
Abstract
As a newly identified checkpoint, T cell immunoreceptor with immunoglobulin and tyrosine-based inhibitory motif (ITIM) domain (TIGIT) is highly expressed on CD4+ T cells, CD8+ T cells, natural killer (NK) cells, regulatory T cells (Tregs), and tumor-infiltrating lymphocytes (TILs). TIGIT has been associated with NK cell exhaustion in vivo and in individuals with various cancers. It not only modulates NK cell survival but also mediates T cell exhaustion. As the primary ligand of TIGIT in humans, CD155 may be the main target for immunotherapy due to its interaction with TIGIT. It has been found that the anti-programmed cell death protein 1 (PD-1) treatment response in cancer immunotherapy is correlated with CD155 but not TIGIT. Anti-TIGIT alone and in combination with anti-PD-1 agents have been tested for cancer immunotherapy. Although two clinical studies on advanced lung cancer had positive results, the TIGIT-targeted antibody, tiragolumab, recently failed in two new trials. In this review, we highlight the current developments on TIGIT for cancer immunotherapy and discuss the characteristics and functions of TIGIT.
Similar content being viewed by others
Introduction
Immune checkpoint inhibitors (ICIs) function by restoring the induction, activation, and expansion of tumor-specific cytotoxic T cells, resulting in durable therapy responses [1,2,3,4]. The Food and Drug Administration (FDA) has approved a number of ICIs targeting programmed cell death protein 1 (PD-1), cytotoxic T lymphocyte antigen-4 (CTLA-4), and lymphocyte activation gene-3 (LAG-3) for the treatment of various cancers, such as non-small cell lung cancer (NSCLC), melanoma, carcinoma of the head and neck, and hematological malignancies [1, 5,6,7,8,9]. There are still a large number of patients who do not respond well to this treatment, and the reason may be low expression of programmed death-ligand 1 (PD-L1), a “cold” tumor microenvironment (TME) characterized by a paucity of effector T cells or a surplus of immune regulatory cells [9,10,11,12]. Furthermore, innate and acquired resistance to such treatment remains a major obstacle [13].
Following the emergence of these well-known ICIs, a novel checkpoint, T cell immunoreceptor with immunoglobulin and tyrosine-based inhibitory motif (ITIM) domain (TIGIT), also known as WUCAM, VSTM3, or VSIG9, has been identified [14,15,16,17]. Clinical trials of anti-TIGIT or anti-TIGIT and anti-PD-1 combinations are being done. Despite being successful in a few clinical trials [18,19,20,21,22], the TIGIT-targeted antibody tiragolumab recently failed to reach the endpoint in patients with advanced lung cancers [23, 24]. In this review, we discuss the characteristics and functions of TIGIT activity. We looked further into the potential reasons why anti-TIGIT experiments in both preclinical and clinical settings failed.
TIGIT: expression, function and the signaling pathway
The expression and function of TIGIT
TIGIT is extensively overexpressed on tumor-infiltrating lymphocytes (TILs) such as CD8+ T cells, CD4+ T cells, regulatory T cells (Tregs), and natural killer (NK) cells across various malignancies [25,26,27,28,29,30,31]. Interestingly, TIGIT was found to be preferentially expressed on the CD16+ NK cell subpopulation in contrast to other PVR-like receptors that are expressed on human NK cells [32]. Research indicated that intratumoral NK and T cells expressed more TIGIT than peripheral blood NK and T cells did [33, 34], TIGIT+ CD4+ T cells are enriched in chronic lymphocytic leukemia (CLL) but not CD8+ T cells [35]. These suggest that the role of TIGIT may vary between solid tumors and hematological cancers. Additionally, in a number of malignancies, the expression of TIGIT was associated with tumor stage, survival, and the TILs component [33, 36,37,38].
Advancements in chimeric antigen receptor T-cell (CAR-T) therapy aim to enhance chemotaxis and infiltration into the TME, while efforts continue to reduce cell exhaustion and treatment-related toxicities [39]. Several CAR-T cell products have been approved for treating hematological malignancies [40,41,42,43]. However, some patients remained poor responders or relapsed with CAR-T therapy [44]. A recent study showed an improvement in CAR-T efficacy with TIGIT inhibition alone based on the sequential analysis of manufactured and infused CAR-T using single-cell RNA and protein expression data [45]. Targeting TIGIT may prevent CAR-T-related relapses and thus promote long-term progression-free survival in mantle cell lymphoma (MCL) patients, according to another study that found that over-expression of TIGIT played a critical role in T-cell suppression associated with CAR-T relapse in mantle cell lymphoma patients [44].
In a typical scenario, the co-expression of co-inhibitory receptors serves as an indicator of immune cell exhaustion, a state characterized by a diminished response to antigen stimulation. For instance, elevated co-expression of TIGIT and T cell immunoglobulin and mucin-domain-containing-3 (TIM-3), on NK cells was found in patients with hepatitis B virus-associated hepatocellular carcinoma (HBV-HCC) [46]. This specific subset of NK cells displayed compromised functionality, as evidenced by a reduction in cytotoxicity, cytokine release, and cell proliferation - all hallmarks of an “exhausted” phenotype [46]. Importantly, these TIGIT+ TIM-3+ NK cells have been implicated in the progression of HBV-HCC, underscoring their significant role in the pathogenesis of this disease [46]. Tumor-infiltrating NK cells (TiNKs) are more dysfunctional compared to circulating human NK cells. Interestingly, while TIGIT+ NK cells demonstrated a higher lytic potential, they paradoxically exhibit lower actual lytic activity against CD155+ major histocompatibility complex (MHC)-class I deficient melanoma cells than their TIGIT- counterparts [34]. An increased percentage of NK lacking CD226 expression and co-expression of TIGIT and CD96 are associated with better survival in AML patients [47].
TIGIT not only influences the survival and exhaustion of NK cells, as observed in animal models and cancer patients, but it also actively mediates T cell depletion. TIGIT functions as a coinhibitory receptor and is typically co-expressed with PD-1 in CD8+ and CD4+ T effector memory cells, where it is associated with T cell exhaustion [36, 48,49,50,51,52,53]. Additionally, TIGIT and PD-1 expression displayed diminished effector functionality of intratumoral T-cells in B-cell non-Hodgkin lymphoma (NHL) and predicted survival in lung squamous cell carcinoma (LUSC) [36, 48]. Therefore, TIGIT and PD-1 may serve as potential predictive indicators for dual-targeting immunotherapy. According to reports, both the quantity of Tregs and the expression of TIGIT were associated with the TILs hypofunction [54]. In addition, TIGIT expression represents a sign of T cell exhaustion in several cancers [55,56,57]. Elevated CD8+ TIGIT+ T cells exhibiting signs of exhaustion have been associated with less favorable clinical outcomes in HBV-HCC [27]. Meanwhile, intratumoral TIGIT+ CD8+ T-cell abundance could serve as an independent prognosticator for clinical outcome and a predictive biomarker [58], and TIGIT and PD-1 expression atlas predicts response to adjuvant chemotherapy and PD-L1 blockade in muscle-invasive bladder cancer [59]. Furthermore, TIGIT-expressing CD4+ T cells represent a tumor-supportive T cell subset in CLL [35] and TIGIT blockade can restore CD8+ T-cell immunity against multiple myeloma (MM) [60]. Blocking TIGIT/CD155 signalling reverses CD8+ T cell exhaustion and enhances the antitumor activity in cervical cancer [56]. Additionally, the secretion of IL-10 from CD8+ T cells can in turn increase TIGIT expression, thereby further exacerbating immunosuppression [61].
Unlike other T cells, the presence of the TIGIT marker on Tregs is linked with consistent FOXP3 activity, nuclear localization of FOXO1, and enhanced suppressive functionality [62]. From a mechanistic perspective, TIGIT functions as a transcriptional target of FOXP3. And the suppression of the PI3K/AKT/mTOR pathway aids in preserving the identity and stability of Tregs, resulting in the nuclear retention of FOXO1 [62]. Additionally, TIGIT+ Tregs form a distinct subset, uniquely defined by their specific capacity to suppress T helper cell (Th)1 and Th17 pro-inflammatory responses, as well as to inhibit the proliferation of effector T cells [63].
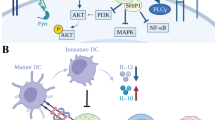
The TIGIT+ CD20+ B cells observed infiltrating tumors and tertiary lymphoid structures (TLS) were found to be significantly correlated with survival rates as well as the response to adjuvant chemotherapy [64]. Besides, TIGIT on memory B cells regulates the immune response by directly influencing T cells and inhibiting DC proinflammatory function. This suppresses Th1, Th2, and Th17 immune responses and CXCR5+ICOS+T cell responses while promoting the immune regulatory function of T cells. TIGIT+ memory B cells are also superior to other B cells at expressing additional inhibitory molecules, including IL-10, TGFβ1, granzyme B, PD-L1, CD39/CD73, and TIM-1 [65]. The mechanisms of immunosuppression mentioned in TIGIT were elucidated in Fig. 1.

Mechanisms of immunosuppression of TIGIT in TME. A. CD155 binds with TIGIT with higher affinity compared to CD226 leading to deactivation of T or NK cells. In addition, CD155 is associated with drug resistance, and tumor cell migration and metastasis. Fap2, secreted from F. nucleatum, triggers inhibition by binding to TIGIT. B. The interaction of CD112 and CD112R, or TIGIT, inhibits proliferation of T-cells and NK cells by reducing IFN-γ production. CD155 increases IL-10 production by binding to TIGIT. C. TIGIT on Treg activates its immunosuppression on Th1, Th17, and effector T cells. D. The binding of TIGIT and CD155 on memory B cells exhibits deactivated DCs and inhibits Th1/17/2. E. The production of IL-10 from DCs inhibits TILs
Generally, the decreased TIGIT expression inhibited tumor growth in mice and activated IFN-γ secretion by NK and CD8+ T cells [66]. In renal and liver allograft patients, a reduction or absence of TIGIT+ memory B cells is linked to heightened donor-specific antibody and T follicular helper cell (TFH) responses while simultaneously being associated with diminished Treg responses [65]. A fascinating study has shed light on the inhibitory role of TIGIT within cells [67]. Making use of chimeric costimulatory switch receptors (CSR), the research successfully managed to transmute coinhibitory signals in T cells into those that advance their activity. The CSR utilized for this purpose was ingeniously constructed by fusing the extracellular domain of TIGIT with the signaling domain of CD28 [67]. Previous research has revealed that the expression of TIGIT could vary, influenced by a multitude of factors [33, 68,69,70,71] (Fig. 2). In addition to its role in infectious disorders, Sumida et al. identified that type 1 interferon (IFN-I) also inhibited the expression of TIGIT in vitro [72]. Notably, chemotherapy was found to reduce the presence of TIGIT on CD8+ T cells in gastric cancer cases [68, 73], while it was observed to increase following microwave ablation [71]. Factors such as glucose deprivation and hypoxic conditions were also identified as triggers for an upsurge in TIGIT expression on tumor cells [70]. Additionally, the C-C motif chemokine ligand 23 (CCL23), typically secreted by macrophages, may play a role in fueling the growth of cancer cells [69, 74]. In an insightful discovery made by Kamat, K., and his team, they noted that CCL23 contributed to the increased expression of TIGIT on CD8+ T cells through a process involving GSK3β phosphorylation [69]. In related findings, dogs diagnosed with metastatic osteosarcoma and treated with inhaled IL-15 showed not only an elevation in activation markers but also a noticeable rise in TIGIT levels [33].

The expression of TIGIT can be influenced by a variety of factors. Notably, Interferon-I (IFN-I), chemotherapy agents, GITR antibodies, and 1α,25-Dihydroxyvitamin D3 (1α,25(OH)(2)D(3)) have been observed to downregulate its expression. In contrast, microwave ablation, glucose deprivation, hypoxic conditions, as well as the presence of CCL23, IL-10, inhaled IL-15, and anti-PD-1/PD-L1 therapies have been found to enhance the expression of TIGIT, thereby contributing to an immunosuppressive TME
The structure of TIGIT
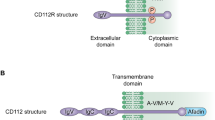
The TIGIT gene, which encodes a transmembrane protein with 244 amino acids, is found on human chromosome 3q13.31 [17]. It was first identified in 2009 by bioinformatics research that sought to identify an inhibitory receptor [14]. Together with CD96 (TACTILE) and CD226 (DNAM-1), it is a member of the immunoglobulin superfamily of receptors, and TIGIT also forms part of the poliovirus receptor (PVR)/nectin family, which includes PVR (CD155) and Nectin-2 (CD112) [75, 76]. Structurally, TIGIT is composed of three parts: an extracellular immunoglobulin variable-set (IgV) domain, a type I transmembrane domain, and a highly conserved intracellular inhibitory domain that contains an ITIM and an immunoglobulin tyrosine tail (ITT) motif, features that are consistent in both mice and humans [14, 77].
The (V/I)(S/T)Q, AX6G, and T(F/Y)P submotifs in the TIGIT IgV domain mediate a ‘lock and key’ trans-interaction with cis-homodimers of PVR [14, 77, 78]. These conserved submotifs were identified in the PVR/nectin family, including CD226, CD96, CD112R, CD155, CD112, and CD113 [77]. Within the intracellular inhibitory domain, ITT-like and ITIM motifs, two conserved inhibitory domains, exert slightly distinct functions. By phosphorylating either the tyrosine residues of the ITIM (Y277) or the ITT-like motif residue (Y233), in mice, the inhibitory activity of TIGIT can be triggered [16]. Whereas in humans, the ITIM motif mediates a weak inhibitory signal [79, 80]. The ITT-like motif is phosphorylated at Tyr225 and binds to cytosolic adapters Grb2 and β-arrestin 2 and is further responsible for the recruitment of the Src homology 2 (SH2) domain containing inositol polyphosphate 5-phosphatase 1 (SHIP1) [56, 81, 82] which impairs tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) auto-ubiquitination to abolish NF-kB and ERK activation [56, 81] and terminates PI3K/MAPK signaling [79, 83], leading to suppression of NK cell and CD8+ T cell function.
The ligands and pathways of TIGIT
TIGIT mainly interacts with four ligands, nectin and nectin-like adhesion molecules, namely CD155 (PVR, or Necl-5), CD112 (PVRL2, Nectin-2), CD113 (PVRL3, Nectin-3), and nectin-4 (PRR4, PVRL4) [83, 84]. In both humans and mice, CD155 acts as the principal ligand for TIGIT [28]. This is primarily due to its superior affinity when compared with CD112 and CD113 [14]. TIGIT and CD226 (DNAM-1) are competitors for the same ligands, CD155 and CD112 [85]. It was discovered that a mutation from Ala to Thr at residue 67 of CD155 increased the binding affinity for TIGIT [86]. Another CD155 receptor with a different affinity from TIGIT is CD96 (TACTILE) [78].
CD155, as the PVR and the fifth member of the nectin-like molecule family, demonstrated different immune efficacy when combined with its ligands CD226, TIGIT and CD96 [83, 87, 88]. By interacting with phosphorylated TIGIT, which was controlled by recruiting SHIP1 and then blocking PI3K/MAPK signaling as previously described (Fig. 3) [83, 89, 90], CD155 exhibits immunosuppressive properties [83]. Conversely, when CD155 binds with CD226, it activates NK cells and T cells [83, 90]. Compared to healthy tissues, malignancies have higher levels of CD155 expression [29, 83, 89, 91, 92]. Follicular dendritic cells, tumor-associated macrophages (TAMs), and other tumor-infiltrating myeloid cells (TIMs) but not tumor-infiltrating lymphocytes (TILs) from multiple tumors have been found to express CD155 in the TME [25, 28, 50, 86, 91]. Intriguingly, a biomarker for predicting a worse prognosis and stage is CD155 high expression [38, 93]. The intrinsic functions of CD155 promote tumor cell migration and metastasis [89, 90]. Targeted TIGIT or CD155 therapy enhances immune cell activation [93]. Kawashima et al. found that in patients who demonstrated a positive response to ICIs, an increase in CD155 expression was observed in tumor cells that survived after TILs. This escalated expression of CD155 led to the suppression of T-cell activation, particularly those expressing TIGIT [25]. Currently, it is possible to use CD155 as an immunotherapy target because of its interaction with activating or inhibiting receptors [92]. In melanoma patients with an inflamed TME, CD155 was associated with ICI resistance, including both primary and acquired resistance [25]. Meanwhile, Jiang, C. et al. discovered that the response to anti-PD-1 is correlated to CD155 expression rather than TIGIT expression [94].

The interaction of TIGIT-related ligands and receptors. PVRL4, CD155, CD112, and CD113 are expressed on tumor cells or APCs as ligands, and CD226, TIGIT, CD96, and CD112R act as receptors on T cells or NK cells. Every receptor here, apart from CD226 which uniquely possesses an ITT-like domain, features an intracellular ITIM domain. TIGIT has an ITT-like motif, and CD96 contains a YXXM motif. TIGIT binds to CD155 with a higher affinity and inhibits the interaction of CD226 and CD155. In humans, the inhibition of TIGIT mainly relies on the phosphorylation of an ITT-like motif and ultimately terminates PI3K/MAPK signaling. However, the downstream function of CD96 has not been clear until now. The arrow’s thickness represents the affinity of ligands and receptors
Research has previously indicated that CD112 predominantly resides at the adherent junctions of epithelial cells and is expressed universally across various cell types, including but not limited to follicular dendritic cells (DCs), monocytes, and multiple cancer cells [50, 95,96,97]. Notably, its association with clinical outcomes may vary based on the type of tumor and where it is expressed [97]. By binding to different ligands, CD112 exerts the opposite function. CD112-CD226 can promote T cell proliferation and NK cell-mediated cytotoxicity [87, 98]. On the contrary, when attached to TIGIT (despite its low affinity) and CD112R (PVRIG), CD112 inhibits proliferation of T-cells and NK cells by reducing interferon-γ (IFN-γ) production and NK cell-mediated cytotoxicity [87, 97]. Moreover, it is noteworthy that TIGIT/CD112R pathways may also be upgraded as potential immunotherapy targets [87].
CD226 is a co-stimulatory receptor that shares common ligands, specifically CD155, with TIGIT and CD96. It is expressed on T cells, NK cells, NK/T cells, B cells, monocytes/macrophages, DCs, megakaryocytes/platelet lineage hematopoietic precursor cells, endothelial cells, and mast cells [75, 99,100,101,102]. In the context of cancer, the interaction between CD226 and CD155 is crucial for activating anti-tumor immune responses. When CD226 expression was low or absent, CD8+ TIL exhibited poor activation and malfunction, respectively [103, 104]. Interestingly, compared to the NK cells in the peripheral blood of healthy donors, those derived from ascites in patients with advanced ovarian cancer (OC) exhibited a decreased expression of the activating receptor CD226 [105], which implies that the anti-tumor functionality of CD226 might rely on both extra- and intra-tumoral heterogeneity, akin to what is seen with CD112 [97]. It was found that CD226 was downregulated in TIGIT+CD8+ follicular lymphoma T cells compared with TIGIT−CD8+ cells [50], which further promoted the immune escape process of tumor cells. As a result, Jin, Z., et al. found that patients with acute myeloid leukemia (AML) tended to have fewer CD226+ and more TIGIT+ γδ T cells [106]. Furthermore, the subset of TIGIT+/CD226− γδ T cells was associated with a better prognosis for AML patients, suggesting TIGIT/CD225 might be a promising therapeutic target [106]. In addition, CD226 expression is associated with clinical outcomes in patients with NSCLC treated with an anti-PD-L1 antibody, and PD-1 reduces phosphorylation of both CD226 and CD28 via its ITIM-containing intracellular domain (ICD) [107]. Without CD226 expression, CD8+ TILs were unable to respond to anti-PD-1, and thus immune checkpoint blockade efficacy was revealed to be ineffective in CD226− mice [103, 107].
The efficacy of immunotherapy is influenced by the equilibrium between CD226 and TIGIT, given their competing interactions with CD155. Using combined immunotherapy that targets PD-1 and the glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR) antibodies, Wang and colleagues exhibited a survival advantage by ameliorating this balance [108]. Mechanistically, CD226 was reactivated by PD-1 inhibition, whereas TIGIT expression was reduced by GITR [108]. The number of infiltrating suppressive Tregs and the clinical effectiveness of immune checkpoint blockade treatment were both negatively correlated with a greater TIGIT/CD226 ratio in Tregs, which indicates higher TIGIT expression and lower CD226 expression [109].
The majority of T cells, NK cells, and NK/T cells express CD96, an Ig-superfamily receptor, and it can serve as a cancer stem cell marker in AML [110,111,112]. The CD8+ T cells co-express CD96 and PD-1 [113, 114]. However, its function remains unclear. CD96 was demonstrated to enhance the activation of NK cells by increasing adhesion to PVR-expressing target cells [115]. Conversely, NK cell function may be directly inhibited by CD96+ NK cells, which is characterized by functional exhaustion and predicts poorer clinical outcomes [116, 117].
Specifically, it was discovered that the novel ligand Nectin-4 (PRR4, PVRL4) only interacts with TIGIT, leading to the inhibition of NK cells [118]. Several cancer cells express Nectin-4 exclusively, as opposed to other nectins [119]. The specific antibody against Nectin-4 enhanced its killing effect on tumor cells in vitro and in vivo [118].
The role of microorganisms in the TME has come under more and more scrutiny in recent years. Fibroblast activation protein 2 (Fap2), which directly interacts with TIGIT in human NK and T cells to cause immunosuppression, has been discovered to be secreted by Fusobacterium nucleatum, a bacterium linked to colorectal cancer (Fig. 1) [120]. This discovery has unveiled a novel target for the therapy of microorganisms.
TIGIT in preclinical studies
Previous studies suggested that reduced tumor burden and survival benefits were found in TIGIT-deficient mice [60, 121]. As such, TIGIT has been identified as a potential star immune checkpoint molecule that inhibits tumor growth and even helps to reduce resistance to immune checkpoint inhibitors in mouse models [25]. In this section, we primarily consolidate the findings from preclinical studies that assess the potential of anti-TIGIT therapy in treating both solid and hematological malignancies. Table 1 presents some of the main preclinical studies on TIGIT.
Solid tumors
The crux of immunotherapy mainly hinges on the activation of T cells, a pivotal process in marshaling the immune response against malignancies. Accordingly, treatment with anti-TIGIT exerts anti-tumor function by activating CD8+ T cells, enhancing the cytotoxicity effects and population of T cells against cancer [66, 122, 123, 133] (Fig. 4). The results from He et al. identified TIGIT blockade to neutralize inhibited T-cell metabolism and IFN-γ production caused by CD155 in gastric cancer cells [26]. Moreover, TIGIT blockade augmented CD8+ T-cell reactions and showed a survival benefit in vivo, which was better when targeting TIGIT and PD-1 [26]. The mechanisms could be that TIGIT blockade reversed the suppressed glucose metabolic activity of T cells, induced apoptosis, and reduced G2/M transit in tumor cells [57]. Interestingly, the blockade of TIGIT specifically affects CD8+ T cells that express high levels of CD226 by enhancing the phosphorylation at tyrosine 322 of CD226 [104], and increasing the population of CD226hiCD8+ T cells improves the effects of anti-TIGIT or anti-PD-1 [104].

Functions of antibodies targeting TIGIT /and PD-1/PD-L1. The left figure shows the natural immunosuppression and the general ‘lock and key’ structure of the binding of TIGIT and CD155. Besides, the interaction between PD-1 and PD-L1 contributes to immunosuppression as well. Thus, the combination of anti-TIGIT and anti-PD-1/PD-L1 or bispecific antibodies targeting TIGIT and PD-1/PD-L1 is promising, as shown in the right figure. This figure is adapted from “T-cell Deactivation vs. Activation”, by BioRender.com (2020). Retrieved from https://app.biorender.com/biorender-templates
Tregs play a major role in the occurrence and development of tumors by restricting adaptive immunity; thus, anti-TIGIT therapies targeting Tregs show great promise [31]. It is reported that anti-TIGIT treatment exerts anti-tumor function by decreasing or depleting FoxP3+ Tregs [28, 123]. Intriguingly, another study revealed that the TIGIT-blocking antibodies with functional Fc binding did not perform function by depleting Tregs but were mediated by “reverse activating signals” through Fcγ receptors (FcγRs) on myeloid cells, which caused the expression of cytokines and chemokines in colorectal cancer [134].
A unique feature of the anti-TIGIT approach is that it functions not only on the effects of the anti-tumor responses of T cells but also of NK cells. NK cell-mediated immunotherapy is under active development [135,136,137,138,139]. Blocking TIGIT may represent a new approach to enhancing NK function [140]. TIGIT inhibition increased CD56dim NK cell activation in response to the OC tumor [105]. Another study showed that anti-TIGIT prevented NK cell exhaustion and improved its immunogenicity in mouse models, resulting in tumor-specific T cell immunity and magnifying effects of combination immunotherapy with PD-1 and PD-L1 [141]. It is surprising that blocking TIGIT maximized the trastuzumab-induced antitumor response by NK cells in breast cancer [32].
Recently, a pre-clinical study explored the mechanisms of BGB-A1217 (Ociperlimab), a new anti-TIGIT monoclonal antibody (mAb) [124]. This study demonstrated that BGB-A1217 competitively binds to TIGIT with high affinity (K(D) = 0.135 nM), resulting in blocking the binding of TIGIT and its ligands, CD155 or CD112 [124]. In addition, BGB-A1217 induced antibody-dependent cellular cytotoxicity (ADCC) against Tregs, boosted the functionality of NK cells and monocytes, and removed TIGIT from T cell surfaces in an Fc-dependent manner [124]. Although TIGIT mAbs have demonstrated potency in pre-clinical models, the dual antibody combination of anti-TIGIT and anti-PD-1 has garnered more attention due to its superior efficacy.
TIGIT and PD-1 are coinhibitory receptors frequently co-expressed on TILs with exhaustion function [51]. The findings that blocking PD-1/PD-L1 signaling increases the expression of TIGIT on TILs and alters the phenotype of TIGIT+ T subsets provide a theoretical basis for the co-blockade strategy [9, 123, 142]. Consistent with this, it was found that blocking TIGIT and PD-1 could increase cytokine production and improve the proliferation of CD4+ and CD8+ T cells in tumors [91, 143, 144]. In vitro studies showed that this double blockade improved the functionality of CD8+ TILs that have no responsiveness to single PD-1 blockades [144]. Besides, the co-blockade also stimulated the CD226 pathway, thereby promoting the T-cell response against tumors [107]. Accordingly, this synergistic treatment could be mitigated by blocking CD226, whose dimerization is disrupted upon direct interaction with TIGIT in a cis configuration [125].
Above all, the most standard measure for evaluating the effectiveness of a treatment is the acquired survival benefit. Hung et al. identified that dual therapy resulted in improved survival rates when compared with control or monotherapy, possibly by activating T cell function and downregulating Tregs in mouse models [126]. It significantly reduced tumor progression, ameliorated the poor response of anti-PD-1 therapy, increased the ratio of cytotoxic to regulatory T cells in tumors, and prolonged survival [142, 145]. CD40, expressed on various antigen-presenting cells (APCs), plays a vital role in preserving T cell functionality, and agonistic CD40 antibodies circumvent the need for assistance from CD4+ T cells, which provides a novel strategy for targeting poor-responsive tumors [127, 146]. Herein, William et al. administered animal models with triple treatment and received significant tumor responses (46% objective response rate (ORR), 71% disease control rate (DCR), and 23% complete responses (CR) [127]. Importantly, the inclusion of TIGIT blockade could potentially overcome any pre-existing or developed resistance to anti-CD40/PD-1 therapy [127]. Another interesting study found that pH-modulating injectable gel (pH(e)-MIG) increased infiltrating CD8+ T cells and thus restored immunosuppression in combination with anti-PD-1 and anti-TIGIT [147]. In addition, the expression of CD96 could be an alternative biomarker for dual blockade efficacy [143]. The introduction of anti-CD96 to dual blockade therapy involving anti-PD-1 and anti-TIGIT resulted in markedly improved responses [113].
Several studies explored the combination of chemotherapy and radiotherapy with anti-TIGIT. On CD8+ T cells, the expression of TIGIT was reduced after chemotherapy [68], whereas radiotherapy increased the expression of TIGIT [71, 128]. It is reported that CD103+ DCs may play a role in that combination therapy [128]. On the contrary, the combination of anti-TIGIT and chemotherapy enhanced CD8+ TIL proliferation and production [68].
A matrix metalloproteinase 2 (MMP-2)-degradable hydrogel that contains doxorubicin (DOX) and blocks of TIGIT has been found to elicit an immunogenic TME and reverse the exhaustion of NK and effector T cells. This led to not only durable localized tumor inhibition but also systemic and long-lasting immune memory responses [148]. As mentioned above, radiotherapy alters the expression of TIGIT and exerts a stronger function in combination with anti-TIGIT or the dual blockade (anti-TIGIT and anti-PD-1). In turn, the use of the anti-TIGIT antibody boosted the efficacy of radiotherapy as well [128]. It was also observed that the dual therapy synergistically extended the accumulation of tumor-infiltrating DCs, which activated CD8+ T cells and altered the morphology of myeloid cells in the TME [71, 128, 149]. Building upon this, the incorporation of a triple therapy regimen comprising radiotherapy, anti-PD-1, and anti-TIGIT has presented promising results in various tumor-bearing models, despite the fact that optimizing the radiotherapy strategy required careful consideration to ensure its effectiveness. It was observed that low-dose radiation reduced the expression of CD155 in TAMs and DCs, leading to a decrease in the percentages of TIGIT+ exhausted T-cells and TIGIT+ Tregs [150, 151].
The TIGIT-Fc fusion protein has received wide attention these years, but the exact effects on tumor immunity remain unclear [129, 152, 153]. It was found that TIGIT-Fc treatment amplified the effects of CD8+ T and NK cells in xenograft mouse models [152]. When combined with anti-PD-L1, it promoted the reactivity and sustained memory of tumor immunity via the development of Th1 in CD4+ T cells [152].
TNFSF14 is a member of the TNF ligand family, which activates lymphoid cells and triggers the apoptosis of various tumor cells. Recently, an engineered fusion protein, TIGIT-Fc-LIGHT, the linkage between the extracellular domain of TIGIT and the extracellular domain of TNFSF14, was reported to show anti-tumor activity in pre-clinical models, especially in those who were resistant to anti-PD-1 treatment [129].
IL-15 attracted attention in three studies [33, 34, 154]. It was found that IL-15 increased the expression of TIGIT and CD226 on TiNKs, augmented NK-cell-mediated melanoma cytotoxicity in vitro, and suppressed tumor metastasis in preclinical models together with TIGIT blockade [34, 154]. Dogs with metastatic osteosarcoma receiving inhaled IL-15 also exhibited upregulation of activation markers and TIGIT [33]. But more in-depth research is needed.
Furthermore, anti-TIGIT in combination with CD112R combinations [155], CD226 agonists [104], anti-hipoxia-inducible factor 1 (HIF-1)α [156], focal adhesion kinase (FAK) inhibitors [157], and DNA methyltransferases (DNMT) inhibitors [158] are potential therapy strategies. Remarkably, the classic nonsteroidal anti-inflammatory drug aspirin was found to elicit an anti-tumor effect by reducing the expression of TIGIT [159].
Li P. et al. demonstrated that vitamin D was correlated with the expression of TIGIT [160]. Subsequent experiments showed that 1α,25(OH)(2)D(3) inhibited the expression of TIGIT by promoting the binding of vitamin D receptors and the promotor region of TIGIT [160]. In addition, CD8+ T cells treated with 1α,25(OH)(2)D(3) increased the functions of production and anti-tumor immunity [160]. Above all, the cytokine production of CD8+ T cells could be intensified by oral 1α,25(OH)(2)D(3) [160], which may serve as a potential therapy.
Since oncolytic virus (OV), especially herpes simplex virus (HSV), adenovirus (ADV), and vaccinia virus (VV), the three engineered viruses, have been evaluated for their effect on cancer treatment in pre-clinical and clinical trials and showed a positive response [161, 162], OV has become a new immunotherapy method for some reasons [130] including the ability to increase tumor-specific effector and memory T cells currently [130, 162, 163]. It is reported that the combination of OV and ICIs was better than monotherapy in melanoma [164] due to the immunosuppressive characteristics of TME [165, 166]. On the strength of the former theoretical result, an engineered HSV that expressed a single-chain fragment variable (scFv) against PD-1 enhanced the anti-tumor immunity [167], Zuo, S. et al. designed a VV carried with scFv targeting TIGIT [130]. This engineered VV worked as a virus, replicated in tumor cells, lysed tumor cells, and secreted the anti-TIGIT scFv [130]. Moreover, the VV helped to summon more activated T cells against the immunosuppression in TME [130].
Hematological tumors
Hematological malignancies form a wide range of tumors that can be categorized as cancers originating in cells of the blood or bone marrow, marked by disturbances and abnormalities in the functioning of the immune system. TIGIT stands out as a highly expressed marker on exhausted NK and T cells, and its high expression was involved in disease progression and the immune escape in various hematological tumors, indicating the promising value of TIGIT blockade treatment [48, 168,169,170,171,172,173]. To further explore its potential application in the treatment of hematological malignancies, anti-TIGIT has been incorporated into several preclinical studies.
In line with solid tumors, high expression levels of TIGIT characterize the exhausted phenotype of T cells, and blocking TIGIT could decrease FoxP3+ Tregs while stimulating the growth of IFN-γ-producing CD4+ T cells and thus confer survival benefits in mouse models [61, 174]. Besides, Mezger et al. concurrently engineered NK-92 cells with knock-out of both CBLB and TIGIT, resulting in enhanced cytotoxicity of NK cells [175]. Also, inhibiting TIGIT/CD155 restored NK cell function in myelodysplastic syndromes (MDS) [176]. As is the case with B cells, TIGIT could curtail B-cell activation and proliferation as a crucial part of an immune shutdown mechanism, which is critical in minimizing immune-induced tissue damage [177]. Contrary to other studies, the research conducted by Tehrani et al. evaluating the combined blocking of TIGIT and PD-1 in CLL did not demonstrate a significant increase in CD8+ T cell proliferation and cytotoxicity compared to single treatments [178].. Considering the infiltration of TIGIT+ cells was associated with the response to TIGIT blockade [174], the heterogeneity of TIGIT expression should be given careful consideration in future research endeavors [168].
Given its pathogenesis, leukemia typically demonstrates a high degree of motility and the ability to metastasize easily, bypassing the substantial mutational burden frequently encountered in solid tumors [179]. To date, numerous studies have indicated that elevated TIGIT expression is associated with a higher risk of relapse and poor outcomes in AML, regardless of whether allogeneic hematopoietic stem cell transplantation (allo-HSCT) has been performed [180,181,182]. In addition, blocking TIGIT could serve as a sentisizer for increasing the anti-leukemic effects in AML [95].
Leukemia-associated macrophages (LAMs) constitute a significant cell population within the tumor microenvironment. Using bone marrow (BM) and blood aspirates from AML patients, Fiedler et al. found that TIGIT+ M2 LAMs seemed to contribute to an intermediate or adverse risk [132]. Remarkably, in vitro studies have demonstrated that the utilization of anti-TIGIT possesses the potential to mediate the phenotypic polarization transition from M2 to M1, a phenomenon scarcely reported within the context of solid tumors [132]. This also led to increased secretion of cytokines and chemokines associated with the M1 type and enhanced the phagocytosis induced by anti-CD47 [132]. Comparing BM and peripheral blood cells (PBCs) between AML patients and healthy donors (HDs), Fiedler and colleagues discovered that TIGIT was specifically expressed on CD56dimCD16+ NK cells from AML [131]. They further demonstrated that blocking TIGIT alone could enhance the killing of AML cells mediated by NK-92, with the effect being amplified when combined with anti-CD39 or A2AR inhibitors [131].
Clinical trials of anti-TIGIT agents
The anti-TIGIT agents registered in clinical trials are listed in Table 2. Phase III clinical trials have been conducted to assess four anti-TIGIT agents: tiragolumab, vibostolimab, domvanalimab, and ociperlimab. Up to this point, the results of three clinical trials evaluating this kind of agent have been published [18,19,20].
Clinical trials in solid tumors
A globle phase II clinical trial (CITYSCAPER, NCT03563716) of tiragolumab, a humanized anti-TIGIT monoclonal antibody, in combination with the PD-L1 inhibitor atezolizumab for 135 PD-L1-positive patients suffering from NSCLC, was first reported at the 2020 American Society of Clinical Oncology (ASCO) meeting [183], and published recently [20]. Regardless of the stage, be it primary or in subsequent update analysis, the combination of tiragolumab with atezolizumab consistently demonstrated significantly improved ORR (primary analysis: 31.3% [95% confidence interval (CI) 19.5–43.2] vs. 16.2% [6.7–25.7] p = 0.031; update analysis [until Aug 16, 2021]: 38.8% [95% CI 26.4–51.2] vs. 20.6% [10.2–30.9] p = 0.013) and progression-free survival (PFS) (primary analysis: 5.4 months [95% CI 4.2–not evaluable] vs. 3.6 months [2.7–4.4], hazard ratio (HR): 0.57 [95% CI 0·37–0.90] p = 0.015) over placebo plus atezolizumab [20]. Remarkably, upon conducting a subgroup analysis based on the expression of PD-L1 (using Tumor Cell Proportion Score, TPS), patients exhibiting high PD-L1 expression levels (TPS ≥50%) demonstrated notably superior benefits, further illuminating the potential of this novel treatment [20]. Moreover, the toxicity was tolerable, with no new adverse effects from the combination [20]. This inspired the continuation of a set of phase III studies known as SKYSCRAPER. The results of the SKYSCRAPER-02 trial (NCT04256421), which evaluated tiragolumab in combination with atezolizumab plus carboplatin and etoposide in small cell lung cancer (SCLC), were reported at the 2022 ASCO annual meeting [23]. Unfortunately, there was no statistical difference in PFS or overall survival (OS) on this trial looking at either the primary analysis set, which included 397 patients, or the full analysis set of 490 patients [23]. Furthermore, the SKYSCRAPER-01 study (NCT04294810), which assessed tiragolumab plus atezolizumab (Tecentriq) vs. atezolizumab alone for patients with PD-L1-high locally advanced or metastatic NSCLC, did not meet the co-primary endpoint of PFS, though OS was immature [21, 24]. Additionally, SKYSCRAPER-03 (NCT04513925) and SKYSCRAPER-07 (NCT04543617) [184] trials are ongoing in patients with locally advanced, unresectable stage III NSCLC and unresectable locally advanced esophageal squamous cell carcinoma (ESCC), respectively.
The results of another study, the MORPHEUS-liver study, were recently updated. This is a phase Ib/II randomized evaluation of tiragolumab in combination with atezolizumab and bevacizumab in patients with unresectable, locally advanced, or metastatic hepatocellular carcinoma. According to the latest information from the 2023 ASCO, a total of 58 patients were enrolled. In the tiragolumab + atezolizumab + bevacizumab arm, the ORR was 42.5% vs. 11.1% in the atezolizumab and bevacizumab control arm. Besides, longer PFS was observed in the treatment arm (11.1 months; 95% CI: 8.2–not evaluable vs. 4.2 months; 95% CI: 1.6–7.4) regardless of the expression of PD-L1, and the toxicity was tolerable. However, it was notable that the 11.1% ORR in the control arm appeared significantly lower than that when used as a first-line therapy [185, 186].
The results of a phase I study on vibostolimab, a mouse and human chimeric anti-TIGIT monoclonal antibody, have just been published [18]. The combination of vibostolimab and pembrolizumab showed promising anti-tumor efficacy and manageable toxicity in patients with advanced solid tumors and anti-PD-1/PD-L1-naive NSCLC [18]. Recently, three phase III trials, including KeyVibe-006 (NCT05298423) [187], KeyVibe-007 (NCT05226598) [188] and KeyVibe-008 (NCT05224141) [189], were started to evaluate the efficacy of different strategies, including vibostolimab for NSCLC [187, 188] or SCLC [189], with the results pending.
Contrary to other anti-TIGIT monoclonal antibodies, domvanalimab (D) is an Fc-silent anti-TIGIT experimental monoclonal antibody that does not interact with effector T cells. The primary results of the phase II study, ARC-7, were presented at the 2022 ASCO meeting. Compared with zimberelimab (Z) single-agent treatment (27%; 95% CI: 15.0–42.8), D-containing arms (including DZ and DZ plus etrumadenant, E) acquired improved ORR (DZ: 41%; 95% CI: 26.3–56.8, EDZ: 40%; 95% CI: 25.7–55.7). Of note, D-containing arms observed significant prolonged PFS (DZ: 12 months; 95% CI: 5.5-not evaluable; HR: 0.55 [95% CI: 0.31–1.0], EDZ: 10.9 months; 95% CI: 4.8-not evaluable; HR: 0.65 [95% CI: 0.37–1.1]) in contrast to Z (5.4 months; 95% CI: 1.8–9.6) in patients with high PD-L1 expression (TPS ≥ 50%), EGFR/ALK wild-type, and metastatic NSCLC after median follow-up of 11.8 months [190]. To substantiate these promising findings, the ARC-10 (NCT04736173), a phase III clinical trial evaluating the efficacy of domvanalimab and zimberelimab combination therapy, has been conducted. In addition, the treatment of NSCLC and upper gastrointestinal tract cancer with D monotherapy or in combination with anti-PD-1/PD-L1 is currently being evaluated in four phase III clinical trials, including ARC-10 (NCT04736173), PACIFIC-8 (NCT05211895) [191], STAR-121 (NCT05502237), and STAR-221 (NCT05568095).
A preclinical study on ociperlimab (BGB-A1217), a humanized monoclonal antibody against TIGIT, elicited its competitive inhibition of TIGIT [124]. Then, the safety and efficacy of ociperlimab plus tislelizumab combination therapy were assessed in a phase I study, AdvanTIG-105 (NCT04047862). The findings were reported to be all-dose-tolerable in advanced solid tumors with promising antitumor activity [192]. On top of that, this combination treatment strategy was also conducted in several other clinical studies, including AdvanTIG-202 (phase II) [193], AdvanTIG-203 (NCT04732494, phase II) [194], AdvanTIG-206 (NCT04948697, phase II) [195], and AdvanTIG-302 (NCT04746924, phase III) [196].
Clinical trials in hematological tumors
In addition to solid tumors, numerous clinical investigations have been focused specifically on hematologic malignancies, as summarized in Table 2. However, none of these studies has yielded definitive results to date. The specific reasons for this lack of conclusive findings remain to be elucidated by researchers in the future.
Potential factors contributing to clinical failures of anti-TIGIT
Based on the immune checkpoint blockades associated with the mechanism of action of TIGIT [1, 26, 66, 197], we summarized a few possible reasons for the failures of early clinical trials in patients with SCLC and NSCLC.
First, controlling the expression of PD-L1, TIGIT, and other biomarkers, as well as the quantity and quality of TILs, circulating tumor cells, and TAMs, can affect tumor invasion, metastasis, and recurrence in various patient populations [198, 199]. There must be enough immune T cells in the patients to be recruited to tumors for immunotherapy. Tumors with T cell exhaustion, called “cold” tumors, vs. tumors with plenty of T cells, named “hot” tumors, may have different reactions to the TIGIT targeting therapy.
Second, phase II results might not accurately represent the TIGIT-blocking therapy’s true effects. TIGIT, which serves as the “braker” in immune checkpoint regulation, together with another “braker”, PD-1/PD-L1, may not be sufficient to activate the immune cells to kill tumor cells, especially in patients with advanced or high-burden tumors. TIGIT cancer immunotherapy may be made more effective by combining it with additional “braker” drugs, such as anti-CTLA-4, anti-vascular endothelial growth factor (VEGF), a triple combination of TIGIT+PD-1/PD-L1+CTLA-4 or TIGIT+PD-1/PD-L1+VEGF, or chemotherapy. Multiple bispecific and trispecific antibodies are currently in clinical development and show promising preliminary therapeutic activity [200].
Conclusion and future perspectives
Immunotherapy, represented by anti-PD-1/PD-L1, has revolutionized the field of oncology. Since the approval of the first immune checkpoint inhibitor, Ipilimumab, by the FDA in 2014, numerous cancers have been included in clinical trials demonstrating significant responses to this form of treatment. However, the reality is harsh. According to a recent statistical report, approximately only half of the patients treated with PD-1/PD-L1 monotherapy exhibited a response, irrespective of their PD-L1 expression and type of cancer [201]. The challenges of sensitivity and resistance consistently obstruct our progress towards eradicating cancer, much like the hurdles we face with chemotherapy and targeted therapy. As a newly implemented immune checkpoint, TIGIT offers an alternative potential solution to this situation, and the enhanced cytotoxicity of combining treatments, including anti-TIGIT therapies, has been observed in several clinical trials [18, 20].
Indeed, as previously mentioned, the failure of the SKYSCRAPER-02 trial seemed to cast a shadow over this “uncertain” regimen. Meanwhile, unlike anti-PD-1/PD-L1 therapies, an effective biomarker for anti-TIGIT treatment is yet to be identified, which further complicates the situation. Therefore, future research should focus on discovering novel biomarkers or different approaches for targeting TIGIT, such as bispecific antibodies, antibody-drug conjugates, and CAR-T cells targeted at TIGIT.
Availability of data and materials
All clinical trials related information was obtained from public databases.
Abbreviations
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- ADV:
-
Adenovirus
- Allo-HSCT:
-
Allogeneic hematopoietic stem cell transplantation
- AML:
-
Acute myeloid leukemia
- APC:
-
Antigen-presenting cell
- ASCO:
-
American Society of Clinical Oncology
- BCNHL:
-
B-Cell Non-Hodgkin Lymphoma
- BM:
-
Bone marrow
- CAR-T:
-
Chimeric antigen receptor T cells
- CCL23:
-
C-C motif chemokine ligand 23
- CI:
-
Confidence interval
- CLL:
-
Chronic lymphocytic leukemia
- CR:
-
Complete response
- CSR:
-
Chimeric costimulatory switch receptors
- CTLA-4:
-
Cytotoxic T lymphocyte antigen-4
- DC:
-
Dendritic cell
- DCR:
-
Disease control rate
- DOX:
-
Doxorubicin
- DLBCL:
-
Diffuse Large B Cell Lymphoma
- DNMT:
-
DNA methyltransferase
- EC:
-
Esophageal Cancer
- ESCC:
-
Esophageal squamous cell carcinoma
- FAK:
-
Focal adhesion kinase
- Fap2:
-
Fibroblast activation protein 2
- FcγR:
-
Fcγ receptors
- FDA:
-
Food and Drug Administration
- GA:
-
Gastric Adenocarcinoma
- GEJ:
-
Gastroesophageal Junction Adenocarcinoma
- GITR:
-
Glucocorticoid-induced tumor necrosis factor receptor-related protein
- HBV-HCC:
-
Hepatitis B virus-associated hepatocellular carcinoma
- HD:
-
Healthy donor
- HIF-1:
-
Hipoxia-inducible factor 1
- HR:
-
Hazard ratio
- HSV:
-
Herpes simplex virus
- ICI:
-
Immune checkpoint inhibitors
- ICD:
-
ITIM-containing intracellular domain
- IFN-γ:
-
Interferon-γ
- IFN-I:
-
Type 1 interferon
- IgV:
-
Immunoglobulin variable-set
- ITIM:
-
Immunoreceptor tyrosine-based inhibitory motif
- ITT:
-
Immunoglobulin tyrosine tail
- LAG-3:
-
Lymphocyte activating gene-3
- LAM:
-
Leukemia-associated macrophage
- LUSC:
-
Lung squamous cell carcinoma
- mAb:
-
Monoclonal antibody
- MCL:
-
Mantle cell lymphoma
- MDS:
-
Myelodysplastic syndromes
- MDSC:
-
Myeloid-derived suppressor cell
- MHC:
-
Major histocompatibility complex
- MM:
-
Multiple myeloma
- MMP-2:
-
Matrix metalloproteinase 2
- NHL:
-
Non-Hodgkin lymphoma
- NK:
-
Natural killer
- NSCLC:
-
Non-small cell lung cancer
- OC:
-
Ovarian cancer
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- OV:
-
Pncolytic viruses
- PBC:
-
Peripheral blood cell
- PDAC:
-
Pancreatic Ductal Adenocarcinoma
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed death-ligand 1
- PFS:
-
Progression free survival
- PVR:
-
Poliovirus receptor
- SCCAC:
-
Squamous Cell Carcinoma of the Anal Canal
- SCCHN:
-
Squamous Cell Carcinoma of Head and Neck
- scFv:
-
Single-chain fragment variable
- SCLC:
-
Small Cell Lung Cancer
- SCT:
-
Stem cell transplantation
- SH2:
-
Src homology 2
- SHIP1:
-
SH2-containing inositol phosphatase 1
- SOX:
-
S-1 plus oxaliplatin
- TAM:
-
Tumor-associated macrophage
- TFH:
-
Targeting follicular helper T cells
- Th:
-
T helper cell
- TIGIT:
-
T-cell immunoreceptor with Ig and ITIM domains
- TIL:
-
Tumor infiltrating lymphocyte
- TIM:
-
Tumor-infiltrating myeloid cell
- TIM-3:
-
T cell immunoglobulin and mucin-domain-containing-3
- TiNK:
-
Tumor-infiltrating NK cell
- TLS:
-
Tertiary lymphoid structure
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple Negative Breast Cancer
- TNF:
-
Tumor necrosis factor
- TPS:
-
Tumor Cell Proportion Score
- TRAF6:
-
TNF receptor-associated factor 6
- Treg:
-
Regulatory T cell
- VEGF:
-
Vascular endothelial growth factor
- VV:
-
Vaccinia virus
References
Wang Y, Zhang H, Liu C, Wang Z, Wu W, Zhang N, et al. Immune checkpoint modulators in cancer immunotherapy: recent advances and emerging concepts. J Hematol Oncol. 2022;15(1):111.
Maritaz C, Broutin S, Chaput N, Marabelle A, Paci A. Immune checkpoint-targeted antibodies: a room for dose and schedule optimization? J Hematol Oncol. 2022;15(1):6.
Weng J, Li S, Zhu Z, Liu Q, Zhang R, Yang Y, et al. Exploring immunotherapy in colorectal cancer. J Hematol Oncol. 2022;15(1):95.
Xu L, Zou C, Zhang S, Chu TSM, Zhang Y, Chen W, et al. Reshaping the systemic tumor immune environment (STIE) and tumor immune microenvironment (TIME) to enhance immunotherapy efficacy in solid tumors. J Hematol Oncol. 2022;15(1):87.
Wu YL, Lu S, Cheng Y, Zhou C, Wang J, Mok T, et al. Nivolumab versus docetaxel in a predominantly Chinese patient population with previously treated advanced NSCLC: CheckMate 078 randomized phase III clinical trial. J Thorac Oncol. 2019;14(5):867–75.
Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, et al. Prolonged survival in stage III melanoma with Ipilimumab adjuvant therapy. N Engl J Med. 2016;375(19):1845–55.
Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–67.
Yang X, Ma L, Zhang X, Huang L, Wei J. Targeting PD-1/PD-L1 pathway in myelodysplastic syndromes and acute myeloid leukemia. Exp Hematol Oncol. 2022;11(1):11.
Wu M, Huang Q, Xie Y, Wu X, Ma H, Zhang Y, et al. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J Hematol Oncol. 2022;15(1):24.
Sabbatino F, Marra A, Liguori L, Scognamiglio G, Fusciello C, Botti G, et al. Resistance to anti-PD-1-based immunotherapy in basal cell carcinoma: a case report and review of the literature. J Immunother Cancer. 2018;6(1):126.
Dai M, Liu M, Yang H, Küçük C, You H. New insights into epigenetic regulation of resistance to PD-1/PD-L1 blockade cancer immunotherapy: mechanisms and therapeutic opportunities. Exp Hematol Oncol. 2022;11(1):101.
Liu Z, Yu X, Xu L, Li Y, Zeng C. Current insight into the regulation of PD-L1 in cancer. Exp Hematol Oncol. 2022;11(1):44.
Shergold AL, Millar R, Nibbs RJB. Understanding and overcoming the resistance of cancer to PD-1/PD-L1 blockade. Pharmacol Res. 2019;145:104258.
Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10(1):48–57.
Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol. 2009;39(3):695–703.
Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol. 2011;41(4):902–15.
Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, et al. GeneCards Version 3: the human gene integrator. Database (Oxford). 2010;2010:baq020.
Niu J, Maurice-Dror C, Lee DH, Kim DW, Nagrial A, Voskoboynik M, et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer(☆). Ann Oncol. 2022;33(2):169–80.
Mettu NB, Ulahannan SV, Bendell JC, Garrido-Laguna I, Strickler JH, Moore KN, et al. A phase 1a/b open-label, dose-escalation study of Etigilimab alone or in combination with Nivolumab in patients with locally advanced or metastatic solid tumors. Clin Cancer Res. 2022;28(5):882–92.
Cho BC, Abreu DR, Hussein M, Cobo M, Patel AJ, Secen N, et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 2022;23(6):781–92.
Recondo G, Mezquita L. Tiragolumab and atezolizumab in patients with PD-L1 positive non-small-cell lung cancer. Lancet Oncol. 2022;23(6):695–7.
Frentzas S, Kao S, Gao R, Zheng H, Rizwan A, Budha N, de la Hoz Pedroza L, Tan W, Meniawy T: AdvanTIG-105: a phase I dose escalation study of the anti-TIGIT monoclonal antibody ociperlimab in combination with tislelizumab in patients with advanced solid tumors. J Immunother Cancer. 2023;11(10).
Rudin CM, Liu SV, Lu S, et al. SKYSCRAPER-02: primary results of a phase III, randomized, double-blind, placebo-controlled study of atezolizumab (atezo) + carboplatin + etoposide (CE) with or without tiragolumab (tira) in patients (pts) with untreated extensive-stage small cell lung cancer (ES-SCLC). J Clin Oncol. 2022;40(17_suppl):LBA8507-LBA.
Roche reports interim results for the phase III SKYSCRAPER-01 study in PD-L1-high metastatic non–small cell lung cancer. News Release. May 11, 2022; Available from: https://bit.ly/39e8QY4.
Kawashima S, Inozume T, Kawazu M, Ueno T, Nagasaki J, Tanji E, et al. TIGIT/CD155 axis mediates resistance to immunotherapy in patients with melanoma with the inflamed tumor microenvironment. J Immunother Cancer. 2021;9(11)
He W, Zhang H, Han F, Chen X, Lin R, Wang W, et al. CD155T/TIGIT signaling regulates CD8(+) T-cell metabolism and promotes tumor progression in human gastric Cancer. Cancer Res. 2017;77(22):6375–88.
Liu X, Li M, Wang X, Dang Z, Jiang Y, Wang X, et al. PD-1(+) TIGIT(+) CD8(+) T cells are associated with pathogenesis and progression of patients with hepatitis B virus-related hepatocellular carcinoma. Cancer Immunol Immunother. 2019;68(12):2041–54.
Ge Z, Peppelenbosch MP, Sprengers D, Kwekkeboom J. TIGIT, the next step towards successful combination immune checkpoint therapy in Cancer. Front Immunol. 2021;12:699895.
Chauvin JM. Zarour HM: TIGIT in cancer immunotherapy. J Immunother Cancer. 2020;8(2)
Weimer P, Wellbrock J, Sturmheit T, Oliveira-Ferrer L, Ding Y, Menzel S, et al. Tissue-Specific Expression of TIGIT, PD-1, TIM-3, and CD39 by γδ T cells in ovarian Cancer. Cells. 2022;11(6)
Tian X, Ning Q, Yu J, Tang S. T-cell immunoglobulin and ITIM domain in cancer immunotherapy: a focus on tumor-infiltrating regulatory T cells. Mol Immunol. 2022;147:62–70.
Xu F, Sunderland A, Zhou Y, Schulick RD, Edil BH, Zhu Y. Blockade of CD112R and TIGIT signaling sensitizes human natural killer cell functions. Cancer Immunol Immunother. 2017;66(10):1367–75.
Judge SJ, Darrow MA, Thorpe SW, Gingrich AA, O'Donnell EF, Bellini AR, et al. Analysis of tumor-infiltrating NK and T cells highlights IL-15 stimulation and TIGIT blockade as a combination immunotherapy strategy for soft tissue sarcomas. J Immunother Cancer. 2020;8(2)
Chauvin JM, Ka M, Pagliano O, Menna C, Ding Q, DeBlasio R, et al. IL15 stimulation with TIGIT blockade reverses CD155-mediated NK-cell dysfunction in melanoma. Clin Cancer Res. 2020;26(20):5520–33.
Catakovic K, Gassner FJ, Ratswohl C, Zaborsky N, Rebhandl S, Schubert M, et al. TIGIT expressing CD4+T cells represent a tumor-supportive T cell subset in chronic lymphocytic leukemia. Oncoimmunology. 2017;7(1):e1371399.
Yang Z, Peng Y, Xu J, Chen P, Zhao Z, Cai Q, et al. PVR/TIGIT and PD-L1/PD-1 expression predicts survival and enlightens combined immunotherapy in lung squamous cell carcinoma. Transl Oncol. 2022;24:101501.
Liu PC, Feng XW, Zhao XM, Ye J, He FL, Liu H, et al. Abnormal expression and clinical significance of surface receptors on natural killer cells in the peripheral blood of patients with non-small cell lung cancer. Neoplasma. 2022;69(4):931–9.
Lee BH, Kim JH, Kang KW, Lee SR, Park Y, Sung HJ, et al. PVR (CD155) expression as a potential prognostic marker in multiple myeloma. Biomedicines. 2022;10(5)
Pan K, Farrukh H, Chittepu V, Xu H, Pan CX, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. 2022;41(1):119.
Ottaviano G, Georgiadis C, Gkazi SA, Syed F, Zhan H, Etuk A, et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci Transl Med. 2022;14(668):eabq3010.
Mailankody S, Devlin SM, Landa J, Nath K, Diamonte C, Carstens EJ, et al. GPRC5D-targeted CAR T cells for myeloma. N Engl J Med. 2022;387(13):1196–206.
Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. Two-year follow-up of KTE-X19 in patients with relapsed or refractory adult B-cell acute lymphoblastic leukemia in ZUMA-3 and its contextualization with SCHOLAR-3, an external historical control study. J Hematol Oncol. 2022;15(1):170.
Zhao W-H, Wang B-Y, Chen L-J, Fu W-J, Xu J, Liu J, et al. Four-year follow-up of LCAR-B38M in relapsed or refractory multiple myeloma: a phase 1, single-arm, open-label, multicenter study in China (LEGEND-2). J Hematol Oncol. 2022;15(1):86.
Jiang VC, Hao D, Jain P, Li Y, Cai Q, Yao Y, et al. TIGIT is the central player in T-cell suppression associated with CAR T-cell relapse in mantle cell lymphoma. Mol Cancer. 2022;21(1):185.
Jackson Z, Hong C, Schauner R, Dropulic B, Caimi PF, de Lima M, et al. Sequential single-cell transcriptional and protein marker profiling reveals TIGIT as a marker of CD19 CAR-T cell dysfunction in patients with non-Hodgkin lymphoma. Cancer Discov. 2022;12(8):1886–903.
Yu L, Liu X, Wang X, Yan F, Wang P, Jiang Y, et al. TIGIT(+) TIM-3(+) NK cells are correlated with NK cell exhaustion and disease progression in patients with hepatitis B virus-related hepatocellular carcinoma. Oncoimmunology. 2021;10(1):1942673.
Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Sanchez-Correa B, Guerrero B, et al. Characterization of the DNAM-1, TIGIT and TACTILE Axis on Circulating NK, NKT-Like and T Cell Subsets in Patients with Acute Myeloid Leukemia. Cancers (Basel). 2020;12(8)
Josefsson SE, Beiske K, Blaker YN, Førsund MS, Holte H, Østenstad B, et al. TIGIT and PD-1 mark Intratumoral T cells with reduced effector function in B-cell non-Hodgkin lymphoma. Cancer Immunol Res. 2019;7(3):355–62.
Xu J, Fang Y, Chen K, Li S, Tang S, Ren Y, et al. Single-cell RNA sequencing reveals the tissue architecture in human high-grade serous ovarian Cancer. Clin Cancer Res. 2022;28(16):3590–602.
Josefsson SE, Huse K, Kolstad A, Beiske K, Pende D, Steen CB, et al. T cells expressing checkpoint receptor TIGIT are enriched in follicular lymphoma tumors and characterized by reversible suppression of T-cell receptor signaling. Clin Cancer Res. 2018;24(4):870–81.
Han HS, Jeong S, Kim H, Kim HD, Kim AR, Kwon M, et al. TOX-expressing terminally exhausted tumor-infiltrating CD8(+) T cells are reinvigorated by co-blockade of PD-1 and TIGIT in bladder cancer. Cancer Lett. 2021;499:137–47.
Wang M, Bu J, Zhou M, Sido J, Lin Y, Liu G, et al. CD8(+)T cells expressing both PD-1 and TIGIT but not CD226 are dysfunctional in acute myeloid leukemia (AML) patients. Clin Immunol. 2018;190:64–73.
Xu L, Liu L, Yao D, Zeng X, Zhang Y, Lai J, et al. PD-1 and TIGIT are highly co-expressed on CD8(+) T cells in AML patient bone marrow. Front Oncol. 2021;11:686156.
Klampatsa A, O'Brien SM, Thompson JC, Rao AS, Stadanlick JE, Martinez MC, et al. Phenotypic and functional analysis of malignant mesothelioma tumor-infiltrating lymphocytes. Oncoimmunology. 2019;8(9):e1638211.
Ostroumov D, Duong S, Wingerath J, Woller N, Manns MP, Timrott K, et al. Transcriptome profiling identifies TIGIT as a marker of T-cell exhaustion in liver Cancer. Hepatology. 2021;73(4):1399–418.
Liu L, Wang A, Liu X, Han S, Sun Y, Zhang J, et al. Blocking TIGIT/CD155 signalling reverses CD8(+) T cell exhaustion and enhances the antitumor activity in cervical cancer. J Transl Med. 2022;20(1):280.
Shao Q, Wang L, Yuan M, Jin X, Chen Z, Wu C. TIGIT induces (CD3+) T cell dysfunction in colorectal Cancer by inhibiting glucose metabolism. Front Immunol. 2021;12:688961.
Liu Z, Zhou Q, Wang Z, Zhang H, Zeng H, Huang Q, et al. Intratumoral TIGIT(+) CD8(+) T-cell infiltration determines poor prognosis and immune evasion in patients with muscle-invasive bladder cancer. J Immunother Cancer. 2020;8(2)
Liu Z, Zeng H, Jin K, Yu Y, You R, Zhang H, et al. TIGIT and PD-1 expression atlas predicts response to adjuvant chemotherapy and PD-L1 blockade in muscle-invasive bladder cancer. Br J Cancer. 2022;126(9):1310–7.
Guillerey C, Harjunpää H, Carrié N, Kassem S, Teo T, Miles K, et al. TIGIT immune checkpoint blockade restores CD8(+) T-cell immunity against multiple myeloma. Blood. 2018;132(16):1689–94.
Minnie SA, Kuns RD, Gartlan KH, Zhang P, Wilkinson AN, Samson L, et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood. 2018;132(16):1675–88.
Lucca LE, Dominguez-Villar M. Modulation of regulatory T cell function and stability by co-inhibitory receptors. Nat Rev Immunol. 2020;20(11):680–93.
Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014;40(4):569–81.
Liu H, Wu J, Xu X, Wang H, Zhang C, Yin S, et al. Peritumoral TIGIT(+)CD20(+) B cell infiltration indicates poor prognosis but favorable adjuvant chemotherapeutic response in gastric cancer. Int Immunopharmacol. 2022;108:108735.
Hasan MM, Nair SS, O'Leary JG, Thompson-Snipes L, Nyarige V, Wang J, et al. Implication of TIGIT(+) human memory B cells in immune regulation. Nat Commun. 2021;12(1):1534.
Zhou XM, Li WQ, Wu YH, Han L, Cao XG, Yang XM, et al. Intrinsic expression of immune checkpoint molecule TIGIT could help tumor growth in vivo by suppressing the function of NK and CD8(+) T cells. Front Immunol. 2018;9:2821.
Hoogi S, Eisenberg V, Mayer S, Shamul A, Barliya T, Cohen CJ. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J Immunother Cancer. 2019;7(1):243.
Tang W, Pan X, Han D, Rong D, Zhang M, Yang L, et al. Clinical significance of CD8(+) T cell immunoreceptor with Ig and ITIM domains(+) in locally advanced gastric cancer treated with SOX regimen after D2 gastrectomy. Oncoimmunology. 2019;8(6):e1593807.
Kamat K, Krishnan V, Dorigo O. Macrophage-derived CCL23 upregulates expression of T-cell exhaustion markers in ovarian cancer. Br J Cancer. 2022;127(6):1026–33.
Davern M, Fitzgerald MC, Buckley CE, Heeran AB, Donlon NE, McGrath J. F OC, Deshpande MR, Hayes C, MacDonald J, Sheppard AD, Reynolds JV, Maher SG, Lynam-Lennon N, Murphy B, Lysaght J: PD-1 and TIGIT blockade differentially affect tumour cell survival under hypoxia and glucose deprived conditions in oesophageal adenocarcinoma; implications for overcoming resistance to PD-1 blockade in hypoxic tumours. Transl Oncol. 2022;19:101381.
Chen Y, Huang H, Li Y, Xiao W, Liu Y, Chen R, et al. TIGIT blockade exerts synergistic effects on microwave ablation against Cancer. Front Immunol. 2022;13:832230.
Sumida TS, Dulberg S, Schupp JC, Lincoln MR, Stillwell HA, Axisa PP, et al. Type I interferon transcriptional network regulates expression of coinhibitory receptors in human T cells. Nat Immunol. 2022;23(4):632–42.
Cai H, Li M, Deng R, Wang M, Shi Y. Advances in molecular biomarkers research and clinical application progress for gastric cancer immunotherapy. Biomarker Res. 2022;10(1):67.
Korbecki J, Kojder K, Simińska D, Bohatyrewicz R, Gutowska I, Chlubek D, et al. CC chemokines in a tumor: a review of pro-Cancer and anti-Cancer properties of the ligands of receptors CCR1, CCR2, CCR3, and CCR4. Int J Mol Sci. 2020;21(21)
Chiang EY, Mellman I. TIGIT-CD226-PVR axis: advancing immune checkpoint blockade for cancer immunotherapy. J Immunother Cancer. 2022;10(4)
Zhou X, Du J, Zhou X, Niu X, Li W, Chen C, et al. Computer-aided design of PVR mutants with enhanced binding affinity to TIGIT. Cell Commun Signal. 2021;19(1):12.
Stengel KF, Harden-Bowles K, Yu X, Rouge L, Yin J, Comps-Agrar L, et al. Structure of TIGIT immunoreceptor bound to poliovirus receptor reveals a cell-cell adhesion and signaling mechanism that requires cis-trans receptor clustering. Proc Natl Acad Sci U S A. 2012;109(14):5399–404.
Deuss FA, Watson GM, Fu Z, Rossjohn J, Berry R. Structural basis for CD96 immune receptor recognition of Nectin-like Protein-5, CD155. Structure. 2019;27(2):219–228.e3.
Liu S, Zhang H, Li M, Hu D, Li C, Ge B, et al. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013;20(3):456–64.
Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A. 2009;106(42):17858–63.
Li M, Xia P, Du Y, Liu S, Huang G, Chen J, et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-γ production of natural killer cells via β-arrestin 2-mediated negative signaling. J Biol Chem. 2014;289(25):17647–57.
Annese T, Tamma R, Ribatti D. Update in TIGIT immune-checkpoint role in Cancer. Front Oncol. 2022;12:871085.
Gao J, Zheng Q, Xin N, Wang W, Zhao C. CD155, an onco-immunologic molecule in human tumors. Cancer Sci. 2017;108(10):1934–8.
Wu B, Zhong C, Lang Q, Liang Z, Zhang Y, Zhao X, et al. Poliovirus receptor (PVR)-like protein cosignaling network: new opportunities for cancer immunotherapy. J Exp Clin Cancer Res. 2021;40(1):267.
Zhang B, Zhao W, Li H, Chen Y, Tian H, Li L, et al. Immunoreceptor TIGIT inhibits the cytotoxicity of human cytokine-induced killer cells by interacting with CD155. Cancer Immunol Immunother. 2016;65(3):305–14.
Matsuo T, Iguchi-Manaka A, Shibuya A, Shibuya K. CD155 mutation (Ala67Thr) increases the binding affinity for and the signaling via an inhibitory immunoreceptor TIGIT. Cancer Sci. 2022;
Sanchez-Correa B, Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Bergua JM, et al. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers (Basel). 2019;11(6)
Li Y, Zhang Y, Cao G, Zheng X, Sun C, Wei H, et al. Blockade of checkpoint receptor PVRIG unleashes anti-tumor immunity of NK cells in murine and human solid tumors. J Hematol Oncol. 2021;14(1):100.
Zhan M, Zhang Z, Zhao X, Zhang Y, Liu T, Lu L, et al. CD155 in tumor progression and targeted therapy. Cancer Lett. 2022;545:215830.
Lupo KB, Matosevic S. CD155 immunoregulation as a target for natural killer cell immunotherapy in glioblastoma. J Hematol Oncol. 2020;13(1):76.
Hansen K, Kumar S, Logronio K, Whelan S, Qurashi S, Cheng HY, et al. COM902, a novel therapeutic antibody targeting TIGIT augments anti-tumor T cell function in combination with PVRIG or PD-1 pathway blockade. Cancer Immunol Immunother. 2021;70(12):3525–40.
Kučan Brlić P, Lenac Roviš T, Cinamon G, Tsukerman P, Mandelboim O, Jonjić S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell Mol Immunol. 2019;16(1):40–52.
Stamm H, Oliveira-Ferrer L, Grossjohann EM, Muschhammer J, Thaden V, Brauneck F, et al. Targeting the TIGIT-PVR immune checkpoint axis as novel therapeutic option in breast cancer. Oncoimmunology. 2019;8(12):e1674605.
Jiang C, Qu X, Ma L, Yi L, Cheng X, Gao X, et al. CD155 expression impairs anti-PD1 therapy response in non-small cell lung cancer. Clin Exp Immunol. 2022;208(2):220–32.
Stamm H, Klingler F, Grossjohann EM, Muschhammer J, Vettorazzi E, Heuser M, et al. Immune checkpoints PVR and PVRL2 are prognostic markers in AML and their blockade represents a new therapeutic option. Oncogene. 2018;37(39):5269–80.
Pende D, Castriconi R, Romagnani P, Spaggiari GM, Marcenaro S, Dondero A, et al. Expression of the DNAM-1 ligands, Nectin-2 (CD112) and poliovirus receptor (CD155), on dendritic cells: relevance for natural killer-dendritic cell interaction. Blood. 2006;107(5):2030–6.
Zeng T, Cao Y, Jin T, Tian Y, Dai C, Xu F. The CD112R/CD112 axis: a breakthrough in cancer immunotherapy. J Exp Clin Cancer Res. 2021;40(1):285.
Pende D, Bottino C, Castriconi R, Cantoni C, Marcenaro S, Rivera P, et al. PVR (CD155) and Nectin-2 (CD112) as ligands of the human DNAM-1 (CD226) activating receptor: involvement in tumor cell lysis. Mol Immunol. 2005;42(4):463–9.
Kojima H, Kanada H, Shimizu S, Kasama E, Shibuya K, Nakauchi H, et al. CD226 mediates platelet and megakaryocytic cell adhesion to vascular endothelial cells. J Biol Chem. 2003;278(38):36748–53.
Reymond N, Imbert AM, Devilard E, Fabre S, Chabannon C, Xerri L, et al. DNAM-1 and PVR regulate monocyte migration through endothelial junctions. J Exp Med. 2004;199(10):1331–41.
Ma D, Sun Y, Lin D, Wang H, Dai B, Zhang X, et al. CD226 is expressed on the megakaryocytic lineage from hematopoietic stem cells/progenitor cells and involved in its polyploidization. Eur J Haematol. 2005;74(3):228–40.
Bachelet I, Munitz A, Mankutad D, Levi-Schaffer F. Mast cell costimulation by CD226/CD112 (DNAM-1/Nectin-2): a novel interface in the allergic process. J Biol Chem. 2006;281(37):27190–6.
Weulersse M, Asrir A, Pichler AC, Lemaitre L, Braun M, Carrié N, et al. Eomes-dependent loss of the co-activating receptor CD226 restrains CD8(+) T cell anti-tumor functions and limits the efficacy of Cancer immunotherapy. Immunity. 2020;53(4):824–839.e10.
Jin HS, Ko M, Choi DS, Kim JH, Lee DH, Kang SH, et al. CD226(hi)CD8(+) T cells are a prerequisite for anti-TIGIT immunotherapy. Cancer Immunol Res. 2020;8(7):912–25.
Maas RJ, Hoogstad-van Evert JS, Van der Meer JM, Mekers V, Rezaeifard S, Korman AJ, et al. TIGIT blockade enhances functionality of peritoneal NK cells with altered expression of DNAM-1/TIGIT/CD96 checkpoint molecules in ovarian cancer. Oncoimmunology. 2020;9(1):1843247.
Jin Z, Lan T, Zhao Y, Du J, Chen J, Lai J, et al. Higher TIGIT(+)CD226(−) γδ T cells in patients with acute myeloid leukemia. Immunol Investig. 2022;51(1):40–50.
Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O'Gorman WE, et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8(+) T cell responses. Immunity. 2022;55(3):512–526.e9.
Wang B, Zhang W, Jankovic V, Golubov J, Poon P, Oswald EM, et al. Skokos D: combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8(+) T cell dysfunction and maintain memory phenotype. Sci Immunol. 2018;3(29)
Fourcade J, Sun Z, Chauvin JM, Ka M, Davar D, Pagliano O, et al. Zarour HM: CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight. 2018;3(14)
Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A. 2007;104(26):11008–13.
Wang PL, O'Farrell S, Clayberger C, Krensky AM. Identification and molecular cloning of tactile. A novel human T cell activation antigen that is a member of the Ig gene superfamily. J Immunol. 1992;148(8):2600–8.
Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. 2017;276(1):112–20.
Mittal D, Lepletier A, Madore J, Aguilera AR, Stannard K, Blake SJ, et al. CD96 is an immune checkpoint that regulates CD8(+) T-cell antitumor function. Cancer Immunol Res. 2019;7(4):559–71.
Wang Y, Wang C, Qiu J, Qu X, Peng J, Lu C, et al. Targeting CD96 overcomes PD-1 blockade resistance by enhancing CD8+ TIL function in cervical cancer. J Immunother Cancer. 2022;10(3)
Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J Immunol. 2004;172(7):3994–8.
Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. 2019;70(1):168–83.
Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. 2014;15(5):431–8.
Reches A, Ophir Y, Stein N, Kol I, Isaacson B, Charpak Amikam Y, et al. Nectin4 is a novel TIGIT ligand which combines checkpoint inhibition and tumor specificity. J Immunother Cancer. 2020;8(1)
Challita-Eid PM, Satpayev D, Yang P, An Z, Morrison K, Shostak Y, et al. Enfortumab Vedotin antibody-drug conjugate targeting Nectin-4 is a highly potent therapeutic agent in multiple preclinical Cancer models. Cancer Res. 2016;76(10):3003–13.
Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42(2):344–55.
Shaw G, Cavalcante L, Giles FJ, Taylor A. Elraglusib (9-ING-41), a selective small-molecule inhibitor of glycogen synthase kinase-3 beta, reduces expression of immune checkpoint molecules PD-1, TIGIT and LAG-3 and enhances CD8(+) T cell cytolytic killing of melanoma cells. J Hematol Oncol. 2022;15(1):134.
Wu Y, Hao X, Wei H, Sun R, Chen Y, Tian Z. Blockade of T-cell receptor with Ig and ITIM domains elicits potent antitumor immunity in naturally occurring HBV-related HCC in mice. Hepatology. 2023;77(3):965–81.
Wu L, Mao L, Liu JF, Chen L, Yu GT, Yang LL, et al. Blockade of TIGIT/CD155 signaling reverses T-cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol Res. 2019;7(10):1700–13.
Chen X, Xue L, Ding X, Zhang J, Jiang L, Liu S, et al. An fc-competent anti-human TIGIT blocking antibody Ociperlimab (BGB-A1217) elicits strong immune responses and potent anti-tumor efficacy in pre-clinical models. Front Immunol. 2022;13:828319.
Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26(6):923–37.
Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. 2018;7(8):e1466769.
Freed-Pastor WA, Lambert LJ, Ely ZA, Pattada NB, Bhutkar A, Eng G, et al. The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell. 2021;39(10):1342–1360.e14.
Zhao K, Jiang L, Si Y, Zhou S, Huang Z, Meng X. TIGIT blockade enhances tumor response to radiotherapy via a CD103 + dendritic cell-dependent mechanism. Cancer Immunol Immunother. 2022;
Yoo KJ, Johannes K, González LE, Patel A, Shuptrine CW, Opheim Z, et al. LIGHT (TNFSF14) Costimulation enhances myeloid cell activation and antitumor immunity in the setting of PD-1/PD-L1 and TIGIT checkpoint blockade. J Immunol. 2022;209(3):510–25.
Zuo S, Wei M, Xu T, Kong L, He B, Wang S, et al. An engineered oncolytic vaccinia virus encoding a single-chain variable fragment against TIGIT induces effective antitumor immunity and synergizes with PD-1 or LAG-3 blockade. J Immunother Cancer. 2021;9(12)
Brauneck F, Seubert E, Wellbrock J, Schulze Zur Wiesch J, Duan Y, Magnus T, et al. Combined blockade of TIGIT and CD39 or A2AR enhances NK-92 cell-mediated cytotoxicity in AML. Int J Mol Sci. 2021;22(23)
Brauneck F, Fischer B, Witt M, Muschhammer J, Oelrich J, da Costa Avelar PH, et al. TIGIT blockade repolarizes AML-associated TIGIT(+) M2 macrophages to an M1 phenotype and increases CD47-mediated phagocytosis. J Immunother Cancer. 2022;10(12)
Zhou Y, Yang D, Yang Q, Lv X, Huang W, Zhou Z, et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat Commun. 2020;11(1):6322.
Han JH, Cai M, Grein J, Perera S, Wang H, Bigler M, et al. Effective anti-tumor response by TIGIT blockade associated with FcγR engagement and myeloid cell activation. Front Immunol. 2020;11:573405.
Liu H, Pan C, Song W, Liu D, Li Z, Zheng L. Novel strategies for immuno-oncology breakthroughs with cell therapy. Biomarker Res. 2021;9(1):62.
Qu T, Li B, Wang Y. Targeting CD47/SIRPα as a therapeutic strategy, where we are and where we are headed. Biomarker Res. 2022;10(1):20.
Zhang L, Meng Y, Feng X, Han Z. CAR-NK cells for cancer immunotherapy: from bench to bedside. Biomarker Res. 2022;10(1):12.
Wang X, Yang X, Yuan X, Wang W, Wang Y. Chimeric antigen receptor-engineered NK cells: new weapons of cancer immunotherapy with great potential. Exp Hematol Oncol. 2022;11(1):85.
Yu J, Sun H, Cao W, Song Y, Jiang Z. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp Hematol Oncol. 2022;11(1):3.
Sanchez-Correa B, Lopez-Sejas N, Duran E, Labella F, Alonso C, Solana R, et al. Modulation of NK cells with checkpoint inhibitors in the context of cancer immunotherapy. Cancer Immunol Immunother. 2019;68(5):861–70.
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–32.
Chiu DK, Yuen VW, Cheu JW, Wei LL, Ting V, Fehlings M, et al. Hepatocellular carcinoma cells up-regulate PVRL1, stabilizing PVR and inhibiting the cytotoxic T-cell response via TIGIT to mediate tumor resistance to PD1 inhibitors in mice. Gastroenterology. 2020;159(2):609–23.
Thibaudin M, Limagne E, Hampe L, Ballot E, Truntzer C, Ghiringhelli F. Targeting PD-L1 and TIGIT could restore intratumoral CD8 T cell function in human colorectal cancer. Cancer Immunol Immunother. 2022;
Ge Z, Zhou G, Campos Carrascosa L, Gausvik E, Boor PPC, Noordam L, et al. JNM IJ, Peppelenbosch MP, Kraan J, Kwekkeboom J, Sprengers D: TIGIT and PD1 co-blockade restores ex vivo functions of human tumor-infiltrating CD8(+) T cells in hepatocellular carcinoma. Cell Mol Gastroenterol Hepatol. 2021;12(2):443–64.
Dixon KO, Schorer M, Nevin J, Etminan Y, Amoozgar Z, Kondo T, et al. Functional anti-TIGIT antibodies regulate development of autoimmunity and antitumor immunity. J Immunol. 2018;200(8):3000–7.
Ribas A, Butterfield LH, Amarnani SN, Dissette VB, Kim D, Meng WS, et al. CD40 cross-linking bypasses the absolute requirement for CD4 T cells during immunization with melanoma antigen gene-modified dendritic cells. Cancer Res. 2001;61(24):8787–93.
Jin HS, Choi DS, Ko M, Kim D, Lee DH, Lee S, et al. Extracellular pH modulating injectable gel for enhancing immune checkpoint inhibitor therapy. J Control Release. 2019;315:65–75.
Tian T, Wang J, Yin F, Wu M, He W, Wang X, et al. Activation of Cascade-like antitumor immune responses through in situ doxorubicin stimulation and blockade of checkpoint Coinhibitory receptor TIGIT. Adv Healthc Mater. 2022;11(1):e2102080.
Yan Y, Huang L, Liu Y, Yi M, Chu Q, Jiao D, et al. Metabolic profiles of regulatory T cells and their adaptations to the tumor microenvironment: implications for antitumor immunity. J Hematol Oncol. 2022;15(1):104.
Barsoumian HB, Sezen D, Menon H, Younes AI, Hu Y, He K, et al. High plus low dose radiation strategy in combination with TIGIT and PD1 blockade to promote systemic antitumor responses. Cancers (Basel). 2022;14(1)
Grapin M, Richard C, Limagne E, Boidot R, Morgand V, Bertaut A, et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: a promising new combination. J Immunother Cancer. 2019;7(1):160.
Shen X, Fu W, Wei Y, Zhu J, Yu Y, Lei C, et al. TIGIT-fc promotes antitumor immunity. Cancer Immunol Res. 2021;9(9):1088–97.
Zhang D, Hu W, Xie J, Zhang Y, Zhou B, Liu X, et al. TIGIT-fc alleviates acute graft-versus-host disease by suppressing CTL activation via promoting the generation of immunoregulatory dendritic cells. Biochim Biophys Acta Mol basis Dis. 2018;1864(9 Pt B):3085–98.
Pietra G, Mingari MC, Moretta L. TIGIT blockade and IL15 in tumor immunotherapy: together is better. Clin Cancer Res. 2020;26(20):5274–5.
Whelan S, Ophir E, Kotturi MF, Levy O, Ganguly S, Leung L, et al. PVRIG and PVRL2 are induced in Cancer and inhibit CD8(+) T-cell function. Cancer Immunol Res. 2019;7(2):257–68.
Fathi M, Bahmanpour S, Barshidi A, Rasouli H, Karoon Kiani F. Mahmoud Salehi Khesht a, Izadi S, Rashidi B, Kermanpour S, Mokhtarian R, Karpisheh V, Hassannia H, Mohammadi H, Jalili a, Jadidi-Niaragh F: simultaneous blockade of TIGIT and HIF-1α induces synergistic anti-tumor effect and decreases the growth and development of cancer cells. Int Immunopharmacol. 2021;101(Pt A):108288.
Ozmadenci D, Shankara Narayanan JS, Andrew J, Ojalill M, Barrie AM, Jiang S, et al. Tumor FAK orchestrates immunosuppression in ovarian cancer via the CD155/TIGIT axis. Proc Natl Acad Sci U S A. 2022;119(17):e2117065119.
Chan IS, Knútsdóttir H, Ramakrishnan G, Padmanaban V, Warrier M, Ramirez JC, et al. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J Cell Biol. 2020;219(9)
Ma B, Duan X, Zhou Q, Liu J, Yang X, Zhang D, et al. Use of aspirin in the prevention of colorectal cancer through TIGIT-CD155 pathway. J Cell Mol Med. 2019;23(7):4514–22.
Li P, Zhu X, Cao G, Wu R, Li K, Yuan W, et al. 1α,25(OH)(2)D(3) reverses exhaustion and enhances antitumor immunity of human cytotoxic T cells. J Immunother Cancer. 2022;10(3)
Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2(4):295–300.
Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–62.
Chon HJ, Lee WS, Yang H, Kong SJ, Lee NK, Moon ES, et al. Tumor microenvironment remodeling by Intratumoral oncolytic vaccinia virus enhances the efficacy of immune-checkpoint blockade. Clin Cancer Res. 2019;25(5):1612–23.
Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene Laherparepvec in combination with Ipilimumab in previously untreated, Unresectable stage IIIB-IV melanoma. J Clin Oncol. 2016;34(22):2619–26.
Zheng M, Huang J, Tong A, Yang H. Oncolytic viruses for Cancer therapy: barriers and recent advances. Mol Ther Oncolytics. 2019;15:234–47.
Tie Y, Tang F, Wei YQ, Wei XW. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. 2022;15(1):61.
Lin C, Ren W, Luo Y, Li S, Chang Y, Li L, et al. Intratumoral delivery of a PD-1-blocking scFv encoded in oncolytic HSV-1 promotes antitumor immunity and synergizes with TIGIT blockade. Cancer Immunol Res. 2020;8(5):632–47.
Li W, Blessin NC, Simon R, Kluth M, Fischer K, Hube-Magg C, et al. Expression of the immune checkpoint receptor TIGIT in Hodgkin's lymphoma. BMC Cancer. 2018;18(1):1209.
Meng F, Li L, Lu F, Yue J, Liu Z, Zhang W, et al. Overexpression of TIGIT in NK and T cells contributes to tumor immune escape in myelodysplastic syndromes. Front Oncol. 2020;10:1595.
Zeng X, Yao D, Liu L, Zhang Y, Lai J, Zhong J, et al. Terminal differentiation of bone marrow NK cells and increased circulation of TIGIT(+) NK cells may be related to poor outcome in acute myeloid leukemia. Asia Pac J Clin Oncol. 2022;18(4):456–64.
Zhang X, Zhang H, Chen L, Feng Z, Gao L, Li Q. TIGIT expression is upregulated in T cells and causes T cell dysfunction independent of PD-1 and Tim-3 in adult B lineage acute lymphoblastic leukemia. Cell Immunol. 2019;344:103958.
Yang ZZ, Kim HJ, Wu H, Jalali S, Tang X, Krull JE, et al. TIGIT expression is associated with T-cell suppression and exhaustion and predicts clinical outcome and anti-PD-1 response in follicular lymphoma. Clin Cancer Res. 2020;26(19):5217–31.
Liu Z, Guo Y, Huang L, Jia Y, Liu H, Peng F, et al. Bone marrow mesenchymal stem cells regulate the dysfunction of NK cells via the T cell immunoglobulin and ITIM domain in patients with myelodysplastic syndromes. Cell Commun Signal. 2022;20(1):169.
Lozano E, Mena MP, Díaz T, Martin-Antonio B, León S, Rodríguez-Lobato LG, et al. Fernández de Larrea C: Nectin-2 expression on malignant plasma cells is associated with better response to TIGIT blockade in multiple myeloma. Clin Cancer Res. 2020;26(17):4688–98.
Ureña-Bailén G, Dobrowolski JM, Hou Y, Dirlam A, Roig-Merino A, Schleicher S, et al. Preclinical evaluation of CRISPR-edited CAR-NK-92 cells for off-the-shelf treatment of AML and B-ALL. Int J Mol Sci. 2022;23(21)
Yue J, Li J, Ma J, Zhai Y, Shen L, Zhang W, et al. Myeloid-derived suppressor cells inhibit natural killer cells in myelodysplastic syndromes through the TIGIT/CD155 pathway. Hematology. 2023;28(1):2166333.
Seiffert M. TIGIT: an immune checkpoint beyond T cells in chronic lymphocytic leukemia. Haematologica. 2023;108(8):1979–81.
Hatefi F, Asgarian-Omran H, Hossein-Nataj H, Akbar A, Shekarriz R, Zaboli E, et al. Combined blockade of PD-1 and TIGIT is not sufficient to improve the function of CD8+ T-cells in chronic lymphocytic leukemia. Asian Pac J Cancer Prev. 2022;23(7):2225–31.
Whiteley AE, Price TT, Cantelli G, Sipkins DA. Leukaemia: a model metastatic disease. Nat Rev Cancer. 2021;21(7):461–75.
Gournay V, Vallet N, Peux V, Vera K, Bordenave J, Lambert M, et al. Peffault de Latour R, Caillat-Zucman S, Socié G, chevalier MF: immune landscape after Allo-HSCT: TIGIT- and CD161-expressing CD4 T cells are associated with subsequent leukemia relapse. Blood. 2022;140(11):1305–21.
Hattori N, Kawaguchi Y, Sasaki Y, Shimada S, Murai S, Abe M, et al. Monitoring TIGIT/DNAM-1 and PVR/PVRL2 immune checkpoint expression levels in allogeneic stem cell transplantation for acute myeloid leukemia. Biol Blood Marrow Transplant. 2019;25(5):861–7.
Huang Y, Zheng H, Zhu Y, Hong Y, Zha J, Lin Z, et al. Loss of CD28 expression associates with severe T-cell exhaustion in acute myeloid leukemia. Front Immunol. 2023;14:1139517.
Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM, et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). J Clin Oncol. 2020;38(15_suppl):9503-9503.
Goodman KA, Xu R-h, Chau I, Chen MH, Cho BC, Shah MA, et al. SKYSCRAPER-07: a phase III, randomized, double-blind, placebo-controlled study of atezolizumab with or without tiragolumab in patients with unresectable ESCC who have not progressed following definitive concurrent chemoradiotherapy. J Clin Oncol. 2022;40(4_suppl):TPS374-TPS374.
D'Alessio A, Fulgenzi CAM, Nishida N, Schönlein M, von Felden J, Schulze K, et al. Preliminary evidence of safety and tolerability of atezolizumab plus bevacizumab in patients with hepatocellular carcinoma and child-Pugh a and B cirrhosis: a real-world study. Hepatology. 2022;76(4):1000–12.
Ando Y, Kawaoka T, Kosaka M, Shirane Y, Johira Y, Miura R, et al. Early tumor response and safety of Atezolizumab plus bevacizumab for patients with Unresectable hepatocellular carcinoma in real-world practice. Cancers (Basel). 2021;13(16)
Jabbour S, Lu B, Fu X, Goksel T, Sugawara S, Song A, et al. 969TiP randomized, phase III study of MK-7684A plus concurrent chemoradiotherapy (cCRT) followed by MK-7684A vs cCRT followed by durvalumab for unresectable, locally advanced, stage III non-small cell lung cancer (NSCLC): KEYVIBE-006. Ann Oncol. 2022;33(7_suppl):S990.
Garassino MC, Felip E, Awad MM, Wang W, Kim SJ, Pietanza MC, et al. A randomized, double-blind, phase 3 trial of MK-7684A plus chemotherapy versus pembrolizumab plus chemotherapy as first-line therapy for metastatic non–small cell lung cancer (NSCLC): KeyVibe-007. J Clin Oncol. 2022;40(16_suppl):TPS9160-TPS9160.
Sands J, Reck M, Navarro A, Chiang AC, Lu S, Peled N, et al. KeyVibe-008: randomized, phase 3 study of first-line vibostolimab plus pembrolizumab plus etoposide/platinum versus atezolizumab plus EP in extensive-stage small cell lung cancer. J Clin Oncol. 2022;40(16_suppl):TPS8606.
Johnson ML, Fox W, Lee Y-G, Lee KH, Ahn HK, Kim Y-C, et al. ARC-7: randomized phase 2 study of domvanalimab + zimberelimab ± etrumadenant versus zimberelimab in first-line, metastatic, PD-L1-high non-small cell lung cancer (NSCLC). J Clin Oncol. 2022;40(36_suppl):397600.
Ozguroglu M, Levy B, Horinouchi H, Yu J, Grainger E, Phuong P, et al. 971TiP phase III trial of durvalumab combined with domvanalimab following concurrent chemoradiotherapy (cCRT) in patients with unresectable stage III NSCLC (PACIFIC-8). Ann Oncol. 2022;33(7_suppl):S991.
Frentzas S, Meniawy T, Kao SC-H, Wang R, Zuo Y, Zheng H, et al. AdvanTIG-105: phase 1 dose-escalation study of anti-TIGIT monoclonal antibody ociperlimab (BGB-A1217) in combination with tislelizumab in patients with advanced solid tumors. J Clin Oncol. 2021;39(15_suppl):2583.
Wu L, Wang P-H, Hsiao S-Y, Chang C-L, Kim HS, Lee J-Y, et al. AdvanTIG-202: a phase 2 study investigating anti-TIGIT monoclonal antibody ociperlimab plus anti-PD-1 monoclonal antibody tislelizumab in patients with previously treated recurrent or metastatic cervical cancer. J Clin Oncol. 2021;39(15_suppl):TPS5595.
Xu R-h, Kim S-B, Tougeron D, Zuo Y, Yang H, Zhang J, et al. AdvanTIG-203: a randomized phase 2 study comparing anti-TIGIT ociperlimab plus tislelizumab versus tislelizumab plus placebo as second-line treatment in patients with advanced or recurrent esophageal squamous cell carcinoma (ESCC) expressing programmed death-ligand 1 (PD-L1). J Clin Oncol. 2021;39(15_suppl):TPS4150.
Fan J, Ren Z, Hsu C, Guo Y, Song T, Wang W, et al. AdvanTIG-206: anti-TIGIT monoclonal antibody (mAb) ociperlimab (BGB-A1217; OCI) plus anti-programmed cell death protein-1 (PD-1) mAb tislelizumab (TIS) plus BAT1706 versus TIS plus BAT1706 as first-line (1L) treatment for advanced hepatocellular carcinoma (HCC). J Clin Oncol. 2022;40(16_suppl):TPS4172.
Socinski MA, Spira AI, Paz-Ares LG, Reck M, Lu S, Nishio M, et al. AdvanTIG-302: anti-TIGIT monoclonal antibody (mAb) ociperlimab (OCI) plus tislelizumab (TIS) versus pembrolizumab (PEM) in programmed death ligand-1 (PD-L1) selected, previously untreated, locally advanced, unresectable or metastatic non-small cell lung cancer (NSCLC). J Clin Oncol. 2021;39(15_suppl):TPS9128.
Liu S, Sun Q, Ren X. Novel strategies for cancer immunotherapy: counter-immunoediting therapy. J Hematol Oncol. 2023;16(1):38.
Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood. 2018;131(16):1809–19.
Zhang J, Liu Z, Cao P, Wang H, Liu H, Hua L, et al. Tumor-associated macrophages regulate the function of cytotoxic T lymphocyte through PD-1/PD-L1 pathway in multiple myeloma. Cancer Med. 2022;
Blanco B, Domínguez-Alonso C, Alvarez-Vallina L. Bispecific immunomodulatory antibodies for Cancer immunotherapy. Clin Cancer Res. 2021;27(20):5457–64.
Zhao B, Zhao H, Zhao J. Efficacy of PD-1/PD-L1 blockade monotherapy in clinical trials. Ther Adv Med Oncol. 2020;12:1758835920937612.
Acknowledgments
Figures in this review were all created with Biorender.com
Funding
This work was supported by the Project of Science and Technology Department of Henan Province, China (LHGJ20190039, SBGJ20202076, recipient JY), and Talent Research Fund of the First Affiliated Hospital of Zhengzhou University, Zhengzhou, China (recipient JY). The funding bodies did not participate in the study design, in data collection, analysis, and interpretation, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
J.Y., Y.S., and S.Z. designed and directed the study, J.Y. and P. Z. wrote the manuscript draft. J.Y., Y.S., S.Z., Z.G and X.L reviewed the manuscript. Z.J. provided resources. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, P., Liu, X., Gu, Z. et al. Targeting TIGIT for cancer immunotherapy: recent advances and future directions. Biomark Res 12, 7 (2024). https://doi.org/10.1186/s40364-023-00543-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-023-00543-z