
Abstract
Background
Cold stress can cause serious abiotic damage that limits the growth, development and yield of rice. Cold tolerance during the booting stage of rice is a key factor that can guarantee a high and stable yield under cold stress. The cold tolerance of rice is controlled by quantitative trait loci (QTLs). Based on the complex genetic basis of cold tolerance in rice, additional efforts are needed to detect reliable QTLs and identify candidate genes. In this study, recombinant inbred lines (RILs) derived from a cross between a cold sensitive variety, Dongnong422, and strongly cold-tolerant variety, Kongyu131, were used to screen for cold-tolerant loci at the booting stage of rice.
Results
A novel major QTL, qPSST6, controlling the percent seed set under cold water treatment (PSST) under the field conditions of 17 °C cold water irrigation was located on the 28.4 cM interval on chromosome 6. Using the combination of bulked-segregant analysis (BSA) and next-generation sequencing (NGS) technology (Seq-BSA), a 1.81 Mb region that contains 269 predicted genes on chromosome 6 was identified as the candidate region of qPSST6. Two genes, LOC_Os06g39740 and LOC_Os06g39750, were annotated as “response to cold” by gene ontology (GO) analysis. qRT-PCR analysis revealed that LOC_Os06g39750 was strongly induced by cold stress. Haplotype analysis also demonstrate a key role of LOC_Os06g39750 in regulating the PSST of rice, suggesting that it was the candidate gene of qPSST6.
Conclusions
The information obtained in this study is useful for gene cloning of qPSST6 and for breeding cold-tolerant varieties of rice using marker assisted selection (MAS).
Similar content being viewed by others

Background
Rice, a staple food crop cultivated worldwide, feeds more than half of the world’s population. Rice originates from tropical and subtropical regions and is more sensitive to low temperature than other crops originating in temperate zones (Wang et al. 2016). In high-latitude or high-altitude regions of Asia, Europe, America and other rice cultivation areas of the world, the temperature is not consistently high enough for the growth of rice. Remarkably, low temperatures that occur frequently during the reproductive stage of rice can cause a fatal yield loss (Jena et al. 2012; Shirasawa et al. 2012). Male sterility, arising from low temperature (lower than 19 °C) during the period of microspore development at the booting stage, is the key reason for the reduction of percent seed set and the resulting loss in yield (Satake and Hayase 1970). Therefore, the breeding of cold-tolerant varieties at the booting stage is an effective method to maintain high and stable yields in rice cultivation regions.
The cold tolerance of rice is a complex trait, and many methods have been established to evaluate and select cold-tolerant varieties of rice (Zhang et al. 2014). A cold-water irrigation system has been developed as a reliable identified method of determining cold tolerance at the booting stage of rice. Rice plants are maintained in a cold deep-water irrigated pool during the entire booting stage, and the spikelet fertility was used to examine the cold tolerance of rice varieties (Shirasawa et al. 2012). This method exposes rice plants in field growth conditions, and its accurate evaluation results are still widely used for selecting cold-tolerant lines and developing cold-tolerant rice varieties (Matsunaga 2005; Jia et al. 2015).
Many studies agree that the cold tolerance of rice is controlled by quantitative trait loci (QTLs) (Andaya and Mackill 2003a; Zhang et al. 2005; Suh et al. 2010; Zhang et al. 2014). QTL mapping is the main approach to excavate and clone cold-tolerant related genes in rice. At the booting stage of rice, a number of QTLs about cold tolerance have been reported, including qCTB3 (Andaya and Mackill 2003b), qPSST-3 (Suh et al. 2010) and qLTB3 (Shirasawa et al. 2012) on chromosome 3, Ctb1 (Saito et al. 2004), Ctb2 (Saito et al. 2001), qCTB-4-1, qCTB-4-2 (Xu et al. 2008), and CTB4a (Zhang et al. 2017) on chromosome 4, and qPSST-7 (Suh et al. 2010) and qCTB7 (Zhou et al. 2010) on chromosome 7. For the different rice materials and research backgrounds, QTLs for cold tolerance at the booting stage were mapped at the various locations on different chromosomes. However, the above QTLs identified by the biparental cross linkage mapping method are labor and time intensive to map the genotypes a large number of individuals in the segregated population and to finely map the target QTL (Salvi and Tuberosa 2005).
A bulked segregant analysis (BSA) is a simple and rapid approach to identify molecular markers tightly linked to the target genes or QTLs (Michelmore et al. 1991). With the rapid development of next-generation sequencing (NGS) technology, the combination of the BSA with NGS strategy (Seq-BSA) is becoming a widely used approach in the mapping of major QTLs and gene identification (Wenger et al. 2010; Takagi et al. 2013). This strategy has been demonstrated in many plants, such as Arabidopsis (Schneeberger et al. 2009), rice (Abe et al. 2012; Wambugu et al. 2017), soybean (Song et al. 2017), wheat (Trick et al. 2012), pigeon pea (Singh et al. 2016), sunflower (Livaja et al. 2013) and sorghum (Han et al. 2015), and it has identified some QTLs and genes for important traits. In the present study, two strategies, traditional QTL mapping and Seq-BSA were employed to identify the genes for cold tolerance at the booting stage in recombinant inbred lines (RILs) derived from a cross between a cold sensitive variety, Dongnong422, and a strongly cold-tolerant variety, Kongyu131. The results contribute to the understanding of the genetic bases for cold tolerance at the booting stage, and future cloning of the candidate gene will facilitate the molecular breeding of cold tolerance in rice.
Methods
Plant materials
Two japonica varieties, “Dongnong422” (DN422) and “Kongyu131 (KY131)” were used as parental lines to develop the RIL population. DN422 is a cold-sensitive variety that was obtained from Northeast Agriculture University. KY131, a strongly cold-tolerant variety, is widely cultivated in the northeast region of China. The mapping populations of 190 F7, F8 and F9 RILs were produced by single seed descent (SSD) from an F2 population of a cross between DN422 and KY131.
Cold tolerance evaluation
An evaluation of cold tolerance was performed on the experimental farm of Northeast Agricultural University, Harbin, China (47°98′N, 128°08′E) in 2014, 2015 and 2016. The parents and RIL populations were grown in a randomized block design with two replications of double row plots, a 2-m row length, a 30-cm row spacing and a 10-cm hill spacing. The 190 RILs were divided into three groups, early maturing, middle-maturing and late-maturing groups, according to their heading dates. DN422 belongs to the late-maturing group, and KY131 belongs to the early maturing group. For the evaluation of cold tolerance, each group was irrigated with 17 °C water in an irrigated pool (25 m × 5 m) independently from the panicle initiation stage (approximately 35 days after transplanting for the early maturing group, approximately 40 days after transplanting for the middle-maturing group, and approximately 45 days after transplanting for the late-maturing group) to the full heading stage. When the auricle of the flag leaf is approximately 5 cm below the auricle of the penultimate leaf on each plant, the pollen should have undergone meiosis, which was indicative of panicle initiation stage (Satake and Hayase 1970). Water at 17 °C was prepared in a storage pool with a cool and warm water mixture measured by the temperature sensors. Flood irrigation was performed from the inlet to the outlet (5 m) of the irrigated pool. The depth of the irrigated water was 18–20 cm, and the irrigated time was from 8 a.m. to 16 p.m. every day.
The percent seed set under the cold-water treatment (PSST), the ratio of the number of fertile seeds in the number of total seeds, was used as the index of cold tolerance. A basic statistical analysis was implemented by the SPSS16.0 software (SPSS Inc., Chicago, IL, USA). The mean data of PSST in RILs over the two replications was used for QTL analysis, and the extreme cold-tolerant and sensitive plants in the RIL population used for Seq-BSA were selected according to the mean data of F7, F8 and F9 RILs.
QTL analysis
One hundred eighty-five polymorphic SSR markers between DN422 and KY131 covering the rice genome were used for genotyping the RIL population. A PCR was performed according to the procedure of Chen et al. (1997), and the PCR products were then separated on a 6% polyacrylamide gel, followed by silver staining.
The MAP function of QTL IciMapping 3.2 (Wang et al. 2012) was used to construct the genetic linkage map of the RIL population, and the Kosambi’s mapping function was used to calculate the genetic distances. QTLs were detected using the inclusive composite interval mapping (ICIM) module of QTL IciMapping 3.2. The threshold of the LOD score for declaring the presence of a significant QTL was determined by the permutation test with 1000 repetitions at P < 0.05. The QTL was named according to the trait and its chromosome location.
Construction of segregating pools and sequencing
For Seq-BSA, two DNA pools were developed by selecting the extreme cold-tolerant and extreme cold-sensitive individuals according to the PSST of the RIL population in the range from 0.50–0.95. The tolerant pool (T-pool) was made by mixing equal amounts of DNA from 20 extreme cold-tolerant RILs with a PSST above 0.90, and the sensitive pool (S-pool) was made by mixing equal amounts of DNA from 20 extreme cold-sensitive RILs with a PSST below 0.63 (Additional file 2: Table S1). The DNA isolated from DN422 and KY131 and the two DNA pools were prepared for sequencing.
Libraries for all the DNA pools were prepared according to the Illumina TruSeq DNA sample Preparation v2 Guide. The DNA libraries were sequenced on Illumina MiSeq platform using MiSeq Reagent Kit v2 (500 cycles) (Illumina Inc., San Diego, CA, USA). The short reads from both parents and the two DNA pools were aligned to Nipponbare reference genome (IRGSP 2005) using the BWA software (Li and Durbin 2009). Reads of the T-pool and S-pool were separately aligned to KY131 and DN422 consensus sequence reads to call SNPs with the SAM tools software (Li and Durbin 2009).
Analysis of the Seq-BSA data
According to the locating results of clean reads among the reference genome, duplicate reads were removed using the Picard tool (http://sourceforge.net/projects/picard/), and GATK software (McKenna et al. 2010) was used to perform the local realignment and base recalibration to insure the accuracy of the SNP detecting. The SNP loci between the test samples and reference genome were obtained using the GATK software according to the best practices on the GATK website (https://www.broadinstitute.org/gatk/guide/best-practices.php). All the SNP loci between the test samples were summarized according to the alignment results of test samples and the reference genome.
The Euclidean distance (ED) and SNP-index were calculated to identify the candidate regions of the genome associated with PSST. The ED algorithm is a method of searching markers with significant differences between the pools according to the sequencing data and evaluating the associated regions between markers and traits (Hill et al. 2013).
The calculation formula of the ED algorithm was as follows:
where Aaa, Caa, Gaa and Taa represent the frequency of bases A, C, G and T in the T-pool, respectively. Aab, Cab, Gab and Tab represent the frequency of bases A, C, G and T in the S-pool, respectively. The depth of each base in different pools and the ED value of each SNP loci were calculated. To eliminate the background noise, the ED value was powered, and the ED5 was used as the associated value (Hill et al. 2013).
The SNP-index association analysis is a method used to calculate genotype frequency differences between two pools (Takagi et al. 2013; Fekih et al. 2013). A SNP-index is the proportion of reads harboring the SNP that are different from the reference sequence. The Δ(SNP-index) of each locus was calculated by subtraction of the SNP-index of the T-pool from that of the S-pool. SNP-index = 0 if the entire short reads contain genomic fragments from DN422; SNP-index = 1 if all the short reads were from KY131. The average of the SNP-index was calculated in a 1 Mb interval using a sliding window analysis with 10 kb. The SNP-index graphs of the T-pool, S-pool and corresponding Δ(SNP-index) graphs were plotted. The statistical confidence intervals of Δ(SNP-index) were calculated under the null hypothesis of no QTLs following the description of Takagi et al. (2013).
qRT-PCR analysis of the candidate genes
Two parents, DN422 and KY131, were used to identify the expression patterns of the putative candidate genes. Samples were collected when they reached the panicle initiation stage after cold water irrigation in 2017. Plants with normal temperature water irrigation were used as the control. The leaves of the two parents were collected at 0, 1, 3, 5, 7, 9, 11 and 13 d, for three repetitions. The samples were placed in liquid nitrogen immediately and then stored at − 80 °C for total RNA isolation.
Total RNA was isolated using the Trizol reagent (Invitrogen) according to the manufacturer’s instructions and was purified using the DNA-free RNA kit. The first-strand cDNA was synthesized using the Fermentas RevertAid First Strand cDNA Synthesis Kit. A pair of primers was designed for each candidate gene using the Primer Premier 5.0 software. The housekeeping gene Actin1 (Os05g36290) was used as the internal control (Siahpoosh et al. 2011). The information of the specific primers is provided in Additional file 2: Table S2. The qRT-PCR reactions were performed using a Roche LightCycler 2.10 with a 2 × SYBR Green I PCR Master Mix. The PCR reaction procedure was as follows: 95 °C for 5 min; 45 cycles of 95 °C for 10 s, 60 °C for 20 s, and 72 °C for 20 s. The mean Ct values of all the biological replicates were normalized with the Ct values of Actin1. The relative expression level was calculated using the 2-ΔΔCt method (Livak and Schmittgen 2001), where ΔΔCt = (Ct, target - Ct, actin)time x - (Ct, target - Ct, actin)time 0.
Sequencing of the nsSNP loci in RILs
Genomic DNA of the 190 RILs was extracted using the CTAB method (Murray and Thompson 1980).The eight SNP loci in LOC_Os06g39750 were sequenced to identify the haplotypes of RIL population. Primers for amplifying the SNP were designed using the Primer Premier 5.0 software, and the sequences of specific primers are provided in Additional file 2: Table S3.
Total 20 μL PCR reaction mixture contained 2 μL of genomic DNA (50 ng/μL), 1.5 μL of forward primer (10 μM), 1.5 μL of reverse primer (10 μM), 5 μL ddH2O, and 10 μL of Pfu Master Mix (CWBio, Inc., Beijing, China) including Taq DNA Polymerase, PCR Buffer, Mg2+ and dNTPs. The PCR reaction was carried out in Eppendorf Mastercycler 5333 thermal cycler, and the procedure was same as the above qRT-PCR. The amplified products were checked by electrophoresis in 1% agarose gel. PCR products were directly sequenced by the BGI Life Tech Co., Ltd.
Results
Phenotypic characterization of the cold tolerance in DN422/KY131 RILs
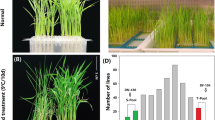
In the present study, two japonica varieties, DN422 and KY131, were crossed to develop the RIL populations for the QTL analysis and the Seq-BSA of cold tolerance. The phenotypic data were collected using the F7, F8 and F9 RILs in 2014, 2015 and 2016, continuously, and the basic statistical analysis of the tested materials is provided in Table 1. Extensive phenotyping data under cold stress and control conditions for heading time, plant height, panicle number, and grain yield per plant were provide in Additional file 2: Table S4 to insights into the cold tolerance of the test materials. The mean PSST values of DN422, KY131 and the RIL population in the 3 years were 0.59, 0.92 and 0.76, respectively, and their mean percent seed set values under normal water irrigation (PSSN) were 0.90, 0.97 and 0.89, respectively. The correlation analysis showed a significantly positive correlation among the PSST in 2014, 2015 and 2016, respectively, and the PSSN had the same pattern (Additional file 2: Table S5). This indicates that the cold-water irrigation was moderate and could differentiate the plants between cold stress and control effectively. The PSST values of the cold-tolerant parent KY131 were extremely significant higher than those of the cold-sensitive DN422 in all 3 years (Fig. 1, Table 1), indicating the stronger cold tolerance of KY131 compared to DN422. Among the RIL populations, the absolute values of skewness and kurtosis for PSST in 3 years were all close to 1 which indicate that the data of PSST were suitable for the QTL analysis (Table 1) (Li et al. 2012).

Panicles of DN422 and KY131 planted under cold water irrigation condition. Scale bars, 3 cm
SSR-based QTL mapping
One thousand SSR markers with a uniform distribution throughout the 12 rice chromosomes were selected to screen the polymorphism between DN422 and KY131. Among them, 158 SSR markers were polymorphic between the two parents and were used to construct the genetic linkage map of the RIL population. The map included 12 chromosomes and covered 2355.3 cM of the rice genome with an average distance of 14.91 cM between markers.
In total, three QTLs for the PSST located on chromosomes 5, 6 and 7 were detected by using the ICIM module of QTL IciMapping 3.2 in 3 years (Table 2). The total phenotypic variation explained (PVE) for all identified QTLs ranged from 9.01 to 47.94%. qPSST5 and qPSST7 were detected in only 1 year, and their PVE were 9.01% and 14.95%, respectively. Their facticity in controlling the PSST of rice is suspicious. Remarkably, qPSST6 in the RM20261-RM20356 interval of chromosome 6 was detected in all 3 years, and the PVE values of the 3 years were 24.30%, 47.94% and 19.11%, respectively, with a mean of 30.45%. In addition, their additive effect values were all negative, indicating that KY131 had a positive effect on increasing the PSST. Thus, qPSST6 is a major QTL for a high PVE and existed stably in the 3 years, suggesting a key role in controlling the PSST of rice. In contrast, six QTLs with small PVE for the PSSN were detected on chromosomes 2, 4, 6, 7, 8, and major QTL was not found (Additional file 2: Table S6).
Sequencing of the parents and extreme pools
Twenty RILs with extreme cold tolerance (PSST ranged from 0.90–0.95) and 20 RILs with extreme cold sensitivity (PSST ranged from 0.50–0.63) were selected to prepare the T-pool and S-pool (Additional file 2: Table S1). Illumina high-throughput sequencing generated 233.62 million raw reads, and 232.30 million clean reads (99.43%) were obtained after filtering. The mean value of Q30 was 91.92%, indicating that most of the bases were high quality. The sequencing depths were 38-fold in DN422, 42-fold in KY131, 31-fold in the T-pool, and 45-fold in the S-pool, which could guarantee the accuracy of the BSA analysis (Additional file 1: Figure S1, Additional file 2: Table S7). There were 336,632 SNPs between DN422 and KY131, which included 36,448 non-synonymous SNPs (nsSNP); there were 184,917 SNPs between the T-pool and the S-pool, which included 18,013 nsSNP. The raw sequencing data were deposited in NCBI (https://www.ncbi.nlm.nih.gov/), and the accession number were SRR6327815, SRR6327816, SRR6327817, SRR6327818 for KY131, DN422, T-pool and S-pool, respectively.
ED and SNP-index analysis
The two analysis methods of Seq-BSA and the ED and SNP-index were used to identify the candidate genome regions associated with PSST. The 184,917 SNPs between the T-pool and the S-pool were used for an association analysis through the two methods. The association threshold of the ED method was 3.82, and six genome regions were significantly correlated with the PSST. The result of the ED association analysis is shown in Fig. 2 and Table 3. The candidate regions had a total size of 23.48 Mb, distributed on chromosomes 4, 5 and 6, and contained 4338 genes with 792 nsSNP loci among them.

Identification of the hot-region for PSST through the ED association analysis method. X-axis represents the position of 12 chromosomes of rice and Y-axis represents the ED. The color dots represent the ED value of every SNP locus. The black lines show the ED value of fitting results, and the red imaginary lines show the association threshold of ED
The SNP-index of the T-pool and S-pool were calculated for each identified SNP in the genome, and an average SNP-index was computed in a 1 Mb interval using a 10-kb sliding window. By combining the information of the SNP-index of the T-pool and S-pool, the Δ(SNP-index) was calculated, and the Δ(SNP-index) trends were visualized by means of a sliding window. Using the association threshold of 0.9532, six genome regions distributed on chromosomes 5 and 6 that were significantly correlated with the PSST were identified. The total length of these regions was 3.68 Mb, and they contained 649 genes with 93 nsSNP loci (Fig. 3, Table 4). By comparing the association results of ED and the SNP-index methods, their intersections of the genome regions were consistent to the association regions of the SNP-index method (Table 4). The 3.68 Mb regions of chromosome 6 by Seq-BSA was the hot region for the PSST of rice.

Identification of the hot-region for PSST through the SNP-index association analysis method. X-axis represents the position of 12 chromosomes of rice and Y-axis represents the SNP-index or Δ (SNP-index). The color dots represent the SNP-index or Δ (SNP-index) value of every SNP locus. The black lines show the SNP-index or Δ (SNP-index) value of fitting results. a: the SNP-index graph of T-pool. b: the SNP-index graph of S-pool. c: the Δ (SNP-index) graph. The red dotted line shows the association threshold value (0.9532)
To narrow the genome region of PSST further, mapping interval of the major QTL qPSST6 was compared to the hot region of Seq-BSA. qPSST6 was detected in the interval between RM20261-RM20356 with a genetic distance of 28.4 cM by using the ICIM module of QTL IciMapping 3.2. RM20261 is located at 21,304,727–21,304,750 bp, and RM20356 is located at 23,654,188–23,654,207 bp on chromosome 6 of the Nipponbare genome. qPSST6 spanned a physical distance of 2.35 Mb and intersected with the hot region of Seq-BSA between 21,840,000 bp and 23,654,188 bp (Fig. 4). Thus, the region containing the PSST locus was narrowed to a region of 1.81 Mb, which contains 269 predicted genes.

Mapping of qPSST6 on chromosome 6 using the methods of QTL analysis and Seq-BSA. a: qPSST6 detected in the RIL population by using the ICIM module of QTL IciMapping 3.2. The LOD scores are shown with three distinct peaks corresponding to qPSST6 in 2014, 2015 and 2016, respectively. b: hot-region for PSST identified by SNP-index method on chromosome 6. The black line shows the Δ (SNP-index) value of fitting results. The red dotted line shows the association threshold value (0.9532), and the black arrows on the x-axis represent the location of hot-region for PSST. c: the intersection of qPSST6 interval identified by QTL analysis and Seq-BSA. The gray region on the chromosome shows the 1.81 Mb intersection of qPSST6
Identification of the candidate genes for PSST
To identify the candidate genes for PSST, the 269 predicted genes were searched in gene ontology (GO) (Ashburner et al. 2000), non-redundant protein (NR) (Deng et al. 2006), Swiss-Prot (Apweiler et al. 2004), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al. 2004) and Cluster of Orthologous Groups of proteins (COG) (Tatusov et al. 2000) databases by the BLAST software (Altschul et al. 1997). The results revealed that among the 269 predicted genes, 212 were successfully annotated. The most enriched terms of biological process ontology were metabolic processes, such as carbohydrate metabolic processes (GO: 0005975), DNA metabolic processes (GO:0006259), cellular macromolecule metabolic processes (GO:0044260), and nucleic acid metabolic processes (GO:0090304). The most enriched terms of molecular function were binding, such as nucleic acid binding (GO:0003676), iron ion binding (GO:0005506), and zinc ion binding, (GO:0008270). The most enriched terms of cellular component were membranes, such as chloroplast outer membranes (GO:0009707), cytoplasmic membrane-bounded vesicles (GO:0016023), and plasma membranes (GO:0005886). All the annotated information is listed in Additional file 2: Table S8. Out of the 212 annotated genes, only two, LOC_Os06g39740 and LOC_Os06g39750, were annotated as the function of “response to cold (GO:0009409)”, suggesting their key roles in regulating cold tolerance in rice. Moreover, 13 predicted genes (4.83%) contained 52 nsSNP loci including three nsSNP in LOC_Os06g39750, but no non-synonymous substitution occurred in LOC_Os06g39740 (Additional file 2: Table S9).
To determine the expression patterns of the two genes annotated as “response to cold” in response to cold stress, a qRT-PCR was performed using the total RNA of the two parents DN422 and KY131 subjected to cold water conditions. Two primers pairs for the qRT-PCR analysis were designed based on the CDS sequences of the two genes (Additional file 2: Table S2). According to the qRT-PCR results (Fig. 5), LOC_Os06g39740 was slightly induced by the cold stress after 3 days in DN422 and after 1 day in KY131. The largest relative expression quantity was nearly 2-fold at day 5 in DN422 and 2.5-fold at day 7 in KY131. The expression patterns of LOC_Os06g39740 have no obvious up or down-regulation for all days at the control conditions in DN422 and KY131. LOC_Os06g39750 exhibits a strong inducement by cold stress at days 5, 7, 9, 11, and 13 after treatment in KY131, and the highest relative expression quantity was above 9-fold at day 9. Compared to KY131, the up-regulated expression of LOC_Os06g39750 under cold stress in DN422 were weak, and the highest relative expression quantity was approximately 2.6-fold at day 7. LOC_Os06g39750 exhibited no obvious regularity in the control condition in both DN422 and KY131. The result of the qRT-PCR suggested that LOC_Os06g39750 was the candidate gene controlling the PSST in rice.

Expression patterns of LOC_Os06g39740 and LOC_Os06g39750 under the conditions of cold water irrigation and CK. Each line with different patterns on the X-axes from left to right indicate the sampling times of 0, 1, 3, 5, 7, 9, 11 and 13 day after cold water irrigating in KY131 and DN422. The Y-axes indicate the relative expression quantity. The error bars indicate SD for the data obtained in three biological replicates
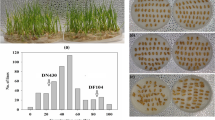
Among the LOC_Os06g39750, eight SNP loci were identified, and they were all in the CDS region of LOC_Os06g39750 (Additional file 2: Table S10). Sequencing of the eight SNP loci in LOC_Os06g39750 revealed that four haplotypes (HapI, HapII, HapIII, and HapIV) existed among the RIL population (Fig. 6, Additional file 2: Table S11). HapI, DN422 genotype, which contains 20 RILs of S-pool and other 24 RILs. HapII, KY131 genotype, which contains 20 RILs of T-pool and other 9 RILs. HapIII contains 92 RILs, which has seven same SNPs to DN422, and only one same SNP to KY131. HapIV contains 25 RILs, which has six same SNPs to DN422, and two same SNPs to KY131. The mean PSST of the 44 HapI and 29 HapII RILs was 0.63 and 0.91, respectively, indicating that RILs with HapII of KY131 genotype usually have a higher mean PSST. Haplotype analysis demonstrate a key role of LOC_Os06g39750 in regulating the PSST of rice once again.

Haplotype distribution of LOC_Os06g39750 in RIL population. Every block represents a RIL, and all RILs are arranged according to their PSST value. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 column represent the RILs with PSST of 0.56~ 0.63, 0.63~ 0.67, 0.68~ 0.70, 0.70~ 0.72, 0.73~ 0.75, 0.75~ 0.77, 0.77~ 0.82, 0.82~ 0.85, 0.85~ 0.90, 0.90~ 0.95 from top block to bottom block, respectively. Yellow, blue, green, and red block represent HapI, HapII, HapIII, and HapIV, respectively
Discussion
Cold stress is one of the major abiotic environmental stresses that significantly affects rice yield. Improving the cold tolerance of rice varieties using cold-tolerant genes is a fast and efficient approach to reduce the yield loss in rice cultivation regions. KY131, a japonica rice, has been widely cultivated in the northeast region of China for nearly 20 years. A prominent characteristic of KY131 is the strong cold tolerance to cope with the frequent low temperatures that occur during the booting stage in this high-latitude region (Yao et al. 2012; Wang et al. 2016). In this study, a RIL population derived from a cross between DN422, a cold sensitive japonica rice, and KY131 was exposed in a cold-water environment over 3 years to acquire the PSST to map cold-tolerant genes. The identification of cold tolerance at the booting stage is more difficult compared to other agronomic traits in rice. Therefore, we divided the RILs into early maturing, middle-maturing and late-maturing groups, and irrigated them independently to evaluate the PSST of the parents and each RIL line. The critical temperature for the cold stress treatment at the booting stage of rice is 17–20 °C (Zhou et al. 2010). The cold-water temperature of 17 °C selected in this study could differentiate the PSST of the two parents significantly, which made the RILs vary widely (Table 1, Fig. 1). All the measures adopted in this study were effective at obtaining accurate PSST phenotypic data.
Conventional methods of fine mapping and map-based cloning of QTLs were very difficult for the development of high-density genetic map and a series of near-isogenic lines, especially the genetic population derived from japonica × japonica or indica × indica crosses. For example, a long time was spent from the preliminary mapping to the cloning of the cold-tolerance gene Ctb1 (Saito et al. 1995; Saito et al. 2001; Saito et al. 2010). Seq-BSA technology provides a powerful, time-saving method to narrow the chromosome interval harboring the target genes/QTLs. Using the strategy of traditional QTL mapping combined with Seq-BSA, Zheng et al. (2016) rapidly mapped and identified a novel broad-spectrum resistance gene to rice blast (Pi65(t)). The Pi65(t) region has been narrowed to 60 Kb, which contains 4 predicted R genes. In the present study, a major QTL qPSST6 that explained 30.45% of the phenotypic variation was initially mapped using the traditional QTL mapping method. Despite the relatively low density of the genetic map containing only 158 SSR markers, the 3 years of phenotypic data of PSST demonstrate a major QTL existing within the 28.4 cM interval between SSR markers RM20261-RM20356. By employing the Seq-BSA method, qPSST6 was quickly delimited to a 1.81 Mb physical interval on chromosome 6 (Fig. 4), and two genes, LOC_Os06g39740 and LOC_Os06g39750, were locked according to their annotated biological function as “responds to cold”. Thus, our study provided a fast and cost-effective strategy to identify the quantitative locus of complex trait variation. Some QTLs about cold tolerance at the booting stage have been reported in recent years. These QTLs were mainly distributed on chromosome 3 (Andaya and Mackill 2003b; Suh et al. 2010; Shirasawa et al. 2012; Zhu et al. 2015; Ulziibat et al. 2016), chromosome 4 (Saito et al. 2004; Saito et al. 2001; Xu et al. 2008; Endo et al. 2016; Zhang et al. 2017), and chromosome 7 (Suh et al. 2010; Zhou et al. 2010). Andaya and Mackill (2003b) and Oh et al. (2004) identified a QTL controlling spikelet fertility (qCTB6) and the days to heading (dth6), respectively, located on chromosome 6, but the two QTLs were approximately 16.4 Mb and 11.9 Mb distant to qPSST6 in this study. Beyond those, we did not find any QTL loci distributed on chromosome 6. Thus, qPSST6 was a new locus for cold tolerance at the booting stage in rice.
Among the 269 predicted genes identified by QTL mapping and Seq-BSA, LOC_Os06g39740 and LOC_Os06g39750 were taken into full consideration as the candidate genes of qPSST6 according to the results of the GO annotation generated by Seq-BSA. The results of the qRT-PCR showed that LOC_Os06g39750 was induced strongly by cold stress in the cold-tolerant parent KY131. Haplotype analysis also demonstrated the RILs with HapII of KY131 genotype usually have a higher mean PSST. These all suggesting that LOC_Os06g39750 was the candidate gene of qPSST6. So far, there has not been any report about LOC_Os06g39750 in rice. By searching the Arabidopsis Information Resource (TAIR, http://www.arabidopsis.org/) database using the gene sequence of LOC_Os06g39750, ten Arabidopsis homologous genes were identified (Additional file 2: Table S12). Eight of the ten genes were the members of the 3-ketoacyl-CoA synthase family involved in the biosynthesis of VLCFA (very long chain fatty acids). Among the ten Arabidopsis homologous genes, At5g43760, At2g26640, At2g16280, At1g25450, At1g01120, and At2g26250 were annotated as functions of “response to cold” in TAIR, indicating that they may participate the regulation of reactions to cold in Arabidopsis. An expression profiling analysis revealed that most of the 3-ketoacyl-CoA synthase (KCS) family genes in Arabidopsis, including the above six genes, responded to cold stress (Joubes et al. 2008). Therefore, it is reasonable to predict that LOC_Os06g39750 is the candidate gene for qPSST6. However, gene clone, genetic transformation, and further studies are needed to functionally validate this conclusion.
Conclusions
The present study using the Dongnong422/Kongyu131 RIL population and a genetic linkage map containing 158 SSR markers identified a novel major QTL qPSST6 for cold tolerance at the booting stage of rice in the 28.4 cM region on chromosome 6. By using the combined Seq-BSA strategy, qPSST6 was narrowed to a region of 1.81 Mb, which contains 269 predicted genes. According to the results of the gene ontology (GO) analysis, LOC_Os06g39740 and LOC_Os06g39750 were annotated as “response to cold”. LOC_Os06g39750 exhibited strong up-regulated expressions at the 5, 7, 9, 11, and 13 day after the cold-water treatment in KY131. Haplotype analysis also demonstrate a key role of LOC_Os06g39750 in regulating the PSST of rice, suggesting that it was the candidate gene of qPSST6. This study provided a fast and cost-effective strategy to identify cold-tolerant genes at the booting stage in rice.
Abbreviations
- BSA:
-
Bulked-segregant analysis
- COG:
-
Cluster of orthologous groups of proteins
- ED:
-
Euclidean distance
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MAS:
-
Marker assisted selection
- NGS:
-
Next-generation sequencing
- NR:
-
Non-redundant protein
- PSSN:
-
Percent seed set under normal water irrigation
- PSST:
-
Percent seed set under cold water treatment
- PVE:
-
Phenotypic variation explained
- QTL:
-
Quantitative trait loci
- RIL:
-
Recombinant inbred line
- S-pool:
-
Sensitive pool
- SSD:
-
Single seed descent
- T-pool:
-
Tolerant pool
References
Abe A, Kosugi S, Yoshida K, natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotech 30:174–178
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Andaya V, Mackill D (2003b) QTLs conferring cold tolerance at the booting stage of rice using recombinant inbred lines from a japonica × indica cross. Theor Appl Genet 106(6):1084–1090
Andaya VC, Mackill DJ (2003a) Mapping of QTLs associated with cold tolerance during the vegetative stage in rice. J Exp Bot 54(392):2579
Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang HZ, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh SL (2004) UniProt: the universal protein knowledgebase. Nucleic Acids Res 32:115–119
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. Nat Genet 25(1):25–29
Chen X, Temnykh S, Xu Y, Cho YG, McCouch SR (1997) Development of a microsatellite framework map providing genome-wide coverage in rice (Oryza sativa L.). Theor Appl Genet 95(4):553–567
Deng YY, Li JQ, Wu SF, Zhu YP, Chen YW, He FC (2006) Integrated nr database in protein annotation system and its localization. Comput Eng 32(5):71–74
Endo T, Chiba B, Wagatsuma K, Saeki K, Ando T, Shomura A, Mizubayashi T, Ueda T, Yamamoto T, Nishio T (2016) Detection of QTLs for cold tolerance of rice cultivar “Kuchum” and effect of QTL pyramiding. Theor Appl Genet 129(3):631
Fekih R, Takagi H, Tamiru M, Abe A, Natsume S, Yaegashi H, Sharma S, Sharma S, Kanzaki H, Matsumura H, Saitoh H, Mitsuoka C, Utsushi H, Saitoh H (2013) MutMap+: genetic mapping and mutant identification without crossing in rice. PLoS One 8(7):e68529
Han YC, Lv P, Hou SL, Li SY, Ji GS, Ma X, Du RH, Liu GQ (2015) Combining next generation sequencing with bulked segregant analysis to fine map a stem moisture locus in Sorghum (Sorghum bicolor L. Moench). PLoS One 10(5):e0127065
Hill JT, Demarest BL, Bisgrove BW, Gorsi B, Su YC, Yost HJ (2013) MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res 23(4):687–697
International Rice Genome Sequencing Project (IRGSP) (2005) The map-based sequence of the rice genome. Nature 436:793–800
Jena KK, Kim SM, Suh JP, Yang CI, Kim YJ (2012) Identification of cold-tolerant breeding lines by quantitative trait loci associated with cold tolerance in rice. Crop Sci 51(2):517–523
Jia Y, Zou DT, Wang JG, Liu HL, Inayat MA, Sha HJ, Zheng HL, Sun J, Zhao HW (2015) Effect of low water temperature at reproductive stage on yield and glutamate metabolism of rice (Oryza sativa L.) in China. Field Crops Res 175:16–25
Joubes J, Raffaele S, Bourdenx B, Garcia C, Laroche-Traineau J, Moreau P, Domergue F, Lessire R (2008) The VLCFA elongase gene family in Arabidopsis thaliana: phylogenetic analysis, 3D modelling and expression profiling. Plant Mol Biol 67:547–566
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori AM (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32:277–280
Li H, Durbin R (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25:1754–1760
Li SH, Wang CS, Chang XP, Jing RL (2012) Genetic dissection of developmental behavior of grain weight in wheat under diverse temperature and water regimes. Genetica 140:393–405
Livaja M, Wang Y, Wieckhorst S, Haseneyer G, Seidel M, Hahn V, Knapp SJ, Taudien S, Schön CC, Bauer E (2013) BSTA: a targeted approach combines bulked segregant analysis with next-generation sequencing and de novo transcriptome assembly for SNP discovery in sunflower. BMC Genomics 14:628
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative pcr and the 2-ΔΔCt method. Methods 25:402–408
Matsunaga K (2005) Establishment of an evaluation method for cold tolerance at the booting stage of rice using deep water irrigation system and development of highly cold-tolerant rice varieties by combining cold tolerance genes. Bull Miyagi Furukawa Agric Exp Stn 4:1–78
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci U S A 88:9828–9832
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucl Acids Res 8:4321–4325
Oh CS, Choi YH, Lee SJ, Yoon DB, Moon HP, Ahn SN (2004) Mapping of quantitative trait loci for cold tolerance in weedy rice. Breeding Sci 54(4):373–380
Saito K, Hayano Y, Kuroki M, Sato Y (2010) Map-based cloning of the rice cold tolerance gene Ctb1. Plant Sci 179:97–102
Saito K, Hayano-Saito Y, Maruyama-Funatsuki W, Sato Y, Kato A (2004) Physical mapping and putative candidate gene identification of a quantitative trait locus Ctb1 for cold tolerance at the booting stage of rice. Theor Appl Genet 109(3):515–522
Saito K, Miura K, Nagano K, Hayano-Saito Y, Araki H, Kato A (2001) Identification of two closely linked quantitative trait loci for cold tolerance on chromosome 4 of rice and their association with anther length. Theor Appl Genet 103(6):862–868
Saito K, Miura K, Nagano K, Hayano-Saito Y, Saito A, Araki H, Kato A (1995) Chromosomal location of quantitative trait loci for cool tolerance at the booting stage in rice variety “Norin-PL8”. Japanese J Breeding 45(3):337–340
Salvi S, Tuberosa R (2005) To clone or not to clone plant QTLs: present and future challenges. Trends Plant Sci 10:297–304
Satake T, Hayase H (1970) Male sterility caused by cooling treatment at the young micro-spore stage in rice plants. V. Estimations of pollen developmental stage and the most sensitive stage to coolness. Proc Crop Sci Soc Jpn 39(4):468–473
Schneeberger K, Ossowski S, Lanz C, Juul T, Petersen AH, Nielsen KL, Jorgensen JE, Weigel D, Andersen SU (2009) SHOREmap: simultaneous mapping and mutation identification by deep sequencing. Nat Methods 6:550–551
Shirasawa S, Endo T, Nakagomi K, Yamaguchi M, Nishio T (2012) Delimitation of a QTL region controlling cold tolerance at booting stage of a cultivar, ‘Lijiangxintuanheigu’, in rice, Oryza sativa L. Theor Appl Genet 124(5):937–946
Siahpoosh MR, Sanchez DH, Schlereth A, Scofield GN, Furbank RT, Dongen JT, Kopka J (2011) Modification of OsSUT1 gene expression modulates the salt response of rice Oryza sativa cv. Taipei 309. Plant Sci 182:101–111
Singh VK, Khan AW, Saxena RK, Kumar V, Kale SM, Sinha P, Chitikineni A, Pazhamala LT, Garg V, Sharma M, Kumar CVS, Parupalli S, Vechalapu S, Patil S, Muniswamy S, Ghanta A, Yamini KN, Dharmaraj PS, Varshney RK (2016) Next-generation sequencing for identification of candidate genes for fusarium wilt and sterility mosaic disease in pigeonpea (Cajanus cajan). Plant Biotech J 14(5):1183–1194
Song J, Li Z, Liu Z, Guo Y, Qiu LJ (2017) Next-generation sequencing from bulked-segregant analysis accelerates the simultaneous identification of two qualitative genes in soybean. Frontiers Plant Sci 8. https://doi.org/10.3389/fpls.2017.00919
Suh JP, Jeung JU, Lee JI, Choi YH, Yea JD, Virk PS, Mackill DJ, Jena KK (2010) Identification and analysis of QTLs controlling cold tolerance at the reproductive stage and validation of effective QTLs in cold-tolerant genotypes of rice (Oryza sativa L.). Theor Appl Genet 120:985–995
Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, Uemura A, Utsushi H, Tamiru M, Takuno S, Innan H, Cano LM, Kamoun S, Terauchi R (2013) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. The. Plant J 74(1):174–183
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36
Trick M, Adamski NM, Mugford SG, Jiang CC, Febrer M, Uauy C (2012) Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol 12:14
Ulziibat B, Ohta H, Fukushima A, Shirasawa S, Kitashiba H, Nishio T (2016) Examination of candidates for the gene of cold tolerance at the booting stage in a delimited QTL region in rice cultivar ‘Lijiangxintuanheigu’. Euphytica 211(3):331–341
Wambugu P, Ndjiondjop MN, Furtado A, Henry R (2017) Sequencing of bulks of segregants allows dissection of genetic control of amylose content in rice. Plant Biotech J. https://doi.org/10.1111/pbi.12752
Wang D, Liu JL, Li CG, Kang HX, Wang Y, Tan XQ, Liu MH, Deng YF, Wang ZL, Liu Y, Zhang DY, Xiao YH, Wang GL (2016) Genome-wide association mapping of cold tolerance genes at the seedling stage in rice. Rice 9(1):61
Wang LZ, Wang LM, Xiang HT, Luo Y, Li R, Li ZJ, Wang CY, Meng Y (2016) Relationship of photosynthetic efficiency and seed-setting rate in two contrasting rice cultivars under chilling stress. Photosynthetica 54(4):581–588
Wenger WJ, Schwartz K, Sherlock G (2010) Bulk segregant analysis by high-throughput sequencing reveals a novel xylose utilization gene from Saccharomyces cerevisiae. PLoS Genet 6:e1000942
Xu LM, Zhou L, Zeng YW, Wang FM, Zhang HL, Shen SQ, Li ZC (2008) Identification and mapping of quantitative trait loci for cold tolerance at the booting stage in a japonica rice near-isogenic line. Plant Sci 174(3):340–347
Yao Y, Miao Y, Huang S, Gao L, Ma X, Zhao G, Gnyp ML (2012) Active canopy sensor-based precision N management strategy for rice. Agron Sustain Dev 32(4):925–933
Zhang Q, Chen QH, Wang SL, Hong YH, Wang ZL (2014) Rice and cold stress: methods for its evaluation and summary of cold tolerance-related quantitative trait loci. Rice 7:24
Zhang ZH, Su L, Li W, Chen W, Zhu YG (2005) A major QTL conferring cold tolerance at the early seedling stage using recombinant inbred lines of rice (Oryza sativa L.). Plant Sci 168(2):527–534
Zhang ZY, Li JJ, Pan YH, Li JL, Zhou L, Shi HL, Zeng YW, Guo HF, Yang SM, Zheng WW, Yu JP, Sun XM, Li GL, Ding YL, Ma L, Shen SQ, Dai LY, Zhang HL, Yang SH, Guo Y, Li ZC (2017) Natural variation in CTB4a enhances rice adaptation to cold habitats. Nat Commun 8:14788
Zheng W, Wang Y, Wang L, Ma ZB, Zhao JM, Wang P, Zhang LX, Liu ZH, Lu XC (2016) Genetic mapping and molecular marker development for Pi65(t), a novel broad-spectrum resistance gene to rice blast using next-generation sequencing. Theor Appl Genet 129(5):1035
Zhou L, Zeng YW, Zheng WW, Tang B, Yang SM, Zhang HL, Li JJ, Li ZC (2010) Fine mapping a QTL qCTB7 for cold tolerance at the booting stage on rice chromosome 7 using a near-isogenic line. Theor Appl Genet 121(5):895
Zhu Y, Chen K, Mi X, Chen T, Ali J, Ye G, Xu JL, Li ZK (2015) Identification and fine mapping of a stably expressed QTL for cold tolerance at the booting stage using an interconnected breeding population in rice. PLoS One 10(12):e0145704
Acknowledgments
We would like to thank the “Young Talents” Project of Northeast Agricultural University for its research funding support, and the National Innovation Center of Regional Technology in Rice for the laboratory and field support.
Funding
This work was supported by the “Young Talents” Project of Northeast Agricultural University (16QC03), National Natural Science Foundation (31701507), Youth Science Foundation of Heilongjiang Province (QC2017015), Major Scientific and Technological Bidding Projects of Heilongjiang Province (GA14B102–02).
Availability of data and materials
All relevant data are provided in Tables and in Supplementary Tables. The raw sequencing data are deposited in NCBI (https://www.ncbi.nlm.nih.gov/).
Author information
Authors and Affiliations
Contributions
SJ and ZDT designed research; YLM performed QTL mapping; WJG analyzed BSA sequence data; LHL, ZHL, and JY irrigated the cold water and got the phenotyping data; XDW and ZMH performed qRT-PCR analysis, FMF analyzed candidate genes; ZHW prepared the T-pool and S-pool; SJ wrote manuscript; ZDT corrected manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Distributions of coverage depth on chromosomes of the sequencing samples. X-axe indicates the 12 chromosomes of rice. Y-axe indicates the log2 value of coverage depth corresponding to chromosome location. (DOC 1130 kb)
Additional file 2:
Table S1. The PSST of T-pool and S-pool. Table S2. Sequences of specific primers for PSST candidate genes. Table S3. Sequences of specific primers for SNP loci in LOC_Os06g39750. Table S4. Heading time, plant height, panicle number, and grain yield per plant of parents and RIL population in 2014, 2015 and 2016. Table S5. Coefficients of correlation (r) among PSST and PSSN in 2014, 2015 and 2016. Table S6. QTLs for PSSN of rice detected in 3 years. Table S7. Summary of Seq-BSA data for each sample. Table S8. Annotation information of predicted genes for PSST. Table S9. nsSNPs between DN422 and KY131 along with their S-pool and T-pool. Table S10. Information of SNP/InDel in LOC_Os06g39740 and LOC_Os06g39750. Table S11. Haplotype of LOC_Os06g39750 in RIL population and mean PSST of each RIL. Table S12. The Arabidopsis homologous genes of LOC_Os06g39750. (XLS 174 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sun, J., Yang, L., Wang, J. et al. Identification of a cold-tolerant locus in rice (Oryza sativa L.) using bulked segregant analysis with a next-generation sequencing strategy. Rice 11, 24 (2018). https://doi.org/10.1186/s12284-018-0218-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-018-0218-1