
Abstract
Background
In crop species, QTL analysis is commonly used for identification of factors contributing to variation of agronomically important traits. As an important pasture species, a large number of QTLs have been reported for perennial ryegrass based on analysis of biparental mapping populations. Further characterisation of those QTLs is, however, essential for utilisation in varietal improvement programs.
Results
A bibliographic survey of perennial ryegrass trait-dissection studies identified a total of 560 QTLs from previously published papers, of which 189, 270 and 101 were classified as morphology-, physiology- and resistance/tolerance-related loci, respectively. The collected dataset permitted a subsequent meta-QTL study and implementation of a cross-species candidate gene identification approach. A meta-QTL analysis based on use of the BioMercator software was performed to identify two consensus regions for pathogen resistance traits. Genes that are candidates for causal polymorphism underpinning perennial ryegrass QTLs were identified through in silico comparative mapping using rice databases, and 7 genes were assigned to the p150/112 reference map. Markers linked to the Lp DGL1, Lp Ph1 and Lp PIPK1 genes were located close to plant size, leaf extension time and heading date-related QTLs, respectively, suggesting that these genes may be functionally associated with important agronomic traits in perennial ryegrass.
Conclusions
Functional markers are valuable for QTL meta-analysis and comparative genomics. Enrichment of such genetic markers may permit further detailed characterisation of QTLs. The outcomes of QTL meta-analysis and comparative genomics studies may be useful for accelerated development of novel perennial ryegrass cultivars with desirable traits.
Similar content being viewed by others

Background
Perennial ryegrass is a native species of Europe, temperate Asia and North Africa and is widely cultivated in temperate regions as a pasture crop[1, 2]. This obligate outbreeding diploid species (2n = 2x = 14) is classified within the Pooideae sub-family of the Poaceae (grass and cereal) family[3]. The Pooideae sub-family contains a broad range of important cereal and forage crop species; hexaploid wheat (Triticum aestivum L.) and barley (Hordeum vulgare L.) are taxonomically classified into the Triticeae tribe; oat (Avena sativa L.) is a representative member of the Aveneae tribe; and perennial ryegrass, tall fescue (Festuca arundinacea Schreb.) and meadow fescue (F. pratensis Huds.) are included in the Poeae tribe[4]. Genome analysis studies have suggested that species in the Pooideae sub-family share a similar chromosomal structure, having been derived from a common ancestor with 7 chromosome pairs[5].
In crop species, agronomically important traits, such as grain number and salinity stress tolerance, are governed by multiple loci with relatively small individual effects, which are known as QTLs[6]. Favorable alleles of QTLs are able to be efficiently introduced into elite cultivars through marker-assisted selection (MAS) technology to generate new varieties with enhanced yield performance and adaptability to environmental conditions[7–9]. During the last decade, a number of molecular genetic studies of perennial ryegrass have been conducted to reveal the genetic basis of herbage quality and productivity. A one-way pseudo-testcross population, designated p150/112, was established through crossing between a multiple heterozygous parent of complex descent (C3) and a doubled haploid parent[10, 11]. Using this population, the first comprehensive genetic linkage map for perennial ryegrass was constructed using simple sequence repeat (SSR), amplified fragment length polymorphism (AFLP) and restriction fragment length polymorphism (RFLP) markers[11, 12]. The seven linkage groups (LGs) were numbered in accordance with conserved synteny with the genetic maps of the Triticeae cereal species[12]. The p150/112 population was also subjected to QTL analyses for plant architecture traits, herbage yield and quality characters, cold tolerance, heading date variation and seed production[13–15]. Following effective use of the p150/112 population, a number of successor mapping populations were developed for QTL identification across a range of common and additional traits[16–20].
The process of QTL meta-analysis was proposed as a means to identify consensus loci reported in numerous distinct studies[21]. The BioMercator software was designed to perform meta-QTL (MQTL) analysis using published data[22]. MQTL analysis has been achieved with the BioMercator software for a wide range of crop species, such as rice (Oryza sativa L.), wheat and soybean (Glycine max L.)[23–25]. Despite complexities of genome structure, 5 and 12 relatively large MQTLs were successfully identified in soybean and hexaploid wheat, respectively[24, 25]. Due to properties of stability under different environmental and genetic backgrounds, such meta-QTLs (MQTLs) are likely to be of particularly high value for breeding activities[24, 26]. The MQTLs identified in the previous studies provide primary targets for fine-structure mapping and gene identification activities[24, 25]. As the number of published trait-dissection studies has increased for perennial ryegrass, so this species has become a viable target for QTL meta-analysis.
Macrosyntenic relationships of genome structure between perennial ryegrass and taxonomically related cereal species, such as rice, wheat and oat have been demonstrated through cross-species mapping of functional genetic markers[12, 27]. By permitting transfer of knowledge from the related species, such colinearity has been used for identification of candidate genes that potentially underpin QTL-containing regions. For instance, co-locations were demonstrated between candidate ortholoci of rice heading date control genes and QTLs for flowering time variation in perennial ryegrass, suggesting functional similarity of these genes between the two species[15, 28, 29]. Similarly, the Lp ABCG5 gene was proposed to contribute to a plant architecture QTL effect in perennial ryegrass, based on a comparative genomics approach between related species, including rice[30]. The value of such an approach for gene identification is currently higher than for map-based cloning strategies in species such perennial ryegrass, as compared to inbreeding species, due to an obligate outbreeding reproductive habit and relatively large genome size[31].
In this study, a bibliographic survey of QTLs that were identified through use of perennial ryegrass-based genetic mapping populations during the last decade is presented. An MQTL analysis for selected loci across a range of functional categories, in concert with comparative analysis with rice QTL databases, was performed. Putative candidate genes were identified and subjected to a genetic linkage analysis with the p150/112 reference mapping population, providing the basis for assessment of QTL co-location in present and future studies.
Methods
Bibliographic survey and QTL categorisation
In order to collate information on perennial ryegrass QTLs, 23 previously published studies were identified, details of which are summarised in Additional file1[2, 13–15, 17, 18, 20, 29, 32–46]. Prefixes relating to nomenclature of the genetic marker classes are explained in Additional file2[47]. QTLs were categorised into 3 functionally related groups (morphology, physiology and resistance/tolerance), and 6–10 sub-groups (e.g. leaf/pseudostem), depending on trait features as previously described in an equivalent study of rice[48].
MQTL analysis for pathogen resistance QTLs
Identification of consensus QTLs was performed using the BioMercator software[22]. The p150/112, NA6 and AU6 maps were initially integrated and then aligned with the WSCF2, MFA, MFB and SB2 x TC1 genetic maps[11, 12, 17, 33, 34, 36, 41, 43, 44]. Locations of QTLs for pathogen resistance were extrapolated onto the consensus map on the basis of common genetic marker positions. Co-location of QTLs was determined on the basis of the Akaike’s information criterion (AIC), and the best fit model was selected for MQTL prediction.
In silico comparative genomic analysis
DNA sequence information was obtained from the NCBI (http://www.ncbi.nlm.nih.gov/), GrainGene (http://www.gramene.org/) and Phytozome (http://www.phytozome.net/) databases. The physical locations of orthologous genes were identified using the Phytozome database. Candidate genes for perennial ryegrass QTLs were identified using the Q-TARO[48] (http://qtaro.abr.affrc.go.jp/) and GRAMENE QTL (http://www.gramene.org/qtl/) databases and by bibliographic search of the NCBI database.
Genetic marker development and linkage analysis
The p150/112 one-way pseudo-testcross mapping population was generated through crossing of a multiply heterozygous genotype to an artificially generated (doubled haploid) homozygote. In this mating design, single nucleotide polymorphisms (SNPs) exhibit segregation patterns of the type AB x AA or AB x CC[10–12]. As a bin-mapping population sub-set, 46 genotypes, which represent individuals with maximal genetic recombination, were selected. Candidate orthologues for rice genes were identified through local BLAST searches. Locus-specific PCR primers were designed using the SequencherTM software version 4.7 for windows (Genecodes) and Oligo Calc program[49]. PCR amplification was performed using ImmolaseTM DNA polymerase (BIOLINE, London UK) following the product instructions. The PCR amplification was examined on a 2.0% (w/v) agarose gel with 0.5 x SYBR® Safe DNA gel staining (Invitrogen). The PCR products were treated with exonuclease I (0.5 U) and shrimp alkaline phosphatase (SAP; 0.5 U) at 37°C for 60 minutes, and enzymes were then deactivated by heat treatment at 85°C for 20 minutes. Sequencing analysis was performed with the BigDyeTM terminator chemistry (Applied Biosystems, at present Applera, Foster City California USA), following the manufacturer’s instructions, and the resulting products were analysed on the ABI 3730xl Prism sequencer (Applied Biosystems). SNPs in the targeted sequence were identified using the SequencherTM software, and genotyping data were scored. A genetic linkage map was constructed using the JoinMAP 3.0 application[50].
Results
Assembly of information on previously identified perennial ryegrass QTLs
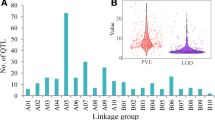
A total of 560 QTLs were described in previously published studies, of which 149 were identified from analysis of perennial ryegrass x Italian ryegrass interspecific hybrid populations[33, 34, 41, 45, 46]. Totals of 189 (34%), 270 (48%) and 101 (18%) were categorized as morphology-, physiology- and resistance/tolerance-related QTLs, respectively (Additional file1: Table S1). Of the 6 sub-classes of morphological QTLs, the proportion of leaf/pseudostem-related loci was 36% (68 QTLs), followed by panicle/flower (24%) and plant mass-related loci (23%). The most consistently reported physiological QTLs were fibre content-related loci. In the resistance/tolerance-related QTL group, 27 hydrate/dehydrate stress, 26 cold stress and 27 crown rust resistance-related loci were found. Totals of 106 and 100 QTLs were located on LGs 1 and 4, respectively, while relatively smaller number of loci were identified on LGs 5 (49 QTLs) and 6 (46 QTLs). This trend was also observed within each trait class (Table1; Additional file3).
The 560 QTLs were classified depending on percentages of phenotypic variances explained (Vp) (Figure1). About 60% of the QTLs displayed a Vp value of less than 15%, while 28 QTLs (5%) explained more than 40% of the phenotypic variance. The average and median values of Vp were 15.8% and 13.1%, respectively.

Distribution of V p values for perennial ryegrass QTLs.
MQTL analysis
A consensus map was constructed based a combination of the p150/112, AU6, NA6, WSCF2, MFA, MFB and SB2 x TC1 maps. Due to insufficiency of common genetic markers, map melding was not performed with linkage maps from the other published studies. Pathogen resistance QTLs were subjected to analysis performed with the BioMercator software. Two MQTLs, designated mqResis-2 and mqResis-6, were identified on LGs 2 and 6 of the consensus map, respectively (Table2). The mqResis-2 MQTL contained a grey leaf spot resistance and three crown rust resistance QTLs, while the mqResis-6 MQTL was a consensus of two grey leaf spot resistance loci and a crown rust resistance locus.
Cross-species candidate gene identification
Information required for the comparative candidate gene identification approach was obtained through the bibliographic survey of QTLs (Additional file 1). Of the 560 QTLs, putative functional markers were identified in the flanking regions of 265 loci. For 212 QTLs, orthologous regions in the rice genome were predicted using sequence information from flanking genetic markers. The candidate regions for 19 QTLs were not, however, located on orthologous chromosomes. For 45 perennial ryegrass QTLs, equivalent QTLs in orthologous regions of the rice genome were identified. A total of 10 rice candidate genes, for which ortholoci may contribute to perennial ryegrass QTL variation, were tentatively recognised (Table3)[51–60].
A total of 6 candidate genes were identified for plant morphogenesis traits. The PSR162-derived marker was located within the confidence interval containing a plant height QTL (Plantheight_C3_1) on perennial ryegrass LG1. A putative ortholocus for PSR162 was identified at the 25.5 Mb location on rice chromosome 5, and the rice EUI1 (elongated uppermost internode1) gene, which is responsible for control of internode length, was found at the closely adjacent 23.7 Mb position of the same chromosome.
A total of 5 herbage yield-related QTLs (Leafwidth_C3_3, Plantheight_C3_3, Tillersize_C3_3, LET_WSCF2_3 and qFG-03-m3) were observed to be linked to the CDO345-derived marker on perennial ryegrass LG3. A candidate CDO345 ortholocus was identified at the 37.9 Mb position on rice chromosome 1, and 3 plant size-related genes (DGL1, GA20ox-2 and Ph1) were observed in the 28.2-38.3 Mb region of the same chromosome.
Leaf length QTLs (qLL_WSC_7 and qLL_p150/112_7) were located on perennial ryegrass LG7, of which the maximum LOD score was observed at the Hd3a locus. On rice chromosome 6, the D3 gene (2.7 Mb) was located closely adjacent to the rice Hd3a gene (2.9 Mb).
A fall growth QTL, qFG-04-f3.2, was closely associated with the CDO460-derived marker. A putative CDO460 ortholocus was identified at the 1.4 Mb position of rice chromosome 1, relatively close (at the 4 Mb coordinate) to the dwarf growth locus OsGA3ox2.
A single candidate gene was identified for nutritive quality traits. Crude protein concentration QTLs (qCP-Sep-03-f2, qCP-Sep-03-m2, qCP-04-m2, qCP-04-f2 and qCP-su-gh-01_2) were identified on perennial ryegrass LG2, linked to the CDO385, CDO418, CDO59, RZ395, CDO1376 or CDO405-derived markers, for which candidate orthologues were located in the 18.5-27.6 Mb interval of rice chromosome 7. A rice phenylalanine biosynthesis gene, PDT, was identified at the 30.0 Mb position of rice chromosome 7. The role of PDT is to control accumulation level of the amino acids phenylalanine and tryptophan.
A single candidate gene was identified for reproductive development traits. Maximum LOD values for heading date variation QTLs (qHD_PxA_4, qHD_WSC_4 and qHD_C3_4) were identified close to the CDO795-derived marker on perennial ryegrass LG4. A heading date locus (OsPIPK 1) was located at the 28.2 Mb position of rice chromosome 3, close to the predicted CDO795 ortholocus (23.1 Mb).
A total of three candidate genes were identified for pathogen resistance traits. QTLs for resistance to powdery mildew (PMR1_INF1_VrnA) and crown rust (qCrownrust04_WI2_MFB3) pathogens were located on perennial ryegrass LG3, in linkage to the LRGA4 and RZ444 loci, respectively. The Pi37 blast resistance locus is located at the 33.1 Mb position on rice chromosome 1, relatively close to putative ortholoci for LRGA4 (20.6 Mb) and RZ444 (35.9 Mb).
The CDO686-derived marker was located in linkage with grey leaf spot resistance QTLs (qGLSGG9gc_1_MFA_6 and qGLSGG9gc_2_MFA_6) on perennial ryegrass LG6. A putative CDO686 ortholocus is located at the 31.8 Mb position of rice chromosome 2, and the rice Pib locus, which is responsible for resistance to rice blast, was observed at the 35.1 Mb coordinate on the same chromosome.
MQTLs were also subjected to the cross-species mapping approach. Sequences orthologous to BCD1184 and Lpest0222, which were located close to mqResis-2, were identified on rice chromosome 2 at the 29.4 Mb and 30.1 Mb positions. A candidate orthologous region for mqResis-6 was identified on rice chromosome 7, through identification of putative CDO497 and RZ273 ortholoci at the 23.7 Mb and 30.7 Mb coordinates, respectively. No equivalent rice QTL was, however, identified for the MQTLs.
Genetic mapping of candidate genes
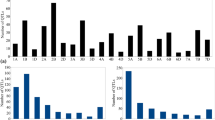
PCR primers for the putative perennial ryegrass ortholoci of the 10 rice candidate genes were designed, and PCR fragments from 9 of the target genes were obtained from the p150/112 C3 (heterozygous) parent (Table4). Successful amplification was not observed when the OsGA3ox2 ortholocus-directed primer pair was used. Direct sequencing analysis identified SNPs in 7 of 9 amplicons, DNA sequence polymorphism being absent from amplicons corresponding to the GA20ox-2 and PDT genes. The p150/112 bin mapping set was genotyped to obtain successful sequencing data from 30–46 individuals. Selected SNP loci from the EUI1, DGL1, Ph1, OsPIPK 1 and D3 gene orthologues were assigned to locations on those LGs that were anticipated on the basis of conserved synteny, while those in Pi37 and Pbi-like sequences were assigned to LGs 4 and 5, respectively (Figures2 and3).

Genetic locations of candidate gene-derived markers and QTLs on LGs 1 and 3 of the p150/112 reference genetic map. Genetic distance (cM) is shown on the left side of the genetic map. Candidate gene-derived markers are represented in red. The positions of the QTL interval and maximum LOD value are shown on the left side of linkage map with the light blue line and red triangle, respectively. The functional markers used for comparative analysis are represented in blue. The LG3 map constructed with the WFC F2 population and QTL intervals for autumn growth (AGC), leaf extension time (LET) and leaf extension rate (LER) is shown on the right side of the LG3 map of the p150/112 map (cited from[44]), and corresponding makers are connected with broken blue lines.

Discussion
QTL meta-analysis in perennial ryegrass
As a pasture crop species, a predominant focus on vegetative yield-related characters has been observed during trait-dissection studies of perennial ryegrass, leading to identification of a large number of leaf/pseudostem and plant mass-related QTLs (Table1). In contrast, as a grain crop, panicle/flower and seed-related traits have received more attention in rice QTL identification activities[48]. In the present study, lower QTL numbers were identified on LGs 5 and 6. In comparison, a meta-study for hexaploid wheat grain yield-related QTLs identified relatively smaller numbers of QTLs were identified on the homoeologous 5, 6 and 7 groups of chromosomes[24], which exhibit extensive macrosynteny with perennial ryegrass LGs 5, 6 and 7[12]. Hence, despite divergent trait-specific biases between perennial ryegrass and hexaploid wheat, a similar chromosomal distribution pattern of QTLs was exhibited (Table1). In a previous study of grain yield under drought stress conditions, conservation of QTL locations between different Poaceae species was observed[26]. As perennial ryegrass and wheat are relatively closely related within the cool-season grasses, the similarity of QTL distribution patterns between these two species suggests that conserved regions corresponding to wheat homoeologous chromosomes 5, 6 and 7 show lower importance than others for agronomic traits, including both vegetative and seed yield characters. Due to a large genome size, the perennial ryegrass genome has not yet been completely sequenced and assembled. Full assembly of genome sequence information from chromosomes that are rich in important QTLs may be more valuable, and should perhaps be prioritised, in comparison to that from other chromosomes.
The bibliographic survey identified putatively conservation of QTL locations under different environmental and across different genetic backgrounds. Plant height QTLs on LG1 were reported in three distinct studies, and heading date QTLs on LGs 4 and 7 were identified with various parental combinations at multiple geographic locations, although further analysis is required to determine whether the common QTLs are controlled by identical genetic factors[13, 15, 18, 29, 32, 42]. The two pathogen resistance MQTLs are also putatively conserved under multiple environmental conditions and genetic backgrounds. Conversely, evidence was also obtained for a relatively large number of QTLs that are either genotype- or environment-specific. QTL analysis studies with two-way pseudo-testcross populations have demonstrated the presence of QTLs only on single parental genetic maps for traits measured under identical environmental conditions[2, 17, 32]. Several studies also subjected single populations to QTL analysis under various environmental conditions, and reported environment-specific QTLs[33, 42, 45, 46]. The p150/112 mapping population was developed for the activities of the International Lolium Genome Initiative (ILGI) and was subjected to QTL analysis for traits such as leaf length, leaf width, and variation for heading date in both Japan and the UK, identifying unique QTLs at the two geographic locations[13, 15]. Leaf length and width QTLs were identified on LGs 5 and 3, respectively under Japanese conditions, while QTLs for both traits were found on LG7 in the UK-based trial. Only a single heading date QTL on LG4 was detected in Japan, while two QTLs on LGs 4 and 7 were found in the UK, probably associated with vernalisation genes (Vrn-1 and Hd3a orthologues, respectively). These results suggest that although stable QTLs may be detected under different environmental and genetic backgrounds, QTL identification largely depends on both genetic and environmental factors in perennial ryegrass.
The frequency distribution of Vp demonstrated in this study (Figure1) was also similar to that obtained from a previous study in rice, in which the mean Vp value was calculated to be c. 13%, based on a sample of 231 QTLs[61]. In both studies, although the distribution range was skewed towards lower Vp values, a considerably small number of QTLs were classed in the 0-5% category. The probable presence of loci of minor effect, which could be excluded from identification due to the requirement for threshold LOD values for QTL detection, was also described for the rice study, and such minor undetected QTLs are also likely to be present in perennial ryegrass. Although F2 and BC1 genetic mapping populations have been generally employed for rice, construction of perennial ryegrass linkage maps has been commonly based on use of one-way and two-way pseudo-testcross strategies, due to the effect of an outbreeding reproductive habit. These crossing formats may not achieve such precise estimation of QTL effects as the F2 and BC1 designs, due to complexity of genetic background[62]. Despite this difference, the distribution patterns of Vp values were largely similar between the two species.
Due to the relatively small sizes of discovery populations (typically in the range from 100–200 genotypes) that have been used for trait-dissection in perennial ryegrass, the magnitudes of QTL effects have probably been over-estimated. Several studies have identified failures to deliver anticipated genetic gains through marker-assisted QTL selection, due apparently to both over-estimation and imprecise estimates of location[63, 64]. The basis of these problems has been extensively discussed, and has in most cases been attributed to the influence of experimental population size, the so-called Beavis effect[65–67]. QTL identification in progressively larger population sets, up to 500–1000 individuals, has been theoretically and empirically demonstrated to enhance the accuracy of QTL effect measurement. Alternatively, more accurate estimates of locus-specific effect are likely to derive from implementation of genome-wide association studies (GWAS). For example, a GWAS for 14 agronomic traits in rice identified six characters associated with colours, grain quality and grain width that exhibited a small number of significant loci with large effects, while the remaining traits were influenced by multiple loci with relatively small effects[68]. Equivalent studies in perennial ryegrass might be anticipated to generate similar results.
The BioMercator software assisted the melding of linkage maps resulting from distinct studies. This process, however, was not fully accomplished in the present study, except for the p150/112, AU6, NA6, WSCF2, MFA, MFB and SB2 x TC1 maps, due to insufficiency of common genetic markers. In previous studies, non-functional DNA-based markers, such as genomic DNA-derived SSR, AFLP and restriction site-associated DNA (RAD) systems, were predominantly used[18, 32, 37]. Such assays are not ideally suited to comparative mapping studies, as multiple locus amplification is often observed for genomic DNA-derived SSR markers, and both AFLPs and RADs are more genotype-specific than functional markers[11, 31, 37]. Enrichment of functional markers is hence essential for a further meta-analysis. A recent study assigned over 700 gene-derived markers to perennial ryegrass LGs with public release of the corresponding information[69]. The outcomes may permit efficient functional marker enrichment in specific chromosomal regions of interest.
Prediction of candidate gene status
Two putative MQTLs were identified for pathogen resistance (Table2). Both mqResis-2 and mqResis-6 were identified as consensus loci containing both grey leaf spot and crown rust resistance QTLs, implying non-specific activities for several pathogens, rather than race-specific resistance QTLs. Through the process of genetic map alignment and MQTL analysis, additional functionally associated markers that are putatively linked to the QTLs were identified. Information from functional markers may support development of novel flanking DNA-based markers for a given target locus based on a comparative genetics approach, enabling candidate gene-based selection and association genetics studies[15, 70]. Although further characterisation is required, both MQTLs and flanking functional markers may be useful for deployment in perennial ryegrass breeding.
Comparative analysis demonstrated close proximity between genetic markers related to the DGL1, Ph1 and OsPIPK 1 ortholoci and the corresponding perennial ryegrass QTLs. This observation suggests that the DGL1 and Ph1 ortholoci are related to, and may provide candidate genes for, the herbage yield-related QTLs on LG3. In a previous study, the CDO795-linked heading date QTLs were suggested to be equivalent to a rice heading date QTL, dth3.3 (Gramene QTL Acc. ID AQFE011)[15, 71]. The physical location of the OsPIPK 1 gene was located in the candidate interval (5.7 Mb) of dth3.3. These results suggest that the perennial ryegrass OsPIPK 1 ortholocus may be related to the heading date QTLs on LG4. For both yield and flowering time traits, plausible evidence for related candidate genes has been obtained.
In contrast, markers linked to the EUI1 and D3 ortholoci were located over 10 cM distant from the maximum LOD values for the target QTLs. In a wide range of plant species, genes causing variation in quantitative traits have been identified to be located within genetic distances of less than 3 cM from the LOD maximum location[72]. It seems, therefore, unlikely that Lp EUI1 and Lp D3 genes are plausible candidates for QTL function. For issues arise for candidate genes associated with disease resistance.
The Pi37 and Pbi genes encode NBS-LRR proteins[51, 59]. Molecular studies have shown a rapid evolutionary rate and limited cross-species synteny of NBS-LRR genes[51, 59, 73, 74]. The comparative approach may not be so effective for such species-specific genes, due to unresolved paralogous relationships between species, and hence accounting for the failure of putative ortholoci to map in regions predicted on the basis of conserved synteny.
Conclusion
In this study, meta-analysis of QTL architecture in perennial ryegrass has permitted evaluation of the range of typical genetic effects across a range of biological trait categories. Additionally, MQTL analysis identified two consensus QTLs for pathogen resistance, as well as putatively linked functional markers. Comparative genetics analysis for a sample of putative candidate genes revealed ortholoci of three rice genes that may plausibly be causally related to QTLs for correlated functions. Enrichment of functional markers may permit further Meta-analysis and comparative approach for those QTLs. Outcomes from those studies may be utilised in the MAS framework for varietal development of perennial ryegrass with desirable traits.
References
Hannaway D, Fransen S, Cropper J, Teel M, Chaney M, Griggs T, Halse R, Hart J, Cheeke P, Hansen D, Klinger R, Lane W: Oregon State University Extension Publication PNW503. Perennial ryegrass (Lolium perenne L.). 1999, Corvallis, Oregon: Oregon State University Extension Publication
Pearson A, Cogan NO, Baillie RC, Hand ML, Bandaranayake CK, Erb S, Wang J, Kearney GA, Gendall AR, Smith KF, Forster JW: Identification of QTLs for morphological traits influencing waterlogging tolerance in perennial ryegrass (Lolium perenne L.). Theor Appl Genet. 2011, 122 (3): 609-622.
Wilkins PW: Breeding perennial ryegrass for agriculture. Euphytica. 1991, 52: 201-214.
Devos KM, Gale MD: Comparative genetics in the grasses. Plant Mol Biol. 1997, 35 (1–2): 3-15.
International, Brachypodium, Initiative: Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010, 463 (7282): 763-768.
Miura K, Ashikari M, Matsuoka M: The role of QTLs in the breeding of high-yielding rice. Trends Plant Sci. 2011, 16 (6): 319-326.
Takeuchi Y, Ebitani T, Yamamoto T, Sato H, Ohta H, Hirabayashi H, Kato H, Ando I, Nemoto H, Imbe T, Yano M: Development of Isogenic Lines of Rice Cultivar Koshihikari with Early and Late Heading by Marker-assisted Selection. Breed Sci. 2006, 56 (4): 405-413.
Collard BC, Mackill DJ: Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc Lond B Biol Sci. 2008, 363 (1491): 557-572.
Takeuchi Y: Developing isogenic lines of Japanese rice cultivar ‘Koshihikari’ with early and late heading. JARQ. 2011, 45 (1): 15-22.
Ritter E, Gebhardt C, Salamini F: Estimation of recombination frequencies and construction of RFLP linkage maps in plants from crosses between heterozygous parents. Genetics. 1990, 125 (3): 645-654.
Jones S, Dupal P, Dumsday L, Hughes J, Forster W: An SSR-based genetic linkage map for perennial ryegrass ( Lolium perenne L.). Theor Appl Genet. 2002, 105 (4): 577-584.
Jones ES, Mahoney NL, Hayward MD, Armstead IP, Jones JG, Humphreys MO, King IP, Kishida T, Yamada T, Balfourier F, Charmet G, Forster JW: An enhanced molecular marker based genetic map of perennial ryegrass (Lolium perenne) reveals comparative relationships with other Poaceae genomes. Genome. 2002, 45 (2): 282-295.
Yamada T, Jones ES, Nomura T, Hisano H, Shimamoto Y, Smith KF, Hayward MD, Forster JW: QTL analysis of morphological, developmental and winter hardiness-associated traits in perennial ryegrass (Lolium perenne L.). Crop Sci. 2004, 44: 925-935.
Cogan NO, Smith KF, Yamada T, Francki MG, Vecchies AC, Jones ES, Spangenberg GC, Forster JW: QTL analysis and comparative genomics of herbage quality traits in perennial ryegrass (Lolium perenne L.). Theor Appl Genet. 2005, 110 (2): 364-380.
Armstead IP, Turner LB, Marshall AH, Humphreys MO, King IP, Thorogood D: Identifying genetic components controlling fertility in the outcrossing grass species perennial ryegrass (Lolium perenne) by quantitative trait loci analysis and comparative genetics. New Phytol. 2008, 178 (3): 559-571.
Faville MJ, Vecchies AC, Schreiber M, Drayton MC, Hughes LJ, Jones ES, Guthridge KM, Smith KF, Sawbridge T, Spangenberg GC, Bryan GT, Forster JW: Functionally associated molecular genetic marker map construction in perennial ryegrass (Lolium perenne L.). Theor Appl Genet. 2004, 110 (1): 12-32.
Dracatos PM, Cogan NO, Dobrowolski MP, Sawbridge TI, Spangenberg GC, Smith KF, Forster JW: Discovery and genetic mapping of single nucleotide polymorphisms in candidate genes for pathogen defence response in perennial ryegrass (Lolium perenne L.). Theor Appl Genet. 2008, 117 (2): 203-219.
Jensen LB, Andersen JR, Frei U, Xing Y, Taylor C, Holm PB, Lubberstedt T: QTL mapping of vernalization response in perennial ryegrass (Lolium perenne L.) reveals co-location with an orthologue of wheat VRN1. Theor Appl Genet. 2005, 110 (3): 527-536.
Anhalt UC, Heslop-Harrison PJ, Byrne S, Guillard A, Barth S: Segregation distortion in Lolium: evidence for genetic effects. Theor Appl Genet. 2008, 117 (2): 297-306.
Anhalt UCM, Heslop-Harrison JS, Piepho HP, Byrne S, Barth S: Quantitative trait loci mapping for biomass yield traits in a Lolium inbred line derived F2 population. Euphytica. 2009, 170: 99-107.
Goffinet B, Gerber S: Quantitative trait loci: a meta-analysis. Genetics. 2000, 155 (1): 463-473.
Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, J J: BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics. 2004, 20 (14): 2324-2326.
Khowaja FS, Norton GJ, Courtois B, Price AH: Improved resolution in the position of drought-related QTLs in a single mapping population of rice by meta-analysis. BMC Genomics. 2009, 10: 276-
Zhang LY, Liu DC, Guo XL, Yang WL, Sun JZ, Wang DW, Zhang A: Genomic distribution of quantitative trait loci for yield and yield-related traits in common wheat. J Integr Plant Biol. 2010, 52 (11): 996-1007.
Zhao-ming Q, Ya-nan S, Qiong W, Chun-yan L, Guo-hua H, Qing-shan C: A meta-analysis of seed protein concentration QTL in soybean. Can J Plant Sci. 2011, 91 (1): 221-230.
Swamy BP, Vikram P, Dixit S, Ahmed HU, Kumar A: Meta-analysis of grain yield QTL identified during agricultural drought in grasses showed consensus. BMC Genomics. 2011, 12: 319-
Paterson AH, Freeling M, Sasaki T: Grains of knowledge: genomics of model cereals. Genome Res. 2005, 15 (12): 1643-1650.
Armstead IP, Skot L, Turner LB, Skot K, Donnison IS, Humphreys MO, King IP: Identification of perennial ryegrass (Lolium perenne (L.)) and meadow fescue (Festuca pratensis (Huds.)) candidate orthologous sequences to the rice Hd1(Se1) and barley HvCO1 CONSTANS-like genes through comparative mapping and microsynteny. New Phytol. 2005, 167 (1): 239-247.
Armstead IP, Turner LB, Farrell M, Skot L, Gomez P, Montoya T, Donnison IS, King IP, Humphreys MO: Synteny between a major heading-date QTL in perennial ryegrass (Lolium perenne L.) and the Hd3 heading-date locus in rice. Theor Appl Genet. 2004, 108 (5): 822-828.
Shinozuka H, Cogan NO, Spangenberg GC, Forster JW: Comparative Genomics in Perennial Ryegrass (Lolium perenne L.): Identification and Characterisation of an Orthologue for the Rice Plant Architecture-Controlling Gene OsABCG5. Int J Plant Genomics. 2011, 291563-
Shinozuka H, Cogan NO, Smith KF, Spangenberg GC, Forster JW: Fine-scale comparative genetic and physical mapping supports map-based cloning strategies for the self-incompatibility loci of perennial ryegrass (Lolium perenne L.). Plant Mol Biol. 2010, 72 (3): 343-355.
Barre P, Moreau L, Mi F, Turner L, Gastal F, Julier B, Ghesquière M: Quantitative trait loci for leaf length in perennial ryegrass (Lolium perenne L.). Grass and Forage Science. 2009, 64: 310-321.
Brown RN, Barker RE, Warnke SE, Cooper LD, Brilman LA, Rouf Mian MA, Jung G, Sim S-C: Identification of quantitative trait loci for seed traits and floral morphology in a field-grown Lolium perenne × Lolium multiflorum mapping population. Plant Breeding. 2010, 129: 29-34.
Curley J, Sim SC, Warnke S, Leong S, Barker R, Jung G: QTL mapping of resistance to gray leaf spot in ryegrass. Theor Appl Genet. 2005, 111 (6): 1107-1117.
Koulman A, Cao M, Faville M, Lane G, Mace W, Rasmussen S: Semi-quantitative and structural metabolic phenotyping by direct infusion ion trap mass spectrometry and its application in genetical metabolomics. Rapid Commun Mass Spectrom. 2009, 23 (15): 2253-2263.
Muylle H, Baert J, Van Bockstaele E, Pertijs J, Roldan-Ruiz I: Four QTLs determine crown rust (Puccinia coronata f. sp. lolii) resistance in a perennial ryegrass (Lolium perenne) population. Heredity (Edinb). 2005, 95 (5): 348-357.
Pfender WF, Saha MC, Johnson EA, Slabaugh MB: Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor Appl Genet. 2011, 122 (8): 1467-1480.
Sartie AM, Matthew C, Easton HS, Faville MJ: Phenotypic and QTL analyses of herbage production-related traits in perennial ryegrass (Lolium perenne L.). Euphytica. 2011, 182 (3): 295-315.
Schejbel B, Jensen LB, Xing Y, Lubberstedt T: QTL analysis of crown rust resistance in perennial ryegrass under conditions of natural and artificial infection. Plant breeding = Zeitschrift fur Pflanzenzuchtung. 2007, 126 (4): 347-352.
Schejbel B, Jensen LB, Xing Y, Lubberstedt T: Mapping of QTL for resistance to powdery mildew and resistance gene analogues in perennial ryegrass. Plant breeding. 2008, 127 (4): 368-375.
Sim S, Diesburg K, Casler M, Jung G: Mapping and Comparative Analysis of QTL for Crown Rust Resistance in an Italian x Perennial Ryegrass Population. Phytopathology. 2007, 97 (6): 767-776.
Studer B, Jensen LB, Hentrup S, Brazauskas G, Kolliker R, Lubberstedt T: Genetic characterisation of seed yield and fertility traits in perennial ryegrass (Lolium perenne L.). Theor Appl Genet. 2008, 117 (5): 781-791.
Turner LB, Cairns AJ, Armstead IP, Ashton J, Skot K, Whittaker D, Humphreys MO: Dissecting the regulation of fructan metabolism in perennial ryegrass (Lolium perenne) with quantitative trait locus mapping. New Phytol. 2006, 169 (1): 45-57.
Turner LB, Cairns AJ, Armstead IP, Thomas H, Humphreys MW, Humphreys MO: Does fructan have a functional role in physiological traits? Investigation by quantitative trait locus mapping. New Phytol. 2008, 179 (3): 765-775.
Xiong Y, Fei S, Arora R, Brummer EC, Baker RE, Jung G, Warnke SE: Identification of quantitative trait loci controlling winter hardiness in an annual perennial ryegrass interspecific hybrid population. Mol Breeding. 2007, 19: 125-136.
Xiong Y, Fei S, Brummer EC, Moore KJ, Baker RE, Jung G, Curley J, Warnke SE: QTL analyses of fiber components and crude protein in an annual x perennial ryegrass interspecific hybrid population. Mol Breeding. 2006, 18: 327-340.
Bian XY, Friedrich A, Bai JR, Baumann U, Hayman DL, Barker SJ, Langridge P: High-resolution mapping of the S and Z loci of Phalaris coerulescens. Genome. 2004, 47 (5): 918-930.
J-i Y, Yamamoto T, Fukuoka S, Uga Y, Hori K, Yano M: Q-TARO: QTL Annotation Rice Online Database. Rice. 2010, 3 (2): 194-203.
Kibbe WA: OligoCalc: an online oligonucleotide properties calculator. Nucl Acids Res. 2007, 35 (suppl 2): W43-W46.
Van Ooijen JW, Voorrips RE: JoinMap® 3.0, Software for the calculation of genetic linkage maps. 2001, Plant Research International, Wageningen
Luo A, Qian Q, Yin H, Liu X, Yin C, Lan Y, Tang J, Tang Z, Cao S, Wang X, Xia K, Fu X, Luo D, Chu C: EUI1, encoding a putative cytochrome P450 monooxygenase, regulates internode elongation by modulating gibberellin responses in rice. Plant Cell Physiol. 2006, 47 (2): 181-191.
Yamada T, Matsuda F, Kasai K, Fukuoka S, Kitamura K, Tozawa Y, Miyagawa H, Wakasa K: Mutation of a rice gene encoding a phenylalanine biosynthetic enzyme results in accumulation of phenylalanine and tryptophan. Plant Cell. 2008, 20 (5): 1316-1329.
Itoh H, Ueguchi-Tanaka M, Sentoku N, Kitano H, Matsuoka M, Kobayashi M: Cloning and functional analysis of two gibberellin 3 beta -hydroxylase genes that are differently expressed during the growth of rice. Proc Natl Acad Sci U S A. 2001, 98 (15): 8909-8914.
Komorisono M, Ueguchi-Tanaka M, Aichi I, Hasegawa Y, Ashikari M, Kitano H, Matsuoka M, Sazuka T: Analysis of the rice mutant dwarf and gladius leaf 1. Aberrant katanin-mediated microtubule organization causes up-regulation of gibberellin biosynthetic genes independently of gibberellin signaling. Plant Physiol. 2005, 138 (4): 1982-1993.
Ashikari M, Sasaki A, Ueguchi-Tanaka M, Itoh H, Nishimura A, Datta S, Ishiyama K, Saito T, Kobayashi M, Khush GS, Kitano H, Matsuoka M: Loss-of-function of a Rice Gibberellin Biosynthetic Gene, GA20 oxidase (GA20ox-2), Led to the Rice ‘Green Revolution’. Breed Sci. 2002, 52: 143-150.
Kovi MR, Zhang Y, Yu S, Yang G, Yan W, Xing Y: Candidacy of a chitin-inducible gibberellin-responsive gene for a major locus affecting plant height in rice that is closely linked to Green Revolution gene sd1. Theor Appl Genet. 2011, 123 (5): 705-714.
Lin F, Chen S, Que Z, Wang L, Liu X, Pan Q: The blast resistance gene Pi37 encodes a nucleotide binding site leucine-rich repeat protein and is a member of a resistance gene cluster on rice chromosome 1. Genetics. 2007, 177 (3): 1871-1880.
Ma H, Xu SP, Luo D, Xu ZH, Xue HW: OsPIPK 1, a rice phosphatidylinositol monophosphate kinase, regulates rice heading by modifying the expression of floral induction genes. Plant Mol Biol. 2004, 54 (2): 295-310.
Wang ZX, Yano M, Yamanouchi U, Iwamoto M, Monna L, Hayasaka H, Katayose Y, Sasaki T: The Pib gene for rice blast resistance belongs to the nucleotide binding and leucine-rich repeat class of plant disease resistance genes. Plant J. 1999, 19 (1): 55-64.
Ishikawa S, Maekawa M, Arite T, Onishi K, Takamure I, Kyozuka J: Suppression of tiller bud activity in tillering dwarf mutants of rice. Plant Cell Physiol. 2005, 46 (1): 79-86.
Yano M, Sasaki T: Genetic and molecular dissection of quantitative traits in rice. Plant Mol Biol. 1997, 35 (1–2): 145-153.
Asíns MJ: Present and future of quantitative trait locus analysis in plant breeding. Plant Breeding. 2002, 121: 281-291.
Bouchez A, Hospital F, Causse M, Gallais A, Charcosset A: Marker-assisted introgression of favorable alleles at quantitative trait loci between maize elite lines. Genetics. 2002, 162 (4): 1945-1959.
Openshaw S, Frascaroli E: QTL detection and marker-assisted selection for complex traits. Proceedings of the 52nd Annual Corn and Sorghum Industry Research Conference: 1997; Washington. 1997, American Seed Trade Association, D.C., 44-53.
Beavis WD: the Proceedings of the Forty-Ninth Annual Corn and Sorghum Industry Research Conference: 1994. The power and deceit of QTL experiments: lessons from comparative QTL studies. 1994, American Seed Trade Association, Chicago, 250-266.
Beavis WD: QTL analyses: power, precision and accuracy. Molecular Dissection of Complex Traits. Edited by: A.H P. 1997, CRC Press, New York
Xu S: Theoretical basis of the Beavis effect. Genetics. 2003, 165 (4): 2259-2268.
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, Li M, Fan D, Guo Y, Wang A, Wang L, Deng L, Li W, Lu Y, Weng Q, Liu K, Huang T, Zhou T, Jing Y, Li W, Lin Z, Buckler ES, Qian Q, Zhang QF, Li J, Han B: Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010, 42 (11): 961-967.
Studer B, Byrne S, Nielsen RO, Panitz F, Bendixen C, Islam MS, Pfeifer M, Lubberstedt T, Asp T: A transcriptome map of perennial ryegrass (Lolium perenne L.). BMC Genomics. 2012, 13: 140-
Andersen JR, Lubberstedt T: Functional markers in plants. Trends Plant Sci. 2003, 8 (11): 554-560.
Thomson MJ, Tai TH, McClung AM, Lai XH, Hinga ME, Lobos KB, Xu Y, Martinez CP, McCouch SR: Mapping quantitative trait loci for yield, yield components and morphological traits in an advanced backcross population between Oryza rufipogon and the Oryza sativa cultivar Jefferson. Theor Appl Genet. 2003, 107 (3): 479-493.
Price AH: Believe it or not, QTLs are accurate!. Trends Plant Sci. 2006, 11 (5): 213-216.
Ratnaparkhe MB, Wang X, Li J, Compton RO, Rainville LK, Lemke C, Kim C, Tang H, Paterson AH: Comparative analysis of peanut NBS-LRR gene clusters suggests evolutionary innovation among duplicated domains and erosion of gene microsynteny. New Phytol. 2011, 192 (1): 164-178.
Zhang X, Feng Y, Cheng H, Tian D, Yang S, Chen JQ: Relative evolutionary rates of NBS-encoding genes revealed by soybean segmental duplication. Mol Genet Genomics. 2011, 285 (1): 79-90.
Acknowledgments
This work was supported by funding from the Victorian Department of Primary Industries and the Dairy Futures Cooperative Research Centre.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors’ contributions
HS co-conceptualised the project, performed the bibliographic survey and experimental work, prepared the tables and figures and the primary drafts of the manuscript, and contributed to finalisation of the text and journal-specific formatting. NC co-conceptualised the project, provided perennial ryegrass sequence information and contributed to computational analysis. GS assisted the project leadership and co-developed the final draft of the manuscript. JF co-conceptualised the project, provided overall project leadership, and co-developed interim and final drafts of the manuscript. All authors have read and approved the final manuscript.
Electronic supplementary material
12863_2012_1056_MOESM1_ESM.xls
Additional file 1:Summary information on QTLs identified through use of perennial ryegrass-based genetic mapping populations. QTLs are designated according to the following nomenclature: trait/date/condition abbreviation_experiment replication number/location_genetic map/population name_LG location_QTL identity (e.g. a or b) for the purpose of locus discrimination, as needed. For analysis type, IM, SIM, CIM, MQM and SMR stand for interval mapping, simple interval mapping, composite interval mapping, multiple QTL mapping and single-marker regression. When multiple parameters (e.g. SIM and CIM) are used for QTL detection, only QTLs identified with the representative parameter are shown. For population type, 1-way, 2-way and F2 stand for one-way pseudo-testcross population, two-way pseudo-testcross population and F2 genetic mapping population types. (XLS 140 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Shinozuka, H., Cogan, N.O., Spangenberg, G.C. et al. Quantitative Trait Locus (QTL) meta-analysis and comparative genomics for candidate gene prediction in perennial ryegrass (Lolium perenne L.). BMC Genet 13, 101 (2012). https://doi.org/10.1186/1471-2156-13-101
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-13-101