
Abstract
Background
Many fungi grow as saprobic organisms and obtain nutrients from a wide range of dead organic materials. Among saprobes, fungal species that grow on wood or in polluted environments have evolved prolific mechanisms for the production of degrading compounds, such as ligninolytic enzymes. These enzymes include arrays of intense redox-potential oxidoreductase, such as laccase, catalase, and peroxidases. The ability to produce ligninolytic enzymes makes a variety of fungal species suitable for application in many industries, including the production of biofuels and antibiotics, bioremediation, and biomedical application as biosensors. However, fungal ligninolytic enzymes are produced naturally in small quantities that may not meet the industrial or market demands. Over the last decade, combined synthetic biology and computational designs have yielded significant results in enhancing the synthesis of natural compounds in fungi.
Main body of the abstract
In this review, we gave insights into different protein engineering methods, including rational, semi-rational, and directed evolution approaches that have been employed to enhance the production of some important ligninolytic enzymes in fungi. We described the role of metabolic pathway engineering to optimize the synthesis of chemical compounds of interest in various fields. We highlighted synthetic biology novel techniques for biosynthetic gene cluster (BGC) activation in fungo and heterologous reconstruction of BGC in microbial cells. We also discussed in detail some recombinant ligninolytic enzymes that have been successfully enhanced and expressed in different heterologous hosts. Finally, we described recent advance in CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas (CRISPR associated) protein systems as the most promising biotechnology for large-scale production of ligninolytic enzymes.
Short conclusion
Aggregation, expression, and regulation of ligninolytic enzymes in fungi require very complex procedures with many interfering factors. Synthetic and computational biology strategies, as explained in this review, are powerful tools that can be combined to solve these puzzles. These integrated strategies can lead to the production of enzymes with special abilities, such as wide substrate specifications, thermo-stability, tolerance to long time storage, and stability in different substrate conditions, such as pH and nutrients.
Similar content being viewed by others

Background
Ligninolytic enzymes (LEs)—also referred to as lignolytic enzymes or lignin modifying enzymes (LMEs)—are proteins that catalyze modifications or degradation of lignin into less complex molecules, to produce CO2 eventually [1, 2]. Many fungal species produce LEs and possess complex enzyme systems for lignin degradation, including laccase (LCC), lignin peroxidase (LiP), manganese peroxidase (MnP), and versatile peroxidase (Vp). These enzymes are characterised by broad specificity in degradation/mineralization of different complex substrates such as wood, paper, animal feeds, pesticides, biofuels, and hydrocarbons [2,3,4,5,6]. The importance of LEs in biotechnological, industrial, and environmental applications—especially in the production of bioenergy or biomaterials, is worldwide recognized [7]. However, many natural products (NPs) are not naturally produced in enough quantities for human or industrial use. Beside NPs, specific fungal enzymes, which either play a role in metabolic pathways or are secreted to carry out their activities in the environment, have grabbed the attention of the Synthetic Biology community.
Metabolic pathway engineering, therefore, has become quickly an important branch of Synthetic Biology, whose goal is to optimize the synthesis of chemical compounds of interest in various fields, from pharmaceutics to biofuel [8]. For instance, the production in Saccharomyces cerevisiae (Desm.) Meyen of the artemisinic acid, a precursor of the antimalarial drug artemisinin—usually extracted from the plant Artemisia annua L.—is still one of the most celebrated successes in Synthetic Biology [9]. Beside plants, filamentous fungi are another primary source of natural products (NPs), which are beneficial to human health. Genes that drive the expression of NPs are, generally, located into small regions of the fungal genome, giving rise to so-called biosynthetic gene clusters (BGCs).
As an engineering science, Synthetic Biology aims at the rational design and model-driven in vivo implementation of biological systems that carry out specific tasks [10]. The first Synthetic Biology artifacts were genetic circuits, more precisely transcriptional networks, able to mimic electronic functions such as oscillations, toggle switches, and Boolean gates [11]. Circuits, initially, were hosted by bacteria—often Escherichia coli—and required the construction of a handful of transcription units i.e. DNA sequences where a promoter is followed, in the order, by a ribosome binding site (RBS), a coding sequence (CDS), and finally, a terminator. In parallel, computational tools were developed to facilitate the design, modeling, and analysis of genetic circuits [12, 13]. The increasing complexity of synthetic gene circuits led to faster and more reliable techniques for DNA assembly, such as the Golden Gate [14] and the isothermal assembly [15] method, where multiple DNA sequences are joined in a single experiment. Signaling and metabolic pathway rewiring and reconstruction in heterologous hosts were among the first Synthetic Biology achievements in eukaryotic cells [16].
In bioinformatics, genome mining refers to the identification of features of an organism through the analysis of its genome. In this context, software has been written ad hoc for the analysis of fungal genomes in order to both trace BGCs associated with known NPs and discover silent BGCs that, once activated, potentially lead to the expression of novel, valuable compounds [17, 18]. Algorithms for the identification of gene clusters into unannotated or partially annotated genomes are very accurate but can detect only BGCs associated with well-defined families of NPs, such as polyketides and RiPPS (ribosomally synthesized and post-translationally modified peptides). Secondary metabolite unique regions finder (SMURF) [19], antibiotics and secondary metabolites analysis shell (antiSMASH) [20, 21], secondary metabolite by inter-pro-scan (SMIPS), and cluster assignment by islands of sites (CASSIS) [22] are popular software for secondary metabolite detection and BGC recognition. More obscure, silent fungal BGCs have been discovered by means of different computational tools. For instance, motif-independent de novo detection algorithm for secondary metabolite biosynthetic gene clusters (MIDDAS-M) [23] has brought to light the BGC of ustiloxin B and MIPS-GS [24] has permitted to decipher the kojic acid BGC in Aspergillus flavus var. oryzae (Ahlb.) Kurtzman, M.J. Smiley, Robnett & Wicklow. These computational works, combined with experimental evidence, have laid the foundation of the MIBiG (Minimum Information about a Biosynthetic Gene cluster) [25] repository and data standard. MIBiG offers a broad collection of building blocks (genes, enzymes, and protein domains) for the in silico design of novel biosynthetic pathways [26, 27].
Synthetic Biology has also put forward novel techniques for BGC activation in fungo and heterologous reconstruction of BGC in microbial cells [28]. Control of single genes or entire pathways in fungo has been achieved through the expression of transcription factors based on bacterial proteins, a technique already largely exploited in mammalian and yeast cells. The Tet-ON system [29] has allowed the synthesis of a novel NP—named fumipyrrole—in Aspergillus fumigatus Fresen [30]. More recently, Rantasalo and co-authors [31] have shown that a synthetic activator, made by the fusion of the DNA-binding bacterial protein Bm3R1 [32] and the viral activation domain VP16 [33], enhances red fluorescence expression in Aspergillus niger Tiegh. and Trichoderma reesei E.G. Simmons.
Genome-based engineering of LEs in fungi is necessary in order to increase their production to meet human demands and industrial standards. To this aim, several approaches such as rational, semi-rational or directed evolution have been employed. However, Synthetic Biology methods and computational tools have not been fully exploited yet for enhancing the production of LEs in fungi or optimizing their synthesis in microbes, such as S. cerevisiae and E. coli. CRISPR (clustered regularly interspaced short palindromic repeats)-Cas (CRISPR associated) protein systems [34] seem the most promising biotechnology for large-scale production of LEs. They have already proved their efficiency in the editing, discovery, and activation of fungal genes and BGCs [35]. Moreover, on the computational side, numerous new algorithms have been written both to optimize CRISPR-Cas systems that edit specific loci in a genome and estimate potential off-target effects, which can lead even to cell death [36].
In this review, after introducing different groups of LEs together with their oxidative mechanisms, we described how to engineer LEs with improved activity, and illustrated general Synthetic Biology techniques both to build synthetic promoters and engineer new proteins (nucleases and transcription factors) for DNA editing and control of gene expression. We showed recent applications of the latter techniques to filamentous fungi and their usage in yeast to enhance the production of LEs. Finally, we discussed how Synthetic Biology methods may improve the use of LEs in biotechnology.
Lignin modifying fungi and ligninolytic consortium
Lignin
Lignin is one of the major structures that strengthen the cellulose and hemicellulose fibres in plant cell wall [37,38,39], as shown in Fig. 1. This peculiar molecule plays an essential role in plant physiology by enhancing the transportation of water and helping plants to resist the attack of pathogens [40]. It is a complex aromatic polymer constituted by heterogenous components of mono-, di-, and non-methoxylated phenylpropaniol moieties with non-hydrolysable linkages between the subunits of p-hydroxycinnamyl alcohols (i.e., the coniferyl, coumaryl and sinapyl alcohols) [41, 42]. Cellulose as an organic polysaccharide polymer consists of a chain of many hundreds or thousands of β-glucose 1–4 cross-linked with D-glucose units. It is an important structural components of plant’s primary cell wall and is also abundant in the cell walls of algae and the fungal class of oomycetes. Moreover, it has been discovered that some bacteria can secret cellulose biomers to form biofilms [39]. Cellulose appears as a crystalline white structure, has a melting point of 260–270 °C with 1.50 g/cm3 density and 162.1406 g/mol molar mass, makes 90% of cotton fibre, about 57% in hemp, and constitutes 40–50% of wood [39]. Hemicellulose, on the other hand, exists as an heteropolymer of organic polysaccharide like arabinoxylans. It usually appears along with cellulose in plant cell wall. Unlike cellulose, hemicellulose is an amorphous structure less strong or resistant to hydrolysis.

adapted from Alcalde [87]
Structural sketch of lignin strengthening cellulose and hemicellulose biomass in the cell wall. Figure
Lignin and cellulose components of plant cell represent the most abundant natural biomass on earth (lignocellulose biomass). The lignin portion of plant biomass is estimated to be produced at a rate of about 200 billion tons/year and it is responsible for larger quantities of carbon fixed in terrestrial ecosystems [42, 43]. Lignin is found in large quantities in woody plants, accounting for about 26–32% dry weight in soft woods and 20–25% in hard woods, and it is estimated to constitute about 15–21% of dry mass in corn stover, and 5–17% in wheat straw [42]. Large chunk of dead lignocellulose components in plant/wood materials are decomposed by microorganisms leading to a fundamental carbon recycling in the biosphere. Although lignin is difficult to degrade for most of microorganisms, due to its complex nature, some fungi have the capability to decay this molecule by producing specific enzymes, which have been studied intensely [43, 44]. Among lignin-decaying fungi, the white-rot basidiomycetes are known to be particularly efficient due to their secretion of vast oxidoreductase enzymes [45].
Lignin modifying fungi
Fungi are more efficient in lignin degradation than other microbes [42]. Wood-decaying basidiomycetes have been divided into two groups, white- and brown-rot basidiomycetes, based on their morphological growth on woods, which reflects their different wood degradation activity. White-rot fungi (WRF) preferentially grow on deciduous trees and gradually degrade cellulose and lignin using high redox-potential oxidoreductive enzymes commonly referred to as LEs. This degradation mechanism results in the typical white colour of the cellulose-enriched wood left behind [46]. On the contrary, brown-rot fungi mostly colonize conifers and primarily degrade hemicellulose and cellulose, leaving a brown coloured residual wood enriched in the darker lignin [47]. However, both white-rot fungi and some brown-rot fungal species, such as Gloeophyllum trabeum (Bourdot & Galzin) Domański, Orloś & Skirg [48], are particularly predisposed to lignin degradation/mineralization due to their production of several oxidoreductase enzymes for conversion of lignin to H2O and CO2 [48,49,50]. Examples of common wood decaying white-rot fungi are Phanerochaete chrysosporium Burds., Armillaria sp., Phlebia sp., Phellinus pini (Brot.) Pilát, Schizophyllum commune Fr., Dichomitus squalens (P. Karst.) D.A. Reid, Trametes versicolor (L.) Lloyd, Daldinia concentrica (Bolton) Ces. & De Not., while the brown-rot fungi include Laetiporus portentosus (Berk.) Rajchenb., Monilinia fructicola (G. Winter) Honey, Fomitopsis lilacinogilva (Berk.) J.E. Wright & J.R. Deschamps, and Postia placenta (Fr.) M.J. Larsen & Lombard.
Production of LEs has not yet been reported in Chytridiomycota [51], whereas reports are increasing from the Ascomycota (mould group) and Mucoromycota [52]. The soft rot fungi (ascomycetes) such as Xylaria, Fusarium, Penicillium, Trichoderma, and Aspergillus species can degrade the softer section of wood that is constituted by a lower proportion of lignin [53]. These fungi have been described to be capable of mineralizing lignin and have shown wood rotting activities, although in lesser rate than in basidiomycetes [54,55,56,57]. In addition, some fungal plant pathogens have been associated with lignin-degrading activities. For instance, extracellular laccase production was found in Botrytis cinerea Pers. a causal agent of soft rot disease in Daucus carota L., Greeneria uvicola (Berk. & M.A. Curtis) Punith, the well-known rot pathogen of wine grape (Vitis vinifera L.), and many others such as Pythium and Fusarium species [58].
Ligninolytic enzyme consortium
Fungi efficiently degrade lignin thanks to their complex ligninolytic bio-refineries with ability to produce different cassettes of ligninolytic secretome (TLS). These are mostly constituted by enzymes known to act as natural oxidative systems (Table 1; Additional file 1: Table S1), which catalyze non-specific reactions leading to depolymerization/degradation of lignin [59]. Many of them are catalogued in Carbohydrate-Active enZymes Database (CAZy) as lignin-degrading auxiliary enzymes (LDA) or commonly as auxiliary activity (AA) enzymes (CAZy; http://www.cazy.org/). The most common LEs in fungi (Fig. 2) are grouped in different families that can be found in https://www.cazypedia.org/:
-
i.
Multicopper oxidase superfamily (AA1), such as laccase (Lcc).
-
ii.
Auxiliary Activity family (AA2), which are peroxidases, including lignin peroxidase (LiP), manganese peroxidase (MnP), and versatile peroxidase (VP).
-
iii.
Peroxidase superfamily, such as dye decolorizing peroxidases (DyP) and heme-thiolate peroxidases (HTPs).
-
iv.
Auxiliary Activity family (AA3), which are glyoxal oxidases (GLOX);
-
v.
Subfamily AA3_1; for example, cellobiose dehydrogenase.
-
vi.
Subfamily AA3_2; for instance, aryl-alcohol oxidase/dehydrogenase, galactose oxidase, glucose dehydrogenase, glucose oxidase, and pyranose dehydrogenase.
-
vii.
Subfamily AA3_3, including alcohol oxidases, such as vanillyl alcohol oxidase and benzoquinone reductase.
-
viii.
Subfamily AA3_4, including pyranose oxidase.
-
ix.
Auxiliary Activity family (AA4), which are vanillyl-alcohol oxidases.

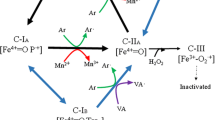
Classification and structure of common fungal ligninolytic enzymes. LiP = Lignin peroxidase, MnP = Manganese peroxidase, VP = Versatile peroxidase, Dyp = Dye-decolorizing peroxidase, AA = Auxiliary activity family
Genes that encode the expression of these enzymes have been widely reported in the genomes of many WRF [60,61,62]. LiP, MnP, and VP are common LEC of class II heme-containing peroxidases [63], which are grouped in AA1-AA3 enzyme family due to their ability to catalyse the oxidation of hydrogen peroxide as co-substrate. These TLS are also very much involved in high-redox catalytic oxidation of complex aromatic hydrocarbons (Additional file 1: Table S2). Peroxidases (LiP, MnP, and VP) generally have a conserved primary, secondary and tertiary architectures including Ca ion binding sites, heme-pocket residues, and four disulfide bridges [64].
The first discovered enzyme from the group of LEs is the LiP, which was independently discovered in 1983 by Tien and Kirk [1] and Glenn et al. [3], from P. chrysosporium. This enzyme was initially named ligninase, but was later reported in some WRF belonging to Trametes, Phanerochaete, Phlebia, and Bjerkandera genera under the name of lignin peroxidase [62]. LiP was found to be able to speed up lignin degradation and oxidation of complex hydrocarbons through a vast electron transfer to a heme cofactor [65]. For example, LiP H8 was found to exhibit catalytic reaction in the presence of redox active substrate containing tryptophan in P. chrysosporium [66]. Similar observation was later made for VP by Ruiz-Dueñas et al. [67]. Both VP and LiP differ from other LEs because of their redox potentials that are estimated to range between 1.4 and 1.5 V during direct oxidation of non-phenolic lignin. These peculiar redox potentials allow VP and LiP to oxidize long chain polycyclic aromatic hydrocarbons through Cα-Cβ ring cleavage and cleave dimeric lignin compound [68]. A key example of LiP action is the oxidation of the aromatic substrate veratryl alcohol (3,4-dimethoxybenzyl alcohol) to veratraldehyde, while VP acts directly on aromatic compounds with Mn oxidizing efficiency, which makes the activity of this enzyme similar to both LiP and MnP [42]. The latter is another example of LE that is commonly produced by wood- and litter-decomposing basidiomycete fungi and has been found involved in catalytic oxidation of phenolic substrates through the oxidation of Mn2+ to Mn3+ [28].
MnP is a LE that has manganese binding sites constituted by three acidic amino acid residues that coordinate Mn2+ (i.e. Glu-35, Glu-39, and Asp-179), as revealed in P. chrysosporium [28, 69]. Chelation of manganese iron Mn3+ by dicarboxylic acid is generally observed in reactions that involve MnPs. A typical example of such reaction is represented by the oxidation of oxalic acid into more stable form. In lignin degradation, chelated Mn3+ is diffused into cell wall lignin through the micropores, which are less accessible for larger enzymes [28, 69]. For MnP it may not be as easy as for LiP and VP to oxidize non-phenolic lignin, due to its lower redox potential, which was estimated around 1.0–1.2 V [68]. However, its action on non-phenolic lignin is generally mediated by MnP-lipid systems of lipid peroxidation, which generates reactive intermediates of peroxyl radicals. Recently, genetic models that encode typical manganese and versatile peroxidases, based on conserved Mn-binding sites and tryptophan as mediator, were reported [60]. In these models, new superfamilies of fungal peroxidases i.e. dye decolorizing peroxidases (DyPs) and HTPs were described. These enzymes, which are not phylogenetically related to LiPs, MnPs or VPs and are also commonly associated with lignin modification/degradation, have been reported in many fungal, bacterial and archaea genomes [70].
Laccase belongs to the group of multicopper oxidase superfamily of LEs and is listed as AA1_1 in the CAZy family [71]. This enzyme is characterized by the presence of four copper atoms and has the potential of catalyzing four different electron transfer reactions via different phenolic compounds, aromatic amines, and dye molecules with 3-dimentional structure and four substrate binding loops as well as four conserved signature sequences, which have amino acids as copper ligands. Laccase has a redox potential ranging from 0.5 to 1.0 V, and usually requires suitable mediator, such as 1-hydroxybenzotriazole and 2,2-azinobis 3-ethylbenz-thiazoline-6-sulfonate (ABTS). This ligninolytic enzyme has a wide range of substrate polymers, from non-phenolics to larger organics. Generally, the laccase-like multicopper oxidase (LMCO) family is phylogenetically divided into sensu-stricto laccases and iron oxidizing Fet3-like ferroxidases, which are very common in formation of pigments and ascorbate oxidases [72]. The LMCO family has been generally associated with morphogenesis and pathogenesis in plants [73].
DyP is a very useful enzyme due to its high redox potentials (1.2–1.5 V), which were found to enable it to oxidize different types of dyes, such as phenolics and carotenoids [74, 75], like other peroxidases. DyP was also found to be able to oxidize veratryl alcohol and non-phenolic lignin at low-level [76]. The first DyP with ability to oxidize Mn2+ was detected in Pleurotus ostreatus (Jacq.) P. Kumm. [77, 78], but the biological role of this enzyme is still not clearly understood. Besides, there have been similar reports on some groups of LEs called nonspecific peroxygenases (UPOs) in different basidiomycetes genera, such as Agrocybe, Marasmius, and Coprinellus [79]. These enzymes belong to the superfamily of versatile HTPs that catalyze the oxidation of aromatic and aliphatic hydrocarbons [80, 81]. UPOs have also been reported with potentials to catalyze the degradation of non-phenolic β-O-4 lignin dimers [68, 82], although their physiological roles have not been described.
Several other fungal enzymes that act as oxidases, according to enzyme commission classification (ECC), have been found associated with lignin and wood degradation, often leading to the evolution of H2O2, as for many LEs, like Mn (III)-mediated oxidation of some organic acids that are secreted by fungi [83]. Glucose-methanol-choline oxidoreductases are examples of oxidase enzymes which are classified in superfamily AA3. These enzymes, in nature, are flavoproteins with flavin-adenine dinucleotide-binding domain and two conserved histidines [84, 85]. Enzymes in superfamily AA3, including cellobiose dehydrogenase (CDH), glucose oxidase (GOX), aryl oxidase (AAO), methanol oxidase, pyranose-2 oxidase (P2O), etc. (Table 1) share similar structures as shown in Fig. 2 and act in similar way, for instance by oxidation of alcohol moiety to aldehydes. Another example of fungal oxidases is represented by the copper radical super-family, which includes glyoxal oxidases (GLOX). These are copper metallo-enzymes characterized by high specificity for oxidation of simple aldehydes into carboxylic acids. GLOX have unusual active site free radical-couple copper similar to those of galactose oxidases. Given their high specificity for aldehydes, GLOX have attracted the interest of researchers to a larger extent compared to other members of copper radical oxidases, and they have also been physiologically linked to LiPs in the fungal species P. chrysosporium, for the conversion of H2O2 in lignin degradation [86].
Fungal LEC has been widely investigated; according to Alcalde [87], the US Department of Energy invested funds in research projects for sequencing of more than 80 fungal genomes, with the aim to identify useful microorganisms for biorefineries of lignocellulosic biomass [88]. These enzymes are involved in a wide range of industrial applications, including the production of renewable organic compounds, such as fermented products, antibiotics, and biopolymers. LEs from fungi can also be very useful for the production of biofuels (bioethanol and biobutanol) and biofuel cells, for biomedical applications (biosensors), bioremediation, and in pulp and textile industries. However, these enzymes are produced naturally in small quantities that may not meet the industrial or market demands. Therefore, there is need for biotechnological enhancement of LEs production in fungi. Recent advancements in computational and synthetic biology systems provide the possibility to manipulate fungal BPs for production of LEs with desired properties in terms of reactivity, specificity, and catalytic potentials.
Engineering new ligninolytic enzymes
The number of already described fungal species was recently estimated in 120,000 out of a total of 2.2–3.8 millions [89]. More than 1000 fungal genomes have been sequenced and annotated. They are available at the Department of Energy Joint Genome Institute [90], the Broad Institute Fungal Genome Initiative (https://www.broadinstitute.org/fungal-genome-initiative), and the FungiDB [91, 92]. The number of genes encoding for NPs, LEs, and other secreted chemicals/enzymes strongly depends on the fungal strain and is highly variable among different genomes, as reported in Wang et al. [93]. Libraries of new ligninolytic enzymes (Fig. 3) are obtained via three main protein engineering methods: (1) Directed evolution (DE) (2) Rational and (3) Semi-rational approaches.

Identification and screening of ligninolytic enzymes in postgenomic era
The DE involves either random recombination of a set of similar sequences (such as gene shuffling) or the introduction of random changes in a single protein sequences, e.g. via error-prone PCR, to engineer improved proteins. The structural information of an enzyme is not necessarily required for DE and variations can occur, at random positions, far from the active site. These changes are often small and can require several rounds of evolution to screen a high number of variants. In fungi, DE has led to the production of LEs characterized by novel features such as tolerance to extreme pH and a prolonged storage time [94, 95]. Moreover, several computational methods such as molecular dynamics, the Monte Carlo algorithm, and ab initio calculations facilitate the study of the interactions between enzymes and ligands/substrates, which helps DE considerably through the identification of residues, and whole regions, as targets for mutagenesis and recombination [96]. Such a computer-driven directed evolution led to the synthesis of laccases with enhanced activity from Pleurotus ostreatus [97, 98]. Moreover, Ouyant and Zhao [99] reported improved laccase activity from the yeast mutant 5E29 with two amino acid substitutions. Directed evolution proved to be a successful technique also for the production of laccases from non-native hosts. Bulter and co-authors [100] expressed, in S. cerevisiae, a laccase from Myceliophthora thermophila (Apinis) Oorschot and improved its catalytic efficiency via 10 DR iterations. Interestingly, the laccase gene that was introduced into the yeast cells included a cleavage site at the C-terminal tail that favoured the protein biosynthesis in the Golgi compartment for packing of mutant into vesicles. Later, Zumárraga et al. [101] further improved this evolved variant through synergistic saturation mutagenesis, which highlighted the importance of the C terminus in ascomycete laccases.
Directed evolution methods have also been adopted for the enhancement of fungal peroxidases/peroxygenases. VP an LE from an oyster mushroom Pleurotus eryngii (DC.) Quel. were successfully expressed with enhanced activities and thermostability in S. cerevisiae by Morawski et al. [102] and Garcia et al. [103]. This enzyme was later was evolved for better oxidative stabilization by Gonzalez-Perez et al. [104]. Directed evolution was also demonstrated by Miyazaki-Imamura et al. [105] to enhance the stability of MnP from a basidiomycete fungus P. chrysosporium in the presence of hydrogen peroxide via an in vitro expression system. Ryu et al. [106, 107] demonstrated enhanced production of LiP from this fungus by yeast surface display. In another study carried out by Molina-Espejaet al. [108], a specific UPO from Agrocybe aegerita (V. Brig.) Singer was successfully expressed in S. cerevisiae via induced mutations in native signal peptides and mature protein, they also used that opportunity to develop tandem yeast-expression system that enables UPO production in P. pastoris. In addition, heterologous expression of CDH from Myriococcum thermophilum (Fergus) Aa. was demonstrated in S. cerevisiae by Sygmund et al. [109]. They however observed best CDH variant in Pichia pastoris (Guillierm.) Phaff via a modification in oxidative and reductive FAD half-reaction.
Rational approach for enzyme engineering is based on the use of protein structures together with biochemical and molecular modelling data for the introduction of site-specific mutagenesis. This approach has a high probability to find beneficial mutations with a consequent reduction in library size. As such, it demands less effort and time. Among rational approaches, molecular evolution has been employed for the production of enzymes with improved catalytic activities and higher tolerance to solvents and temperature [110]. Molecular evolution pursues an enhancement of fungal enzyme activity via molecular modifications (e.g. direct site mutations) driven by structural considerations. This technique has been adopted to increase the oxidative capacity of some LEs such as laccases, which degrade non-phenolic substrates, on bulky phenolic molecules [111, 112].
Semi-rational design, on the other hand, combines the use of rational and random protein design to create smaller and smarter libraries from biochemical and/or structural data [113]. Site-saturation mutagenesis is a procedure used in semi-rational design to modify hotspot residues in an enzyme. Here, one or more amino acids are replaced, at their positions, by all other possible amino acids. CASTing (combinatorial active site saturation testing) is a particular semi-rational approach that employs information derived from structural data for the identification of amino acids in particular regions (like an active site of a protein sequence) that are then mutated randomly or via site-saturation mutagenesis [114]. A different semi-rational approach, which involves the usage of focused directed evolution methods—such as combined saturation and domain mutagenesis—has been exploited to obtain enzymes with 3- to eightfold higher catalytic efficiencies [115].
Synthetic Biology in fungo: gene editing and transcription activation
Synthetic biology in fungi involves the redesigning/engineering of fungal genomes to enhance their performance or give them new abilities. This demands to construct new biological parts, devices, and circuits in fungi. Many computational tools (such as those listed in Additional file 1: Table S3) are now available for use in pathway construction/enhancement in fungi, in this way, fungi can be harnessed to tackle issues in medicine, industry, human environment, and agriculture. In principle, synthetic gene circuits shall be orthogonal to their host cells i.e. they shall not interfere with the molecular processes of their chassis (the organism into which they are placed). Wanka and co-authors [116] achieved orthogonality in Aspergillus species by using the bacterial Tet-OFF system [117]. Here, the chimeric activator TetR-VP16 binds its operators on the DNA only in the absence of doxycyline, a derivative of the antibiotic tetracycline. The endogenous genes targeted in this work (racA and gfaA in A. niger; pabaA in A. fumigatus) were first deleted from and then re-inserted into the fungal genome as parts of “expression cassettes”, where they laid downstream of a minimal promoter preceded by seven copies of the tet operator, as showed in a previous work by Belli’ and co-authors [118]. Overall, these three genes were downregulated in proportion to the concentration of doxycycline in the growth medium.
Synthetic promoters made by merging sequences from different promoters (even belonging to diverse organisms) are referred to as hybrid and were already used before the advent of the Synthetic Biology era—for instance, see Carver et al. [119]. Even though the hybrid promoters guarantee orthogonality, they demand several genomic manipulations and are limited by the small number of well-characterized transcription factor-operator pairs. Transcription Activator-like Effectors (TALEs), which were discovered in plant-pathogenic bacteria belonging to the genus Xanthomonas, represent a tool for assembling novel transcription factor able to bind a large number of DNA sequences and, therefore, control the activity of endogenous promoters [120]. The main feature of TALEs is their modular DNA-binding domain that is made, in natural configuration, of 13 up to 28 repeats, most of which containing 34 amino acids (the last repeat is an exception and is only 20 amino acids long). The amino acid at position 13 determines the nucleotide that is recognized and bound by each repeat. For this reason, it is termed as base-specific residue [121].
A synthetic DNA-binding domain is assembled by joining as many repeats as there are nucleotides in the target sequence. Each repeat, usually, carries at position 13 an Asparagine to bind a guanine; a Glycine to bind a thymine; an Isoleucine to bind an adenine; or an Aspartic Acid to bind a cytosine. As a constraint, only DNA sequences preceded by a thymine or a cytosine are bound by TALEs-based proteins. Despite their overall great potential [122], TAL effectors have been used, in fungi, mainly for gene editing in the form of Transcription activator-like effector nucleases (TALENs or TAL effector nucleases) upon fusion to FokI nuclease [123, 124].
CRISPR-Cas systems [125] have rapidly become a preferred solution to operate modifications also on fungal genomes. CRISPR-Cas proteins are a component of the immune system of bacteria and archaea [126] that triggers the degradation of invading DNAs. The CRISPR array, along the prokaryotic chromosome, is made of spacers (i.e. sequences from previously encountered DNA intruders) separated by repeats. Upon infection, the CRISPR array is, first, transcribed into a long pre-crRNA (pre-CRISPR RNA) molecule that is, then, processed into shorter crRNAs, each containing a spacer and a repeat (in some cases, a crRNA is bound to another short RNA sequence, termed tracrRNA—transactivating RNA). The spacer binds, by complementary base-pairing, the target DNA in the proximity of the protospacer adjacent motif (PAM)—a short hallmark that consists of 3–4 nucleotides. The repeat can be seen as a handle where the corresponding Cas protein—a nuclease—binds. Overall, the Cas:crRNA complex recognize, binds, and cuts—with a double-strand break—a foreign DNA sequence.
Among the six types of CRISPR-Cas systems so far discovered, type II and type V are the most popular in biotechnology because they require a single Cas protein to cleave the DNA: Cas9 [127] and Cas12a [128], respectively (to be precise, Cas12a belongs to the subtype A of type V). In order to carry out genomic modification, CRISPR-Cas systems are an easier solution than TALENs. CRISPR-Cas demand to express a short crRNA sequence (up to about 100 nucleotides) instead of engineering a complicated, modular DNA-binding domain. Moreover, TAL effectors require to be attached to a nuclease domain that, in contrast, is naturally present in the CRISPR associated proteins. Recently, applications of CRISPR-Cas9 to filamentous fungi have focused, mainly, on Aspergillus. In particular, efficient gene knockout has been reported in A. niger [129], A. fumigatus [130], and A. flavus var. oryzae [131]. The latter work described also the first CRISPR-Cas9-based gene editing in Nodulisporium sp. and Sporormiella minima (Auersw.) S.I. Ahmed & Cain.
Another interesting work in A. fumigatus showed how to utilize CRISPR-Cas9 to activate, in a non-productive strain of this fungus, the BGC for the synthesis of the antibiotic trypacidin [132]. Shi and co-authors [133] exploited CRISPR-Cas9 to rewire metabolic pathways in Fusarium fujikuroi Nirenberg and change, as a consequence, the product profile of the gibberellic acids. CRISPR-Cas9 has also been used to engineer BPs in many fungi [134]. The only work where CRISPR-Cas9 was used to improve the production of a ligninolytic enzyme, cellulase, is a multiplexed genome engineering in Myceliophthora by Liu and co-authors [135]. Here, enhanced cellulase production (about 5 folds) and activity (up to over 13 folds) resulted, with respect to the wild-type strain, after the deletion of four selected genes: cre1, res1, gh1-1, and alp-1. Other previous research papers on CRISPR-Cas9-based genome editing in filamentous fungi have been reviewed in [136].
CRISPR-Cas12a systems have been employed in fungal genomic manipulation only over the last two years. Also in this case, most of the experiments have been done in Aspergillus. The first proof of the working of LbCas12a (i.e. from Lachnospiraceae bacterium) in filamentous fungi—A. nidulans (Eidam) G. Winter and A. niger—was given in [137]. A later work, again in A. nidulans, was the first to use a nuclease-deficient version of a CRISPR associated protein—here LbCas12a again—to construct a synthetic activator and trigger gene expression in fungo [138]. The catalytically dead protein dLbCas12a was fused to the strong VPR synthetic activation domain [139] and tested, initially, on an exogenous gene encoding for a fluorescent protein. The successful production of a fluorescence signal led Roux and co-authors [138] to address the action of the synthetic activator to the endogenous micA gene, which permitted an increased synthesis of microperfuranone. Finally, dLbCas12a-VPR allowed the activation of a previously unknown BGC, whose product turned out to be dehydromicroperfuranone. Kwon and co-authors [140], in contrast, tested a different version of Cas12a (FnCas12 from Francisella novicida and AsCas12a from Acidaminococcus sp. BV3L6) together with Cas9 (the usual one, from Streptococcus pyogenes) on single-gene and multiplex-gene targeting in Thermothelomyces thermophilus (Apinis) Y. Marín, Stchigel, Guarro & Cano. Every CRISPR-Cas system proved to be active in this particular filamentous fungus with FnCas12a showing higher resilience than the other two CRISPR associated proteins.
CRISPR-Cas systems have not been used for the enhancement of LE expression in fungi yet. Probably, the main reason has been the lack of sequenced genomes since template genomes from different fungi show extensive variations and, therefore, do not allow for precise DNA targeting. However, recent progress in next-gen sequencing technology could facilitate fungal genomic engineering and Synthetic Biology in the incoming years.
Heterologous expression of recombinant ligninolytic enzymes
Expression of recombinant proteins in hosts that can be cultivated easily has been encouraged to reduce the production cost. Ligninolytic enzymes that are produced by many basidiomycete fungi, such as Myceliophthora thermophila, P. chrysosporium, Pleurotus sp., Trametes trogii Berk., and Trametes versicolor have been expressed in some ascomycete fungi. Record et al. [141] established over-expression of laccase lac1 gene from the white rot fungus Pycnoporus cinnabarinus (Jacq.) P. Karst. (commonly referred to as Cinnabar polypore) in Aspergillus niger. The lac1 gene was placed downstream of the strong constitutive promoter of glyceraldehyde-3-phosphate dehydrogenase, which provoked an 80-fold increase in the synthesis of the recombinant laccase. In a later study, Kiiskinen et al. [142] reported the production of Melanocarpus albomyces (Cooney & R. Emers.) Arx laccase lcc in Trichoderma reesei under the strong cellobiohydrolase cbh promoter. In the same year, Hoshida et al. [143] described the use of A. flavus var. oryzae and S. cerevisiae as hosts for overexpressing Pycnoporus coccineus (Fr.) Bondartsev & Singer laccase lcc1. Laccase synthesis was driven by the maltose inducible promoter amyB in A. flavus var. oryzae, whereas the galactose inducible GAL10 promoter was employed in S. cerevisiae.
Yeasts (methylotrophs or non-methylotrophs) have post-translational modifications. Moreover, they are fast growing, have simple morphology, produce biomass, and are safe [144]. Yeasts have been established as suitable hosts for the heterologous expression of recombinant proteins [145, 146]. Examples of yeast genera commonly adopted as a chassis for the synthesis of heterologous proteins are Saccharomyces and Yarrowia, among non-methylotroph yeasts, together with P. pastoris and Hansenula polymorpha (methylotroph yeasts). Many wild-type and mutant strains of various Saccharomyces species (e.g. S. cerevisiae, S. uvaruium, and S. boulardi) are now available for research and industrial applications. Compared to other fungi and higher eukaryotes in general, the yeast genome is easier to modify by exploiting homologous recombination (Fig. 4).

General overview on the approaches for optimizing enzyme expression in yeast
Moreover, many constitutive and strong synthetic promoters have been characterized in several yeast species like Kluyveromyces lactis, P. pastoris, Yarrowia lipolytica, Pichia methalonic, Cryptococcus sp., and S. cerevisiae [147]. These promoters have been used to improve in yeasts the expression of different LEs from higher fungi [148]. Apart from using strong synthetic promoters, enzyme production in yeast can be enhanced by means of multicopy plasmids. These plasmids can dramatically increase the copies of a transcription unit carrying the enzyme CDS into yeast cells [145]. Episomal shuttle vectors replicate in the host cell autonomously and are commonly utilized for the expression of heterologous genes in yeast [149]. These plasmids are called shuttle because they can propagate in both yeast and bacteria. The gene of interest is inserted into the episomal shuttle vector in vitro. Afterwards, bacterial—usually E. coli—and yeast cells are transformed with the newly assembled plasmid. Escherichia coli serves as a storage means, whereas yeast provides the machinery necessary for gene expression. However, E. coli can also be used, besides yeast and filamentous fungi, as an expression system for several kinds of LEs, as witnessed by the numerous works listed in Table 2.
Conclusion
Ligninolytic enzymes produced by fungi have been extensively exploited in a number of industries due to their versatile application, which derives from their lignin-based compounds catalytic oxidation properties. The large variety of lignin modifying fungal species and related ligninolytic enzyme consortium described in this review have been used in the production of biofuels, antibiotics, and fermented products, as well as in bioremediation and biomedical application as biosensors. The essential prerequisite for a successful use of fungal ligninolytic enzymes is their production in large quantities. Over the last decade, combined synthetic biology and computational designs have yielded significant results in enhancing the synthesis of natural compounds in fungi. Application of CRISPR-Cas system has shown great efficiency in discovery, activation, and editing of fungal genes/BGCs, and it holds a potential headway for production or enhancement of ligninolytic enzymes in large-scale. In addition, many newly available algorithms have been used to optimize CRISPR-Cas systems for editing specific loci or estimating potential off-target effects if used in fungal genomes. Further research could be directed towards improving the production of ligninolytic secretomes (or new ligninolytic genes) with desired attributes such as thermal, catalytic, or substrate adaptability. Various bacteria such as E. coli and Bacillus subtilis and filamentous fungi such as Aspergillus and Trichoderma species have been used as synthetic hosts for the expression of some LEs from white rot fungi. Yeasts such as S. cerevisiae, P. pastoris, K. lactis, and Y. lipolytica have been identified with good minicellulosomes, thus being good candidates for synthetic production of ligninolytic secretomes. Several studies have demonstrated heterologous expression in yeasts of ligninolytic enzymes from higher fungi. These re-engineered yeasts show adequate enzyme secretion with high tither values that do not affect negatively their cellular physiology. Yeast transformants in most cases showed better tolerance to oxidative stress, after the experiment, and ability to produce recombinant LEs in multiple folds, having better specification, quality, and reactivity as compared to the wild type. In addition, recombinant LEs can be easily characterized with molecular structure and studied for iron trafficking/hierarchy distribution in the cell.
Abbreviations
- AAO:
-
Aryl-alcohol oxidase
- ABTS:
-
1-Hydroxybenzotriazole and 2,2-azinobis 3-ethylbenz-thiazoline-6-sulfonate
- antiSMASH:
-
Antibiotics and secondary metabolites analysis shell
- AA:
-
Auxiliary activity
- BGCs:
-
Biosynthetic gene clusters
- BPs:
-
Biosynthetic pathways
- BQR:
-
Benzoquinone reductase
- CAD:
-
Computer-aided design
- CASSIS:
-
Cluster assignment by islands of sites
- CASTing:
-
Combinatorial active site saturation testing
- CAZy:
-
Carbohydrate active enzymes
- CB:
-
Combinatorial biosynthesis
- CDH:
-
Cellobiose hydrogenase
- CE:
-
Chimeric enzymes
- CDS:
-
Coding sequence
- CPO:
-
Chloroperoxidase
- CRISPR-Cas:
-
Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated protein
- CU:
-
Codon usage
- DE:
-
Directed evolution
- DyP:
-
Dye-decolorizing peroxidase
- ECC:
-
Enzyme Commission classification
- Eps:
-
Enzymatic pathways
- ER:
-
Enzyme resurrection
- GAO:
-
Galactose oxidase
- GLOX:
-
Glyoxal oxidase
- GMMs:
-
Genome-scale metabolic models
- GOX:
-
Glucose oxidase
- Hex:
-
Heterologous expression
- HTPs:
-
Heme thiolate peroxidases
- LB:
-
Lignocellulose biorefinery
- LCC:
-
Laccase
- LDA:
-
Lignin-degrading auxiliary enzymes
- LEs:
-
Ligninolytic enzymes
- LEC:
-
Ligninolytic enzyme consortium
- LiP:
-
Lignin peroxidase
- LMCO:
-
Laccase-like multicopper oxidase
- LMEs:
-
Lignin-modifying enzymes
- MF:
-
Metabolic flux
- MIBiG:
-
Minimum Information about Biosynthetic Gene cluster
- MnP:
-
Manganese peroxidase
- MOX:
-
Methanol oxidase
- MIDDAS-M:
-
Motif-independent de novo detection algorithm for secondary metabolite biosynthetic gene clusters
- NPs:
-
Natural products
- P2O:
-
Pyranose 2-oxidase
- RiPPs:
-
Ribosomally synthesized and post-translationally modified peptides
- RBS:
-
Ribosome binding site
- SBP:
-
Silent biosynthetic pathways
- SMIPS:
-
Secondary metabolite by inter-pro-scan
- SMURF:
-
Secondary metabolite unique regions finder
- TLS:
-
The ligninolytic secretome
- TALENs:
-
Transcription Activator-like Effector Nucleases
- UPOs:
-
Nonspecific peroxygenases
- VAO:
-
Vanillyl-alcohol oxidase
- Vp:
-
Versatile peroxidase
- WRF:
-
White-rot fungi
References
Tien M, Kirk TK. Lignin-degrading enzyme from the hymenomycete Phanerochaete chrysosporium Burds. Science. 1983;221:661–3.
Blanchette RA, Krueger EW, Haight JE, Akhtar M, Akin DE. Cell wall alterations in loblolly pine wood decayed by the white-rot fungus Ceriporiopsis subvermispora. J Biotechnol. 1997;53:203–13. https://doi.org/10.1016/S0168-1656(97)01674-X.
Glenn JK, Morgan MA, Mayfield MB, Kuwahara M, Gold MH. An extracellular H2O2-requiring enzyme preparation involved in lignin biodegradation by the white rot basidiomycete Phanerochaete chrysosporium. Biochem Biophy Res Co. 1983;114(3):1077–83. https://doi.org/10.1016/0006-291X(83)90672-1.
Asemoloye MD, Ahmad R, Jonathan SG. Transcriptomic responses of catalase, peroxidase and laccase encoding genes and enzymatic activities of oil spill inhabiting rhizospheric fungal strains. Environ Pollut. 2017;235:55–64. https://doi.org/10.1016/j.envpol.2017.12.042.
Asemoloye MD, Tosi S, Daccò C, Wang X, Xu S, Marchisio MA, Gao W, Jonathan SG, Pecoraro L. Hydrocarbon degradation and enzyme activities of Aspergillus oryzae and Mucor irregularis isolated from Nigerian crude oil-polluted sites. Microorganisms. 2020;8(12):1912. https://doi.org/10.3390/microorganisms8121912.
Wang H, Hunag L, Yanzhen L, et al. Characterization and application of a novel laccase derived from Bacillus amyloliquefaciens. International J Biol Macromol. 2020;150:982–90.
Rodríguez Couto S, Toca Herrera JL. Industrial and biotechnological applications of laccases: a review. Biotechnol Adv. 2006;24(5):500–13. https://doi.org/10.1016/j.biotechadv.2006.04.003.
Choi KR, Jang WD, Yang D, Cho JS, Park D, Lee SY. Systems Metabolic Engineering Strategies: Integrating Systems and Synthetic Biology with Metabolic Engineering. Trends in Biotechnology. 2019;1–21. http://doi.org/https://doi.org/10.1016/j.tibtech.2019.01.003.
Ro DK, Paradise EM, Ouellet M, et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440(7086):940–3. https://doi.org/10.1038/nature04640.
Endy D. Foundations for engineering biology. Nature. 2005;438(7067):449–53. https://doi.org/10.1038/nature04342.
Hasty J, McMillen D, Collins JJ. Engineered gene circuits. Nature. 2002;420(6912):224–30. https://doi.org/10.1038/nature01257.
Marchisio MA, Stelling J. Computational design tools for synthetic biology. Cur Opin Biotechnol. 2009;20(4):479–85. https://doi.org/10.1016/j.copbio.2009.08.007.
MacDonald JT, Barnes C, Kitney RI, Freemont PS, Stan GBV. Computational design approaches and tools for synthetic biology. Integrative Biology. 2011;3(2):97–108. https://doi.org/10.1039/c0ib00077a.
Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE. 2008;3(11):e3647. https://doi.org/10.1371/journal.pone.0003647.
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–5. https://doi.org/10.1038/nmeth.1318.
Khalil AS, Collins JJ. Synthetic biology: applications come of age. Nat Rev Genet. 2010;11(5):367–79. https://doi.org/10.1038/nrg2775.
Medema MH, Fischbach MA. Computational approaches to natural product discovery. Nat Chem Biol. 2015;11:639–48. https://doi.org/10.1038/nchembio.1884.
van der Lee TAJ, Medema MH. Computational strategies for genome-based natural product discovery and engineering in fungi Fungal Gen. Biol. 2016;89:29–36.
Khaldi N, Seifuddin FT, Turner G, et al. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47:736–41.
Medema MH, Blin K, Cimermancic P, et al. AntiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2013;9:w339–46. https://doi.org/10.1093/nar/gkr466.
Blin K, Shaw S, Steinke K, et al. antiSMASH 50: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019;47(W1):W81–7. https://doi.org/10.1093/nar/gkz310.
Wolf T, Shelest V, Nath N, Shelest E. CASSIS and SMIPS: promoter-based prediction of secondary metabolite gene clusters in eukaryotic genomes. Bioinformatics. 2016;32(8):1138–43. https://doi.org/10.1093/bioinformatics/btv713.
Umemura M, Koike H, Nagano N, et al. MIDDAS-M: motif-independent de novo detection of secondary metabolite gene clusters through the integration of genome sequencing and transcriptome data. PLoS ONE. 2013;8(12):e84028–110. https://doi.org/10.1371/journal.pone.0084028.
Takeda I, Umemura M, Koike H, Asai K, Machida M. Motif-independent prediction of a secondary metabolism gene cluster using comparative genomics: application to sequenced genomes of Aspergillus and ten other filamentous fungal species. DNA Res. 2014;21:447–57. https://doi.org/10.1093/dnares/dsu010.
Kautsar SA, Blin K, Shaw S, et al. MIBiG 2.0: a repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2019;47:W81–5. https://doi.org/10.1093/nar/gkz882.
Medema MH, van Raaphorst R, Takano E, Breitling R. Computational tools for the synthetic design of biochemical pathways. Nat Rev. 2012;10:191–202. https://doi.org/10.1038/nrmicro2717.
Carbonell P, Parutto P, Baudier C, Junot C, Faulon JL. Retropath: automated pipeline for embedded metabolic circuits. ACS Synth Biol. 2014;3(8):565–77. https://doi.org/10.1021/sb4001273.
Skellam E. Strategies for engineering natural product biosynthesis in fungi. Tre Biotechnol. 2019;37:4. https://doi.org/10.1016/j.tibtech.2018.09.003.
Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci. 1992;89(12):5547–51.
Macheleidt J, Scherlach K, Neuwirth T, et al. Transcriptome analysis of cyclic AMP-dependent protein kinase A-regulated genes reveals the production of the novel natural compound fumipyrrole by Aspergillus fumigatus. Mol Microbiol. 2015;96(1):148–62. https://doi.org/10.1111/mmi.12926.
Rantasalo A, Landowski CP, Kuivanen J, et al. A universal gene expression system for fungi. Nucleic Acids Res. 2018;42:e48–15. https://doi.org/10.1093/nar/gky558.
Palmer CN, Axen E, Hughes V, Wolf CR. The repressor protein, Bm3R1, mediates an adaptive response to toxic fatty acids in Bacillus megaterium. J Biolo Chem. 1998;273(29):18109–16. https://doi.org/10.1074/jbc.273.29.18109.
Louvion JF, Havaux-Copf B, Picard D. Fusion of GAL4-VP16 to a steroid-binding domain provides a tool for gratuitous induction of galactose-responsive genes in yeast. Gene. 1993;131(1):129–34. https://doi.org/10.1016/0378-1119(93)90681-r.
Zhang S, Guo F, Yan W, et al. Recent Advances of CRISPR/Cas9-Based Genetic Engineering and Transcriptional Regulation in Industrial Biology. Front Bioeng Biotechnol. 2020;7:585–611. https://doi.org/10.3389/fbioe.2019.00459.
Martins-Santana L, Nora LC, Sanches-Medeiros A, Lovate GL, Cassiano MHA, Silva-Rocha R. Systems and synthetic biology approaches to engineer fungi for fine chemical production. Front Bioeng Biotechnol. 2018;6:831–918. https://doi.org/10.3389/fbioe.2018.00117.
Cui Y. Review of CRISPR/Cas9 sgRNA design tools. Interdiscipl Sci Comput Life Sci. 2018;10(2):455–65. https://doi.org/10.1007/s12539-018-0298-z.
Li H, Dai M, Dai S, Dong X. Current status and environment impact of direct straw return in China’s cropland—a review. Ecotox Environ Safe. 2018;159:293–300.
Liu H, Ou X, Yuan J, Yan X. Experience of producing natural gas from corn straw in China. Resour Conserv Recycl. 2018;135:216–24.
Li X, Xia J, Zhu X, Bilal M, Tan Z, Shi H. Construction and characterization of bifunctional cellulases: Caldicellulosiruptor-sourced endoglucanase, CBM, and exoglucanase for efficient degradation of lignocellulose. Biochem Eng J. 2019;151:107363.
Moura JC, Bonine CA, de Oliveira F, Viana J, Dornelas MC, Mazzafera P. Abiotic and biotic stresses and changes in the lignin content and composition in plants. J Integr Plant Biol. 2010;52:360–76. https://doi.org/10.1111/j.1744-7909.2010.00892.x.
Bilal M, Iqbal HMN. Lignin peroxidase immobilization on Ca-alginate beads and its dye degradation performance in a packed bed reactor system. Biocatal Agricult Biotechnol. 2019;20:101205.
Mäkelä MR, Bredeweg EL, Magnuson JK, Baker SE, de Vries RP, Hildén K. Fungal ligninolytic enzymes and their applications. Microbiol. Spect. 2016;4(6):FUNK-0017-2016. doi:https://doi.org/10.1128/microbiolspec. FUNK-0017-2016.
Zhang S, Xiao J, Wang G, Chen G. Enzymatic hydrolysis of lignin by ligninolytic enzymes and analysis of the hydrolyzed lignin products. Biores Technol. 2020;304:122975.
Kirk TK, Farrell RL. Enzymatic “combustion”: the microbial degradation of lignin. Annu Rev Microbiol. 1987;41:465–505.
Plácido J, Capareda S. Ligninolytic enzymes: a biotechnological alternative for bioethanol production. Biores Bioproc. 2015;2:23. https://doi.org/10.1186/s40643-015-0049-5.
Manavalan T, Manavalan A, Heese K. Characterization of lignocellulolytic enzymes from whiterot fungi. Curr Microbiol. 2015;70(4):485–98.
Brischke C, Alfredsen G. Wood-water relationships and their role for wood susceptibility to fungal decay. Appl Microbiol Biotechnol. 2020;104:3781–95. https://doi.org/10.1007/s00253-020-10479-1.
Kaffenberger JT, Schilling JS. Using a grass substrate to compare decay among two clades of brown rot fungi. Appl Microbiol Biotechnol. 2013;97(19):8831–40. https://doi.org/10.1007/s00253-013-5142-0.
Sigoillot J, Berrin J, Bey M, et al. Fungal strategies for lignin degradation. Adv Bot Res. 2012;61:263–308.
Spina F, Gea M, Bicchi C, Cordero C, Schilir T, Varese GC. Ecofriendly laccases treatment to challenge micropollutants issue in municipal wastewaters. Environ Pol. 2020;257:113579.
Singh AD, Kumar SR. Ligninolytic fungal laccases and their biotechnological applications. Appl Biochem Biotech. 2010;160:1760–88.
Fukasawa Y, Osono T, Takeda H. Wood decomposing abilities of diverse lignicolous fungi on nondecayed and decayed beech wood. Mycologia. 2011;103:474–82.
Nagy LG, Riley R, Tritt A, Adam C, Daum C, Floudas D. Comparative genomics of early diverging mushroom-forming fungi provides insights into the origins of lignocellulose decay capabilities. Mol Biol Evol. 2016;3:959–70. https://doi.org/10.1093/molbev/msv337.
Betts WB, Dart RK. Printed in Great Britain 241 3 the degradation of lignin-related compounds by Aspergillus flavus. J Gen Microbiol. 1988;134(241):3–2420.
Liers C, Ullrich R, Steffen KT, Hatakka A, Hofrichter M. Mineralization of 14C-labelled synthetic lignin and extracellular enzyme activities of the wood-colonizing ascomycetes Xylaria hypoxylon and Xylaria polymorpha. Appl Microbiol Biotechnol. 2006;69:573–9. https://doi.org/10.1007/s00253-005-0010-1.
Asemoloye MD, Ahmad R, Jonathan SG. Synergistic action of rhizospheric fungi with Megathyrsus maximus root speeds up hydrocarbon degradation kinetics in oil polluted soil. Chemosphere. 2017;184:1–12.
Asemoloye MD, Jonathan SG, Jayeola AA, Ahmad R. Mediational influence of spent mushroom compost on phytoremediation of black-oil hydrocarbon polluted soil and response of Megathyrsus maximus Jacq. J Environ Manage. 2017;200:253–62.
Solairaj D, Ngea N, Legrand G, Yang Q, Zhang H. Isolation of pathogenic fungi causing postharvest decay in table grapes and in vivo biocontrol activity of selected yeasts against them. Physiol Mol Plant Pathol. 2020;110:101478. https://doi.org/10.1016/j.pmpp.2020.101478.
Lombard V, Golaconda RH, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(D1):D490–5. https://doi.org/10.1093/nar/gkt1178.
Floudas D, Binder M, Riley R, et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336:1715–9. https://doi.org/10.1126/science.1221748.
Ruiz-Dueñas FJ, Lundell T, Floudas D, et al. Lignin-degrading peroxidases in Polyporales: an evolutionary survey based on 10 sequenced genomes. Mycologia. 2013;105:1428–44. https://doi.org/10.3852/13-059.
Riley R, Salamov AA, Brown DW, et al. Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc Natl Acad Sci USA. 2014;111:9923–8. https://doi.org/10.1073/pnas.1400592111.
Hatakka A, Hammel KE. Fungal biodegradation of lignocelluloses. In: Esser K, Hofrichter M, editors. The mycota: a comprehensive treatise on fungi as experimental systems for basic and applied research industrial applications, vol. X. 2nd ed. Berlin: Springer; 2010. p. 319–40.
Martínez AT. Molecular biology and structure-function of lignin-degrading heme peroxidases. Enzyme Microb Technol. 2002;30:425–44. https://doi.org/10.1016/S0141-0229(01)00521-X.
Doyle WA, Blodig W, Veitch NC, Piontek K, Smith AT. Two substrate interaction sites in lignin peroxidase revealed by site-directed mutagenesis. Biochemistry. 1998;37:15097–105. https://doi.org/10.1021/bi981633h.
Choinowski T, Blodig W, Winterhalter KH, Piontek K. The crystal structure of lignin peroxidase at 1.70 Å resolution reveals a hydroxy group on the cβ of tryptophan 171: a novel radical site formed during the redox cycle. J Mol Biol. 1999;286:809–27. https://doi.org/10.1006/jmbi.1998.2507.
Ruiz-Dueñas FJ, Morales M, García E, Miki Y, Martínez MJ, Martínez AT. Substrate oxidation sites in versatile peroxidase and other basidiomycete peroxidases. J Exp Bot. 2009;6:441–52. https://doi.org/10.1093/jxb/ern261.
Hofrichter M, Ullrich R, Pecyna MJ, Liers C, Lundell T. New and classic families of secreted fungal heme peroxidases. Appl Microbiol Biotechnol. 2010;87:871–97. https://doi.org/10.1007/s00253-010-2633-0.
Kojiro K, Miki T, Sugimoto H, Nakajima M, Kanayama K. Microspores and mesospores in the cell wall of dry wood. J Wood Sci. 2010;56:107–11. https://doi.org/10.1007/s10086-009-1063-z.
Colpa DI, Fraaije MW, van Bloois E. DyP-type peroxidases: a promising and versatile class of enzymes. J Ind Microbiol Biotechnol. 2014;41:1–7. https://doi.org/10.1007/s10295-013-1371-6.
Hoegger PJ, Kilaru S, James TY, Thacker JR, Kües U. Phylogenetic comparison and classification of laccase and related multicopper oxidase protein sequences. FEBS J. 2006;273:2308–26. https://doi.org/10.1111/j.1742-4658.2006.05247.x.
Kües U, Rühl M. Multiple multi-copper oxidase gene families in basidiomycetes: what for? Curr Genomics. 2011;12:72–94. https://doi.org/10.2174/138920211795564377.
Thurston CF. The structure and function of fungal laccases. Microbiol. 1994;140:19–26. https://doi.org/10.1099/13500872-140-1-19.
Liers C, Aranda E, Strittmatter E, et al. Phenol oxidation by DyP-type peroxidases in comparison to fungal and plant peroxidases. J Mol Catal B Enzym. 2014;03:41–6. https://doi.org/10.1016/j.molcatb.2013.09.025.
Liers C, Bobeth C, Pecyna M, Ullrich R, Hofrichter M. DyPlike peroxidases of the jelly fungus Auricularia auricula-judae oxidize nonphenolic lignin model compounds and high-redox potential dyes. Appl Microbiol Biotechnol. 2010;85:1869–79. https://doi.org/10.1007/s00253-009-2173-7.
Liers C, Pecyna MJ, Kellner H. Substrate oxidation by dye-decolorizing peroxidases (DyPs) from wood- and litter-degrading agaricomycetes compared to other fungal and plant heme-peroxidases. Appl Microbiol Biotechnol. 2013;97:5839–49. https://doi.org/10.1007/s00253-012-4521-2.
Sugano Y, Nakano R, Sasaki K, Shoda M. Efficient heterologous expression in Aspergillus oryzae of a unique dye-decolorizing peroxidase, DyP, of Geotrichum candidum Dec 1. Appl Environ Microbiol. 2000;66:1754–8. https://doi.org/10.1128/AEM.66.4.1754-1758.2000.
Fernández-Fueyo E, Linde D, Almendral D, López-Lucendo MF, Ruiz-Dueñas FJ, Martínez AT. Description of the first fungal dyedecolorizing peroxidase oxidizing manganese(II). Appl Microbiol Biotechnol. 2015;99:8927–42. https://doi.org/10.1007/s00253-015-6665-3.
Kinne M, Poraj-Kobielska M, Ullrich R, et al. Oxidative cleavage of non-phenolic β-O-4 lignin model dimers by an extracellular aromatic peroxygenase. Holzforschung. 2011;65:673–9. https://doi.org/10.1515/hf.2011.057.
Ullrich R, Hofrichter M. The haloperoxidase of the agaric fungus Agrocybe aegerita hydroxylates toluene and naphthalene. FEBS Lett. 2005;579:6247–50. https://doi.org/10.1016/j.febslet.2005.10.014.
Hofrichter M, Martínez AT, del Río JC. Regioselective oxygenation of fatty acids, fatty alcohols and other aliphatic compounds by a basidiomycete heme-thiolate peroxidase. Arch Biochem Biophys. 2011;514:33–43. https://doi.org/10.1016/j.abb.2011.08.001.
Hofrichter M, Kellner H, Pecyna MJ, Ullrich R. Fungal unspecific peroxygenases: heme-thiolate proteins that combine peroxidase and cytochrome p450 properties. Adv Exp Med Biol. 2015;851:341–68. https://doi.org/10.1007/978-3-319-16009-2_13.
Giardina P, Faraco V, Pezzella C, Piscitelli A, Vanhulle S, Sannia G. Laccases: a never-ending story. Cell Mol Life Sci. 2010;67:369–85. https://doi.org/10.1007/s00018-009-0169-1.
Hernández-Ortega A, Ferreira P, Martínez AT. Fungal arylalcohol oxidase: a peroxide-producing flavoenzyme involved in lignin degradation. Appl Microbiol Biotechnol. 2012;93:1395–410. https://doi.org/10.1007/s00253-011-3836-8.
Wongnate T, Surawatanawong P, Visitsatthawong S, Sucharitakul J, Scrutton NS, Chaiyen P. Proton-coupled electron transfer and adduct configuration are important for C4a-hydroperoxyflavin formation and stabilization in a flavoenzyme. J Am Chem Soc. 2014;136:241–53. https://doi.org/10.1021/ja4088055.
Kersten PJ, Kirk TK. Involvement of a new enzyme, glyoxal oxidase, in extracellular H2O2 production by Phanerochaete chrysosporium. J Bacteriol. 1987;169:2195–201.
Alcalde M. Engineering the ligninolytic enzyme consortium Trend. Biotechnol. 2014;8:8. https://doi.org/10.1016/j.tibtech.2014.12.007.
Bozell JJ, Petersen GR. Technology development for the production of biobased products from biorefinery carbohydrates—the US Department of Energy’s ‘Top 10’ revisited. Green Chem. 2010;12:539–54.
Hawksworth DL, Lücking R. Fungal diversity revis-ited: 22 to 38 million species. Microbiol Spectr. 2017;5:120. https://doi.org/10.1128/microbiolspec.FUNK-0052-2016.
Nordberg H, Cantor M, Dusheyko S, et al. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2014;42(Database issue):D26–31. https://doi.org/10.1093/nar/gkt1069.
Stajich JE, Harris T, Brunk BP, et al. FungiDB: an integrated functional genomics database for fungi. Nucleic Acids Res. 2011;40(D1):D675–81. https://doi.org/10.1093/nar/gkr918.
Basenko E, Pulman J, Shanmugasundram A, et al. FungiDB: an integrated bioinformatic resource for fungi and oomycetes. J Fungi. 2018;4(1):39–28. https://doi.org/10.3390/jof4010039.
Wang X, Zhang X, Liu L. et al. Genomic and transcriptomic analysis of the endophytic fungus Pestalotiopsis fici reveals its lifestyle and high potential for synthesis of natural products. BMC Genomics 2015;16:28. https://doi.org/https://doi.org/10.1186/s12864-014-1190-9.
Gupta RD, Tawfik DS. Directed enzyme evolution via small and effective neutral drift libraries. Nat Met. 2008;5:939–42.
Goldsmith M, Tawfik DS. Enzyme engineering by targeted libraries. Method Enzymol. 2013;523:257–83.
Cimermancic P, Medema MH, Claesen J, et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell. 2014;158:412–21. https://doi.org/10.1016/j.cell.2014.06.034.
Festa G, Autore F, Fraternali F, Giardina P, Sannia G. Development of new laccases by directed evolution: functional and computational analyses. Proteins. 2008;72(1):25–34. https://doi.org/10.1002/prot.21889.
Garcia-Ruiz E, Gonzalez-Perez D, Ruiz-Dueñas FJ, Martínez AT, Alcalde M. Directed evolution of a temperature-, peroxide- and alkaline pH-tolerant versatile peroxidase. Biochem J. 2012;441(1):487–98. https://doi.org/10.1042/BJ20111199.
Ouyang F, Zhao M. Enhanced catalytic efficiency of CotA-laccase by DNA shuffling. Bioengineered. 2019;10(1):182–9. https://doi.org/10.1080/21655979.2019.1621134.
Bulter T, Alcalde M, Sieber V, Meinhold P, Schlachtbauer C, Arnold FH. Functional expression of a fungal laccase in Saccharomyces cerevisiae by directed evolution [published correction appears. Appl Environ Microbiol. 2003;69(2):987–95. https://doi.org/10.1128/aem.69.2.987-995.2003.
Zumárraga M, Vaz Domínguez C, Camarero S, et al. Combinatorial saturation mutagenesis of the Myceliophthora thermophila laccase T2 mutant: the connection between the C-terminal plug and the conserved 509VSG511tripeptide. Comb Chem High-Throughput Screen. 2008;11:807–16.
Morawski B, Lin Z, Cirino P, Joo H, Bandara G, Arnold FH. Functional expression of horseradish peroxidase in Saccharomyces cerevisiae and Pichia pastoris. Prot Eng Design Sel. 2000;13(5):377–84. https://doi.org/10.1093/protein/13.5.377.
García-Ruiz E, Maté D, Ballesteros A, Martinez AT, Alcalde M. Evolving thermostability in mutant libraries of ligninolytic oxidoreductases expressed in yeast. Microb Cell Fact. 2010;9:17. https://doi.org/10.1186/1475-2859-9-17.
Gonzalez-Perez D, Garcia-Ruiz E, Ruiz-Dueñas FJ, Martinez AT, Alcalde M. Structural determinants of oxidativestabilization in an evolved versatile peroxidase. ACS Catal. 2014;4:3891–901.
Miyazaki-Imamura C, Oohira K, Kitagawa R, Nakano H, Yamane T, Takahashi H. Improvement of H2O2 stability of manganese peroxidase by combinatorial mutagenesis and high-throughput screening using in vitro expression with protein disulfide isomerase. Protein Eng. 2003;16(6):423–8. https://doi.org/10.1093/protein/gzg054.
Ryu K, Hwang SY, Kim KH, Kang JH, Lee EK. Functionality improvement of fungal lignin peroxidase by DNA shuffling for 2,4-dichlorophenol degradability and H2O2 stability. J Biotechnol. 2008;133(1):110–5. https://doi.org/10.1016/j.jbiotec.2007.09.008.
Ryu K, Kang JH, Wang L, Lee EK. Expression in yeast of secreted lignin peroxidase with improved 2,4-dichlorophenol degradability by DNA shuffling. J Biotechnol. 2008;135(3):241–6. https://doi.org/10.1016/j.jbiotec.2008.04.007.
Molina-Espeja P, Garcia-Ruiz E, Gonzalez-Perez D, Ullrich R, Hofrichter M, Alcalde M. Directed evolution of unspecific peroxygenase from Agrocybe aegerita. Appl Environ Microbiol. 2014;80(11):3496–507. https://doi.org/10.1128/AEM.00490-14.
Sygmund C, Santner P, Krondorfer I, et al. Semi-rational engineering of cellobiose dehydrogenase for improved hydrogen peroxide production. Microb Cell Fact. 2013;12:38. https://doi.org/10.1186/1475-2859-12-38.
Mate DM, Alcalde M. Laccase engineering: from rational design to directed evolution. Biotechnol Adv. 2015;33:25–40.
Madzak C, Mimmi MC, Caminade E, et al. Shifting the optimal pH of activity for a laccase from the fungus Trametes versicolor by structure-based mutagenesis. Prot Eng Des Sel. 2006;9:77–84.
Galli C, Gentili P, Jolivalt C, Madzak C, Vadalà R. How is the reactivity of laccase affected by single-point mutations? Engineering laccase for improved activity towards sterically demanding substrates. Appl Microbiol Biotechnol. 2011;91:123–31.
Chica RA, Doucet N, Pelletier JN. Semi rational approaches to engineering enzyme activity: combining the benefits of directed evolution and rational design. Curr Opin Biotechnol. 2005;16:378–84.
Reetz MT, Wang L, Bocola M. Directed evolution of enantioselective enzymes: iterative cycles of CASTing for probing protein-sequence space. Angew Chem. 2006;45:1236–41.
Andberg M, Hakulinen N, Auer S, et al. Essential role of the C-terminus in Melanocarpus albomyces laccase for enzyme production, catalytic properties and structure. FEBS J. 2009;276:6285–300.
Wanka F, Cairns T, Boecker S, et al. Tet-on, or Tet-off, that is the question: Advanced conditional gene expression in Aspergillus. Fungal Genet Biol. 2016;89(C):72–83. https://doi.org/10.1016/j.fgb.2015.11.003.
Gossen M, Freundlieb S, Bender G, Müller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–9. https://doi.org/10.1126/science.7792603.
Bellí G, Garí E, Piedrafita L, Aldea M, Herrero E. An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic Acids Res. 1998;26(4):942–7.
Carver LA, Jackiw V, Bradfield CA. The 90-kDa heat shock protein is essential for Ah receptor signaling in a yeast expression system. J Biol Chem. 1994;269(48):30109–12.
Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333(6051):1843–6. https://doi.org/10.1126/science.1204094.
Lange O, Binder A, Lahaye T. From dead leaf, to new life: TALeffectors as tools for synthetic biology. Plant J. 2014;78(5):753–71. https://doi.org/10.1111/tpj.12431.
Perez-Quintero AL, Szurek B. A decade decoded: spies and hackers in the history of TAL effectors research. Annu Rev Phytopathol. 2019;57(1):459–81. https://doi.org/10.1146/annurev-phyto-082718-100026.
Arazoe T, Ogawa T, Miyoshi K, et al. Tailor-made TALEN system for highly efficient targeted gene replacement in the rice blast fungus. Biotechnol Bioeng. 2015;112:1335–42. https://doi.org/10.1002/bit.25559.
Mizutani O, Arazoe T, Toshida K, et al. Detailed analysis of targeted gene mutations caused by the platinum-fungal TALENs in Aspergillus oryzae RIB40 strain and a ligD disruptant. J Biosci Bioeng. 2017;123:287–93. https://doi.org/10.1016/j.jbiosc.2016.09.014.
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169(12):5429–33. https://doi.org/10.1128/jb.169.12.5429-5433.1987.
Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–12. https://doi.org/10.1126/science.1138140.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–21. https://doi.org/10.1126/science.1225829.
Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163(3):759–71. https://doi.org/10.1016/j.cell.2015.09.038.
van Leeuwe TM, Arentshorst M, Ernst T, Alazi E, Punt PJ, Ram AFJ. Efficient marker free CRISPR/Cas9 genome editing for functional analysis of gene families in filamentous fungi. Fungal Biol Biotechnol 2019;1–13.
Zhang C, Meng X, Wei X, Lu L. Highly efficient CRISPR mutagenesis by microhomology-mediated end joining in Aspergillus fumigatus. Fungal Genet Biol. 2016;86(C):47–57. https://doi.org/10.1016/j.fgb.2015.12.007.
Zheng YM, Lin F, Zhou G, et al. Development of a versatile and conventional technique for gene disruption in filamentous fungi based on CRISPR–Cas9 technology. Sci Rep. 2017;7:9250. https://doi.org/10.1038/s41598-017-10052-3.
Weber J, Valiante V, Nødvig CS, et al. Functional reconstitution of a fungal natural product gene cluster by advanced genome editing. ACS Synth Biol. 2017;6(1):62–8. https://doi.org/10.1021/acssynbio.6b00203.
Shi TQ, Gao J, Wang WJ, et al. CRISPR/Cas9-based genome editing in the filamentous fungus Fusarium fujikuroi and its application in strain engineering for gibberellic acid production. ACS Synth Biol. 2019. https://doi.org/10.1021/acssynbio.8b00478.
Kim H, Ji C, Je H, Kim J, Kang H. mpCRISTAR: Multiple plasmid approach for CRISPR/Cas9 and TAR-mediated multiplexed refactoring of natural product biosynthetic gene clusters. ACS Synth Biol. 2019. https://doi.org/10.1021/acssynbio.9b00382.
Liu Q, Gao R, Li J, Lin L, Zhao J, Sun W, Tian C. Development of a genome-editing CRISPR/Cas9 system in thermophilic fungal Myceliophthora species and its application to hyper-cellulase production strain engineering. Biotechnol Biofuels. 2017. https://doi.org/10.1186/s13068-016-0693-9.
Shi TQ, Liu G, Yu J, et al. CRISPR/Cas9-based genome editing of the filamentous fungi: the state of the art. Appl Microbiol Biotechnol. 2017;1:7435–43. https://doi.org/10.1007/s00253-017-8497-9.
Vanegas KG, Jarczynska ZD, Strucko T, Mortensen UH. Cpf1 enables fast and efficient genome editing in Aspergilli. Fungal Biol Biotechnol. 2019. https://doi.org/10.1186/s40694-019-0069-6.
Roux I, Woodcraft C, Hu J, Wolters R, Gilchrist CLM, Chooi YH. CRISPR-mediated activation of biosynthetic gene clusters for bioactive molecule discovery in filamentous fungi. ACS Synth Biol. 2020. https://doi.org/10.1021/acssynbio.0c00197.
Beerli RR, Dreier B, Barbas CF. Positive and negative regulation of endogenous genes by designed transcription factors. Proc Natl Acad Sci. 2000;97(4):1495–500.
Kwon MJ, Schütze T, Spohner S, Haefner S, Meyer V. Practical guidance for the implementation of the CRISPR genome editing tool in filamentous fungi. Fungal Biol Biotechnol. 2019. https://doi.org/10.1186/s40694-019-0079-4.
Record E, Punt PJ, Chamkha M, et al. Expression of the Pycnoporus cinnabarinus laccase gene in Aspergillus niger and characterization of the recombinant enzyme. Eur J Biochem. 2002;269:602–9.
Kiiskinen LL, Kruus K, Bailey M, Ylosmaki E, Siika-aho M, Saloheimo M. Expression of Melanocarpus albomyces laccase in Trichoderma reesei and characterization of the purified enzyme. Microbiol. 2004;150:3065–74.
Hoshida H, Fujita T, Murata K, Kubo K, Akada R. Copper-dependent production of a Pycnoporus coccineus extracellular laccase in Aspergillus oryzae and Saccharomyces cerevisiae. Biosci Biotechnol Biochem. 2005;69:1090–7.
Baghban R, Farajnia S, Ghasemi Y, Mortazavi M, Zarghami N, Samadi N. New developments in Pichia pastoris expression system, review and update. Curr Pharm Biotechnol. 2018;19:451–67.
Vieira Gomes AM, Souza CT, Silva CL, Mendonça BF, Parachin NS. Comparison of yeasts as hosts for recombinant protein production. Microorganisms. 2018;6(2):38.
Vieira SM, da Rocha SLG, da Neves-Ferreira AG, Almeida RV, Perales J. Heterologous expression of the antimyotoxic protein DM64 in Pichia pastoris. PLoS Negl Trop Dis. 2017;11:1–20.
Machens F, Balazadeh S, Mueller-Roeber B, Messerschmidt K. Synthetic promoters and transcription factors for heterologous protein expression in Saccharomyces cerevisiae. Front Bioeng Biotechnol. 2017;5:1–11.
Antosova Z, Sychrova H. Yeast hosts for the production of recombinant laccases: a review. Mol Biotechnol. 2015. https://doi.org/10.1007/s12033-015-9910-1.
Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122(1):19–27.
Mate DM, Gonzalez-Perez D, Falk M, et al. Blood tolerant laccase by directed evolution. ChemBiol. 2013;20:223–37.
Mate DM, Gonzalez-Perez D, Kittl R, Ludwig R, Alcalde M. Functional expression of a blood tolerant laccase in Pichia pastoris. BMC Biotechnol. 2013;13:38.
Yang J, Xu X, Ng TB, Lin J, Ye X. Laccase gene family in Cerrena sp. HYB07: sequences, heterologous expression and transcriptional analysis. Molecules. 2016;21:1017.
Arana-Cuenca A, Tellez-Jurado A, Yague S, et al. Delignification of Pinus radiata kraft pulp by treatment with a yeast genetically modified to produce laccases. For Syst. 2010;19:234–40.
Zhang J, Ma F, Zhang X, Geng A. Directed evolution of a homodimeric laccase from Cerrena unicolor BBP6 by random mutagenesis and in vivo assembly. Int J Mol Sci. 2018;19:2989.
Tian YS, Xu H, Peng RH, Yao QH, Wang RT. Heterologous expression and characterization of laccase 2 from Coprinopsis cinerea capable of decolourizing different recalcitrant dyes. Biotechnol Biotechnol Equip. 2014;28:248–58.
Lin YQ, Zhang Z, Tian YS, et al. Purification and characterization of a novel laccase from Coprinus cinereus and decolorization of different chemically dyes. Mol Biol Rep. 2013;40:1487–94.
Gu C, Zheng F, Long L, Wang J, Ding S. Engineering the expression and characterization of two novel laccase isoenzymes from Coprinus comatus in Pichia pastoris by fusing an additional ten amino acids tag at N-terminus. PLoS ONE. 2014;9:e93912.
Zhuo R, Ma L, Fan F, et al. Decolorization of different dyes by a newly isolated white-rot fungi strain Ganoderma sp. En3 and cloning and functional analysis of its laccase gene. J Haz Mat. 2011;192:855–73.
Zhuo R, He F, Zhang X, Yang Y. Characterization of a yeast recombinant laccase rLAC-EN3-1 and its application in decolorizing synthetic dye with the coexistence of metal ions and organic solvents. Biochem Engg J. 2015;93:63–72.
Lu CX, Wang HY, Luo YM, Guo L. An efficient system for pre-delignification of gramineous biofuel feedstock in vitro: application of a laccase from Pycnoporus sanguineus H275. Proc Biochem. 2010;45:1141–7.
Ji L, Shen Y, Xu L, Peng B, Xiao Y, Bao X. Enhanced resistance of Saccharomyces cerevisiae to vanillin by expression of lacA from Trametes sp. AH28-2. Biores technol. 2011;102:8105–9.
Nakanishi A, Bae JG, Fukai K, et al. Effect of pretreatment of hydrothermally processed rice straw with laccase-displaying yeast on ethanol fermentation. Appl Microbiol Biotechnol. 2012;94:939–48.
Yang Y, Fan FF, Zhuo R, et al. Expression of the laccase gene from a white rot fungus in Pichia pastoris can enhance the resistance of this yeast to H2O2-mediated oxidative stress by stimulating the glutathione-based antioxidative system. Appl Environ Microbiol. 2012;78:5845–54.
Fan F, Zhuo R, Sun S, Wan X, Jiang M, Zhang X, Yang Y. Cloning and functional analysis of a new laccase gene from Trametes sp. 48424 which had the high yield of laccase and strong ability for decolorizing different dyes. Biores Technol. 2011;102:3126–37.
Theerachat M, Emond S, Cambon E, et al. Engineering and production of laccase from Trametes versicolor in the yeast Yarrowia lipolytica. Biores technol. 2012;125:267–74.
Li Q, Ge L, Cai J, Pei J, Xie J, Zhao L. Comparison of two laccases from Trametes versicolor for application in the decolorization of dyes. J Microbiol Biotechnol. 2014;24:545–55.
Li Q, Pei J, Zhao L, Xie J, Cao F, Wang G. Overexpression and characterization of laccase from Trametes versicolor in Pichia pastoris. Prikl Biokhim Mikrobiol. 2014;50:163–70.
Bertrand B, Trejo-Hern M, Andez R, et al. Functional expression, production, and biochemical characterization of a laccase using yeast surface display technology. Fungal Biol. 2016;120:1609–22.
Miele A, Giardina P, Sannia G, Faraco V. Random mutants of a Pleurotus ostreatus laccase as new biocatalysts for industrial effluents bioremediation. J Appl Microbiol. 2010;108:998–1006.
Behrens CJ, Linke D, Allister AB, Zelena K, Berger RG. Variants of PpuLcc, a multi-dye decolorizing laccase from Pleurotus pulmonarius expressed in Pichia pastoris. Protein Expr Purif. 2017;137:34–42.
Lu CX, Wang HY, Luo YM, Guo L. An efficient system for pre-delignification of gramineous biofuel feedstock in vitro: application of a laccase from Pycnoporus sanguineus H275. Process Biochem. 2010;45:1141–7.
Song Q, Deng X, Song RQ. Expression of Pleurotus ostreatus laccase gene in Pichia pastoris and its degradation of corn stover lignin. Microorganisms. 2020;8:601.
Kittl R, Mueangtoom K, Gonaus C. A chloride tolerant laccase from the plant pathogen ascomycete Botrytis aclada expressed at high levels in Pichia pastoris. J Biotechnol. 2012;157:304–14.
Bao WH, Peng RH, Zhang Z, et al. Expression, characterization and 2,4,6-trichlorophenol degradation of laccase from Monilinia fructigena. Mol Biol Rep. 2012;39(4):3871–7. https://doi.org/10.1007/s11033-011-1166-7.
Kalyani D, Tiwari MK. A highly efficient recombinant laccase from the yeast Yarrowia lipolytica and its application in the hydrolysis of biomass. PLoS ONE. 2015;10:e0120156.
Wu J, Kim KS, Lee JH, Lee YC. Cloning, expression in Escherichia coli, and enzymatic properties of laccase from Aeromonas hydrophila WL-11. J Environ Sci. 2010;22:635–40.
Guan ZB, Song CM, Zhang N, et al. Overexpression, characterization, and dye-decolorizing ability of a thermostable, pH-stable, and organic solvent-tolerant laccase from Bacillus pumilus W. J Mol Cat B. 2014;101:1–6.
Ghasemi Y, Yarahmadi E, Ghoshoon MB. Cloning, expression and purification of laccase gene from Bacillus subtilis in Escherichia coli. Minerva Biotech. 2014;26:295–300.
Ihssen J, Reiss R, Luchsinger R, et al. Biochemical properties and yields of diverse bacterial laccase-like multicopper oxidases expressed in Escherichia coli. Sci Rep. 2015;5:10465. https://doi.org/10.1038/srep10465.
Li T, Huang L, Li Y, et al. The heterologous expression, characterization, and application of a novel laccase from Bacillus velezensis. Sci Total Environ. 2020;713:202.
Liu Y, Huang L, Guo W, Jia L, Fu Y, Gui S, Lu FF. Cloning, expression, and characterization of a thermostable andpH-stable laccase from Klebsiella pneumoniae and its application to dye decolorization. Process Biochem. 2017;53(2017):125–34.
Yadav D, Ranjan B, Mchunu N, et al. Secretory expression of recombinant small laccase from Streptomyces coelicolor A3(2) in Pichia pastoris. Int J Biol Macromol. 2018;108:642–9. https://doi.org/10.1016/j.ijbiomac.2017.11.169.
Feng H, Zhang D, Sun Y, et al. Expression and characterization of a recombinant laccase with alkalistable and thermostable properties from Streptomyces griseorubens JSD-1. Appl Biochem Biotechnol. 2015;176:547–62.
Liu H, Cheng Y, Du B, et al. Overexpression of a novel thermostable and chloridetolerant laccase from Thermus thermophilus SG05JP17–16 in Pichia pastoris and its application in synthetic dye decolorization. PLoS ONE. 2015;10:33.
Kim H, Lee S, Park H, Jeon S. Expression, refolding, and characterization of a small laccase from Thermus thermophilus HJ6. Protein Expr Purif. 2015;114:37–43.
Fathi-Roudsari M, Behmanesh M, Salmanian AH, Sadeghizadeh M, Khajeh K. Substrate-dependent expression of laccase in genetically modified Escherichia coli: design and construction of an inducible phenol-degrading system. Prep Biochem Biotechnol. 2013;43:456–67.
Mathews SL, Smithson CE, Grunden AM. Purification and characterization of a recombinant laccase-like multi-copper oxidase from Paenibacillus glucanolyticus SLM1. J Appl Microbiol. 2016;121:1335–45.
Kumar M, Mishra A, Singh SS, Srivastava S, Thakur IS. Expression and characterization of novel laccase gene from Pandoraea sp. ISTKB and its application. Int J Biol Macromol. 2018;115:308–16.
Uthandi S, Saad B, Humbard MA, Maupin-Furlow JA. LccA, an archaeal laccase secreted as a highly stable glycoprotein into the extracellular medium by Haloferax volcanii. Appl Environ Microbiol. 2010;76:733–43.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
MDA, MAM, VKG, and LP were all involved in the concept and design of this review. They jointly partake in data acquisition, review and analysis. MDA drafted the manuscript while all authors contributed in reviews and approval of the final paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Authors declare no competing interest on this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Fungal ligninolytic enzymes that are involved in the degradation of lignin, pesticides, drugs or hydrocarbons and their mediators. Table S2. Fungal Ligninolytic enzymes and their reactions in mediator containing media. Table S3. Example of some computation tools that can be used for pathway construction in fungi.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Asemoloye, M.D., Marchisio, M.A., Gupta, V.K. et al. Genome-based engineering of ligninolytic enzymes in fungi. Microb Cell Fact 20, 20 (2021). https://doi.org/10.1186/s12934-021-01510-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-021-01510-9