
Abstract
Wild species of rice (genus Oryza) contain many useful genes but a vast majority of these genes remain untapped to date because it is often difficult to transfer these genes into cultivated rice (Oryza sativa L.). Chromosome segment substitution lines (CSSLs) and backcross inbred lines (BILs) are powerful tools for identifying these naturally occurring, favorable alleles in unadapted germplasm. In this paper, we present an overview of the research involving CSSLs and BILs in the introgression of quantitative trait loci (QTLs) associated with the improved performance of rice including resistance to various biotic and abiotic stresses, and even high yield from wild relatives of rice and other unadapted germplasm into the genetic background of adapted rice cultivars. The CSSLs can be used to dissect quantitative traits into the component genetic factors and evaluate gene action as single factors (monogenic loci). CSSLs have the potential to uncover new alleles from the unadapted, non-productive wild rice accessions, develop genome-wide genetic stocks, and clone genes identified in QTL studies for functional genomics research. Recent development of high-density single-nucleotide polymorphism (SNP) arrays in rice and availability of custom-designed medium- and low-density SNP arrays will enhance the CSSL development process with smaller marker-defined segment introgressions from unadapted germplasm.
Similar content being viewed by others


Introduction
Rice (Oryza sativa L.) is a major cereal crop and a primary source of food for more than half of the world’s human population (Khush 2005; www.csrees.usda.gov/newsroom/impact/2009/nri/02171_rice_gene.html). It is grown worldwide under a wide range of agro-climatic conditions. Rice productivity is affected by many abiotic and biotic stresses. Some of the major biotic stresses include diseases, such as bacterial leaf blight, blast, sheath blight, tungro virus, and insect pests, such as brown planthopper, green leafhopper, and stem borer. Similarly, abiotic stresses, such as drought, cold, salinity, acidity, iron toxicity, and submergence under water, severely affect rice production. Furthermore, changes in disease pathogen races and insect biotypes are continual threats to rice production. The genetic variability for some important traits including resistance to diseases, pests, and abiotic stresses are limited in the cultivated rice germplasm. During the course of domestication of cultivated rice from wild relatives, there was a significant reduction in genetic diversity (Brar and Khush 2003) as desirable agronomic traits were selected, leaving the number of alleles in cultivated rice reduced by 50–60% compared to wild rice (Sun et al. 2001). This emphasizes the importance of broadening the gene pool in rice by introgression of new genes from diverse sources.
In this paper, we review research illustrating the usefulness of chromosome segment substitution lines (CSSLs) and backcross inbred lines (BILs) for identifying new alleles of genes controlling agronomically important traits in rice wild relatives and their transfer into a cultivated rice genetic background to improve productivity, CSSL and BIL development strategies, and progress in developing CSSLs as genome-wide genetic stocks for gene cloning and functional genomics studies.
Potential of wild rice species
Wild species of rice have evolved in a wide range of environments over millions of years (Stebbins 1981). The Oryza wild species have either 2n = 24 or 2n = 48 chromosomes represented by the AA, BB, CC, BBCC, CCDD, EE, FF, GG, HHJJ, or HHKK genomes (Brar and Khush 1997; Jena and Khush 2000; Lu et al. 2010; Vaughan 1994). The geographical distribution of wild species with the AA genome is shown in Fig. 1. The AA genome is known to exist in Asia (Oryza nivara and Oryza rufipogon), Africa (African cultivated rice or Oryza glaberrima, Oryza barthii, and Oryza longistaminata), Australia (Oryza meridionalis), and Latin America (Oryza glumaepatula) (Vaughan et al. 2005).

Geographic distribution of AA genome wild Oryza species (Lu et al. 2010).
Wild species of rice are a reservoir of many valuable genes which could be exploited for improving elite cultivars (Brar and Khush 1997). Almost all the wild rice species have been reported to contain some valuable traits (Table 1; Jena and Khush 2000). Although O. glaberrima is a cultivated species, mainly grown in Africa, it bears numerous interesting genes that can be used to improve O. sativa, including resistance to nematodes (both Heterodera and Meloidogyne genus) and insects (African gall midge; Brar and Khush 2006), viruses (rice yellow mottle virus, rice stripe necrosis virus; Ndjiondjop et al. 1999; Gutiérrez et al. 2010), and tolerance to drought stress (Ndjiondjop et al. 2010; Furuya et al. 1994). The first report of transfer of an agronomically important gene from a wild species to cultivated rice was the introgression of grassy stunt virus resistance from O. nivara (Khush et al. 1977). Other noteworthy reports include the transfer of cytoplasmic male sterility (CMS) from O. sativa f. spontanea (Lin and Yuan 1980) and the Xa-21 gene for bacterial blight resistance from O. longistaminata (Khush et al. 1990). In these cases, the donor Oryza species were of the same genome class (AA) as cultivated rice (O. sativa). More recently, successful introgression of traits from non-AA genome species include the transfer of blast resistance from Oryza minuta (CCDD) and Oryza australiensis (EE) into O. sativa (Brar and Khush 2002; Fu et al. 2008; Jena and Khush 2000). Also, there are several more recent successful gene transfers from wild AA genome Oryza species into cultivated rice (Table 2).
Wild species contain novel yield-enhancing genes
In addition to being a largely untapped reservoir of genes for resistance to various biotic or abiotic stresses, wild species also contain genes that can enhance productivity and yield in the background of adapted cultivars. In general, wild species have smaller fruit (seeds), produce fewer seeds that often shatter, and other undesirable traits compared to cultivars, and thus appear to be poor as donors for enhancing yield. However, an early study in oats (Avena spp.) showed the transgressive segregants had about 20% yield increase over the recurrent parent (Lawrence and Frey 1975), and another study revealed that the yield of cultivated oats, A. sativa, was increased by 4–7% through crossing with wild species, Avena sterilis (Frey et al. 1984). In tomato (Lycopersicon esculentum), lines derived from crosses with the wild species, Lycopersicon chmielewskii, which has small green fruit, led to progenies with large red fruit and increased fruit weight (Rick 1974). Using advanced backcross lines or introgression lines, novel alleles from the wild progenitor of cultivated rice, O. rufipogon (IRGC 105491) were simultaneously identified, mapped, and introgressed into the genetic background of several adapted cultivars (McCouch et al. 2007). Alleles associated with the yield-enhancing traits from this O. rufipogon accession were incorporated into the genetic background of the well-known indica cultivar IR64 (Cheema et al. 2008; Septiningsih et al. 2003), the US tropical japonica cultivar Jefferson (Thomson et al. 2003), the indica V20B maintainer line of a widely used CMS line ‘V20A’ (Xiao et al. 1998), and the Korean temperate japonica cultivar Hwaeongbyeo (Xie et al. 2006). Alleles associated with the yield-related traits, such as spikelet number, grain weight, and panicle length also were identified in this O. rufipogon accession using a backcross inbred line population derived from a cross with Zhenshan 97B, an indica cultivar (Yu, personal communication). Similarly, alleles associated with the improved traits related to yield from two other O. rufipogon accessions were introgressed into the Chinese cultivars TeQing (Tan et al. 2007) and Guichao 2 (Tian et al. 2006). Yield trials of selected Jefferson/O. rufipogon near-isogenic lines (NILs) revealed yield enhancing QTLs when compared to the donor parent (Kimball et al. 2009). These studies suggest that additional novel alleles associated with high productivity and yield may be found in other accessions of O. rufipogon, as well as other wild Oryza species.
Challenges of transferring genes from unadapted germplasm or wild species
Most of the world’s rice is produced from inbred varieties which have been developed almost exclusively from crosses between the accessions of the same subpopulation or between related subpopulations (tropical japonica × temperate japonica, indica × aus; Lu et al. 2005; Ni et al. 2002; Ali et al. 2011; in review). This is because it is difficult to obtain a random array of fertile recombinants from crosses between the indica × japonica varietal groups (in other words O. sativa ssp. indica × O. sativa ssp. japonica), largely due to the prevalence of subpopulation incompatibilities (Harushima et al. 2002; Oka 1988; Sano 1993; Win et al. 2009). The long-term consequence of restricting crossing and population development to within subpopulations is a limited pool of genetic variation available to rice breeders for identifying new and useful combinations of genes. Over time, this has compounded the problems associated with genetic bottlenecks that were primarily created during the domestication process and led to the cryptic form of genetic erosion that dramatically slows the rate of genetic gain (McCouch et al. 2007).
Inter-specific breeding offers a way of expanding the gene pool of cultivated rice and enlarging the range of genetic variation available for plant improvement (McCouch et al. 2007). Wide hybridization has been used for many years to introduce qualitative characters from wild species into elite breeding materials, such as disease and insect resistance, and male sterility (Brar and Khush 1997; Dalmacio et al. 1995). However, several incompatibility barriers, such as low crossing success, increased sterility, and limited recombination between the chromosomes of wild and cultivated species seriously hampered the transfer of useful genes (Brar and Khush 1986; Fu et al. 2008; Miura et al. 2008). Recent advances in tissue culture and genomics helped the production of wide hybrids between distantly related species. A major challenge is the difficulty in identifying genes from wild or unadapted materials that are likely to enhance the performance of elite cultivars without disrupting favorable gene complexes or allele combinations. A substantial “pre-breeding” research can provide a platform to identify and transfer favorable alleles from wild and unadapted sources into elite rice cultivars (McCouch et al. 2007). Often, the genetic potential of an individual is obscured by the presence of a few deleterious alleles that confound the breeders’ ability to accurately determine its breeding value. Today, with the advent of quality sequence information, genetic maps and molecular markers, it is possible to identify regions of the genome associated with specific components of a phenotype and determine which parent contributes the favorable allele(s) at a particular locus. This information is very helpful for selecting which genes or components of quantitative trait variation to introduce from a wild or exotic gene pool into an elite cultivar.
CSSL development strategies
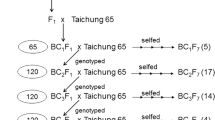
In the crossing program to develop rice CSSLs, the recurrent parent, which is usually an adapted cultivar or germplasm accession, is used as the female, while the donor parent, usually a wild species or unadapted germplasm accession, is used as the male in order to avoid cytoplasmic sterility in subsequent generations. A typical backcross scheme to develop CSSLs is shown in Fig. 2. A set of polymorphic markers (either SSRs or SNPs) is used in the selection of backcross lines during the backcrossing process. For backcrossing, either the F1 hybrid plant(s) or the recurrent parent can be used as female, depending on the fertility of the F1s. Next, a set of 50–60 randomly chosen BC1F1 individuals is backcrossed again to generate BC2F1s. A set of 10–12 individuals from each of the BC2F1 families (50–60 BC1founder lines) is selected for the first-round of genotyping with DNA markers. About 150–200 selected BC2F1 individuals (see Fig. 3 showing introgressed segments) are used for the next round of backcrossing to generate BC3F1s. About 120–150 BC3F1 individuals are selected for selfing after genotyping a second time from 400 to 500 BC3F1 individuals, to develop homozygous lines or for the next round of backcrossing to generate BC4F1 plants. During each generation of selection, one must identify individuals that carry the introgression(s) of interest along each chromosome. Graphical genotyping software programs, such as GGT (van Berloo 2008) or CSSL Finder (http://mapdisto.free.fr/CSSLFinder), are very useful tools to select desired backcross progenies based on their genotypic content. The third or last round of selection is made after genotyping of BC3F2 or BC4F2 individuals. At this stage, each individual generally contains only one marker defined-target segment and all the selected individuals together cover the whole donor genome in an overlapping manner as shown in Fig. 4. In order to obtain adequate seed, 10 plants are selfed from each of the selected BC4F2 families carrying the desired traits and target introgressed segments. These introgression lines are suitable for phenotypic evaluation of the “trait(s) of interest”, and once they are fixed, may be evaluated in multiple environments.

A backcross scheme for developing chromosome segment substitution lines (CSSL) utilizing molecular markers for selecting the progeny each generation.

Graphical genotypes of 20 BC2F1s showing introgressed segments from O. rufipogon in the background of IR64 based on 384-SNP assays. The heterozygous segments are purple and the homozygous IR64 segments are light blue while yellow represents missing segments. Genotypes were created in GGT (van Berloo 2008). The consensus genotype identifies the homozygous IR64 background across all lines.

The graphical genotypes for a set of 105 CSSLs with donor segments from O. rufipogon (IRGC105491) in the background of Zhenshan 97B, an indica hybrid rice maintainer line. The CSSLs are arranged vertically in order of their chromosome substitution segments, with purple and yellow indicating homozygous and heterozygous wild alleles, respectively. Genotypes were created in GGT (van Berloo 2008). The consensus genotype identifies the homozygous Zhenshen 97B background across all CSSLs.
A useful tool for developing rice CSSLs is the rice universal core genetic map (Orjuela et al. 2010). This map can easily be used to select polymorphic SSR markers between the parents in a specific cross or crosses, and provide a platform for making comparisons across a wide range of maps derived from crosses involving parents belonging to diverse groups. Markers on the universal core map of rice provide a set of uniformly distributed polymorphisms that readily distinguish cultivars and wild accessions, especially within the AA genome.
Use of 384-SNP assays substantially increases the efficiency of genotyping the backcross lines (Tung et al. 2010). Using multiple 384-SNP assays, a large number of SNP markers can be employed for genotyping which will allow more precise identification of the genome-wide introgression lines. The SNP-linked genes/QTLs on the introgressed segments can also be mapped and cloned with relative ease, due to the small size of the substituted chromosome segments in the CSSLs, and the known physical locations of SNPs (Zhu et al. 2009). Even larger numbers of polymorphic SNP markers with uniform distribution over the 12 rice chromosomes can be chosen from the newly constructed high density 44K-SNP array or 1 million SNP array (a future endeavor) after genotyping the parental lines (Tung et al. 2010). Using saturated maps of SNPs would help in identifying the double recombination events that cannot be revealed by lower density maps. Identification of these double recombination events is critical as they can result in false interpretation of the results.
Indica–japonica inter-subspecific CSSLs
In addition to being an effective tool for introgressing valuable genes from wild species to cultivated rice and subsequently transferring genes to commercial cultivars, CSSLs have been used to introgress genes from one subspecies to another in rice, especially between the varietal groups, indica into japonica or japonica into indica. Because of the incompatibility barriers encountered, some researchers have chosen to develop CSSLs to introgress chromosome segments containing genes of interest from either indica or japonica cultivars into the desired genetic background (Harushima et al. 2002; Oka 1988; Sano 1993) even though limited success was achieved because of high sterility or hybrid breakdown (Miura et al. 2008; Win et al. 2009). Reported successful intra-specific CSSLs include ‘IR24’, an indica donor parent, introgressed into ‘Asominori’, a japonica recurrent parent, in which alleles for some important quantitative traits were mapped including eating quality (Wan et al. 2004), grain dimension and chalkiness (Wan et al. 2005; Wang et al. 2007), grain length (Wan et al. 2006), and the sterility gene, S31 (Zhao et al. 2007). Similarly, CSSLs with donor segments from Kasalath, an Indica landrace from India, in the genetic background of the Japonica cultivar, Koshihikari, have been used to introgress and map QTLs affecting heading date (Ebitani et al. 2005), cadmium concentration in brown rice (Ishikawa et al. 2005), increased panicle number per plant, increased grain number per panicle, and increased root mass (Maduoka et al. 2008), among others. Other examples of CSSLs containing donor segments from an indica cultivar in a japonica genetic background include: Koshihikari (japonica recurrent parent) × Nona Bokra (indica donor parent) developed to map QTLs for salt tolerance and heading date (Takai et al. 2007), and Sasanishiki (japonica, recurrent parent) × Habataki (high yielding indica donor) which was used to dissect and map QTLs for panicle architecture (Ando et al. 2008).
With similar objectives as described above, CSSLs have also been developed in the background of an indica cultivar with donor segments from japonica varieties. These populations include HJX74 (indica, recurrent parent) × four japonica donors (Suyunuo, IRAT261, Lemont, and JAPAR9) developed to map QTLs for different traits including days to heading and grain length (Xi et al. 2006); Zhenshen 97B (indica recurrent parent) × Nipponbare (japonica donor) to map QTL alleles enhancing the culturability of indica rice (Zhao et al. 2009); and ‘93–11’ (indica recurrent parent) × Nipponbare (Japonica donor) to detect QTLs for panicle number per plant and grain yield under low nitrogen and phosphorus conditions (Wang et al. 2009) and seed shattering, grain length, and grain width (Zhu et al. 2009).
Backcross inbred lines, an alternative tool to CSSLs for gene introgression
Like CSSLs, populations of BILs can be used to introgress useful genes from wild species into the genetic background of cultivated rice. Wehrhahn and Allard (1965) demonstrated the effects of individual QTLs in wheat (Tritcum aestivum) that can be measured by using backcross inbred lines. BILs are characterized by a low proportion of donor parent in each line and they are well suited for mapping inter-specific variation. Since each line carries only a small fraction of the wild species genome, most of the fertility problems can be eliminated and yield-associated traits can be measured (Eshed and Zamir 1995). BIL development, like CSSL development, involves crossing the wild species accession containing genes of interest to a cultivated parent. The F1 hybrid is backcrossed with the cultivated parent and the BC1F1 plants (50–75 founder lines) produced are backcrossed with the cultivated parent to generate BC2F1 plants. The BC2F1 plants representing all the founder lines are generally used for developing BILs. A single BC2F1 seed selected from each of the approximately 200 lines composing the population is selfed to produce BC2F2 plants. If sterility is a major problem, these lines may need to be backcrossed an additional generation, to the BC3F1 before being selfed. Following the single-seed descent method, the BC2:3F2 plants are advanced to BC2:3F6 to create BILs. At this stage, these BILs are nearly homozygous and may be genotyped with molecular markers. Based on genotype, populations of approximately 100 BILs containing one or more introgressed wild segments may be selected to represent the genome of the wild species donor. If any regions of the genome are not represented in the population, additional backcrosses can be made to recover the missing wild segment(s) in the cultivated parent background. A set of ∼100 BILs comprise a library of BILs, similar in nature to the aforementioned CSSLs, but each line often has several homozygous donor segments randomly distributed in the genetic background of the cultivated (recurrent) parent, as compared to the CSSLs, where each line generally carries a single, defined introgression segment from the divergent (wild) donor genome. The BIL libraries are not developed with the concept of whole donor genome coverage unlike CSSL libraries which are basically designed to recover the whole donor genome. Ideally, CSSLs are a collection of NILs because each CSSL carries a single donor segment in the near-isogenic background of the recurrent parent genotype (Yamamoto and Yano 2008). But in reality, in CSSL populations generated after three or four rounds of backcrossing, some individual lines still may contain more than the one targeted segment (Fig. 4). In this case, through additional backcrossing of these individuals, lines with purely a single segment introgression could be generated in order to obtain a final set of single introgressions, thus these could also be referred to as NILs.
For the purposes of genetic analysis, the advantages of using BIL or CSSL populations include (1) the presence of a high genetic and morphological similarity between lines that enables more precise estimates of quantitative traits and (2) the opportunity to study QTL × environment interactions more accurately because lines are homozygous, making it possible to evaluate phenotypic variation collected from multiple replications across years and environments (Jeuken and Lindhaut 2004). A practical advantage of BILs or CSSLs for commercial breeding purposes is the low percentage of the wild species parent genome, which allows the transfer of an interesting trait into a commercial cultivar to be relatively straightforward and rapid. As with CSSLs, the development of a complete set of BILs is labor- and time-intensive, and the marker evaluation is relatively expensive. For genetic studies involving epistasis, individual BILs or CSSLs may be crossed to identify interacting loci.
There are a number of reports of using BILs to introgress QTLs from the wild rice species into cultivated rice, some of which are: alleles from O. rufipogon associated with the increased number of grains (Tian et al. 2006) and improved drought tolerance (Zhang et al. 2006a, b) in the background of the indica cultivar Guichao 2, alleles associated with increased yield and its components in the background of the indica cultivar IR64 (Cheema et al. 2008), alleles associated with early flowering (Maas et al., 2010) in the background of the tropical japonica cultivar Jefferson, alleles from O. glumaepatula increased yield in the background of indica cultivar BG90-2 (Rangel et al. 2008), and alleles from O. glaberrima improved grain size in the background of indica male sterile line V20A (Li et al. 2004; Table 2). There are also reports of introgression between the subspecies, indica and japonica, such as alleles from the indica donor parent ‘Kasalath’ increased tiller number and salt tolerance (Takehisa et al. 2004) and low temperature germination ability (Miura et al. 2001) in the background of the temperate japonica cultivar ‘Nipponbare’.
CSSL libraries in rice currently under development
Currently, over two dozen CSSL or BIL libraries in rice are being developed at different research institutions around the world (Table 2). As a collaboration between the University of Arizona, United States Department of Agriculture-Agricultural Research Service (USDA-ARS), and the University of Arkansas, it is anticipated that four BIL libraries will be developed using both O. glaberrima and O. barthii donors and two US rice cultivars, LaGrue, a long-grain tropical japonica adapted to the southern region of the USA and M-202, a medium grain temperate japonica adapted to California, as recurrent parents (Table 2; Eizenga and Sanchez, personal communication). At USDA-ARS, Stuttgart, AR, under the RiceCAP program, three BILs libraries are being developed using O. nivara and O. meridionalis as donors and two US tropical japonica cultivars, Bengal and Lemont as recipient parents with a view to map sheath blight and blast resistance genes (Table 2; Eizenga, personal communication).
At Huazhong Agricultural University in China, backcrossing programs are underway to develop 14 CSSL/IL libraries using seven AA genome wild Oryza species accessions that have BAC-end sequence as part of the Oryza Map Alignment project (Wing et al. 2007) as donors and two indica cultivars as recurrent parents, Zhenshan 97B, a hybrid rice maintainer line, and 93–11, a restorer line sequenced by the Beijing Genomics Institute (Table 2; Yu, personal communication). The donor accessions belong to the AA-genome wild species, O. barthii, O. glumaepatula, O. meridionalis, O. nivara, and O. rufipogon. The objective of generating these introgression lines is to develop a resource platform for identification of QTLs and gene discovery in these wild species which have the potential to improve the performance of cultivated rice. Under this program, a set of 105 CSSLs with donor segments from O. rufipogon (IRGC105491) in the background of Zhenshan 97B has been developed (Fig. 4). A companion mapping population (BC2F5) derived from the same cross combination was also developed and evaluated in the field. More than 50% of the alleles derived from the wild parent contributed to the increase of yield-related traits, such as spikelet number per panicle, grain weight, and panicle length, confirming earlier reports using the same donor in five different recurrent parent backgrounds (McCouch et al. 2007). In addition, a set of ILs (BC3F4) carrying donor segments from O. glaberrima in the genetic background of Zhenshen 97B are being developed with the objective of improving drought tolerance and grain quality.
The International Center for Tropical Agriculture and Institut de Recherche pour le Développement (CIAT/IRD) rice genetics and genomics group lead a Generation Challenge Project (GCP) that is developing four libraries of CSSLs with the wild species, O. barthii, O. glumaepatula, O. meridionalis, and O. rufipogon as donors, all sharing the same genetic background of the tropical japonica cultivar Curinga. The GCP-associated partners with this effort are Cornell University (USA), Fedearroz (Colombia), Embrapa-CNPAF (Brazil), and AfricaRice (Benin; see http://www.generationcp.org/arm/ARM06/day_2/Lorieux_part_1.pdf; http://www.generationcp.org/arm/ARM06/day_2/Lorieux_part_2.pdf; Lorieux, personnel communication). Development of introgresion lines from the O. sativa × O. glumaepatula interspecific cross (Rangel et al. 2008) was also undertaken in the GCP initiative.
Six CSSL libraries are being constructed through collaboration between Cornell University, USDA-ARS Stuttgart, Arkansas and the University of Arkansas (Tung et al. 2010) using three diverse O. rufipogon/O. nivara accessions as donors and two O. sativa recurrent parents, IR64, an indica developed by the International Rice Research Institute (IRRI) in the Philippines, and Cybonnet, a long-grain tropical japonica cultivar adapted to the southern USA (Table 2).
As these introgression lines are developed, the CSSL libraries will be evaluated under field and/or greenhouse conditions for different agronomic and quality parameters. This was recently illustrated by Gutiérrez et al. (2010) where the CSSL library of MG12, an O. glaberrima accession, in the background of the tropical japonica cultivar, Caiapo, was used to identify QTLs for rice stripe necrosis virus resistance and yield components. Selected introgression lines will be used as breeding lines to transfer various traits of interest into the new cultivars, for cloning genes, and for functional genomics research.
Potential of CSSLs for allele discovery
The development of CSSLs in O. sativa represents a powerful approach for broadening the genetic variation in the cultivated rice gene pool and better utilizing the genetic potential of wild rice germplasm resources. CSSLs contain marker-defined chromosome segment(s) from agriculturally unadapted donor plants in the background of an adapted cultivar. A population of CSSLs ideally covers the entire donor genome and these lines have been referred to as introgression lines (Eshed and Zamir 1995), contig lines (Ghesquiere et al. 1997), NILs (Tanksley and McCouch 1997) and are collectively referred to as an exotic library (Zamir 2001). CSSL development involves a series of backcrosses to the recurrent (recipient) parent until the recurrent parent’s genome is fully recovered and provides a method of broadening the genetic variation of plant materials for plant breeding (Tanksley and McCouch 1997; Zamir 2001).
CSSLs are also effective tools for the detection of genes and QTLs controlling various traits from wild or unadapted germplasm. As differences between CSSLs and their parental lines must be due to the genes and QTLs located in the introgressed regions (Aida et al. 1998; Eshed and Zamir 1994), CSSLs are considered excellent starting materials for dissecting complex traits into a set of monogenic loci (Zhu et al. 2009). Through additional backcrossing and selfing of CSSLs, NILs can be generated for fine mapping genes, as well as, for precise estimation of the effect of each gene after testing in multiple environments.
In tomato, CSSLs covering the entire genome were used to identify alleles from a wild species that were associated with the increased total soluble solids and green fruit weight (Eshed and Zamir 1995). Similarly, Knorff et al. (2004) reported using introgression lines to identify an allele originating from an exotic barley accession that was associated with early flowering in cultivated barley (Hordeum vulgare). At Kyushu University in Japan, a series of CSSLs in rice were developed using accessions of O. glaberrima and several wild rice species (O. glumaepatula, O. meridionalis, O. nivara, O. rufipogon) as donor parents in the background of O. sativa cv. Taichung 65 (Doi et al. 1997; Kurakazu et al. 2001; Sobrizal and Yoshimura 2002; Matsushita et al. 2003) with the objective of uncovering new, naturally occurring quantitative trait variation. Using these introgression lines, alleles associated with a number of desirable traits were identified, such as awn character (Matsushita et al. 2003), seed shattering (Sanchez et al. 2001), blast resistance (Jeung et al. 2003), green leafhopper resistance (Fujita et al. 2004), bacterial blight resistance (Ram et al. 2010a), and brown planthopper resistance (Deen et al. 2010). At CIAT, using introgression lines of O. glaberrima cv. MG12 (IRGC103544) in the background of the O. sativa tropical japonica cultivar Caiapo, a rice stripe necrosis virus resistance locus was identified (Table 2; Gutiérrez et al. 2010).
Backcross introgression lines are often the only type of segregating population that can be derived between cultivated and wild species. This is because a strong sterility barrier often separates the species and does not allow one to derive classical segregating populations such as F2, BC, DH (doubled haploid), and RILs (recombinant inbred lines) from the interspecific F1 hybrids. Although sterility prevents developing populations through selfing, the backcrossing used to develop CSSLs and other kinds of ILs enables the recovery of fertility.
Most agronomic traits of interest to breeders are genetically complex, governed by multiple genes or QTLs that interact with each other, as well as the environment. In the genetic analysis of such quantitative traits, CSSLs are a powerful tool as they can be used to systematically detect QTLs with small additive effects that are masked by QTLs with larger effects in primary mapping populations, such as F2 populations or RILs (Ebitani et al. 2005; Keurentjes et al. 2007). Because QTL analysis is based on a statistical calculation, it is difficult to identify the precise location of an individual QTL, even using a large population and saturating the QTL region with many markers. On the contrary, NILs, which carry only one target QTL in a unique genetic background, facilitate the dissection, cloning, and comprehensive analysis of target QTLs. CSSLs, which represent a genome-wide collection of NILs, are very useful as precursors for the fine mapping of QTLs.
CSSLs as genetic stocks for functional genomics research
The most common types of genetic stocks for functional genomics studies include genome-wide insertion and deletion mutants which have been developed in rice by several groups (Guiderdoni et al. 2003; Leung et al. 2003; Upadhyaya et al. 2003). The major limitation of using mutants for functional genomics research is the absence of detectable (visible) phenotypes for most mutants even when the disrupted gene(s) in a mutant line is known. Also, it remains an unanswered question as to whether the mutant stocks with single gene mutations are suitable for functional genomics studies of complex phenotypes, which are controlled by many genes and influenced by the environment (Li et al. 2005). Most rice traits, such as grain yield, resistance to abiotic stresses, and quality parameters are quantitative in nature. Understanding the genetic basis of complex phenotypes in rice is a major challenge. Over the past two decades, researchers worldwide have dissected quantitative trait variation in rice and other crops by QTL mapping and thousands of QTLs influencing the quantitative traits have been identified (Ni et al. 2009). There are many QTLs affecting specific quantitative traits and they are widely distributed in the rice genome but a few QTLs are detectable in a single mapping population and environment (Li et al. 2005). With the completion of the rice genome sequence, QTL cloning, while still time consuming and labor intensive, has become increasingly routine in rice where genes underlying QTL are isolated based on CSSLs/NILs and characterized on a regular basis (Shomura et al. 2008; Wei et al. 2010; Zhang et al. 2006a, b).
Although artificially induced variation, such as the mutants mentioned earlier, have been used historically for genetic studies in plant species, the development of new, cost-effective genomics, and re-sequencing technologies have made it possible to directly investigate the functional significance of natural variation in plants (Gilad et al. 2009; Morozova and Marra 2008; Mitchell-Olds and Schmitt 2006). In combination with these new genomic technologies, the availability of genome-wide libraries of introgression lines (i.e., CSSLs) represent particularly useful resources for assessing the phenotypic consequences associated with diverse donor alleles in specific genetic backgrounds, and subsequently for map-based cloning of genes (Ahn et al. 2002a, b; Doi et al. 2002; Koornneef et al. 2004; Sobrizal et al. 1996). Because the CSSLs could be used to dissect complex quantitative traits into a set of monogenic loci and to assign phenotypic values to different alleles at the locus of interest (Xi et al. 2006), they offer biologists access to essential tools that can be used to determine the genetic and functional basis of trait(s) down to the base pair level.
Application of CSSLs or introgression lines to breeding schemes
When CSSL libraries are generated in the genetic background of an adapted, elite cultivar, it provides plant breeders with an opportunity to select superior introgression lines possessing particular traits of interest. The best performing introgression lines may be recommended as new cultivars or used as parents in a breeding program to transfer the introgressed segments with superior performing genes into new cultivars under development. At IRRI, genes for resistance to brown planthopper, bacterial blight, blast, grassy stunt virus and tungro virus, and genes for tolerance to acid soil and cytoplasmic male sterility have been introgressed from different AA genome Oryza species, as well as, from distantly related genomes of wild species into elite breeding lines (Brar and Khush 2002; Multani et al. 2003). In some instances, selected lines carrying the introgressed genes have been released as commercial cultivars. Also, in some cases favorable alleles for multiple traits were detected in the same chromosome regions, thus CSSLs with these chromosome segments are potentially useful breeding materials for simultaneously improving multiple traits.
After the discovery of interesting traits in introgression lines (ILs), desirable QTLs can be combined through crossing of IL-QTLs to pyramid into a common genetic background with the aid of molecular markers (Ashikari and Matsuoka 2006; Xi et al. 2006). The validity of this approach has been demonstrated by pyramiding two QTLs involved in plant height and grain number (Ashikari et al. 2005). QTL pyramiding into the Koshihikari (temperate japonica cultivar) genetic background showed increased grain production (+23%) and reduced plant height (−20%) when compared to Koshihikari without the introgression from Kasalath, the indica donor parent. This illustrates that breeders can produce new cultivars with interesting traits by combining introgression lines carrying the desired QTLs using marker assisted selection.
Summary and conclusion
There is no doubt that wild Oryza species are reservoirs of genes that confer many valuable qualitative and quantitative traits which, if transferred to the cultivated rice, would broaden the gene pool and improve productivity. There are thousands of wild species accessions collected from a range of geographic, climatic, and ecological conditions. To date, this wealth of germplasm is largely untapped owing to the difficulty of identifying agronomically important genes and transferring these genes into cultivated rice without any linkage drag or deleterious traits. CSSLs and BILs have proven to be very useful tools for identifying and transferring desirable genes from wild relatives into the genetic background of adapted cultivars. These populations are excellent materials for dissecting complex quantitative traits into the component variation as a set of monogenic loci, thus providing a platform for discovery of new QTL alleles. Individual CSSLs with one or more introgressed QTLs in the background of elite rice cultivars could be used as parents to transfer genes of interest into new commercial cultivars or could be used directly as commercial cultivars. Many introgressed genes or QTL alleles in the genetic background of an adapted rice cultivar show improved performance over the initial recurrent parent. Introgression lines with genes (QTLs) of interest can be readily used to pyramid these genes and create transgressive variants, thus improving multiple traits in a single breeding line.
Individual introgression lines having desirable trait variation can be used for fine mapping of QTLs with the ultimate aim of cloning the gene(s) and characterizing its function. Although about two dozen of the CSSL/BIL libraries have been developed in rice, representing several different O. sativa backgrounds and wild donors, the range of natural variation represented by the wild gene pool remains largely unexplored. A collaborative effort to evaluate these resources in different environments would allow the rice community to gain valuable insights into the breeding value of alleles from diverse wild species and to understand how diverse wild introgressions affect phenotypic performance across different elite genetic backgrounds. As pre-breeding material, the CSSLs represent a valuable resource for broadening the genetic base of high yielding rice cultivars around the world, and provide new sources of resistance to biotic or abiotic stresses. It is interesting to note that introgressions from wild species often carry ‘gain of function’ alleles in cultivated backgrounds. Thus, RNAi technology could be effectively used to knockout candidate genes underlying QTLs in selected lines to validate their function.
Most of the donors and recurrent parents involved in current CSSL and BIL development efforts have been re-sequenced using second-generation sequencing technology (McCouch SR, Cornell University, personnel communication and Wing RA, University of Arizona, personnel communication) and/or genotyped using the 44,100 SNP array described by Tung et al. (2010). Therefore, a large number of well-distributed polymorphic SNP markers can be readily selected for genotyping assays and used to develop smaller, 384-SNP assays that can be economically used to genotype the backcross progenies, allowing rapid selection of lines to advance during the backcrossing process. High-throughput SNP assays will enhance the efficiency of the CSSL development process, and allow for the selection of introgression lines with smaller marker-defined segments for genome-wide coverage in the CSSL library. SNP markers across the introgressed donor regions will facilitate rapid fine mapping and cloning of genes underlying target QTLs, aided by the availability of physical maps of 17 Oryza species (representing the 10 genome types) developed by the Oryza Map Alignment Project (Wing et al. 2007; Kim et al. 2008; http://www.omap.org). In an era where sequencing technology has essentially solved the problem of high throughput genotyping, the value of genetic resources, such as CSSLs is underscored because they link natural variation with breeding potential, promote more effective use of the wild gene pool link and simultaneously help geneticists explore the fundamental relationship between sequence variation and phenotypic diversity. Thus, the public availability of introgression libraries enhances the value of the genetic resources available in our in situ and ex situ germplasm collections and leads to more efficient development of improved rice varieties for agricultural producers and consumers around the world.
References
Ahn SN, Kwon SJ, Suh JP, Kang KH, Kim HJ, Hwang HG, et al. Introgression of Oryza grandiglumis chromatin into rice affecting plant height and grain length. Rice Genet Newsl. 2002a;19:12.
Ahn SN, Suh JP, Oh CS, Lee SJ, Suh HS. Development of introgression lines of weedy rice in the background of Tongil-type rice. Rice Genet Newsl. 2002b;19:14.
Aida Y, Tsunematsu H, Doi K, Yoshimura A. Development of a series of introgression lines of japonica in the background of indica rice. Rice Genet Newsl. 1998;14:41–2.
Ali ML, McClung AM, Jia MH, Kimball JA, McCouch SR, Eizenga GC. A “Rice Diversity Panel” evaluated for genetic and agro-morphological diversity between subpopulations and its geographic distribution. Crop Sci. (2011; in review).
Amante-Bordeos A, Sitch LA, Nelson R, Dalmaeio RD, Oliva NP, Aswidinnoor H, et al. Transfer of bacterial blight and blast resistance from the tetraploid wild rice Oryza minuta to cultivated rice, Oryza sativa. Theor Appl Genet. 1992;84:345–54.
Ando T, Yamamoto T, Shimizu T, Ma XF, Shomura A, Takeuchi Y, et al. Genetic dissection and pyramiding of quantitative traits for panicle architecture by using chromosome segment substitution lines in rice. Theor Appl Genet. 2008;116:881–90.
Angeles-Shim RB, Asano K, Takashi T, Kitano H, Ashikari M. Mapping of the glabrous gene in rice using CSSLs derived from the cross Oryza sativa subsp. Japonica cv. Koshihikari × O. glaberrima. In: Proc 6th Int Rice Genet Symp, Manila, Philippines, 16-19 Nov; 2009. [CD-ROM].
Ashikari M, Matsuoka M. Identification, isolation and pyramiding of quantitative trait loci for rice breeding. Trends Plant Sci. 2006;11:344–50.
Ashikari M et al. Cytokinin oxidase regulates rice grain production. Science. 2005;309:741–5.
Brar DS, Khush GS. Wide hybridization and chromosome manipulation in cereals. In: Evans DH, Sharp WR, Amirato PV, editors. Handbook of plant cell culture, vol. 4. Techniques and applications. New York: MacMillan Publ; 1986. p. 221–63.
Brar DS, Khush GS. Alien gene introgression in rice. Plant Mol Biol. 1997;35:35–47.
Brar DS, Khush GS. Transferring genes from wild species into rice. In: Kang MS, editor. Quanttitative genetics, genomics and plant breeding. Wallingford (UK): CAB International; 2002. p. 197–217.
Brar DS, Khush GS. Utilization of wild species of genus Oryza in rice improvement. In: Nanda JS, Sharma SD, editors. Monograph on genus Oryza. Enfield: Science Publ; 2003. p. 283–309.
Brar DS, Khush GS. Cytogenetic manipulation and germplasm enhancement of rice. In: Singh RJ, Jauhar PP, editors. Genetic resources, chromosome engineering and crop improvement. Boca Raton: CRC Press; 2006. p. 115–58.
Cheema KK, Bains NS, Mangat GS, Das A, Vikal Y, Brar DS, et al. Development of high yielding IR64 × Oryza rufipogon (Griff.) introgression lines and identification of introgressed alien chromosome segments using markers. Euphytica. 2008;160:401–9.
Chen J, Bughio HUR, Chen D-Z, Liu G-J, Zheng K-L. Development of chromosomal segment substitution lines from a backcross recombinant inbred population of interspecific rice cross. Rice Sci. 2006;13:15–21.
Chen X-R, Yang K-S, Fu J-R, Zhu C-I, Peng X-S, He X-P. Identification and genetic analysis of fertility restoration ability in Dongxiang wild rice (Oryza rufipogon). Rice Sci. 2008;15:21–8.
Chen Z, Hu F, Xu P, Li J, Deng X, Zhou J, et al. QTL analysis for hybrid sterility and plant height in interspecific populations derived from a wild rice relative, O. longistaminata. Breed Sci. 2009;59:441–5.
Chen J, Huang D-R, Wang L, Liu GJ, Zhuang JY. Identificatiion of quantitative trait loci for resistance to whitebacked planthopper, Sogatella furcifera, from an interspecific cross Oryza sativa × O. rufipogon. Breed Sci. 2010;60:153–9.
Chaudhary RC, Khush GS. Breeding rice varieties for resistance against Chilo spp. of stem borers in Asia and Africa. Insect Sci Appl. 1990;11:659–69.
Chu YE. Genetic basis of crossing barriers between Oryza-perennis subsp. barthii and its related taxa. Evolution. 1970;24:135–44.
Chu YE, Oka HI. Introgression across isolating barriers in wild and cultivated Oryza species. Evolution. 1970;24:344–55.
Dalmacio RD, Brar DS, Ishii T, Sitch TA, Virmani SS, Khush GS. Identification and transfer of a new cytoplasmic male sterility source from Oryza perennis into indica rice (Oryza sativa). Euphytica. 1995;82:221–5.
Deen R, Ramesh K, Gautam SK, Rao YK, Lakshmi VJ, Viraktamath BC, et al. Identification of new gene for BPH resistance introgressed form O. rufipogon. Rice Genet Newsl. 2010;25:70.
Devadath S. A strain of Oryza barthii, an African wild rice immune to bacterial blight of rice. Curr Sci India. 1983;52:27–8.
Doi K, Iwata N, Yoshimura A. The construction of chromosome substitution lines of African rice (Oryza glaberrima Steud.) in the background of japonica rice (O. sativa L.). Rice Genet Newsl. 1997;14:39–41.
Doi K, Sobrizal, Ikeda K, Sanchez PL, Kurakazu T, Nagai Y, Yoshimura A. Developing and evaluating rice chromosome segment substitution lines. In: Proc Int Rice Res Conf, Beijing, China, 16–19 Sept; 2002. p. 289–296.
Ebitani T, Takeuchi Y, Nonoue Y, Yamamoto T, Takeuchi K, Yano M. Construction and evaluation of chromosome segment substitution lines carrying overlapping chromosome segments of indica rice cultivar ‘Kasalath’ in a genetic background of japonica elite cultivar ‘Koshihikari’. Breed Sci. 2005;55:65–73.
Eshed Y, Zamir D. Introgression from Lycopersicon pennellii can improve the soluble solids yield of tomato hybrids. Theor Appl Genet. 1994;88:891–7.
Eshed Y, Zamir D. An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics. 1995;141:1147–62.
Frey KJ, Cox TS, Rodgers DM, Bramel-Cox P. Increasing cereal yields with genes from wild and weedy species. In: Genetics, new frontiers. Chopra VL et al. editors. Proc XV Int Congr Genet, vol. IV. 1984. p. 51–68.
Fu X-L, Lu Y-G, Liu X-D, Li JQ. Progress on transfering elite genes from non-AA genome wild rice into Oryza sativa through interspecific hybridization. Rice Sci. 2008;15:79–87.
Fu Q, Zhang P, Tan L, Zhu Z, Ma D, Fu Y, et al. Analysis of QTLs for yield-related traits in Yuanjiang common wild rice (Oryza rufipogon Griff.). J Genet Genomics. 2010;37:147–57.
Fujita D, Doi K, Yoshimura A, Yasui H. Mapping new resistance gene for green rice leafhopper introgressed from Oryza rufipogon Griff. into cultivated rice, Oryza sativa L. Rice Genet Newsl. 2003;20:79.
Fujita D, Doi K, Yoshimura A, Yasui H. Introgression of a resistance gene for green leafhopper from Oryza nivara into cultivated rice, Oryza sativa L. Rice Genet Newsl. 2004;21:64.
Furuya A, Itoh R, Ishii R. Mechanisms of different responses of leaf photosynthesis in African rice (Oryza glaberrima Steud.) and rice (Oryza sativa L) to low leaf water potential. Jpn J Crop Sci. 1994;63:625–31.
Ghesquiere A, Sequier J, Second G, Lorieux M. First steps toward a rational use of African rice, Oryza glaberrima, in rice breeding: a contig line concept. Euphytica. 1997;96:31–9.
Gilad Y, Pritchard JK, Thornton K. Characterizing natural variation using next-generation sequencing technologies. Trends Genet. 2009;25:463–71.
Guiderdoni E, Piffanelli P, Sallaud C, Gay C, Larmande P, et al. Generation, characterization, and evaluation of the Genoplante rice insertion line library. Proc 1st Int Symp Rice Functional Genomics, Shangai, China. 19–21 Nov. 2003. p. 16.
Gutiérrez AG, Carabalí SJ, Giraldo OX, Martínez CP, Correa F, Prado G, et al. Identification of a rice stripe necrosis virus resistance locus and yield component QTLs using Oryza sativa × O. glaberrima introgression lines. BMC Plant Biol. 2010;10(1):6.
Harushima Y, Nakagahra M, Yano M, Sasaki T, Kurata N. Diverse variation of reproductive barriers in three intraspecific rice crosses. Genetics. 2002;160:312–22.
Heinrichs EA, Viajante VD, Romena AM. Resistance of wild rices, Oryza spp. to the whorl maggot Hydrellia-Philippina Ferino (Diptera, Ephydridae). Environ Entomol. 1985;14:404–7.
Heuer S, Miezan KM. Assessing hybrid sterility in Oryza glaberrima × O. sativa hybrid progenies by PCR marker analysis and crossing with wide compatibility varieties. Theor Appl Genet. 2003;107:902–9.
Hoan NT, Sarma NP, Siddiq EA. Identification and characterization of new sources of cytoplasmic male sterility in rice. Plant Breed. 1997;116:547–51.
Ishikawa S, Ae N, Yano M. Chromosomal regions with quantitative trait loci controlling cadmium concentration in brown rice (Oryza sativa). New Phytol. 2005;168:345–50.
Jena KK, Khush GS. Exploitation of species in rice improvement: opportunities, achievements and future challenges. In: Nanda JS, editor. Rice breeding and genetic: research priorities and challenges. Enfield: Science Publ; 2000. p. 269–84.
Jeung JU, Han SS, Cho YC, Hwang HG, Choi HC, Moon HP, et al. Identification of a new source of resistance to blast isolates of Korea in an alien introgression line of rice. Rice Genet Newsl. 2003;20:92.
Jeuken MJW, Lindhaut P. The development of lettuce backcross inbred lines (BILs) for exploitation of the Lactuca salina (wild lettuce) germplasm. Theor Appl Genet. 2004;109:394–401.
Jin FX, Kwon SJ, Kang KH, Jeong OY, Le LH, Yon DB, et al. Introgression for grain traits from Oryza sativa. Rice Genet Newsl. 2004;21:15.
Jungtsung W, Heinrichs EA, Medrano FG. Resistance of wild rices, Oryza spp. to the brown planthopper, Nilaparvata-Lugens (Homoptera, Delphacidae). Environ Entomol. 1986;15:648–53.
Kang JW, Suh JP, Kim DM, Oh CS, Oh JM, Ahn SN. QTL Mapping of agronomic traits in an advanced backcross population from a cross between Oryza sativa L. cv. Milyang 23 and O. glaberrima. Korean J Breed Sci. 2008;40:243–9.
Keurentjes JJB, Bentsink L, Alonso-Blanco C, Hanhart CJ, Blankestijn-DeVries H, Effgen S, et al. Development of a near isogenic line population of Arabidopsis thaliana and comparison of mapping power with a recombinant inbred line population. Genetics. 2007;175:891–905.
Khush GS. What will it take to feed 5.0 million rice consumers in 2030. Plant Mol Biol. 2005;59:1–6.
Khush GS, Brar DS. Biotechnology for rice breeding: progress and potential impact. In: Int Rice Commission, XX Session (Bangkok, Thailand: FAO). 2002.
Khush GS, Ling KC, Aquino RC, Aguiero VM. Breeding for resistance to grassy stunt in rice. Proc 3rd Int Congr Soc Adv Breed Res Asia and Oceania (SABRAO). Plant Breed Papers. 1977;1(4b):3–9.
Khush GS, Bacalangco E, Ogawa T. A new gene for resistance to bacterial blight from O. longistaminata. Rice Genet Newsl. 1990;7:121–2.
Kim H, Hurwitz B, Yu Y, Collura K, Gill N, SanMiguel P, et al. Construction, alignment and analysis of twelve framework physical maps that represent the ten genome types of the genus Oryza. Genome Biol. 2008;9:R45.
Kimball JA, Moon S, McCouch SR. McClung AM. Oryza rufipogon introgressions improve yield in the U.S. cultivar Jefferson. In: Proc 6th Int Rice Genet Symp, Manila, Philippines. 16–19 Nov. 2009. [CD-ROM].
Knorff MV, Wang H, Leon J, Pillen K. Development of candidate introgression lines using an exotic barley accession (Hordeum vulgare ssp. spontaneum) as donor. Theor Appl Genet. 2004;109:1736–45.
Kobayashi N, Ikeda R, Domingo IT, Vaughan DA. Resistance to infection of rice tungro viruses and vector resistance in wild species of rice (Oryza spp.). Jpn J Breed. 1993;43:377–87.
Kobayashi N, Ikeda R, Vaughan DA. Screening wild-species of rice (Oryza spp.) for resistance to rice tungro disease. Jarq-Jpn Agr Res Q. 1994;28:230–6.
Koornneef M, Alonso-Blanco C, Vreugdenhil D. Naturally occuring genetic variation in Arabidopsis thaliana. Annu Rev Plant Biol. 2004;55:141–72.
Kurakazu T, Sobrizal, Ikeda K, Sanchez PL, Doi K, Angeles ER, et al. Oryza meridionalis chromosomal segment introgression lines in cultivated rice, O. sativa L. Rice Genet Newsl. 2001;18:81–2.
Lawrence PK, Frey KJ. Backcross variability for grain yield in oat species crosses (Avena sativa L. × Avena sterilis L.). Euphytica. 1975;24:77–85.
Leung H, Wu J, Lei C, Wu C, Baraoidan M, et al. Forward and reverse genetics using chemicals and irradiation-induced mutants of Indica rice IR64A. Proc 1st Int Symp Rice Functional Genomics. Shangai, China. 19–21 Nov. 2003. p. 20.
Lee SJ, Suh CS, Oh CS, McCouch SR, Ahn SN. Development of introgression lines in the background of japonica rice. Rice Genet Newsl. 2003;20:105.
Li D, Sun C, Fu Y, Li C, Zhu Z, Chen L, et al. Identification and mapping of genes for improving yield from Chinese common wild rice (O. rufipogon Griff.) using advanced backcross QTL analysis. Chinese Sci Bull. 2002;47:1533–7.
Li J, Xiao J, Grandillo S, Jiang L, Wan Y, Deng Q, et al. QTL detection for rice grain quality traits using an interspecific backcross population derived from cultivated Asian (O. sativa L.) and African (O. glaberrima Steud.) rice. Genome. 2004;47:697–704.
Li Z-K, Fu B-Y, Gao Y-M, Xu J-L, Ali J, Lafitte HR, et al. Genome-wide introgression lines and their use in genetic and molecular dissection of complex phenotypes in rice (Oryza sativa L.). Plant Mol Biol. 2005;59:33–52.
Lin SC, Yuan LP. A mass screeing method for testing grassy stunt disease of rice. Hybrid rice breeding in China. In: Innovative approaches to rice improvement. Int Rice Res Inst, Manila, Philippines.
Linh L-H, Jin F-S, Kang K-H, Lee YT, Kwon S-J, Ahn S-N. Mapping quantitative trait loci for heading date and awn length using an advanced backcross line from a cross between Oryza sativa and O. minuta. Breed Sci. 2006;56:341–9.
Liu G, Lu G, Zeng L, Wang G-L. Two broad-spectrum blast resistance genes, Pi9(t) and Pi2(t), are physically linked on rice chromosome 6. Mol Genet Genomics. 2002;267:472–80.
Lu H, Redus MA, Coburn JR, Rutger JN, McCouch SR, Tai TH. Population structure and breeding patterns of 145 US rice cultivars based on SSR marker analysis. Crop Sci. 2005;45:66–76.
Lu B-R, Jackson M, Vaughn D. Wild rice taxonomy. In: Rice knowledge bank, Int Rice Res Inst, Philippines. 2010. Available from: http://www.knowledgebank.irri.org/extension/index.php/wild-rice-taxonomy. (verified 13 Dec 2010).
Maduoka Y, Kashiwagi T, Hirotsu N, Ishimaru K. Indian rice ‘Kasalath’ contains genes that improve traits of Japanese premium rice ‘Koshihikari’. Theor Appl Genet. 2008;116:603–12.
Maas LF, McClung A, McCouch S. Dissection of a QTL reveals an adaptive, interacting gene complex associated with transgressive variation for flowering time in rice. Theor Appl Genet. 2010;120:895–908.
Matsushita S, Kurakazu T, Sobrizal, Doi K, Yoshimura A. Mapping genes for awn in rice using Oryza meridionalis introgression lines. Rice Genet Newsl. 2003;20:17.
McCouch SR, Sweeney M, Li J, Jiang H, Thomson M, Septiningsih E, et al. Through the genetic bottleneck: O. rufipogon as a source of trait-enhancing alleles for O. sativa. Euphytica. 2007;154:317–39.
Mitchell-Olds T, Schmitt J. Genetic mechanisms and evolutionary significance of natural variation in Arabidopsis. Nature. 2006;441:947–52.
Miura K, Lin SY, Yano M, Nagamine T. Mapping quantitative trait loci controlling low temperature germinability in rice (Oryza sativa L.). Breed Sci. 2001;51:293–9.
Miura K, Yamamota E, Morinaka Y, Takashi T, Kitano H, Matsuoka M, et al. The hybrid breakdown 1(t) locus induces interspecific hybrid breakdown between rice Oryza sativa cv. Koshihikari and its wild relative O. nivara. Breed Sci. 2008;58:99–105.
Morozova O, Marra MA. Application of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255–64.
Multani DS, Khush GS, delos Reyes BG, Brar DS. Alien gene introgression and development of monosomic alien addition line from Oryza latifolia Desv. to rice, Oryza sativa L. Theor Appl Genet. 2003;107:395–405.
Nayar NM. Prevalence of self-incompatibility in Oryza barthii Cheval: its bearing on evolution of rice and related taxa. Genetica. 1968;38:521–7.
Ni J, Colowit PM, Mackill DJ. Evaluation of genetic diversity in rice subspecies using microsatellite markers. Crop Sci. 2002;42:601–7.
Ni J, Pujar A, Youens-Clark K, Yap I, Jaiswal P, Tecle I, Tung CW, Ren L, Spooner W, Wei X, Avraham S, Ware D, Stein L, McCouch SR. Gramene QTL database: Development, content and applications. Database: J Biol Databases and Curation. 2009. doi:10.1093/database/bap005.
Ndjiondjop MM, Fargette D, Fauquet C, Ghesquiere A. The genetic basis of high resistance to rice yellow mottle virus (RYMV) in cultivars of two cultivated rice species. Plant Dis. 1999;83:931–5.
Ndjiondjop MN, Manneh B, Cissoko M, Drame NK, Kakai GK, Bocco R, et al. Drought resistance in an interspecific backcross population of rice (Oryza spp.) derived from the cross WAB56-104 (O. sativa) × CG14 (O. glaberrima). Plant Sci. 2010;179:364–73.
Oka HI. In: Oka HI, editor. Origin of cultivated rice. Tokyo: Elsevier Science/Japan Scientific Societies Press; 1988. p. 254.
Orjuela J, Garavito A, Bouniol M, Arbelaez JD, Moreno L, Kimball J, et al. A universal core genetic map for rice. Theor Appl Genet. 2010;120:563–72.
Prasad B, Eizenga GC. Rice sheath blight disease resistance identified in Oryza spp. accessions. Plant Dis. 2008;92:1503–9.
Rahman ML, Chu SH, Choi MS, Qiao YL, Jiang W, Piao R, et al. Identification of QTLs for some agronomic traits in rice using an introgression line from Oryza minuta. Mol Cells. 2007;24:16–26.
Ram T, Majumder ND, Padmavathi G, Mishra B. Improving rice for broad-spectrum resistance to blast and salinity tolerance by introgressing genes from O. rufipogon. Int Rice Res Notes. 2005;30:17–9.
Ram T, Laha GS, Gautam SK, Deen R, Madhav MS, Brar DS, et al. Identification of new gene introgressed from Oryza brachyantha with broad-spectrum resistance to bacterial blight of rice in India. Rice Genet Newsl. 2010a;25:57.
Ram T, Deen R, Gautam SK, Ramesh K, Rao YK, Brar DS. Identification of new genes for brown planthopper resistance in rice introgressed from O. glaberrima and O. minuta. Rice Genet Newsl. 2010b;25:67.
Rangel PN, Brondani RPV, Rangel PHN, Brondani C. Agronomic and molecular characterization of introgression lines from the interspecific cross Oryza sativa (BG90-2) × Oryza glumaepatula (RS-16). Genet Mol Res. 2008;7:184–95.
Rick CM. High soluble-solids content in large fruited tomato lines derived from a wild green-fruited species. Hilgardia. 1974;42:493.
Sakamoto W, Kadowaki K, Tano S, Yabuno T. Analysis of mitochondrial DNAs from Oryza-glaberrima and its cytoplasmic substituted line for Oryza-sativa associated with cytoplasmic male-sterility. Jpn J Genet. 1990;65:1–6.
Sano Y. Constraints in using wild relatives in breeding: lack of basic knowledge on crop gene pools. In: Buxton DR, Shibles R, Forsberg RA, Blad BL, Asay KH, Paulsen GM, Wilson RF, editors. Int Crop Sci I, vol 1. Madison: CSSA; 1993. p. 437–43.
Sanchez PL, Sobrizal, Ikeda K, Yasui H, Yoshimura A. RFLP mapping of genes controlling heading date found in Oryza glumaepatula Steud. introgression lines in rice. Rice Genet Newsl. 2001;18:57.
Sanchez PL, Kurakazu T, Hirata C, Sobrizal, Yoshimura A. Identification and mapping of seed shattering genes using introgression lines from wild rice species. Rice Genet Newsl. 2002;19:78.
Septiningsih EM, Prasetiyono J, Lubis E, Tai TH, Tjubaryat T, Moeljopawiro S, et al. Identification of quantitative trait loci for yield and yield components in an advanced backcross population derived from the Oryza sativa variety IR64 and the wild relative O. rufipogon. Theor Appl Genet. 2003;107:1419–32.
Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H, Konishi S, et al. Deletion in a gene associated with grain size increased yields during rice domestication. Nat Genet. 2008;40:1023–8.
Sobrizal, Ikeda K, Sanchez PL, Doi K, Angeles ER, Khush GS, et al. Development of Oryza glumaepatula introgression lines in rice, O. sativa L. Rice Genet Newsl. 1996;16:107–8.
Sobrizal, Yoshimura A. Mapping of genes for slender kernel using Oryza glumaepatula introgression lines in rice. Rice Genet Newsl. 2002;19:40.
Stebbins GL. Coevolution of grasses and herbivores. Ann Mo Bot Garden. 1981;68:75–86.
Suh JP, Roh JH, Cho YC, Han SS, Kim YG, Jena KK. The Pi40 gene for durable resistance to rice blast and molecular analysis of Pi40-advanced backcross breeding lines. Phytopath. 2009;99:243–50.
Sun CQ, Wang XK, Li ZC, Yoshimura A, Iwata N. Comparison of the genetic diversity of common wild rice (Oryza rufipogon Griff.) and cultivated rice (O. sativa L.) using RFLP markers. Theor Appl Genet. 2001;102:157–62.
Takai T, Nonoue Y, Yamamoto S, Yamaonuchi U, Matsubara K, Liang ZW, et al. Development of chromosome segment substitution lines derived from backcross between indica donor rice cultivar ‘Nona Bokra’ and japonica recipient cultivar ‘Koshihikari’. Breed Sci. 2007;57(3):257–61.
Takehisa H, Shimodate T, Fukuta Y, Ueda T, Yano M, Yamaya T, et al. Identification of quantitative trait loci for plant growth of rice in paddy field flooded with salt water. Field Crops Res. 2004;89:85–95.
Tan L, Liu F, Xue W, Wang G, Ye S, Zhu Z, et al. Development of Oryza rufipogon and O. sativa introgression lines and assessment for yield-related quantitative trait loci. J Integr Plant Biol. 2007;49:871–84.
Tanksley SD, McCouch SR. Seed maps and molecular maps: unlocking genetic potential from the wild. Science. 1997;277:1063–6.
Thomson MJ, Tai TH, McClung AM, Lai X-H, Hinga ME, Lobos KB, et al. Mapping quantitative trait loci for yield, yield components and morphological traits in an advanced backcross population between Oryza rufipogon and Oryza sativa cultivar Jefferson. Theor Appl Genet. 2003;107:479–93.
Tian F, Li DJ, Fu Q, Zhu ZF, Fu YC, Wang XK, et al. Construction of introgression lines carrying wild rice (Oryza rufipogon Griff.) segments in cultivated rice (Oryza sativa L.) background and characterization of introgressed segments associated with yield-related traits. Theor Appl Genet. 2006;112:570–80.
Tseng ST, Oster JJ. Registration of 87-Y-550, a rice germplasm line resistant to stem rot disease. Crop Sci. 1994;34:314.
Tung C-W, Zhao K, Wright M, Ali ML, Jung J, Kimball J, et al. Development of a research platform for dissecting phenotype-genotype assocations in rice (Oryza spp.). Rice. 2010. doi:10.1007/s12284-010-9056-5.
Upadhyaya NM, Zhu Q, Eamens A, Margis M, and Ramm K, et al. Ac/Ds for insertional mutagenesis in rice. Proc 1st Int Symp Rice Functional Genomics. Shangai, China. 19–21 Nov. 2003. p. 20.
Vales M. Study of complete resistance to Pyricularia-oryzae Cav. of Oryza sativa × Oryza longistaminata hybrids and their Oryza longistaminata parent. Agron Trop. 1985;40:148–56.
van Berloo R. GGT 2.0: versatile software for visualization and analysis of genetic data. J Hered. 2008;99:232–6.
Vaughan DA. The wild relatives of rice: a genetic resources handbook. Los Banos: IRRI; 1994. p. 137.
Vaughan DA, Kadowaki KI, Kaga A, Tomooka N. On the phlylogeny and biogeography of the genus Oryza. Breed Sci. 2005;55:113–22.
Velusamy R, Kumar MG, Edward YSJT. Mechanisms of resistance to the brown planthopper Nilaparvata-Lugens in wild-rice (Oryza spp.) cultivars. Entomol Exp Appl. 1995;74:245–51.
Wan XY, Wan JM, Su CC, Wang CM, Shen WB, Li JM, et al. QTL detection for eating quality of cooked rice in a population of chromosome segment substitution lines. Theor Appl Genet. 2004;110:71–9.
Wan XY, Wan JM, Weng JF, Jiang L, Bi JC, Wang CM, et al. Stability of QTLs for rice grain dimension and endosperm chalkiness charactaeristics across eight environments. Theor Appl Genet. 2005;110:1334–46.
Wan XY, Wan JM, Jiang L, Wang JK, Zhai HQ, Weng JF, et al. QTL analysis for rice grain length and fine mapping of an identified QTL with stable and major effects. Theor Appl Genet. 2006;112:1258–70.
Wang J, Wan X, Li H, Pfeiffer WH, Crouch J, Wan J. Application of identified QTL-marker associations in rice quality improvement through a design-breeding approach. Theor Appl Genet. 2007;115:87–100.
Wang Y, Sun YJ, Chen DY, Yu SB. Analysis of quantitative trait loci in response to nitrogen and phosphorus deficiency in rice using chromosomal segment substitution lines. Acta Agron Sin. 2009;35:580–7.
Wehrhahn C, Allard W. The detection and measurement of the effects of individual genes involved in the inheritance of a quantitative character in wheat. Genetics. 1965;135:1175–86.
Wei X, Junfeng X, Guo H, Jiang L, Chen S, Yu C, et al. DTHS suppress flowering in rice influencing plant height and yield potential simultaneously. Plant Physiol. 2010;153:1747–58.
Win KT, Kubo T, Miyazaki Y, Doi K, Yamagata Y, Yoshimura A. Identification of two loci causing F1 pollen sterility in inter- and intraspecific crosses of rice. Breed Sci. 2009;59:411–8.
Win KT, Yamagata Y, Miyazaki Y, Doi K, Yasui H, Yoshimura A. Independent evolution of a new allele of a F1 pollen sterility gene S27 encoding mitochondrial ribosomal protein L27 in Oryza nivara. Theor Appl Genet. 2010. doi:10.1007/s00122-00-1454-y.
Wing RA, Kim HR, Goicoexhea JL, Yu Y, Kudrna D, Zuccolo A, et al. The Oryza Map Alignment Project (OMAP): a new resource for 700 comparative genome studies within Oryza. In: Upadhyaya NM, editor. Rice functional genomics: challenges, progress and prospects. New York: Springer; 2007. p. 395–409.
Xi ZY, He FH, Zeng RZ, Zhang ZM, Ding XH, Li W-T, et al. Development of wide population of chromosome segment substitution lines in the genetic background of an elite cultivar of rice (Oryza sativa L.). Genome. 2006;49:476–84.
Xiao J, Li J, Grandillo S, Ahn SN, Yuan L, Tanksley SD, et al. Identification of trait-improving quantitative trait loci alleles from a wild rice relative, Oryza rufipogon. Genetics. 1998;150:899–909.
Xie X, Song MH, Jin F, Ahn SN, Suh JP, Hwang H-G, et al. Fine mapping of a grain weight quantitative trait locus on rice chromosome 8 using near-isogenic lines derived from a cross between Oryza sativa and Oryza rufipogon. Theor Appl Genet. 2006;113:885–94.
Yamamoto T, Yano M. Detection and molecular cloning of genes underlying quantitative phenotypic variation in rice. In: Hirano H-Y et al. editors. Rice Biology in the Genomics Era. Biotechnology in Agriculture and Forestry. 2008; 62:295–308.
Zamir D. Improving plant breeding with exotic genetic libraries. Nat Rev Genet. 2001;2:983–9.
Zhang X, Zhou S, Fu Y, Su Z, Wang X, Sun C. Identification of drought tolerant introgression line derived from Dongxiang common wild rice (O. rufipogon Griff.). Plant Mol Biol. 2006a;62:247–59.
Zhang Y, Luo L, Xu C, Zhang Q, Xing Y. Quantitative trait loci for panicle size, heading date and plant height co-segregating in trait-performance derived near isogenic lines of rice (Oryza sativa). Theor Appl Genet. 2006b;113:361–8.
Zhao ZG, Jian L, Zhang WW, Yu CY, Zhu SS, Xie K, et al. Fine mapping of S31, a gene responsible for hybrid embryo-sac abortion in rice (Oryza sativa L.). Planta. 2007;226:1087–96.
Zhao L, Zhou H, Lu L, Liu L, Li X, Lin Y, et al. Identification of quantitative trait loci controlling rice mature seed culturability using chromosomal segment substitution lines. Plant Cell Rep. 2009;28:247–56.
Zhu W, Lin J, Yang D, Zhao L, Zhang Y, Zhu Z, et al. Development of chromosome segment substitution lines derived from backcross between two sequenced rice cultivars, Indica recipient 93–11 and Japonica donor Nipponbare. Plant Mol Biol Rep. 2009;27:126–31.
Acknowledgments
Funding from the National Science Foundation PRGP award 0606461 to G.C. Eizenga is gratefully acknowledged for supporting the postdoctoral fellowship of ML Ali and the CSSL development referenced by Tung et al. (2010). PL Sanchez was supported with funding from the National Science Foundation award 0822284. Grant support from the National Natural Science Foundation of China (30620120431, 30971750) is acknowledged for supporting the CSSLs reported by S-B Yu. The Generation Challenge Program is acknowledged for its support of the cultivated rice × wild Oryza species CSSL development reported by M Lorieux. The authors thank SR McCouch and TH Tai for their critical reviews of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ali, M.L., Sanchez, P.L., Yu, Sb. et al. Chromosome Segment Substitution Lines: A Powerful Tool for the Introgression of Valuable Genes from Oryza Wild Species into Cultivated Rice (O. sativa). Rice 3, 218–234 (2010). https://doi.org/10.1007/s12284-010-9058-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12284-010-9058-3