
Abstract
Immunotherapy using PD-1 and CTLA4 inhibitors to stimulate T cell immunity has achieved significant clinical success. However, only a portion of patients benefit from T cell-based immunotherapy. Macrophages, the most abundant type of innate immune cells in the body, play an important role in eliminating tumor cells and infectious microbes. The phagocytic check point protein CD47 inhibits the phagocytic activity of macrophages through binding to SIRPα expressed on macrophages. Blockade of the interaction between CD47 and SIRPα could restore phagocytic activity and eliminate tumor cells in vitro and in vivo. In this manuscript, we review the mechanism of action and development status of agents (antibodies targeting CD47 and SIRPα, SIRPα-Fc fusion proteins, and bi-specific antibodies) that block CD47/SIRPα interaction in preclinical studies and in the clinical setting. In addition, small molecules, mRNA, and CAR-T/M that target the CD47/SIRPα axis are also reviewed in this article.
Similar content being viewed by others
Introduction
Tumor cells evade immune destruction by transmitting inhibitory signals to lymphocytes and myeloid cells [1]. Blockade of these inhibitory molecules, which include CTLA4, PD-1, and PD-ligand 1 (PD-L1), could restore T cell function and promote elimination of tumor cells. Immune checkpoint inhibitors (ICIs) have improved outcomes for patients with multiple types of cancers. However, many patients do not respond to this type of immunotherapy [2, 3]. In some cases, these therapeutic agents have been associated with disease progression [4, 5], the cause of which is currently being investigated. Therefore, drugs that act on a novel class of targets to immobilize a broader immune cell population are needed to improve upon current therapeutic options.
Macrophages [6] are typically the first dedicated innate immune cells to detect the presence of infectious pathogens or tumor cells. Macrophages are derived from monocyte precursors that circulate in the blood and migrate into tissues, after which they differentiate into tissue macrophages such as Kupffer cells in the liver, alveolar macrophages in the lung, and microglia in the brain. Circulating monocytes and resident tissue macrophages can directly kill tumor cells via phagocytosis (innate immune response) and can activate the adaptive immune response. However, these immune responses can be inhibited by ligand binding to inhibitory receptors expressed on the macrophage cell surface [7]. Signal regulatory protein alpha (SIRPα) is a transmembrane protein expressed on all myeloid cells, including monocytes, macrophages, and neutrophils. SIRPα contains immunoreceptor tyrosine-based inhibition motifs (ITIMs) that can be phosphorylated, resulting in recruitment of inhibitory molecules such as Src homology 2 (SH2) domain-containing protein tyrosine phosphatase (SHP)-1 and SHP-2 [8]. Binding of CD47 to SIRPα triggers coupling of SIRPα to these phosphatases, resulting in inhibition of phagocytic activity [9, 10]. CD47 is ubiquitously expressed on many types of cells to prevent phagocytosis by phagocytes. However, tumor cells overexpress CD47 to evade the immune system through inhibition of myeloid cell-mediated elimination [11]. Inhibition of CD47-SIRPα interaction restored the phagocytic activity of phagocytes in vitro and in vivo [12,13,14,15]. Targeting the CD47/SIRPα axis has become a promising strategy to promote tumor elimination through innate immunity. This review focuses on development, safety, and efficacy of agents that target the CD47/SIRPα axis in preclinical and clinical studies.
CD47/SIRPα: the molecules and biology
SIRPα [16, 17], also named SHPS-1 or CD172a, is a transmembrane glycoprotein mainly expressed on neurons and myeloid cells that is particular enriched on macrophages. Human SIRPα is coded by the SHPS-1 gene located at human chromosome 20p13. The open reading frame region is composed of eight exons, including a signal peptide, extracellular domain, a transmembrane segment, and three parts of one cytoplasmic domain. The extracellular domain consists of three Ig-like regions, an NH2-terminal immunoglobulin (Ig) variable (V) region (domain 1, D1), and two Ig constant (C) regions (domain 2 and 3). The cytoplasmic region contains two immunoreceptor tyrosine based inhibitory motifs (ITIMs) and a proline-rich region (YYYY), which bind to Src homology (SH2) domain-containing molecules.
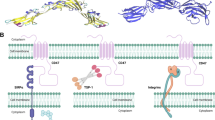
CD47 is a 52 kD transmembrane glycoprotein belonging to the immunoglobulin superfamily. Human CD47 is encoded by the CD47 gene located at the q13.12 region of chromosome 3. Human CD47 contains an NH2-terminal Ig variable-like extracellular domain (ECD), a 5-transmembrane spanning helical bundle domain, and a short intracellular COOH-terminal domain (CTD) [18]. CD47 is an essential component of the innate immune system, and binding of its extracellular domain with its ligands αVβ3, SIRPα, and thrombospondin-1 (Tsp-1) activates different signaling pathways that control cell proliferation and differentiation, angiogenesis, and immune regulation. The CTD is alternatively spliced and can exist as four isoforms, ranging from 4 to 36 residues. A schematic diagram of the compositions of CD47 and SIRPα proteins are shown in Fig. 1.

Diagram of composition of CD47 and SIRPα protein. The upper panel is the SIRPα protein with signal region (1–30), Ig-like V-type (32–137), Ig-like C1-type1 (148–247), C1-type2 (254–348), transmembrane (helical, 374–394), and cytoplasmic region (365–504) with four short spacers for SH2-binding. The lower is the CD47 protein with signal region (1–18), extracellular domain (19–141), five transmembrane region (142–289, Helical 1 to Helical 5), and cytoplasmic region (290–323)
The interaction of the NH2-terminal IgV domain of CD47 with the D1 region of SIRPα promotes phosphorylation of tyrosine residues (Fig. 2). The phosphorylated ITIM recruits and activates protein tyrosine phosphatases (PTPase), including the Src homology region 2 (SH2)-domain-containing phosphatase-1 (SHP-1) and 2 (SHP-2). The interaction of the phosphatase SH2-domains with the phosphorylated ITIM of SIRPα disrupts its auto-inhibitory activity towards the PTPase domain, resulting in enzymatic activity. Dephosphorylation of phosphotyrosine residues on a variety of proximal substrates counterbalances activation of signaling pathways that depend on tyrosine phosphorylation, thereby restricting phagocytic function. CD47 was first identified as a “marker of self” on murine red blood cells, and was shown to interact with SIRPα to prevent phagocytosis of red blood cells by macrophages in the spleen [19]. Studies have shown that CD47 is broadly expressed on many types of normal cells and tissues. A study by Jaiswal demonstrated that CD47 was upregulated on circulating hematopoietic stem cells and leukemia cells, which prevented phagocytosis of these cells [11]. Targeting CD47 with anti-CD47 antibodies stimulated macrophage phagocytosis of AML cells in vitro and showed therapeutic efficacy against AML in mouse models [20]. In addition, CD47 was also overexpressed on hematologic [19, 21,22,23] and solid [24,25,26] malignancies, and treatment with agents that block CD47-SIRPα interaction stimulated macrophage phagocytosis in vitro and anti-tumor responses in vivo.

Biology of CD47/SIRPα interaction. CD47 binds to SIRPα to transmit inhibitory signals to macrophages and to inhibit or lessen phagocytic activity through uncoupling of receptor binding and signal transduction. Therapeutic agents block the interaction between CD47 and SIRPα to remove the inhibitory signal and restore the phagocytic activity
Therapeutic strategy
The critical role of the CD47/SIRPα axis in the innate immune response suggests that these two proteins may be attractive therapeutic targets. Antagonists targeting the innate immune checkpoint CD47/SIRPα pathway are currently in clinical development. These antagonists include 1) monoclonal antibodies targeting CD47 or SIRPα, 2) SIRPα-Fc fusion proteins, 3) bispecific antibodies (BsAb), 4) small molecules to down-regulate CD47 on tumor cells, 5) RNAi and, 6) CD47-chimeric antigen receptor-T cell/Macrophages.
Monoclonal antibodies and fc fusion proteins
Three types of agents targeted to the CD47/SIRPα axis were developed: antibodies, SIRPα-Fc fusion proteins targeted to CD47, and antibodies targeted to SIRPα. The mechanisms of CD47-SIRPα blocking agents are summarized in Fig. 3. Agents targeted to CD47 should block the CD47-SIRPα interaction to remove the anti-phagocytic signal and restore the phagocytic activity of macrophages [27]. In addition, engagement of FcRs to limit activity is considered to be necessary for agents targeted to CD47 [28]. In addition, anti-SIRPα antibodies using inert Fc to prevent toxicity resulting from SIRPα expressed on myeloid immune cell perhaps have therapeutic potential.

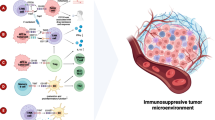
Mechanism of action of interruption of the CD47/SIRPα axis. Three mechanisms can be used to inhibit the CD47/SIRPα interaction. Phagocytosis (the most important mechanism of action): blocks the CD47/SIRPα interaction to remove inhibitory signals and promote phagocytosis of tumor cells; Antigen presentation: antiCD47 antibody connects tumor cells and SIRPα+ DCs to promote antigen presentation; Apoptosis: some antiCD47 antibodies could induce tumor cell poptosis
The second therapeutic mechanism is bridging innate and acquired immunity [29]. Tumor cells are recognized, taken up by antigen presenting cells (APCs, such as dendritic cells and macrophages), and presented to naive T cells, resulting in T cell activation. Antibodies targeted to CD47 could induce direct killing of tumor cells by inhibiting protein kinase A via Giα, resulting in clustering of CD47 in the membrane, and caspase-independent programmed cell death [30].
Anti-CD47 monoclonal antibodies
More than ten anti-CD47 antibodies are in different phases of clinical development (Table 1). All of these antibodies are based on human IgG4-Fc, except AO-176, which is based on human IgG2-Fc [31]. Clinical study results of these antibodies were as follows (ordered by clinical trial developmental state).
Magrolimab (Hu5F9-G4)
Magrolimab, previously known as Hu5F9-G4, was the first anti-CD47 antibody to enter clinical trials, and is currently in Phase III development. The clinical trials for magrolimab are listed in the Table 1. In a preclinical study [32], combined treatment with magrolimab and rituximab showed significant clearance of Raji cells in vitro and elimination of AML tumor cells in vivo. Magrolimab blocks the CD47/SIRPα interaction and the anti-phagocytic signal on macrophages, and binding of rituximab to FcRs initiates pro-phagocytic signaling. Activation of pro-phagocytic signaling is a beneficial effect of anti-CD47 antibody treatment. Magrolimab caused significant hemagglutination and phagocytosis of RBCs in vitro, which indicated potential toxicity. Non-human primate pharmacokinetic and toxicology studies showed dose-dependent anemia. To overcome treatment-related anemia and thrombocytopenia, a low priming dose was given to stimulate production of new RBCs and to facilitate tolerance of subsequent higher maintenance doses. The same strategy (1 mg/kg priming dose on day 1) effectively controlled anemia during subsequent infusion of magrolimab in a clinical trial. The saturation concentration (receptor occupancy) on circulating white and red cells was 30 mg/kg [33]. In a phase Ib [34] (NCT02953509) study evaluating relapsed or refractory non-Hodgkin’s lymphoma, patients with diffuse large B-cell lymphoma (DLBCL) or follicular lymphoma were treated with magrolimab in combination with rituximab. The results showed that 50% of the patients had an objective (i.e., complete or partial) response, with 36% having a complete response. Specifically, the rates of objective response and complete response were 40 and 33%, respectively, among patients with DLBCL, and 71 and 43%, respectively, among those with follicular lymphoma. Calreticulin (CRT) is a member of the endoplasmic reticulum lectin chaperone family of proteins that plays important biological roles in Ca2+ homeostasis (in the endoplasmic reticulum, ER), integrin-dependent cell adhesion (in the cytoplasm), and immune response activation (on the plasma membrane) [35]. CRT translocates from the ER to the cell surface during immunogenic cell death in response to various stress factors such as chemotherapy, irradiation, photodynamic therapy, and cytokines. CRT on the surface of stressed or dying cells acts as an “eat me” signal by binding to LRP1 on macrophages [36].
Azacitidine is a chemotherapeutic drug that could induce ICD through translocation of CRT to the cell surface. Interaction of CRT with LRP1 transmits “eat me” signals to macrophages and promotes phagocytosis of dying cells [37]. Combined treatment with magrolimab and azacitidine in a preclinical study resulted in significantly increased phagocytic activity in vitro, and elimination of HL60 in vivo [38]. Preliminary data from a trial (NCT04778397) of magrolimab combined with azacitidine for treatment of AML resulted in a 65% OR and a 40% CR in 34 patients. Transient on-target anemia was observed, and 56% of patients with AML became red blood cell transfusion independent in response to this therapy. In patients with TP53-mutant AML, 15/21 (71%) achieved an OR and 10 (48%) achieved CR [39]. However, this study was halted by the FDA due to imbalance in investigator-reported suspected unexpected serious adverse reactions (SUSARs). Both magrolimab [33] and azacitidine [40] induce anaemia, neutropenia and thrombocytopenia. The combination of magrolimab and azacidine may increase hematological toxicity.
Ligufalimab (AK117)
Ligufalimab (AK117) is a humanized IgG4 antibody against CD47 from Akeso Biopharma that has completed a phase I trial in Australia, and is currently in phase II trials in China and Australia. In pre-clinical studies, Ligufalimab was comparable to magrolimab with regard to EC50 values and induction of phagocytic activity on Raji and Jurkat cells. However, Ligufalimab did not induce hemagglutination at concentrations up to 1050 μg/ml, while magrolimab induced strong hemagglutination at 1.44 μg/ml (unpublished data by our lab). In a clinical trial (NCT04349969) [41] evaluating treatment of R/R advanced or metastatic solid or lymphoid tumors, Ligufalimab did not induce symptoms of drug-related anemia at doses up to 45 mg/kg, and no priming dose was required. A CD47 receptor binding study on peripheral T cells in patients dosed with 3 mg/kg of Ligufalimab showed nearly 100% binding. No Dose-limiting toxicity (DLTs) occurred in subjects receiving up to 45 mg/kg QW (quaque week) of Ligufalimab. Combination with bispecific antibody AK104 (PD1-CTLA4), AK112 (PD1-VEGF).
Lemzoparlimab (TJC4)
Lemzoparlimab (TJC4), from I-MAB, is a human IgG4 antibody targeted to CD47 that was screened using a phage display system. The crystal structure of the TJC4/CD47 complex (straighter head-to-head orientation) showed that TJC4 binds to a different epitope than does magrolimab (tilted head-to-head orientation) [42]. In addition, TJC4 showed minimal binding to RBC and no hemagglutination was observed at 100 μg/ml in vitro. No significant erythrocyte toxicity was observed in cynomolgus monkeys dosed with 10–100 mg/kg QW. A phase I trial (NCT03934814) [43] evaluating treatment of R/R advanced solid tumors and lymphoma with TJC4 alone, or in combination with pembrolizumab or rituximab, is ongoing. No DLT or SAE (severe adverse events) were observed, and treatment-associated anemia occurred in 30% of patients (6 of 20, 2 receiving 3 mg/kg, 2 receiving 10 mg/kg, 1 receiving 20 mg/kg, and 1 receiving 30 mg/kg). Anti-drug antibody (ADA) events occurred in 25% of patients, but there were no concerns regarding safety or pharmacokinetics (PK). Maximal saturation of peripheral T cells was achieved at a dose of 20 mg/kg administered weekly.
AO176
AO176, developed by Arch Oncology, is a humanized IgG2 subclass anti-CD47 antibody [31]. AO176 was shown to bind to integrin-β1 expressed on tumor cells, but not on RBC. Interestingly, AO176 blocked the CD47/SIRPα interaction to stimulate phagocytosis of tumor cells, and also directly killed tumor cells (non-ADCC). A tolerability and hematologic study on Cynomolgus monkeys showed no anemia. In phase I/II clinical trials [44], Grade 3 TRAE (treatment-related adverse events) were observed in > 10% of patients. In addition, DLT was observed at 20 mg/kg. Studies evaluating AO176 monotherapy and combination therapy with paclitaxel are ongoing.
CC-90002
CC-90002 is a humanized monoclonal IgG4 CD47 antibody. An early clinical trial (NCT02641002) that evaluated treatment of Acute Myeloid Leukemia (AML) and high-risk myelodysplastic syndrome (MDS) was terminated due to poor activity and safety profiles [45, 46]. Another phase I trial (NCT02367196) evaluated treatment of advanced solid (alone) and hematologic malignancies (in combination with rituximab). In a combination trial with patients with R/R NHL [47], the ORR (overall response rate) was 13% with 25% achieving stable disease. However, 50% had anemia (of any grade), 33% had thrombocytopenia, and DLTs were observed in 3 subjects (1 subject infusion-related reaction at 15 mg/kg Q2W and 2 subjects had grade 3 thrombocytopenia requiring platelet transfusion occurring at 30 mg/kg).
SGN-CD47M
SGN-CD47M, developed by Seagen (Seattle Genetics), is a CD47 targeting probody drug conjugate (PDCs). Probody therapeutics [48] are antibody prodrugs designed to remain inactive until proteolytically cleaved and activated in the tumor microenvironment. Probody drug conjugates [49] can be activated by multiple proteases in the tumor microenvironment, but remain inactive in the circulation and in normal tissues. The efficacy of PDCs depends on multiple factors including binding affinity and specificity for the antigen, efficiency of cleavage in the tumor microenvironment, lack of cleavage in normal tissues, and internalization efficiency. Studies focused on PDCs are in the early clinical stage, and safety and efficacy have yet to be determined. Clinical trial (NCT03957096) of SGN-CD47M for treatment of advanced solid tumors was terminated based on portfolio prioritization.
Other drugs in early clinical development for safety and dosing evaluation are listed in Table 1.
SIRPα-fc
Among the 10 allelic variants of SIRPα, SIRPα V1 and V2 are the most prevalent variants [50]. The affinity of wild type SIRPα binding to CD47 is in the micromolar range, which is 1000-fold weaker than that of anti-CD47 antibodies. Soluble SIRPα binding to CD47 on tumor cells could block inhibitory signals and enhance phagocytic activity. Six SIRPα-fusion proteins are currently in phase I or phase II clinical trials (Table 2, ordered by clinical trial developmental state).
Evorpacept (ALX148)
Evorpacept is comprised of SIRPα variant 1 domain 1 (V1D1) and inactive human IgG1-Fc [51]. Evorpacept (CV1) was selected from mutant libraries and includes 9 mutations (V6I, A271, I31F, E47V, K53R, E54S, H56P, L66T, V92I), which resulted in a 50,000-fold increase in affinity compared with that of wild type SIRPα. Preclinical data showed that evorpacept augmented macrophage antitumor activity in vitro and in vivo in combination with tumor-opsonizing antibodies (trastuzumab, obinutuzumab and cetuximab). However, no phagocytosis or antitumor activity was observed following treatment with evorpacept alone. A trial [52] in which patients with NHL received evorpacept alone at 10 mg/kg or at 15 mg/kg in combination with rituximab resulted in ORRs of 40.9 and 63.6%, respectively. The CD47 receptor occupancy on RBC and CD4 T cells was approximately 90% at 10–15 mg/kg. A phase I study [53] of evorpacept in combination with pembrolizumab, trastuzumab, or zanidatamab, and/or chemotherapeutic agents, evaluating treatment of advanced solid malignancy is ongoing. Preliminary results showed anti-cancer activity of evorpacept in combination with pembrolizumab (AP) and/or chemotherapy (5FU + platinum) in patients with second line or greater HNSCC (head and neck squamous cell carcinoma) with prior platinum therapy. The ORR in patients with checkpoint inhibitor-naïve HNSCC (n = 10) treated with AP was 40%, but 0% in patients with HNSCC who had previously received checkpoint inhibitors (n = 10). A phase II study of evorpacept in combination with pembrolizumab for treatment of HNSCC was recently initiated. Treatment with evorpacept in combination with transtuzumab, ramucirumab, and paclitaxel (TRP) showed favorable tolerability and demonstrated objective response in patients with HER2-positive gastric/gastroesophageal cancer. The maximum tolerated dose was not reached in this study. The maximum administered dose was15 mg/kg QW with 22.2% grade 3 or above TARE. The ORR of evorpacept (10 or 15 mg/kg, QW) in combination with TRP in patients with second line HER2 positive gastric/GEJ (gastroesophageal junction) cancer was 72.2%.
TTI-621 and TTI-622
TT1–621 and TT1–622 are constructed from the wild type SIRPα variant 2 domain 1 (V2D1) fused to human IgG-Fc with IgG1 and IgG4 backbones, respectively. Preclinical data demonstrated binding of TTI-621/TTI-622 to cancer cells but only minimal binding to RBCs [54]. However, TTI-621 caused significant anemia in monkeys, presumably due to activation of NK cells by wild type IgG1 with intact Fc function. Both molecules augment tumor-cell killing mediated by macrophages and T cells (phagocytosis by macrophages and presentation of tumor antigens to CD8 T cells to stimulate cytotoxicity, respectively) and both exhibited enhanced activity when used in combination in a preclinical study. Monotherapy using TTI-621 (NCT02663518) or TTI-622 (NCT03530683) against R/R lymphoma induced transient anemia and thrombocytopenia, with recovery within 7 days. The CD47 receptor occupancy on normal peripheral T cells was 60% at a dose of 2 mg/kg. In addition, TTI-622 was well tolerated at 18 mg/kg and the ORR of TTI-622 during treatment of R/R lymphomas was 33%. The ORR for TTI-621 was 18–29% at up to 2 mg/kg and two dosing levels (0.2 and 2.0 mg/kg) will be evaluated in phase Ib/II [55].
IMM01
IMM01 is V2D1 with an N80A mutation and IMM0306 is IMM01-fused to an IgG1 anti-CD20 antibody (rituximab). Both IMM01 and IMM0306 are based on wild type IgG1-Fc. IMM01 does not bind to human erythrocytes, avoiding “antigenic sink.” Monotherapy using IMM01 was administered to 14 patients with R/R lymphoma [56]. Transient platelet count decreased after 2 h and returned to baseline at 24 to 48 h following the first infusion. One patient experienced grade 3 platelet count decrease. Preliminary results showed anti-tumor activity at 1.0 mg/kg. In preclinical in vivo study, IMM01 showed strong synergistic anti-tumor activity combined with rituximab (antiCD20 antibody), imatinib (tyrosine kinase inhibitor, TKI). Clinical trials are ongoing of IMM01 combined with rituximab, imatinib in China.
HX009
HX009 (Table 3) is wild type SIRPα V2D1 fused to the C terminal of an IgG4 anti-PD1 antibody. A phase I clinical trial (NCT04097769) showed that HX009 was well-tolerated at 7.5 mg/kg with no DLT [57] when used to treat patients with advanced solid tumors. Antitumor activity was seen at 1 mg/kg and 5 mg/kg cohorts with objective responses in multiple tumor types (gallbladder adenocarcinoma (1 mg/kg), triple negative breast cancer (5 mg/kg), metastatic squamous cell carcinoma of head and neck (5 mg/kg)). Phase I/II trial in Chinese patients with relapsed/refractory lymphoma is ongoing.
Antibodies targeting SIRPα on myeloid cells
Anti-SIRPα antibodies induce weak or no phagocytic activity alone, but induce significantly increased phagocytic activity when combined with opsonizing antibodies (rituximab, cetuximab) [58,59,60]. Several issues with anti-SIRPα antibodies are important to consider. First, since SIRPα is expressed on myeloid cells, anti-SIRPα antibodies using inactive human IgG-Fc to avoid Fc effector-mediated toxicity on these immune cells may be advantageous [60]. Second, SIRPγ expressed on T and NK cell shares 74.37% amino acid similarity with the extracellular domain with SIRPα [61]. SIRPγ on T cells binds to CD47 on APCs to mediate cell-cell adhesion and enhances antigen presentation, resulting in T cell proliferation and cytokine secretion [62]. Development of a SIRPα targeting antibody with specificity toward SIRPα to avoid interference with the interaction between CD47 and SIRPγ may preserve T cell activity. Third, antibody internalization could lead to rapid clearance of antibody in vivo. Higher doses or multiple dosing is required to ensure that levels remain therapeutically relevant [63, 64]. Internalization of SIRPα decreases the inhibitory signal and may enhance the ability of antibodies to restore phagocytic activity [59, 65]. Finally, Although SIRPα has ten known variants, V1, V2, and V8 are the most prominent (over 90%) haplotypes in the human population. Antibodies targeting all three of these variants may be more potent than those that target a single variant [66, 67].
Antibodies targeting SIRPα currently being evaluated are listed in the Table 4. Trials evaluating treatment with SIRPα antibodies in combination with immunotherapies are in the early clinical stage of development, and include OSE-172 [68] from OSE Immunotherapeutics (co-developed with Boehringer Ingelheim), CC-95251 from Celgene, and FSI-189 from Gilead [59].
Bi-specific molecules
Bi-specific molecules bind two targets or two distinct epitopes of one target. The antigen binding sites of bi-specific molecules could consist of two antibodies or proteins (ligand or receptor), or could consist of one antibody and one protein. Bi-specific antibodies can bind two target antigens in-cis and in-trans. Rational design of bi-specific molecules based on biological activity may result in distinct effects or improved efficacy when compared to combination treatments. Four bispecific antibodies, catumaxomab [69] (withdrawn in 2017), blinatumomab [70], emicizumab [71], and amivantamab-vmjw [72] have been approved by EMA or FDA.
Bi-specific molecules can be constructed from CD47 targeting antibodies or SIRPα and other antigen-targeting molecules (Fig. 4). Target antigens could include: A) tumor associated cell surface antigens (PD-L1, CD20, CD19, MSLN (Mesothelin), Claudin18.2, and Her2), B) immune checkpoint proteins (PD-1, CD40, 41BB), and C) cytokines or receptors (CSF-2 receptor, VEGF). Reduced affinity for CD47 and increased affinity for the second target may reduce toxicity and enhance efficacy. For type A antibodies, IgG1-Fc was selected to enhance antibody-mediated killing of tumor cells (ADCC, ADCP, and CDC). However, inactivated Fc is preferred when used in type B bi-specific molecules. Use of CD47-targeting biologics has shown significant clinical efficacy for treatment of R/R AML, NHL, and MDS. The combination of Hu5F9 and rituximab showed particular promise as a treatment approach for R/R NHL [34]. Several CD47-related BsAb are in early-stage clinical trials. NI1701 [73], which targets CD47 and CD19, is an IgG-like BsAb constructed using modified knobs-into-hole technology [74], and contains an IgG1-Fc. Preclinical data showed that NI1701 [75] selectively binds to CD47 and CD19 co-expressing cells, but interacts poorly with normal healthy cells (CD47+CD19−), resulting in avoidance of normal cells acting as sinks for binding of the antibodies, and reduced toxicity. In vitro and in vivo studies showed that NI1701 more potently killed tumor cells than did anti-CD47 and anti-CD19 antibodies alone, or in combination. A BsAb named NI1801 [73] targeted to CD47 and MSLN showed similar preclinical activity as NI1701. IMM0306, a bispecific antibody fusion protein targeted to CD20 (rituximab) and CD47 (SIRPα) with wild type IgG1-Fc [76], is in a phase I trial evaluating treatment of R/R CD20-positive B-cell non-Hodgkin’s lymphoma. SL-172154 [77] is a fusion protein targeted to CD47 with SIRPα and CD40 with CD40L, and is in a phase I trial for treatment of solid tumors. A BsAb, HX009, comprised of the extracellular region of SIRPα V2D fused with an anti-PD1 antibody (HX008) was evaluated in a phase I trial. The study showed that HX009 blocked both CD47/SIRPα and PD-1/PDL-1 interactions, and interacted with CD47 on tumor cells and PD1 on T cells to help present tumor antigens to T cells, resulting in activation of the innate and acquired immune responses. Several BsAbs have been developed to inhibit CD47/SIRPα and PD-1/PD-L1 interactions. One such BsAb, IBI322 [78], was designed as a selective CD47 binding CD47/PD-L1 bispecific antibody. IBI322 showed no negative hematological effects in cynomolgus monkeys, but the binding affinity of IBI322 to cynomolgus CD47 was not disclosed, and IBI322 showed dose-dependent binding to RBC. Other BsAb antibodies listed in Table 2 are in the preclinical proof-of-concept stage.

Types of bis-specific molecules targeted CD47 and other molecules. Bispecific Ab-type 1: KIH format, targeted to CD47 (yellow) and other tumor associated antigens (CD19, CD20, and MSLN, blue); Bi-specific Ab-type 2: KIH format, targeted to CD47 (yellow) and immune checkpoint molecules (PD-1, CD40, and 41BB, green); Bi-specific Ab-type 3: SIRPα (cyan) fusion to N-terminus of H-chain of IgG (targeting tumor associated antigen, blue); Bi-specific Ab-type 4: ligand/receptor to modulate TME (GM-CSF, and VEGFR2, pink) fusion to the C-terminus of the H-chain of IgG (yellow); Bi-specific Ab-type 4: SIRPα fusion to the N-terminus of IgG-Fc and ligand/receptor to modulate TME (GM-CSF, VEGFR2, pink) fusion to the C-terminus
Engineered T cells and macrophages
T cells with chimeric antigen receptors (CARs) showed promising therapeutic efficacy against hematologic malignancies, and several CAR-T therapeutics have been approved [79,80,81,82,83]. However, low treatment response rates of solid tumors to CAR-T treatment were observed. Golubovskaya et al. [84] showed that CD47-CAR-T cells effectively killed ovarian, pancreatic, and other cancer cells, and induced production of high levels of IL-2, which correlated with expression of CD47 antigens. Treatment with CD47-CAR-T cells may be a novel strategy for treating different types of cancers. Huyen [85] designed a third generation of CD47-CAR-T cell that could effectively kill lung cancer cells (A549) and inhibit lung cancer cell metastasis. A dual CAR-T targeting CD47 and TAG-72 (tumor-associated glycoprotein 72) generated by Shu [86] showed promising results against ovarian cancer in preclinical experiments. An Anti-PD-L1 (A12) CAR-T with the ability to secrete anti-CD47 VHH (variable heavy domain of heavy chain antibodies or nanobodies) (A4), developed by Xie [87], represented a novel strategy for cancer treatment. An A12-A4 CAR-T showed better anti-cancer activity than an A12 CAR-T plus soluble A4 in C57BL/6 PD-L1-KO mice bearing B16F10 cells. Each of these CD47-CAR-T cells are in the preclinical stage, and efficacy and safety should be further investigated.
Development of CAR-M1 (M1: classically activated macrophages) is an emerging therapeutic strategy [88], and several studies of engineered CAR-M cells [89,90,91] showed tumor cell elimination activity in vitro and in vivo. These studies showed that CAR-M induced phagocytosis and induced M2 to M1 polarization through secretion of pro-inflammatory factors and chemokines [90]. CD47 is ubiquitously expressed on the surfaces of multiple hematopoietic and solid tumor cells. However, adverse events due to cytokine secretion by macrophages during immune checkpoint activation [92, 93] and CAR-T [94, 95] treatment was common. Technological improvements for preparation and production of CAR-M are needed to offer scalable and reproducible manufacturing processes.
Small molecules, peptides, and microRNA
RRx-001 [96] is an anticancer agent designed to induce M2 to M1 polarization and to promote recovery of phagocytic activity of macrophages toward tumor cells. The anti-phagocytic inhibitory signal was removed or reduced through downregulation of both CD47 and SIRPα gene expression on tumor cells and macrophages, respectively. Elimination of tumor cells was shown in in vitro and in vivo. Phase III clinical trials (NCT03699956, NCT02489903) against small cell lung cancer [97, 98] are ongoing.
D4–2 [99], a macrocyclic peptide targeted to mouse SIRPα was designed to inhibit the interaction between CD47 and SIRPα and promote macrophage-mediated phagocytosis of tumor cells when combined with rituximab. PKHB1 [100], a TSP-1-derived CD47 agonist peptide, induced cell death (CRT exposure and DAMP release) in chronic lymphocytic leukemia cells.
MicroRNAs (miRNAs), which are 20–22 nucleotides in length, play important roles in cancer pathogenesis and progression since they can repress the target gene at the translational level by directly binding to the 3’untranslated regions (3’UTRs) [101].
Overexpression miR-378a [102] in mice peritoneal macrophages downregulates SIRPα mRNA expression. Phagocytosis of Ishikawa cells by macrophages-miR-378a and macrophages was carried out in vitro. Phagocytic index in macrophages-miR-378a group is 3 times than that in macrophages group.
Zhao [103] reported that miR-200a inhibited the expression of CD47 by directly targeting the 3’UTR of the CD47 mRNA. MicroRNA 200a suppressed nasopharyngeal carcinoma (NPC) cell proliferation, migration, and invasion, and promoted phagocytosis of NPC cells by macrophages through down-regulation of CD47 expression on NPC cells.
MicroRNA 708 [104] was directly targeted CD47 and resulted in downregulation of CD47 on T cell acute lymphoblastic leukemia cell line. MicroRNA 708 expression in the T-ALL cell line was sufficient to promote phagocytosis by macrophages in vitro, and inhibited tumor engraftment in vivo.
Conclusions and future perspectives
Following the clinical success of therapeutic antibodies targeting T cell checkpoint molecules, combination therapies using checkpoint inhibitors with other agents have been a major theme of clinical oncology studies. Specifically, inhibitors of the CD47/SIRPα pathway have emerged as promising therapeutic candidates. Overexpression on tumor cells makes CD47 an ideal target for cancer therapy. Antibodies targeting CD47 showed promising results against MDS and AML [105,106,107]. However, side effects such as anemia, hyperbilirubinemia, thrombocytopenia, and lymphopenia induced by CD47-targeting molecules are of specific concern and need to be addressed with development of new therapeutic agents.
In addition, the therapeutic effects of agents targeting the CD47/SIRPα axis on solid tumors are limited. Additional therapeutic strategies, including combination therapy and bi-specific antibodies, may be promising. Combination therapy with opsonizing antibodies, immune checkpoint inhibitors, chemotherapeutic agents to activate FcR on macrophages, and T cell sensitizers that induce immunogenic cell death to stimulate a more potent immunological effect all have potential. Agents targeted to the CD47-SIRPα axis should not only block the CD47/SIRPα interaction, but also activate signaling on macrophages (FcγR, CRT expression). Other agents including small molecules, mRNA, and CAR-T/M that block the CD47/SIRPα interaction are also in development. Many promising strategies targeting the CD47-SIRPα axis are in development and offer a great deal of hope to patients with cancer.
Availability of data and materials
Not applicable.
Abbreviations
- ADA:
-
Anti-drug antibody
- ADCC:
-
Antibody Dependent Cellular Cytotoxicity
- ADCP:
-
Antibody Dependent Cellular Phagocytosis
- ADCs:
-
Antibody Drug Conjugates
- AML:
-
Acute Myeloid Leukemia
- PTPase:
-
Protein Tyrosine Phosphatases
- BiAb:
-
Bispecific Antibody
- CAR:
-
Chimeric Antigen Receptor
- CR:
-
Complete response Rate
- CRT:
-
Calreticulin
- CTLA-4:
-
Cytotoxic T Lymphocyte-associated Antigen-4
- DLBCL:
-
Diffuse Large B-Cell Lymphoma
- DLT:
-
Dose-Limited Toxicity
- DAMP:
-
Damage Associated Molecular Patterns
- GEJ cancer:
-
Gastroesophageal junction cancer
- ICIs:
-
Immune Checkpoint Inhibitors
- ITIMs:
-
Immunoreceptor Tyrosine-based Inhibition Motifs
- MDS:
-
Myelodysplastic Syndrome
- MSLN:
-
Mesothelin
- NHL:
-
non-Hodgkin’s Lymphoma
- ORR:
-
Overall Response Rate
- PD-1:
-
Programmed cell Death1
- PDCs:
-
Probody Drug Conjugates
- PD-L1:
-
Programmed cell Death Ligand 1
- PK:
-
Pharmacokinetics
- SH2:
-
Src Homology 2
- SIRPα:
-
Signal regulatory protein alpha
- TME:
-
Tumor Micro-Environment
- TRAE:
-
Treatment-related adverse event
- VEGF:
-
Vascular endothelial growth factor
References
Kuol N, Stojanovska L, Nurgali K, Apostolopoulos V. The mechanisms tumor cells utilize to evade the host's immune system. Maturitas. 2017;105:8–15.
Cogdill AP, Andrews MC, Wargo JA. Hallmarks of response to immune checkpoint blockade. Br J Cancer. 2017;117(1):1–7.
Okwundu N, Grossman D, Hu-Lieskovan S, Grossmann KF, Swami U. The dark side of immunotherapy. Ann Transl Med. 2021;9(12):1041–55.
Zhang D, Zhang Y, Huang Y, Kong L, Yu J. Hyper-progressive disease in a patient with advanced non-small cell lung cancer on immune checkpoint inhibitor therapy: a case report and literature review. Lung Cancer. 2020;139:18–21.
Kubota Y, Yoshimura K, Hamada K, Hirasawa Y, Shida M, Taniguchi M, et al. Rare Nivolumab-associated super hyper progressive disease in patients with advanced gastric Cancer. In Vivo. 2021;35(3):1865–75.
Ziegler-Heitbrock L. Macrophage: origin, activation and polarization. In: Mantovani AS, Biswas K, editors. Macrophages: biology and role in the pathology of diseases. New York: Springer; 2014. p. 11–95.
Feng M, Jiang W, Kim BYS, Zhang CC, Fu Y, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. 2019;19(10):568–86.
Fujioka Y, Matozaki T, Noguchi T, Iwamatsu A, Yamao T, Takahashi N, et al. A novel membrane glycoprotein, SHPS-1, that binds the SH2-domain-containing protein tyrosine phosphatase SHP-2 in response to mitogens and cell adhesion. Mol Cell Biol. 1996;16(12):6887–99.
van den Berg TK, van der Schoot CE. Innate immune ‘self’ recognition: a role for CD47–SIRPα interactions in hematopoietic stem cell transplantation. Trends Immunol. 2008;29(5):203–6.
Barclay AN, Brown MH. The SIRP family of receptors and immune regulation. Nat Rev Immunol. 2006;6(6):457–64.
Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is Upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–85.
Xiao Z, Chung H, Banan B, Manning PT, Ott KC, Lin S, et al. Antibody mediated therapy targeting CD47 inhibits tumor progression of hepatocellular carcinoma. Cancer Lett. 2015;360(2):302–9.
Michaels AD, Newhook TE, Adair SJ, Morioka S, Goudreau BJ, Nagdas S, et al. CD47 blockade as an adjuvant immunotherapy for Resectable pancreatic Cancer. Clin Cancer Res. 2018;24(6):1415–25.
Kaur S, Singh SP, Elkahloun AG, Wu W, Abu-Asab MS, Roberts DD. CD47-dependent immunomodulatory and angiogenic activities of extracellular vesicles produced by T cells. Matrix Biol. 2014;37:49–59.
Goto H, Kojima Y, Matsuda K, Kariya R, Taura M, Kuwahara K, et al. Efficacy of anti-CD47 antibody-mediated phagocytosis with macrophages against primary effusion lymphoma. Eur J Cancer. 2014;50(10):1836–46.
Alvey CM, Spinler KR, Irianto J, Pfeifer CR, Hayes B, Xia Y, et al. SIRPA-inhibited, marrow-derived macrophages engorge, accumulate, and differentiate in antibody-targeted regression of solid tumors. Curr Biol. 2017;27(14):2065–77.
Barclay AN, van den Berg TK. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32(1):25–50.
Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11(3):130–5.
Oldenborg P, Zheleznyak A, Fang Y, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288(5473):2051–4.
Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286–99.
Kim D, Wang J, Willingham SB, Martin R, Wernig G, Weissman IL. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia. 2012;26(12):2538–45.
Chao MP, Tang C, Pachynski RK, Chin R, Majeti R, Weissman IL. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood. 2011;118(18):4890–901.
Chao MP, Alizadeh AA, Tang C, Jan M, Weissman-Tsukamoto R, Zhao F, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011;71(4):1374–84.
Lee TK, Cheung VC, Lu P, Lau EYT, Ma S, Tang KH, et al. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology. 2014;60(1):179–91.
Steinert G, Schölch S, Niemietz T, Iwata N, García SA, Behrens B, et al. Immune escape and survival mechanisms in circulating tumor cells of colorectal Cancer. Cancer Res. 2014;74(6):1694–704.
Brightwell RM, Grzankowski KS, Lele S, Eng K, Arshad M, Chen H, et al. The CD47 “don't eat me signal” is highly expressed in human ovarian cancer. Gynecol Oncol. 2016;143(2):393–7.
Gholamin S, Mitra SS, Feroze AH, Liu J, Kahn SA, Zhang M, et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med. 2017;9(381):f2968–80.
Jain S, Van Scoyk A, Morgan EA, Matthews A, Stevenson K, Newton G, et al. Targeted inhibition of CD47-SIRPα requires fc-FcγR interactions to maximize activity in T-cell lymphomas. Blood. 2019;134(17):1430–40.
Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, et al. CD47 blockade triggers T cell–mediated destruction of immunogenic tumors. Nat Med. 2015;21(10):1209–15.
Manna PP, Frazier WA. CD47 mediates killing of breast tumor cells via Gi-dependent inhibition of protein kinase a. Cancer Res. 2004;64(3):1026–36.
Puro RJ, Bouchlaka MN, Hiebsch RR, Capoccia BJ, Donio MJ, Manning PT, et al. Development of AO-176, a next-generation humanized anti-CD47 antibody with novel anticancer properties and negligible red blood cell binding. Mol Cancer Ther. 2020;19(3):835–46.
Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L, et al. Pre-clinical development of a humanized anti-CD47 antibody with anti-Cancer therapeutic potential. PLoS One. 2015;10(9):e137345–67.
Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D, et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J Clin Oncol. 2019;37(12):946–53.
Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. New Engl J Med. 2018;379(18):1711–21.
Krause KH, Michalak M. Calreticulin Cell. 1997;88(4):439–43.
Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123(2):321–34.
Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med. 2010;2(63):63–94.
Feng D, Gip P, McKenna KM, Zhao F, Mata O, Choi TS, et al. Combination treatment with 5F9 and Azacitidine enhances phagocytic elimination of acute myeloid leukemia. Blood. 2018;132(Suppl 1):2729.
Sallman D, Asch A, Kambhampati S, Malki MA, Zeidner J, Donnellan W, et al. The first-in-class anti-CD47 antibody Magrolimab in combination with Azacitidine is well tolerated and effective in AML patients: phase 1b results. Clin Lymphoma Myeloma Leukemia. 2021;21:S290.
Santini V, Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Silverman LR, List A, et al. Management and supportive care measures for adverse events in patients with myelodysplastic syndromes treated with azacitidine*. Eur J Haematol. 2010;85(2):130–8.
Gan HK, Coward J, Mislang ARA, Cosman R, Nagrial A, Jin X, et al. Safety of AK117, an anti-CD47 monoclonal antibody, in patients with advanced or metastatic solid tumors in a phase I study. J Clin Oncol. 2021;39(15_suppl):2630.
Guo TB, Wang Z, And L F, Zang J. A differentiated CD47 therapeutic antibody recognizing a novel epitope and sparing erythrocytes and platelets. 2017, Poster presentation on EACR 2017, https://www.i-mabbiopharma.com/userfiles/images/2019-10-30/1pdf.pdf
Berlin J, Harb W, Adjei A, Xing Y, Swiecicki P, Seetharam M, et al. A first-in-human study of lemzoparlimab, a differentiated anti-CD47 antibody, in subjects with relapsed/refractory malignancy: initial monotherapy results. J Immunother Cancer. 2020;8(Suppl 3):A410.
Burris HA III, Spira AI, Taylor MH, Yeku OO, Liu JF, Munster PN, et al. A first-in-human study of AO-176, a highly differentiated anti-CD47 antibody, in patients with advanced solid tumors. J Clin Oncol. 2021;39(15_suppl):2516.
Zeidan AM, DeAngelo DJ, Palmer J, Seet CS, Tallman MS, Wei X, et al. Phase 1 study of anti-CD47 monoclonal antibody CC-90002 in patients with relapsed/refractory acute myeloid leukemia and high-risk myelodysplastic syndromes. Ann Hematol. 2022;101(3):557–69.
Zeidan AM, DeAngelo DJ, Palmer JM, Seet CS, Tallman MS, Wei X, et al. A phase I study of CC-90002, a monoclonal antibody targeting CD47, in patients with relapsed and/or refractory (R/R) acute myeloid leukemia (AML) and high-risk Myelodysplastic syndromes (MDS): final results. Blood. 2019;134(Supplement_1):1320.
Abrisqueta P, Sancho J, Cordoba R, Persky DO, Andreadis C, Huntington SF, et al. Anti-CD47 antibody, CC-90002, in combination with rituximab in subjects with relapsed and/or refractory non-Hodgkin lymphoma (R/R NHL). Blood. 2019;134:4089.
Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EEM, Liang TW, et al. Tumor-specific activation of an EGFR-targeting Probody enhances therapeutic index. Sci Transl Med. 2013;5(207):144–207.
Chomet M, Schreurs M, Nguyen M, Howng B, Villanueva R, Krimm M, et al. The tumor targeting performance of anti-CD166 Probody drug conjugate CX-2009 and its parental derivatives as monitored by89 Zr-immuno-PET in xenograft bearing mice. Theranostics. 2020;10(13):5815–28.
Hatherley D, Lea SM, Johnson S, Barclay AN. Polymorphisms in the human inhibitory signal-regulatory protein α do not affect binding to its ligand CD47. J Biol Chem. 2014;289(14):10024–8.
Weiskopf K, Ring AM, Ho CCM, Volkmer J, Levin AM, Volkmer AK, et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341(6141):88–91.
Kim TM, Lakhani N, Gainor J, Kamdar M, Fanning P, Squifflet P, et al. A phase 1 study of ALX148, a CD47 blocker, in combination with rituximab in patients with non-Hodgkin lymphoma. Blood. 2019;134(Suppl 1):1953.
Chow LQM, Gainor JF, Lakhani NJ, Lee KW, Chung HC, Lee J, et al. A phase I study of ALX148, a CD47 blocker, in combination with standard anticancer antibodies and chemotherapy regimens in patients with advanced malignancy. J Clin Oncol. 2020;38(15_suppl):3056.
Petrova PS, Viller NN, Wong M, Pang X, Lin GHY, Dodge K, et al. TTI-621 (SIRPαFc): a CD47-blocking innate immune checkpoint inhibitor with broad antitumor activity and minimal erythrocyte binding. Clin Cancer Res. 2017;23(4):1068–79.
Ansell SM, Maris MB, Lesokhin AM, Chen RW, Flinn IW, Sawas A, et al. Phase I study of the CD47 blocker TTI-621 in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2021;27(8):2190–9.
Sun M, Qi J, Zheng W, Song L, Jiang B, Wang Z, et al. Preliminary results of a first-in-human phase I dtudy of IMM01, SIRPα fc protein in patients with relapsed or refractory lymphoma. J Clin Oncol. 2021;39(15_suppl):2550.
Roohullah A, Ganju V, Zhang F, Zhang L, Yu T, Wilkinson K, et al. First-in-human phase 1 dose escalation study of HX009, a novel recombinant humanized anti-PD-1 and CD47 bispecific antibody, in patients with advanced malignancies. J Clin Oncol. 2021;39(15_suppl):2517.
Ring NG, Herndler-Brandstetter D, Weiskopf K, Shan L, Volkmer J, George BM, et al. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc Natl Acad Sci. 2017;114(49):E10578–85.
Liu J, Xavy S, Mihardja S, Chen S, Sompalli K, Feng D, et al. Targeting macrophage checkpoint inhibitor SIRPα for anticancer therapy. JCI Insight. 2020;5(12):e134728–40.
Kuo TC, Chen A, Harrabi O, Sockolosky JT, Zhang A, Sangalang E, et al. Targeting the myeloid checkpoint receptor SIRPalpha potentiates innate and adaptive immune responses to promote anti-tumor activity. J Hematol Oncol. 2020;13(1):160.
Brooke G, Holbrook JD, Brown MH, Barclay AN. Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J Immunol. 2004;173(4):2562–70.
Dehmani S, Nerriere-Daguin V, Neel M, Elain-Duret N, Heslan JM, Belarif L, et al. SIRPgamma-CD47 interaction positively regulates the activation of human T cells in situation of chronic stimulation. Front Immunol. 2021;12:732530.
Amano J, Masuyama N, Hirota Y, Tanaka Y, Igawa Y, Shiokawa R, et al. Antigen-dependent internalization is related to rapid elimination from plasma of humanized anti-HM1.24 monoclonal antibody. Drug Metab Dispos. 2010;38(12):2339–46.
Ovacik M, Lin K. Tutorial on monoclonal antibody pharmacokinetics and its considerations in early development. Clin Transl Sci. 2018;11(6):540–52.
Andrejeva G, Capoccia BJ, Hiebsch RR, Donio MJ, Darwech IM, Puro RJ, et al. Novel SIRPalpha antibodies that induce single-agent phagocytosis of tumor cells while preserving T cells. J Immunol. 2021;206(4):712–21.
Voets E, Paradé M, Lutje Hulsik D, Spijkers S, Janssen W, Rens J, et al. Functional characterization of the selective pan-allele anti-SIRPα antibody ADU-1805 that blocks the SIRPα–CD47 innate immune checkpoint. J Immunother Cancer. 2019;7(1):340.
Sim J, Sockolosky JT, Sangalang E, Izquierdo S, Pedersen D, Harriman W, et al. Discovery of high affinity, pan-allelic, and pan-mammalian reactive antibodies against the myeloid checkpoint receptor SIRPα. MAbs. 2019;11(6):1036–52.
Champiat S, Cassier PA, Kotecki N, Korakis I, Vinceneux A, Jungels C, et al. Safety, pharmacokinetics, efficacy, and preliminary biomarker data of first-in-class BI 765063, a selective SIRPα inhibitor: results of monotherapy dose escalation in phase 1 study in patients with advanced solid tumors. J Clin Oncol. 2021;39(15_suppl):2623.
Sebastian M, Kuemmel A, Schmidt M, Schmittel A. Catumaxomab: a bispecific trifunctional antibody. Drugs Today (Barc). 2009;45(8):589–97.
Pulte ED, Vallejo J, Przepiorka D, Nie L, Farrell AT, Goldberg KB, et al. FDA supplemental approval: Blinatumomab for treatment of relapsed and refractory precursor B-cell acute lymphoblastic leukemia. Oncologist. 2018;23(11):1366–71.
Scott LJ, Kim ES. Emicizumab-kxwh: first global approval. Drugs. 2018;78(2):269–74.
Syed YY. Amivantamab: First Approval. Drugs. 2021;81(11):1349–53.
Dheilly E, Moine V, Broyer L, Salgado-Pires S, Johnson Z, Papaioannou A, et al. Selective blockade of the ubiquitous checkpoint receptor CD47 is enabled by dual-targeting Bispecific antibodies. Mol Ther. 2017;25(2):523–33.
Fischer N, Elson G, Magistrelli G, Dheilly E, Fouque N, Laurendon A, et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat Commun. 2015;6:6113.
Buatois V, Johnson Z, Salgado-Pires S, Papaioannou A, Hatterer E, Chauchet X, et al. Preclinical development of a Bispecific antibody that safely and effectively targets CD19 and CD47 for the treatment of B-cell lymphoma and leukemia. Mol Cancer Ther. 2018;17(8):1739–51.
Yu J, Song Y, Tian W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J Hematol Oncol. 2020;13(1):45–54.
de Silva S, Fromm G, Shuptrine CW, Johannes K, Patel A, Yoo KJ, et al. CD40 enhances type I interferon responses downstream of CD47 blockade, bridging innate and adaptive immunity. Cancer Immunol Res. 2020;8(2):230–45.
Wang Y, Ni H, Zhou S, He K, Gao Y, Wu W, et al. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol Immunother. 2021;70(2):365–76.
Reagan PM, Friedberg JW. Axicabtagene ciloleucel and brexucabtagene autoleucel in relapsed and refractory diffuse large B-cell and mantle cell lymphomas. Future Oncol. 2021;17(11):1269–83.
Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, et al. FDA approval summary: Axicabtagene Ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res. 2019;25(6):1702–8.
Mullard A. FDA approves first BCMA-targeted therapeutic. Nat Rev Drug Discov. 2020;19(10):659.
Prasad V. Tisagenlecleucel — the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol. 2018;15(1):11–2.
Liu Y, Chen X, Han W, Zhang Y. Tisagenlecleucel, an approved anti-CD19 chimeric antigen receptor T-cell therapy for the treatment of leukemia. Drug Today. 2017;53(11):597.
Golubovskaya V, Berahovich R, Zhou H, Xu S, Harto H, Li L, et al. CD47-CAR-T cells effectively kill target Cancer cells and block pancreatic tumor growth. Cancers. 2017;9(10):139–53.
La HT, Tran DBT, Tran HM, Nguyen LT. Third-generation anti-CD47-specific CAR-T cells effectively kill Cancer cells and reduce the genes expression in lung Cancer cell metastasis. J Immunol Res. 2021;2021:1–13.
Shu R, Evtimov VJ, Hammett MV, Nguyen NN, Zhuang J, Hudson PJ, et al. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol Ther Oncolytics. 2021;20:325–41.
Xie YJ, Dougan M, Ingram JR, Pishesha N, Fang T, Momin N, et al. Improved antitumor efficacy of chimeric antigen receptor T cells that secrete single-domain antibody fragments. Cancer Immunol Res. 2020;8(4):518–29.
Chen Y, Yu Z, Tan X, Jiang H, Xu Z, Fang Y, et al. CAR-macrophage: a new immunotherapy candidate against solid tumors. Biomed Pharmacother. 2021;139:111605–11.
Zhang W, Liu L, Su H, Liu Q, Shen J, Dai H, et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Brit J Cancer. 2019;121(10):837–45.
Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38(8):947–53.
Zhang L, Tian L, Dai X, Yu H, Wang J, Lei A, et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J Hematol Oncol. 2020;13(1):153–7.
Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56–69.
Rotz SJ, Leino D, Szabo S, Mangino JL, Turpin BK, Pressey JG. Severe cytokine release syndrome in a patient receiving PD-1-directed therapy. Pediatr Blood Cancer. 2017;64(12):e26642.
Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24(6):731–8.
Hao Z, Li R, Meng L, Han Z, Hong Z. Macrophage, the potential key mediator in CAR-T related CRS. Exp Hematol Oncol. 2020;9(1):15–26.
Cabrales P. RRx-001 acts as a dual small molecule checkpoint inhibitor by Downregulating CD47 on Cancer cells and SIRP-α on monocytes/macrophages. Transl Oncol. 2019;12(4):626–32.
Tomita Y, Oronsky B, Abrouk N, Cabrales P, Reid TR, Lee M, et al. In small cell lung cancer patients treated with RRx-001, a downregulator of CD47, decreased expression of PD-L1 on circulating tumor cells significantly correlates with clinical benefit. Transl Lung Cancer Res. 2021;10(1):274–8.
Oronsky B, Reid TR, Larson C, Caroen S, Quinn M, Burbano E, et al. REPLATINUM phase III randomized study: RRx-001 + platinum doublet versus platinum doublet in third-line small cell lung cancer. Future Oncol. 2019;15(30):3427–33.
Hazama D, Yin Y, Murata Y, Matsuda M, Okamoto T, Tanaka D, et al. Macrocyclic peptide-mediated blockade of the CD47-SIRPα interaction as a potential Cancer immunotherapy. Cell Chem Biol. 2020;27(9):1181–91.
Uscanga Palomeque AC, Calvillo Rodríguez KM, Gómez Morales L, Lardé E, Denèfle T, Caballero Hernández D, et al. CD47 agonist peptidePKHB1 induces immunogenic cell death in T-cell acute lymphoblastic leukemia cells. Cancer Sci. 2019;110(1):256–68.
O'Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne). 2018;9:402.
Chen W, Li X, Wang J, Song N, Zhu A, Jia L. miR-378a modulates macrophage phagocytosis and differentiation through targeting CD47-SIRPα Axis in atherosclerosis. Scand J Immunol. 2019;90(1):e12766.
Zhao Y, Yu X, Tang H, Han R, Wang X, Wang J, et al. MicroRNA-200a promotes phagocytosis of macrophages and suppresses cell proliferation, migration, and invasion in nasopharyngeal carcinoma by targeting CD47. Biomed Res Int. 2020;2020:3723781.
Huang W, Wang W, Fang K, Chen Z, Sun Y, Han C, et al. MIR-708 promotes phagocytosis to eradicate T-ALL cells by targeting CD47. Mol Cancer. 2018;17(1):12–7.
Haddad F, Daver N. Targeting CD47/SIRPa in acute myeloid leukemia and Myelodysplastic syndrome: preclinical and clinical developments of Magrolimab. J Immunother Precis Oncol. 2021;4(2):67–71.
Sallman DA, Donnellan WB, Asch AS, Lee DJ, Al Malki M, Marcucci G, et al. The first-in-class anti-CD47 antibody Hu5F9-G4 is active and well tolerated alone or with azacitidine in AML and MDS patients: initial phase 1b results. J Clin Oncol. 2019;37(15_suppl):7009.
Sallman DA, Al Malki M, Asch AS, Lee DJ, Kambhampati S, Donnellan WB, et al. Tolerability and efficacy of the first-in-class anti-CD47 antibody magrolimab combined with azacitidine in MDS and AML patients: phase Ib results. J Clin Oncol. 2020;38(15_suppl):7507.
Acknowledgments
The coauthors thank members in Department of Antibody Discovery, Akeso Biopharma. Jimmy Zeng in Faculty of Health sciences in University of MACAU.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
TQ and BL designed the study. TQ drafted the manuscript. All authors were involved in manuscript preparation and revisions. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This is not applicable for this review.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qu, T., Li, B. & Wang, Y. Targeting CD47/SIRPα as a therapeutic strategy, where we are and where we are headed. Biomark Res 10, 20 (2022). https://doi.org/10.1186/s40364-022-00373-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-022-00373-5