
Abstract
The morphogenic potential of five Gentiana species of medicinal and ornamental value, i.e.G. cruciata, G. kurroo, G. lutea, G. septemfida, and G. tibetica, was compared using agarose-bead leaf mesophyll protoplast culture. Modified Murashige and Skoog medium containing 2.0 mg l−1 1-naphthaleneacetic acid and 0.1 mg l−1 thidiazuron ensured the highest cell division frequency during both protoplast and protoplast-derived cell culture or callus formation. Indirect plant regeneration mainly via the somatic embryogenesis pathway was achieved for G. kurroo and G. tibetica with the greatest efficiency occurring with MS medium supplemented with 1.0 mg l−1 kinetin, 0.5 mg l−1 gibberellic acid, and 80 mg l−1 adenine sulfate. A considerable percentage of autopolyploid and aneuploid cells was detected in callus lines obtained from protoplasts using flow cytometry. The presence of polyploid cells in calli resulted in the regeneration of 85 % polyploid plants of G. kurroo and 14 % polyploids of G. tibetica. Chromosome counting and stomatal characteristic confirmed the ploidy variation among regenerants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plants belonging to the genus Gentiana L. are the source of important pharmacologically active phytochemicals having anti-inflammatory, antipyretic, antimicrobial, cyto- and hepatoprotective, analgesic, stomachic and gastroprotective properties (Singh 2008), and have thus been frequently used in traditional herbal medicine and in pharmacy. Of the 360 species of gentian, Gentiana lutea L. is widely used for pharmaceutical purposes in Europe, G. kurroo Royle in India (Behera and Raina 2012) and Pakistan (Qureshi et al. 2007), and G. tibetica King in China (Zhao et al. 2010). Conversely, because of their very decorative flowers, gentians are also used as ornamental plants, either as cut and potted flowers or in rock and naturalistic gardens (Takahata et al. 1995). Gentiana cruciata L. and G. septemfida Pall owing to their their blue and violet flowers, and ease of cultivation are popular for average garden conditions (Köhlein 1991).
The majority of Gentiana species are rare or even face possible extinction because of their exploitation by man. However, modern biotechnological techniques combined with in vitro cultures offer attractive alternative approaches to traditional cultivation techniques resulting both in the rapid micropropagation of valuable gentian species (Momčilović et al. 1997; Hosokawa et al. 1998), and providing an efficient source of secondary metabolites (Tiwari et al. 2007; Cai et al. 2009; Hayta et al. 2011). Moreover, this technique enables modification of gentian genome for introduction of some desirable characteristics (Mishiba et al. 2006; Nakatsuka et al. 2010) and the conservation of gentian biodiversity (Tanaka et al. 2004; Suzuki et al. 2005; Mikuła et al. 2008). An important prerequisite for the aforementioned use of in vitro cultures is the development of an efficient plant regeneration system from a suitable explant type that expresses a high degree of morphogenic potential.
Of the various regeneration systems available, the protoplast-to-plant system deserves special attention as it forms the basis of protoclonal variation, somatic hybridization or direct genetic transformation by DNA uptake. So far, plants of the genus Gentiana have been regenerated only from green leaf mesophyll protoplasts of G. scabra (Takahata and Jomori 1989), G. triflora (Jomori et al. 1995) and G. scabra x G. triflora (Nakano et al. 1995), from callus protoplasts of G. crassicaulis (Meng et al. 1996) and from cell suspension protoplasts of G. kurroo (Fiuk and Rybczyński 2007; Wójcik and Rybczyński 2015) and G. macrophylla (Hu et al. 2015). Although leaves are the most widely accessible plant material for protoplast isolation, cultures derived from leaf mesophyll protoplasts of gentians are usually associated with low frequency of morphogenetic processes. Some findings of Nakano et al. (1995) and Takahata et al. (1995) indicated that thidiazuron (TDZ) was a more effective cytokinin than benzylaminopurine (BAP) in cultures of mesophyll protoplasts of G. triflora and G. triflora × G. scabra. Recently, we succeeded in regenerating plants from leaf mesophyll protoplasts of G. decumbens using TDZ in media used for protoplast culture and callus proliferation (Tomiczak et al. 2015). However, only tetraploid plants were regenerated, indicating that culture conditions applied are able to disrupt cell division and promote endoreduplication (Tomiczak et al. 2015).
Thus, the aims of this study were (1) to compare the morphogenic potential of five other Gentiana species (G. cruciata, G. kurroo, G. lutea, G. septemfida and G. tibetica) in leaf mesophyll protoplast cultures and (2) to assess the ploidy stability of protocalli and plants obtained from protoplasts. Here the genome size of G. kurroo is estimated, to the best of our knowledge, for the first time.
Materials and methods
Plant material
Seeds of Gentiana species were obtained from the PAS Botanical Garden-CBDC in Powsin, in Poland (G. cruciata) and from the Botanic Garden in Pruhonice, in the Czech Republic (G. kurroo, G. lutea, G. septemfida, G. tibetica). The seeds were sterilized with 70 % (v/v) ethanol for 30 s and then with 20 % (v/v) “Domestos” for 15 min, followed by three rinses in sterile water. Germination was carried out in a Petri dish, on half-strength Murashige and Skoog medium (MS; Murashige and Skoog 1962) supplemented with 0.5 mg l−1 gibberellic acid (GA3), 10 g l−1 sucrose and 7 g l−1 agar, pH 5.8. Plantlets derived from seedlings were transferred to glass jars containing half-strength MS medium with 15 g l−1 sucrose and 8 g l−1 agar and were cultured in a phytotron at 21 ± 1 °C under 16-h day length. Illumination was provided by 100 µM m−2 s−1 daylight fluorescent tubes.
Protoplast isolation
For protoplast isolation, young but fully expanded leaves harvested from in vitro-grown plantlets were used. The leaf abaxial epidermis was peeled off and 1 g of leaf tissue was incubated in 10 ml of cell and protoplast wash solution (CPW; Frearson et al. 1973) supplemented with 0.33 M mannitol (pH 5.8) for 30 min to induce cell plasmolysis. Cell wall digestion was carried out for 3–4 h in the dark at 26 °C in filter-sterilized enzyme solution consisting of 1.0 % (w/v) Cellulase Onozuka R-10 (Yakult Honsha Co., Ltd, Japan) and 0.5 % (w/v) Macerozyme R-10 (Yakult Honsha Co., LTD, Japan) dissolved in CPW with the addition of 0.5 M mannitol and 5 mM 2-(N-Morpholino)ethanesulfonic acid (MES), pH 5.8. In order to improve the release of protoplasts, gentle agitation on a rotary shaker (50 rpm) was employed. The released protoplasts were separated from the debris by filtering through a 45 µm nylon sieve and centrifuged at 180 rpm for 10 min. In the case of G. cruciata, G. kurroo, G. lutea, and G. septemfida the protoplast pellet was rinsed three times with CPW solution supplemented with 0.5 M mannitol and collected each time by centrifugation at 180 rpm for 10 min. To rinse pellet of G. tibetica protoplasts, the CPW solution was replaced by wash solution (WS) containing 1.0 mM CaCl2, 0.5 M mannitol, and 5 mM MES, pH 5.8. The yield of protoplasts was estimated with the use of a Bürker counting chamber and expressed as the number of protoplasts obtained from 1 g of fresh weight of gentian leaves. Protoplast diameter was measured using “analySIS® FIVE” software (Olympus). To assess their viability, protoplasts were stained with 0.01 % (w/v) fluorescein diacetate (FDA) solution (Larkin 1976) and observed through an epifluorescence microscope (Vanox AHBT3, Olympus, 100× magnification). For each species, mean yield, diameter and viability were calculated on the basis of results obtained from 6 independent isolations, and 10 microscopic fields of view per isolation.
Protoplast culture
Protoplasts were cultured at a density of 1.0 × 105 cells per ml in 100-µl agarose beads, composed of protoplast culture medium (PCM) solidified with 0.8 % (w/v) Sea Plaque® Agarose (Cambrex Bio Science Rockland Inc., USA). The PCM medium contained full MS macro- and micronutrients except for NH4NO3, MS vitamins, 30 g l−1 glucose, 3.0 g l−1 glutamine, 0.5 M mannitol, and appropriate plant growth regulators (PGRs), namely: α-naphthaleneacetic acid (NAA) in combination with TDZ or BAP (Table 1). From 12 to 14 beads were formed per 50-mm Petri dish and submerged in 5.0 ml of the liquid medium. Cultures were incubated at either 21 or 26 °C in the dark. Cell wall regeneration was monitored for the first week of culture by staining the protoplasts with 0.001 % (w/v) Calcofluor White (Fluorescent Brightener 28, Sigma-Aldrich) solution. Seven days after culture initiation, the plating efficiency was calculated. Protoplast-derived cells, which had divided at least once, were scored using an inverted light microscope (IMT-2, Olympus, 100× magnification) from 10 fields of view per each agarose bead culture. Plating efficiency was expressed as the number of cells that divided at least once per total number of plated protoplasts multiplied by 100. To prevent cultures from browning, liquid PCM medium was refreshed every week by replacing half the volume (2.5 ml) of the current medium with the same volume of fresh medium. To stimulate further cell division and colony formation, the mannitol concentration of the freshly added medium was gradually reduced after 4 weeks of the protoplast culture; firstly to 0.33 M (in fifth and sixth week), then to 0.17 M (in seventh and eighth week) and finally to zero (in the following weeks).
Callus culture and plant regeneration
Visible protoplast-derived microcalli (about 1–2 mm in diameter) were transferred with agarose beads to 90-mm Petri dish containing 0.8 % agar-solidified MS callus proliferation medium (CPM) supplemented with 30 mg l−1 sucrose and various PGRs (Table 1). Cultures were maintained in the dark at 26 °C. The effect of 4 different PGR combinations on the degree of callus proliferation was estimated after 4 weeks of culture.
Calli lines obtained after 4–8 weeks were placed on 0.8 % agar-solidified MS plant regeneration medium (PRM; Table 1) and cultured under growth cabinet conditions (21 °C, 16/8 h photoperiod). The number of regenerating somatic embryos and/or shoots was estimated after 3 months of culture. Somatic embryos and/or shoots regenerating from callus tissue were transferred to glass jars containing agar-solidified half-strength MS medium supplemented with 15 g l−1 sucrose for further growth and rooting.
Flow cytometry
Randomly chosen proliferating calli (7 per species) and young leaves of all regenerated plants of G. kurroo (20) and G. tibetica (70) were analyzed using flow cytometry. About 6-month-old seed-derived plants (over 20 per species) that were the source of the protoplasts constituted the control. Petunia hybrida cv. P × Pc6 (2.85 pg/2C; Marie and Brown 1993) and Pisum sativum L. ‘Set’ (9.11 pg/2C; Sliwinska et al. 2005) served as an internal standards for the estimation of the total nuclear DNA content of G. kurroo and G. tibetica plants, respectively. Plant material was chopped with a sharp razor blade in a Petri dish containing 1.0 ml of nuclei-isolation buffer (0.1 M Tris, 2.5 mM MgCl2 × 6H2O, 85 mM NaCl, 0.1 % Triton X-100; pH 7.0) supplemented with 50 µg ml−1 of RNase A and 50 µg ml−1 of propidium iodide (PI). The suspension was subsequently passed through a 50-µm mesh nylon filter. For each sample, 3000–7000 nuclei were analyzed immediately following preparation using a Partec CCA (Munster, Germany) flow cytometer. Linear amplification was used for most of the samples; only for the analyses of calli of G. kurroo was logarithmic amplification applied because of the highly elevated endopolyploidy present in the cells of this plant material. PI fluorescence histograms were analyzed using DPAC V.2.2 software (Partec, GmbH, Münster, Germany). For callus samples, the mean C-value (mean ploidy; µ) was calculated (Lemontey et al. 2000). The nuclear DNA content of plants was calculated using the linear relationship between the ratio of the G0/G1 peak positions Gentiana/internal standard, on the fluorescence intensity histogram. Furthermore, the 1Cx-value (DNA content of a monoploid genome with chromosome number x) was calculated.
Chromosome counting
Roots of G. kurroo and G. tibetica regenerants and control plants were used for evaluation of the number of chromosomes. Root tips were pretreated with 2 mM 8-hydroxyquinoline for 2 h at 21 °C and for the following 2 h at 4 °C, and then were fixed in an absolute ethanol: glacial acetic acid (3:1, v/v) for at least 24 h. After macerating for 50 min in 5 M HCl at 21 °C, root tips were stained in Schiff’s reagent (Sigma-Aldrich) for 2 h in the dark and then rinsed three times with potassium metabisulfite solution (225 mM K2S2O5, 5 mM HCl). Metaphase plates were prepared by gently squashing of root-tip meristems in a drop of 45 % (v/v) acetic acid on a microscopic slide. Observations were carried out using a Vanox AHBT3 microscope (Olympus) at 1000× magnification. Chromosomes were counted for at least 10 different, well-spread metaphase plates per each plant.
Stomatal guard cell length and frequency
The size and frequency of stomata were estimated for the abaxial epidermis of the youngest pair of fully expanded leaves of G. kurroo and G. tibetica plants grown in vitro. Observations were performed using a Vanox AHBT3 light microscope. The guard cell length of all stomata visible in 10 randomly selected fields of view per each leaf was measured using “analySIS® FIVE” software. Also, all stomata from 10 randomly selected microscopic fields of view per leaf were counted. The stomatal frequency was expressed as the number of stomata per 1 mm2 area of the leaf blade.
Statistical analysis
Each in vitro culture experiment was repeated three times. Three Petri dishes per each combination of culture conditions represented each replicate. Statistical analyses were performed with the help of Statistica ver. 6.0 (StatSoft Polska Sp. z o.o., Poland). One- or two-way analysis of variance was performed. Means were compared using Tukey’s honestly significant difference (HSD) test, at the 0.05 level of probability. The correlation between mean size of protoplasts from different species and their yield was computed using Pearson product-moment correlation coefficient.
Results
Protoplast isolation and culture of five Gentiana species
Protoplasts were successfully isolated from leaf mesophyll tissue of all five Gentiana species. All protoplasts were spherical in shape and mostly rich in chloroplasts (Fig. 1a). Their mean size ranged from 24.23 (G. kurroo) to 36.96 µm in diameter (G. cruciata) (Table 2). The size was negatively correlated with the yield of protoplasts (Pearson correlation coefficient r = −0.91), which varied from 3.46 × 105 protoplasts per 1 g of fresh leaf tissue of G. septemfida to 11.41 × 105 protoplasts from 1 g of G. kurroo leaf material (Table 2). The protoplast viability after isolation, as revealed by FDA staining (Fig. 1b) was similar for all species and reached 80–90 % (Table 2).

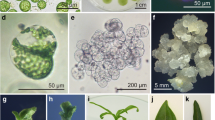
Plant regeneration in green leaf mesophyll protoplast cultures of Gentiana species: a freshly isolated protoplasts of G. tibetica and b their viability revealed by FDA staining, c regeneration of the cell wall following 5 days of protoplast culture, and emphasized by treatment with Calcofluor White. Note the formation of a cell plate during first cell division, d three daughter cells formed as a result of the first and second division of protoplast-derived cell after 7 days of culture, e multicellular aggregate of G. kurroo formed after 5 and f 9 weeks of protoplast culture, respectively, g small minicalli of G. tibetica formed inside agarose beads with spontaneously regenerated roots (arrows), h magnification of spontaneously regenerated root, i callus tissue arising from single agarose bead, j somatic embryo of G. tibetica regenerated after 6 weeks of callus culture on PRM1 medium, k shoots of G. kurroo regenerated after 8 weeks of callus culture on PRM2 medium, l regenerated plant of G. tibetica
Within the first week of culture in agarose beads, protoplasts of all studied species, except for G. lutea completed the regeneration of the cell wall and started to divide (Fig. 1c–d). Protoplasts of G. lutea burst following 48 h of culture on both PCM1 and PCM2 medium.
For the remaining four species studied, culturing of protoplasts on PCM1 medium containing 0.1 mg l−1 TDZ resulted in higher plating efficiency than on PCM2 supplemented with 1 mg l−1 BAP (Table 3). Furthermore, the percentage of cell division noted at 26 °C was, on average, twice as much as at 21 °C (Table 3). Of all the species tested, G. septemfida was generally distinguished in that it demonstrated greatest plating efficiency at day 7 of culture.
Despite the weekly addition of the fresh medium, cultures of G. cruciata and G. septemfida protoplasts turned brown and died within 2 weeks of culture. Protoplast-derived cells of G. kurroo and G. tibetica continued to divide and formed multicellular aggregates after 5–9 weeks on either PCM1 or PCM2 (Fig. 1e–f). After 10–12 weeks of culture, visible microcalli measuring of 1–2 mm in diameter were obtained. In addition, spontaneous rhizogenesis was observed at times in G. tibetica cultures maintained on both media (Fig. 1g–h).
Callus culture and plant regeneration
Callus proliferation was achieved for all agarose beads transferred onto CPM media (Fig. 1i). The formation of callus tissue of both species, G. kurroo and G. tibetica, was faster when protoplasts were previously cultured on medium supplemented with TDZ (i.e. PCM1), than when cultured on the medium containing BAP (i.e. PCM2). The presence of TDZ in CPM1 medium promoted rapid growth of protoplast-derived callus as well (Online Resource 1). Effective callus proliferation of both G. kurroo and G. tibetica was also achieved on CPM3 medium supplemented with dicamba, NAA, BAP, and adenine sulfate.
The process of plant regeneration from protoplast-derived calli was mostly via somatic embryogenesis (Fig. 1j). For both species, PRM3 medium was the most effective in inducing somatic embryogenesis, but only from calli maintained previously on CPM1 and CPM3 media. The greatest number of G. kurroo somatic embryos per agarose bead (1.08) was recorded for those regenerated on PRM3 medium from callus cultured previously on CPM1 medium, whereas for G. tibetica the best embryogenic response (0.55 somatic embryos regenerated per agarose bead) was achieved for callus maintained on CPM3 medium. In a few G. kurroo callus cultures subjected to PRM2 medium, the regeneration of shoots was also observed (Fig. 1k, Online Resource 2).
Most of mature somatic embryos of both species easily developed into plantlets on half-strength MS medium with 2 % (w/v) sucrose. On the same medium, most of regenerated shoots of G. kurroo also rooted. Most of regenerated plants (Fig. 1l) grew vigorously on full strength MS medium.
Cell ploidy of protoplast-derived calli
Well over 90 % of the nuclei of leaves from seedlings of both Gentiana species investigated contained 2C DNA, only a few having 4C nuclei (Figs. 2a, 3a). Conversely, all G. kurroo protoplast-derived calli analyzed cytometrically exhibited mixoploidy, containing, beside 2C and 4C nuclei, some endopolyploid nuclei with DNA content above 4C (Fig. 2). Nuclei with DNA content 2C, 4C, 6C, 8C, and 16C were detected, and in some calli a few nuclei with 32C were present. In three of five calli, 4C nuclei predominated, the remaining two having 8C nuclei. The mean C-value for calli of this species varied from 4.87 to 11.22. In the case of protoplast-derived calli of G. tibetica, the degree of mixoploidy was lower than for G. kurroo, and very few endopolyploid nuclei were detected (Fig. 3). However, surprisingly, in some calli, nuclei with DNA content lower than 2C (about 1.4C) occurred. Consequently, the mean C-value for G. tibetica callus cells varied from 1.48 to 2.51.

Histograms of relative nuclear DNA content obtained after flow cytometric analysis of nuclei isolated from G. kurroo leaves and calli: a leaf of a seed-derived control plant, b–f protoplast-derived 4-month-old callus cultures. Note that cells with 2C DNA content are absent in the samples (c), (e) and (f). µ—mean C-value (mean ploidy)

Histograms of relative nuclear DNA content obtained after flow cytometric analysis of nuclei isolated from G. tibetica leaves and calli: a leaf of a seed-derived control plant, b–f protoplast-derived 4-month-old callus cultures. Note the presence of cells with relative nuclear DNA content below 2C in the samples (b), (c) and (d). µ—mean C-value (mean ploidy)
DNA content and chromosome number of regenerated plants
Flow cytometry and chromosome counting revealed that control seed-derived plants of G. kurroo possessed 3.32 pg/2C DNA and 26 chromosomes (Fig. 4a; Table 4). Control plants of G. tibetica possessed 6.91 pg/2C DNA and 52 chromosomes (Fig. 4d; Table 4).

Chromosome counting of G. kurroo and G. tibetica plants: a 26 mitotic metaphase chromosomes in a root-tip cell of a seed-derived parent plant of G. kurroo and d 52 chromosomes in a root-tip cell of a seed-derived parent plant of G. tibetica, b protoplast-derived regenerants of G. kurroo with 26 and c 52 chromosomes, e protoplast-derived regenerants of G. tibetica with 52 and f about 100 chromosomes
In 20 protoplast-derived plants of G. kurroo, 10.0 % possessed a genome and a chromosome number equal in size to that of the controls (Fig. 4b; Table 4), whereas in 85 %, nuclear DNA content and number of chromosomes had doubled (Fig. 4c; Table 4). One of G. kurroo regenerants was identified as a possible aneuploid having a 2C DNA content of 8.75 pg, below that expected for the hexaploid (assuming that 1Cx of this species is equal to about 1.7 pg, hexaploid DNA content would be about 10 pg). The chromosome number of this plant could not be assessed because of its inability to root and because it died rapidly in vitro. Of the 70 regenerants of G. tibetica also about 85 % had the same nuclear DNA content and number of chromosomes as control plants (Fig. 4e; Table 4), whereas the remaining protoplast-derived plants had their DNA content and twice as many chromosomes as the control (Fig. 4f; Table 4).
Plant morphology and stomatal guard cell length and frequency
Two plants regenerated from protoplasts of G. kurroo, which had the same nuclear DNA content and chromosome number as control plants, possessed a similar number of stomata per mm2 of leaf blade area (Table 4). Regenerants with doubled DNA content and chromosome number, with the exception of leaf blades (Fig. 5b) which were wider than those of parent plants (Fig. 5a), showed no visible morphological differences under in vitro conditions. However, they had two-fold fewer stomata on the abaxial leaf surface than control plants (Table 4). Also, in comparison to the controls, the stomata were slightly smaller in regenerants with the same nuclear DNA content, but significantly larger in plants with doubled DNA content (Table 4). The stomata were not analyzed for an aneuploid G. kurroo regenerant, however severe leaf blade malformations such as swelling and crinkling were observed in this plant (Fig. 5c).

Leaf morphology of G. kurroo and G. tibetica plants: a seed-derived parent plant of G. kurroo, b protoplast-derived regenerant of G. kurroo with doubled DNA content and chromosome number, c aneuploid protoplast-derived regenerant of G. kurroo, d seed-derived parent plant of G. tibetica, e protoplast-derived regenerant of G. tibetica with doubled DNA content and chromosome number
Similar relationships between ploidy level and the number and length of stomata were detected for G. tibetica protoplast-derived plants (Table 4). However, the regenerants with octoploid DNA content were dwarf and possessed thicker and wider leaves (Fig. 5e) than control plants and regenerants with tetraploid DNA content (Fig. 5d). They also developed shorter and thicker roots.
Discussion
Assessment of the morphogenic potential in protoplast culture, followed by the development of the protoplast-to-plant system, are the first steps in researching somatic hybridization or direct genetic transformation in species of interest. In this work, for the first time, leaf mesophyll protoplasts were isolated for five different gentian species of high medicinal and ornamental importance. Early response of protoplasts to culture conditions was compared for G. cruciata, G. kurroo, G. septemfida and G. tibetica. Regeneration potential and ploidy stability were evaluated for G. kurroo and G. tibetica.
The influence of culture conditions on protoplast-derived cell viability and divisions
Leaf mesophyll tissue of in vitro grown gentians is a suitable plant material for routine isolation of viable protoplasts. The physical and chemical conditions used for protoplast release allowed us to obtain yields of G. kurroo protoplasts similar to those of G. decumbens (Tomiczak et al. 2015) and G. crassicaulis (Meng et al. 1996), whereas the yields of protoplasts for other species was comparable to those that of G. acaulis (Jomori et al. 1995), G. triflora, and G. triflora × G. scabra (Nakano et al. 1995). A large number of factors related to plant material used, including plant genotype, age, and preculture conditions, influence the yield of released protoplast (Wright 1985; Grèzes et al. 1994; Hu et al. 2015). Our study indicates that yield of protoplasts can be also affected by protoplast size, thereby the cell size of source material.
Despite their relatively great viability directly after isolation, protoplasts of G. lutea did not survive the first 48 h in culture. Probably the concentration and/or the type of osmoticum, or the concentration of macro- and micronutrients in the protoplast culture medium were inadequate. Takahata et al. (1995) referred to the use of half-strength MS gellan gum-solidified medium supplemented with 1.5 % sucrose and 0.25 M mannitol for cell suspension-derived G. lutea protoplasts. However, to our best knowledge, no further reports on their regeneration into entire plants have ever been published. Furthermore, in our experience, leaves of G. lutea provide explants that respond poorly and have very low morphogenic potential (Fiuk and Rybczyński 2008b).
Influenced by the discovery of Nakano et al. (1995) and Takahata et al. (1995) that TDZ is a more effective cytokinin than BAP for culturing mesophyll protoplast of G. triflora and G. triflora × G. scabra, as well as by our previous work on G. decumbens (Tomiczak et al. 2015), we compared the influence of TDZ and BAP on cell division efficiency in protoplast cultures of G. cruciata, G. kurroo, G. septemfida, and G. tibetica. TDZ, a substituted phenylurea compound, induces a diverse array of cultural responses ranging from callus formation to regeneration of shoots and somatic embryos (Murthy et al. 1998). For four gentian species studied in this work, a greater percentage of dividing cells was obtained when protoplasts were cultured on medium containing TDZ than when grown on medium supplemented with BAP.
Browning of the protoplast cultures of G. cruciata and G. septemfida, which was observed within 2 weeks from culture initiation, is thought to be caused by oxidation of phenolic compounds released from plant cells into the culture medium (Zhu et al. 1997). This phenomenon was also described by Takahata and Jomori (1989); Jomori et al. (1995); Kunitake et al. (1995) and Nakano et al. (1995), and indicates that it is one of the major problems in protoplast cultures of Gentianaceae. In the case of G. cruciata and G. septemfida, weekly medium refreshment was insufficient to prevent cell browning. Whether the addition of activated charcoal or polyvinylpyrrolidone (PVP) is beneficial for these species, as in Eustoma grandiflorum (Kunitake et al. 1995) or Vitis vinifera (Zhu et al. 1997), would be worth investigating. Another possibly valuable modification would be conditioned (Chen et al. 2004; Komai et al. 2006) or nurse type of protoplast culture (Zhou et al. 2005).
The influence of culture conditions on callus proliferation and plant regeneration
The positive effect of TDZ on proliferation of protoplast-derived callus of G. kurroo and G. tibetica was manifested in two ways. Firstly, a greater volume of callus tissue was obtained from agarose beads cultured previously on TDZ-supplemented PCM1 medium, than from beads cultured on PCM2 containing BAP. Very similar effects were observed during callus formation derived from mesophyll protoplasts of G. decumbens (Tomiczak et al. 2015). Secondly, it transpired that CPM1 medium is the most appropriate for callus proliferation of G. kurroo and is almost as good as CPM3 for G. tibetica. CPM3 medium containing 1.0 mg l−1 dicamba, 2.0 mg l−1 BAP, 0.1 mg l−1 NAA and 80 mg l−1 adenine sulfate is an universal medium typically used for the initiation and maintenance of cell suspension cultures of G. tibetica, G. cruciata and G. pannonica (Mikuła et al. 2002, 2005).
Publications involving the development of a protoplast-to-plant system for Gentiana mesophyll protoplasts available pre-2015 described only one way of plant regeneration i.e. shoot organogenesis (Takahata and Jomori 1989; Nakano et al. 1995). Regeneration of shoots was achieved on media containing high concentrations (5–10 mg l−1) of cytokinins (TDZ or BAP) in combination with 0.1 mg l−1 NAA (Nakano et al. 1995) or 1.0 mg l−1 IAA (Takahata and Jomori 1989). Recently, we succeeded in regenerating plants from leaf mesophyll protoplasts of G. decumbens by indirect somatic embryogenesis (Tomiczak et al. 2015). PRM3 medium containing 1.0 mg l−1 kinetin, 0.5 mg l−1 GA3, 80 mg l−1 adenine sulfate, which led to the formation of somatic embryos from protoplast-derived tissues of G. decumbens, was the same medium as was widely used for the induction of somatic embryogenesis from callus and cell suspensions of G. pannonica, G. cruciata, G. tibetica (Mikuła and Rybczyński 2001; Mikuła et al. 2002), and G. kurroo (Fiuk and Rybczyński 2008a), as well as from protoplasts of G. kurroo cell suspensions (Fiuk and Rybczyński 2007). The greatest number of somatic embryos regenerated from mesophyll protoplast-derived callus of G. tibetica and G. kurroo was also achieved by using this medium. Shoot organogenesis was also observed in G. kurroo cultures, but only on PRM2 medium supplemented with a high concentration (6.0 mg l−1) of BAP. Interestingly, single somatic embryos were obtained on PRM1 medium, containing 8.0 mg l−1 TDZ. However, as reviewed by Murthy et al. (1998), both traditional cytokinin and auxin-type responses to TDZ in tissue culture medium have been reported. Induction of highly morphogenic callus followed by regeneration of shoots and somatic embryos has been documented for a wide variety of plant species and for different explant types (Tegeder et al. 1995; Gill and Ozias-Akins 1999; Zhang et al. 2001; Mithila et al. 2003; Jones et al. 2007; Thomas 2007).
Ploidy of cells of callus and regenerated plants
Young leaves of Gentiana seem to provide adequate explant material for initiation of cytogenetically stable cell cultures, as they do not contain endopolyploid cells. Most gentian leaf mesophyll cell nuclei become arrested in the G1/G0 phase of the cell cycle (2C DNA content), only a small fraction of G2 nuclei were detected (Mikuła et al. 2008; Fiuk et al. 2010). However, the conditions for protoplast isolation and culture, as well as an indirect method of plant regeneration (through long-term callus phase), can induce somaclonal variation (known as protoclonal variation in this case), causing numerical and structural chromosome changes, and consequently leading to cell aneuploidy and/or polyploidy (Karp et al. 1982; Karp 1994). This variation is especially evident in protoplast cultures, where a high frequency of errors occurs during mitosis. It was confirmed in the present experiment that extensive variation in cell ploidy occurs in protoplast-derived callus. This variation was especially evident in G. kurroo; apart from endoreduplication, resulting in the presence of 8C and 16C cells and an increased proportion of 4C cells, other chromosome disturbances occur, precipitating the formation of aneuploid cells having a DNA content ranging from 5C to 6C. Also, in the tetraploid species G. tibetica, probably some errors occurred during early divisions, with the result that a considerable proportion of cells had a DNA content of less than 2C. Similarly, in protoplast cultures of potato, endoreduplication, polyploidy, and aneuploidy occurred very early during the initial stages of protoplast development and callus induction. An increase in the nuclear DNA content and chromosome number was observed as early as the first 2 weeks of protoplast culture. This process continued in the callus and finally, when regeneration occurred, led to polyploidy or aneuploidy in regenerated plants (Sree Ramulu et al. 1984, 1989). The proportion of polyploid/aneuploid regenerants to true-to-type plants can vary from single aneuploid and polyploid regenerated plants like it was noted for Pelargonium × hortorum (Nassour et al. 2003) or Eustoma grandiflorum (Lindsay et al. 1994), through almost half of the regenerants having a ploidy value different from that of the mother plants (Fragaria × ananassa; Nyman and Wallin 1992), to only polyploid plants being obtained from protoplast culture (G. decumbens; Tomiczak et al. 2015). In the present research, over 85 % of G. tibetica regenerants possessed a genome of the same size as that of control plants, and only 14 % were polyploid. There was no regeneration from the cells having a DNA content lower than 2C, as also observed with other species (Elmaghrabi and Ochatt 2006). However, in G. kurroo, the majority of regenerated plants (85 %) contained double the amount of DNA as control plants. This was probably due to the high proportion of 4C cells and the low proportion, or even lack of 2C nuclei in the calli of this species (as shown in Fig. 2c, d, f). By contrast, Nontaswatsri and Fukai (2005) reported a greater ability to regenerate in diploid cells compared with tetraploids and octoploids, resulting in preferential regeneration of diploid plants from mixoploid calli of Dianthus hybrid. The greater totipotency in vitro of diploid cells was also confirmed for Cucumis sativus (Kubaláková et al. 1996), Pisum sativum (Ochatt et al. 2000), and Medicago truncatula (Elmaghrabi and Ochatt 2006). The presence of aneuploid cells (5–6C) in some calli of G. kurroo in the present study resulted in the regeneration of a single plant, which, however, showed abnormal morphology and did not survive. In our previous studies on G. kurroo, about 30–37 % polyploids (tetraploids and hexaploids) was regenerated from cell suspension-derived protoplasts (Fiuk and Rybczyński 2007), and 14 % tetraploids from hypocotyl-derived cell suspension (Fiuk and Rybczyński 2008a).
According to the literature, G. kurroo is a diploid species having 26 chromosomes (Kaur et al. 2009; Yuan et al. 1998), and G. tibetica is a tetraploid possessing 52 chromosomes (Mishiba et al. 2009; Yuan et al. 1998). In our study, the chromosome number of control plants of both species was consistent with previously reported data, and regenerants with double the amount of DNA also possessed twice the number of chromosomes. Furthermore, we observed the typical correlation that length of stomata increased and the number of stomata decreased with increasing ploidy, as was previously reported for G. decumbens (Tomiczak et al. 2015) and for many other genera and species (reviewed by Knight and Beaulieu 2008). On the other hand, the relationships between ploidy and morphology of gentians regenerated both in this and a previous study (Tomiczak et al. 2015) comply with the general statement that there is an optimum for ploidy increase, beyond which the individuals become dwarf or less vigorous (Müntzing 1936). Gentian plants with a ploidy level close to 6x and 8x exhibited dwarf stature, thick and wrinkled leaves, as well as shorter and thicker roots than diploid and tetraploid plants. This can indicate that hexaploidy and octoploidy are beyond optimal values for Gentiana species.
Conclusions
In conclusion, we have compared the morphogenic potential of five Gentiana species in green leaf mesophyll protoplast culture and succeeded in the indirect plant regeneration of plants of G. kurroo and G. tibetica. Polyploidy detected amongst regenerants of both species had its origin in the various ploidy levels of protoplast-derived callus cells. The protoplast-to-plant system developed in this study may provide the prerequisite for creating novel genotypes of these valuable medicinal species by means of genetic manipulations and somatic cell hybridization.
References
Behera MC, Raina R (2012) Gentiana kurroo—a critically endangered bitter herb. Int J Med Aromat Plants 2(1):22–29
Cai YF, Liu YL, Liu ZH, Zhang F, Xiang FN, Xia GM (2009) High frequency embryogenesis and regeneration of plants with high content of gentiopicroside from the Chinese medicinal plant Gentiana straminea Maxim. In Vitro Cell Dev Biol Plant 45:730–739
Chen L-P, Zhang M-F, Xiao Q-B, Wu J-G, Hirata Y (2004) Plant regeneration from hypocotyl protoplasts of red cabbage (Brassica oleracea) by using nurse cultures. Plant Cell Tiss Org Cult 77:133–138
Elmaghrabi A, Ochatt S (2006) Isoenzymes and flow cytometry for the assessment of true-to-typeness of calluses and cell suspensions of barrel medic prior to regeneration. Plant Cell Tiss Org Cult 85:31–43
Fiuk A, Rybczyński JJ (2007) The effect of several factors on somatic embryogenesis and plant regeneration in protoplast cultures of Gentiana kurroo (Royle). Plant Cell Tissue Org Cult 91:263–271
Fiuk A, Rybczyński JJ (2008a) Factors influencing efficiency of somatic embryogenesis of Gentiana kurroo (Royle) cell suspension. Plant Biotechnol Rep 2:33–39
Fiuk A, Rybczyński JJ (2008b) Genotype and plant growth regulator-dependent response of somatic embryogenesis from Gentiana leaf explants. In Vitro Cell Dev Biol Plant 44:90–99
Fiuk A, Bednarek PT, Rybczyński JJ (2010) Flow cytometry, HPLC-RP, and metAFLP analyses to assess genetic variability in somatic embryo-derived plantlets of Gentiana pannonica Scop. Plant Mol Biol Rep 28:413–420
Frearson EM, Power JB, Cocking EC (1973) The isolation, culture and regeneration of Petunia leaf protoplasts. Dev Biol 33:130–137
Gill R, Ozias-Akins P (1999) Thidiazuron-induced highly morphogenic callus and high frequency regeneration of fertile peanut (Arachis hypogaea L.) plants. In Vitro Cell Dev Biol Plant 35:445–450
Grèzes J, Thomas D, Thomasset B (1994) Factors influencing protoplast isolation from Coffea arabica cells. Plant Cell Tissue Org Cult 36:91–97
Hayta S, Akgun IH, Ganzera M, Bedir E, Gurel A (2011) Shoot proliferation and HPLC-determination of iridoid glycosides in clones of Gentiana cruciata L. Plant Cell Tissue Org Cult 107:175–180
Hosokawa K, Oikawa Y, Yamamura S (1998) Mass propagation of ornamental gentian in liquid medium. Plant Cell Rep 17:747–751
Hu X, Yin Y, He T (2015) Plant regeneration from protoplasts of Gentiana macrophylla Pall. using agar-pool culture. Plant Cell Tissue Org Cult 121:345–351
Jomori H, Takahata Y, Kaizuma N (1995) Plant regeneration from leaf-derived calli of gentians and their protoplast culture. Acta Hort 392:81–86
Jones MPA, Yi Z, Murch SJ, Saxena PK (2007) Thidiazuron-induced regeneration of Echinacea purpurea L.: micropropagation in solid and liquid culture systems. Plant Cell Rep 26:13–19
Karp A (1994) Origin, causes and uses of variation in plant tissue cultures. In: Vasil IK, Thorpe TA (eds) Plant, cell and tissue culture. Kluwer Academic Publishers, Dordrecht, pp 139–151
Karp A, Nelson RS, Thomas E, Bright SWJ (1982) Chromosome variation in protoplast-derived potato plants. Theor Appl Genet 63:265–272
Kaur R, Panwar N, Saxena B, Raina R, Bharadwaj SV (2009) Genetic stability in long-term micropropagated plants of Gentiana kurroo—an endangered medicinal plant. J New Seeds 10:236–244
Knight CA, Beaulieu JM (2008) Genome size scaling through phenotype space. Ann Bot 101:759–766
Köhlein F (1991) Gentians. Timber Press, Portland
Komai F, Morohashi H, Horita M (2006) Application of nurse culture for plant regeneration from protoplasts of Lilium japonicum Thunb. In Vitro Cell Dev Biol Plant 42:252–255
Kubaláková M, Doležel J, Lebeda A (1996) Ploidy instability of embryogenic cucumber (Cucumis sativus L.) callus culture. Biol Plant 38:475–480
Kunitake H, Nakashima T, Mori K, Tanaka M, Mii M (1995) Plant regeneration from mesophyll protoplasts of lisianthus (Eustoma grandiflorum) by adding activated charcoal into protoplast culture medium. Plant Cell Tissue Org Cult 43:59–65
Larkin PJ (1976) Purification and viability determinations of plant protoplasts. Planta 128:213–216
Lemontey C, Mousset-Déclas C, Munier-Jolain N, Boutin JP (2000) Maternal genotype influences pea seed size by controlling both mitotic activity during early embryogenesis and final endoreduplication level/cotyledon cell size in mature seed. J Exp Bot 51:167–175
Lindsay GC, Hopping ME, O’Brien IEW (1994) Detection of protoplast-derived DNA tetraploid Lisianthus (Eustoma grandiflorum) plants by leaf and flower characteristics and by flow cytometry. Plant Cell Tissue Org Cult 38:53–55
Marie D, Brown SC (1993) A cytometric exercise in plant DNA histograms, with 2C values for 70 species. Biol Cell 78:41–51
Meng Y, Gao Y, Jia J (1996) Plant regeneration from protoplasts isolated from callus of Gentiana crassicaulis. Plant Cell Rep 16:88–91
Mikuła A, Rybczyński JJ (2001) Somatic embryogenesis of Gentiana genus I. The effect of the preculture treatment and primary explant origin on somatic embryogenesis of Gentiana cruciata (L.), G. pannonica (Scop.), and G. tibetica (King). Acta Physiol Plant 23:15–25
Mikuła A, Skierski J, Rybczyński JJ (2002) Somatic embryogenesis of Gentiana genus III. Characterization of three-year-old embryogenic suspensions of G. pannonica originated from various seedling explants. Acta Physiol Plant 24:311–322
Mikuła A, Rybczyński JJ, Skierski J, Latkowska MJ, Fiuk A (2005) Somatic embryogenesis of Gentiana genus IV: Characterization of Gentiana cruciata and Gentiana tibetica embryogenic cell suspensions. In: Hvolsef-Eide AK, Preil W (eds) Liquid culture systems for in vitro plant propagation. Springer, Dordrecht, pp 345–358
Mikuła A, Olas M, Sliwinska E, Rybczyński JJ (2008) Cryopreservation by encapsulation of Gentiana spp cell suspensions maintains regrowth, embryogenic competence and DNA content. CryoLetters 29:409–418
Mishiba K, Nishihara M, Abe Y, Nakatsuka T, Kawamura H, Kodama K, Takesawa T, Abe J, Yamamura S (2006) Production of dwarf potted gentian using wild-type Agrobacterium rhizogenes. Plant Biotechnol 23:33–38
Mishiba K, Yamane K, Nakatsuka T, Nakano Y, Yamamura S, Abe J, Kawamura H, Takahata Y, Nishihara M (2009) Genetic relationships in the genus Gentiana based on chloroplast DNA sequence data and nuclear DNA content. Breed Sci 59:119–127
Mithila J, Hall JC, Victor JMR, Saxena PK (2003) Thidiazuron induces shoot organogenesis at low concentrations and somatic embryogenesis at high concentrations on leaf and petiole explants of African violet (Saintpaulia ionantha Wendl.). Plant Cell Rep 21:408–414
Momčilović I, Grubišić D, Nešković M (1997) Micropropagation of four Gentiana species (G. lutea, G. cruciata, G. purpurea and G. acaulis). Plant Cell Tissue Org Cult 49:141–144
Müntzing A (1936) The evolutionary significance of autopolyploidy. Hereditas 21:263–378
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Murthy BNS, Murch SJ, Saxena PK (1998) Thidiazuron: a potent regulator of in vitro plant morphogenesis. In Vitro Cell Dev Biol Plant 34:267–275
Nakano M, Hosokawa K, Oomiya T, Yamamura S (1995) Plant regeneration from protoplasts of Gentiana by embedding protoplasts in gellan gum. Plant Cell Tiss Org Cult 41:221–227
Nakatsuka T, Mishiba K, Kubota A, Abe Y, Yamamura S, Nakamura N, Tanaka Y, Nishihara M (2010) Genetic engineering of novel flower colour by suppression of anthocyanin modification genes in gentian. J Plant Physiol 167:231–237
Nassour M, Chassériaux G, Dorion N (2003) Optimization of protoplast-to-plant system for Pelargonium × hortorum ‘Alain’ and genetic stability of the regenerated plants. Plant Sci 165:121–128
Nontaswatsri C, Fukai S (2005) Regenerative callus of Dianthus ‘Telstar Scarlet’ showing mixoploidy produce diploid plants. Plant Cell Tiss Org Cult 83:351–355
Nyman M, Wallin A (1992) Improved culture technique for strawberry (Fragaria × ananassa Duch.) protoplasts and the determination of DNA content in protoplast derived plants. Plant Cell Tiss Org Cult 30:127–133
Ochatt SJ, Mousset-Declas C, Rancillac M (2000) Fertile pea plants regenerate from protoplasts when calluses have not undergone endoreduplication. Plant Sci 156:177–183
Qureshi RA, Gilani SA, Ashraf M (2007) Ethnobotanical studies with special reference to plant phenology at Sudhan Gali and Ganga Chotti Hills (District Bagh, A.K.). EJEAFChe 6(7):2207–2215
Singh A (2008) Phytochemicals of Gentianaceae: a review of pharmacological properties. Int J Pharm Sci Nanotechnol 1(1):33–36
Sliwinska E, Zielinska E, Jedrzejczyk I (2005) Are seeds suitable for flow cytometric estimation of plant genome? Cytometry 64A(2):72–79
Sree Ramulu K, Dijkhuis P, Roest S, Bokelmann GS, De Groot B (1984) Early occurence of genetic instability in protoplast cultures of potato. Plant Sci Lett 36:79–86
Sree Ramulu K, Dijkhuis P, Roest S (1989) Patterns of phenotypic and chromosome variation in plants derived from protoplast cultures of monohaploid, dihaploid and diploid genotypes and in somatic hybrids of potato. Plant Sci 60:101–110
Suzuki M, Akihama T, Ishikawa M (2005) Cryopreservation of encapsulated gentian axillary buds following 2 step-preculture with sucrose and desiccation. Plant Cell Tiss Org Cult 83:115–121
Takahata Y, Jomori H (1989) Plant regeneration from mesophyll protoplasts of gentiana (Gentiana scabra Bungei). Plant Tiss Cult Lett 6:19–21
Takahata Y, Jomori H, Miyano S, Kunitake H, Mii M (1995) Regeneration of plants from protoplasts of Gentiana species (Gentian). In: Bajaj YPS (ed) Biotechnology in agriculture and forestry, vol 34., Plant protoplasts and genetic engineering VISpringer-Verlag, Berlin, pp 57–62
Tanaka D, Niimo T, Isuzugawa K, Hikage T, Uemura M (2004) Cryopreservation of shoot apieces of in vitro grown gentian plants: comparison of vitrification and encapsulation-vitrification protocols. CryoLetters 25:167–176
Tegeder M, Gebhardt D, Scheider O, Pickardt T (1995) Thidiazuron-induced plant regeneration from protoplasts of Vicia faba cv. Mythos. Plant Cell Rep 15:164–169
Thomas TD (2007) Pretreatment in thidiazuron improves the in vitro shoot induction from leaves in Curculigo orchioides Gaertn., an endangered medicinal plant. Acta Physiol Plant 29:455–461
Tiwari RK, Trivedi M, Guang ZC, Guo G-Q, Zheng G-C (2007) Genetic transformation of Gentiana macrophylla with Agrobacterium rhizogenes: growth and production of secoiridoid glucoside gentiopicroside in transformed hairy root cultures. Plant Cell Rep 26:199–210
Tomiczak K, Mikuła A, Sliwinska A, Rybczynski JJ (2015) Autotetraploid plant regeneration by indirect somatic embryogenesis from leaf mesophyll protoplasts of diploid Gentiana decumbens L.f. In Vitro Cell Dev Biol Plant 51:350–359
Wójcik A, Rybczyński JJ (2015) Electroporation and morphogenic potential of Gentiana kurroo (Royle) embryogenic cell suspension protoplasts. BioTechnologia 96:19–29
Wright DC (1985) Factors affecting isolation of protoplasts from leaves of grape (Vitis vinifera). Plant Cell Tiss Org Cult 4:95–100
Yuan Y-M, Küpfer P, Zeltner L (1998) Chromosomal evolution of Gentiana and Jaeschkea (Gentianaceae), with further documentation of chromosome data for 35 species from western China. Plant Syst Evol 210:231–247
Zhang C-L, Chen D-F, Elliott MC, Slater A (2001) Thidiazuron-induced organogenesis and somatic embryogenesis in sugar beet (Beta vulgaris L.). In Vitro Cell Dev Biol Plant 37:305–310
Zhao ZL, Dorje G, Wang ZT (2010) Identification of medicinal plants used as Tibetan Traditional Medicine Jie-Ji. J Ethnopharm 132:122–126
Zhou J, Wang B, Zhu L (2005) Conditioned culture for protoplasts isolated from chrysanthemum: an efficient approach. Colloids Surf B 45:113–119
Zhu YM, Hoshino Y, Nakano M, Takahashi E, Mii M (1997) Highly efficient system of plant regeneration from protoplasts of grapevine (Vitis vinifera L.) through somatic embryogenesis by using embryogenic callus and activated charcoal. Plant Sci 123:151–157
Acknowledgments
The authors wish to thank Aleksandra Chomacka for her technical assistance in flow cytometric analysis. This work was supported by the Polish Ministry of Science and Higher Education (Grant No. 3P04C 037 23).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tomiczak, K., Sliwinska, E. & Rybczyński, J.J. Comparison of the morphogenic potential of five Gentiana species in leaf mesophyll protoplast culture and ploidy stability of regenerated calli and plants. Plant Cell Tiss Organ Cult 126, 319–331 (2016). https://doi.org/10.1007/s11240-016-1000-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-016-1000-y