
Abstract
Natural killer (NK) cells are unique innate immune cells and manifest rapid and potent cytotoxicity for cancer immunotherapy and pathogen removal without the requirement of prior sensitization or recognition of peptide antigens. Distinguish from the T lymphocyte-based cythotherapy with toxic side effects, chimeric antigen receptor-transduced NK (CAR-NK) cells are adequate to simultaneously improve efficacy and control adverse effects including acute cytokine release syndrome (CRS), neurotoxicity and graft-versus-host disease (GVHD). Moreover, considering the inherent properties of NK cells, the CAR-NK cells are “off-the-shelf” product satisfying the clinical demand for large-scale manufacture for cancer immunotherapy attribute to the cytotoxic effect via both NK cell receptor-dependent and CAR-dependent signaling cascades. In this review, we mainly focus on the latest updates of CAR-NK cell-based tactics, together with the opportunities and challenges for cancer immunotherapies, which represent the paradigm for boosting the immune system to enhance antitumor responses and ultimately eliminate malignancies. Collectively, we summarize and highlight the auspicious improvement in CAR-NK cells and will benefit the large-scale preclinical and clinical investigations in adoptive immunotherapy.
Similar content being viewed by others

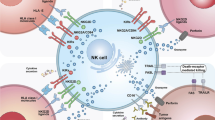
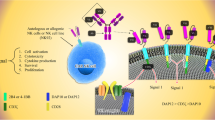
Introduction
Anticancer immunotherapies, including adoptive cytotherapy and checkpoint inhibitors, have present as novel pillars with oncology management [1, 2]. For decades, pioneering investigators have devoted to verify the interactions between the responses of the human immune system and numerous cancers or invaders such as bacteria and viruses, which collectively accelerate the development of clinically effective cancer immunotherapy [3, 4]. However, those reported “immune enhancement” strategies have a series of disadvantages such as rare objective responses and concomitant immune-related adverse events (irAEs), which could be largely alleviated by the termed “immune normalization” (e.g., PD-1/PD-L1) with more beneficial cancer response-to-toxicity profile [4].
Current studies have indicated the diversity of cancer immunotherapies together with the potentially combined strategies in multiple indications [5, 6]. For instance, we and other investigators have reported the successful generation of T lymphocyte-mediated tactics including chimeric antigen receptor-modified T (CAR-T) cells and T cell receptor-engineered T (TCR-T) cells as well as tumor-infiltrating lymphocytes (TILs) and regulatory T (Treg) cells in eliminating malignant hematologic tumors (e.g., B acute lymphoblastic leukemia) and metastatic solid tumors (e.g., HBV-related hepatocellular carcinoma, head and neck squamous cell carcinoma) processed by antigen-presenting cells (APCs) and fine-tuned by co-stimulatory or co-inhibitory signals [6,7,8,9,10,11,12]. However, the adoptive T cell-based immunotherapy severely constrained by the major limitation of the rapidly declined cellular viability and function, together with the requirement of concurrent administration of the adjuvant drugs after transplantation [13]. Moreover, due to the alteration in genetic mutation and cell-surface biomarker expression and the resultant off-target effects, tumor escape has become a common but intractable outcome of malignant transformation and identification of more optimal candidates or personalized neoantigens seems boundless [14,15,16]. Collectively, the autologous T lymphocytes, including the classical T cell receptor-engineered T (TCR-T) cells and CAR-transduced T (CAR-T) cells, are labor-intensive to manufacture and logistically challenging to personalized deliver to inpatients.
In consequence, state-of-the-art renewal has turned to rediscover the immune recognition and eradication of tumor cells by comminating with immune checkpoint blockade (e.g., CTLA4, PD-1/PD-L1), and in particular, to harness the innate immune response with moderate cytotoxicity and reduced adverse effects [17, 18]. Of the indicated innate immune cells such as macrophages (Mø) and dendritic cells (DCs), autologous or allogeneic NK cells are adequate to fulfill the biofunction of combating malignant tumors and pathogenic microorganisms via paracrine effects (e.g., IFN-γ, GM-CSF), antibody-dependent cell-mediated cytotoxicity (ADCC) and direct cytolytic effect dispense with preliminary antigen presentation as well as manipulating other immune contextures to recognize and attack cancer cells [1, 5, 19,20,21]. However, the heterogeneous tumor cells with genetic or epigenetic variations are also sufficient to elude the immunological surveillance and even reversely suppress NK cell cytotoxicity by interdicting the corresponding activating receptors [5, 22, 23]. Considering the deficiency of CAR-T and non-gene-edited NK cells, CAR-NK cells have been recognized as novel therapeutic options aiming at reducing the incidence of relapse and attaining complete remission. Of note, considerable progresses have been achieved in an increasing number of therapeutic dimensions ranging from preclinical studies to clinical practices [24].
Therefore, in this review article, we mainly summarize the key elements and current advances of CAR-NK cell-based immunotherapy including cell sources, novel target selection, design of CAR construction, mode of CAR transduction, and ultimately discuss the opportunities and challenges of adoptive CAR-NK cell-based cancer immunotherapy. Collectively, the CAR-NK cell-mediated cytotherapy has constituted a promising area of cellular immunotherapy innovation.
Cell sources for CAR-NK cells
NK cell lines
Generally, considering the difficulty in isolating, purifying and expanding primary NK cells as well as the inefficiency in transducing CAR constructs, the well-established NK cell lines with indefinite expansion capacity have been used in clinical practice [25]. Notably, the representative IL-2 depend NK-92 cell line and the NK-92MI derivation exhibit splendid advantages of easy expansion, cultivation and activation in the context of lymphodepletion, together with sustainable and reliable cytotoxicity after infusion against leukemia cells (Fig. 1) [24]. For example, Boyiadzis and the colleagues conducted a phase 1 clinical trial of NK92 cell-based adoptive immunotherapy in patients with refractory and relapsed acute myeloid leukemia (AML) and confirmed the feasibility, safety and strong anti-leukemia activity [26]. Simultaneously, it’s noteworthy that NK92 cells are originated from patients with non-Hodgkin’s lymphoma and thus require irradiation prior to infusion to eliminate risks of malignant transformation and the accompanied chromosomal abnormalities [27]. Another preclinical study upon AML immunotherapy by Kloess et al verified that engineered CD123-CAR-NK-92 cells showed higher levels of granzyme and interleukin secretion and preferable cytotoxic activities over the primary human donor-derived CD123-CAR-NK cells, while revealed significant side effects against nonmalignant cells as well [28]. Additionally, Binyamin et al. found that NK-92 cells revealed enhanced ADCC by blocking the inhibitory receptors (e.g., KIR2DL1, KIR3DL1) and combining with rituximab [29].

The schematic diagram of the sources of NK cells
Another classical NK cell line YT with the prostate cancer cell antigen PSMA transduction and shp-2 (PTPN11) deletion has been indicated with enhanced cytotoxicity [30]. Meanwhile, the leukemic cell line YT has also been proved with spontaneous cytotoxicity against B lymphoma and specifical lytic effect upon AML by targeting CD80+/CD86+ B lymphoblastoid cells and CD33+ leukemia cells, respectively [31, 32]. Despite the increasing references of CAR-NK cell-based cancer immunotherapy, yet most of the current studies are preclinical. However, the observations of the existing studies are favor of novel treatment concepts employing CAR-NK cell lines with potent degranulation and selective cytotoxicity in malignancies [33].
Peripheral blood-derived NK (PB-NK) cells
In peripheral blood, NK cells account for a proportion of 5–20% of leukocytes, which are divided into the dominating CD56dimCD16high subset (85–95%) and the minimal CD56brightCD16low/neg subset (5–15%) [5, 34]. Besides, PB-NK cells express a wide range of active receptors and thus hold potential as splendid sources for adoptive CAR-NK cell generation (Fig. 1) [21, 35]. In general, resting PB-NK cells reveal a tremendous proliferative CD3−CD56bright cellular phenotype and are capable of secreting immunomodulatory cytokines, while the CD3−CD56dim counterpart possesses highly cytotoxicity and poor proliferation in response to cytokine stimulation (e.g., IL-2) [36].
To date, autogenous and allogeneic donor-derived PB-NK cells and CAR-PB-NK cells have been most effective in the treatment of acute leukemia (clinically effective doses ranging from 1 × 106/kg to 9.3 × 106/kg) whereas with relatively minimal activity against solid tumors [37,38,39]. Recently, a preclinical study by Quintarelli et al confirmed that CD19-CAR-transduced PB-NK cells were sufficient to mediate robust cytotoxicity against B-cell precursor acute lymphocytic leukemia (Bcp-ALL) and maintain the function of all “native” NK coreceptors after genetic modification [40].
Umbilical cord blood-derived NK (UC-NK) cells
Differ from PB-NK cells, NK cells in umbilical cord blood (UC-NK) only account for a proportion of approximately 5% of total mononuclear cells (TNCs) but offer unique alloreactive advantages for adoptive immunotherapy and boost the potential as a third-party product for extensive clinical scalability (Fig. 1) [36, 41]. In spite of the higher percentage of naïve NK cell population in circulating umbilical cord blood, yet most of the UC-NK cells were adequate to differentiate into functionally mature and active effector cells and thus the successful acquisition of functional competence after ex vivo co-stimulation with cytokine cocktails (e.g., IL-2, IL-7, IL-12, IL-15, IL-18) [36, 42]. For example, Xing et al reported the low cytolytic activity of resident UC-NK cells in vivo attribute to impaired lytic immunological synapse formation as well as the enhancement by IL-2 stimulation during ex vivo expansion and activation [43]. Collectively, the existing literatures indicate that UC-NK cells are phenotypically and functionally immature but are capable of maturation [42].
Of note, an increasing number of investigators have turned to UC-NK cells for generating preclinically or clinically tested CAR-NK cells [44]. For instance, a fist-in-human phase 1/2 clinical trial identified the feasibility of lympho-depleted CAR-UC-NK cells for the treatment of recurrent and refractory CD19+ B-cell lymphoma including seven cases with complete remissions without causing major toxic effects [45]. Despite the once reported generation of over 100 engineered CAR-NK cell doses from one cord blood unit, yet the UC-NK cells have noteworthy limitations in cell mass for large-scale adoptive immunotherapy but with high level of the inhibitory receptor NKG2A expression and poor in vitro cytotoxicity [36, 42, 46].
Placental blood-derived NK (P-NK) cells
In spite of the rare content of P-NK cells (< 2%) in TNCs, yet placental blood is more abundant compared to the aforementioned adult peripheral blood and umbilical cord blood (Fig. 1). Meanwhile, current strategies have indicated the high-efficient generation of clinical-grade CD3−CD56+ NK cells (an average of nearly 1.0 billion NK cells per donor) with remarkably increased antitumor cytolytic activity from placenta perfusate [38]. Compared to UC-NK cells, the derived P-NK cells are largely similar to UC-NK cells phenotypically and functionally, but display distinct microRNA expression profiles, immunophenotypes and superiority in killing a wide range of cancer cell lines in vitro and thus hold potential for CAR-P-NK cell-based immunotherapeutic development [38, 47]. Notably, Guo et al very recently raised the possibility of enhancing cytotoxicity of P-NK cells via CRISPR/Cas9-induced CBLB ablation [48].
Stem cell-derived NK cells
Currently, prospective studies have also indicated the feasibility of deriving mature NK cells from CD34+ hematopoietic stem/progenitor cells (HSPCs). In a preclinical study upon AML xenograft model, Cany et al reported the proof-of-concept safety and efficiency of targeting bone marrow-residing leukemia cells via the CCR6/CCL20 and CXCR3/CXCL10–11 axis in NOD/SCID/IL2Rgnull mice [49].
Human pluripotent stem cells (hPSCs), including human induced pluripotent stem cells (hiPSCs) and human embryonic stem cells (hESCs), possess self-renewal and multi-lineage differentiation potential [50,51,52]. During the past decades, we and other investigators have reported the generation of progenitor cells and functional cells from hPSCs including mesenchymal stem/stromal cells (MSCs), megakaryocytes (MKs) and NK cells [50, 53]. Notably, differ from other cell sources with dominating limitations in NK cell survival and proliferation, hPSC-NK cells can be manufactured from the standardized hPSC population and thus satisfy the clinical demands for large-scale, homogeneous CAR-NK cell products (Fig. 1) [54, 55]. Meanwhile, considering the relatively low efficiency of CAR transduction into primary NK cells, the deficiency of adult PB-NK cells and perinatal UC-NK cells and P-NK cells in cellular activity against solid tumors might be overcome by the genetically modified CAR-hPSC-NK cells via viral or non-viral strategies as hypothesized by pioneering investigators [54, 56].
Moreover, it is considered that CARs can be easily and conveniently delivered into hPSCs by utilizing the non-viral transgenic strategy [57, 58]. For example, Ni and the colleagues reported the integration of chimeric receptor CD4ζ into hPSC-derived NK cells (CD4ζ-hESC-NK cells, CD4ζ-hiPSC-NK cells) with improved efficacy upon human immunodeficiency virus (HIV)/AIDS [57]. Of note, Li et al recently highlighted the feasibility of transducing CAR constructs with conventional NK cell-specific intracellular activating domains into iPSC-NK cells (CAR+ iPSC-NK cells) for the further optimization of tumor-specific recognition and cytotoxicity [59].
Memory-like NK (ML-NK) cells
Cytokine-induced ML-NK cells with the dominant NKG2A checkpoint expression, phenotypically distinct from the in vivo conventional NK cells, have been considered safe and sufficient to induce remissions in patients with AML, which are recognized as new avenues to facilitate CAR-NK cell therapeutics (Fig. 1) [60, 61]. Therefore, we and other investigators presume that CAR-ML-NK cells possess more potent and flexible response to a variety of cancer cell-associated triggers compared to the conventional NK cells [62, 63]. It’s noteworthy that the differentiated memory-like CAR-NK cells displayed elevated activating receptors against myeloid leukemia and prolonged survival in vivo dispense with the typical KIR-KIR ligand interactions [61, 64]. Meanwhile, numerous preclinical data have demonstrated the superior degranulation and IFN-γ-associated response of CAR-NK cells as well as enhanced cancer cell killing and ADCC effect against tumor cells [65, 66].
Notably, peripheral blood-derived ML-NKs with a truncated CD-19-CAR transduction were phenotypically and functionally mature and manifested significantly raised IFN-γ secretion and degranulation, broaden recognition and specific killing against NK-resistant lymphoma compared to the conventional CAR-NK cells or no-specific NK cells [63]. Moreover, Foltz et al reported that the CAR-NK cells could even survive and persist in vivo for over 2 months following adoptive transfer in the immune compatible environment, which would be significant improvement of the short lifespan of conventional NK cells.
Targets for CAR-NK cells
Aiming to generate novel CAR-NK cell-based cancer therapeutics, the consideration of tumor-specific surface antigens and the costimulatory molecules is the first-line decision to be made by investigators [55]. Direct transfer of CAR structures involved in CAR-T (e.g., CD19, CD3ζ, 4-1BB, CD28) into NK cells is the predominantly initial CAR-NK cell-based studies (Fig. 2, Table 1) [111, 112]. For instance, there are a series of activating receptors such as TNF-related apoptosis-inducing ligands (TRAILs), co-stimulatory receptors (e.g., CD244, CD137) and the well-established subsets (e.g., FcgRIIIa, FasL, NKG2D, NKp44, NKp46), which are capable of provoking cytolytic programs via intra-cytoplasmic ITAMs (e.g., 2B4, 41BB) [59, 113, 114]. Significant efforts have been made to enhance CAR-NK cell responses against surface antigens by multiple targeted activation such as CD19, CD20, CD22, CD276, CD138, CS1, HER-2, NKG2D and GD 2 [5, 67, 69, 115, 116].

The overview of the constructs and targets of CAR-NK cells
Nowadays, several groups further indicated the re-designment of CAR-NK cells with NK cell signaling-associated domains to enhance the antitumor efficacy by improving cytotoxicity and INF-γ secretion such as DAP-10, DAP-12, 2B 4 [117]. Nevertheless, due to the rare tumor-specific cell-surface antigens, CAR-NK cells also endure the main disadvantages of requiring extracellular surface expression of specific targets on cancer cells, which thus restrict the broadness and specificity of CAR-NK cell application [4, 118]. To overcome the short lifespan and transient cytotoxic activity of CAR-NK cells, Zhang et al recently transduced the homodimers and heterodimers of ErbB2/HER2-specific CARs with CD3ζ and composite CD28ζ signaling domains into NK-92/63.z cells to induce long-lasting endogenous cytotoxicity against immunocompetent glioblastoma (Fig. 2, Table 1) [87].
Construction of CARs
Generally, the CARs are engineered receptor proteins to enable NK cells with novel ability to target cancer cell-specific antigen proteins, which are composed of an intracellular activating signaling domain, a transmembrane region and an extracellular antigen binding domain (Fig. 2, Table 1) [55]. The intracellular activating signaling domains (e.g., CD137, CD28) mainly mediate the activation and cytotoxicity of CAR-NK cells, while the extracellular antigen binding domains (e.g., the single-chain variable fragments) recognize the specific antigen of tumor cells [21]. For example, Quintarelli and the colleagues reported the successful design of retroviral plasmid carrying the cassettes of a second-generation CD19-CAR construct and transduced into PB-NK cells for Bcp-ALL management [40]. Simultaneously, another preclinical study reported the splendid efficiency of CAR-NK-92 and CAR-NK-92MI cells with the second- or third-generation CARs targeting CD3ζ and CD5 domains against mouse model of T-cell malignancies, respectively [69, 119]. Very recently, Daher et al took advantage of the fourth-generation “armored” CARs for generating CAR-UC-NK cells by targeting the cytokine-inducible SH2-containing (CIS) protein, which efficiently boosted NK cell antitumor activity against lymphoma xenografts and resulted in enhanced aerobic glycolysis [41, 120].
Therewith, due to the potentially excessive cytokine secretion of CAR-NKs, there is a possibility that the fourth-generation of CARs might cause unanticipated toxicity and should reinsert suicide genes (Fig. 2, Table 1) [121]. Collectively, the genetically engineered CAR-NK cells contain a typically extracellular antigen-binding domain, a hinge and transmembrane region and the concomitant intracellular costimulatory domains from receptors, which represents the paradigmatic design of utilizing engineered NK cells for effective attack of cancer cells [4, 122]. Of note, the choice and design of the NK cell activating receptor (e.g., NKG2D, DAP10) and remaining domains of the CAR construct, including the aforementioned transmembrane domains (e.g., CD28), co-stimulatory domains and signaling domains (e.g., CD3ζ), together with the NK cell subtypes, should be taken into incorporated consideration [55].
CAR transduction
CAR-transduced cytotherapy was initially advocated using autologous T lymphocytes (CAR-T) and obtained great success in treating hematological malignancies whereas marginal success in facing solid tumors largely attributes to the highly immunosuppressive cancer microenvironment [9, 122, 123]. Advances in genome editing technique and the applicability of the approach have vastly accelerated the development of designer CAR-NK cell therapy products, which are currently being tested in both preclinical studies and clinical trials [55].
Retrovirus
For decades, a plethora of groups have reported the successful transduction of CAR constructs into expanded NK cells by utilizing a single round of retroviral vector-based method ranging from 27 to 52%, and in particular, an even higher efficient transduction (approximately 70%) has been achieved by Imamura and the colleagues for IL-15 expression [124, 125]. Strikingly, Daher and the colleagues compared the retrovirus transfection of iC9.CAR19.CD28-z-2A-IL-15 (with IL-15) with CAR19.CD28-z (without IL-15) into UC-NK cells, and confirmed the feasibility of high-efficient CAR-NK cell generation and the persistence of in vivo antitumor activity in xenogeneic lymphoma models [41]. Furthermore, Suerth et al systematically compared different retroviral pseudotypes, retroviral vector systems and transduction protocols, and verified that the highest (up to 60%) transduction levels of CAR-19 expression cassette were achieved with α-retroviral plasmids into primary human NK cells [126]. Simultaneously, the retrovirus-based gene-delivery vehicles also have main obstacles to widespread clinical applications including the immunogenicity and insertional mutagenesis as well [58].
Lentivirus
As with retrovirus, lentiviral also have been widely used by preclinical studies for CAR vector transduction into NK cells such as UC-NK cells, PB-NK cells, iPSC-NK cells and ML-NK cells. For example, talented investigators utilized the scFvs-based CD33-CAR and CD19-CAR lacking the immunoreceptor tyrosine-based activation motif (ITAM) domains to manufacture the second-generation CARs with CD3ζ and CD137 intracellular domains [63, 127, 128]. Simultaneously, Oelsner and other investigators verified that gene-modified CAR-NK-92 cells after lentiviral-mediated gene transfer displayed stable and homogeneous CD-19-and CD20-specific CAR expression, high and specific ADCC against lymphoma and leukemia cells [5, 33, 129].
Nonviral-mediated transfection
To overcome the major barriers of low-efficient exogenous gene transfer into the primary NK cells, a certain number of research groups turn to the nonviral-mediated transfection such as lipofection and electroporation [58, 130]. Of them, the nonviral plasmids are emerging as promising alternatives and facilitate clinical CAR-NK cell-based cancer immunotherapy [58]. Compared to the aforementioned virus-mediated strategies, the nonviral methods revealed transient and rapid expression of CAR-conjunct genes as well as less variability and apoptosis, whereas the generated CAR-NK cells showed declined expression level of transgenes due to the non-integrated property [58]. Of note, the current progresses of transfection technologies, including the DNA transposon systems composed of the PiggyBac (PB) and the sleeping beauty (SB) subsets, are sufficient to deliver CAR structures into the genomes of primary NK cells or iPSCs with long-lasting transgene expression [131].
Collectively, the transposon systems for high-efficient CAR-NK cell generation have splendid advantages such as increased biosafety, rapid and persistent transgene expression, low immunogenicity, cost-effect and capacity for large gene fragment (> 100 kb) transduction, which make them attractive options for CAR-NK cell manufacturing [130]. Nevertheless, considering the dissatisfactory efficiency in current transduction and the potential advances in CAR construct design, the next-generation of CAR-NK cells might cause fewer cytotoxicity and even ultimately eliminate the demands for suicide genes or safety switches [121].
CAR-NKs in cancer immunotherapy
Nowadays, CAR-NK cell-mediated immunotherapy has grown exponentially and emerged as an alternative treatment option for patients with metastatic malignancies (Fig. 3, Table 2). Despite the manifestation of CAR-NK cells in cancer immunotherapy has been extensively explored, yet most of the applications in numerous tumor models are relatively restricted and mainly staying at the preclinical stage [5, 24]. Clinically approved second-generation CAR-NK cells usually contain the CD3ζ domain in combination with either a 4-1BB (Kymriah®) or CD28 (Yescarta®) co-stimulatory domain, and prominently focus on CD19+ lymphoid-derived hematologic malignancies [55].

The distribution of CAR-NK cell-based clinical trials worldwide
Hematological malignancies
Similar to CAR-T-based immunotherapy, CAR-NK cells were initially introduced to combat with hematological malignancies such as lymphoma, myeloma and leukemia (Fig. 4, Table 2) [121, 122]. Current studies have indicated the preferable efficiency of CD19-CAR-NK cells in combating lymphoid malignancies over CAR-T-based cellular immunotherapy, which largely attribute to the aforementioned merits [55, 132]. For example, a preclinical study took advantage of the UC-NK cells and the fourth-generation iCas9.CAR-19-CD28-ζ-IL15 plasmid with a suicide gene for Raji lymphoma xenograft model treatment, the CD19-CAR-UC-NK cells exhibited preferable antitumor efficacy and enhanced persistence over the non-transduced UC-NK cells [46].

The dissection of CAR-NK cell-based clinical trials A-B. The status (A) and phase (B) of CAR-NK cell-based clinical trials. C. The detailed distribution of CAR-NK cell-based clinical trials in cancer immunotherapy
Simultaneously, the first large-scale trial of HLA-mismatched CAR-UC-NK cell-based immunotherapy (ClinicalTrials.gov number: NCT03056339) in combination with lymphodepleting chemotherapy for 11 patients with CD19+ CLL and B cell lymphoid tumors has shown safety and inspiring clinical outcomes (response rate: 73%; complete remission rate: 64%) within 30 days [45]. However, as commented by Karadimitris, CAR-UC-NK cells could also result in high rate of non-durable clinical response, which suggested the favorable initial toxicity and efficacy but uncertain durability of clinical manifestation of CAR-NK-based leukemia immunotherapy [133].
Metastatic solid tumors
In recent years, CAR-T cell immunotherapy has revealed promising therapeutic manifestation in a series of hematologic malignancies, yet also with considerable drawbacks and limitations in metastatic solid tumor management as well (Fig. 4, Table 2) [55, 134]. Considering the intrinsic and advantaged properties, including substantially cytolytic ability, non-MHC-restricted recognition, natural infiltration in tumor tissues and convenience for their preparation as well as minimal untoward effects (e.g., CRS, GvHD and neurotoxicity), engineered CAR-NK cells are supposed as promising therapeutic option for solid tumor administration in clinical practice [54, 134, 135]. Currently, CAR-NK cells have been preclinically tested in multiple solid tumors including breast cancer, ovarian cancer, pancreatic cancer, colon cancer, glioblastoma, hepatocellular carcinoma, head and neck squamous cell carcinoma (HNSCC) [90, 117, 136, 137]. For example, numerous preclinical studies have confirmed the efficacy of CAR-NK cells upon CXCL12/SDF-1α secreting glioblastoma and epithelial cell adhesion molecule (EpCAM) positive colorectal cancer cells in xenograft model by targeting EGFRvIII via intravenous infusion [80, 138].
However, there are very limited clinical data existing on the potential of CAR-NK cells in solid tumor treatment (Fig. 4, Table 2) [117]. For instance, the outcomes of three phase I/II clinical studies of allogeneic ROBO-1-CAR-NK-92 cell-based cellular immunotherapy in pancreatic ductal adenocarcinoma (PDAC) and relative solid tumors with ROBO-1 expression for enrolled patients in China (NCT03940820, NCT03941457, NCT03931720) ulteriorly indicated the feasibility for non-hematological neoplasm treatment with CAR-NK cells [117, 135, 139]. Another phase II trial of PD-L1-CAR-NK cell-based immunotherapy combined with IL-15 agonist (N-803) and pembrolizumab (NCT04847466) is currently launched in the United States. Therefore, integration of the CAR-NK cells and antitumor drugs is promising for further altering immunosuppressive tumor microenvironment (TME) and administrating the aggressiveness and the metastatic ability of the resistant tumors [117].
Conclusion and perspective
The heterogeneous NK cell population is advantaged immune cells with powerful cytotoxic activity and plays a unique role in both innate and adoptive immune responses, while the signatures could be eluded by tumor microenvironment [118, 140,141,142]. NK cells engineered with CAR expression (CAR-NK cells) have been celebrated as a landmark breakthrough of anti-tumor immunotherapy by discerning germline-encoded cell surface receptors between healthy and cancer tissues [143, 144]. Differ from NK cells and CAR-T cells, the genetically modified NK (CAR-NK) cells have superiority in further augmenting the specificity and cytotoxicity of adoptive NK cells without causing adverse effects including GvHD, CRS or immune cell- associated neurotoxicity syndrome (ICANS) [5, 60, 143, 145]. Thus, CAR-NK cells with favorable cytotoxicity, short lifespan and low manufacturing costs have been considered as promising alternatives to engineered CAR-T cells [143].
Despite the encouraging progress of CAR-NK cell-based immunotherapy, the discontented response-to-toxicity ratio and the insufficiently broad spectrum of indications further limit the application of these therapies [4]. Moreover, compared to the updates of CAR-T-based immunotherapy, the number of clinical trials and the detailed information of CAR-NK cell infusion in patients, including the in vivo spatio-temporal metabolism and host factors in the microenvironment that reversely contribute to CAR-NK cells, have not been extensively investigated [60]. Generally, compared to CAR-T cells, CAR-NK cell-based adoptive immunotherapy is also restricted to the major limitations before large-scale practical application such as insufficient capacity in proliferation and activation in vivo and durability, together with the obstacle in preparation of CAR-NK cells (e.g., low genetic transfection efficiency, low proportion of NK cells in blood, and limited amplification efficiency) [133, 146]. Conversely, CAR-NK cells have various merits over CAR-T cells due to their unique biological characteristics [117]. On the one hand, CAR-NK cells can be conveniently prepared from a wider range of autogenous and allogeneic sources without causing severe adverse reaction (e.g., aGVHD, CRS) by CAR-T-based implantation. On the other hand, CAR-NK cells are adequate for cell immunosurveillance dispense with pre-sensitization and thus have more flexible killing capacity upon both hematologic and solid tumor cells over CAR-T cells via both the CAR-dependent manner and CAR-independent intrinsic mechanisms, which thus provide alternative strategies for conquering tumor escape and varied adverse events (e.g., “on-target, off-tumor toxicity” during CAR-T application) [5, 112, 117, 147].
Nevertheless, the success of CAR-NK cells in the administration of multiple malignancies provides proof-of-principle for harnessing the immune system therapeutically, yet the short lifespan (2 weeks) and in vivo kinetics (e.g., proliferation rate, ageing) of CAR-NKs also narrows the therapeutic window and the resultant short duration of responses after infusion [122, 148, 149]. Therefore, the ex vivo expansion and activation of primary NK cells as well as the storage and shipping of NK cells for large-scale CAR-NK cell generation are prerequisites for ensuring the safety and effectiveness in vivo. For instance, a series of methodologies have been continuously developed for the persistence and activation of primary NK cells such as feeder cell stimulation (e.g., the irradiated PBMCs, K562-mb15-41BBL cells, EBV-LCLs) [150,151,152,153], cytokine cocktail (e.g., IL-2, IL15, 1 L-18) [14, 35] and physicochemical irritation (e.g., bioreactors with an assorted bag) [154]. Of note, it is great important to improve the freezing and shipping conditions (e.g., refrigeration, liquid nitrogen, drikold) for sustaining primary NK cell vitality, which will finally help CAR-NK cells necessitate adaptation of cancer immunotherapy under GMP conditions [5, 155].
Therefore, before large-scale application in cancer immunotherapy, a cohort of central issues in basic research and clinical practice of CAR-NK cells need to be improved. Firstly, many healthy tissues of the body also express cancer-associated surface antigens on tumor cells, which might cause potential off tumor or on target toxicity of CAR-NK cells. Current pioneering studies have suggested the engineering solution by inserting suicide genes or transducing genes encoding cytokines into the CAR-transduced effectors to prolong the in vivo persistence, respectively [45, 118, 156]. Secondly, the lessons learned from modified T cells (e.g., CAR-T, TCR-T) and NK cells in the administration of malignancies are worthy to be further exploited in CAR-NK cell-mediated cancer immunotherapy in future such as the optimization of large-scale NK cell-expansion approaches, antigen targets including the chimeric co-stimulatory converting receptors (CCCRs), structures with nanobodies and delivery efficiency as well as the selection of ideal patient populations in clinical trials [91, 111, 117]. Likewise, the potential tumor escape from CAR-NK cell cytotoxicity by shedding pivotal ligands and even via immunosuppressive tumor microenvironment should also take into consideration [14, 140, 157]. In particular, the pivotal signaling pathways and inhibitory checkpoints (e.g., CIS) that orchestrate CAR-NK cell “fitness” (e.g., effector function, survival and differentiation) and biofunction in tumor microenvironments have not been well defined [41, 120, 158]. Thirdly, despite CAR-NK cells are considered as “off-the-shelf” cancer immunotherapy, there’s still a long way before the large-scale generation of clinical-grade products under good manufacturing practice (GMP) and convenient to universally save the lives of inpatients with standard supervision. For example, Gaddy and Broxmeyer reported the functional maturation subset (adult-like NK activity) and the phenotypic maturation subset (adult-like CD3−CD56+CD16+ or CD3−CD56+CD16− phenotype) of UC-NK cells by IL-2, IL-12 or IL-15 stimulation [42]. As to the recommended parameters of cell products, the manufactured CAR-NK cells should contain mostly CD3−CD56+ cells (≥90%), minimally CD3+ (≤0.2%) and CD14+ (≤5%) cells, together with the removal of endotoxin, mycoplasma and bacterial contaminations as well as contamination of co-cultured cells (≤1%) [159, 160]. Fourthly, the schedule of CAR-NK cell-based cancer immunotherapy, including the dosage, duration, kinetics and the concurrent interactions with endogenous immune cells as well as the underlying mechanisms of CAR-NK cell function, is awaiting to be optimized according to large-scale clinical trials [5, 161]. For instance, the molecular understanding of the biofunction of the natural cytotoxicity receptors (NCRs) (e.g., NKp30, NKp44, NKp46) and multidimensional immune correlations (e.g., NKG2A+, CD8α+) in cancer immunosurveillance, and in particular, the ontogenic development and maturational signals of NK cells is instrumental to exploring novel access points to combat malignancies [42, 60, 144, 162].
Moreover, the comprehensive treatment composed of a plethora of methods, including traditional therapy (e.g., surgery, radiotherapy, chemotherapy), non-cellular immunotherapy (e.g., CTLA-4, mRNA vaccine) and cellular immunotherapy (e.g., CAR-T, TCR-T, γδT, ML-NK cells) as well as auxiliary methods (TriKEs, ROCK engagers, TriNKETs) and immune-checkpoint inhibitors (e.g., CTLA-4, PD-1/PD-L1), are under clinical investigation to augment the longevity and cytotoxicity of CAR-NK cells [146, 163]. For example, a latest study upon combined approach suggested that antitumor activity and metabolic fitness of armored CAR-NK cells with IL-15 secretion could be enhanced by targeting a cytokine checkpoint, which represented an important milestone in the exploration of the next-generation cancer immunotherapy [41, 117, 164, 165]. Another study conducted combined CAR-T infusion after NKG2D.ζ-NK cell administration and found improved anti-cancer activity and tumor infiltration [55]. Collectively, the pioneering preclinical and clinical studies have suggested the multifaceted opportunities and challenges of allogeneic CAR-NK cells, which are recognized as pivotal and promising “off-the-shelf” product in the next-generation cellular immunotherapies targeting recurrent and refractory malignancies.
Availability of data and materials
All data are included in this published article. Meanwhile, the datasets involved in the current study are available from the corresponding author on reasonable request.
Abbreviations
- CAR-NK:
-
Chimeric antigen receptor-transduced NK
- CRS:
-
Cytokine release syndrome
- GVHD:
-
Graft-versus-host disease
- irAEs:
-
Immune-related adverse events
- CAR-T:
-
Chimeric antigen receptor-modified T
- TCR-T:
-
T cell receptor-engineered T
- TILs:
-
Tumor-infiltrating lymphocytes
- APCs:
-
Antigen-presenting cells
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- AML:
-
Acute myeloid leukemia
- LSCs:
-
Leukemia stem cells
- PB-NK:
-
Peripheral blood-derived NK
- Bcp-ALL:
-
B-cell precursor acute lymphocytic leukemia
- UC-NK:
-
Umbilical cord blood-derived NK
- P-NK:
-
Placental blood-derived NK
- hPSCs:
-
Human pluripotent stem cells
- hiPSCs:
-
Human induced pluripotent stem cells
- hESCs:
-
Human embryonic stem cells
- MSCs:
-
Mesenchymal stem/stromal cells
- EpCAM:
-
Epithelial cell adhesion molecule
- ICANS:
-
Immune cell- associated neurotoxicity syndrome
- GMP:
-
Good manufacturing practice
- NCRs:
-
Natural cytotoxicity receptors
References
Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70(2):86–104.
O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–67.
Abbott M, Ustoyev Y. Cancer and the immune system: the history and background of immunotherapy. Semin Oncol Nurs. 2019;35(5):150923.
Sanmamed MF, Chen L. A paradigm shift in Cancer immunotherapy: from enhancement to normalization. Cell. 2018;175(2):313–26.
Zhang L, Liu M, Yang S, Wang J, Feng X, Han Z. Natural killer cells: of-the-shelf cytotherapy for cancer immunosurveillance. Am J Cancer Res. 2021;11(4):1770–91.
Pan J, Tan Y, Deng B, Tong C, Hua L, Ling Z, et al. Frequent occurrence of CD19-negative relapse after CD19 CAR T and consolidation therapy in 14 TP53-mutated r/r B-ALL children. Leukemia. 2020;34(12):3382–7.
Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z, et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia. 2019;33(12):2854–66.
Pan J, Tan Y, Wang G, Deng B, Ling Z, Song W, et al. Donor-derived CD7 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia: first-in-human, Phase I trial. J Clin Oncol. 2021;39(30):3340–51. https://doi.org/10.1200/JCO.21.00389.
Zhao L, Cao YJ. Engineered T cell therapy for Cancer in the clinic. Front Immunol. 2019;10:2250.
Liu Q, Tian Y, Li Y, Zhang W, Cai W, Liu Y, et al. In vivo therapeutic effects of affinity-improved-TCR engineered T-cells on HBV-related hepatocellular carcinoma. J Immunother Cancer. 2020;8(2):e001748. https://doi.org/10.1136/jitc-2020-001748.
Wei T, Leisegang M, Xia M, Kiyotani K, Li N, Zeng C, et al. Generation of neoantigen-specific T cells for adoptive cell transfer for treating head and neck squamous cell carcinoma. Oncoimmunology. 2021;10(1):1929726.
Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109–18.
Wallace A, Kapoor V, Sun J, Mrass P, Weninger W, Heitjan DF, et al. Transforming growth factor-beta receptor blockade augments the effectiveness of adoptive T-cell therapy of established solid cancers. Clin Cancer Res. 2008;14(12):3966–74.
Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17(9):1025–36.
Steven A, Fisher SA, Robinson BW. Immunotherapy for lung cancer. Respirology. 2016;21(5):821–33.
Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. 2019;19(10):568–86.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125(9):3335–7.
Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39(1):11–26.
Vivier E. What is natural in natural killer cells? Immunol Lett. 2006;107(1):1–7.
Xia J, Minamino S, Kuwabara K. CAR-expressing NK cells for cancer therapy: a new hope. Biosci Trends. 2020;14(5):354–9.
Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature. 2007;447(7143):482–6.
Urosevic M, Dummer R. Human leukocyte antigen-G and cancer immunoediting. Cancer Res. 2008;68(3):627–30.
Xu J, Niu T. Natural killer cell-based immunotherapy for acute myeloid leukemia. J Hematol Oncol. 2020;13(1):167.
Zeng J, Tang SY, Toh LL, Wang S. Generation of “off-the-shelf” natural killer cells from peripheral blood cell-derived induced pluripotent stem cells. Stem Cell Reports. 2017;9(6):1796–812.
Boyiadzis M, Agha M, Redner RL, Sehgal A, Im A, Hou JZ, et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy. 2017;19(10):1225–32.
Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. 2017;25(8):1769–81.
Kloess S, Oberschmidt O, Dahlke J, Vu XK, Neudoerfl C, Kloos A, et al. Preclinical assessment of suitable natural killer cell sources for chimeric antigen receptor natural killer-based “off-the-shelf” acute myeloid leukemia immunotherapies. Hum Gene Ther. 2019;30(4):381–401.
Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol. 2008;180(9):6392–401.
Subrakova VG, Kulemzin SV, Belovezhets TN, Chikaev AN, Chikaev NA, Koval OA, et al. shp-2 gene knockout upregulates CAR-driven cytotoxicity of YT NK cells. Vavilovskii Zhurnal Genet Selektsii. 2020;24(1):80–6.
Teng JM, Liu XR, Mills GB, Dupont B. CD28-mediated cytotoxicity by the human leukemic NK cell line YT involves tyrosine phosphorylation, activation of phosphatidylinositol 3-kinase, and protein kinase C. J Immunol. 1996;156(9):3222–32.
Schirrmann T, Pecher G. Specific targeting of CD33(+) leukemia cells by a natural killer cell line modified with a chimeric receptor. Leuk Res. 2005;29(3):301–6.
Oelsner S, Friede ME, Zhang C, Wagner J, Badura S, Bader P, et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy. 2017;19(2):235–49.
Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: a promising cellular immunotherapy for cancer. EBioMedicine. 2020;59:102975.
Liu M, Meng Y, Zhang L, Han Z, Feng X. High-efficient generation of natural killer cells from peripheral blood with preferable cell vitality and enhanced cytotoxicity by combination of IL-2, IL-15 and IL-18. Biochem Biophys Res Commun. 2021;534:149–56.
Mehta RS, Shpall EJ, Rezvani K. Cord blood as a source of natural killer cells. Front Med (Lausanne). 2015;2:93.
Handgretinger R, Lang P, Andre MC. Exploitation of natural killer cells for the treatment of acute leukemia. Blood. 2016;127(26):3341–9.
Kang L, Voskinarian-Berse V, Law E, Reddin T, Bhatia M, Hariri A, et al. Characterization and ex vivo expansion of human placenta-derived natural killer cells for cancer immunotherapy. Front Immunol. 2013;4:101.
Meyer-Monard S, Passweg J, Siegler U, Kalberer C, Koehl U, Rovo A, et al. Clinical-grade purification of natural killer cells in haploidentical hematopoietic stem cell transplantation. Transfusion. 2009;49(2):362–71.
Quintarelli C, Sivori S, Caruso S, Carlomagno S, Falco M, Boffa I, et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B-cell precursor acute lymphoblastic leukemia. Leukemia. 2020;34(4):1102–15.
Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137(5):624–36.
Gaddy J, Broxmeyer HE. Cord blood CD16+56- cells with low lytic activity are possible precursors of mature natural killer cells. Cell Immunol. 1997;180(2):132–42.
Xing D, Ramsay AG, Gribben JG, Decker WK, Burks JK, Munsell M, et al. Cord blood natural killer cells exhibit impaired lytic immunological synapse formation that is reversed with IL-2 exvivo expansion. J Immunother. 2010;33(7):684–96.
Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, Maas F, et al. Successful transfer of umbilical cord blood CD34(+) hematopoietic stem and progenitor-derived NK cells in older acute myeloid leukemia patients. Clin Cancer Res. 2017;23(15):4107–18.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53.
Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–31.
Huntington ND, Vosshenrich CA, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol. 2007;7(9):703–14.
Guo X, Mahlakoiv T, Ye Q, Somanchi S, He S, Rana H, et al. CBLB ablation with CRISPR/Cas9 enhances cytotoxicity of human placental stem cell-derived NK cells for cancer immunotherapy. J Immunother Cancer. 2021;9(3):e001975. https://doi.org/10.1136/jitc-2020-001975.
Cany J, van der Waart AB, Tordoir M, Franssen GM, Hangalapura BN, de Vries J, et al. Natural killer cells generated from cord blood hematopoietic progenitor cells efficiently target bone marrow-residing human leukemia cells in NOD/SCID/IL2Rg(null) mice. Plos One. 2013;8(6):e64384.
Zhang L, Wei Y, Chi Y, Liu D, Yang S, Han Z, et al. Two-step generation of mesenchymal stem/stromal cells from human pluripotent stem cells with reinforced efficacy upon osteoarthritis rabbits by HA hydrogel. Cell Biosci. 2021;11(1):6.
Zhang L, Wang H, Liu C, Wu Q, Su P, Wu D, et al. MSX2 initiates and accelerates mesenchymal stem/stromal cell specification of hPSCs by regulating TWIST1 and PRAME. Stem Cell Reports. 2018;11(2):497–513.
Wu Q, Zhang L, Su P, Lei X, Liu X, Wang H, et al. MSX2 mediates entry of human pluripotent stem cells into mesendoderm by simultaneously suppressing SOX2 and activating NODAL signaling. Cell Res. 2015;25(12):1314–32.
Zhang L, Liu C, Wang H, Wu D, Su P, Wang M, et al. Thrombopoietin knock-in augments platelet generation from human embryonic stem cells. Stem Cell Res Ther. 2018;9(1):194.
Hermanson DL, Kaufman DS. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front Immunol. 2015;6:195.
Morgan MA, Buning H, Sauer M, Schambach A. Use of cell and genome modification technologies to generate improved “off-the-shelf” CAR T and CAR NK cells. Front Immunol. 2020;11:1965.
Childs RW, Carlsten M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat Rev Drug Discov. 2015;14(7):487–98.
Ni Z, Knorr DA, Bendzick L, Allred J, Kaufman DS. Expression of chimeric receptor CD4zeta by natural killer cells derived from human pluripotent stem cells improves in vitro activity but does not enhance suppression of HIV infection in vivo. Stem Cells. 2014;32(4):1021–31.
Vargas JE, Chicaybam L, Stein RT, Tanuri A, Delgado-Canedo A, Bonamino MH. Retroviral vectors and transposons for stable gene therapy: advances, current challenges and perspectives. J Transl Med. 2016;14(1):288.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23(2):181–192 e185.
Berrien-Elliott MM, Cashen AF, Cubitt CC, Neal CC, Wong P, Wagner JA, et al. Multidimensional analyses of donor memory-like NK cells reveal new associations with response after adoptive immunotherapy for leukemia. Cancer Discov. 2020;10(12):1854–71.
Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8(357):357ra123.
Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, et al. Cytokine activation induces human memory-like NK cells. Blood. 2012;120(24):4751–60.
Gang M, Marin ND, Wong P, Neal CC, Marsala L, Foster M, et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood. 2020;136(20):2308–18.
Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A. 2009;106(6):1915–9.
Sarhan D, Cichocki F, Zhang B, Yingst A, Spellman SR, Cooley S, et al. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. 2016;76(19):5696–706.
Sarhan D, Hippen KL, Lemire A, Hying S, Luo X, Lenvik T, et al. Adaptive NK cells resist regulatory T-cell suppression driven by IL37. Cancer Immunol Res. 2018;6(7):766–75.
Romanski A, Uherek C, Bug G, Seifried E, Klingemann H, Wels WS, et al. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J Cell Mol Med. 2016;20(7):1287–94.
You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, et al. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. 2019;9(1):64–78.
Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019;12(1):49.
Oelsner S, Waldmann A, Billmeier A, Roder J, Lindner A, Ullrich E, et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int J Cancer. 2019;145(7):1935–45.
Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8(6):1083–9.
Kloss S, Oberschmidt O, Morgan M, Dahlke J, Arseniev L, Huppert V, et al. Optimization of human NK cell manufacturing: fully automated separation, improved ex vivo expansion using IL-21 with autologous feeder cells, and generation of anti-CD123-CAR-expressing effector cells. Hum Gene Ther. 2017;28(10):897–913.
Pinz KG, Yakaboski E, Jares A, Liu H, Firor AE, Chen KH, et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget. 2017;8(68):112783–96.
Schonfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23(2):330–8.
Sahm C, Schonfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. 2012;61(9):1451–61.
Hu Z. Tissue factor as a new target for CAR-NK cell immunotherapy of triple-negative breast cancer. Sci Rep. 2020;10(1):2815.
Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget. 2016;7(19):27764–77.
Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73(6):1777–86.
Boissel L, Betancur M, Wels WS, Tuncer H, Klingemann H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res. 2009;33(9):1255–9.
Zhang Q, Zhang H, Ding J, Liu H, Li H, Li H, et al. Combination therapy with EpCAM-CAR-NK-92 cells and Regorafenib against human colorectal Cancer models. J Immunol Res. 2018;2018:4263520.
Shiozawa M, Chang CH, Huang YC, Chen YC, Chi MS, Hao HC, et al. Pharmacologically upregulated carcinoembryonic antigen-expression enhances the cytolytic activity of genetically-modified chimeric antigen receptor NK-92MI against colorectal cancer cells. BMC Immunol. 2018;19(1):27.
Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal Cancer patients. Mol Ther. 2019;27(6):1114–25.
Kailayangiri S, Altvater B, Spurny C, Jamitzky S, Schelhaas S, Jacobs AH, et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology. 2017;6(1):e1250050.
Wu X, Huang S. HER2-specific chimeric antigen receptor-engineered natural killer cells combined with apatinib for the treatment of gastric cancer. Bull Cancer. 2019;106(11):946–58.
Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. 2015;5:11483.
Wang J, Lupo KB, Chambers AM, Matosevic S. Purinergic targeting enhances immunotherapy of CD73(+) solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J Immunother Cancer. 2018;6(1):136.
Zhang C, Burger MC, Jennewein L, Genssler S, Schonfeld K, Zeiner P, et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J Natl Cancer Inst. 2016;108(5). https://doi.org/10.1093/jnci/djv375.
Qu Y, Bi JZ. Killing effect of Robo1 targeted chimeric antigen receptor modified NK92 cells against glioma and neuroblastoma cells. Zhonghua Yi Xue Za Zhi. 2018;98(11):860–6.
Huang Y, Zeng J, Liu T, Xu Q, Song X, Zeng J. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of anti-GPC3 chimeric antigen receptor-modified natural killer cells against hepatocellular Cancer cells in vitro. Cancer Manag Res. 2020;12:3247–55.
Liu B, Liu ZZ, Zhou ML, Lin JW, Chen XM, Li Z, et al. Development of cMETspecific chimeric antigen receptorengineered natural killer cells with cytotoxic effects on human liver cancer HepG2 cells. Mol Med Rep. 2019;20(3):2823–31.
Lu C, Guo C, Chen H, Zhang H, Zhi L, Lv T, et al. A novel chimeric PD1-NKG2D-41BB receptor enhances antitumor activity of NK92 cells against human lung cancer H1299 cells by triggering pyroptosis. Mol Immunol. 2020;122:200–6.
Yang S, Cao B, Zhou G, Zhu L, Wang L, Zhang L, et al. Targeting B7-H3 immune checkpoint with chimeric antigen receptor-engineered natural killer cells exhibits potent cytotoxicity against non-small cell lung Cancer. Front Pharmacol. 2020;11:1089.
Zhang G, Liu R, Zhu X, Wang L, Ma J, Han H, et al. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol Cell Biol. 2013;91(10):615–24.
Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8(2):297–310.
Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–27.
Ng YY, Du Z, Zhang X, Chng WJ, Wang S. CXCR4 and anti-BCMA CAR co-modified natural killer cells suppress multiple myeloma progression in a xenograft mouse model. Cancer Gene Ther. 2021. https://doi.org/10.1038/s41417-021-00365-x.
Esser R, Muller T, Stefes D, Kloess S, Seidel D, Gillies SD, et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. 2012;16(3):569–81.
Elahi R, Heidary AH, Hadiloo K, Esmaeilzadeh A. Chimeric antigen receptor-engineered natural killer (CAR NK) cells in Cancer treatment; recent advances and future prospects. Stem Cell Rev Rep. 2021;17(6):2081–106.
Ao X, Yang Y, Li W, Tan Y, Guo W, Ao L, et al. Anti-alphaFR CAR-engineered NK-92 cells display potent cytotoxicity against alphaFR-positive ovarian Cancer. J Immunother. 2019;42(8):284–96.
Kruschinski A, Moosmann A, Poschke I, Norell H, Chmielewski M, Seliger B, et al. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc Natl Acad Sci U S A. 2008;105(45):17481–6.
Cao B, Liu M, Wang L, Liang B, Feng Y, Chen X, et al. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem Biophys Res Commun. 2020;524(1):96–102.
Ueda T, Kumagai A, Iriguchi S, Yasui Y, Miyasaka T, Nakagoshi K, et al. Non-clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti-glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020;111(5):1478–90.
Ng YY, Tay JCK, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Ther Oncolytics. 2020;16:75–85.
Batchu RB, Gruzdyn OV, Tavva PS, Kolli BK, Dachepalli R, Weaver DW, et al. Engraftment of mesothelin chimeric antigen receptor using a hybrid sleeping beauty/minicircle vector into NK-92MI cells for treatment of pancreatic cancer. Surgery. 2019;166(4):503–8.
Li C, Yang N, Li H, Wang Z. Robo1-specific chimeric antigen receptor natural killer cell therapy for pancreatic ductal adenocarcinoma with liver metastasis. J Cancer Res Ther. 2020;16(2):393–6.
Montagner IM, Penna A, Fracasso G, Carpanese D, Dalla Pieta A, Barbieri V, et al. Anti-PSMA CAR-engineered NK-92 cells: an off-the-shelf cell therapy for prostate cancer. Cells. 2020;9(6):1382. https://doi.org/10.3390/cells9061382.
Zhang Q, Tian K, Xu J, Zhang H, Li L, Fu Q, et al. Synergistic effects of Cabozantinib and EGFR-specific CAR-NK-92 cells in renal cell carcinoma. J Immunol Res. 2017;2017:6915912.
Topfer K, Cartellieri M, Michen S, Wiedemuth R, Muller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. 2015;194(7):3201–12.
Jan CI, Huang SW, Canoll P, Bruce JN, Lin YC, Pan CM, et al. Targeting human leukocyte antigen G with chimeric antigen receptors of natural killer cells convert immunosuppression to ablate solid tumors. J Immunother Cancer. 2021;9(10):e003050. https://doi.org/10.1136/jitc-2021-003050.
Muller T, Uherek C, Maki G, Chow KU, Schimpf A, Klingemann HG, et al. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother. 2008;57(3):411–23.
Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, et al. CAR-T design: elements and their synergistic function. EBioMedicine. 2020;58:102931.
Zhang Y, Li Y, Cao W, Wang F, Xie X, Li Y, et al. Single-cell analysis of target antigens of CAR-T reveals a potential landscape of “on-target, off-tumor toxicity”. Front Immunol. 2021;12:799206.
Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev. 2006;214:73–91.
Zamai L, Del Zotto G, Buccella F, Gabrielli S, Canonico B, Artico M, et al. Understanding the synergy of NKp46 and co-activating signals in various NK cell subpopulations: paving the way for more successful NK-cell-based immunotherapy. Cells. 2020;9(3):753. https://doi.org/10.3390/cells9030753.
Genssler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology. 2016;5(4):e1119354.
Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–83.
Wrona E, Borowiec M, Potemski P. CAR-NK Cells in the Treatment of Solid Tumors. Int J Mol Sci. 2021;22(11):5899. https://doi.org/10.3390/ijms22115899.
Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, et al. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol. 2015;6:21.
Chen KH, Wada M, Pinz KG, Liu H, Lin KW, Jares A, et al. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia. 2017;31(10):2151–60.
Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. 2016;17(7):816–24.
Daher M, Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 2021;11(1):45–58.
Siegler EL, Zhu Y, Wang P, Yang L. Off-the-shelf CAR-NK cells for cancer immunotherapy. Cell Stem Cell. 2018;23(2):160–1.
Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. 2017;168(4):724–40.
Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SM, Coustan-Smith E, et al. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood. 2014;124(7):1081–8.
Guven H, Konstantinidis KV, Alici E, Aints A, Abedi-Valugerdi M, Christensson B, et al. Efficient gene transfer into primary human natural killer cells by retroviral transduction. Exp Hematol. 2005;33(11):1320–8.
Suerth JD, Morgan MA, Kloess S, Heckl D, Neudorfl C, Falk CS, et al. Efficient generation of gene-modified human natural killer cells via alpharetroviral vectors. J Mol Med (Berl). 2016;94(1):83–93.
Love PE, Hayes SM. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb Perspect Biol. 2010;2(6):a002485.
Wilcox RA, Tamada K, Strome SE, Chen L. Signaling through NK cell-associated CD137 promotes both helper function for CD8+ cytolytic T cells and responsiveness to IL-2 but not cytolytic activity. J Immunol. 2002;169(8):4230–6.
Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, et al. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology. 2013;2(10):e26527.
Rostovskaya M, Fu J, Obst M, Baer I, Weidlich S, Wang H, et al. Transposon-mediated BAC transgenesis in human ES cells. Nucleic Acids Res. 2012;40(19):e150.
Kim A, Pyykko I. Size matters: versatile use of PiggyBac transposons as a genetic manipulation tool. Mol Cell Biochem. 2011;354(1–2):301–9.
Marofi F, Saleh MM, Rahman HS, Suksatan W, Al-Gazally ME, Abdelbasset WK, et al. CAR-engineered NK cells; a promising therapeutic option for treatment of hematological malignancies. Stem Cell Res Ther. 2021;12(1):374.
Karadimitris A. Cord blood CAR-NK cells: favorable initial efficacy and toxicity but durability of clinical responses not yet clear. Cancer Cell. 2020;37(4):426–7.
Marofi F, Rahman HS, Thangavelu L, Dorofeev A, Bayas-Morejon F, Shirafkan N, et al. Renaissance of armored immune effector cells, CAR-NK cells, brings the higher hope for successful cancer therapy. Stem Cell Res Ther. 2021;12(1):200.
Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy. 2011;13(1):98–107.
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–32.
Lee MY, Robbins Y, Sievers C, Friedman J, Abdul Sater H, Clavijo PE, et al. Chimeric antigen receptor engineered NK cellular immunotherapy overcomes the selection of T-cell escape variant cancer cells. J Immunother Cancer. 2021;9(3):e002128. https://doi.org/10.1136/jitc-2020-002128.
Muller N, Michen S, Tietze S, Topfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1alpha-secreting glioblastoma. J Immunother. 2015;38(5):197–210.
Yang Y, Lim O, Kim TM, Ahn YO, Choi H, Chung H, et al. Phase I study of random healthy donor-derived allogeneic natural killer cell therapy in patients with malignant lymphoma or advanced solid tumors. Cancer Immunol Res. 2016;4(3):215–24.
Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol. 2014;44(6):1582–92.
Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front Immunol. 2019;10:3038.
Baba Y, Nomoto D, Okadome K, Ishimoto T, Iwatsuki M, Miyamoto Y, et al. Tumor immune microenvironment and immune checkpoint inhibitors in esophageal squamous cell carcinoma. Cancer Sci. 2020;111(9):3132–41.
Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. 2014;3:e28147.
Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013;34(4):182–91.
Cummins KD, Gill S. Chimeric antigen receptor T-cell therapy for acute myeloid leukemia: how close to reality? Haematologica. 2019;104(7):1302–8.
Daher M, Melo Garcia L, Li Y, Rezvani K. CAR-NK cells: the next wave of cellular therapy for cancer. Clin Transl Immunology. 2021;10(4):e1274.
Lu SJ, Feng Q. CAR-NK cells from engineered pluripotent stem cells: off-the-shelf therapeutics for all patients. Stem Cells Transl Med. 2021;10(Suppl 2):S10–7.
Bjorklund AT, Carlsten M, Sohlberg E, Liu LL, Clancy T, Karimi M, et al. Complete remission with reduction of high-risk clones following Haploidentical NK-cell therapy against MDS and AML. Clin Cancer Res. 2018;24(8):1834–44.
Zhang Y, Wallace DL, de Lara CM, Ghattas H, Asquith B, Worth A, et al. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology. 2007;121(2):258–65.
Koepsell SA, Miller JS, McKenna DH Jr. Natural killer cells: a review of manufacturing and clinical utility. Transfusion. 2013;53(2):404–10.
Miller JS, Oelkers S, Verfaillie C, McGlave P. Role of monocytes in the expansion of human activated natural killer cells. Blood. 1992;80(9):2221–9.
Vasu S, Berg M, Davidson-Moncada J, Tian X, Cullis H, Childs RW. A novel method to expand large numbers of CD56(+) natural killer cells from a minute fraction of selectively accessed cryopreserved cord blood for immunotherapy after transplantation. Cytotherapy. 2015;17(11):1582–93.
Lapteva N, Durett AG, Sun J, Rollins LA, Huye LL, Fang J, et al. Large-scale ex vivo expansion and characterization of natural killer cells for clinical applications. Cytotherapy. 2012;14(9):1131–43.
Sutlu T, Stellan B, Gilljam M, Quezada HC, Nahi H, Gahrton G, et al. Clinical-grade, large-scale, feeder-free expansion of highly active human natural killer cells for adoptive immunotherapy using an automated bioreactor. Cytotherapy. 2010;12(8):1044–55.
Becker PS, Suck G, Nowakowska P, Ullrich E, Seifried E, Bader P, et al. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol Immunother. 2016;65(4):477–84.
Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, et al. IL15 Trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res. 2016;22(14):3440–50.
Hilpert J, Grosse-Hovest L, Grunebach F, Buechele C, Nuebling T, Raum T, et al. Comprehensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses. J Immunol. 2012;189(3):1360–71.
Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell. 2020;27(2):224–237 e226.
Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009;69(9):4010–7.
Ojo EO, Sharma AA, Liu R, Moreton S, Checkley-Luttge MA, Gupta K, et al. Membrane bound IL-21 based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci Rep. 2019;9(1):14916.
Pahl JHW, Koch J, Gotz JJ, Arnold A, Reusch U, Gantke T, et al. CD16A activation of NK cells promotes NK cell proliferation and memory-like cytotoxicity against Cancer cells. Cancer Immunol Res. 2018;6(5):517–27.
Barrow AD, Martin CJ, Colonna M. The natural cytotoxicity receptors in health and disease. Front Immunol. 2019;10:909.
Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18(2):85–100.
Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr Opin Immunol. 2018;51:146–53.
Gurney M, O’Dwyer M. Realizing Innate Potential: CAR-NK Cell Therapies for Acute Myeloid Leukemia. Cancers (Basel). 2021;13(7):1568. https://doi.org/10.3390/cancers13071568.
Acknowledgements
The coauthors thank the members in Key Laboratory of Molecular Diagnostics and Precision Medicine for Surgical Oncology in Gansu Province of Gansu Provincial Hospital, NHC Key Laboratory of Diagnosis and Therapy of Gastrointestinal Tumor, The First Affiliated Hospital of Shandong First Medical University, Key Laboratory of Radiation Technology and Biophysics in Hefei Institute of Physical Science in Chinese Academy of Sciences, and State Key Laboratory of Experimental Hematology & National Clinical Research Center for Blood Disease, Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College for their technical support.
Funding
This work was supported by grants from the project Youth Fund supported by Shandong Provincial Natural Science Foundation (ZR2020QC097), National Natural Science Foundation of China (81330015), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019PT320005), Science and technology projects of Guizhou Province (QKH-J-ZK [2021]-107), Project funded by China Postdoctoral Science Foundation (2019 M661033), Natural Science Foundation of Hebei Province (H2020206403), Natural Science Foundation of Tianjin City (19JCQNJC12500), Major Project of Fundamental Research Funds of the Central Public Welfare Scientific Research Institutes of the Chinese Academy of Medical Sciences (2018PT31048, 2019PT310013), Jiangxi Provincial Key New Product Incubation Program Funded by Technical Innovation Guidance Program of Shangrao (2020G002), Natural Science Foundation of Jiangxi Province (20212BAB216073), Key project funded by Department of Science and Technology of Shangrao City (2020AB002).
Author information
Authors and Affiliations
Contributions
L.Z., Y.M., X.F. and Z.H.: collection and assembly of data, manuscript writing; X.F. and Z.H.: helped with collection and assembly of data; L.Z.: data analysis and interpretation, manuscript writing; L.Z.: conception and design, revision, final approval of manuscript. All coauthors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare there’s no competing interests and all authors consent to publish the data.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, L., Meng, Y., Feng, X. et al. CAR-NK cells for cancer immunotherapy: from bench to bedside. Biomark Res 10, 12 (2022). https://doi.org/10.1186/s40364-022-00364-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-022-00364-6