
Abstract
The paucity of sequence information flanking the simple sequence repeat (SSR) motifs identified especially in the transcript sequences has been limiting factor in the development of SSR markers for plant genome analysis as well as breeding applications. To overcome this and enhance the genic SSR marker repertoire in chickpea, the draft genome sequence of kabuli chickpea (CDC Frontier) and publicly available transcript sequences consisting of in silico identified SSR motifs were deployed in the present study. In this direction, the 300 bp sequence flanking the SSR motifs were retrieved by aligning 566 SSR containing transcripts of ICCV 2 available in public domain on the reference chickpea genome. A set of 202 novel genic SSRs were developed from a set of 507 primer pairs designed, based on in silico amplification of single locus and having no similarity to the publicly available SSR markers. Further, 40 genic SSRs equally distributed on chickpea genome were validated on a select set of 44 chickpea genotypes (including 41 Cicer arietinum and 3 Cicer reticulatum), out of which 25 were reported to be polymorphic. The polymorphism information content (PIC) value of 25 polymorphic genic SSRs ranged from 0.11 to 0.77 and number of alleles varied from 2 to 9. Clear demarcation among founder lines of multi-parent advanced generation inter-cross (MAGIC) population developed at ICRISAT and near-isogenic nature of JG 11 and JG11 + demonstrates the usefulness of these markers in chickpea diversity analysis and breeding studies. Further, genic polymorphic SSRs reported between parental lines of 16 different mapping populations along with the novel SSRs can be deployed for trait mapping and breeding applications in chickpea.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chickpea (Cicer arietinum L.) is the second most important grain legume widely cultivated throughout tropical and sub-tropical regions of the world (Gaur et al. 2012). It is a self-pollinated crop with basic chromosome number eight (2n = 2x = 16) and genome size of 738 Mb (Varshney et al. 2013b). Globally it is cultivated on 11.55 million ha with an annual production of 10.46 million tons and average productivity of 905 kg/ha (FAOSTAT data 2013). Based on the market class, two types of cultivated chickpeas viz. desi and kabuli are grown worldwide. Desi types are cultivated mainly in Indian sub-continent while kabuli types are grown in the Mediterranean region including Southern Europe, Western Asia and Northern Africa (Moreno and Cubero 1978).
Chickpea besides being rich and important source of protein, vitamins and essential minerals for human diet also enhances soil fertility by fixing atmospheric nitrogen through symbiotic association. Chickpea production is affected by several abiotic stresses like drought, heat, salinity and biotic stresses like Fusarium wilt, Ascochyta blight, Botrytis grey mould and dry root rot. Despite its economic importance, the climate changing scenarios in recent times are further aggravating impact of production constraints which kept the average productivity less than one ton per hectare for last six decades (Thudi et al. 2014). Nevertheless, the recent advances in genomics coupled with the availability of the genome sequence for several important crop plants (Michael and Jackson 2013) including chickpea (Varshney et al. 2013b) provide an opportunity to translate the knowledge to breed superior variety with enhanced resistance/tolerance to biotic and abiotic stresses in several crop species including chickpea (Gaur et al. 2012; Varshney and Tuberosa 2013a, b).
Molecular markers have become indispensable tools in genomics-assisted breeding (Varshney et al. 2007) for crop improvement. Among several kinds of molecular markers, the simple sequence repeat (SSR) markers are considered as the markers of choice owing to high reproducibility and co-dominant nature (Gupta and Varshney 2000). Further, genic SSRs (derived from cDNA/expressed sequence tags (ESTs) are, more robust for gene discovery and to study variation in transcribed regions and genes of known-function, very useful in plant genome analysis and crop improvement (Varshney et al. 2005, 2007). In addition, genic SSRs derived from ESTs with homology to candidate genes are good targets for genetic mapping and aligning genome linkage maps across distantly related species for comparative analysis (Holton et al. 2002).
In self-pollinated species like chickpea, with narrow genetic base, harnessing the potential of genomics assisted breeding (GAB) has been limited owing to availability of a limited number of SSR markers until 2006 (Varshney et al. 2012). For instance, only 255 SSRs (22 sequence tagged microsatellites, STMS markers from Huttel et al. 1999; 218 SSRs from Winter et al. 1999; 15 SSRs from Udupa and Baum 2003 and 108 expressed sequence tag (EST)-SSRs Buhariwalla et al. 2005) were available for chickpea genetics and breeding applications. Nevertheless, as a result of recent efforts,The resulting matrix was used to the chickpea SSR marker repertoire has been increased from a few hundred to several thousands. Among recent studies, SSRs derived from microsatellite enriched libraries (Nayak et al. 2010), bacterial artificial chromosome (BAC)—end sequence (BES-SSRs) are noteworthy. Similarly genic SSRs were developed in a number of studies (Choudhary et al. 2008; Varshney et al. 2009; Gujaria et al. 2011; Hiremath et al. 2011). In many of the studies, SSR motifs were identified however, paucity of flanking sequences restricted the primers to be designed for these SSRs. For instance, 26,252 SSRs from tentative unique sequences (TUSs) (Hiremath et al. 2011), 6,845 BES-SSRs (Thudi et al. 2011), 3,728 EST-SSRs (Varshney et al. 2009), 1,269 transcription factor gene-derived microsatellite (TFGMS) (Kujur et al. 2013), were identified. However, primer pairs could be designed for as low as 2.7 % and a maximum for 87 % of SSRs in the specified cases. This low level of primer designing ability may be attributed to the absence of sufficient sequence information flanking the SSR repeat motifs.
In a recent study (Agarwal et al. 2012), transcript derived SSRs were identified, however, for 273 SSR motifs primers could not designed due to lack for sufficient flanking sequence information. Nevertheless, the constraint of designing the primers for SSR motifs without flanking sequence has been overcome in the present study because of the availability of reference chickpea genome sequence (Varshney et al. 2013b). Although in silico genome-wide survey is useful for the identification of large number of SSRs, identification of polymorphic markers is time consuming under laboratory conditions. In view of above, the present study demonstrates the utility of chickpea genome sequence for designing the primers for genic SSRs. Furthermore, this study also validates a select set of SSRs in 44 chickpea genotypes.
Materials and methods
Plant material and DNA isolation
Forty-four chickpea genotypes representing the members of primary gene pool including 41 genotypes of C. arietinum (cultivated) and 3 genotypes of C. reticulatum (wild) were used in the present study. The origin, pedigree and salient features of the chickpea genotypes are listed in Table 1.
Genomic DNA from all 44 genotypes was extracted at ICRISAT from about 80 mg of fresh leaves collected from 15 to 20 day-old seedlings employing high-throughput DNA isolation method suggested by Cuc et al. (2008). DNA was normalized to 5 ng/µl after checking the quality on 0.8 % agarose gel.
Primer designing
Previous transcriptome study by Agarwal et al. (2012) on kabuli cultivar of chickpea, ICCV 2 identified 43,389 transcripts which contained 566 transcripts having in silico identified genic polymorphic SSRs (here on regarded as in silico genic SSRs). For the present study the above transcripts available in the public domain (CTDB v 1.0, http://www.nipgr.res.in/ctdb.html) were utilized for designing primers and further validated a few of these markers on a set of 44 chickpea genotypes mentioned above.
The genomic loci of the in silico genic SSRs were identified by aligning the transcripts containing these SSRs on the publicly available chickpea genome (Varshney et al. 2013b). SSR motifs showing precise alignment with the genome with at least a 300 bp flanking sequences were selected. The SSR motifs along with respective flanking sequences were then used for designing primers employing batch Primer3web version 4.0.0 (http://probes.pw.usda.gov/batchprimer3/). In order to remove the redundancy, the designed primers were subjected to Blast against publicly available SSR markers (Winter et al. 1999; Lichtenzveig et al. 2005; Gaur et al. 2011; Sethy et al. 2006; Buhariwalla et al. 2005; Choudhary et al. 2006, 2008; Varshney et al. 2009; Nayak et al. 2010; Hiremath et al. 2011; Thudi et al. 2011; Kujur et al. 2013). The primers showing hits with an expected product size and covering the SSR motif were considered to be redundant.
Polymerase chain reaction (PCR)
For amplification of SSRs, PCR was carried out in a 10 μl reaction volume containing 10 ng DNA, 10X PCR buffer, 2.5 mM MgCl2, 2 mM dNTPs, 0.1 U Taq DNA polymerase (Sib-Enzyme, Novosibrisk, Russia), 2 pmol primer using a thermocycler (Bio-Rad, Berkeley, CA, USA). PCR amplification was carried out using touchdown cycles involving initial denaturation for 4 min at 94 °C; followed by 10 cycles of 94 °C for 20 s, 65 °C for 20 s and 72 °C for 30 s; (touchdown from 65 to 55 °C with 1 °C decrease in each cycle for 20 s)then by 35 cycles of 94 °C for 30 s; 55 °C for 50 s; and 72 °C for 30 s, a final extension of 20 min at 72 °C and left at 4 °C until further use.
Capillary electrophoresis
Equal volumes of PCR products of four different markers labelled with different fluorescent dyes (FAM- VIC- NED- and PET) were pooled along with 7 μl of formamide (Applied Biosystems, CA, USA), 0.05 μl of the GeneScanTM 500 LIZ Size Standard (Applied Biosystems, CA, USA) and 2.95 μl of distilled water. The pooled PCR amplicons were denatured and size fractioned using capillary electrophoresis on ABI 3730 DNA Genetic Analyzer (Applied Biosystems, CA, USA) at ICRISAT. Allele calling was done using GeneMapper 4software (Applied Biosystems, CA, USA) by using the internal LIZ-500 size standard.
Data analysis
To estimate the genetic diversity the amplified DNA fragments were either scored as presence (1) or absence (0) for each primer-genotype combination. The resulting matrix was used to compute Jaccard’s similarity coefficients (Sneath and Sokal 1973) and UPGMA (Unweighted Pair Group Method with Arithmetic means) cluster analysis using computer package NTSYSpc (Numerical Taxonomy and Multivariate Analysis System) version 2.1 (Rohlf 2000) to determine the genetic relationship. Grouping of genotypes was further done by partitioning the variance of the data-sets using principal component analysis (PCA). Polymorphism information content (PIC) was calculated using the following formula
where Pi is the proportion of the population carrying the ith allele, calculated for each SSR locus.
Results
Identification of novel genic SSRs
A total of 566 unique transcripts containing 623 in silico identified genic SSRs were downloaded from Chickpea Transcriptome Database (CTDB v 1.0, http://www.nipgr.res.in/ctdb.html). A total of 608 hits were obtained as a result of BlastN search of 566 unique transcripts against CDC Frontier genome of which 559 were unique hits. Further, the genome coordinates with a minimum of 300 bp flanking sequences on either side of genic SSR motifs were identified for 526 genic SSR motifs. These sequences were extracted using customised perl script and then subjected to primer design using Batch Primer3. As a result primer pairs could be designed for 507 SSR motifs.
In order to determine the uniqueness and develop novel, robust genic SSR markers for applications in crop improvement the following in silico analysis was adopted:
-
1.
All 507 primers designed were subjected to BlastN analysis against the reference chickpea genome to identify primer pairs that amplify single genomic locus.
-
2.
Primer pairs designed for SSR motifs with >10 di-nucleotide repeats and >5 tri-nucleotide repeats were considered.
-
3.
Primer pairs were blasted against available marker repertoire.
As a result of BlastN analysis, out of 507 primer pairs, 7 primer pairs showed multiple hits (>9 hits) on the reference genome and therefore 500 were considered for further studies. These 500 primer pairs included primers for 239 genic SSRs out of a total of 273 genic SSR motifs targeted, which were not reported in earlier study (Fig. 1). Although primer pairs were designed for all 273 genic SSR motifs targeted, 34 primer pairs did not flank the desired repeat motif. Hence, the primer pairs for 239 genic SSRs were tested for novelty by doing Blast against available marker repertoire. Flanking sequence of 300 bp on either side of SSR motif from chickpea genome were obtained and subjected to BlastN against the publicly available primers. Forward primers resulted in 142 hits while reverse primers gave 128 hits. However, there were only 32 unique hits that were shown by both forward and reverse primers. We therefore considered only 32 out of 239 SSRs as redundant. BAC-end sequences (46,270) reported by Thudi et al. (2011) were also used to further find out the redundant SSRs. These BAC-end sequences were also subjected to BlastN against the primers designed in this study and the hits were checked on the basis of alignment length including the SSR motif covered. Sequences showing hits with BAC-end sequences including the desired SSR motif including flanking region that could produce primers were discarded. We got 11 such genic SSRs containing sequences. However, only 5 out of these 11 were not a subset of 32 sequences which showed hits with the existing primers set. A total of only 37 primers (both forward and reverse primers) had unique hits with existing markers. Thus, primer pairs designed for 202 in silico genic SSRs were identified as novel. The details of all primer pairs designed including the genome coordinates of the repeat motifs are available in Supplementary Table 1.

Schematic workflow of identification of novel genic SSRs. The results of alignment of SSR containing transcripts on reference chickpea genome and identification of novel genic SSRs are depicted
Validation and characterisation of SSRs
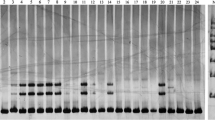
Among 202 novel in silico genic SSRs, 40 evenly distributed SSRs across eight pseudo molecules of chickpea were selected for validation. Of these 40 markers, 15 markers were monomorphic on a set of 44 chickpea genotypes that were genotyped. In total 95 alleles were produced by 25 genic polymorphic SSR markers. The number of alleles per locus ranged from 2 to 9 with an average of 3.8 alleles (Table 2). Polymorphism information content (PIC) ranged from 0.11 to 0.77 with an average of 0.42. The major allele frequency ranged from 0.34 to 0.93. The gene diversity ranged from 0.12 to 0.80.
A maximum of 9 (ICCV 2 × JG 11 and ICCV 2 × JG 62) and a minimum of 1 polymorphic marker (C 214 × WR 315) were identified for 16 different parental combinations used to generate segregating mapping populations for various traits (Supplementary Table 2).
Of the 25 SSR markers used in the study, 9 amplified genotype specific alleles for seven genotypes (Table 3). These SSRs could be used as molecular tags to identify the specific genotypes and these markers can be used to identify the seed lot for specific genotypes. The two wild chickpea species accessions (PI 489777 and IG 72953) could be identified using four different genic SSRs. Two genic SSRs (CakTSSR01526, CakTSSR00621) were specific to IG 72953, and the other two (CakTSSR01652, CakTSSR04407) were specific to PI 489777. Arerti (a desi variety of Ethiopian origin), Chefe (kabuli), ICC 1882 (a desi cultivar and parent of mapping population segregating for drought), ICCV 04112 (desi) and ICCV 97105 (desi) varieties were amplified for unique alleles using one SSR marker (CakTSSR00763, CakTSSR04052, CakTSSR00621, CakTSSR03039 and CakTSSR03970) for each genotype respectively.
Genetic diversity analysis
In order to understand the genetic diversity among the 44 chickpea genotypes, a UPGMA (Unweighted Pair Group Method with Arithmetic Averages) dendrogram was constructed. Among 44 chickpea genotypes used in the present study, IG 72953 and PI 489777 had least similarity with rest of the genotypes analysed (Fig. 2). While remaining 42 genotyped were grouped into two major clusters namely Cluster I and Cluster II. Cluster I contained 18 genotypes and Cluster II contained 24 genotypes. The pairwise genetic similarity among the accessions are given in Supplementary Table 3.

Dendrogram showing hierarchical clustering (NTSYSpc) of 44 chickpea genotypes based on 95 alleles generated by 25 SSRs in chickpea
Among 18 genotypes grouped in Cluster I, 12 were desi types (Annigeri-1, ICCV 04516, ICCV 05107, ICC 3137, ICCV 03112, ICC 506, ICCV 04112, ICC 995, ICCV 05530, Vijay, Arerti and Ejere) and 6 were kabuli (ICCV 95423, KAK 2, ICCV 2, Chefe and ICC 6263, ICC 8261) types. Further, among 24 genotypes clustered in Cluster II included wild type, IG 72933 (C. reticulatum accession) and the remaining 23 were cultivated desi type (C. arietinum accessions) (Fig. 2).
The diversity within eight desi founder parents (ICCV 10, ICCV 00108, JG 16, JAKI 9218, JG 130, ICC 4958, JG 11, ICCV 97105) used to develop first multi-parent advanced generation inter-cross (MAGIC) population in chickpea was revealed using the markers developed in the present study. The MAGIC parents were uniformly distributed in cluster II and revealed considerable variation. The genetic similarity within MAGIC parents ranged from 0.40 (between ICCV 00108 and ICCV 10) to 0.80 (between JAKI 9218 and JG 130) (Supplementary Table 3).
The PCA analysis was performed to further validate the results reflected from dendrogram. PCA was utilized to derive a 2-dimensional scatter plot of individuals, such that the geometrical distances among individuals in the plot reflect the genetic distances among them with minimal distortion (Fig. 3). The 2D PCA plot broadly depicted two Clusters (Cluster I and II). However, 2 out of 3 wild chickpea species accessions, originated from Turkey, IG 72953 and PI 489777, did not fall into any of the two clusters and fall solitarily into two different places. Cluster I consisted of 26 genotypes, all of which are desi types and a wild accession (IG 72933). Cluster I also represented a leading variety JG 11 and it’s MABC derived line JG 11 + for root trait QTL (Varshney et al. 2013a). It also represented all the 8 MAGIC founder parents (desi type) used to develop MAGIC population. The other Cluster (Cluster II) was represented by 16 genotypes, of which 10 genotypes are desi and 6 are of kabuli type. This group shows similarity to a part of Cluster I of dendrogram and consisted of two genotypes of Ethiopian origin namely, Ejere and Arerti.

Two-dimensional plot of PCA analysis (NTSYSpc) showing genetic diversity of 44 genotypes using 95 alleles from 25 SSR loci
Discussion
Genic SSRs have been the marker of choice and are more robust as compared to non-genic SSRs in genetic and QTL mapping experiments. Genic SSRs are not prone to recombination between marker and trait associated with it as they lie within the gene of interest which also limits the identification of false positives in marker-assisted selection program (Gupta et al. 2010). Another important feature of the genic SSR markers is that, unlike genomic SSRs, they are transferable among related species and genera (Varshney et al. 2005). Recent advances in next generation sequencing technologies changed the sequencing scenarios which led to availability of draft genomes and transcriptome of several crop plants including chickpea (Varshney et al. 2013b; Varshney et al. 2009; Hiremath et al. 2011). The availability of chickpea genome made it possible to identify several thousands of SSRs in the genome using MISA (Thiel et al. 2003). However, classifying these into genic and genomic SSRs will be a difficult task. Nevertheless, several transcriptomic studies in recent past lead to identification of SSRs motifs in the transcriptome (Varshney et al. 2009; Hiremath et al. 2011; Agarwal et al. 2012). However, genic SSR markers could be developed for only a few SSR motifs. In the study done by Agarwal et al. (2012), although 623 genic SSRs were reported, primer pairs could be designed only for 350 SSRs and for the remaining 273 in silico genic SSRs primers could not be designed. In order to design the primers for SSR motifs lacking the flanking sequence in transcripts, 300 bp flanking sequence was obtained after aligning the transcripts on genome. The reason for considering such a long stretch of sequence for primer designing was to identify the novel SSRs) by subjecting the publicly available primers to Blast against these flanking sequences.
In the present study, primers for the mentioned 273 genic SSR motifs were designed utilizing the available chickpea draft genome. Also, the novelty of these SSRs were confirmed. Further, a subset of uniformly distributed genic SSRs (40 markers) on the genome was validated for their usefulness in genetic diversity analysis. In silico identification of SSRs reduces the effort that goes in validation of number of markers for their polymorphism. These polymorphic markers are used to study the genetic diversity and relationship among the germplasm available, which will be helpful in chickpea improvement programs.
In this study, we report primer pairs for a set of 202 novel, non-redundant genic SSRs. The criterion adopted for removing the redundancy further avoids overestimation of the EST-SSRs in chickpea. We also evaluated and proved the authenticity of 25 in silico identified genic SSRs. Similar work was also reported in castor bean (Qiu et al. 2010), pepper (Kong et al. 2012) and date palm (Zhao et al. 2012), where the available EST sequences were used for identification of genic polymorphic SSRs followed by validation of some of these markers. In this study, 44 chickpea genotypes of kabuli, desi and wild type representing cultivated chickpea breeding lines, wild relatives and parents of mapping populations were exploited for diversity study using 25 genic SSR markers. In the present study we obtained PIC values in the range of 0.11–0.77 using only 25 SSRs in a set of 44 genotypes as compared to PIC values of 0.27 to 0.50, 0.37 to 0.91, 0.46 to 0.97 and 0.40 to 0.80 reported by Keneni et al. (2012), Sefera et al. (2011), Upadhyaya et al. (2008) and Choudhary et al. (2012) respectively using more number of genotypes with more number of markers. The polymorphic markers identified among parental genotypes of 16 different mapping populations segregating for different biotic (Helicoverpa, Ascochyta blight, Fusarium wilt) and abiotic stresses (drought etc.) can be used in marker assisted breeding programs.
Two of the reported markers (CakTSSR04062 and CakTSSR04214) were identified in the “QTL-hotspot” region located on CaLG04 (Varshney et al. 2014) which harbour QTLs for several drought tolerance related traits. The markers can be used in trait mapping and reducing the QTL region. As very few SSR markers are available for marker-assisted backcrossing (MABC; Varshney et al. 2012) and polymorphism of these markers in several genetic backgrounds is very low (Thudi et al. 2014), additional markers in this region will enable introgression of this region into different genic backgrounds. Other genic SSRs reported for the parental crosses can be useful in a marker-assisted selection program like background selection and trait mapping studies. Similar diversity studies were carried out by Keneni et al. (2012) in Ethiopian chickpea accessions of different geographical origins using only 33 previously identified polymorphic SSR markers with a PIC value ranging from 0.27 to 0.50 and 3.36 bands per marker advocating the usefulness of those markers in germplasm characterization. SSR markers like CakTSSR00621 with high PIC value (0.77) and the markers signature alleles like CakTSSR01996 and CakTSSR03248 can be used to distinguish the accessions from each other signifying the relevance of these markers to generate specific genetic fingerprints for each genotype useful for genetic purity analysis.
High genetic distance between a wild and cultivated accession and a low genetic distance between two cultivated accessions was an expected outcome, for instance, molecular analysis revealed a high genetic distance (0.90) between parents IG 72953 (wild) and Annigeri-1 (parents of 2 different populations segregating for Helicoverpa resistance and drought avoidance root traits). Similar trend of genetic distance was observed in chickpea accession by Choudhary et al. (2012). Interestingly, the cluster analysis revealed that, Pusa 362 and JAKI 9218 genotypes are closely related (with almost 100 % similarity) with this set of markers followed by 90 % similarity (0.90) between ICCV 00108 and JG 74. Also, this study reconfirms that JG 11 + is a derived near isogenic line (NIL) of JG 11 with a genetic similarity of 89 %. This analysis also substantiates the diversity of selected chickpea MAGIC parents as revealed by dendrogram.
Cluster analysis separated all chickpea accessions into two major clusters with a wild accession IG 72953 not being part of either of the two clusters. PCA was in accordance with cluster analysis as IG 72953 out lied distantly with the other wild accession PI 489777 and not falling in either of the two clusters. The grouping of all the kabuli cultivars into Cluster II (Fig. 3) highlights the utility of these markers in discriminating kabuli genotypes with that of desi genotypes. Similarly, other desi accessions were intervened by breeding lines (desi) suggesting that breeding lines tend to cluster with kabuli and desi genotypes with the exception of a couple of desi genotypes grouped with the kabuli genotypes. Therefore, it can be concluded that kabuli and desi genotypes can be distinguished to a good extent exploiting SSR based diversity studies and this is in accordance with the findings of Upadhyaya et al. (2008), Sefera et al. (2011). However, the accessions originating from Turkey did not cluster together, which could be due to geographical separation. On the other hand, the accessions from Ethiopia were present in the same cluster despite having different pedigree and representing both desi (2) and kabuli (1) types.
The present study has resulted in identification of novel genic SSRs using the available chickpea genome. The strategy used here can also be deployed in other crops where there is scarcity of genic SSRs. The study has also avoided overestimation of chickpea genic SSRs by avoiding the duplicity with the already available SSRs in the public domain. Not only have these markers added to the existing repertoire of SSRs but also demonstrated their applicability in diversity studies, trait mapping, saturating genetic maps and in MABC programs for chickpea improvement.
References
Agarwal G, Jhanwar S, Priya P, Singh VK, Saxena MS, Parida SK, Garg R, Tyagi AK, Jain M (2012) Comparative analysis of kabuli chickpea transcriptome with desi and wild chickpea provides a rich resource for development of functional markers. PLoS ONE 7:e52443
Buhariwalla HK, Jayashree B, Eshwar K, Crouch JH (2005) Development of ESTs from chickpea roots and their use in diversity analysis of the Cicer genus. BMC Plant Biol 5:16
Choudhary S, Sethy NK, Shokeen BA, Bhatia S (2006) Development of sequence-tagged microsatellite site markers for chickpea (Cicer arietinum L). Mol Ecol Notes 6:93–95
Choudhary S, Sethy NK, Shokeen BA, Bhatia S (2008) Development of chickpea EST-SSR markers and analysis of allelic variation across related species. Theor Appl Genet 118:591–608
Choudhary P, Khanna SM, Jain PK, Bharadwaj C, Kumar J, Lakhera PC, Srinivasan R (2012) Genetic structure and diversity analysis of the primary gene pool of chickpea using SSR markers. Genet Mol Res 11:891–905
Cuc LM, Mace E, Crouch J, Quang VD, Long TD, Varshney RK (2008) Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (Arachis hypogaea L.). BMC Plant Biol 8:55
FAOSTAT data (2013) http://faostat.fao.org/site/339/default. aspx
Gaur R, Sethy NK, Choudhary S, Shokeen B, Gupta V, Bhatia S (2011) Advancing the STMS genomic resources for defining new locations on the intraspecific genetic linkage map of chickpea (Cicer arietinum L.). BMC Genom 12:117
Gaur PM, Jukanti AK, Varshney RK (2012) Impact of genomic technologies on chickpea breeding strategies. Agronomy 2:199–221
Gujaria N, Kumar A, Dauthal P, Dubey A, Hiremath P, BhanuPrakash A, Farmer A, Bhide M, Shah T, Gaur PM, Upadhyaya HD, Bhatia S, Cook DR, May GD, Varshney RK (2011) Development and use of genic molecular markers (GMMs) for construction of a transcript map of chickpea (Cicer arietinum L.). Theor Appl Genet 122:1577–1589
Gupta PK, Varshney RK (2000) The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat. Euphytica 113:163–185
Gupta PK, Langridge P, Mir RR (2010) Marker-assisted wheat breeding: present status and future possibilities. Mol Breed 26:145–161
Hiremath PJ, Farmer A, Cannon SB, Woodward J, Kudapa H, Tuteja R, Kumar A, BhanuPrakash A, Mulaosmanovic B, Gujaria N, Krishnamurthy L, Gaur PM, KaviKishor P, Shah T, Srinivasan S, Lohse M, Xiao Y, Town CD, Cook DR, May GD, Varshney RK (2011) Large-scale transcriptome analysis in chickpea (Cicer arietinum L.) an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnol J 9:922–931
Holton TA, Christopher JT, McClure L, Harker N, Henry RJ (2002) Identification and mapping of polymorphic SSR markers from expressed sequences of barley and wheat. Mol Breed 9:63–71
Hüttel B, Winter P, Weising K (1999) Sequence tagged microsatellite site markers for chickpea (Cicer arietinum L.). Genome 42:210–217
Keneni G, Bekele E, Imtiaz M, Dagne K, Getu E, Assefa F (2012) Genetic diversity and population structure of Ethiopian chickpea (Cicer arietinum L.) germplasm accessions from different geographical origins as revealed by microsatellite markers. Plant Mol Biol Rep 30:654–665
Kong Q, Zhang G, Chen W, Zhang Z, Zou X (2012) Identification and development of polymorphic EST-SSR markers by sequence alignment in pepper, Capsicum annuum (Solanaceae). Am J Bot 99:e59–e61
Kujur A, Bajaj D, Saxena MS, Tripathi S, Upadhyaya HD, Gowda CLL, Singh S, Jain M, Tyagi AK, Parida SK (2013) Functionally relevant microsatellite markers from chickpea transcription factor genes for efficient genotyping applications and trait association mapping. DNA Res 20:355–374
Lichtenzveig J, Scheuring C, Dodge J, Abbo S, Zhang HB (2005) Construction of BAC and BIBAC libraries and their applications for generation of SSR markers for genome analysis of chickpea, Cicer arietinum L. Theor Appl Genet 110:492–510
Michael TP, Jackson S (2013) The first 50 plant genomes. The Plant Genome 6:2. doi:10.3835/plantgenome2013.03.0001in
Moreno M, Cubero JI (1978) Variation in Cicer arietinum L. Euphytica 27:465–485
Nayak SN, Zhu H, Varghese N, Datta S, Choi H-K, Horres R, Jüngling R, Singh J, KaviKishor PB, Sivaramakrihnan S, Hoisington DA, Kahl G, Winter P, Cook DR, Varshney RK (2010) Integration of novel SSR and gene-based SNP marker loci in the chickpea genetic map and establishment of new anchor points with Medicago truncatula genome. Theor Appl Genet 120:1415–1441
Qiu L, Yang C, Tian B, Yang JB, Liu A (2010) Exploiting EST databases for the development and characterization of EST-SSR markers in castor bean (Ricinus communis L.). BMC Plant Biol 10:278
Rohlf FJ (2000) NTSYSpc Version 2.1: Numerical taxonomy and multivariate analysis system. Exeter Publications, New York
Sefera T, Abebie B, Gaur PM, Assefa K, Varshney RK (2011) Characterisation and genetic diversity analysis of selected chickpea cultivars of nine countries using simple sequence repeat (SSR) markers. Crop Pasture Sci 62:177–187
Sethy NK, Shokeen B, Edwards KJ, Bhatia S (2006) Development of microsatellite markers and analysis of intraspecific genetic variability in chickpea (Cicer arietinum L.). Theor Appl Genet 112:1416–1428
Sneath PHA, Sokal RR (1973) Numerical taxonomy. The principles and practice of numerical classification. W.H. Freeman and Company, San Francisco
Thiel T, Michalek W, Varshney R, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Thudi M, Bohra A, Nayak SN, Varghese N, Shah TM, Penmetsa RV, Thirunavukkarasu N, Gudipati S, Gaur PM, Kulwal PL, Upadhyaya HD, KaviKishor PB, Winter P, Kahl G, Town CD, Kilian A, Cook DR, Varshney RK (2011) Novel SSR markers from BAC-end sequences, DArT arrays and a comprehensive genetic map with 1,291 marker loci for chickpea (Cicer arietinum L.). PLoS ONE 6:e27275
Thudi M, Gaur PM, Krishnamurthy L, Mir RR, Kudapa H, Fikre A, Kimurto P, Tripathi S, Soren KR, Mulwa R, Bharadwaj C, Datta S, Chaturvedi SK, Varshney RK (2014) Genomics-assisted breeding for drought tolerance in chickpea. Funct Plant Biol 41:1178–1190
Udupa SM, Baum M (2003) Genetic dissection of pathotype specific resistance to Ascochyta blight disease in chickpea (Cicer arietinum L.) using microsatellite markers. Theor Appl Genet 106:1196–1202
Upadhyaya HD, Dwivedi SL, Baum M, Varshney RK, Udupa SM, Gowda CLL, Hoisington DA, Singh S (2008) Genetic structure, diversity, and allelic richness in composite collection and reference set in chickpea (Cicer arietinum L.). BMC Plant Biol 8:106
Varshney RK, Graner A, Sorrells ME (2005) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23:48–55
Varshney RK, Hoisington DA, Upadhyaya HD, Gaur PM, Nigam SN, Saxena K, Vadez V, Sethy NK, Bhatia S, Aruna R, Gowda MVC, Singh NK (2007) Molecular genetics and breeding of grain legume crops for the semi-arid tropics. In: Varshney RK, Tuberosa R (eds) Genomics-assisted crop improvement. Genomics applications in crops, vol II. Springer, Dordrecht, pp 207–242
Varshney RK, Nayak SN, May GD, Jackson SA (2009) Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol 27:522–530
Varshney RK, Kudapa H, Roorkiwal M, Thudi M, Pandey MK, Saxena RK, Chamarthi SK, Sabbavarapu MM, Mallikarjuna N, Upadhyaya H, Gaur PM, Krishnamurthy L, Saxena K, Nigam SN, Pande S (2012) Advances in genomics research and molecular breeding applications in SAT legume crops by using next generation sequencing and high-throughput genotyping technologies. J Biosci 37:811–820
Varshney RK, Tuberosa R (2013a) Genomics applications in crop breeding for biotic stress tolerance, an introduction. In: Varshney RK, Tuberosa R (eds) Translational Genomics for Crop Breeding: Improvement for Abiotic Stress, Quality and Yield Improvement, vol II. Wiley Blackwell, Hoboken, pp 1–9
Varshney RK, Tuberosa R (2013b) Genomics applications in crop breeding for biotic stress tolerance, an introduction. In: Varshney RK, Tuberosa R (eds) Translational Genomics for Crop Breeding: Biotic Stress, vol I. Wiley Blackwell, Hoboken, pp 1–9
Varshney RK, Gaur PM, Chamarthi SK, Krishnamurthy L, Tripathi S, Kashiwagi J, Samineni S, Singh VK, Thudi M, Jaganathan D (2013a) Fast-track introgression of “QTL-hotspot” for root traits and other drought tolerance traits in JG 11, an elite and leading variety of chickpea (Cicer arietinum L.). Plant Genome 6:3 doi: 10.3835/plantgenome2013.07.0022
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Tar’an B, Millan T, Zhang X, Ramsay LD, Iwata A, Wang Y, Nelson W, Farmer AD, Gaur PM, Soderlund C, Penmetsa RV, Xu C, Bharti AK, He W, Winter P, Zhao S, Hane JK, Carrasquilla-Garcia N, Condie JA, Upadhyaya HD, Luo MC, Thudi M, Gowda CLL, Singh NP, Lichtenzveiz J, Gali KK, Rubio J, Nadarajan N, Dolezel J, Bansal KC, Xu X, Edwards D, Zhang G, Kahl G, Gil J, Singh KB, Datta SK, Jackson SA, Wang J, Cook DR (2013b) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31:240–246
Varshney RK, Thudi M, Nayak SN, Gaur PM, Kashiwagi J, Krishnamurthy L, Jaganathan D, Koppolu J, Bohra A, Tripathi S, Rathore A, Jukanti AK, Jayalakshmi V, Vemula A, Singh S, Yasin M, Sheshshayee MS, Viswanatha KP (2014) Genetic dissection of drought tolerance in chickpea (Cicer arietinum L.). Theor Appl Genet 127:445–462
Winter P, Pfaf T, Udupa SM (1999) Characterization and mapping of sequence-tagged microsatellite sites in the chickpea (Cicer arietinum L.) genome. Mol Gen Genet 262:90–101
Zhao Y, Williams R, Prakash CS, He G (2012) Identification and characterization of gene-based SSR markers in date palm (Phoenix dactylifera L.). BMC Plant Biol 12:237
Acknowledgments
The authors are thankful to KVSS Prasad, K Eshwar and Sri Swathi for technical help. This work has been under taken as part of the CGIAR Research Program on Grain Legumes. ICRISAT is a member of CGIAR Consortium.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Agarwal, G., Sabbavarapu, M.M., Singh, V.K. et al. Identification of a non-redundant set of 202 in silico SSR markers and applicability of a select set in chickpea (Cicer arietinum L.). Euphytica 205, 381–394 (2015). https://doi.org/10.1007/s10681-015-1394-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-015-1394-3