
Abstract
Background
Acetoin (AC) and 2,3-butanediol (2,3-BD) as highly promising bio-based platform chemicals have received more attentions due to their wide range of applications. However, the non-efficient substrate conversion and mutually transition between AC and 2,3-BD in their natural producing strains not only led to a low selectivity but also increase the difficulty of downstream purification. Therefore, synthetic engineering of more suitable strains should be a reliable strategy to selectively produce AC and 2,3-BD, respectively.
Results
In this study, the respective AC (alsS and alsD) and 2,3-BD biosynthesis pathway genes (alsS, alsD, and bdhA) derived from Bacillus subtilis 168 were successfully expressed in non-natural AC and 2,3-BD producing Corynebacterium crenatum, and generated recombinant strains, C. crenatum SD and C. crenatum SDA, were proved to produce 9.86 g L−1 of AC and 17.08 g L−1 of 2,3-BD, respectively. To further increase AC and 2,3-BD selectivity, the AC reducing gene (butA) and lactic acid dehydrogenase gene (ldh) in C. crenatum were then deleted. Finally, C. crenatumΔbutAΔldh SD produced 76.93 g L−1 AC in one-step biocatalysis with the yield of 0.67 mol mol−1. Meanwhile, after eliminating the lactic acid production and enhancing 2,3-butanediol dehydrogenase activity, C. crenatumΔldh SDA synthesized 88.83 g L−1 of 2,3-BD with the yield of 0.80 mol mol−1.
Conclusions
The synthetically engineered C. crenatumΔbutAΔldh SD and C. crenatumΔldh SDA in this study were proved as an efficient microbial cell factory for selective AC and 2,3-BD production. Based on the insights from this study, further synthetic engineering of C. crenatum for AC and 2,3-BD production is suggested.
Similar content being viewed by others
Background
As important platform compounds, acetoin (AC) and 2,3-butanediol (2,3-BD) are widely used in food, medicine and chemical industries [1,2,3,4]. In addition, AC and 2,3-BD production by microbial fermentation and biotransformation have drawn attentions since the environment crisis [5].
In recent years, consolidate efforts have been made towards increasing AC and 2,3-BD production by inactivation of competitive pathways or overexpression of key genes of AC and 2,3-BD biosynthesis pathways [6]. Nielsen et al. strengthened the expression of α-acetolactate synthase (ALS), α-acetolactate decarboxylase (ALDC) and 2,3-butanediol dehydrogenase (BDH) to increase the titer of AC (0.87 g L−1) and 2,3-BD (1.12 g L−1) in Escherichia coli [7]. The similar strategy of overexpressing ALS and ALDC was also applied to Bacillus subtilis and Saccharomyces cerevisiae, in which the productivity of AC from glucose was increased by approximately 62.9% and 58%, respectively [8, 9]. By further expressing the homogenous BDH1 in engineered S. cerevisiae, 2,3-BD was further increased to 96.2 g L−1 [10]. On the other hand, Ji et al. increased the yield of 2,3-BD to 0.48 g g−1 by reducing the biosynthesis of lactic acid and acetic acid in Klebsiella oxytoca [11]. Wang et al. knocked out bdhA, pta and acoA in B. subtilis and increased the yield of AC to 0.49 g g−1 [12]. More recently, Erian et al. increased the 2,3-BD titer about 2.4-fold by eliminating the lactate formation in engineered E. coli [13]. Meanwhile, strains of non-natural AC and 2,3-BD producing strains were metabolically engineered by expressing AC and 2,3-BD biosynthesis pathways derived from Serratia marcescens, Klebsiella pneumoniae, Enterobacter and S. cerevisiae [3, 5, 14]. Took the advantages of photosynthesis by converting light energy to chemical energy, Oliver et al. introduced a novel synthetic pathway into Cyanobacteria, and successfully produced AC and 2,3-BD using CO2 [15]. In addition, Corynebacterium glutamicum was also engineered for 2,3-BD production by expressing gene cluster of 2,3-BD biosynthetic pathway from Lactococcus lactis [16] or the budABC cluster from K. pneumoniae [17], which synthesized 6.3 and 18.9 g L−1 of 2,3-BD, respectively. After multistage modification, C. glutamicum could also be used to produce optically pure d-(−)-AC [18]. However, these AC and 2,3-BD producing strains also have some drawbacks because by-products, such as lactic acid and acetic acid, and mutually transition between AC and 2,3-BD not only lead to a low yield but also increase the difficulty of products purification.
Compared to AC and 2,3-BD fermentation, AC and 2,3-BD biocatalysis generally present a higher yield and selectivity [19, 20]. Efficiently mutual transformation of AC and 2,3-BD were realized by regenerating the intracellular NAD+/NADH levels in E. coli and B. subtilis [21, 22]. However, AC and 2,3-BD as substrates are both expensive and thus are not feasible for commercialization strategies by biocatalysis. Thus, to better meet industrial production needs, we urged to find more suitable host strains and more efficient biocatalyst processes. In this study, we aimed to develop one-step bioconversion from glucose to selective AC and 2,3-BD production, and chose to engineer non-natural AC and 2,3-BD producing Corynebacterium crenatum SYPA5-5 as the host since the following reasons: C. crenatum, genetically close to C. glutamicum, has the advantages of growing fast, non-sporulation, and easy gene editing [23]. In addition, the BDH identified in C. crenatum was demonstrated that catalyze both AC and diacetyl (DA) for 2,3-BD formation [24]. Therefore, synthetic engineering C. crenatum SYPA5-5 should be a reliable strategy to selectively produce AC and 2,3-BD, respectively.
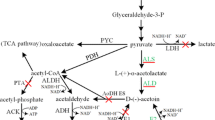
In this work, AC (alsS and alsD) and 2,3-BD biosynthesis pathway genes (alsS, alsD, and bdhA) from B. subtilis 168 were overexpressed in C. crenatumΔldh and C. crenatumΔbutAΔldh, respectively, in which glucose was specifically transformed into AC or 2,3-BD in one-step biocatalysis (Fig. 1). This work provides a simple and efficient bioconversion process to selectively convert glucose to high value-added chemicals of AC and 2,3-BD by engineered C. crenatum as microbial cell factory.

AC and 2,3-BD pathway construction in C. crenatum. The blue arrow indicate AC and 2,3-BD synthetic pathways from B. subtilis 168. The red cross marks indicate the disrupted pathways. GAPDH (glyceraldehyde-3-phosphate dehydrogenase), PEPC (phosphoenolpyruvate carboxylase), MDH (malic dehydrogenase), FUM (fumarase), SDH (succinic dehydrogenase)
Results and discussion
Overexpression of ALS, ALDC, and AR/BDH in C. crenatum
Here, an artificial operon including alsS, alsD, and bdhA from B. subtilis was selected to overexpress in C. crenatum to investigate its effect on AC and 2,3-BD production. The confirmed recombinants were named C. crenatum/pXMJ19-alsS (C. crenatum S), C. crenatum/pXMJ19-alsD (C. crenatum D), C. crenatum/pXMJ19-bdhA (C. crenatum A), C. crenatum/pXMJ19-alsSD (C. crenatum SD), and C. crenatum/pXMJ19-alsSD-bdhA (C. crenatum SDA), respectively.
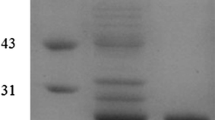
The SDS-PAGE was used to confirm the expression of ALS (AHAS), ALDC, and AR/BDH in C. crenatum recombinants (Additional file 1: Figure S1). As expected, ALS (AHAS), ALDC, and AR/BDH on gel demonstrated their respective molecular mass about 60, 29, and 38 kDa, respectively. Enzyme activity assays also confirmed the functional expression of ALS (AHAS), ALDC, and AR/BDH in C. crenatum transformants. As shown in Table 1, the wild type strain showed negligible acetohydroxyacid synthase (AHAS) activity, which might be inhibited by the anabolism of l-valine and l-leucine in C. crenatum [25, 26]. Meanwhile, no ALDC activity was detected in C. crenatum WT. After enhanced expression of ALS and ALDC, the enzyme activities in C. crenatum SD and C. crenatum SDA was increased to 2.22/2.32 and 8.08/5.68 U mg−1, respectively. However, it should be noted that the lower increased activities than that of only ALS or ALDC expression probably due to lower efficiency in the tandem co-expression driven by one promoter for two or three genes. On the other hand, compared to C. crenatum WT, C. crenatum SDA showed a high AR and BDH activities (0.32 and 0.10 U mg−1) comparable to C. crenatum A (0.38 and 0.19 U mg−1). In addition, although AR/BDH was a bidirectional reversible enzyme [24, 27], the heterogenous AR/BDH from B. subtilis present a higher AR activity than BDH activity in C. crenatum A and C. crenatum SDA. With ALS, ALDC, and (or) AR/BDH, the recombinant C. crenatum can catalyze the pyruvate to either AC or DA, thus allowing the respective conversion of glucose to AC and 2,3-BD.
Production of AC and 2,3-BD by shake flask fermentation of recombinant C. crenatum
Batch fermentations with 120 g L−1 glucose as the sole carbon source were first performed in shack flask to investigate the metabolic impact of ALS, ALDC, and BDH expression. Since C. crenatum is a facultative anaerobe, cell growth under different dissolved oxygen condition often leads to generate different products [28, 29]. Previous study has demonstrated that C. crenatum shows a fast growth rate of biomass when using glucose as the substrate, and is diffusely used in the yielding of sundry amino acids such as l-glutamic acid and l-arginine under sufficient oxygen supply conditions [28]. Therefore, the by-product l-arginine was also determined at the end of fermentation in this study. The results are shown in Table 2.
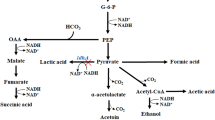
As expected, C. crenatum WT yield 23.6 g L−1 l-arginine, and no AC and 2,3-BD were detected in the fermentation broth. Theoretically, overexpression of alsS, alsD, and bdhA gene, respectively, cannot produce AC and 2,3-BD in C. crenatum. However, except for little AC and 2,3-BD producing, C. crenatum S also produced 2.17 g L−1 of unexpected DA, which could not be accumulated in B. subtilis [30]. This might be due to the spontaneous non-enzymatic decarboxylation reaction of α-acetolactate under natural oxidative aerobic conditions [31]. In addition, our previous work has also proved that the BDH from C. crenatum could catalyze DA to AC and then convert to 2,3-BD [24]. This explains why the chain reaction was got through in C. crenatum S without expression of ALDC. Moreover, it has been reported that DA is a bacteriostatic food additive that can inhibit bacteria growth [3]. Thus, C. crenatum S consumed only about 36 g L−1 glucose and then suspended the fermentation while accumulated comparable amount of DA, leading to a low efficiency of the multiple-step reaction from pyruvate to AC and 2,3-BD.
On the contrary, after introducing the alsSD operon with one promoter into C. crenatum, co-expression of ALS and ALDC effectively converted pyruvate to 13.59 g L−1 AC and 10.61 g L−1 2,3-BD, respectively. To enhance 2,3-BD production, bdhA was further expressed in C. crenatum SD and the generated C. crenatum SDA dramatically increased 2,3-BD titer by 43%. These results indicated that ALS and ALDC are both important for AC and 2,3-BD biosynthesis in C. crenatum. In addition, enhanced heterogeneous BDH activity was confirmed the positive effect on 2,3-BD biosynthesis. However, C. crenatum SD and C. crenatum SDA still produced comparable amount of l-arginine as by-product, resulting in a low yield of AC and 2,3-BD. Therefore, l-arginine biosynthesis must be decreased.
Bioconversion of glucose to AC and 2,3-BD by recombinants C. crenatum resting cells
l-Arginine as growth factor is necessary for C. crenatum growth, which cannot be directly depressed by gene knock-out. l-arginine production by C. crenatum needs high level of dissolved oxygen, while there is no strict demand on dissolved oxygen for the acid-butanediol fermentation. Therefore, we attempted to produce AC and 2,3-BD using resting cell bioconversion with glucose as substrate by C. crenatum SD and C. crenatum SDA, respectively.
As demonstrated in Fig. 2, no l-arginine was detected during the resting cell bioconversion process (data not shown). C. crenatum SD converted 100 g L−1 glucose to 9.86 g L−1 AC and 12.76 g L−1 2,3-BD, respectively. Similarly, C. crenatum SDA further accumulated 2,3-BD to 17.08 g L−1 with a high 2,3-BD/AC ratio. However, the content of organic acids, especially lactic acid, were also detected in both C. crenatum SD and C. crenatum SDA, which was consistent with previous study that C. crenatum can produce various organic acids such as succinic acid, acetic acid and lactic acid in the case of limited dissolved oxygen [29]. As the accumulation of organic acids increases the consumption of carbon sources [32, 33], the synthetic pathways of organic acids may competitively inhibit the accumulation of AC and 2,3-BD.

Resting cell bioconversion analysis of C. crenatum and the recombinant strains in shack flask. Recombinant C. crenatum (100 µL) was transferred to 10 mL LBG medium and incubated for approximately 12 h at 30 °C, 180 r min−1. The cultured bacterial solution (3 mL) was transferred into 30 mL of fermentation medium at the same culture condition for 24 h. The cultured cells were harvested by centrifugation at 8000 r min−1 for 10 min and then resuspended in resting cell bioconversion medium (containing 100 g L−1 glucose) for resting cell bioconversion. Formed metabolites and organic acid during the bioconversion of resting cells are shown. The results of resting cell bioconversion kinetics are shown as the mean ± standard of three replicates. +: overexpression, Δ: knockout
Construction of ldh and butA blocked recombinant C. crenatum
Although resting cell bioconversion reduced l-arginine formation, some pyruvate was still converted to lactic acid. In addition, unexpected high yield of 2,3-BD was still produced by C. crenatum SD. To further decrease by-products and enhance the AC and 2,3-BD selectivity, blocking the competitive synthesis pathways in C. crenatum was considered to be necessity. The homologous lactate dehydrogenase (ldh) and butanol dehydrogenase (butA) genes in C. crenatum were knocked out to increase respective AC and 2,3-BD selectivity. The suicide vectors pK18-Δldh (Additional file 2: Figure S2) and pK18-ΔbutA (Additional file 3: Figure S3), respectively, was constructed and transformed into C. crenatum. The mutant strains C. crenatumΔldh and C. crenatumΔbutA were finally obtained after two rounds of homologous recombination. Then, pK18-Δldh was further introduced into C. crenatumΔbutA and generated C. crenatumΔbutAΔldh. All of mutant strains were verified by PCR using the upstream and downstream primers of ldh and butA (Additional file 4: Figure S4).
LDH and AR/BDH activities of C. crenatum mutants were determined, and the results were shown in Table 3. LDH activity was reduced by 83–88% after blocking of ldh gene. Meanwhile, AR and BDH activities were both reduced by 62–82% and 83–87%, while butA gene was knocked out. The results indicated that the AC reduction and lactic acid biosynthesis pathway were depressed by knocking out butA and ldh genes. By comparing the dry cell weights of C. crenatum WT and the knockout strains (Additional file 5: Table S1), it was found that the knockout of ldh and butA genes had little effect on the growth of the bacteria.
Then, the recombinant C. crenatumΔbutA, C. crenatumΔldh, and C. crenatumΔbutAΔldh were subjected to resting cell and the bioconversion results were shown in Fig. 2. As expected, after knocking out ldh gene, the acetic and succinic acid production were slightly increased, while the lactic acid production dramatically decreased by about 85% in C. crenatumΔldh and C. crenatumΔbutAΔldh. Moreover, compared with C. crenatum WT and C. crenatumΔldh, there is no significant effect on biocatalysis after butA delection.
Efficient one-step bioconversion of glucose to AC by recombinant C. crenatumΔbutAΔldh/pXMJ19-alsSD resting cell
To construct the one-step bioconversion of glucose to AC, plasmid pXMJ19-alsSD was then transformed into ldh and butA deletion strain to construct C. crenatumΔbutAΔldh/pXMJ19-alsSD (C. crenatumΔbutAΔldh SD). C. crenatumΔbutAΔldh SD consumed 94 g L−1 glucose after 60 h with an uptake rate of 1.57 g L−1 h−1, which was significantly higher (2.18-fold and 2.34-fold, respectively) than that of C. crenatum WT and C. crenatumΔbutAΔldh, indicating that the expression of heterologous AC biosynthesis pathway strongly promoted glucose for AC production. Consequently, AC production dramatically increased from 9.86 to 23.56 g L−1, about 2.39-fold than before. Meanwhile, only a small amount of 2,3-BD (1.28 g L−1) and lactic acid (1.83 g L−1) were detected, resulting in an AC molar yield of up to 0.51 mol mol−1. Consistence with that of C. crenatumΔbutAΔldh, acetic acid and succinic acid has hardly changed (Fig. 2).
Efficient one-step bioconversion of glucose to 2,3-BD by recombinant C. crenatumΔldh/pXMJ19-alsSD-bdhA resting cell
To further extend the products chain for 2,3-BD production, pXMJ19-alsSD-bdhA was introduced into C. crenatumΔldh, resulting in C. crenatumΔldh/pXMJ19-alsSD-bdhA (C. crenatumΔldh SDA) which co-expresses both homologous and heterologous AR/BDH. Generally, compared with C. crenatum SDA as the positive control, C. crenatumΔldh SDA resting cell converted more glucose to 2,3-BD. After 60 h, C. crenatumΔldh SDA consumed 95 g L−1 glucose with an uptake rate of 1.58 g L−1 h−1. With overexpression of AR/BDH and depressing LDH, 2,3-BD production increased from 17.08 to 25.93 g L−1, about 52% higher than the control. Meanwhile, lactic acid was decreased about 95%, leading to a high 2,3-BD molar yield of 0.55 mol mol−1 (Fig. 2). It should be noted that the bioconversion for 2,3-BD production was still accompanied by certain AC accumulation because of the reversible reaction of AR/BDH. Moreover, 2,3-BD and lactic acid are both NADH-dependent products. Therefore, depressing LDH also releases additional reducing equivalent, which further releases the constraint of co-enzyme poll on 2,3-BD synthesis. However, the yields of acetic acid (1.6 g L−1) and succinic acid (1.1 g L−1) were slightly higher than C. crenatum SDA.
Repeated batch resting cell bioconversion for AC and 2,3-BD production in 5 L bioreactor
To investigate the long-term stability and performance of recombinant C. crenatum, repeated batch resting cell bioconversion were performed in 5 L bioreactors, and the results are shown in Fig. 3. Generally, the resting cell bioconversion was repeated for 3 cycles with 100 g L−1 of glucose as the substrate. In the first two batch conversions, AC production was stably increased by C. crenatumΔbutAΔldh SD. However, AC productivity of the third batch has gradually declined. Finally, after total 60 h of bioconversion, 76.93 g L−1 of AC with the yield of 0.67 mol mol−1 was produced by C. crenatumΔbutAΔldh SD (Fig. 3a). Meanwhile, after eliminating the lactic acid production and enhancing 2,3-butanediol dehydrogenase activity, C. crenatumΔldh SDA also synthesized 88.83 g L−1 of 2,3-BD with the yield of 0.80 mol mol−1 (Fig. 3b). It should be noted that both average AC and 2,3-BD productivity throughout the repeated batch conversion process reached 1.28–1.48 g L−1 h−1, which increased ~ 3.5-fold compared to flask batch biocatalysis. However, significant decrease of enzyme activities was observed during the third process (data not shown). Therefore, further strategies should be developed to maintain the cell viability and stability during the whole bioconversion process.

Resting cell bioconversion analysis of recombinant C. crenatum in 5 L bioreactor. Recombinant C. crenatum (200 µL) was transferred to 20 mL LBG medium and incubated for 24 h at 180 r min−1, 30 °C. After the 10% cultures were transferred and cultured in 200 mL seed medium for 18 h, the whole cultures were transferred to 5 L bioreactor containing 2 L of fermentation medium, and incubated for 24 h at 30 °C, 600 r min−1. 4 L recombinant C. crenatum cultures were collected and resuspended in 2 L resting cell bioconversion medium containing 100 g L−1 glucose. Resting cell bioconversion was performed in batches at 30 °C, 250 r min−1. Consumed glucose, formed metabolites, and organic acid during the bioconversion of resting cells are shown. The results of resting cell bioconversion kinetics are shown as the mean ± standard of three replicates
Comparison with other study
Currently, microbial fermentation is still the main method for producing AC and 2,3-BD from various substrates. However, its relatively long fermentation period and low productivity and substrate conversion rate are currently obstructed the industrial application [18, 34, 35]. Biocatalysis, as a highly efficient and environmentally friendly methods, can significantly improve substrate utilization rate and product yield. As shown in Table 4, previous studies have demonstrated that engineered E. coli and B. subtilis can be used as host for biocatalytic synthesis of AC and 2,3-BD, in which the highest yield of AC and 2,3-BD can be reached to 0.98 mol mol−1 and 0.96 mol mol−1. However, most of these biocatalysts cases were carried out using AC or 2,3-BD as a substrate, which are not suitable for industrial production at all. Although 2,3-BD and AC bioconversion from glucose were achieved by K. pneumoniae and B. subtilis, two step batch strategy still cannot selectively separate mixed (2S,3S)-2,3-BD and (3S)-AC [41]. In this study, we developed a new mono-bioconversion system using C. crenatum as the only host for AC and 2,3-BD production directly from glucose. After depressing competition pathways and overexpressing AC and 2,3-BD biosynthesis genes from B. subtilis, one-step biosynthesis of AC and 2,3-BD, respectively, were achieved in C. crenatumΔbutAΔldh SD and C. crenatumΔldh SDA without additional complex nutrients.
Compared to usual 100–120 g L−1 AC and 2,3-BD produced by microbial fermentation, further improvements of C. crenatum are necessary for industrial application. Although depressing ldh and butA and enhancing AC or 2,3-BD biosynthesis pathway activity, C. crenatumΔbutAΔldh SD and C. crenatumΔldh SDA showed a higher AC and 2,3-BD selectivity, respectively, some pyruvate still converted to acetic and succinic acid. Therefore, increased AC and 2,3-BD production can be realized by disruption of the acetate and succinate biosynthesis pathway, as demonstrated in K. oxytoca and B. subtilis [11, 12]. Moreover, modification of key enzymes is critical for promoting cell metabolism. Previously, we have relieved the feedback inhibition of l-arginine by site-directed mutation of the key enzyme (NAGK) of C. creantum, and overexpressed the l-arginine operon, which effectively increased the yield of l-arginine by 41.7% [42] and 29% [43]. Therefore, the site-specific mutagenesis of butanol dehydrogenase (butA) gene might be further improved the AC and 2,3-BD yield and selectivity in C. creantum. In addition, although the repeated batch biocatalysis showed a high average productivity for AC and 2,3-BD production, the catalytic efficiency significantly deceased after only two batches, which can be further improved by cell immobilization to increase the cell viability and stability [44, 45]. On the other hand, process engineering, including buffer optimization, substrate concentration optimization, multiple biocatalysis strategies, and high cell density et al., can further improve AC and 2,3-BD production for commercial development. These synthetic and process engineering strategies can be applied to together to develop an efficient microbial cell factory for selective AC and 2,3-BD production in C. creantum.
Conclusion
In this study, we successfully engineered C. crenatum SYPA5-5 to overexpress alsS, alsD and/or bdhA from B. subtilis 168 for selective AC and 2,3-BD biosynthesis from glucose. After depressing competition pathways, recombinant C. crenatumΔbutAΔldh SD and C. crenatumΔldh SDA further increased AC and 2,3-BD production to 76.93 g L−1 and 88.83 g L−1, respectively. Overall, respective non-natural AC and 2,3-BD biosynthesis pathway constructed in this study efficiently reduced the by-products accumulation and improved AC and 2,3-BD selectivity in C. crenatum. The optimal selection of AC and 2,3-BD biosynthesis strategies with further metabolic engineering should lead to the development of a promising microbial cell factory for AC and 2,3-BD production.
Materials and methods
Microorganisms and plasmids
All strains, plasmids and primers involved are shown in Additional file 6: Table S2 and Table 5, respectively. B. subtilis 168, E. coli BL21 and JM109, C. crenatum SYPA5-5 were stored in our laboratory. E. coli BL21 and JM109 were host strains of recombinant plasmid pXMJ19 and pk18, respectively. pXMJ19 is the shuttle expression vector of E. coli and Corynebacterium. pK18mobsacB carrying sacB gene is used for gene integration and knockout in C. crenatum [46].
Chemicals, mediums and cultivation conditions
FastPure Gel DNA Extraction Mini Kit, FastPure Plasmid Mini Kit and FastPure Bacteria DNA Isolation Mini Kit were all purchased from Vazyme (Nanjing, China). Antibiotics, restriction endonucleases and other tool enzymes such as high-fidelity 2× ExTaq DNA polymerase all purchased from Shenggong Biological (Shanghai, China). All other reagents of analysis grade or higher quality were obtained from Wuxi Reagent Company.
Luria–Bertani (LB) medium was used to culture E. coli. LBG (LB + 0.5% glucose) medium was used for preincubation of C. crenatum. C. crenatum competent medium for electroporation: LB medium was supplemented with 3% glycine and 0.1% tween 80. Shake flask fermentation medium (g L−1): (NH4)2SO4 40, yeast extract 8, KH2PO4 1.5, KCl 1, MnSO4·H2O 0.02, FeSO4·7H2O 0.02, MgSO4·7H2O 0.5, CaCO3 30. Seed medium of 5 L bioreactor (g L−1): yeast extract 20, MgSO4·7H2O 0.5, (NH4)2SO4 20, KH2PO4 1.5. Fermentation medium of 5 L bioreactor (g L−1): (NH4)2SO4 20, yeast extract 20, KH2PO4 1.5, MnSO4·H2O 0.02, FeSO4·7H2O 0.02, MgSO4·7H2O 0.5. Resting cell bioconversion medium (g L−1): K2HPO4·3H2O 0.5, KH2PO4 0.5, MnSO4·H2O 4.2, MgSO4·7H2O 0.5, FeSO4·7H2O 6. E. coli was cultured at 37 °C, 180 r min−1. C. crenatum was cultured at 30 °C, 180 r min−1 or 250 r min−1.
Construction of ΔbutA and Δldh fusion fragments
The butA gene fragment was obtained by PCR using the C. crenatum SYPA5-5 genome as template and PbutA1F and PbutA2R as primers. The PCR product was connected to pMD18-T cloning vector and sent to Shenggong Biological (Shanghai) for sequencing. Firstly, PbutA1F, PbutA3R and PbutA2R, PbutA4F primers were used for the first round of PCR using the C. crenatum SYPA5-5 genome as template to obtain two PCR products. Secondly, the deleted gene fragment ΔbutA was obtained by fusing the two PCR products in the overlap-extension PCR and PbutA1F and PbutA2R were used (Additional file 7: Figure S5). The method for obtaining the deleted gene fragment Δldh was the same as above (Additional file 8: Figure S6).
Construction of recombinant C. crenatumΔbutAΔldh
The deletion gene fragments ΔbutA and Δldh were ligated to the pK18mobsacB suicide plasmid, respectively. Recombinant plasmids pK18-ΔbutA and pK18-Δldh were constructed in E. coli JM109. The plasmid pK18-ΔbutA was electroporated into C. crenatum according to the reported method [47]. The construction method of the ldh gene deletion strain was the same as above. The gene-deleted strains C. crenatumΔbutA and C. crenatumΔldh were obtained, respectively. The constructed homologous integration plasmid pK18-Δldh was electroporated into C. crenatumΔbutA, and after two homologous recombination, butA and ldh double gene deletion type recombinant strain C. crenatumΔbutAΔldh were obtained.
Crude enzyme extraction and related enzyme activity assay
Recombinant C. crenatum cells were harvested by centrifugation at 8000 r min−1, 4 °C. Then cells were washed using 50 mM Tris–HCl buffer (pH 7.0). The washed cells were suspended in 5 mL of Tris–HCl buffer and lysozyme was added to treat the cell wall for 3–4 h. Cell disruption was accomplished by sonication for 30 min under ice bath conditions. Cell debris was removed by centrifugation at 4 °C, 12,000 r min−1 for 30 min and the supernatant was harvested for measurement of intracellular enzyme activity. Protein concentration in supernatant was determined by Bradford method [48]. ALS/AHAS, ALDC [49, 50], AR/BDH [51] and LDH [52] enzyme activities were determined according to the reported enzyme activity assay.
Fermentation and resting cell bioconversion
The bacterial solution was inoculated into 10 mL LBG medium at a ratio of 1% and incubated for about 12 h. 3 mL bacterial liquid was transferred to 30 mL fermentation medium and cultured for 24 h. The cells in the fermentation medium were harvested and suspended in resting cell bioconversion medium containing 100 g L−1 glucose for resting cell bioconversion. 30 g L−1 CaCO3 was added during transformation to neutralize the organic acids produced. When resting cell bioconversion was performed in 5 L bioreactor, 4 L recombinant C. crenatum cultures were collected and resuspended in 2 L resting cell bioconversion medium. Resting cell bioconversion was performed in three batches at 30 °C, 250 r min−1. The converted solution was kept at pH 7.0 by feeding 50% NH3·H2O automatically. After the end of each batch of transformation, cells were harvested by centrifugation and then washed 3 times using Tris–HCl buffer. Resting cell bioconversion was continued by resuspending the cells in 2 L resting cell bioconversion medium containing 100 g L−1 glucose.
Parameter measurement and analysis
For biomass analysis, the absorbance of cell cultures were measured at 562 nm with distilled water as the blank control. The dry cell weight (DCW) can be converted by using the equation (1 OD562 = 0.375 g L−1 DCW). The concentration of glucose in conversion solution and fermentation broth was determined using SBA-40E biosensor analyzer. The concentration of AC and 2,3-BD in conversion solution and fermentation broth was measured using Agilent gas chromatography [53]. Amino acids [28] and organic acids [54] were analyzed following the descriptions of previous reports.
Availability of data and materials
All data involved in this study, if not found in this article or additional material, may be obtained from the corresponding author.
Abbreviations
- AC:
-
acetoin
- 2,3-BD:
-
2,3-butanediol
- ALS/AHAS:
-
α-acetolactate synthase
- ALDC:
-
α-acetolactate decarboxylase
- AR/BDH:
-
2,3-butanediol dehydrogenase
- DA:
-
diacetyl
- LDH:
-
lactate dehydrogenase
- DCW:
-
dry cell weight
References
Xiao Z, Xu P. Acetoin metabolism in bacteria. Crit Rev Microbiol. 2007;33(2):127–40.
Zijun X, Jian RL. Generation of acetoin and its derivatives in foods. J Agric Food Chem. 2014;62(28):6487–97.
Celińska E, Grajek W. Biotechnological production of 2,3-butanediol—current state and prospects. Biotechnol Adv. 2009;27(6):715–25.
Werpy TA, Holladay JE, White JF. Top value added chemicals from biomass: I. Results of screening for potential candidates from sugars and synthesis gas. Synthetic Fuels. 2004.
Xiao Z, Lu JR. Strategies for enhancing fermentative production of acetoin: a review. Biotechnol Adv. 2014;32(2):492–503.
Renna MC, Najimudin N, Winik LR, et al. Regulation of the Bacillus subtilis alsS, alsD, and alsR genes involved in post-exponential-phase production of acetoin. J Bacteriol. 1993;175(12):3863–75.
Nielsen DR, Yoon S, Yuan CJ, et al. Metabolic engineering of acetoin and meso-2,3-butanediol biosynthesis in E. coli. Biotechnol J. 2010;5(3):274–84.
Zhang X, Zhang R, Bao T, et al. Moderate expression of the transcriptional regulator ALsR enhances acetoin production by Bacillus subtilis. J Ind Microbiol Biotechnol. 2013;40(9):1067–76.
Bae SJ, Kim S, Hahn JS. Efficient production of acetoin in Saccharomyces cerevisiae by disruption of 2,3-butanediol dehydrogenase and expression of NADH oxidase. Sci Rep. 2016;6:27667.
Soo-Jung K, Seung-Oh S, Yong-Su J, et al. Production of 2,3-butanediol by engineered Saccharomyces cerevisiae. Bioresour Technol. 2013;146(10):274–81.
Ji X, Huang H, Li S, et al. Enhanced 2,3-butanediol production by altering the mixed acid fermentation pathway in Klebsiella oxytoca. Biotechnol Lett. 2008;30(4):731–4.
Wang M, Fu J, Zhang X, et al. Metabolic engineering of Bacillus subtilis for enhanced production of acetoin. Biotechnol Lett. 2012;34(10):1877–85.
Erian AM, Gibisch M, Pflügl S. Engineered E. coli W enables efficient 2,3-butanediol production from glucose and sugar beet molasses using defined minimal medium as economic basis. Microb Cell Fact. 2018;17(1):190.
Ng CY, Jung MY, Lee J, et al. Production of 2,3-butanediol in Saccharomyces cerevisiae by in silico aided metabolic engineering. Microb Cell Fact. 2012;11(1):68.
Oliver JWK, Machado IMP, Hisanari Y, et al. Cyanobacterial conversion of carbon dioxide to 2,3-butanediol. PNAS. 2013;110(4):1249–54.
Radoš D, Carvalho AL, Wieschalka S, et al. Engineering Corynebacterium glutamicum for the production of 2,3-butanediol. Microb Cell Fact. 2015;14(1):171.
Yang J, Kim B, Kim H, et al. Industrial production of 2,3-butanediol from the engineered Corynebacterium glutamicum. Appl Biochem Biotechnol. 2015;176(8):2303–13.
Mao Y, Jing F, Ran T, et al. Systematic metabolic engineering of Corynebacterium glutamicum for industrial-level production of optically pure d-(−)-Acetoin. Green Chem. 2017;19(23):10–1039.
Fessner W. Systems biocatalysis: development and engineering of cell-free “artificial metabolisms” for preparative multi-enzymatic synthesis. New Biotechnol. 2015;32(6):658–64.
Andrea S, Marcella P, Maura M, et al. Biodiesel production from triolein and short chain alcohols through biocatalysis. J Biotechnol. 2005;119(3):291–9.
Zijun X, Chuanjuan L, Chao G, et al. A novel whole-cell biocatalyst with NAD+ regeneration for production of chiral chemicals. PLoS ONE. 2010;5(1):e8860.
Bao T, Zhang X, Rao Z, et al. Efficient whole-cell biocatalyst for acetoin production with NAD+ regeneration system through homologous co-expression of 2,3-butanediol dehydrogenase and NADH oxidase in engineered Bacillus subtilis. PLoS ONE. 2014;9(7):e102951.
Dou W, Xu M, Cai D, et al. Improvement of l-arginine production by overexpression of a bifunctional ornithine acetyltransferase in Corynebacterium crenatum. Appl Biochem Biotechnol. 2011;165(3–4):845–55.
Zhao X, Zhang X, Rao Z, et al. Identification and characterization of a novel 2,3-butanediol dehydrogenase/acetoin reductase from Corynebacterium crenatum SYPA5-5. Lett Appl Microbiol. 2015;61(6):573–9.
Xu M, Rao Z, Yang J, et al. Heterologous and homologous expression of the arginine biosynthetic arg C–H cluster from Corynebacterium crenatum for improvement of l-arginine production. J Ind Microbiol Biotechnol. 2012;39(3):495–502.
Veronika E, Miroslav P, Jirí H, et al. Feedback-resistant acetohydroxy acid synthase increases valine production in Corynebacterium glutamicum. Appl Environ Microbiol. 2005;71(1):207–13.
Xian Z, Teng B, Zhiming R, et al. Two-stage pH control strategy based on the pH preference of acetoin reductase regulates acetoin and 2,3-butanediol distribution in Bacillus subtilis. PLoS ONE. 2014;9(3):e91187.
Xu H, Dou W, Xu H, et al. A two-stage oxygen supply strategy for enhanced l-arginine production by Corynebacterium crenatum based on metabolic fluxes analysis. Biochem Eng J. 2009;43(1):41–51.
Inui M, Suda M, Okino S, et al. Transcriptional profiling of Corynebacterium glutamicum metabolism during organic acid production under oxygen deprivation conditions. Microbiology. 2007;153(8):2491–504.
López JM, Thomas B, Rehbein H. Acetoin degradation in Bacillus subtilis by direct oxidative cleavage. Eur J Biochem. 1975;57(2):425–30.
Mink R, Sommer S, Kölling R, et al. Time course of diacetyl formation during vinification with Saccharomyces cerevisiae and Oenococcus oeni co-cultivation. Aust J Grape Wine Res. 2014;20(2):194–8.
Chen XJ, Jiang ST, Xing-Jiang LI, et al. Metabolic regulation of succinic acid and lactic acid production by Corynebacterium crenatum. Food Sci. 2013;63(1):39–44.
Yamauchi Y, Hirasawa T, Nishii M, et al. Enhanced acetic acid and succinic acid production under microaerobic conditions by Corynebacterium glutamicum harboring Escherichia coli transhydrogenase gene pntAB. J Gen Appl Microbiol. 2014;60(3):112–8.
Wang D, Zhou J, Chen C, et al. R-acetoin accumulation and dissimilation in Klebsiella pneumoniae. J Ind Microbiol Biotechnol. 2015;42(8):1105–15.
Bai F, Dai L, Fan J, et al. Erratum to: engineered Serratia marcescens for efficient (3R)-acetoin and (2R,3R)-2,3-butanediol production. J Ind Microbiol Biotechnol. 2015;42(6):977.
Gao J, Xu YY, Li FW, et al. Production of S-acetoin from diacetyl by Escherichia coli transformant cells that express the diacetyl reductase gene of Paenibacillus polymyxa ZJ-9. Lett Appl Microbiol. 2013;57(4):274–81.
Guo Z, Zhao X, He Y, et al. Efficient (3R)-acetoin production from meso-2,3-butanediol using a new whole-cell biocatalyst with co-expression of meso-2,3-butanediol dehydrogenase, NADH oxidase, and Vitreoscilla hemoglobin. J Microbiol Biotechnol. 2017;27(1):92–100.
He Y, Chen F, Sun M, et al. Efficient (3S)-acetoin and (2S,3S)-2,3-butanediol production from meso-2,3-butanediol using whole-cell biocatalysis. Molecules. 2018;23(3):691.
Wang Y, Li L, Ma C, et al. Engineering of cofactor regeneration enhances (2S,3S)-2,3-butanediol production from diacetyl. Sci Rep. 2013;3:2643.
Samuel N, Bao T, Zhang X, et al. Optimized whole cell biocatalyst from acetoin to 2,3-butanediol through coexpression of acetoin reductase with NADH regeneration systems in engineered Bacillus subtilis. J Chem Technol Biotechnol. 2017;92:2477–87.
Liu Z, Qin J, Gao C, et al. Production of (2S,3S)-2,3-butanediol and (3S)-acetoin from glucose using resting cells of Klebsiella pneumonia and Bacillus subtilis. Bioresour Technol. 2011;102(22):10741–4.
Meijuan X, Zhiming R, Wenfang D, et al. Site-directed mutagenesis and feedback-resistant N-acetyl-l-glutamate kinase (NAGK) increase Corynebacterium crenatum l-arginine production. Amino Acids. 2012;43(1):255–66.
Man Z, Xu M, Rao Z, et al. Systems pathway engineering of Corynebacterium crenatum for improved l-arginine production. Sci Rep. 2016;6:28629.
Wei G, Ma W, Zhang A, et al. Enhancing catalytic stability and cadaverine tolerance by whole-cell immobilization and the addition of cell protectant during cadaverine production. Appl Microbiol Biotechnol. 2018;102(18):7837–47.
Ma W, Liu L, Chen H, et al. Micropatterned immobilization of membrane-mimicking polymer and peptides for regulation of cell behaviors in vitro. RSC Adv. 2018;8(37):20836–50.
Schäfer A, Tauch A, Jäger W, et al. Small mobilizable muti-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994;145(1):69–73.
Tauch A, Kirchner O, Löffler B, et al. Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr Microbiol. 2002;45(5):362–7.
Bradford MM. A rapid method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72(s1–2):248–54.
Shota A, Zhen L, Liao JC. Acetolactate synthase from Bacillus subtilis serves as a 2-ketoisovalerate decarboxylase for isobutanol biosynthesis in Escherichia coli. Appl Environ Microbiol. 2009;75(19):6306.
Holtzclaw WD, Chapman LF. Degradative acetolactate synthase of Bacillus subtilis: purification and properties. J Bacteriol. 1975;121(3):917–22.
Gonzalez E, Fernandez MR, Larroy C, et al. Characterization and functional role of Saccharomyces cerevisiae 2,3-butanediol dehydrogenase. Chem Biol Interact. 2001;130–132(1–3):425–34.
Jiang GR, Nikolova S, Clark DP. Regulation of the ldhA gene, encoding the fermentative lactate dehydrogenase of Escherichia coli. Microbiology. 2001;147(9):2437–46.
Xiao Z, Wang X, Huang Y, et al. Thermophilic fermentation of acetoin and 2,3-butanediol by a novel Geobacillus strain. Biotechnol Biofuels. 2012;5(1):88.
Wieschalka S, Blombach B, Eikmanns BJ. Engineering Corynebacterium glutamicum for the production of pyruvate. Appl Microbiol Biotechnol. 2012;94(2):449–59.
Acknowledgements
We thank for the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions, 111 Project, the Opening Foundation of Beijing Key Laboratory of Biomass Waste Resource Utilization, Jiangsu province “Collaborative Innovation Center for Advanced Industrial Fermentation” industry development program.
Funding
This work was supported by the National Natural Science Foundation of China (Nos.: 31500065, 21778024, 31870066), the National Key Research and Development Program of China (2018YFA0900304), Program of the Key Laboratory of Industrial Biotechnology, Ministry of Education, China (KLIB-KF201902), the Science and Technology Innovation Team Foundation of Ningxia hui autonomous region (KJT2017001), the Key Research and Development Program of Ningxia hui autonomous region (2017BY069, 2019BCH01002), the General Project of Beijing Municipal Education Commission (SQKM201311417004), the National First-class Discipline Program of Light Industry Technology and Engineering (LITE2018-06).
Author information
Authors and Affiliations
Contributions
ZMR and XZ designed the experiment protocol. RMH, XZ and XJZ executed experimental work and analyzed data. XFL, YXZ and MCZ analyzed data. XZ, RMH, and TB wrote the draft and revision of the manuscript. TWY and MJX helped to revise and proofread the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Figure S1.
SDS-PAGE analysis of ALS, ALDC, and BDH in recombinant C. crenatum. The following samples and markers are shown: M: Protein marker; Lane 1: whole cell protein of C. crenatum WT; Lane 2: whole cell protein of C. crenatum S; Lane 3: whole cell protein of C. crenatum D; Lane 4: whole cell protein of C. crenatum A; Lane 5: whole cell protein of C. crenatum SD; Lane 6: C. crenatum SDA.
Additional file 2: Figure S2.
Identification of pK18-Δldh by enzyme digestion. The following samples and markers are shown: M1: λDNA/HindIII marker; M2: DL2000 marker; Lane 1: pK18-Δldh digested with EcoRI; Lane 2: pK18-Δldh digested with EcoRI and HindIII.
Additional file 3: Figure S3.
Identification of pK18-ΔbutA by enzyme digestion. The following samples and markers are shown: M1: λDNA/HindIII marker; M2: DL2000 marker; Lane 1: pK18-ΔbutA digested with EcoRI; Lane 2: pK18-ΔbutA digested with EcoRI and HindIII.
Additional file 4: Figure S4.
A: PCR identification of ldh gene knockout strains and recovery strains. M: DL2000 marker. Lane 1: ldh gene recovery strain. Lane 2 and 3: ldh gene knockout strains. B: PCR identification of butA gene knockout strains and recovery strains. M: DL2000 marker. Lane 1 and 4: butA gene recovery strains. Lane 2 and 3: butA gene knockout strains. C: PCR identification of ldh gene and butA knockout strain. M: DL2000 marker. Lane 1: PCR amplification of ΔbutA. Lane 2: PCR amplification of Δldh.
Additional file 5: Table S1.
Dry cell weight of C. crenatumΔldh, C. crenatumΔbutA and C. crenatumΔbutAΔldh. Results are shown as the mean ± standard of three replicates.
Additional file 6: Table S2.
Strains and plasmids used in this study. Kanamycin resistance is labeled as KmR, and chloramphenicol resistance is labeled as CmR.
Additional file 7: Figure S5.
PCR amplification of the knockout fragment ΔbutA.
Additional file 8: Figure S6.
PCR amplification of the knockout fragment Δldh.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, X., Han, R., Bao, T. et al. Synthetic engineering of Corynebacterium crenatum to selectively produce acetoin or 2,3-butanediol by one step bioconversion method. Microb Cell Fact 18, 128 (2019). https://doi.org/10.1186/s12934-019-1183-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-019-1183-0