
Abstract
Background
Simao pine is one of the primary economic tree species for resin and timber production in southwest China. The exploitation and utilization of Simao pine are constrained by the relatively lacking of genetic information. Construction a fine genetic linkage map and detecting quantitative trait locis (QTLs) for growth-related traits is a prerequisite section of Simao Pine's molecular breeding program.
Results
In our study, a high-resolution Simao pine genetic map employed specific locus amplified fragment sequencing (SLAF-seq) technology and based on an F1 pseudo-testcross population has been constructed. There were 11,544 SNPs assigned to 12 linkage groups (LGs), and the total length of the map was 2,062.85 cM with a mean distance of 0.37 cM between markers. According to the phenotypic variation analysis for three consecutive years, a total of seventeen QTLs for four traits were detected. Among 17 QTLs, there were six for plant height (Dh.16.1, Dh16.2, Dh17.1, Dh18.1–3), five for basal diameter (Dbd.17.1–5), four for needle length (Dnl17.1–3, Dnl18.1) and two for needle diameter (Dnd17.1 and Dnd18.1) respectively. These QTLs individually explained phenotypic variance from 11.0–16.3%, and the logarithm of odds (LOD) value ranged from 2.52 to 3.87.
Conclusions
In our study, a fine genetic map of Simao pine applied the technology of SLAF-seq has been constructed for the first time. Based on the map, a total of 17 QTLs for four growth-related traits were identified. It provides helpful information for genomic studies and marker-assisted selection (MAS) in Simao pine.
Similar content being viewed by others


Background
Pinus kesiya Royle ex Gordon var. langbianensis (A. Chev.) Gaussen (2n = 24) [64], also called Simao/Szemao pine in China, is a geographic variant of Pinus kesiya [54, 55], mainly distributed in the humid and sub-humid mountainous areas in southwest China [30, 74]. Compared with other conifer tree species, it has unique rapid growth characteristics that its branches can grow 2 or more rounds a year [41, 66]. It is one of the most important timber and resin production tree species in Yunnan Province [5]. Simao pine is not only a high commercial value plant for its higher turpentine output [13, 57] but also essential tree species in the forest ecosystem for the function of biological carbon sequestration and water conservation [6, 29, 58, 73]. However, its exploitation and utilization are constrained by relatively lacking genetic information [5].
Highly saturated genetic linkage map construction is a useful tool for QTL mapping and MAS [19, 71, 72]. During the last two decades, a large number of genetic maps for perennial woody plants had constructed [18, 26, 52], various QTLs associated with essential traits had been identified based on these maps [12], [42] [36]. However, a great majority of them were low saturated frame-work maps that lowered the degree of accuracy of QTL mapping [28, 31]. Single nucleotide polymorphism (SNP) is a sort of molecular marker technique developed by high-throughput sequencing. It is convenient, abundant, highly polymorphic and commonly used in genetic map construction [34, 45, 46]. In pace with the rapid development technology of next-generation sequencing (NGS), the technology of SLAF-seq becomes one of the popular methods for SNP markers development and high-resolution genetic map construction [9, 44, 69]. Until now, various plants genetic linkage maps had established by SLAF-seq, and it greatly heighten the efficiency and the degree of QTL mapping accuracy [11, 37, 68, 70].
Construction of genetic linkage map for Simao pine will provide helpful information for genomic studies and facilitate the breeding applications. Growth-related traits were important economic traits for woody tree breeding, and detecting QTLs for these traits is a crucial section in the molecular breeding program for Simao Pine. Therefore, we employed the SLAF-seq technology to actualize the fast SNPs development and a high-density linkage map will be constructed. The QTLs linked to growth-related traits will be identified based on the genetic linkage map. It will provide a powerful tool for future detection of other economic characteristics QTLs and MAS in Simao pine breeding.
Results
Analysis of sequencing data and SLAF markers
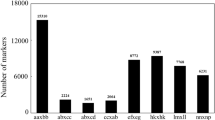
The Simao pine SLAF libraries were constructed successfully. A total of 461.41 M reads (guanine-cytosine of 40.23% and Q30 of 92.25%) were obtained. The number of reads for the maternal and paternal parents was 22,947,158 and 18,534,664, the mean for the F1 individual was 4,659,126 (Table 1). After filtering out the low-quality reads, the number of SLAFs for the two parents and average in F1 progeny was 535,598, 482,851, and 375,315. The average depth of the SLAFs for the maternal and paternal parent was 10.48-fold and 9.77-fold, and the average for each F1 individual was 3.47-fold (Table 1). A total of 633,086 high-quality SLAFs were obtained. Among these markers, 239,790 were polymorphic markers (37.88%), 385,976 were non-polymorphic markers (60.97%), and 7,320 were repetitive markers (1.15%). Finally, 140,485 polymorphic SLAFs of 8 segregation patterns were achieved (Fig. 1). After removing the markers showing aa × bb segregation pattern, 97,891 (15.46%) polymorphic SLAFs will be used to further Simao pine genetic map construction.

Segregation pattern of polymorphic SLAF markers. The x-axis represents the segregating pattern numbers and the y-axis indicates the genotypes of markers
Construction and evaluation of genetic linkage map
After discarding the unsuitable markers, a total of 5,643 SLAFs were used successfully for the linkage map construction. Among them, 11,544 SNP markers were detected (Table 2). Based on these markers, we constructed a high saturated genetic linkage map covering 2062.85 cM, comprising 12 LGs, and a mean distance of 0.37 cM (Fig. 2, Table 2). The genetic length of individual LGs varied from 147.38 (LG4) to 194.85 cM (LG9) with a mean of 171.90 cM. Among 12 LGs, LG8 was the largest linkage group (523 SLAFs), while LG7 was the smallest group (367 SLAFs). The average number of markers for each LG was 470. For the density, LG5 was the densest linkage group with the minimum marker distance (0.32 cM), whereas LG1, LG3, and LG7 were the lowest density linkage groups (0.40 cM). The max gap in the map was 11.17 cM located in LG2 and LG3.

The high-density linkage map of Simao pine. A black bar indicates a SLAF marker. The x-axis represents the linkage group number and the y-axis indicates the genetic distance (cM) within each linkage group
Three approaches were used for detecting the quality of Simao pine genetic map. (1) The markers integrity analysis showed that each individual mapped marker's complete degree was 99.99% (Fig. 3), the average depth for parents was more than five times of the offspring (Table 3). It suggested that genotyping was accurate and the mapping population was suitable for further analysis. (2) The result of Haplotype maps analysis revealed that most of the recombination blocks were distinctly defined (Supplementary Fig. 1). It suggested that the constructed high saturated genetic map was adaptive for subsequent genetic analysis. (3) The analysis of Heat maps showed that the markers were well ordered in most linkage groups, indicated that the constructed Simao pine genetic map with high accuracy (Supplementary Fig. 2).

The integrity distribution map of all individuals. The x-axis represents the 100 individuals and y-axis represents the complete degree of mapped markers
Phenotypic variation analysis
The 3 years phenotypic data and statistical values for growth-related traits were summarized (Table 4). The results showed that the four traits were normal distribution for three years (Fig. 4). And a relatively higher degree of genetic variation was found. The CV of plant height was 21.74%, 14.80% and 12.07% during 2016, 2017 and 2018. The CV of basal diameter was 25.21% (2016), 23.42% (2017), and 22.33% (2018) respectively. The CV of needle length was 21.69% (2017) and 18.89% (2018). The CV of needle diameter was 25.96% (2017) and 21.44% (2018). The Pearson correlations analyses showed the significant correlation among four traits (Table 5).

Frequency distributions of growth traits in mapping population during three consecutive years
QTL mapping
Using the constructed map and analyzing the data of phenotypic characteristics in the mapping population, 17 QTLs linked to 4 traits were identified (Table 6, Supplementary Fig. 3). The individual QTL explained the phenotypic variation varied from 11.0–16.3%, and the LOD value ranged from 2.52 to 3.87. There were 6 plant height QTLs detected in the map. In 2016, two plant height QTLs were located on LG3 (Dh16.1) and LG10 (Dh16.2) explained 12.7% and 12.4% of the phenotypic variance. In 2017, only one plant height QTL located on LG9 (Dh17.1) explained 15.5% of the phenotypic variation was detected. In 2018, the other three plant height QTLs were identified which located on LG11 (Dh18.1, Dh18.2), and LG12 (Dh18.3), and explained 16.3%, 16.1% and 13.8% of the phenotypic variation, respectively. A total of 5 QTLs associated with basal diameter were detected on LG3 (Ddb17.1, 11.6%), LG4 (Ddb17.2, 12.7%) and LG6 (Ddb17.3, 11.6%; Ddb17.4, 13.1%; Ddb17.5, 11.4%), respectively. Four needle length QTLs were identified which located on LG1 (Dnl17.1, 12.1%; Dnl17.2, 11.0%; Dnl17.3, 12.1%) and LG12 (Dnl18.1, 13.5%), respectively. There were two needle diameter QTLs located on LG4 (Dnb17.1 and Dnb18.1) explained 12.9% and 13.1% of the phenotypic variation were detected.
Discussion
Construction of the high-resolution map for Simao pine will provide helpful information for genomic studies and facilitate the breeding applications. In our study, a fine genetic map of Simao pine applied the technology of SLAF-seq has been constructed. It contained 12 LGs and 11,544 SNPs spanned 2,062.85 cM with a mean marker distance of 0.37 cM, representing a significant improvement over the previous linkage maps in coniferous plants [8, 10, 14, 15, 39, 60]. As we know, this was one of the highest saturated genetic maps to date in coniferous tree species. Furthermore, a total of 17 QTLs for four growth-related traits were identified based on the constructed genetic map, and these QTLs were valuable resources for genetic breeding and MAS in Simao pine.
An appropriate mapping population laid a solid foundation for the genetic map construction [75]. It's hard for perennial woody trees to get Backcross (BC), Recombination Inbred Lines (RILs) and F2 populations in the short term because of the long generation constraints. The pseudo-testcross strategy has been put forward that the F1 population was created to replace the other populations [20]. This strategy has been successfully applied to various forestry trees, especially non-model and un-sequenced species [32, 35, 47, 61, 62, 65]. In this report, nine F1 populations were obtained by artificial hybridization, based on the analysis of field phenotypic characteristics variation among populations and genetic similarity coefficient among parents, superior clones SM11 (high resin content) and JG1 (fast growth) were chosen for maternal and paternal parents. The F1 hybrid population was applied as the mapping population for map construction in our study. The obvious variation will present in the segregation population due to the significant difference in the resin content and the parents' growth speed, which could facilitate QTL mapping for these traits.
Molecular markers were powerful tools for genetic map construction [2]. The mainstream molecular markers for genetic linkage map construction of heterozygous perennial forest tree species included SNP, simple sequence repeat (SSR), inter-simple sequence repeat (ISSR), amplified fragment length polymorphism (AFLP) and random amplified polymorphic DNA (RAPD) et al. [7, 21, 28, 31, 38, 56]. Among these markers, SNP was thought of as one of the ideal markers for genetic map construction for the merits of abundance, fast and covering the whole genome [3, 16]. Significant changes have taken place in genetic map construction with the development of high-throughput sequencing technology [15]. Recently, SLAF-seq technique has become one of the most popular SNP marker development assays [45]. A high-density genetic linkage map for Simao pine had been successfully constructed by using this approach. It indicated that SNP markers could be efficiently applied in constructing a genetic linkage map of Simao pine.
High-quality genetic maps can increase the accuracy of QTL mapping [28, 31, 32, 35]. The number of markers in the genetic map is one of the essential indicators to evaluate its quality. A genetic map with a large number of markers has the characteristics of suitable distance and high-resolution [70]. This constructed genetic map was the first map that contained over ten thousand SNP markers in coniferous tree species. It supported that we have built a high-quality genetic map for Simao Pine. Moreover, other three approaches were used for evaluating the quality of Simao pine genetic map. All results indicated that the current high-accuracy map would provide sufficient information for QTL mapping.
Growth-related traits were important economic traits for woody tree breeding, and detecting QTLs for growth-related traits is an introductory section in the molecular breeding program for Simao Pine. In this study, a total of 17 QTLs for four growth-related traits were identified. The individual QTL explained the phenotypic variation varied from 11.0% to16.3%, and it indicated that several significant useful genes might control the growth-related traits of Simao pine (Rönnberg et al., 2005; [28, 31]. In the four traits, only the QTLs for plant height were consistently detected during the three years, but QTLs for the other three traits were not consecutively expressed. It suggested that different genes/QTLs might influence Simao pine's growth-related traits in different seasons/ages or that the QTLs stabilization varied by the effect of environmental change. In agreement with previous studies in the woody tree, growth-related traits were mainly quantitative traits, which dominated by involved genes and easily affected by the environment, probably changes as the tree matures [27, 67]. In other words, the multi-environment QTL analysis is more accurate than a single-environment experiment for the heterozygous perennial woody tree growth-related traits QTL mapping [17]. Thus, to eliminate interference brought by the environment and improve QTLs accuracy, the multi-environment QTL test in other domains using different backgrounds for Simao pine must be carried out in the future [1, 28, 31].
The size of the mapping population decided the success of genetic mapping and QTL analysis, and the influence of missing genotypes is more obvious in small-sized populations than in large one. In general, mapping population consisting of 50–250 individuals may be sufficient to construct the initial skeletal linkage map [24], [4]. However, a larger population size is needed for high resolution or fine mapping [40, 54, 55]. In our study, a main limitation of the Simao pine genetic linkage map is that it is smaller mapping population size (100 individuals), it may be provided fragmented linkage groups and inaccurate locus order for the genetic linkage map and affected the accuracy of the QTL mapping [40]. It is well known that artificially controlled pollination of conifers trees is more difficult and the number of hybrid offspring is less than other tree species too [23]. To our knowledge, high-density genetic maps which constructed with the small size mapping populations have been successfully used for QTL fine mapping [61, 62, 75]. However, for further improving the accuracy of the map and QTLs, the size of Simao pine mapping population must be increased in the future.
Conclusions
We report the first high-density genetic map for Simao pine. The map was constructed using an F1 population and was based on SNP markers developed by using the SLAF-seq approach, which allowed the efficient development of a large number of markers in a short time. A total of 17 QTLs for growth-related traits were identified based on the constructed genetic map. The results of this study will provide a platform for map-based gene isolation and molecular breeding for Simao pine.
Methods
Mapping population and DNA extraction
According to factorial mating design, eleven superior clones with the good characters of rapid growth and high resin content were selected as the hybrid parents from 105 clones. Among them, five clones as the male parents (superior clone JG1, NR7, LC3, ZY1, PW2) and the other six clones for the female parents (PW12, LC9, JG7, JD5, PW3, SM11). In the spring of 2014, a total of 30 hybridized combinations of artificially controlled pollination were conducted. Two years later, a total of 9 full-sib families were obtained and grown at the farm of Pu'er city institute of forestry sciences (N 22◦ 47′/E 100◦ 59′) by harvesting, sowing and culturing the seedlings. Proceed to the next step, family 9 was selected as the mapping population for Simao pine genetic linkage map construction by analyzing population phenotypic variation and parent's genetic similarity coefficient [57]. The parents for the mapping population were superior clone SM11 (maternal, with the characteristic of high resin content) and JG1 (paternal, with the aspect of rapid growth). Fresh and young healthy needles from parents and mapping population (100 hybrid individuals) were collected and frozen in liquid nitrogen at once. The genomic DNA was isolated using the improved cetyl trimethyl ammonium bromide (CTAB) method [53]. The permission of the plant materials collection was approved by Southwest Forestry University (project number: 2018Y31). The plant species was identified by Assoc. Prof. Jianghua Liu (College of Forestry, Southwest Forestry University), and the voucher specimens were stored at the Key Laboratory for Forest Resources Conservation and Utilization in the Southwest Mountains of China Ministry of Education, Southwest Forestry University, Kunming, China.
SLAF library establishment and sequencing
The similar experiment procedure of high-throughput sequencing and establishment of the SLAF library for the mapping population was performed according to the previous study by Zhang et al. [68] with minor modified. Briefly, two different steps were applied. First, all of the genomic DNA for SLAF library construction were digested by a single enzyme Hae III (New England Biolabs, NEB, USA). Second, only the SLAF fragments in which the length ranging from 414 to 464 bp will be excised and diluted for pair-end sequenced by Illumina HiSeq 2500 platform (Illumina, Inc; San Diego, CA, USA).
Analysing and genotyping for sequence data
The SLAF-seq data grouping and SNP genotyping were the same as Wang et al. [56]. After discarding the low-quality reads, the remaining reads with more than 90% similarity will gather in the same SLAF locus. As Simao pine is a diploid plant, so only the SLAF has 2 to 4 alleles that will designate as the potential and polymorphic marker. The aa × bb segregation pattern markers will not be used to construct the genetic map, as the mapping population is obtaining by a cross between two heterozygote parents of Simao pine [22, 25].
Linkage map construction and evaluation
The HighMap software [22] with the cross-pollination (CP) option was utilized for Simao pine genetic linkage map construction. The estimation parameters were set for a minimum LOD threshold of 5.0 for linkage groups and a maximum recombination fraction of 0.4, and the map distance in centi-Morgans was calculated with the Kosambi mapping algorithm [51]. After linkage grouping, the maximum likelihood method was used to order the SLAFs markers in all LGs [50], [37]. The SMOOTH algorithm was utilized to put correct genotyping errors [49]. To evaluate the quality of the constructed Simao pine genetic map, the analysis of mapped markers integrity, construction of haplotype maps and heat maps for each LG were carried out [33, 59, 63].
Growth-related traits assessment and QTL analysis
Growth-related traits, including plant height, basal diameter, needle length, and needle diameter of the progenies are determined during three consecutive years. The plant height and needle length were measured by a line tape, while the basal diameter and needle diameter were measured with the vernier caliper in December from 2016–2018. The phenotypic variation analysis, including coefficients of variation (CV) and the correlation coefficients between all investigated traits, was performed with software SPSS 20.0. The QTLs underlying the growth-related traits were implemented in MapQTL 6.0 software and the interval mapping method [48]. The 95% Bayesian credible interval method was used to calculate the confidence intervals for all QTLs [43]. One thousand permutations decided the threshold value. According to the permutations, the minimum LOD score of 2.5 was conducted in our study. The percentage of phenotypic variance explained of each detected QTL was achieved based on the phenotypic variance in the population [65].
Availability of data and materials
The data generated or analyzed in this study are included in this article and its supplementary information files. Other materials that support the findings of this study are available from the corresponding author on reasonable request.
Not applicable.
Abbreviations
- QTL:
-
Quantitative trait loci
- LG:
-
Linage group
- SLAF-seq:
-
Specific locus amplified fragment sequencing
- LOD:
-
Logarithm of odds
- MAS:
-
Marker-assisted selection
- SNP:
-
Single nucleotide polymorphism
- NGS:
-
Next-generation sequencing
- CTAB:
-
Cetyl trimethyl ammonium bromide
- CP:
-
Cross-pollination
- CV:
-
Coefficients of variation
- BC:
-
Backcross
- RILs:
-
Recombination Inbred Lines
- SSR:
-
Simple sequence repeat
- ISSR:
-
Inter-simple sequence repeat
- AFLP:
-
Amplified fragment length polymorphism
- RAPD:
-
Random amplified polymorphic DNA
References
Adhikari L, Lindstrom OM, Markham J, Missaoui AM. Dissecting Key Adaptation Traits in the Polyploid Perennial Medicago sativa Using GBS-SNP Mapping. Front Plant Sci. 2018;9:934. https://doi.org/10.3389/fpls.2018.0093.
Alsaleh A, Baloch FS, Derya M, et al. Genetic Linkage Map of Anatolian Durum Wheat Derived from a Cross of Kunduru-1149 × Cham1. Plant Mol Bio Rep. 2015;33:209–20. https://doi.org/10.1007/s11105-014-0749-6.
Baird NA, Etter PD, Atwood TS, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE. 2008;3: e3376. https://doi.org/10.1371/journal.pone.0003376.
Bernhardsson C, Vidalis A, Wang X, et al. An Ultra-Dense Haploid Genetic Map for Evaluating the Highly Fragmented Genome Assembly of Norway Spruce ( Picea abies ). G3-Genes Genomes Genetics. 2019;9(5):g3.200840.2018.
Cai N. H, Xu Y. L, Wang D. W, Chen S, Li G.Q. Identification and characterization of microsatellite markers in pinus kesiya var. langbianensis (pinaceae). Appli Plant Sci. 2017;5:1600126. https://doi.org/10.3732/apps.1600126.
Cao XL, Zhang Y. Analysis on the China certified emission reductions, economic value and its sensitivity of Pinus kesiya var. langbianensis afforestation project in Yunnan province. Ecol Environ Sci. 2017;26:234–42. https://doi.org/10.16258/j.cnki.1674-5906.2017.02.008.
Casasoli M, Mattioni C, Cherubini M, Villani F. A genetic linkage map of European chestnut (Castanea sativa Mill.) based on RAPD, ISSR and isozyme markers. Theor Appl Genet. 2001;102:1190–9. https://doi.org/10.1007/s00122-001-0553-1.
Chancerel E, Lepoittevin C, Le P, G., et al. Development and implementation of a highly-multiplexed SNP array for genetic mapping in maritime pine and comparative mapping with loblolly pine. BMC Genomics. 2011;12:368. https://doi.org/10.1186/1471-2164-12-368.
Chang YQ, Ding J, Xu YH, Li D, Zhang WJ, Li L, Song J. SLAF-based high-density genetic map construction and QTL mapping for major economic traits in sea urchin Strongylocentrotus intermedius. Sci Rep. 2018;8:820. https://doi.org/10.1038/s41598-017-18768-y.
Chen MM, Feng F, Sui X, et al. Genetic linkage maps of Pinus koraiensis Sieb. et Zucc. based on AFLP markers. Afric J Biotech. 2010;9:5659–64. https://doi.org/10.1186/1471-2105-11-441.
Conson ARO, Taniguti CH, Amadeu RR, Andreotti IAA, de Souza LM, et al. High-resolution genetic map and QTL analysis of growth-related traits of hevea brasiliensis cultivated under suboptimal temperature and humidity conditions. Front Plant Sci. 2018;9:1255. https://doi.org/10.3389/fpls.2018.01255.
Devey ME, Groom KA, Nolan MF, Bell JC, Dudzinski MJ, Old KM, Matheson AC, Moran GF. Detection and verification of quantitative trait loci for resistance to Dothistroma needle blight in Pinus radiata. Theor Appl Genet. 2004;108:1056–63. https://doi.org/10.1007/s00122-003-1471-1.
Dong JX, Guo HJ, Li P, Zhao YF, Yun XH. Oleoresin resources and development capacity in Yunnan. J Northwest For Uni. 2009;24:157–60. https://doi.org/10.1007/978-1-4020-9623-5.
Echt CS, Saha S, Krutovsky KV, et al. An annotated genetic map of loblolly pine based on microsatellite and cDNA markers. BMC Genet. 2011;12:17. https://doi.org/10.1186/1471-2156-12-17.
Eckert AJ, Pande B, Ersoz ES, et al. High-throughput genotyping and mapping of single nucleotide polymorphisms in loblolly pine (Pinus taeda L.). Tree Genet Genom. 2009;5:225–34. https://doi.org/10.1007/s11295-008-0183-8.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE. 2011;6: e19379. https://doi.org/10.1371/journal.pone.0019379.
El-Soda M, Malosetti M, Zwaan BJ, Koornneef M, Aarts GM. Genotype × environment interaction QTL mapping in plants: lessons from Arabidopsis. Trends Plant Sci. 2014;19:390–8. https://doi.org/10.1016/j.tplants.2014.01.001.
Freeman JS, Potts BM, Shepherd M. Parental and consensus linkage maps of Eucalyptus globulus using AFLP and microsatellite markers. Silvae Genet. 2006;55:202–17. https://doi.org/10.1134/s1022795406010145.
Gao W, Qu JB, Zhang JX, Sonnenberg A, Chen Q, Zhang Y, Huang CY. A genetic linkage map of Pleurotus tuoliensis integrated with physical mapping of the de novo sequenced genome and the mating type loci. BMC Genomics. 2018;19:18. https://doi.org/10.1186/s12864-017-4421-z.
Grattapaglia D, Sederoff R. Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics. 1994;137:1121–37. https://doi.org/10.1101/gad.8.15.1853.
Gulsen O, Uzun A, Canan I, et al. A new citrus linkage map based on SRAP, SSR, ISSR, POGP, RGA and RAPD markers. Euphytica. 2010;173:265–77. https://doi.org/10.1007/s10681-010-0146-7.
He Y. X, Yuan W. J, Dong M. F, et al. The first genetic map in sweet osmanthus (osmanthus fragrans lour.) using specific locus amplified fragment sequencing. Front Plant Sci. 2017;8:1621. https://doi.org/10.3389/fpls.2017.01621.
Hirao T, Matsunaga K, Hirakawa H, et al. Construction of genetic linkage map and identification of a novel major locus for resistance to pine wood nematode in Japanese black pine (Pinus thunbergii). BMC Plant Biol. 2019;19(1):424. https://doi.org/10.1186/s12870-019-2045-y.
Howad W, Yamamoto T, Dirlewanger E, Testolin R, Cosson P, Cipriani G, Monforte AJ, Georgi L, Abbott AG, Arús P. Mapping with a few plants: using selective mapping for microsatellite saturation of the Prunus reference map. Genetics. 2005;171(3):1305–9. https://doi.org/10.1534/genetics.105.043661.
Jansen J. Construction of linkage maps in full-sib families of diploid outbreeding species by minimizing the number of recombinations in hidden inheritance vectors. Genetics. 2005;170:2013–25. https://doi.org/10.1534/genetics.105.041822.
Jiang TB, Zhou BR, Gao FL, Guo BZ. Genetic linkage maps of white birches (Betula platyphylla Suk. and B. pendula Roth) based on RAPD and AFLP markers. Mol Breed. 2011;27:347–56. https://doi.org/10.1007/s11032-010-9436-y.
Kenis K, Keulemans J. Study of tree architecture of apple (Malus × domestics Borkh.) by QTL analysis of growth traits. Mol Breed. 2007;19:193–208. https://doi.org/10.1007/s11032-006-9022-5.
Li B, Tian L, Zhang J. Y, Huang L, Han F. X, Yan S. R, Wang L. Z, Zheng H. K, Sun J. M. Construction of a high-density genetic map based on large-scale markers developed by specific length amplified fragment sequencing (SLAF-seq) and its application to QTL analysis for isoflavone content in glycine max. Bmc Genomics. 2014;15:1086. https://doi.org/10.1186/1471-2164-15-1086.
Li SF, Su JR, Lang XD, Huang XB, Miao YC, Yang LH. Genetic variation and early selection analysis of open-pollinated families of Pinus kesiya var. langbianensis. For Res. 2017;30:929–35. https://doi.org/10.13275/j.cnki.lykxyj.2017.06.00.
Li SF, Su JR, Lang XD, Liu WD, Ou GL. Positive relationship between species richness and aboveground biomass across forest strata in a primary Pinus kesiya forest. Sci Rep. 2018;8:2227. https://doi.org/10.1038/s41598-018-20165-y.
Li Y, Wang DW, Li ZQ, et al. A Molecular Genetic Linkage Map of Eucommia ulmoides and Quantitative Trait Loci (QTL) Analysis for Growth Traits. Int J Mol Sci. 2014;15:2053–74. https://doi.org/10.3390/ijms15022053.
Liu C, Fan BJ, Cao ZM, et al. Development of a high-density genetic linkage map and identification of flowering time QTLs in adzuki bean (Vigna angularis). Sci Rep. 2016;6:39523. https://doi.org/10.1038/srep39523.
Liu HL, Cao FL, Yin TM, Chen YN. A Highly dense genetic map for Ginkgo biloba constructed Using sequence-based markers. Front Plant Sci. 2017;8:1041. https://doi.org/10.3389/fpls.2017.01041.
Liu J, Huang SM, Sun MY, Liu SY, Liu YM, Wang WX, Zhang XR, Wang HZ, Hua W. An improved allele-specific PCR primer design method for SNP marker analysis and its application. Plant Methods. 2012;8:34. https://doi.org/10.1186/1746-4811-8-34.
Liu ZH, Bao DG, Liu DL, et al. Construction of a Genetic Linkage Map and QTL Analysis of Fruit-related Traits in an F1 Red Fuji × Hongrou Apple Hybrid. Open Life Sci. 2016;11:487–97. https://doi.org/10.1515/biol-2016-0063.
Lowry D. B, Taylor S, Bonnette J, Aspinwall M. J, Asmus A, Keitt T. H, Tobias C. M, Juenger T. E. QTLs for biomass and developmental traits in switchgrass (Panicum virgatum). BioEnergy Res. 2015;8:1856–67. https://doi.org/10.1007/s12155-015-9629-7.
Luo C, Shu B, Yao Q, Wu H, Xu W, Wang S. Construction of a high-density genetic map based on large-scale marker development in mango using Specific-Locus Amplified Fragment Sequencing (SLAF-seq). Front Plant Sci. 2016;7:1310. https://doi.org/10.3389/fpls.2016.0131.
Mortaza K, Salih K, Ziya ME, Nergiz C. In silico polymorphic novel SSR marker development and the first SSR-based genetic linkage map in pistachio. Tree Genet Genom. 2018;14:45. https://doi.org/10.1007/s10681-015-1394-3.
Neves L. G, Davis J. M, Barbazuk W. B, et al. A high-density gene map of loblolly pine (Pinus taeda L.) based on exome sequence capture genotyping. G3. 2014;4:29–37. https://doi.org/10.1534/g3.113.008714.
Nunziata SO, Weisrock DW. Estimation of contemporary effective population size and population declines using RAD sequence data. Heredity. 2018;120(3):196–207. https://doi.org/10.1038/s41437-017-0037-y.
Ou GL, Wang JF, Xu H, Chen KY, Zheng HM, Zhang B, Sun XL, Xu TT, Xiao YF. Incorporating topographic factors in nonlinear mixed-effects models for aboveground biomass of natural Simao pine in Yunnan. China J For Res. 2016;27:119–31. https://doi.org/10.1007/s11676-015-0143-8.
Rönnberg-Wästljung AC, Glynn C, Weih M. QTL analysis of drought tolerance and growth for a Salix dasyclados × Salix viminalis hybrid in contrasting water regimes. Theor Appl Genet. 2005;110:537–49. https://doi.org/10.1007/s00122-004-1866-7.
Sen S, Churchill GA. A statistical framework for quantitative trait mapping. Genetics. 2001;159:371–87. https://doi.org/10.1089/10906570152742353.
Shang JL, Li N, Li NN, Xu YY, Ma SW, Wang JM. Construction of a high-density genetic map for watermelon (Citrullus lanatus L.) based on large-scale SNP discovery by specific length amplified fragment sequencing (SLAF-seq). Sci Hortic. 2016;203:38–46. https://doi.org/10.1016/j.scienta.2016.03.007.
Sun XW, Liu DY, Zhang XF, Li WB, Liu H, Hong WG, et al. SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE. 2013;8: e58700. https://doi.org/10.1371/journal.pone.0058700.
Troggio M, Malacarne G, Coppola G, Segala C, Cartwright DA, Pindo M, et al. A dense single nucleotide polymorphism-based genetic linkage map of grapevine (Vitis vinifera L.) anchoring Pinot Noir bacterial artificial chromosome contigs. Genetics. 2007;176:2637–50. https://doi.org/10.1534/genetics.106.067462.
Ukrainetz NK, Ritland K, Mansfield SD. An AFLP linkage map for Douglas-fir based upon multiple full-sib families. Tree Genet Genom. 2008;4:181–91. https://doi.org/10.1007/s11295-007-0099-8.
Van Ooijen, J. W. (2009) Map QTL 6: software for the mapping of quantitative trait loci in experimental populations of diploid species. Wageningen, Netherlands. https://www.kyazma.nl/index.php/mc.MapQTL.
Van OH, Stam P, Visser RG, Van Eck HJ. SMOOTH: a statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theor Appl Genet. 2005;112:187–94. https://doi.org/10.1007/s00122-005-0124-y.
Van OJ. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet Res. 2011;93:343–9. https://doi.org/10.1017/S0016672311000279.
Vinod KK. Kosambi and the genetic mapping function. Resonance. 2011;16:540–50. https://doi.org/10.1007/s12045-011-0060-x.
Wang DW, Li Y, Li L, Wei YC, Li ZQ. The first genetic linkage map of Eucommia ulmoides. J Genet. 2014;93:13–20. https://doi.org/10.1007/s12041-014-0322-y.
Wang DW, Shen BQ, Gong HD. Genetic diversity of Simao pine in China revealed by SRAP markers. Peer J. 2019;7: e6529. https://doi.org/10.7717/peerj.6529.
Wang HF, Lencinas MV, Friedman CR, Zhu ZX, Qiu JX. Understory plant diversity assessment of Szemao pine (Pinus kesiya var. langbianensis) plantations in Yunnan. China Collectanea Botanica. 2012;31:51–65. https://doi.org/10.1371/journal.pone.0135946.
Wang H, Smith KP, Combs E, et al. Effect of population size and unbalanced data sets on QTL detection using genome-wide association mapping in barley breeding germplasm. Theor Appl Genet. 2012;124:111–24. https://doi.org/10.1007/s00122-011-1691-8.
Wang L, Li XG, Wang L, Xue HB, Wu J, Yin H, Zhang SL. Construction of a high-density genetic linkage map in pear (Pyrus communis × Pyrus pyrifolia nakai) using SSRs and SNPs developed by SLAF-seq. Sci Hortic. 2017;218:198–204. https://doi.org/10.1016/j.scienta.2017.02.015.
Wang Y, Yuan XL, Hua M, Li J, Wang J. Transcriptome and gene expression analysis revealed mechanisms for producing high oleoresin yields from Simao pine (Pinus kesiya var. langbianensis). Plant Omics J. 2018;11:42–9. https://doi.org/10.21475/poj.11.01.18.pne1085.
Wen QZ, Zhao YF, Chen XM, Yang ZX, Ai JL, Yang XS. Dynamic study on the values for ecological service function of Pinus kesiya forest in China. For Res. 2010;23:671–7. https://doi.org/10.3724/sp.j.1238.2010.00474.
West MA, Van LH, Kozik A, et al. High-density haplotyping with microarray-based expression and single feature polymorphism markers in Arabidopsis. Genome Res. 2006;16:787–95. https://doi.org/10.1101/gr.5011206.
Westbrook JW, Chhatre VE, Wu LS, et al. A consensus genetic map for pinus taeda and pinus elliottii and extent of linkage disequilibrium in two genotype-phenotype discovery populations of pinus taeda. G3. 2015;5:1685. https://doi.org/10.1534/g3.115.019588.
Wu J, T Li L, Li M, et al. High-density genetic linkage map construction and identification of fruit-related QTLs in pear using SNP and SSR markers. J Exp Bot. 2014;65(20):5771–81. https://doi.org/10.1093/jxb/eru311.
Wu J, Li LT, Li M, Khan A, Li XG, Chen H, Yin H, Zhang SL. High-density genetic linkage map construction and identification of fruit-related QTLs in pear using SNP and SSR markers. J Experi Bot. 2014;65:5771–81. https://doi.org/10.1093/jxb/eru311.
Wu J, Zhao Q, Zhang L, et al. QTL Mapping of fiber-related traits based on a high-density genetic map in flax (Linum usitatissimum L.). Fron.t Plant Sci. 2018;9:885. https://doi.org/10.3389/fpls.2018.0088.
Wu LY, Zhao WS. Study on Chromosome’ s Karyotypes of Different Populations of Pinus kesiya var. langbianensis (A. Chev.) Gaussen. Yunnan For Sci Tech. 1999;87:32–5. https://doi.org/10.16473/j.cnki.xblykx1972.1999.02.006.
Xia Z. Q, Zhang S. K, Wen M. F, Lu C, Sun Y. F, Zou M. L, Wang W. Q. Construction of an ultrahigh-density genetic linkage map for Jatropha curcas L. and identification of QTL for fruit yield. Biotech Biofuels. 2018;11:3. https://doi.org/10.1186/s13068-017-1004-9.
Xu MY, Deng GX, Ling WG. Exploration on the utilization of Pinus khasya seeds. Seed. 2012;31:95–101. https://doi.org/10.16590/j.cnki.1001-4705.2012.08.079.
Yang HX, Liu TY, Liu CX, Zhao FC, Huang SW. QTL detection for growth traits in Pinus elliottii var. elliottii and P. caribaea var. hondurensis. For Sci Practice. 2013;15:196–205. https://doi.org/10.1007/s11632-013-0306-7.
Zhang J, Zhang QX, Cheng TR, Yang WR, Pan HT, Zhong JJ, Huang L, Liu EZ. High-density genetic map construction and identification of a locus controlling weeping trait in an ornamental woody plant (Prunus mume Sieb. et Zucc). DNA Res. 2015;22:183–91. https://doi.org/10.1093/dnares/dsv003.
Zhang YX, Wang LH, Xin HG, Li DH, Ma CX, Ding X, Hong WG, Zhang XR. Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC Plant Biol. 2013;13:141. https://doi.org/10.1186/1471-2229-13-141.
Zhang Z, Shang H. H, Shi Y. Z, Huang L, Li J. W, Ge Q, Gong G.W, et al. Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to Quantitative Trait Loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum.). BMC Plant Biol. 2016;1:79. https://doi.org/10.1186/s12870-016-0741-4.
Zhao XX, Huang K, Zhang XQ, Wang JP, Yan DF, Li J, Tang L, Li XL, Shi TW. Construction of high-density genetic linkage map and identification of flowering-time QTLs in orchardgrass using SSRs and SLAF-seq. Sci Rep. 2016;6:29345. https://doi.org/10.1038/srep29345.
Zhao Z. Q, Gu H. H, Sheng X. G, Yu H. F, Wang J. S, Huang L, Wang D. Genome-wide single-nucleotide polymorphisms discovery and high-density genetic map construction in cauli flower using specific-locus amplified fragment sequencing. Front Plan.t Sci. 2016;7:334. https://doi.org/10.3389/fpls.2016.00334.
Zhu YF, Chen SY, Hao JB, Wu T. Genetic analysis of superior clones and specific marker of elite clones of Pinus kesiya var. langbianensis. J West China For Sci. 2016;45:141–6. https://doi.org/10.16473/j.cnki.xblykx1972.2016.04.024.
Zhu YF, Chen SY, Hao JB, Wu T. Genetic analysis of superior clones and identification of specific marker of elite clones of Pinus kesiya. Agri Sci Tech. 2017;18:2524–7. https://doi.org/10.16175/j.cnki.1009-4229,2017.12.073.
Zhu Y. F, Yin Y. F, Yang K. Q, et al. Construction of a high-density genetic map using specific length amplified fragment markers and identification of a quantitative trait locus for anthracnose resistance in walnut (Juglans regia L.). Bmc Genomics. 2015;16:614. https://doi.org/10.1186/s12864-015-1822-8.
Acknowledgements
The authors thank Dr. Zong Dan and Ph.d Shen Bingqi for excellent technical assistance.
Funding
The study was funded by National Natural Science Foundation of China [Grants 31500536] and Special Funds for local scientific and Technological Development Guided by the Central Government [Grants 219001]. Authors declare that none of the funding bodies have any role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
DW, AD and HG conceived and designed the study. DW, CS, SL and NC performed the experiments. CH and HT analyzed the data, DW, CS and LY wrote the manuscript. LY, HG revised the whole manuscript and helped in preparing the tables and references. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figures 1. Haplotype maps for 12 LGs. Figures 2. Heat maps for 12 LGs. Figures 3. 17 growth-related traits QTLs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, D., Yang, L., Shi, C. et al. QTL mapping for growth-related traits by constructing the first genetic linkage map in Simao pine. BMC Plant Biol 22, 48 (2022). https://doi.org/10.1186/s12870-022-03425-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03425-y