
Abstract
Two phylogenetically divergent genes of the new family of dye-decolorizing peroxidases (DyPs) were found during comparison of the four DyP genes identified in the Pleurotus ostreatus genome with over 200 DyP genes from other basidiomycete genomes. The heterologously expressed enzymes (Pleos-DyP1 and Pleos-DyP4, following the genome nomenclature) efficiently oxidize anthraquinoid dyes (such as Reactive Blue 19), which are characteristic DyP substrates, as well as low redox-potential dyes (such as 2,2-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid)) and substituted phenols. However, only Pleos-DyP4 oxidizes the high redox-potential dye Reactive Black 5, at the same time that it displays high thermal and pH stability. Unexpectedly, both enzymes also oxidize Mn2+ to Mn3+, albeit with very different catalytic efficiencies. Pleos-DyP4 presents a Mn2+ turnover (56 s−1) nearly in the same order of the two other Mn2+-oxidizing peroxidase families identified in the P. ostreatus genome: manganese peroxidases (100 s−1 average turnover) and versatile peroxidases (145 s−1 average turnover), whose genes were also heterologously expressed. Oxidation of Mn2+ has been reported for an Amycolatopsis DyP (24 s−1) and claimed for other bacterial DyPs, albeit with lower activities, but this is the first time that Mn2+ oxidation is reported for a fungal DyP. Interestingly, Pleos-DyP4 (together with ligninolytic peroxidases) is detected in the secretome of P. ostreatus grown on different lignocellulosic substrates. It is suggested that generation of Mn3+ oxidizers plays a role in the P. ostreatus white-rot lifestyle since three different families of Mn2+-oxidizing peroxidase genes are present in its genome being expressed during lignocellulose degradation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Peroxidases (EC1.11.1) represent a large group of oxidoreductases that use hydrogen peroxide as the final electron acceptor. Heme peroxidases were classified by Welinder (1992) into two superfamilies, animal and nonanimal peroxidases, the second one being divided into three classes according to their origin. Class I peroxidases come from the prokaryotic lineage, and classes II and III are secretory peroxidases from fungi and plants, respectively. The discovery of new peroxidase types in the last few years resulted in two novel (super)families of secreted fungal peroxidases: hemethiolate peroxidases (HTP), in which chloroperoxidases (EC 1.11.1.10) and unspecific peroxygenases (EC 1.11.2.1) are included, and dye-decolorizing peroxidases (DyPs; EC 1.11.1.19) (Hofrichter et al. 2010; Ruiz-Dueñas and Martínez 2010; Sugano et al. 2007; Sugano 2009).
DyPs constitute a group of enzymes phylogenetically unrelated to the catalase-peroxidase superfamily, where the classical nonanimal peroxidases—including lignin peroxidase (LiP; 1.11.1.14), manganese peroxidase (MnP; EC 1.11.1.13), and versatile peroxidase (VP; EC 1.11.1.16)—are currently classified (Zámocký et al. 2015). From a structural point of view, DyPs are members of the functionally diverse CDE superfamily (including chlorite dismutases, DyP-type enzymes, and proteins related to Escherichia coli EfeB) characterized by the presence of a ferredoxin-like core of β-sheets (Goblirsch et al. 2011). The first DyP was discovered in a culture of the fungus Bjerkandera adusta (initially described as Geotrichum candidum) (Kim and Shoda 1999; Sugano et al. 1999). Despite only seven fungal and eleven bacterial DyPs have been purified and characterized to date (Linde et al. 2015b), 233 DyP sequences from fungal, bacterial and archaeal genomes, and other sources are deposited in PeroxiBase (http://peroxibase.toulouse.inra.fr; March 2015 data), confirming their ubiquitous distribution (Colpa et al. 2014; Sugano 2009).
The range of biological processes in which peroxidases are involved includes defense, immune response, pathogenicity, detoxification, and biomass degradation (Torres and Ayala 2010). The natural ecophysiological role of DyPs still remains unclear, but these enzymes offer attractive properties for biotechnological purposes, being able to oxidize anthraquinoid and other dye types (Kim and Shoda 1999; Liers et al. 2010), carotenoids (Scheibner et al. 2008; Zelena et al. 2009a), and phenolic compounds (Liers et al. 2014; Linde et al. 2014; Sugano et al. 2000), among others. Moreover, the ability of DyPs to oxidize lignin-related compounds has been reported (Ahmad et al. 2011; Brown et al. 2012; Roberts et al. 2011; Salvachúa et al. 2013), although their catalytic efficiency on veratryl alcohol (Liers et al. 2013; Salvachúa et al. 2013) and nonphenolic lignin model dimers (Liers et al. 2010) is very low compared with that of basidiomycete LiPs (Linde et al. 2015b).
According to genomic data, LiPs, MnPs, and VPs are exclusive of typical white-rot (ligninolytic) basidiomycetes (most of them in the order Polyporales) (Floudas et al. 2012, 2015; Ruiz-Dueñas et al. 2013). Even though DyP genes do not show a so clear distribution, they are significantly more abundant in the genomes of lignin-degrading basidiomycetes (white-rot fungi) than in those of cellulose-degrading basidiomycetes (brown-rot fungi) according to the above genomic studies. This fact, together with their ability to oxidize aromatic compounds, suggests that DyPs could be involved in degradation of lignin compounds and derived soil organic matter. Moreover, the ability to oxidize Mn2+, a typical substrate of MnPs and VPs (Ruiz-Dueñas et al. 2009), has been reported for bacterial DyPs from Rhodococcus josti, Amycolatopsis sp., and Pseudomonas fluorescens (Brown et al. 2012; Rahmanpour and Bugg 2015; Roberts et al. 2011). This pointed to possible involvement in lignin degradation via Mn3+-initiated lipid peroxidation reactions, as proposed for MnP (Bao et al. 1994).
Pleurotus ostreatus, a white-rot fungus in the order Agaricales, is the second most consumed edible mushroom worldwide (Sánchez 2010). Its genome includes a total of 17 peroxidase genes: nine class II peroxidase genes (of the MnP and VP families) and one class I peroxidase gene, as well as four and three genes from the DyP and HTP (super)families, respectively (Ruiz-Dueñas et al. 2011). The catalytic properties of P. ostreatus VPs and MnPs have been thoroughly investigated after heterologous expression, and their secretion in lignocellulose media has been confirmed (Fernández-Fueyo et al. 2014b, d). However, a single report on P. ostreatus DyPs has been published to date, describing the Pleos-DyP1-encoding gene (Faraco et al. 2007).
In the present study, we identified the DyPs and other peroxidases secreted by P. ostreatus when grown on lignocellulosic substrates by using nanoflow liquid chromatography coupled to tandem mass spectrometry (nLC-MS/MS). Simultaneously, four DyP genes were identified in the P. ostreatus genome and two of them (presenting different evolutionary histories) were heterologously expressed in E. coli as active soluble enzymes, and their catalytic properties compared with two VPs and two MnPs from the same fungus. Unexpectedly, these P. ostreatus DyPs were able to oxidize Mn2+, a characteristic that had not been reported before for any other fungal DyPs.
Material and methods
P. ostreatus strains and genome
Dikaryotic P. ostreatus N001 (CECT20600), maintained in 2 % malt extract agar (MEA), was used in this study. The genomic sequences of monokaryons PC9 (CECT20311) and PC15 (CECT20312), isolated from the above dikaryon (Larraya et al. 1999), are available (http://genome.jgi-psf.org/PleosPC9_1 and http://genome.jgi-psf.org/PleosPC15_2). They were sequenced in a JGI project coordinated by A.G. Pisabarro (Public University of Navarre, Spain).
DyP screening in the P. ostreatus genome
The inventory of DyP coding genes in the P. ostreatus genome was obtained by the following: (i) screening the automatically annotated genomes; (ii) revising and manually curating the positions of introns and the C- and N-termini, the latter using SignalP 4.1 for predicting signal peptides; (iii) comparing the predicted amino acid sequences with related peroxidases, after multiple alignment with MEGA5 (Tamura et al. 2011); and (iv) confirming the presence of characteristic residues at the heme pocket and substrate oxidation sites. These residues were identified after homology modeling at the SWISS-MODEL server using the crystal structures of Auricularia auricula-judae (PDB 4W7J) and B. adusta (PDB 2D3Q) DyPs as templates. Finally, the revised sequences were compared with all the DyP sequences (a total of 218) from the 114 basidiomycete (Agaricomycotina) genomes currently (7 December 2014) sequenced and annotated at JGI, which are available at the MycoCosm portal (http://genome.jgi-psf.org/programs/fungi). A phylogram—also including available GenBank sequences from DyPs of A. auricula-judae (JQ650250), Mycetinis scorodonius (CS490662 and CS490657), Ganoderma lucidum (ADN05763), Termitomyces albuminosus (AAM21606), and an unidentified Polyporaceae species (AAB58908)—was constructed with MEGA5 using MUSCLE alignment and maximal likelihood clustering.
Gene synthesis
The mature protein coding sequences of two DyP (JGI protein ID 62271 and 1069077 from PC15, corresponding to Pleos-DyP1 and Pleos-DyP4, respectively), two MnP (1089546 and 1041740 from PC15, corresponding to Pleos-MnP3 and Pleos-MnP6, respectively), and two VP genes (137757 from PC9, corresponding to Pleos-VP1 and 1113241 from PC15, corresponding to Pleos-VP2) from the P. ostreatus genomes (http://genome.jgi-psf.org/PleosPC9_1 and http://genome.jgi-psf.org/PleosPC15_2) were synthesized by ATG:biosynthetics (Merzhausen, Germany), after verifying that all the codons had previously been used for expressing other genes in the E. coli strains mentioned below, and substituting them when required. The optimized cDNA sequences of Pleos-DyP1 (KP973935), Pleos-DyP4 (KP973936), Pleos-MnP3 (KP973939), Pleos-MnP6 (KP973940), Pleos-VP1 (KP973937), and Pleos-VP2 (KP973938) were deposited at GenBank.
DyP production in E. coli
The above two DyP coding sequences were cloned in the pET23a vector (Novagen, Darmstadt, Germany), and the resulting plasmids (pET23a-62271 and pET23a-1069077) were used for expression in E. coli BL21(DE3)pLysS (Novagen, Darmstadt, Germany). Cells were grown for 3 h at 37 °C in Terrific Broth, induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and grown further for 48 h at 16 °C in the presence of 20 μM of hemin. Cells were harvested by centrifugation at 8000 rpm for 10 min at 4 °C. Bacteria were resuspended in 300 mL lysis buffer (20 mM Tris-HCl pH 8.0 containing 1 mM EDTA, and 5 mM DTT), supplemented with lysozyme (Sigma-Aldrich, Steinheim, Germany) (2 mg.mL−1) and DNase I (Roche Diagnostics, Mannheim, Germany). After 1 h of incubation, cells were sonicated, and debris was removed by centrifugation at 20,000 rpm for 4 h. The supernatants were concentrated with Amicon (Millipore, Darmstadt, Germany) of 10-kDa cutoff, and dialyzed against 20 mM acetate, pH 4.3. Insoluble material was eliminated (15,000 rpm for 30 min) and the solution further dialyzed (10 mM Tris, pH 7.5).
DyPs were purified using an ÄKTA high-performance liquid chromatography (HPLC) system (GE Healthcare Bio-Sciences AB, Uppsala, Sweden), in three consecutive steps. The first separation was performed on a 6-mL Resource™ Q cartridge (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) using 10 mM Tris (pH 7.5) at a flow rate of 2 mL.min−1. After 15 mL, the retained proteins were eluted with a 0–0.35 M NaCl gradient in 50 mL, followed by 0.5–1 M NaCl gradient in 10 mL, and 1 M NaCl in 15 mL. Peroxidase activity was followed by 2,2-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) oxidation in the presence of H2O2, as described below. The appropriate fractions were pooled, concentrated, and dialyzed against 10 mM Tris (pH 7.5), and loaded into a Mono Q high-resolution 5/5 column (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) at a flow rate of 1 mL.min−1 (same buffer) using a 0–0.3 M NaCl gradient in 40 mL, followed by 1 M NaCl in 10 mL. Finally, size-exclusion chromatography in a Superdex-75 HR 10/30 column (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) with 10 mM tartrate (pH 5), containing 100 mM NaCl, at 0.2 mL.min−1, was performed. Pleos-DyP1 and Pleos-DyP4 purification was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in 12 % gels stained with Coomassie Brilliant Blue R-250 (Sigma). Electronic absorption spectra of the purified enzymes were recorded with an Agilent 8453 (Agilent Technologies, Santa Clara, USA) diode array UV-visible spectrophotometer.
MnP and VP production in E. coli
The Pleos-MnP3 (PC15 1089546), Pleos-MnP6 (PC15 1041740), Pleos-VP1 (PC9 137757), and Pleos-VP2 (PC15 1113241) coding sequences were cloned in the pFLAG1 (International Biotechnologies Inc., Kodak, CT, USA) or pET23a (+) (Novagen, Darmstadt, Germany) vectors, and the resulting plasmids (pFLAG1-1089546, pET23a-1041740, pFLAG1-137757, and pFLAG1-1113241) were used for expression in E. coli K-12 W3110 (pFLAG1 plasmids) and BL21(DE3)pLysS (pET23a plasmids). Cells were grown for 3 h in Terrific Broth, induced with 1 mM IPTG, and grown further for 4 h. The apoenzyme accumulated in inclusion bodies, as observed by SDS-PAGE, and was solubilized with 8 M urea.
In vitro refolding was performed using 0.16 M urea, 5 mM Ca2+, 20 μM hemin, 0.5 mM oxidized glutathione, 0.1 mM dithiothreitol, and 0.1 mg.mL−1 protein, at pH 9.5 (Pérez-Boada et al. 2002). For Pleos-MnP6 optimized refolding was obtained using 0.1 M urea, 5 mM Ca2+, 20 μM hemin, 1.5 mM oxidized glutathione, 0.1 mM dithiothreitol, and 0.1 mg.mL−1 protein, at pH 8. Enzymes were purified by Resource-Q chromatography using a 0–0.3 M NaCl gradient (2 mL.min−1, 20 min) in 10 mM tartrate (pH 5.5) containing 1 mM CaCl2.
Peroxidase kinetic constants and optimal pH
The optimal pH for substrate oxidation by the two P. ostreatus DyPs was determined using saturating concentrations of (i) Reactive Black 5 (RB5; 15 μM), Reactive Blue 19 (RB19; 200 μM), ABTS (5 mM), and 2,6-dimethoxyphenol (DMP; 60 mM) in 0.1 M Britton-Robinson (B&R) buffer (pH 2–5.5); and (ii) Mn2+ (6 mM) in 0.1 mM tartrate buffer (pH 2–6), and the assays described below.
The kinetic constants of the P. ostreatus DyPs, MnPs, and VPs were estimated from the absorbance changes observed during substrate oxidation in 0.1 M tartrate (at optimal pH values) at 25 °C in a Thermo BioMate 5 spectrophotometer (Thermo Fisher Scientific, Waltham, USA). The reactions were initiated by addition of H2O2 to a final concentration of 0.25 mM. Oxidation of Mn2+ was followed at pH 4.5 by monitoring formation of Mn3+-tartrate complex (ε 238 6.5 mM−1.cm−1). RB19 and ABTS oxidation were assayed at pH 3.5 and monitored by RB19 disappearance (ε 595 10 mM−1.cm−1) and formation of ABTS cation radical (ε 436 29.3 mM−1.cm−1). RB5 and DMP oxidation were assayed at pH 3 and monitored by RB5 disappearance (ε 598 30 mM−1.cm−1) and dimeric coerulignone (ε 469 55 mM−1.cm−1) formation, respectively. Means and standard errors for Michaelis constant (K m) and enzyme turnover (k cat) values were obtained by nonlinear least-squares fitting to the Michaelis-Menten model. Fitting of these constants to the normalized equation v = (k cat/K m)[S]/(1 + [S]/K m) yielded the catalytic efficiency values (k cat/K m) with their corresponding standard errors.
Studies on DyP pH and thermal inactivation
To study the effect of incubation at different pH values on the activity of the two P. ostreatus DyPs (0.01 μM), the enzymes were dissolved in 0.1 M B&R buffer in the range of pH 2–9, and kept at 4 °C for different time periods. Activity was determined by oxidation of ABTS (5 mM) under the conditions described above. Residual activities were measured after 1 min (to evaluate the initial survival of the enzyme at each pH value) and 1, 4, 24 and 120 h incubation. The highest activity after 1 min (at any pH) was taken as 100 % activity, and the percentages of residual activity at the different times and pH conditions were calculated according to this maximal value.
In a similar way, to study the effect of incubation at different temperatures, the enzymes in 10 mM tartrate (pH 5) were kept for 10 min in the temperature range of 25–85 °C. Residual activity was determined at 25 °C, as described above, and that obtained after 25 °C incubation was taken as 100 %. Temperature stability was finally presented as 10-min T 50 value, i.e., that temperature at which 50 % of the activity was lost after incubation for the above time period.
Secretomic analyses of P. ostreatus peroxidases
Secretomic studies were performed with dikaryotic P. ostreatus N001 in glucose and (two different) lignocellulose culture media. Glucose cultures were grown in 1 L shaken (200 rpm) flasks with 200 mL of HAT medium (Spinnler et al. 1994) containing 10 g glucose, 0.2 g KH2PO4, 0.5 g MgSO4·7H2O, 1 g casamino acids, 1 g yeast extract, 0.368 g ammonium tartrate, and 1 L of distilled water. Inocula consisted of 15 mL of homogenized actively growing mycelium from M7GY (Castanera et al. 2012) liquid cultures incubated at 200 rpm. Lignocellulose cultures were grown on 10 g of milled wheat straw or small poplar chips (particle size < 4 mm) soaked with 70 mL of distilled water in 1 L flasks (stationary cultures). Inocula consisted of 15 mL of homogenized mycelium from M7GY cultures. All the above cultures were maintained at 25 °C.
Total extracellular proteins in the filtrates of cultures grown for 21 days in the above media were freeze-dried, resuspended in 10 mM tartrate (pH 5), impurities removed by a short PAGE run, and stained by Colloidal Blue Kit (Invitrogen, Thermo Fisher Scientific, Waltham, USA). The protein band was cut and destained using 50 mM ammonium bicarbonate in 50 % acetonitrile (ACN), reduced with 10 mM dithiothreitol for 30 min at 56 °C, alkylated with 55 mM iodoacetamide in the dark for 30 min at 24 °C, and digested with 12.5 ng.μL−1 trypsin in 50 mm ammonium bicarbonate, overnight at 30 °C. Peptides were extracted at 37 °C using 100 % ACN and then 0.5 % trifluoroacetic acid, dried, cleaned using ZipTip with 0.6 μL C18 resin (Millipore), and reconstituted in 5 μL of 0.1 % formic acid in 2 % ACN.
Tryptic peptides were analyzed in an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Waltham, USA) coupled to a nanoEasy HPLC equipment (Proxeon). Peptides were first trapped onto a C18-A1 ASY-Column 2 cm precolumn (Thermo Fisher Scientific, Waltham, USA) and then eluted onto a Biosphere C18 column (75 μm inner diameter, 15 cm long, and 3 μm particle size) (NanoSeparations) using a 130-min gradient from 0–45 % buffer-B (buffer-A: 0.1 % formic acid in 2 % ACN; buffer B: 0.1 % formic acid in pure ACN) at a flow rate of 250 nL.min−1. Mass spectra were acquired in the positive ion mode and data-dependent manner selecting the 20 most intense ions for fragmentation using collision-induced dissociation (CID). MS spectra (m/z 300–1600) were acquired in the Orbitrap with a target value of 1,000,000 at a resolution of 30,000 (at m/z 400), and MS2 spectra were acquired in the linear ion trap with a target value of 10,000 and normalized collision energy of 35 %. Precursor ion charge state screening and monoisotopic precursor selection were enabled. Singly charged ions and unassigned charge states were rejected. Dynamic exclusion was enabled with a repeat count of 1 and exclusion duration of 30 s.
Acquired spectra were searched against the P. ostreatus PC9 and PC15 genomic databases, downloaded from JGI (http://genome.jgi-psf.org/programs/fungi), using Sequest search engine through Proteome Discoverer (version 1.4). As for the search parameters, precursor and fragment mass tolerance were set to 10 ppm and 0.8 Da, respectively. Carbamidomethylation of cysteines was set as a fixed modification, and oxidation of methionines was set as a dynamic modification. Two missed cleavages were allowed. Identified peptides were validated using Percolator algorithm with a q value threshold of 0.01.
Results
DyP gene analysis in the P. ostreatus genome
Four DyP coding genes were identified and manually annotated in the genomes of two P. ostreatus monokaryons (PC9 and PC15). The alleles from these two monokaryons are 99.0 % (Pleos-DyP1), 99.8 % (Pleos-DyP2), 98.7 % (Pleos-DyP3), and 98.2 % (Pleos-DyP4) identical at the protein level, and the gene models from the genome of P. ostreatus PC15 v2.0 (http://genome.jgi-psf.org/PleosPC15_2) are analyzed here. Pleos-DyP1 (protein ID 62271, scaffold 09:213879-216064), Pleos-DyP2 (protein ID 1092668, scaffold 04:657069-659105), and Pleos-DyP3 (protein ID 52170, scaffold 07:3196067-3198039) are constituted by ten translated exons giving rise to proteins of 516, 511, and 493 total amino acids, respectively, while Pleos-DyP4 (protein ID 1069077, scaffold 11:2524348-2526415) is formed by only eight exons that codify a 504 amino acid protein (small introns of 50–89 nucleotides separate the above exons in the four genes). Signal peptides of 19 amino acids in Pleos-DyP1 and Pleos-DyP3 (we manually curated the latter gene at the JGI portal to include this signal peptide), and 23 amino acids in Pleos-DyP2 were identified (no signal peptide was predicted for Pleos-DyP4) resulting in mature sequences of less than 500 amino acids for Pleos-DyP1 (498), Pleos-DyP2 (488), and Pleos-DyP3 (474) while mature Pleos-DyP4 would be formed by 504 amino acids (Fig. 1). The first three enzymes showed 63–74 % sequence identity among them (from 280–352 aligned residues) (Table S1 in the Supplementary Material). However, their individual sequence identities with Pleos-DyP4 were always below 40 % (and only 176–191 residues could be aligned) suggesting a divergent evolutionary origin of the latter protein.

Alignment of the four DyPs from the P. ostreatus genome (Pleos-DyP1 to Pleos-DyP4; JGI protein ID 62271, 1092668, 52170, and 1069077, respectively) and the best known A. auricula-judae DyP (Aurau-DyP1, GenBank JQ650250). Numbering (right) corresponds to the predicted mature proteins (numbering of underlined signal peptide, when present, is indicated on the left). Highlighted residues include the following: (i) proximal histidine (magenta) and aspartic acid (red); (ii) distal-side arginine (cyan) and aspartic acid (red), the latter forming part of the GXXDG motif (red box) together with two conserved glycines (olive), a leucine, and an aromatic residue (two positions before) corresponding to a phenylalanine (gray) that is substituted by tryptophan (yellow) or tyrosine (green) in two of the sequences; and (iii) four aromatic residues exposed to the solvent, corresponding to three conserved tryptophans (yellow), and one tyrosine (green) that is substituted by phenylalanine (gray) in one of the sequences. Alignment was produced with ClustalW2 (European Bioinformatics Institute, Hinxton, UK), and symbols below the sequences indicate full conservation of the same (asterisk) or equivalent residues (colon) and partial residue conservation (dot)
DyP molecular models
For the structural-functional classification of the P. ostreatus peroxidases, molecular models of the deduced mature proteins were prepared using related crystal structures as templates. These models confirmed the presence of important residues for DyP catalysis (Linde et al. 2015b) including as follows: (i) proximal histidine (His-361 and His-334 in Pleos-DyP1 and Pleos-DyP4, respectively) acting as the fifth ligand of the heme iron and H-bonded aspartic acid (Asp-445 and Asp-433 in Pleos-DyP1 and Pleos-DyP4, respectively); and (ii) second aspartic acid (Asp-218 and Asp-196 in Pleos-DyP1 and Pleos-DyP4, respectively), forming a part of a conserved GXXDG motif (Colpa et al. 2014), and neighbor arginine (Arg-382 and Arg-360 in Pleos-DyP1 and Pleos-DyP4, respectively) at the opposite side of the heme, helping the heterolytic cleavage of H2O2 to form compound I (the above residues are marked on the Fig. 1 alignment, together with other relevant residues mentioned below).
The Pleos-DyP4 molecular model showed several significant differences with respect to the other three DyP models. They are illustrated in Fig. 2 showing the whole molecular models for Pleos-DyP1 and Pleos-DyP4. The Pleos-DyP4 molecular model (Fig. 2a) shows four extra loops (arrows) that, together with the 15-residue longer C-terminal tail (Fig. 2b), result in a longer mature protein, despite its 42-residue shorter N-terminal tail. Four aromatic residues are exposed to the solvent in Pleos-DyP1 (Fig. 2a) including Trp-427, homologous to A. auricula-judae catalytic Trp-377 (Linde et al. 2015a), together with two other tryptophans and one surface tyrosine. The latter residue is substituted by a phenylalanine in Pleos-DyP4 that, however, conserves the putative catalytic Trp-405 and the two other tryptophan residues (Fig. 2b). The same surface aromatic residues found in Pleos-DyP1 are present in Pleos-DyP2 and Pleos-DyP3 (Fig. 1).

Molecular models of Pleos-DyP1 (a) and Pleos-DyP4 (b). The ribbon representations include 3–4 exposed aromatic residues (as CPK-colored spheres), the heme cofactor (as CPK-colored sticks), and some extra loops in Pleos-DyP4 (arrows in b). The two enzymes were modeled using related crystal structures as template (some N- and C-terminal residues are not included in the models)
After annotating the DyP-encoding genes in the P. ostreatus genome, and analyzing the predicted sequences and molecular structures of the corresponding enzymes, two of them (Pleos-DyP1 and Pleos-DyP4) were selected for heterologous expression and biochemical characterization, due to their divergent evolutionary origin.
DyP production, purification, and electronic absorption spectra
For heterologous expression and purification, large-scale (9 L) cultures of E. coli transformants were grown under conditions resulting in Pleos-DyP1 and Pleos-DyP4 production as active proteins. For enzyme recovery, cells were lysed, centrifuged, and the supernatants concentrated and dialyzed. The purification process required three different chromatographic steps. As illustrated for Pleos-DyP4 (Fig. 3), SDS-PAGE monitoring of the purification process showed that the whole soluble fraction from the E. coli cultures (lane 1) was considerably enriched in the DyP (55.3 kDa) band by Resource Q chromatography (lane 2), but a number of other proteins remained. Most of these contaminating proteins were removed by Mono Q (lane 3), and, using a final Sephadex 75 step, electrophoretically homogeneous DyP was obtained (lane 4) with purification yields of 6.3 mg Pleos-DyP1 and 11.7 mg Pleos-DyP4 per E. coli culture (9 L), and Reinheitzahl (Rz) A 405/A 280 ratio of 1.9 and 2.2, respectively.

SDS-PAGE during purification of Pleos-DyP1 (55.3 kDa) to attain electrophoretic homogeneity. Lanes 1 to 4 correspond to the whole soluble fraction from E. coli culture and protein samples after Resource Q, Mono Q, and Sephadex 75 purification steps (molecular mass standards are included)
The electronic absorption spectra of the two enzymes were nearly identical, and those of Pleos-DyP4 are shown in Fig. 4. The resting state spectrum at physiological pH 3 shows the main Soret band at 405 nm, and two small charge-transfer maxima at 504 and 636 nm, confirming proper incorporation of the cofactor by E. coli (Fig. 4a, black line). The two enzymes were able to form reactive compound I, as revealed by the spectral changes after H2O2 addition, including reduced intensity of the Soret band and disappearance of the above charge-transfer maxima being substituted by several small peaks (Fig. 4a, red line). Compound II seemed to be quickly self-reduced since the corresponding spectrum could not be subsequently observed, and compound I directly returned to the initial spectrum (resting state). However, as reported for other fungal DyPs (Linde et al. 2015a), and illustrated in Fig. 4b for Pleos-DyP4, a compound II-like spectrum was observed when the resting enzyme at pH 7 (black line) reacted with H2O2 resulting in Soret band displacement from 403 nm (as found for resting DyP at pH 7) to 414 nm and disappearance of the 504 nm maximum, among other changes (green line).

Electronic absorption spectra of Pleos-DyP4. a Compound I spectrum (red) obtained 2 s after addition of 2 eq. of H2O2 to DyP resting state (black) in 10 mM tartrate, pH 3, showing the main Soret band at 404 nm, and disappearance of the maxima at 504 and 636 nm. b Compound II-like spectrum (green) obtained 2 s after addition of 2 eq. of H2O2 to DyP resting state (black) in 10 mM Tris-HCl, pH 7, showing the main Soret band displacement from 403 to 414 nm, and disappearance of the maximum at 504 nm. Spectra were obtained with an Agilent 8453 diode array UV-visible spectrophotometer
For comparison of catalytic properties with DyPs, two MnPs and two VPs from the P. ostreatus genome were produced as inclusion bodies, in vitro activated, purified to electrophoretic homogeneity, and characterized, as described elsewhere (Fernández-Fueyo et al. 2014d) (Fig. S1 in the Supplementary Material).
DyP catalytic properties compared with other P. ostreatus peroxidases
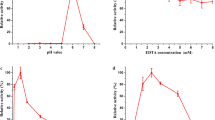
Before analyzing their kinetic constants, the optimal pH for oxidation of the different substrates (at saturating concentrations) was determined for Pleos-DyP1 and Pleos-DyP4 (Fig. 5a, b, respectively). Although Pleos-DyP1 showed higher pH optimum for RB19 oxidation than Pleos-DyP4, the general pH profiles were similar for both enzymes, with an optimum at pH 3–4 for the phenolic and dye substrates, and a higher optimal pH (4.5) for Mn2+, suggesting the involvement of deprotonated acidic residues in the latter activity. In general, no activity was detected over pH 4 in the case of RB5, and over pH 6 for the rest of the DyP substrates. The optimal pH for P. ostreatus MnP and VP reactions has been reported elsewhere (Fernández-Fueyo et al. 2014d) being also characterized by their acidic optima.

Optimal pH values for the oxidation of ABTS (5 mM), DMP (60 mM), RB5 (15 μM), and RB19 (200 μM) in 0.1 M B&R buffer, and Mn2+ (6 mM) 0.1 M tartrate buffer by Pleos-DyP1 (a) and Pleos-DyP4 (b) from the P. ostreatus genome
Table 1 shows the kinetic constants of Pleos-DyP1 and Pleos-DyP4 (expressed in E. coli as active proteins) on five representative substrates, compared with Pleos-MnP3, Pleos-MnP6, Pleos-VP1, and Pleos-VP2 (also expressed in E. coli but in vitro activated). VA oxidation was also assayed, but, although this nonphenolic substrate was oxidized by VPs, no activity was detected for any of the DyPs and MnPs. The three peroxidase families (DyPs, MnPs, and VPs) differed in their catalytic properties, although the low redox-potential dye ABTS could be efficiently oxidized by all of them. Significant differences were also observed between the two P. ostreatus DyPs, as described below.
Only Pleos-DyP4 was able to oxidize the high redox-potential RB5, showing a K m similar to that of VPs (~5 μM) but with a 2–4-fold lower k cat (MnPs show no activity on this compound). In the case of RB19, a typical DyP substrate that we show is also oxidized by VPs; Pleos-DyP1 presented k cat values similar to Pleos-VP1 and Pleos-VP2, while the Pleos-DyP4 k cat is up to 30-folds higher (again MnPs had no activity on this compound). Regarding DMP oxidation, Pleos-DyP4 was up to 1000-folds more efficient than Pleos-DyP1, due to its lower (250-fold) K m, and a higher (fourfold) k cat. Pleos-DyP4 was the most efficient enzyme on this phenolic substrate, which is also oxidized not only by VPs but also by the P. ostreatus MnPs that belong to the short MnP subfamily (Fernández-Fueyo et al. 2014a). Unexpectedly, the two DyPs from the P. ostreatus genome also exhibited Mn2+-oxidizing activity, which was especially significant for Pleos-DyP4 (near 200 s−1.mM−1). Both DyPs exhibit lower affinity for Mn2+ than the MnPs and VPs, the difference being more dramatic for Pleos-DyP1 with a K m value 2–3 orders of magnitude higher (mM range). However, the Pleos-DyP4 K m for Mn2+ is only 2–3-folds higher than those of Pleos-MnP3, Pleos-MnP6, and Pleos-VP1.
The above results show that Pleos-DyP4 always has higher (1.3–1000-fold) k cat values than Pleos-DyP1, suggesting that some differences in the heme environment provide this enzyme with a higher redox potential. However, better fitting of the corresponding substrates at the oxidation site can also result in faster electron transfer in Pleos-DyP4. For Mn2+ and DMP, significantly lower (9–250-fold) K m values than those of Pleos-DyP1 were also obtained, indicating a better binding of the two substrates by Pleos-DyP4.
pH and thermal stability of DyP isoenzymes
The pH and thermal stabilities of the two P. ostreatus DyPs described above were estimated by incubating the enzymes (0.01 μM concentration) at different pHs (pH 2–9 range) and temperatures (25–85 °C range) and determining the remaining activity. The P. ostreatus DyPs are extremely stable at pH 3, maintaining more than 80 % of the initial activity after 24 h incubation (Fig. 6a). However, at pH 2, strong differences appeared between the two enzymes, with Pleos-DyP4 still maintaining over 80 % of the initial activity while Pleos-DyP1 was very quickly (1 min) fully inactivated. On the other side, both DyPs were also extremely stable at basic pH, maintaining over 80 % of the initial activity after 24 h of incubation at pH 9.

pH and thermal stabilities of Pleos-DyP1 (green) and Pleos-DyP4 (red) from the P. ostreatus genome. Residual activities were estimated after 24 h incubation in the range of pH 2–9 (in B&R buffer, 4 °C) (a) and at different temperatures in the range of 25–70 °C (in 0.01 M tartrate, pH 5) (b), and referred to the highest activity measured immediately after adding the enzyme
Comparison of the thermal stability of the two DyPs, measured as the residual activity after 10-min incubation (Fig. 6b), showed that Pleos-DyP4 was 100 % stable in the 60–70 °C range, while Pleos-DyP1 was fully inactivated under the same conditions. From the thermal inactivation profiles, 10-min T 50 values of 52 and 73 °C were estimated for Pleos-DyP1 and Pleos-DyP4, respectively. The above differences are even higher than those observed for the pH stability.
Secretomic analysis of peroxidases in glucose, straw, and wood cultures
The secretomes of dikaryotic P. ostreatus grown on poplar chips, wheat straw, and glucose (HSA) medium were analyzed by nLC-MS/MS of the total tryptic peptides obtained. Different DyP, MnP, and VP proteins were identified (Table 2) from the presence of unique peptides (at least two per protein and sample) whose sequences were identified by comparison with the available P. ostreatus genome.
Pleos-DyP4, Pleos-VP1, Pleos-VP2, Pleos-MnP3, and Pleos-MnP6 were identified on both poplar chips and wheat straw, with Pleos-VP3 appearing as a sixth peroxidase on poplar chips. In both cases, Pleos-VP1 and Pleos-MnP3 were the most abundant peroxidases, as revealed by the semiquantitative PSM value, although the abundance order on poplar chips (Pleos-VP1 followed by MnP3) was inverted on wheat straw (Pleos-MnP3 followed by Pleos-VP1). On the other hand, in the HSA medium, only Pleos-DyP4 and Pleos-VP1 were identified.
Pleos-DyP4 was the unique peroxidase of the DyP family identified in the three cultures analyzed, being the 115, 49, and 34 most abundant protein when the whole secretome was analyzed on poplar chips, wheat straw, and HSA cultures, respectively (data not shown). On the other hand, no Pleos-DyP1, Pleos-DyP2, and Pleos-DyP3 peptides were detected in any of the samples.
Discussion
Identification of phylogenetically divergent DyPs in the P. ostreatus genome
Since their discovery in B. adusta (Kim and Shoda 1999), DyPs have been purified and characterized from six additional fungal species, namely A. auricula-judae, Exidia glandulosa, Irpex lacteus, Mycena epipterygia, M. scorodonius, and Thermomyces albuminosus (Johjima et al. 2003; Liers et al. 2010, 2013; Salvachúa et al. 2013; Scheibner et al. 2008; Zelena et al. 2009b). Among them, those of B. adusta (Sugano et al. 1999), A. auricula-judae (Liers et al. 2010), M. scorodonius (Scheibner et al. 2008), T. albuminosus (Johjima et al. 2003), and I. lacteus (most probably corresponding to AAB58908 directly submitted to GenBank by Han, 1996, as unidentified Polyporaceae peroxidase sequence) (Linde et al. 2015b) had been cloned and sequenced, together with G. lucidum (Kung et al. 2014) and P. ostreatus (Faraco et al. 2007) DyPs. Among the different basidiomycete DyPs, only those of B. adusta and A. auricula-judae have been characterized from a structural and mechanistic point of view (Linde et al. 2014, 2015a; Strittmatter et al. 2013; Sugano 2009; Yoshida et al. 2011, 2012). However, the availability of sequenced genomes is strongly increasing the number of known DyP sequences in fungi, and up to 37 DyP genes were reported by Floudas et al. (2012) after analyzing 32 fungal genomes of different taxonomic groups. The present study contributes to our knowledge on fungal DyPs by mining the sequenced genome of P. ostreatus for enzymes of this new peroxidase family. The molecular models obtained for their structural-functional classification confirm conservation of residues typical of DyPs at both sides of the heme cofactor (Linde et al. 2015b), as well as the surface tryptophan residue recently identified as responsible for high-turnover substrate oxidation by long-range electron transfer (Linde et al. 2015a).
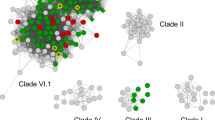
To establish the evolutionary histories of the four P. ostreatus DyPs, a comparison of all the DyP sequences (a total of 218) from Agaricomycotina genomes available to date at the MycoCosm portal (http://genome.jgi-psf.org/programs/fungi) was performed, also including the available DyP sequences (from GenBank) of the above basidiomycete species. The phylogram obtained (Fig. 7) revealed that the P. ostreatus DyPs correspond to two largely divergent evolutionary groups. Pleos-DyP1, Pleos-DyP2, and Pleos-DyP3 (together with a few more Agaricales DyPs) belong to phylogram cluster I, which includes all the identified DyPs of fungi from the orders Auriculariales, Gomphales, and Sebacinales. Cluster I also includes all the other basidiomycete DyP characterized to date, from A. auricula-judae, B. adusta, I. lacteus, M. scorodonius, T. albuminosus, and E. glandulosa (the latter corresponding to one of the sequences of the unpublished genome in the Auriculariales subcluster). In contrast, Pleos-DyP4 belongs to a large subcluster of Agaricales DyPs in the more basal cluster III, predominantly constituted by sequences from Agaricales and Polyporales (the latter including an hypothetical DyP cloned from G. lucidum). Therefore, Pleos-DyP4 is the first basidiomycete DyP from a cluster different from cluster I to be characterized. Interestingly, while Agaricales present an average of 1.6 DyP genes per genome, which are often located in separate evolutionary clusters (as found in P. ostreatus), the opposite tendency was observed in other basidiomycete orders, e.g., in Auriculariales with an average of 10.0 DyP genes per genome, all of them belonging to the same evolutionary cluster.

Maximal likelihood phylogram showing the position of the four P. ostreatus DyPs with respect to 218 DyP sequences identified in 64 Agaricomycotina genomes (among the 114 available at the JGI MycoCosm Portal on 7 December 2014; 45 of them lacking DyP genes and five corresponding to a second sequenced strain). Six GenBank sequences from DyPs of A. auricula-judae (Aurau-DyP, JQ650250), M. scorodonius (Mycsc-DyP1, CS490662; and Mycsc-DyP2, CS490657), G. lucidum (Ganlu-DyP, ADN05763), unidentified Polyporaceae species (Polsp-DyP, AAB58908), and T. albuminosus (Teral-DyP, AAM21606) are also included, as well as B. adusta DyP (Bjead-DyP, BAA77283; corresponding to JGI B. adusta genome protein ID 72253), and their positions indicated on the tree together with those of the P. ostreatus DyPs. Subclusters from species of the orders Agaricales and Polyporales are widespread through the tree, with P. ostreatus DyPs in clusters I (Pleos-DyP1 to Pleos-DyP3) and III (Pleos-DyP4). Although with lower gene numbers, subclusters from Geastrales, Corticiales, and Cantharellales are also widespread. In contrast, DyPs from other orders are restricted to specific clusters: (i) Auriculariales, Gomphales, and Sebacinales to cluster I; (ii) Hymenochaetales to cluster II; (iii) Atheliales and Russulales to cluster III; and (iv) Boletales to cluster VI. No DyP genes were found in Dacrymycetales, Gloeophylliales, or Trechisporales with 3, 2, and 2 genomes, respectively (order adscription for the different species is from Index Fungorum, http://www.indexfungorum.org). Colored triangles on the phylogram show the position of DyPs from the above 14 orders, with the total gene numbers (including six from GenBank) followed by the number of genomes indicated in the legend for each order. Agaricomycotina DyP data are from published (Branco et al. 2015; Eastwood et al. 2011; Fernández-Fueyo et al. 2012; Floudas et al. 2012, 2015; Hane et al. 2014; Hori et al. 2014; Janbon et al. 2014; Kohler et al. 2015; Martin et al. 2008; Morin et al. 2012; Ohm et al. 2010; Riley et al. 2014; Rineau et al. 2012; Ruiz-Dueñas et al. 2013) and unpublished genomes available at the JGI MycoCosm Portal (the latter with permission of the project PIs)
Heterologous expression of active DyP
An evaluation of the biochemical diversity of the large number of DyP sequences available in basidiomycete genomes is hampered by the difficulties for their heterologous expression, compared with bacterial DyPs that are easily produced in prokaryotic hosts. Similar studies on class II peroxidases have been largely based on E. coli expression followed by in vitro activation of the peroxidase protein recovered from inclusion bodies using optimized protocols (Doyle and Smith 1996; Miki et al. 2009; Pérez-Boada et al. 2002).
A similar protocol has been recently developed by Linde et al. (2014) for the in vitro activation of an E. coli-expressed A. auricula-judae DyP, and different variants from site-directed mutagenesis (Linde et al. 2015a). Unfortunately, this protocol is not of general application to other basidiomycete DyPs, most probably because of their structural heterogeneity (e.g., Pleos-DyP4 shows less than 40 % identity with the other P. ostreatus DyPs).
In the present study, we followed a different approach consisting in optimization of a method for DyP expression as active soluble forms by adding hemin to the E. coli culture medium and decreasing the growth temperature to 16 °C after IPTG induction. Similar procedures have been used in the heterologous expression of some bacterial DyPs (Ogola et al. 2009; Santos et al. 2014). Following this strategy, we avoided a costly and time-consuming refolding protocol, although the final purification was more complicated and required three chromatographic steps.
Remarkable biochemical properties of P. ostreatus DyPs
In agreement with their divergent evolutionary origin, Pleos-DyP1 and Pleos-DyP4 significantly differed in their kinetic and stability properties. Differences in isoenzyme stabilities have been recently reported for P. ostreatus MnPs and VPs (Fernández-Fueyo et al. 2014d) and related to differential gene expression under environmental conditions (Fernández-Fueyo et al. 2014b). Similar or even larger differences were observed here, with Pleos-DyP4 (i) being over 90 % stable after 24 h at pH 2 (while Pleos-DyP1 was fully inactivated in 1 min) and (ii) showing over 20 °C higher 10-min T 50 values (73 °C for Pleos-DyP4 compared with only 52 °C for Pleos-DyP1).
Compared with other peroxidases, Pleos-DyP4 is more thermostable than previously described fungal DyPs (Linde et al. 2014; Salvachúa et al. 2013). However, a recently described bacterial DyP retains 90 % activity after 30 min at 80 °C (Yu et al. 2014). Compared with other enzymes, Pleos-DyP4 has higher thermal stability than the peroxidase from the African oil palm tree (Elaeis guineensis) that, with a 10-min T 50 of 69 °C, is one of the most thermostable peroxidases described to date (Rodriguez et al. 2002). Both P. ostreatus DyPs are stable at pH 3, in agreement with their maximal activity at acidic pH, and the high stability of Pleos-DyP4 at pH 2 is similar to that recently reported for a remarkably stable extralong MnP from Ceriporiopsis subvermispora (Fernández-Fueyo et al. 2014c).
Concerning catalytic properties, Pleos-DyP4 showed high turnover numbers for all the substrates assayed, except for ABTS that was similarly oxidized by the two DyPs, suggesting higher redox-potential than Pleos-DyP1, or faster electron transfer due to better substrate fitting at the oxidation site (in agreement with the lack of activity of the latter enzyme on the recalcitrant dye RB5).
However, the most surprisingly catalytic property of Pleos-DyP4 (and of Pleos-DyP1 in lower degree) is its ability to oxidize Mn2+ to Mn3+, a reaction that is seen as characteristic of MnP and VP among fungal peroxidases (Ruiz-Dueñas et al. 2009). The Pleos-DyP4 turnover for Mn2+ (56 s−1) was in the same order of those found for the two MnPs (63 and 125 s−1) and the two VPs (79 and 185 s−1) from the P. ostreatus genome, analyzed for comparison. The Pleos-DyP4 affinity for Mn2+ was also similar (only 2–3-fold higher K m) to those found for three of the four MnPs/VPs analyzed (the exception being Pleos-VP2 that has a fivefold lower K m). In contrast, Pleos-DyP1 has a very low affinity for Mn2+, which contributed to its very low catalytic efficiency (50-fold lower than Pleos-DyP4). Interestingly, oxidation of Mn2+ has been reported for four bacterial DyPs (Brown et al. 2012; Rahmanpour and Bugg 2015; Santos et al. 2014; Singh et al. 2013), albeit with lower turnover (up to 21 s−1 for Amycolatopsis DyP) than MnPs and VPs. However, this is the first time that Mn2+ oxidation is reported for a fungal DyP.
The present study also showed that VPs are able to oxidize RB19, an anthraquinoid dye whose oxidation is often presented as a unique characteristic of DyPs (Sugano et al. 2006). Although they belong to different superfamilies, a significant convergence in catalytic properties has been produced between some DyPs (such as Pleos-DyP4) in the CDE superfamily (Goblirsch et al. 2011) and VPs in the catalase-peroxidase superfamily (Zámocký et al. 2015). These enzymes not only adapted very different folds to provide the heme cofactor with an adequate environment, as discussed by Linde et al. (2015b), but they also converged in oxidation of a common range of substrates (from low redox-potential compounds to recalcitrant dyes and Mn2+). The specific site for Mn2+ oxidation is well characterized in MnPs and VPs (Gold et al. 2000; Ruiz-Dueñas et al. 2009), but, although some crystal structures have been reported for bacterial DyPs containing Mn2+ (Singh et al. 2013), more studies are required for a better characterization.
DyP role in nature: secretomic and other studies
The secretomic studies performed show that Pleos-DyP4, and several VPs and MnPs are part of the P. ostreatus extracellular proteome during growth on lignocellulosic materials (poplar chips or wheat straw) as the sole C (and N) source, as well as in submerged glucose cultures. This agrees with transcriptomic studies showing widespread expression of DyP genes in forest soils (Kellner et al. 2014). Paradoxically, Pleos-DyP4, identified in the different secretomes, is the only P. ostreatus DyP that lacks a typical signal peptide. A similar situation has been reported for other basidiomycete oxidoreductases involved in lignocellulose decay, such as Gloeophyllum trabeum methanol oxidase (Daniel et al. 2007), suggesting an alternative secretion mechanism (or enzyme release by hyphal autolysis).
Some controversy exists on the natural role of DyPs, which was not reported when the first (fungal) DyP was described as representing a new peroxidase family being able to oxidize substituted anthraquinoid dyes (Kim and Shoda 1999; Sugano et al. 2007). Subsequent studies on some bacterial DyPs described them as lignin-degrading enzymes (Ahmad et al. 2011; Brown and Chang 2014; Rahmanpour and Bugg 2015). However, the evidence is scarce and largely based on modification of uncharacterized lignocellulosic materials and oxidation of phenolic dimers (Ahmad et al. 2010, 2011; Brown et al. 2012; Rahmanpour and Bugg 2015). Even if some fungal, DyPs are also able to oxidize veratryl alcohol and nonphenolic lignin model dimers (representing the main substructures in the polymer) (Liers et al. 2010, 2013; Linde et al. 2014); their activity is too low to play a relevant role in the initial attack to the lignin polymer in nature (Linde et al. 2015b).
The present study reveals the evolutionary and biochemical diversity of DyPs in the P. ostreatus genome. Moreover, it shows that one of them (Pleos-DyP4) shares catalytic properties with VP, whose ligninolytic ability has been recently demonstrated (Fernández-Fueyo et al. 2014d), including oxidation of recalcitrant RB5 and Mn2+. A rather unique property of P. ostreatus, evidenced in the present study, is the secretion of three different peroxidase types being able to oxidize Mn2+. Their similarities with ligninolytic peroxidases, together with their simultaneous secretion in lignocellulose cultures and presence in natural habitats, suggest that DyPs could participate in lignocellulose degradation by oxidizing lignin-derived compounds or modified lignin, contributing to the genesis of lignin-derived organic matter in soils.
References
Ahmad M, Taylor CR, Pink D, Burton K, Eastwood D, Bending GD, Bugg TDH (2010) Development of novel assays for lignin degradation: comparative analysis of bacterial and fungal lignin degraders. Mol Biosys 6:815–821
Ahmad M, Roberts JN, Hardiman EM, Singh R, Eltis LD, Bugg TDH (2011) Identification of DypB from Rhodococcus jostii RHA1 as a lignin peroxidase. Biochemistry 50:5096–5107
Bao WL, Fukushima Y, Jensen KA, Moen MA, Hammel KE (1994) Oxidative degradation of non-phenolic lignin during lipid peroxidation by fungal manganese peroxidase. FEBS Lett 354:297–300
Branco S, Gladieux P, Ellison CE, Kuo A, LaButti K, Lipzen A, Grigoriev IV, Liao HL, Vilgalys R, Peay KG, Taylor JW, Bruns TD (2015) Genetic isolation between two recently diverged populations of a symbiotic fungus. Mol Ecol. doi:10.1111/mec.13132
Brown ME, Chang MCY (2014) Exploring bacterial lignin degradation. Curr Opin Chem Biol 19:1–7
Brown ME, Barros T, Chang MCY (2012) Identification and characterization of a multifunctional dye peroxidase from a lignin-reactive bacterium. ACS Chem Biol 7:2074–2081
Castanera R, Pérez G, Omarini A, Alfaro M, Pisabarro AG, Faraco V, Amore A, Ramírez L (2012) Transcriptional and enzymatic profiling of Pleurotus ostreatus laccase genes in submerged and solid-state fermentation cultures. Appl Environ Microbiol 78:4037–4045
Colpa DI, Fraaije MW, van Bloois E (2014) DyP-type peroxidases: a promising and versatile class of enzymes. J Ind Microbiol Biotechnol 41:1–7
Daniel G, Volc J, Filonova L, Plihal O, Kubátová E, Halada P (2007) Characteristics of Gloeophyllum trabeum alcohol oxidase, an extracellular source of H2O2 in brown rot decay of wood. Appl Environ Microbiol 73:6241–6253
Doyle WA, Smith AT (1996) Expression of lignin peroxidase H8 in Escherichia coli: folding and activation of the recombinant enzyme with Ca2+ and haem. Biochem J 315:15–19
Eastwood DC, Floudas D, Binder M, Majcherczyk A, Schneider P, Aerts A, Asiegbu FO, Baker SE, Barry K, Bendiksby M, Blumentritt M, Coutinho PM, Cullen D, de Vries RP, Gathman A, Goodell B, Henrissat B, Ihrmark K, Kauserud H, Kohler A, LaButti K, Lapidus A, Lavin JL, Lee YH, Lindquist E, Lilly W, Lucas S, Morin E, Murat C, Oguiza JA, Park J, Pisabarro AG, Riley R, Rosling A, Salamov A, Schmidt O, Schmutz J, Skrede I, Stenlid J, Wiebenga A, Xie XF, Kües U, Hibbett DS, Hoffmeister D, Högberg N, Martin F, Grigoriev IV, Watkinson SC (2011) The plant cell wall-decomposing machinery underlies the functional diversity of forest fungi. Science 333:762–765
Faraco V, Piscitelli A, Sannia G, Giardina P (2007) Identification of a new member of the dye-decolorizing peroxidase family from Pleurotus ostreatus. World J Microbiol Biotechnol 23:889–893
Fernández-Fueyo E, Ruiz-Dueñas FJ, Ferreira P, Floudas D, Hibbett DS, Canessa P, Larrondo L, James TY, Seelenfreund D, Lobos S, Polanco R, Tello M, Honda Y, Watanabe T, Watanabe T, Ryu JS, Kubicek CP, Schmoll M, Gaskell J, Hammel KE, St. John FJ, Vanden Wymelenberg A, Sabat G, Bondurant SS, Syed K, Yadav J, Doddapaneni H, Subramanian V, Lavín JL, Oguiza JA, Perez G, Pisabarro AG, Ramírez L, Santoyo F, Master E, Coutinho PM, Henrissat B, Lombard V, Magnuson JK, Kües U, Hori C, Igarashi K, Samejima M, Held BW, Barry K, LaButti K, Lapidus A, Lindquist E, Lucas S, Riley R, Salamov A, Hoffmeister D, Schwenk D, Hadar Y, Yarden O, de Vries RP, Wiebenga A, Stenlid J, Eastwood DC, Grigoriev IV, Berka R, Blanchette RA, Kersten P, Martínez AT, Vicuña R, Cullen D (2012) Comparative genomics of Ceriporiopsis subvermispora and Phanerochaete chrysosporium provide insight into selective ligninolysis. Proc Natl Acad Sci U S A 109:5458–5463
Fernández-Fueyo E, Acebes S, Ruiz-Dueñas FJ, Martínez MJ, Romero A, Medrano FJ, Guallar V, Martínez AT (2014a) Structural implications of the C-terminal tail in the catalytic and stability properties of manganese peroxidases from ligninolytic fungi. Acta Crystallogr D Biol Crystallogr 70:3253–3265
Fernández-Fueyo E, Castanera R, Ruiz-Dueñas FJ, López-Lucendo MF, Ramírez L, Pisabarro AG, Martínez AT (2014b) Ligninolytic peroxidase gene expression by Pleurotus ostreatus: differential regulation in lignocellulose medium and effect of temperature and pH. Fungal Genet Biol 72:150–161
Fernández-Fueyo E, Ruiz-Dueñas FJ, Martínez AT (2014c) Engineering a fungal peroxidase that degrades lignin at very acidic pH. Biotechnol Biofuels 7:114
Fernández-Fueyo E, Ruiz-Dueñas FJ, Martínez MJ, Romero A, Hammel KE, Medrano FJ, Martínez AT (2014d) Ligninolytic peroxidase genes in the oyster mushroom genome: heterologous expression, molecular structure, catalytic and stability properties and lignin-degrading ability. Biotechnol Biofuels 7:2
Floudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B, Martínez AT, Otillar R, Spatafora JW, Yadav JS, Aerts A, Benoit I, Boyd A, Carlson A, Copeland A, Coutinho PM, de Vries RP, Ferreira P, Findley K, Foster B, Gaskell J, Glotzer D, Górecki P, Heitman J, Hesse C, Hori C, Igarashi K, Jurgens JA, Kallen N, Kersten P, Kohler A, Kües U, Kumar TKA, Kuo A, LaButti K, Larrondo LF, Lindquist E, Ling A, Lombard V, Lucas S, Lundell T, Martin R, McLaughlin DJ, Morgenstern I, Morin E, Murat C, Nolan M, Ohm RA, Patyshakuliyeva A, Rokas A, Ruiz-Dueñas FJ, Sabat G, Salamov A, Samejima M, Schmutz J, Slot JC, St. John F, Stenlid J, Sun H, Sun S, Syed K, Tsang A, Wiebenga A, Young D, Pisabarro A, Eastwood DC, Martin F, Cullen D, Grigoriev IV, Hibbett DS (2012) The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336:1715–1719
Floudas D, Held BW, Riley R, Nagy LG, Koehler G, Ransdell AS, Younus H, Chow J, Chiniqui J, Lipzen A, Tritt A, Sun H, Haridas S, LaButti K, Ohm RA, Kües U, Blanchette RA, Grigoriev IV, Minto RE, Hibbett DS (2015) Evolution of novel wood decay mechanisms in Agaricales revealed by the genome sequences of Fistulina hepatica and Cylindrobasidium torrendii. Fungal Genet Biol 76:78–92
Goblirsch B, Kurker RC, Streit BR, Wilmot CM, Dubois JL (2011) Chlorite dismutases, DyPs, and EfeB: 3 microbial heme enzyme families comprise the CDE structural superfamily. J Mol Biol 408:379–398
Gold MH, Youngs HL, Gelpke MD (2000) Manganese peroxidase. Met Ions Biol Syst 37:559–586
Hane JK, Anderson JP, Williams AH, Sperschneider J, Singh KB (2014) Genome sequencing and comparative genomics of the broad host-range pathogen Rhizoctonia solani AG8. PLoS Genet 10(5), e1004281. doi:10.1371/journal.pgen.10042
Hofrichter M, Ullrich R, Pecyna MJ, Liers C, Lundell T (2010) New and classic families of secreted fungal heme peroxidases. Appl Microbiol Biotechnol 87:871–897
Hori C, Ishida T, Igarashi K, Samejima M, Suzuki H, Master E, Ferreira P, Ruiz-Dueñas FJ, Held B, Canessa P, Larrondo LF, Schmoll M, Druzhinina IS, Kubicek CP, Gaskell J, Kersten P, St. John F, Glasner JD, Sabat G, Bond-Durant SS, Khajamohiddin S, Yadav J, Mgbeahuruike AC, Kovalchuk A, Asiegbu FO, Lackner G, Hoffmeister D, Rencoret J, Gutiérrez A, Sun H, Lindquist E, Barry K, Riley R, Grigoriev IV, Henrissat B, Kües U, Berka RM, Martínez AT, Covert SF, Blanchette RA, Cullen D (2014) Analysis of the Phlebiopsis gigantea genome, transcriptome and secretome gives insight into its pioneer colonization strategies of wood. PLoS Genet 10(12), e1004759. doi:10.1371/journal.pgen.1004759
Janbon G, Ormerod KL, Paulet D, Byrnes EJ, Yadav V, Chatterjee G, Mullapudi N, Hon CC, Billmyre RB, Brunel F, Bahn YS, Chen WD, Chen Y, Chow EWL, Coppee JY, Floyd-Averette A, Gaillardin C, Gerik KJ, Goldberg J, Gonzalez-Hilarion S, Gujja S, Hamlin JL, Hsueh YP, Ianiri G, Jones S, Kodira CD, Kozubowski L, Lam W, Marra M, Mesner LD, Mieczkowski PA, Moyrand F, Nielsen K, Proux C, Rossignol T, Schein JE, Sun S, Wollschlaeger C, Wood IA, Zeng QD, Neuveglise C, Newlon CS, Perfect JR, Lodge JK, Idnurm A, Stajich JE, Kronstad JW, Sanyal K, Heitman J, Fraser JA, Cuomo CA, Dietrich FS (2014) Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. PLoS Genet 10:4
Johjima T, Ohkuma M, Kudo T (2003) Isolation and cDNA cloning of novel hydrogen peroxide-dependent phenol oxidase from the basidiomycete Termitomyces albuminosus. Appl Microbiol Biotechnol 61:220–225
Kellner H, Luis P, Pecyna MJ, Barbi F, Kapturska D, Krüger D, Zak DR, Marmeisse R, Vandenbol M, Hofrichter M (2014) Widespread occurrence of expressed fungal secretory peroxidases in forest soils. PLoS ONE 9:4
Kim SJ, Shoda M (1999) Purification and characterization of a novel peroxidase from Geotrichum candidum Dec 1 involved in decolorization of dyes. Appl Environ Microbiol 65:1029–1035
Kohler A, Kuo A, Nagy LG, Morin E, Barry KW, Buscot F, Canbäck B, Choi C, Cichocki N, Clum A, Colpaert J, Copeland A, Costa MD, Dore J, Floudas D, Gay G, Girlanda M, Henrissat B, Herrmann S, Hess J, Högberg N, Johansson T, Khouja HR, LaButti K, Lahrmann U, Levasseur A, Lindquist EA, Lipzen A, Marmeisse R, Martino E, Murat C, Ngan CY, Nehls U, Plett JM, Pringle A, Ohm RA, Perotto S, Peter M, Riley R, Rineau F, Ruytinx J, Salamov A, Shah F, Sun H, Tarkka M, Tritt A, Veneault-Fourrey C, Zuccaro A, Mycorrhizal Genomics Initiative C, Tunlid A, Grigoriev IV, Hibbett DS, Martin F (2015) Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat Genet 47:410–415
Kung CP, Wu YR, Chuang HW (2014) Expression of a dye-decolorizing peroxidase results in hypersensitive response to cadmium stress through reducing the ROS signal in Arabidopsis. Environ Exp Bot 101:47–55
Larraya LM, Pérez G, Peñas MM, Baars JJP, Mikosch TSP, Pisabarro AG, Ramírez L (1999) Molecular karyotype of the white rot fungus Pleurotus ostreatus. Appl Environ Microbiol 65:3413–3417
Liers C, Bobeth C, Pecyna M, Ullrich R, Hofrichter M (2010) DyP-like peroxidases of the jelly fungus Auricularia auricula-judae oxidize nonphenolic lignin model compounds and high-redox potential dyes. Appl Microbiol Biotechnol 85:1869–1879
Liers C, Pecyna MJ, Kellner H, Worrich A, Zorn H, Steffen KT, Hofrichter M, Ullrich R (2013) Substrate oxidation by dye-decolorizing peroxidases (DyPs) from wood- and litter-degrading agaricomycetes compared to other fungal and plant heme-peroxidases. Appl Microbiol Biotechnol 87:5839–5849
Liers C, Aranda E, Strittmatter E, Piontek K, Plattner DA, Zorn H, Ullrich R, Hofrichter M (2014) Phenol oxidation by DyP-type peroxidases in comparison to fungal and plant peroxidases. J Mol Catal B Enzym 103:41–46
Linde D, Coscolín C, Liers C, Hofrichter M, Martínez AT, Ruiz-Dueñas FJ (2014) Heterologous expression and physicochemical characterization of a fungal dye-decolorizing peroxidase from Auricularia auricula-judae. Protein Expr Purif 103:28–37
Linde D, Pogni R, Cañellas M, Lucas F, Guallar V, Baratto MC, Sinicropi A, Sáez-Jiménez V, Coscolín C, Romero A, Medrano FJ, Ruiz-Dueñas FJ, Martínez AT (2015a) Catalytic surface radical in dye-decolorizing peroxidase: a computational, spectroscopic and directed mutagenesis study. Biochem J 466:253–262
Linde D, Ruiz-Dueñas FJ, Fernández-Fueyo E, Guallar V, Hammel KE, Pogni R, Martínez AT (2015b) Basidiomycete DyPs: genomic diversity, structural-functional aspects, reaction mechanism and environmental significance. Arch Biochem Biophys. doi:10.1016/j.abb.2015.01.018
Martin F, Aerts A, Ahren D, Brun A, Danchin EGJ, Duchaussoy F, Gibon J, Kohler A, Lindquist E, Pereda V, Salamov A, Shapiro HJ, Wuyts J, Blaudez D, Buée M, Brokstein P, Canbäck B, Cohen D, Courty PE, Coutinho PM, Delaruelle C, Detter JC, Deveau A, DiFazio S, Duplessis S, Fraissinet-Tachet L, Lucic E, Frey-Klett P, Fourrey C, Feussner I, Gay G, Grimwood J, Hoegger PJ, Jain P, Kilaru S, Labbe J, Lin YC, Legue V, Le Tacon F, Marmeisse R, Melayah D, Montanini B, Muratet M, Nehls U, Niculita-Hirzel H, Oudot-Le Secq MP, Peter M, Quesneville H, Rajashekar B, Reich M, Rouhier N, Schmutz J, Yin T, Chalot M, Henrissat B, Kües U, Lucas S, Van de Peer Y, Podila GK, Polle A, Pukkila PJ, Richardson PM, Rouze P, Sanders IR, Stajich JE, Tunlid A, Tuskan G, Grigoriev IV (2008) The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 452:88–92
Miki Y, Morales M, Ruiz-Dueñas FJ, Martínez MJ, Wariishi H, Martínez AT (2009) Escherichia coli expression and in vitro activation of a unique ligninolytic peroxidase that has a catalytic tyrosine residue. Protein Expr Purif 68:208–214
Morin E, Kohler A, Baker AR, Foulongne-Oriol M, Lombard V, Nagy LG, Ohm RA, Patyshakuliyeva A, Brun A, Aerts AL, Bailey AM, Billette C, Coutinho PM, Deakin G, Doddapaneni H, Floudas D, Grimwood J, Hildén K, Kües U, LaButti KM, Lapidus A, Lindquist EA, Lucas SM, Murat C, Riley RW, Salamov AA, Schmutz J, Subramanian V, Wösten HAB, Xu JP, Eastwood DC, Foster GD, Sonnenberg ASM, Cullen D, de Vries RP, Lundell T, Hibbett DS, Henrissat B, Burton KS, Kerrigan RW, Challen MP, Grigoriev IV, Martin F (2012) Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc Natl Acad Sci U S A 109:17501–17506
Ogola HJO, Kamiike T, Hashimoto N, Ashida H, Ishikawa T, Shibata H, Sawa Y (2009) Molecular characterization of a novel peroxidase from the cyanobacterium Anabaena sp strain PCC 7120. Appl Environ Microbiol 75:7509–7518
Ohm RA, de Jong JF, Lugones LG, Aerts A, Kothe E, Stajich JE, de Vries RP, Record E, Levasseur A, Baker SE, Bartholomew KA, Coutinho PM, Erdmann S, Fowler TJ, Gathman AC, Lombard V, Henrissat B, Knabe N, Kües U, Lilly WW, Lindquist E, Lucas S, Magnuson JK, Piumi F, Raudaskoski M, Salamov A, Schmutz J, Schwarze FWMR, vanKuyk PA, Horton JS, Grigoriev IV, Wösten HAB (2010) Genome sequence of the model mushroom Schizophyllum commune. Nat Biotechnol 28:957–963
Pérez-Boada M, Doyle WA, Ruiz-Dueñas FJ, Martínez MJ, Martínez AT, Smith AT (2002) Expression of Pleurotus eryngii versatile peroxidase in Escherichia coli and optimisation of in vitro folding. Enzym Microb Technol 30:518–524
Rahmanpour R, Bugg TDH (2015) Characterisation of Dyp-type peroxidases from Pseudomonas fluorescens Pf-5: oxidation of Mn(II) and polymeric lignin by Dyp1B. Arch Biochem Biophys. doi:10.1016/j.abb.2014.12.022
Riley R, Salamov AA, Brown DW, Nagy LG, Floudas D, Held BW, Levasseur A, Lombard V, Morin E, Otillar R, Lindquist EA, Sun H, LaButti KM, Schmutz J, Jabbour D, Luo H, Baker SE, Pisabarro AG, Walton JD, Blanchette RA, Henrissat B, Martin F, Cullen D, Hibbett DS, Grigoriev IV (2014) Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc Natl Acad Sci U S A 111:9923–9928
Rineau F, Roth D, Shah F, Smits M, Johansson T, Canbäck B, Olsen PB, Persson P, Grell MN, Lindquist E, Grigoriev IV, Lange L, Tunlid A (2012) The ectomycorrhizal fungus Paxillus involutus converts organic matter in plant litter using a trimmed brown-rot mechanism involving Fenton chemistry. Environ Microbiol 14:1477–1487
Roberts JN, Singh R, Grigg JC, Murphy MEP, Bugg TDH, Eltis LD (2011) Characterization of dye-decolorizing peroxidases from Rhodococcus jostii RHA1. Biochemistry 50:5108–5119
Rodriguez A, Pina DG, Yelamos B, Leon JJC, Zhadan GG, Villar E, Gavilanes F, Roig MG, Sakharov IY, Shnyrov VL (2002) Thermal stability of peroxidase from the African oil palm tree Elaeis guineensis. Eur J Biochem 269:2584–2590
Ruiz-Dueñas FJ, Martínez AT (2010) Structural and functional features of peroxidases with a potential as industrial biocatalysts. In: Torres E, Ayala M (eds) Biocatalysts based on heme peroxidases. Springer, Berlin, pp 37–59
Ruiz-Dueñas FJ, Morales M, García E, Miki Y, Martínez MJ, Martínez AT (2009) Substrate oxidation sites in versatile peroxidase and other basidiomycete peroxidases. J Exp Bot 60:441–452
Ruiz-Dueñas FJ, Fernández E, Martínez MJ, Martínez AT (2011) Pleurotus ostreatus heme peroxidases: an in silico analysis from the genome sequence to the enzyme molecular structure. C R Biol 334:795–805
Ruiz-Dueñas FJ, Lundell T, Floudas D, Nagy LG, Barrasa JM, Hibbett DS, Martínez AT (2013) Lignin-degrading peroxidases in Polyporales: an evolutionary survey based on ten sequenced genomes. Mycologia 105:1428–1444
Salvachúa D, Prieto A, Martínez AT, Martínez MJ (2013) Characterization of a novel dye-decolorizing peroxidase (DyP)-type enzyme from Irpex lacteus and its application in enzymatic hydrolysis of wheat straw. Appl Environ Microbiol 79:4316–4324
Sánchez C (2010) Cultivation of Pleurotus ostreatus and other edible mushrooms. Appl Microbiol Biotechnol 85:1321–1337
Santos A, Mendes S, Brissos V, Martins LO (2014) New dye-decolorizing peroxidases from Bacillus subtilis and Pseudomonas putida MET94: towards biotechnological applications. Appl Microbiol Biotechnol 98:2053–2065
Scheibner M, Hulsdau B, Zelena K, Nimtz M, de Boer L, Berger RG, Zorn H (2008) Novel peroxidases of Marasmius scorodonius degrade β-carotene. Appl Microbiol Biotechnol 77:1241–1250
Singh R, Grigg JC, Qin W, Kadla JF, Murphy MEP, Eltis LD (2013) Improved manganese-oxidizing activity of DypB, a peroxidase from a lignolytic bacterium. ACS Chem Biol 8:700–706
Spinnler HE, de Jong E, Mauvais G, Semon E, Le Quéré J-L (1994) Production of halogenated compounds by Bjerkandera adusta. Appl Microbiol Biotechnol 42:212–221
Strittmatter E, Liers C, Ullrich R, Wachter S, Hofrichter M, Plattner DA, Piontek K (2013) First crystal structure of a fungal high-redox potential dye-decolorizing peroxidase: substrate interaction sites and long-range electron transfer. J Biol Chem 288:4095–4102
Sugano Y (2009) DyP-type peroxidases comprise a novel heme peroxidase family. Cell Mol Life Sci 66:1387–1403
Sugano Y, Sasaki K, Shoda M (1999) cDNA cloning and genetic analysis of a novel decolorizing enzyme, peroxidase gene dyp from Geotrichum candidum Dec 1. J Biosci Bioeng 87:411–417
Sugano Y, Nakano R, Sasaki K, Shoda M (2000) Efficient heterologous expression in Aspergillus oryzae of a unique dye decolorizing peroxidase, DyP, of Geotrichum candidum Dec 1. Appl Environ Microbiol 66:1754–1758
Sugano Y, Matsushima Y, Shoda M (2006) Complete decolorization of the anthraquinone dye Reactive blue 5 by the concerted action of two peroxidases from Thanatephorus cucumeris Dec 1. Appl Microbiol Biotechnol 73:862–871
Sugano Y, Muramatsu R, Ichiyanagi A, Sato T, Shoda M (2007) DyP, a unique dye-decolorizing peroxidase, represents a novel heme peroxidase family. Asp171 replaces the distal histidine of classical peroxidases. J Biol Chem 282:36652–36658
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Torres E, Ayala M (2010) Biocatalysis based on heme peroxidases. Springer, Berlin
Welinder KG (1992) Superfamily of plant, fungal and bacterial peroxidases. Curr Opin Struct Biol 2:388–393
Yoshida T, Tsuge H, Konno H, Hisabori T, Sugano Y (2011) The catalytic mechanism of dye-decolorizing peroxidase DyP may require the swinging movement of an aspartic acid residue. FEBS J 278:2387–2394
Yoshida T, Tsuge H, Hisabori T, Sugano Y (2012) Crystal structures of dye-decolorizing peroxidase with ascorbic acid and 2,6-dimethoxyphenol. FEBS Lett 586:4351–4356
Yu WN, Liu WN, Huang HQ, Zheng F, Wang XY, Wu YY, Li KJ, Xie XM, Jin Y (2014) Application of a novel alkali-tolerant thermostable DyP-type peroxidase from Saccharomonospora viridis DSM 43017 in biobleaching of eucalyptus kraft pulp. PLoS ONE 9, e110319
Zámocký M, Hofbauer S, Schaffner I, Gasselhuber B, Nicolussi A, Soudi M, Pirker KF, Furtmüller PG, Obinger C (2015) Independent evolution of four heme peroxidase superfamilies. Arch Biochem Biophys. doi:10.1016/j.abb.2014.12.025
Zelena K, Hardebusch B, Hulsdau B, Berger RG, Zorn H (2009a) Generation of norisoprenoid flavors from carotenoids by fungal peroxidases. J Agric Food Chem 57:9951–9955
Zelena K, Zorn H, Nimtz M, Berger RG (2009b) Heterologous expression of the msp2 gene from Marasmius scorodonius. Arch Microbiol 191:397–402
Acknowledgments
This work was supported by the HIPOP (BIO2011-26694) project of the Spanish Ministry of Economy and Competitiveness (MINECO), and the INDOX (KBBE-2013-7-613549) European project. Antonio G. Pisabarro (Public University of Navarre, Pamplona, Spain) is acknowledged for coordinating the JGI project for sequencing the P. ostreatus genome. E.F.-F. acknowledges a Junta de Ampliación de Estudios fellowship of CSIC, co-funded by the European Social Fund, and F.J.R.-D. acknowledges a Ramón y Cajal contract of the Spanish MINECO.
We also thank Daniel Lindner, David Fitzpatrick, David S. Hibbett, Eric Record, Francis Martin, Gregory Bonito, Igor Grigoriev, In-Geol Choi, Jon K. Magnuson, Joseph Spatafora, László Nagy, Laurie Connell, Mary C. Aime, Minou Nowrousian, Otto Miettinen, and Steven Singer for the permission to use (in Fig. 7 preparation) not published genome data of Agaricus bisporus var. burnettii, Antrodia sinuosa, Aporpium caryae, Armillaria mellea, Artolenzites elegans, Auriscalpium vulgare, Boletus edulis, Calocera cornea, Calocera viscosa, Cerrena unicolor, Clavicorona pyxidata, Clavulina sp., Cortinarius glaucopus, Cryptococcus vishniacii, Daedalea quercina, Dendrothele bispora, Dioszegia cryoxerica, Exidia glandulosa, Fibulorhizoctonia sp., Gymnopus androsaceus, Gyrodon lividus, Laetiporus sulphureus var. sulphureus, Lentinus tigrinus, Macrolepiota fuliginosa, Neolentinus lepideus. Piloderma croceum, Polyporus arcularius, Pycnoporus coccineus, Pycnoporus sanguineus, Ramaria rubella, Scleroderma citrinum, Sistotremastrum niveocremeum, Sistotremastrum suecicum, Trametes ljubarskyi, Trichaptum abietinum, Tricholoma matsutake, Peniophora sp., Guyanagaster necrorhiza, Gymnopilus chrysopellus, Leucogyrophana mollusca, Panellus stipticus, Schizopora paradoxa, Obba rivulosa, Rhizopogon vinicolor, and Trichosporon oleaginosus, produced by the US Department of Energy Joint Genome Institute; a DOE Office of Science User Facility supported under Contract No. DE-AC02-05CH11231.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 208 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fernández-Fueyo, E., Linde, D., Almendral, D. et al. Description of the first fungal dye-decolorizing peroxidase oxidizing manganese(II). Appl Microbiol Biotechnol 99, 8927–8942 (2015). https://doi.org/10.1007/s00253-015-6665-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6665-3