
Abstract
Primary mitochondrial disease (PMD) is a group of complex genetic disorders that arise due to pathogenic variants in nuclear or mitochondrial genomes. Although PMD is one of the most prevalent inborn errors of metabolism, it often exhibits marked phenotypic variation and can therefore be difficult to recognise. Current treatment for PMD revolves around supportive and preventive approaches, with few disease-specific therapies available. However, over the last decade there has been considerable progress in our understanding of both the genetics and pathophysiology of PMD. This has resulted in the development of a plethora of new pharmacological and non-pharmacological therapies at varying stages of development. Many of these therapies are currently undergoing clinical trials. This review summarises the latest emerging therapies that may become mainstream treatment in the coming years. It is distinct from other recent reviews in the field by comprehensively addressing both pharmacological non-pharmacological therapy from both a bench and a bedside perspective. We highlight the current and developing therapeutic landscape in novel pharmacological treatment, dietary supplementation, exercise training, device use, mitochondrial donation, tissue replacement gene therapy, hypoxic therapy and mitochondrial base editing.
Similar content being viewed by others

Novel pharmacological agents are under development to treat mitochondrial disease. |
There is an emerging field of non-pharmacological treatments that have not been comprehensively reviewed in the literature. |
Rigorous randomised control trials with objective patient-centred primary outcomes are required to establish evidence-based guidelines. |
1 Introduction
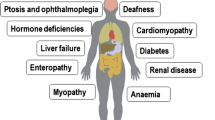
Primary mitochondrial diseases (PMDs) are clinically diverse metabolic disorders arising from pathogenic variants in genes that affect mitochondrial function [1]. These pathogenic variants generally impair mitochondrial structure and function by disrupting multiple mitochondrial processes such as oxidative phosphorylation, ion transport across the mitochondrial membrane and mitochondrial fission and fusion [2]. PMDs are caused by a vast spectrum of pathogenic variants in both nuclear (nDNA) and mitochondrial DNA (mtDNA) [3]. mtDNA is a 16.5 Kb, double-stranded, circular molecule of DNA that is distinct from nDNA and present in multiple copies throughout the cell’s cytoplasm. Typically, individuals have only one mtDNA genotype (inherited maternally), and all mitochondrial genomes are genetically identical. However, wild-type and mutated maternal alleles can coexist, a situation known as heteroplasmy [3]. When a tissue-specific threshold is exceeded, the severity of symptoms in patients with PMD is often directly associated with the level of heteroplasmy in affected tissues [4]. Nuclear DNA (nDNA) by contrast, is inherited from both parents and resides in strands in a cell’s nucleus. nDNA is also substantially larger in size (3 billon base pairs with 20,000 known genes). PMDs are clinically challenging to diagnose and treat as they are highly heterogeneous regardless of genetic aetiology [4]. PMD can affect almost any organ at any age. However, with the exception of Leber’s Hereditary Optic Neuropathy (LHON), PMDs rarely causes disease isolated to a single tissue. Indeed there are a number of well characterised syndromic presentations of PMD (and perhaps more cases with non-syndromic multiple organ involvement) that present throughout the age spectrum [5]. At present, the prevalence of PMD in the UK population is estimated at 1 in 4,300 (adults and children) [6]. However, this prevalence rate can vary substantially between different ethnic populations [7]. Current treatment of PMD involves symptomatic management with a limited therapeutic arsenal of specific disease-modifying therapies [8, 9]. However, in recent years there has been a plethora of emerging pharmacological and non-pharmacological treatment [8, 9]. The purpose of this review is to summarise the current and emerging therapeutic landscape in PMD. The article is distinguishable from other recent reviews in the field by focusing on many of the non-pharmacological treatments in development, from both a basic science and a clinical perspective.
2 Current Therapy
A common challenge faced in rare genetic diseases such as PMD is a historic paucity of disease-specific treatment [8,9,10]. The management of PMD conditions remains a poorly researched area and there is little expert advice available for the treatment of specific aspects of mitochondrial disease [8,9,10]. The involvement of several organs can pose additional patient management challenges for clinicians [8,9,10]. The current mainstay of treatment involves the optimisation of conventional organ-specific care and regular health surveillance for potential complications of PMD [8,9,10]. There have been multiple attempts to standardise the diagnosis and treatment of PMD both nationally and internationally [11,12,13]. Several guidelines and consensus recommendations have been generated by clinical experts from the UK (https://www.newcastle-mitochondria.com/) and the USA, with the aim of providing guidance to healthcare professionals in the management of patients with mitochondrial disease [11,12,13].
Although few in number, some disease-specific therapies for PMD are available [8,9,10]. For instance, mitochondrial disease patients with a cofactor deficiency (i.e. thiamine, riboflavin, biotin or niacin) respond to cofactor treatment [14, 15]. The genes implicated in these genetic cofactor deficiencies are summarised in Table 1 [16,17,18,19,20]. Beyond these deficiency syndromes, there is limited clinical use of disease-specific treatment. Controversially in 2015, the European Medicines Agency (EMA) licenced the synthetic antioxidant idebenone for the treatment of LHON in patients over 12 years of age [22]. However, the UK-based National Institute for Health and Care Excellence (NICE) prevented the provision of idebenone through the NHS, given the expense of the drug and low-quality evidence for its efficacy (further discussed in section 5.2) [21]. Long-term survival in patients with mitochondrial neurogastrointestinal encephalomyopathy has been increased by allogeneic hematopoietic stem cell transplantation [22]. While not universally accepted, there is emerging evidence that multiple agents can prevent and treat stroke-like episodes in MELAS, with L-arginine now recommended in North American (Mitochondrial Medicine Society) consensus guidelines [11]. Additionally, taurine supplementation has been shown in a small phase 3 randomised controlled trial (RCT) to prevent stroke-like episodes in MELAS [23]. However, further studies in larger patient cohorts are required before taurine supplementation can be recommended.
3 Emerging Therapies
In recent years there has been an increase in the development of novel pharmacological and non-pharmacological treatment [8,9,10]. To review this landscape, we searched the MEDLINE database via PubMed, using the following keywords: ‘mitochondrial disorder(s)’, ‘mitochondrial disease(s)’, ‘treatment(s)’ and ‘therapy’ (with no search limits). We supplemented this review by searchingClinicalTrials.gov for ‘mitochondrial disorders’ (up until 10 August 2020) (with no search limits). From our search, we discovered that there has been an exponential growth in the number of clinical trials registered to start since 2000 (Fig. 1), with a total of 232 studies conducted (of which 182 were interventional). The most common subcategories identified in the treatment of PMD included pharmacological (N = 105 trials) and dietary supplements (N = 15 trials) (Fig. 1, Online Supplementary Material (OSM)).

The cumulative number and subcategories of clinical trials registered at ClinicalTrials.gov using the search term ‘mitochondrial disorders’ up until 10/08/2020.
4 Emerging Pharmacological Treatment
The translation of modern genomic technology into clinical practice and research has facilitated major advances in the development of treatment for rare genetic diseases [24]. Accordingly, there has been an exponential expansion in the development of novel pharmacological treatment for PMD (Table 2), facilitated by regulatory and financial incentives aimed at increasing investment in orphan drug discovery [8,9,10, 25]. Many developing therapies may also be of potential use in a range of other neurological diseases [26]. It is encouraging that many of these newly developed compounds are now undergoing clinical trials in humans. To date, 131 clinical trials involving a pharmacological intervention in the treatment of mitochondrial disease have been registered publicly on Clinicaltrials.gov (Fig. 1). The specific preclinical mechanisms of these newly developed therapies can be categorised into four major groups: (1) increasing the cellular concentration of mitochondria and nicotinamide adenine dinucleotide+ (NAD+), (2) protecting mitochondria from damage, (3) improving mitochondrial function and (4) restoring mtDNA homeostasis [27,28,29,30].
4.1 Increasing the Cellular Concentration of Mitochondrial NAD+ and Mitochondria
Numerous novel pharmacological agents have been developed that aim to increase the cellular concentration of NAD+ and the number of mitochondria [31]. NAD+ is an oxidising cofactor that accepts electrons in redox reactions [31]. This allows electrons to be carried from one reaction to another, a fundamental process required for oxidative metabolism [31]. Higher concentrations of both mitochondria and NAD+ in cells may increase oxidative phosphorylation in diseased tissue, ultimately improving symptoms and disease progression in PMD [31]. Acipimox, bezafibrate, omaveloxolone and REN001 aim to treat mitochondrial disease by increasing the cellular mitochondrial concentration. In contrast, KL1333 is purported to increase the cellular concentration of NAD+.
Acipimox (5-carboxyl-2-methyl pyrazine 1-oxide) is a niacin derivative and nicotinic acid analogue with activity as a hypolipidaemic agent [32,33,34]. The drug is currently used for the treatment of hyperlipidaemia in non- insulin-requiring diabetes mellitus [32,33,34]. Acipimox has been shown to have a direct effect on human skeletal muscle mitochondrial function in patients with diabetes [35]. Acipimox lowers plasma free fatty acids by inhibiting lipolysis through indirectly inhibiting hormone-sensitive lipase [36]. This in turn promotes an increase in NAD+, sirtuin 1 (SIRT1) activation and enhanced mitochondrial gene expression [37]. Although acipimox has a favourable side-effect profile, it can cause facial flushing, which may limit patient retention (and blinding) in clinical trials [32]. An ongoing double-blinded, placebo-controlled RCT of the efficacy of acipimox in patients with mitochondrial myopathy aims to repurpose acipimox as a potent stimulator of mitochondrial biogenesis.
Bezafibrate is a fibrate drug that increases mitochondrial biogenesis [38]. It was originally licensed in 1978 to treat hyperlipidaemia and therefore has long-term evidence of a favourable side-effect profile [39]. An open‐label, non‐randomised trial in patients with mitochondrial myopathy (m.3243A>G point pathogenic variant, n = 6) demonstrated that bezafibrate decreased the number of complex IV‐deficient muscle fibres and improved cardiac function, with no significant adverse effects reported [29]. However, this improvement was accompanied by an increase in serum biomarkers of PMD (e.g. GDF15 [growth and differentiation factor 15]), raising concerns about long-term treatment effects in patients with PMD [38]. A further phase 2 RCT is required to further ascertain the efficacy and safety of bezafibrate.
REN001, a selective and orally bioavailable peroxisome proliferator-activated receptor delta (PPARδ) agonist, is currently being trialled as a treatment for patients with fatty acid oxidation (FAO) disorders and mitochondrial myopathies [40]. This compound was previously used for prevention of respiratory failure and immobilisation-induced skeletal muscle atrophy [40]. PPARδ regulates genes involved in several cellular metabolic processes including glucose homeostasis, fatty acid synthesis and storage, and fatty acid mobilization and metabolism [41]. PPARδ is expressed in liver, skeletal muscle and adipose tissue [41]. In animal models, PPARδ increases the number of muscle fibres with high mitochondrial content and improves FAO [41]. The premise for treatment in patients with PMD is that this selective PPARδ agonist may ameliorate the cellular energy deficit via increasing the proportion of wild-type mtDNA [42]. The other potential benefits include increasing FAO and OXPHOS activity, thereby enhancing mitochondrial ATP-generating capacity and boosting mitochondrial biogenesis [59]. An open-label study to evaluate the tolerability of this drug over 12 weeks was paused due to the COVID-19 pandemic (NCT03862846).
Omaveloxolone is an oleanolic triterpenoid that prevents nuclear factor erythroid 2-related factor 2 (Nrf2) degradation, inhibiting the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pro-inflammatory pathway [43]. This inhibition has been shown to improve oxidative phosphorylation and mitochondrial biogenesis in patients with mitochondrial myopathy (n = 53) [43]. A phase 2 double-blind, dose-ranging, placebo-controlled RCT of omaveloxolone in 53 patients with mitochondrial myopathy failed to meet its primary outcome (peak workload assessed via an incremental cycling exercise test) [44]. However, omaveloxolone treatment resulted in a lower heart rate and blood lactate level during a constant workload (submaximal) exercise test, suggesting some improvement in mitochondrial function [44]. Further clinical trials with methodical refinement may therefore be required to ascertain the efficacy of omaveloxolone.
KL1333 is a substrate for NAD(P)H:dehydrogenase [quinone]1, which produces nicotinamide adenine. KL1333 then transfers these electrons to the mitochondrial electron transport system, directly promoting ATP production [45]. The elevated level of NAD+ leads to the activation of mitochondrial biogenesis pathways SIRT1, 5’-adenosine monophosphate-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), thereby improving mitochondrial function [45]. Given these beneficial effects on mitochondrial function, a double-blinded, parallel-group, placebo-controlled, phase 1 RCT to assess the pharmacokinetics and pharmacodynamics of this compound in patients with PMD has recently been launched (NCT03056209).
4.2 Protecting Mitochondria from Damage
Protecting functioning mitochondria from damage that could ultimately reduce their ability to perform oxidative phosphorylation is an emerging mechanism for potential treatment [8,9,10]. There are multiple mechanisms to potentially protect mitochondria including increasing the production of endogenous antioxidants (cysteamine), reducing the production of toxic reactive oxygen species (ROS) (KH17 and elamipretide), and acting as a potent synthetic antioxidant to absorb free radicals before they damage mitochondria (EPI-743 and idebenone). ROS are reactive compounds that contain oxygen in its reduced chemically reactive form (O−2) [46]. These compounds are a by-product of normal metabolism of oxygen but during metabolic stress (e.g. during illness) ROS levels can dramatically increase, this process can damage proteins from intracellular organelles including mitochondria in a process known as oxidative stress [46]. Some compounds have also been designed with a dual mechanism. For example, KH176 protects mitochondria by acting as both a synthetic antioxidant and a redox modulator.
Cysteamine is a US Food and Drug Administration (FDA)-approved treatment for nephropathic cystinosis that has the potential to be repurposed for the treatment of PMD [47]. Its primary mechanism is to increase glutathione biosynthesis (a potent antioxidant). In vivo (C elegans and zebrafish) and in vitro (human fibroblast) experiments have shown that cysteamine improves mitochondrial function by protecting complex I and IV from damage [48]. However, a phase 2 open-label, dose-escalating study assessing the safety, tolerability and efficacy of cysteamine treatment in PMD (n = 36) was terminated due to a lack of efficacy, leaving the repurposing of cysteine in jeopardy (NCT02023866).
KH176 is a potent intracellular reduction-oxidation‐modulating compound developed to treat PMD [49]. The compound also acts as a synthetic antioxidant (removing oxidative stress). Through this duel mechanism, KH176 can reduce both ROS production and absorption. KH176 has demonstrated safety in a phase 1 trial [50]. A subsequent phase 2 double-blind, placebo-controlled, crossover RCT in 18 patients with m.3243A>G pathogenic variants demonstrated that the medication was safe and tolerated (KHENERGY Study) (NCT02909400) [51]. However, this study was exploratory, and therefore lacked any defined primary and secondary outcomes. A further phase 3 trial is currently being conducted to ascertain the efficacy and safety of KH176 (NCT02909400).
Elamipretide is a tetrapeptide that targets the mitochondria, stabilizing cardiolipin whilst reducing the production of ROS [52]. A phase 1/2 multicentre, double-blind, placebo-controlled RCT investigating elamipretide for the treatment of mitochondrial myopathy in 36 patients (MMPOWER study) demonstrated safety and efficacy (increase in 6-min walk test distance (6MWTD)) [53]. However, a follow-up phase 3 study (n = 218 patients) (MMPOWER-3, NCT03323749) did not meet its primary end-point (increase in 6MWTD), leaving the efficacy of elamipretide unclear.
EPI-743 (also known as vatiquinone) is a novel small-molecule catalytic, 2-electron transfer NAD(P)H quinone dehydrogenase 1 (NQO1) cofactor that decreases the production of intracellular glutathione [54]. The drug is a para-benzoquinone analogue of CoQ10 with up to 10,000 times its antioxidant potency [54]. A 2012 phase 2A open-label trial of EPI-743 for children with Leigh syndrome (n = 10) demonstrated a statistically significant objective neurological improvement in the Newcastle Paediatric Mitochondrial Disease Scale (NPMDS), with no significant drug-related adverse events [55]. Based on these results, further phase 1/2 clinical trials are currently being conducted (Table 2). EPI-743 has been used in randomized, double-blind, placebo-controlled clinical trials in children with Leigh syndrome (NCT01721733 and NCT02352896). Enrolled patients first participated in a 6-month placebo-controlled phase, which was followed by a 30-month extension phase to assess the long-term drug safety and impact on disease severity [56]. The results of this study were reported at the United Mitochondrial Disease Foundation Annual Meeting, Seattle, Washington, USA, 2016. However, despite evidence of EPI-743 reducing hospital admission, the study is yet to be published [56]. This highlights the need to publish trial results rapidly and transparently. Of note, the FDA recently granted approval for a study to investigate the safety and efficacy of the drug in 94 children with terminal genetically confirmed inherited respiratory chain diseases (NCT01370447). This study is scheduled to report in December 2021.
Idebenone is a short-chain hydrosoluble quinone synthetic analog of CoQ10 that acts as an electron carrier in the respiratory chain [57, 58]. The drug was originally developed to treat CoQ10 deficiency but was unsuccessful in this regard and has recently been repurposed and licensed to treat patients with LHON [59]. A double-blind, phase 2 RCT (RHODOS) demonstrated limited efficacy, whereby no significant difference in the primary outcome (visual acuity) was observed compared to placebo treatment [59]. However, subgroup analysis demonstrated an improvement in discordant visual acuity [59]. Two phase 4 clinical trials are currently being conducted to further understand the benefits of idebenone (NCT02774005 and NCT02771379) (Table 2). Further studies are also underway to investigate the efficacy of idebenone in other PMDs (Table 2). As previously mentioned, although idebenone has been licensed by the EMA, NICE recommended that it should not be available for prescription through the NHS in the UK [22]. This is a consequence of the high cost of treatment relative to the lack of robust evidence demonstrating efficacy. Additionally, NICE also cited a lack of long-term follow up in the original clinical trial. NICE also expressed additional concern at the exclusion of a patient from analysis resulting in a substantial demographic difference between the control and treatment group [22]. The current situation regarding the prescription of idebenone will likely be repeated in other emerging disease-specific treatments for PMD in the NHS and other public health systems worldwide. Orphan drugs in PMD have high development costs. When combined with the rarity of PMD, high drug prices are likely. Therefore, if the evidence for efficacy is not robust, NICE are unlikely to allow public prescription, and, in turn, prevent long-term follow-up studies that can elucidate efficacy.
4.3 Improving Mitochondrial Function
Various pharmacological agents have been developed that improve the efficiency of oxidative phosphorylation in diseased mitochondria as opposed to increasing their number [8,9,10]. ABI-009 and dichloroacetate are two medications from this class that are currently undergoing clinical trials.
ABI-009 (known as nab-sirolimus) is a nanoparticle undergoing phase 2a open-label trials evaluating safety, tolerability and efficacy in the treatment of Leigh or Leigh-like syndrome (NCT03747328). ABI-009 is composed of the antibiotic rapamycin bound to nanoparticle albumin. The compound is reported to improve mitochondrial function by increasing amino acid catabolism, reducing glycolysis [29]. The reduction in glycolytic intermediates increases mitochondrial oxidative phosphorylation by an unknown mechanism [29]. Preclinical evidence in a Leigh syndrome murine model has demonstrated that ABI-009 reduces neuroinflammation and the size of brains lesions, delaying the onset of neurological symptoms [29]. However, the validity of preclinical evidence is limited as the Leigh syndrome murine model reflects only one genotype (NDUFS4). This contrasts with Leigh syndrome in humans, which is caused by many different genetic variants involving both mtDNA and nDNA [60].
Dichloroacetate (DCA) is a drug that stimulates the pyruvate dehydrogenase (PDH) complex (a common site of pathological metabolic dysfunction) by altering its phosphorylation state and thus promoting complex stability [18]. Three clinical trials have been conducted for DCA in mitochondrial genetic disease [19, 61, 62]. Two of these trials showed an improvement in post-prandial and exercise blood lactate compared to placebo. However, these studies did not show any improvement in patient quality of life (assessed through global assessment of treatment efficacy scores) or aerobic metabolism function (assed through a 1-h treadmill test) [61, 62]. However, a third trial (conducted in older subjects) was terminated due to drug-associated neuropathy [19]. A phase 3 quadruple blinded RCT is currently being conducted to investigate the efficacy of DCA in pyruvate dehydrogenase complex deficiency (PDCD) (NCT02616484).
4.4 Restoring mtDNA Homeostasis
Loss of function pathogenic variants in the synthesis of mtDNA result in a reduction in mitochondrial oxidative phosphorylation. This results in numerous PMDs including mitochondrial depletion syndrome (MDS) and mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), for which there is emerging pharmacological treatment.
In mitochondrial depletion syndrome (MDS), there is a reduction in the production of mtDNA. This is due to an autosomal recessive loss-of-function pathogenic variant, resulting in defective thymidine kinase 2 (TK2) activity. TK2 is a critical enzyme in mtDNA synthesis through its phosphorylation of precursor nucleosides (deoxycytidine and deoxythymidine). Pathogenic variants result in a reduced pool of deoxynucleoside triphosphates, which are required for mtDNA replication and maintenance. A retrospective analysis of 92 patients with genetically confirmed TK2 deficiency revealed three myopathic phenotypes with divergent survival: infantile-onset, childhood-onset and late-onset myopathies [63]. Oral supplementary deoxynucleoside administration is an emerging therapy in MDS that bypasses the defective enzymatic pathway [64, 65]. Preclinical evidence in murine models has demonstrated that deoxynucleoside treatment is both tolerable and effective in restoring mtDNA copy number and respiratory chain enzyme activity, resulting in a prolonged lifespan [65]. A combined phase 1/2 open-label compassionate-use study (n = 16) of oral deoxynucleoside to treat TK2-deficient patients demonstrated a favourable side-effect profile and improved survival and motor function [46]. A further combined phase 1/2 trial (n = 20 patients) is currently being conducted to expand on this work (NCT03639701).
MNGIE is an autosomal recessive disorder caused by loss-of-function pathogenic variants in the gene that encodes for thymidine phosphorylase (TYMP), a critical enzyme in mtDNA synthesis [66]. The disease presents in patients aged in their 20s and is characterised by progressive gastrointestinal dysmotility, neuropathy and leukoencephalopathy [66]. There is currently no disease-specific therapy. Erythrocyte-encapsulated thymidine phosphorylase (EE-TP) is an emerging clinical therapy. EE TP is intravenously infused into a patient and aims to replace defective thymidine phosphorylase activity [66]. A phase 2, multicentre, multiple-dose, open-label trial is currently being conducted to assess the medication’s safety, tolerability, pharmacodynamics and efficacy [66].
5 Dietary Supplements as Emerging Treatment in Mitochondrial Disease
A mainstay in the treatment of many inborn errors of metabolism is dietary manipulation. This may include dietary restriction strategies or supplementation that aim to limit toxic effects, maximise residual enzyme function or utilise alternative metabolic pathways [67, 68]. While dietary restriction has not found favour in managing mitochondrial disease, dietary supplements have been purported to be effective through a range of cellular mechanisms [67]. There are numerous theoretical reasons why certain supplements may improve metabolic functions. For example, creatine could potentially be used as an alternative energy source boosting mitochondrial ATP production [69]. Other supplements including antioxidants (vitamin C, vitamin E and alpha-lipoic acid) and electron acceptors (CoQ10 or carthine) may remove ROS from cells, improving mitochondrial function. Supplements could also bypass a cellular defect (e.g. a deficiency in the activity of complexes I, II or III in the electron transport chain) [69,70,71]. The potential benefits of dietary supplementation are demonstrated by an open-label trial (NCT03973203) in five patients with adult-onset mitochondrial myopathy. In this trial, dietary supplement with niacin (i.e. an NAD+ booster) increased muscle strength and mitochondrial biogenesis of affected patients when compared to healthy matched controls [72]. Despite their theoretical potential, there is a paucity of robust evidence to support the routine use of dietary supplements in all types of mitochondrial diseases.
Although there may be a potential clinical benefit of dietary supplementation use, there is a lack of high-quality evidence demonstrating their efficacy [10, 12]. The majority of favourable evidence available is from low-quality studies that lack robust designs to effectively demonstrate that the intervention studied was responsible for the reported benefit (e.g. case reports, retrospective studies and small underpowered non-RCTs) (Table 3) [73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106]. Another issue that has been encountered in dietary interventional trials is the heterogeneity in the composition, combination, dosing regimens and duration of treatment [12]. Generating high-quality evidence for the use of dietary supplements through RCTs is challenging. Studies are often underpowered with highly variable designs that lack standardization. This makes comparing study results difficult and prevents meta-analysis [70]. Recruitment to clinical trials is also difficult as supplements are commonly used by patients, excluding their inclusion into the study [70]. Additionally, some patients are reluctant to join a study that might assign them to a placebo group where they will be required to temporarily stop their usual supplement use [70]. Further work is required to build a more rigorous evidence base through larger multicentre RCTs.
The lack of high quality evidence means that there are no regulatory approved dietary supplements to treat, mitigate or cure PMD in either the EU or the USA [107]. Despite these legalities, dietary supplements are frequently used for this purpose either as monotherapy or as polytherapy (known as a mitochondrial cocktail) due to the theoretical synergistic benefit [67]. However, there is no known standardisation nationally or internationally, with their clinical use often dependent on centre or physician preference as opposed to evidence-based guidelines [108].
5.1 Diet as an Emerging Therapy
The neurological manifestations of mitochondrial disease can make both food consumption and ingestion more difficult [73]. In addition to this, the diet of patients with mitochondrial disease tends to be deficient in protein, calcium and fluid [109, 110]. These dietary deficiencies mean that patients are at higher risk of malnutrition than the general population [109, 110]. Therefore, individualised dietary counselling and monitoring is a simple approach to address these effectively. The role of dietary adaptation in PMD can also include several other strategies depending on the clinical features and genetic predisposition. Among them, the three most investigated diets are the ketogenic diet (KD), low-residue diet (LRD) and high-nitrate diet [111,112,113]. However, despite the potential offered by these diets in improving symptoms of mitochondrial function in PMD, there have been few disease-specific clinical trials. This is likely due to challenges in patient recruitment and retention, given the restrictive nature of dietary interventional trials.
The KD involves replacing the majority of a patient’s dietary consumption of carbohydrates with fat (defined as < 20 g of carbohydrate per day) [114]. This reduction in carbohydrate places the body into a metabolic state of ketosis [114]. The KD may have multiple benefits in PMD. The first benefit is that the KD may better control intractable epilepsy regardless of the underlying neurological or metabolic cause [114]. The specific mechanism of how the KD reduces epileptic activity is unclear [114]. There have been numerous RCTs demonstrating that the KD has efficacy in treating non-disease-specific intractable epilepsy [115]. However, a 2020 Cochrane review concluded that the current evidence base is inconclusive [115]. The review cited not only a lack of studies but criticised the majority of the RTCs for both a lack of power but also a short follow-up [115]. However, numerous observational studies have demonstrated a benefit of the KD in reducing intractable epilepsy in PMD [111, 116, 117]. Furthermore, a Swedish cohort study in paediatric patients with PDCD (n = 19) demonstrated that a KD not only reduced epileptic seizures, but also improved ataxia, sleep disturbance, speech/language development, social functioning and frequency of hospitalizations [118]. Additionally, the state of ketosis may be beneficial to mitochondrial function; there is preclinical evidence from in vivo (using cultured human cell lines) and in vitro (murine models) that the state of ketosis improves mitochondrial function (Table 4) [119,120,121,122]. However, no study in man has been conducted to asses if this could translate into clinical practice. To establish if the KD is an effective treatment for PMD-specific (and non-specific) intractable epilepsy and improving in vivo mitochondrial function, a large RCT is required. This trial should not only assess an epileptic control cohort, but also include objective assessments of mitochondrial and global neurological function. This could potentially be a sub-arm of a larger definitive trial to establish the efficacy of the KD in treating intractable epilepsy.
The second diet investigated in MD is a low residue diet (LRD; a diet with less than 10 g fibre per day) [123]. Mitochondrial dysfunction in the smooth muscle of the gastrointestinal tract causes various symptoms including abdominal pain, bloating and severe constipation, frequently leading to hospital admission [112]. Therefore, in contrast to mainstream dietary advice, the LRD requires patients to reduce their intake of dietary fibre. The diet was initially designed for people with inflammatory bowel disease to prevent the obstruction of inflamed bowel because fibre provides resistance to the defective peristaltic action of diseased smooth muscle [112]. A phase 2 feasibility study of the efficacy and acceptability of a LRD in adult patients with PMD has been completed with the results due to be published imminently (NCT03388528).
There is also evidence that a high-nitrate diet can benefit the function of mitochondria [113]. Nitrate is an inorganic anion that is found in high quantities in root vegetables [113]. Once ingested, it is converted into bioactive nitrogen oxides, which reduces the concentration of ATP/ADP translocase improving basal mitochondrial function [113]. A double-blind crossover trial in healthy participants demonstrated that a high-nitrate diet improved oxidative phosphorylation and reduced oxygen demand during exercise in harvested muscle [113]. To date, there have been no studies extending this finding to patients with PMD, leaving this a ripe area of research in PMD despite the likely challenges of patient recruitment and retention. Dietary supplementation with nitrate supplements could be an alternative therapeutic avenue.
6 Device Use as Emerging Treatment
Bioengineering has resulted in the development of multiple devices to treat various neurological diseases with many now approved by the FDA [124]. There are two promising devices under development to treat PMD: near-infrared light-emitting diode (NIR-LED) and transcranial direct current stimulation (TDCS) (Table 5) [125,126,127]. These devices have the potential to open another valuable therapeutic front in both treating the symptoms of PMD and also altering the underlying pathophysiology. Like many therapies for PMD, there is currently a lack of evidence to assess their efficacy, meaning that rigorous clinical trials are needed to realise this potential.
NIR-LED uses low-energy lasers or light-emitting diodes to radiate tissue with light at near infrared range (630–1,000 nm) of the electromagnetic spectrum [125]. There is in vivo and in vitro preclinical evidence that the technology improves mitochondrial function [125]. The NIR light radiates the mitochondrial photoacceptor molecule cytochrome C oxidase, thereby activating it. Once activated, cytochrome C oxidase increases the expression of mitochondrial proteins involved in antioxidant protection and energy production [125]. To date, there has only been one open-label phase 1/2 clinical trial assessing the therapeutic benefit of using NIR-LED to treat LHON (n = 4 patients) (NCT01389817). The study intended to assess the N95 retinal ganglion cell peak as its primary outcome. However, it was terminated due to difficulties obtaining this measurement in LHON patients. Further studies are therefore required that utilise more refined measurable outcomes.
TDCS is a neuromodulatory technique that involves delivery of a constant direct current to the cortex of the brain via electrodes placed on the skin of the head [128]. This current alters the resting membrane potential of cortical neurons, resulting in neuronal depolarisation or hyperpolarisation [128, 129]. The resulting decrease in firing neurons has been shown to have therapeutic potential in patients with resistant epilepsy and status epilepticus with emerging preclinical and clinical evidence [128, 129]. In a double-blind, sham-controlled RCT, TDCS reduced seizure frequency in patients with refractory focal epilepsy (n = 70) [129]. Two case reports have also utilised this technology for treatment of POLG‐related mitochondrial epilepsy [126, 127], with conflicting findings: one study reported efficacy in control status epilepticus while the other observed no benefit [126, 127].
7 Exercise as a Treatment
Progressive proximal skeletal myopathy is a prominent clinical feature of many different PMDs. Impaired mitochondrial oxidative phosphorylation due to respiratory chain defects frequently result in symptoms of exercise intolerance, muscle weakness and muscle fatigue even with minimal exertion [130]. As well as a reduced aerobic capacity, patients with PMD are less physically active compared to matched controls, consequently exacerbating muscle deconditioning and function [131].
Exercise training involves frequently repeated bouts of skeletal muscle contraction. Aerobic exercise training is widely accepted as a potent stimulator of mitochondrial biogenesis, therefore representing a potential therapeutic intervention for mitochondrial disease [87, 88]. Indeed, it is firmly established that aerobic exercise training in patients with PMD results in a number of metabolic and physiological adaptations including an increase in oxygen consumption and work capacity, mediated by an increase in oxygen extraction [132,133,134,135,136,137,138,139,140,141]. Importantly, these improvements have been shown to translate into clinically meaningful increases in quality of life and self-reported activities of daily living [134, 136, 141]. Other significant benefits include enhanced magnetic resonance spectroscopy (MRS) recovery kinetics and a decrease in blood lactate at rest and with exercise, suggesting reduced anaerobic metabolism [132, 136, 142].
At a molecular genetic level, one of the first exercise interventional studies in patients with PMD raised concerns of a potential selective amplification of mitochondrial pathogenic variant deletion [143]. However, an increase in mDNA heteroplasmy has not been replicated by this group or others [134, 137]. Further, skeletal muscle biopsy studies in patients with mitochondrial disease have demonstrated an aerobic exercise training-induced improvement in skeletal muscle oxidative capacity and mitochondrial biogenesis reflected by an increase in respiratory chain complex activities and mitochondrial volume [132, 134, 141, 144].
Resistance exercise training has also been investigated as a potential intervention for patients with mitochondrial myopathy caused by sporadic mtDNA point pathogenic variants or single large-scale mDNA deletions. It was postulated that the myofiber damage-induced activation of muscle stem cells (satellite cells; predominantly containing wild-type mtDNA) may consequently reduce mtDNA mutant load (i.e. the mitochondrial DNA shifting theory) [145,146,147]. Strength training has been shown to effectively increase muscle strength1,2 despite no significant effect on mutated mtDNA, muscle fibre area or mtDNA copy number2. Importantly, strength training benefits translate to an enhanced aerobic capacity (p = 0.06), mediated largely by an increased peak oxygen extraction (a-vO2 difference) [148].
The abundance of evidence suggests that exercise training is efficacious, well tolerated and safe; no studies report clinical adverse events or detrimental effects on muscle (serum creatine kinase levels), perceived fatigue or self-reported activities of daily living [132, 134, 136, 137, 141, 143, 147, 148]. Our recently conducted systematic review and meta-analysis to determine the effect of exercise across a range of outcomes in patients with neuromuscular disorders, which includes mitochondrial disease, also supports these findings [149]. While further research is required to fully elucidate the mechanisms of the potential therapeutic benefit of exercise training, these results encourage the translation of exercise treatment into the clinical care of patients with mitochondrial disease [150]. A combination of progressively prescribed aerobic and strength exercise training is recommended; however, it should be noted that this is limited to those who are physically capable, highlighting the necessity for the development of pharmaceutical drugs that mimic the effect of exercise [12]. A summary of the literature is presented in Table 6 [151].
8 Mitochondrial Replacement Therapy
The severity and intensity of PMD means that preventing genetic transmission is highly desirable. Current preventative therapies include voluntary childlessness, adoption, oocyte donation, prenatal testing and preimplantation genetic diagnosis (PGD) [152]. There are numerous considerations for each technique. Voluntary childlessness, adoption and oocyte donation may be personally or culturally unacceptable [152]. Prenatal testing is only suitable for women who have a low risk of transmission of mtDNA disease and who would be willing to terminate an affected fetus [153]. There is also a procedural risk to the viability of the pregnancy with a miscarriage rate of 1–2% following chorionic villus biopsy [153]. PGD involves testing in vitro-generated embryos for a pathogenic variant before subsequent implantation. This means the technique is limited by the number of embryos available with pathogenic variant levels that are unlikely to result in severe PMD [152]. Additionally, PGD is a modified version of in vitro fertilization and therefore has associated risks, namely increased risk of multiple and ectopic pregnancies [153]. In contrast to Mendelian genetic disorders, PGD and prenatal testing face unique limitations in PMD. Hetroplasmy (variable levels of mutated mtDNA) and a highly heterogeneous disease phenotype make it impossible to predict PMD severity in any subsequent children. This highlights the need to prevent transmission of mtDNA but allow the transmission of nDNA.
Over the last decade, and following a change in UK law, it has become possible to offer mitochondrial replacement therapy (MRT), otherwise known as mitochondrial donation, as a means of preventing transmission of mtDNA disease [152, 154]. In the UK, mitochondrial donation can be offered to women with homoplasmic or high-level heteroplasmic mtDNA variants when there is an expectation of transmitting severe disease and preimplantation genetic diagnosis is not considered suitable [119, 120]. Two slightly different forms of MRT can be employed, pronuclear transfer (PNT) or maternal spindle transfer (MST). PNT involves the in vitro fertilization of oocytes from both the intending mother and donor with sperm from the intending father [152, 154]. The pronuclei are then removed from the donor oocyte and replaced with the pronuclei from the intending mother’s fertilised oocyte [152, 154]. In the alternative approach, MST, the metaphase II spindles from the donor oocyte are removed and replaced with those from the intending mother’s oocyte. The resulting oocyte is then fertilised in vitro with the intending father’s sperm [152, 154]. In both techniques the resulting foetus has the nuclear DNA of both parents but the mitochondrial DNA of the donor [152, 154]. MRT has produced viable embryos and subsequent offspring in murine, primates and man [155,156,157]. However, mice conceived through MST MRT have been shown to have defective biochemical processes (increased production of ROS, defective proteostasis and insulin resistance), resulting in mice that aged prematurely [158]. These studies used inbred conplastic murine strains, a contrasting situation to the high genetic diversity observed in the human population. There are some other possible technical limitations, with some 20% of stem cell lines derived from embryos after PNT reverting to the maternal mtDNA genotype. The cause for this reversion remains unknown, but it only appears to occur following prolonged culture in an unrestricted environment [158].
Despite the legal and ethical challenges posed by MRT, in 2015 the UK became the first country in the world to create the legislative framework required to allow clinical trials to assess the long-term efficacy and safety of the technology in humans [159]. Multiple children have now been born through MRT globally in unregulated environments with no current reported developmental concerns [160]. A clinical trial of the first 75 children born via MRT is currently being conducted at the Wellcome Centre for Mitochondrial Research at the Newcastle University. The study aims to identify if there are any long-term developmental concerns from the technique in humans.
9 Other Therapeutic Options
There are various other exciting clinical therapies currently being developed including cell replacement, gene therapy, mitochondrial augmentation therapy (MAT), hypoxic therapy and mtDNA base editing [129, 161, 162, 162, 163].
Gene therapy is an emerging experimental technique using vectors (e.g. viruses) to replace pathogenic genes with the respective functional wild types [164]. As PMD is caused by both pathogenic variants in mtDNA and nDNA, there are two targets for genes therapy [164]. Various in vivo and in vitro preclinical studies have successfully replaced mutated nDNA and mtDNA using adeno-associated viruses (AAVs) and CRISPR/Cas9 in a variety of PMDs [130, 131, 133, 134]. Although much of the literature is preclinical, the technology has now moved into early-stage clinical trials in LHON. A phase 1 clinical trial of nine patients demonstrated that recombinant AAV vector ND4 (rAAV2) gene therapy was safe and tolerated in a long-term follow-up [165]. A further phase 1 study is currently investigating the safety and efficacy of an AAV vector gene therapy of LHON in 30 patients (NCT02064569), whilst a phase 1/2 study is currently investigating the safety and efficacy of scAAV2-P1ND4v2 in 30 patients (NCT02161380).
Cell-replacement therapy involves restoring the loss of function of a diseased tissue by replacing the defective cells with effective cells [166]. In PMD, a patient’s cells are replaced with cells from another individual with unaffected nDNA or mtDNA leading to the production of functioning mitochondrial proteins [22, 167]. Cell-replacement therapy is only likely to be effective in treating PMD when the disease presents as a consequence of defective mitochondria in a specific replaceable tissue (e.g. the liver or bone marrow), and not disorders that affect tissues systemically. Hematopoietic stem cell transplantation has been shown to increases long-term survival in patients with mitochondrial neurogastrointestinal encephalomyopathy [22]. Cell-replacement therapy via liver transplantation has been shown to improve multiple symptoms in ethylmalonic encephalopathy due to pathogenic variants in ETHE1 [167]. As cell-replacement therapy is likely to be a disease-specific therapy in PMD, further research is required to conduct intervention specific trials in specific diseases.
MAT is another emerging treatment for PMD that has recently entered early-stage clinical trials. In MAT, human mitochondria are extracted from white blood cells derived from a patient’s mothers’ serum [163]. A patient is then subsequently treated with filgrastim to stimulate the release of CD34+ hematopoietic stem cells. These stem cells can then be removed by apheresis and co-cultured in vitro with the donor’s extracted mitochondria [163]. The stem cells can then be returned to the affected individual by infusion [163]. The FDA recently granted fast-track, orphan drug and rare paediatric disease designations to the treatment. This facilitated the development of an open-label combined phase 1 and 2 clinical trial (NCT03384420) (n = 7) investigating the efficacy of the treatment in Pearson’s syndrome (a rare and severe neonatal mitochondrial deletion syndrome) due to report in 2021.
There is emerging preclinical in vivo and in vitro experimental evidence that stimulating a hypoxic response may benefit mitochondrial function in mitochondrial disease [168]. Models of PMD were created by treating human cells lines and zebrafish models with respiratory chain (RC) inhibitors [168]. When these models were treated with a hypoxia-inducible factor they demonstrated increased growth when compared to non-treated controls [168]. Additionally, in vivo murine models of Leigh syndrome (Ndufs4 KO) demonstrated improved survival and locomotor activity after continuously breathing 11% oxygen [168]. Although these experiments are promising they need to be validated in more complex clinically relevant models of human PMD [168]. Despite the potential benefit offered by hypoxic therapy, it is likely to be highly challenging to organically create a hypoxic response in humans. Given that patients with Leigh syndrome may already have an impaired respiratory drive due to brainstem lesions, it would be highly impractical and potentially dangerous for patients to continuously breath air with a depleted oxygen content (supplied by a breathing device). However, the hypoxic response may be better exploited as a pharmacological target.
Manipulating the mitochondrial genome to remove pathogenic variants is of interest to clinicians and researchers, but a feasible method is still elusive [1]. Several targeted gene-editing techniques such as anti-sense peptide nucleic acids (endonucleases, TALENS (transcription activator-like effector nucleases), zinc-finger nucleases and Crispr-Cas9) have recently emerged in the laboratory to do this, but none of them are anywhere near human trials [1]. Even in the laboratory settings, there are challenges to overcome, namely the impermeability of the inner mitochondrial membrane and the lack of selective molecules to cause timely degradation of mutant mtDNA [1]. Thus, the manipulation of mtDNA to date has been limited to the targeted destruction of the mitochondrial genome by designer nucleases [169]. The use of RNA-free DddA-derived cytosine base editors promise the potential to enable CRISPR-free mitochondrial base editing [169]. Although this technology offers the exciting prospect of being able to remove mutations from the mitochondrial genome, this preclinical approach is yet to be used even in basic animal models.
10 Conclusion
The rarity of PMD along with its clinical and scientific complexity makes developing treatment an enormous challenge, with only one disease-specific pharmacological agent in current clinical practice. However, there have been major recent advances in the fundamental understanding of PMD through the establishment of national cohorts and the widespread use of gene agnostic next-generation sequencing approaches. This has resulted in a truly exciting emerging therapeutic arsenal. To develop effective treatment and establish a more concrete evidence base for existing clinical guidelines, extensive high-powered comprehensive clinical trials must now be conducted. Given the heterogeneity and rarity of PMD, these trials will need multicentre collaboration and detailed phenotype-specific subgroup analysis. This is a major challenge given that some of the most beneficial therapies (i.e. exercise, diet, supplements and off-patent repurposed medications) have limited commercial incentive. This highlights the role that charities and governments must play in funding the development of an evidence-base that will enable translation of treatment from the bench to bedside in PMD. Historically, clinical trials have typically used biomarkers as primary outcomes that are not relevant to clinicians or to patients’ lives. Many of these biomarkers have been used due to their ability to allow statistical analysis and not because they can capture the benefit of a treatment on a patient’s quality of life. This highlights the importance of co-designing clinical trials, based on what matters to patients. Any future trials of value should utilise established international cohorts of patients with PMD and employ internationally agreed outcome measures that are clinically meaningful and patient relevant. This will not only aid the recruitment of patients but provide an international infrastructure to assess the efficacy of treatment.
References
Russell OM, Gorman GS, Lightowlers RN, Turnbull DM. Mitochondrial diseases: hope for the future. Cell. 2020;181:168–88.
Wallace DC, Fan W. The pathophysiology of mitochondrial disease as modeled in the mouse. Genes Dev. 2009;23:1714–36.
Garagnani P, Pirazzini C, Giuliani C, Candela M, Brigidi P, Sevini F, et al. The Three Genetics (Nuclear DNA, Mitochondrial DNA, and Gut Microbiome) of Longevity in Humans Considered as Metaorganisms. Biomed Res Int [Internet]. 2014 [cited 2020 Aug 26];2014. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4017728/
Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–42.
Liang C, Ahmad K, Sue CM. The broadening spectrum of mitochondrial disease: shifts in the diagnostic paradigm. Biochim Biophys Acta. 2014;1840:1360–7.
Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77:753–9.
Hakonen AH, Davidzon G, Salemi R, Bindoff LA, Van Goethem G, DiMauro S, et al. Abundance of the POLG disease mutations in Europe, Australia, New Zealand, and the United States explained by single ancient European founders. Eur J Hum Genet. 2007;15:779–83.
Zhang L, Zhang Z, Khan A, Zheng H, Yuan C, Jiang H. Advances in drug therapy for mitochondrial diseases. Ann Transl Med [Internet]. 2020 [cited 2020 Aug 14];8. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6995731/
Hirano M, Emmanuele V, Quinzii CM. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018;62:467–81.
Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012;CD004426.
Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015;17:689–701.
Parikh S, Goldstein A, Karaa A, Koenig MK, Anselm I, Brunel-Guitton C, et al. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2017;2:19.
McCormick EM, Lott MT, Dulik MC, Shen L, Attimonelli M, Vitale O, et al. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum Mutat. 2020;2:2.
Distelmaier F, Haack TB, Wortmann SB, Mayr JA, Prokisch H. Treatable mitochondrial diseases: cofactor metabolism and beyond. Brain Oxf Acad. 2017;140:e11–e11.
Mancuso M, Orsucci D, Filosto M, Simoncini C, Siciliano G. Drugs and mitochondrial diseases: 40 queries and answers. Expert Opin Pharmacother. 2012;13:527–43.
Melegh B, Trombitás K. Valproate treatment induces lipid globule accumulation with ultrastructural abnormalities of mitochondria in skeletal muscle. Neuropediatrics. 1997;28:257–61.
Brecht M, Richardson M, Taranath A, Grist S, Thorburn D, Bratkovic D. Leigh syndrome caused by the MT-ND5 m.13513G>A mutation: a case presenting with WPW-like conduction defect, cardiomyopathy, hypertension and hyponatraemia. JIMD Rep. 2015;19:95–100.
Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev. 2008;60:1478–87.
Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66:324–30.
Kanabus M, Heales SJ, Rahman S. Development of pharmacological strategies for mitochondrial disorders. Br J Pharmacol. 2014;171:1798–817.
Idebenone for treating visual impairment in adults and young people with Leber’s hereditary optic neuropathy - NHS England - Citizen Space [Internet]. [cited 2020 Sep 16]. Available from: https://www.engage.england.nhs.uk/consultation/idebenone-for-treating-visual-impairment/
Halter J, Schüpbach W, Casali C, Elhasid R, Fay K, Hammans S, et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant. 2011;46:330–7.
Ohsawa Y, Hagiwara H, Nishimatsu S, Hirakawa A, Kamimura N, Ohtsubo H, et al. Taurine supplementation for prevention of stroke-like episodes in MELAS: a multicentre, open-label, 52-week phase III trial. J Neurol Neurosurg Psychiatry. 2019;90:529–36.
Posey JE. Genome sequencing and implications for rare disorders. Orphanet J Rare Dis. 2019;14:153.
Sharma A, Jacob A, Tandon M, Kumar D. Orphan drug: development trends and strategies. J Pharm Bioallied Sci. 2010;2:290–9.
Murphy MP, Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov. 2018;17:865–86.
Whitaker RM, Corum D, Beeson CC, Schnellmann RG. Mitochondrial biogenesis as a pharmacological target: a new approach to acute and chronic diseases. Annu Rev Pharmacol Toxicol. 2016;56:229–49.
Civiletto G, Dogan SA, Cerutti R, Fagiolari G, Moggio M, Lamperti C, et al. Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol Med. 2018;10:2.
Johnson SC, Yanos ME, Kayser E-B, Quintana A, Sangesland M, Castanza A, et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. 2013;342:1524–8.
McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 2018;27(439–449):e5.
Cantó C, Menzies K, Auwerx J. NAD+ metabolism and the control of energy homeostasis—a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53.
O’Kane MJ, Trinick TR, Tynan MB, Trimble ER, Nicholls DP. A comparison of acipimox and nicotinic acid in type 2b hyperlipidaemia. Br J Clin Pharmacol. 1992;33:451–3.
Christie AW, McCormick DK, Emmison N, Kraemer FB, Alberti KG, Yeaman SJ. Mechanism of anti-lipolytic action of acipimox in isolated rat adipocytes. Diabetologia. 1996;39:45–53.
Pike NB. Flushing out the role of GPR109A (HM74A) in the clinical efficacy of nicotinic acid. J Clin Invest. 2005;115:3400–3.
Koev D, Zlateva S, Susic M, Babic D, Profozic V, Skrabalo Z, et al. Improvement of lipoprotein lipid composition in type II diabetic patients with concomitant hyperlipoproteinemia by acipimox treatment. Results of a multicenter trial. Diabetes Care. 1993;16:1285–90.
Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–70.
Houtkooper RH, Auwerx J. Exploring the therapeutic space around NAD+. J Cell Biol. 2012;199:205–9.
Steele H, Gomez‐Duran A, Pyle A, Hopton S, Newman J, Stefanetti RJ, et al. Metabolic effects of bezafibrate in mitochondrial disease. EMBO Mol Med [Internet]. 2020 [cited 2020 Aug 18];12. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7059007/
Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation. American Heart Association; 2000;102:21–7.
Oliver WR, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci USA. 2001;98:5306–11.
Wang Y-X, Zhang C-L, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294.
Bastin J, Aubey F, Rötig A, Munnich A, Djouadi F. Activation of peroxisome proliferator-activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients’ cells lacking its components. J Clin Endocrinol Metab. 2008;93:1433–41.
Reisman SA, Gahir SS, Lee CY, Proksch JW, Sakamoto M, Ward KW. Pharmacokinetics and pharmacodynamics of the novel Nrf2 activator omaveloxolone in primates. Drug Des Devel Ther. 2019;13:1259–70.
Madsen KL, Buch AE, Cohen BH, Falk MJ, Goldsberry A, Goldstein A, et al. Safety and efficacy of omaveloxolone in patients with mitochondrial myopathy. Neurology. 2020;94:e687–98.
Seo K-S, Kim J-H, Min K-N, Moon J-A, Roh T-C, Lee M-J, et al. KL1333, a novel NAD+ modulator, improves energy metabolism and mitochondrial dysfunction in MELAS fibroblasts. Front Neurol. 2018;9:552.
Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–50.
Ariceta G, Giordano V, Santos F. Effects of long-term cysteamine treatment in patients with cystinosis. Pediatr Nephrol. 2019;34:571–8.
Guha S, Konkwo C, Lavorato M, Mathew ND, Peng M, Ostrovsky J, et al. Pre-clinical evaluation of cysteamine bitartrate as a therapeutic agent for mitochondrial respiratory chain disease. Hum Mol Genet. 2019;28:1837–52.
Beyrath J, Pellegrini M, Renkema H, Houben L, Pecheritsyna S, van Zandvoort P, et al. KH176 safeguards mitochondrial diseased cells from redox stress-induced cell death by interacting with the thioredoxin system/peroxiredoxin enzyme machinery. Sci Rep. 2018;8:6577.
Koene S, Spaans E, Van Bortel L, Van Lancker G, Delafontaine B, Badilini F, et al. KH176 under development for rare mitochondrial disease: a first in man randomized controlled clinical trial in healthy male volunteers. Orphanet J Rare Dis [Internet]. 2017 [cited 2020 Aug 26];12. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5644106/
Janssen MCH, Koene S, de Laat P, Hemelaar P, Pickkers P, Spaans E, et al. The KHENERGY study: safety and efficacy of KH176 in mitochondrial m.3243A>G spectrum disorders. Clin Pharmacol Ther. 2019;105:101–11.
Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171:2029–50.
Karaa A, Haas R, Goldstein A, Vockley J, Weaver WD, Cohen BH. Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology. 2018;90:e1212–21.
Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab. 2012;105:91–102.
Martinelli D, Catteruccia M, Piemonte F, Pastore A, Tozzi G, Dionisi-Vici C, et al. EPI-743 reverses the progression of the pediatric mitochondrial disease–genetically defined Leigh syndrome. Mol Genet Metab. 2012;107:383–8.
Enns GM, Cohen BH. Clinical Trials in Mitochondrial Disease: An Update on EPI-743 and RP103. Journal of Inborn Errors of Metabolism and Screening [Internet]. SAGE PublicationsSage CA: Los Angeles, CA; 2017 [cited 2020 Sep 14]; Available from: https://journals.sagepub.com/https://doi.org/10.1177/2326409817733013
Leonardi A, Crasci L, Panico A, Pignatello R. Antioxidant activity of idebenone-loaded neutral and cationic solid-lipid nanoparticles. Pharm Dev Technol. 2015;20:716–23.
Giorgio V, Petronilli V, Ghelli A, Carelli V, Rugolo M, Lenaz G, et al. The effects of idebenone on mitochondrial bioenergetics. Biochim Biophys Acta. 2012;1817:363–9.
Klopstock T, Yu-Wai-Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain. 2011;134:2677–86.
Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008;7:312–20.
Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519–31.
Duncan GE, Perkins LA, Theriaque DW, Neiberger RE, Stacpoole PW. Dichloroacetate therapy attenuates the blood lactate response to submaximal exercise in patients with defects in mitochondrial energy metabolism. J Clin Endocrinol Metab. 2004;89:1733–8.
Garone C, Taylor RW, Nascimento A, Poulton J, Fratter C, Domínguez-González C, et al. Retrospective natural history of thymidine kinase 2 deficiency. J Med Genet. 2018;55:515–21.
Lopez-Gomez C, Hewan H, Sierra C, Akman HO, Sanchez-Quintero MJ, Juanola-Falgarona M, et al. Bioavailability and cytosolic kinases modulate response to deoxynucleoside therapy in TK2 deficiency. EBioMedicine. 2019;46:356–67.
Lopez-Gomez C, Levy RJ, Sanchez-Quintero MJ, Juanola-Falgarona M, Barca E, Garcia-Diaz B, et al. Deoxycytidine and deoxythymidine treatment for thymidine kinase 2 deficiency. Ann Neurol. 2017;81:641–52.
Bax BE, Levene M, Bain MD, Fairbanks LD, Filosto M, Kalkan Uçar S, et al. Erythrocyte encapsulated thymidine phosphorylase for the treatment of patients with mitochondrial neurogastrointestinal encephalomyopathy: study protocol for a multi-centre, multiple dose, open label trial. J Clin Med. 2019;8:2.
Goldstein A, Wolfe LA. The elusive magic pill: finding effective therapies for mitochondrial disorders. Neurotherapeutics. 2013;10:320–8.
Camp KM, Krotoski D, Parisi MA, Gwinn KA, Cohen BH, Cox CS, et al. Nutritional interventions in primary mitochondrial disorders: developing an evidence base. Mol Genet Metab. 2016;119:187–206.
Hayashi G, Cortopassi G. Oxidative stress in inherited mitochondrial diseases. Free Radic Biol Med. 2015;88:10–7.
Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol. 2009;11:414–30.
Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018;27:529–47.
Pirinen E, Auranen M, Khan NA, Brilhante V, Urho N, Pessia A, et al. Niacin cures systemic NAD+ deficiency and improves muscle performance in adult-onset mitochondrial myopathy. Cell Metab. 2020;31(1078–1090):e5.
Barbiroli B, Medori R, Tritschler HJ, Klopstock T, Seibel P, Reichmann H, et al. Lipoic (thioctic) acid increases brain energy availability and skeletal muscle performance as shown by in vivo 31P-MRS in a patient with mitochondrial cytopathy. J Neurol. 1995;242:472–7.
Koga Y, Ishibashi M, Ueki I, Yatsuga S, Fukiyama R, Akita Y, et al. Effects of L-arginine on the acute phase of strokes in three patients with MELAS. Neurology. 2002;58:827–8.
Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Tanabe Y, et al. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64:710–2.
Koga Y, Akita Y, Junko N, Yatsuga S, Povalko N, Fukiyama R, et al. Endothelial dysfunction in MELAS improved by l-arginine supplementation. Neurology. 2006;66:1766–9.
Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Katayama K, et al. MELAS and l-arginine therapy. Mitochondrion. 2007;7:133–9.
Gimenes AC, Bravo DM, Nápolis LM, Mello MT, Oliveira ASB, Neder JA, et al. Effect of L-carnitine on exercise performance in patients with mitochondrial myopathy. Braz J Med Biol Res. 2015;48:354–62.
Campos Y, Huertas R, Lorenzo G, Bautista J, Gutiérrez E, Aparicio M, et al. Plasma carnitine insufficiency and effectiveness of L-carnitine therapy in patients with mitochondrial myopathy. Muscle Nerve. 1993;16:150–3.
El-Hattab AW, Hsu JW, Emrick LT, Wong LJC, Craigen WJ, Jahoor F, et al. Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol Genet Metab. 2012;105:607–14.
El-Hattab AW, Emrick LT, Hsu JW, Chanprasert S, Almannai M, Craigen WJ, et al. Impaired nitric oxide production in children with MELAS syndrome and the effect of arginine and citrulline supplementation. Mol Genet Metab. 2016;117:407–12.
Musumeci O, Naini A, Slonim AE, Skavin N, Hadjigeorgiou GL, Krawiecki N, et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology. 2001;56:849–55.
Chen RS, Huang CC, Chu NS. Coenzyme Q10 treatment in mitochondrial encephalomyopathies. Short-term double-blind, crossover study. Eur Neurol. 1997;37:212–8.
Bresolin N, Doriguzzi C, Ponzetto C, Angelini C, Moroni I, Castelli E, et al. Ubidecarenone in the treatment of mitochondrial myopathies: a multi-center double-blind trial. J Neurol Sci. 1990;100:70–8.
Chan A, Reichmann H, Kögel A, Beck A, Gold R. Metabolic changes in patients with mitochondrial myopathies and effects of coenzyme Q10 therapy. J Neurol. 1998;245:681–5.
Gold R, Seibel P, Reinelt G, Schindler R, Landwehr P, Beck A, et al. Phosphorus magnetic resonance spectroscopy in the evaluation of mitochondrial myopathies: results of a 6-month therapy study with coenzyme Q. Eur Neurol. 1996;36:191–6.
Tarnopolsky MA, Roy BD, MacDonald JR. A randomized, controlled trial of creatine monohydrate in patients with mitochondrial cytopathies. Muscle Nerve. 1997;20:1502–9.
Komura K, Hobbiebrunken E, Wilichowski EKG, Hanefeld FA. Effectiveness of creatine monohydrate in mitochondrial encephalomyopathies. Pediatr Neurol. 2003;28:53–8.
Klopstock T, Querner V, Schmidt F, Gekeler F, Walter M, Hartard M, et al. A placebo-controlled crossover trial of creatine in mitochondrial diseases. Neurology. 2000;55:1748–51.
Kornblum C, Schröder R, Müller K, Vorgerd M, Eggers J, Bogdanow M, et al. Creatine has no beneficial effect on skeletal muscle energy metabolism in patients with single mitochondrial DNA deletions: a placebo-controlled, double-blind 31P-MRS crossover study. Eur J Neurol. 2005;12:300–9.
Quijada-Fraile P, O’Callaghan M, Martín-Hernández E, Montero R, Garcia-Cazorla À, de Aragón AM, et al. Follow-up of folinic acid supplementation for patients with cerebral folate deficiency and Kearns-Sayre syndrome. Orphanet J Rare Dis [Internet]. 2014 [cited 2020 Aug 18];9. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4302586/
Pineda M, Ormazabal A, López-Gallardo E, Nascimento A, Solano A, Herrero MD, et al. Cerebral folate deficiency and leukoencephalopathy caused by a mitochondrial DNA deletion. Ann Neurol. 2006;59:394–8.
Majamaa K, Rusanen H, Remes AM, Pyhtinen J, Hassinen IE. Increase of blood NAD+ and attenuation of lactacidemia during nicotinamide treatment of a patient with the MELAS syndrome. Life Sci. 1996;58:691–9.
Bernsen PL, Gabreëls FJ, Ruitenbeek W, Hamburger HL. Treatment of complex I deficiency with riboflavin. J Neurol Sci. 1993;118:181–7.
Ogle RF, Christodoulou J, Fagan E, Blok RB, Kirby DM, Seller KL, et al. Mitochondrial myopathy with tRNA(Leu(UUR)) mutation and complex I deficiency responsive to riboflavin. J Pediatr. 1997;130:138–45.
Garone C, Donati MA, Sacchini M, Garcia-Diaz B, Bruno C, Calvo S, et al. Mitochondrial encephalomyopathy due to a novel mutation in ACAD9. JAMA Neurol. 2013;70:1177–9.
Lou HC. Correction of increased plasma pyruvate and plasma lactate levels using large doses of thiamine in patients with kearns-sayre syndrome. Arch Neurol. 1981;38:469–469.
Rodriguez MC, MacDonald JR, Mahoney DJ, Parise G, Beal MF, Tarnopolsky MA. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve. 2007;35:235–42.
Bernsen PL, Gabreëls FJ, Ruitenbeek W, Sengers RC, Stadhouders AM, Renier WO. Successful treatment of pure myopathy, associated with complex I deficiency, with riboflavin and carnitine. Arch Neurol. 1991;48:334–8.
Marriage BJ, Clandinin MT, Macdonald IM, Glerum DM. Cofactor treatment improves ATP synthetic capacity in patients with oxidative phosphorylation disorders. Mol Genet Metab. 2004;81:263–72.
Artuch R, Vilaseca MA, Pineda M. Biochemical monitoring of the treatment in paediatric patients with mitochondrial disease. J Inherit Metab Dis. 1998;21:837–45.
Pl P. The treatment of mitochondrial myopathies and encephalomyopathies. Biochim Biophys Acta. 1995;1271:275–80.
Matthews PM, Ford B, Dandurand RJ, Eidelman DH, O’Connor D, Sherwin A, et al. Coenzyme Q10 with multiple vitamins is generally ineffective in treatment of mitochondrial disease. Neurology. 1993;43:884–90.
Ramaekers VT, Weis J, Sequeira JM, Quadros EV, Blau N. Mitochondrial complex I encephalomyopathy and cerebral 5-methyltetrahydrofolate deficiency. Neuropediatrics. 2007;38:184–7.
Argov Z, Bank WJ, Maris J, Eleff S, Kennaway NG, Olson RE, et al. Treatment of mitochondrial myopathy due to complex III deficiency with vitamins K3 and C: a 31P-NMR follow-up study. Ann Neurol. 1986;19:598–602.
Toscano A, Fazio MC, Vita G, Cannavó S, Bresolin N, Bet L, et al. Early-onset cerebellar ataxia, myoclonus and hypogonadism in a case of mitochondrial complex III deficiency treated with vitamins K3 and C. J Neurol. 1995;242:203–9.
Starr RR. Too little, too late: ineffective regulation of dietary supplements in the United States. Am J Public Health. 2015;105:478–85.
Marriage B, Clandinin MT, Glerum DM. Nutritional cofactor treatment in mitochondrial disorders. J Am Diet Assoc. 2003;103:1029–38.
de Laat P, Zweers HEE, Knuijt S, Smeitink JM, Wanten GJA, Janssen MCH. Dysphagia, malnutrition and gastrointestinal problems in patients with mitochondrial disease caused by the m3243A>G mutation. Neth J Med. 2015;73:30–6.
Zweers H, Janssen MCH, Leij S, Wanten G. Patients with mitochondrial disease have an inadequate nutritional intake. JPEN J Parenter Enteral Nutr. 2018;42:581–6.
Martikainen MH, Päivärinta M, Jääskeläinen S, Majamaa K. Successful treatment of POLG-related mitochondrial epilepsy with antiepileptic drugs and low glycaemic index diet. Epileptic Disord. 2012;14:438–41.
Finsterer J, Frank M. Gastrointestinal manifestations of mitochondrial disorders: a systematic review. Therap Adv Gastroenterol. 2017;10:142–54.
Larsen FJ, Schiffer TA, Borniquel S, Sahlin K, Ekblom B, Lundberg JO, et al. Dietary inorganic nitrate improves mitochondrial efficiency in humans. Cell Metab. 2011;13:149–59.
Ludwig DS. The ketogenic diet: evidence for optimism but high-quality research needed. J Nutr. 2020;150:1354–9.
Martin-McGill KJ, Bresnahan R, Levy RG, Cooper PN. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst Rev. 2020;6:1903.
Lee YM, Kang HC, Lee JS, Kim SH, Kim EY, Lee SK, et al. Mitochondrial respiratory chain defects: underlying etiology in various epileptic conditions. Epilepsia. 2008;49:685–90.
Kang H-C, Lee Y-M, Kim HD, Lee JS, Slama A. Safe and effective use of the ketogenic diet in children with epilepsy and mitochondrial respiratory chain complex defects. Epilepsia. 2007;48:82–8.
Sofou K, Dahlin M, Hallböök T, Lindefeldt M, Viggedal G, Darin N. Ketogenic diet in pyruvate dehydrogenase complex deficiency: short- and long-term outcomes. J Inherit Metab Dis. 2017;40:237–45.
Santra S, Gilkerson RW, Davidson M, Schon EA. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann Neurol. 2004;56:662–9.
Ahola-Erkkilä S, Carroll CJ, Peltola-Mjösund K, Tulkki V, Mattila I, Seppänen-Laakso T, et al. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum Mol Genet. 2010;19:1974–84.
Frey S, Geffroy G, Desquiret-Dumas V, Gueguen N, Bris C, Belal S, et al. The addition of ketone bodies alleviates mitochondrial dysfunction by restoring complex I assembly in a MELAS cellular model. Biochim Biophys Acta Mol Basis Dis. 2017;1863:284–91.
Ahola S, Auranen M, Isohanni P, Niemisalo S, Urho N, Buzkova J, et al. Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol Med. 2016;8:1234–47.
Chiba M, Yoshida T, Komatsu M. From low-residue diets to plant-based diets in inflammatory bowel disease. Dig Dis Sci. 2014;59:3129–30.
Anderson L, Antkowiak P, Asefa A, Ballard A, Bansal T, Bello A, et al. FDA regulation of neurological and physical medicine devices: access to safe and effective neurotechnologies for all Americans. Neuron. 2016;92:943–8.
Eells JT, Wong-Riley MTT, VerHoeve J, Henry M, Buchman EV, Kane MP, et al. Mitochondrial signal transduction in accelerated wound and retinal healing by near-infrared light therapy. Mitochondrion. 2004;4:559–67.
Ng YS, van Ruiten H, Lai HM, Scott R, Ramesh V, Horridge K, et al. The adjunctive application of transcranial direct current stimulation in the management of de novo refractory epilepsia partialis continua in adolescent-onset POLG-related mitochondrial disease. Epilepsia Open. 2018;3:103–8.
Marquardt L, Eichele T, Bindoff LA, Olberg HK, Veiby G, Eichele H, et al. No effect of electrical transcranial direct current stimulation adjunct treatment for epilepsia partialis continua in POLG disease. Epilepsy Behav Rep [Internet]. 2019 [cited 2020 Aug 19];12. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6849077/
Dhamne SC, Ekstein D, Zhuo Z, Gersner R, Zurakowski D, Loddenkemper T, et al. Acute seizure suppression by transcranial direct current stimulation in rats. Ann Clin Transl Neurol. 2015;2:843–56.
Yang D, Wang Q, Xu C, Fang F, Fan J, Li L, et al. Transcranial direct current stimulation reduces seizure frequency in patients with refractory focal epilepsy: a randomized, double-blind, sham-controlled, and three-arm parallel multicenter study. Brain Stimul. 2020;13:109–16.
Taivassalo T, Jensen TD, Kennaway N, DiMauro S, Vissing J, Haller RG. The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain. 2003;126:413–23.
Apabhai S, Gorman GS, Sutton L, Elson JL, Plötz T, Turnbull DM, et al. Habitual physical activity in mitochondrial disease. PLoS ONE. 2011;6:e22294.
Taivassalo T, Shoubridge EA, Chen J, Kennaway NG, DiMauro S, Arnold DL, et al. Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic effects. Ann Neurol. 2001;50:133–41.
Trenell MI, Sue CM, Kemp GJ, Sachinwalla T, Thompson CH. Aerobic exercise and muscle metabolism in patients with mitochondrial myopathy. Muscle Nerve. 2006;33:524–31.
Jeppesen TD, Schwartz M, Olsen DB, Wibrand F, Krag T, Dunø M, et al. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain. 2006;129:3402–12.
Bates MGD, Newman JH, Jakovljevic DG, Hollingsworth KG, Alston CL, Zalewski P, et al. Defining cardiac adaptations and safety of endurance training in patients with m.3243A>G-related mitochondrial disease. Int J Cardiol. 2013;168:3599–608.
Taivassalo T, De Stefano N, Argov Z, Matthews PM, Chen J, Genge A, et al. Effects of aerobic training in patients with mitochondrial myopathies. Neurology. 1998;50:1055–60.
Taivassalo T, Gardner JL, Taylor RW, Schaefer AM, Newman J, Barron MJ, et al. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain. 2006;129:3391–401.
Cejudo P, Bautista J, Montemayor T, Villagómez R, Jiménez L, Ortega F, et al. Exercise training in mitochondrial myopathy: a randomized controlled trial. Muscle Nerve. 2005;32:342–50.
Porcelli S, Marzorati M, Morandi L, Grassi B. Home-based aerobic exercise training improves skeletal muscle oxidative metabolism in patients with metabolic myopathies. J Appl Physiol. 2016;121:699–708.
Newman J, Galna B, Jakovljevic DG, Bates MG, Schaefer AM, McFarland R, et al. Preliminary evaluation of clinician rated outcome measures in mitochondrial disease. J Neuromuscul Dis. 2015;2:151–5.
Jeppesen TD, Dunø M, Schwartz M, Krag T, Rafiq J, Wibrand F, et al. Short- and long-term effects of endurance training in patients with mitochondrial myopathy. Eur J Neurol. 2009;16:1336–9.
Siciliano G, Manca ML, Renna M, Prontera C, Mercuri A, Murri L. Effects of aerobic training on lactate and catecholaminergic exercise responses in mitochondrial myopathies. Neuromuscul Disord. 2000;10:40–5.
Taivassalo T, De Stefano N, Chen J, Karpati G, Arnold DL, Argov Z. Short-term aerobic training response in chronic myopathies. Muscle Nerve. 1999;22:1239–43.
Adhihetty PJ, Taivassalo T, Haller RG, Walkinshaw DR, Hood DA. The effect of training on the expression of mitochondrial biogenesis- and apoptosis-related proteins in skeletal muscle of patients with mtDNA defects. Am J Physiol Endocrinol Metab. 2007;293:E672-680.
Fu K, Hartlen R, Johns T, Genge A, Karpati G, Shoubridge EA. A novel heteroplasmic tRNAleu(CUN) mtDNA point mutation in a sporadic patient with mitochondrial encephalomyopathy segregates rapidly in skeletal muscle and suggests an approach to therapy. Hum Mol Genet. 1996;5:1835–40.
Shoubridge EA, Johns T, Karpati G. Complete restoration of a wild-type mtDNA genotype in regenerating muscle fibres in a patient with a tRNA point mutation and mitochondrial encephalomyopathy. Hum Mol Genet. 1997;6:2239–42.
Taivassalo T, Fu K, Johns T, Arnold D, Karpati G, Shoubridge EA. Gene shifting: a novel therapy for mitochondrial myopathy. Hum Mol Genet. 1999;8:1047–52.
Murphy JL, Blakely EL, Schaefer AM, He L, Wyrick P, Haller RG, et al. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain. 2008;131:2832–40.
Stefanetti RJ, Blain A, Jimenez-Moreno C, Errington L, Ng YS, McFarland R, et al. Measuring the effects of exercise in neuromuscular disorders: a systematic review and meta-analyses. Wellcome Open Res [Internet]. 2020 [cited 2020 Nov 30];5. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7331112/
Stefanetti RJ, Blain A, Jimenez-Moreno C, Errington L, Ng YS, McFarland R, et al. Measuring the effects of exercise in neuromuscular disorders: a systematic review and meta-analyses. Wellcome Open Res [Internet]. 2020 [cited 2020 Sep 17];5. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7331112/
Siciliano G, Simoncini C, Gerfo AL, Orsucci D, Ricci G, Mancuso M. Effects of aerobic training on exercise-related oxidative stress in mitochondrial myopathies. Neuromuscul Disord. 2012;22:S172–7.
Gorman GS, McFarland R, Stewart J, Feeney C, Turnbull DM. Mitochondrial donation: from test tube to clinic. Lancet. 2018;392:1191–2.
Nesbitt V, Alston CL, Blakely EL, Fratter C, Feeney CL, Poulton J, et al. A national perspective on prenatal testing for mitochondrial disease. Eur J Hum Genet. 2014;22:1255–9.
Cree L, Loi P. Mitochondrial replacement: from basic research to assisted reproductive technology portfolio tool-technicalities and possible risks. Mol Hum Reprod. 2015;21:3–10.
Sato A, Kono T, Nakada K, Ishikawa K, Inoue S-I, Yonekawa H, et al. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc Natl Acad Sci USA. 2005;102:16765–70.
Cree LM, Samuels DC, de Sousa Lopes SC, Rajasimha HK, Wonnapinij P, Mann JR, et al. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet. 2008;40:249–54.
Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J, et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–72.
Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, Sánchez-Cabo F, Torroja C, Acín-Pérez R, et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature. 2016;535:561–5.
Castro RJ. Mitochondrial replacement therapy: the UK and US regulatory landscapes. J Law Biosci. 2016;3:726–35.
Zhang J, Liu H, Luo S, Lu Z, Chávez-Badiola A, Liu Z, et al. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod Biomed Online. 2017;34:361–8.
Bouaita A, Augustin S, Lechauve C, Cwerman-Thibault H, Bénit P, Simonutti M, et al. Downregulation of apoptosis-inducing factor in Harlequin mice induces progressive and severe optic atrophy which is durably prevented by AAV2-AIF1 gene therapy. Brain. 2012;135:35–52.
Romero-Moya D, Santos-Ocaña C, Castaño J, Garrabou G, Rodríguez-Gómez JA, Ruiz-Bonilla V, et al. Genetic rescue of mitochondrial and skeletal muscle impairment in an induced pluripotent stem cells model of coenzyme Q10 deficiency. Stem Cells. 2017;35:1687–703.
Lightowlers RN, Chrzanowska-Lightowlers ZM, Russell OM. Mitochondrial transplantation-a possible therapeutic for mitochondrial dysfunction? Mitochondrial transfer is a potential cure for many diseases but proof of efficacy and safety is still lacking. EMBO Rep. 2020;2:e50964.
Slone J, Huang T. The special considerations of gene therapy for mitochondrial diseases. NPJ Genom Med. 2020;5:7.
Vignal C, Uretsky S, Fitoussi S, Galy A, Blouin L, Girmens J-F, et al. Safety of rAAV2/2-ND4 gene therapy for leber hereditary optic neuropathy. Ophthalmology. 2018;125:945–7.
Lindvall O, Björklund A. Cell replacement therapy: helping the brain to repair itself. NeuroRx. 2004;1:379–81.
Dionisi-Vici C, Diodato D, Torre G, Picca S, Pariante R, Giuseppe Picardo S, et al. Liver transplant in ethylmalonic encephalopathy: a new treatment for an otherwise fatal disease. Brain. 2016;139:1045–51.
Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, et al. Hypoxia as a therapy for mitochondrial disease. Science. 2016;352:54–61.
Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020;583:631–7.
Author information
Authors and Affiliations
Contributions
All authors drafted and critically revised the manuscript.
Corresponding author
Ethics declarations
Author contributions
All authors drafted and critically revised the manuscript.
Conflicts of interest and Funding
RM receives research support from Wellcome, Medical Research Council (UK), Lily Foundation, UMDF and the Ryan Stanford Appeal. Minovia Therapeutics and Reneo Ltd have paid consultancy fees to Newcastle University for services provided by RM. AL is a Clinical Research Fellow funded through Wellcome’s financial support for the Wellcome Centre for Mitochondrial Research. AL’s research is also funded by UMDF and the Lily Foundation.
Ethics approval
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Tinker, R.J., Lim, A.Z., Stefanetti, R.J. et al. Current and Emerging Clinical Treatment in Mitochondrial Disease. Mol Diagn Ther 25, 181–206 (2021). https://doi.org/10.1007/s40291-020-00510-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-020-00510-6