
Abstract
Background
Stripe rust (also called yellow rust) is a common and serious fungal disease of wheat (Triticum aestivum L.) caused by Puccinia striiformis f. sp. tritici. The narrow genetic basis of modern wheat cultivars and rapid evolution of the rust pathogen have been responsible for periodic and devastating epidemics of wheat rust diseases. In this study, we conducted a genome-wide association study with 44,059 single nucleotide polymorphism markers to identify loci associated with resistance to stripe rust in 244 Sichuan wheat accessions, including 79 landraces and 165 cultivars, in six environments.
Results
In all the field assessments, 24 accessions displayed stable high resistance to stripe rust. Significant correlations among environments were observed for both infection (IT) and disease severity (DS), and high heritability levels were found for both IT and DS. Using mixed linear models, 12 quantitative trait loci (QTLs) significantly associated with IT and/or DS were identified. Two QTLs were mapped on chromosomes 5AS and 5AL and were distant from previously identified stripe rust resistance genes or QTL regions, indicating that they may be novel resistance loci.
Conclusions
Our results revealed that resistance alleles to stripe rust were accumulated in Sichuan wheat germplasm, implying direct or indirect selection for improved stripe rust resistance in elite wheat breeding programs. The identified stable QTLs or favorable alleles could be important chromosome regions in Sichuan wheat that controlled the resistance to stripe rust. These markers can be used molecular marker-assisted breeding of Sichuan wheat cultivars, and will be useful in the ongoing effort to develop new wheat cultivars with strong resistance to stripe rust.
Similar content being viewed by others


Background
Wheat is an important food crop worldwide. However, wheat stripe rust caused by Puccinia striiformis f. sp. tritici (Pst) seriously threatens wheat production. Stripe rust is an airborne fungal disease, and Pst spores can be spread rapidly over long distances by the wind. Many wheat cultivars have become susceptible because newly developed races in the Pst population are virulent against the resistance genes in wheat cultivars. Stripe rust is a devastating disease in the cool, temperate wheat growing regions of the world, such as Asia, Europe, North America, South America, the Middle East and Africa [1]. China has the largest region in the world where stripe rust is endemic. The major endemic areas of stripe rust are the winter-wheat growing regions in the Northwest, Southwest and North of China, and the spring-wheat growing regions in Northwest China [2]. Sichuan Province in Southwest China is a hotspot for Pst to overwinter and oversummer. The Pst races CYR32 and CYR33 were detected in 1994 and 1997, and developed to predominant epidemic virulence levels in 2000 and 2007, respectively. This resulted in the loss of resistance to stripe rust in many cultivars, including Kangyin 655, Suwon 11 and Fan 6, as well as their derived cultivars [3]. Some cultivars retained resistance to CYR32 and CYR33 (e.g., cultivars with the gene Yr24/26, including Guinong 22 and its derived cultivars) and were planted as the main varieties in major regions of Sichuan Province. However, a new virulent pathogen, CYR34, appeared in 2009. In 2015, it became the main source of virulence against the Yr24/26-containing cultivars [4]. The variability of a pathogen is closely related to the evolution of host varieties. Breeding wheat cultivars for resistance to stripe rust is the most efficient, safe and environmentally sound strategy to control this disease.
Stripe rust resistance genes can be classified as race-specific and non-race-specific based on their effectiveness against different Pst races. A race-specific resistance gene confers resistance to a single race or many races, but is ineffective against other races. This resistance occurs in all growth stages, from seedling to adult stage. Therefore, it can be considered as seedling or all-stage resistance (ASR). Wheat cultivars with race-specific resistance genes have high-level resistance but may become susceptible when virulent races appear. In contrast, a non-race-specific resistance gene confers resistance to all races, Genes conferring adult-plant resistance (APR) are usually non-race-specific. High-temperature adult-plant resistance is a type of APR that is more effective at higher temperatures [5]. Because it is non-race specific, APR is usually durable, but often incomplete, and the resistance level is lower than that of ASR. Combining APR and ASR genes is the optimal method to develop cultivars with adequate durable resistance to stripe rust [6,7,8].
The use of a limited number of genetic stocks has decreased the genetic variation level in wheat breeding. Reduced diversity has become a bottleneck for stripe rust resistance improvement. To broaden the genetic basis of wheat cultivars, many resistance genes have been identified in wheat landraces and cultivars [9,10,11,12,13,14,15,16,17,18,19,20,21,22,23]. Wheat landraces have been shaped by traditional cultures and local cropping systems during long-term agricultural development. They adapt to different environments and collectively have a high diversity level and stable heritability [24]. Compared with cultivars, landraces have a greater diversity of genes that respond to abiotic and biotic stresses. Therefore, wheat landraces are valuable resources for stripe rust resistance breeding. The objectives of the present study were to (1) evaluate 244 Sichuan wheat accessions for resistance against Pst at the adult-plant stage in multiple years and field locations in Sichuan Province; (2) assess the genetic diversity, population structure and linkage disequilibrium (LD) pattern of the Sichuan wheat collection using the 55 K single nucleotide polymorphism (SNP) microarray; and (3) identify new stripe rust resistance loci using the genome-wide association study (GWAS) method.
Results
Phenotypic variation and diversity of stripe rust resistance
The results of a correlation analysis revealed significant correlations among the six environments when IT and DS were separately analyzed at the adult-plant stage (Table 1). We identified 52 accessions that showed stable resistance to stripe rust in the six test environments. Among them, 24 accessions showed stable high-level resistance to stripe rust, including 12 landraces and 12 cultivars (Additional file 1). To reduce the environmental impacts on the stripe rust response, the best linear unbiased prediction (BLUP) values were calculated using a linear model with IT or DS in the six environments (Additional file 1). Based on the BLUP values, the IT distribution revealed that 53 landraces (67.09% of 79 landraces) exhibited stripe rust resistance; among them, 17 (21.52% of 79 landraces) landraces were highly resistant. For cultivars, 82 (49.70% of 165 cultivars) accessions showed resistance to stripe rust, and 33 (20.00% of 165 cultivars) cultivars were highly resistant. However, 26 (32.91% of 79 landraces) landraces and 83 (50.30% of 165 landraces) cultivars were susceptible to stripe rust (Fig. 1a; Additional file 2). Similarly, the DS distribution revealed that there were more resistant landraces than resistant cultivars (Fig. 1b; Additional file 2). The IT and DS data are summarized in Table 2. The mean IT and DS values of the landraces were lower than those of the cultivars in each test environment. When the environmental impact was removed from the BLUP values, the landraces still had higher resistance levels than did the cultivars. The mean IT values in landraces and cultivars were 2.16 and 2.52, respectively, and the mean DS values of landraces and cultivars were 23.23 and 37.15, respectively. Both IT and DS exhibited high heritability levels (0.91 and 0.90, respectively).

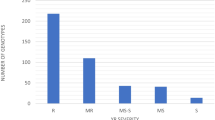
The distribution of infection type (IT) and disease severity (DS) for stripe rust based on BLUP values. a exhibited the IT distribution. b exhibited the DS distribution. IT, infection type; DS, disease severity; HR, highly resistance; MR, moderately resistance; MS, moderately susceptible; S, susceptible; The data are supplied in Additional file 2
The 165 cultivars were collected from our institute and several other universities in Sichuan Province, and they were classified into four periods on the basis when they were bred. Periods 1, 2, 3, and 4 were from 1997 to 2001, 2002 to 2006, 2007 to 2011 and 2012 to 2016, respectively. There were 16, 55, 43, and 51 cultivars bred in periods 1, 2, 3, and 4, respectively (Additional file 1). The fewest cultivars were bred in period 1. Based on the analyses of BLUP values for IT, the cultivars bred in periods 2 (IT = 2.35) and 3 (IT = 2.35) displayed the highest resistance against stripe rust, while the cultivars bred in period 1 showed the lowest resistance (IT = 3.31). The cultivars bred in period 4 (IT = 2.61) showed moderate resistance. The Shannon–Weaver diversity index (H′) values were 0.80, 0.96, 0.98, and 0.98 for breeding periods 1, 2, 3, and 4, respectively (Table 3).
Genetic diversity analysis
A total of 44,059 SNP markers were obtained for the 244 accessions based on the criteria of missing values ≤10% and minor allele frequency (MAF) ≥ 5%. Among them, 16,026, 16,770 and 11,263 SNP markers were mapped on sub-genomes A, B and D, respectively (Table 4). The map lengths of sub-genomes A, B and D were 4930.66, 5177.04 and 3947.75 million base pairs (Mb), respectively, and the average marker densities for the three sub-genomes were 3.3, 3.3 and 2.9 markers per Mb, respectively. Chromosome 6A had the highest marker density at 4.0 markers per Mb, while chromosome 4D had the least markers (944) and had the lowest marker density at 1.9 markers per Mb. Among the three sub-genomes, sub-genome B showed the lowest major allele frequency (0.695), highest gene diversity (0.396), and highest polymorphism information content index (PIC) (0.312) values. Sub-genome D exhibited the highest major allele frequency (0.719), lowest gene diversity (0.369), and lowest PIC (0.293) values. Among the chromosomes, chromosome 4A showed the highest major allele frequency (0.778), lowest gene diversity (0.316), and lowest PIC (0.259) values, while chromosome 5B exhibited the lowest major allele frequency (0.665), greatest gene diversity (0.422), and highest PIC (0.329) values.
There were significant differences in genetic diversity between landraces and cultivars (Fig. 2; Additional file 3). The major allele frequencies were significantly higher in landraces than in cultivars among the three sub-genomes and 21 chromosomes, except for chromosome 2A. The gene diversity and PIC values of cultivars were significantly higher than those of landraces among the three sub-genomes and 21 chromosomes, except for the gene diversity of chromosome 2A.

The genetic diversity between landraces and cultivars using SNP markers. SNP, single nucleotide polymorphism; PIC, polymorphism information content index; The data are supplied in Additional file 3
There were differences in genetic diversity among the four breeding periods. The cultivars bred in periods 2 and 3 exhibited the highest gene diversity (0.34) and PIC (0.27) values. The cultivars bred in period 1 had the lowest gene diversity (0.30) and the lowest PIC (0.24) values. The cultivars bred in period 4 had medium diversity (0.31) and PIC (0.25) values (Table 3).
Population structure, kinship and LD analyses
The population structure (Q-matrix) was calculated using the 44,059 SNP markers for 244 accessions based on the delta K (ΔK) method of Bayesian clustering. The accessions were classified into two sub-populations, 1 (SP1) and 2 (SP2) (Fig. 3). In total, 78 and 166 accessions were included in SP1 and SP2, respectively. All the landraces were classified in SP1, except AS661599 and AS1679, which were included in SP2. All the cultivars were classified in SP2, except for one cultivar Xifu14, which was included in SP1. According to the mean BLUP-based IT and DS values, the landraces in SP1 had higher resistance levels (IT = 2.14, DS = 22.50) than the cultivars in SP2 (IT = 2.53, DS = 37.41) (Fig. 3; Additional file 4). In addition, the cultivars in SP2 showed higher genetic diversity (PIC = 0.27, gene diversity = 0.33) and higher phenotypic diversity indexes (H′ = 0.99 for IT and H′ = 0.75 for DS). The landraces in SP1 showed lower genetic diversity (PIC = 0.15, gene diversity = 0.19) and lower phenotypic diversity indexes (H′ = 0.84 for IT and H′ = 0.58 for DS) (Additional file 4).

Population structure of 244 Sichuan wheat accessions. a Hierarchical clustering based on Ward method with K-matrix indicated the 244 accessions could be divided into three clusters (cluster 1, cluster 2 and cluster 3). b Heat map of the IBS (identity by state) relative K-matrix. c The population structure of 244 accessions with Bayesian clustering analysis. Two colors stand for 2 different compositions. Different color area of each line represented different proportion of composition. The Sub-population 1 (SP1) mainly showed as green color. The Sub-population 2 (SP2) mainly showed as red color. d Heat map of IT and DS (IT, infection type; DS, disease severity) with BLUP (best linear unbiased prediction) values across six environments in the field. Blue to yellow to red lines show resistance to intermediate to susceptibility to stripe rust of the corresponding accessions. e Percentage memberships of accessions from two subpopulations based on Q-matrix. The landraces showed as blue color and cultivars showed as red color. The red dashed divided the K-matrix into three parts according the hierarchical clustering and the black dashed divided the items into two parts according the Bayesian clustering
To understand the genetic relationships among the 244 accessions, an identity-by-state relative kinship matrix (K-matrix) was estimated. The heat map of the K-matrix is shown in Fig. 3. Unlike the Bayesian clustering analysis with the Q-matrix, the hierarchical clustering based on the Ward method with the K-matrix divided the 244 accessions being divided into three clusters. There were 54, 25, and 165 accessions in clusters 1, 2, and 3, respectively. As shown in Fig. 3, the landraces classified as SP1 by Bayesian clustering were further classified as clusters 1 and 2 by hierarchical clustering. Relatively, cluster 2 comprised cultivars, except for cultivars Yumai 1 and Xifu 14, which were assigned to cluster 1.
The pairwise measure of LD was estimated based on the allele frequency correlations (r2) between significant pairs of intra-chromosomal SNP markers with physical distances (Fig. 4). The half-decay distance was 2.12 Mb when the LD declined to 50% (r2 = 0.65) of its initial value. We defined the significant associated loci on the same chromosome within the 2.12-Mb genomic region as being in the same QTL block.

Genome-wide average linkage disequilibrium (LD) decay over physical distances based on 44,059 SNP markers. The red curve represents the model fits to LD decay. The blue line represents the half LD decay distance was 2.12 Mb when the LD declined to 50% (r2 = 0.65) of its initial value. Mb, million base pairs
Genome-wide association analyses
Based on the mixed linear model analysis with Q + K as covariates using 44,059 SNP markers, GWASs were performed for the IT and DS of stripe rust against the Pst populations within each of the six environments at the adult-plant stage. There were 7 and 12 high-confidence loci associated with IT and DS at P < 0.001, and they were located on the long arms of chromosomes 1B, 3D, 5A, 5B, and 7B and the short arms of chromosomes 1A, 5A, 6A, 6B, and 7A (Table 5). The loci linked to AX-111488534 were associated with both IT and DS, and the phenotypic variation explained (PVE) was up to 13.6 and 17%. Additionally, we detected 18 high-confidence loci associated with resistance to stripe rust. The high-confidence loci in the six test environments are displayed as Manhattan plots with P values across 21 wheat chromosomes in Fig. 5. Furthermore, 12 QTLs were identified according to the half-decay distance 2.12 Mb. Chromosomes 1BL, 5A and 5BL contained two QTLs each. Also, one QTL was identified on each of chromosomes 1AS, 3DL, 6AS, 6BS, 7AS, and 7BL. Two of the QTLs were potentially novel based on their unique chromosomal locations when referenced to the consensus [25] and physical [26] maps.

The P values of associated loci with infection type (IT) and disease severity (DS) displayed as Manhattan plots. The associated loci with IT and DS in six environments were displayed as Manhattan plots with P values across 21 wheat chromosomes. The significant associated locus was considered as –log10(P) > 3 which upper the blue lines. IT, infection type; DS, disease severity. CZ15 = Chongzhou 2015, CZ16 = Chongzhou 2016, MY16 = Mianyang 2016, CZ17 = Chongzhou 2017, MY17 = Mianyang 2017, WJ17 = Wenjiang 2017
Favorable allele analyses
There were 18 SNP markers significantly associated with stripe rust in at least three environments. Of them, 12 favorable alleles were identified based on significant differences in IT and DS between the accessions with and without favorable alleles as detected by t-test. According to the total frequency calculation, the landraces contained more favorable alleles than did the cultivars (Table 6; Additional file 5). The correlation analyses revealed a significant negative correlation between favorable allele number and the reaction against stripe rust (Additional file 6). The materials with more favorable alleles, and they exhibited higher resistance to stripe rust. In contrast, those with fewer favorable alleles had weaker resistance (or higher susceptibility). These results support the use of a combination of several loci for wheat disease resistance breeding.
Discussion
Phenotypic and genetic diversity
To identify the stripe rust resistance genes in our materials, we used a uniform mixture of seven Pst races to infect 244 accessions in the field. The seven Pst races were the predominant races in China at present, of which CYR34 (V26) is the latest race virulent to Yr24 (= Yr26 and YrCH42). In total, 12 landraces (15.19% of 79 landraces) and 12 (7.27% of 165 cultivars) cultivars showed stable high resistance to stripe rust in all the test environments. A higher percentage of landraces than cultivars exhibited resistance to stripe rust. The BLUP values were calculated to reduce the environmental impact. The mean IT and DS values also revealed that the landraces had higher resistance than did the cultivars, independent of the environment and when using BLUP values (Table 2). In addition, the frequency of favorable alleles was higher in landraces than in cultivars (Table 6; Additional file 5). Thus, the landraces contained abundant resistance genes and were identified as the elite germplasms for stripe rust resistance breeding. The wheat landraces are the outcome of natural selection, and reportedly have rich elite genes and a good affinity for hybridization. Chinese Spring is a well-known landrace derived from China (Sichuan Province) that is used as the international reference (IWGSC, http://www.wheatgenome.org/). The APR and pleiotropic gene Lr34/Yr18 has maintained stable resistance to multiple fungal pathogens for a long time. It was derived from Chinese landraces and is widely used in modern wheat cultivars [14, 16, 25, 27,28,29]. In addition, many stripe rust resistance genes have been identified in landraces, such as Yr45 [17], Yr52 [18], Yr53 [30], Yr59 [31], Yr62 [32], Yr64 [33], Yr65 [33], Yr72 [34] and Yr79 [22] as well as many other genes and QTLs [25].
Although more landraces than cultivars were resistant to stripe rust, the cultivars from Sichuan Province showed good resistance. In total, 82 (49.70% of 165 cultivars) cultivars showed resistance against stripe rust and maintained resistance for several years, as a result of the use of resistance genes in breeding. In addition, genotype analyses revealed that the cultivars had higher genetic diversity and more phenotypic variation than did the landraces, as assessed by the 44,059 SNP markers (Fig. 2; Additional file 3). We classified the materials into four periods according to breeding years. The analyses of phenotype and genotype showed that the cultivars bred in period 1 had the lowest H′ (0.80), resistance (IT = 3.31), gene diversity (0.30), and PIC (0.24) values. While those bred in periods 2–4 showed significantly higher resistance, H′, gene diversity, and PIC values (Table 3). The phenotypic diversity index also increased in more recent breeding years. The cultivars in period 1 were bred between 1997 and 2001, when were few superior stocks for breeding. Consequently, fewer accessions were bred in those years. The limited breeding stocks resulted in less diversity and a narrow genetic background, which caused a breeding bottleneck. We speculated about the reasons for the significant differences between period 1 and the other three periods. One possible explanation is the ‘Cooperative China/International Maize and Wheat Improvement Centre shuttle Breeding Program’, which was signed in 1987 [35]. Sichuan Province was one of three designated shuttle breeding areas, but it took about 10 years to exchange information and germplasms to prepare for breeding. Therefore, the breeding of new cultivars containing foreign germplasm in their pedigrees started in about 2000 in Sichuan Province [36]. This would explain the obvious differences in stripe rust resistance, phenotypic variation, and genetic diversity between period 1 and the other periods (periods 2–4) (Table 3). The introduction of new resources is an efficient strategy to improve genetic diversity and broaden the genetic basis. Thus, landraces, as a major resource for resistance to stripe rust, are elite germplasms for wheat breeding.
Population structure and genetic relationships
Whether the population structure was established by a Bayesian model-based clustering or a Ward method-based genetic relationship with the K-matrix, the cultivars were mainly classified in the same cluster and the landraces were mainly classified in other clusters (Fig. 3). The significant differences in phenotypic variation and genetic diversity supported the classification based on the Q-matrix, which demonstrated the relatively distant genetic relationships between landraces and cultivars. The accessions in SP1 had higher resistance to stripe rust, lower phenotypic diversity and lower genetic diversity values than accessions in SP2 (Additional file 4). Compared with Bayesian clustering, the hierarchical clustering classified the cultivars into one cluster, cluster 3, but further divided the landraces into two clusters (clusters 1 and 2). The hierarchical clustering also indicated the differences in genotyping between landraces and cultivars (Fig. 3). These results indicated that the landraces in this study are the elite germplasms that can be used to broaden the genetic background and improve genetic diversity.
Comparison of high-confidence loci with reported yellow rust resistance (Yr) genes and QTLs
In total, 18 high-confidence loci were detected. Based on the half-decay distance of 2.12 Mb, we classified the 18 high-confidence loci into 12 QTLs, which were located on 1AS, 1BL, 3DL, 5AS, 5AL, 5BL, 6AS, 6BS, 7AS and 7BL. The QTLs were named as follows: Qyrsicau-1AS, Qyrsicau-1BL, Qyrsicau-3DL, Qyrsicau-5AS, Qyrsicau-5AL, Qyrsicau-5BL, Qyrsicau-6AS, Qyrsicau-6BS, Qyrsicau-7AS, and Qyrsicau-7BL (Table 5).
Qyrsicau-1AS was located at 55.9 Mb in the distal region of 1AS, which was covered by QYr.sgi-1A.1 [37]. Qyrsicau-1BL.1 and Qyrsicau-1BL.2 were located at 670 Mb and 681 Mb, respectively. The marker AX-111488534 linked to Qyrsicau-1BL.2 explained up to 17.0% PVE and was associated with both IT and DS. There were four reported Yr genes and more than 10 QTLs near this region. Qyrsicau-1BL.1 was close to Yr29 and overlapped with Qyrsun-1BL [38]. Additionally, Qyrsicau-1BL.2 was very close to QYr.tam-1B, which was linked to XwPt-668,027 [39] according to the physical map IWGSC RefSeq v1.0 [26].
In group 3 chromosomes, only one QTL (Qyrsicau-3DL) was located at 3DL. The associated marker AX-109329567 was located at 595 Mb, and the PVE ranged from 8.0 to 10.1%. Yr71 on 3DL was identified as an APR gene and was located 1.8 cM from marker Xgwm114b/KASP_8306/KASP_17207/KASP_16434 [40], which was close to Qyrsicau-3DL.
There were four QTLs located in group 5 chromosomes, two on chromosome 5A and two on chromosome 5B. Qyrsicau-5AS was located at the distal region of 5AS (Fig. 6). There were two QTLs reported at 5AS. The QTL linked with wsnp_Ex_c807_1586396-5AS [41] was identified as an APR and was ~ 80 Mb from Qyrsicau-5AS. Another QTL, QYr.cau-5AS [42] was a major QTL that exhibited a high PVE, but Qyrsicau-5AS was a minor QTL with a PVE of 7.1 to 8.1%. Two previously reported QTLs at 5AS were different from Qyrsicau-5AS identified in our research, suggesting that Qyrsicau-5AS is a novel QTL. Another QTL on 5A, Qyrsicau-5AL (Fig. 6), was identified as a potential novel QTL located between QYrtb.pau-5A [43] and QYR5-5A [44] on the consensus map [25]. Another two QTLs were located at the middle region of 5BL. Qyrsicau-5BL.1 had a PVE of 9.4%, and Qyrsicau-5BL.2 had a PVE of 8.8%. Qyrsicau-5BL.1 was mapped at 554 Mb on 5BL and was very close to the QTL [41] linked to marker wsnp_Ex_c2582_4804223. Another QTL on 5BL was Qyrsicau-5BL.2, which was covered by QYr.tem-5B.2 [45].

The position of the potentially novel QTLs on the chromosome in this study. The chromosome lengths were all standardized to the same relative length. QTLs marked as red color on the left side of chromosomes were the potentially new QTLs in this study. The reported genes and QTLs were marked as black color and mapped on the left and right side of the chromosomes separately which referred Wang and Chen (2017) [25]
Two QTLs were located in group 6 chromosomes. Qyrsicau-6AS was located at the end of 6AS and covered by QYr.wgp-6AS [46]. Qyrsicau-6BS was located at the distal end of 6BS and was very close to previously reported QYr.ufs-6B [37].
The group 7 chromosomes contained 2 QTLs. Qyrsicau-7AS was mapped at the 81-Mb position on chromosome 7AS and was covered by the known QYr.inra-7A [47] and QYr.sun-7A [48] based on the consensus map [25]. Qyrsicau-7BL was associated with AX-110518451, which was covered by QYr.caas-7BL.2 [49], and the PVE ranged from 6.4 to 11.3%.
Finally, six QTLs were covered by reported genes or QTLs. Four QTLs were very close to reported genes or QTLs, and two QTLs were identified as potential novel QTLs. Because of the lack of markers in common with the consensus map, and because only the rough locations of some reported genes and QTLs are known, these QTLs need to be analyzed in more detail. Allelism tests and fine mapping are effective ways to identify real novel resistance genes in future studies.
Analyses of putative candidate genes in two potential novel QTLs
Based on the Chinese Spring reference RefSeq v1.0 (IWGSC) and RefSeq Annotation v1.1 [26], 12 and 36 genes were identified within the Qyrsicau-5AS and Qyrsicau-5AL regions, respectively. By referencing the disease resistance-related signaling pathway, resistance-related protein family, and genes reported previously, nine candidate genes (Additional file 7) related to stripe rust resistance were identified, which were homologous to those in Arabidopsis (Arabidopsis thaliana), rice (Oryza sativa L. ssp. japonica) and maize (Zea mays).
One putative candidate gene, TraesCS5A02G079700, was detected in Qyrsicau-5AS and orthologous to Os03g0144800 in rice (O. sativa subsp. japonica) and MUR3 (xyloglucan galactosyltransferase) in Arabidopsis. Both these genes belong to the glycosyltransferase 47 family and are involved in the salicylic acid-mediated signaling pathway [50]. Salicylic acid is a plant hormone and that functions as a signaling molecule in systemic immunity in response to pathogen infection [51].
Eight presumptive candidate genes (TraesCS5A02G364700, TraesCS5A02G365300, TraesCS5A02G365600, TraesCS5A02G365700, TraesCS5A02G365800, TraesCS5A02G367100, TraesCS5A02G367700 and TraesCS5A02G367900) were present in Qyrsicau-5AL. TraesCS5A02G364700, TraesCS5A02G365300, TraesCS5A02G365600 and TraesCS5A02G365700 were all homologous to the wall-associated receptor kinase WAK and to At5g49770 in Arabidopsis. WAK belongs to the serine/threonine (Ser/Thr) protein kinase family, and it is involved in defense responses to fungal infections and to salicylic acid [52, 53]. The other gene, At5g49770, also belongs to the Ser/Thr protein kinase family. This gene is a leucine-rich repeat receptor-like protein kinase, which may be related to disease resistance [54,55,56]. The TraesCS5A02G365800 orthologous genes, KDEL-tailed cysteine endopeptidase CEP1, cysteine proteinase RD21A, and senescence-specific cysteine protease SAG12, which all belong to the peptidase C1 family and are involved in defense responses to fungal infections [57,58,59]. Furthermore, cysteine protease plays an important role in immunity and is involved in elicitor-stimulated programmed cell death [60]. The symptoms in infected wheat include the appearance of hypersensitive flecks or necrosis, which are characteristics of programmed cell death. TraesCS5A02G367100 is orthologous with the Arabidopsis genes BAM1 (derived from barley any meristem 1), BAM2, BAM3 receptor-like kinases, and RLK5 (receptor-like protein kinase 5), the rice gene FON1 (floral organ number 1), and the maize gene TD1 (thick tassel dwarf 1). All of these genes belong to the Ser/Thr protein kinase family and contain a disease resistance-related domain (leucine-rich repeat receptor-like protein kinases). TraesCS5A02G367700 is homologous to the rice gene Os12g0486900, and the Arabidopsis genes At4g00960, LRK10L-2.4, and PR5K (pathogenesis-related protein 5-like receptor kinase). They also belong to the Ser/Thr protein kinase family. In particular, the gene LRK10L-2.4 is a leaf rust resistance locus and a receptor-like gene in Arabidopsis [61]. TraesCS5A02G367900 is aligned with rice gene Os03g0670100 and the Arabidopsis gene TGD3 (trigalactosyldiacylglycerol 3), which contain ATP-binding cassette (ABC) transporters. We considered this gene as a candidate because Yr18/Lr34 conferring APR also contains an ABC transporter [62].
The candidate gene analyses were limited by the use of the Chinese Spring reference genome because dispensable genes or variations exist among individual genotypes within and between species. However, the collinear alignment analyses have provided us with important clues to identify candidate genes in our accessions. We will study these putative candidate genes by reverse genetics in future studies.
Conclusions
To use multiple resistance loci to breed for stable and durable resistance to stripe rust, more and new resistance loci need to be discovered. Wheat landraces are elite germplasms for the exploration of new resistance genes. Here, we identified 24 accessions harboring resistance genes that showed stable high-level resistance in six test environments. The GWAS results revealed 12 QTLs associated with 18 SNP markers. Among them, there were 12 favorable alleles for stripe rust resistance and two potentially novel loci. Finally, we predicted nine candidate genes related to stripe rust resistance. Our study provides SNP markers associated with resistance loci that will be useful for marker-assisted selection in wheat breeding.
Methods
Plant materials
A collection of 244 Sichuan wheat accessions was assembled mainly from germplasm bank accessions collected and stored at the Triticeae Research Institute, Sichuan Agricultural University (germplasm numbers abbreviated AS) and the Chinese Crop Germplasm Resources Bank (germplasm numbers abbreviated ZM), China. We analyzed a total of 79 Sichuan wheat landraces and 165 commercial varieties (Additional file 1), which had been derived from different breeding units (Sichuan Academy of Agricultural Sciences, Mianyang Academy of Agricultural Sciences, Neijiang Academy of Agricultural Sciences, Chengdu Institute of Biology, Chinese Academy of Sciences, Sichuan Agricultural University and Southwest University of Science and Technology) in Sichuan Province since 1997. The 165 cultivars were bred between 1997 and 2016. We classified them into four 5-years breeding periods according to the time they were bred. Periods 1–4 were from 1997 to 2001, 2002 to 2006, 2007 to 2011, and 2012 to 2016, respectively.
Phenotyping for stripe rust resistance in field environments
The 244 accessions were tested for field stripe rust responses at the adult stage after artificial inoculation in the following six environments in Sichuan: Chongzhou (30°33′N 103°39′E) in 2015, 2016 and 2017; Mianyang (31°23′N 104°49′E) in 2016 and 2017, and Wenjiang (30°43′N 103°52′E) in 2017, referred to as CZ15, CZ16, MY16, CZ17, MY17 and WJ17, respectively. In all test environments, 20 seeds of each accession were planted in rows 2-m in length spaced 30-cm apart, with individual plants spaced 10-cm apart. Susceptible wheat varieties SY95–71 and Taichung 29 were planted every 20 rows and around each plot as spreader rows to increase the uniformity of the stripe rust inoculum across the trials. The accessions were inoculated with urediniospores of uniformly mixed Pst isolates (CYR32, CYR33, CYR34, G22–14, Su11–4, Su11–5, and Su11–7) (Table 7) [4, 42, 63,64,65,66] when plants had developed to the shooting stage in January. The Pst isolates were the prevalent races in China. The stripe rust reaction response collection was initiated when SY95–71 and Taichung 29 displayed DS levels of up to 80%. The stripe rust response was evaluated three times, once per week. We scored IT using the 0–4 scale described by Stakman et al. (1962) [67], as follows: highly resistant (HR, 0–1), moderately resistant (MR, 2), moderately susceptible (MS, 3), and susceptible (S, 4). The DS was scored as percentage of infected leaf area (0, 5, 10, 20, 40, 60, 80%, or 100%) according to the rules for Monitoring and Forecast of wheat stripe rust (National Standard of the People’s Republic of China, GB/T 15795–2011).
Phenotypic data analyses
To eliminate the environmental impact on stripe rust, we used a linear model with random effects for variance components to calculate the BLUP values with the lme4 package in R [68]. Based on the BLUP values, the analysis of variance and a correlation analysis with the Pearson’s method were computed using SPSS 20.0 (IBM Corp., Armonk, NY, USA). The broad-sense heritability (H2) estimates for IT and DS were calculated across six test environments using the lme4 package [68] with the formula H2 = VG/(VG + VE), where VG and VE represent the genotypic and environmental variances, respectively [69]. The phenotypic variations were confirmed by the value range, average values, standard deviation (STDEV), and coefficient of variation (CV) of all traits in six environments and BLUP values. The H′ was calculated for IT and DS using BLUP values [70].
Genotyping and molecular diversity analyses
For each accession, genomic DNA was extracted from mixed leaves of five 1-week-old seedlings using a plant DNA kit (Biofit Co., Chengdu, China). The 244 accessions were genotyped using the 55 K SNP microarray (Affymetrix Axiom Wheat55K) at the China Golden Marker Biotechnology Company Ltd. (Beijing, China). Markers with missing values of ≤10% and MAFs of ≥5% were selected for the linkage analysis. Statistical analyses of PIC, major allele frequency, and gene diversity were performed using the software POWERMARKER v3.25 [71] to determine genetic diversity. These indices of genetic diversity were used to compare the extent of molecular diversity among different sub-genomes and chromosomes. The same comparisons were performed between landraces and cultivars, as well as among different classifications.
Population structure, kinship and LD analyses
To analyze the population structure (Q-matrix), Bayesian model-based clustering was performed in STRUCTURE v2.3.4 using 44,059 SNP markers (missing ≤10% and MAF ≥ 5%) with the ΔK method [72, 73]. In total, five independent STRUCTURE runs were performed with K values from 2 to 10 using the admixture model with 100,000 replicates for burn-in length and 100,000 replicates for Markov chain Monte Carlo iterations. The optimal K value was chosen based on the ΔK method [73], which was implemented using the web-based informatics tool STRUCTURE HARVESTER [74].
The identity-by-state relative K-matrix was estimated between pairs of accessions as a measure of relatedness. Heat maps were generated with the pheatmap R package v1.0.8 [75] based on the K-matrix. The pairwise measure of LD was estimated as squared allele frequency correlation (r2) values between pairs of intra-chromosomal markers with known chromosomal positions. Significant pair-wise markers were chosen using the criteria pDiseq < 0.001 and r2 > 0.1. The LD decay plot and half-decay distance were generated from the r2 value and the distance between markers using ggplot2 package with R [76]. All the high-confidence associated loci that were included in the half-decay distance regions of the same chromosome were defined as the same QTL block. The K-matrix and LD were analyzed using TASSEL v5.2.38 [77].
Genome-wide association analyses
A GWAS was performed on 244 wheat accessions with the software TASSEL v5.2.38 based on a mixed linear model with Q and K as covariates [77,78,79]. The marker-trait associations for responses to stripe rust were identified from 44,059 SNP markers using IT and DS values collected from six test environments. The significant association loci were considered if the p-value < 0.001. The associated loci were visualized with a Manhattan plot using ggplot2 package with R [76]. To obtain confident marker-trait associations, highly associated loci detected in at least three environments were selected for further analyses. The statistical analysis for favorable alleles was performed using SPSS 20.0 (IBM Corp).
Analyses of high-confidence significant associated resistance loci
To determine whether our associated loci were novel, we compared the locations of QTLs in this study with those of previously reported Yr genes and QTLs based on an integrated map. This map, included the 78 permanently named Yr genes, 67 temporarily designated Yr genes and 327 QTLs, and was constructed by Wang and Chen (2017) [25] using the software BioMercator v4.2 [80]. Some correlations between QTLs in this study and reported Yr genes or QTLs could not be determined because the associated markers were absent from the consensus map. The physical position comparison was carried out using the Chinese Spring reference (IWGSC RefSeq v1.0) [26] with BLAST+ v2.7.1 [81].
Analyses of presumptive candidate genes
Here, we identified the gene sequences of potential novel QTLs based on the Chinese Spring reference genome (IWGSC RefSeq v1.0) and RefSeq Annotation v1.1 [26]. Collinear alignment with A. thaliana, Brachypodium distachyon, O. sativa L. ssp. japonica, Z. mays L. and Hordeum vulgare was carried out to determine candidate genes. The candidate genes were obtained using online BLAST at the EnsemblPlants website (https://plants.ensembl.org/Multi/Tools/Blast?db=core) with default parameters and using DIAMOND blastx [82] (E value < 10− 10 and bitscore > 60) based on the SWISS-PROT database [83].
Abbreviations
- ABC:
-
ATP-binding cassette
- APR:
-
Adult plant resistance
- ASR:
-
All-stage resistance
- BLUP:
-
Best linear unbiased prediction
- CV:
-
Coefficient of variation
- DS:
-
Disease severity
- GWAS:
-
Genome-wide association study
- HR:
-
Highly resistance
- H‘ :
-
Shannon-Weaver diversity index.
- H 2 :
-
Broad sense heritability
- IT:
-
Infection type
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- Mb:
-
Million base pairs
- MR:
-
Moderately resistance
- MS:
-
Moderately susceptible
- PIC:
-
Polymorphism information content index
- Pst :
-
Puccinia striiformis f. sp. tritici
- PVE:
-
Phenotypic variation explained
- QTL:
-
Quantitative trait loci
- S:
-
Susceptible
- SNP:
-
Single nucleotide polymorphism
- SP:
-
Sub-population
- STDEV:
-
Standard deviation
- Yr :
-
Yellow rust resistance
References
Wellings CR. Global status of stripe rust: a review of historical and current threats. Euphytica. 2011;179(1):129–41.
Zeng SM, Luo Y. Long-distance spread and interregional epidemics of wheat stripe rust in China. Plant Dis. 2006;90(8):980–8.
Chen WQ, Wu LR, Liu TG, Xu SC, Jin SL, Peng YL, et al. Race dynamics, diversity, and virulence evolution in Puccinia striiformis f. sp. tritici, the causal agent of wheat stripe rust in China from 2003 to 2007. Plant Dis. 2009;93(11):1093–101.
Liu B, Liu T, Zhang Z, Jia Q, Wang B, Gao L, et al. Discovery and pathogenicity of CYR34, a new race of Puccinia striiformis f. sp. tritici in China. Acta Phytopathol Sin. 2017;47:681–7.
Chen XM. High-temperature adult-plant resistance, key for sustainable control of stripe rust. Am J Plant Sci. 2013;4(03):608–27.
Chen XM, Line RF. Gene action in wheat cultivars for durable, high-temperature, adult-plant resistance and interaction with race-specific, seedling resistance to Puccinia striiformis. Phytopathology. 1995;85:567–72.
Singh RP, Huerta-Espino J, William HM. Genetics and breeding for durable resistance to leaf and stripe rusts in wheat. Turk J Agric For. 2005;29(2):121–7.
Chen XM. Integration of cultivar resistance and fungicide application for control of wheat stripe rust. Can J Plant Pathol. 2014;36(3):311–26.
Wang L, Ma J, Zhou R, Wang X, Jia J. Molecular tagging of the yellow rust resistance gene Yr10 in common wheat, PI 178383 (Triticum aestivum L.). Euphytica. 2002;124(1):71–3.
Singh RP, Nelson JC, Sorrells ME. Mapping Yr28 and other genes for resistance to stripe rust in wheat. Crop Sci. 2000;40(4):1148–55.
Herrera-Foessel SA, Lagudah ES, Huerta-Espino J, Hayden MJ, Bariana HS, Singh D, et al. New slow-rusting leaf rust and stripe rust resistance genes Lr67 and Yr46 in wheat are pleiotropic or closely linked. Theor Appl Genet. 2011;122(1):239–49.
Dong Z, Hegarty JM, Zhang J, Zhang W, Chao S, Chen X, et al. Validation and characterization of a QTL for adult plant resistance to stripe rust on wheat chromosome arm 6BS (Yr78). Theor Appl Genet. 2017;130(10):2127–37.
Lupton FGH, Macer RCF. Inheritance of resistance to yellow rust (Puccinia glumarum Erikss. & Henn.) in seven varieties of wheat. Trans Br Mycol Soc. 1962;45(1):21–45.
McIntosh RA. Close genetic linkage of genes conferring adult-plant resistance to leaf rust and stripe rust in wheat. Plant Pathol. 1992;41(5):523–7.
Reif JC, Zhang P, Dreisigacker S, Warburton ML, van Ginkel M, Hoisington D, et al. Wheat genetic diversity trends during domestication and breeding. Theor Appl Genet. 2015;110(5):859–64.
Kolmer JA, Singh RP, Garvin DF, Viccars L, William HM, Huerta-Espino J, et al. Analysis of the Lr34/Yr18 rust resistance region in wheat germplasm. Crop Sci. 2008;48(5):1841–52.
Li Q, Chen XM, Wang MN, Jing JX. Yr45, a new wheat gene for stripe rust resistance on the long arm of chromosome 3D. Theor Appl Genet. 2011;122(1):189–97.
Ren RS, Wang MN, Chen XM, Zhang ZJ. Characterization and molecular mapping of Yr52 for high-temperature adult-plant resistance to stripe rust in spring wheat germplasm PI 183527. Theor Appl Genet. 2012;125(5):847–57.
Moore JW, Herrera-Foessel S, Lan C, Schnippenkoetter W, Ayliffe M, Huerta-Espino J, et al. A recently evolved hexose transporter variant confers resistance to multiple pathogens in wheat. Nat Genet. 2015;47(12):1494–8.
Kandel JS, Krishnan V, Jiwan D, Chen X, Skinner DZ, See DR. Mapping genes for resistance to stripe rust in spring wheat landrace PI 480035. PLoS One. 2017;12(5):e0177898.
Nsabiyera V, Bariana HS, Qureshi N, Wong D, Hayden MJ, Bansal UK. Characterisation and mapping of adult plant stripe rust resistance in wheat accession Aus27284. Theor Appl Genet. 2018;131(7):1459–67.
Feng J, Wang M, See DR, Chao S, Zheng YL, Chen X. Characterization of novel gene Yr79 and four additional QTL for all-stage and high-temperature adult-plant resistance to stripe rust in spring wheat PI 182103. Phytopathology. 2018;108(6):737–47.
Qureshi N, Bariana H, Kumran VV, Muruga S, Forrest KL, Hayden MJ, et al. A new leaf rust resistance gene Lr79 mapped in chromosome 3BL from the durum wheat landrace Aus26582. Theor Appl Genet. 2018;131(5):1091–8.
Jaradat AA. Wheat landraces: genetic resources for sustenance and sustainability. Usda-ars. 2011:1–20.
Wang M, Chen X. Stripe rust: Stripe rust resistance. Dordrecht: Springer; 2017. p. 353–558.
Appels R, Eversole K, Feuillet C, Keller B, Rogers J, Stein N, et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018;361(6403):eaar7191.
Singh RP. Genetic association of leaf rust resistance gene Lr34 with adult plant resistance to stripe rust in bread wheat. Phytopathology. 1992;82:835–8.
Yang WX, Yang FP, Liang D, He ZH, Shang XW, Xia XC. Molecular characterization of slow-rusting genes Lr34/Yr18 in Chinese wheat cultivars. Acta Agronom Sinica. 2008;34:1109–13.
Wu L, Xia X, Rosewarne GM, Zhu H, Li S, Zhang Z, et al. Stripe rust resistance gene Yr18 and its suppressor gene in Chinese wheat landraces. Plant Breed. 2015;134(6):634–40.
Xu LS, Wang MN, Cheng P, Kang ZS, Hulbert SH, Chen XM. Molecular mapping of Yr53, a new gene for stripe rust resistance in durum wheat accession PI 480148 and its transfer to common wheat. Theor Appl Genet. 2013;126(2):523–33.
Zhou XL, Wang MN, Chen XM, Lu Y, Kang ZS, Jing JX. Identification of Yr59 conferring high-temperature adult-plant resistance to stripe rust in wheat germplasm PI 178759. Theor Appl Genet. 2014;127(4):935–45.
Lu Y, Wang M, Chen X, See D, Chao S, Jing J. Mapping of Yr62 and a small-effect QTL for high-temperature adult-plant resistance to stripe rust in spring wheat PI 192252. Theor Appl Genet. 2014;127(6):1449–59.
Cheng P, Xu LS, Wang MN, See DR, Chen XM. Molecular mapping of genes Yr64 and Yr65 for stripe rust resistance in hexaploid derivatives of durum wheat accessions PI 331260 and PI 480016. Theor Appl Genet. 2014;127(10):2267–77.
Chhetri M. Molecular mapping and genetic characterization of rust resistance in wheat. PhD thesis. Australia: The University of Sydney; 2015.
Zhonghu H, Rajaram S. China/CIMMYT collaboration on wheat breeding and germplasm exchange: results of 10 years of shuttle breeding (1984–1994). In: CIMMYT; 1997.
Zou Y, Yang W, Zhu H, Yang E, Pu Z, Wu L, et al. Utilization of CIMMYT germplasm and breeding technologies in wheat improvement in Sichuan, China. Southwest China J Agric Sci. 2007;20(2):183–90.
Prins R, Pretorius ZA, Bender CM, Lehmensiek A. QTL mapping of stripe, leaf and stem rust resistance genes in a Kariega × avocet S doubled haploid wheat population. Mol Breed. 2011;27(2):259–70.
Bansal UK, Kazi AG, Singh B, Hare RA, Bariana HS. Mapping of durable stripe rust resistance in a durum wheat cultivar Wollaroi. Mol Breed. 2014;33(1):51–9.
Basnet BR, Singh RP, Ibrahim AMH, Herrera-Foessel SA, Huerta-Espino J, Lan C, et al. Characterization of Yr54 and other genes associated with adult plant resistance to yellow rust and leaf rust in common wheat Quaiu 3. Mol Breed. 2014;33(2):385–99.
Bariana H, Forrest K, Qureshi N, Miah H, Hayden M, Bansal U. Adult plant stripe rust resistance gene Yr71 maps close to Lr24 in chromosome 3D of common wheat. Mol Breed. 2016;36(7):98.
Zegeye H, Rasheed A, Makdis F, Badebo A, Ogbonnaya FC. Genome-wide association mapping for seedling and adult plant resistance to stripe rust in synthetic hexaploid wheat. PLoS One. 2014;9(8):e105593.
Quan W, Hou G, Chen J, Du Z, Lin F, Guo Y, et al. Mapping of QTL lengthening the latent period of Puccinia striiformis in winter wheat at the tillering growth stage. Eur J Plant Pathol. 2013;136(4):715–27.
Chhuneja P, Kaur S, Garg T, Ghai M, Kaur S, Prashar M, et al. Mapping of adult plant stripe rust resistance genes in diploid a genome wheat species and their transfer to bread wheat. Theor Appl Genet. 2008;116(3):313–24.
Boukhatem N, Baret PV, Mingeot D, Jacquemin JM. Quantitative trait loci for resistance against yellow rust in two wheat-derived recombinant inbred line populations. Theor Appl Genet. 2002;104(1):111–8.
Feng J, Zuo LL, Zhang ZY, Lin RM, Cao YY, Xu SC. Quantitative trait loci for temperature-sensitive resistance to Puccinia striiformis f. sp. tritici in wheat cultivar Flinor. Euphytica. 2011;178(3):321–9.
Lin F, Chen XM. Quantitative trait loci for non-race-specific, high-temperature adult-plant resistance to stripe rust in wheat cultivar express. Theor Appl Genet. 2009;118(4):631–42.
Dedryver F, Paillard S, Mallard S, Robert O, Trottet M, Nègre S, et al. Characterization of genetic components involved in durable resistance to stripe rust in the bread wheat ‘Renan’. Phytopathology. 2009;99(8):968–73.
Zwart RS, Thompson JP, Milgate AW, Bansal UK, Williamson PM, Raman H, et al. QTL mapping of multiple foliar disease and root-lesion nematode resistances in wheat. Mol Breed. 2010;26(1):107–24.
Ren Y, He Z, Li J, Lillemo M, Wu L, Bai B, et al. QTL mapping of adult-plant resistance to stripe rust in a population derived from common wheat cultivars Naxos and Shanghai 3/catbird. Theor Appl Genet. 2012;125(6):1211–21.
Blume YB, Krasylenko YA, Yemets AI. The role of the plant cytoskeleton in phytohormone signaling under abiotic and biotic stresses. Mech Plant Horm Signaling Stress. 2017:127–85.
Pieterse CM, Leon-Reyes A, Van der Ent S, Van Wees SC. Networking by small-molecule hormones in plant immunity. Nat Chem Biol. 2009;5(5):308–16.
Brutus A, Sicilia F, Macone A, Cervone F, De Lorenzo G. A domain swap approach reveals a role of the plant wall-associated kinase 1 (WAK1) as a receptor of oligogalacturonides. Proc Natl Acad Sci U S A. 2010;107(20):9452–7.
Zuo W, Chao Q, Zhang N, Ye J, Tan G, Li B, et al. A maize wall-associated kinase confers quantitative resistance to head smut. Nat Genet. 2015;47(2):151–7.
Thapa G, Gunupuru LR, Heir JG, Kahla A, Mullins E, Doohan FM. A pathogen-responsive leucine rich receptor like kinase contributes to Fusarium resistance in cereals. Front Plant Sci. 2018;9:867.
Sharma S, Kumar R, Mendu V, Singh K, Upadhyay SK. Genomic dissection and expression profiling revealed functional divergence in Triticum aestivum leucine rich repeat receptor like kinases (TaLRRKs). Front Plant Sci. 2016;7:1374.
Cao A, Xing L, Wang X, Yang X, Wang W, Sun Y, et al. Serine/threonine kinase gene Stpk-V, a key member of powdery mildew resistance gene Pm21, confers powdery mildew resistance in wheat. Proc Natl Acad Sci U S A. 2011;108(19):7727–32.
Höwing T, Huesmann C, Hoefle C, Nagel MK, Isono E, Huckelhoven R, et al. Endoplasmic reticulum KDEL-tailed cysteine endopeptidase 1 of Arabidopsis (AtCEP1) is involved in pathogen defense. Front Plant Sci. 2014;5:58.
Mukherjee AK, Carp MJ, Zuchman R, Ziv T, Horwitz BA, Gepstein S. Proteomics of the response of Arabidopsis thaliana to infection with Alternaria brassicicola. J Proteome. 2010;73(4):709–20.
Shindo T, Misas-Villamil JC, Hörger AC, Song J, van der Hoorn RA. A role in immunity for Arabidopsis cysteine protease RD21, the ortholog of the tomato immune protease C14. PLoS One. 2012;7(1):e29317.
Lampl N, Alkan N, Davydov O, Fluhr R. Set-point control of RD21 protease activity by AtSerpin1 controls cell death in Arabidopsis. Plant J. 2013;74(3):498–510.
Shiu SH, Bleecker AB. Expansion of the receptor-like kinase/Pelle gene family and receptor-like proteins in Arabidopsis. Plant Physiol. 2003;132(2):530–43.
Krattinger SG, Lagudah ES, Spielmeyer W, Singh RP, Huerta-Espino J, McFadden H, et al. A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science. 2009;323(5919):1360–3.
Liu TG, Peng YL, Chen WQ, Zhang ZY. First detection of virulence in Puccinia striiformis f. sp. tritici in China to resistance genes Yr24 (= Yr26) present in wheat cultivar Chuanmai 42. Plant Dis 2010; 94(9):1163–1163.
Wu JH, Wang QL, Chen XM, Wang MJ, Mu JM, Lv XN, et al. Stripe rust resistance in wheat breeding lines developed for Central Shaanxi, an overwintering region for Puccinia striiformis f. sp. tritici in China. Can J Plant Pathol. 2016;38(3):317–24.
Han DJ, Wang QL, Chen XM, Zeng QD, Wu JH, Xue WB, et al. Emerging Yr26-virulent races of Puccinia striiformis f. tritici are threatening wheat production in the Sichuan Basin, China. Plant Dis. 2015;99(6):754–60.
Zhang B, Jai Q, Huang J, Cao S, Sun Z, Luo H, et al. Trends and toxicity analysis of new strains G22-9 and G22-14 in Puccinia striiformis f. sp. Tritici. Acta Agric Boreali-Occidentalis Sin. 2015;24(7):125–30.
Stakman EC, Stewart DM, Loegering WQ. Identification of physiologic races of Puccinia graminis var. tritici. USDA-ARS bulletin E617. 1962.
Bates D, Maechler M, Bolker B, Walker S. lme4: linear mixed-effects models using Eigen and S4. R package version. 2014;1(7):1–23.
Smith SE, Kuehl RO, Ray IM, Hui R, Soleri D. Evaluation of simple methods for estimating broad-sense heritability in stands of randomly planted genotypes. Crop Sci. 1998;38:1125–9.
Li R, Zeng Y, Xu J, Wang Q, Wu F, Cao M, et al. Genetic variation for maize root architecture in response to drought stress at the seedling stage. Breed Sci. 2015;65(4):298–307.
Liu K, Muse SV. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21(9):2128–9.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–20.
Earl DA. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genet Resour. 2012;4(2):359–61.
Kolde R pheatmap: Pretty Heatmaps. R Package Version 1.0.8. https://CRAN.R–project.org/package=pheatmap. 2015.
Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer; 2016.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–5.
Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38:203–8.
Zhang Z, Ersoz E, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA, et al. Mixed linear model approach adapted for genome-wide association studies. Nat Genet. 2010;42:355–60.
Sosnowski O, De Oliveira Y, Moreau L, Charcosset A, Joets J, Jorge V, et al. BioMercator. Un Logiciel Dédié à la Compilation de Cartes Génétiques et la Meta-Analyse de QTLs. http://prodinra.inra.fr/record/255679. 2012.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10(1):421.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12(1):59–60.
Bairoch A, Apweiler R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. 1999;27:49–54.
Acknowledgments
The authors thank Prof. Qiu-Zheng Jia (Plant Protection Research Institute, Gansu Academy of Agricultural Sciences, Lanzhou, P. R. China) for providing the stripe rust isolates. Prof. Li-Hui Li and Xiu-Quan Li (Chinese Academy of Agricultural Sciences) for plant materials (Sichuan wheat landraces) support.
Funding
This work was supported by the National Key Research and Development Program of China (2017YFD0100900, 2016YFD0100100, 2016YFD0102000). The funders had no role in the study design, collection, analysis and interpretation of data, or in the writing of the report or decision to submit the article for publication.
Availability of data and materials
The datasets supporting the results of this publication are included within the article and its Additional files.
Author information
Authors and Affiliations
Contributions
XLY carried out the experiment, analysed the data, and drafted the manuscript; JL1 carried out the phenotypic evaluation and extracted the DNA; YC carried out the analyses of association mapping; FJY, LL, CY, YQW, YW and JL2 carried out the phenotypic evaluation; JRW contributed to revise the manuscript and manage plant materials; QTJ, WL, JM and YMW participated in the field experiment; YLZ participated in the design of the experiments; GYC formulated the questions, designed and carried out the experiment, analysed the data and revised the manuscript. All authors have reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
The 244 wheat accessions used in this study and the evaluation of their infection type (IT) and disease Severity (DS) in six environments. (XLSX 36 kb)
Additional file 2:
The distribution of infection type (IT) and disease severity (DS) for stripe rust between landraces and cultivars in adult-plant stage based on BLUP values. (XLSX 10 kb)
Additional file 3:
Summary of genetic diversity between landraces and cultivars with 44,059 SNP markers. (XLSX 10 kb)
Additional file 4:
The phenotypic and genetic diversity analyses of 244 wheat accessions in two sub-populations. (XLSX 9 kb)
Additional file 5:
The distribution of the favorable alleles in 244 wheat accessions. (XLSX 32 kb)
Additional file 6:
The Pearson correlation analyses between favorable allele number and the stripe rust response. (XLSX 9 kb)
Additional file 7:
The putative candidate genes of two potentially novel QTLs. (XLSX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ye, X., Li, J., Cheng, Y. et al. Genome-wide association study of resistance to stripe rust (Puccinia striiformis f. sp. tritici) in Sichuan wheat. BMC Plant Biol 19, 147 (2019). https://doi.org/10.1186/s12870-019-1764-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1764-4