
Abstract
X-linked inhibitor of apoptosis protein (XIAP), survivin, and BRUCE are members of the inhibitor-of-apoptosis protein (IAP) family known for their inhibitory effects on caspase activity and dysregulation of these molecules has widely been shown to cause embryonic defects and to promote tumorigenesis in human. Besides the anti-apoptotic functions, recent discoveries have revealed that XIAP, survivin, and BRUCE also exhibit regulatory functions for autophagy in cells. As the role of autophagy in human diseases has already been discussed extensively in different reviews; in this review, we will discuss the emerging autophagic role of XIAP, survivin, and BRUCE in cancer cells. We also provide an update on the anti-apoptotic functions and the roles in maintaining DNA integrity of these molecules. Second mitochondria-derived activator of caspases (Smac) is a pro-apoptotic protein and IAPs are the molecular targets of various Smac mimetics currently under clinical trials. Better understanding on the functions of XIAP, survivin, and BRUCE can enable us to predict possible side effects of these drugs and to design a more “patient-specific” clinical trial for Smac mimetics in the future.
Similar content being viewed by others
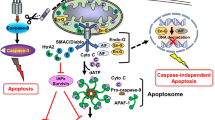


Introduction
Apoptosis is a cellular process highly regulated by different pro-apoptotic and anti-apoptotic proteins, like members of the inhibitor-of-apoptosis protein (IAP) family and the Bcl-2 family. Currently, there are eight IAP family members in human - cIAP1, cIAP2, ML-IAP/Livin, Ts-IAP/ILP-2, NIAP, XIAP, survivin, and BRUCE. Structurally, IAP family members are characterized by the presence of at least one Baculoviral IAP Repeat (BIR) domain (Table 1) and it has widely been demonstrated that the presence of the BIR domain is crucial for IAPs to inhibit the activity of different caspases through physical interactions. As IAP family members regulate a variety of cellular physiological processes [1,2,3] and dysregulations (i.e. mostly upregulation) of these molecules are known to promote tumorigenesis, tumor metastasis, and anti-cancer therapy resistance in human [4,5,6,7,8,9], it is important to understand the biology of different IAP family members and the mechanism underlying the dysregulation of these molecules in cancer cells. Although some of the IAP family members have already been known for more than two decades and several anti-cancer small-molecule Smac mimetics (i.e. a class of IAPs-targeting compounds) have been developed and reached clinical trials [10,11,12], scientists still not yet fully understand their molecular functions in cancer cells.
Autophagy is currently one of the hottest topics in cancer research. Despite intensive research has been conducted in the past decade to better understand the process of autophagy [13,14,15,16,17,18,19,20,21,22,23], the detailed regulatory mechanism and cellular effects are still not yet fully understood. Generally, autophagy is a dynamic catabolic process used for removing unnecessary or dysfunctional proteins and organelles in cells. Pathologically, dysregulation of autophagy promotes tumorigenesis and upregulation of autophagy has widely been shown to provide extra survival signals in both normal and cancer cells exposed to various internal and external stresses [14,15,16,17,18,19,20,21,22]. For example, hypoxia-induced autophagy process might contribute to the resistance to chemotherapeutic agent, cisplatin, in non-small cell lung cancer [19]. The process of apoptosis and autophagy was believed to be mutually exclusive; however, emerging evidence suggests that they are inter-connected and inter-regulated at the molecular level (e.g. through Bcl-2) in cells. In the following sections, we will discuss the lately discovered autophagic role of the well-known anti-apoptotic molecules, XIAP, survivin, and BRUCE.
XIAP as a regulator of apoptosis and necroptosis
XIAP, discovered in 1996, contains three BIR domains (BIR1, BIR2, and BIR3) and a single Really Interesting New Gene (RING) finger domain (Fig. 1). As an apoptosis inhibitor, the caspase-3 and -7 inhibiting activity has been localized to the BIR2 domain and the BIR3 domain of XIAP is responsible for the inhibition of caspase-9 [24, 25]. In contrast, the RING domain of XIAP exhibits E3 ubiquitin ligase activity and this activity is required for the XIAP-mediated cancer cell migration [26,27,28]. Besides interacting with caspase-9 and caspase-3, XIAP also directly or indirectly interacts with different IAPs and Smac [also known as direct inhibitor of apoptosis-binding protein with low pI (DIABLO)] [29,30,31,32]. The RING finger domain of XIAP is capable of interacting with the BIR2 and BIR3 domain of cIAP2 and this XIAP-cIAP2 complexation upregulates the protein stability of cIAP2 in glioblastoma cells [33]. On the other hand, formation of the survivin-XIAP complex prevents XIAP undergoing polyubiquitination and the subsequent proteasomal degradation, thereby stabilizing XIAP in cancer cells [29]. In contrast, Smac is a known pro-apoptotic molecule and formation of the Smac-XIAP complex prevents XIAP binding to different caspases and promotes cellular apoptosis [30,31,32]. A recent study by Caballero-Lopez et al. reveals that XIAP binds to the pro-apoptotic molecule, FAS-associated factor 1 (FAF1), leads to the polyubiquitination and degradation of this molecule, and consequently inhibits FAF1-mediated cell death in cancer cells [34]. However, the effects of the E3 ubiquitin ligase activity of XIAP seems not to be “pro-apoptotic molecule specific” as XIAP also stimulates ubiquitin proteasome system (UPS)-mediated degradation of the anti-apoptotic molecule, Bcl-2, to promote apoptosis upon the formation of an XIAP- apoptosis related protein in TGF-β signaling pathway (ARTS)-Bcl-2 ternary complex [35]. These findings are indeed interesting because they suggest that even though XIAP exhibits both anti-apoptotic and pro-apoptotic activities, the anti-apoptotic activity seems to be prominent as overexpression of XIAP has widely been demonstrated to promote cells survival and tumorigenesis, whereas, downregulation of this molecule promotes cancer cells death. Besides apoptosis, cells can also undergo a specific type of programmed self-destruction called necroptosis. Necroptosis is a form of programmed cell death mediated by receptor-interacting kinase 1 (RIPK1), RIPK3, and mixed lineage kinase domain-like protein (MLKL). It is now clear that that XIAP also plays an important role in regulating necroptosis in innate immune cells [36,37,38]. For example, loss of XIAP has been shown to promote the switch from tumor necrosis factor-α (TNFα; at high concentrations)-induced apoptosis to RIPK3-dependent necroptosis in mouse neutrophils [37].

Identified binding partners of XIAP
XIAP as a controversial autophagy modulator
Despite XIAP was originally discovered as an inhibitor of caspases and apoptosis, a number of studies suggest that XIAP is an autophagy modulator. An inverse correlation in the expression between XIAP and a known autophagy-related molecule, microtubule-associated protein light chain 3 (LC3), in hepatocellular carcinoma tissue specimens has been reported in the past [39]. The most direct evidence supporting its role as an autophagy negative-regulator came from a study by Huang et al. In this study, XIAP was shown to be capable of inhibiting autophagy via a XIAP-Mouse double minute 2 homolog (Mdm2)-p53 signaling pathway in the wild-type p53 (p53WT)-expressing HCT116 cancer cells, but not in the p53−/− HCT116 cancer cells [40]. Bone morphogenetic protein receptor 2 (BMPR2) is a growth factor receptor and downregulation of BMPR2 by siRNA was demonstrated to induce autophagy in chondrosarcoma cells, again, via the XIAP-Mdm2-p53 signaling pathway [41]. Recent studies further reveal that direct or indirect inhibitions/downregulations of XIAP can promote the induction of cellular autophagy. For example, the microRNA miR-23a was found to be a negative regulator of XIAP (i.e. downregulates the expression) and overexpression of miR-23a was shown to upregulate the endogenous autophagic levels of breast cancer cells in a XIAP-dependent manner (Fig. 2) [42]. Embelin (2,5-dihydroxy-3-undecyl-2,5-cyclohexadiene-1,4-dione) is a natural compound isolated from Embelia ribes [43]. Lee et al. showed that inhibiting XIAP by embelin induced autophagy in the human oral Ca9–22 squamous carcinoma cells in vitro [44]. Furthermore, it has been demonstrated that adenovirus vector-mediated XIAP-associated factor 1 (XAF1) expression induces autophagy and autophagic cell death via Beclin-1 upregulation in gastric cancer cells [45]. Of note, XAF1 is a known XIAP molecular antagonist that negatively modulates the caspase inhibitory function of XIAP through physical interactions and the subsequent redistribution of XIAP from the cytoplasm to the nucleus [46].

Schematic diagram showing the interactions between XIAP, survivin, BRUCE, and other molecules in the regulation of cellular autophagy
XIAP has also been suggested as an autophagy upregulator. Even though targeting IAPs including XIAP, cIAP1, and cIAP2 by a Smac mimetic, APG-1387, was shown to induce autophagy and cell death in human ovarian cancer cells [47]; contrary, addition of a different Smac mimetic, LCL161 (a drug known to target cIAP1, cIAP2, and XIAP), at high dose was shown to inhibit the fusion between autophagosome and lysosome in mouse embryonic cells (MEFs) [48]. Downregulations of cIAP2 and XIAP by siRNA were demonstrated to induce similar cellular phenotypes in MEFs [48], further suggesting that XIAP can act as an autophagy suppressor, despite the detailed molecular mechanism remains to be determined. Noticeably, XIAP and cIAP1 have also been suggested to positively-regulate the expression of Beclin 1, which is a protein crucial for the biogenesis of autophagosome during canonical autophagy, via an nuclear factor-κB (NFκB)-signaling pathway [49]. Thus, XIAP seems to exhibit differential autophagic roles in different cells under different circumstances.
Survivin as an apoptosis inhibitor and a mitosis positive regulator
Survivin, discovered in 1997, is the smallest member of the IAP family proteins and it contains only a single BIR domain. Similar to other IAP family members, survivin is believed or has been demonstrated to be an apoptosis negative-regulator [50]. For example, Chandele et al. showed that survivin inhibited caspase-9 activity and promoted staurosporine-resistance in human SK-N-MC neuroblastoma cells [51]. A purified recombinant human survivin protein expressed in E. coli was shown capable of binding to caspase-3 and caspase-7 in solution [52]. Furthermore, activation of caspase-3 and induction of apoptosis were widely observed in cancer cells with survivin downregulations or inhibitions [53,54,55,56,57,58,59]. As aforementioned, Smac is a negative-regulator of XIAP and it promotes caspase activation and apoptosis through formation of the XIAP-Smac protein complex. As an anti-apoptotic molecule, survivin binds to Smac and consequently prevents this molecule from binding onto XIAP, resulting in the inhibition of caspase-9 and caspase-3 [60,61,62]. In addition, it has been shown that survivin negatively modulates the activation of caspase-independent apoptosis through regulation of the nuclear translocation of apoptosis-inducing factor (AIF) [63].
Unlike other IAP family members, survivin also plays an important role in mitosis. At the molecular level, survivin forms the chromosomal passenger complex (CPC) with inner centromere protein (INCENP), borealin (also known as Dasra), and Aurora B kinase and proper formation (and localization) of the CPC during M phase of the cell cycle are both crucial for the completion of mitosis [64, 65]. Interestingly, a recent study revealed that the survivin homodimer interacts with myosin II to regulate cytokinesis [66]. Therefore, survivin is widely accepted as a multi-functions protein, which is capable of inhibiting caspase-dependent and -independent apoptosis through both direct and indirect modulations and promoting mitosis through formation of the CPC in cancer cells.
Survivin negatively modulates autophagy
Emerging evidence indicates that survivin is a negative regulator of autophagy. For example, the small molecule survivin suppressant, YM155, was shown to induce the death of salivary adenoid cystic carcinoma, breast cancer, and the Bcl-xL silenced glioma cells in an autophagy-dependent manner [67,68,69]. Despite autophagy upregulation is known to promote homologous recombination and DNA repair in cells under genotoxic stress [70, 71], Cheng et al. demonstrated YM155 also induces autophagy-dependent DNA damage in breast cancer cells regardless to the expression of p53 and caspase-3 [68]. Moreover, delivery of a survivin promoter-driven antisense survivin-expressing plasmid DNA was shown to induce apoptosis and autophagy in A549, MDA-MB-231, and PANC-1 cancer cells in vitro [58]. Conversely, survivin overexpression inhibits autophagy. For example, chemokine (C-C motif) ligand 2 (CCL2, also known as MCP1) was found to protect human PC3 prostate cancer cells from undergoing autophagic death via PI3K/AKT-dependent survivin upregulations (Fig. 2) [72].
Mechanistically, survivin suppresses autophagy possibly through interference with the development of autophagosome in cells [73]. It has been demonstrated autophagy related protein 5 (ATG5) interacts with survivin to displace Aurora B kinase from survivin in the nucleus in MDA-MB-231 breast cancer cells treated with DNA-damaging agents [74]. Interestingly, we recently discovered that survivin inhibits the conjugation between autophagy related protein 12 (ATG12) and ATG5 (i.e. the formation of ATG12-ATG5 conjugate) through physical interactions with both ATG12 (i.e. ATG12-survivin complexation) and ATG5 (i.e. ATG5-survivin complexation) [75]. We also found that survivin binds to ATG12-ATG5 conjugate (i.e. ATG12-ATG5-survivin complexation) and inhibits the formation of ATG12-ATG5-ATG16L1 in human cancer and mouse embryonic fibroblast cells (Fig. 2) [75]. Besides inhibiting the conjugation and complexation between ATG12, ATG5, and ATG16L1, survivin also negatively modulates the protein stability of autophagy related protein 7 (ATG7; a protein that facilitates LC3 lipidation) in part through an heat shock protein 27 (Hsp27) dependent mechanism [75]. Given that successful formation of the ATG12-ATG5-ATG16L1 protein complex is crucial for the elongation of autophagophore during canonical autophagy, inhibiting the formation of this protein complex shall block the autophagic flux in cells (Fig. 2).
It is worth noting that the translation of survivin mRNA transcripts is positively regulated by the AKT/mTOR signaling pathway and targeting this signaling pathway by small molecule inhibitor, rapamycin, has been shown to induce autophagy in cells [76,77,78,79,80]. Furthermore, as mentioned, XIAP inhibits autophagy via an XIAP-Mdm2-p53 signaling pathway in p53WT-expressing cancer cells. Thus, survivin may inhibit autophagy in part through interference with the XIAP-Mdm2-p53 pathway in p53WT-expressing cells. Collectively, even though the detailed mechanistic role of XIAP and survivin on autophagy regulation remains to be fully elucidated, especially in p53−/− and p53mutant expressing cells; however, it is clear that XIAP and survivin are not solely an apoptosis inhibitor but a dual/multi-functions protein, which participates in both apoptosis, mitosis, and autophagy regulations in cells.
BRUCE mediates homologous recombination and autophagosome-lysosome fusion
BIR repeat containing ubiquitin-conjugating enzyme (BRUCE, also known as Apollon) was discovered in 1998 as a member of IAPs family [81]. Structurally, it contains a single BIR domain and a single Ubiquitin-conjugating enzymes (UBC) domain (i.e. exhibits E2/E3 ubiquitin ligase activity) [82, 83]. Mechanistically, BRUCE inhibits apoptosis through physical interactions with DIABLO/Smac and caspase-9 and promotes their degradation through protein ubiquitination [84, 85]. Like survivin, BRUCE was also found to exhibit caspase inhibitory unrelated functions in cells. Breast cancer susceptibility gene C terminus-repeat inhibitor of human telomerase repeat transcriptase expression 1 (BRIT1) is an early double damage response factor. During DNA damage, BRIT1 is recruited to the phosphated-H2AX (γ-H2AX) attached DNA double-strand breaks and subsequently to facilitate DNA repair. Downregulation of BRUCE was shown to inhibit the ataxia-telangiectasia mutated and RAD3-related (ATR)-signaling pathway and to impair BRIT1 deubiquitinationin in U2OS cells. As demonstrated by Ge et al., the presence of BRUCE is crucial during DNA replication and the DNA double-strand breaks repair [86, 87]. Besides acting as an apoptosis inhibitor, a study by Kikuchi et al. showed that BRUCE also regulates mitosis through modulating the ubiquitylation and protein stability of cyclin A [88].
Recent evidence suggests that BRUCE may play a role in the formation of autolysosome (autophagosome-lysosome fusion). As described in the above sections, autophagosome and autolysosome formations are medicated by both sequential activations and complex formations between different ATG family proteins. Among these ATG family proteins, Autophagy related protein 8 (ATG8) family proteins such as LC3, GABA type A receptor-associated protein (GABARAP), and GABARAP-LIKE 1 (GABARAPL1/GEC1) govern the fusion between autophagosome and lysosome (i.e. formation of autolysosome) [89]. An interesting study by Ebner et al. revealed that BRUCE physically interacts with syntaxin 17 (STX17), GABARAP, and GABARAPL1, to promote autophagosome-lysosome fusion in mammalian cells independent of its catalytic function (Fig. 2) [90].
IAPs as “pro-survival autophagy” guardians?
Upregulation of autophagy has been shown to promote the survival of cancer and cancer-related cells treated with a variety of therapeutics including tamoxifen, paclitaxel, epirubicin, and azacytidine [14,15,16,17,18]. Autophagy is also known to assist homologous recombination, which is a type of DNA repair mechanisms, in cells treated with DNA damaging agents and UV radiation [91,92,93]. As aforementioned, overexpression of IAPs has widely been demonstrated to inhibit chemotherapeutic/targeted therapeutic drugs induced apoptosis in cancer cells. Therefore, it is unclear on the reason of having IAPs such as XIAP and survivin as autophagy suppressors, given that upregulation of autophagy and IAPs should both promote the survival of cancer cells, especially under cellular stressful conditions. Perhaps the main function of XIAP, survivin, and BRUCE on autophagy is not to largely promote or suppress this process, but to fine tune and to maintain the level of autophagy within certain “pro-survival” ranges. Despite upregulation of autophagy is widely believed to promote DNA repair (like homologous recombination), a few studies showed that excessive activation of autophagy causes DNA damage in cells. For example, it has been demonstrated that targeting cathepsin S (CTSS) induces autophagy, leading to the autophagy-dependent reactive oxygen species (ROS) production and DNA damage in OEC-M1 cells [94]. A study by Chen et al. showed that upregulation of autophagy decreases the intracellular pool of deoxyribonucleotide triphosphate (dNTP) in Huh-7 cells treated with Earle’s balanced salt solution (EBSS) or rapamycin [95]. We also demonstrated that downregulating survivin by YM155 and siRNA induces autophagy-dependent DNA damage and cell death in human cancer cells [68, 75]. So clearly, excessive autophagy (passing certain thresholds) can cause genomic instability, and by altering the expression, post-translational modification, and subcellular-localization of XIAP, survivin, and BRUCE, cells can precisely regulate the autophagy level to maintain their survival under stressful conditions. However, if XIAP, survivin, and BRUCE are three of the “guardians” of the “pro-survival autophagy” (via fine tuning the autophagic level of cells), then why contradicting results were frequently reported regarding to the role of the “induced autophagy” (i.e. autophagy-promoted survival cell or autophagy-induced cell death) in cells treated with agents targeting XIAP and BRUCE? As most IAPs can directly or indirectly interact with multiple molecules, which regulate different molecular and cellular processes like DNA repair and mitosis, the observed “resulting autophagic effects” probably were not solely caused by the direct protein-protein interaction effects of these IAPs on different autophagy core molecules, but were results of the dynamic crosstalk between different IAPs-involved molecular and cellular processes (Fig. 3). As the “weight” of each of these processes varies under different cellular environments or treatments, the autophagic outcome can be completely different. Therefore, besides understanding the direct effects of XIAP, survivin, and BRUCE on various autophagic/apoptotic/mitotic components, it is also important to understand the dynamic interactions between the autophagic process and the surrounding molecular environments within the cell.

The dynamic autophagic environments model
Conclusion and future directions
IAP family members are traditionally classified as caspase inhibitors with negative-modulating effects on cellular apoptosis. However, emerging evidence suggest that these molecules can also regulate cellular autophagy. It is not surprising that the anti-apoptotic molecules XIAP, survivin, and BRUCE are capable of modulating autophagy, given that the well-studied anti-apoptotic molecule, Bcl-2, is also known to be an apoptosis-autophagy dual modulator (i.e. inhibits Beclin 1-dependent autophagy) in cells [96]. As mitosis, apoptosis, and autophagy are inter-connected, XIAP, survivin, BRUCE, and Bcl-2 may act as bridging molecules that control the dynamics and the balance between these cellular processes. For example, cancer cells can upregulate autophagy to produce the “minimal” energy needed for their survival under serum deprivation. In addition, cancer cells can also temporarily halt mitosis, probably to spare energy, under serum deprivation. In fact, we found in a previous study that serum deprivation decreases the complexation between survivin and ATG12/ATG5 (possibly to upregulate autophagy), but not caspase-3 (concurrently maintains apoptosis inhibition), in human cancer cells [75]. However, it is still unclear on how cancer cells regulate the expression and protein-protein interaction (i.e. binding-target switch) of these IAPs to inter-regulate apoptosis, mitosis, and autophagy under different circumstances (like under hypoxia and nutrient deprivation). Thus, further investigations are needed to understand the differential regulations of these IAPs at the molecular level in cancer and non-cancerous cells. As various Smac mimetics (IAP antagonists) are currently in different phases of clinical trial and pre-clinical development (Table 2) (Fig. 4) [12, 97,98,99,100,101,102,103,104], better understanding on the functions of IAPs (e.g. XIAP, survivin, and BRUCE) can enable us to predict possible side effects of the drugs and to design a more “patient-specific” clinical trial for Smac mimetics in the future.

The chemical structure of different Smac mimetics developed for cancer treatments
Availability of data and materials
Not applicable.
Abbreviations
- ATG:
-
Autophagy related gene/protein
- BIR:
-
Baculovirus inhibitor of apoptosis protein repeat
- BRIT1:
-
Breast cancer susceptibility gene C terminus-repeat inhibitor of human telomerase repeat transcriptase expression 1
- BRUCE:
-
BIR repeat-containing ubiquitin-conjugating enzyme
- cIAP1:
-
Cellular inhibitor of apoptosis protein 1
- cIAP2:
-
Cellular inhibitor of apoptosis protein 2
- DIABLO:
-
Direct inhibitor of apoptosis-binding protein with low pI
- GABARAP:
-
GABA type A receptor-associated protein
- IAP:
-
Inhibitor-of-apoptosis protein
- LC3:
-
Microtubule-associated protein light chain 3
- ML-IAP:
-
Melanoma inhibitor of apoptosis protein
- NIAP:
-
Neuronal apoptosis inhibitory protein
- Smac:
-
Second mitochondrial activator of caspases
- Ts-IAP:
-
Testis-specific inhibitor of apoptosis protein
- XIAP:
-
X-linked inhibitor of apoptosis protein
References
Jiang Y, et al. Essential role for survivin in early brain development. J Neurosci. 2005;25(30):6962–70.
Olayioye MA, et al. XIAP-deficiency leads to delayed lobuloalveolar development in the mammary gland. Cell Death Differ. 2005;12(1):87–90.
Arroyo J, et al. XIAP protein is induced by placenta growth factor (PLGF) and decreased during preeclampsia in trophoblast cells. Syst Biol Reprod Med. 2014;60(5):263–73.
Cheung CH, et al. Survivin counteracts the therapeutic effect of microtubule de-stabilizers by stabilizing tubulin polymers. Mol Cancer. 2009;8:43.
Evans MK, et al. X-linked inhibitor of apoptosis protein mediates tumor cell resistance to antibody-dependent cellular cytotoxicity. Cell Death Dis. 2016;7(1):e2073.
Chu XY, et al. Overexpression of survivin is correlated with increased invasion and metastasis of colorectal cancer. J Surg Oncol. 2012;105(6):520–8.
Huang W-T, et al. HDAC2 and HDAC5 Up-regulations modulate survivin and miR-125a-5p expressions and promote hormone therapy resistance in estrogen receptor positive breast cancer cells. Front Pharmacol. 2017;8:902.
Guo H, et al. Expression and clinical significance of Apollon in renal carcinoma. Oncol Lett. 2016;12(6):5129–35.
Li R, et al. Expression and clinical significance of Apollon in esophageal squamous cell carcinoma. Mol Med Rep. 2016;14(3):1933–40.
Welsh K, et al. Characterization of potent SMAC Mimetics that sensitize Cancer cells to TNF family-induced apoptosis. PLoS One. 2016;11(9):e0161952.
Lu J, et al. Therapeutic potential and molecular mechanism of a novel, potent, nonpeptide, Smac mimetic SM-164 in combination with TRAIL for Cancer treatment. Mol Cancer Ther. 2011;10(5):902.
Chen Z, et al. The SMAC mimetic APG-1387 sensitizes immune-mediated cell apoptosis in hepatocellular carcinoma. Front Pharmacol. 2018;9:1298.
Guimaraes DS, Gomes MD. Expression, purification, and characterization of the TRIM49 protein. Protein Expr Purif. 2018;143:57–61.
Nagelkerke A, et al. LAMP3 is involved in tamoxifen resistance in breast cancer cells through the modulation of autophagy. Endocr Relat Cancer. 2014;21(1):101–12.
Lee MH, et al. MTA1 is a novel regulator of autophagy that induces tamoxifen resistance in breast cancer cells. Autophagy. 2018;14(5):812–24.
Zhang SF, et al. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy. 2015;11(2):225–38.
Sun WL, et al. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy. 2011;7(9):1035–44.
Romano A, et al. Proteomic analysis reveals autophagy as pro-survival pathway elicited by long-term exposure with 5-Azacitidine in high-risk Myelodysplasia. Front Pharmacol. 2017;8:204.
Wu HM, et al. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci Rep. 2015;5:12291.
Steiger-Barraissoul S, Rami A. Serum deprivation induced autophagy and predominantly an AIF-dependent apoptosis in hippocampal HT22 neurons. Apoptosis. 2009;14(11):1274–88.
Degenhardt K, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64.
Mizumura K, Choi AMK, Ryter SW. Emerging role of selective autophagy in human diseases. Front Pharmacol. 2014;5:244.
Hu YF, et al. Expressions and clinical significance of autophagy-related markers Beclin1, LC3, and EGFR in human cervical squamous cell carcinoma. Onco Targets Ther. 2015;8:2243–9.
Deveraux QL, et al. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388(6639):300–4.
Riedl SJ, et al. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104(5):791–800.
Schile AJ, Garcia-Fernandez M, Steller H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008;22(16):2256–66.
Nakatani Y, et al. Regulation of ubiquitin transfer by XIAP, a dimeric RING E3 ligase. Biochem J. 2013;450(3):629–38.
Liu J, et al. E3 ligase activity of XIAP RING domain is required for XIAP-mediated Cancer cell migration, but not for its RhoGDI binding activity. PLoS One. 2012;7(4):e35682.
Dohi T, et al. An IAP-IAP complex inhibits apoptosis. J Biol Chem. 2004;279(33):34087–90.
Srinivasula SM, et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410(6824):112–6.
Liu ZH, et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408(6815):1004–8.
Saito A, et al. Interaction between XIAP and Smac/DIABLO in the mouse brain after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2003;23(9):1010–9.
Yang W, et al. Distinctive effects of the cellular inhibitor of apoptosis protein c-IAP2 through stabilization by XIAP in glioblastoma multiforme cells. Cell Cycle. 2014;13(6):992–1005.
Caballero-Lopez MJ, et al. XIAP interacts with and regulates the activity of FAF1. Biochim Biophys Acta-Mol Cell Res. 2017;1864(7):1335–48.
Edison N, et al. Degradation of Bcl-2 by XIAP and ARTS promotes apoptosis. Cell Rep. 2017;21(2):442–54.
Yabal M, et al. XIAP restricts TNF- and RIP3-dependent cell death and Inflammasome activation. Cell Rep. 2014;7(6):1796–808.
Wicki S, et al. Loss of XIAP facilitates switch to TNFα-induced necroptosis in mouse neutrophils. Cell Death Dis. 2016;7:e2422.
Lawlor KE, et al. XIAP loss triggers RIPK3- and Caspase-8-driven IL-1β activation and cell death as a consequence of TLR-MyD88-induced cIAP1-TRAF2 degradation. Cell Rep. 2017;20(3):668–82.
Wu WY, et al. Clinical significance of autophagic protein LC3 levels and its correlation with XIAP expression in hepatocellular carcinoma. Med Oncol. 2014;31(8):108.
Huang X, et al. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J. 2013;32(16):2204–16.
Jiao G, et al. BMPR2 inhibition induced apoptosis and autophagy via destabilization of XIAP in human chondrosarcoma cells. Cell Death Dis. 2014;5:e1571.
Chen P, et al. MiR-23a modulates X-linked inhibitor of apoptosis-mediated autophagy in human luminal breast cancer cell lines. Oncotarget. 2017;8(46):80709–21.
Hao K, Ali M, Siddiqui AW. New compounds from the seeds of Embelia ribes Burm. Pharmazie. 2005;60(1):69–71.
Lee YJ, et al. XIAP inhibitor embelin induces autophagic and apoptotic cell death in human oral squamous cell carcinoma cells. Environ Toxicol. 2017;32(11):2371–8.
Sun PH, et al. The XAF1 tumor suppressor induces autophagic cell death via upregulation of Beclin-1 and inhibition of Akt pathway. Cancer Lett. 2011;310(2):170–80.
Liston P, et al. Identification of XAF1 as an antagonist of XIAP anti-Caspase activity. Nat Cell Biol. 2001;3:128.
Li BX, et al. Novel smac mimetic APG-1387 elicits ovarian cancer cell killing through TNF-alpha, Ripoptosome and autophagy mediated cell death pathway. J Exp Clin Cancer Res. 2018;37(1):53.
Gradzka S, et al. Inhibitor of apoptosis proteins are required for effective fusion of autophagosomes with lysosomes. Cell Death Dis. 2018;9(5):529.
Lin F, et al. XIAP and cIAP1 amplifications induce Beclin 1-dependent autophagy through NFκB activation. Hum Mol Genet. 2015;24(10):2899–913.
Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3(8):917–21.
Chandele A, et al. Upregulation of survivin in G2/M cells and inhibition of caspase 9 activity enhances resistance in staurosporine-induced apoptosis. Neoplasia. 2004;6(1):29–40.
Shin S, et al. An anti-apoptotic protein human Survivin is a direct inhibitor of Caspase-3 and -7. Biochemistry. 2001;40(4):1117–23.
Montazeri Aliabadi H, et al. Induction of apoptosis by Survivin silencing through siRNA delivery in a human breast Cancer cell line. Mol Pharm. 2011;8(5):1821–30.
Uchida H, et al. Adenovirus-mediated transfer of siRNA against Survivin induced apoptosis and attenuated tumor cell growth in vitro and in vivo. Mol Ther. 2004;10(1):162–71.
Raviv Z, et al. Methyl jasmonate down-regulates survivin expression and sensitizes colon carcinoma cells towards TRAIL-induced cytotoxicity. Br J Pharmacol. 2011;164(5):1433–44.
Cheung CHA, et al. A cell-permeable dominant-negative survivin protein induces apoptosis and sensitizes prostate cancer cells to TNF-α therapy. Cancer Cell Int. 2010;10:36.
Tsai SL, et al. Cloning, expression, and purification of the recombinant pro-apoptotic dominant-negative survivin T34A-C84A protein in Escherichia coli. Protein Expr Purif. 2019;160:73–83.
Lin K-Y, et al. Delivery of a survivin promoter-driven antisense survivin-expressing plasmid DNA as a cancer therapeutic: a proof-of-concept study. OncoTargets Ther. 2016;9:2601–13.
Roy K, et al. Competitive inhibition of survivin using a cell-permeable recombinant protein induces cancer-specific apoptosis in colon cancer model. Int J Nanomedicine. 2015;10:1019–43.
Ceballos-Cancino G, et al. Regulation of mitochondrial Smac/DIABLO-selective release by survivin. Oncogene. 2007;26(54):7569–75.
Song Z, Yao X, Wu M. Direct interaction between Survivin and Smac/DIABLO is essential for the anti-apoptotic activity of Survivin during Taxol-induced apoptosis. J Biol Chem. 2003;278(25):23130–40.
Cristian OP, Manuel GRJ, Jaime RM. Protein–protein recognition as a first step towards the inhibition of XIAP and Survivin anti-apoptotic proteins. J Mol Recognit. 2008;21(3):190–204.
Pavlyukov MS, et al. Survivin monomer plays an essential role in apoptosis regulation. J Biol Chem. 2011;286(26):23296–307.
Vader G, et al. Survivin mediates targeting of the chromosomal passenger complex to the centromere and midbody. EMBO Rep. 2006;7(1):85–92.
Klein UR, Nigg EA, Gruneberg U. Centromere targeting of the chromosomal passenger complex requires a ternary subcomplex of Borealin, Survivin, and the N-terminal domain of INCENP. Mol Biol Cell. 2006;17(6):2547–58.
Babkoff A, et al. A direct interaction between survivin and myosin II is required for cytokinesis. J Cell Sci. 2019;132(14):jcs233130.
Wang YF, et al. Induction of autophagy-dependent cell death by the survivin suppressant YM155 in salivary adenoid cystic carcinoma. Apoptosis. 2014;19(4):748–58.
Cheng SM, et al. YM155 down-regulates survivin and XIAP, modulates autophagy and induces autophagy-dependent DNA damage in breast cancer cells. Br J Pharmacol. 2015;172(1):214–34.
Jane EP, et al. Survivin inhibitor YM155 induces mitochondrial dysfunction, autophagy, DNA damage and apoptosis in Bcl-xL silenced glioma cell lines. Mol Carcinog. 2017;56(4):1251–65.
Liu EY, et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci. 2015;112(3):773.
Lin W, et al. Autophagy confers DNA damage repair pathways to protect the hematopoietic system from nuclear radiation injury. Sci Rep. 2015;5:12362.
Roca H, Varsos Z, Pienta KJ. CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation. J Biol Chem. 2008;283(36):25057–73.
Humphry NJ, Wheatley SP. Survivin inhibits excessive autophagy in cancer cells but does so independently of its interaction with LC3. Biol Open. 2018;7(10):bio037374.
Maskey D, et al. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun. 2013;4:2130.
Lin T-Y, et al. BIRC5/Survivin is a novel ATG12–ATG5 conjugate interactor and an autophagy-induced DNA damage suppressor in human cancer and mouse embryonic fibroblast cells, Autophagy. 2019;1–18. https://doi.org/10.1080/15548627.2019.1671643.
Huang WT, et al. HDAC2 and HDAC5 up-regulations modulate Survivin and miR-125a-5p expressions and promote hormone therapy resistance in estrogen receptor positive breast cancer cells. Front Pharmacol. 2017;8:902.
Anandharaj A, Cinghu S, Park WY. Rapamycin-mediated mTOR inhibition attenuates survivin and sensitizes glioblastoma cells to radiation therapy. Acta Biochim Biophys Sin Shanghai. 2011;43(4):292–300.
Guo R, et al. Involvement of mTOR and survivin inhibition in tamoxifen-induced apoptosis in human hepatoblastoma cell line HepG2. Biomed Pharmacother. 2010;64(4):249–53.
Kim YC, Guan K-L. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32.
Tanemura M, et al. Rapamycin causes Upregulation of autophagy and impairs islets function both in vitro and in vivo. Am J Transplant. 2012;12(1):102–14.
Hauser H-P, et al. A Giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J Cell Biol. 1998;141(6):1415.
Chen Z, et al. A human IAP-family gene, Apollon, expressed in human brain Cancer cells. Biochem Biophys Res Commun. 1999;264(3):847–54.
Bartke T, et al. Dual role of BRUCE as an antiapoptotic IAP and a chimeric E2/E3 ubiquitin ligase. Mol Cell. 2004;14(6):801–11.
Hao YY, et al. Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat Cell Biol. 2004;6(9):849–60.
Qiu XB, Goldberg AL. The membrane-associated inhibitor of apoptosis protein, BRUCE/Apollon, antagonizes both the precursor and mature forms of Smac and caspase-9. J Biol Chem. 2005;280(1):174–82.
Ge C, et al. The BRUCE-ATR signaling Axis is required for accurate DNA replication and suppression of liver cancer development. Hepatology. 2019;69(6):2608–22.
Ge C, et al. BRUCE regulates DNA double-strand break response by promoting USP8 deubiquitination of BRIT1. Proc Natl Acad Sci. 2015;112(11):E1210.
Kikuchi R, et al. APOLLON protein promotes early mitotic CYCLIN a degradation independent of the spindle assembly checkpoint. J Biol Chem. 2014;289(6):3457–67.
Nguyen TN, et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome–lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. 2016;215(6):857.
Ebner P, et al. The IAP family member BRUCE regulates autophagosome–lysosome fusion. Nat Commun. 2018;9(1):599.
Liu EY, et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci U S A. 2015;112(3):773–8.
Chen S, et al. RAD6 promotes homologous recombination repair by activating the autophagy-mediated degradation of heterochromatin protein HP1. Mol Cell Biol. 2015;35(2):406.
Gomes LR, Menck CFM, Leandro GS. Autophagy roles in the modulation of DNA repair pathways. Int J Mol Sci. 2017;18(11)2351.
Huang CC, et al. Autophagy-regulated ROS from xanthine oxidase acts as an early effector for triggering late mitochondria-dependent apoptosis in cathepsin S-targeted tumor cells. PLoS One. 2015;10(6):e0128045.
Chen W, et al. Reciprocal regulation of autophagy and dNTP pools in human cancer cells. Autophagy. 2014;10(7):1272–84.
Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–39.
Thibault B, et al. DEBIO 1143, an IAP inhibitor, reverses carboplatin resistance in ovarian cancer cells and triggers apoptotic or necroptotic cell death. Sci Rep. 2018;8(1):17862.
Runckel K, et al. The SMAC mimetic LCL-161 displays antitumor activity in preclinical models of rituximab-resistant B-cell lymphoma. Blood Adv. 2018;2(23):3516–25.
Kadletz L, et al. AZD5582, an IAP antagonist that leads to apoptosis in head and neck squamous cell carcinoma cell lines and is eligible for combination with irradiation. Acta Otolaryngol. 2017;137(3):320–5.
Hennessy EJ, et al. Discovery of a novel class of Dimeric Smac Mimetics as potent IAP antagonists resulting in a clinical candidate for the treatment of Cancer (AZD5582). J Med Chem. 2013;56(24):9897–919.
Brunckhorst MK, et al. AT-406, an orally active antagonist of multiple inhibitor of apoptosis proteins, inhibits progression of human ovarian cancer. Cancer Biol Ther. 2012;13(9):804–11.
Ding R, et al. WX20120108, a novel IAP antagonist, induces tumor cell autophagy via activating ROS-FOXO pathway. Acta Pharmacol Sin. 2019;40(11):1466–79.
Shekhar TM, et al. Smac mimetics LCL161 and GDC-0152 inhibit osteosarcoma growth and metastasis in mice. BMC Cancer. 2019;19(1):924.
Kamata E, et al. Synergistic effects of a Smac mimetic with doxorubicin against human osteosarcoma. Anticancer Res. 2017;37(11):6097–106.
Acknowledgements
Not applicable.
Funding
This work was financially supported by the Ministry of Science and Technology of Taiwan [MOST 108–2320-B-006 − 024].
Author information
Authors and Affiliations
Contributions
Literature research: T-Y Lin, SM Cheng, Y-C Chang, CHA Cheung; Figure and table preparation: T-Y Lin, Y-C Chang, CHA Cheung; Manuscript writing and proofreading: CHA Cheung, E Leung. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cheung, C.H.A., Chang, YC., Lin, TY. et al. Anti-apoptotic proteins in the autophagic world: an update on functions of XIAP, Survivin, and BRUCE. J Biomed Sci 27, 31 (2020). https://doi.org/10.1186/s12929-020-0627-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-020-0627-5