
Abstract
Tyrosine kinases are implicated in tumorigenesis and progression, and have emerged as major targets for drug discovery. Tyrosine kinase inhibitors (TKIs) inhibit corresponding kinases from phosphorylating tyrosine residues of their substrates and then block the activation of downstream signaling pathways. Over the past 20 years, multiple robust and well-tolerated TKIs with single or multiple targets including EGFR, ALK, ROS1, HER2, NTRK, VEGFR, RET, MET, MEK, FGFR, PDGFR, and KIT have been developed, contributing to the realization of precision cancer medicine based on individual patient’s genetic alteration features. TKIs have dramatically improved patients’ survival and quality of life, and shifted treatment paradigm of various solid tumors. In this article, we summarized the developing history of TKIs for treatment of solid tumors, aiming to provide up-to-date evidence for clinical decision-making and insight for future studies.
Similar content being viewed by others


Introduction
According to GLOBOCAN 2018, an estimated 18.1 million new cancer cases and 9.6 million cancer deaths occurred in 2018 worldwide [1]. Targeted agents are superior to traditional chemotherapeutic ones in selectivity, efficacy, and safety by acting on specific targets involved in proliferation and differentiation of cancer cells with minimal activity on normal cells.
At least 58 receptor tyrosine kinases (RTKs) and 32 non-receptor tyrosine kinases (NRTKs) have been found so far [2]. RTKs and NRTKs function by catalyzing the transfer of a phosphoryl group from a nucleoside triphosphate donor to the hydroxyl group of tyrosine residues on protein substrates and then triggering the activation of downstream signaling cascades [3]. Abnormal activation of tyrosine kinases due to mutations, translocations, or amplifications is implicated in tumorigenesis, progression, invasion, and metastasis of malignancies. In addition, wild-type tyrosine kinases can also function as critical nodes for pathway activation in cancer. As such, tyrosine kinases have emerged as major targets for drug discovery [4, 5]. A tyrosine kinase inhibitor (TKI) is designed to inhibit the corresponding kinase from playing its role of catalyzing phosphorylation [6]. Since US Food and Drug Administration (FDA) approved imatinib for the treatment of chronic myeloid leukemia in 2001, multiple potent and well-tolerated TKIs—targets including EGFR, ALK, ROS1, HER2, NTRK, VEGFR, RET, MET, MEK, FGFR, PDGFR, and KIT—have been emerging and contributing to the significant progress in cancer treatment. Besides TKIs with one target, some TKIs block a broader range of targets, such as VEGFR-associated multi-targeted TKIs. Noted that some of the multi-targeted TKIs were initially designed to be highly selective, but they turned out to cover other unexpected targets as well [7, 8].
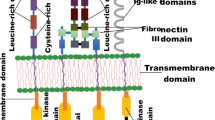
In this article, we summarized the developing history of TKIs for treatment of solid tumors in the past 20 years (2001–2020). And we presented a schematic summary of the approved TKIs for different targets in Fig. 1.

A schematic summary of the approved TKIs in 2001–2020. NMPA National Medical Products Administration, MHLW Ministry of Health, Labor and Welfare, FDA Food and Drug Administration
EGFR-TKIs
The epidermal growth factor receptor (EGFR), also called HER1, belongs to ErbB family which is composed of four structure-related RTKs: HER1-4. EGFR is a transmembrane glycoprotein with tyrosine kinase activity in its endo-domain. The activation of EGFR can initiate several crucial signal cascades including RAS/RAF/MEK/ERK, PI3K/AKT/mTOR, and STAT pathways [9, 10]. EGFR-sensitizing mutations (i.e., exons 19 deletions and exon 21 L858R substitution) occur in 17.3% of Western and 45.7% of Asian patients with lung adenocarcinoma [11, 12]. To date, EGFR-TKIs are relatively in depth researched with four generations being developed and have been playing irreplaceable roles in the treatment of EGFR-mutant NSCLC patients [13]. Table 1 summarizes advances of EGFR-TKIs.
First- and second-generation EGFR-TKIs for EGFR-sensitizing mutations
First-generation reversible EGFR-TKIs (e.g., gefitinib [14, 15], erlotinib [16] and icotinib [17, 18]) have yielded significant survival benefits for patients with advanced NSCLC harboring EGFR-sensitizing mutations. Additionally, efforts to investigate them in adjuvant setting have also been made [19,20,21]. Second-generation EGFR-TKIs (e.g., afatinib, dacomitinib) bind irreversibly to EGFR and typically belong to pan-HER inhibitors. Dacomitinib yielded an improved median progression-free survival (mPFS) (14.7 vs 9.2 months; hazard ratio (HR) 0.59; p < 0.0001) and median overall survival (mOS) (34.1 vs 26.8 months; HR 0.76; p = 0.044) compared to gefitinib in first-line treatment of advanced EGFR-mutant NSCLC [22, 23]. However, both afatinib and dacomitinib have increased toxicities, which may limit their use in clinical practice.
Third-generation EGFR-TKIs
Approximately 50% of resistance to first- and second-generation EGFR-TKIs are due to EGFR T790M mutation, in which the significantly bulkier methionine residue replaces the small polar threonine at position 790 of EGFR exon 20. As a gatekeeper to the adenosine triphosphate (ATP) binding pocket of EGFR, T790M could cause conformational change resulting in the development of steric hindrance; it could also increase the ATP affinity; all of these reduce binding ability and access of first- and second-generation EGFR inhibitors to the EGFR ATP binding pocket [24].
Osimertinib is a third-generation irreversible EGFR-TKI that inhibits both EGFR-sensitizing and EGFR T790M mutations, and was initially approved for NSCLC with EGFR T790M mutation [25]. Later, it also demonstrated superiority over gefitinib or erlotinib in the first-line treatment of EGFR-mutant NSCLC [26, 27]. Along with its favorable safety, osimertinib is likely to surpass other approaches in the standard of care [28]. In addition, osimertinib as adjuvant therapy for stage IB–IIIA EGFR-mutant NSCLC after complete tumor resection also achieved meaningful survival results [29].
Up to 40% of NSCLC patients with EGFR mutation develop central nervous system (CNS) metastases on or after first- or second-generation EGFR-TKIs treatment due to their poor penetration of the blood–brain barrier (BBB). Notably, osimertinib demonstrated favorable efficacy for patients harboring CNS metastases with or without prior EGFR-TKIs treatment [30] or those with EGFR T790M mutation [25]. Besides, EGFR-mutated NSCLC patients with leptomeningeal metastases could also benefit from osimertinib after progression on previous EGFR-TKIs [31].
Almonertinib, another third-generation EGFR-TKI, was approved by China National Medical Products Administration (NMPA). In its phase II trial, an objective response rate (ORR) of 68.9% was observed in patients with previously treated EGFR T790M-positive NSCLC along with a CNS ORR of 60.9% [32]. Other third-generation EGFR-TKIs including furmonertinib (AST2818) [33, 34], lazertinib (YH25448) [35], BPI-7711 [36], and nazartinib (EGF816) [37] have shown promising efficacies and acceptable safeties in advanced NSCLC with EGFR T790M mutation. In a phase IIb trial of furmonertinib, an ORR of 73.6% was observed in patients with EGFR T790M mutated NSCLC [34]. In safety analysis, skin and gastrointestinal disorders as well as interstitial lung disease (ILD) related to furmonertinib seem to be less common than osimertinib [33].
Strategies for resistance of osimertinib
Long-term responses of third-generation EGFR-TKIs are often compromised by acquired resistant mutations, with EGFR exon 20 C797S mutation as the predominant cause [10]. The prime therapeutic strategy after resistance of osimertinib remains unclear. Though patients harboring C797S in trans with T790M (at different alleles) may respond to the combination of first- and third-generation EGFR-TKIs [38, 39], patients harboring C797S in cis with T790M (at the same allele), which is more common, are likely to show no response [40]. Fourth-generation EGFR-TKIs are under development to overcome osimertinib resistance mediated by EGFR-dependent mutation mechanisms, such as EAI045 against T790M and C797S mutations [41], and TQB3804 against osimertinib-resistant EGFR triple mutant (d746-750/T790M/C797S, L858R/T790M/C797S) or double mutant (d746-750/T790M, L858R/T790M) [42]. In addition, the combination of brigatinib with cetuximab has showed preliminary efficacy in patients with EGFR/T790M/cis-C797S triple mutation [43].
TKIs for rare EGFR mutations
Targeted therapy for rare EGFR mutations remains an unmet need in NSCLC. Osimertinib showed efficacy against NSCLC with uncommon mutations including L861Q, G719X, or S768I substitutions [44]. Several agents such as TAK-788 (mobocertinib), poziotinib, and tarloxotinib are under investigations for a refractory variant: EGFR exon 20 insertion. A phase I/II study of TAK-788 demonstrated an ORR of 54% in previously treated NSCLC patients harboring EGFR exon 20 insertions [45] and was granted a breakthrough therapy designation by FDA. Poziotinib failed to meet its primary endpoint (ORR 14.8%), but induced tumor reduction in 65% of NSCLC patients with EGFR exon 20 insertion mutants in a phase II trial [46].
Summary
EGFR-TKIs are effective treatment approaches for EGFR-sensiting-mutant NSCLC. T790M mutation has been the most common mechanism of resistance to first- or second-generation EGFR-TKIs, which fortunately can benefit from third-generation EGFR-TKIs. Novel inhibitors for uncommon EGFR mutations have been emerging. Besides, fourth-generation EGFR-TKIs are under development for resistance of third-generation ones caused by C797S mutation. Moreover, combination treatments have been under investigations. Unlike the concern of toxicities with EGFR-TKIs in combination with the programmed death ligand 1 (PD-L1) antibody [47], combining EGFR-TKIs with anti-VEGF antibody (e.g., ramucirumab) or chemotherapy has shown survival benefit in patients with EGFR mutations.
ALK-TKIs
The fusion gene of echinoderm microtubule-associated protein-like 4 and anaplastic lymphocyte kinase (EML4-ALK) occurs in 3–5% of NSCLC [48]. It is called “diamond mutation” for the exceedingly prolonged survival benefit from sequential treatment of potent ALK-TKIs [49].
First- and second-generation ALK-TKIs
First-generation ALK-TKI crizotinib, which targets ALK, ROS1, and c-MET, showed superiority for ALK-positive NSCLC over chemotherapy [50]. However, its unsatisfactory PFS benefits and limited control of brain metastases pushed the development of second-generation ALK-TKIs (ceritinib, alectinib and brigatinib) which are characterized as higher selectivity and CNS penetration, and they are generally effective after failure of crizotinib [51]. As a new ALK-TKI, ensartinib potently inhibits wild-type ALK and common crizotinib-resistant mutations, demonstrating an ORR of 52% in patients who were progressed on crizotinib [52, 53].
Second-generation ALK-TKIs have shown favorable efficacies after progression on crizotinib in clinical practice. A mOS of 89.6 months has been reported in 84 ALK-positive NSCLC patients by the sequencing treatment of second-generation ALK-TKIs after crizotinib resistance in a real-world setting [54]. Moreover, attempt to set second-generation ALK-TKIs as first-line therapy in ALK-positive NSCLC has achieved surprising outcomes, and its standard place has been established. Alectinib is considered as a preferred choice, while ceritinib and brigatinib can serve as other recommended options. In a phase III study of alectinib, it significantly prolonged mPFS compared to crizotinib in treatment-naïve advanced ALK-positive NSCLC patients (34.8 vs 10.9 months; HR 0.43) [55, 56]. Despite the superiority of ceritinib over crizotinib predicted by adjusted indirect comparison in front-line setting [57], no comparative study has been prospectively conducted yet. For brigatinib, a superior mPFS over crizotinib was also observed in the first-line setting (24.0 vs 11.0 months; HR 0.49; p < 0.0001) [58, 59]. It has received an approval from both European Medicines Agency (EMA) and FDA as a first-line approach for metastatic ALK-positive NSCLC. Second-generation ALK-TKIs such as alectinib and brigatinib also demonstrated CNS benefits over crizotinib [59, 60]. Other second-generation ALK-TKIs including WX-0593 [61] and CT707 [62] are under clinical investigations and have produced promising outcomes.
Third-generation ALK-TKIs
Generally, 56% of patients treated with second-generation ALK-TKIs develop acquired resistance due to secondary ALK mutations [63]. When such resistance occurs, lorlatinib, a third-generation ALK/ ROS1-TKI with potency against most known ALK mutations, is a therapeutic option. In a phase II trial, lorlatinib demonstrated meaningful activity as both first-line and subsequent therapies for ALK-rearranged NSCLC, with an ORR of 90% in treatment-naïve patients, 47% in patients with one previous ALK-TKI treatment, and 38.7% in patients with two or more previous ALK-TKIs treatment [64]. In respect of safety profile, apart from common adverse events (AEs) such as hypercholesterolemia and hypertriglyceridemia, grade ≥ 3 neurological toxicity including peripheral neuropathy (2%), cognitive effect (1%), and dizziness (1%) should be taken with caution. With complex mechanisms of resistance to lorlatinib being identified, future tailored approaches for such patients are warranted [65]. Another third-generation ALK-TKI CT-3505 is under investigation (ChiCTR1900025619).
Summary
Treatment strategies for ALK-rearranged NSCLC patients have advanced considerably with the development of crizotinib and newer generations of ALK-TKIs. Acquisition of resistance to ALK-TKIs ultimately occurs; the best sequencing strategy of first- to third-generation ALK-TKIs warrants further investigations. Additionally, combination treatment of ALK-TKIs and immune checkpoint inhibitors (ICIs) is associated with higher morbidity in many cases, combination with other classes of agents are ongoing.
ROS1-TKIs
ROS proto-oncogene 1 (ROS1) rearrangements are identified in 1–2% of NSCLC and in several other malignancies such as cholangiocarcinoma (CCA), glioblastoma, or colorectal cancer [66]. Since kinase domains of ROS1 and ALK share similar amino acid residues, crizotinib, ceritinib, and lorlatinib have also shown clinical benefits in NSCLC patients with ROS1 fusion. Crizotinib and entrectinib are the two recommended first-line options. Crizotinib was approved with an ORR of 72% [67]. Entrectinib was simultaneously approved for the treatment of ROS1-rearranged metastatic NSCLC and NTRK gene fusion-positive solid tumors. In ROS1 fusion-positive NSCLC patients, entrectinib demonstrated an ORR of 77% and an intracranial ORR of 55%, along with an acceptable safety profile [68].
When resistance to first-line therapies occurs, lorlatinib and repotrectinib are alternative options. Lorlatinib demonstrated clinical activity in advanced NSCLC patients with ROS1 fusion, including those who are ROS1-TKI-naïve (ORR 62%), crizotinib pretreated (35%), and those with CNS metastases (first-line ORR 64%; second line 50%) [69]. In a phase I study, repotrectinib showed activity both in ROS1-TKI-naïve (ORR 82%) and ROS1-TKI-treated patients (39%) [70]. DS-6051b is potent against ROS1 G2032R (a predominant mechanism of resistance to crizotinib [71]) and other crizotinib-resistant ROS1 mutations and has demonstrated clinical efficacy [72, 73]. Table 2 summarizes advances of ALK/ROS1-TKIs.
HER2-TKIs
Human epidermal growth factor receptor 2 (HER2) is a member of ErbB family and shares similar structure with EGFR. Positive-HER2 was reported in 15–20% of invasive breast cancer and considered to be associated with poor differentiation, rapid cell proliferation, lymph node involvement, and resistance to certain types of chemotherapy [74, 75]. The outcome of patients with HER2-positive breast cancer has been significantly improved in the era of targeted therapy. Four TKIs are available for HER2-positive metastatic breast cancer (MBC), namely lapatinib, neratinib, pyrotinib, and tucatinib. Table 3 summarizes advances of HER2-TKIs.
Lapatinib is a reversible EGFR/HER2-TKI, which was first approved in combination with capecitabine for patients with HER2-positive MBC who have failed trastuzumab-based therapy [76]. Later lapatinib plus letrozole obtained another approval as a first-line therapeutic option for the post-menopausal MBC patients with co-expressing hormone receptors and HER2 [77, 78].
Neratinib is an irreversible pan-ErbB inhibitor. The role of neratinib in conferring synergic effect and overcoming resistance of trastuzumab has been identified [75]. In a phase III trial, one-year neratinib after trastuzumab-based adjuvant treatment for early-stage HER2 positive breast cancer reduced invasive disease-free survival events (116 vs 163 events; HR 0.73; p = 0.0083) without increasing risk of toxicity, which established its first approval [79]. Recently, it received its second approval in combination with capecitabine for adult patients with HER2-positive MBC after two or more prior anti-HER2 treatment. A phase III study showed neratinib plus capecitabine significantly reduced the risk of disease progression or death (HR 0.76; p = 0.006) and delayed symptomatic CNS metastasis (p = 0.043) compared with lapatinib plus capecitabine [80].
Pyrotinib, another irreversible pan-ErbB inhibitor, was approved by NMPA based on a phase II study, in which pyrotinib plus capecitabine combination showed superior efficacy in previously treated HER2-positive MBC patients compared to lapatinib plus capecitabine combination [81]. Subsequently, its phase III trial only recruited HER2-positive MBC patients pretreated with taxane and trastuzumab and showed significantly longer PFS in pyrotinib plus capecitabine group than lapatinib plus capecitabine group (12.5 vs 6.8 months; HR 0.39; p < 0.0001) [82]. In addition, pyrotinib also demonstrated promising efficacy in chemotherapy-treated NSCLC patients harboring HER2 exon 20 mutation with an ORR of 30% [83].
Tucatinib, a highly selective HER2-TKI, was approved in combination with trastuzumab and capecitabine for previously treated HER2-positive MBC. The tucatinib combination group revealed improved 1-year PFS (33.1 vs 12.3%; HR 0.54; p < 0.001) and 2-year OS (44.9 vs 26.6%; HR 0.66; p = 0.005) compared to placebo plus trastuzumab and capecitabine group. In terms of safety, grade ≥ 3 diarrhea (12.9 vs 8.6%), elevated alanine aminotransferase (ALT) (5.4 vs 0.5%), and elevated aspartate aminotransferase (AST) (4.5 vs 0.5%) were more common in the tucatinib combination group [84].
Brain metastases occur in 30–50% of HER2-positive MBC, which is tricky with limited evidence-based therapeutic options. Though efficacy of HER2-TKI as a single agent was moderate, combinations of HER2-TKI with capecitabine offered survival benefit for HER2-positive patients with brain metastasis [85, 86]. Lapatinib, neratinib, and tucatinib all had successful attempt in this area [87,88,89].
Intrinsic and acquired resistance of HER2-TKIs has been investigated. Several genes and pathways (including EGFR family, PI3K/Akt/mTOR, RAS/RAF/MEK/MAPK, autophagy, tumor metabolism, low PTEN, PIK3CA mutations, etc.) have been reported to be associated with lapatinib resistance [90] and may provide inspiration for future HER2-TKI development.
TRK inhibitors
Tropomyosin receptor kinase (TRK) refers to the neurotrophin receptor tyrosine kinase genes NTRK1/2/3 and their respective encoding neurotrophin protein receptors TRKA/B/C. TRK signal pathways play crucial roles in neuronal development and differentiation. Fusions involving NTRK1/2/3 are the most common mechanisms of oncogenic TRK activation, which are found across a wide variety of malignancies independent of tumor lineage and patients’ age. Rare tumors are reported to have a higher TRK fusion frequency than common tumors [91, 92]. TRK inhibition provides a prime example of the basket trial for targeted therapy, wherein same genomic-altered cancers are treated with one matched therapeutic agent regardless of tumor histology [92,93,94].
First-generation TRK inhibitors
Larotrectinib and entrectinib are two approved first-generation TRK inhibitors for adult and pediatric (12 years of age and older) patients with solid tumors harboring NTRK gene fusions which are unresectable, resistant, or lack of satisfactory standard therapy.
Larotrectinib is a selective inhibitor of TRKA/B/C which obtained its approval based on the combined analysis of three phase I/II trials involving 17 unique TRK fusion-positive tumor types [95]. In an expanded data set, patients treated with larotrectinib achieved an ORR of 79%, with manageable toxicities: the most common grade 3–4 TRAEs included increased ALT (3%), anemia (2%), and decreased neutrophil count (2%) [96]. Later analysis showed patients who had received more lines of treatment tend to have less effective response to larotrectinib; response rate dropped more sharply as the Eastern Cooperative Oncology Group (ECOG) performance status (PS) got worse, but a ECOG PS score of 1–2 still can benefit from larotrectinib treatment [97].
Entrectinib is a multi-kinase inhibitor targeting TRK, ROS1, and ALK. The pooled analysis revealed an ORR of 57.4% in patients with TRK fusion across 10 tumor types [98]. In a recent study, entrectinib produced favorable responses in children and adolescents with refractory CNS and extracranial solid tumors harboring NTRK, ROS1, or ALK fusions, as well as those with ALK-mutated neuroblastoma [99].
Other multi-kinase inhibitors including crizotinib, cabozantinib, ponatinib, nintedanib, lestaurtinib, altiratinib, foretinib, merestinib, MGCD516, PLX7486, DS-6051b, and TSR-011 have varying degrees of activity against TRKA/B/C in vitro, but their clinical activities have not been as robust as those of larotrectinib and entrectinib [93, 100].
Second-generation TRK inhibitors
On-target or off-target mechanisms would disappointedly result in resistance to first-generation TRK inhibitors. On-target resistance mechanisms mainly refer to NTRK kinase domain mutations involving amino acid substitutions of the solvent front, the gatekeeper residue, or the xDFG motif [100].
Two major developing second-generation TRK inhibitors selitrectinib (LOXO-195) and repotrectinib (TPX0005) are designed to overcome the acquired on-target resistance of first-generation ones and possess enhanced activities against wild-type TRKA/B/C. Selitrectinib (LOXO-195) selectively targets multiple TRK kinase domain mutations including solvent front and xDFG substitutions [101]. The largest data set of LOXO-195 till now enrolled 31 TRK-fusion patients with 11 cancer types progressing or being intolerant to at least one prior TRK inhibitor: the ORR was 54% in patients with on-target TRK mutations [102]. Repotrectinib (TPX-0005), another next-generation ROS1/TRK/ALK inhibitor, has shown promising anti-tumor activity, a confirmed partial response (reduced by 82%) in an entrectinib-resistant patient with a salivary gland tumor and a tumor regression (reduced by 33%) in a patient with larotrectinib-resistant cholangiocarcinoma were reported [103].
Mechanisms of off-target resistance to first- or second-generation TRK inhibitors include KRAS and BRAF V600E mutations, MET amplifications, IGF1R activation, etc. The convergent activation of mitogen-activated protein kinase (MAPK) pathway was also proposed to mediate the resistance of TRK inhibition. Second-generation TRK inhibitors are ineffective against off-target resistance, whereas a single targeted agent for off-target mutation or combined with TRK inhibition might re-established disease control in this situation. For example, simultaneous inhibition of TRK and MEK (belongs to MAPK pathway) was found to successfully manage some off-target TRK resistance in vitro and vivo [104, 105].
Table 4 summarizes advances of TRK inhibitors.
VEGFR-associated multi-targeted TKIs
The vascular endothelial growth factor (VEGF) family is composed of VEGF-A/B/C/D/E and placental growth factor. VEGF-A, also called VEGF, is the master regulator of angiogenesis. The binding of VEGF to VEGFR-2 plays a key role in stimulating the proliferation and migration of endothelial cells as well as regulating vascular permeability [106]. In recent years, VEGFR-associated multi-targeted TKIs have emerged as potent anti-tumor weapons against multiple solid tumors [107].
Applications of VEGFR-associated multi-targeted TKIs in hepatocellular carcinoma (HCC)
The potency of VEGFR-associated multi-targeted TKIs was supported by robust evidence in HCC [108]. Sorafenib, targeting VEGFR, PDGFR, FGFR, and other signaling targets, is recommended for front-line therapy for unresectable HCC [109, 110]. When compared with sorafenib, lenvatinib demonstrated a superior mPFS (7.4 vs 3.7 months; p < 0.0001) and a non-inferior mOS (13.6 vs 12.3 months; HR 0.92). Lenvatinib produced fewer grade ≥ 3 palmar–plantar erythrodysaesthesia but with higher incidence of hypertension and proteinuria [111]. More recently, the combination of lenvatinib with anti-programmed cell death protein-1 (PD-1) antibody pembrolizumab showed encouraging anti-tumor activity in patients with untreated/unresectable HCC with an ORR of 36%, a mPFS of 8.6 months, and a mOS of 22.0 months [112]. This combination has been granted a breakthrough therapy designation by FDA. In a phase II/III trial involving advanced HCC, another VEGFR-associated multi-targeted TKI donatinib achieved a superior OS over sorafenib (12.1 vs 10.3 months; HR 0.83; p = 0.0363) [113]. In addition, several other VEGFR-associated multi-targeted TKIs including regorafenib [114], cabozantinib [115], and apatinib [116] are applied as subsequent-line therapies of HCC.
Applications of VEGFR-associated multi-targeted TKIs in renal cell carcinoma (RCC), lung cancer, and other cancer type
RCC is another cancer type deriving great benefit from VEGFR-associated multi-targeted TKIs. Sorafenib, sunitinib, pazopanib, cabozantinib, the combination of axitinib and pembrolizumab/avelumab were successively approved as first-line treatment options for metastatic RCC.
Anlotinib has yielded favorable outcomes in lung cancer and was approved for third-line or further-line therapy for both NSCLC [117] and SCLC [118] by NMPA. In its phase II trial for patients with relapsed SCLC, a mPFS of 4.1 months was reported [119]. Similarly, apatinib presented a mPFS of 5.4 months in patients with extensive-stage SCLC after failure of two or more lines of chemotherapy [120].
Furthermore, VEGFR-associated multi-targeted TKIs also demonstrated survival benefits in patients with thyroid carcinoma, soft tissue sarcoma (STS), and other solid malignancies [121] (see Table 5).
Anti-angiogenesis and PD-1/PD-L1 inhibition
Preclinical and clinical studies suggested that the combination of anti-angiogenesis inhibitors with ICIs could provide superior anti-tumor activity over either single agent. VEGFR inhibitors might potentially improve immunotherapeutic activity of PD-1/PD-L1 antibodies by enhancing tumor infiltration of immune cells and reducing immunosuppressive effects of myeloid-derived suppressor cells [122]. Investigational combinations of VEGFR-associated multi-targeted TKIs and anti-PD-1/PD-L1 antibodies are summarized in Table 6.
RET-TKIs
The rearranged during transfection (RET) tyrosine kinase plays a role in transducing signals involving cell growth and differentiation. RET alterations (i.e., RET fusions and point mutations) are implicated in the pathogenesis of lung, thyroid, and other cancers. RET fusions were found in 10–20% of papillary thyroid cancers and 1–2% of NSCLC, while RET point mutations occur in approximately 60–90% of advanced medullary thyroid cancers (MTC) [123]. Table 7 summarizes advances of RET-TKIs.
Before any selective RET inhibitor becomes available, chemotherapy, multi-targeted TKIs, and clinical trials are common choices for RET-altered cancer patients. Multi-targeted TKIs including cabozantinib and vandetanib have been clinically used in RET-driven lung and thyroid cancers, but their insufficient inhibition of RET and off-target toxicities limited broader application [124]. Likewise, NSCLC patients with RET rearrangements have minimal response to immunotherapy (ORR 6%) [112].
Currently, two selective RET-TKIs shed light in this area. Selpercatinib (LOXO-292) is the first approved RET-TKI with applications for advanced RET fusion-positive NSCLC, thyroid cancer, and RET-mutant MTC. In the treatment of RET fusion-positive NSCLCs, selpercatinib presented an ORR of 85% in patient who were systemic treatment-naïve ones, 64% in previously treated patients, 91% in patients with CNS metastases [125]. In the treatment of RET-altered thyroid cancers, the ORRs were 73% and 69%, respectively, in treatment-naïve and previously treated RET-mutant MTCs patients, and 79% in previously treated thyroid cancers patients with RET fusion. In general, selpercatinib was well tolerated, with only 2% of 531 patients discontinuing treatment due to TRAEs [126].
Pralsetinib (BLU-667) recently obtained a rolling new drug application (NDA) submission for RET fusion-positive NSCLC. It demonstrated promising clinical efficacy regardless of RET fusion genotype or prior therapies status. The ORR was 73% for treatment-naïve NSCLC patients harboring RET fusion and 61% for platinum-exposed patients. It is well tolerated with most TRAEs being grade 1–2, including increased AST (31%), anemia (22%), increased ALT (21%), constipation (21%), and hypertension (20%) [127]. A broad range of anti-tumor activity of pralsetinib on multiple advanced RET fusion-positive solid tumors in addition to NSCLC has also been reported; tumor type includes papillary thyroid cancers, undifferentiated thyroid cancer, pancreatic cancer, colon cancer, etc.[128].
Up-to-date evidence of resistance mechanism to selective RET inhibitor remains limited. RET G810 solvent front mutation represents a recurrent mechanism of resistance to selpercatinib and should be considered when developing more potent or next-generation RET-TKIs [129].
MET-TKIs
The mesenchymal–epithelial transition factor (MET) is also called c-MET or hepatocyte growth factor receptor (HGFR). The binding of MET to its ligand HGF activates various signaling pathways and plays a role in cellular proliferation, motility, migration, and invasion [130,131,132]. Identifying potential patients sensitive to MET inhibitors by detection of MET exon14 skipping alterations (METex14) or MET amplification has made some progress in recent years. Table 8 summarizes advances of MET-TKIs.
Targeting MET as the primary oncogenic event of NSCLC
METex14 occur in approximately 3% of lung adenocarcinoma, 2% of other lung neoplasms, and rare in other tumors [133]. Intriguingly, crizotinib was initially developed as a MET inhibitor and later on made great achievements in ALK and ROS1 inhibition. But it still showed meaningful activity against MET amplification and METex14 [134]. In a recent study, crizotinib demonstrated an ORR of 32% in NSCLC patients with METex14-mutation [135]. Other multi-targeted TKIs such as cabozantinib, merestinib, glesatinib, and sitravatinib also showed meaningful activities against MET [136].
Apart from multi-targeted TKIs, selective MET inhibitors like tepotinib, camaptinib, and savolitinib have emerged with promising survival benefits. Tepotinib has received a breakthrough therapy designation by FDA for treatment of metastatic NSCLC after failure of platinum-based therapy with an ORR of 47% and a mPFS of 11 months. 27% of patients experienced grade 3–4 AEs, with peripheral edema being the most common AE (7%) [137,138,139]. Recently, tepotinib was approved by Japanese Ministry of Health, Labor and Welfare (MHLW) for the treatment of unresectable, advanced or recurrent NSCLC with METex14 mutation, making it the first approved MET-TKI worldwide.
Another MET inhibitor, capmatinib, was approved for the treatment of adult NSCLC patients with METex14 mutation regardless of treatment history. In a phase II study, the efficacy of capmatinib was evaluated in advanced NSCLC patients with METex14 mutation or MET amplification across 6 cohorts. The ORRs were 41% and 68% among cohort 4 (previously treated METex14 mutation) and cohort 5b (treatment-naïve METex14 mutation), respectively. Its safety profile was acceptable across all cohorts (n = 315), with peripheral edema (49.2%), nausea (43.2%), and vomiting (28.3%) as the most common AEs [140]. Other cohorts also demonstrated the efficacy of capmatinib in advanced NSCLC with high-level MET amplification [141, 142].
Savolitinib (also called volitinib) demonstrated promising anti-tumor activity and manageable toxicity in pulmonary sarcomatoid carcinoma (PSC) and other types of NSCLC with METex14-mutation, with an ORR of 47.5%. Notably, 14.3% of patients discontinued savolitinib due to TRAEs, with liver injury and hypersensitivity being the most common AEs (each 2.9%) [143].
Targeting MET as the secondary event of EGFR-TKI resistant NSCLC
MET amplification is an important resistant mechanism of EGFR-TKIs for NSCLC treatment, accounting for 6.25–22%. More importantly, this patient population is unlikely to respond well to osimertinib or other third-generation EGFR-TKIs. Preclinical and clinical data suggest the combination of EGFR-TKIs with MET-TKIs could be an alternative option to overcome MET-driven acquired resistance of NSCLCs who have progressed on a previous EGFR-TKI [144, 145]. For instance, tepotinib plus gefitinib significantly prolonged mPFS (19.3 vs 5.5 months; HR 0.18), mOS (37.3 vs 13.1 months; HR 0.08), and ORR (75.0 vs 42.9%; OR 4.00) compared to chemotherapy for such patient population. In terms of safety, tepotinib plus gefitinib combination treatment significantly increased grade ≥ 3 amylase and lipase, while anemia, neutrophil, or white blood cell count decrease was less common compared to chemotherapy [146]. In a phase Ib/II trial, capmatinib plus gefitinib yielded an ORR of 47% in EGFR-mutant NSCLC patients with high MET amplification. The most common grade 3–4 AEs were also elevated amylase and lipase levels [147, 148]. Now with increasing use of osimertinib in the front-line treatment of EGFR-mutant NSCLC, combining MET-TKIs with osimertinib has also been explored. Savolitinib plus osimertinib presented an ORR of 48% (with or without a previous third-generation EGFR-TKI) along with acceptable tolerability [149]. The exciting results suggest it may be necessary to identify MET status before starting osimertinib treatment in patients who failed previous former-generation EGFR-TKI treatment.
Fluorescence in situ hybridization (FISH), next generation sequencing (NGS), immunohistochemistry (IHC), and droplet digital PCR (ddPCR) are methods to detect MET-mediated resistance, each with its own advantages and disadvantages. The results of different testing methods do not overlap completely, and a single assay might overlook suitable patients. Therefore, applying more than one method is recommended in future clinical practice and scientific research. Besides, other biomarkers like phosphorylated MET (p-MET) should be explored to help predict response and tailor treatment.[144, 149].
MEK-TKIs
The classic mitogen-activated protein kinase (MAPK) pathway—RAS/RAF/MEK/ERK—is critical in signal transduction, whose dysregulation is implicated in one third of all malignancies. RAS and RAF mutations are implicated in a great portion of malignancies: BRAF V600 mutation is found in 40–60% of melanomas and 10–12% of metastatic colorectal cancer (mCRC), KRAS or NRAS in 55% of mCRC, and KRAS in 20–30% of lung adenocarcinoma [150]. Although the mutation of MEK, also called mitogen-activated protein kinase kinase (MAPKK), is not frequently identified in solid tumors, it is a central and critical component that lies downstream of RAS and RAF, and upstream of ERK for transduction. Currently, MEK serves as a hotspot target for the treatment of RAS/RAF mutant cancers [151].
MEK inhibitors function mainly by non-ATP-competitively blocking the phosphorylation of tyrosine and serine/threonine domain of its downstream ERK [152]. To date, four MEK inhibitors–trametinib, cobimetinib, binimetinib, and selumetinib, as single agent or in combination with BRAF inhibitors–have been approved for melanoma/NSCLC/neurofibromas [153,154,155,156,157,158]. In the treatment of BRAF V600 mutant melanoma, the combination of MEK and BRAF inhibition achieved better outcome than used alone, with manageable safety profiles and lower rates of hypoproliferative skin toxicities and musculoskeletal complaints than BRAF inhibitors, and has become the standard of therapy [157, 159]. In addition, in the treatment of BRAF V600 mutant mCRC, the addition of binimetinib to the doublet inhibition of BRAF and EGFR (encorafenib and cetuximab) showed promising results in a phase III trial [160].
The advances of MEK-TKIs are summarized in Table 9.
FGFR-TKIs
The fibroblast growth factor (FGF) pathway is implicated in tumor growth and angiogenesis [161, 162]. Most FGFR-TKIs approved belong to multi-targeted TKIs (Table 5). Meanwhile, several FGFR inhibitors have achieved applications for certain cancers recently, such as erdafitinib for urothelial carcinoma and pemigatinib for CCA.
Mutations and fusions in FGFR2/3 occur in 20% of patients with urothelial carcinoma [163]. The FGFR1-4 TKI erdafitinib has been approved for the treatment of adult patients with previously treated locally advanced or metastatic urothelial carcinoma harboring susceptible FGFR2/3 mutation with an ORR of 40%. The response was more favorable compared to antibody–drug conjugates such as enfortumab vedotin or sacituzumab govitecan (ORRs of 33–34%) [164] and pembrolizumab (20.1%) as second-line therapy for advanced urothelial carcinoma [165]. 59% of patients who had undergone previous immunotherapy responded to erdafitinib treatment. Nevertheless, it should be noted that nearly half of the patients experienced at least one grade ≥ 3 AE, among which hyponatremia (11%), stomatitis (10%), and asthenia (7%) were most common [166]. Other pan-FGFR inhibitors are under development; for example, infigratinib (BGJ 398) has produced an ORR of 25.4% in the treatment of FGFR3-mutated urothelial carcinoma [164].
In addition to urothelial malignancies, FGFR2 alterations are also implicated in CCA. A FGFR1-3 TKI pemigatinib has been approved for the treatment of locally advanced or metastatic CCA harboring FGFR2 fusions or rearrangements with an ORR of 35.5% [167]. Several other FGFR-TKIs (e.g., futibatinib, infigratinib [168]) have shown promising results for CCA.
Table 10 summarizes advances of FGFR-TKIs.
KIT
KIT proto-oncogene takes part in fertility, homeostasis, and melanogenesis, while the dysregulation of KIT has been found to participate in the occurrence of leukemia, gastrointestinal stromal tumor (GIST), melanoma, and other cancers [169]. KIT usually presents in multi-targeted TKIs as an inconspicuous target (Table 5) since a single selective KIT-TKI failed to cure most cancers. However, the aberrant activation of KIT is particularly responsible for the tumorigenesis of GIST, making it a pivotal target in this disease entity. Besides, KIT inhibition also showed efficacy in KIT-positive melanoma.
PDGFR
Platelet-derived growth factor (PDGF) is a family of a multi-functional polypeptide involved in cellular growth, proliferation, differentiation, and angiogenesis. PDGFR is found to play a crucial role in angiogenesis by promoting the maturation of new blood vessels and up-regulating the expression of VEGF [170]. Most VEGFR-associated multi-kinase inhibitors target PDGFR as well to augment anti-angiogenesis effect and suppress tumor growth (Table 5). Additionally, the inhibition of PDGFR plays an important role specifically in the treatment of GIST (Table 11).
KIT- and PDGFR-TKIs in GIST
GIST generally resists to conventional chemotherapy. Fortunately, since GIST has high frequency of KIT and PDGFR mutation (approximately 80% of GISTs harbor KIT mutation, 10% involve PDGFR mutations), KIT and PDGFR inhibition has been recognized as the primary therapeutic modality for unresectable or metastatic GIST [171]. Table 8 summarizes advances of KIT/PDGFR TKIs.
Imatinib remains as first-line treatment of KIT-positive unresectable GIST. Though more than half of GISTs respond to imatinib, resistance inevitably occurs. Sunitinib and regorafenib are the standard second- and third-line treatment for advanced GIST, respectively [172, 173]. Sunitinib greatly improved median time to tumor progression than placebo (27.3 vs 6.3 weeks; HR 0.33; p < 0.0001) in patients with advanced GIST after failure of imatinib, but with a low ORR of 6.8% [174]. Studies indicated the inconsistent activity of sunitinib in imatinib-resistant populations, with higher response in patients harboring ATP-binding pocket mutations than those with mutations in KIT activation loop [175].
Recently, two selective TKIs targeting KIT and PDGFRA mutants, avapritinib and ripretinib, were approved as fourth-line treatment for GIST. A phase I trial of avapritinib demonstrated an ORR of 86% in GIST patients with PDGFRA exon18 mutation and an ORR of 22% in those who have failed ≥ third-line treatment. Toxicity was generally manageable with anemia, fatigue, hypophosphatemia, hyperbilirubinemia, neutropenia, and diarrhea being the most common grade 3–4 AEs [176]. A phase III trial of ripretinib demonstrated an improved mPFS (6.3 vs 1.0 months; HR 0.15; p < 0.0001) and mOS (15.1 vs 6.6 months; HR 0.36; p = 0.0004) compared to placebo [177, 178]. Besides, in a phase I study, an investigational KIT inhibitor PLX9486 alone or in combination with pexidartinib presented preliminary efficacy against resistant GIST [179].
KIT inhibition in Melanoma
KIT mutations occur in 35–40% of mucosal and acral melanoma, and 28% of melanomas on chronically sun-damaged skin [180]. Imatinib and nilotinib demonstrated ORRs of 17–30% and disease control rates (DCRs) of 35–57% in metastatic melanoma patients with KIT mutation/amplification [181,182,183]. However, most of the reported response only had short duration, and no further phase III trials have been conducted. Until now, none KIT-TKI has received an approval for KIT-mutant melanoma.
Other tyrosine kinase
The insulin-like growth factor 1 receptor (IGF-1R) is a RTK involved in the growth and survival of normal and neoplastic cells; however, multiple trials of IGF-1R inhibitors failed to show definitive clinical benefit [184]. For example, a phase III trial of IGF-1R inhibitor linsitinib for patients with advanced adrenal corticocarcinoma failed to prolong either PFS or OS compared to placebo [185].
Bruton's tyrosine kinase (BTK), an intracellular NRTK, plays a crucial role in B-cell antigen receptor (BCR) signaling pathway. The application of BTK inhibitors, such as ibrutinib, acalabrutinib, and zanubrutinib, is considered as a breakthrough in B-cell-related hematological malignancies and autoimmune diseases, but with limited positive finding in solid tumors [186]. Nevertheless, increasing knowledge of off-target effects of BTK inhibitors and B-cells’ role in proliferation of solid tumors still encourages further but careful exploration of BTK inhibitors in solid malignancies, either as single agent or in combination with other treatment strategies like ICIs [187, 188].
Prospects and conclusions
Twenty years have passed since the approval of the pioneer TKI imatinib for chronic myeloid leukemia in 2001 which was deemed as the beginning of targeted therapeutic era. Increasing numbers of TKIs for tough-to-inhibit oncogenic targets are available for clinical use, providing precise targeted therapy options based on individual patients’ genetic alteration features. TKIs have dramatically improved patients’ survival and quality of life, and shifted cancer treatment paradigm.
Despite numerous advances, therapeutic responses of TKIs vary widely in individual patients and across patient populations, depending on multiple factors such as potency and selectivity of TKIs, variability of drug metabolism and pharmacokinetics profiles among individuals, tumor biology including tumor heterogeneity, and tumor microenvironment, etc.[189]. Drug resistance (de novo or acquired resistance) inevitably develops. Mechanisms of acquired resistance to TKIs could be generally categorized into three classes: i) on-target mutations, like EGFR-T790M to first-/second-generation EGFR-TKIs; ii) off-target mutations: parallel, downstream or alternative pathways activation, like EGFR-independent resistant mechanisms caused by MET/HER2 amplification, HGF overexpression, etc.; iii) histological transformation into another type such as neuroendocrine or mesenchymal tumor [190].
How to solve and prevent drug resistance will be the key issue for future TKIs development. Besides, to improve safety profile and patient’s compliance, it’s also critical to develop more selective TKIs since multi-targeted TKIs might cause unnecessary off-target toxicities by the inhibition of irrelevant targets. Next-generation TKIs are designed to overcome on-target resistance and serve as therapeutic options after progression of former-generation ones. They are generally equipped with enhanced therapeutic efficacy and selectivity, some with better penetration to BBB and CNS responses, and even are recommended or have potential to take place the standard of care. But for other off-target mechanisms, original compounds combined with targeted agents for newly discovered aberration might work. Besides, a series of clinical studies are exploring TKI combination treatment with antibodies or other inhibitors involving different mechanisms to amplify efficacy.
Future collaborative efforts are expected to enhance understanding of resistance mechanism of TKIs; to develop more potent, selective, and better BBB-penetrated TKIs or next-generation TKIs; and to discover more effective and low toxic combinational therapy and sequency. These attempts will help overcome resistance and bring further survival benefit and better quality of life for patients with solid tumors in the future.
Availability of data and materials
All data generated or analyzed during this study are included.
Abbreviations
- TKI:
-
Tyrosine kinase inhibitors
- ATP:
-
Adenosine triphosphate
- RTK:
-
Receptor tyrosine kinases
- NRTK:
-
Non-receptor tyrosine kinases
- EGFR:
-
Epidermal growth factor receptor
- ALK:
-
Anaplastic lymphocyte kinase
- HER2:
-
Human epidermal growth factor receptor 2
- TRK:
-
Tropomyosin receptor kinase
- VEGFR:
-
Vascular endothelial growth factor receptor
- RET:
-
Rearranged during transfection
- MET:
-
Mesenchymal–epithelial transition factor
- PDGFR:
-
Platelet-derived growth factor receptor
- Non-MSI-H :
-
Non-microsatellite instability-high
- FGFR:
-
Fibroblast growth factor receptor
- mPFS:
-
Median progression-free survival
- mOS:
-
Median overall survival
- USA:
-
The United States of America
- ORR:
-
Overall response rate
- DCR:
-
Disease control rate
- HR:
-
Hazard ratio
- AEs:
-
Adverse events
- TRAE:
-
Treatment-related adverse events
- FDA:
-
Food and Drug Administration
- NMPA:
-
National Medical Products Administration
- EMA:
-
European Medicines Agency
- CNS:
-
Central nervous system
- BBB:
-
Blood–brain barrier
- ILD:
-
Interstitial lung disease
- NSCLC:
-
Non-small cell lung cancer
- SqCC:
-
Squamous cell carcinoma
- MBC:
-
Metastatic breast cancer
- MHLW:
-
Ministry of Health, Labor and Welfare
- NTRK:
-
Neurotrophin receptor tyrosine kinase
- ECOG:
-
Eastern Cooperative Oncology Group
- PS:
-
Performance status
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- PD-1:
-
Programmed cell death protein-1
- PD-L1:
-
Programmed death ligand 1
- ICIs:
-
Immune checkpoint inhibitors
- SCLC:
-
Small cell lung cancer
- mRCC:
-
Metastatic renal cell carcinoma
- HCC:
-
Hepatocellular carcinoma
- DTC:
-
Differentiated thyroid cancer
- MTC:
-
Medullary thyroid cancer
- CRC:
-
Colorectal cancer
- GIST:
-
Gastrointestinal stromal tumor
- STS:
-
Soft tissue sarcoma
- NET:
-
Neuroendocrine tumors
- NPC:
-
Nasopharyngeal carcinoma
- FLT3:
-
Fetal liver tyrosine kinase receptor 3
- RCC:
-
Renal cell carcinoma
- EC:
-
Endometrial cancer
- HGFR:
-
Hepatocyte growth factor receptor
- METex14:
-
MET exon14 skipping alterations
- PSC:
-
Pulmonary sarcomatoid carcinoma
- CCA:
-
Cholangiocarcinoma
- NDA:
-
New drug application
- IGF-1R:
-
Insulin-like growth factor 1 receptor
- BTK:
-
Bruton's tyrosine kinase
- BCR:
-
B-cell antigen receptor
References
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19(49):5548–57.
Zámečníkova A. Novel approaches to the development of tyrosine kinase inhibitors and their role in the fight against cancer. Expert Opin Drug Discov. 2014;9(1):77–92.
Drake JM, Lee JK, Witte ON. Clinical targeting of mutated and wild-type protein tyrosine kinases in cancer. Mol Cell Biol. 2014;34(10):1722–32.
Jiao Q, Bi L, Ren Y, et al. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. 2018;17(1):36.
Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28–39.
Krug M, Hilgeroth A. Recent advances in the development of multi-kinase inhibitors. Mini Rev Med Chem. 2008;8(13):1312–27.
Broekman F, Giovannetti E, Peters G. Tyrosine kinase inhibitors: multi-targeted or single-targeted? World J Clin Oncol. 2011;2(2):80–93.
Ayati A, Moghimi S, Salarinejad S, et al. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg Chem. 2020;99:103811.
Le T, Gerber DE. Newer-generation EGFR inhibitors in lung cancer: how are they best used? Cancers (Basel). 2019;11(3):366.
Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361(10):958–67.
Shi Y, Au JS-K, Thongprasert S, et al. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J Thorac Oncol. 2014;9(2):154–62.
Zhang C, Leighl NB, Wu Y-L, et al. Emerging therapies for non-small cell lung cancer. J Hematol Oncol. 2019;12(1):45.
Mok TS, Wu Y-L, Thongprasert S, et al. Gefitinib or Carboplatin-Paclitaxel in Pulmonary Adenocarcinoma. N Engl J Med. 2009;361(10):947–57.
Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–8.
Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–46.
Shi Y, Zhang L, Liu X, et al. Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): a randomised, double-blind phase 3 non-inferiority trial. Lancet Oncol. 2013;14(10):953–61.
Shi YK, Wang L, Han BH, et al. First-line icotinib versus cisplatin/pemetrexed plus pemetrexed maintenance therapy for patients with advanced EGFR mutation-positive lung adenocarcinoma (CONVINCE): a phase 3, open-label, randomized study. Ann Oncol. 2017;28(10):2443–50.
Zhong W-Z, Wang Q, Mao W-M, et al. Gefitinib versus vinorelbine plus cisplatin as adjuvant treatment for stage II–IIIA (N1–N2) EGFR-mutant NSCLC (ADJUVANT/CTONG1104): a randomised, open-label, phase 3 study. Lancet Oncol. 2018;19(1):139–48.
Yue D, Xu S, Wang Q, et al. Erlotinib versus vinorelbine plus cisplatin as adjuvant therapy in Chinese patients with stage IIIA EGFR mutation-positive non-small-cell lung cancer (EVAN): a randomised, open-label, phase 2 trial. Lancet Respir Med. 2018;6(11):863–73.
Liu Y, Hao X, Liu D, et al. Icotinib as adjuvant treatment for stage II-IIIA lung adenocarcinoma patients with EGFR mutation (ICWIP study): study protocol for a randomised controlled trial. Cancer Manag Res. 2020;12:4633–43.
Wu YL, Cheng Y, Zhou X, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18(11):1454–66.
Mok TS, Cheng Y, Zhou X, et al. Improvement in overall survival in a randomized study that compared dacomitinib with gefitinib in patients with advanced non-small-cell lung cancer and EGFR-activating mutations. J Clin Oncol. 2018;36(22):2244–50.
Lim SM, Syn NL, Cho BC, et al. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat Rev. 2018;65:1–10.
Mok TS, Wu Y-L, Ahn M-J, et al. Osimertinib or platinum-pemetrexed in EGFR T790M—positive lung cancer. N Engl J Med. 2016;376(7):629–40.
Soria J, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113–25.
Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382(1):41–50.
Mezquita L, Varga A, Planchard D. Safety of osimertinib in EGFR-mutated non-small cell lung cancer. Expert Opin Drug Saf. 2018;17(12):1239–48.
Herbst RS, Tsuboi M, John T, et al. Osimertinib as adjuvant therapy in patients (pts) with stage IB–IIIA EGFR mutation positive (EGFRm) NSCLC after complete tumor resection: ADAURA. J Clin Oncol. 2020;38(18_suppl):LBA5-LBA.
Reungwetwattana T, Nakagawa K, Cho BC, et al. CNS response to osimertinib versus standard epidermal growth factor receptor tyrosine kinase inhibitors in patients with untreated EGFR-mutated advanced non-small-cell lung cancer. J Clin Oncol. 2018;36(33):3290–7.
Yang JCH, Kim S-W, Kim D-W, et al. Osimertinib in patients with epidermal growth factor receptor mutation-positive non-small-cell lung cancer and leptomeningeal metastases: the BLOOM study. J Clin Oncol. 2019;38(6):538–47.
Lu S, Wang Q, Zhang G, et al. A multicenter, open-label, single-arm, phase II study: the third generation EGFR tyrosine kinase inhibitor almonertinib for pretreated EGFR T790M-positive locally advanced or metastatic non-small cell lung cancer (APOLLO). In Presented at AACR Annual Meeting. 2020; Abstract# CT190.
Shi Y, Zhang S, Hu X, et al. Safety, clinical activity, and pharmacokinetics of alflutinib (AST2818) in patients with advanced NSCLC with EGFR T790M mutation. J Thorac Oncol. 2020;15(6):1015–26.
Shi Y, Hu X, Zhang S, et al. Efficacy and safety of alflutinib (AST2818) in patients with T790M mutation-positive NSCLC: A phase IIb multicenter single-arm study. J Clin Oncol. 2020;38(15_suppl):9602.
Ahn M, Han J, Lee KH, et al. Lazertinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: results from the dose escalation and dose expansion parts of a first-in-human, open-label, multicentre, phase 1–2 study. Lancet Oncol. 2019;20(12):1681–90.
Shi Y, Fang J, Shu Y, et al. OA01.08 a phase I study to evaluate safety and antitumor activity of BPI-7711 in EGFRM+/T790M+ advanced or recurrent NSCLC patients. J Thorac Oncol. 2019;14(11):S1126–7.
Tan DSW, Leighl NB, Riely GJ, et al. Safety and efficacy of nazartinib (EGF816) in adults with EGFR-mutant non-small-cell lung carcinoma: a multicentre, open-label, phase 1 study. Lancet Respir Med. 2020;8(6):561–72.
Wang Z, Yang J, Huang J, et al. Lung adenocarcinoma harboring EGFR T790M and in trans C797S responds to combination therapy of first- and third-generation EGFR TKIs and shifts allelic configuration at resistance. J Thorac Oncol. 2017;12(11):1723–7.
Arulananda S, Do H, Musafer A, et al. Combination osimertinib and gefitinib in C797S and T790M EGFR-mutated non-small cell lung cancer. J Thorac Oncol. 2017;12(11):1728–32.
Leonetti A, Sharma S, Minari R, et al. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121(9):725–37.
Wang S, Song Y, Liu D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017;385:51–4.
Liu X, Zhang X, Yang L, et al. Abstract 1320: preclinical evaluation of TQB3804, a potent EGFR C797S inhibitor. Cancer Res. 2019;79(13 Supplement):1320.
Wang Y, Yang N, Zhang Y, et al. Effective treatment of lung adenocarcinoma harboring EGFR-activating mutation, T790M, and cis-C797S triple mutations by brigatinib and cetuximab combination therapy. J Thorac Oncol. 2020;15:1369–75.
Cho JH, Lim SH, An HJ, et al. Osimertinib for patients with non-small-cell lung cancer harboring uncommon EGFR mutations: a multicenter, open-label, phase II trial (KCSG-LU15-09). J Clin Oncol. 2020;38(5):488–95.
Janne PA, Neal JW, Camidge DR, et al. Antitumor activity of TAK-788 in NSCLC with EGFR exon 20 insertions. J Clin Oncol. 2019;37_suppl(15):9007.
Le X, Goldman JW, Clarke JM, et al. Poziotinib shows activity and durability of responses in subgroups of previously treated EGFR exon 20 NSCLC patients. J Clin Oncol. 2020;38(15_suppl):9514.
Ahn MJ, Yang J, Yu H, et al. 136O: Osimertinib combined with durvalumab in EGFR-mutant non-small cell lung cancer: results from the TATTON phase Ib trial. J Thorac Oncol. 2016;11(4):S115.
Chia P, Mitchell P, Dobrovic A, et al. Prevalence and natural history of ALK positive non-small-cell lung cancer and the clinical impact of targeted therapy with ALK inhibitors. Clin Epidemiol. 2014;6:423–32.
McCusker MG, Russo A, Scilla KA, et al. How I treat ALK-positive non-small cell lung cancer. ESMO Open. 2019;4(Suppl 2):e000524.
Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–77.
Shaw AT, Solomon BJ, Besse B, et al. ALK resistance mutations and efficacy of lorlatinib in advanced anaplastic lymphoma kinase-positive non-small-cell lung cancer. J Clin Oncol. 2019;37(16):1370–9.
Yang Y, Zhou J, Zhou J, et al. Efficacy, safety, and biomarker analysis of ensartinib in crizotinib-resistant, ALK-positive non-small-cell lung cancer: a multicentre, phase 2 trial. Lancet Respir Med. 2020;8(1):45–53.
Horn L, Infante JR, Reckamp KL, et al. Ensartinib (X-396) in ALK-positive non-small cell lung cancer: results from a first-in-human phase I/II. Multicenter Study Clin Cancer Res. 2018;24(12):2771–9.
Duruisseaux M, Besse B, Cadranel J, et al. Overall survival with crizotinib and next-generation ALK inhibitors in ALK-positive non-small-cell lung cancer (IFCT-1302 CLINALK): a French nationwide cohort retrospective study. Oncotarget. 2017;8(13):21903–17.
Camidge DR, Dziadziuszko R, Peters S, et al. Updated efficacy and safety data and impact of the EML4-ALK fusion variant on the efficacy of alectinib in untreated ALK-positive advanced non-small cell lung cancer in the global phase III ALEX study. J Thorac Oncol. 2019;14(7):1233–43.
Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med. 2017;377(9):829–38.
Li J, Knoll S, Bocharova I, et al. Comparative efficacy of first-line ceritinib and crizotinib in advanced or metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer: an adjusted indirect comparison with external controls. Curr Med Res Opin. 2019;35(1):105–11.
Camidge DR, Kim HR, Ahn MJ, et al. Brigatinib versus crizotinib in ALK-positive non-small-cell lung cancer. N Engl J Med. 2018;379(21):2027–39.
Camidge R, Kim HR, Ahn MJ, et al. Brigatinib vs crizotinib in patients with ALK inhibitor-naive advanced ALK+ NSCLC: Updated results from the phase III ALTA-1L trial. Ann Oncol. 2019;30:ix195–6.
Gadgeel S, Peters S, Mok T, et al. Alectinib versus crizotinib in treatment-naive anaplastic lymphoma kinase-positive (ALK+) non-small-cell lung cancer: CNS efficacy results from the ALEX study. Ann Oncol. 2018;29(11):2214–22.
Shi YK, Fang J, Zhang S, et al. Safety and efficacy of WX-0593 in ALK-positive or ROS1-positive non-small cell lung cancer. Ann Oncol. 2019;30:v607–8.
Shi YK, Hao XZ, Xing P, et al. Phase I study of safety and pharmacokinetics for CT-707 in ALK-positive advanced non-small cell lung cancer. Ann Oncol. 2017;28:x132.
Gainor JF, Dardaei L, Yoda S, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6(10):1118–33.
Solomon BJ, Besse B, Bauer TM, et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol. 2018;19(12):1654–67.
Recondo G, Mezquita L, Facchinetti F, et al. Diverse resistance mechanisms to the third-generation ALK inhibitor lorlatinib in ALK-rearranged lung cancer. Clin Cancer Res. 2020;26(1):242–55.
Lin Jessica JSAT. Recent advances in targeting ROS1 in lung cancer. J Thorac Oncol. 2017;12(11):1611–25.
Shaw A, Ou S, Bang Y, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371(21):1963–71.
Drilon A, Siena S, Dziadziuszko R, et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21(2):261–70.
Shaw A, Solomon B, Chiari R, et al. Lorlatinib in advanced ROS1-positive non-small-cell lung cancer: a multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2019;20(12):1691–701.
Cho BC, Drilon AE, Doebele RC, et al. Safety and preliminary clinical activity of repotrectinib in patients with advanced ROS1 fusion-positive non-small cell lung cancer (TRIDENT-1 study). J Clin Oncol. 2019;37(15_suppl):9011.
Gainor JF, Tseng D, Yoda S, et al. Patterns of metastatic spread and mechanisms of resistance to crizotinib in ROS1-positive non-small-cell lung cancer. JCO Precis Oncol. 2017;1:1–13.
Fujiwara Y, Takeda M, Yamamoto N, et al. Safety and pharmacokinetics of DS-6051b in Japanese patients with non-small cell lung cancer harboring ROS1 fusions: a phase I study. Oncotarget. 2018;9(34):23729.
Katayama R, Gong B, Togashi N, et al. The new-generation selective ROS1/NTRK inhibitor DS-6051b overcomes crizotinib resistant ROS1-G2032R mutation in preclinical models. Nat Commun. 2019;10(1):3604.
Hj B. The distinctive nature of HER2-positive breast cancers. N Engl J Med. 2005;353(16):1652–4.
Wang M, Hu Y, Yu T, et al. Pan-HER-targeted approach for cancer therapy: mechanisms, recent advances and clinical prospect. Cancer Lett. 2018;439:113–30.
Geyer C, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–43.
Xuhong JC, Qi XW, Zhang Y, et al. Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer. Am J Cancer Res. 2019;9(10):2103–19.
Johnston S Jr, Pivot JPX, et al. lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor–positive metastatic breast cancer. J Clin Oncol. 2009;27(33):5538–46.
Martin M, Holmes F, Ejlertsen B, et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688–700.
Saura C, Oliveira M, Feng Y-H, et al. Neratinib + capecitabine versus lapatinib + capecitabine in patients with HER2+ metastatic breast cancer previously treated with ≥ 2 HER2-directed regimens: findings from the multinational, randomized, phase III NALA trial. J Clin Oncol. 2019;37(15_suppl):1002.
Ma F, Li Q, Chen S, et al. Phase I study and biomarker analysis of pyrotinib, a novel irreversible Pan-ErbB receptor tyrosine kinase inhibitor, in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol. 2017;35(27):3105–12.
Xu B, Yan M, Ma F, et al. Pyrotinib or lapatinib plus capecitabine for HER2+ metastatic breast cancer (PHOEBE): a randomized phase III trial. J Clin Oncol. 2020;38(15_suppl):1003.
Zhou C, Li X, Wang Q, et al. Pyrotinib in HER2-mutant advanced lung adenocarcinoma after platinum-based chemotherapy: a multicenter, open-label, single-arm, phase II study. J Clin Oncol. 2020;38:2753–61.
Murthy RK, Loi S, Okines A, et al. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N Engl J Med. 2020;382(7):597–609.
Stirrups R. Neratinib and capecitabine for breast cancer brain metastases. Lancet Oncol. 2019;20(4):e197.
Nasrazadani A, Brufsky A. Neratinib: the emergence of a new player in the management of HER2+ breast cancer brain metastasis. Future Oncol. 2020;16(7):247–54.
Bachelot T, Romieu G, Campone M, et al. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): a single-group phase 2 study. Lancet Oncol. 2013;14(1):64–71.
Freedman RA, Gelman RS, Anders CK, et al. TBCRC 022: a phase II trial of neratinib and capecitabine for patients with human epidermal growth factor receptor 2-positive breast cancer and brain metastases. J Clin Oncol. 2019;37(13):1081–9.
Lin NU, Murthy RK, Anders CK, et al. Tucatinib versus placebo added to trastuzumab and capecitabine for patients with previously treated HER2+ metastatic breast cancer with brain metastases (HER2CLIMB). J Clin Oncol. 2020;38(15_suppl):1005.
Rimawi M, De Angelis C, Contreras A, et al. Low PTEN levels and PIK3CA mutations predict resistance to neoadjuvant lapatinib and trastuzumab without chemotherapy in patients with HER2 over-expressing breast cancer. Breast Cancer Res Treat. 2018;167(3):731–40.
Hyman DM, Laetsch TW, Kummar S, et al. The efficacy of larotrectinib (LOXO-101), a selective tropomyosin receptor kinase (TRK) inhibitor, in adult and pediatric TRK fusion cancers. J Clin Oncol. 2017;35(18_suppl):LBA2501-LBA.
Chen Y, Chi P. Basket trial of TRK inhibitors demonstrates efficacy in TRK fusion-positive cancers. J Hematol Oncol. 2018;11(1):78.
Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15(12):731–47.
Vaishnavi A, Le A, Doebele R. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5(1):25–34.
Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378(8):731–9.
Hong DS, DuBois SG, Kummar S, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020;21(4):531–40.
Drilon A, van Tilburg CM, Farago AF, et al. Abstract CT199: Larotrectinib in TRK fusion cancer patients: outcomes by prior therapy and performance status. Cancer Res. 2020;80(16 Supplement):CT199.
Demetri GD, Paz-Ares L, Farago AF, et al. Efficacy and safety of entrectinib in patients with NTRK fusion-positive tumours: Pooled analysis of STARTRK-2, STARTRK-1, and ALKA-372-001. Ann Oncol. 2018;29:ix175.
Robinson GW, Gajjar AJ, Gauvain KM, et al. (2009) Phase 1/1B trial to assess the activity of entrectinib in children and adolescents with recurrent or refractory solid tumors including central nervous system (CNS) tumors. J Clin Oncol. 2019;37(15_suppl):10009.
Drilon A. TRK inhibitors in TRK fusion-positive cancers. Ann Oncol. 2019a;30:viii23–30.
Drilon A, Nagasubramanian R, Blake JF, et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 2017;7(9):963–72.
Hyman D, Kummar S, Farago A, et al. Abstract CT127: phase I and expanded access experience of LOXO-195 (BAY 2731954), a selective next-generation TRK inhibitor (TRKi). Cancer Res. 2019;79(13 Supplement):127.
Drilon A, Zhai D, Deng W, et al. Abstract 442: Repotrectinib, a next generation TRK inhibitor, overcomes TRK resistance mutations including solvent front, gatekeeper and compound mutations. Cancer Res. 2019;79(13 Supplement):442.
Drilon A. TRK inhibitors in TRK fusion-positive cancers. Ann Oncol. 2019b;30 Suppl 8:viii23–30.
Cocco E, Schram AM, Kulick A, et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat Med. 2019;25(9):1422–7.
Ferrara N, Adamis AP. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov. 2016;15(6):385–403.
Qin S, Li A, Yi M, et al. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J Hematol Oncol. 2019;12(1):27.
Medavaram S, Zhang YJE. Emerging therapies in advanced hepatocellular carcinoma. Exp Hematol Oncol. 2018;7:17.
Llovet J, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90.
Cheng A, Kang Y, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34.
Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–73.
Zhu AX, Finn RS, Ikeda M, et al. A phase Ib study of lenvatinib (LEN) plus pembrolizumab (PEMBRO) in unresectable hepatocellular carcinoma (uHCC). J Clin Oncol. 2020;38(15_suppl):4519.
Bi F, Qin S, Gu S, et al. Donafenib versus sorafenib as first-line therapy in advanced hepatocellular carcinoma: An open-label, randomized, multicenter phase II/III trial. J Clin Oncol. 2020;38(15_suppl):4506.
Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66.
Abou-Alfa G, Meyer T, Cheng A, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med. 2018;379(1):54–63.
Li Q, Qin S, Gu S, et al. Apatinib as second-line therapy in Chinese patients with advanced hepatocellular carcinoma: a randomized, placebo-controlled, double-blind, phase III study. J Clin Oncol. 2020;38(15):4507.
Han B, Li K, Wang Q, et al. Effect of anlotinib as a third-line or further treatment on overall survival of patients with advanced non-small cell lung cancer: the ALTER 0303 phase 3 randomized clinical trial. JAMA Oncol. 2018;4(11):1569–75.
Cheng Y, Wang Q, Li K, et al. OA13.03 anlotinib as third-line or further-line treatment in relapsed SCLC: a multicentre, randomized, double-blind phase 2 trial. J Thorac Oncol. 2018;13(10):S351–2.
Wu D, Nie J, Hu W, et al. A phase II study of anlotinib in 45 patients with relapsed small cell lung cancer. Int J Cancer. 2020. https://doi.org/10.1002/ijc.33161.
Liu Y, Hu X, Jiang J, et al. A prospective study of apatinib in patients with extensive-stage small cell lung cancer after failure of two or more lines of chemotherapy. Oncologist. 2020;25(5):e833–42.
Poddubskaya E, Baranova M, Allina D, et al. Personalized prescription of tyrosine kinase inhibitors in unresectable metastatic cholangiocarcinoma. Exp Hematol Oncol. 2018;7:21.
Kato Y, Tabata K, Kimura T, et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS One. 2019;14(2):e0212513.
Subbiah V, Velcheti V, Tuch B, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol. 2018;29(8):1869–76.
Drilon A, Lin JJ, Filleron T, et al. Frequency of brain metastases and multikinase inhibitor outcomes in patients with RET-rearranged lung cancers. J Thorac Oncol. 2018;13(10):1595–601.
Drilon A, Oxnard GR, Tan DSW, et al. Efficacy of selpercatinib in RET fusion-positive non-small-cell lung cancer. N Engl J Med. 2020;383(9):813–24.
Wirth LJ, Sherman E, Robinson B, et al. Efficacy of selpercatinib in RET-altered thyroid cancers. N Engl J Med. 2020;383(9):825–35.
Gainor JFCG, Kim D-W, et al. Registrational dataset from the phase I/II ARROW trial of pralsetinib (BLU-667) in patients (pts) with advanced RET fusion+ non-small cell lung cancer (NSCLC). J Clin Oncol. 2020;38(15_suppl):9515.
Subbiah V, Hu MIN, Gainor JF, et al. Clinical activity of the RET inhibitor pralsetinib (BLU-667) in patients with RET fusion+ solid tumors. J Clin Oncol. 2020;38(15_suppl):109.
Solomon BJ, Tan L, Lin JJ, et al. RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven malignancies. J Thorac Oncol. 2020;15(4):541–9.
Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3(1 Suppl):S7–19.
Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. 2013;31(8):1089–96.
Comoglio PM, Trusolino L, Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. 2018;18(6):341–58.
Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–9.
Camidge DR, Otterson GA, Clark JW, et al. Crizotinib in patients (pts) with MET-amplified non-small cell lung cancer (NSCLC): Updated safety and efficacy findings from a phase 1 trial. J Clin Oncol. 2018;36(15_suppl):9062.
Drilon A, Clark J, Weiss J, et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Med. 2020;26(1):47–51.
Shabnam R, Dy GK. MET inhibition in non-small cell lung cancer. EMJ. 2018;4(1):100–11.
Le X, Felip E, Veillon R, et al. Primary efficacy and biomarker analyses from the VISION study of tepotinib in patients (pts) with non-small cell lung cancer (NSCLC) with METex14 skipping. J Clin Oncol. 2020;38(15_suppl):9556.
Paik PK, Veillon R, Cortot AB, et al. Phase II study of tepotinib in NSCLC patients with METex14 mutations. J Clin Oncol. 2019;37(15_suppl):9005.
Paik PK, Felip E, Veillon R, et al. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N Engl J Med. 2020;383:931–43.
Wolf J, Seto T, Han J-Y, et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. J Clin Oncol. 2019;37(15_suppl):9004.
Wolf J, Overbeck TR, Han J-Y, et al. Capmatinib in patients with high-level MET-amplified advanced non–small cell lung cancer (NSCLC): results from the phase 2 GEOMETRY mono-1 study. J Clin Oncol. 2020;38(15_suppl):9509.
Groen HJM, Akerley WL, Souquet PJ, et al. Capmatinib in patients with METex14-mutated or high-level MET-amplified advanced non-small-cell lung cancer (NSCLC): results from cohort 6 of the phase 2 GEOMETRY mono-1 study. J Clin Oncol. 2020;38(15_suppl):9520.
Lu S, Fang J, Li X, et al. Phase II study of savolitinib in patients (pts) with pulmonary sarcomatoid carcinoma (PSC) and other types of non-small cell lung cancer (NSCLC) harboring MET exon 14 skipping mutations (METex14+). J Clin Oncol. 2020;38(15_suppl):9519.
Wang Q, Yang S, Wang K, et al. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J Hematol Oncol. 2019;12(1):63.
Zhang Z, Yang S, Wang Q. Impact of MET alterations on targeted therapy with EGFR-tyrosine kinase inhibitors for EGFR-mutant lung cancer. Biomark Res. 2019;7:27.
Park K, Zhou J, Kim DW, et al. Tepotinib plus gefitinib in patients with MET-amplified EGFR-mutant NSCLC: long-term outcomes of the INSIGHT study. Ann Oncol. 2019;30:159.
Ko B, Halmos B. Capmatinib and gefitinib combination therapy: will EGFR-mutated MET-dysregulated NSCLC “capitulate”? Trans Lung Cancer Res. 2018;7:S321–5.
Wu YL, Zhang L, Kim DW, et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J Clin Oncol. 2018;36(31):3101–9.
Sequist LV, Han J-Y, Ahn M-J, et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020;21(3):373–86.
Martinelli E, Morgillo F, Troiani T, et al. Cancer resistance to therapies against the EGFR-RAS-RAF pathway: the role of MEK. Cancer Treat Rev. 2017;53:61–9.
Cheng Y, Tian H. Current development status of MEK inhibitors. Molecules. 2017;22(10):1551.
Roskoski R Jr. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66(2):105–43.
Robert C, Flaherty KT, Hersey P, et al. METRIC phase III study: Efficacy of trametinib (T), a potent and selective MEK inhibitor (MEKi), in progression-free survival (PFS) and overall survival (OS), compared with chemotherapy (C) in patients (pts) with BRAFV600E/K mutant advanced or metastatic melanoma (MM). J Clin Oncol. 2012;30(18_suppl):LBA8509.
Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2014;372(1):30–9.
Subbiah V, Kreitman R, Wainberg Z, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol. 2018;36(1):7–13.
Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76.
Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19(10):1315–27.
Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020;382(15):1430–42.
Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28(7):1631–9.
Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med. 2019;381(17):1632–43.
Haugsten EM, Wiedlocha A, Olsnes S, et al. Roles of fibroblast growth factor receptors in carcinogenesis. Mol Cancer Res. 2010;8(11):1439–52.
Zarrabi K, Paroya A, Wu SJJ, et al. Emerging therapeutic agents for genitourinary cancers. J Hematol Oncol. 2019;12(1):89.
Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015;15(1):25–41.
Pal SK, Rosenberg JE, Hoffman-Censits JH, et al. Efficacy of BGJ398, a fibroblast growth factor receptor 1–3 inhibitor, in patients with previously treated advanced urothelial carcinoma with FGFR3 alterations. Cancer Discov. 2018;8(7):812.
Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376(11):1015–26.
Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381(4):338–48.
Vogel A, Sahai V, Hollebecque A, et al. FIGHT-202: a phase II study of pemigatinib in patients (pts) with previously treated locally advanced or metastatic cholangiocarcinoma (CCA). Ann Oncol. 2019;30:v876.
Javle M, Kelley RK, Roychowdhury S, et al. AB051. P-19. A phase II study of infigratinib (BGJ398) in previously-treated advanced cholangiocarcinoma containing FGFR2 fusions. Hepatobiliary Surg Nutr. 2019;8(Suppl 1):AB051.
Abbaspour Babaei M, Kamalidehghan B, Saleem M, et al. Receptor tyrosine kinase (c-Kit) inhibitors: a potential therapeutic target in cancer cells. Drug Des Devel Ther. 2016;10:2443–59.
De Falco S. Antiangiogenesis therapy: an update after the first decade. Korean J Intern Med. 2014;29(1):1–11.
Mei L, Du W, Idowu M, et al. Advances and challenges on management of gastrointestinal stromal tumors. Front Oncol. 2018;8:135.
Goodman VL, Rock EP, Dagher R, et al. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13(5):1367–73.
Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302.
Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–38.
Guo T, Hajdu M, Agaram NP, et al. Mechanisms of sunitinib resistance in gastrointestinal stromal tumors harboring KITAY502-3ins mutation: an in vitro mutagenesis screen for drug resistance. Clin Cancer Res. 2009;15(22):6862–70.
Heinrich MC, Jones RL, Mehren MV, et al. Clinical activity of avapritinib in ≥ fourth-line (4L+) and PDGFRA Exon 18 gastrointestinal stromal tumors (GIST). J Clin Oncol. 2019;37(15_suppl):11022.
von Mehren M, Serrano C, Bauer S, et al. INVICTUS: a phase III, interventional, double-blind, placebo-controlled study to assess the safety and efficacy of ripretinib as ≥ 4th-line therapy in patients with advanced gastrointestinal stromal tumors (GIST) who have received treatment with prior anticancer therapies. Ann Oncol. 2019;30:v925–6.
Blay J-Y, Serrano C, Heinrich MC, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(7):923–34.
Wagner AJ, Tap WD, Shields AF, et al. A phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of PLX9486 alone and in combination (combo) with the KIT inhibitors pexidartinib (pexi) or sunitinib (su) in patients (Pts) with advanced solid tumors and gastrointestinal stromal tumor (GIST). J Clin Oncol. 2018;36(15_suppl):11509.
Curtin JA, Busam K, Pinkel D, et al. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340–6.
Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305(22):2327–34.
Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31(26):3182–90.
Guo J, Carvajal RD, Dummer R, et al. Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: final results from the global, single-arm, phase II TEAM trial. Ann Oncol. 2017;28(6):1380–7.
Beckwith H, Yee D. Minireview: were the IGF signaling inhibitors all bad? Mol Endocrinol. 2015;29(11):1549–57.
Fassnacht M, Berruti A, Baudin E, et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. Lancet Oncol. 2015;16(4):426–35.
Tempero M, Oh D, Macarulla T, et al. Ibrutinib in combination with nab-paclitaxel and gemcitabine as first-line treatment for patients with metastatic pancreatic adenocarcinoma: results from the phase 3 RESOLVE study. Ann Oncol. 2019;30:iv126.
Molina-Cerrillo J, Alonso-Gordoa T, Gajate P, et al. Bruton’s tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treat Rev. 2017;58:41–50.
Campbell R, Chong G, Hawkes EA. Novel indications for Bruton’s tyrosine kinase inhibitors, beyond hematological malignancies. J Clin Med. 2018;7(4):62.
Bhullar KS, Lagarón NO, McGowan EM, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer. 2018;17(1):48.
Neel DS, Bivona TG. Resistance is futile: overcoming resistance to targeted therapies in lung adenocarcinoma. NPJ Precis Oncol. 2017;1(1):3.
Nakagawa K, Garon EB, Seto T, et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(12):1655–69.
Yang JC, Wu YL, Schuler M, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16(2):141–51.
Park K, Tan EH, O’Byrne K, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016;17(5):577–89.
Paz-Ares L, Tan EH, O’Byrne K, et al. Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: overall survival data from the phase IIb LUX-Lung 7 trial. Ann Oncol. 2017;28(2):270–7.
Soria J-C, Felip E, Cobo M, et al. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol. 2015;16(8):897–907.
Yang JC, Shih JY, Su WC, et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trial. Lancet Oncol. 2012;13(5):539–48.
Suda K, Nishino M, Koga T, et al. Abstract 2200: potent in vitro activity of Tarloxotinib for EGFR C797S and other mutations refractory to current EGFR tyrosine kinase inhibitors. Cancer Res. 2019;79(13 Supplement):2200.
Soria J-C, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. The Lancet. 2017;389(10072):917–29.
Lim SM, Kim HR, Lee J-S, et al. Open-label, multicenter, phase II study of ceritinib in patients with non-small-cell lung cancer harboring ROS1 rearrangement. J Clin Oncol. 2017;35(23):2613–8.
Drilon A, Siena S, Ou S, et al. Safety and antitumor activity of the multitargeted Pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase i trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017;7(4):400–9.
Papadopoulos KP, Gandhi L, Janne PA, et al. First-in-human study of DS-6051b in patients (pts) with advanced solid tumors (AST) conducted in the US. J Clin Oncol. 2018;36(15_suppl):2514.
Wang Y, Jiang T, Qin Z, et al. HER2 exon 20 insertions in non-small-cell lung cancer are sensitive to the irreversible pan-HER receptor tyrosine kinase inhibitor pyrotinib. Ann Oncol. 2019;30(3):447–55.
Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–34.
Brose M, Nutting C, Jarzab B, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384(9940):319–28.
Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–13.
Thornton K, Kim G, Maher VE, et al. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: US Food and Drug Administration drug approval summary. Clin Cancer Res. 2012;18(14):3722–30.
Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303–12.
Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med. 2015;372(7):621–30.
Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16(15):1473–82.
Makker V, Rasco D, Vogelzang NJ, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: an interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019;20(5):711–8.
Elisei R, Schlumberger M, Müller S, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31(29):3639–46.
Choueiri T, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17(7):917–27.
Motzer R, Hutson T, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–24.
Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1116–27.
Motzer RJ, Penkov K, Haanen J, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1103–15.
Chi Y, Yao Y, Wang S, et al. Anlotinib for metastasis soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(15_suppl):11503.
Li J, Qin S, Xu RH, et al. Effect of fruquintinib vs placebo on overall survival in patients with previously treated metastatic colorectal cancer: the FRESCO randomized clinical trial. JAMA. 2018;319(24):2486–96.
Li J, Qin S, Xu J, et al. Randomized, double-blind, placebo-controlled phase III trial of apatinib in patients with chemotherapy-refractory advanced or metastatic adenocarcinoma of the stomach or gastroesophageal junction. J Clin Oncol. 2016;34(13):1448–54.
Xu J, Shen L, Zhou Z, et al. Efficacy and safety of surufatinib in patients with well-differentiated advanced extrapancreatic neuroendocrine tumors (NETs): results from the randomized phase III study (SANET-ep). Ann Oncol. 2019;30:v911.
Kim JW, Hafez N, Soliman HH, et al. Preliminary efficacy data of platinum-pretreated small cell lung cancer (SCLC) cohort of NCI 9881 study: a phase II study of cediranib in combination with olaparib in advanced solid tumors. J Clin Oncol. 2020;38(15_suppl):9065.
Taylor MH, Lee C-H, Makker V, et al. Phase IB/II trial of lenvatinib plus pembrolizumab in patients with advanced renal cell carcinoma, endometrial cancer, and other selected advanced solid tumors. J Clin Oncol. 2020;38(11):1154–63.
Wang J, Fan Y, Zhao J, et al. Abstract CT083: Camrelizumab plus apatinib in extensive-stage small-cell lung cancer (PASSION): A multicenter, two-stage, phase 2 trial. Cancer Res. 2020;80(16 Supplement):CT083.
Choueiri TK, Motzer RJ, Rini BI, et al. Updated efficacy results from the JAVELIN Renal 101 trial: first-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann Oncol. 2020;31(8):1030–9.
Sheng X, Yan X, Chi Z, et al. Overall survival and biomarker analysis of a phase Ib combination study of toripalimab, a humanized IgG4 mAb against programmed death-1 (PD-1) with axitinib in patients with metastatic mucosal melanoma. J Clin Oncol. 2020;38(15_suppl):10007.
Cousin S, Bellera CA, Guégan JP, et al. REGOMUNE: a phase II study of regorafenib plus avelumab in solid tumors—results of the non-MSI-H metastatic colorectal cancer (mCRC) cohort. J Clin Oncol. 2020;38(15_suppl):4019.
Lu M, Cao Y, Gong J, et al. Abstract CT142: A phase I trial of surufatinib plus toripalimab in patients with advanced solid tumor. Cancer Res. 2020;80(16 Supplement)):CT142.
Shah MH, Sherman EJ, Robinson B, et al. Selpercatinib (LOXO-292) in patients with RET-mutant medullary thyroid cancer. J Clin Oncol. 2020;38(15_suppl):3594.
Goto K, Oxnard GR, Tan DS-W, et al. Selpercatinib (LOXO-292) in patients with RET-fusion+ non-small cell lung cancer. J Clin Oncol. 2020;38(15_suppl):3584.
Acknowledgements
Not applicable
Funding
This work was supported by China National Major Project for New Drug Innovation (Grant No. 2017ZX09304015) and Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS) (Grant No. 2016-I2M-1-001).
Author information
Authors and Affiliations
Contributions
YKS concepted and designed the manuscript. YKS, LLH, and SYJ drafted and revised the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors approved the final manuscript for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, L., Jiang, S. & Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J Hematol Oncol 13, 143 (2020). https://doi.org/10.1186/s13045-020-00977-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00977-0