
Abstract
Mitochondrial diseases (MD) are a heterogeneous group of multisystem disorders involving metabolic errors. MD are characterized by extremely heterogeneous symptoms, ranging from organ-specific to multisystem dysfunction with different clinical courses. Most primary MD are autosomal recessive but maternal inheritance (from mtDNA), autosomal dominant, and X-linked inheritance is also known. Mitochondria are unique energy-generating cellular organelles designed to survive and contain their own unique genetic coding material, a circular mtDNA fragment of approximately 16,000 base pairs. The mitochondrial genetic system incorporates closely interacting bi-genomic factors encoded by the nuclear and mitochondrial genomes. Understanding the dynamics of mitochondrial genetics supporting mitochondrial biogenesis is especially important for the development of strategies for the treatment of rare and difficult-to-diagnose diseases. Gene therapy is one of the methods for correcting mitochondrial disorders.
Graphical Abstract

Similar content being viewed by others

Introduction
MD—is a heterogeneous group of disorders, caused by mutations in genes, the role of which is crucial for mitochondrial physiology, and for function of the electron transport chain (ETC) and oxidative phosphorylation (OXPHOS), in particular. Causative mutations in MD can occur both in nuclear (n) or mitochondrial (mt) DNA and thus, with exception of rare cases of non-inherited mutations occurring de novo, MD are characterized by monogenic autosomal, X-linked or maternal inheritance [1]. As mitochondria play a key role in metabolism and are involved in a wide spectrum of cellular functions, the mitochondrial dysfunction leads to both tissue-specific and systemic disorders [2]. MD can affect almost all types of tissues, to greater extent the nervous, muscle and retinal [3].
There are still not so many effective ways to enhance mitochondrial function in terms of evidence-based medicine. Currently, drugs targeting MD are primarily antioxidant or metabolic therapy [4], e.g. dichloroacetate (DCA), arginine, coenzyme Q10, idebenone, etc. and their efficiency seems to be very modest [5,6,7].
Gene therapy (GT) is one of the revolutionary strategies for curing hereditary diseases on which the greatest hopes are so far pinned in this century. The mind-boggling successes shown by the GT drugs for the treatment of spinal muscular atrophy and Leber's optic atrophy are truly impressive and give a reason to hope for further progress. Of special interest is the rapid development of genome-editing systems, particularly Clustered regularly interspaced short palindromic repeats (CRISPR) / CRISPR-associated protein (Cas), which make it possible to precisely repair unwanted mutations [8].
However, because of some aspects of mitochondrial biology, there is a row of stumbling blocks on the way of GT applications in MD. In this review, we focused on the current status including success and difficulties in GT of MD. We critically discuss the possibilities and difficulties of using genome editing systems. Most of all we focused on GT approaches already tested in vivo.
Basic mitochondrial biology
Mitochondria are organelles responsible for multiple functions in the cells, which consist of two membranes—outer and inner. The inner mitochondrial membrane, containing anchored complexes of the ETC, is the main structure for the mitochondrial respiration and maintenance of the mitochondrial transmembrane potential. Mitochondrial membrane potential is crucial for regulating cellular life and death processes and also serves as a proton motive force in OXPHOS, which generates ATP [9]. Mitochondrial matrix contains cyclic mitochondrial DNA (mtDNA). The transcription and translation of mtDNA differs from the nuclear system and requires more than 100 nuclear-encoded proteins transported into mitochondria by a complex protein transport system [10, 11]. Mitochondria are signaling hubs that integrate the catabolic and anabolic metabolism and regulate cell growth, differentiation, vitality, and death [12]. Mitochondria cannot be synthesised de novo; therefore, they are subjected to constant quality control and regeneration through budding of mitochondrial-derived vesicles and mitophagy [13, 14]. Another important mechanism maintaining stable state of mitochondria is mitochondrial fission and fusion. In brief, mitochondrial fission is essential for growing and dividing cells to populate them with adequate numbers of mitochondria. On the other hand, mitochondrial fusion enables partly impaired mitochondria to physically join together and utilize each other’s protein machinery compensating their own limitations (so called cross-complementation) and maximizing oxidative capacity in response to stress [15].
The mitochondrial genetic system incorporates closely interacting bi-genomic factors encoded by the nuclear and mitochondrial genomes. Genes involved in mitochondrial function are unevenly distributed between the nuclear and mitochondrial genome. Dramatic shift manifests itself in about 1,500 nuclear and just 37 mitochondrial genes. Moreover, circular mtDNA does not contain introns and makes up 1% of the whole cellular DNA. At the same time, mammalian mtDNA is present in thousands of copies per cell and it is characterized by high levels of heterogeneity. It encodes mRNAs for 13 polypeptides of the OXPHOS system, as well as 2 rRNAs and 22 tRNAs for their translation [16]. Four out of five complexes of the ETC and OXPHOS (I, III, IV & V) are composed of subunits, encoded both by the nuclear and mtDNA, while only one (II) is encoded solely by nuclear genome [17].
The mitochondrial genome has a higher mutation rate (about 100–1000 fold), than the nuclear genome [18]. The pathological mutations of mitochondria are survived due to inter-mitochondrial exchange of nutrients and functional complementation taking place through mitochondrial fusion [19, 20]. It leads to heterogeneity of population of mtDNA in single cell or mitochondria and as a result mitochondria become heteroplasmic. There are ~ 1,000 mtDNA molecules in a cell, and in the case of mutations the wild-type mtDNA can compensate for the presence of mutant mtDNA, up to threshold levels, which are usually relatively high, 70–95 [17, 21,22,23].
Clinical features of MD
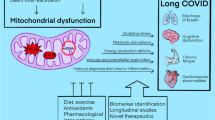
Mitochondrial and nuclear mutations lead to ~ 40 clinical forms of mitochondrial diseases (MD) with clear molecular-genetical and biochemical dysfunction of mitochondria. More than 30 of these MD have a mutation in nuclear DNA and more than 10 syndromes and diseases induced by mutations in mtDNA [24]. MD usually affects children, but the age of MD onset, as well as patient’s lifetime, varies a lot. The most common clinical signs of MD include neurological (epilepsy, ataxia, cognitive deficits), sensory (hearing loss, blindness) and muscular (myopathy) symptoms, but the clinical picture can be very different, manifesting in pathology of many other organs (diabetes, liver disease, kidney dysfunction, infertility, arrythmias etc.) (Fig. 1). Some symptoms of MD are prone to manifest together which allows to unite them into syndromes, such as MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes) or MIDD (maternally inherited diabetes and deafness). Some constellations of symptoms and signs are caused by mutations in different genes and, vice versa, the same mutations can cause different syndromes. Most often, in nearly one-third of the cases, MD are caused by disturbances of the respiratory chain complex I [25,26,27].

Overview of the MD in the clinical and pharmacological contexts. The patients with MD are characterized by involvement of different organs and tissues which results on various symptoms and their combinations. Of the most typical clinical signs are neurological, sensory and muscular symptoms. All these symptoms are caused by defective work of mitochondrial respiration as a result of mutations in nDNA or mtDNA. Classical pharmacological methods to compensate inadequate functioning of mitochondria rely on reinforcement of mitochondrial metabolic cascades and reduce of toxic agents such as lactate and ROS. For instance, thiamine, lipoic acid and dichloracetate are shown to activate pyruvate dehydrogenase resulting in decrease of lactate accumulation due to turning pyruvate into another metabolite acetyl-CoA. Succinate, riboflavin, and CoQ10 promote ETC donating electrons or restoring the function of complexes I and II. Some compounds such as idebenone, N-acetylcysteine and lipoic acid have the ability to reduce ROS production or inactivate them
Standard treatment options for MD
With a limited base of evidence and little data from randomized trials, the treatment of MD remains anecdotal. Moreover, of prescribed treatments the most are considered medical foods [5]. Interventions are mostly focused on vitamin-based and cofactor-based mitochondrial therapies intended to promote critical enzymatic reactions, reduce oxidative stress, and scavenge toxic acyl coenzyme A (acyl CoA) molecules, which accumulate in mitochondrial disease [28]. Using cocktails of vitamins and co-factors is more justified when factors in question are decreased either due to deficiency or defect in their transport [29,30,31]. Additionally, patients struggling with MD are recommended to diet and lifestyle changes [32, 33].
However, only the small cohorts of patients respond to this treatment since all the methods relying on metabolic therapy are poorly effective because of their non-selectiveness, low mitochondrial hitting and low overlap of their action with disease-causing mechanisms [34, 35]. Standard pharmacological interventions are reviewed in [5, 28, 35]. Some of them are presented in the Fig. 1.
Methods of gene therapy (GT)
The idea to treat genetic disorders on the genome level as an alternative to the classical pharmacotherapy has been prospering for decades [36]. Actually, it began in the 60s of the previous century, just after several discoveries showed that foreign DNA could be inserted and expressed into mammalian cells [37, 38]. These findings opened up brand-new avenues for medicine, finally resulting in the development of the conception of GT.
GT is “the introduction, removal, or change in the content of a person’s genetic code with the goal of treating or curing a disease” (according to American Society of Gene and Cell Therapy definition) [39]. Classically, GT is based on the delivery of the correct copy of a gene to compensate for the function of the mutated one. It can be either ex vivo treatment of cells (usually for blood cells) collected from patient or in vivo interventions used as systemic or local administration of GT agents.
Physically GT drugs might be viral (adenoviruses, adeno-associated viruses) or non-viral (plasmids, biomaterial particle-based delivery vectors) vectors, containing functional DNA/RNA sequence [40]. Depending on GT strategy these sequences may result in different effects in the cell, to wit: (1) provide the proper version of mutated gene replacing it functionally; (2) modify expression of the mutated gene; (3) change the original sequence of the mutated gene to improve its function.
Replacement therapy is probably most straightforward method of treating monogenic disorders. Once in the cell, the exogenous copy of the correct gene becomes the template for the synthesis of the correct protein, resulting in disease attenuation. Unfortunately, for some monogenic diseases, GT may not be as simple as gene replacement for the disease-causing gene because of dominant disease traits, large gene sizes and immune rejection are a few of the challenges that face gene replacement. In this respect some approaches rely on the use of siRNA (small interfering RNA), shRNA (short hairpin RNA) and miRNA (microRNA) silencing the mutated gene by altering its expression on mRNA level. The last strategy is known as antisense therapy but it is appliable only when causative mutation is displayed as dominant negative or gain-of-function [41].
However, as a matter of fact, the silver bullet for the treatment of monogenic diseases is GT, based on genome editing technologies. It includes approaches based on gene repair involving the correction of an existing mutation to restore the expression of the correct version of the protein [42], Sheila [43]. For precise editing of genome, a few technologies were proposed. Probably the most promising advances in this field are related to CRISPR (Clustered Regularly-Interspaced Short Palindromic Repeats)-Cas9 (CRISPR associated protein 9), the system, which discovery was awarded by 2020 Nobel prize in chemistry. Several CRISPR/Cas9 based in vivo interventions have already reached the first clinical trials [44, 45].
Zinc fingers nucleases (ZFN) and transcription activator-like effector nucleases (TALEN)
The programmable nucleases ZFN and TALEN contain a variable DNA-binding amino acid sequence and a constant endonuclease domain [46, 47]. Changing the DNA-binding domain by genetic engineering methods, one may target ZFN and TALEN to different regions of the genome, in which nuclease creates double-stranded breaks. To repair these breaks the cell recruits standard DNA repair systems, resulting in homological or non-homological recombination. ZFN and TALEN are recognized as the most efficient genome-editing systems, more precise than CRISPR/Cas9 [48, 49]. However, application of these systems is complicated by difficulties with design of target specific DNA-binding domains: adjacent amino acids influence each other, and the result of this interaction is difficult to predict.
CRISPR/Cas9
In contrast to ZFN and TALEN, the CRISPR/Cas9 system implements the RNA-mediated recognition of the target DNA. In brief, Cas (Cas9 or others) nucleases are guided by single guide RNA (sgRNA) sequence responsible for specific complementary recognition of target DNA. Endonuclease Cas9 generates a double-stranded break in the DNA molecule, 3–4 nucleotides before motif adjacent to the protospacer (pam-site) [50]. Some modified forms of Cas9 are able to introduce break in only one of the two strands.
Although CRISPR/Cas9 generates more off-target events than TALEN and ZFN, the accuracy of CRISPR/Cas rapidly improves, giving rise to hopes for high clinical efficacy and safety in the future [51].
Detailed description of the ZFN, TALEN and CRISPR/Cas systems can be found in [42, 52].
GT in the focus of MD
Because of some aspects of mitochondrial biology GT of MD is challenging. First of all, mitochondria are characterized by complicated permeability to all non-mitochondrial proteins, RNA and DNA. Besides that, mitochondrial systems of DNA replication and reparation and even genetic code are different from nuclear ones. Altogether, these hallmarks may not influence the application of GT in MD caused by mutations of nDNA, but trouble treatment of diseases associated with mutations of the mitochondrial genome. Moreover, a cell may contain thousands of mitochondria, each with its own multiple DNA copies and regulatory circuits of gene expression. Finally, for the effective mitochondrial targeting GT-vector must be delivered to all affected tissues. Below we attempt to consider all of these obstacles and current advances on the way of their overcoming.
Delivery
As mentioned above, the brain, retina and skeletal muscles are the organs most often affected in MD. Multisystem damage can also involve the liver, gastrointestinal tract, pancreas, kidneys, and others. Generation of new GT approach should solve the problems of delivery to these target tissues. Both viral and non-viral delivery systems have been proposed for MD GT. The advantages of non-viral vectors are attracting many researchers to explore the promising delivery system among polymers, lipids, peptides, inorganic materials, and their combinations. The range of application of non-viral vectors is wider due to the possibility of delivering both coding genetic constructs, short, modified oligonucleotides, and large genome editing complexes.
Biodegradable particles demonstrate a perfect safety profile and give the opportunity for repeated delivery. Noncationic liposomes, amphiphile carriers, dequalinium-based liposome-like vesicles (DQAsomes) and others were used for mitochondria targeting in vitro [53, 54]. Current research is focused on increasing transfection efficiency while lowering cytotoxicity [55].
The most popular viral vectors are recombinant adeno-associated viruses (rAAV). They are used in a majority of preclinical and clinical trials for genetic diseases including MD due to their safety profile and widespread biodistribution [56, 57]. Different natural and engineered AAV serotypes effectively deliver DNA constructs into the target tissues and provide long-term expression with a low rate of integration events. Among all known for the time AAV vectors AAV2, AAV5, AAV6, AAV9, AAVrh.10, AAVrh.74 are the most popular serotypes for gene transfer into the brain, eye and skeletal muscles. A detailed review of AAV tropism discovered in NHP studies is presented in Table 1. Natural tropism of viral vectors allows to use of systemic delivery to reach the target site. But other routes of administration that are restricted to the damaged organs become even more popular. Recent research revealed that intracerebrospinal injections of AAV9 or AAVrh10 lead to 10–100 times higher brain expression level than after intravenous injection [58]. Direct gene transfer to the eye during intravitreal (IVT), suprachoroidal (SC), or subretinal (SR) injections demonstrate robust transgene expression in target cells. SR administration was most efficient in targeting the retina and photoreceptors [59]. Percutaneous transendocardial delivery helps to transduce heart tissue by various AAV serotypes [60, 61]. Additional technologies can promote transduction. For example, MRI-guided focused ultrasound (MRIgFUS) helps AAV6 to pass BBB and transduce neuronal cells without accumulation in the liver [62]. New serotypes of AAV are intensively tested in preclinical trials. They are designed to increase the specificity and effectiveness of delivery. The rapid evolution of AAV9 resulted in new variants AAV-PHP.eB and 9P03-33 with enhanced tropism to the central nervous system and lowered liver transduction [63, 64].
Advances in AAV-vector based therapy of MD are summarized in the recent review [65].
Mitochondrial transfer of GT components
Deficiency in mitochondrial proteins or RNA could be compensated only by direct mitochondrial targeting of gene therapeutic agents. Considering that, inner membrane of mitochondria is mainly impermeable and amino acids or nucleotide resides should be delivered to the matrix of mitochondria using special natural or artificial transporters.
Proteins
Mitochondrial proteins, encoded in the nuclear genome, could be transferred to the mitochondria by N-terminal presequences that serve as mitochondrial targeting sequences (MTSs) and are cleaved upon import [85, 86]. These sequences form amphipathic helices that vary largely in primary sequence but are characterized by a length of about 15–60 residues, a net charge of + 3 to + 6, the absence of negatively charged residues and a high content of hydroxylated amino acids [86, 87]. Rational design methods have also been utilized for the creation of new viral capsids with penetrating peptides or mitochondria localization signals [88, 89].
RNA
Mitochondrial import of RNA is a sophisticated and not entirely clear process. Mammals harbor 3 specific types of non-coding RNA transferring into mitochondria after synthesis in the nucleus [90]. In addition, some authors [91, 92] reproduced the mechanism of mitochondrial transfer of tRNA in mammals which was previously identified only for yeasts [93]. This yeast-specific translocation of tRNA is mediated by a protein-import-like mechanism and its crucial components are F- and D-hairpins in RNA structure [94].
The ability for transfer other types of RNA into mitochondria is still under question. mRNA can only be localized on the outer layer [95, 96], but does not pass through the mitochondrial membrane. This surface mitochondrial localization is guided by specific mitochondrial targeting sequences in 5ʹ prime and 3’UTR. Being translated from mitochondrial surface-anchored mRNA, mitochondrial proteins are immediately transported into mitochondria. This was shown, in particular, for the expressed in the nucleus mitochondrial ATP6 gene (provided with MTS) and the 3′UTR of the nuclear SOD2 (superoxide dismutase 2) gene 3′UTR of [97].
Most studies of mitochondrial RNA transfer conclude that RNA can be translocated only by engaging protein import mechanisms [98]. In this way, after extramitochondrial assembly the ribonucleoprotein complex can be translocated into mitochondria [99]. In terms of GT of MD, this opens avenues for mitochondrial delivery of RNA-based therapeutic agents such as CRISPR/Cas or antireplicative machines such as FD-RNA (see below).
DNA
Several strategies of direct mitochondrial gene delivery were proposed. This might be DNA delivery by MTS-harbouring vectors, modified delivery conditions, reversed charge, etc. [100]. For instance, the use of mitochondrially-targeted AAV was shown to be effective in the mitochondrial transfer of mutant human ND6 gene in murine mitochondria [101].
Another interesting approach for targeting mitochondria was proposed by Yasuzaki et al. In the procedure, called “hydrodynamic injection”, a large volume of naked plasmid DNA is rapidly injected, resulting in plasmids-to-mitochondrial transfer [102].
Yamada et al. described “MITO-Porter”, a liposome-based carrier that introduces cargos into mitochondria via a membrane fusion mechanism. The system consists of high-density octaarginine-modified liposomes which can escape from macropinosomes efficiently to the cytosol keeping the encapsulated compounds intact. Upon release from the macropinosomes, MITO-Porter then binds to the mitochondrial membrane via electrostatic interactions, which induce fusion between the MITO-Porter and the mitochondrion [103]. MITO-Porter-based systems were shown to be effective in the mitochondrial transfer of DNA as well as for proteins and RNA[104,105,106,107,107].
Detailed description of the advances in mitochondria-targeted therapeutics delivery is presented in [53, 54, 108].
GT of diseases caused by nDNA mutations
In general, GT of MD caused by nDNA mutations completely corresponds GT strategies used in the treatment of not-mitochondrial monogenic diseases. In the case of these disorders there is no need to overcome difficulties determined by peculiarities of mitochondrial biology such as low mitochondrial transfer of GT-components, mismatch of genetic code between mitochondrion and nucleus and others (the problems with mtDNA are discussed in the "GT of diseases caused by mtDNA mutations" section in details).
The most prevalent strategy for the treatment of MD, associated with altered nucleus-encoded genes is the simple transport of copies of proper DNA into the affected cells. Fortunately, there are various animal models of MD [109] in which such an approach was tested (summarized in Table 1).
TYMP
Mitochondrial neurogastrointestinal encephalopathy (MNGIE) syndrome is a rare autosomal recessive MD, caused by mutation in TYMP, the nuclear gene encoding the enzyme thymidine phosphorylase (TP) [110]. In patients, TP dysfunction causes accumulation of the nucleosides—thymidine and deoxyuridine, which interferes with mtDNA replication [111, 112].
In MNGIE murine model with double Tymp/Upp1 knockout, Torres-Torronteras et al. demonstrated, that administration of haematopoietic donor cells with TYMP coding sequence transduced by lentiviral vector, allows to restore TP activity and to decrease metabolic abnormalities [113]. Later, the same group evaluated in vivo approaches of TYMP replacement therapy via systemic administration of AAV containing the TYMP coding sequence transcriptionally targeted to the liver [114, 115]. In both studies, AAV-mediated GT appeared to be effective in the correction of biochemical abnormalities.
Ferran Vila-Julià et al. enhanced clinical phenotype in the MNGIE mice by administration of thymidine and deoxyuridine and then tested several doses of AAV8 vector carrying the human TYMP coding sequence under the control of different liver-specific promoters (TBG, AAT, or HLP). In this study different gene therapeutic agents ameliorated both biochemical and clinical phenotype in a dose-dependent manner [116].
Slc25a46
In another study Li Yang et al. tested GT in mice carrying a knockout of the mitochondrial fusion-fission-related gene solute carrier family 25 member 46 (Slc25a46). They showed AAV-Slc25a46 treatment was able to rescue the premature death in the Slc25a46-/- mice, restoring mitochondrial complex activities, normalizing mitochondrial morphology, and improving electrical conductivity of the nervous system, as well as attenuating neurodegeneration and neuroinflammation [117].
OPA1
Several successful gene therapeutic approaches based on AAV-mediated delivery of OPA1 gene were proposed. OPA1 encodes a mitochondrial enzyme called dynamin-like 120 kDa protein. This is a large mitochondrial GTPase with crucial roles in membrane dynamics and cell survival [118]. OPA1 mutations affect mitochondrial fusion, energy metabolism, control of apoptosis, calcium clearance and maintenance of mitochondrial genome integrity. OPA1 mutations can cause dominant Optic Atrophy (DOA), a neuro-ophthalmic condition characterized by bilateral degeneration of the optic nerves, causing insidious visual loss, typically starting during the first decade of life [119, 120].
In a murine model of DOA, caused by mutation c.2708_2711delTTAG in Opa1 gene, IVT injection of AAV2 carrying the human OPA1 cDNA under the control of the cytomegalovirus promoter resulted in the prevention of retinal degeneration [121].
OPA1 GT was also assessed in non-genetic models. Rotenone is a lipophilic, naturally occurring compound acting as a strong inhibitor of complex I of the mitochondrial respiratory chain [122]. Using the murine model of rotenone-induced optic neuropathy, Maloney et al. the reported therapeutic benefit of OPA1 isoform 1 and 7 delivered by AAV-2 via IVT route [123].
In another study AAV-mediated OPA1 delivery was used to ameliorate retinal damage caused by ischemia–reperfusion (I/R). AAV–Opa1-ΔS1, IVT injected 25 days before retinal I/R injury, significantly prevented I/R-induced retinal thinning and the cell loss in the ganglion cells layer, reducing levels of markers of apoptosis and necrosis [124].
Finally, AAV-mediated delivery of OPA1 was tested to attenuate neurological condition in a rat model of cerebral focal I/R. It was shown that intracranial injection of AAV-OPA1-v1ΔS1 markedly improved cerebral I/R-induced motor function damage, attenuated brain infarct volume, neuronal apoptosis, mitochondrial bioenergetics deficits, oxidative stress, and restored the morphology of mitochondrial cristae and mitochondrial length. It also preserved the mitochondrial integrity and reinforced the mtDNA content and expression of mitochondrial biogenesis factors in ischemic rats [125].
NDUFS4
Mutations in NDUFS4 gene, encoding NADH dehydrogenase (ubiquinone) iron-sulfur protein 4, result in compromised activity of mitochondrial complex I, causing Leigh syndrome- the most common infantile mitochondrial encephalopathy [126]. Several studies have shown that administration of human NDUFS4 coding sequence by AAV2/9 and/or AAV-PHP.B vectors improved clinical phenotype and prolonged the lifespan in Ndufs4 − / − mice representing a model of Leigh syndrome [127,128,129].
Fdxr
Li Yang et al. have utilized a mouse model carrying a p.Arg389Gln mutation of the mitochondrial Ferredoxin Reductase gene (Fdxr) and used it to test neurotropic AAV-PHP.B vector loaded with the mouse Fdxr cDNA sequence. They observed that the AAV vector was effectively transduced in the central nervous system and all peripheral organs, and AAV-Fdxr treatment reversed almost all the symptoms of the mutants. This therapy improved the electrical conductivity of the sciatic nerves, prevented optic nerve atrophy, improved mobility, and restored mitochondrial complex function [130].
ETHE1
Ethylmalonic encephalopathy (EE) is a severe monogenic disorder caused by mutations of the nuclear gene ETHE1, encoding a ubiquitous mitochondrial protein sulfur dioxygenase (SDO) (note: EE is not classified as MD) [131], involved in the detoxification of H2S [132, 133] showed that AAV2/8-mediated, ETHE1-gene transfer to the liver of a genetically, metabolically, and clinically faithful EE mouse model resulted in the full restoration of SDO activity, correction of plasma thiosulfate, a biomarker reflecting the accumulation of H2S, and spectacular clinical improvement [133].
Descriptions of GT approaches based on the replacement of mutated mitochondrial proteins encoded in the nucleus are summarized in Table 2.
TK2
Autosomal recessive human thymidine kinase 2 (TK2) mutations cause TK2 deficiency, which typically manifests as a progressive and fatal mitochondrial myopathy in infants and children (note: not classified as MD). Normally, TK2 localizes to the mitochondria and specifically phosphorylates thymidine, deoxycytidine, and deoxyuridine, required for mtDNA synthesis. Decrease in TK2 function leads to mtDNA depletion resulting in mitochondrial dysfunction [134].
Krishnan S. et al. showed attenuation of mtDNA depletion in TK2-deficient mice transgenic with nucleoside kinase from Drosophila melanogaster (Dm-dNK). In these Dm-dNK±Tk2−/− the lifespan was expanded for 3 weeks to at least 20 months and the reduction of subcutaneous and visceral fat was the only visible difference compared with wild type mice [135].
TK2-caused mitochondrial defects in murine model was also compensated via gene therapeutic approach based on AAV9 delivery of human TK2 cDNA. This intervention enhanced replacement therapy with pyrimidine deoxynucleosides delaying disease onset and extending lifespan in mice expressing mutant TK2 (Tk2KI). Furthermore, sequential treatment of Tk2KI mice with AAV9 first followed by AAV2 at different ages enabled to reduce the viral dose while further prolonging the lifespan [136].
GT of diseases caused by mtDNA mutations
In contrast to nDNA, targeting mtDNA mutations determines the necessity to search for original strategies taking into account unique traits of mitochondrial genome organization. If one is aimed to replace mutated gene encoded by mtDNA, such factors as difficulties with mitochondrial transfer of GT components and mitochondrial genetic code must be considered. In the case of gene editing strategies, circularity of mtDNA, differences in nDNA and mtDNA reparation mechanisms (DSBs in mtDNA cannot be repaired) and once again selectiveness of mitochondrial permeability may trouble applicability of standard methods. However, for diseases caused by mutations in mtDNA, several methods, including allotopic expression, direct mitochondrial transfer, replacement of mitochondrial proteins by orthologs, base edition were proposed and successfully tested in vivo ("Allotopic expression of mitochondrial genes", "Replacement of mitochondrial proteins by orthologs", "Direct mitochondrial transfer of mitochondrial genes" sections and Fig. 2). Interestingly, unique mechanisms of mtDNA reparation enable to use nuleases to selectively remove mutant mtDNA ("Shift of heteroplasmy" section and Fig. 3).

Basic approaches for replacement of causative for MD genes with the use of allotopic expression or direct mitochondrial transfer. In order to compensate the function of altered mitochondrial gene, the delivery of correct forms of DNA or mRNA is used. The sequence is transferred in the form of plasmid or delivered by AAV, LNP or in. 1. AAV vectors are able to deliver single-stranded (ss)DNA in nucleus where it further replicates to form double-stranded (ds)DNA, template for synthesis of the proper version of mitochondrial protein. To guide this protein into mitochondria coding ssDNA must contain mitochondrial targeting sequence (MTS). 2. AAV-MTS vectors carrying MTS on its surface are able to directly target mitochondria to provide mitochondrial transfer of ssDNA and subsequent synthesis of proper protein with the use of mitochondrial apparatus. 3. dsDNA can be delivered by being packed in lipid nanoparticles (LNP). As the part of LNP dsDNA apparently can be targeted directly into mitochondria or allotopically expressed in the nucleus, 4. The protein’s coding sequence is delivered into the mitochondria in the form of plasmid by hydrodynamic injection

Representation of the basic strategies allowing for decrease of the mutant mtDNA content. The ratio between mutant and wildtype mtDNA (heteroplasmy) determines the severity of mitochondrial dysfunction. One way (left) for shifting heteroplasmy is based on the introduction of double strand breaks (DSBs). To introduce DSBs the nuclease machineries (TALEN, ZFN or CRISPR/Cas) targeted on mutant loci are used. Since all the linear dsDNA molecules are normally being eliminated, the levels of mutant mtDNA decrease. Another way (right) to shift heteroplasmy is to complicate mtDNA replication and hence decrease its content against wild type mtDNA copies. Peptide nucleic acid oligomers (PNC) and F-/D-hairpins containing RNA (FD-RNA) can directly pair with high affinity to the sequence of interest and interrupt further replication of this region. Both ways lead to shift of heteroplasmy resulting in attenuation of mitochondrial dysfunction
Allotopic expression of mitochondrial genes
Allotopic expression stands for “mitochondrial gene transfer to the nucleus”. Before the transfer, mitochondrial genes should be re-coded into nuclear code and re-targeted to mitochondria by addition of MTS coding sequence. Interestingly, MTS can direct both translated protein and mRNA to mitochondria. In the second case translation occurs on the outer layer of mitochondria but with the use of classic cytoplasmic mRNA (Detailed description in review [137]). Notably, polypeptides encoded by mtDNA are highly hydrophobic, this may cause problems such as aggregation of the expressed polypeptides in cytoplasm and triggering immune response by over-expressed hydrophobic subunits in the cytoplasm [138]. For instance, mitochondrial proteins encoded in nDNA require chaperones to keep them soluble in the cytosol [139].
The strategy of allotopic expression as a roundabout route for GT of MD was proposed in 2002 by G. Manfredi et al. [140] to treat 8993 T– > G mutation of mtDNA MTATP6 gene causing impaired mitochondrial ATP synthesis in two related MD: neuropathy, ataxia, and retinitis pigmentosa and maternally inherited Leigh syndrome. The authors transfected wild-type mitochondrial MTATP6 gene (recoded to be compatible with the universal genetic code and appended to MTS and carboxy-terminal FLAG epitope tag) in the nucleus of wild-type and mutant human cells. After transfection of wild-type human cells, the precursor polypeptide was expressed, imported into, and processed within mitochondria, and incorporated into complex V. Allotopic expression of stably transfected constructs in cytoplasmic hybrids (cybrids) homoplasmic with respect to the 8993 T– > G mutation showed a significantly improved recovery after growth in the selective medium as well as a significant increase in ATP synthesis [141].
In vivo delivery of allotopically expressed genes was successfully tested in several in vivo studies in rodents. Qi et al. have developed an approach for modeling of LHON in mice by translocation of a nuclear version of the human ND4 subunit of complex I (naturally encoded in mitochondrial genome) carrying human mutation Arg340His into the mitochondria of rodent RGCs due to AAV-2 vector. This amino acid substitution caused by AG-to-A transition at nucleotide 11778 in mtDNA is responsible for half of the cases of Leber hereditary optic neuropathy (LHON), a disease that causes blindness in young adults [142, 143]. Allotopic expression of mutant human ND4 in the mouse visual system disrupted mitochondrial cytoarchitecture, elevated reactive oxygen species, induced swelling of the optic nerve head, and induced apoptosis, with a progressive demise of ganglion cells in the retina and their axons comprising the optic nerve [144].
S. Ellouze et al. created a similar model of LHON representing Arg340His mutation of ND4 in rats [145]. The authors expressed mutant human mitochondrial gene ND4 in the retinal cells of rats via electroporation and showed development of retinal abnormalities similar to the classic course of LHON [145]. The treatment induced visual abnormalities and degeneration of retinal ganglion cells, which were 40% less abundant in treated eyes than in control eyes. A subsequent electroporation with wild-type ND4 prevented both retinal ganglion cells loss and the impairment of visual function [145].
J.Guy et al. allotopically expressed wild-type human ND4 complex I subunit in mice, showing its effective mitochondrial delivery and safety for the mouse visual system [146]. Human ND4 was properly processed and imported into the mitochondria of RGCs and axons of mouse optic nerve after IVT injection. The expression of normal human ND4 in murine mitochondria did not induce the loss of RGCs, ATP synthesis, or pattern electroretinography amplitude, suggesting that allotopic ND4 may be safe for the treatment of patients with LHON.
The only clinically tested approach of treatment of MD was based on allotopical expression—nuclear transfer of ND4 coding sequence by IVT application of AAV2. Re-coded in the “universal” genetic code ND4 with appended targeting sequence derived from the P1 isoform of subunit c of the ATP synthase (ATPc) for import into the mitochondrion (rAAV2-ND4), it was shown to be effective in LHON patients [147,148,149]. IVT injections of AAV2/2-ND4 led to progressively improved visual acuity in Phase 3 clinical trials [150].
Moreover, there is evidence that mitochondrial tRNAs can also be expressed allotopically. Using F- и D-hairpins-dependent mechanism of transport of tRNA, MERRF syndrome [91] as well as MELAS syndrome (A3243G mutation of mitochondrial tRNA) [92] were corrected in human cell models.
Replacement of mitochondrial proteins by orthologs
Another elegant GT strategy is based on the use of small orthologs of human ETC components, much more compact and encoded by single non-mammalian genes. For instance, transkingdom GT for complex I diseases was proposed for NDI1, yeast ortholog of human NADH dehydrogenase. In the S. cerevisiae mitochondria, the NDI1 enzyme catalyses electron transfer from NADH to Q in the matrix compartment and is the main entry point into the respiratory chain, just as in complex I. The yeast NDI1 gene is composed of 1539 bp which is predicted to encode a precursor polypeptide of 513 amino acid residues. The first 26 amino acid residues of the N-terminal serve as signal sequence for import into mitochondria [151]. In a few studies NDI1 gene of Saccharomyces cerevisiae was successfully introduced into mammalian cell lines. The expressed protein was correctly targeted to the matrix side of the inner mitochondrial membranes and was fully functional and restored the NADH oxidase activity to the complex I-deficient cells. The transduced cells were more resistant to complex I inhibitors and diminished production of reactive oxygen species induced by rotenone [151,152,153,154,155].
Chadderton et al. treated mice with rotenone-induced mitochondrial dysfunction by intraocular injection of a nuclear NDI1. In this study, recombinant AAV2 expressing NDI1 (AAV-NDI1) was shown to protect RGCs in a rotenone-induced murine model of LHON, significantly reducing RGC death by 1.5-fold and optic nerve atrophy by 1.4-fold [156]. Authors suggest a clear advantage of using the NDI1 gene over allotopic expression of mammalian mitochondrial complex I subunit genes as a therapy for LHON and that a single gene may provide benefit to all LHON patients with a complex I deficiency irrespective of which complex I subunit gene is causative of this debilitating retinopathy.
Direct mitochondrial transfer of mitochondrial genes
Hong Yu et al. recapitulated LHON-associated phenotype by mitochondrial transfer of the mutant human NADH ubiquinone oxidoreductase subunit VI gene with T14484C mutation (hND6T14484C) in retinal ganglion cells of adult mice. In brief, via IVT injection of mitochondrial-targeted AAV, hND6T14484C was directly delivered into mitochondria causing retinal thinning, apoptosis in RGCs and visual loss. Immunostaining of the retinal slices showed sufficient expression of GT-components colocalizing with a mitochondrion-specific dye MitoTracker Deep Red [101]. This work brightly demonstrates that in vivo methods of direct delivery of gene therapeutic agents into mitochondria work.
Yasuzaki et al. demonstrated effective transfer of exogenous DNA to mitochondria in skeletal muscle of rats following hydrodynamic limb vein injection. In brief, in the hydrodynamic limb vein injection procedure a tourniquet is used to limit the delivery area to one limb per injection and naked pDNA is rapidly injected into the vein in the anterograde direction [157]. It was shown that after hydrodynamic injection of pcDNA3.1 ( +)-luc plasmid, plasmid DNA had been localized in isolated mitochondria of the limbs [102].
Shift of heteroplasmy
Heteroplasmy, the amazing feature of mitochondria, is another key to the treatment of MD. Heteroplasmy is a dynamically changing result of co-existence of diverse types of mtDNA in one organism. One way to shift heteroplasmy from mutant to wild type of mitochondria is to eliminate mutated mtDNA without any precise editing. This approach involves targeting mutation resulting in non-repairable disruption or inhibition of replication in mutant mtDNA. As a result, wild type mtDNA copies begin to prevail which leads to attenuation of mitochondrial dysfunction. Notably, each mitochondrion most often contains both mutant and wild type mtDNA [158] so elimination of mutant mtDNA could scarcely cause mitochondrial death [159].
Antireplicative machines
The first approach to shift heteroplasmy was the application of peptide nucleic acid (PNA) oligomers interrupting replication of mutant mtDNA [160]. These small molecules, where the individual nucleobases are linked to an achiral peptide backbone [161], have greater affinity than equivalent oligodeoxynucleotide when pairing with single stranded complementary DNA. Moreover, PNA is not charged and have has increased cellular and, apparently mitochondrial permeability, especially after small modifications [162]. The basic principle of PNA-dependent heteroplasmy shift is in inhibition of mtDNA replication after strong binding with mutant-specific mtDNA sequence (Fig. 3). This mechanism is feasible because mtDNA is single-stranded during much of its replication [163].
Similarly, there were attempts to design specific antireplicative RNA molecules, complementary to mutant mtDNA [164]. To guide these RNAs into mitochondria they were added by F- and D-hairpins, the structures providing efficient mitochondrial transfer (see "Mitochondrial transfer of GT components" section). The resulting FD-RNAs were successfully tested in Kearns–Sayre syndrome, caused by 4.9 kb-lenght deletion in the mitochondrial genome [165] and in cybrid cells carrying pathogenic A13514G point mutation in the mtDNA ND5 gene [166].
Nucleases
Apparently, in contrast to mitochondria of plants [167], and probably fishes [168], mammalian mitochondria not only cannot repair double strand breaks (DSBs), but also have systems of rapid degradation of double stranded linear DNA molecules [169,170,171,172]. Since double-stranded breaks cause degradation of mtDNA, the nucleases can be used to eliminate mtDNA molecules (Fig. 3). For the first time, the approach based on the use of high-specific nucleases to shift heteroplasmy was proposed in 2001. Srivastava and colleagues showed that bacterial enzyme PstI endonuclease which has two restriction sites in the human genome can decrease the levels of mutant mtDNA being expressed in mammalian cells [173]. One of these restriction sites is specific for patients with neuropathy, ataxia and retinitis pigmentosa (NARP), as the T8399G mutation creates a unique restriction site that is not present in wild-type human mtDNA. However, not surprisingly, the only application of PstI is limited by patients carrying this particular mutation. The same limitations are specific to all non-programmable nucleases.
TALEN and ZFN
Minczuk et al. developed an approach for routing engineered ZFNs to mitochondria, constructing chimeric enzymes targeted to specific mtDNA sequences. A basic hallmark of their approach was an augmentation of mitochondrial routing by adding MTS and nuclear export signal (NES) domains instead of only MTS to basic mtDNA targeted amino acids sequence of ZFP [174]. To test the efficacy of site-specific alteration of mtDNA the authors combined a ZFP with the easily assayed DNA-modifying activity of hDNMT3a methylase. Expression of the mutation-specific chimeric methylase resulted in the selective methylation of cytosines adjacent to the mutation site. Interestingly, targeted mtDNA methylation itself can be an additional option to fight MD and mitochondrial dysfunction [175].
The next step was the design of mitochondria-targeted TALEN (mito-TALEN). In 2013 Bacman and colleagues showed irreversible elimination of mtDNA carrying major deletion m.8483_13459del4977 or pathogenic point mutation m.14459G > A in patient-derived cell cultures [176]. The similar strategy was next successfully tested in animal tissue cultures [177, 178], murine oocytes [179] and even plants [180,181,182].
For mitochondrial targeting, a few modifications of TALEN were proposed. Naoki Yahata and colleagues showed a therapeutic shift of heteroplasmy in human induced pluripotent stem cells (iPSCs) carrying m.13513G > A mutation of mtDNA after treatment by G13513A-mpTALEN conjugated with MTS (ATP5B7 or Cox8) [183]. Another modification, the replacement of nuclease domain FokI by the smaller one I-TevI resulted in increased mitochondrial permeability of TALE-I-TevI complex. This modified version was shown to shift heteroplasmy in patient-derived cybrids harboring different levels of the m.8344A > G mtDNA point mutation, associated with myoclonic epilepsy with ragged-red fibers (MERRF) [184].
Moreover, mitoTALEN was also used for the revelation of causative mechanisms leading to a “common deletion”, the most frequent aberrancy of the mitochondrial genome, involving a 4,977-bp region flanked by 13-bp repeats [185].
Finally, programmable nucleases were also tested in vivo in animal models. MitoTALEN, delivered by AAV9 vector was shown to effectively reduce the levels of mutant mtDNA and recovery of tRNAAla in a murine model of heteroplasmic MD-associated mutation [186]. Similarly, in mice with heteroplasmic pathogenic mutation m.5024C > T displaying typical characteristics of classic mitochondrial disease [187], AAV delivery of mitochondrially targeted zinc finger-nucleases (mtZFNs) induced elimination of mutant mtDNA and attenuation of biochemical phenotypes [188].
CRISPR/Cas
Unfortunately, there are still not enough studies testing CRISPR/Cas9-based approach for shift of heteroplasmy and results of existing are mostly controversial [189]. Whereas some authors reported CRISPR/Cas9 might be efficiently translocated into mitochondria [99, 168], others report its failure [190]. Possible reasons for the low effectiveness could be related to the low permeability of Cas9 nuclease. Because of its large size and low positive charge, Cas9, even containing MTS, poorly penetrates into mitochondria, and, having penetrated, has a destructive effect on them [191].
Moreover, some concerns about unfeasibility of RNA component of CRISPR/Cas9 to transfer into mitochondria questioned the applicability of this approach even more. To overcome these limitations, some authors adapted CRISPR/Cas components by appending the fragments of “mitochondria recognizable” motifs to RNA and proteins.
In general, these are the same strategies which were used to gain mitochondrial permeability of ZFN, TALEN and antireplicative machines. For instance, Loutre et al. designed CRISPR/Cas9 addressed to mitochondria (mito-CRISPR/Cas9) by adding COX8A-derived MTS to Cas9 and hairpin structures to sgRNA (FD-RNA). This system effectively shifted heteroplasmy in Kearns Sayre Syndrome cybrids. Interestingly, in this study, it turned out, that sgRNA molecules lacking the import determinants also penetrated to mitochondria, suggesting that own hairpins of sgRNA can substitute them [192]. Similar strategy was utilized by Hussain and colleagues by adding the RNA transport-derived stem loop element (RP-loop), naturally guiding RNA into mitochondria, to the sgRNA [193]. In combination with MTS-harboring Cas9, this complex was effectively targeted to mitochondria resulting in the decrease of mtDNA copy number in the cells with 11205G variant in their ND4 sequence [193].
Antón et al. [191] conducted comparative trials of different modified Cas and revealed the most effective mitochondrial import for SaCas9 (Staphylococcus aureus Cas9), LbCas12a (Lachnospiraceae bacterium Cas12) and AsCas12a versions (Acidaminococcus sp. Cas12). Whereas Cas9 comprises two nuclease domains which generate blunt-ended DSBs of DNA [194, 195]. Cas12a, contains a single endonuclease domain that cleaves the two DNA strands in turn, resulting in a staggered DSB with a 5′ overhang [196]. Additionally, in this work MTS derived from Su9 (Neurospora crassa ATPase subunit 9) and mammalian ATG4D were shown to be more efficient to guide Cas isoforms to mitochondria [191].
In general, mito-CRISPR/Cas is a rapidly improving technology. Thus, CRISPR/Cas is a promising tool for shift of heteroplasmy, especially because of its adjustability and simplicity. However, to date therapeutic applicability of mito-CRISPR/Cas for mtDNA editing needs to be confirmed in further studies.
Other nucleases
Other nucleases which have been tested for shift of heteroplasmy are summarized in Table 3. All listed nucleases are non-programmable.
mtDNA editing
As previously described, genome editing systems can be successfully delivered into mitochondria. Theoretically, as mitochondria are not able to repair DSBs in mtDNA, editing of the mitochondrial genome with the use of classic methods based on homological reparation seems scarcely feasible. Nevertheless, Bian and colleagues provided in vitro and in vivo evidences that mtDNA could be repaired through the mito-CRISPR/Cas9 system via homological recombination. In brief, an exogenous single-stranded DNA with short homologous arm was imported in the mitochondria of human cells. Moreover, the construction was knocked into the targeting loci of zebrafish, and this mutagenesis was steadily transmitted to F1 generation [168]. Obviously, the ability and versatility of homological reparation of mtDNA after DSBs needs to be confirmed in further studies, but to date this is one of the most promising strategies for the treatment of MD.
Meanwhile, another powerful approach to repair mtDNA mutations was proposed. Apparently, the novel strategy of programmable base edition without DSBs provides the precise tool to introduce petite changes in mtDNA [199,200,201]. Base edition is a form of genome editing that enables direct, irreversible conversion of one base pair to another at a target genomic locus without requiring double-stranded DNA cleavage, homology-directed repair processes, or donor DNA templates [202]. For this conversion two base editors could be used: cytosine base editor, which chemically converts a cytosine–guanine (C–G) base pair into a thymine–adenine (T–A) and adenine base editor, chemically transforming A–T to G–C base pairs. Base editors require one of the classical genome editing systems (CRISPR, TALE etc.) for guiding to target loci. Because deaminases can operate only on single-stranded nucleic acids [203] CRISPR, as machinery able to unwind double-stranded DNA, is usually utilized for base edition.
However, difficulties with mitochondrial addressing of sgRNA aimed Mok et al. to engineer non-CRISPR system which is able to operate with unwound dsDNA. The authors fused MTS-containing TALE and DddA, toxicity-lack derivative of interbacterial toxin catalyzing the deamination of cytidines within dsDNA. The resulting non-toxic RNA-free DddA-derived cytosine base editors was shown to catalyse C•G-to-T•A conversions in human mtDNA with high target specificity and product purity. This system was challenged for editing 5 mitochondrial genes: MT-ND1, MT-ND2, MT-ND4, MT-ND5 and MT-ATP8 in HEK293T cells. Depending on the sequence, the conversion efficiency of CG to TA reached 50% [204].
The same DddA-TALE machinery was further utilized for the development of the murine models of MD by Lee and colleagues. Two mutations of the ND5 gene were successfully recapitulated into the mice: silent mutation, m.G12918A causing multiple human MD, and m.C12336T that incorporates a premature stop codon at the 199th position of the ND5 protein. Notably, it was also shown that mtDNA heteroplasmy induced by DdCBEs in one-cell stage zygotes can be maintained throughout the development and differentiation and transmitted to the next generation [205].
Thus, although quite laborious, the DddA-TALE method provides high efficiency of mitochondrial genome editing. The main GT approaches for the treatment of MD are presented in Fig. 4.

Multiple GT approaches for the treatment of MD. Existing approaches enable targeting both nuclear(n) or mitochondrial(mt) DNA. In general, possible interventions may be divided into the following strategies: (1) Reparation of mutant genes (including base edition); (2) Elimination of mutant mtDNA; (3) Replacement (including allotopic expression).1. Reparation of mutant nuclear genes can be performed with the use of programmed nucleases (TALEN, ZFN, CRISPR/Cas). A modified version of CRISPR (CRISPR-deaminase) can be utilized for base edition (introduction of single nucleotide substitutions) in nuclear genome whereas modified version of TALEN (TALEN-deaminase) can be utilized for base edition in the mitochondrial genome. 2. In order to eliminate mutant mtDNA, double-stranded breaks induced by programmable nucleases TALEN, ZFN, CRISPR/Cas, can be used. Another option is application of antireplicative machines FD-RNA or peptide nucleic acids (PNA).3. The function of altered gene may also be compensated by its wild type copy delivered into mitochondria or nucleus. In the case of replacement of mitochondrial genes, sequence can be allotopically expressed in the nucleus, but only after reprogramming in mitochondrial genetic code and appending mitochondrial-targeting sequence
Perspectives and difficulties
Unfortunately, the issues of the treatment of MD are still mostly up-in-the-air. Although only emerging, GT is the most promising avenue to treat MD, especially on the background of modest clinical efficacy of classical pharmacotherapy. In terms of applicability, modern GT approaches are quite close to successful treatment of those MD which are caused by nDNA mutations. However, nDNA mutations are found only in 15% of all patients struggling MD, whereas the prevalent majority of patients carry mutations in the mitochondrial genome [206].
Because of the major diversity of mutations associated with MD, all the emerging GT strategies could take an important place. To date some of them are close to the clinical application, but some–are still in the beginning stages of development. Largely, the strategies for a shift of heteroplasmy and edition of mtDNA are far from therapeutic implementation, although move by leaps and bounds. At the same time, because of its more solid background, the replacement GT approach apparently can advance to clinical application in the near future. Already, GT based on AAV-mediated delivery of the ND4 with MTS for the treatment of LHON caused by a mutation in the mitochondrial genome has reached an advanced stage of clinical trials.
Additionally, there are also a few possible strategies we did not focus on in this paper. In this review, we did not include approaches based on antisense oligonucleotides (ASN). ASN is another promising strategy for manipulating mutant genes, but it is not GT in terms of FDA. Because of its ability to complementarily bind to a target site in pre-mRNA ASN may serve to reprogram or disrupt splicing of mutant genes resulting in a therapeutic effect [207]. Currently, ASN based therapies have been approved for the treatment of spinal muscular atrophy [208] and Duchenne muscular dystrophy [209]. This approach was also tested for silencing mtDNA [210,211,212] as well as splice correction or reducing the inclusion of a non-productive exon of OPA1 [213, 214], disclosing its potential in treating MD.
Finally, interesting approaches based on the delivery of anti-apoptotic genes were proposed. For instance, recently Wassmer et al. utilized a murine model expressing mutant ND4 to test the potential of delivery of X-linked inhibitor of apoptosis (XIAP) to prevent retinal ganglion cell apoptosis and reduce disease progression. Authors reported significant amelioration of the disease course protecting the nerve fiber layer and optic nerve architecture [215]. XIAP is an inhibitor of caspases and apoptosis, and possible autophagy modulator [216]. As apoptosis and autophagy are key events caused by mitochondrial dysfunction, XIAP-based GT is a promising strategy for the treatment of MD, both nDNA and mtDNA-associated.
The remarkable thing in the issues of GT approaches for MD is that many data obtained are controversial. Although many breakthrough studies are reported there are even more studies reporting negative results when attempting reproduce them. In order to present state-of-the-art picture of mitochondrial GT we mostly focused on positive reports though mentioning some opposite results. Herein, we close our review with some critical concerns to not be misleading. In particular, even though some authors have demonstrated beneficial allotopic expression of mitochondrial genes, several lines of evidence demonstrate that the results showing import of allotopically expressed proteins are indeed artefactual [217]. The same is with technologies relying on mitochondrial import of RNA as the general view in the mitochondrial community is still that RNAs cannot enter mitochondria. In this regard, the feasibility of DSB-based editing of mitochondrial genome seems to be in especial need to be further challenged as it also does not correspond to existing view of mtDNA reparation. We believe that further studies may shed light on disputable questions of mitochondrial biology and open up brand new landscapes in the treatment of MD.
Availability of data and materials
Not applicable.
Change history
08 February 2023
A Correction to this paper has been published: https://doi.org/10.1186/s12967-023-03915-z
Abbreviations
- MD:
-
Mitochondrial diseases
- OXPHOS:
-
Oxidative phosphorylation
- mtDNA:
-
Mitochondrial DNA
- DCA:
-
Dichloroacetate
- GT:
-
Gene therapy
- siRNA:
-
Small interfering RNA
- shRNA:
-
Short hairpin RNA
- miRNA:
-
MicroRNA
- CRISPR/Cas:
-
Clustered regularly interspaced short palindromic repeats / CRISPR-associated protein
- ETC:
-
Electron transport chain
- MELAS:
-
Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes
- MIDD:
-
Maternally inherited diabetes and deafness
- ZFN:
-
Zinc fingers nucleases
- TALEN:
-
Transcription activator-like effector nucleases
- DQAsomes:
-
Dequalinium-based liposome-like vesicles
- rAAV:
-
Recombinant adeno-associated viruses
- IVT:
-
Intravitreal
- SC:
-
Suprachoroidal
- SR:
-
Subretinal
- MRIgFUS:
-
Magnetic resonance-guide focused ultrasound
- MTSs:
-
Mitochondrial targeting sequences
- SOD2 :
-
Superoxide dismutase 2 gene
- TYMP :
-
Thymidine phosphorylase gene
- Slc25a46 :
-
Mitochondrial fusion-fission-related gene solute carrier family 25 member 46
- OPA1 :
-
Dynamin-like 120 kDa protein
- NDUFS4 :
-
NADH dehydrogenase (ubiquinone) iron-sulfur protein 4 gene
- Fdxr :
-
Ferredoxin reductase gene
- ETHE1 :
-
Ethylmalonic encephalopathy gene
- SDO:
-
Sulfur dioxygenase
- LHON:
-
Leber hereditary optic neuropathy
- RGC:
-
Retinal ganglion cells
- NARP:
-
Neuropathy, ataxia and retinitis pigmentosa
References
Stenton SL, Prokisch H. Genetics of mitochondrial diseases: identifying mutations to help diagnosis. EBioMedicine. 2020;56:102784.
Angelova PR. Sources and triggers of oxidative damage in neurodegeneration. Free Radical Biol Med. 2021;173:52–63.
Kanungo S, Morton J, Neelakantan M, Ching K, Saeedian J, Goldstein A. Mitochondrial disorders. Ann Transl Med. 2018;6:475–475.
Angelova PR, Esteras N, Abramov AY. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: finding ways for prevention. Med Res Rev. 2021;41:770–84.
Avula S, Parikh S, Demarest S, Kurz J, Gropman A. Treatment of mitochondrial disorders. Curr Treat Options Neurol. 2014;16:292.
Ganetzky RD, Falk MJ. 8-year retrospective analysis of intravenous arginine therapy for acute metabolic strokes in pediatric mitochondrial disease. Mol Genet Metab. 2018;123:301–8.
Ikawa M, Povalko N, Koga Y. Arginine therapy in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Curr Opin Clin Nutr Metab Care. 2020;23:17–22.
Wu S-S, Li Q-C, Yin C-Q, Xue W, Song C-Q. Advances in CRISPR/Cas-based gene therapy in human genetic diseases. Theranostics. 2020;10:4374–82.
Skulachev VP. Mitochondrial physiology and pathology; concepts of programmed death of organelles, cells and organisms. Mol Aspects Med. 1999;20:139–84.
Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–67.
Kadenbach B. Introduction to mitochondrial oxidative phosphorylation. In: Kadenbach B, editor. Mitochondrial oxidative phosphorylation. New York: Springer; 2012.
Liu YJ, McIntyre RL, Janssens GE, Houtkooper RH. Mitochondrial fission and fusion: a dynamic role in aging and potential target for age-related disease. Mech Ageing Dev. 2020;186:111212.
Quirós PM, Langer T, López-Otín C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 2015;16:345–59.
Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/parkin mitophagy. Trends Cell Biol. 2016;26:733–44.
Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5.
D’Souza AR, Minczuk M. Mitochondrial transcription and translation: overview. Essays Biochem. 2018;62:309–20.
Filograna R, Mennuni M, Alsina D, Larsson N. Mitochondrial DNA copy number in human disease: the more the better? FEBS Lett. 2021;595:976–1002.
Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol. 2013;5:a021220–a021220.
Shtolz N, Mishmar D. The mitochondrial genome–on selective constraints and signatures at the organism, cell, and single mitochondrion levels. Front Ecol Evol. 2019;7:342.
Schon EA, Gilkerson RW. Functional complementation of mitochondrial DNAs: Mobilizing mitochondrial genetics against dysfunction. Biochim Biophys Acta. 2010;1800:245–9.
Thorburn DR, Dahl H-HM. Mitochondrial disorders: genetics, counseling, prenatal diagnosis and reproductive options. Am J Med Genet. 2001;106:102–14.
DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–68.
Gardner JL, Craven L, Turnbull DM, Taylor RW. Experimental strategies towards treating mitochondrial DNA disorders. Biosci Rep. 2007;27:139–50.
Chinnery PF, et al. Primary mitochondrial disorders overview. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, et al., editors. Genereviews®. Seattle, WA: University of Washington; 1993.
Kirby DM, Crawford M, Cleary MA, Dahl H-HM, Dennett X, Thorburn DR. Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology. 1999;52:1255–1255.
Fassone E, Rahman S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J Med Genet. 2012;49:578–90.
Abramov AY, Angelova PR. Cellular mechanisms of complex I-associated pathology. Biochem Soc Trans. 2019;47:1963–9.
Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R, Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol. 2009;11:414–30.
Prietsch V, Lindner M, Zschocke J, Nyhan WL, Hoffmann GF. Emergency management of inherited metabolic diseases. J Inherit Metab Dis. 2002;25:531–46.
Barshop BA, Naviaux RK, McGowan KA, Levine F, Nyhan WL, Loupis-Geller A, et al. Chronic treatment of mitochondrial disease patients with dichloroacetate. Mol Genet Metab. 2004;83:138–49.
Khan NA, Govindaraj P, Meena AK, Thangaraj K. Mitochondrial disorders: challenges in diagnosis & treatment. Indian J Med Res. 2015;141:13.
Morava E, Rodenburg R, van Essen HZ, De Vries M, Smeitink J. Dietary intervention and oxidative phosphorylation capacity. J Inherit Metab Dis. 2006;29:589–589.
Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. 2006;60:223–35.
Viscomi C, Bottani E, Zeviani M. Emerging concepts in the therapy of mitochondrial disease. Biochim Biophys Acta. 2015;1847:544–57.
Tinker RJ, Lim AZ, Stefanetti RJ, McFarland R. Current and emerging clinical treatment in mitochondrial disease. Mol Diagn Ther. 2021;25:181–206.
Friedmann T. A brief history of gene therapy. Nat Genet. 1992;2:93–8.
Borenfreund E, Bendich A. A study of the penetration of mammalian cells by deoxyribonucleic acids. J Biophys Biochem Cytol. 1961;9:81–91.
Szybalska EH, Szybalski W. Genetics of human cell lines, IV. DNA-mediated heritable transformation of a biochemical trait. Proc Natl Acad Sci USA. 1962;48:2026–34.
American Society of Gene & Cell Therapy. Gene and Cell Therapy FAQ’s. Waukesha, Wisconsin, USA: American Society of Gene & Cell Therapy. 2019. https://www.asgct.org/education/more-resources/gene-and-cell-therapy-faqs
Rao RC, Zacks DN. Cell and gene therapy. In: Casaroli-Marano RP, Zarbin MA, editors. Developments in ophthalmology. S. Karger AG: Basel; 2014. p. 167–77.
Crudele JM, Chamberlain JS. AAV-based gene therapies for the muscular dystrophies. Hum Mol Genet. 2019;28:R102–7.
Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Sig Transduct Target Ther. 2020;5:1.
Maestro S, Weber ND, Zabaleta N, Aldabe R, Gonzalez-Aseguinolaza G. Novel vectors and approaches for gene therapy in liver diseases. JHEP Reports. 2021;3:100300.
First CRISPR therapy dosed. Nat Biotechnol. 2020;38:382–382. https://doi.org/10.1038/s41587-020-0493-4
Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385:493–502.
Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA. 1996;93:1156–60.
Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–61.
Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–41.
Hendel A, Fine EJ, Bao G, Porteus MH. Quantifying on- and off-target genome editing. Trends Biotechnol. 2015;33:132–40.
Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096.
Doman JL, Raguram A, Newby GA, Liu DR. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat Biotechnol. 2020;38:620–8.
Wijshake T, Baker DJ, van de Sluis B. Endonucleases: new tools to edit the mouse genome. Biochim Biophys Acta. 2014;1842:1942–50.
Mileshina D, Niazi AK, Weber-Lotfi F, Gualberto J, Dietrich A. Mitochondrial genetic manipulation. In: Li X-Q, Donnelly DJ, Jensen TG, editors. Somatic genome manipulation. New York, NY: Springer; 2015. p. 275–321.
Jang Y, Ahn SR, Shim J, Lim K. Engineering genetic systems for treating mitochondrial diseases. Pharmaceutics. 2021;13:810.
Zu H, Gao D. Non-viral vectors in gene therapy: recent development, challenges, and prospects. AAPS J. 2021;23:78.
Bottani E, Lamperti C, Prigione A, Tiranti V, Persico N, Brunetti D. Therapeutic approaches to treat mitochondrial diseases: “one-size-fits-all” and “precision medicine” strategies. Pharmaceutics. 2020;12:1083.
Slone J, Huang T. The special considerations of gene therapy for mitochondrial diseases. npj Genom Med. 2020;5:7.
Ballon DJ, Rosenberg JB, Fung EK, Nikolopoulou A, Kothari P, De BP, et al. Quantitative whole-body imaging of I-124-labeled adeno-associated viral vector biodistribution in nonhuman primates. Hum Gene Ther. 2020;31:1237–59.
Daily JL, Burstein SR, Keiser NW, Cobb SR. A non-human primate biodistribution study comparing multiple routes of administration of AAV9. Mol Ther. 2021;29:171–2.
Bish LT, Sleeper MM, Brainard B, Cole S, Russell N, Withnall E, et al. Percutaneous transendocardial delivery of self-complementary adeno-associated virus 6 achieves global cardiac gene transfer in canines. Mol Ther. 2008;16:1953–9.
Gao G, Bish LT, Sleeper MM, Mu X, Sun L, Lou Y, et al. Transendocardial delivery of AAV6 results in highly efficient and global cardiac gene transfer in rhesus macaques. Hum Gene Ther. 2011;22:979–84.
Weber-Adrian D, Kofoed RH, Silburt J, Noroozian Z, Shah K, Burgess A, et al. Systemic AAV6-synapsin-GFP administration results in lower liver biodistribution, compared to AAV1&2 and AAV9, with neuronal expression following ultrasound-mediated brain delivery. Sci Rep. 2021;11:1934.
Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu W-L, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 2017;20:1172–9.
Nonnenmacher M, Wang W, Child MA, Ren X-Q, Huang C, Ren AZ, et al. Rapid evolution of blood-brain-barrier-penetrating AAV capsids by RNA-driven biopanning. Mol Ther Methods Clin Dev. 2021;20:366–78.
Hanaford AR, Cho Y-J, Nakai H. AAV-vector based gene therapy for mitochondrial disease: progress and future perspectives. Orphanet J Rare Dis. 2022;17:217.
Mori S, Takeuchi T, Enomoto Y, Kondo K, Sato K, Ono F, et al. Biodistribution of a low dose of intravenously administered AAV-2, 10, and 11 vectors to cynomolgus monkeys. Jpn J Infect Dis. 2006;59:285–93.
Herzog CD, Bishop KM, Brown L, Wilson A, Kordower JH, Bartus RT. Gene transfer provides a practical means for safe, long-term, targeted delivery of biologically active neurotrophic factor proteins for neurodegenerative diseases. Drug Deliv and Transl Res. 2011;1:361–82.
Salegio EA, Kells AP, Richardson RM, Hadaczek P, Forsayeth J, Bringas J, et al. Magnetic resonance imaging-guided delivery of adeno-associated virus type 2 to the primate brain for the treatment of lysosomal storage disorders. Hum Gene Ther. 2010;21:1093–103.
Spronck E, Vallès A, Lampen M, Montenegro-Miranda P, Keskin S, Heijink L, et al. Intrastriatal administration of AAV5-miHTT in non-human primates and rats is well tolerated and results in miHTT transgene expression in key areas of huntington disease pathology. Brain Sci. 2021;11:129.
Nathwani AC, Rosales C, McIntosh J, Rastegarlari G, Nathwani D, Raj D, et al. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther. 2011;19:876–85.
Hordeaux J, Hinderer C, Buza EL, Louboutin J-P, Jahan T, Bell P, et al. Safe and sustained expression of human iduronidase after intrathecal administration of adeno-associated virus serotype 9 in infant rhesus monkeys. Hum Gene Ther. 2019;30:957–66.
Bevan AK, Duque S, Foust KD, Morales PR, Braun L, Schmelzer L, et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther. 2011;19:1971–80.
Rosenberg JB, Sondhi D, Rubin DG, Monette S, Chen A, Cram S, et al. Comparative efficacy and safety of multiple routes of direct CNS administration of adeno-associated virus gene transfer vector serotype rh.10 expressing the human arylsulfatase a cDNA to nonhuman primates. Hum Gene Ther Clin Dev. 2014;25:164–77.
Yang B, Li S, Wang H, Guo Y, Gessler DJ, Cao C, et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol Ther. 2014;22:1299–309.
Hocquemiller M, Hemsley KM, Douglass ML, Tamang SJ, Neumann D, King BM, et al. AAVrh10 vector corrects disease pathology in MPS IIIA mice and achieves widespread distribution of SGSH in large animal brains. Mol Ther Methods Clin Dev. 2020;17:174–87.
Rosenberg JB, Kaplitt MG, De BP, Chen A, Flagiello T, Salami C, et al. AAVrh.10-mediated APOE2 central nervous system gene therapy for APOE4-associated Alzheimer’s disease. Hum Gene Ther Clin Dev. 2018;29:24–47.
Frederick A, Sullivan J, Liu L, Adamowicz M, Lukason M, Raymer J, et al. Engineered capsids for efficient gene delivery to the retina and cornea. Hum Gene Ther. 2020;31:756–74.
Pavlou M, Schön C, Occelli LM, Rossi A, Meumann N, Boyd RF, et al. Novel AAV capsids for intravitreal gene therapy of photoreceptor disorders. EMBO Mol Med. 2021;13:e13392.
Vandenberghe LH, Bell P, Maguire AM, Xiao R, Hopkins TB, Grant R, et al. AAV9 targets cone photoreceptors in the nonhuman primate retina. PLoS ONE. 2013;8:e53463.
Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE, et al. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006;99:e3-9.
Potter RA, Griffin DA, Sondergaard PC, Johnson RW, Pozsgai ER, Heller KN, et al. Systemic delivery of dysferlin overlap vectors provides long-term gene expression and functional improvement for dysferlinopathy. Hum Gene Ther. 2018;29:749–62.
Gao G, Lu Y, Calcedo R, Grant RL, Bell P, Wang L, et al. Biology of AAV serotype vectors in liver-directed gene transfer to nonhuman primates. Mol Ther. 2006;13:77–87.
Li S, Ling C, Zhong L, Li M, Su Q, He R, et al. Efficient and targeted transduction of nonhuman primate liver with systemically delivered optimized AAV3B vectors. Mol Ther. 2015;23:1867–76.
Gao G, Wang Q, Calcedo R, Mays L, Bell P, Wang L, et al. Adeno-associated virus-mediated gene transfer to nonhuman primate liver can elicit destructive transgene-specific T cell responses. Hum Gene Ther. 2009;20:930–42.
Calvo SE, Julien O, Clauser KR, Shen H, Kamer KJ, Wells JA, et al. Comparative analysis of mitochondrial n-termini from mouse, human, and yeast. Mol Cell Proteomics. 2017;16:512–23.
Backes S, Herrmann JM. Protein translocation into the intermembrane space and matrix of mitochondria: mechanisms and driving forces. Front Mol Biosci. 2017;4:83.
Vögtle F-N, Wortelkamp S, Zahedi RP, Becker D, Leidhold C, Gevaert K, et al. Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell. 2009;139:428–39.
Meng Y, Sun D, Qin Y, Dong X, Luo G, Liu Y. Cell-penetrating peptides enhance the transduction of adeno-associated virus serotype 9 in the central nervous system. Mol Ther Methods Clin Dev. 2021;21:28–41.
Yu H, Koilkonda RD, Chou T-H, Porciatti V, Ozdemir SS, Chiodo V, et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc Natl Acad Sci USA. 2012;109:e1238.
Jeandard D, Smirnova A, Tarassov I, Barrey E, Smirnov A, Entelis N. Import of non-coding RNAs into human mitochondria: a critical review and emerging approaches. Cells. 2019;8:286.
Kolesnikova OA, Entelis NS, Jacquin-Becker C, Goltzene F, Chrzanowska-Lightowlers ZM, Lightowlers RN, et al. Nuclear DNA-encoded tRNAs targeted into mitochondria can rescue a mitochondrial DNA mutation associated with the MERRF syndrome in cultured human cells. Hum Mol Genet. 2004;13:2519–34.
Karicheva OZ, Kolesnikova OA, Schirtz T, Vysokikh MY, Mager-Heckel A-M, Lombès A, et al. Correction of the consequences of mitochondrial 3243A>G mutation in the MT-TL1 gene causing the MELAS syndrome by tRNA import into mitochondria. Nucleic Acids Res. 2011;39:8173–86.
Martin RP, Schneller JM, Stahl AJ, Dirheimer G. Import of nuclear deoxyribonucleic acid coded lysine-accepting transfer ribonucleic acid (anticodon C-U-U) into yeast mitochondria. Biochemistry. 1979;18:4600–5.
Kolesnikova O, Kazakova H, Comte C, Steinberg S, Kamenski P, Martin RP, et al. Selection of RNA aptamers imported into yeast and human mitochondria. RNA. 2010;16:926–41.
Golani-Armon A, Arava Y. Localization of nuclear-encoded mRNAs to mitochondria outer surface. Biochemistry (Mosc). 2016;81:1038–43.
Michaud M, Maréchal-Drouard L, Duchêne A-M. Targeting of cytosolic mRNA to mitochondria: naked RNA can bind to the mitochondrial surface. Biochimie. 2014;100:159–66.
Kaltimbacher V, Bonnet C, Lecoeuvre G, Forster V, Sahel J-A, Corral-Debrinski M. mRNA localization to the mitochondrial surface allows the efficient translocation inside the organelle of a nuclear recoded ATP6 protein. RNA. 2006;12:1408–17.
Sieber F, Placido A, El Farouk-Ameqrane S, Duchêne A-M, Maréchal-Drouard L. A protein shuttle system to target RNA into mitochondria. Nucleic Acids Res. 2011;39:e96–e96.
Jo A, Ham S, Lee GH, Lee Y-I, Kim S, Lee Y-S, et al. Efficient mitochondrial genome editing by CRISPR/Cas9. Biomed Res Int. 2015;2015:305716.
Liew SS, Qin X, Zhou J, Li L, Huang W, Yao SQ. Smart design of nanomaterials for mitochondria-targeted nanotherapeutics. Angew Chem Int Ed. 2021;60:2232–56.
Yu H, Sant DW, Wang G, Guy J. Mitochondrial transfer of the mutant human ND6T14484C gene causes visual loss and optic neuropathy. Trans Vis Sci Tech. 2020;9:1.
Yasuzaki Y, Yamada Y, Kanefuji T, Harashima H. Localization of exogenous DNA to mitochondria in skeletal muscle following hydrodynamic limb vein injection. J Control Release. 2013;172:805–11.
Yamada Y, Akita H, Kamiya H, Kogure K, Yamamoto T, Shinohara Y, et al. MITO-Porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim Biophys Acta. 2008;1778:423–32.
Yamada Y, Harashima H. Delivery of bioactive molecules to the mitochondrial genome using a membrane-fusing, liposome-based carrier DF-MITO-Porter. Biomaterials. 2012;33:1589–95.
Yamada Y, Kawamura E, Harashima H. Mitochondrial-targeted DNA delivery using a DF-MITO-Porter, an innovative nano carrier with cytoplasmic and mitochondrial fusogenic envelopes. J Nanopart Res. 2012;14:1013.
Yamada Y, Harashima H. MITO-porter for mitochondrial delivery and mitochondrial functional analysis. In: Singh H, Sheu S-S, editors. Pharmacology of mitochondria. Cham: Springer International Publishing; 2016. p. 457–72.
Yamada Y, Maruyama M, Kita T, Usami S, Kitajiri S, Harashima H. The use of a MITO-Porter to deliver exogenous therapeutic RNA to a mitochondrial disease’s cell with a A1555G mutation in the mitochondrial 12S rRNA gene results in an increase in mitochondrial respiratory activity. Mitochondrion. 2020;55:134–44.
Jang Y, Lim K. Recent advances in mitochondria-targeted gene delivery. Molecules. 2018;23:2316.
Wallace DC. Mouse models for mitochondrial disease. Am J Med Genet. 2001;106:71–93.
Hirano M, Silvestri G, Blake DM, Lombes A, Minetti C, Bonilla E, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology. 1994;44:721–7.
Spinazzola A, Marti R, Nishino I, Andreu AL, Naini A, Tadesse S, et al. Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem. 2002;277:4128–33.
Martí R, Nishigaki Y, Hirano M. Elevated plasma deoxyuridine in patients with thymidine phosphorylase deficiency. Biochem Biophys Res Commun. 2003;303:14–8.
Torres-Torronteras J, Gómez A, Eixarch H, Palenzuela L, Pizzorno G, Hirano M, et al. Hematopoietic gene therapy restores thymidine phosphorylase activity in a cell culture and a murine model of MNGIE. Gene Ther. 2011;18:795–806.
Torres-Torronteras J, Viscomi C, Cabrera-Pérez R, Cámara Y, Di Meo I, Barquinero J, et al. Gene therapy using a liver-targeted AAV vector restores nucleoside and nucleotide homeostasis in a murine model of MNGIE. Mol Ther. 2014;22:901–7.
Torres-Torronteras J, Cabrera-Pérez R, Vila-Julià F, Viscomi C, Cámara Y, Hirano M, et al. Long-term sustained effect of liver-targeted adeno-associated virus gene therapy for mitochondrial neurogastrointestinal encephalomyopathy. Hum Gene Ther. 2018;29:708–18.
Vila-Julià F, Cabrera-Pérez R, Cámara Y, Molina-Berenguer M, Lope-Piedrafita S, Hirano M, et al. Efficacy of adeno-associated virus gene therapy in a MNGIE murine model enhanced by chronic exposure to nucleosides. eBioMedicine. 2020;62:103133.
Yang L, Slone J, Li Z, Lou X, Hu Y-C, Queme LF, et al. Systemic administration of AAV-Slc25a46 mitigates mitochondrial neuropathy in Slc25a46-/- mice. Hum Mol Genet. 2020;29:649–61.
Civiletto G, Varanita T, Cerutti R, Gorletta T, Barbaro S, Marchet S, et al. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 2015;21:845–54.
Delettre C, Lenaers G, Pelloquin L, Belenguer P, Hamel CP. OPA1 (Kjer type) dominant optic atrophy: a novel mitochondrial disease. Mol Genet Metab. 2002;75:97–107.
Lenaers G, Hamel C, Delettre C, Amati-Bonneau P, Procaccio V, Bonneau D, et al. Dominant optic atrophy. Orphanet J Rare Dis. 2012;7:46.
Sarzi E, Seveno M, Piro-Mégy C, Elzière L, Quilès M, Péquignot M, et al. OPA1 gene therapy prevents retinal ganglion cell loss in a dominant optic atrophy mouse model. Sci Rep. 2018;8:2468.
Palmer G, Horgan DJ, Tisdale H, Singer TP, Beinert H. Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase. XIV. Location of the sites of inhibition of rotenone, barbiturates, and piericidin by means of electron paramagnetic resonance spectroscopy. J Biol Chem. 1968;243:844–7.
Maloney DM, Chadderton N, Millington-Ward S, Palfi A, Shortall C, O’Byrne JJ, et al. Optimized OPA1 Isoforms 1 and 7 provide therapeutic benefit in models of mitochondrial dysfunction. Front Neurosci. 2020;14:571479.
Sun Y, Xue W, Song Z, Huang K, Zheng L. Restoration of Opa1-long isoform inhibits retinal injury-induced neurodegeneration. J Mol Med. 2016;94:335–46.
Lai Y, Lin P, Chen M, Zhang Y, Chen J, Zheng M, et al. Restoration of L-OPA1 alleviates acute ischemic stroke injury in rats via inhibiting neuronal apoptosis and preserving mitochondrial function. Redox Biol. 2020;34:101503.
Budde SMS, van den Heuvel LPWJ, Smeets RJP, Skladal D, Mayr JA, Boelen C, et al. Clinical heterogeneity in patients with mutations in the NDUFS4 gene of mitochondrial complex I. J Inherit Metab Dis. 2003;26:813–5.
Di Meo I, Marchet S, Lamperti C, Zeviani M, Viscomi C. AAV9-based gene therapy partially ameliorates the clinical phenotype of a mouse model of Leigh syndrome. Gene Ther. 2017;24:661–7.
Silva-Pinheiro P, Cerutti R, Luna-Sanchez M, Zeviani M, Viscomi C. A single intravenous injection of AAV-PHP.B-hNDUFS4 ameliorates the phenotype of Ndufs4 Mice. Mol Ther Methods Clin Dev. 2020;17:1071–8.
Reynaud-Dulaurier R, Benegiamo G, Marrocco E, Al-Tannir R, Surace EM, Auwerx J, et al. Gene replacement therapy provides benefit in an adult mouse model of Leigh syndrome. Brain. 2020;143:1686–96.
Yang L, Slone J, Zou W, Queme LF, Jankowski MP, Yin F, et al. Systemic delivery of AAV-Fdxr mitigates the phenotypes of mitochondrial disorders in Fdxr mutant Mice. Mol Ther Methods Clin Dev. 2020;18:84–97.
Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med. 2009;15:200–5.
Hildebrandt TM, Grieshaber MK. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008;275:3352–61.
Di Meo I, Auricchio A, Lamperti C, Burlina A, Viscomi C, Zeviani M. Effective AAV-mediated gene therapy in a mouse model of ethylmalonic encephalopathy. EMBO Mol Med. 2012;4:1008–14.
Tyynismaa H, Sun R, Ahola-Erkkila S, Almusa H, Poyhonen R, Korpela M, et al. Thymidine kinase 2 mutations in autosomal recessive progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Hum Mol Genet. 2012;21:66–75.
Krishnan S, Paredes JA, Zhou X, Kuiper RV, Hultenby K, Curbo S, et al. Long term expression of drosophila melanogaster nucleoside kinase in thymidine kinase 2-deficient Mice with no lethal effects caused by nucleotide pool imbalances. J Biol Chem. 2014;289:32835–44.
Lopez-Gomez C, Sanchez-Quintero MJ, Lee EJ, Kleiner G, Tadesse S, Xie J, et al. Synergistic deoxynucleoside and gene therapies for thymidine kinase 2 deficiency. Ann Neurol. 2021;90:640–52.
Artika IM. Allotopic expression of mitochondrial genes: basic strategy and progress. Genes Dis. 2020;7:578–84.
Marella M, Seo BB, Thomas BB, Matsuno-Yagi A, Yagi T. Successful amelioration of mitochondrial optic neuropathy using the yeast NDI1 gene in a rat animal model. PLoS ONE. 2010;5:e11472.
Fan ACY, Bhangoo MK, Young JC. Hsp90 functions in the targeting and outer membrane translocation steps of Tom70-mediated mitochondrial import. J Biol Chem. 2006;281:33313–24.
Turnbull DM, Lightowlers RN. A roundabout route to gene therapy. Nat Genet. 2002;30:345–6.
Manfredi G, Fu J, Ojaimi J, Sadlock JE, Kwong JQ, Guy J, et al. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002;30:394–9.
Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–30.
Brown MD, Torroni A, Reckord CL, Wallace DC. Phylogenetic analysis of Leber’s hereditary optic neuropathy mitochondrial DNA’s indicates multiple independent occurrences of the common mutations. Hum Mutat. 1995;6:311–25.
Qi X, Sun L, Lewin AS, Hauswirth WW, Guy J. The mutant human ND4 subunit of complex I induces optic neuropathy in the mouse. Invest Ophthalmol Vis Sci. 2007;48:1–10.
Ellouze S, Augustin S, Bouaita A, Bonnet C, Simonutti M, Forster V, et al. Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. Am J Hum Genet. 2008;83:373–87.
Guy J, Qi X, Koilkonda RD, Arguello T, Chou T-H, Ruggeri M, et al. Efficiency and safety of AAV-mediated gene delivery of the human ND4 complex I subunit in the mouse visual system. Invest Ophthalmol Vis Sci. 2009;50:4205.
Guy J, Feuer WJ, Davis JL, Porciatti V, Gonzalez PJ, Koilkonda RD, et al. Gene therapy for Leber hereditary optic neuropathy: low- and medium-dose visual results. Ophthalmology. 2017;124:1621–34.
Finsterer J, Zarrouk-Mahjoub S. Re: Guy et al.: gene therapy for Leber hereditary optic neuropathy: low-and medium-dose visual results ( ophthalmology. 2017;124:1621–1634). Ophthalmology. 2018;125:e14-5.
Wan X, Pei H, Zhao M, Yang S, Hu W, He H, et al. Efficacy and safety of rAAV2-ND4 treatment for Leber’s hereditary optic neuropathy. Sci Rep. 2016;6:21587.
Newman NJ, Yu-Wai-Man P, Carelli V, Biousse V, Moster ML, Vignal-Clermont C, et al. Intravitreal gene therapy vs. natural history in patients with Leber hereditary optic neuropathy carrying the m.11778G>A ND4 mutation: systematic review and indirect comparison. Front Neurol. 2021;12:662838.
Yagi T, Seo BB, Nakamaru-Ogiso E, Marella M, Barber-Singh J, Yamashita T, et al. Possibility of transkingdom gene therapy for Complex I diseases. Biochim Biophys Acta. 2006;1757:708–14.
Seo BB, Kitajima-Ihara T, Chan EK, Scheffler IE, Matsuno-Yagi A, Yagi T. Molecular remedy of complex I defects: rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc Natl Acad Sci U S A. 1998;95:9167–71.
Seo BB, Matsuno-Yagi A, Yagi T. Modulation of oxidative phosphorylation of human kidney 293 cells by transfection with the internal rotenone-insensitive NADH-quinone oxidoreductase (NDI1) gene of Saccharomyces cerevisiae. Biochim Biophys Acta. 1999;1412:56–65.
Seo BB, Nakamaru-Ogiso E, Flotte TR, Yagi T, Matsuno-Yagi A. A single-subunit NADH-quinone oxidoreductase renders resistance to mammalian nerve cells against complex I inhibition. Mol Ther. 2002;6:336–41.
Bai Y, Hájek P, Chomyn A, Chan E, Seo BB, Matsuno-Yagi A, et al. Lack of complex I activity in human cells carrying a mutation in MtDNA-encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH-quinone oxidoreductase (NDI1) gene. J Biol Chem. 2001;276:38808–13.
Chadderton N, Palfi A, Millington-Ward S, Gobbo O, Overlack N, Carrigan M, et al. Intravitreal delivery of AAV-NDI1 provides functional benefit in a murine model of Leber hereditary optic neuropathy. Eur J Hum Genet. 2013;21:62–8.
Hagstrom JE, Hegge J, Zhang G, Noble M, Budker V, Lewis DL, et al. A facile nonviral method for delivering genes and siRNAs to skeletal muscle of mammalian limbs. Mol Ther. 2004;10:386–98.
Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–42.
Jackson CB, Turnbull DM, Minczuk M, Gammage PA. Therapeutic manipulation of mtDNA Heteroplasmy: a Shifting Perspective. Trends Mol Med. 2020;26:698–709.
Taylor RW, Chinnery PF, Turnbull DM, Lightowlers RN. Selective inhibition of mutant human mitochondrial DNA replication in vitro by peptide nucleic acids. Nat Genet. 1997;15:212–5.
Egholm M, Buchardt O, Nielsen PE, Berg RH. Peptide nucleic acids (PNA). Oligonucleotide analogs with an achiral peptide backbone. J Am Chem Soc. 1992;114:1895–7.
Muratovska A. Targeting peptide nucleic acid (PNA) oligomers to mitochondria within cells by conjugation to lipophilic cations: implications for mitochondrial DNA replication, expression and disease. Nucleic Acids Res. 2001;29:1852–63.
Clayton DA. Transcription and replication of mitochondrial DNA. Hum Reprod. 2000;15(Suppl 2):11–7.
Loutre R, Heckel A-M, Jeandard D, Tarassov I, Entelis N. Anti-replicative recombinant 5S rRNA molecules can modulate the mtDNA heteroplasmy in a glucose-dependent manner. PLoS ONE. 2018;13:e0199258.
Comte C, Tonin Y, Heckel-Mager A-M, Boucheham A, Smirnov A, Auré K, et al. Mitochondrial targeting of recombinant RNAs modulates the level of a heteroplasmic mutation in human mitochondrial DNA associated with Kearns Sayre Syndrome. Nucleic Acids Res. 2013;41:418–33.
Tonin Y, Heckel A-M, Vysokikh M, Dovydenko I, Meschaninova M, Rötig A, et al. Modeling of antigenomic therapy of mitochondrial diseases by mitochondrially addressed RNA targeting a pathogenic point mutation in mitochondrial DNA. J Biol Chem. 2014;289:13323–34.
Arimura S. Effects of mitoTALENs-directed double-strand breaks on plant mitochondrial genomes. Genes. 2021;12:153.
Bian W-P, Chen Y-L, Luo J-J, Wang C, Xie S-L, Pei D-S. Knock-in strategy for editing human and zebrafish mitochondrial DNA using mito-CRISPR/Cas9 system. ACS Synth Biol. 2019;8:621–32.
Moraes CT. What regulates mitochondrial DNA copy number in animal cells? Trends Genet. 2001;17:199–205.
Peeva V, Blei D, Trombly G, Corsi S, Szukszto MJ, Rebelo-Guiomar P, et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat Commun. 2018;9:1727.
Moretton A, Morel F, Macao B, Lachaume P, Ishak L, Lefebvre M, et al. Selective mitochondrial DNA degradation following double-strand breaks. PLoS ONE. 2017;12:e0176795.
Nissanka N, Bacman SR, Plastini MJ, Moraes CT. The mitochondrial DNA polymerase gamma degrades linear DNA fragments precluding the formation of deletions. Nat Commun. 2018;9:2491.
Srivastava S. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum Mol Genet. 2001;10:3093–9.
Minczuk M, Papworth MA, Kolasinska P, Murphy MP, Klug A. Sequence-specific modification of mitochondrial DNA using a chimeric zinc finger methylase. Proc Natl Acad Sci USA. 2006;103:19689–94.
D’Aquila P, Ronchetti D, Gallo Cantafio ME, Todoerti K, Taiana E, Fabiani F, et al. epigenetic regulation of mitochondrial quality control genes in multiple myeloma: a sequenom massARRAY pilot investigation on HMCLs. JCM. 2021;10:1295.
Bacman SR, Williams SL, Pinto M, Peralta S, Moraes CT. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat Med. 2013;19:1111–3.
Hashimoto M, Bacman SR, Peralta S, Falk MJ, Chomyn A, Chan DC, et al. MitoTALEN: a general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol Ther. 2015;23:1592–9.
Yang Y, Wu H, Kang X, Liang Y, Lan T, Li T, et al. Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell. 2018;9:283–97.
Reddy P, Ocampo A, Suzuki K, Luo J, Bacman SR, Williams SL, et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell. 2015;161:459–69.
Kazama T, Okuno M, Watari Y, Yanase S, Koizuka C, Tsuruta Y, et al. Curing cytoplasmic male sterility via TALEN-mediated mitochondrial genome editing. Nat Plants. 2019;5:722–30.
Arimura S, Ayabe H, Sugaya H, Okuno M, Tamura Y, Tsuruta Y, et al. Targeted gene disruption of ATP synthases 6–1 and 6–2 in the mitochondrial genome of Arabidopsis thaliana by mitoTALENs. Plant J. 2020;104:1459–71.
Omukai S, Arimura S, Toriyama K, Kazama T. Disruption of mitochondrial open reading frame 352 partially restores pollen development in cytoplasmic male sterile rice. Plant Physiol. 2021;187:236–46.
Yahata N, Matsumoto Y, Omi M, Yamamoto N, Hata R. TALEN-mediated shift of mitochondrial DNA heteroplasmy in MELAS-iPSCs with m.13513G>A mutation. Sci Rep. 2017;7:15557.
Pereira CV, Bacman SR, Arguello T, Zekonyte U, Williams SL, Edgell DR, et al. mitoTev-TALE: a monomeric DNA editing enzyme to reduce mutant mitochondrial DNA levels. EMBO Mol Med. 2018;10:e8084.
Phillips AF, Millet AR, Tigano M, Dubois SM, Crimmins H, Babin L, et al. Single-molecule analysis of mtDNA replication uncovers the basis of the common deletion. Mol Cell. 2017;65:527-538.e6.
Bacman SR, Kauppila JHK, Pereira CV, Nissanka N, Miranda M, Pinto M, et al. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat Med. 2018;24:1696–700.
Kauppila JHK, Baines HL, Bratic A, Simard M-L, Freyer C, Mourier A, et al. A phenotype-driven approach to generate mouse models with pathogenic mtDNA mutations causing mitochondrial disease. Cell Rep. 2016;16:2980–90.
Gammage PA, Viscomi C, Simard M-L, Costa ASH, Gaude E, Powell CA, et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat Med. 2018;24:1691–5.
Pereira CV, Moraes CT. Current strategies towards therapeutic manipulation of mtDNA heteroplasmy. Front Biosci (Landmark Ed). 2017;22:991–1010.
Gammage PA, Moraes CT, Minczuk M. Mitochondrial genome engineering: the revolution may not be CRISPR-Ized. Trends Genet. 2018;34:101–10.
Antón Z, Mullally G, Ford H, van der Kamp MW, Szczelkun MD, Lane JD. Mitochondrial import, health and mtDNA copy number variability using type II and type V CRISPR effectors. J Cell Sci. 2020;133:jcs.248468.
Loutre R, Heckel A-M, Smirnova A, Entelis N, Tarassov I. Can Mitochondrial DNA be CRISPRized: pro and contra. IUBMB Life. 2018;70:1233–9.
Hussain S-RA, Yalvac ME, Khoo B, Eckardt S, McLaughlin KJ. Adapting CRISPR/Cas9 system for targeting mitochondrial genome. Front Genet. 2021;12:627050.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–40.
Swarts DC. Making the cut(s): how Cas12a cleaves target and non-target DNA. Biochem Soc Trans. 2019;47:1499–510.
Tanaka M, Borgeld H-J, Zhang J, Muramatsu S, Gong J-S, Yoneda M, et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease smai into mitochondria. J Biomed Sci. 2002;9:534–41.
Bayona-Bafaluy MP, Blits B, Battersby BJ, Shoubridge EA, Moraes CT. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc Natl Acad Sci USA. 2005;102:14392–7.
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–4.
Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016;353:aaf8729.
Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–71.
Komor AC, Zhao KT, Packer MS, Gaudelli NM, Waterbury AL, Koblan LW, et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T: a base editors with higher efficiency and product purity. Sci Adv. 2017;3:eaao4774.
Salter JD, Smith HC. Modeling the embrace of a mutator: APOBEC selection of nucleic acid ligands. Trends Biochem Sci. 2018;43:606–22.
Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020;583:631–7.
Lee H, Lee S, Baek G, Kim A, Kang B-C, Seo H, et al. Mitochondrial DNA editing in mice with DddA-TALE fusion deaminases. Nat Commun. 2021;12:1190.
Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77:753–9.
Sridharan K, Gogtay NJ. Therapeutic nucleic acids: current clinical status. Br J Clin Pharmacol. 2016;82:659–72.
Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378:625–35.
Kesselheim AS, Avorn J. Approving a problematic muscular dystrophy drug: implications for FDA policy. JAMA. 2016;316:2357–8.
Furukawa R, Yamada Y, Kawamura E, Harashima H. Mitochondrial delivery of antisense RNA by MITO-porter results in mitochondrial RNA knockdown, and has a functional impact on mitochondria. Biomaterials. 2015;57:107–15.
Kawamura E, Hibino M, Harashima H, Yamada Y. Targeted mitochondrial delivery of antisense RNA-containing nanoparticles by a MITO-Porter for safe and efficient mitochondrial gene silencing. Mitochondrion. 2019;49:178–88.
Cerrato CP, Kivijärvi T, Tozzi R, Lehto T, Gestin M, Langel Ü. Intracellular delivery of therapeutic antisense oligonucleotides targeting mRNA coding mitochondrial proteins by cell-penetrating peptides. J Mater Chem B. 2020;8:10825–36.
Venkatesh A, Ali S, Oh RS, Sonntag D, Li Z, McKenty T, et al. Antisense oligonucleotide mediated increase in OPA1 improves mitochondrial function in fibroblasts derived from patients with autosomal dominant optic atrophy (ADOA). Invest Ophthalmol Vis Sci. 2021;62:1482–1482.
Bonifert T, Gonzalez Menendez I, Battke F, Theurer Y, Synofzik M, Schöls L, et al. Antisense oligonucleotide mediated splice correction of a deep intronic mutation in OPA1. Mol Ther Nucleic Acids. 2016;5:e390.
Wassmer SJ, De Repentigny Y, Sheppard D, Lagali PS, Fang L, Coupland SG, et al. XIAP protects retinal ganglion cells in the mutant ND4 mouse model of Leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2020;61:49.
Cheung CHA, Chang Y-C, Lin T-Y, Cheng SM, Leung E. Anti-apoptotic proteins in the autophagic world: an update on functions of XIAP, Survivin, and BRUCE. J Biomed Sci. 2020;27:31.
Perales-Clemente E, Fernández-Silva P, Acín-Pérez R, Pérez-Martos A, Enríquez JA. Allotopic expression of mitochondrial-encoded genes in mammals: achieved goal, undemonstrated mechanism or impossible task? Nucleic Acids Res. 2011;39:225–34.
Acknowledgements
Not applicable.
Funding
The reported study was supported by the Ministry of Science and Higher Education of the Russian Federation (Agreement No. 075–15-2021–1346) and the State Task of the Laboratory of Genome Editing for Biomedicine and Animal Health of the Belgorod State National Research University (Agreement No. FZWG-2021–0016). P.R.A. is funded by CO Research Trust, UK.
Author information
Authors and Affiliations
Contributions
VOS—main idea and conceptualization, project conception, literature search, analysis and interpretation, writing original draft, visualization; MVK—literature search, analysis and interpretation, writing original draft, validation; MYS—literature search, analysis and interpretation, writing original draft; AEB—conceptualization, literature search, writing original draft; TVE—literature search, writing original draft; MVK—formal analysis, review & editing; MVP—formal analysis, review & editing; AVD—main idea and conceptualization, funding acquisition; PRA—supervision, review & editing, validation. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: affiliations of author Vladislav O. Soldatov were incorrectly published - Vladislav O. Soldatov1,2,6,8*. It should be Vladislav O. Soldatov1,2,6*.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Soldatov, V.O., Kubekina, M.V., Skorkina, M.Y. et al. Current advances in gene therapy of mitochondrial diseases. J Transl Med 20, 562 (2022). https://doi.org/10.1186/s12967-022-03685-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-022-03685-0