IL-1α signaling is a potential therapeutic target against PDAC cells and inflammatory CAFs in the tumor microenvironment. Elyada et al. used single-cell transcriptomics to examine CAF heterogeneity associated with PDAC and identified a novel CAF population characterized by high expression levels of major histocompatibility complex class II [32]. These antigen-presenting CAFs can present antigens to CD4-positive T lymphocytes.
Costa et al. used multicolor flow cytometry to identify four subtypes of CAFs (CAF-S1, 2, 3, and 4) associated with breast cancer [30] that show differential expression of CAF marker molecules such as α-SMA, caveolin-1 (Cav-1), FAP, and PDGFRβ. CAF subset maps confirmed that CAF-S2 is enriched in luminal-type breast cancer, whereas CAF-S1 and CAF-S4 accumulate in triple-negative breast cancer. The CAF-S1 type attracts CD4 + CD25+ T lymphocytes and retains them through OX40L, PD-L2, and JAM2. CAF-S1 cells promote the inhibition of T effector proliferation by regulatory T cells (Treg). Mechanistically, Costa et al. identified dipeptidyl peptidase 4, a FAP-dimerization counterpart, as a key player in CAF-S1-mediated Treg activation [30, 33]. Givel et al. identified four subpopulations of CAFs in mesenchymal-type high-grade serous ovarian cancer (HGSOC) according to α-SMA, CD29 (integrin β-1), FAP, and FSP1 expression levels [34]. CAF-S2 and CAF-S3 are defined as non-activated CAFs because they have low levels of α-SMA, whereas CAF-S1 and CAF-S4 are considered activated CAFs with high α-SMA expression levels. FOXP3-positive T lymphocytes are enriched exclusively in the CAF-S1 population, which differs from the CAF-S4 subtype in mesenchymal-type HGSOC. The chemokine CXCL12β is expressed at higher levels in CAF-S1 than in CAF-S4, which is associated with the poor prognosis of HGSOC [34, 35]. The CXCL12β isoform is regulated by microRNA (miR)-141/200a; the miR-200 family members miR-141 and miR-200a are responsible for the downregulation of CXCL12β in CAF-S4, whereas miR141/200a promote the specific accumulation of CACL12β in the CAF-S1 subpopulation, inducing the infiltration of CD4 + CD25+ T lymphocytes [34]. Taken together, these data indicate that the antagonistic effects of CAFs on the malignant phenotype may be related to the existence of subpopulations of CAFs with opposing functions.
Recent PDAC studies challenged the concept of tumor-promoting CAFs based on data showing increased tumor growth and aggressiveness following eradication of α-SMA-expressing CAFs and/or targeting of the desmoplastic response induced by the Hh signaling pathway [36, 37]. PDAC lesions with approximately 80% depletion of α-SMA-positive interstitial myofibroblasts show an activated EMT program associated with increased numbers of cancer stem cells (CSCs) and upregulation of EMT-related transcription factors such as Snail, Slug, and Twist. Clinically, lower CAF numbers are correlated with decreased survival in patients with PDAC [36]. Although sonic hedgehog (Shh) ligand and downstream signaling are induced de novo in preneoplastic lesions linked to pancreatic intraepithelial neoplasia and increase significantly during PDAC progression as the stromal compartment enlarges [38], a Shh-depleted PDAC mouse model showed Slug and Zeb1 upregulation leading to poorly differentiated histology [37]. Mizutani et al. identified Meflin as a functional marker of tumor-retarding CAFs in PDAC [39]. Meflin, a glycosylphosphatidylinositol-anchored protein, is a marker of mesenchymal stem cells (MSCs) and maintains their undifferentiated state [40]. In situ hybridization analysis revealed an inverse correlation between α-SMA and Meflin expression in PDAC-associated CAFs. Kaplan-Meier survival analyses showed that high expression levels of Meflin in surgically resected human PDAC tissues are positively correlated with better prognosis, and Meflin-high PDAC displays a more differentiated pathohistology than the Meflin-low group. This suggests that the phenotype of Meflin-high CAFs is distinct from that of tumor-promoting CAFs with high α-SMA expression levels. The PDAC-associated stroma in Meflin-KO genetically engineered model mice shows straighter and wider collagen structures than those of tumors in Meflin-WT PDAC model mice. Lineage-tracing experiments indicate that Meflin-lineage stromal cells contain α-SMA-positive CAFs, which downregulate Meflin and upregulate α-SMA in response to TGF-β and tumor stiffness. This explains why the stromagenic switch, in which tumor-restricting CAFs with high Meflin expression generate tumor-promoting CAFs, contributes to CAF heterogeneity during tumor progression.
Crosstalk between tumor cells and CAFs
Increasing evidence suggests that CAFs contribute to collective cell migration and invasion by remodeling the ECM to create tracks for tumor cell migration and/or by expressing different cadherins that enable cells to retain adhesion while controlling front/rear polarization of the leading cells [41,42,43]. Labernadie et al. showed that CAFs increase the invasive potential of tumor cells through N-cadherin upregulation [42]. Intercellular physical force is transmitted between cancer cells and CAFs by a heterophilic adhesion complex involving E-cadherin at the cancer cell membrane and N-cadherin at the CAF membrane. This heterotypic cancer cell-CAF interaction triggers β-catenin recruitment, α-catenin/vinculin interaction, and actin remodeling, allowing CAFs to exert an intercellular physical force on cancer cells and promote cooperative tumor invasion [42].
CAFs contribute to the ‘education’ of carcinoma cells into an invasive and metastatic phenotype [9, 44]. TGF-β and CXCL12 secreted by CAFs enhances the metastatic potential of breast cancer cells undergoing incomplete EMT. Although developmental cells undergo complete EMT during embryogenesis, which is characterized by the cadherin switch, tumor cells express both epithelial and mesenchymal markers [epithelial/mesenchymal (E/M) hybrid phenotype] concurrently, which is defined as “partial EMT” in the process of invasion and distant metastasis [45, 46]. Indeed, circulating tumor cells that survive in the bloodstream show an E/M hybrid phenotype, become resistant to anoikis, and exit the bloodstream more efficiently [46, 47]. CAFs stimulate the invasion of E/M hybrid-type breast cancer cells, which are associated with epithelial-type cancer cell clusters, leading to collective invasion of both epithelial and E/M hybrid tumor cell clusters [48, 49]. Chen et al. recently reported that the epithelial-to-mesenchymal plasticity of lung cancer cells established from a patient-derived xenograft (PDX) is enhanced in the presence of CAFs under three-dimensional culture [50, 51]. CAFs antagonize the oncogenic transcriptional factor SOX2 to restore the formation of luminal structures and promote invasion. Stromal cell-derived factor 1 promotes EMT and increases the stemness of lung squamous cancer cells (LSCCs). Most LSCCs express E-cadherin, and only a small population is positive for vimentin [50]. This finding suggests that spheroids derived from PDX are heterogeneous. The presence of tumor cells positive for both E-cadherin and vimentin suggests that partial EMT occurs in the original tumor, PDX model, and spheroids [46].
CAFs play an important role in the establishment of the omental tumor microenvironment in ovarian cancer. Omental fibroblasts contribute to the creation of a pre-metastatic niche, and influence tropism for the omentum and the metastatic colonization of ovarian tumor cells [52]. Ovarian cancer-derived lysophosphatidic acid and exosomes promote the differentiation of adipose-derived MSCs into CAFs [52,53,54], which are characterized by the expression of α-SMA, FAP, FSP1, and PDGFR, by activating TGF-β-related signaling pathways [6,7,8]. Furthermore, ovarian cancer cells reprogram normal omental fibroblasts into CAFs by upregulating miR-155 and downregulating miR-31 and miR-214 [55]. This action promotes tumor proliferation by increasing the secretion of CCL5. Ovarian cancer-derived TGF-β is involved in stimulating the production of various tumor-promoting factors including IL-6, CXCL12, and VEGF-A in the metastatic tumor microenvironment [56]. Omental dissemination induced by this cascade is driven by overexpression of HOXA9 in ovarian cancer cells. CAF-derived TGF-α promotes the metastatic colonization of ovarian cancer cells via the activation of the Akt, epidermal growth factor receptor (EGFR), and extracellular signal-regulated kinase (ERK)-1/2 signaling pathways [57]. Metastasizing ovarian cancer cells can activate p38α MAPK signaling in omental CAFs, and CAF-derived p38α MAPK-regulated cytokines and chemokines, including IL-6, CCL5, and CXCL10, induce glycogen metabolism in cancer cells via glycolysis, which mediates energy production and promotes the aggressiveness of ovarian cancer cells [58]. Furthermore, the differential expression patterns of monocarboxylate transporters (MCT) in cancer cells and CAFs contribute to metabolic symbiosis, in which CAFs depend on aerobic glycolysis and secrete lactate via MCT4 [9, 59, 60]. This “reverse Warburg effect” enables MCT1-positive CSCs to play a fundamental role in maintaining the hierarchy in tumor cellular society unlike MCT4-positive CAFs [59]. In addition, CAFs tend to exhibit robust activity regarding aerobic glycolysis as well as Atg5/7-dependent selective autophagy because of the loss of Cav-1 expression [9, 61, 62]. Such stromal autophagy generates building blocks from recycled free amino acids, fatty acids, and nucleotides, which can be directly utilized by tumor cells to sustain growth and maintain cellular viability. Therefore, CAFs evolve with ovarian cancer cells in the intraperitoneal metastatic microenvironment and govern the metastatic cascade, including the adhesion, proliferation, invasion, and colonization of metastatic sites [52].
Podoplanin-positive CAFs drive tumor progression in a xenograft model, and podoplanin expression in CAFs predicts a poor outcome in patients with lung adenocarcinoma [63, 64]. However, CAFs positive for podoplanin are more frequent in poorly differentiated adenocarcinoma. Clinical cases characterized by the presence of podoplanin-expressing CAFs display a poor response to EGFR tyrosine kinase inhibitors (EGFR-TKIs) in patients with lung adenocarcinoma harboring constitutively active mutations of EGFR [65]. By contrast, knockdown of podoplanin makes CAFs susceptible to EGFR-TKIs [66]. Direct contact between cancer cells and CAFs is necessary for acquired resistance to EGFR-TKIs.
Significance of EGFR signaling in CAFs
The epidermal growth factor receptor (EGFR) belongs to the ErbB family of receptor tyrosine kinases (RTKs) and exhibits critical functions in the epithelial cell physiology [67]. Ligand-dependent activation of EGFR transduces multiple signaling pathways such as PI3K/Akt and Ras/MAPK pathways [68]. Canonical EGFR signaling is essential for several cellular functions including differentiation, proliferation and survival [67]. Notably, increased EGFR expression is positively correlated with reduced recurrence-free and overall survival periods in several kinds of malignancy [69].
Grasset et al. demonstrated that collective invasion of squamous cancer cells (SCCs) is driven by the matrix-dependent mechano-sensitization of EGF signaling [70] (Fig. 2a). Increasing evidence suggests a connection between mechanotransduction and receptor tyrosine kinase (RTK) signaling pathways. RTKs are activated by dimerization and are involved in integrin-mediated mechanotransduction signaling, which promotes tumor progression [72]. Induction of collagen crosslinking results in stiffness of the ECM, promotes focal adhesion kinase (FAK) expression, increases phosphoinositide 3-kinase activity, and promotes the invasion of oncogene-initiated epithelial cells. By contrast, suppression of integrin signaling inhibits the invasion of a premalignant epithelium into a stiffened, crosslinked stroma. Cell-to-ECM adhesion favors EGFR-dependent cancer proliferation [73]. Because RTKs interact exclusively with active integrins, the composition of the ECM determines the type of RTK/integrin interaction occurring at the cellular membrane. This selectivity may change the intracellular location or conformation of EGFR, thereby changing the accessibility of the receptor intracellular domain to downstream signaling molecules. One of the downstream proteins is FAK, which is targeted to sites of integrin/RTK complex formation and is essential for the transmission of motility signals from EGFR [73, 74]. Furthermore, the EGFR gene is amplified, overexpressed, or mutated in SCCs, such as head and neck squamous cell carcinoma (HNSCC) [75, 76]. In the clinical setting, EGFR amplification predicts sensitivity to gefitinib in HNSCC [76]. EGFR activation and expression levels are positively correlated with poor prognosis of breast cancer and HNSCC independently from anticancer therapeutics [77]. Grasset et al. identified an association between EGFR activity and stromal stiffness during collective cellular migration [70]. The degree of EGFR signaling is positively correlated with collective cell migration (Fig. 2a). The L-type calcium channel Cav1.1 is a critical regulatory element during the collective invasion of squamous cell carcinoma and acts downstream of ECM stiffness and EGFR signaling both in vitro and in vivo. The L-type calcium channel Cav1.1 is a critical regulator of SCC collective migration in response to stromal stiffness and EGFR signaling activation (Fig. 2b), and calcium channel blockers, which are widely used for the treatment of arrhythmia and hypertension, are promising therapeutic agents against SCC invasion and metastasis. EGFR blockage induces EMT and CAF activation in HNSCC [78], and the calcium channel antagonists verapamil and diltiazem reduce resistance to EGFR-targeting treatments [70]. This is an example of drug repositioning, namely, screening for the anticancer therapeutic effects of conventionally administered medications for non-malignant disorders [59, 62]. Increased rigidity in the tumor stroma favors EGFR activity and results in the calcium-dependent regulation of Cdc42 small GTPase activity in tumor cells. This signaling route is critical for HNSCC cell invasion into the stiffened stroma. Although myosin light chain (MLC) kinase, an important regulator of actomyosin contractility in cancer cells [79], does not play a role in collective SCC migration, Cdc42 finely regulates the actomyosin-dependent remodeling of the ECM by CAFs [70]. PDX models developed in verapamil- or diltiazem-treated mice show reduced levels of phosphorylated MLC2 and a decrease in the number of filopodia, which regulate tumor cell invasion [51, 70] (Fig. 2b).

Collective migration of squamous cancer cells (SCCs) is driven by the matrix-dependent mechano-sensitization of EGF signaling. a Collective migration of epithelial tumor cells is a tissue-driven process dictated by ECM remodeling, which is orchestrated by CAFs. CAFs lead the way for cancer cells by digging tracks within the ECM that SCCs use to invade [70, 71]. b Calcium ions are intracellular second messengers that modulate actomyosin contractility. ECM stiffness and activation of the EGFR signaling pathway increase intracellular calcium concentration mediated by Cav1.1 expression in SCCs [70]. EGF stimulation promotes myosin light chain 2 (MLC2) phosphorylation only when SCCs are exposed to ECM stiffness. Note that ‘p’ indicates phosphorylation
More recently, Gao et al. have demonstrated that CAFs associated with HGSOC contribute to the formation of heterotypic spheroids in the malignant ascites [80]. Those CAFs-centered spheroids recruits floating ovarian cancer cells, resulting in the formation of ‘metastatic units’ at early stages of transcoelomic metastasis [80, 81]. Mechanistically, floating ovarian cancer cells drive the production and secretion of EGF by CAFs located at the center region of the spheroids. This consequently promotes ITGA5 (integrin α5) expression in tumor cells, which in turn further enhances the tumor-stromal interaction inside the heterotypic spheroids [80]. That is why EGF is expected to the promising therapeutic target to prevent the peritoneal dissemination of HGSOC. Indeed, it has been shown that a neutralizing anti-EGF antibody can suppress the formation of spheroid in ascites mediated by the attenuated expression of ITGA5 in floating ovarian cancer cells, leading to the prolonged survival period [80].
Significance of canonical Wnt signaling in CAFs
Signal activation due to the Wnt family of secreted glycolipoproteins is one of the crucial machineries underlying the cellular polarity, proliferation, and cell fate determination during the embryonic development and tissue homeostasis [82]. In the absence of Wnt ligand, cytoplasmic β-catenin is degraded constantly by Axin complex, which is composed of Axin, APC, casein kinase 1 (CK1), and glycogen synthase kinase 3 (GSK3) [82]. CD44, c-Myc, Axin2, and Cyclin D1 are the typical target molecules regulated by the nuclear translocation of β-catenin. Interestingly, reactive oxygen species (ROS) activates canonical β-catenin-dependent Wnt signal pathway [83, 84], which is responsible for up-regulation of c-Myc at the invasive front enriched in cancer stem-like cells [85, 86].
Ferrari et al. recently reported that Dickkopf (DKK)-3, which activates the canonical Wnt signaling pathway, is highly expressed in CAFs in breast, colon, and ovarian cancer stroma [87]. DKK1 expression is downregulated in fibroblasts in pachydermoperiostosis (PDP), a rare chronic inflammatory disease characterized by unique skin and bone phenotypes associated with loss-of-function mutation of the HPGD gene, thereby increasing the proliferation capacity of PDP-associated fibroblasts [88, 89]. In contrast to DKK1, DKK2, and DKK4, which suppress Wnt/β-catenin signal transduction, DKK3 does not interact with LDL-receptor-related protein (LRP) 5/6 and therefore cannot fulfill the bona fide antagonistic role of the DKK family in the canonical Wnt signaling pathway [90, 91]. Instead, DKK3 decreases the stability of Kremen, a Wnt negative regulator, resulting in increased LRP6 membrane localization, which in turn stabilizes both β-catenin and Yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) levels [87, 91]. Of note, heat-shock factor 1 (HSF1) interacts with the enhancer and promoter regions of the Dkk3 locus, and contributes to the upregulation of DKK3 in CAFs. HSF1 promotes tumor growth by inducing cytokines such as CXCL12 and TGF-β [92]. Although β-catenin-mediated Wnt signaling is dispensable for the function of CAFs in remodeling the ECM and promoting tumor cell proliferation and invasion, DKK3-driven YAP activation is necessary to induce a tumor-promoting phenotype [87]. Absence of DKK3 in DKK3-null normal fibroblasts and CAFs is associated with decreased YAP/TAZ and β-catenin activity. By contrast, depletion of DKK3 leads to the concomitant upregulation of Kremen, LRP6 inactivation, and destabilization of both β-catenin and YAP/TAZ in CAFs. This DKK3-mediated localization and stabilization of YAP/TAZ in the nucleus is independent from the Hippo pathway, in which phosphorylated YAP (Ser127) plays a central role [93]. Thus, DKK3 is expected to mediate the crosstalk between Wnt/β-catenin signaling and YAP/TAZ.
Periostin, which is abundantly produced and secreted by CAFs in HNSCC, promotes the CSC phenotype via the canonical Wnt/β-catenin signaling pathway [94]. Periostin is highly expressed in the tumor stroma compared with cancer cells and promotes tumor progression and metastasis in HNSCC [94, 95]. Yu et al. showed that periostin secreted by CAFs is a potential ligand for protein tyrosine kinase 7 (PTK7), which is frequently upregulated in HNSCC tissues and is correlated with Wnt/β-catenin pathway activation and poor clinical outcome in HNSCC patients [94]. CAF-derived periostin is highly likely to bind to PTK7 on the cancer cell membrane and transduces signals to disheveled protein through the cell surface receptor LRP6; this induces the phosphorylation of GSK-3β and the hypophosphorylation of β-catenin, which causes β-catenin to translocate into the nucleus, suggesting that the periostin-PTK7 axis activates the canonical Wnt signaling pathway [94]. The periostin-PTK7 axis promotes tumorigenesis, lung metastasis, and chemoresistance mediated by β-catenin expression in HNSCC. Thus, treatment with a PTK7 neutralizing antibody increases the therapeutic efficacy of erlotinib, a small-molecule TKI effective for the treatment of metastatic and/or recurrent HNSCC, by downregulating β-catenin [94, 96].
Significance of Hippo signaling in CAFs
Hippo signal pathway is an evolutionary well-conserved regulator of organ size, which is first discovered in Drosophila. Central to this signaling is a kinase cascade leading from the tumor suppressor Hippo (Mst1/ Mst2 in mammals) to the oncogenic Yki (YAP/TAZ in mammals), which is a transcriptional coactivator of target genes involved in cell proliferation and survival [93]. The major target transcription factors regulated by YAP and TAZ are the four proteins of the TEA-domain-containing (TEAD) family (TEAD1-TEAD4). While Mst1/2 is downregulated in several kinds of carcinomas, TAZ has been reported to be upregulated in invasive breast cancer [97].
The Hippo pathway is activated by stromal stiffness in solid tumor tissues, and a growing body of evidence suggests that the transcriptional factor YAP is activated in CAFs [13, 98, 99]. YAP/TAZ is activated in response to mechanical stress and perturbation of the actin cytoskeleton [100]. YAP/TAZ activation by mechanical stimuli in cells is influenced by Rho-GTPase, Rho-associated protein kinase (ROCK), and the integrity of the actomyosin cytoskeleton in a manner largely independent from the large tumor suppressor kinase [99]. Pathohistological analysis of normal murine mammary tissues and PyMT-driven breast tumors shows nuclear accumulation of YAP in the stroma of both adenoma and carcinoma lesions [98]. YAP activation in the stroma is further enhanced in the peripheral tumor regions of advanced carcinomas such as breast cancer and squamous cell carcinoma. YAP controls the expression of several cytoskeletal regulators including ANLN, connective tissue growth factor (CTGF), and diaphanous homolog 3 (DIAPH3), and then regulates the expression levels of MYL9/myosin light chain (MLC)-2. Matrix stiffening promotes YAP activation, thereby establishing a positive-feedback loop that helps maintain the CAF phenotype [98]. Increased interstitial fluid pressure (IFP) blocks the delivery of therapeutic agents, whereas reduced tumor IFP improves the uptake of chemotherapeutic drugs [101, 102]. Therefore, lowering tumor interstitial hypertension, which acts as a barrier against tumor transvascular transport, has been proposed as a general strategy to increase tumor uptake as well as the therapeutic effects of anticancer drugs. Blocking the tyrosine kinase PDGFRβ increases the susceptibility to conventional chemotherapy in a xenograft model of anaplastic thyroid carcinoma [101].
YAP activation serves as an independent prognostic marker for the overall survival of PDAC patients and its association with liver metastasis [103]. Hyaluronic acid (HA) is the major determinant of elevated IFP in PDAC. In addition, the presence of α-SMA-positive myofibroblasts increases the number of fibrotic foci and promotes contraction of the interstitial space [8]. The increased number of blood vessels together with increased hydraulic conductivity or the relative ease with which fluid moves across the vessel wall is responsible for an irregular and increased influx of fluid into the tumor stroma. Increased IFP is frequently reported in solid tumors such as breast carcinoma, glioblastoma, and malignant melanoma [104,105,106]. Enzymatic degradation of HA results in the rapid reduction of IFP and the appearance of widely patent functioning vessels in the tumor microenvironment [102]. Removing these barriers permits high concentrations of chemotherapy agents to reach PDAC tissues, which improves survival and reveals an unappreciated sensitivity of the disease to conventional cytotoxic agents (Fig. 3). In the clinical setting, the combination of gemcitabine and PEGPH20 has attracted attention for the treatment of stage IV PDAC because of its effect on HA degradation in the tumor stroma [102, 107].

Degradation of hyaluronic acid renders chemoresistant tumors sensitive to anticancer drugs by attenuating the collapse of blood vessels. The stroma associated with pancreatic ductal adenocarcinoma (PDAC) shows a robust and complex desmoplasia, with notable hyaluronic acid (HA) content and the collapse of blood vessels in response to high interstitial fluid pressure (IFP). Stromal stiffness is positively correlated with the Hippo pathway. PEGPH20 contributes to the enzymic degradation of HA and decreases the degree of stromal stiffness [102]. The attenuated IFP broadens the lumen of tumor vessels, thereby increasing the efficacy of drug delivery. A clinical trial of PEGPH20 in advanced solid tumors is ongoing [107]
Significance of TGF-β signaling in CAFs
TGF-β signal pathway contributes to the maintenance of tissue homeostasis and prevention of incipient malignancy by regulating not only cellular adhesion, differentiation, proliferation and survival, but also the microenvironment [108]. Injured epithelial tissue is gradually repaired by the formation of granulation tissues composed of α-SMA-positive myofibroblasts, macrophages, platelets, newly formed blood vessels and ECM [9] (Fig. 1). Pathological forms of TGF-β signal pathway drive tumor growth and invasive phenotype, evasion of immune surveillance, and distant metastasis including cancer cell dissemination [108]. As with the wound-healing process, tumor-derived TGF-β is likely to recruit other stromal cell types characterized by CAFs and osteoclasts, which are enriched at the invasive front and at the bone metastatic disease, respectively.
The dependence of myofibroblastic CAFs on autocrine TGF-β signaling remained unclear until a study demonstrated that the establishment of self-sustaining CXCL12 and TGF-β autocrine signaling pathways results in the formation of tumor-promoting CAFs during breast cancer progression [109]. The two autocrine signaling pathways triggered by CXCL12 and TGF-β may promote CAF differentiation associated with increased α-SMA expression levels [109]. TGF-β and CXCL12 secreted by cancer cells upregulate CXCR4 and stabilize the Smad-dependent TGF-β pathway.
Loss of Cav-1 in the tumor stroma activates TGF-β signaling in CAFs [9, 110]. Activation of the TGF-β ligand by proteolytic cleavage promotes its interaction with specific receptors. TGF-β binds to TGF-β receptor type II, and promotes the formation of a hetero-oligomeric complex with TGF-β receptor type I, resulting in the activation of the TGF-β receptor kinase. The TGF-β receptor in turn phosphorylates serine/threonine residues in downstream target effectors such as Smad proteins. The activated TGF-β receptor complex initiates several downstream cascades, including the canonical Smad2/3 signaling pathway and non-canonical pathways, such as TGF-β-activated kinase-mediated p38- or JNK-signaling [111, 112]. Activation of TGF-β signaling leads to EMT in cancer cells, which express high levels of matrix metalloproteinase (MMP) [113, 114]. Discoidin domain receptor 2 upregulates MMP2 and MT1-MMP in a manner dependent on the ERK2/SNAIL1 axis in hepatocellular carcinoma [114, 115].
Ligand-initiated activation of the Smad-independent TGF-β pathway triggers GSK-3β phosphorylation by c-Abl and PKC-δ non-receptor kinases. Phosphorylation of GSK-3β at serine 9 (Ser9) causes its inhibition, which in turn allows Snail1 to enter the nucleus. Nuclear accumulation of Snail1 leads to acquisition of the myofibroblastic phenotype with stimulation of α-SMA and type I collagen instead of VE-cadherin. Inhibition of c-Abl activity with imatinib allows GSK-3β to phosphorylate Snail1, which targets it for proteasomal degradation and effectively abolishes the acquisition of the myofibroblastic phenotype and the fibrotic response. Rottlerin and imatinib abrogate EndoMT by inhibiting PKC-δ and c-Abl, respectively [116] (Fig. 4).

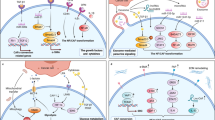
A non-Smad TGF-β signaling pathway is involved in tissue fibrosis by promoting endothelial-mesenchymal transition (EndoMT). Following ligand-initiated activation of the Smad-independent TGF-β pathway, GSK-3β is phosphorylated by c-Abl and PKC-δ non-receptor kinases. Phosphorylation of GSK-3β at serine 9 (Ser9) causes its inhibition, which allows Snail1 to enter the nucleus. Nuclear accumulation of Snail1 leads to acquisition of the myofibroblast phenotype characterized by stimulation of α-SMA and type I collagen instead of VE-cadherin. The specific inhibition of c-Abl activity with imatinib allows GSK-3β to phosphorylate Snail1, which targets it for proteasomal degradation, thereby abolishing the acquisition of the myofibroblastic phenotype and the fibrotic response. Rottlerin and imatinib inhibit PKC-δ and c-Abl, respectively, which is why both agents abrogate EndoMT [116]. Note that ‘p’ indicates phosphorylation. Black arrows indicate induction or promotion, whereas blue arrows indicate inhibition
Mammary fibroblasts in which Cav-1 is depleted show CAF phenotypes, such as the conversion of fibroblasts to myofibroblasts and enhanced TGF-β signaling [117]. Cav-1 directly inhibits TGF-β signaling. Mechanistically, Cav-1 interacts with the TGF-β receptor type 1, inducing its degradation, and suppresses TGF-β-dependent Smad2 phosphorylation and nuclear translocation [118]. Activation of the TGF-β signaling pathway is sufficient to downregulate Cav-1 expression [119], thereby forming a positive-feedback loop involving Cav-1 expression levels and TGF-β signaling activity. Significant downregulation of stromal Cav-1 is responsible for the metabolic reprogramming of CAFs, which is characterized by the induction of aerobic glycolysis (also referred to as “reverse Warburg effect”) and autophagy in the tumor-associated stroma. This results in the stromal production of energy-rich metabolites including L-lactate, pyruvate, and ketone bodies, as well as chemical building blocks such as amino acids (glutamine), nucleotides, and fatty acids [59, 120]. These recycled nutrients are then transferred to adjacent epithelial tumor cells, thereby fueling cancer progression in a paracrine fashion. Because activation of TGF-β signaling attenuates mitochondrial metabolism, and enhances aerobic glycolysis and autophagy (especially mitophagy, in which old dysfunctional mitochondria undergo degradation) [62, 110], CAFs that secrete TGF-β ligands in an autocrine manner can proliferate independently of angiogenesis. Cancer cell-induced ROS promote the loss of stromal Cav-1 in fibroblasts via autophagy and activate hypoxia-inducible factor α (HIF1-α) under ROS-induced pseudohypoxic conditions [9, 59, 121]. This phenomenon is termed metabolic symbiosis [9, 59]. Enhanced expression levels of MCT4 and BNIP3 in CAFs are responsible for the activation of aerobic glycolysis via metabolic symbiosis and mitophagy, respectively [9, 59, 110].
Significance of JAK/STAT signaling in CAFs
JAKs are non-receptor tyrosine kinases mediating signal transduction which is involved in cellular proliferation and survival. The seven mammalian STAT family contain the tyrosine residue near the C-terminus which is phosphorylated by JAK family in the presence of growth factors, interleukins and interferons (IFN) [122]. This phosphorylation allows STATs to form the dimer via the interaction with a conserved SH2 domain. Remarkably, there are several cytokines with distinct, and sometimes opposing, functions are likely to activate the same STAT protein [123, 124]. For a typical instance, IL-6, a pro-inflammatory cytokine which utilizes gp130, promotes the activation of STAT3. In contrast, IL-10, which is a potent anti-inflammatory cytokine, does not utilize gp130 but promotes STAT3 phosphorylation.
Actomyosin contractility plays a key role in tumor cell migration, affecting both the tumor cells themselves and the remodeling of the ECM by tumor fibroblasts to permit cell migration [71]. CAFs remodel the ECM using contractile forces and proteolytic activity, thereby generating tracks for the migration of tumor cells as collective strands led by a fibroblast [43]. Force-mediated matrix remodeling largely depends on integrins α3 and α5, as well as Rho-mediated regulation of MLC activity in fibroblasts. However, these factors are not required in cancer cells. Instead, tumor cells depend on Cdc42 and myotonic dystrophy kinase-related Cdc42-binding protein kinase (MRCK)-mediated regulation of MLC to follow the tracks generated by fibroblasts in the ECM. Force-mediated matrix remodeling by CAFs depends on actomyosin contractility modulated by the ROCK signaling pathway [43, 125]. Rab21-positive vesicles preferentially localize to the areas of cell contraction, and both integrin α5 and Rab21 are required for MLC phosphorylation [125]. Rab21 delivers integrin α5 to the cellular membrane, where it signals to the contractile machinery. At least three Rab proteins, including 5a, 11b, and 21 subtypes, are needed in CAFs for their ability to promote SCC invasion. Depletion of these Rab proteins does not affect the ability of SCC cells to invade the ECM previously remodeled by CAFs. This can be attributed to the fact that the ‘following’ SCC cells do not remodel the matrix, and matrix remodeling pathways are therefore dispensable in SCC cells. Cytokine signaling through GP130-IL6ST and JAK1 stimulates actomyosin-mediated contractility in cancer cells and in the tumor-associated stroma [71]. GP130-IL6ST signaling to JAK1 drives actomyosin-mediated contractility in CAFs and promotes matrix remodeling. JAK1 signaling regulates actomyosin contractility by regulating the level of phosphorylated-MLC2 in both melanoma cells and CAFs, the latter of which lead SCC invasion. Pro-inflammatory cytokines, such as IL-6 and LIF, are aberrantly expressed by CAFs in the tumor microenvironment and induce chemoresistance as well as EMT [126, 127]. The axis involving the cytokine oncostatin M (OSM) acts through GP130-IL6ST, JAK1, and ROCK to drive actomyosin contractility and matrix remodeling by CAFs for the collective invasion of SCCs [71]. OSM induces fibrotic changes in the lungs and liver, and promotes EMT and the myofibroblastic phenotype via the JAK/STAT axis, thereby predisposing to cancer development [128, 129]. JAK/STAT signaling may involve a Rho guanine nucleotide exchange factor, ARHGEF1, to activate RhoA to the GTP-bound state as in vascular smooth muscle cells. This is supported by data showing that basal RhoA activity in CAFs is sensitive to inhibition of JAK, and OSM activates RhoA in a JAK-dependent manner [71, 130]. Unlike melanoma cells, in which GP130-IL6ST/JAK1-ROCK signaling is required for cancer cell migration, this signaling pathway is not necessary in tumor cells, whereas it is required in CAFs for ECM remodeling leading to the collective invasion of SCCs [71]. Therapeutic agents, including blocking antibodies against cytokines, such as IL-6, or small molecule inhibitors of JAK kinase or STAT activity, could be useful agents to block invasion and metastasis in malignant diseases. IL-6 receptor blockage inhibits lung metastasis of breast cancer cells by suppressing IL-6-induced JAK/STAT signaling [131]. Furthermore, an anti-IL-6 neutralizing antibody named siltuximab inhibits non-small cell lung cancer progression [132].
In an analysis of epigenetic alterations, Albrengues et al. demonstrated that aberrant DNA methylation maintains the phenotype of tumor-promoting CAFs via the JAK/STAT cascade [133]. JAK1/STAT3 signaling is constitutively activated in CAFs, partly because STAT3 acetylation induced by CBP/p300 leads to the epigenetic-dependent loss of expression of Src homology phosphatase-1 (SHP-1), which is a negative regulator of JAK/STAT signaling. SHP-1, also known as tyrosine-protein phosphatase non-receptor type 6, dephosphorylates several tyrosine kinases including JAK1 [134]. Silencing of SHP-1 by DNA methyltransferase 1-mediated promoter hypermethylation leads to the sustained constitutive phosphorylation of JAK1 kinase and the STAT3 transcription factor, which maintain the contractile and pro-invasive properties of CAFs [133]. Pharmacological inhibition with both 5-azacytidine and ruxolitinib results in the long-term abrogation of JAK1/STAT3 phosphorylation and rescues the expression of SHP-1, thereby inhibiting the tumor-promoting invasive phenotype of CAFs. Given that genetic mutations are rare in CAFs, further investigations are warranted to identify epigenetic abnormalities in the cancer-associated stroma [9, 135, 136].
Conclusion
CAFs contribute to the formation and maintenance of the tumor microenvironment in cooperation with tumor cells by activating several signaling cascades including the EGFR, JAK/STAT, TGF-β, and Wnt pathways. In addition, stromal stiffness leads to drug resistance and poor prognosis of cancer patients. Given that α-SMA-positive activated fibroblasts form a senescence-associated secretory phenotype loop in response to treatment with HDAC inhibitors [137, 138], re-education of the tumor stroma could be a promising therapeutic strategy. Treatment with chemotherapy and/or radiotherapy eradicates responsive diseases. However, survival of CAFs is associated with minimal residual disease. The surviving CAFs acquire innate and adaptive resistance to therapy, which is accompanied by stromal inflammation and increased ECM accumulation, leading to iatrogenic tumor stiffness and the development of chemoresistant tumors [9]. Hirata et al. indicated that CAFs associated with BRAF-mutant malignant melanoma are activated in response to PLX4720, a selective BRAF inhibitor. PLX4720 paradoxically activates ERK/MAPK signaling in residual disease, promotes collagen production and matrix remodeling, and promotes MLC phosphorylation [139]. This iatrogenic activation of CAFs is responsible for the FAK-dependent persistent survival of melanoma cells. The ability of melanoma-associated fibroblasts to confer PLX4720 tolerance largely depends on both FAK and integrin β1 in melanoma cells. Furthermore, stiffness of the fibronectin-rich stroma is sufficient to abrogate the effects of BRAF inhibition. PDX models indicate that dual inhibition of BRAF and FAK inhibits ERK/MAPK re-activation in the tumor stroma, which facilitates the efficient therapeutic control of BRAF-mutant melanoma [51, 139]. As such, the tumor microenvironment mainly composed of CAFs determines the dynamic phenotype and plasticity of cancer cells in cooperation with intrinsic genetic/epigenetic alterations [140]. The degree of cancer cell differentiation may be largely controlled by the “stromagenic switch”, which results in CAF heterogeneity. In addition, α-SMA-negative and PDGFRβ-positive CAF subpopulations contribute to the malignant potential of tumor cells by interacting with integrin α11 [141, 142]. Of note, α-SMA-negative inflammatory CAFs secrete high levels of pro-inflammatory cytokines such as IL-6, IL-11, and LIF, and activate the JAK1/STAT3 cascade [24]. In verity, several molecular machineries underlying invasive/metastatic phenotype and therapy-resistance driven by CAFs have been uncovered. Surprisingly enough, there exist tumor-restricting CAF populations which have been shown to inhibit tumor growth and progression [143, 144]. Accumulating evidence demonstrates that activation of Hh signal pathway in CAFs suppresses the growth of tumors mediated by bone morphogenetic protein (BMP) signaling in cancer cells, which strongly suggests the presence of CAF populations with tumor-suppressive functions. Taken together, the existence of several potential CAF markers suggests that further investigation is warranted to identify the pathophysiological functions of these molecules.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- α-SMA:
-
α-smooth muscle actin
- CAFs:
-
Cancer-associated fibroblasts
- Cav-1:
-
Caveolin-1
- CSCs:
-
Cancer stem cells
- CXCL12:
-
C-X-C motif chemokine 12
- CXCR4:
-
C-X-C chemokine receptor type 4
- DKK:
-
Dickkopf
- ECM:
-
Extracellular matrix
- EGFR:
-
Epithelial growth factor receptor
- EMT:
-
Epithelial-mesenchymal transition
- EndoMT:
-
Endothelial-mesenchymal transition
- ERK:
-
Extracellular signal-regulated kinase
- FAK:
-
Focal adhesion kinase
- FAP:
-
Fibroblast activation protein
- FSP1:
-
Fibroblast-specific protein 1
- GSK-3β:
-
Glycogen synthase kinase 3β
- HA:
-
Hyaluronic acid
- HGSOC:
-
High-grade serous ovarian cancer
- Hh:
-
Hedgehog
- HNSCC:
-
Head and neck squamous cell carcinoma
- HSF1:
-
Heat-shock factor 1
- IFP:
-
Interstitial fluid pressure
- IL:
-
Interleukin
- JAK:
-
Janus kinase
- LIF:
-
Leukemia inhibitory factor
- LRP:
-
LDL-receptor-related protein
- MCT:
-
Monocarboxylate transporter
- miR:
-
microRNA
- MLC:
-
Myosin light chain
- MMP:
-
Matrix metalloproteinase
- MSCs:
-
Mesenchymal stem cells
- OSM:
-
Oncostatin M
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDGFR:
-
Platelet-derived growth factor receptor
- PD-L1:
-
Programmed cell death-1 ligand-1
- PDX:
-
Patient-derived xenograft
- PTK7:
-
Protein tyrosine kinase 7
- ROCK:
-
Rho-associated protein kinase
- ROS:
-
Reactive oxygen species
- RTK:
-
Receptor tyrosine kinase
- SCC:
-
Squamous cancer cell
- Shh:
-
Sonic hedgehog
- SHP-1:
-
Src homology phosphatase-1
- STAT:
-
Signal transducer and activator of transcription
- TAZ:
-
Transcriptional coactivator with PDZ-binding motif
- TKI:
-
Tyrosine kinase inhibitor
- TGF-β:
-
Transforming growth factor-β
- Treg:
-
Regulatory T cell
- YAP:
-
Yes-associated protein
References
Parsonage G, Filer AD, Haworth O, Nash GB, Rainger GE, Salmon M, Buckley CD. A stromal address code defined by fibroblasts. Trends Immunol. 2005;26(3):150–6.
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3(5):349–63.
Watt FM, Fujiwara H. Cell-extracellular matrix interactions in normal and diseased skin. Cold Spring Harb Perspect Biol. 2011;3(4):a005124. https://doi.org/10.1101/cshperspect.a005124.
Driskell RR, Watt FM. Understanding fibroblast heterogeneity in the skin. Trends Cell Biol. 2015;25(2):92–9.
Lynch MD, Watt FM. Fibroblast heterogeneity: implications for human disease. J Clin Invest. 2018;128(1):26–35.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–98.
LeBleu VS, Kalluri R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Model Mech. 2018;11(4):dmm029447. https://doi.org/10.1242/dmm.029447.
Yoshida GJ, Azuma A, Miura Y, Orimo A. Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. Int J Mol Sci. 2019;20(9):2256. https://doi.org/10.3390/ijms20092256.
Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016;30(9):1002–19.
Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–9.
Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9(8):628–38.
Foster CT, Gualdrini F, Treisman R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017;31(23–24):2361–75.
Foster DS, Jones RE, Ransom RC, Longaker MT, Norton JA. The evolving relationship of wound healing and tumor stroma. JCI Insight. 2018;3(18):e99911. https://doi.org/10.1172/jci.insight.99911.
Darby IA, Laverdet B, Bonte F, Desmouliere A. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol. 2014;7:301–11.
Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107(5):1761–7.
Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers (Basel). 2015;7(4):2443–58.
Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99(9):1375–9.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8.
Rasanen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res. 2010;316(17):2713–22.
Han Y, Zhang Y, Jia T, Sun Y. Molecular mechanism underlying the tumor-promoting functions of carcinoma-associated fibroblasts. Tumour Biol. 2015;36(3):1385–94.
Cremasco V, Astarita JL, Grauel AL, Keerthivasan S, MacIsaac K, Woodruff MC, Wu M, Spel L, Santoro S, Amoozgar Z, et al. FAP delineates heterogeneous and functionally divergent stromal cells in immune-excluded breast tumors. Cancer Immunol Res. 2018;6(12):1472–85.
Zhao H, Peehl DM. Tumor-promoting phenotype of CD90hi prostate cancer-associated fibroblasts. Prostate. 2009;69(9):991–1000.
Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214(3):579–96.
Kobayashi H, Enomoto A, Woods SL, Burt AD, Takahashi M, Worthley DL. Cancer-associated fibroblasts in gastrointestinal cancer. Nat Rev Gastroenterol Hepatol. 2019;16(5):282–95.
Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. 2016;99(Pt B):186–96.
Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016;13(5):273–90.
Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50(12):165.
Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–7.
Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, Sirven P, Magagna I, Fuhrmann L, Bernard C, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast Cancer. Cancer Cell. 2018;33(3):463–79 e410.
Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, Tuveson DA. IL1-induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9(2):282–301.
Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting Cancer-associated fibroblasts. Cancer Discov. 2019;9(8):1102–23.
Wonganu B, Berger BW. A specific, transmembrane interface regulates fibroblast activation protein (FAP) homodimerization, trafficking and exopeptidase activity. Biochim Biophys Acta. 2016;1858(8):1876–82.
Givel AM, Kieffer Y, Scholer-Dahirel A, Sirven P, Cardon M, Pelon F, Magagna I, Gentric G, Costa A, Bonneau C, et al. miR200-regulated CXCL12beta promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat Commun. 2018;9(1):1056.
Popple A, Durrant LG, Spendlove I, Rolland P, Scott IV, Deen S, Ramage JM. The chemokine, CXCL12, is an independent predictor of poor survival in ovarian cancer. Br J Cancer. 2012;106(7):1306–13.
Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–34.
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–47.
Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo C, Yajnik V, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–6.
Mizutani Y, Kobayashi H, Iida T, Asai N, Masamune A, Hara A, Esaki N, Ushida K, Mii S, Shiraki Y, et al. Meflin-positive Cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019;79(20):5367–81.
Maeda K, Enomoto A, Hara A, Asai N, Kobayashi T, Horinouchi A, Maruyama S, Ishikawa Y, Nishiyama T, Kiyoi H, et al. Identification of Meflin as a potential marker for Mesenchymal stromal cells. Sci Rep. 2016;6:22288.
Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59(19):5002–11.
Labernadie A, Kato T, Brugues A, Serra-Picamal X, Derzsi S, Arwert E, Weston A, Gonzalez-Tarrago V, Elosegui-Artola A, Albertazzi L, et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol. 2017;19(3):224–37.
Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9(12):1392–400.
Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: an essential role in the tumor microenvironment. Oncol Lett. 2017;14(3):2611–20.
Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–29.
Saitoh M. Involvement of partial EMT in cancer progression. J Biochem. 2018;164(4):257–64.
Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339(6119):580–4.
Pearson GW. Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis. J Clin Med. 2019;8(5):646. https://doi.org/10.3390/jcm8050646.
Yang C, Cao M, Liu Y, He Y, Du Y, Zhang G, Gao F. Inducible formation of leader cells driven by CD44 switching gives rise to collective invasion and metastases in luminal breast carcinomas. Oncogene.;38(46):7113–32. https://doi.org/10.1038/s41388-019-0899-y. Epub 2019 Aug 15.
Chen S, Giannakou A, Wyman S, Gruzas J, Golas J, Zhong W, Loreth C, Sridharan L, Yamin TT, Damelin M, et al. Cancer-associated fibroblasts suppress SOX2-induced dysplasia in a lung squamous cancer coculture. Proc Natl Acad Sci U S A. 2018;115(50):E11671–80.
Yoshida GJ. Applications of patient-derived tumor xenograft models and tumor organoids. J Hematol Oncol. 2020;13(1):4.
Motohara T, Masuda K, Morotti M, Zheng Y, El-Sahhar S, Chong KY, Wietek N, Alsaadi A, Karaminejadranjbar M, Hu Z, et al. An evolving story of the metastatic voyage of ovarian cancer cells: cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene. 2019;38(16):2885–98.
Jeon ES, Moon HJ, Lee MJ, Song HY, Kim YM, Cho M, Suh DS, Yoon MS, Chang CL, Jung JS, et al. Cancer-derived lysophosphatidic acid stimulates differentiation of human mesenchymal stem cells to myofibroblast-like cells. Stem Cells. 2008;26(3):789–97.
Cho JA, Park H, Lim EH, Kim KH, Choi JS, Lee JH, Shin JW, Lee KW. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol Oncol. 2011;123(2):379–86.
Mitra AK, Zillhardt M, Hua Y, Tiwari P, Murmann AE, Peter ME, Lengyel E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012;2(12):1100–8.
Ko SY, Barengo N, Ladanyi A, Lee JS, Marini F, Lengyel E, Naora H. HOXA9 promotes ovarian cancer growth by stimulating cancer-associated fibroblasts. J Clin Invest. 2012;122(10):3603–17.
Lau TS, Chan LK, Wong EC, Hui CW, Sneddon K, Cheung TH, Yim SF, Lee JH, Yeung CS, Chung TK, et al. A loop of cancer-stroma-cancer interaction promotes peritoneal metastasis of ovarian cancer via TNFalpha-TGFalpha-EGFR. Oncogene. 2017;36(25):3576–87.
Curtis M, Kenny HA, Ashcroft B, Mukherjee A, Johnson A, Zhang Y, Helou Y, Batlle R, Liu X, Gutierrez N, et al. Fibroblasts mobilize tumor cell glycogen to promote proliferation and metastasis. Cell Metab. 2019;29(1):141–55 e149.
Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015;34:111.
Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8(23):3984–4001.
Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, Wang C, Pavlides S, Martinez-Cantarin MP, Capozza F, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9(17):3515–33.
Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J Hematol Oncol. 2017;10(1):67.
Kawase A, Ishii G, Nagai K, Ito T, Nagano T, Murata Y, Hishida T, Nishimura M, Yoshida J, Suzuki K, et al. Podoplanin expression by cancer associated fibroblasts predicts poor prognosis of lung adenocarcinoma. Int J Cancer. 2008;123(5):1053–9.
Hoshino A, Ishii G, Ito T, Aoyagi K, Ohtaki Y, Nagai K, Sasaki H, Ochiai A. Podoplanin-positive fibroblasts enhance lung adenocarcinoma tumor formation: podoplanin in fibroblast functions for tumor progression. Cancer Res. 2011;71(14):4769–79.
Yoshida T, Ishii G, Goto K, Yoh K, Niho S, Umemura S, Matsumoto S, Ohmatsu H, Nagai K, Ohe Y, et al. Solid predominant histology predicts EGFR tyrosine kinase inhibitor response in patients with EGFR mutation-positive lung adenocarcinoma. J Cancer Res Clin Oncol. 2013;139(10):1691–700.
Yoshida T, Ishii G, Goto K, Neri S, Hashimoto H, Yoh K, Niho S, Umemura S, Matsumoto S, Ohmatsu H, et al. Podoplanin-positive cancer-associated fibroblasts in the tumor microenvironment induce primary resistance to EGFR-TKIs in lung adenocarcinoma with EGFR mutation. Clin Cancer Res. 2015;21(3):642–51.
Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12(1):3–20.
Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–34.
Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl 4):S9–15.
Grasset EM, Bertero T, Bozec A, Friard J, Bourget I, Pisano S, Lecacheur M, Maiel M, Bailleux C, Emelyanov A, et al. Matrix stiffening and EGFR cooperate to promote the collective invasion of Cancer cells. Cancer Res. 2018;78(18):5229–42.
Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, Hooper S, Mitter R, Feral CC, Cook M, et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell. 2011;20(2):229–45.
Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906.
Yarwood SJ, Woodgett JR. Extracellular matrix composition determines the transcriptional response to epidermal growth factor receptor activation. Proc Natl Acad Sci U S A. 2001;98(8):4472–7.
Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2(5):249–56.
Gullick WJ. Prevalence of aberrant expression of the epidermal growth factor receptor in human cancers. Br Med Bull. 1991;47(1):87–98.
Erjala K, Sundvall M, Junttila TT, Zhang N, Savisalo M, Mali P, Kulmala J, Pulkkinen J, Grenman R, Elenius K. Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin Cancer Res. 2006;12(13):4103–11.
Ciardiello F, Tortora G. Epidermal growth factor receptor (EGFR) as a target in cancer therapy: understanding the role of receptor expression and other molecular determinants that could influence the response to anti-EGFR drugs. Eur J Cancer. 2003;39(10):1348–54.
Schmitz S, Bindea G, Albu RI, Mlecnik B, Machiels JP. Cetuximab promotes epithelial to mesenchymal transition and cancer associated fibroblasts in patients with head and neck cancer. Oncotarget. 2015;6(33):34288–99.
Kassianidou E, Hughes JH, Kumar S. Activation of ROCK and MLCK tunes regional stress fiber formation and mechanics via preferential myosin light chain phosphorylation. Mol Biol Cell. 2017;28(26):3832–43.
Gao Q, Yang Z, Xu S, Li X, Yang X, Jin P, Liu Y, Zhou X, Zhang T, Gong C, et al. Heterotypic CAF-tumor spheroids promote early peritoneal metastatis of ovarian cancer. J Exp Med. 2019;216(3):688–703.
Han Q, Huang B, Huang Z, Cai J, Gong L, Zhang Y, Jiang J, Dong W, Wang Z. Tumor cellfibroblast heterotypic aggregates in malignant ascites of patients with ovarian cancer. Int J Mol Med. 2019;44(6):2245–55.
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26.
Funato Y, Miki H. Redox regulation of Wnt signalling via nucleoredoxin. Free Radic Res. 2010;44(4):379–88.
Yoshida GJ, Saya H. Inversed relationship between CD44 variant and c-Myc due to oxidative stress-induced canonical Wnt activation. Biochem Biophys Res Commun. 2014;443(2):622–7.
Yoshida GJ. The heterogeneity of cancer stem-like cells at the invasive front. Cancer Cell Int. 2017;17:23.
Yoshida GJ. Emerging roles of Myc in stem cell biology and novel tumor therapies. J Exp Clin Cancer Res. 2018;37(1):173.
Ferrari N, Ranftl R, Chicherova I, Slaven ND, Moeendarbary E, Farrugia AJ, Lam M, Semiannikova M, Westergaard MCW, Tchou J, et al. Dickkopf-3 links HSF1 and YAP/TAZ signalling to control aggressive behaviours in cancer-associated fibroblasts. Nat Commun. 2019;10(1):130.
Kabashima K, Sakabe J, Yoshiki R, Tabata Y, Kohno K, Tokura Y. Involvement of Wnt signaling in dermal fibroblasts. Am J Pathol. 2010;176(2):721–32.
Yoshida GJ, Saya H, Zouboulis CC. Three-dimensional culture of sebaceous gland cells revealing the role of prostaglandin E2-induced activation of canonical Wnt signaling. Biochem Biophys Res Commun. 2013;438(4):640–6.
Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature. 2001;411(6835):321–5.
Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, et al. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature. 2002;417(6889):664–7.
Scherz-Shouval R, Santagata S, Mendillo ML, Sholl LM, Ben-Aharon I, Beck AH, Dias-Santagata D, Koeva M, Stemmer SM, Whitesell L, et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158(3):564–78.
Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505.
Yu B, Wu K, Wang X, Zhang J, Wang L, Jiang Y, Zhu X, Chen W, Yan M. Periostin secreted by cancer-associated fibroblasts promotes cancer stemness in head and neck cancer by activating protein tyrosine kinase 7. Cell Death Dis. 2018;9(11):1082.
Qin X, Yan M, Zhang J, Wang X, Shen Z, Lv Z, Li Z, Wei W, Chen W. TGFbeta3-mediated induction of Periostin facilitates head and neck cancer growth and is associated with metastasis. Sci Rep. 2016;6:20587.
Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22(1):77–85.
Zhao B, Li L, Guan KL. Hippo signaling at a glance. J Cell Sci. 2010;123(Pt 23):4001–6.
Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15(6):637–46.
Zanconato F, Battilana G, Cordenonsi M, Piccolo S. YAP/TAZ as therapeutic targets in cancer. Curr Opin Pharmacol. 2016;29:26–33.
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–83.
Pietras K, Rubin K, Sjoblom T, Buchdunger E, Sjoquist M, Heldin CH, Ostman A. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002;62(19):5476–84.
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–29.
Salcedo Allende MT, Zeron-Medina J, Hernandez J, Macarulla T, Balsells J, Merino X, Allende H, Tabernero J, Ramon YCS. Overexpression of yes associated protein 1, an independent prognostic marker in patients with pancreatic ductal adenocarcinoma, correlated with liver metastasis and poor prognosis. Pancreas. 2017;46(7):913–20.
Nathanson SD, Nelson L. Interstitial fluid pressure in breast cancer, benign breast conditions, and breast parenchyma. Ann Surg Oncol. 1994;1(4):333–8.
Boucher Y, Salehi H, Witwer B, Harsh GR, Jain RK. Interstitial fluid pressure in intracranial tumours in patients and in rodents. Br J Cancer. 1997;75(6):829–36.
Simonsen TG, Gaustad JV, Leinaas MN, Rofstad EK. High interstitial fluid pressure is associated with tumor-line specific vascular abnormalities in human melanoma xenografts. PLoS One. 2012;7(6):e40006.
Infante JR, Korn RL, Rosen LS, LoRusso P, Dychter SS, Zhu J, Maneval DC, Jiang P, Shepard HM, Frost G, et al. Phase 1 trials of PEGylated recombinant human hyaluronidase PH20 in patients with advanced solid tumours. Br J Cancer. 2018;118(2):e3.
Massague J. TGFbeta in Cancer. Cell. 2008;134(2):215–30.
Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg RA, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107(46):20009–14.
Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, Howell A, Aquila S, Ando S, Martinez-Outschoorn U, et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-beta drives tumor growth: connecting TGF-beta signaling with "Warburg-like" cancer metabolism and L-lactate production. Cell Cycle. 2012;11(16):3019–35.
Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700.
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–84.
Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119(6):1429–37.
Yoshida GJ. Emerging role of epithelial-mesenchymal transition in hepatic cancer. J Exp Clin Cancer Res. 2016;35(1):141.
Xie B, Lin W, Ye J, Wang X, Zhang B, Xiong S, Li H, Tan G. DDR2 facilitates hepatocellular carcinoma invasion and metastasis via activating ERK signaling and stabilizing SNAIL1. J Exp Clin Cancer Res. 2015;34:101.
Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179(3):1074–80.
Sotgia F, Del Galdo F, Casimiro MC, Bonuccelli G, Mercier I, Whitaker-Menezes D, Daumer KM, Zhou J, Wang C, Katiyar S, et al. Caveolin-1−/− null mammary stromal fibroblasts share characteristics with human breast cancer-associated fibroblasts. Am J Pathol. 2009;174(3):746–61.
Razani B, Zhang XL, Bitzer M, von Gersdorff G, Bottinger EP, Lisanti MP. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J Biol Chem. 2001;276(9):6727–38.
Igarashi J, Shoji K, Hashimoto T, Moriue T, Yoneda K, Takamura T, Yamashita T, Kubota Y, Kosaka H. Transforming growth factor-beta1 downregulates caveolin-1 expression and enhances sphingosine 1-phosphate signaling in cultured vascular endothelial cells. Am J Physiol Cell Physiol. 2009;297(5):C1263–74.
Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells "fuel" cancer cell mitochondria. Cell Metab. 2012;15(1):4–5.
Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, Frank PG, Casimiro MC, Wang C, Pestell RG, et al. The autophagic tumor stroma model of cancer: role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9(17):3485–505.
Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117(Pt 8):1281–3.
Delgoffe GM, Murray PJ, Vignali DA. Interpreting mixed signals: the cell's cytokine conundrum. Curr Opin Immunol. 2011;23(5):632–8.
Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178(5):2623–9.
Hooper S, Gaggioli C, Sahai E. A chemical biology screen reveals a role for Rab21-mediated control of actomyosin contractility in fibroblast-driven cancer invasion. Br J Cancer. 2010;102(2):392–402.
Shintani Y, Fujiwara A, Kimura T, Kawamura T, Funaki S, Minami M, Okumura M. IL-6 secreted from Cancer-associated fibroblasts mediates Chemoresistance in NSCLC by increasing epithelial-Mesenchymal transition signaling. J Thorac Oncol. 2016;11(9):1482–92.
Qiao Y, Zhang C, Li A, Wang D, Luo Z, Ping Y, Zhou B, Liu S, Li H, Yue D, et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene. 2018;37(7):873–83.
Mozaffarian A, Brewer AW, Trueblood ES, Luzina IG, Todd NW, Atamas SP, Arnett HA. Mechanisms of oncostatin M-induced pulmonary inflammation and fibrosis. J Immunol. 2008;181(10):7243–53.
Nightingale J, Patel S, Suzuki N, Buxton R, Takagi KI, Suzuki J, Sumi Y, Imaizumi A, Mason RM, Zhang Z. Oncostatin M, a cytokine released by activated mononuclear cells, induces epithelial cell-myofibroblast transdifferentiation via Jak/Stat pathway activation. J Am Soc Nephrol. 2004;15(1):21–32.
Guilluy C, Bregeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, Henrion D, Scalbert E, Bril A, Torres RM, et al. The rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med. 2010;16(2):183–90.
Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, Daly L, Wels J, Theilen T, Granitto S, Zhang X, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia. 2013;15(7):848–62.
Song L, Smith MA, Doshi P, Sasser K, Fulp W, Altiok S, Haura EB. Antitumor efficacy of the anti-interleukin-6 (IL-6) antibody siltuximab in mouse xenograft models of lung cancer. J Thorac Oncol. 2014;9(7):974–82.
Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, Philippe C, Herraiz Serrano C, Benamar S, Croce O, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun. 2015;6:10204.
Sengupta TK, Schmitt EM, Ivashkiv LB. Inhibition of cytokines and JAK-STAT activation by distinct signaling pathways. Proc Natl Acad Sci U S A. 1996;93(18):9499–504.
Hu M, Yao J, Cai L, Bachman KE, van den Brule F, Velculescu V, Polyak K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37(8):899–905.
Mishra R, Haldar S, Suchanti S, Bhowmick NA. Epigenetic changes in fibroblasts drive cancer metabolism and differentiation. Endocr Relat Cancer. 2019;26(12):R673–88. https://doi.org/10.1530/ERC-19-0347.
Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica-Worms D, Stewart SA. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012;72(9):2251–61.
Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673–91.
Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, Larkin J, Marais R, Sahai E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell. 2015;27(4):574–88.
Bissell MJ, Labarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7(1):17–23.
Navab R, Strumpf D, To C, Pasko E, Kim KS, Park CJ, Hai J, Liu J, Jonkman J, Barczyk M, et al. Integrin alpha11beta1 regulates cancer stromal stiffness and promotes tumorigenicity and metastasis in non-small cell lung cancer. Oncogene. 2016;35(15):1899–908.
Primac I, Maquoi E, Blacher S, Heljasvaara R, Van Deun J, Smeland HY, Canale A, Louis T, Stuhr L, Sounni NE, et al. Stromal integrin alpha11 regulates PDGFR-beta signaling and promotes breast cancer progression. J Clin Invest. 2019;130:4609–28.
Shin K, Lim A, Zhao C, Sahoo D, Pan Y, Spiekerkoetter E, Liao JC, Beachy PA. Hedgehog signaling restrains bladder cancer progression by eliciting stromal production of urothelial differentiation factors. Cancer Cell. 2014;26(4):521–33.
Gerling M, Buller NV, Kirn LM, Joost S, Frings O, Englert B, Bergstrom A, Kuiper RV, Blaas L, Wielenga MC, et al. Stromal hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat Commun. 2016;7:12321.
Acknowledgments
I would like to thank Dr. Enomoto (Department of Pathology, Nagoya University Graduate School of Medicine) for advice and useful comments that helped finalize this review article.
Funding
This review article was financially supported by the Japan Society for the Promotion of Science (19 K23898).
Author information
Authors and Affiliations
Contributions
GJY searched the literature and wrote the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
GJY agrees to the content of this review paper, and to being the corresponding author.
Competing interests
There are no competing interests to be addressed.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yoshida, G.J. Regulation of heterogeneous cancer-associated fibroblasts: the molecular pathology of activated signaling pathways. J Exp Clin Cancer Res 39, 112 (2020). https://doi.org/10.1186/s13046-020-01611-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-020-01611-0