
Abstract
Volatile organic compounds (VOCs) in plants are involved in aroma and pest resistance. These compounds form a complex mixture whose composition is specific to species and often to varieties. Despite their importance as essential factors that determine peach fruit quality, understanding of molecular, genetic, and physiological mechanisms underlying aroma formation is limited. The aim of this study was the identification in peach of quantitative trait loci (QTLs) for fruit VOCs to understand their genetic basis using an F1 population of 126 seedlings deriving from the cross between “Bolero” (B) and “OroA” (O), two peach cultivars differing in their aroma profile. Dense single nucleotide polymorphism (SNP) and SSR maps covering the eight linkage groups of the peach genome were constructed by genotyping with the International Peach SNP Consortium peach SNP array v1, and data for 23 VOCs with high or unknown “odor activity value” were obtained by gas chromatography–mass spectrometry analysis of fruit essential oil in the years 2007 and 2008. A total of 72 QTLs were identified, most consistent in both years. QTLs were identified for the 23 VOCs studied, including three major QTLs for nonanal, linalool, and for p-menth-1-en-9-al stable in both years. Collocations between candidate genes and major QTLs were identified taking advantage of the peach genome sequence: genes encoding two putative terpene synthases and one lipoxygenase (Lox) might be involved in the biosynthesis of linalool and p-menth-1-en-9-al, and nonanal, respectively. Implications for marker-assisted selection and future research on the subject are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Understanding the genetic and molecular bases of volatile organic compounds (VOCs) emitted by the peach fruit helps to describe the peach aroma, an essential component of fruit quality. Furthermore, specific volatile molecules implicated in aroma also contribute to pest resistance (Laothawornkitkul et al. 2009). Knowledge of the mechanisms underlying VOC production could also be useful for the food and fragrance industry (Tholl and Rose 2006).
Genetic dissection of aroma in plants is difficult due to the polygenic nature of the trait, the number of compounds involved, and the analytical methods used in VOC quantification. Improved genotyping capacity (Agarwal et al. 2008) and VOC quantification methods (Tholl and Rose 2006) are now enabling the application of quantitative trait loci (QTL) analysis to aroma components. Several studies have addressed the identification of QTLs for VOCs in Rosaceae species as apple (Zini et al. 2005; Dunemann et al. 2009; Rowan et al. 2009a), rose (Spiller et al. 2010), and strawberry (Zorrilla-Fontanesi et al. 2012) and in other plant species including tomato (Saliba-Colombani et al. 2001; Tadmor et al. 2002; Mathieu et al. 2009; Zanor et al. 2009), grape (Doligez et al. 2006), melon (Obando-Ulloa et al. 2008), rice (Amarawathi et al. 2008), eucalypt (Henery et al. 2007; O’Reilly-Wapstra et al. 2011), and tobacco (Julio et al. 2006). Conversely, no QTLs underlying peach fruit aroma have been reported.
Aroma formation in peach fruit is a dynamic process involving variations in the concentrations of VOCs depending on ripening stage and pre- and post-harvest conditions and genotype. Peach VOCs have been found in skin and flesh, where around 100 compounds have been identified (Aubert et al. 2003). Of these, about 25 determine the typical peach aroma (Eduardo et al. 2010): particularly γ- and δ-decalactone play a key role in association with C6 compounds, aldehydes, aliphatic alcohols, esters, norisoprenoids, phenylpropanoids, and terpenes (Horvat et al. 1990). Key genes associated with aroma production have been described in few plants, including strawberry and apple (Schwab et al. 2008; Mathieu et al. 2009; Battilana et al. 2011; Dunemann et al. 2011; Zorrilla-Fontanesi et al. 2012), but little is known in peach.
Peach breeding programs have traditionally focused on traits such as fruit size, appearance, firmness, post-harvest behavior, productivity, resistance to biotic and abiotic stress, and ease of tree training. In strawberry and “Greek basil” few generations of breeding were sufficient to induce the loss of some VOCs, when these were not positively selected (Olbricht et al. 2008; Koutsos et al. 2009).
Peach breeding is hampered by a juvenile phase of 2 to 3 years, a short blossoming time, and the reduced genetic variability of elite germplasm (Scorza et al. 1985). Despite these constraints, small genome size and high level of homozygosity make peach a reference species for the identification, characterization, and cloning of important genes in the Rosaceae (Shulaev et al. 2008). A range of genomic tools, including molecular markers, genetic maps, EST libraries, the genome sequence, and recently a 9,000 SNP array have been developed for this species (Abbott et al. 2008; Arús et al. 2012; Verde et al. 2012; http://www.rosaceae.org/peach/genome). Because of the high degree of transferability of molecular markers among Rosaceae (Gasic et al. 2009), peach breeding programs can be facilitated by combining desirable alleles from different accessions or other Prunus species (Dirlewanger et al. 2004a). Linkage maps from peach varietal crosses have been developed (Dirlewanger et al. 1998; Lu et al. 1998; Sosinski et al. 1998; Yamamoto et al. 2001; Gillen and Bliss 2005; Blenda et al. 2007; Eduardo et al. 2011), along with maps deriving from crosses between almond, peach, apricot, cherry, Prunus davidiana, Prunus cerasifera, and Prunus ferganensis (Dirlewanger et al. 1996, 2003, 2004b; Dettori et al. 2001; Aranzana et al. 2003; Foulongne et al. 2003a). Moreover, in these species, the results of several QTL studies have been reported (Abbott et al. 1998; Dirlewanger et al. 1999; Quarta et al. 2000; Etienne et al. 2002; Foulongne et al. 2003b; Quilot et al. 2004; Ogundiwin et al. 2009; Eduardo et al. 2011). These studies have often been limited due to the low marker density and the lack of markers in some linkage groups. To circumvent these problems, the International Peach SNP Consortium (IPSC) developed an Illumina 9,000 SNP array that can be applied to the analysis of both biparental populations and germplasm collections (Verde et al. 2012).
In this paper, we report the first identification of peach genomic regions controlling the variability of fruit VOCs. QTL analysis was performed in a cross between two peach cultivars differing in their aroma profiles, “Bolero” (B) and “OroA” (O) (Eduardo et al. 2011), integrating genotyping information obtained with the 9,000 peach SNP array and phenotypic data from gas chromatography–mass spectrometry (GC-MS) analysis of fruit essential oil. Candidate genes for three major VOC QTLs are proposed, based on sequence analysis of the corresponding genomic regions.
Materials and methods
Plant material
The progeny assessed in this study is an F1 comprising 126 seedlings obtained from a cross between the melting peach cultivar Bolero (B) and the non-melting peach cultivar OroA (O), chosen for their distinct fruit flavor (Eduardo et al. 2011). Trees located in the CRPV (fruit trees research unit “Mario Neri”) orchards in Castel S. Pietro (Emilia Romagna, northern Italy) were planted on their own roots, with spacing of 1 m within and 4 m between rows and trained as slender spindle (one stem with short lateral scaffolds). Pruning was performed yearly and standard cultural practices applied. Fruits were thinned before pit hardening to a load of only 30–40 fruits per tree according to vigor, in order to allow a full expression of fruit size not limited by competition.
Aroma compound analysis
Phenotypic evaluation was repeated over 2 years (2007 and 2008). Each year, 20 fruits per tree were harvested at commercial maturity based on visual color change and manual evaluation of firmness. A slice was cut from each of the ten most uniform fruits obtaining 150 g total of fruit pieces (comprising flesh and skin), to which 200 ml of dichloromethane (Sigma, Italy) was added immediately after cutting. Dichloromethane allows to elute the hydrophobic molecules and to avoid oxidation processes (Tava et al. 2009). Samples were stored at 4 °C in the dark until steam distillation was performed.
Samples were steam-distilled for 1 h at 100 °C in a Clevenger apparatus with distilled water followed by solvent separation at room temperature of the volatile organic fraction, with 20 ml dichloromethane and 50 ml diethyl ether. One milliliter of each 3-methyl cyclohexanone (0.161 mg ml−1) and 4-nonanol (0.077 mg ml−1) was added as internal standards before the distillation started. The organic phase was concentrated in a rotary evaporator using a heating bath at 37 °C, dried over anhydrous NaSO4 to reach a final volume of 100 μl, and stored at −20 °C until chromatographic analysis.
Gas chromatography–flame ionization detector (GC-FID) analysis was carried out using a Perkin Elmer Clarus 500 gas chromatograph equipped with a 30 m × 0.32 mm i.d. Elite-5MS (5 % diphenyl/95 % dimethyl polysiloxane) capillary column (0.5 μm film thickness). Samples (0.5 μl) were injected with a column temperature program of 40 °C for 3 min, increasing to 300 °C at 4 °C/min and finally held at this last temperature for 5 min. Injector and detector were set at 300 and 320 °C, respectively. The carrier gas was helium with a head pressure of 12.0 psi.
GC-MS analyses were carried out using a Perkin Elmer Clarus 500 gas chromatograph equipped with a Clarus 500 mass spectrometer, using the same capillary column and chromatographic conditions as for the GC-FID analyses. Mass spectra were acquired over the 40–500 amu range at 1 scan/s, with ionizing electron energy of 70 eV and ion source at 200 °C. The transfer line was set at 300 °C and the carrier gas was helium at 1.0 ml/min. All measurements were performed in triplicate.
The identification of the essential oil components was performed using their retention indices; mass spectra by comparison with the NIST database mass spectral library (NIST/EPA/NIH Mass Spectral Database, Version 2.1, Perkin-Elmer Instrument LLC), published mass spectra 21, and authentic reference compounds (Sigma Aldrich, Italy). Retention indices were calculated using an n-alkane series (C6 to C32) under the same chromatographic conditions. Quantification was conducted using internal standards equivalents and response factors calculated from standard curves prepared with authentic compounds. A calibration factor curve, simulating compound behavior and recovery rate during distillation process, concentration, and column run, was obtained for the principal classes of compounds that better describe peach aroma. The calibration curve was obtained performing a triple series of distillations of authentic reference compounds at different concentrations (0.5×, 1×, 2×, 3×, and 4×) while the internal standards were kept at the same concentration (1×) in all samples. A calibration curve and equation were obtained for aldehydes, C6 compounds, esters, lactones, long chain hydrocarbons, benzaldehyde, phenylacetaldehyde, eugenol, and β-ionone. Quantification for acids, terpenes, and miscellaneous compounds was performed as direct comparison with internal standards peaks.
Using GC-FID and GC-MS analysis, we identified 49 VOCs in 2007 and 57 in 2008 (Eduardo et al. 2010). Only the 23 VOCs that were identified in both years and with a high or unknown “odor activity value” (OAV) according to Eduardo et al. (2010) were considered in QTL analysis.
Data analysis
R version 2.7.1 [R Development Core Team (2008). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org] was used to produce histograms of the different traits and to calculate the correlation coefficients between traits and between years.
SNP genotyping
The BxO F1 progeny was genotyped using the recently developed Illumina 9,000 SNP array v1 for peach (Verde et al. 2012) to further saturate the genetic map described in Eduardo et al. (2011). Four SNPs located in candidate genes were also genotyped using KASPar technology (see candidate gene mapping section).
For 9,000 SNP array genotyping, DNA was extracted with the DNAeasy 96 Plant kit (Qiagen) and diluted to 50 ng/μl. Genotyping was performed at IASMA Research and Innovation Centre (San Michele all’Adige, Italy) following the manufacturer’s recommendations as described in Verde et al. (2012). SNP data were scored using GenomeStudio Data Analysis software (Illumina Inc.) using a GenCall threshold of 0.15. SNPs with GenTrain score < 0.6 and those showing severe segregation distortion (χ2 test, p < 10−6) and more than 1 % of missing data were excluded from further analyses.
Linkage mapping
Genetic linkage analysis and map constructions were performed with JoinMap 4.1 (Van Ooijen 2011). Three maps (B map, O map, and BxO map) were produced for the BxO cross, one for each parent and one for the linkage groups that could be integrated (double pseudo-test cross strategy). Grouping was performed using independence LOD with groups presenting a LOD score between 4 and 10. Map construction was performed using the multipoint maximum likelihood based algorithm. Linkage groups nomenclature was according to the Prunus reference map (TxE) (Dirlewanger et al. 2004a).
QTL analysis
QTL analysis was carried out by MAPQTL software v.6.0 (Van Ooijen 2009) using the B, O, and BxO maps described in the linkage mapping results. After a Shapiro–Wilk test, we found eight VOCs presenting normal distributions in one (E-2-hexenyl acetate, nonanal, 6-pentyl-α-pyrone, γ-decalactone, γ-dodecalactone, eugenol, unidentified1, and unidentified2) or both years (E-β-damascenone); therefore, we decided to analyze QTLs using both interval mapping (IM) and the non-parametric Kruskal–Wallis rank-sum test (K-W). A QTL was considered significant when presented LOD >3 in the IM or a stringent significance level of p < 0.005 in the K-W analysis, as suggested by Van Ooijen (2009). In one case, we also considered a QTL that showed significance in one year and levels very close to significance in the other year (e.g., the phenylacetaldehyde QTL detected on linkage group B6 in year 2008 with LOD 2.63). In the present study, a major QTL is defined as explaining more than 20 % of the phenotypic variability in both years.
Candidate gene mapping
SNPs were identified in the sequences of the candidate genes based on B and O genomic resequencing information (International PeachGenome Initiative, The genome sequence of peach, a key diploid tree species, reveals unique patterns of genetic diversity, domestication and genome evolution, unpublished). SNPs identified in the terpene synthases candidate genes ppa003380m and ppa002670m were genotyped using KASPar technology (KBiosciences, UK) and integrated into BxO genetic maps (B, O, and BxO). SNP assays were designed by KBioscience (UK) and genotyping was carried out at the PTP Genomics Platform following the standard KASPar protocol on an ABI 7900 Real Time PCR system (Applied Biosystems, Italy).
Phylogenetic analysis of peach LOX proteins
Peach protein sequences annotated as lipoxygenases in the peach genome were downloaded from the Phytozome database (http://www.phytozome.org). LOX protein sequences from other plant species were retrieved from Han et al. (2011). Multiple protein sequence alignment and phylogenetic analysis was performed using MEGA4.0 software (http://www.megasoftware.net/).
Results
Linkage maps
Out of 8,144 SNPs from the 9,000 peach array, 2,744 did not pass the filtering criteria and 3,652 were homozygous in both parents; therefore, they were discarded from the analysis. A total of 1,453 SNPs in B, 229 SNPs in O, and 66 SNPs in both B and O were detected as heterozygous. We combined this dataset with the available genotypes of the SSR markers described in Eduardo et al. (2011), and with the genotypes obtained for the four SNPs from the terpene synthases candidate genes ppa003380m and ppa002670m. Preliminary linkage analysis showed a very high number of markers that cosegregated in clusters. To reduce the datasets (B, O, and BxO) for QTL analysis, we included in the final maps one marker for each cluster. The selected datasets recover the maximum number of recombination events that could be detected with the available genotypes.
The B, O, and BxO linkage maps are reported in Supplementary Materials (SM1). The B map is composed of 231 markers distributed in nine linkage groups (B1.1, B1.2, B2, B3, B4, B5, B6, B7, and B8) spanning a total genetic distance of 405 cM and covering 67 % of the physical distance of the peach genome (scaffolds 1 to 8). The groups B1.1 and B1.2 both correspond to G1 from the Prunus reference map, but it was not possible to integrate them in the same group. Interestingly, in the case of B3, there were two SSR markers segregating, while no SNPs could be found. The two SSR markers that form the B3 map, MA031 and AMPA101, are located in scaffolds 14 and 10, respectively, in the peach genome sequence.
The O map is composed of 87 markers distributed in five linkage groups (O1, O2, O3, O5, and O7) spanning a total genetic distance of 228.5 cM and covering 17 % of the physical distance of the peach genome (scaffolds 1 to 8). No heterozygous markers were found for linkage groups corresponding to G4, G6, and G8 of the Prunus reference map.
The BxO map is composed of 123 markers distributed in three linkage groups (BxO2, BxO5, and BxO7) spanning a genetic distance of 199.6 cM and covering 27 % of the physical distance of the peach genome (scaffolds 1 to 8). Given the availability of only two markers segregating in both parents (one SSR and a cluster of SNPs), B1 and O1 maps could not be properly integrated.
In general, marker order was in agreement with the peach genome sequence v1.0 (www.rosaceae.org) with few exceptions. We only found ten small inversions of adjacent SNP markers and two SNP markers located in B2 (IGA_461045 and IGA_458918) that are physically located on scaffold 4 in the peach genome. In the case of the latter two SNPs, this inconsistency is explained by a mis-assembly in the peach genome sequence (International PeachGenome Initiative, The genome sequence of peach, a key diploid tree species, reveals unique patterns of genetic diversity, domestication and genome evolution, unpublished).
Compared to the SSR-based maps previously produced for the BxO cross (Eduardo et al. 2011), these newly constructed linkage maps considerably increase marker saturation and genome coverage, providing an ideal basis for QTL dissection in this intraspecific population.
Distribution and correlations of fruit VOCs in two parents and in their F1 progeny
In this paper, we focus our analysis on 23 fruit VOCs that could be identified in both years of analysis (2007 and 2008) and representing high-impact compounds in peach. Fifteen VOCs, hexyl acetate, Z-3-hexenyl acetate, nonanal, γ-octalactone, 6-pentyl-α-pyrone, γ-decalactone, δ-decalactone, γ-dodecalactone, 3-methylbutanoic acid, benzaldehyde, phenylacetaldehyde, eugenol, linalool, E-β-damascenone, β-ionone, and E,E-2,4-decadienal, have already been described as high-impact compounds in peach due to their OAV (Eduardo et al. 2010). Eight more VOCs, E-2-hexenyl acetate, E-3-nonen-2-one, chavicol, p-menth-1-en-9-al, squalene, unidentified1, and unidentified2, were included in the analysis, since their odor thresholds were not available and OAVs could not be estimated and they could not be excluded as high-impact compounds participating to peach aroma.
Levels of VOCs in 2007 mature fruit of the parents B and O, as well as the 2007 and 2008 mean, maximum, minimum, and standard deviations of 126 seedlings of the BxO F1 progeny, are reported in Supplementary Materials (SM2). In 2008, fruits from O were not available, but data from 2007 indicate a large variation in VOC values between parents. For example, 3-methylbutanoic acid was two orders of magnitude higher in B (3,550.4 μg/kg of fresh weight) compared to O (31.3 μg/kg of fresh weight). Similar values between parents were found for hexyl acetate, nonanal, γ-octalactone, and benzaldehyde, with differences lower than 25 %. In the F1 progeny, esters and compounds derived from leucine and phenylalanine were lower in 2008, while C9 compounds, γ-dodecalactone and β-ionone, were higher. Correlation of the concentrations of the same VOCs between years was low (Table 1), with only nonanal and linalool showing significant r values (Spearman) of 0.29 and 0.40, respectively. p-Menth-1-en-9-al also showed a positive, although not significant, value of 0.23.
Correlations between different VOCs were significant in both years for several compounds. This was the case for the three esters, hexyl acetate, Z-3-hexenyl acetate, and E-2-hexenyl acetate. Nonanal values were correlated with those of linalool and p-menth-1-en-9-al. E-3-nonen-2-one was correlated with lactones (except δ-decalactone), chavicol, and E,E-2,4-decadienal. Lactones concentrations were correlated with each other, as well as with those of chavicol, E,E-2,4-decadienal, and two unidentified compounds. 3-Methylbutanoic acid correlated with chavicol and β-ionone values, while chavicol with E,E-2,4-decadienal and the unidentified number 1. The concentrations of the two unidentified compounds were highly correlated with each other.
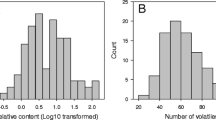
The distribution of concentration values of each VOC in the F1 progeny was similar in the 2 years (2007 data are reported in Fig. 1). The distribution of concentration values in the F1 progeny was different depending on the VOC considered: some VOCs were present in all progenies, e.g., nonanal, while others were found only in some seedlings, such as linalool. Furthermore, VOCs with a bimodal distribution were noted (the case of p-menth-1-en-9-al), while others, as benzaldehyde and nonanal, had a distribution skewed towards low or high values.

The distributions of VOCs values quantified in 2007 in mature fruits of the F1 population derived from the cross “Bolero” × “OroA” (BxO) are reported for the 23 VOCs analyzed in this study. Arrows indicate the position of values detected for the two parents of the cross. On the vertical axis, the frequency of given class for 119 individuals is reported. VOC concentrations are indicated as micrograms per kilogram of fresh weight
In the same year, transgressive segregation, i.e., progenies with values not included in the interval defined by the two parents, was observed for all VOCs, except for δ-decalactone and 3-methylbutanoic acid.
QTL analysis
A total of 72 QTLs were detected using IM and K-W analysis on the three maps B, O, and BxO (Table 2). When QTLs for a specific VOC were detected in the same chromosomal regions (with overlapping confidence intervals) in both years, they were considered to coincide. On this basis, the initial 72 QTLs can be summarized into 43 unique QTLs for 23 different VOCs: among them 29 (>67 %) were consistent over the 2 years of phenotypic analysis and 14 were evident only in 1 of the 2 years. Between one and four QTLs were detected for each VOC. Out of the total 72 QTLs, 40 were detected with both IM and K-W analysis, while three QTLs were only detected using IM. A total of 29 QTLs were only detected using K-W: of these, 13 had a LOD score in the IM between 2 and 3, and 16 had a LOD score in the IM lower than 2. Six QTLs were detected using the integrated BxO map but not in the B and O parental maps. QTLs were mapped to linkage groups 4, 5, 6, and 7 on the B map; 2, 3, and 7 on the O map; and 2, 5, and 7 on the BxO map (Fig. 2). Most of them (52) were detected using the B map, 8 using the O map, and 12 using the BxO map.

QTL location. Map location of fruit VOC QTLs detected in the BxO progeny determined using K-W and IM (2007 and 2008 data). Genetic distances (in centimolar) and markers names are shown, respectively, on the left and on the right of each linkage group. Linkage groups according to the nomenclature of Prunus chromosomes are indicated on the top specifying the parent genotype (B for Bolero, O for OroA). QTLs are represented by block vertical bars positioned at the left of each linkage group. Thin lines correspond to LOD-2 and black bars to LOD-1 confidence interval. The asterisk marks the K-W significance. Only linkage groups with QTLs are presented. VOCs names are the same used in the text or in the tables and are followed by the year in which they were detected
QTLs in the B map were grouped in three main clusters. The first cluster is located at the beginning of B4, where major QTLs for nonanal (belonging to the aldehyde class), linalool, and p-menth-1-en-9-al (belonging to the terpene class) were identified in both years. Also in this cluster, a minor QTL for benzaldehyde was identified in both years, but significant in K-W analysis only. The second cluster is located at the end of B4, where QTLs were identified for one ester (E-2-hexenyl acetate), one C9 compound (E-3-nonen-2-one), four lactones (γ-octalactone, γ-decalactone, γ-dodecalactone, and 6-penthyl-α-pyrone), one phenylalanine-derived compound (chavicol), one terpenic compound (E-β-damascenone), and the two unidentified compounds. All of them were detected in both years except for γ-dodecalactone. The third cluster of QTLs is located in B6, where QTLs were detected for one C9 compound (E-3-nonen-2-one), four lactones (γ-decalactone, δ-decalactone, γ-dodecalactone, and 6-penthyl-α-pyrone), one leucine-derived compound (3-methylbutanoic acid), three phenylalanine-derived compounds (benzaldehyde, phenylacetaldehyde and chavicol), one terpenic compound (squalene), and one C13 norisoprenoid (β-ionone) and E,E-2,4-decadienal. All of them were detected in both years except for γ-decalactone, benzaldehyde, squalene, and β-ionone. Also in the B map, a repeatable QTL for phenylacetaldehyde was detected at the very south of B7 and three minor QTLs detected only in 1 year, one for β-ionone in B7 and two in B5 for benzaldehyde and eugenol.
In the O map, repeatable QTLs were detected for 6-penthyl-α-pyrone in O2 and for hexyl acetate in O7. QTLs were detected in O3 for γ-octalactone, γ-decalactone, γ-dodecalactone, and chavicol, but only for 1 year.
In the BxO map, repeatable QTLs were detected for 6-penthyl-α-pyrone in BxO2, for benzaldehyde in BxO5, and for phenylacetaldehyde and for E-β-damascenone in BxO7. Non-repeatable QTLs were detected in BxO2 for Z-3-hexenyl acetate and E-3-nonen-2-one, in BxO5 for eugenol and in BxO7 for δ-decalactone.
Candidate genes and QTL collocations
The detected QTLs were compared to the map position of fruit quality and aroma-related genes present in the Prunus reference map (Illa et al. 2011) and annotated in the peach genome sequence (www.rosaceae.org) to identify potential candidate genes.
We identified two terpene synthases (ppa002670m and ppa003380m) located in tandem within 7 kb of each other on scaffold 4 and potentially involved in the major QTLs for linalool and p-menth-1-en-9-al. In addition, a lipoxygenase (Lox) gene (ppa001082m) was identified as putatively implicated in the major QTL for nonanal at the beginning of G4. The reader is referred to the “Discussion” for further details regarding terpene synthase and Lox genes as candidates.
To obtain more evidence for these collocations, we searched for SNPs in candidate gene sequences, using B and O genomic resequencing information (International PeachGenome Initiative, The genome sequence of peach, a key diploid tree species, reveals unique patterns of genetic diversity, domestication and genome evolution, unpublished). Two SNPs in each terpene synthase candidate gene were identified, genotyped in the BxO population, and included in the map dataset. Three out of the four SNPs mapped (SNPs S4_1420538 and S4_1420689 in ppa002670m; SNP S4_1413937 in ppa003380m) caused amino acid variation in the N-terminal region of the protein sequence. Positions, allele variations, annotations, and amino acid changes of the SNPs are reported in Supplementary Materials (SM3). QTL analysis supports the possible involvement of these terpene synthase genes in controlling linalool accumulation, as they both co-map with the QTL peak (Table 2).
Candidate gene ppa001082m belongs to the Lox multigene family. The lypoxigenase pathway has been shown to yield aldehydes from fatty acids (Schwab et al. 2008). To gain further insight into the possible involvement of this CG in nonanal accumulation, a phylogenetic analysis of the peach LOX family was conducted. In the peach genome reference sequence, the Lox gene family comprises 15 annotated genes. Phylogenetic analysis of inferred amino acid sequences along with other known plant LOXs grouped seven and eight peach Lox genes within the 13-LOX and 9-LOX groups, respectively. Results are reported in Supplementary Materials (SM4). Peach LOX ppa001082m, ppa001085m, ppa001064m, and ppa000968m, ppa001634m, ppa001631m and ppa001316m clustered within the 9-LOX group. Conversely, peach LOXs ppa026489m, ppa001207m, ppa001216m, ppa017962m, ppa001016m, ppa001287m, ppa001293m, and ppa001311m clustered within the 13-LOXs group. Hence, the candidate gene ppa001082m was part of the 9-LOX group. Neither SNPs nor deletion/insertion polymorphisms could be identified in the 5′- and 3′-UTRs, and in the coding sequence of this Lox gene.
Discussion
Linkage mapping
Genotyping with the IPSC 9,000 SNP array has allowed to greatly and efficiently increase saturation and coverage of the SSR-based maps previously developed for the BxO population (Eduardo et al. 2011). After testing 344 Prunus SSRs, only 81 and 47 were heterozygous in B and O, respectively. The phenomenon of low coverage of the genome and lack of polymorphic markers in some linkage groups was interpreted as an indication of extensive genome homozygosity, suggesting a case of “identity by descent” within the genomes of B and O (Eduardo et al. 2011). Although there are still some linkage groups where no markers could be mapped (e.g., in O), the use of SNP markers allowed us to recover the new linkage group B6 (59.2 cM) and create integrated maps BxO2, BxO5, and BxO7, which were not present in our previous study (Eduardo et al. 2011). Lack of markers on O4, O6, and O8 may be due to the already mentioned identity by descent or ascertainment biases in the SNPs represented in the array (Nielsen et al. 2004; Albrechtsen et al. 2010). Availability of genomic resequencing information from B and O (International Peach Genome Initiative 2012) will help to discriminate between these possibilities.
In some cases, we found regions covered by SSR markers and not by SNPs and vice versa. For example, two SSRs were mapped on B3, but no SNPs were positioned on this linkage group. These SSRs (MA031a, AMPA101) were previously assigned to linkage group 3 by genetic mapping (Howad et al. 2005; Dirlewanger et al. 2004b), and in the peach genome sequence they are associated to scaffolds 10 and 14. In the peach 9,000 SNP array, there are only 19 SNPs that are not located in the eight main scaffolds corresponding to the eight chromosomes (Verde et al. 2012). These considerations may help to explain the particular situation of B3. In the case of the distal part of BxO5, coverage with SSR markers only may reflect the ascertainment bias of the SNP panel used for the peach 9,000 array. Surprisingly, the map of linkage group B6 includes a high number of SNPs, but only one SSR. Here again, genomic resequencing information from B and O (International PeachGenome Initiative, The genome sequence of peach, a key diploid tree species, reveals unique patterns of genetic diversity, domestication and genome evolution, unpublished) may help to understand the distribution of polymorphisms along this linkage group and the lack of coverage from SSR markers. In conclusion, the use of both SNP and SSR markers seems appropriate to obtain maximum coverage in peach intraspecific maps.
Peach fruit VOCs correlations, segregation, and QTL analysis
Statistical analyses of fruit VOC levels revealed low correlations of VOC values between years, a variety of patterns of inheritance, a number of VOCs presenting transgressive segregation in the progeny with respect to the parents, major QTLs for some VOCs, and clustering of QTLs underpinning VOCs from the same chemical family. Similar observations have been reported in apple (Dunemann et al. 2009; Rowan et al. 2009b), strawberry (Olbricht et al. 2008; Zorrilla-Fontanesi et al. 2012), rose (Spiller et al. 2010), and in non Rosaceae species, such as tomato (Tadmor et al. 2002; Zanor et al. 2009), grape (Doligez et al. 2006), and eucalypt (O’Reilly-Wapstra et al. 2011).
The low correlations observed for fruit VOC concentrations between years are consistent with previous reports and most probably reflect the high variation in environmental conditions of the field. Dunemann et al. (2009) showed that apple parental cultivars had varying quantities of VOCs in three consecutive years. In strawberry, some VOCs were highly variable depending on the time of harvest, while others were constant (Olbricht et al. 2008). Despite this year-to-year variability, >67 % of the QTLs in the present study were recovered in both years. This is in line with results recently reported for a similar study in strawberry, where 50 % of QTLs were reproducible over 2 or 3 years of phenotypic analysis (Zorrilla-Fontanesi et al. 2012). The fact that correlations are low, but significant, is due to the fact that although there are some individuals that present very different phenotypes in the different years, most of the individuals present a correlation strong enough to produce significant QTLs. A possible explanation for this is that different individuals are affected by the environment in different ways indicating the presence of GxE interaction. Another possible explanation is that there are small QTLs that are on the limit of detection, and depending on environmental effects, they are significant in 1 year and not in the other.
Interestingly, a significant correlation was found between different VOCs from the same biochemical pathway in both years. For example, the three esters, hexyl acetate, Z-3-hexenyl acetate, and E-2-hexenyl acetate, all deriving from the lipoxygenase pathway (Schwab et al. 2008), showed significant pairwise correlations in 2007 and 2008. Lactones were also correlated with each other. Although the pathway or the enzymes involved in the biosynthesis of lactones have not been clarified (Schwab et al. 2008), a role for epoxide hydrolases has been proposed (Schottler and Boland 1996) suggesting that a common mechanism may underlie the synthesis of these compounds. The concentrations of the two unidentified compounds were also highly correlated: considering their highly similar mass spectra (Eduardo et al. 2010) and their slight difference in elution time, it is likely they are isomers. The correlations are consistent with the existence of few genomic regions controlling the levels of most of the investigated VOCs and with the clustering of QTLs for the same compound class. A similar pattern has been observed in apple and was explained as the result of tight linkage between distinct loci or pleiotropic effects of a single locus (Dunemann et al. 2009). Clustering of QTLs controlling VOCs with similar chemical structure was also found in Arabidopsis (Lisec et al. 2008), tomato (Zanor et al. 2009), and very recently in strawberry (Zorrilla-Fontanesi et al. 2012). In our case, it remains to be established whether the observed QTL clusters correspond to a tight linkage between distinct loci, to genes acting upstream in the respective VOC metabolic pathways or are due to other pleiotropic effects. For example, the QTLs detected at the south end of the linkage group 4 could be due to pleiotropic effects of a major maturity date QTL mapped in the same position as it has been proposed for other fruit quality traits (Eduardo et al. 2011).
Transgressive segregation was common for the VOCs analyzed except for δ-decalactone and 3-methylbutanoic acid. In tomato, transgressive segregation was also observed for VOCs but not for primary metabolites (Zanor et al. 2009). The variety of patterns of inheritance found in this work was also described in VOC analyses in strawberry (Olbricht et al. 2008; Zorrilla-Fontanesi et al. 2012) and apple (Dunemann et al. 2009). The case of linalool, where approximately 50 % of the individuals accumulate no linalool or very low levels (less than 10 % of the maximum value), was interpreted as the result of mixed qualitative and quantitative inheritance. In species as Mentha and Thymus, it has been suggested that monoterpene composition was influenced by a combination of Mendelian inheritance and complex epistatic interactions, and in other species, major gene control was suggested for different terpenes (O’Reilly-Wapstra et al. 2011). Similarly, mixed qualitative and quantitative inheritance was proposed by other authors for the genetic control of external color of peach fruit, when considering some progenies segregating 1:1 for partial red or no red to full red (Beckman and Sherman 2003; Beckman et al. 2005) and others with a 3:1 segregation. These results could be interpreted as deriving from the segregation of a single gene. This could also be the case for the major linalool QTL on B4, as individuals that were homozygous for the closest marker showed no linalool or very low levels of it. Thus, a locus mapping in the distal region of linkage group 4 is proposed to control presence or absence of linalool and additional genetic and/or environmental factors condition variation of concentration among linalool-producing seedlings.
Collocation of major QTLs and candidate genes
Linalool is an acyclic alcohol monoterpene contributing to the aroma of fruits and leaves of several plant species, including citrus fruits (Kelebek and Selli 2011; Eduardo et al. 2010). In peach, the presence of linalool can discriminate peach cultivars by their origin (Wang et al. 2009). In this work, a major QTL for linalool was detected in the distal part of linkage group 4. In this same region, two terpene synthases have been identified cosegregating with this QTL. Despite the substantial degree of amino acid sequence similarity among plant monoterpene and sesquiterpene synthases, similarity may depend more on taxonomic affinities of the species from which the gene was isolated rather than from the type of products formed (Degenhardt et al. 2009). Because of this, it is very difficult to predict the final product of a terpene synthase gene simply based on its sequence. Both terpene synthases present in the peach genome scaffold 4 (ppa002670m and ppa003380m) exhibit high similarities with linalool synthases, but significant levels of similarity are also detected with nerolidol, limonene, and 1,8-cineole synthases (data not shown). Linalool synthase is a terpene synthase and it was the first floral enzyme responsible for scent to be isolated and characterized (Cseke et al. 1998). In basil, a 1-bp insertion in the (R)-linalool synthase gene, resulting in a frameshift mutation, causes differences in the amount and types of terpenes produced by different cultivars (Iijima et al. 2004). Similar results were obtained in snapdragon flowers, where two nearly identical terpene synthases catalyze the formation of nerolidol and linalool, respectively (Nagegowda et al. 2008).
p-Menth-1-en-9-al was first isolated in Rosa damascena, and, as other oxygenated p-menthanes, was proposed to be a product of the photoxidation of limonene (Overton 1971). Limonene is a cyclic terpene possessing a strong smell of oranges; therefore, both terpene synthases mentioned above could also be candidate genes for the major p-menth-1-en-9-al QTL.
Nonanal is an aldehyde reported as an odor active compound in orange (Kelebek and Selli 2011). In the same genomic region hosting the monoterpene QTLs in the distal part of linkage group 4, a major QTL affecting nonanal concentration collocated with a 9-Lox candidate gene (ppa001082m). LOX activities have been identified in a wide range of organisms, tissues, and developmental stages and have been implicated in a variety of processes including regulation of plant development, fruit ripening, plant senescence, disease, and pest resistance (Kolomiets et al. 2001; Zhang et al. 2006; Chen et al. 2004). The role of the LOX pathway in the biosynthesis of fruit aroma compounds has already been reported for tomato (Kausch and Handa 1997), olive (Salas et al. 2000), bell pepper (Matsui et al. 1997), apple (Rowan et al. 1999), citrus fruit (Gomi et al. 2003), and strawberry (Perez et al. 1999). LOX proteins are classified into two groups depending on their ability to oxygenate the carbon chain at position 9 (9-LOX) or 13 (13-LOX). Although the direct involvement of LOX has not yet been demonstrated in the case of nonanal production, 9-LOX can use linolenic and linoleic acids as substrates for the synthesis of structurally similar volatile aldehydes, such as 3Z, 6Z-nonadienal (Schwab et al. 2008).
Implication for aroma MAS in peach
The results presented in this paper support the use of MAS for some VOCs. For example, selection of the appropriate allelic configurations of molecular markers at the distal region of linkage group 4 may assist identification of seedlings lacking or producing linalool and exhibiting higher or lower nonanal and p-menth-1-en-9-al concentrations. However, transferability of the identified markers to other populations should be verified. Analyses of other crosses segregating for other VOCs, compared to the ones considered here, will also be important for a better understanding of the genetic basis of peach fruit aroma. Our results suggest that it will be difficult to predict segregation patterns based on parental concentrations values, although an exception has been reported recently: Brandi et al. (2011) showed that the presence of norisoprenoids depends on carotenoid concentration and is thus associated with flesh color. VOC QTL analysis could be carried out in the progeny of a cross between yellow and white flesh cultivars to verify these results. If this is true, efficient MAS for flesh color could also be used to select plants with high or low levels of norisoprenoids. An additional target is the further dissection of lactone accumulation. An interesting starting material for this could be the cultivar “Romagna Big” that was shown to lack lactones in the fruit (Eduardo et al. 2010).
Conclusions
We report for the first time the identification of QTLs for VOCs in peach. To this end, we used the recently developed Illumina 9,000 SNP array platform to produce dense genetic maps, substantially increasing genome coverage compared to the existing SSR-based maps of the BxO cross (Eduardo et al. 2011). Despite being based on a single biparental population and in spite of the complexity of this trait, the results provide a first insight into the genetic determinants of volatile composition in this important fruit species. Three major QTLs for three different VOCs were mapped in the same chromosomal region, and candidate genes were identified by analysis of gene annotations from the corresponding genomic sequence. Importance of this region in VOC variation can be validated analyzing other peach populations and germplasm collections and using different techniques for volatile quantification. These data could open opportunities for aroma MAS in peach. Further work is also required to establish the relevance of linalool, nonanal, and p-menth-1-en-9-al in peach aroma perception. The significance of these and other VOCs could be assessed by means of sensory experiments, using panel tests to determine their involvement in the perception of peach aroma by humans.
References
Abbott AG, Rajapakse S, Sosinski B, Lu ZX, Sossey-Alaoui K, Gannavarapu M, Reighard G, Ballard RE, Baird WV, Scorza R, Callahan A (1998) Construction of saturated linkage maps of peach crosses segregating for characters controlling fruit quality, tree architecture and pest resistance. Fourth International Peach Symposium 1–2:41–49
Abbott AG, Arús P, Scorza R (2008) Genetic engineering and genomics. In: Layne DR, Bassi D (eds) The peach: botany, production and uses. CABI, Cambridge, pp 85–105
Agarwal M, Shrivastava N, Padh H (2008) Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep 27:617–631
Albrechtsen A, Nielsen FC, Nielsen R (2010) Ascertainment biases in SNP chips affect measures of population divergence. Mol Biol Evol 11:2534–2547
Amarawathi Y, Singh R, Singh AK, Singh VP, Mohapatra T, Sharma TR, Singh NK (2008) Mapping of quantitative trait loci for basmati quality traits in rice (Oryza sativa L.). Mol Breeding 21:49–65
Aranzana MJ, Pineda A, Cosson P, Dirlewanger E, Ascasibar J, Cipriani G, Ryder CD, Testolin R, Abbott A, King GJ, Iezzoni AF, Arús P (2003) A set of simple-sequence repeat (SSR) markers covering the Prunus genome. Theor Appl Genet 106:819–825
Arús P, Verde I, Sosinski B, Zhebentyayeva T, Abbott AG (2012) The peach genome. Tree Genet Genomes. doi:10.1007/s11295-012-0493-8
Aubert C, Gunata Z, Ambid C, Baumes R (2003) Changes in physicochemical characteristics and volatile constituents of yellow- and white-fleshed nectarines during maturation and artificial ripening. J Agr Food Chem 51:3083–3091
Battilana J, Emanuelli F, Gambino G, Gribaudo I, Gasperi F, Boss PK, Grando MS (2011) Functional effect of grapevine 1-deoxy-D-xylulose 5-phosphate synthase substitution K284N on Muscat flavour formation. J Exp Bot 62(15):5497–5508
Beckman TG, Alcazar JR, Sherman WB, Werner DJ (2005) Evidence for qualitative suppression of red skin color in peach. Hortscience 40:523–524
Beckman TG, Sherman WB (2003) Probable qualitative inheritance of full red skin color in peach. Hortscience 38:1184–1185
Blenda AV, Verde I, Georgi LL, Reighard GL, Forrest SD, Munoz-Torres M, Baird WV, Abbott AG (2007) Construction of a genetic linkage map and identification of molecular markers in peach rootstocks for response to peach tree short life syndrome. Tree Genet Genomes 3:341–350
Brandi F, Bar E, Mourgues F, Horváth G, Turcsi E, Giuliano G, Liverani A, Tartarini S, Lewinsohn E, Rosati C (2011) Study of ‘Redhaven’ peach and its white-fleshed mutant suggests a key role of CCD4 carotenoid dioxygenase in carotenoid and norisoprenoid volatile metabolism. BMC Plant Biol 11:24
Chen GP, Hackett R, Walker D, Taylor A, Lin ZF, Grierson D (2004) Identification of a specific isoform of tomato lipoxygenase (TomloxC) involved in the generation of fatty acid-derived flavor compounds. Plant Physiol 136:2641–2651
Cseke L, Dudareva N, Pichersky E (1998) Structure and evolution of linalool synthase. Mol Biol Evol 15:1491–1498
Degenhardt J, Köllner TG, Gershenzon J (2009) Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 70:1621–1637
Dettori MT, Quarta R, Verde I (2001) A peach linkage map integrating RFLPs, SSRs, RAPDs, and morphological markers. Genome 44:783–790
Dirlewanger E, Pascal T, Zuger C, Kervella J (1996) Analysis of molecular markers associated with powdery mildew resistance genes in peach (Prunus persica (L) Batsch) × Prunus davidiana hybrids. Theor Appl Genet 93:909–919
Dirlewanger E, Pronier V, Parvery C, Rothan C, Guye A, Monet R (1998) Genetic linkage map of peach [Prunus persica (L.) Batsch] using morphological and molecular markers. Theor Appl Genet 97:888–895
Dirlewanger E, Moing A, Rothan C, Svanella L, Pronier V, Guye A, Plomion C, Monet R (1999) Mapping QTLs controlling fruit quality in peach (Prunus persica (L.) Batsch). Theor Appl Genet 98:18–31
Dirlewanger E, Cosson P, Poizat C, Laigret F, Aranzana MJ, Arús P, Dettori MT, Verde I, Quarta R (2003) Synteny within the Prunus genomes detected by molecular markers. In: Janick J (ed), Genetics and breeding of tree fruits and nuts, pp 177–187
Dirlewanger E, Graziano E, Joobeur T, Garriga-Caldere F, Cosson P, Howad W, Arús P (2004a) Comparative mapping and marker-assisted selection in Rosaceae fruit crops. P Natl Acad Sci USA 101:9891–9896
Dirlewanger E, Cosson P, Howad W, Capdeville G, Bosselut N, Claverie M, Voisin R, Poizat C, Lafargue B, Baron O, Laigret F, Kleinhentz M, Arús P, Esmenjaud D (2004b) Microsatellite genetic linkage maps of myrobalan plum and an almond-peach hybrid-location of root-knot nematode resistance genes. Theor Appl Genet 109:827–838
Doligez A, Audiot E, Baumes R, This P (2006) QTLs for muscat flavor and monoterpenic odorant content in grapevine (Vitis vinifera L.). Mol Breeding 18:109–125
Dunemann F, Ulrich D, Boudichevskaia A, Grafe C, Weber WE (2009) QTL mapping of aroma compounds analysed by headspace solid-phase microextraction gas chromatography in the apple progeny ‘Discovery’ x ‘Prima’. Mol Breeding 23:501–521
Dunemann F, Ulrich D, Malysheva-Otto L, Weber WE, Longhi Velasco R, Costa F (2011) Functional allelic diversity of the apple alcohol acyl-transferase gene MdAAT1 associated with fruit ester volatile contents in apple cultivars. Mol Breeding 29:609–625
Eduardo I, Chietera G, Bassi D, Rossini L, Vecchietti A (2010) Identification of key odor volatile compounds in the essential oil of nine peach accessions. J Sci Food Agr 90:1146–1154
Eduardo I, Pacheco I, Chietera G, Bassi D, Pozzi C, Vecchietti A, Rossini L (2011) QTL analysis of fruit quality traits in two peach intraspecific populations and importance of maturity date pleiotropic effect. Tree Genet Genomes 7(2):323–335
Etienne C, Rothan C, Moing A, Plomion C, Bodenes C, Svanella-Dumas L, Cosson P, Pronier V, Monet R, Dirlewanger E (2002) Candidate genes and QTLs for sugar and organic acid content in peach [Prunus persica (L.) Batsch]. Theor Appl Genet 105:145–159
Foulongne M, Pascal T, Arús P, Kervella J (2003a) The potential of Prunus davidiana for introgression into peach [Prunus persica (L.) Batsch] assessed by comparative mapping. Theor Appl Genet 107:227–238
Foulongne M, Pascal T, Pfeiffer F, Kervella J (2003b) QTLs for powdery mildew resistance in peach × Prunus davidiana crosses: consistency across generations and environments. Mol Breeding 12:33–50
Gasic K, Han YP, Kertbundit S, Shulaev V, Iezzoni AF, Stover EW, Bell RL, Wisniewski ME, Korban SS (2009) Characteristics and transferability of new apple EST-derived SSRs to other Rosaceae species. Mol Breeding 23:397–411
Gillen AM, Bliss FA (2005) Identification and mapping of markers linked to the Mi gene for root-knot nematode resistance in peach. J Am Soc Hortic Sci 130:24–33
Gomi K, Yamasaki Y, Yamamoto H, Akimitsu K (2003) Characterization of a hydroperoxide lyase gene and effect of C6-volatiles on expression of genes of the oxylipin metabolism in Citrus. J Plant Physiol 160:1219–1231
Han M, Zhang T, Zhao C, Zhi J (2011) Regulation of the expression of lipoxygenase genes in Prunus persica fruit ripening. Acta Physiol Plant 33:1345–1352
Henery ML, Moran GF, Wallis IR, Foley WJ (2007) Identification of quantitative trait loci influencing foliar concentrations of terpenes and formylated phloroglucinol compounds in Eucalyptus nitens. New Phytol 176:82–95
Horvat RJ, Chapman GW, Robertson JA, Meredith FI, Scorza R, Callahan AM, Morgens P (1990) Comparison of the volatile compounds from several commercial peach cultivars. J Agr Food Chem 38:234–237
Howad W, Yamamoto T, Dirlewanger E, Testolin R, Cosson P, Cipriani G, Monforte AJ, Georgi L, Abbott AG, Arus P (2005) Mapping with a few plants: using selective mapping for microsatellite saturation of the Prunus reference map. Genetics 171:1305–1309
Iijima Y, Davidovich-Rikanati R, Fridman E, Gang DR, Bar E, Lewinsohn E, Pichersky E (2004) The biochemical and molecular basis for the divergent patterns in the biosynthesis of terpenes and phenylpropenes in the peltate glands of three cultivars of basil. Plant Physiol 136:3724–3736
Illa E, Eduardo I, Audergon JM, Barale F, Dirlewanger E, Li X, Moing A, Lambert P, Le Dantec L, Gao Z, Poëssel JL, Pozzi C, Rossini L, Vecchietti A, Arús P, Howad W (2011) Saturating the Prunus (stone fruits) genome with candidate genes for fruit quality. Mol Breeding 28(4):667–682
Julio E, Denoyes-Rothan B, Verrier JL, de Borne FD (2006) Detection of QTLs linked to leaf and smoke properties in Nicotiana tabacum based on a study of 114 recombinant inbred lines. Mol Breeding 18:69–91
Kausch KD, Handa AK (1997) Molecular cloning of a ripening-specific lipoxygenase and its expression during wild-type and mutant tomato fruit development. Plant Physiol 113:1041–1050
Kelebek H, Selli S (2011) Determination of volatile, phenolic, organic acid and sugar components in a Turkish cv. Dortyol (Citrus sinensis L. Osbeck) orange juice. J Sci Food Agr 91:1855–1862
Koutsos TV, Chatzopoulou PS, Katsiotis ST (2009) Effects of individual selection on agronomical and morphological traits and essential oil of a “Greek basil” population. Euphytica 170:365–370
Kolomiets MV, Hannapel DJ, Chen H, Tymeson M, Gladon RJ (2001) Lipoxygenase is involved in the control of potato tuber development. Plant Cell 13:613–626
Laothawornkitkul J, Taylor JE, Paul ND, Hewitt CN (2009) Biogenic volatile organic compounds in the Earth system. New Phytol 183:27–51
Lisec J, Meyer RC, Steinfath M, Redestig H, Becher M, Witucka-Wall H, Fiehn O, Torjek O, Selbig J, Altmann T, Willmitzer L (2008) Identification of metabolic and biomass QTL in Arabidopsis thaliana in a parallel analysis of RIL and IL populations. Plant J 53:960–972
Lu ZX, Sosinski B, Reighard GL, Baird WV, Abbott AG (1998) Construction of a genetic linkage map and identification of AFLP markers for resistance to root-knot nematodes in peach rootstocks. Genome 41:199–207
Mathieu S, Cin VD, Fei ZJ, Li H, Bliss P, Taylor MG, Klee HJ, Tieman DM (2009) Flavour compounds in tomato fruits: identification of loci and potential pathways affecting volatile composition. J Exp Bot 60:325–337
Matsui K, Shibata Y, Tateba H, Hatanaka A, Kajiwara T (1997) Changes of lipoxygenase and fatty acid hydroperoxide lyase activities in bell pepper fruits during maturation. Biosci Biotech Bioch 61:199–201
Nagegowda DA, Gutensohn M, Wilkerson CG, Dudareva N (2008) Two nearly identical terpene synthases catalyze the formation of nerolidol and linalool in snapdragon flowers. Plant J 55:224–239
Nielsen R, Hubisz MJ, Clark AG (2004) Reconstituting the frequency spectrum of ascertained single-nucleotide polymorphism data. Genetics 168:2373–2382
Obando-Ulloa JM, Ruiz J, Monforte AJ, Fernandez-Trujillo JP (2008) Aroma profile of a collection of near-isogenic lines of melon (Cucumis melo L.). Food Chem 118:815–822
Ogundiwin EA, Peace CP, Gradziel TM, Parfitt DE, Bliss FA, Crisosto CH (2009) A fruit quality gene map of Prunus. BMC Genomics 10:587
Olbricht K, Grafe C, Weiss K, Ulrich D (2008) Inheritance of aroma compounds in a model population of Fragaria × ananassa Duch. Plant Breeding 127:87–93
O’Reilly-Wapstra JM, Freeman JS, Davies NW, Vaillancourt RE, Fitzgerald H, Potts BM (2011) Quantitative trait loci for foliar terpenes in a global eucalypt species. Tree Genet Genomes 7:485–498
Overton KH (1971) Terpenoids and steroids. A specialistic periodical report. Chemical Society
Perez AG, Sanz C, Olias R, Olias JM (1999) Lipoxygenase and hydroperoxide lyase activities in ripening strawberry fruits. J Agr Food Chem 47:249–253
Quarta R, Dettori MT, Sartori A, Verde I (2000) Genetic linkage map and QTL analysis in peach. Proceedings of the XXV International Horticultural Congress Pt 11:233–241
Quilot B, Wu BH, Kervella J, Genard M, Foulongne M, Moreau K (2004) QTL analysis of quality traits in an advanced backcross between Prunus persica cultivars and the wild relative species P. davidiana. Theor Appl Genet 109:884–897
Rowan DD, Allen JM, Fielder S, Hunt MB (1999) Biosynthesis of straight-chain ester volatiles in Red Delicious and Granny Smith apples using deuterium-labeled precursors. J Agr Food Chem 47:2553–2562
Rowan DD, Hunt MB, Alspach PA, Whitworth CJ, Oraguzie NC (2009a) Heritability and genetic and phenotypic correlations of apple (Malus × domestica) fruit volatiles in a genetically diverse breeding population. J Agr Food Chem 57:7944–7952
Rowan DD, Hunt MB, Dimouro A, Alspach PA, Weskett R, Volz RK, Gardiner SE, Chagne D (2009b) Profiling fruit volatiles in the progeny of a ‘Royal Gala’ × ‘Granny Smith’ apple (Malus × domestica) cross. J Agr Food Chem 57:7953–7961
Salas JJ, Sanchez J, Ramli US, Manaf AM, Williams M, Harwood JL (2000) Biochemistry of lipid metabolism in olive and other oil fruits. Prog Lipid Res 39:151–180
Saliba-Colombani V, Causse M, Langlois D, Philouze J, Buret M (2001) Genetic analysis of organoleptic quality in fresh market tomato. 1. Mapping QTLs for physical and chemical traits. Theor Appl Genet 102:259–272
Schottler M, Boland W (1996) Biosynthesis of dodecano-4-lactone in ripening fruits: Crucial role of an epoxide-hydrolase in enantioselective generation of aroma components of the nectarine (Prunus persica var nucipersica) and the strawberry (Fragaria ananassa). Helv Chim Acta 79:1488–1496
Schwab W, Davidovich-Rikanati R, Lewinsohn E (2008) Biosynthesis of plant-derived flavor compounds. Plant J 54:712–732
Scorza R, Mehlenbacher SA, Lightner GW (1985) Inbreeding and coancestry of freestone peach cultivars of the Eastern-United-States and implications for peach germplasm improvement. J Am Soc Hortic Sci 110:547–552
Shulaev V, Korban SS, Sosinski B, Abbott AG, Aldwinckle HS, Folta KM, Iezzoni A, Main D, Arús P, Dandekar AM, Lewers K, Brown SK, Davis TM, Gardiner SE, Potter D, Veilleux RE (2008) Multiple models for Rosaceae genomics. Plant Physiol 147:985–1003
Sosinski B, Sossey-Alaoui K, Rajapakse S, Glassmoyer K, Ballard RE, Abbott AG, Lu ZX, Baird WV, Reighard G, Tabb A, Scorza R (1998) Use of AFLP and RFLP markers to create a combined linkage map in peach [Prunus persica (L.) Batsch] for use in marker assisted selection. Fourth International Peach Symposium 1–2:61–68
Spiller M, Berger RG, Debener T (2010) Genetic dissection of scent metabolic profiles in diploid rose populations. Theor Appl Genet 120:1461–1471
Tadmor Y, Fridman E, Gur A, Larkov O, Lastochkin E, Ravid U, Zamir D, Lewinsohn E (2002) Identification of malodorous, a wild species allele affecting tomato aroma that was selected against during domestication. J Agr Food Chem 50:2005–2009
Tava A, Ramella D, Grecchi M, Aceto P, Paoletti R, Piano E (2009) Volatile constituents of Trifolium pratense and T. repens from N.E. Italian alpine pastures. Nat Prod Commun 4:835–838
Tholl D, Rose USR (2006) Detection and identification of floral scent compounds. Biol Floral Scent 3–25
Van Ooijen JW (2009) MapQTL® 6, Software for the mapping of quantitative trait loci in experimental populations of diploid species. Kyazma BV, Wageningen
Van Ooijen JW (2011) Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet Res 93:343–349
Verde I, Bassil N, Scalabrin S, Gilmore B, Lawley CT et al (2012) Development and evaluation of a 9 K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS One 7(4):e35668
Wang YJ, Yang CX, Li SH, Yang L, Wang YN, Zhao JB, Jiang Q (2009) Volatile characteristics of 50 peaches and nectarines evaluated by HP-SPME with GC-MS. Food Chem 116:356–364
Yamamoto T, Shimada T, Imai T, Yaegaki H, Haji T, Matsuta N, Yamaguchi M, Hayashi T (2001) Characterization of morphological traits based on a genetic linkage map in peach. Breeding Sci 51:271–278
Zanor MI, Rambla JL, Chaib J, Steppa A, Medina A, Granell A, Fernie AR, Causse M (2009) Metabolic characterization of loci affecting sensory attributes in tomato allows an assessment of the influence of the levels of primary metabolites and volatile organic contents. J Exp Bot 60:2139–2154
Zhang B, Chen KS, Bowen J, Allan A, Espley R, Karunairetnam S, Ferguson I (2006) Differential expression within the LOX gene family in ripening kiwifruit. J Exp Bot 57:3825–3836
Zini E, Biasioli F, Gasperi F, Mott D, Aprea E, Mark TD, Patocchi A, Gessler C, Komjanc M (2005) QTL mapping of volatile compounds in ripe apples detected by proton transfer reaction-mass spectrometry. Euphytica 145:269–279
Zorrilla-Fontanesi Y, Rambla JL, Cabeza A, Medina JJ, Sánchez-Sevilla JF, Valpuesta V, Botella MA, Granell A, Amaya I (2012) Genetic analysis of strawberry fruit aroma and identification of O-methyltransferase FaOMT as the locus controlling natural variation in mesifurane content. Plant Physiol. doi:10.1104/pp. 111.188318
Acknowledgments
This work was supported by a grant from Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) to PTP (Progetto industriale “Sviluppo di sistemi integrati per la caratterizzazione, il miglioramento, la selezione e la propagazione di razze animali e vegetali. Utilizzazione di sistemi molecolari avanzati per la diagnostica dei prodotti agrari”) and by an Italian grant to DB funded by private and public agencies “MAS.PES: apricot and peach breeding by molecular-assisted selection”. Construction of the linkage map was partly funded under the EU seventh Framework Programme by the FruitBreedomics project no. 265582: “Integrated approach for increasing breeding efficiency in fruit tree crops.” GC was supported by two grants from MIUR, PRIN project numbers 2005074520, 20074AX5CA. RP was supported by MIUR FIRB grant RBIP06CTBR. The authors wish to thank C. Buscaroli, L. Castellari, S. Foschi, M. Lama, E. Olivi, and M. Rizzo for technical assistance in field and lab operations, as well as S. Biffani for advice on statistical analyses and F. Salamini, A. Fricano, J. L. Williams for critical reading of the manuscript.
Disclaimer
The views expressed in this work are the sole responsibility of the authors and do not necessarily reflect the views of the European Commission.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Abbott
Authors’ contributions IE constructed the genetic map, conducted QTL analysis and drafted the manuscript. GC performed GC-MS analysis of volatile compounds, analysed data, helped to draft the manuscript. IPC participated in construction of the genetic map, performed statistical analyses, helped to draft the manuscript. DB assessed the potential of the plant material, developed the population, helped to draft the manuscript. RP analyzed data, identified and mapped candidate genes, performed the LOX phylogenetic analysis, and helped to draft the manuscript. MT analysed SNP data, contributed to the revision of the manuscript. EB performed SNP genotyping and contributed to SNP data analysis. LR managed and coordinated construction of the linkage map, analysed data, helped to draft the manuscript. AV managed and coordinated the study, participated in its design, analyzed data, helped to draft the manuscript. CP conceived and designed the study, managed its start up, helped to draft the manuscript. All authors read and approved the final manuscript.
Alberto Vecchietti and Carlo Pozzi share senior authorship.
Supplementary materials
Below is the link to the electronic supplementary material.
SM 1
B, O, and BxO linkage maps. Genetic distances (in centiMorgan, cM) and markers names are shown, respectively, on the left and on the right of each linkage group. Linkage groups according to the nomenclature of Prunus chromosomes are indicated on the top specifying the parent genotype (B, O, and BxO in the case of integrated maps) (PDF 33 kb)
SM 2
Quantification of fruit VOCs. Quantifications of VOCs in mature fruit of both parents in 2007 and the mean, max, min, and standard deviations (SD) of the F1 BxO progeny in 2007 and 2008. Values are expressed in micrograms per kilogram of fresh weight (PDF 66 kb)
SM 3
SNPs summary. Positions, allele variations, annotations, and amino acid changes of the SNPs mapped in the F1 BxO progeny (XLS 34 kb)
SM 4
LOX phylogenetic tree. Phylogenetic tree of amino acid sequences of known plant LOXs and peach annotated LOXs. For Prunus persica LOXs accession is the name of transcript model found on the reference genome: ppa000968m, ppa001016m, ppa001064m, ppa001082m, ppa001085m, ppa001112m, ppa001207m, ppa001216m, ppa001287m, ppa001293m, ppa001311m, ppa001316m, ppa001631m, ppa001634m, ppa002308m, ppa017962m, ppa026489m. Accession number of other known plant LOXs are as follows. Actinidia delicionsa: AdLox2, DQ497797; Arabidopsis thaliana: AtLox1, AAA32827; AtLox2, AAA32749; AtLox3, AT1G17420; AtLox4, AT1G72520; AtLox5, AT3G22400, AtLox6, AT1G67560; Lycopersicon esculentum: TomLoxA, AAA53184, TomLoxB, AAA53183, TomLoxC, AAB65766, TomLoxD, AAB65767; TomLoxE, AAG21691; Prunus dulcis: PdLox, CAD10779; Carica papaya: CpLox, AAR84664; Camellia sinensis: CsLox1, ABW75772; Cucumis sativus: CusLox, CAB83038; Fragaria x ananassa: FaLox, CAE17327; Sesbania rostrata: SrLox, CAC43237; Nicotiana attenuata: NaLox1, AAP83134; NaLox2, AAP83137; NaLox3, AAP83138; Solanum tuberosum: PotLx1, AAB67858; StLoxH2, CAA65268; StLoxH3, CAA65269; Oryza sativa: OsLox1, CAA457338, OsLox2, A53054; Glycine max: GmLox1, AAA33986; GmLox2, AAA33987; GmLox3, CAA31664; GmLox4, P38417; GmLox5, AAB67732; GmvLxC, AAA96817; GmLox7, AAC49159; Hordeum vulgare: HvLoxA, AAA64893; HvLoxB, AAB60715 (PDF 269 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Eduardo, I., Chietera, G., Pirona, R. et al. Genetic dissection of aroma volatile compounds from the essential oil of peach fruit: QTL analysis and identification of candidate genes using dense SNP maps. Tree Genetics & Genomes 9, 189–204 (2013). https://doi.org/10.1007/s11295-012-0546-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-012-0546-z