
Abstract
Cytosolic DNA sensing, the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway, is an important novel role in the immune system. Multiple STING agonists were developed for cancer therapy study with great results achieved in pre-clinical work. Recent progress in the mechanical understanding of STING pathway in IFN production and T cell priming, indicates its promising role for cancer immunotherapy. STING agonists co-administrated with other cancer immunotherapies, including cancer vaccines, immune checkpoint inhibitors such as anti-programmed death 1 and cytotoxic T lymphocyte-associated antigen 4 antibodies, and adoptive T cell transfer therapies, would hold a promise of treating medium and advanced cancers. Despite the applications of STING agonists in cancer immunotherapy, lots of obstacles remain for further study. In this review, we mainly examine the biological characters, current applications, challenges, and future directions of cGAS-STING in cancer immunotherapy.
Similar content being viewed by others

Background
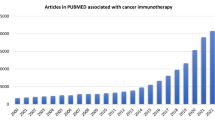
Cancer is one of the major lethal diseases worldwide, with a high morbidity of 18.1 million estimated new diagnosed cases and mortality of 9.6 million deaths in 2018 reported in the Global cancer statistics [1]. Cancer immunotherapy has made a great breakthrough in oncology, and the discovery of immune checkpoint inhibitors (ICIs) was awarded the 2018 Nobel Prize. Although the application of anti-cytotoxic T lymphocyte antigen 4 (CTLA-4) and anti-programmed death 1 (PD-1) therapies has yielded impressive clinical efficacy, response to these methods only presents in a fraction of patients, and recent evidence has suggested some drug-resistant and lethal cases [2, 3].
The stimulator of interferon genes (STING) is a novel player with pleiotropic effects in the field of the immune system. The discovery of STING as a 42-kDa “dimeric adaptor protein” in 2008 quickly expanded the fields of immunology research as well as cancer immunotherapy [4]. The STING-targeted treatment is a novel candidate for anti-tumor immunotherapy and agents such as ADU-S100(MIW815) (NCT02675439), MK-1454(NCT03010176), and E7766(NCT04144140) have been approved for clinical trials to test their capability of mediating cancer progression in human beings. The understanding of the activated STING pathway has made much progress in antitumor responses necessarily via tumor microenvironment (TME) heating-up by interferon (IFN) secretion and lymphocyte infiltration, which is an excitingly promising direction for cancer immunotherapy (Fig. 1). Several excellent reviews showed unique perspectives on the cyclic GMP-AMP synthase (cGAS)-STING pathway, which identify the structural biology of STING protein, its role in the immune system, as well as the regulation and function of it in DNA sensing [5,6,7]. In this review, we focus on the basis of the application and pharmacological effect of STING agonists as antitumor therapy, the application of STING in antitumor immunotherapy, its limitations, and some feasible suggestions in the use of STING agonists.

Timeline of the understanding of STING pathway and its role in cancer immunotherapy. Abbreviation: DMXAA: dimethyloxoxanthenyl acetic acid; FAA: Flavone 8-acetic acid; IRF: interferon regulatory factor; TBK1: TANK-binding kinase 1
Basis of STING signaling pathway
cGAS-STING pathway
The cGAS-STING pathway is the central cellular cytosolic double-stranded DNA (dsDNA) sensor, allowing innate immune to respond to infections, inflammation, and cancer [8, 9]. Both intrinsic and extrinsic self-DNA sensing can contribute to its activation. It is clear that the STING pathway is more than just important in pathogen detection, but also plays an important role in the detection of rather the self-DNA released from tumor cells and dying cells [10]. It was also reported that the mitochondrial DNA (mtDNA) instability promoted the escape of mtDNA into the cytosol and activated the antiviral immunity via the cGAS-STING pathway [11].
The upstream dsDNA interacts with enzyme cGAS in a sequence-independent way [12, 13], promoting a conformational change of cGAS to catalyze the formation of 2′,3′-cyclic GMP-AMP (cGAMP), a cyclic dinucleotide (CDN) from ATP and GTP, containing the phosphodiester linkages of both 2′–5′ and 3′–5′ [14]. The cGAS activation as well as cGAMP synthase activate protein STING, in which the STING undergoes endoplasmic reticulum (ER)-to-Golgi trafficking and tetramer formation via a higher-order oligomerization [15] (Fig. 2). Palmitoylation of STING in Golgi is proposed for TANK binding kinase 1 (TBK1) as well as interferon regulatory factor 3 (IRF3) recruitment. The STING tetramerization induces recruitment and activation of TBK1 dimers, and TBK1 transphosphorylate STING at its C-terminal domains for IRF3 activation [16]. The IRF3 then displaces to the nucleus and induces immune-stimulated genes (ISG) and type I IFN expression [13]. The NF-κB signaling can also be activated by STING (Fig. 2).

cGAS-STING pathway. Exogenous DNA from dying cell, tumor cell, virus and bacteria, and endogenous DNA leakage from mitochondria, interact with the cytosolic DNA sensor cGAS in a sequence-independent way, promoting a conformational change of cGAS to catalyze the formation of 2′,3′-cyclic GMP-AMP (cGAMP). The cGAS activation as well as cGAMP synthase activate protein STING, in which the STING undergoes endoplasmic reticulum (ER)-to-Golgi trafficking and tetramer formation via a higher-order oligomerization. Palmitoylation of STING in Golgi is proposed for TANK binding kinase 1 (TBK1) as well as interferon regulatory factor 3 (IRF3) recruitment. The STING tetramerization induces recruitment and activation of TBK1 dimers, and TBK1 transphosphorylates STING at its C-terminal domains for IRF3 activation. The IRF3 then displaces to the nucleus and induces immune-stimulated genes and type I IFN expression. The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling can also be activated by STING. Abbreviations: cGAMP, 2′,3′-cyclic GMP-AMP; ER, endoplasmic reticulum; IRF3, interferon regulatory factor 3; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; TBK1, TANK-binding kinase 1
Biology and expression of protein STING and its pathway
The structure of protein human STING (hSTING) comprises a N-terminal trans-membrane domain with four helices (aa 1-154), an acidic C-terminal tail (aa 342-379), and a central globular domain (aa 155-341) separated the former two [17]. The mouse STING (mSTING) presents 81% similarity and 68% amino acid identity with hSTING, and the different sequence alleles were reported [18]. STING is present variously in different tissues, and its expressions in the skeletal muscle, brain, kidney, small intestine, colon, and liver were poorly found [4].
STING-deficiency has been reported to correlate with cancer incidence. In six cancerous melanoma cell lines (G361, MeWo, SK-MEL-5, SK-MEL-2, SK-MEL-28, and WM115), STING expression was not detectable or significantly inhibited [19]. Several colorectal adenocarcinoma human cell lines have described low or defective STING pathway activity, which was correlated with poorer Dukes’ stage [20]. Also, the STING silencing was observed in KRAS-mutated lung cancer, with the loss of the tumor suppressor gene LKB1 [21]. Further studies of a co-culture of tumor-immune cells revealed that a downregulated cGAS-STING pathway could induce cancer resistance to immune effectors [22]. Their study also showed the relationship between the decreased intratumoral CD8+ T cell infiltration and downregulated cGAS-STING pathway mediated via the reduction of the expressions of the downstream IFN-I targeted genes such as chemokine (C-X-C motif) ligands 10 (CXCL10) [22]. Surviving cancer cells tend to harbor deficiencies in the cGAS-STING pathway under selective pressure [23].
cGAS-STING pathway in cancer-immunity cycle
The activation of the cGAS-STING pathway plays a crucial role in both tumor cells and immune cells as an innate immune sensor, which could regulate multiple steps in cancer-immunity cycle. This cytosolic DNA sensing has been well-characterized, which can induce IFN production and arouse host immune responses mediated by infiltration of immune cells such as T cells and natural killer (NK) cells [24, 25]. Activation of the cGAS-STING pathway in tumor cells may pose an obstacle to the progression of early neoplastic cells by upregulating type I IFNs or other inflammatory genes (Fig. 3a). Importantly, the cGAS-STING pathway has also been robustly linked to the induction of cancer cell senescence [26], thereby mediating the oncosuppressive effects. The capability of cGAS-STING signaling to promote senescence is dependent on the secretion of the chemokines, pro-inflammatory cytokines, growth factors, and proteases, which are components of the senescence-associated secretory phenotype (SASP) [26,27,28]. These immune-stimulatory factors can either contribute to the tumor control in a tumor-cell autonomous manner or arouse immune cells against tumors [26, 29, 30].

Role of STING pathway in tumor suppression. a cGAS-STING pathway and cancer-immunity cycle. cGAS-STING pathway functions as a tumor suppressor induced by DNA damage. Cytosolic DNA generated from different sources of DNA damage could be sensed by enzyme cGAS in a tumor cell. The cGAS then activates STING to upregulate type I IFN expression, which mediates tumor-suppressive effects. In addition, the cGAS-STING signaling allows the crosstalk between the tumor cells and immune cells nearby. Tumor-derived cGAMP or tumor-derived DNA could activate the activation of DCs, which activates the cGAS-STING pathway and promote immune cells against tumors. b cGAS-STING in innate immune sensing and spontaneous anti-tumor T cell responses. Tumor-derived DNA can induce cGAS-STING pathway activation of APCs and upregulate expression of type I IFNs, which increases its lymph node-homing capability and spontaneous T cells. Abbreviations: cGAMP, 2′,3′-cyclic GMP-AMP; CTL, cytotoxic T lymphocytes; IFN, interferon; NK cells, natural killer cells; SASP, senescence-associated secretory phenotype; TME, tumor microenvironment
Pre-clinical studies have demonstrated the activation of STING pathway in host immune cells is vital for IFN-β production [31], and in turn, the STING pathway and type I IFN signaling are revealed necessary for productive CD8+ T cell cross-priming via Batf3-lineage dendritic cells (DCs) [32] (Fig. 3b). Two major hypotheses have been prompted for the DC activation by cancer cells: tumor-derived DNA activates the DCs [31], or tumor-derived cGAMP directly activates the STING pathway via protein STING [25], thereby leading to the production of type I IFNs. The type I IFN signaling is important for CD8α+ DC survival and antigen retention, which enhances DC’s cross-presentation [33]. Besides, type I IFNs have been reported capable of upregulating the expression of CCR7, MIP-3beta, and Th-1 chemokines, which reinforces the capability of lymph node-homing [34]. Through gene expression profiling studies of tumor biopsies, type I IFN signaling was found correlated with adaptive T cell responses against tumor-specific antigens [35, 36]. In addition, basic experiments in type I IFNR−/− mouse models or mice with the absence of the downstream transcription factor Stat1, suggested significantly reduced tumor antigen specific T cell responses in vivo [36, 37]. Further studies will be needed to unveil how the tumor and host-immune cGAS-STING signaling cooperates to promote tumor suppression.
The pharmacological effect of STING agonists as antitumor therapy
STING-nucleotidic agonists
Due to the correlation between deficiencies in cGAS-STING pathway and surviving cancer cells, as well as the importance of cGAS-STING in the regulation of cancer-immunity cycle, STING agonists were developed to mimic this activation to enhance anti-cancer effects (Table 1). CDNs have been verified as the mediators of cGAS-STING pathway in the immune system. Their antitumor modulation was discovered first in c[di-GMP], in which the tumor progression of human colon cancer cells was inhibited [44]. The endogenous cGAS production 2′,3′-cGAMP showed reduced tumor size and prolonged survival in mice bearing colon adenocarcinoma CT26 tumors [45]. Intratumoral injection of 2′,3′-cGAMP in the B16F10 mouse model also significantly delayed tumor growth and reduced lung metastases [40]. The results of their experiments in the mouse melanoma model demonstrated that intratumoral injection of cGAMP enforced anti-cancer CD8+ T cell responses, and this ability could be further enhanced when both PD-1 and CTLA-4 were blocked. Their further studies showed this immune response depended on the production of type I IFNs from endothelial cells in TME, indicating the strategy of targeting tumor endothelial cells for melanoma immunotherapy. Another study enhanced STING activation of the tumor and lymph nodes by using the cGAMP-based nanoparticles, which enhanced the cytosolic delivery of cGAMP by promoting its endosomal escape and triggered the formation of a “hot” type TME with enriched T cell infiltration [46]. This novel therapy presented marked efficacy combined with ICIs. Further, the STING-activating nanoparticles also showed the potential to induce immunological memory against cancers. Indeed, these cured mice in the rechallenge experiments rejected the tumors in the contralateral flank [46]. Theses therapeutic benefits highlight the importance of STING signaling in anti-cancer immunity in tumors.
Beyond naturally derived CDNs, synthetic CDNs with better properties were developed. The anti-tumor compound dithio CDN (ML RR-S2 CDN, also known as ADU-S100 or MIW815) showed a high binding affinity to hSTING alleles [41]. This CDN analog showed marked antitumor efficacy in various cancer mouse models, which made it become the first STING agonist entering clinical trials in advanced metastatic solid tumors or lymphomas, with the first results reported in 2018, at the Society for ImmunoTherapy of Cancer meeting [47]. The inclusion criteria included 18-years old or older patients with advanced/metastasis solid tumors or lymphomas, Eastern Cooperative Oncology Group performance status of 0-1, and two or more cutaneous or subcutaneous neoplastic lesions accessible for biopsy, with one that could be injected. This phase I study enrolled 41 patients heavily pretreated before: 3 (7.3%) patients had received at least one prior-line treatment, 34 (82.9%) patients had received at least two prior treatments, and 22 (53.7%) had exposed to the ICIs therapy prior. During treatment, 35 of them discontinued because of disease progression (n = 26), physician or patient decision (n = 8), and death (n = 1) [47]. Dose-limiting toxicities were not reported, and the common adverse events were mainly including pyrexia, pain at the injection site, and headache. Based on Evaluation Criteria in Solid Tumors, partial response was observed in two patients (Merkel cell carcinoma, anti-PD-1 antibody-naïve; parotid gland adenocarcinoma, PD1 antibody-refractory). Treatment continued for more than 6 months in four patients. Follow-up clinical trials of combinations of this compound with ICIs are ongoing, which will be presented in Table 2.
STING-non-CDN agonists
A growing body of evidence showed the pharmacological function of non-CDN agents in cGAS-STING activation. The initiated agonist targeting the STING pathway is dimethyloxoxanthenyl acetic acid (DMXAA) [48]. Actually, DMXAA was first used as an anti-angiogenesis drug. However, the treatment of DMXAA failed the phase III trials in non-small cell lung cancer patients with no significant benefit brought [49]. The fact is that DXMAA is actually a competitive mSTING agonist with strong affinity, but not for hSTING [50]. Conlon and colleagues [50] found DMXAA and STING interacted restrictedly in mice, but too poor in human to promote type I IFN production.
The design of agent amidobenzimidazole (ABZI) represented a new breakthrough of STING agonist in immune-modifying cancer treatment [43]. This novel STING agonist was reported with significantly enhanced binding affinity using the 4-carbon butane linker (di-ABZI) for dimerization. The evaluation of STING activity was identified by IFN-β, and di-ABZI showed lower EC50 concentration than cGAMP. Treatment of di-ABZI in mice with subcutaneous CT-26 tumor-induced tumor regression and survival increase, and specially, 80% treated animals remained tumor-free until the end of this study. To our knowledge, this molecular is the initiated non-CDN agonist with competitive antitumor efficacy and hSTING selectivity.
Applications of STING pathway in cancer immunotherapy
STING agonist as a cancer vaccine adjuvant
Appropriate adjuvants play an essential role in tolerance overcome and tumor-specific immunity enhancement, and innate immunity activation is able to boost antigen-presenting cell (APC) activation, which facilities the immunogenicity of tumor-associated antigens (TAAs) [51]. STINGVAX is regarded as the first designed STING-based cancer vaccine, containing both the cancer cells secreting granulocyte-macrophage colony-stimulating factor (GM-CSF) and CDNs [52]. The STINGVAX injection in the contralateral part of the B16 transplanted melanoma, significantly inhibited the tumor size with a dose-dependent effect. The combined STINGVAX enhanced T cell infiltration in tumor tissues compared with the vaccine of single GM-CSF-secreting cancer cells. Besides, several tumor-bearing mice models demonstrated the strong antitumor effect of STINGVAX. Feasibility of STING-based cancer vaccine was verified later in mice bearing pancreatic cancer and melanoma [53, 54]. In addition, recently, Miao et al. identified and designed an effective STING-dependent cyclic lipid nanoparticles (LNP), as the adjuvant of antigen-specific mRNA vaccine delivery [55]. This research team developed multiple synthetical lipid structures using a one-step three-component reaction method, in which the lipids with a cyclic amino head group could activate the STING pathway. In the mouse model, the application of this combinatorial LNP demonstrated marked survival advantage, which showed a promising role of STING agonists in antitumor therapy [55].
STING agonist combined with ICIs
Several STING agonists have been used as an anti-cancer therapy in clinical trials, and the STING agonists/ICIs combinations were also developed (Table 2). cGAS-STING agonists are ideal partners for ICIs. Firstly, the STING signaling and type I IFNs play crucial roles in spontaneous T cell responses via the cross-present of CD8α+ DCs [37], which promotes the intratumoral T cell infiltration. Importantly, high densities of adaptive immune cells (CD3+, CD8+, GZMB+, and CD45RO+ cells) represent favorable prognosis and positive clinical results for cancer patients [56]. Cytotoxic lymphocyte (CTL) infiltration is also regarded as an indicator for optimal response to ICIs. Besides, cGAS-STING pathway agonists can also increase antigen-presenting molecules such as Tap1, Tap2, and MHC-I with IFN upregulation, which may enhance the tumor immune surveillance [57]. In addition, STING agonists can increase tumor cells’ sensitivity to immune NK cells and CTLs [22]. Indeed, NLRX1 and NLRC3 proteins that could downregulate STING-mediated IFN-I signaling were increased in resistant tumor cells [22].
STING agonist combined with anti-CTLA-4 immunotherapy
The threshold of T lymphocyte activation can be reduced by the application of anti-CTLA-4 therapy [58]. Evidence suggests that the integrity of the STING pathway is essential to the optimal effect of CTLA-4-based immunotherapy [59]. Under the treatment of ionizing radiation combined with an-CTLA-4 therapy, Shane’s group found that STING absence prevented abscopal tumor regression, and deficient STING significantly impaired CD8+ T cell infiltration in the tumor tissues. Ager’s group also made a relevant study [38]. Their result showed the administration of combined anti-CTLA-4 therapy (9H10), anti-PD-1 therapy (RMP114), and agonistic anti-4-1-BB therapy (3H3), induced bilateral tumor regression in 40% of mice while the STING agonist CDG added, markedly inhibited the bilateral tumors in 75% of these mice. Therefore, the combination of CDG and ICIs effectively enhanced the antitumor effect.
STING agonist combined with anti-PD-1/programmed death-ligand 1 (PD-L1) immunotherapy
The cGAMP/antigenic peptide nanosatellite vaccine SatVax, combined with anti-PD-L1 therapy in the xenograft model, showed elevated E7-specific CD8+ CTLs, and decreased ratio of CD8+ Tim3+ and CD8+ PD-1+ T cells [22]. This successful combination led to significant tumor control, with four completely tumor-free mice of five animals. Reduced and delayed tumor growth was also showed in the B16 melanoma mouse model, treated by the co-administration of CDN-based poly beta-amino ester (PBAE-CDN) nanoparticles and anti-PD-1 therapy [60]. In addition, this combined therapy provided the mice with protection to tumor rechallenge [52]. Another advantage of this combined treatment is that the application of anti-PD-1/PD-L1 blockers can neutralize the STING agonist’s immunosuppressive effect. The upregulation of PD-L1 expression was reported in the cGAS-STING activation [57].
STING pathway as a prognostic predictive biomarker in oncolytic immunotherapy
Oncolytic viruses have been regarded as a versatile platform to treat cancer. They are viral vectors that can kill tumor cells selectively and also have the potential to amplify the immune response and enhance anti-tumor effects [61]. The oncolytic immunotherapy talimogene laherparepvec has demonstrated a therapeutic benefit in patients with advanced melanoma in a phase III clinical trial [62]. The integrity of the cGAS-STING pathway is critical for response to the invasion from multiple pathogens and tumors, while cancers including melanoma and colon cancers are common with its deficiency [19, 20], as mentioned above. Based on these findings, it is reasonable to select the oncolytic immunotherapy to treat STING-loss cancers. In the melanoma mouse model, the herpes simplex virus type 1 (HSV-1) with γ34.5 gene deficiency was used to test its effect on cancer [19]. Under normal conditions, the existence of HSV-1Δγ34.5 could activate the STING signaling pathway effectively and help the host to clear its infection. Interestingly, in their study, the STING-deficiency melanoma cells were observed susceptibility to the virus infection while cancer cells with intact STING pathway grew rapidly. Additionally, a similar observation was also found in STING-loss mice with ovarian cancer [63]. Given that the STING deficiency correlates with an improved prognosis with oncolytic virus treatment, with further in vivo and clinical trials, it may represent a prognostic/predictive biomarker for oncolytic immunotherapy in cancer patients.
STING agonist combined with CAR-T therapy
Engineered T cell has the ability to recognize the targeted antigen of tumor cells with the single-chain variable fragment domain, through transferring gene encoding chimeric antigen receptor (CAR) [64]. The CAR-T therapy is successful in several hematological diseases, but its application in solid tumors is limited [65]. The immune killing of CAR-T cells can be escaped mainly due to immunosuppressive TME and tumor heterogeneity [66, 67]. A new implantable bioactive device has been tested its property to deliver the CAR-modified T cells to the surfaces of tumors [68]. Although the delivery of this novel carrier promoted T cell expansion and a temporary tumor regression, antigen-negative tumors could not be eliminated completely. They further found that ribonucleic acid export 1 (RAE1)-high tumor cells were destroyed and the cells with RAE1 loss/low expression still survived. Thus, cyclic di-GMP (cdGMP), a STING agonist, was applied. Combination therapy of cdGMP and CAR-T cells led to a significant activation of the host APCs and lymphocyte responses, which eradicated tumors completely in four of ten pancreatic-bearing mice, with longer survival [68]. Meanwhile, after re-injection of tumor cells, these four tumor-free mice inhibited tumor growth to measurable mass. This CAR-T/cdGMP presented durable antitumor ability. The mechanisms how the corelease of STING agonists and CAR-T cells activates the host immunity remain to be fully elucidated.
The challenge of STING-targeted immunotherapy against cancer:an emerging pro-tumor role of cGAS-STING
Undoubtedly, STING agonists showed impressive potential in antitumor immunity. However, emerging evidence suggested the pro-tumor roles of the cGAS-STING pathway, from tumor initiation and development, to metastasis [69,70,71,72] (Fig. 4), which makes the application of STING agonists in the clinic remains a lot to challenge. First, different from acute STING-induced SASP, chronic SASP-correlated inflammation relates to malignant behaviors such as immune-suppression and oncogene-driven senescence evasion [26, 73]. Similarly, high chromosome instability (CIN) tumors generated micronuclei, and its rupture could release DNA to the cytosol, enhancing the sense of cGAS-STING. This regulation was reported related to the secretion of the pro-inflammatory cytokines by activation of NF-κB signaling and metastasis [74] (Fig. 4). When malignancies tolerate the long-term use of STING agonists, and lose the cell-cycle regulators downstream, inflammatory processes are able to function their pro-tumor effects. Apart from the intrinsic cGAS-STING activation in malignancies, metastasis could also be induced in a cancer cell non-autonomous manner. cGAMP, particularly, was reported to transfer from the tumor cells through gap junctions to astrocytes, promoting NF-κB and IFN signaling and inducing brain metastasis ultimately [69]. STING upregulation was also correlated with increased infiltration of regulatory T cells [70], and immune-regulatory enzyme indoleamine 2,3-dioxygenase (IDO), which can mediate tumor immune evasion and inhibit T cell proliferation [75]. Thus, chronic cGAS-STING activation may promote tumor metastasis which needs to be overcome. An important unanswered question existed is how these cancer cells change the STING’s downstream circuitry to mediate metastasis. One hypothesis supports that the precise control of levels of STING expression may be involved in this alteration [76]. In this study, the relationship between STING signaling magnitude and the apoptotic programs in T cells and macrophages was demonstrated. To overcome this challenge, further examinations are warranted to unveil the molecular requirements and regulations that function in metastatic promotion or suppression downstream of cGAS-STING cascade.

Regulation of cGAS-STING pathway in tumor promotion. The cGAS-STING pathway could exert its pro-tumor role in metastatic tumor settings. STING functions as a platform for different tumorigenic programs. High chromosome instability (CIN) tumors generated micronuclei, and its rupture could release DNA to the cytosol, enhancing the sense of cGAS-STING. Low tumor antigenicity and cytoplasmic chromatin chronic activation contribute to malignancy formation, through the activation of the cGAS-STING pathway. Chronic activation could also downregulate the expression of type I IFNs, upregulate noncanonical NF-κB signaling, and promote tumor metastasis. Indoleamine 2,3-dioxygenase (IDO) and anti-inflammatory cytokines released from the tumor induces the formation of an immunosuppressive TME. STING may also promote immune evasion and tumor metastasis by PD-L1 upregulation and autophagy induction. In addition, cGAMP, particularly, was reported to transfer from the tumor cells through gap junctions to astrocytes, promoting NF-κB and IFN signaling and inducing brain metastasis ultimately. Abbreviations: cGAMP, 2′,3′-cyclic GMP-AMP; IDO, indoleamine 2,3-dioxygenase; PD-L1, programmed death-ligand 1; TME, tumor microenvironment
Beyond the pro-tumor chronic inflammation and CIN, the ER stress response as well as autophagy also serve as a barrier to the anti-tumor effect of STING agonists. They play a part in the advanced tumor progression by making tumor cells survive under stressful conditions [77]. The ER stress response enables disseminating tumor cells to exert the potentials of immune evasion [78]. cGAS-STING signaling was also revealed able to cooperate with the autophagy-ER stress responses to promote tumor progression [79] (Fig. 4). Additionally, metabolic programs may also be exploited by TME to induce immune suppression. The uptake of glucose or other nutrients in intratumoral T cells was restricted by tumor milieu, which results in ER stress and activation of the response of IRE1α–XBP1 unfolded protein. Chronic IRE1α–XBP1 activation makes T cells into a dysfunctional state, with reduced antitumor effects [77]. Further investigations are needed to discover the molecular hierarchy of ER stress and the cGAS-STING pathway, as well as how they interact to promote tumorigenesis.
Another major challenge is how to carefully select patients to enhance clinical response to STING agonists. The preliminary clinical phase 1 trial of STING agonist (MK-1454) in solid tumors and lymphomas has shown only a modest clinical response using single-agent treatment with no marked activity seen, and the co-administration of it with ICIs in advanced cancers showed only partial responses [80]. Therefore, novel STING agonist ABZI was developed by systemic administrated to patients [43]. Its safety remains to be considered, and hemodynamics of patients using such drugs need to be closely monitored. To determine which patients STING agonists might benefit is necessary. Different CIN state in primary and metastatic tumors indicates the active CIN might become a biomarker to predict personalized administrations. In fact, it is metastatic but not primary tumors, correlate with increased CIN, chronic STING activation, as well as poor patient prognosis [74]. Therefore, effective biomarkers and selection schemes are required to identify the patients who might benefit from STING agonists.
In addition, there may have technical limitations of the bulk RNA sequencing since the relationship between patient outcomes and cGAS-STING RNA levels is incongruent across different tumors. For example, poor patient survival was revealed with downregulated cGAS-STING signaling in a subset of human tumors [28, 72]. While in patients with colorectal cancer, high STING expression has also been demonstrated related to poor prognosis [81]. Contaminating stromal cells may mask the actual expression of cGAS-STING, or this difference reflects both pro-tumor and anti-tumor roles of cGA-STING in specific tumor types.
Implications of the cGAS-STING pathway in cancer therapy
A comprehensive mechanistic understanding of the biologic STING signaling may greatly help to develop agents to potently activate it while reducing its immune-suppressive effects. For example, several feedback loops were found to attenuate STING activation including autophagy induction, AIM2 inflammasome activation, and autocrine IFN signaling [82, 83]. Compounds designed to impede one or more loops might greatly interact with the STING agonists. Additionally, a recent study demonstrated STIM1 anchors the STING to the ER membrane as a calcium sensor. In fact, deficiency of STIM1 can lead to spontaneous activation of STING with type I IFN production. This makes the STIM1-targeted inhibitor a potential strategy for future therapies [84]. Although the application of STING agonists is exciting, the recent correlation between the cGAS-STING pathway and metastasis also suggests the prospect for STING inhibition in late-stage cancers. Importantly, a recent covalent STING palmitoylation inhibitor has been discovered to attenuate metastasis in its therapeutic interventions [85]. Therefore, a personalized method of the use of STING agonists and antagonists may be helpful.
Another implication for selectively promoting desired outputs is to increase the local concentration of type I IFNs. One method is the tumor-targeted monoclonal antibodies combined with IFN-β. For example, a method of coupling either anti-EGFR or anti-Her2 monoclonal antibodies to IFN-β can result in tumor regression in tumor-bearing mice [86]. In addition, the type I IFNR−/− mice lost the antitumor effect, indicating the importance of host immune cell priming [86]. Thus, transient expression of low IFN-β doses in TME may elevate tumor adaptive immune response.
It is also critical to understanding the STING-based TME changes in pre-clinical experiments and clinical trials, so that less side effects may be produced in cancer treatment. Most STING agonists have not encountered pro-tumor effects because just a few doses of treatment could result in a burst of type I IFN production to activate anti-tumor immune system [83]. Therefore, selecting an appropriate dose is vital. For instance, in mouse models, the ADU-S100 injection with low, single dose appears to produce effective tumor-associated T cell responses, while high repetitive doses may impair both T cell response and immune memory formation. Based on this, lower doses might be more helpful because of their ability to generate adaptive immune responses [87]. In addition, given the PD-L1 upregulation observed in STING activation, the combined therapy of STING agonists with anti-PD-1/PD-L1 antibodies would be extremely helpful for anti-cancer therapies.
Conclusion remarks
The burgeoning interest in the STING pathway, using patients’ own immunity to eradicate tumors, is extremely appealing and several STING agonists were developed for cancer treatment with promising pre-clinical results. As a critical immune sensor, the STING pathway plays an important role in tumor control through tumor-derived DNA sensing and T cell priming. This induces T cell presentation in tumors, making the STING pathway a promising strategy combined with ICI therapy. STING agonists can elevate the efficacy of therapies from cancer vaccine, to ICIs to CAR-T immunotherapies. However, emerging pro-tumor roles have been described and a greater understanding of STING-associated TME and biologic mechanism is needed. We believe cGAS-STING pathway manipulation might become a promising strategy combined with cancer immunotherapy.
Availability of data and materials
Not applicable.
Abbreviations
- ABZ:
-
Amidobenzimidazole
- APC:
-
Antigen-presenting cell
- AACR 2017:
-
American Association for Cancer Research 2017 Conference on Tumor Immunology and Immunotherapy
- cGAS-STING:
-
Cyclic GMP-AMP synthase-stimulator of interferon genes
- CTLA-4:
-
Cytotoxic T lymphocyte antigen 4
- cGAMP:
-
2′,3′-cyclic GMP-AMP
- CDN:
-
Cyclic dinucleotide
- CTL:
-
Cytotoxic lymphocyte
- CAR:
-
Chimeric antigen receptor
- cdGMP:
-
Cyclic di-GMP
- CIN:
-
Chromosome instability
- dsDNA:
-
Double-stranded DNA
- DMXAA:
-
Dimethyloxoxanthenyl acetic acid
- DCs:
-
Dendritic cells
- ER:
-
Endoplasmic reticulum
- FAA:
-
Flavone 8-acetic acid
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- hSTING:
-
Human STING
- HSV-1:
-
Herpes simplex virus type 1
- ICIs:
-
Immune checkpoint inhibitors
- IFNs:
-
Interferons
- IRF3:
-
Interferon regulatory factor 3
- IDO:
-
Indoleamine 2,3-dioxygenase
- IT:
-
Intratumoral
- IV:
-
Intravenous
- IP:
-
Intraperitoneal
- LNP:
-
Lipid nanoparticles
- mtDNA:
-
Mitochondrial DNA
- mSTING:
-
Mouse STING
- NK cells:
-
Natural killer cells
- PD-1:
-
Programmed death 1
- PBAE-CDN:
-
CDN-based poly beta-amino ester
- RAE1:
-
Ribonucleic acid export 1
- SITC 2018:
-
Society for the Immunotherapy of Cancer 2018 Annual Meeting
- SASP:
-
Senescence-associated secretory phenotype
- TME:
-
Tumor microenvironment
- TBK1:
-
TANK binding kinase 1
- TAAs:
-
Tumor-associated antigens
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118(1):9–16.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–23.
Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling (vol 455, pg 674, 2008). Nature. 2008;456(7219):274.
Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760–70.
Berger G, Marloye M, Lawler SE. Pharmacological modulation of the STING pathway for cancer immunotherapy. Trends Mol Med. 2019;25(5):412–27.
Kwon J, Bakhoum SF. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020;10(1):26–39.
Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nature Immunology. 2013;14(1):19–26.
Dhanwani R, Takahashi M, Sharma S. Cytosolic sensing of immuno-stimulatory DNA, the enemy within. Curr Opin Immunol. 2018;50:82–7.
Chen Q, Sun LJ, Chen ZJJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142–9.
West AP, Khoury-Hanold W, Staron M, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553.
Civril F, Deimling T, Mann CCD, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498(7454):332.
Sun LJ, Wu JX, Du FH, Chen X, Chen ZJJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–91.
Ablasser A, Goldeck M, Cavlar T, et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498(7454):380.
Shang GJ, Zhang CG, Chen ZJJ, Bai XC, Zhang XW. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature. 2019;567(7748):389.
Zhang CG, Shang GJ, Gui X, Zhang XW, Bai XC, Chen ZJJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature. 2019;567(7748):394.
Gao P, Ascano M, Zillinger T, et al. Structure-function analysis of STING activation by c[G(2',5') pA(3',5')p] and targeting by antiviral DMXAA. Cell. 2013;154(4):748–62.
Diner EJ, Burdette DL, Wilson SC, et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Reports. 2013;3(5):1355–61.
Xia TL, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016;76(22):6747–59.
Xia TL, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016;14(2):282–97.
Kitapma S, Ivanova E, Guo S, et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 2019;9(1):34–45.
Tan YS, Sansanaphongpricha K, Xie YY, et al. Mitigating SOX2-potentiated immune escape of head and neck squamous cell carcinoma with a STING-inducing nanosatellite vaccine. Clin Cancer Res. 2018;24(17):4242–55.
Li A, Yi M, Qin S, Song Y, Chu Q, Wu K. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J Hematol Oncol. 2019;12(1):35.
Harlin H, Meng Y, Peterson AC, et al. Chemokine expression in melanoma metastases associated with CD8(+) T-cell recruitment. Cancer Res. 2009;69(7):3077–85.
Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity. 2018;49(4):754.
Dou ZX, Ghosh K, Vizioli MG, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550(7676):402–6.
Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. Plos Biol. 2008;6(12):2853–68.
Yang H, Wang HZ, Ren JY, Chen Q, Chen ZJJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA. 2017;114(23):E4612–20.
Gluck S, Guey B, Gulen MF, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19(9):1061.
Ranoa DRE, Widau RC, Mallon S, et al. STING promotes homeostasis via regulation of cell proliferation and chromosomal stability. Cancer Res. 2019;79(7):1465–79.
Woo SR, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–42.
Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013;34(2):67–73.
Liu HP, Zhang HP, Wu XY, et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature. 2018;563(7729):131.
Parlato S, Santini SM, Lapenta C, et al. Expression of CCR-7, MIP-3beta, and Th-1 chemokines in type I IFN-induced monocyte-derived dendritic cells: importance for the rapid acquisition of potent migratory and functional activities. Blood. 2001;98(10):3022–9.
Bertucci F, Ueno NT, Finetti P, et al. Gene expression profiles of inflammatory breast cancer: correlation with response to neoadjuvant chemotherapy and metastasis-free survival. Ann Oncol. 2014;25(2):358–65.
Fuertes MB, Kacha AK, Kline J, et al. Host type I IFN signals are required for antitumor CD8(+) T cell responses through CD8 alpha(+) dendritic cells. J Exp Med. 2011;208(10):2005–16.
Diamond MS, Kinder M, Matsushita H, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003.
Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res. 2017;5(8):676–84.
Tang CHA, Zundell JA, Ranatunga S, et al. Agonist-mediated activation of STING induces apoptosis in malignant B cells. Cancer Res. 2016;76(8):2137–52.
Demaria O, De Gassart A, Coso S, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci USA. 2015;112(50):15408–13.
Corrales L, Glickman LH, McWhirter SM, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11(7):1018–30.
McKeage MJ, Reck M, Jameson MB, et al. Phase II study of ASA404 (vadimezan, 5,6-dimethylxanthenone-4-acetic acid/DMXAA) 1800 mg/m(2) combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Lung Cancer. 2009;65(2):192–7.
Ramanjulu JM, Pesiridis GS, Yang J, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564(7736):439–43.
Karaolis DKR, Cheng KR, Lipsky M, et al. 3 ',5 '-Cyclic diguanylic acid (c-di-GMP) inhibits basal and growth factor-stimulated human colon cancer cell proliferation. Biochem Biophys Res Commun. 2005;329(1):40–5.
Li T, Cheng H, Yuan H, et al. Antitumor activity of cGAMP via stimulation of cGAS-cGAMP-STING-IRF3 mediated innate immune response. Sci Rep. 2016;6:19049.
Shae D, Becker KW, Christov P, et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol. 2019;14(3):269.
Berry S, Giraldo N, Nguyen P, et al. Correction to: 33rd Annual Meeting & Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2018). J Immunother Cancer. 2019;7(1):46.
Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, Barber GN, Arai H, Taguchi T. Activation of STING requires palmitoylation at the Golgi. Nat Commun. 2016;7:11932.
Lara PN, Douillard JY, Nakagawa K, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29(22):2965–71.
Conlon J, Burdette DL, Sharma S, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol. 2013;190(10):5216–25.
Dubensky TW, Reed SG. Adjuvants for cancer vaccines. Semin Immunol. 2010;22(3):155–61.
Fu J, Kanne DB, Leong M, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283.
Kinkead HL, Hopkins A, Lutz E, et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. Jci Insight. 2018;3(20):e122857.
Wang ZL, Celis E. STING activator c-di-GMP enhances the anti-tumor effects of peptide vaccines in melanoma-bearing mice. Cancer Immunol Immunother. 2015;64(8):1057–66.
Miao L, Li LX, Huang YX, et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat Biotechnol. 2019;37(10):1174.
Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–4.
Grabosch S, Bulatovic M, Zeng FTZ, et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene. 2019;38(13):2380–93.
Ok CY, Young KH. Checkpoint inhibitors in hematological malignancies. J Hematol Oncol. 2017;10(1):103.
Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Reenberg RAG. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466.
Wilson DR, Sen R, Sunshine JC, Pardoll DM, Green JJ, Kim YJ. Biodegradable STING agonist nanoparticles for enhanced cancer immunotherapy. Nanomed Nanotechnol Biol Med. 2018;14(2):237–46.
Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC, Nelson BH. Oncolytic viruses as engineering platforms for combination immunotherapy (vol 18, pg 419, 2018). Nat Rev Cancer. 2018;18(8):526.
Andtbacka RHI, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–U98.
de Queiroz NMGP, Xia TL, Konno H, Barber GN. Ovarian cancer cells commonly exhibit defective STING signaling which affects sensitivity to viral oncolysis. Mol Cancer Res. 2019;17(4):974–86.
Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma-subunit or zeta-subunit of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993;90(2):720–4.
Pang YY, Hou XY, Yang CS, Liu YQ, Jiang G. Advances on chimeric antigen receptor-modified T-cell therapy for oncotherapy. Mol Cancer. 2018;17(1):91.
Newick K, O'Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med. 2017;68:139–52.
Zhang EH, Gu JY, Xu HM. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol Cancer. 2018;17(1):7.
Smith TT, Moffett HF, Stephan SB, et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. Journal of Clinical Investigation. 2017;127(6):2176–91.
Chen Q, Boire A, Jin X, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533(7604):493.
Ding L, Huang XF, Dong GJ, et al. Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-jun/CCL22 signal. Biochim Biophys Acta-Mol Basis Dis. 2015;1852(11):2494–503.
Lemos H, Mohamed E, Huang L, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016;76(8):2076–81.
Song SS, Peng PK, Tang ZQ, et al. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci Rep. 2017;7:39858.
Toso A, Revandkar A, Di Mitri D, et al. Enhancing chemotherapy efficacy in pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9(1):75–89.
Bakhoum SF, Ngo B, Laughney AM, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553(7689):467.
Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37(3):193–207.
Gulen MF, Koch U, Haag SM, et al. Signalling strength determines proapoptotic functions of STING. Nat Commun. 2017;8(1):427.
Song M, Sandoval TA, Chae CS, et al. IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. 2018;562(7727):423–8.
Pommier A, Anaparthy N, Memos N, et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science. 2018;360(6394):1202.
Terai H, Kitajima S, Potter DS, et al. ER stress signaling promotes the survival of cancer “persister cells” tolerant to EGFR tyrosine kinase inhibitors. Cancer Res. 2018;78(4):1044–57.
Harrington KJ, Brody J, Ingham M, et al. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann Oncol. 2018;29:712.
An X, Zhu YY, Zheng TS, et al. An analysis of the expression and association with immune cell infiltration of the cGAS/STING pathway in pan-cancer. Mol Ther Nucleic Acids. 2019;14:80–9.
Corrales L, Woo SR, Williams JB, McWhirter SM, Dubensky TW, Gajewski TF. Antagonism of the STING pathway via activation of the AIM2 inflammasome by intracellular DNA. J Immunol. 2016;196(7):3191–8.
Flood BA, Higgs EF, Li SY, Luke JJ, Gajewski TF. STING pathway agonism as a cancer therapeutic. Immunol Rev. 2019;290(1):24–38.
Srikanth S, Woo JS, Wu BB, et al. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol. 2019;20(2):152.
Haag SM, Gulen MF, Reymond L, et al. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559(7713):269–73.
Yang XM, Zhang XM, Fu ML, et al. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell. 2014;25(1):37–48.
Sivick KE, Desbien AL, Glickman LH, et al. Magnitude of therapeutic STING activation determines CD8(+) T cell-mediated anti-tumor immunity. Cell Rep. 2018;25(11):3074.
Acknowledgements
Not applicable.
Funding
This study was supported in part by a grant from the National Natural Science Foundation of China (81802255), Shanghai Pujiang Program (17PJD036) and a grant from the Shanghai Municipal Commission of Health and Family Planning Program (20174Y0131), National Key Research and Development Project (2016YFC0902300), Major Disease Clinical Skills Enhancement Program of a 3-year action plan for promoting clinical skills and clinical innovation in municipal hospitals, Shanghai Shen Kang Hospital Development Center Clinical Research Plan of SHDC (16CR1001A), “Dream Tutor” Outstanding Young Talents Program (fkyq1901), Key Disciplines of Shanghai Pulmonary Hospital (2017ZZ02012), a grant of Shanghai Science and Technology Commission (16JC1405900).
Author information
Authors and Affiliations
Contributions
MJ, PC, LW, WL, BC, YL, HW, SZ, LY, YH, and CZ contributed to the manuscript design, writing, and editing. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiang, M., Chen, P., Wang, L. et al. cGAS-STING, an important pathway in cancer immunotherapy. J Hematol Oncol 13, 81 (2020). https://doi.org/10.1186/s13045-020-00916-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00916-z