
Abstract
Thermal modification is a well-established commercial technology for improving the dimensional stability and durability of timber. Numerous reviews of thermally modified timber (TMT) are to be found in the scientific literature, but until now a review of the influence of cell wall moisture content during the modification process on the properties of TMT has been lacking. This paper reviews the current state of knowledge regarding the hygroscopic and dimensional behaviour of TMT modified under dry (cell wall at nearly zero moisture content) and wet (cell wall contains moisture) conditions. After an overview of the topic area, the review explores the literature on the thermal degradation of the polysaccharidic and lignin components of the cell wall, as well as the role of extractives. The properties of TMT modified under wet and dry conditions are compared including mass loss, hygroscopic behaviour and dimensional stability. The role of hydroxyl groups in determining the hygroscopicity is discussed, as well as the importance of considering the mobility of the cell wall polymers and crosslinking when interpreting sorption behaviour. TMT produced under wet processing conditions exhibits behaviour that changes when the wood is subjected to water leaching post-treatment, which includes further weight loss, changes in sorption behaviour and dimensional stability, but without any further change in accessible hydroxyl (OH) content. This raises serious questions regarding the role that OH groups play in sorption behaviour.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Background
The commercial production of thermally modified timber (TMT) usually takes place between temperatures of 160 and 240 °C. This results in a material that is darker in colour [1], has improved dimensional stability and microbial resistance compared to unmodified wood, but there is a significant reduction in strength, especially fracture resistance. The reduction in ductility makes the use of TMT problematic for many load-bearing applications [2]. The chemistry of thermal degradation, particularly of the lignin, is different below about 150 °C [3, 4], and thermal modification is herein defined as being above this temperature, although irreversible changes also occur to wood heated below this temperature [5, 6]. Thermal modification processes do not employ temperatures above 240 °C, since the degradation of the wood becomes too severe to make the process commercially useful.
The first reports of the thermal modification of wood date back as far as 1915 to the work of Harry Tiemann, a kiln specialist at the Forest Products Laboratory in Madison Wisconsin, who heated air-dry wood in superheated steam at 150 °C, finding a reduction in hygroscopicity of the TMT. In the 1930s and 1940s, the Forest Products Laboratory continued this research [7, 8], resulting in the granting of a patent [9]. Early ventures to exploit this technology took place in the USA, involving the heating of wood which was submerged under molten metal at temperatures between 160 to 260 °C (Staybwood) [8]. Perhaps unsurprisingly, this was not a commercial success. Research and development continued into the 1970s and 1980s, with further attempts at commercialisation in Germany [10, 11], but again without success. However, in the 1990s there was renewed interest in thermal modification and the commercial landscape was finally favourable to the exploitation of this technology. This renewed interest, in Europe especially, was driven by environmental concerns and the introduction of legislation, such as the European Biocides Directive. As a response, there were commercial processes developed in France (rétification), Finland (Thermowood), Germany (Menz Holz) and the Netherlands (Plato Wood) [12]. In the twenty-first century, TMT is finding increasing application in exterior cladding, decking and joinery applications and is a well-established commercial technology, with European production of TMT now exceeding 500,000 m3 [13].
Description of process conditions
The primary purpose of this review is to examine the differences in properties of wood that has been thermally modified under ‘wet’ and ‘dry’ conditions. Initial stages of dry thermal modification involve an increase in temperature (ramp) during which the wood loses sorbed water. Alternatively, the heating can take place under conditions designed to keep the water in the wood cell wall, in a wet process. Wet conditions are herein defined as being thermal modification where the cell wall of the wood still contains moisture during the process, whereas dry conditions (HTdry) are defined as those in which the wood has been dried to close to zero moisture content before the thermal modification process. In HTdry processes, the timber can be heated under an inert gas blanket of nitrogen, in a vacuum, under superheated steam or in ambient air. The presence of water vapour or water in the thermal modification process affects the heat transfer process and the chemistry taking place in the wood. The types of thermal modification process differ, depending on time and temperature of treatment (including ramp-up, hold and ramp-down stages), treatment atmosphere (inert gas, air, vacuum), open or closed systems, wood species (with important differences between hardwoods and softwoods) and ‘wet’ or ‘dry’ systems. Wet conditions can be formally divided into processes which use saturated steam as the heating medium (hygrothermal or HTwet) and those in which the wood is completely immersed in water during the process (hydrothermal or HDTwet). HTwet conditions produce degradation products that remain in the cell wall until a subsequent leaching step is performed. Apart from being used as one of the thermal modification stages in the Plato process [14], HDTwet treatments are mainly used to extract cell wall degradation products for biorefinery or torrefaction processes and are not the main subject of this review [15] and much higher weight losses are recorded with HDTwet compared to HTwet processes [16]. The technical details of the different thermal modification processes have been discussed in considerable detail in many reviews of the topic [2, 12, 17,18,19,20], and only a brief overview of the most pertinent differences is given herein. Despite a considerable amount of attention being given to reviewing this subject in the peer review literature, we were not able to locate any review of the differences observed in the properties of wood modified under wet and dry conditions, hence the need for the current work.
The different conditions under which thermal modification is performed are categorised as shown in Fig. 1.

Different thermal conditions. Thermal modification is defined as the temperature region from 150 °C to 240 °C. Above 240 °C is defined as thermal degradation, although degradation processes actually take place throughout the temperature range. Below 150 °C is defined as thermal treatment or ageing
In addition to the options presented in Fig. 1, there is the possibility of running the modification plant with a closed vessel or an open system. With closed processes, there is a build-up of degradation products and steam in the vessel creating a pressurised system. With an open system, it is possible to employ a continuous flow, where the degradation products and exhaust gases are vented to the atmosphere (subject to effective emissions controls) or a re-circulating system, where the hot gases are returned to the autoclave, with the condensable fraction being removed. Open flow systems are invariably operated at atmospheric pressure, although a re-circulating system can be employed with a pressurised vessel. With a pressurised system, pressure is applied with the addition of nitrogen or steam (saturated or superheated steam, depending upon the temperature and pressure conditions). Careful control of pressure is required in a closed system to modify the wood under constant saturated or superheated steam conditions.
Willems and Altgen [21] give a detailed description of the differences between dry and wet thermal treatment processes by defining them in terms of water activity against temperature (Fig. 2). Heating at high temperatures (over 200 °C) and low water activity, results in dry thermolysis, with the pressure making little contribution to the variables and the main controlling factor being the process temperature. Wet processes operate at lower temperatures (160–180 °C) in saturated steam, with the necessary moist conditions ensured by control of temperature and pressure, although pressure is the most important parameter when determining mass loss (before and after post-treatment leaching) [22, 23]. Saturated steam is in equilibrium with liquid water at the same temperature and pressure, whereas superheated steam is dry water vapour that has been heated above its saturation point. The degradation of wood is also mainly controlled by the pressure when nitrogen rather than steam is used [11].

Definitions of different moisture conditions in thermal modification according to [21]
The symbol aw is the water activity (the ratio of the vapour pressure of water in a material to the vapour pressure of pure water at the same temperature).
Although air is included for completeness’ sake; in reality the atmospheric systems are operated so that there is a build-up of steam in the vessel from the wood moisture and driving off of the water of constitution so that the thermal modification operates under essentially non-oxidative conditions (although saturated steam conditions will not be realised). The presence of air results in oxidative degradative processes taking place, and, in the absence of a steam blanket, fire is a risk. Vacuum systems require a means of heating the wood during the modification cycle, and this can be achieved using heated plattens or by initiating a thermal degradation reaction in air and then applying a vacuum.
Property changes
A summary of the main properties of thermally modified timber is given in Table 1.
Thermal modification results in weight loss, and there is usually a change in macroscopic dimensions of the wood accompanying this. This weight loss is related to the time and temperature of treatment under dry conditions [12], and there may be an associated reduction in volume of the wood. Loss of weight is dependent on wood species, with hardwoods exhibiting higher weight loss than softwoods under identical conditions [24], and is dependent on whether dry or wet conditions are employed [25]. The weight loss after modification is often used as an indicator of other properties of the TMT, but an accumulation of water-soluble degradation products can occur, which often leads to an underestimate of actual weight loss due to thermal modification [26]. When wood is heated under saturated steam (HTwet) conditions, this can (depending on the exact process conditions) result in a greater loss of weight, starting at a lower temperature, compared with heating under dry, but otherwise identical conditions. This higher weight loss with HTwet processes may not be observed immediately after treatment, but is always seen when a post-treatment water-leaching step is performed post-treatment [23, 27,28,29]. The weight loss is usually associated with a reduction in apparent density, although this depends upon treatment conditions and wood species [30]. Weight loss can also be correlated with other properties, such as hygroscopicity, but a leaching step may be necessary to reveal this relationship [31].
Hygrothermal treatment results in both irreversible and reversible changes to the wood, the latter being erased by exposure to high RH or liquid water [6]. There is a volume decrease which occurs during the heat treatment, but additional reductions in volume (and weight) may also be observed during subsequent leaching cycles. In part, this reduction in volume can be directly attributed to the loss of degraded cell wall material, but there is also the likelihood that there are rearrangements in molecular structure within the cell wall. These changes in structure can be ‘frozen’ at the end of the thermal modification process, and water-soaking restores the mobility of the structure, allowing for an annealing process with associated changes in hygroscopicity and dimensional behaviour [32]. Apart from the extremes of 0% or 100% RH, there have been relatively few studies where the effect of relative humidity during thermal modification has been investigated, but this is an area of considerable interest [5].
Degradation processes
Thermal degradation is accompanied by the emission of water, carbon dioxide, methanol, acetic acid and formic acid [33], with non-volatile degradation products often remaining in the wood. The degradation products arise from the breakdown of cell wall polymers and from the wood extractives. Wood extractives can also migrate to the surface if they are mobile under the thermal modification conditions. Thermal degradation of the amorphous polysaccharide content occurs, mainly associated with the hemicelluloses, which is also facilitated by the presence of water [12, 34]. The most mobile parts of the polymer network are also the most accessible to water and have void volume around them when water-saturated, which also makes them the most susceptible to degradation under HTwet conditions. Degradation of carbohydrates under HTdry conditions is likely to lead to dehydration products, such as furfurals, which have the potential to polymerise and form insoluble hydrophobic materials in the cell wall, which are less susceptible to leaching. The formation of furfurals under hydrolysis conditions is not favoured. Thermal degradation of the hemicelluloses commences at lower temperatures compared to lignin [4]. Because thermal modification results in the degradation of cell wall polymers, there should be no swelling associated with the production of TMT. The hemicelluloses display chain mobility when moisture is present, and their removal means that this property is reduced in TMT. The mobility of the amorphous part of cell wall polymer network is also restricted if crosslinking takes place. The wood polymer matrix of thermally modified wood then acts as a stiff, elastic, brittle material [35, 36]. This change in mobility affects the mechanosorptive properties of the material. For example, it is known that the sorption hysteresis is affected by the extent of crosslinking of hydrogels [37].
The degradation products may escape from the cell wall during the thermal process by migration, evolution of volatiles or leaching in wet conditions, which will result in a reduction in cell wall volume. This may be recorded as a reduction in macroscopic volume if the lumen dimensions do not increase, but if the lumen increases in size, there may be no recorded change in macroscopic dimensions. If the degradation products remain inside the cell wall, there may be no change in cell wall dimensions until they are removed in a subsequent leaching stage. Changes in dimension also require mobility of the cell wall matrix macromolecules, and this may be restricted if thermal modification takes place in dry conditions.
The presence of water in the cell wall has an important influence upon the degradation processes taking place during thermal modification. As noted, water molecules in the cell wall in a wet thermal modification process increase the mobility of the cell wall polymers and also potentially of the degradation products during the process. The presence of hydronium ions in the water can also catalyse the hydrolysis of glycosidic bonds and of acetyl groups in the hemicelluloses. The weight loss is attributed mainly to loss of hemicelluloses, which are considered to be the most thermally labile of the wood cell wall polymers and is greater for hardwoods compared to softwoods treated under identical conditions. Degradation of hemicelluloses has been observed at temperatures as low as 100 °C, when wood was heated for extended periods of time (48 h) [38] and indirect evidence for thermal degradation at temperatures as low as 64 °C under wet conditions has been obtained using dynamic mechanical analysis [39]. Wood will slowly undergo irreversible changes even at room temperature and the study of wood ageing and attempts to mimic or accelerate these effects for use in musical instruments continue to elicit interest [6].
There is also a smaller reduction in amorphous cellulose content, but a relative proportional increase in the lignin content. However, some caution is advised when determining the lignin content of thermally modified wood, since the breakdown products of hemicelluloses could contribute to char formation when analysing residual lignin by acid digestion techniques [40]. Although the hemicelluloses are considered the most thermally labile component, there are also reactions that take place in the lignin network, which include chain scission and/or crosslinking. Whether the overall crosslink density of the lignin increases as a result of thermal modification is not known.
There is a decrease in the O/C ratio which is accompanied by degradation of the less ordered carbohydrate regions, hemicellulose depolymerisation accompanied by the generation of acetic acid, furfural products, aldehydes and lignin breakdown/modification [41,42,43,44,45,46]. Hemicellulose (acetic acid, furfural, methyl furfural, hydroxy methyl furfural) and lignin (acetic acid, guaiacol, syringol, vanillin, syringaldehyde, acetovanillone, acetosyringone) degradation products are found in increasing amounts as treatment temperature is increased [25] or with increase in treatment pressure in closed systems [23]. The presence of water facilitates the cleavage of hemicellulosic acetyl groups; furthermore, the use of closed systems ensures that the released acetic acid remains, as well as any formic acid that may be produced [16, 23, 47]. These acidic conditions catalyse the depolymerisation of polysaccharides. The decrease in O/C ratio correlates with other properties, such as changes in microbial durability and EMC [48,49,50], and is also related to the reduction in the number of polar sites (mainly OH groups) in the TMT, with a concomitant decrease in surface free-energy [51] and changes in wettability [52].
TMT contains stable free-radicals, which can be detected by electron spin resonance (ESR) [21, 53]. The ESR signal is invariably relatively broad without fine structure, with a magnitude that increases with temperature of treatment and correlates with mass loss. The source of stable radicals may be found in the conjugated structure of thermally modified lignin associated with semiquinone radicals and diamagnetic quinhydrones [54].
The degradation reactions taking place within the wood cell wall during thermal modification are extremely complex, and for this reason, many studies have been performed on isolated well wall polymers. Although this may provide useful information, it is certain that the behaviour of the isolated polymers will be different from that when they are located in the cell wall. For example, it has been shown that lignin inhibits the thermal degradation of polysaccharides in pine wood flour [55] and isolated cellulose [56, 57]. The following sections mainly detail the current level of understanding of those changes that take place in the cell wall.
Chemical changes
Polysaccharides
For the purposes of this review, the polysaccharides are treated as one group, but are classified according to whether they are amorphous or crystalline. The crystalline polysaccharides primarily comprise the cellulose in the microfibrils, whereas the amorphous content is mainly, but not exclusively, associated with the hemicelluloses. This review is not concerned with the details of the morphology of the elementary fibrils and how they combine to form aggregated structures, which is covered in detail elsewhere [58, 59]. However, the issue of polysaccharide chain accessibility and mobility is of crucial significance when discussing the role of water in the degradation processes and the properties of TMT.
Cellulose has a major reinforcing structural function in the cell wall, and chains of cellulose were originally supposed to be aggregated into units of 36, but are now thought to be created in groups of 18 or 24 to form elementary microfibrils in the wood cell wall [58]. The shape of the microfibril cross section is unknown, but various structures can be envisaged, which present different hydrophobic and hydrophilic faces for interaction with the matrix. There is an amorphous and crystalline component to the cellulose, with the amorphous part being accessible to water molecules and also susceptible to thermal degradation. The proportion of accessible OH groups which are available to water depends upon the dimensions of the microfibril aggregates, with deuterium exchange indicating that 28% are exchangeable [58]. Cellulose is composed solely of glucopyranose units, linked with a regular β-(1,4) linear bonding motif, with no branching or substitution of the chains. The degree of polymerisation (DP) of cellulose is very high (at least 104), and the crystalline core of the microfibril is not accessible to water, as well as being stable under the conditions employed in timber thermal modification. Microfibrils cluster together to form aggregates which have different dimensions in various plant species. There is some evidence suggesting that microfibrillar aggregates in the cell wall of conifers may be up to 10–20 nm across [60].
The size of the cellulose crystal in wood influences the thermal degradation, with greater crystal size correlating with higher thermal stability [61]. It was found that the interfibrillar distance increased when wood was heated in saturated steam (HTwet) at 150 °C or 180 °C [62]. There is also a change in both the degree of crystallinity and the width of the cellulose crystal as a result of heat treatment, and differences in behaviour have been noted between HTwet and HTdry conditions. In one (albeit, preliminary) study, wood powder was heated in dry conditions in air for 20 h, at a range of temperatures from 120 to 220 °C or steam heated for 10 min at the same temperatures [63]. It was found that under HTwet conditions, there was a slight increase in the degree of crystallinity as temperature was increased, but a decrease under HTdry conditions. Meanwhile, the width of the cellulose crystal increased under HTdry conditions as temperature of treatment was increased, but decreased with the HTwet samples. Differences in behaviour were also noted between wet and dry treatment, when heated in water under pressure or in an oven in air at 180, 200 or 220 °C [64]. In all cases, there was an increase in degree of crystallinity (taking into account the sample weight loss) with time of heating; however, as heating was continued, the proportion of crystallinity fell. In these experiments, it was found that the width of the cellulose crystal increased in all cases (greater for HTwet compared to HTdry) until wood samples were exposed to extended heating times. Based on the determined activation energies, it was concluded that crystallisation occurred more readily under HTwet compared to HTdry conditions. Other studies have shown that the width of the cellulose crystal [65] and the crystallinity index [66, 67] increase, fall and then increase again [68], as the modification temperature rises, up to a maximum temperature. In order to determine the crystalline content, it is necessary to separate the crystalline part of the signal from the amorphous component in the scattering, with contributions not just from amorphous cellulose, but hemicelluloses and lignin, which is a non-trivial problem [69]. To circumvent these problems, the use of XRD experiments in combination with crystallographic texture analysis was used to show that cellulose reordering was found after thermal modification at 180 °C in moist air, in conjunction with a decrease in moderately ordered or amorphous regions [70]. Although there is some disagreement, the majority of studies indicate an increase in both crystallinity and width of crystal up to a maximum temperature or time of heating, which is facilitated by using HTwet conditions, followed by a decrease in these two parameters. It is apparent that further work is required to establish the behaviour with certainty. Apart from direct measurement using X-ray diffraction, crystallinity can be inferred using NMR or IR techniques. Determination of the crystallinity index using 13C CPMAS NMR is only indicative, and accurate data require methods in which the peaks from hemicelluloses and lignin can be reliably identified and removed [53]. Similar problems with interpretation are found when IR spectroscopy is used.
The hemicelluloses are thought to have a much lower DP than cellulose, but the DP is not known with certainty since they are easily depolymerised during extraction. They are composed of more than one sugar, branched and may be substituted by acetyl groups to varying extents. They also contain carboxylic functional groups associated with glucuronic and galacturonic moieties, which may participate in crosslinking reactions if calcium ions are present [71] and can form ester bonds with the lignin. They have a role as interfacial coupling agents between the predominantly hydrophilic surface of the cellulose microfibrils and the hydrophobic lignin. They can tether adjacent cellulose microfibrils or form loops which penetrate the lignin/hemicellulose matrix. There is some evidence that hemicelluloses are present within and between microfibrils in the primary cell wall of higher plants [72], and it has been speculated that this may also be the case for the secondary wall [73]. Bonding between the surface of the cellulose microfibrils and the hemicelluloses is exclusively by hydrogen bonding, which is facilitated by the β-(1,4) backbone of the hemicelluloses, allowing for an efficient steric fit [35, 74, 75]. However, the different orientation of the OH groups in the non-glycosidic residues reduces the hydrogen bonding interaction compared to a backbone consisting entirely of β-D-glucose units. Although xylose, the monomer of the backbone of xylans, is a pentose, the orientation of the OH groups of the sugar is identical to that of glucose, which facilitates H-bonding interactions [76]. Hardwoods (angiosperms) generally have a higher hemicellulose content, with a greater proportion of pentoses and greater extent of acetylation, compared to softwoods (gymnosperms).
Xylans (glucuronoxylans) are bound to the cellulose microfibril surface in angiosperm and gymnosperm cell walls [59, 77]. Xylans have a homo-polymeric backbone composed of xylose units and are the most abundant hemicellulose in hardwoods. In hardwood xylans, units of 4-O-methylglucuronic acid are bonded to the xylose backbone via α-(1,2)-glycosidic linkages and there are a few irregular and short side branches bonded to the C3 of the backbone. The distribution of substitution on xylans is not random but exhibits patterns related to cell wall type and plant species [71]. Hardwood xylans have a ratio of substitution of acetyl groups (xyl:acet) of 0.5–0.6 [78], consistent with alternative xylose units being acetylated, as is characteristic of dicotyledons [79]. This form of substitution means that the xylans have to adopt a twofold helical screw configuration, with the result that all the decorations are on one side, which thereby forms an unsubstituted xylan surface. This is able to dock with the hydrophilic surface of the microfibril, giving an apparent microfibril diameter of 3.5–4.2 nm [80]. This shielding of the hydrophilic surface creates a hydrophobic surface to interact with the lignin or prevent aggregation of adjacent microfibrils. Softwood xylans differ from hardwoods by having a lack of acetyl groups and with arabinofuranose units linked by α-(1,3)-glycosidic linkages to the xylose backbone, located two xylosyl residues apart. They also have a higher proportion of 4-O-methylglucuronic acid, which is located on every sixth xylosyl residue and fewer branches compared with hardwood xylans [81]. The pattern of substitution also allows for a distorted twofold helical screw configuration, facilitating interaction with the hydrophilic face of the microfibril. In this way, all of the branch points are facing outwards from the microfibril surface and are able to interact with the matrix. Removal of side groups enables a more extended structure, allowing for close interchain interactions and the formation of extended crystalline regions in the aggregated xylans [82].
The dominant hemicelluloses in softwoods are the galactoglucomannans, which have a hetero-polymeric backbone comprised of glucose and mannose units in a 3:1 ratio [78] with α-D-galactose residues linked to the backbone via the 6-C(OH) at approximately every 10th mannopyranosyl unit in spruce. Galactoglucomannans are acetylated, with a degree of mannose acetylation of 0.28–0.37 at the C2 and C3 positions in spruce [83]. Whereas xylans are bound to the faces of the cellulose microfibrils, much of the lignin and acetylated galactoglucomannans are located in the matrix between the macrofibrils [84]. Hardwoods contain only minor amounts of glucomannans, which are partially acetylated.
The degradation of polysaccharides in water/steam environments is referred to as autohydrolysis. Under autohydrolysis conditions, hemicelluloses and amorphous cellulose break down to a mixture of mainly oligomeric saccharides, under mild conditions, while higher temperatures yield higher levels of hydrolysis and a higher proportion of monosaccharides, as well as degradation products. Elevated levels of degradation products, such as furfurals, are found as the degradation temperature increases [73, 85]. The depolymerisation reaction in autohydrolysis involves the reaction of water with the glycosidic linkage between sugar monomers, being initiated by protonation of the glycosidic or ring oxygen to form a carbonium cation, which then reacts with a water molecule [78] (Fig. 3). This reaction is accelerated in acidic conditions and the de-esterification of hemicelluloses to produce acetic acid promotes acidic conditions (pH 2–4) [47, 86]. Since hardwoods contain a higher proportion of acetyl groups in the hemicelluloses, they degrade at lower temperatures. Furthermore, the xylans of hardwoods are acetylated, whereas those of softwoods are not, which would suggest that degradation reactions at the microfibrillar surface will occur more readily at the microfibril–matrix interface in hardwoods.

Potential routes for acid catalysed polysaccharide depolymerisation [78]
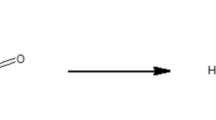
Furfural (2-furaldehyde) and HMF (5-hydroxymethyl-2-furaldehyde) are formed in increasing amounts with an increase in process temperature and acid concentration during the thermal degradation of monosaccharides under acidic conditions [87]. HMF is formed by the thermal degradation of hexoses, and furfural is mainly formed by the degradation of pentoses [88]. Both furfural and HMF have been detected in TMT [89]. Under the appropriate acidic conditions, furfural can further break down to formic acid and HMF can break down into a mixture of formic and levulinic acids (Fig. 4). Levulinic acid has been esterified with ethanol in the presence of a lignin-furfural-based catalyst and would presumably react in a similar manner to any available alcohol-OH groups in the wood cell wall [90]. However, the production of levulinic acid in the thermal modification of wood has not been reported and probably requires a stronger acid than acetic acid as a catalyst [91].

Formation of 2-furaldehyde (furfural) from pentoses and 5-hydroxymethyl-2-furaldehyde (HMF) from hexoses; further heating can produce formic acid or levulinic acid, but this is unlikely under the conditions of wood thermal modification
It is known that thermal degradation products accumulate in the wood under HTwet conditions at elevated pressure. The degradation of glucomannan with steam heat treatment of Picea abies, involving breakdown of glucuronic acid and loss of mannose units in the backbone, has been recorded [3]. Higher quantities of water-soluble sugars are found in the leachate of TMT produced by HTwet processes compared to HTdry, although the amount of acetic acid found in the HTwet TMT is lower [29]. Acetic acid does not form an azeotrope with water, and it was presumed that the difference was due to loss of acetic acid as a vapour, although why this is greater for the HTwet compared to HTdry process is not clear. Elevated moisture levels in the wood will contribute to higher levels of acetic acid production, and it would be expected that this would be seen in the leachate [14]. Lower levels of furfural were found in the HTwet TMT leachate, which was attributed to reduced production of these degradation products under wet conditions. This is because formation of furfural is a dehydration reaction, which is suppressed by the presence of water (Fig. 4). The formation of furfural can result in higher levels of lignin being recorded in TMT, when measured using sulphuric acid digestion assays [40, 92]. The furan-based polymers derived from the hemicellulose decomposition, under acidic conditions, may be involved in reactions with the more thermally resistant lignin, with the potential for participating in lignin-lignin condensation reactions, resulting in crosslinking [93].
There are covalent links between the hemicelluloses and the lignin—forming the lignin carbohydrate complex, with ether, ester and glycosidic linkages between the two components [94]. During the autohydrolysis of wood, xylan oligomers are extracted in the early stages, whereas xylan bound to lignin is found as the autohydrolysis reaction proceeds [95]. The existence of these covalent bonds between the lignin and hemicelluloses places limitations on hemicellulose removal during autohydrolysis reactions [96]. Four different types of linkage between the hemicelluloses and lignin are proposed in the literature, namely: benzyl ethers, benzyl esters, γ-esters and phenyl glycosidic [97, 98]. Depending on the treatment temperature, esters will be hydrolysed under autohydrolysis conditions, but ethers are generally more resistant [99].
Lignin
Lignin forms part of the matrix the wood cell wall and is a crosslinked amorphous network polymer comprised of phenyl propane units [78]. There are important differences in lignin isolated from softwoods (gymnosperms) and hardwoods (angiosperms), with hardwood lignins having significantly higher proportions of syringyl units. Syringyl components confer higher thermal stability on isolated lignin [100], and guaiacyl units appear to be preferentially involved with the condensation reactions [101]. Although lignin is considered to be more resistant to thermal degradation, evidenced by the relative increase in lignin content of TMT, there are nonetheless chemical changes that take place, depending on the conditions employed for the thermal treatment. Lignin thermal degradation is considered to start at 120 °C [102] and as the temperature of treatment is increased, the lignin becomes partially extractable by acetone up to a maximum temperature [103]. Loss of aromatic C = O at temperatures of 160 °C and above has been recorded [3, 66].
The literature on the autohydrolysis of lignin is extensive, due to the potential use of this as a pre-treatment for lignocellulosic biomass [86]. Lignin remaining in wood that is subjected to autohydrolysis has been reported to have a higher syringyl/guaiacyl (S/G) ratio (hardwood), higher molecular weight, fewer β-O-4 linkages, but an increase in condensed C–C linkages and a reduction in alcoholic OH groups, as well as a higher proportion of phenolic OH groups due to a reduction in OCH3 groups, which is also associated with the potential to form more condensed structures [104, 105], although -OCH3 groups are claimed to be stable up to 220 °C (with a heating time of 1 h) [38]. Increases in Klason lignin content following thermal modification are attributable to the reduction in polysaccharide content, but also condensation reactions between lignin and polysaccharide breakdown products [106]. It has been shown that alkali metal chlorides have a major effect upon the thermal degradation of lignin model compounds [107], which may have implications regarding any differences in HTwet and HTdry processes, where there is the potential for such salts to be leached out of the wood in wet thermal treatments, especially hydrothermal conditions. The presence of acidic residues does not catalyse the degradation of lignin [108].
One approach to understanding molecular changes taking place in situ is to employ cross-polarised magic angle spinning nuclear magnetic resonance spectroscopy (13C CPMAS NMR). The use of 13C CPMAS NMR has been employed to understand the changes occurring at a molecular level during the thermal modification of wood, which has shown an increase in phenolic OH content, assigned to the increase in a peak at 117 ppm, as well as an increase in the proportion of methylene bridges connecting phenolic nuclei, attributed to a peak at 29 ppm. However, 13C NMR signals at 125–135 ppm and 35 ppm which were noted in degraded holocellulose fractions were thought to be due to the formation of carbonaceous material in the polysaccharide component, in disagreement with previous interpretation as being due to lignin cross-linking and methylene bridges [109]. Later studies indicated that these signals were possibly associated with the LCC [101]. In one study [110], thermal modification did not change the signal pattern of 13C CP-MAS NMR. However, when pulse saturation transfer magic angle spinning NMR was used (13C PST-MAS NMR), this showed a decrease in the lignin -OCH3 signal with heating temperature, which was interpreted as the loss of small molecules with signals that overlapped that of lignin -OCH3. Sivonen et al. [53] reported changes in the 13C CPMAS NMR, on thermally modified Pinus sylvestris. They noted that the problem with 13C CPMAS NMR is the quantification of the data as well as frequent overlap of the peaks from the different components present in the wood, including the extractives. This is a common issue with many spectroscopic techniques applied to wood, potentially resulting in misinterpretation.
Apart from self-condensation reactions, lignin has the capability to react with furfural under acidic conditions. In acidic media, there is higher electron density located at the C2 and C6 positions of the phenylpropanoid units of the lignin, which can react with the electrophilic carbonyl carbon of the furfural (Fig. 5) [90, 93].

PCA analysis of FTIR spectra of dry wood thermally modified in air or under a nitrogen atmosphere revealed the formation of aldehydic and ether linkages in the wood, which was attributed to crosslink formation in the lignin with polysaccharidic degradation products [45]. Crosslinking within the lignin framework may also be facilitated by the production of formaldehyde during lignin degradation under acidic hydrolysis conditions [111]. Formaldehyde can also be produced by the thermal degradation of unsaturated fatty acids, which are produced by oxidative scission of triglyceride extractives in the wood, although it has not been reported as a VOC associated with TMT [112]; its absence in TMT suggests that it has already participated in reactions or been lost from the wood during thermal processing.
It is widely accepted that acidic degradation of biomass does cleave a proportion of aryl-ether linkages in lignin, but the mechanism is not fully understood. Such studies are complicated due to the unpredictable (and to an extent, unknown) structure of the lignin, plus the strongly heterogenous nature of reactions within the lignocellulosic cell wall and for this reason attention has been given to investigating model compounds representing various structural motifs. Two major pieces of work were conducted in this field in the 1970s and 1980s. Lundquist and coworkers [113] published a series of studies investigating the acid-catalysed cleavage of β-O-4 linkages in lignin. Yasuda and coworkers [114, 115] also conducted in-depth investigations into the sulphuric acid catalysed degradation of lignin, with the work of both groups being extensively reviewed by Sturgeon et al. [116] and Yokoyama [117]. Ionic mechanisms have been proposed to explain the acid-catalysed (O.2 M HCl in dioxane) cleavage of non-phenolic model dimers via an enol-ether intermediate [113]. Yokoyama [117] summarises the accepted mechanisms for β-O-4 cleavage based upon the studies of Lundquist and co-workers (Fig. 6).

Mechanisms of lignin β-O-4 cleavage under thermolysis via an enol ether intermediate [117]
The primary reaction of the β-O-4 linkage (I) in acid catalysed cleavage involves the formation of the benzyl cation intermediate (II) via reaction pathway (A). Under favourable conditions, this can be attacked by another lignin moiety to form product (III) (pathway (B)), increasing the crosslinking density. Loss of a proton, through pathways (C) and (D) forms the enol ethers (IV) or (V), respectively, which are susceptible to acidolysis, resulting in cleavage. Both cleavage reactions (E), (F) require the presence of water, although the first step (A) in the reaction involves the elimination of water and will be less favoured under ‘wet’ conditions. These reaction pathways are dependent of the type of acid used in the reaction (HBr, HCl, H2SO4), and their applicability to thermolysis reactions of wood, where the main acid generated is acetic acid, is unknown. Furthermore, Yokoyama [117] find discrepancies in the reaction rate that are not wholly explained by these accepted mechanisms. Rather, they propose that the β-O-4 cleavage reaction goes via a rearrangement of the carbo-cation intermediate, rather than an enol ether (Fig. 7). Cleavage of the β-O-4 linkage will potentially produce soluble fragments; however, repolymerisation of the lignin fragments via C–C linkages is a likely outcome [105], and there is competition between these two reactions. In this context, it is interesting to note that the use of carbocation scavengers during the autohydrolysis pre-treatment of lignocellulosic biomass can improve enzymatic digestibility of the cellulose [86]. The self-condensation of lignin via diphenyl methane C–C condensation reactions is less favoured under HTdry conditions [38].

Mechanism of β-O-4 cleavage under thermolysis via a carbo-cation intermediate [117]
Another proposed mechanism involving homolytic cleavage via quinone methide intermediates is considered unlikely [116]. Studies of the cleavage of β-aryl-ether linkages in lignin model compounds in the presence of 0.2 M H2SO4 at 150 °C have shown that those model compounds containing phenolic moieties reacted much faster [116]. They also propose an additional reaction for the carbocation, where it undergoes self-condensation via electrophilic aromatic substitution to give the product (III) (Fig. 8). Sturgeon et al. [116] did not detect any formaldehyde in their reactions and speculated that any formaldehyde that was released took part in polymerisation reactions to form char.

Mechanism of self-condensation of lignin under thermolysis
Extractives
The extractives in wood are either removed from the wood or degraded during the thermal modification process. Fats and waxes migrate along flow pathways, such as ray parenchyma at temperatures as low as 100 °C, whereas resin acids are detectable up to 100 °C [118]. Most of the native extractives are progressively lost or degraded as the process temperature increases, but new extractable compounds caused by thermal degradation of carbohydrate and to a lesser extent, lignin are formed [89, 119, 120]. In one study, extractives were shown to provide a small, but detectable, protective effect on the wood polymers during thermal degradation [55]. In contrast, other workers have found that the presence of extractives in wood resulted in lower thermal stability [61, 121], although this may be attributable to the degradation of extractives during thermal modification. New extractives are formed during the thermal modification process, and degraded residues of extractives may contribute to cell wall bulking and will affect sorption properties, possibly by masking accessible OH groups or may contribute to increased hygroscopicity if they contain sorption sites (more likely to occur with thermal modification under oxidative conditions). The importance of accessibility of OH content in controlling hygroscopicity is currently the subject of much debate in the literature and is considered in more detail in the following sections.
Hydroxyl (OH) content
The thermal modification of wood results in a reduction in accessible hydroxyl content. This can be directly determined by the use of hydrogen–deuterium exchange (HDX), by exposure of the TMT to liquid D2O or D2O vapour or by the use of spectroscopic techniques, such as near infra-red (NIR). With HDX, the hydrogen atoms of the accessible OH groups are exchanged for deuterium using D2O (liquid or vapour), the wood samples are dried and the mass increase due to HDX exchange is then determined. Alternatively, the exchange of OH groups can be measured by the intensity (with suitable corrections) of the O-D stretching band at 2500 cm−1 in the IR region [122,123,124,125]. It is vitally important that the HDX wood is not re-exposed to H2O before weighing; otherwise, re-exchange can rapidly occur [126].
Previous studies of cellulose deuteration have shown that the rate and extent of exchange depend upon the crystallinity of the cellulose sample, temperature and relative pressure of the D2O vapour, mode of deuteration (liquid or vapour) and preparation history of the sample [123]. Atalla et al. [127] used immersion in liquid D2O and Raman spectroscopy to show that drying of isolated native cellulose microfibrils resulted in a lower level of OH accessibility compared to never-dried microfibrils. This was attributed to hornification on drying. The exposure of deuterated samples to H2O will re-exchange the OD groups to OH, but if these regions are no longer accessible, some OD will remain. Thus, drying of an HDX sample in the absence of water molecules followed by exposure to H2O will reveal if any hornification has occurred. This was the approach adopted by Suchy et al. [123] who deuterium-exchanged native wood samples using liquid D2O and then dried the samples under controlled conditions. Depending on the drying conditions employed, subsequent exposure to H2O revealed that there were some residual OD groups remaining, although the effect was much more pronounced when wood pulp was examined using the same experimental protocol. However, it should be noted that in such accessibility experiments, it is vitally important that sufficient time is allowed during the H2O-soaking phase to ensure that slowly re-protonating OD groups are fully exchanged. It is known that the measured accessible OH content is affected by the mode of exposure of the wood to D2O and the drying protocols adopted [125]; Thybring et al. found that when wood samples were dried from the native state, the hydroxyl accessibility was not affected when determined by exposure to liquid D2O. However, if the wood samples were instead exposed to D2O vapour, the measured OH accessibility was reduced by differing amounts, depending on the drying protocol adopted [125]. This effect could be erased by subsequent exposure to liquid water, which (by definition) eliminated hornification upon drying as an explanation. The issue of hornification is discussed further in a later section. The use of HDX is potentially a useful technique for measuring accessible OH content, but needs to be carefully undertaken and interpreted.
When cellulose is exposed to D2O vapour, a two-stage exchange process is observed. Over a relatively short time frame (depending on sample size and morphology), there is exchange with the non-crystalline parts of the cellulose, with a much slower exchange presumably occurring with the crystalline regions. However, it is thought that the cores of the crystalline regions are totally inaccessible to H–D exchange processes [122]. It has also been shown experimentally that the OH(3) group of the C3-atom of the glucopyranose unit of cellulose on the microfibril surface cannot be deuterated, even though it is available to hydrogen bond with water molecules [128]. The lack of exchange of 1H for 2H of the OH(3) is thought to be due to the relative stability of the OH(3) – O(5) hydrogen bond. Although this has not been studied, this may also apply to the OH(3) of sugar residues of the backbone in hemicelluloses in close proximity with the microfibril. According to Kulasinski et al. [129], approximately 15% of OH groups of glucomannans and xylans were not accessible to deuterium exchange, when exposed to D2O vapour. But these experiments were performed on cast films of isolated hemicelluloses, which may produce hornified or otherwise inaccessible crystalline regions in the structure, which are not present when the hemicelluloses are located within the cell wall.
The earliest example of the use of HDX to determine the accessible OH content of TMT was reported by Phuong et al. [130], who thermally treated Styrax tonkinensis wood under dry conditions. TMT specimens were dried at 105 °C and then submerged in D2O in a vial which was then sealed. Exposure was for two days to achieve full exchange. Dried HDX samples were then ignited in a platinum crucible in an oxygen atmosphere and the released D2O determined using NMR. The number of accessible OH groups in unmodified wood was determined to be 6.8(± 0.2) mmol/g, with a reduction thereof with heat treatment. This technique is relatively time-consuming, and the advent of dynamic vapour sorption apparatus has (potentially) simplified the procedure of determining accessible OH content through HDX experiments, although the experimental protocol adopted is crucial to obtaining reproducible results. In DVS experiments, the water reservoir is filled with D2O and the wood is dried using an appropriate protocol and then exposed to a D2O vapour stream, with the final weight difference being recorded. Rautkari et al. [131] conducted the first such DVS study using multiple exposure cycles, which found that there was a poor correlation between the EMC of the wood at constant RH and the accessible OH content. However, subsequent work in this area has questioned the use of multiple sorption cycles in the D2O exposure step, and the use of a single cycle of sufficiently long exposure has instead been recommended [124, 125, 132], as well as changes in other experimental details, especially the drying protocol. The finding that the mode of drying from the native (never-dried) state also affects the apparent accessible OH content when wood samples are exposed to D2O vapour also adds to the potential sources of error [125]. In order to resolve this issue, Uimonen et al. [133] conducted a study to determine what DVS experimental parameters were required to obtain reliable data in HDX experiments. The variables studied included sample weight (minimum 10 mg), length of exposure to D2O vapour at 95% RH (10 h) and rate of mass change (dm/dt = 0.0005% min−1) of the drying steps (dm/dt = 0.002% min−1 was used previously). An initial wetting and drying stage was also recommended in order to release any potentially trapped solvents. There are still possible sources of error arising from the estimation of dry mass within the DVS [134], and although the HDX experiments using a modified protocol produced results with repeatable results, a higher drying temperature in the DVS in order to measure the dry weight was found to produce an increase in accessible OH content by + 0.5 mmol OH groups per gram of dry wood mass [135]. Further development is apparently required to produce reliably accurate OH accessibility data using a DVS apparatus in HDX experiments, but this remains a very convenient method for determination of accessible OH groups once reliability can be established.
The use of deuterium exchange has the advantage that accessible OH content can be directly measured, albeit taking into account the caveats presented in the preceding paragraph. There are also different spectroscopic techniques that can be applied to studying the OH content of TMT, such as FTIR [103, 136], NIR[137]. Mitsui et al. [137] used NIR to observe changes in OH content of TMT, concluding that the degradation reactions commenced in the amorphous polysaccharide regions before progressing to the crystalline regions and that the phenolic OH groups were not degraded. The use of NIR or FTIR requires the use of peak fitting to spectra that often exhibit indistinct and merged absorption peaks. This makes the quantitative evaluation of OH content subject to error. OH content can be determined by acetylation, followed by aminolysis with pyrrolidine and GC analysis [138]. This method relies upon the assumption that all of the H2O accessible OH groups are also accessible by acetylation.
Changes in physical properties
Hygroscopicity
Thermal modification reduces the amount of bound water in the wood cell wall [139, 140]. However, the behaviour of HTdry and HTwet wood is different. The behaviour observed depends upon whether the EMC of the wood is measured directly after post-treatment or if there is exposure to liquid water or high levels of moisture after the thermal modification. Although dry thermal modification of wood results in a reduction in EMC, the effects are partially reversible, in contrast to TMT produced by wet thermal processes (under thermal modification conditions where the RH is 100%). In the case of the hygroscopicity of dry thermally modified wood, exposure to high RH levels or liquid water can partially reverse the initial reduction in hygroscopicity [5, 32, 141,142,143,144,145,146]. The explanation for this behaviour was due to stress remaining in the wood after the dry thermal modification stage. When dry wood is heated, the amorphous region remains in a stressed state, which is relaxed by subsequent exposure to high moisture levels. When wood is modified under HTwet conditions, the amorphous content remains in a plasticised state and stress relaxation occurs during the thermal modification process [141]. This partial reversibility of the behaviour is also noted when thermal treatment is undertaken at an intermediate RH. Endo et al. [5] found that although the EMC of wood was reduced by thermal treatment at intermediate humidities, this reduction was effectively eliminated by exposing the wood to saturated water vapour at 25 °C. This was found for wood heated at 120 °C for two days at a range of different humidities. The reversible effect was observed at 0% treatment humidity and increased up to 60% treatment RH, reducing thereafter and becoming zero at a treatment RH of 95%. Borrega and Kärenlampi [147] heated wood at 150 °C or 170 °C in a pressure vessel at a range of treatment RHs up to saturation. They found that thermal modification at an intermediate RH produced TMT which had markedly reduced hygroscopicity compared to heating at 100% RH, in agreement with the experiments conducted at 120 °C [5]. This behaviour was attributed to the formation of irreversible H-bonds in the structure at intermediate RH, but as Obataya et al. [6] point out, these experiments did not examine the hygroscopicity after exposure to saturated water vapour and therefore did not take account of the reversible behaviour. By definition, hornification is not reversible. As noted previously, in a hygrothermal modification experiment performed at 120 °C at different RHs, wood samples heated at a RH of 100% exhibited reduced EMC at the lower end of the hygroscopic range, whereas at the highest end of the range, the EMC exceeded that of unheated wood. This was attributed to the presence of water-soluble and deliquescent degradation products that were formed in the cell wall during the hygrothermal process. After a leaching experiment, the EMC at higher RH was reduced. This change in behaviour after leaching was not found when such treatments were performed at RHs below 80%, where only minor mass losses were noted with the post-treatment leaching step [6]
The hygroscopicity properties of wood are undoubtedly linked in some way to the OH groups in the cell wall polymer matrix. This is often assumed to be because the OH groups are considered to be primary sorption sites and models involving close association of water molecules with the cell wall OH groups are invoked. Thermal modification results in a reduction of OH content (mainly due to hemicellulose degradation); hence, the EMC at any given RH would be expected to be lower compared to unmodified wood, assuming the primary sorption sites model to be the correct explanation for hygroscopic behaviour. This simple relationship between OH content and EMC, was called into doubt by the work of Rautkari et al. [131] who found that there was no direct correlation between OH content, as measured by HDX and the EMC of TMT. This led to the conclusion that there were additional mechanisms responsible for the reduction in EMC, other than the accessible OH content. However, as discussed, later work has shown that these data were adversely affected by the measurement protocol employed. More recent work [148] has shown that there is a linear relationship between the EMC at 25 °C and 93% RH and the accessible OH content was determined gravimetrically using DVS, using an improved method of measurement. Furthermore, the regression slope for HTdry TMT is much greater than that for HTwet TMT, because the EMC of the HTdry TMT is lower for the same accessible OH content. An accessibility of 1.01 H2O molecules per accessible OH group was determined for unmodified Scots pine (Pinus sylvestris). Willems [149] has suggested that each sorption site in the wood is actually comprised of a pair of OH groups which may be associated with a single or a pair of water molecules. In this model, the sorption isotherm at RHs below 50% is composed of strongly bound water monomers and at higher RH, the sorbed water is composed of loosely bound dimers. The elimination of a sorption site therefore requires the loss of only one OH group of an associated pair; however, if there is an opportunity for a molecular rearrangement to occur, new such-associating pairs can be created. Such a situation occurs during any HTwet process, where molecular mobility is facilitated during the thermal modification process or it can occur during a post-treatment annealing stage in the presence of liquid water or at high moisture levels. Using such a model only requires the number of accessible OH groups to be considered. Altgen et al. [148] used this model to suggest that thermal modification created single OH sites that were not able to act as sorption sites; however, under HTwet conditions, relaxation of the polymer network occurred allowing for the formation of new (albeit fewer overall) sorption site OH pairs. When wood was modified under HTdry conditions, the initial configuration of the polymer network left unpaired OH groups, subsequent annealing allowed new pairs to be formed and consequently an increase in hygroscopicity. This idea was further developed by Willems et al. [135] who measured the accessible OH content of the same HTdry sample before and after a post-treatment water saturation. They found that there was a slight decrease in OH content, but that the EMC increased significantly after the post-treatment water soaking experiment. The change in OH content was attributed to loss of degraded cell wall material during the water-soaking phase, but the important point was that the increase in EMC when exposed to 93% RH (from 8.45 to 9.72%) was not accompanied by an increase in OH content. It was also found that the accessible OH content did not increase when the sample was soaked in DMSO post-treatment, although the EMC was significantly higher after water exchange, drying in a desiccator over silica gel and then exposure to 93% RH (EMC = 11.31%). This is a very interesting observation, which was used in support of the hypothesis that the enhanced polymer mobility during the water-saturation phase allowed for the creation of new sorption site OH pairs; hence, there was an increased EMC noted in subsequent sorption experiments, even though the OH accessibility had not changed. However, there are other explanations for this behaviour apart from invoking the hypothesis of sorption site pairs or OH accessibility. These alternatives are discussed in more detail in a following section.
Apart from the destruction of sorption sites, other reasons that have been suggested for the reduction in hygroscopicity are linked to a degradation of the hemicelluloses with concomitant reductions in cell wall polymer mobility or to crosslinking reactions within the cell wall. In the case of crosslinking, any reduction in hygroscopicity would most likely be permanent. However, if the reduction is caused by the formation of H-bonding networks within the cell wall [147], then this could be reversed if the hydrogen bonds within such regions were broken (not invoking hornification as a mechanism). This structural rearrangement would occur when there was sufficient molecular mobility to allow for this, implying that a threshold moisture content is required. Regions of the network may be unable to relax to a lower energy configuration because they are in a glassy state and there is insufficient void volume to allow for molecular reconfiguration. This would lead to reduced EMC when exposed to water vapour at RH less than 95% or thereabouts. However, upon water soaking or exposure to high RH levels, a non-equilibrium configuration of the cell wall polymer matrix could then relax to a lower energy state due to the enhanced chain mobility conferred by the presence of water in the cell wall [141]. If linked to impermanent H-bonding networks, this suggests that measurements of accessible OH content using D2O would change before and after the water-soaking stage. But this has not been observed [135]. The loss of mobile cell wall components during thermal modification will result in a cell wall that is in a higher energy stressed state under HTdry conditions, which can then relax when subsequently exposed to higher levels of moisture. If wood is modified under wet conditions, then the continuing chain mobility that occurs throughout the wet thermal modification process allows for a lower energy state to be realised during the process. Hence, there is little further change in EMC observed in sorption isotherm experiments following any subsequent water exposure cycle [32, 135, 141]. This would not affect the accessible OH content.
Altgen et al. [29] found that the EMC and maximum swelling of HTdry TMT were lower than those of HTwet for the same weight loss. They attributed the reduction in hygroscopicity to the loss of accessible OH groups during thermal modification, mainly due to loss of hemicelluloses. However, the enhanced reduction in EMC of the HTdry TMT suggested an additional mechanism, which was thought to be related to increased levels of crosslinking in the cell wall resulting in an increase in the matrix stiffness. With HTwet, the presence of water in the cell wall favours the cleavage of the amorphous cell wall polymers, resulting in a decrease in chain length and an increase in free volume allowing for polymer rearrangement. In contrast, HTdry encourages the formation of crosslinks within the cell wall, increasing matrix stiffness and reducing polymer mobility. Stiffening of the hemicellulose matrix takes place during water removal in any case, which elevates the glass transition temperature of dry hemicellulose to over 200 °C [150]. Assuming that more extensive crosslinks are formed under HTdry conditions, the consequent reduction in polymer mobility is not sufficient to prevent the rearrangements necessary for new OH sorption site pairs to be formed, although the rate at which such rearrangements occur would be slower. If the crosslinking is confined to the amorphous polysaccharide domain, then it is possible that overall swelling in solvents, such as DMSO, may be unaffected.
Diffusion of water through the polymer matrix of the wood cell wall is dependent upon the molecular dynamics of the polymers in the non-crystalline regions. The diffusion process is dependent upon whether these polymers are in a glassy or rubbery state [151, 152]. Swelling of the amorphous regions of the cell wall is associated with an increase in the free-volume (and hence chain mobility) of the molecules. This ‘softening’ of the structure facilitates the transport of water molecules and is also associated with a reduction in the modulus of the cell wall. Mechanical and sorptive properties are inextricably linked. Loss of the hemicellulose component through thermal degradation will change the relationship between elastic modulus and MC, by removing components that exhibit higher molecular mobility in the presence of water molecules. It is well known that the elastic modulus of wood decreases as the cell wall MC increases. As water is introduced into the cell wall, the density of the hemicellulose component reduces, along with the elastic modulus. Simultaneously, the molecular mobility of the hemicellulose chains increases in line with the creation of free volume within the matrix. However, the elastic modulus of the lignin increases up to about 10% MC and 2.5% MC for the LCC. At low MC, the water molecules predominantly form hydrogen bonds with the cell wall polymers, whereas above approximately 10% MC, the water molecules increasingly self-associate into ‘nano-droplets’ [153]. The model that is described by Willems [149] does not include the formation of such water clusters (other than dimers), nor consider the chain mobility of the amorphous parts of the cell wall polymer matrix.
Dimensional stability
Post-modification, the wood is often subjected to wet/dry cycling in order to determine the dimensional stability. This can result in leaching of decomposition residues, but also facilitate molecular rearrangements within the cell wall. Loss of degraded cell wall material by diffusion is much more likely under conditions where water is present and leaching occurs during thermal modification, but some degraded material can still remain in the cell wall until a subsequent leaching stage. The remaining degradation products act as bulking agents inside the cell wall and when they are removed, there is a consequently a reduction in the volume of the cell wall, which is usually reflected as a reduction in the dry wood volume at a macroscopic level. The water-saturated volume of the wood may be reduced as a result of thermal modification, but this is not always observed [26]. If the dry volume is reduced and the water-saturated volume remains the same, then this results in a higher overall volume change and hence lower-dimensional stability. If crosslinking of the polymer network occurs, there is a restriction of swelling, leading to a reduced water-saturated volume; but if the thermal modification takes place in wood that is in a water-swollen state, there is subsequently unlikely to be any observed reduction in water-swollen volume as a consequence. By contrast, crosslinking of wood thermally modified in the dry state would result in restricted swelling when subsequently water-saturated.
There are two main methods for determining the dimensional stability of wood. Wood samples can be exposed to varying conditions of relative humidity, with dimensions of samples determined after the wood has reached equilibrium at a specified atmospheric RH and temperature. The difference in dimensions as a sample is transferred between conditions of high and low RH which is then expressed in a formula to give the dimensional stability. This is the principle behind the European Standard EN 1910:2016. The use of measurement methods which rely upon determining changes in dimensions of wood samples when exposed to conditions of high and low RH is perfectly reasonable for the determination of solid wood, wood-based laminates and composites, where the wood has not been modified. However, if the wood has been modified by a physical, chemical or biological process, it may be that such a modification is not permanent when this material is used under service conditions. For such situations, it is advisable to either include a water-leaching stage (such as EN 84) prior to the determination of dimensional stability or to determine dimensional stability by using a water soak/oven drying (WS/OD) test, as described by Rowell and Ellis [154]. In this test, the wood sample is vacuum impregnated with water and then left to soak for a specified interval before the water saturated external dimensions are determined. The wood samples are then dried in an oven, and the oven-dry dimensions and weight are then determined. In most cases, the oven-dry dimensions are determined after modification, before the first water soaking cycle. It is necessary to complete a number of cycles to determine the stability of the dimensional stabilisation effect (if any).
When water enters the cell wall of wood, the cell wall swells, which is reflected in macroscopic dimensional increases of the wood. One of the reasons for producing TMT on a commercial scale is to create a more dimensionally stable product, which consequently has superior performance in exterior applications. An understanding of these properties is therefore of great importance for product improvement.
Improved dimensional stability can be achieved by:
-
The presence of bulking agents within the cell wall. This will increase the dry dimensions of the wood. The water saturated dimensions are unlikely to be affected, meaning that the difference between the dry and wet volume is reduced, resulting in improved dimensional stability [2]. Thermal modification will not add bulking agents to the cell wall, and hence, there will be no increase in dry dimensions of the wood; on the contrary, a decrease in volume is usually observed (although this may only occur after a subsequent leaching stage post-treatment). However, thermally degraded components may remain in the cell wall, where they have a bulking property, and this filled space is not available to water molecules. The interaction with absorbed water will depend upon whether the bulking agent is hydrophobic or hydrophilic; if the latter happens, then the cell wall will be able to accommodate larger amounts of water compared to hydrophobic bulking agents. If the degraded components are subsequently leached out, the cell wall will lose these bulking agents, resulting in a reduction in dry volume.
-
Removal of OH groups, which will reduce the hydrophilicity of the wood [155]. This will change the free energy of mixing between the wood polymer network and the penetrating water molecules, which is related to the interaction parameter of the Flory–Huggins theory [156]. Loss of OH groups is also related to changes in hygroscopicity and surface energy (wetting). This loss also results in a reduction in primary sorption sites, but the importance of this compared to changes in the free energy of mixing is debatable.
-
Increased crosslinking within the cell wall polymers, which restricts the ability of the cell wall matrix to swell to accommodate sorbed water molecules; increased elastic modulus of the cell wall requires a greater internal swelling force (related to free energy of mixing) to achieve the same moisture content. Changing the crosslink density also affects the configurational entropy of the polymer network on swelling and the glass transition temperature [157]. This crosslinking can occur by the formation of covalent bonds due to the reaction of degradation products (such as furfurals) or due to hornification within the polymer network, if this occurs with lignified materials [147]. However, crosslinking is not solely associated with the lignin matrix.
-
Changes in the mobility of the polymer network. Removal, degradation or crosslinking of the more mobile hemicelluloses will produce a stiffer cell wall matrix, requiring a greater ‘internal pressure’ to be generated to produce the same volume increase as with the unmodified equivalent. This relationship between elastic strain of the matrix and free energy of mixing is considered in a later section.
Although details vary, most research data are reported in terms of a percentage swelling (or shrinkage) coefficient (S), as follows:
where Vwet is the volume of the wood under water-saturated conditions (or high humidity) and Vdry is the volume of wood under oven-dry (or low humidity) conditions.
The differences in swelling behaviour between modified and unmodified wood can be reported in terms of an anti-shrink efficiency (ASE):
where Sunmod is the swelling coefficient of the unmodified wood and Smod is the swelling coefficient of the modified wood. As the swelling coefficient of the modified wood approaches zero, the ASE approaches 100%.
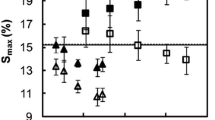
The result of a dimensional stability experiment using over-dry/water-saturated conditions is shown in Fig. 9a. This represents a study of unmodified wood over three cycles, where there is no loss of material from the cell wall.

Influence of bulking agent, crosslinking and loss of cell wall material on dimensional behaviour (see text for explanation)
In Fig. 9b, the presence of a bulking agent in the cell wall swells the wood by an amount Vb and consequently the wood exhibits dimensional stability because the difference in Vwet and Vdry is smaller than it was in Fig. 9a. In this case, the bulking agent is non-leachable and consequently the dimensional stability is permanent, whereas in Fig. 9c, the bulking agent is leached out over the three wet/dry cycles and the dimensional stability is lost. In Fig. 9d, the swelling of the wood is restrained due to crosslinking in the cell wall, but this situation will only occur if the crosslinking does not involve any bulking due to the presence of the crosslinking agent. In Fig. 9e, there is crosslinking of the cell wall and the crosslinking agent also occupies cell wall volume. In Fig. 9f, there is loss of material from the cell wall, causing a reduction in Vdry and Vwet is lower due to crosslinking in the cell wall. An increase in Vdry requires the impregnation of the cell wall with an external bulking agent (not occurring with thermal modification). A combination of restriction of swelling by crosslinking and gradual loss of cell wall material is shown in Fig. 9g. Loss of cell wall material and reduction in crosslinking are illustrated in Fig. 9h, and loss of cell wall material with no additional restraint upon swelling is shown in Fig. 9i.
It has long been known that there are important differences in swelling behaviour between TMT modified under wet and dry conditions and that leaching of the wood after thermal modification reduces the ASE [7]. It was also reported that ‘heating wood in water-saturated atmospheres has no permanent effect upon the swelling and shrinking’ [7]. The possibility of crosslinking occurring during thermal modification was first posited as a mechanism explaining the reduction in hygroscopicity and improved dimensional stabilisation by Stamm and Hansen in 1937 [7]. They thought that the presence of excess water in the cell wall during thermal modification prevented crosslinking reactions occurring between adjacent OH groups, whereas the absence of moisture allowed for these dehydration reactions to occur, thereby forming ether crosslinks. This neatly explained the difference in swelling behaviour between TMT heated under HTdry and HTwet conditions. However, the existence of ether crosslinks was later tested by the use of swelling liquids on TMT, which showed that thermal modification only reduced the swelling in water [158]. This indicated that crosslinking could not be the explanation for the improved dimensional stability.
Seborg et al. [158] conducted studies where they heated wood in pressure bombs under dry conditions, where the wood had previously been equilibrated at 97% RH and where such equilibrated wood also had additional water placed in the bomb before it was sealed. After thermal modification, the specimens were leached with hot water to remove soluble degradation products. They did not observe any significant differences in dimensional stability or hygroscopicity between samples heated under dry or moist conditions. However, the samples were heated at 300 °C, considerably higher than the temperatures employed for commercial thermal modification reactions.
Wood heated under saturated steam in an initially dry state showed different properties compared to wood that contained moisture (conditioned at 65% or 95% RH at 20 °C) prior to modification [159]. Samples which contained moisture prior to thermal treatment showed higher mass losses, but larger swelling coefficients (and hence reduced ASE) compared to the samples which were oven-dried prior to thermal modification (180 °C, 10 bar). However, the S% was lower than the reference sample for all of the thermal modified samples. Only one water-soak/oven-dry cycle was employed in these experiments.
Other work has shown that wood thermally modified under HTwet conditions shows a reduction in dimensional stability over a number of water-soak/oven-dry cycles, which is related to loss of decomposition residues by leaching [160]. Čermák et al. [144] noted that there was a gradual decrease in dimensional stability with each wetting cycle, especially between the first and subsequent cycles. This loss of degradation products is accompanied by a decrease in water-saturated volume in dimensional stability experiments during the first cycle, but this relationship breaks down during subsequent cycles [146]. When the degradation products are removed from HTwet TMT, their bulking of the cell wall is also lost and the dimensional stability deteriorates and is less stable than unmodified wood [32]. After water-leaching, the dimensional stability of the HTwet TMT was worse than that of the unmodified wood, with the S% increasing with severity of treatment. Before leaching, the HTwet TMT exhibited an S% which decreased slightly with severity of treatment. With HTdry TMT, the dimensional stability was better than the controls and the S% decreased with severity of treatment. Although the dimensional stability decreased after leaching, it was still better than the control samples and still showed the same relationship with severity of treatment. The difference between these behaviours before and after leaching is mainly related to the loss of cell wall degradation products after leaching, which resulted in a decrease in weight and a decrease in both dry and wet dimensions of the TMT. By plotting the change in wet and dry dimensions against the loss of water-soluble compounds, it was shown that the relationship was nonlinear for the HTwet TMT oven-dry dimensions. In addition, the difference between the wet and dry dimensions increased with severity of treatment, leading to the increase in S%. This showed that there must be a mechanism other than the removal of hemicelluloses which reduced the swelling of the cell wall, but hornification was rejected as a possible explanation. It was thought that the behaviour could be explained by the impact of thermal modification on the cell wall polymer matrix stiffness. In the case of HTwet, the presence of water in the cell wall during the modification process facilitated the cleavage of bonds, reducing the crosslink density, whereas in the case the HTdry, there were additional bonds created, which reduced the polymer mobility. This indicates that there is no crosslinking occurring in HTwet TMT, whereas this is a possibility with HTdry TMT [21].
Obataya and Higashihara [146] found that Vwet of HTdry heated wood was lower than that of HTwet heated wood immediately after treatment, but this was reversed after leaching, with a large reduction of Vwet for the wet thermally treated wood, but no change in Vwet of the dry heat-treated wood. The swelling of HTwet TMT was much larger than that of HTdry and untreated wood, because the reduction in dry volume was much greater for the HTwet TMT. The large reduction in wet volume after leaching was attributed to two possible mechanisms:
-
The formation of decomposition residues in the cell wall of the steamed wood, which acted as a bulking agent, which leached after the first cycle. After leaching, a ‘hydrophobic structure’ remained in the cell wall, which restricted access to water molecules and resulted in a reduced swelling when saturated with water. However, when saturated with aprotic solvents, such as dimethyl sulphoxide (DMSO), the hydrophobic structure did not limit access and the saturated volume was consequently not reduced. Although this excess swelling by DMSO is also observed with unmodified wood, the fact that it is also observed with thermally modified wood makes crosslinking an unlikely explanation.
-
The cell wall lumen may be expanded by steaming accompanied by a plasticisation of the cell wall polymer matrix. This expansion is reversed when water-soluble decomposition residues are lost, during the water-leaching stage.
As noted previously, the existence of crosslinks formed during the thermal modification process can be tested by the use of liquids which are known to swell the wood more than water [161]. If the TMT is swollen more than it is when immersed in water and if it returns to its original swelling behaviour after solvent exchange back to water, it may be concluded that the dimensional stabilisation mechanism is not crosslinking via covalent bonds. Seborg et al. [158] found that swelling of TMT (dry heated in air) using sodium hydroxide solution, morpholine or pyridine, all resulted in larger dimensional changes than observed with water saturation. This was taken as proof that no crosslinks were formed during the thermal modification process. Rapp et al.[162] found that the maximum tangential swelling of thermally modified wood decreased with increasing treatment temperature. However, soaking in aqueous 2% KOH resulted in increased swelling. For this reason, the formation of crosslinking ether bonds in the cell wall matrix during thermal modification was ruled out, because ether bonds are not broken under these conditions. Obataya and Higashihara [146] noted that the swelling of TMT (dry and wet heated) in DMSO was greater than that observed with water soaking, again indicating the absence of crosslink formation during the thermal modification process. When the DMSO was fully exchanged for water, a return to the original swelling behaviour was seen. These data show that covalent crosslinks are not responsible for the restrictions on swelling exhibited by HTdry TMT. Nonetheless, it remains a possibility that hydrogen bonded networks are formed between regions of microfibril where hemicelluloses have been lost through degradation or through the participation of hemicelluloses where the substituents have been lost in the thermal treatment process. These regions of hydrogen-bonding networks are not accessible to water molecules, but can be disrupted by cell wall swelling agents, such as DMSO. Upon removal of the DMSO through solvent exchange with water, these hydrogen bonding networks are re-created when the wood is dried and the restrictions on swelling in water continue to be observed. However, if this were the case, additional OH groups would become accessible in solvents, such as DMSO, which could be revealed by using HDX experiments under suitably controlled conditions.
The formation of irreversible hydrogen-bonding networks (hornification) is a well-known phenomenon in wood pulps, where the matrix substances have been largely removed [163]. As noted previously, although hornification has also been invoked to explain the changes in hygroscopicity observed when wood is thermally modified [147], the presence of lignin and hemicelluloses in the regions between the cellulose microfibrils limits their ability to aggregate on drying [125]. Although permanent H-bonding structures are not formed, there is nonetheless, apparently some potential for aggregation within the wood cell wall polymer structure, evidenced by the existence of apparently inaccessible OD groups within dried wood, which has previously been exchanged with D2O [123]. However, this observation is more likely to be linked to insufficient water-soaking time being allowed for slowly re-protonating OD groups.
It has been shown that drying and re-wetting of Pinus radiata through several cycles result in an accumulation of conformational changes of the cellulose crystallites, increasing the density of packing thereof. These changes have been attributed to the formation of hydrogen bonding regions between the surfaces of microfibrils during dehydration [164]. Evidence for this can be seen from an increase in the width of the cellulose crystal. An alternative explanation invokes the decrease in compressive matrix strain upon the cellulose crystal, as the matrix softens when water molecules penetrate the structure. However, this effect has been shown to be insufficient to account for the observed crystallinity changes in the wood cell wall [165]. The ability of the cell wall polymers to form aggregated structures during wood drying, resulting in the formation of inaccessible OH content, is partially reversible [164] and is highly dependent upon the mode of drying employed [125], but these aggregated structures may be more stable when produced by thermal modification. Thermal degradation of the lignin and, more importantly, the hemicelluloses may reduce the limitation upon forming aggregated structures within the cellulose component and with proximal xylan chain backbones [82]. This aggregation phenomenon is facilitated by the presence of moisture in the cell wall [64, 147], suggesting that HTwet conditions should give better dimensional stability compared to HTdry conditions, which is not observed.
Discussion
To summarise, wood thermally treated under wet conditions (hygrothermally) exhibits:
-
Higher treatment mass losses during processing and in post-treatment water-leaching compared to HTdry TMT
-
Worse dimensional stability (higher S%) compared to untreated wood and HTdry TMT, which is observed particularly after the first water-soak/oven dry sample
-
Higher EMC than HTdry TMT at the same RH and temperature
Water-soaking after thermal treatment will remove degradation agents that are acting to bulk the cell wall, with a concomitant reduction in Vdry. Vwet may be unaffected, and as a consequence, there will be an increase in S%. This is more of an issue for HTwet TMT, where dimensional stability is invariably inferior to that of unmodified wood.
There is a reduction in accessible OH content in TMT as a result of thermal treatment, but for HTwet conditions, the hygroscopicity of the wood is higher than it is for the TMT produced under HTdry conditions, for the same accessible OH content. However, after the post-treatment water-soaking phase, the EMC of the HTdry TMT increases, but the accessible OH content remains essentially unchanged. This is a very important observation and requires more research.
Dry process conditions are thought to be more favourable for the formation of crosslinking reactions [92]; this is because the essentially anhydrous conditions favour the formation of furfural derivatives from the polysaccharide content and these degradation products are able to participate in reactions with the lignin and remaining polysaccharide content [93]. The significantly reduced swelling of HTdry processed wood compared to HTwet processed wood when the material is water-saturated which is considered to support the idea that there are more crosslinks in HTdry TMT [135]. However, it is also necessary to take into account the observations of swelling of TMT in swelling solvents, such as DMSO, where it is shown that thermal treatment does not affect the swelling behaviour, which suggests that chemical crosslinking within the lignin network is unlikely to be the reason for the observed behaviour [146]. The potential formation of crosslinks under HTdry conditions, explains the improved dimensional stability of the TMT compared to HTwet or unmodified, but simultaneously there has to be sufficient mobility to allow for the creation of new OH sorption site pairs when the TMT is subjected to a post-treatment water-soak phase, even though the total OH content remains unchanged (if this explanation is correct). Similar considerations apply to thermally induced hornification (which can be ruled out) or to the formation of non-permanent H-bonding networks associated with aggregation. Crosslinking between the hemicellulose degradation products remains a possibility, however.
Is there an alternative explanation to the observed change in swelling behaviour that does not require the consideration of crosslinking as a mechanism? The swelling/shrinking behaviour of wood in the presence of water or moisture can be approximated to that of a gel [166], with the classical approach to the swelling behaviour of gels being given by the Flory–Rehner theory, describing the balance between the free energy of mixing and the elastic free energy [167].
The incorporation of water molecules within the wood structure creates what can be described as an internal swelling pressure, which is resisted by the matrix, with elastic energy being stored in the polymer network. Constraints on swelling at a molecular level are provided by physical and chemical crosslinks within the matrix. At a macroscopic level, constraints also arise from the presence of the S1 and S3 cell walls and neighbouring cells. The loss of mobile components and changes in crosslink density will change the response of the matrix to the swelling forces [168]. The elastic response of the matrix is related to the mobility of the macromolecular components [167]. The dynamic behaviour of the cell wall matrix when water molecules enter the structure is controlled by the crosslink density and also by the number of available configurations of the matrix molecules. Removal of more mobile components will change both the free energy of mixing and the elastic response of the matrix.
The swelling behaviour of wood is defined by the interplay of all these factors, with an equilibrium state being realised when free energies of mixing and elastic strain are equal. According to the Flory–Rehner theory, changes in the OH density in the matrix will affect the Gibbs free energy of mixing, with the elastic Gibbs free energy affected by the crosslink density or overall chain mobility. This means that, in principle, changes in the free energy of mixing alone could influence the swelling behaviour of the polymer system, without the necessity to invoke changes in the crosslink density or considering OH primary sorption sites as an explanation. It is more likely that changes in the free energy of mixing and of the elastic component are both responsible and these could be altered simply by the removal of the more mobile cell wall molecular components (especially hemicelluloses, without any change in crosslink density). Swelling with different solvents, such as DMSO systems, may not necessarily be affected.
How then can differences between HTwet and HTdry conditions be explained? There is undoubtedly a potential for greater crosslink formation within the cell wall structure under HTdry conditions. Formation of furfural derivatives under anhydrous conditions [88] and reaction with lignin may occur, but this requires acidic conditions and it is a possibility that reaction within the hemicellulosic mass is more likely and that crosslinking is confined there. This may be less influential upon the overall swelling compared to crosslinking within the lignin. This may explain the differences in swelling between water and, for example, DMSO. By contrast, β-O-4 cleavage within the lignin requires water to be present [117], suggesting that depolymerisation of lignin will occur, although there are also competing polymerisation reactions. Reaction under HTwet conditions favours the formation of hygroscopic and water-soluble degradation products from the amorphous polysaccharide components [146], which initially provides some dimensional stability by bulking, but not after leaching. The degraded hemicellulosic mass in HTdry treated wood is more highly crosslinked, not hygroscopic and non-leachable, thereby contributing to cell wall bulking and reducing EMC.
In summary, the potential reasons for larger MC reduction by HTdry compared to HTwet are as follows:
-
1.
Covalent crosslinks in cell wall matrix formed during HTdry cause swelling restraint; however, the evidence that there is strong swelling in other liquids (e.g. DMSO), appears to invalidate this as an explanation as it would be expected that any such restraint by covalent bonds would function in any solvent.
-
2.
Reduced swelling restraint during HTwet by hydrolytic cleavage of covalent bonds, while “natural” swelling restraint of the wood cell wall, is preserved during HTdry. Note that during HTdry, reduction of water-saturated dimensions is larger than the reduction of dry dimensions; hence, there must be some mechanism other than removal of cell wall polymers.
-
3.
Formation of resin-like, crosslinked and hydrophobic compounds in the cell wall during HTdry. This is different from (1) as this mechanism would not rely on the modification of the residual cell wall matrix polymers or any interaction between cell wall polymers and such resin-like structures. It is suggested that such reactions are confined to the amorphous polysaccharide components. This is similar to the effect of in situ cure of melamine formaldehyde resin within the wood: (a) resin occupies space in the cell wall and (b) the low moisture uptake of the pure resin results in decreasing MC ratio of the modified wood with increasing RH, which looks similar to the effect of crosslinking.
-
4.
Formation of aggregated structures by drying is stronger at elevated temperatures and while such drying effect may be partially cancelled by water-saturation, some of this aggregation may be permanent. Although this sounds very plausible, how does this work exactly? Irreversible hydrogen bonding should result in a lower OH accessibility, but a more or less uniform decrease in OH accessibility with decreasing O/C ratio has been found.
Conclusions
There are substantial differences in the properties of thermally modified timber (TMT) that is modified under dry or wet conditions, including differences in weight loss, sorption behaviour and dimensional stability. Indeed, the dimensional stability of TMT modified under HTwet conditions can actually be worse than unmodified wood, whereas the reduced hygroscopicity of TMT modified under HTdry conditions is at least partially reversed by a post-treatment water-soaking cycle. Thermal modification under dry conditions is thought to be more favourable to the formation of crosslinks (especially within the lignin network), but the swelling behaviour of HTdry modified TMT with solvents, such as DMSO, does not appear to support the hypothesis that crosslink density has increased. However, there is a reduction in both dry volume and water-saturated volume with HTdry TMT, and the reduction in water-saturated volume is greater, resulting in higher-dimensional stability. This observation requires more than simply the removal of cell wall polymers to explain the results. If increased crosslinking is not the mechanism, then it is necessary to seek additional explanations. A reduction in hydroxyl (OH) content is often invoked as an explanation for the reduction in equilibrium moisture content (EMC) of TMT. However, recent results have shown that changes in EMC are observed after a post-treatment water-saturation cycle, but without any associated change in OH content. Further work is needed to investigate this interesting observation further. Changes in cell wall polymer mobility undoubtedly occur as a result of thermal modification, and the interrelationship between polymer mobility, sorption and swelling is an area that requires further exploration as a potential explanation for the changes in behaviour, including annealing effects that occur after post-treatment water-saturation cycles. Crosslinking may be invoked as a potential mechanism for preventing leaching of polysaccharide degradation components which are formed in HTdry conditions. Such crosslinking reactions would not affect the swelling behaviour associated with solvents such as DMSO, but the presence of a hydrophobic non-leachable mass of material entangled within the cell wall structure would limit accessibility by water molecules. Further work is needed to clarify which mechanism is operative/dominant, but if the main modification is confined to the amorphous polysaccharide component of the matrix, then the formation of aggregated structures within the cell wall would appear to be the most likely explanation given the current state of knowledge. Finally, the dividing line between ‘dry’ and ‘wet’ conditions needs to be more clearly defined, with further experiments exploring the relationship between thermal modification under different conditions of relative humidity and pre-conditioning of the wood. The control of moisture content within the cell wall is an important area of research that allows for the possibility of exercising fine control over the thermal modification process to allow for the enhancement of properties of interest, whilst ensuring that the negative impacts of thermal modification (e.g. embrittlement) are minimised. Much improved commercial products are potentially available if this subject is studied in more depth.
References
Bekhta P, Niemz P (2003) Effect of high temperature on the change in color, dimensional stability and mechanical properties of spruce wood. Holzforschung 57:539–546
Hill CAS (2006) Wood modification: Chemical, thermal and other processes. John Wiley & Sons Ltd, Chichester
Yin Y, Berglund L, Salmén L (2011) Effect of Steam Treatment on the Properties of Wood Cell Walls. Biomacromol 12:194–202. https://doi.org/10.1021/bm101144m
Ganne-Chédeville C, Jääskeläinen A-S, Froidevaux J et al (2012) Natural and artificial ageing of spruce wood as observed by FTIR-ATR and UVRR spectroscopy. Holzforschung 66:163–170. https://doi.org/10.1515/HF.2011.148
Endo K, Obataya E, Zeniya N, Matsuo M (2016) Effects of heating humidity on the physical properties of hydrothermally treated spruce wood. Wood Sci Technol 50:1161–1179. https://doi.org/10.1007/s00226-016-0822-4
Obataya E, Zeniya N, Endo-Ujiie K (2019) Effects of water-soluble extractives on the moisture sorption properties of spruce wood hygrothermally treated at 120°C and different humidity levels. Wood Mat Sci Eng. https://doi.org/10.1080/17480272.2019.1635642
Stamm AJ, Hansen LA (1937) Minimizing Wood Shrinkage and Swelling: Effect of Heating in Various Gases. Ind Eng Chem 29:831–833. https://doi.org/10.1021/ie50331a021
Stamm AJ, Burr HK, Kline AA (1946) Staybwood—Heat-Stabilized Wood. Ind Eng Chem 38:630–634. https://doi.org/10.1021/ie50438a027
Stamm AJ (1942) Antishrink treatment for wood. US Patent No. 2 296 316
Burmester A (1973) Effects of heat-pressure treatments of semi-dry wood on its dimensional stability. Holz Als Roh-Und Werkstoff 31:237–243
Giebeler E (1983) Dimensional stabilisation of wood by moisture-heat-pressure-treatment. Holz Als Roh-und Werkst 41:87–94
Militz H, Altgen M (2014) Processes and Properties of Thermally Modified Wood Manufactured in Europe In Deterioration and Protection of Sustainable Biomaterials. Am Chem Soc 16:269–285
Jones D, Sandberg D, Goli G, Todaro L (2019) Wood Modification in Europe: a state-of-the-art about processes, products and applications. Firenze University Press
Tjeerdsma BF, Militz H (2005) Chemical changes in hydrothermal treated wood: FTIR analysis of combined hydrothermal and dry heat-treated wood. Holz Als Roh-und Werkst 63:102–111. https://doi.org/10.1007/s00107-004-0532-8
Leppänen K, Spetz P, Pranovich A et al (2011) Pressurized hot water extraction of Norway spruce hemicelluloses using a flow-through system. Wood Sci Technol 45:223–236. https://doi.org/10.1007/s00226-010-0320-z
Sundqvist B, Karlsson O, Westermark U (2006) Determination of formic-acid and acetic acid concentrations formed during hydrothermal treatment of birch wood and its relation to colour, strength and hardness. Wood Sci Technol 40:549–561. https://doi.org/10.1007/s00226-006-0071-z
Sandberg D, Kutnar A (2016) Thermally modified timber: Recent developments in Europe and North America. Wood Fiber Sci 48:28–39
Esteves BM, Pereira HM (2009) Wood modification by heat treatment: a review. BioResources 4:370–404
Pelaez-Samaniego MR, Yadama V, Lowell E, Espinoza-Herrera R (2013) A review of wood thermal pretreatments to improve wood composite properties. Wood Sci Technol 47:1285–1319. https://doi.org/10.1007/s00226-013-0574-3
Willems W, Mai C, Militz H (2013) Thermal wood modification chemistry analysed using van Krevelen’s representation. International Wood Products Journal 4:166–171
Willems W, Altgen M (2020) Hygrothermolytic wood modification. process description and treatment level characterisation. Wood Mat Sci Eng 15:213–222. https://doi.org/10.1080/17480272.2019.1570970
Willems W, Altgen M, Militz H (2015) Comparison of EMC and durability of heat treated wood from high versus low water vapour pressure reactor systems. International Wood Products Journal 6:21–26
Altgen M, Willems W, Militz H (2016) Wood degradation affected by process conditions during thermal modification of European beech in a high-pressure reactor system. European Journal of Wood and Wood Products 74:653–662. https://doi.org/10.1007/s00107-016-1045-y
MacLean J (1951) Rate of disintegration of wood under different heating conditions. Proceedings of the American Wood Preservers Association 47:155–169
Candelier K, Chaouch M, Dumarçay S et al (2011) Utilization of thermodesorption coupled to GC–MS to study stability of different wood species to thermodegradation. J Anal Appl Pyrol 92:376–383. https://doi.org/10.1016/j.jaap.2011.07.010
Wentzel M, Altgen M, Militz H (2018) Analyzing reversible changes in hygroscopicity of thermally modified eucalypt wood from open and closed reactor systems. Wood Sci Technol 52:889–907. https://doi.org/10.1007/s00226-018-1012-3
Obataya E, Higashihara T, Tomita B (2002) Hygroscopicity of heat-treated wood III. Effect of steaming on the hygroscopicity of wood. Mokuzai Gakkaishi 48:348–355
Obataya E, Shibutani S, Hanata K, Doi S (2006) Effects of high temperature kiln drying on the practical performances of Japanese cedar wood (Cryptomeria japonica) I: changes in hygroscopicity due to heating. J Wood Sci 52:33–38. https://doi.org/10.1007/s10086-005-0716-9
Altgen M, Hofmann T, Militz H (2016) Wood moisture content during the thermal modification process affects the improvement in hygroscopicity of Scots pine sapwood. Wood Sci Technol 50:1181–1195. https://doi.org/10.1007/s00226-016-0845-x
Pfriem A, Zauer M, Wagenführ A (2009) Alteration of the pore structure of spruce (Picea abies (L.) Karst.) and maple (Acer pseudoplatanus L.) due to thermal treatment as determined by helium pycnometry and mercury intrusion porosimetry. Holzforschung 63:94–98
Willems W, Altgen M (2019) Hygrothermolytic wood modification. process description and treatment level characterisation. Wood Mat Sci Eng 15:213–222. https://doi.org/10.1080/17480272.2019.1570970
Altgen M, Militz H (2016) Influence of process conditions on hygroscopicity and mechanical properties of thermally modified wood in a closed reactor system. Holzforschung 70:971–979
DeGroot W, Pan W-P, Rahman M, Richards G (1988) First chemical events in pyrolysis of wood. J Anal Appl Pyrol 13:221–231
Li T, Cheng D, Avramidis S et al (2017) Response of hygroscopicity to heat treatment and its relation to durability of thermally modified wood. Constr Build Mater 144:671–676. https://doi.org/10.1016/j.conbuildmat.2017.03.218
Hughes M, Hill C, Pfriem A (2015) The toughness of hygrothermally modified wood – a review. Hfsg. 69:851–862. https://doi.org/10.1515/hf-2014-0184
Willems W (2016) Glassy state of wood polymers in native and thermally modified wood: effects on long-term material performance in service. International Wood Products Journal 7:71–75. https://doi.org/10.1080/20426445.2016.1160589
Ruike M, Inoue T, Takada S et al (1999) Water sorption and drying behaviour of crosslinked dextrans. Biosci Biotechnol Biochem. 63:271–275. https://doi.org/10.1271/bbb.63.271
Funaoka M, Kako T, Abe I (1990) Condensation of lignin during heating of wood. Wood Sci Technol 24:277–288
Placet V, Passard J, Perré P (2008) Viscoelastic properties of wood across the grain measured under water-saturated conditions up to 135 °C: evidence of thermal degradation. J Mater Sci 43:3210–3217. https://doi.org/10.1007/s10853-008-2546-9
González-Peña MM, Curling SF, Hale MDC (2009) On the effect of heat on the chemical composition and dimensions of thermally-modified wood. Polym Degrad Stabil 94:2184–2193. https://doi.org/10.1016/j.polymdegradstab.2009.09.003
Boonstra MJ, Tjeerdsma B (2006) Chemical analysis of heat treated softwoods. Holz Als Roh-und Werkst 64:204–211. https://doi.org/10.1007/s00107-005-0078-4
Boonstra MJ, Van Acker J, Tjeerdsma BF, Kegel EV (2007) Strength properties of thermally modified softwoods and its relation to polymeric structural wood constituents. Ann For Sci 64:679–690. https://doi.org/10.1051/forest:2007048
Bryne LE, Lausmaa J, Ernstsson M et al (2010) Ageing of modified wood. Part 2: Determination of surface composition of acetylated, furfurylated and thermally modified wood by XPS and ToF-SIMS. Holzforschung 64:305–313. https://doi.org/10.1515/hf.2010.062
Candelier K, Dumarçay S, Pétrissans A et al (2013) Comparison of chemical composition and decay durability of heat treated wood cured under different inert atmospheres: Nitrogen or vacuum. Polym Degrad Stab 98:677–681. https://doi.org/10.1016/j.polymdegradstab.2012.10.022
Chien Y-C, Yang T-C, Hung K-C et al (2018) Effects of heat treatment on the chemical compositions and thermal decomposition kinetics of Japanese cedar and beech wood. Polym Degrad Stab 158:220–227. https://doi.org/10.1016/j.polymdegradstab.2018.11.003
Brito J, Sila F, Leao M, Almeida G (2008) Chemical composition changes in eucalyptus and pinus woods submitted to heat treatment. Biores Technol 99:8545–8548. https://doi.org/10.1016/j.biortech.2008.03.069
Garrote G, Domínguez H, Parajó JC (2001) Study on the deacetylation of hemicelluloses during the hydrothermal processing of Eucalyptus wood. Holz als Roh- und Werkstoff 59:53–59. https://doi.org/10.1007/s001070050473
Chaouch M, Dumarçay S, Pétrissans A et al (2013) Effect of heat treatment intensity on some conferred properties of different European softwood and hardwood species. Wood Sci Technol 47:663–673. https://doi.org/10.1007/s00226-013-0533-z
Willems W, Gérardin P, Militz H (2013) The average carbon oxidation state of thermally modified wood as a marker for its decay resistance against Basidiomycetes. Polym Degrad Stab 98:2140–2145. https://doi.org/10.1016/j.polymdegradstab.2013.09.003
Willems W (2014) The water vapor sorption mechanism and its hysteresis in wood: the water/void mixture postulate. Wood Sci Technol 48:499–518. https://doi.org/10.1007/s00226-014-0617-4
Gérardin P, Petric M, Petrissans M et al (2007) Evolution of wood surface free energy after heat treatment. Polym Degrad Stabil 92:653–657. https://doi.org/10.1016/j.polymdegradstab.2007.01.016
Wang W, Zhu Y, Cao JZ, Sun WJ (2015) Correlation between dynamic wetting behavior and chemical components of thermally modified wood. Appl Surf Sci 324:332–338. https://doi.org/10.1016/j.apsusc.2014.10.139
Sivonen H, Maunu SL, Sundholm F et al (2002) Magnetic resonance studies of thermally modified wood. Holzforschung 56:648–654
Patil SV, Argyropoulos DS (2017) Stable Organic Radicals in Lignin: A Review. Chemsuschem 10:3284–3303. https://doi.org/10.1002/cssc.201700869
Chen Y, Tshabalala MA, Gao J et al (2014) Thermal behavior of extracted and delignified pine wood flour. Thermochim Acta 591:40–44. https://doi.org/10.1016/j.tca.2014.06.012
Nair SS, Yan N (2015) Effect of high residual lignin on the thermal stability of nanofibrils and its enhanced mechanical performance in aqueous environments. Cellulose 22:3137–3150. https://doi.org/10.1007/s10570-015-0737-5
Vänskä E, Vihelä T, Peresin MS et al (2016) Residual lignin inhibits thermal degradation of cellulosic fiber sheets. Cellulose 23:199–212. https://doi.org/10.1007/s10570-015-0791-z
Fernandes AN, Thomas LH, Altaner CM et al (2011) Nanostructure of cellulose microfibrils in spruce wood. Proc Natl Acad Sci 108:E1195–E1203. https://doi.org/10.1073/pnas.1108942108
Cosgrove DJ, Jarvis MC (2012) Comparative structure and biomechanics of plant primary and secondary cell walls. Front Plant Sci. https://doi.org/10.3389/fpls.2012.00204
Donaldson L (2007) Cellulose microfibril aggregates and their size variation with cell wall type. Wood Sci Technol 41:443–460. https://doi.org/10.1007/s00226-006-0121-6
Poletto M, Zattera AJ, Santana RMC (2012) Thermal decomposition of wood: Kinetics and degradation mechanisms. Biores Technol 126:7–12. https://doi.org/10.1016/j.biortech.2012.08.133
Kymäläinen M, Ben Mlouka S, Belt T et al (2018) Chemical, water vapour sorption and ultrastructural analysis of Scots pine wood thermally modified in high-pressure reactor under saturated steam. J Mater Sci 53:3027–3037. https://doi.org/10.1007/s10853-017-1714-1
Dwianto F, Tanaka F, Inoue M, Norimoto M (1996) Crystallinity changes of wood by heat or steam treatment. Wood research: bulletin of the Wood Research Institute Kyoto University 83:47–49
Bhuiyan MTR, Hirai N, Sobue N (2000) Changes of crystallinity in wood cellulose by heat treatment under dried and moist conditions. J Wood Sci 46:431–436
Andersson S, Serimaa R, Vaananen T et al (2005) X-ray scattering studies of thermally modified Scots pine (Pinus sylvestris L.). Holzforschung 59:422–427
Yin J, Yuan T, Lu Y et al (2017) Effect of compression combined with steam treatment on the porosity, chemical compositon and cellulose crystalline structure of wood cell walls. Carbohyd Polym 155:163–172. https://doi.org/10.1016/j.carbpol.2016.08.013
Wang X, Wu Z, Fang L, et al (2015) Changes of Chemical Composition, Crystallinity and Fourier Transform Infrared Spectra of Eucalypt pellita Wood under Different Vacuum Heat Treatment Temperatures. Forest Products Journal. 65;346–351. https://doi.org/https://doi.org/10.13073/FPJ-D-13-00099
Cheng XY, Li XJ, Xu K, et al (2017) Effect of Thermal Treatment on Functional Groups and Degree of Cellulose Crystallinity of Eucalyptus Wood ( Eucalyptus grandis × Eucalyptus urophylla ) Forest Products Journal. 67:135–140 https://doi.org/https://doi.org/10.13073/FPJ-D-15-00075
Thygesen A, Oddershede J, Lilholt H et al (2005) On the determination of crystallinity and cellulose content in plant fibres. Cellulose 12:563–576. https://doi.org/10.1007/s10570-005-9001-8
Olek W, Bonarski JT (2014) Effects of thermal modification on wood ultrastructure analyzed with crystallographic texture. Holzforschung 68:721–726. https://doi.org/10.1515/hf-2013-0165
Pereira CS, Silveira RL, Dupree P, Skaf MS (2017) Effects of Xylan Side-Chain Substitutions on Xylan-Cellulose Interactions and Implications for Thermal Pretreatment of Cellulosic Biomass. Biomacromol 18:1311–1321. https://doi.org/10.1021/acs.biomac.7b00067
Pauly M, Albersheim P, Darvill A, York WS (1999) Molecular domains of the cellulose/xyloglucan network in the cell walls of higher plants. Plant J 20:629–639. https://doi.org/10.1046/j.1365-313X.1999.00630.x
Sella Kapu N, Trajano HL (2014) Review of hemicellulose hydrolysis in softwoods and bamboo. Biofuels, Bioprod Biorefin 8:857–870. https://doi.org/10.1002/bbb.1517
Åkerholm M, Salmén L (2001) Interactions between wood polymers studied by dynamic FT-IR spectroscopy. Polymer 42:963–969. https://doi.org/10.1016/S0032-3861(00)00434-1
Hinterstoisser B, Åkerholm M, Salmén L (2001) Effect of fiber orientation in dynamic FTIR study on native cellulose. Carbohyd Res 334:27–37. https://doi.org/10.1016/S0008-6215(01)00167-7
Busse-Wicher M, Grantham NJ, Lyczakowski JJ et al (2016) Xylan decoration patterns and the plant secondary cell wall molecular architecture. Biochem Soc Trans 44:74–78. https://doi.org/10.1042/BST20150183
Terrett OM, Lyczakowski JJ, Yu L et al (2019) Molecular architecture of softwood revealed by solid-state NMR. Nat Commun 10:4978. https://doi.org/10.1038/s41467-019-12979-9
Fengel D, Wegener G (1983) Wood Chemistry, Ultrastructure. Reactions, De Gruyter, Berlin and New York
Busse-Wicher M, Gomes TCF, Tryfona T et al (2014) The pattern of xylan acetylation suggests xylan may interact with cellulose microfibrils as a twofold helical screw in the secondary plant cell wall of Arabidopsis thaliana. Plant J 79:492–506. https://doi.org/10.1111/tpj.12575
Thomas LH, Forsyth VT, Martel A et al (2014) Structure and spacing of cellulose microfibrils in woody cell walls of dicots. Cellulose 21:3887–3895. https://doi.org/10.1007/s10570-014-0431-z
Busse-Wicher M, Li A, Silveira RL et al (2016) Evolution of Xylan Substitution Patterns in Gymnosperms and Angiosperms: Implications for Xylan Interaction with Cellulose. Plant Physiol 171:2418–2431. https://doi.org/10.1104/pp.16.00539
Bosmans TJ, Stépán AM, Toriz G et al (2014) Assembly of Debranched Xylan from Solution and on Nanocellulosic Surfaces. Biomacromol 15:924–930. https://doi.org/10.1021/bm4017868
Hannuksela T, Hervé du Penhoat C (2004) NMR structural determination of dissolved O-acetylated galactoglucomannan isolated from spruce thermomechanical pulp. Carbohyd Res 339:301–312. https://doi.org/10.1016/j.carres.2003.10.025
Thomas LH, Martel A, Grillo I, Jarvis MC (2020) Hemicellulose binding and the spacing of cellulose microfibrils in spruce wood. Cellulose 27:4249–4254. https://doi.org/10.1007/s10570-020-03091-z
Xue L, Zhao Z, Zhang Y et al (2016) Analysis of gas hromatographcy-mass spectrometry coupled with dynamic headspace sampling on volatile organic compounds of heat-treated poplar at high temperatures. BioResources 11:3550–3560
Pielhop T, Larrazábal GO, Studer MH et al (2015) Lignin repolymerisation in spruce autohydrolysis pretreatment increases cellulase deactivation. Green Chem 17:3521–3532. https://doi.org/10.1039/C4GC02381A
Chen R, Wang Y-Z, Liao Q et al (2013) Hydrolysates of lignocellulosic materials for biohydrogen production. BMB Reports 46:244–251. https://doi.org/10.5483/BMBRep.2013.46.5.038
Karlsson O, Torniainen P, Dagbro O et al (2012) Presence of water-soluble compounds in thermally modified wood: carbonydrates and furfurals. BioResources 7:3679–3689
Peters J, Fischer K, Fischer S (2008) Characterisation of emissions from thermally modified wood and their reduction by chemical treatment. BioResources 3:491–502
Zainol MM, Amin NAS, Asmadi M (2019) Kinetics and thermodynamic analysis of levulinic acid esterification using lignin-furfural carbon cryogel catalyst. Renewable Energy 130:547–557. https://doi.org/10.1016/j.renene.2018.06.085
Rackemann D, Doherty W (2012) A review on the production of levulinic acid and furanics from sugars. International Sugar Journal 114:30–36
Altgen M, Uimonen T, Rautkari L (2018) The effect of de- and re-polymerization during heat-treatment on the mechanical behavior of Scots pine sapwood under quasi-static load. Polym Degrad Stab 147:197–205. https://doi.org/10.1016/j.polymdegradstab.2017.12.007
Dongre P, Driscoll M, Amidon T, Bujanovic B (2015) Lignin-Furfural Based Adhesives Energies 8:7897–7914. https://doi.org/10.3390/en8087897
Koshijima T, Watanabe T (2003) Association Between Lignin and Carbohydrates in Wood and Other Plant Tissues. Springer, Berlin Heidelberg, Berlin, Heidelberg
Chen X, Lawoko M, van Heiningen A (2010) Kinetics and mechanism of autohydrolysis of hardwoods. Biores Technol 101:7812–7819. https://doi.org/10.1016/j.biortech.2010.05.006
Tunc M, Lawoko M (2010) van Heiningen A Understanding the limitations of removal of hemicelluloses during autohydrolysis of a mixture of southern hardwoods. BioResources 5:356–371
Tarasov D, Leitch M, Fatehi P (2018) Lignin–carbohydrate complexes: properties, applications, analyses and methods of extraction: a review. Biotechnol Biofuels 11:269. https://doi.org/10.1186/s13068-018-1262-1
Giummarella N, Balakshin M, Koutaniemi S et al (2019) Nativity of lignin carbohydrate bonds substantiated by biomimetic synthesis. J Exp Bot 70:5591–5601. https://doi.org/10.1093/jxb/erz324
Giummarella N, Pu Y, Ragauskas AJ, Lawoko M (2019) A critical review on the analysis of lignin carbohydrate bonds. Green Chem 21:1573–1595. https://doi.org/10.1039/C8GC03606C
Gao S, Zhao J, Wang X et al (2018) Lignin Structure and Solvent Effects on the Selective Removal of Condensed Units and Enrichment of S-Type Lignin. Polymers 10:967. https://doi.org/10.3390/polym10090967
Brosse N, El Hage R, Chaouch M et al (2010) Investigation of the chemical modifications of beech wood lignin during heat treatment. Polym Degrad Stabil 95:1721–1726. https://doi.org/10.1016/j.polymdegradstab.2010.05.018
Fenner RA, Lephardt JO (1981) Examination of the thermal decomposition of kraft pine lignin by Fourier transform infrared evolved gas analysis. J Agric Food Chem 29:846–849. https://doi.org/10.1021/jf00106a042
Nuopponen M, Vuorinen T, Jämsä S, Viitaniemi P (2005) Thermal Modifications in Softwood Studied by FT-IR and UV Resonance Raman Spectroscopies. J Wood Chem Technol 24:13–26
Wikberg H, Maunu S (2004) Characterisation of thermally modified hard- and softwoods by 13C CPMAS NMR. Carbohyd Polym 58:461–466. https://doi.org/10.1016/j.carbpol.2004.08.008
Wang P, Fu Y, Shao Z et al (2016) Structural changes to aspen wood lignin during autohydrolysis pretreatment. BioResources 11:4086–4103
Rousset P, Lapierre C, Pollet B et al (2009) Effect of severe thermal treatment on spruce and beech wood lignins. Ann For Sci. https://doi.org/10.1051/forest/2008078
Jiang X, Lu Q, Hu B et al (2019) Influence of inherent alkali metal chlorides on pyrolysis mechanism of a lignin model dimer based on DFT study. J Therm Anal Calorim 137:151–160. https://doi.org/10.1007/s10973-018-7920-5
Ibbett R, Gaddipati S, Davies S et al (2011) The mechanisms of hydrothermal deconstruction of lignocellulose: New insights from thermal-analytical and complementary studies. Biores Technol 102:9272–9278. https://doi.org/10.1016/j.biortech.2011.06.044
Inari GN, Mounguengui S, Dumarçay S et al (2007) Evidence of char formation during wood heat treatment by mild pyrolysis. Polym Degrad Stab 92:997–1002. https://doi.org/10.1016/j.polymdegradstab.2007.03.003
Nishida M, Tanaka T, Miki T et al (2019) Integrated analysis of modified Japanese cypress using solid-state NMR spectra and nuclear magnetic relaxation times. Cellulose 26:3625–3642. https://doi.org/10.1007/s10570-019-02330-2
Wan G, Frazier CE (2017) Lignin Acidolysis Predicts Formaldehyde Generation in Pine Wood. ACS Sustainable Chem Eng 5:4830–4836. https://doi.org/10.1021/acssuschemeng.7b00264
Pohleven J, Burnard M, Kutnar A (2019) Volatile organic compounds emitted from untreated and thermally modified wood – a review. WFS. 51:231–254. https://doi.org/https://doi.org/10.22382/wfs-2019-023
Lundquist K, Lundgren R (1972) Acid degradation of lignin. VII. Cleavage of ether bonds. Acta Chem Scand 26:2005–2023
Yasuda S, Fukushima K, Kakehi A (2001) Formation and chemical structures of acid-soluble lignin I: sulfuric acid treatment time and acid-soluble lignin content of hardwood. J Wood Sci 47:69–72. https://doi.org/10.1007/BF00776648
Matsushita Y, Kakehi A, Miyawaki S, Yasuda S (2004) Formation and chemical structures of acid-soluble lignin II: reaction of aromatic nuclei model compounds with xylan in the presence of a counterpart for condensation and behavior of lignin model compounds with guaiacyl and syringyl nuclei in 72% sulfuric acid. J Wood Sci 50:136–141. https://doi.org/10.1007/s10086-003-0543-9
Sturgeon MR, Kim S, Lawrence K et al (2014) A Mechanistic Investigation of Acid-Catalyzed Cleavage of Aryl-Ether Linkages: Implications for Lignin Depolymerization in Acidic Environments. ACS Sustainable Chem Eng 2:472–485. https://doi.org/10.1021/sc400384w
Yokoyama T (2015) Revisiting the Mechanism of β- O -4 Bond Cleavage During Acidolysis of Lignin. Part 6: A Review. J Wood Chem Technol 35:27–42. https://doi.org/10.1080/02773813.2014.881375
Nuopponen M, Vuorinen T, Jamsa S, Viitaniemi P (2003) The effects of a heat treatment on the behaviour of extractives in softwood studied by FTIR spectroscopic methods. Wood Sci Technol 37:109–115. https://doi.org/10.1007/s00226-003-0178-4
Esteves B, Graça J, Pereira H (2008) Extractive composition and summative chemical analysis of thermally treated eucalypt wood. Hfsg. 62:344–351. https://doi.org/10.1515/hf.2008.057
Esteves B, Videira R, Pereira H (2011) Chemistry and ecotoxicity of heat-treated pine wood extractives. Wood Sci Technol 45:661–676. https://doi.org/10.1007/s00226-010-0356-0
Shebani AN, van Reenen AJ, Meincken M (2008) The effect of wood extractives on the thermal stability of different wood species. Thermochim Acta 471:43–50. https://doi.org/10.1016/j.tca.2008.02.020
Hofstetter K, Hinterstoisser B, Salmén L (2006) Moisture uptake in native cellulose – the roles of different hydrogen bonds: a dynamic FT-IR study using Deuterium exchange. Cellulose 13:131–145. https://doi.org/10.1007/s10570-006-9055-2
Suchy M, Virtanen J, Kontturi E, Vuorinen T (2010) Impact of Drying on Wood Ultrastructure Observed by Deuterium Exchange and Photoacoustic FT-IR Spectroscopy. Biomacromol 11:515–520. https://doi.org/10.1021/bm901268j
Pönni R, Rautkari L, Hill CAS, Vuorinen T (2014) Accessibility of hydroxyl groups in birch kraft pulps quantified by deuterium exchange in D2O vapor. Cellulose 21:1217–1226. https://doi.org/10.1007/s10570-014-0166-x
Thybring EE, Thygesen LG, Burgert I (2017) Hydroxyl accessibility in wood cell walls as affected by drying and re-wetting procedures. Cellulose 24:2375–2384. https://doi.org/10.1007/s10570-017-1278-x
Tarmian A, Burgert I, Thybring EE (2017) Hydroxyl accessibility in wood by deuterium exchange and ATR-FTIR spectroscopy: methodological uncertainties. Wood Sci Technol 51:845–853. https://doi.org/10.1007/s00226-017-0922-9
Atalla RS, Crowley MF, Himmel ME, Atalla RH (2014) Irreversible transformations of native celluloses, upon exposure to elevated temperatures. Carbohyd Polym 100:2–8. https://doi.org/10.1016/j.carbpol.2013.06.007
Lindh EL, Bergenstråhle-Wohlert M, Terenzi C et al (2016) Non-exchanging hydroxyl groups on the surface of cellulose fibrils: The role of interaction with water. Carbohyd Res 434:136–142. https://doi.org/10.1016/j.carres.2016.09.006
Kulasinski K, Salmén L, Derome D, Carmeliet J (2016) Moisture adsorption of glucomannan and xylan hemicelluloses. Cellulose 23:1629–1637. https://doi.org/10.1007/s10570-016-0944-8
Phuong LX, Takayama M, Shida S et al (2007) Determination of the accessible hydroxyl groups in heat-treated Styrax tonkinensis (Pierre) Craib ex Hartwich wood by hydrogen-deuterium exchange and H-2 NMR spectroscopy. Holzforschung 61:488–491. https://doi.org/10.1515/hf2007.086
Rautkari L, Hill CAS, Curling S et al (2013) What is the role of the accessibility of wood hydroxyl groups in controlling moisture content? J Mater Sci 48:6352–6356. https://doi.org/10.1007/s10853-013-7434-2
Kymäläinen M, Rautkari L, Hill CAS (2015) Sorption behaviour of torrefied wood and charcoal determined by dynamic vapour sorption. J Mater Sci 50:7673–7680. https://doi.org/10.1007/s10853-015-9332-2
Uimonen T, Hautamäki S, Altgen M et al (2020) Dynamic vapour sorption protocols for the quantification of accessible hydroxyl groups in wood. Holzforschung 74:412–419. https://doi.org/10.1515/hf-2019-0058
Glass SV, Boardman CR, Thybring EE, Zelinka SL (2018) Quantifying and reducing errors in equilibrium moisture content measurements with dynamic vapor sorption (DVS) experiments. Wood Sci Technol 52:909–927. https://doi.org/10.1007/s00226-018-1007-0
Willems W, Altgen M, Rautkari L (2020) A molecular model for reversible and irreversible hygroscopicity changes by thermal wood modification. Holzforschung 74:420–425. https://doi.org/10.1515/hf-2019-0057
Esteves B, Velez Marques A, Domingos I, Pereira H (2013) Chemical changes of heat treated pine and eucalypt wood monitored by FTIR. Maderas Ciencia y tecnologÃ\-a. 15:245–258
Mitsui K, Inagaki T, Tsuchikawa S (2008) Monitoring of hydroxyl groups in wood during heat treatment using NIR spectroscopy. Biomacromol 9:286–288. https://doi.org/10.1021/bm7008069
Windeisen E, Strobel C, Wegener G (2007) Chemical changes during the production of thermo-treated beech wood. Wood Sci Technol 41:523–536. https://doi.org/10.1007/s00226-007-0146-5
Kekkonen PM, Ylisassi A, Telkki V-V (2014) Absorption of Water in Thermally Modified Pine Wood As Studied by Nuclear Magnetic Resonance. The Journal of Physical Chemistry C 118:2146–2153. https://doi.org/10.1021/jp411199r
Cai C, Javed MA, Komulainen S et al (2020) Effect of natural weathering on water absorption and pore size distribution in thermally modified wood determined by nuclear magnetic resonance. Cellulose 27:4235–4247. https://doi.org/10.1007/s10570-020-03093-x
Obataya E, Tomita B (2002) Hygroscopicity of heat-treated wood II. Reversible and irreversible reductions in the hygroscopicity of wood due to heating. Mokuzai Gakkaishi 48:288–295
Hill C, Ramsay J, Keating B et al (2012) The water vapour sorption properties of thermally modified and densified wood. J Mater Sci 47:3191–3197. https://doi.org/10.1007/s10853-011-6154-8
Majka J, Czajkowski Ł, Olek W (2016) Effects of cyclic changes in relative humidity on the sorption hysteresis of thermally modified spruce wood. BioResources 11:5265–6275
Čermák P, Rautkari L, Horacek P et al (2015) Analysis of Dimensional Stability of Thermally Modified Wood Affected by Re-Wetting Cycles. BioResources 10:3242–3253
Tarmian A, Mastouri A (2019) Changes in moisture exclusion efficiency and crystallinity of thermally modified wood with aging. iForest. 12:92–97. https://doi.org/10.3832/ifor2723-011
Obataya E, Higashihara T (2017) Reversible and irreversible dimensional changes of heat-treated wood during alternate wetting and drying. Wood Sci Technol 51:739–749. https://doi.org/10.1007/s00226-017-0918-5
Borrega M, Kärenlampi P (2010) Hygroscopicity of heat-treated Norway spruce (Picea abies) wood. European Journal of Wood and Wood Products 68:233–235. https://doi.org/10.1007/s00107-009-0371-8
Altgen M, Willems W, Hosseinpourpia R, Rautkari L (2018) Hydroxyl accessibility and dimensional changes of Scots pine sapwood affected by alterations in the cell wall ultrastructure during heat-treatment. Polym Degrad Stab 152:244–252. https://doi.org/10.1016/j.polymdegradstab.2018.05.005
Willems W (2018) Hygroscopic wood moisture: single and dimerized water molecules at hydroxyl-pair sites? Wood Sci Technol 52:777–791. https://doi.org/10.1007/s00226-018-0998-x
Salmén L, Olsson A-M (1998) Interaction between hemicelluloses, lignin and cellulose: Structure-property relationships. J Pulp Pap Sci 24:99–103
Hill CAS, Keating BA, Jalaludin Z, Mahrdt E (2012) A rheological description of the water vapour sorption kinetics behaviour of wood invoking a model using a canonical assembly of Kelvin-Voigt elements and a possible link with sorption hysteresis. Holzforschung 66:35–47. https://doi.org/10.1515/HF.2011.115
Jakes JE, Hunt CG, Zelinka SL et al (2019) Effects of Moisture on Diffusion in Unmodified Wood Cell Walls: A Phenomenological Polymer Science Approach. Forests 10:1084. https://doi.org/10.3390/f10121084
Youssefian S, Jakes JE, Rahbar N (2017) Variation of Nanostructures, Molecular Interactions and Anisotropic Elastic Moduli of Lignocellulosic Cell Walls with Moisture. Sci Rep 7:2054. https://doi.org/10.1038/s41598-017-02288-w
Rowell RM, Ellis WD (1978) Determination of the dimensional stabilization of wood using the water-soak method. Wood Fiber Sci 10:104–111
Thybring EE, Piqueras S, Tarmian A, Burgert I (2020) Water accessibility to hydroxyls confined in solid wood cell walls. Cellulose 27:5617–5627. https://doi.org/10.1007/s10570-020-03182-x
Flory P (1953) Phase equilibria in polymer systems: swelling of network structures. In: Principles of Polymer Chemistry. Cornell University, Ithaca, NY, p 688
Sasaki T, Uchida T, Sakurai K (2006) Effect of crosslink on the characteristic length of glass transition of network polymers: Glass Transition of Network Polymers. J Polym Sci B Polym Phys 44:1958–1966. https://doi.org/10.1002/polb.20852
Seborg M, Tarkow H, Stamm AJ (1953) Effect of heat upon the dimensional stabilization of wood. Journal of the Forest Products Research Society 3:59–67
Rautkari L, Hill CAS (2014) Effect of initial moisture content on the anti-swelling efficiency of thermally modified Scots pine sapwood treated in a high-pressure reactor under saturated steam. Holzforschung 68:323–326. https://doi.org/10.1515/hf-2013-0078
Biziks V, Andersons B, Sansonetti E et al (2015) One-stage thermo-hydro treatment (THT) of hardwoods: an analysis of form stability after five soaking-drying cycles. Holzforschung 69:563–571. https://doi.org/10.1515/hf-2014-0083
Mantanis GI, Young RA, Rowell RM (1994) Swelling of Wood. Part II. Swelling in Organic Liquids Holzforschung 48:480–490. https://doi.org/10.1515/hfsg.1994.48.6.480
Rapp AO, Brischke C, Welzbacher CR, Jazayeri L (2008) Increased resistance of thermally modified Norway spruce timber (TMT) against brown rot decay by Oligoporus placenta - Study on the mode of protective action. Wood Res 53:13–25
Crawshaw J, Cameron RE (2000) A small angle X-ray scattering study of pore structure in Tencel ® cellulose fibres and the effects of physical treatments. Polymer 41:4691–4698. https://doi.org/10.1016/S0032-3861(99)00502-9
Hill SJ, Kirby NM, Mudie ST et al (2010) Effect of drying and rewetting of wood on cellulose molecular packing. Holzforschung. https://doi.org/10.1515/hf.2010.065
Abe K, Yamamoto H (2005) Mechanical interaction between cellulose microfibril and matrix substance in wood cell wall determined by X-ray diffraction. J Wood Sci 51:334–338. https://doi.org/10.1007/s10086-004-0667-6
Barkas WW (1942) Wood water relationships—VII. Swelling pressure and sorption hysteresis in gels. Trans Faraday Soc 38:194–209. https://doi.org/10.1039/TF9423800194
Flory PJ, Rehner J (1943) Statistical Mechanics of Cross-Linked Polymer Networks II. Swelling The Journal of Chemical Physics 11:521–526. https://doi.org/10.1063/1.1723792
Ganji F, Vasheghani-farahani S, Vasheghani-farahani E (2010) Theoretical description of hydrogel swelling: a review. Iran Polym J 19:375–398
Acknowledgements
The authors wish to acknowledge financial support from the Academy of Finland (Grant No. 309881). The authors of this review also wish to express gratitude to the referees for giving up their time to undertake their reviews and for the helpful comments which have been used to make improvements to this paper.
Author information
Authors and Affiliations
Contributions
Callum Hill, wrote the main body of the text.
Lauri Rautkari designed paper concept and structure, rewrote text and additional comments.
Michael Altgen designed paper concept and structure, re-wrote text, additional comments and conclusions.
Corresponding author
Ethics declarations
Conflicts of interest
The authors of this work declare that they have no conflicts of interest or competing interests.
Additional information
Handling Editor: Stephen Eichhorn.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hill, C., Altgen, M. & Rautkari, L. Thermal modification of wood—a review: chemical changes and hygroscopicity. J Mater Sci 56, 6581–6614 (2021). https://doi.org/10.1007/s10853-020-05722-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-020-05722-z