
Abstract
Natural killer (NK) cells are powerful immune effectors, modulating their anti-tumor function through a balance activating and inhibitor ligands on their cell surface. Though still emerging, cancer immunotherapies utilizing NK cells are proving promising as a modality for the treatment of a number of solid tumors, including glioblastoma (GBM) and other gliomas, but are often limited due to complex immunosuppression associated with the GBM tumor microenvironment which includes overexpression of inhibitory receptors on GBM cells. CD155, or poliovirus receptor (PVR), has recently emerged as a pro-tumorigenic antigen, overexpressed on GBM and contributing to increased GBM migration and aggressiveness. CD155 has also been established as an immunomodulatory receptor, able to both activate NK cells through interactions with CD226 (DNAM-1) and CD96 and inhibit them through interaction with TIGIT. However, NK cell TIGIT expression has been shown to be upregulated in cancer, establishing CD155 as a predominantly inhibitory receptor within the context of GBM and other solid tumors, and rendering it of interest as a potential target for antigen-specific NK cell-based immunotherapy. This review will explore the function of CD155 within GBM as it relates to tumor migration and NK cell immunoregulation, as well as pre-clinical and clinical targeting of CD155/TIGIT and the potential that this pathway holds for the development of emerging NK cell-based immunotherapies.
Similar content being viewed by others


Introduction
Among the multiple elements contributing to the aggressive pathology of glioblastoma (GBM)—the most malignant brain tumor which currently stands with no curative treatment—is the emergence of CD155 as a pro-tumorigenic antigen [1,2,3]. A cell adhesion molecule of the immunoglobulin (Ig) superfamily, CD155 is a type I transmembrane glycoprotein that was first described as a poliovirus receptor (PVR) [4]. Though its expression can be detected at low levels on epithelial and endothelial cells in a variety of tissues, its overexpression on malignant cells has been associated with poor prognosis in patients with breast cancer [5], lung adenocarcinoma [6], pancreatic cancer [7], cholangiocarcinoma [8], melanoma [9], and various soft tissue tumors [10].
High-grade malignant gliomas, including GBM (grade IV), are associated with overexpression of CD155 [11], which was shown to contribute to cancer cell dispersal [1]. The receptor’s adhesive capacity has a well-established role in promoting migration and invasiveness of tumor cells [2]. Though CD155 has been shown to regulate certain immune cell responses such as graft-versus-host-disease [12], its role as a pro-tumorigenic antigen has received increased attention as of late. A dose-escalation trial of a recombinant nonpathogenic polio–rhinovirus chimera (PVSRIPO) delivered intratumorally to patients with grade IV glioma resulted in longer survival of treated patients at 24 and 36 months compared to patients treated historically [13].
CD155 exerts its functions by interacting with multiple ligands. Engagement of CD155 with ligands including CD226 (DNAM-1) and CD96 has been demonstrated to drive anti-tumor immune responses, particularly those by NK cells [14]. NK cells, moreover, express T cell immunoreceptor with Ig and ITIM domains (TIGIT), an immunoglobulin superfamily receptor, whose ligands include CD155, CD112, and CD113 [15]. TIGIT—which competes with DNAM-1 for binding to CD115—interacts with these receptors resulting in inhibition of NK cell anti-tumor function including impaired granule polarization and IFN-γ production [16, 17] and shows higher binding affinity for CD155 than CD112 [18]. Blockade of TIGIT on NK cells has resulted in restoration of powerful NK cell effector function in vivo and reversal of their functional exhaustion [19]. Partly because the expression of TIGIT is higher on NK cells compared to other lymphocytes [20], its role as an immune checkpoint within the CD155-TIGIT axis is receiving considerable attention [21, 22].
In GBM, TIGIT has been targeted in combination with PD-1 as a strategy to overcome adaptive resistance to single checkpoint blockade [23] while its overexpression on tumor-infiltrating immune cells correlates to their functional exhaustion [24]. Less is known about the prognostic significance of TIGIT in GBM, although evidence that it correlates negatively with patient survival, at least for low-grade glioma, has been suggested [23].
Despite demonstrated evidence that supports targeting the CD155-TIGIT axis as an immunotherapeutic strategy for solid tumors including GBM, the complexity of the pathway, the multiple related ligands, and receptors involved as well as its mobilization of immune responses by not just NK cells has caused many questions to remain open. Here, we present an evidence-based discussion on efforts aimed at understanding and exploiting CD155 as a target for immunotherapy of GBM mediated by NK cells.
Expression and function of CD155 in GBM
CD155 is a cell surface receptor which belongs to the nectin and nectin-like family of immunoglobulin-like molecules that function as the receptor for poliovirus [4]. CD155 is overexpressed on GBM [1, 2] and other solid tumors, including melanoma [9], breast cancer [5], lung adenocarcinoma [6], pancreatic cancer [7], and a variety of soft tissue tumors [10]. In the context of GBM, Sloan et al. were among the first to describe the overexpression of CD155 in GBM using the U87-MG malignant glioma cell line and demonstrate that it plays a role in GBM invasiveness [2]. Upregulation of both membrane-bound and soluble CD155 in U87MG glioblastoma cells was subsequently reported by other groups [25]. Thompson et al. showed that a variety of low-grade and malignant pediatric brain tumors also overexpress CD155 and that targeting of CD155 on these tumors using an oncolytic virus could inhibit cellular proliferation of GBM [26]. Further confirming these findings have been large-scale IHC studies which have indicated the prevalence of CD155 in GBM and other gliomas [11, 27, 28].
The prognostic value of CD155 overexpression has been more extensively characterized for tumors other than GBM so far. Bevelacqua and colleagues measured overexpression of CD155 on melanoma cell lines WM35, A375, and M14 and samples from patients, and related such upregulation to increases in cancer cell migration and invasiveness [9]. Elsewhere, overexpression of CD155 was shown to correlate to a poorer prognostic outlook in patients with breast cancer [5], soft tissue sarcoma [10], lung adenocarcinoma, pancreatic cancer, malignant glioma, and colorectal carcinoma [2, 6, 7, 29]. This has been largely attributed to CD155’s demonstrated roles in the promotion of tumor invasion [1, 2, 30]. These findings have fueled interest in understanding the immunosuppressive role of CD155 in cancer via its interactions with its various ligands.
It has been established that CD155 and other nectin-like molecules, such as CD112 and CD113, play key roles in cell adhesion and migration [31]. CD155 interacts with a number of different ligands which are primarily present on immune cells, in particular cytotoxic T cells and NK cells. These interactions are both complex and varied and result in sometimes divergent immunoregulatory stimuli. CD155 ligands include inhibitory receptor TIGIT and activating receptors DNAM-1 and CD96 [32,33,34]. CD155 modulates the immunoregulation of T and NK cells through interactions with TIGIT, DNAM-1, and CD96, with, in particular, interactions between TIGIT and CD155 thought to result in severe immunosuppression of NK and cytotoxic T cells in the tumor microenvironment (TME) [35]. CD155 has also been shown to trans-interact with nectin-3, thus promoting cell migration through colocalization to epithelial cell junctions [36, 37]. In addition, CD155 overexpression has been linked to increases in cell proliferation, pitting CD155 as a key driver of cancer migration [2, 9, 11, 29].
In solid tumors, including gliomas, CD155 is recruited to the leading edge of the tumor where it co-localizes with actin and αv-integrin, compounds known to mediate cellular adhesion to the extracellular matrix (ECM) and to other cells [2, 38]. Following cellular adhesion, CD155 has been shown to increase activation of tyrosine kinases Src and FAK, which are recruited to the focal adhesions [2]. FAK is phosphorylated by Src, subsequently activating downstream molecules paxillin and p130Cas, which induce disassembly of focal adhesions, ultimately allowing for adhesion turnover and tumor migration [1]. Through this pathway, CD155 inhibits formation of mature focal adhesions and induces tumor cell migration in vitro and in primary brain tissues [1]. CD155 further mediates tumor progression by encouraging tumor cell growth through shortening of the G0/G1 phase of the cell cycle [3]. Heightened CD155 expression has additionally been associated with increase in VEGF expression and induction of angiogenesis, recognized markers of tumor invasiveness [7].
A number of groups have reported that CD155 also plays a key role in the invasiveness of GBM. Sloan and colleagues have shown that CD155 is upregulated in the GBM cell line U87MG, and knockdown of CD155 significantly inhibited chemoattractant-induced cell migration in transwell studies, suggesting CD155 has a role in tumor progression through metastasis and dispersal [2]. Upregulation of CD155 in GBM has also been shown to influence lymphocyte populations within the TME. Using surgical resection samples and matched blood from GBM donors, Lucca et al. showed that TIGIT+/DNAM-1− tumor-infiltrating lymphocyte populations within GBM, which exhibited enhanced in vitro activation following TIGIT blockade, increased their presence from 25 to 60%, while such lymphocytes were typically absent within normal brain tissue [39]. While DNAM-1+ tumor-infiltrating lymphocytes were also observed at higher levels than in normal brain tissue, DNAM-1 was typically co-expressed with TIGIT, likely disrupting DNAM-1 homodimerization and rendering DNAM-1 nonfunctional. Further, the authors reported increases in peripheral TIGIT+/DNAM-1− lymphocytes along with the localized increase of TIGIT+/DNAM-1− lymphocytes within the TME, indicating that CD155 upregulation could affect expression of CD155 ligands on local and peripheral lymphocyte populations, though the exact mechanism has yet to be elucidated [39]. NK cell killing of neuroblastoma has also been shown to be mediated through CD155-DNAM-1 interactions, suggesting expression levels of CD155, TIGIT, and DNAM-1 play important roles in the regulation of activation and cytotoxic functions of NK cells in vivo, a concept which holds potentially significant clinical implications [40]. Collectively, these findings suggest that CD155/TIGIT appears to be a key interaction to consider when targeting immunomodulating pathways in GBM.
NK immune regulation through CD155/TIGIT
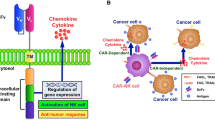
In addition to cancer-driven immunometabolic reprogramming which impairs NK cell effector function in the solid tumor TME [41, 42], a number of immunosuppressive pathways in solid tumors that specifically affect NK cells involve various nectin-like molecules (NECL), such as CD155 (PVR) and CD112, which are expressed at low levels on normal cells but are highly upregulated in solid tumors, including GBM [2, 15]. CD155 and CD112 have been linked to tumor progression and migration in primary tumors and have also been shown to have an immunomodulatory role through interaction with DNAM-1, TIGIT, and CD96 on NK cells (Fig. 1) [2, 3, 43, 44]. While the link between upregulation of CD155 and tumor progression in GBM and other tumors has rendered CD155 a checkpoint target of potential interest, the related downregulation of DNAM-1 on NK cells in the TME within the context of CD155/TIGIT activity, coupled with its relatively lower affinity for CD155 and CD112 compared to that of TIGIT, limits NK cell activation and colors these pathways with additional complexity [40, 45].

Diagram of CD155/CD112-mediated immunoregulatory pathway on NK cells. Interaction between TIGIT and CD155/CD112 induces inhibition of NK cell function via the phosphorylation of the ITIM domain and recruitment of SHIP1. This, in turn, inhibits MAPK, PI3K, and NF-κβ signaling. Interaction between DNAM-1/CD96 and CD155/CD112 provides an activating stimulus to NK cells. Upon binding, DNAM-1 relocates to lipid rafts where it homodimerizes and crosslinks with LFA-1 receptors. These cause phosphorylation of the Tyr322 residue of DNAM-1 resulting in NK cell cytolytic activity, cytokine secretion, and degranulation
Among the ligands that interact with CD155, TIGIT is of particular interest to the development of NK cell therapies [46] owing to its overexpression on NK cells in various cancers [20]. Due to its strong affinity for CD155, TIGIT is capable of interfering with positive DNAM-1 interactions and inducing NK cell inhibition. In addition, its role and function have been characterized extensively, making it a particularly intriguing target for immunotherapies. The TIGIT receptor on NK cells consists of an Ig-like extracellular domain along with a cytoplasmic domain consisting of an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoglobulin tail tyrosine (ITT) motif [18, 33, 47, 48]. TIGIT binds with high affinity to CD155, higher than that of DNAM-1, and to CD112, but with lower affinity, competitively inhibiting NK cell activation and functions [19, 49, 50]. While the exact intracellular mechanism is not completely understood, the ITIM domain of the TIGIT receptor plays a key role in NK cell inhibition, as it does in other inhibitory NK receptors (e.g., inhibitory KIRs, CD94/NKG2A) [44, 51, 52]. More specifically, following engagement of TIGIT with CD155, the ITIM domain of the TIGIT receptor is phosphorylated intracellularly, likely by a Src family kinase [53,54,55]. This is often achieved through crosslinking of an activating receptor to bring the Src kinase within close proximity of the ITIM domain [54]. ITIM phosphorylation then induces recruitment of SH2 receptors, SHIP-1, SHP-1, and SHP-2 [56]. Crosslinking of the ITIM domain with an activating receptor, often containing an ITAM domain, at cellular synapses allows these phosphatases to dephosphorylate molecules involved in NK cell activation, inhibiting NK cell cytolytic responses [56, 57]. Through this engagement of TIGIT with CD155 or CD112, NK cell cytolytic function is effectively diminished via reduction in IFN-γ secretion and granule polarization, ultimately interfering with the release of cytolytic granules by NK cells [16, 35]. Due to the high affinity with which TIGIT and CD155 bind, blockade of TIGIT has proven efficacious in enhancing NK cell immunotherapy [19].
Additionally, several other receptors expressed on NK cells interact with CD155 and CD112, including DNAM-1 and CD96. However, these receptors have been shown to have stimulatory functions in NK cells. DNAM-1 is an adhesion receptor containing two extracellular Ig-like domains attached to a cytoplasmic tail of three tyrosine residues. Unlike many other activating receptors, which induce activation through phosphorylation of intracellular ITAM domains, upon binding to CD155/CD112, homodimerization of DNAM-1 or crosslinking of DNAM-1 with other receptors activates NK cell cytolytic functions [32]. Specifically, activated DNAM-1 relocates to lipid rafts along the NK cell surface, binding with the actin cytoskeleton [58]. Here, the Ser329 residue located within the DNAM-1 intracellular domain is phosphorylated by protein kinase C (PKC) allowing for homodimerization of DNAM-1 molecules and crosslinking between DNAM-1 and LFA-1 receptors [59]. These interactions between DNAM-1 and LFA-1 have been shown to induce NK cell education [60, 61]. LFA-1 recruits Fyn Src kinase to phosphorylate the Tyr322 residue of DNAM-1, triggering phosphorylation of SLP76 and Vav1, and, subsequently, NK cell cytolysis, cytokine secretion, and degranulation via PLCg2 activation [60, 62, 63]. However, co-activation of other NK cell receptors is also required to prevent c-CbI inhibition of Vav1 and allow for DNAM-1-induced NK cell activation [64].
CD96, similarly to DNAM-1, has been implicated in the activation of human NK cell cytolytic functions [65]. While CD96 contains inhibitory intracellular ITIM domains, much like TIGIT, it also expresses a YXXM motif, conserved on many NK cell activating receptors, such as NKG2D, thus triggering activation upon phosphorylation [66]. This suggests that CD96 may play a regulatory role in NK cells, through either activation or inhibition in response to different cells, but this remains poorly understood. Nonetheless, CD96 has been shown to potentiate NK cell activation through cellular adhesion and interaction with CD155, but the exact mechanism and extent of activation induced by CD96 remains to be elucidated [65].
Current immunotherapies targeting the CD155/TIGIT axis
With novel advances in activation strategies for NK cells, the development of new NK cell sources, as well as advancements in genetic engineering toward improved targeting approaches for various cancers [67,68,69,70,71], NK cell therapies have emerged as a potentially promising platform for treating difficult-to-cure solid tumors, including GBM [1, 2, 69, 72,73,74,75] and other gliomas [26]. Additionally, preclinical trials with combination therapies, utilizing donor-derived NK cells with checkpoint inhibitor antibodies and other molecules, have proven efficacious in reducing tumor-induced immunosuppression in GBM and enhancing NK cell responses [76, 77]. Apart from the clinical use of native, unmodified NK cells, therapies with engineered NK cells have been utilized in multiple clinical and preclinical studies to target a number of ligands which are overexpressed in GBM, including HER2 [78], IL-13Rα2 [79], EGFR [80], EGFRvIII [80], CSPG4 [77], and CD133 [81]. Despite emerging as a known immunosuppressor in GBM, however, CD155 has yet to be significantly targeted with NK cell therapies [72]. Burger et al. provide a thorough discussion of current NK cell therapies targeting important ligands on GBM and their implications for NK cell-based immunotherapies [72].
Nonetheless, together with the expanding body of work demonstrating CD155’s roles in tumor progression, evidence about its upregulation in GBM, and strong interaction with inhibitory TIGIT receptor on NK cells, targeting of the CD155/TIGIT axis is growing as a potentially powerful strategy to elicit substantial NK-mediated anti-tumor responses. However, with immunotherapies targeting CD155 and TIGIT within GBM being relatively new, clinical examples are limited. In fact, there are no active clinical trials utilizing NK or T cells to target CD155 on GBM, but there have been multiple studies targeting other ligands on GBM and malignant gliomas. Lee et al., for instance, observed that unmodified NK cells were able to exert significant cytolytic functions, including inhibition of GBM metastases, when adoptively transferred into a U87MG xenograft model in NSG mice [82]. Clinically, significant responses of autologous blood-derived NK cells to treat malignant gliomas have also been reported. In an early clinical study, NK cells were harvested from male and female patients and expanded using irradiated HFWT cells in RHAMα medium supplemented with autologous plasma and rhIL-2 for 21 days prior to infusion. Patients received one or more courses of treatment, consisting of weekly intra-tumoral and intravenous injections of NK cells for 3 weeks, and tumor responses were measured via MRI imaging. These treatments resulted in two of nine patients recording positive clinical responses, namely a reduction in tumor volume by more than 50%, which persisted for longer than 4 weeks [83]. In a current, separate phase 1 trial at M.D. Anderson Cancer Center, autologous, ex vivo-expanded, native NK cells are adoptively transferred into patients intravenously weekly for 1 to 3 weeks to target recurrent medulloblastoma and ependymoma and determine the side effects and maximum tolerated dose of this treatment (NCT02271711).
Adoptively-transferred NK cell therapies have also been combined with other therapeutics to improve NK cell anti-tumor effects in GBM. Wu et al. reported that stimulation with IFN-γ induced upregulation of NKG2D and NCR ligands on brain tumors, enhancing cell lysis by NK cells using surgical samples of two GBM tumors and two astrocytomas obtained from resected tumor tissue of human patients [84]. Elsewhere, blockade or depletion of immunosuppressive compounds, cytokines, and ligands has been shown to inhibit immunosuppression and enhance NK cell activation against GBM [85, 86]. Administration of recombinant antibodies targeting GD2 and inducing NK activation via IL-2 has also proven efficacious in treating neuroblastoma, further demonstrating that NK cell cytolytic functions can be enhanced in various ways to treat GBM and other brain tumors [87, 88].
There are also a number of active clinical trials targeting CD155 or TIGIT within GBM, albeit without NK cells. Results from a recent dose-escalation study treating patients with WHO grade IV malignant glioma through intra-tumoral delivery of the recombinant nonpathogenic polio–rhinovirus chimera (PVSRIPO) [13] showed promising anti-tumor responses. The study evaluated 7 doses of PVSRIPO in 61 patients and resulted in an increase in survival rate of 21% at 24 and 36 months when compared with historical controls (14% at 24 months and 4% at 36 months), with patients surviving for more than 70 months after infusion [13]. Several therapies have also been described that target the CD155/TIGIT axis in other tumor types, primarily utilizing checkpoint inhibitors and often targeting one or multiple other ligands in combination with CD155/TIGIT (Table 1).
Inhibition of CD155 has also been shown to result in a reduction in tumor dispersion and invasiveness, as well as a reduction in secretion and activity of matrix metalloproteinase-2 (MMP-2) [89]. MMP-2 is a secreted endopeptidase which degrades ECM components and contributes to in vivo invasiveness of GBM [89]. Therapeutically, MMP-2 inhibition has been well established as contributing to the reduction of invasion of GBM cells [90, 91]. Therefore, CD155 inhibition alone can reduce production of MMP-2 and may be beneficial in slowing progression of GBM in vivo [2, 89]. However, clinical studies have so far mostly focused on inhibition of TIGIT, as CD155 interacts with other activation receptors on cells including NK cells (DNAM-1) and can positively stimulate immune responses in vivo. Inhibition of TIGIT strongly enhances NK cell function, including proliferation and production of pro-inflammatory cytokines [92]. Combinatorial checkpoint inhibition therapies targeting TIGIT and PD-1 on NK cells have also proven effective in the treatment of GBM [23]. Li et al. demonstrate that dual-targeting of the CD155/TIGIT and PD-1/PD-L1 axes in NK and T cells improved effector cell-mediated antitumor immunity in solid tumors [93]. In their study, triple blockade of PD-1, TIGIT, and CD96 significantly inhibited tumor growth in a B16F10 melanoma BALB/c WT mouse model [93]. Separately, Zhang et al. reported that TIGIT blockade could reverse TME-induced exhaustion of NK cells and restore NK cell anti-tumor functions [19]. Their study measured functional exhaustion of NK cells, extracted from tumor-bearing mice, through the expression of IFN-γ, TNF, CD107a, and cytotoxic molecule TRAIL, noting a reduction in expression of these markers which was restored through TIGIT blockade. Elsewhere, Johnston et al. reported that TIGIT blockade alone could enhance effector function of CD8+ T cells after adoptive transfer but did not measure this effect in NK cells [94].
Engineering NK cells to overcome TIGIT-induced inhibition
The immunomodulatory role of CD155/CD112, through its interaction with both DNAM-1 and TIGIT on NK cells and its heightened expression on GBM, represent an opportunity for advances in immunotherapy treatments. By targeting this axis in NK cells, through engineered cell therapies and combinatorial antibody/cell therapy approaches, it is possible to suppress CD155-induced inhibition and enhance the natural cytolytic functions of NK cells. Recent advances in chimeric antigen receptor (CAR)-NK therapies have allowed engineered NK cells to find their way to the clinic [95], where they have predominantly targeted CD19 on B cell malignancies [74, 96, 97], but also a growing number of solid tumor ligands, such as HER2 and MUC1 [98,99,100].
Engineered NK cells have also been under active investigation as immunotherapeutic effectors against GBM [80, 101]. Indeed, several preclinical studies utilizing CAR-engineered NK cells have targeted ligands on GBM, although they have not specifically targeted TIGIT/CD155 induced immunosuppression. A phase 1 clinical trial aimed at treating GBM patients with CAR-NK-92 cells engineered with a second-generation CAR targeting HER2 (NCT03383978) [102] is currently underway at Johann Wolfgang Goethe University Hospital. In this dose-escalation study, between 1 × 107 and 1 × 108 NK-92 cells have been adoptively transferred into treated subjects, with the aim of identifying optimal dose ranges for single and repeated injections.
Mueller et al. engineered an NK cell line (YTS) to express a CAR targeting EGFRvIII on glioblastoma, as well as a CXCR4 chemokine receptor [101]. While NK cells expressing EGFRvIII-CARs alone effectively delayed tumor growth, the combined EGFRvIII/CXCR4 targeted therapies significantly increased survival in mice with U87MG xenografts over EGFRvIII-CAR NK cells or controls [101]. Han et al. engineered a variety of NK cells (cell lines NK-92 and NKL, and primary NK cells) with CARs targeting EGFR and EGFRvIII and noted an enhanced cytolytic ability and cytokine secretion of these engineered NK cells in response GBM cells [80]. Elsewhere, enhanced cytolytic function of cetuximab-based CAR-engineered NK-92 cells, targeting both EGFR and EGFRvIII on GBM with a single ligand, has been reported. The dual antigen-targeting approach was used to prevent immune escape, a feature previously described for GBM [103]. Other ligands expressed highly on GBM have also been investigated through CAR-NK therapies, including Erb2 and HER2 [78]. While no studies have been reported targeting the CD155/TIGIT axis with engineered NK cells, the emerging role CD155 in GBM progression and immunoregulation makes it an increasingly attractive target for this space.
Clinical outlook and future directions
It is becoming evident that targeting the TIGIT/CD155 axis through cellular engineering or with antibodies holds tremendous potential to induce significant anti-GBM responses. The demonstrated role of this axis in regulating NK cell immune responses makes NK cells an attractive immunotherapeutic proposition in GBM immunotherapy via this pathway. NK cell-based therapies alone have proven effective in treating a variety of cancers [5,6,7, 9, 10, 104], and recent targeting of GBM with NK cell therapies has proven promising [78, 80, 101, 103]. CD155 represents a potentially significant target with implications both in the regulation of immune cells and tumor progression, proliferation, and invasion [89, 92]. Though there is great complexity, including functional divergence, of the many pathways and ligands involved in the various CD155 signaling interactions, clinical trials targeting CD155 within GBM have already proven efficacious, with intratumoral delivery of a recombinant nonpathogenic polio-rhinovirus chimera (PVSRIPO) yielding a 3-year survival rate of 21% as compared to historical 3-year survival rates of 4% [13]. Therefore, utilizing NK cell therapies to target the CD155/TIGIT axis can be exploited as a strategy for treating GBM toward improved clinical outcomes. The use of combinatorial therapies targeting CD155 together with other ligands overexpressed in the GBM TME will likely yield the most robust and durable clinical responses. This is already evident by the fact that most of the current clinical therapies targeting CD155 also target other checkpoints (PD-1/PDL-1, CXCR4, etc.) present within solid tumors. Ultimately, the complexity of the TME of solid cancers, particularly that of GBM, is likely to require concomitant targeting of multiple ligands and cytokines for therapeutic effect and a better understanding of how the various interactions CD155 functions along guide effector functions of NK cells.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Sloan KE, Stewart JK, Treloar AF, Matthews RT, Jay DG. CD155/PVR enhances glioma cell dispersal by regulating adhesion signaling and focal adhesion dynamics. Cancer Res. 2005;65(23):10930–7.
Sloan KE, Eustace BK, Stewart JK, Zehetmeier C, Torella C, Simeone M, et al. CD155/PVR plays a key role in cell motility during tumor cell invasion and migration. BMC Cancer. 2004;4:73.
Kono T, Imai Y, Yasuda S, Ohmori K, Fukui H, Ichikawa K, et al. The CD155/poliovirus receptor enhances the proliferation of ras-mutated cells: CD155 enhances ras-mutated cell proliferation. Int J Cancer. 2008;122(2):317–24.
Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56(5):855–65.
Li Y-C, Zhou Q, Song Q-K, Wang R-B, Lyu S, Guan X, et al. Overexpression of an immune checkpoint (CD155) in breast cancer associated with prognostic significance and exhausted tumor-infiltrating lymphocytes: a cohort study. J Immunol Res. 2020;3948928:1–9.
Nakai R, Maniwa Y, Tanaka Y, Nishio W, Yoshimura M, Okita Y, et al. Overexpression of Necl-5 correlates with unfavorable prognosis in patients with lung adenocarcinoma. Cancer Science. 2010;101(5):1326–30.
Nishiwada S, Sho M, Yasuda S, Shimada K, Yamato I, Akahori T, et al. Clinical significance of CD155 expression in human pancreatic cancer. Anticancer Res. 2015;35(4):2287–97.
Huang D-W, Huang M, Lin X-S, Huang Q. CD155 expression and its correlation with clinicopathologic characteristics, angiogenesis, and prognosis in human cholangiocarcinoma. Onco Targets Ther. 2017;10:3817–25.
Bevelacqua V, Bevelacqua Y, Candido S, Skarmoutsou E, Amoroso A, Guarneri C, et al. Nectin like-5 overexpression correlates with the malignant phenotype in cutaneous melanoma. Oncotarget. 2012;3(8):882–92.
Atsumi S, Matsumine A, Toyoda H, Niimi R, Iino T, Sudo A. Prognostic significance of CD155 mRNA expression in soft tissue sarcomas. Oncology Letters. 2013;5(6):1771–6.
Merrill MK, Bernhardt G, Sampson JH, Wikstrand CJ, Bigner DD, Gromeier M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro-Oncology. 2004;6(3):208–17.
Seth S, Ravens I, Lee C-W, Glage S, Bleich A, Forster R, et al. Absence of CD155 aggravates acute graft-versus-host disease. Proc Nat Acad Sci. 2011;108(10):E32–3.
Desjardins A, Gromeier M, Herndon JE, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018;379:150–61.
Tahara-Hanaoka S. Functional characterization of DNAM-1 (CD226) interaction with its ligands PVR (CD155) and nectin-2 (PRR-2/CD112). Int Immunol. 2004;16(4):533–8.
Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol. 2015;15(4):243–54.
Liu S, Zhang H, Li M, Hu D, Li C, Ge B, et al. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013;20(3):456–64.
Sanchez-Correa B, Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Bergua JM, et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers (Basel) [Internet]. 2019 [cited 2020 Apr 6];11. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6628015/.
Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Nat Acad Sci. 2009;106(42):17858–63.
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19:723–32.
Wang F, Hou H, Wu S, Tang Q, Liu W, Huang M, et al. TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals. Eur J Immunol. 2015;45(10):2886–97.
Sanchez-Correa B, Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Bergua JM, et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers. 2019;11(6):877.
Mahnke K, Enk AH. TIGIT-CD155 interactions in melanoma: a novel co-inhibitory pathway with potential for clinical intervention. J Invest Dermatol. 2016;136(1):9–11.
Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. OncoImmunology. 2018;7(8):e1466769.
Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. 2018;24(17):4175–86.
Iguchi-Manaka A, Okumura G, Kojima H, Cho Y, Hirochika R, Bando H, et al. Increased soluble CD155 in the serum of cancer patients. Shiku H, editor. PLoS ONE. 2016;11(4):e0152982.
Thompson EM, Brown M, Dobrikova E, Ramaswamy V, Taylor MD, McLendon R, et al. Poliovirus Receptor (CD155) Expression in pediatric brain tumors mediates oncolysis of medulloblastoma and pleomorphic xanthoastrocytoma. J Neuropathol Exp Neurol. 2018;77(8):696–702.
Chandramohan V, Bryant JD, Piao H, Keir ST, Lipp ES, Lefaivre M, et al. Validation of an immunohistochemistry assay for detection of CD155, the poliovirus receptor, in malignant gliomas. Arch Pathol Lab Med. 2017;141(12):1697–704.
Gromeier M, Lachmann S, Rosenfeld MR, Gutin PH, Wimmer E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc Nat Acad Sci. 2000;97(12):6803–8.
Masson D, Jarry A, Baury B, Blanchardie P, Laboisse C, Lustenberger P, et al. Overexpression of the CD155 gene in human colorectal carcinoma. Gut. 2001;49(2):236–40.
Morimoto K, Satoh-Yamaguchi K, Hamaguchi A, Inoue Y, Takeuchi M, Okada M, et al. Interaction of cancer cells with platelets mediated by Necl-5/poliovirus receptor enhances cancer cell metastasis to the lungs. Oncogene. 2008;27(3):264–73.
Takai Y, Irie K, Shimizu K, Sakisaka T, Ikeda W. Nectins and nectin-like molecules: roles in cell adhesion, migration, and polarization. Cancer Sci. 2003;94(8):655–67.
Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity. 1996;4(6):573–81.
Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10(1):48–57.
Wang PL, O’Farrell S, Clayberger C, Krensky AM. Identification and molecular cloning of tactile. A novel human T cell activation antigen that is a member of the Ig gene superfamily. J Immunol. 1992;148(8):2600–8.
Li M, Xia P, Du Y, Liu S, Huang G, Chen J, et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-γ production of natural killer cells via β-arrestin 2-mediated negative signaling. J Biol Chem. 2014;289(25):17647–57.
Ikeda W, Kakunaga S, Itoh S, Shingai T, Takekuni K, Satoh K, et al. Tage4/Nectin-like Molecule-5 heterophilically trans-interacts with cell adhesion molecule Nectin-3 and enhances cell migration. J Biol Chem. 2003;278(30):28167–72.
Mueller S, Wimmer E. Recruitment of nectin-3 to cell-cell junctions through trans-heterophilic interaction with CD155, a vitronectin and poliovirus receptor that localizes to α v β 3 integrin-containing membrane microdomains. J Biol Chem. 2003;278(33):31251–60.
Marshall JF, Hart IR. The role of αv-integrins in tumour progression and metastasis. Seminar Cancer Biol. 1996;7(3):129–38.
Lucca LE, Lerner BA, Park C, DeBartolo D, Harnett B, Kumar VP, et al. Differential expression of the T-cell inhibitor TIGIT in glioblastoma and MS. Neurol Neuroimmunol Neuroinflamm. 2020;7(3):e712.
Castriconi R, Dondero A, Corrias MV, Lanino E, Pende D, Moretta L, et al. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: critical role of DNAX accessory molecule-1–poliovirus receptor interaction. Cancer Res. 2004;64(24):9180–4.
Chambers AM, Lupo KB, Matosevic S. Tumor microenvironment-induced immunometabolic reprogramming of natural killer cells. Front Immunol. 2018;9:2517.
Matosevic S, Chambers AM. Immunometabolic dysfunction of natural killer cells mediated by the hypoxia-CD73 axis in solid tumors. Front Mol Biosci. 2019;6:60.
Lakshmikanth T, Burke S, Ali TH, Kimpfler S, Ursini F, Ruggeri L, et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J Clin Invest. 2009;119(5):1251–63.
Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R, et al. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR: innate immunity. Eur J Immunol. 2013;43(8):2138–50.
Sanchez-Correa B, Gayoso I, Bergua JM, Casado JG, Morgado S, Solana R, et al. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol Cell Biol. 2012;90(1):109–15.
Dao TN, Matosevic S. Immunometabolic responses of natural killer cells to inhibitory tumor microenvironment checkpoints. Immunometabolism. 2019;1(1):e190003.
Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol. 2011;41(4):902–15.
Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol. 2009;39(3):695–703.
Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. 2017;276(1):112–20.
de Andrade LF, Smyth MJ, Martinet L. DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol Cell Biol. 2014;92(3):237–44.
Colonna M, Navarro F, Bellón T, Llano M, García P, Samaridis J, et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med. 1997;186(11):1809–18.
Anderson SK, Ortaldo JR, McVicar DW. The ever-expanding Ly49 gene family: repertoire and signaling. Immunol Rev. 2001;181:79–89.
Newman DK, Hamilton C, Newman PJ. Inhibition of antigen-receptor signaling by Platelet Endothelial Cell Adhesion Molecule-1 (CD31) requires functional ITIMs, SHP-2, and p56lck. Blood. 2001;97(8):2351–7.
Verbrugge A. Differential contribution of the immunoreceptor tyrosine-based inhibitory motifs of human leukocyte-associated Ig-like receptor-1 to inhibitory function and phosphatase recruitment. International Immunology. 2003;15(11):1349–58.
Xu R, Abramson J, Fridkin M, Pecht I. SH2 domain-containing inositol polyphosphate 5′-phosphatase is the main mediator of the inhibitory action of the mast cell function-associated antigen. J Immunol. 2001;167(11):6394–402.
Bléry M, Delon J, Trautmann A, Cambiaggi A, Olcese L, Biassoni R, et al. Reconstituted killer cell inhibitory receptors for major histocompatibility complex class I molecules control mast cell activation induced via immunoreceptor tyrosine-based activation motifs. J Biol Chem. 1997;272(14):8989–96.
Vyas YM, Maniar H, Lyddane CE, Sadelain M, Dupont B. Ligand binding to inhibitory killer cell Ig-like receptors induce colocalization with Src homology domain 2-containing protein tyrosine phosphatase 1 and interruption of ongoing activation signals. J Immunol. 2004;173(3):1571–8.
Ralston KJ, Hird SL, Zhang X, Scott JL, Jin B, Thorne RF, et al. The LFA-1-associated molecule PTA-1 (CD226) on T cells forms a dynamic molecular complex with protein 4.1G and human discs large. J Biol Chem. 2004;279(32):33816–28.
Shibuya A, Lanier LL, Phillips JH. Protein kinase C is involved in the regulation of both signaling and adhesion mediated by DNAX accessory molecule-1 receptor. J Immunol. 1998;161(4):1671.
Shibuya K, Lanier LL, Phillips JH, Ochs HD, Shimizu K, Nakayama E, et al. Physical and functional association of LFA-1 with DNAM-1 adhesion molecule. Immunity. 1999;11(5):615–23.
Enqvist M, Ask EH, Forslund E, Carlsten M, Abrahamsen G, Béziat V, et al. Coordinated expression of DNAM-1 and LFA-1 in educated NK cells. JI. 2015;194(9):4518–27.
Bryceson YT, March ME, Ljunggren H-G, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. 2006;107(1):159–66.
Kim HS, Long EO. Complementary phosphorylation sites in the adaptor protein SLP-76 promote synergistic activation of natural killer cells. Science Signl. 2012;5(232):ra49–ra49.
Kim HS, Das A, Gross CC, Bryceson YT, Long EO. Synergistic signals for natural cytotoxicity are required to overcome inhibition by c-Cbl ubiquitin ligase. Immunity. 2010;32(2):175–86.
Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J Immunol. 2004;172(7):3994–8.
Georgiev H, Ravens I, Papadogianni G, Bernhardt G. Coming of age: CD96 emerges as modulator of immune responses. Front Immunol. 2018;9:1072.
Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol. 2013;10(3):230–52.
Fang F, Xiao W, Tian Z. NK cell-based immunotherapy for cancer. Semin Immunol. 2017;31:37–54.
Lupo KB, Matosevic S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers. 2019;11(6):769.
Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. 2017;25(8):1769–81.
Miller JS, Lanier LL. Natural killer cells in cancer immunotherapy. Annu Rev Cancer Biol. 2019;3:77–103.
Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, Senft C, et al. CAR-engineered NK cells for the treatment of glioblastoma: turning innate effectors into precision tools for cancer immunotherapy. Front Immunol. 2019;10:2683.
Cantoni C, Grauwet K, Pietra G, Parodi M, Mingari MC, Maria AD, et al. Role of NK cells in immunotherapy and virotherapy of solid tumors. Immunotherapy. 2015;7(8):861–82.
Rezvani K, Rouce RH. The application of natural killer cell immunotherapy for the treatment of cancer. Front Immunol. 2015;6:578.
Kuramitsu S, Yamamichi A, Ohka F, Motomura K, Hara M, Natsume A. Adoptive immunotherapy for the treatment of glioblastoma: progress and possibilities. Immunotherapy. 2016;8(12):1393–404.
Höring E, Podlech O, Silkenstedt B, Rota IA, Adamopoulou E, Naumann U. The histone deacetylase inhibitor trichostatin a promotes apoptosis and antitumor immunity in glioblastoma cells. Anticancer Res. 2013;33(4):1351–60.
Kmiecik J, Gras Navarro A, Poli A, Planagumà JP, Zimmer J, Chekenya M. Combining NK cells and mAb9.2.27 to combat NG2-dependent and anti-inflammatory signals in glioblastoma. OncoImmunology. 2014;3(1):e27185.
Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. JNCI: J Natl Cancer Inst. 2016;108(5):djv375.
Hegde M, Corder A, Grada Z, Byrd TT, Chow KK, Brawley VS, et al. A bispecific chimeric antigen receptor targeting antigen escape variants in glioblastoma. Cytotherapy. 2014;16(4):S28.
Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma Stem Cells. Sci Rep. 2015;5:11483.
Schmohl JU, Gleason MK, Dougherty PR, Miller JS, Vallera DA. Heterodimeric bispecific single chain variable fragments (scFv) killer engagers (BiKEs) enhance NK-cell activity against CD133+ colorectal cancer cells. Targ Oncol. 2016;11(3):353–61.
Lee SJ, Kang WY, Yoon Y, Jin JY, Song HJ, Her JH, et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer. 2015;15:1011.
Ishikawa E, Tsuboi K, Saijo K, Harada H, Takano S, Nose T, et al. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res. 2004;24(3b):1861–71.
Wu A, Wiesner S, Xiao J, Ericson K, Chen W, Hall WA, et al. Expression of MHC I and NK ligands on human CD133+ glioma cells: possible targets of immunotherapy. J Neurooncol. 2007;83(2):121–31.
Huang BY, Zhan YP, Zong WJ, Yu CJ, Li JF, Qu YM, et al. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. Ulasov I, editor. PLoS ONE. 2015;10(8):e0134715.
Friese MA, Wischhusen J, Wick W, Weiler M, Eisele G, Steinle A, et al. RNA Interference targeting transforming growth factor-β enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004;64(20):7596–603.
Delgado DC, Hank JA, Kolesar J, Lorentzen D, Gan J, Seo S, et al. Genotypes of NK cell KIR receptors, their ligands, and Fc receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Research. 2010;70(23):9554–61.
Yang RK, Kalogriopoulos NA, Rakhmilevich AL, Ranheim EA, Seo S, Kim K, et al. Intratumoral treatment of smaller mouse neuroblastoma tumors with a recombinant protein consisting of IL-2 linked to the Hu14.18 antibody increases intratumoral CD8+ T and NK cells and improves survival. Cancer Immunol Immunother. 2013;62(8):1303–13.
Enloe BM, Jay DG. Inhibition of Necl-5 (CD155/PVR) reduces glioblastoma dispersal and decreases MMP-2 expression and activity. J Neurooncol. 2011;102(2):225–35.
Noha M, Yoshida D, Watanabe K, Teramoto A. Suppression of cell invasion on human malignant glioma cell lines by a novel matrix-metalloproteinase inhibitor SI-27: in vitro study. J Neurooncol. 2000;48(3):217–23.
Uhm JH, Dooley NP, Villemure J-G, Yong VW. Glioma invasionin vitro: regulation by matrix metalloprotease-2 and protein kinase C. Clin Exp Metast. 1996;14(5):421–33.
Zhang B, Zhao W, Li H, Chen Y, Tian H, Li L, et al. Immunoreceptor TIGIT inhibits the cytotoxicity of human cytokine-induced killer cells by interacting with CD155. Cancer Immunol Immunother. 2016;65(3):305–14.
Li X-Y, Das I, Lepletier A, Addala V, Bald T, Stannard K, et al. CD155 loss enhances tumor suppression via combined host and tumor-intrinsic mechanisms. J Clin Invest. 2018;128(6):2613–25.
Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8 + T cell effector function. Cancer Cell. 2014;26(6):923–37.
Matosevic S. Viral and nonviral engineering of natural killer cells as emerging adoptive cancer immunotherapies. J Immunol Res. 2018;2018:4054815.
Li Y, Yin J, Li T, Huang S, Yan H, Leavenworth J, et al. NK cell-based cancer immunotherapy: from basic biology to clinical application. Sci China Life Sci. 2015;58(12):1233–45.
Baggio L, Laureano ÁM, Silla LMDR, Lee DA. Natural killer cell adoptive immunotherapy: coming of age. Clin Immunol. 2017;177:3–11..
Kloess S, Kretschmer A, Stahl L, Fricke S, Koehl U. CAR-expressing natural killer cells for cancer retargeting. Transfus Med Hemother. 2019;46(1):4–13.
Schönfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Molecular Therapy. 2015;23(2):330–8.
Li Q, Wang Y, Lin M, Xia L, Bao Y, Sun X, et al. Abstract A014: phase I clinical trial with PD-1/MUC1 CAR-pNK92 immunotherapy. Cancer Immunol Res. 2019;7(2):A014.
Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 Improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma. J Immunother. 2015;38(5):197–210.
Nowakowska P, Romanski A, Miller N, Odendahl M, Bonig H, Zhang C, et al. Clinical grade manufacturing of genetically modified, CAR-expressing NK-92 cells for the treatment of ErbB2-positive malignancies. Cancer Immunol Immunother. 2018;67(1):25–38.
Genßler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. OncoImmunology. 2016;5(4):e1119354.
Wang J, Lupo KB, Chambers AM, Matosevic S. Purinergic targeting enhances immunotherapy of CD73+ solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J ImmunoTher Cancer. 2018;6(1):136.
Acknowledgements
Not applicable.
Funding
Support from the Purdue Center for Cancer Research Jim and Diann Robbers Cancer Research Grant for New Investigators Award is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
KBL and SM researched the literature, wrote the draft, and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lupo, K.B., Matosevic, S. CD155 immunoregulation as a target for natural killer cell immunotherapy in glioblastoma. J Hematol Oncol 13, 76 (2020). https://doi.org/10.1186/s13045-020-00913-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00913-2