
Abstract
Maize is one of the world’s most important crops and a model for grass genome research. Long terminal repeat (LTR) retrotransposons comprise most of the maize genome; their ability to produce new copies makes them efficient high-throughput genetic markers. Inter-retrotransposon-amplified polymorphisms (IRAPs) were used to study the genetic diversity of maize germplasm. Five LTR retrotransposons (Huck, Tekay, Opie, Ji, and Grande) were chosen, based on their large number of copies in the maize genome, whereas polymerase chain reaction primers were designed based on consensus LTR sequences. The LTR primers showed high quality and reproducible DNA fingerprints, with a total of 677 bands including 392 polymorphic bands showing 58% polymorphism between maize hybrid lines. These markers were used to identify genetic similarities among all lines of maize. Analysis of genetic similarity was carried out based on polymorphic amplicon profiles and genetic similarity phylogeny analysis. This diversity was expected to display ecogeographical patterns of variation and local adaptation. The clustering method showed that the varieties were grouped into three clusters differing in ecogeographical origin. Each of these clusters comprised divergent hybrids with convergent characters. The clusters reflected the differences among maize hybrids and were in accordance with their pedigree. The IRAP technique is an efficient high-throughput genetic marker-generating method.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
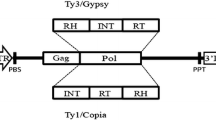
The long terminal repeat (LTR) retrotransposons (RLX) [1, 2] are a large class of transposable elements that propagate in the genome by a “copy-and-paste” mechanism that is essentially identical to the intracellular phase of retrovirus replication [1,2,3,4,5], in contrast to the “cut-and-paste” mobility of DNA transposons. The RLX lifecycle involves transcription of an integrated copy, reverse transcription of the transcript into cDNA, and integration of the new copy. Because the RLX mother copy remains part of the chromosome and the daughter copies integrate at new loci, the precise insertion points for the daughter are unlikely to be identical in lines diverging by descent. Complete understanding of the genome and the relationship between genotype and phenotype requires knowledge of both the role and function of the genes as well as of the repetitive component, particularly regarding RLX dynamics [3]. Most eukaryotic genomes comprise over 70% repetitive DNA, with gene numbers, ranging from 10,000 to 50,000, showing much less variation at the monoploid level [4,5,6,7]. Particularly in higher plants, RLXs compose more than half of the repetitive DNA; they not only facilitate homologous recombination, but also can undergo intra- and inter-RLX recombination that is part of their dynamism [4, 8,9,10]. The RLXs are generally dispersed throughout genome, displaying relatively high structural diversity [11,12,13,14,15]. Retroelements have been suggested as an important creative force in genome evolution, driving processes such as mutation, recombination, genome expansion, and adaptation of an organism to changing environmental conditions [3, 14, 16].
All these properties make RLXs excellent sources of efficient high-throughput genetic markers in eukaryotic, especially plant, genomes [17]. The presence or absence of RLX insertions at particular loci can be surveyed by polymerase chain reaction (PCR)-based genotyping methods [17,18,19,20]. The RLX-based genetic marker techniques rely on PCR amplification between features such as the LTR that are conserved in RLXs, or between these features and other dispersed and conserved motifs in the genome. The methods include retrotransposon-based insertion polymorphism (RBIP) [21], sequence-specific amplified polymorphism (S-SAP) [22], inter-retrotransposon-amplified polymorphism (IRAP) [19], retrotransposon-microsatellite-amplified polymorphism (REMAP), and inter-primer binding site (iPBS) amplification [17,18,19, 23,24,25,26,27].
These retrotransposon-based high-throughput genetic DNA fingerprinting methods are both highly informative and polymorphic, even in areas of chromosomes showing low levels of inter-genic recombination and therefore haplotypes with few genic single-nucleotide polymorphisms (SNPs); RLX markers are consistent with geographical and morphological data. The stability of retrotransposon integration sites and recombination events allows them to be used as molecular genetic markers in genetic map construction [28,29,30,31,32]. Retrotransposon markers have also been widely used to assess genetic diversity in many species [33,34,35,36,37,38,39]. Given that plant retrotransposons are stress-activated [15, 40], their role in generating ecogeographical patterns of genomic diversity is of particular interest. Retrotransposon markers have been applied successfully to the analysis of genetic diversity in various genera and species, such as apple, rice, sunflower, grapevine, flax, and alfalfa [38, 41, 42].
Maize (Zea mays L.) is a very special species among the cereal crops, because of its high phenotypic and genomic diversity [43]. Maize is important worldwide as a food and feed crop, and also as an energy crop, due to its high biomass potential. Moreover, it has long been used as a model organism for plant biology. Maize was the first eukaryote in which transposable elements (TEs) were discovered, during the mid-twentieth century, by the Barbara McClintock, bringing her the Nobel Prize [44]. TEs and, particularly, retrotransposons, comprise most of the maize genome; 95% of maize TEs are RLXs [43, 45, 46]. Through their copy number variation, rearrangements, and polymorphic loci, the TEs contribute most of the genome variation between maize lines, where amplification of a few retrotransposon families is the major cause of “genomic obesity” [47, 48]. High-throughput sequencing methodologies have demonstrated that some families of TEs show considerable transcriptional activity.
In the present study, we developed and applied a high-throughput IRAP technique for five RLX families to detect genetic polymorphisms among maize germplasm. These families (Opie, Ji, Cinful, Huck, and Grande) comprise a large fraction of the maize genome [49,50,51,52,53,54], up to 25% of the total. The main goal was to find efficient and high-throughput retrotransposon markers for diversity analyses and to assess the polymorphism of these markers among maize genotypes originating from different ecogeographical origins. The IRAP genetic markers developed were used to compare the genetic variability among maize cultivars and breeding lines differing in ecogeographical origin to detect correlations between phenotypic characters and retrotransposon markers.
Materials and methods
Plant material and DNA extraction
Grains of maize lines and hybrids were kindly provided by the Maize Research Section, Agricultural Research Center (ARC) and U.S. Department of Agriculture (USDA). The names of these hybrids are listed in Table 1 and Supplemental Data 1. Further data on the genotypes can be found on the National Germplasm Resources Laboratory homepage (https://npgsweb.ars-grin.gov/gringlobal/taxonomygenus.aspx?id=13020).
DNA was isolated from leaves of 10-day-old seedlings of the 16 hybrids using the cetyltrimethylammonium bromide (CTAB) extraction protocol with modifications (CTAB solution: 2% CTAB, 1.5 M sodium chloride (NaCl), 20 mM ethylenediaminetetraacetic acid trisodium salt (Na3EDTA), and 0.1 M (N-[2-hydroxyethyl]piperazine-Nʹ-[2-ethanesulfonic acid]) (HEPES), pH 5.3), as described (https://primerdigital.com/dna.html), with RNase A treatment. A detailed protocol for DNA isolation was deposited at protocols.io [55]. The DNA samples were diluted in 1 × TE buffer. The DNA quality was checked electrophoretically, as well as spectrophotometrically with a Nanodrop apparatus (Thermo Fisher Scientific Inc., Waltham, MA, USA).
TE sequence sources and PCR primer design
Thirty LTR primers were designed, based on the most abundant RLX groups in maize (Cinful1 (AC231746), Huck1 (AC230001), Ji (DQ002406), Opie (AY664413), Grande (AY664416.1:70909-83340), and Tekay (AF050455)). The RLX sequences were obtained from the TRansposable Elements Platform (TREP) database (https://botserv2.uzh.ch/kelldata/trep-db/), and analysis of homologous sequences was performed on the output of the National Center for Biotechnology Information (NCBI) search results. For a given family of retrotransposons, their LTRs showed sequence variability, but certain regions were relatively conserved. For each family, the sequence accessions were aligned and conservation assessed with the multiple alignment procedure of MULTALIN [56]. The conserved segments of the LTR of the retrotransposons were used for the design of PCR primers, which was carried out with the program FastPCR (https://primerdigital.com/fastpcr.html) [57,58,59]. Several inverted primers at both ends of the LTRs of each retrotransposon to compare the efficiency and reproducibility of amplification were designed. The sequences of the primers are shown in Table 2. None of the primers chosen formed self-dimers, and all showed high PCR efficiency for IRAP fingerprinting. The chosen primers matched the motifs sufficiently conserved in the retrotransposons to allow amplification of the great majority of targets in the genome.
PCR analysis
IRAP analysis was conducted according to Kalendar and Schulman [19], using 30 primers from the LTRs of the five RLXs. The PCRs were performed in 25-µL reaction mixtures containing 25 ng genomic DNA, 1 × DreamTaq buffer, 200 mM deoxyribonucleotide triphosphate (dNTP), 400 nM primer and 1 U DreamTaq DNA Polymerase (Thermo Fisher Scientific). The amplifications were performed in the MasterCycler Gradient (Eppendorf AG, Hamburg, Germany). The PCR reaction program consisted of 1 cycle at 95 °C, 2 min; 30 cycles of 95 °C for 15 s, 60 °C for 20 s, 72 °C for 40 s; and a final elongation at 72 °C for 2 min.
The PCR products were separated by electrophoresis at 70 V for 8–10 h in a 1.4% agarose gel (Wide Range; SERVA Electrophoresis GmbH, Heidelberg, Germany) with 1 × Tris-HEPES-EDTA (THE) electrophoresis buffer [18]. The Thermo Scientific GeneRuler DNA Ladder Mix, 100–10,000 base pairs (bp), #SM0332, was used as a standard. The gels were stained with ethidium bromide (EtBr) and scanned, using an FLA-5100 imaging system (FUJi Photo Film GmbH; now FUJIFILM Europe GmbH, Heidelberg, Germany) with a resolution of 50 µm.
Data analysis
From the IRAP fingerprint profiles, all clear bands were scored at each band position for each primer in all samples. Polymorphic bands (PBs) of the same size were assumed to represent a single locus. The presence or absence of a fragment of a given length was recorded in binary code. The gels were scored of a total of 677 PBs for the samples. Based on the primary data, the level of genetic diversity as defined by Nei was determined, using Arlequin software [60]. The method applied was based on cluster analysis expressing the relationships of the hybrids examined as a distance percentage in a cluster tree and similarity matrix. The data were analyzed, using NTSYSpc software (Numerical Taxonomy and Multivariate Analysis System) version 2.11 (https://www.exetersoftware.com/cat/ntsyspc/ntsyspc.html).
The primary genetic data were bootstrapped with SEQBOOT (https://csbf.stanford.edu/phylip/seqboot.html), after which the pairwise genetic distances were calculated, using the Genetic Distance Matrix Program (GENDIST) (https://www.bablokb.de/gendist/). Both programs are from PHYLIP (Phylogeny Inference Package) software package (https://evolution.gs.washington.edu/phylip.html). The ability of IRAP markers to reveal genetic relationships among all the maize accessions was evaluated phylogenetically by neighbor-joining (NJ), for which an algorithm was constructed, using PAUP software [61]. Support for the tree was determined by performing 1000 bootstrap operations on the dataset generated by distance analysis. To determine the partitioning of the IRAP genetic variation into inter- and intrapopulation variance components, analysis of molecular variance (AMOVA) was conducted with the program Genetic Analysis in Excel (GenAlex) 6.5 [62].
Summary statistics related to the number of bands generated by each genotype (NTI) and for each group only, including the number of PBs, percentage of polymorphic loci (PPL%), number of private bands (NPB), Shannon’s Information Index (I), genetic differentiation index (PhiPT) among populations, Nei’s genetic distance (D), and Nei’s genetic identity (IN), were calculated using GenAlex 6.5. Genetic distance, using minimum Jaccard coefficients, was calculated with Factor Analysis of Mixed Data (FAMD) 1.31. A dendrogram for the studied genotypes was constructed, based on the maximum likelihood method [63], using MEGA X software [64].
Results and discussion
In silico PCR analysis of the maize genome
We performed in silico IRAP analysis, using FastPCR software for the maize (B73 RefGen_v4) (https://www.ncbi.nlm.nih.gov/genome/12) and sorghum (Sorghum bicolor (L.) Moench) (Sorghum_bicolor_NCBIv3) (https://www.ncbi.nlm.nih.gov/genome/108) genomes, using a single LTR primer corresponding to a sequence highly conserved in the RLXs examined. For in silico IRAP analysis, we applied the default options, since the length of potential PCR products varied from 50 to 3000 bp and allowed a single mismatch within the 3′-termini of the LTR primer. The results of the in silico IRAP analysis are represented in Table 3. As expected, no amplicons were predicted for the Sorghum bicolor genome, due to high divergence of the RLX sequences in the maize genome from those in Sorghum bicolor.
The number of in silico IRAP amplicons perfectly correlated with the results in the wet-lab IRAP experiments. The LTR primers that produced many in silico IRAP amplicons (over 50 per maize genome) also showed a strong background of multiple and overlapping IRAP amplicons in the experiments. For example, LTR primers 4309, 4310, 4311, and 4312 for the Ji RLX family produced the highest number of IRAP amplicons in the in silico analysis. As in the PCR experiments, we observed a strong background over the entire range of PCR fragments. For this reason, the IRAP amplicons with very high numbers in the PCR experiments could not be adequately resolved on an agarose gel, and these primers were not used further to analyze maize lines. LTR primer 4331 from Cinful1 yielded over 389 IRAP amplicons in in silico analysis of the B73 line; in the PCR experiment, it also resulted in a fairly strong background over the entire range of PCR fragments. Similarly, LTR primer 4326 from Tekay produced over 150 IRAP amplicons in in silico analysis, while LTR primers 4317, 4318, and 4319 from the Opie RLX generated 92, 81, and 167 IRAP amplicons, respectively. Another important result from the in silico IRAP analysis is that it did not matter in which direction the primers were oriented at each terminus of the LTR; in either case, similar results were obtained. The number of IRAP amplicons was dependent only on the copy number of a particular RLX and how conserved the LTR region was for which the primers were designed.
Diversity assay among Egyptian maize hybrids
The maize genome is composed of a diverse group of RLXs that are major sources of genetic variations. The selection of effective LTR primers must consider the abundance and distribution of the RLX family. In this study, the LTR primers used were designed for five different LTR retrotransposon families belonging to the high-copy classes gypsy and copia. LTR primers are usually designed to complement areas as close as possible to the 5′ or 3′ ends of the LTR, which is the most conservative part, and to minimize the amplification of long LTR fragments. The effectiveness of IRAP amplification was directly dependents not only on the total number of copies of the element, but also on the degree of LTR regional conservation. Screening for single primers resulted in the selection of 30 LTR primers for IRAP (Fig. 1). All selected LTR primers yielded 20–60 scorable bands and showed high-quality reproducible DNA fingerprints.

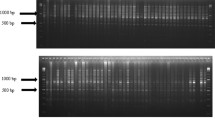
Agarose gel electrophoresis patterns of IRAP amplicons obtained with primers 4306 (Huck), 4317 (Opie), 4309 (Ji), and 4312 (Ji). Maize genotype numbers (H1–H16) reported in Supporting Information 1. M—Thermo Scientific GeneRuler DNA Ladder Mix (100–10,000 bp)
Overall, 677 amplicons were scored as effective, using all LTR primers from the five RLX families, of which 392 were polymorphic, with a mean polymorphism of 58%. A total of 40 unique IRAP amplicons were generated, including 22 positive and 18 negative unique bands, using 20 primers. Primers 4319 (Opie) and 4324 (Grande) produced the highest number of unique bands (7 and 6, respectively). The Grande RLX showed the highest number of unique bands (13), while the Huck LTR primers produced the smallest number (3). These results were expected and corresponded to the result of in silico IRAP analysis. The maize genome is relatively large (2182.61 Mb), and consists of about 50% RLX sequences. The size of the genome, with the main part comprising RLXs, increased the efficiency of the RLX-based methods and resulted in a high percentage of polymorphism, using a single primer. Due to the ability of retrotransposons to integrate into a multitude of loci in the genome, they constituted informative molecular markers for the plant species. The pattern obtained was related to the copy number and the size of the RLX family. The PCR products and the polymorphism resulted from the amplification of hundreds to many thousands of target sites in the genome. These polymorphisms functioned as means of identification, in detecting genetic erosion, and in revealing genetic relationships.
The LTR primers revealed different levels of polymorphism among the maize lines examined. Tables 4 and 5 showed that the LTR primers applied in 16 maize hybrids produced 677 bands; 392 of these were PBs and 285 were monomorphic bands. LTR primer 4304 (Huck) produced the highest number of bands (58) and primer 4325 (Tekay) the lowest number (19). The highest percentage of PBs was produced by primer 4310 (93%) for the Ji RLX and the lowest percentage by Opie LTR primer 4318 (26%). The number of polymorphic amplicons per primer ranged from 8 with 4318 (Opie) to 33 with 4306 (Huck). On average, the number of amplicons per primer throughout the 16 genotypes was 34 and for polymorphic amplicons were 20%. Various levels of polymorphism among primers were detected that ranged from 93% for primer 4310 (Ji) to 26% for primer 4318 (Opie) and were also recorded for each TE family. The primers based on the Grande elements showed the highest percentage of polymorphism (75%), compared with those based on the Ji, Huck, Opie, and Tekay elements, which showed total polymorphism of 55%, 53%, 48%, and 63%, respectively.
IRAP markers based on the Ji family
Five LTR primers for the Ji RLX were tested in 16 maize hybrids, four of which (4309–4312) revealed variability among maize hybrids (Table 4). The bands ranged from 100 to 2000 bp. The total number of bands generated from the Ji element-based primers was 149; 82 were PBs with a mean polymorphism of 55%. Unique bands, either positive or negative (present or absent), characterized the maize hybrids. Five positive unique bands of 680, 2550, 1120, 410, and 500 bp characterized hybrids H-10, H-7, and H-16, whereas only one negative band of 298 bp, resulting from primer 4309, characterized H-11. Each band distinguished its respective hybrid and could be used as a fingerprint.
IRAP markers based on the Huck family
Five LTR primers for the Huck RLX were tested, four of which (4303–4307) revealed variability among the maize hybrids (Table 4). A total of 181 number of bands were generated by the Huck-based primers, and ranged from 160 to 5100 bp; 96 PBs and 85 monomorphic bands were generated, with a mean of 53% polymorphic. The Huck-based LTR primers characterized H-15 and H-11 with two unique bands of 1301 and 1600 bp, respectively, while a single unique negative band of 2400 bp was generated from primer 4303 and characterized H-5 (Table 5). These bands could be used to fingerprint their respective genotypes.
IRAP markers based on the Opie family
Seven LTR primers for the Opie RLX were tested in maize hybrids, of which primers 4315–4319 were suitable for scoring amplicons. A total of 126 bands were evaluated, of which 61 were PBs with a mean polymorphism of 48% (Table 4). The Opie LTR primers detected five positive unique bands with molecular sizes of 560, 720, 730, 1,031, and 2050 bp for hybrids H-16, H-7, H-14, H-16, and H-13, respectively. LTR primer 4317 detected one unique negative band of 720 bp for hybrid H-9, whereas primer 4319 distinguished H-13 with five unique negative bands with molecular sizes of 1080, 1090, 1400, 1510, and 1800 bp.
IRAP markers based on the Grande element
Five LTR primers for the Grande RLX were tested in the maize hybrids, four of which (4320–4324) enabled molecular genetic evaluation. In all, 112 bands were detected, using Grande LTR primers, of which 84 were PBs with a mean polymorphism of 75%; the bands ranged from 250 to 3100 bp (Table 4). Six unique positive bands with molecular sizes of 705, 1200, (1400, 650), (1020, 1650), and 1031 bp characterized H-14, H-7, H-16, H-7, and H-8, respectively. In contrast, six unique negative bands characterized H-15, H-11, H-10, and H-16 with molecular sizes of 490 (610, 805), 250, and 795 bp, respectively (Table 5).
IRAP markers based on the Tekay element
Six LTR primers for the Tekay RLX were tested in the 16 hybrids, four of which gave rise to suitable amplification for scoring (4325–4328). A total of 109 bands were generated from the four primers against the 16 maize hybrids, and ranged from 400 to 4100 bp. The number of PBs was 69, with a mean polymorphism percentage of 63% (Table 4). Three positive unique bands of 2900, 560, and 750 bp characterized H-5, H-16, and H-6, respectively, while four unique negative bands were detected with molecular sizes of 1040, 1590, and (1180, 2400) that characterized H-5, H-16, and H-1, respectively (Table 5).
Identifying maize hybrids with unique IRAP markers
Althogether (Table 5) 40 unique IRAP markers were generated from the 20 primers used, including 22 positive and 18 negative unique bands. Primers 4319 and 4324 produced the highest number of unique bands (7 and 6 bands, respectively). In contrast, the Grande RLX showed the highest number of unique bands (13), while the Huck RLX showed the smallest number of bands (3). The Ji, Opie, and Tekay RLXs showed 6, 11, and 7 unique bands, respectively. Table 5 shows that primer 4324 clearly separated five hybrid lines and LTR primer 4322 distinguished three hybrids, while LTR primers 4309, 4310, 4303, 4315, 4317, and 4327 distinguished only two hybrid lines. LTR primers 4312, 4304, 4319, 4320, 4323, 4325, 4326, and 4328 characterized one hybrid each, while primers 4311, 4306, 4307, and 4318 could not separate any lines (Table 5).
The IRAP analysis used in this study succeeded in demonstrating positive and negative unique markers that aided in genotype discrimination. In all, 16 out of the 20 primers used revealed 40 unique IRAP markers, of which four (4311, 4306, 4307, 4318) did not produce any unique bands. The results revealed that some IRAP bands represented markers that are restricted to some hybrids. No unique bands were detected in hybrids H-2, H-3, H-4, and H-12, while other hybrid lines (H-7 and H-16) were identified by several primers. These unique IRAP bands could have several potential uses for the study of maize genetic resources and for determining intellectual property rights for maize varieties.
Genetic relationships among maize hybrids
Understanding the relationships among genotypes within and between species has valuable applications in crop improvement programs. For this task, we selected two standard maize hybrids (A619 x A632 and B73 x Mo17) and their parental inbred lines (A619, A632, B73, Mo17) to evaluate the effectiveness of the IRAP method. Single-seed-derived DNA samples exclusively were used in this analysis. The bands shared between a hybrid and its parent inbred lines (Fig. 2) are clearly visible and also reflect by their decreased brightness the allelic dosage in the hybrid compared with the parent. For example, primer 4317 (Opie) yielded about 45 bands that could be detected well for all these maize lines, of which 25 were polymorphic (45%). Similar for primer 4320 (Grande), about 32 bands were well detected for all these maize lines, of which 16 were polymorphic (50%).

Agarose gel electrophoresis patterns of IRAP amplicons obtained with primers 4317 (Opie) and 4320 (Grande) for individual seeds maize parental inbred lines (A619, A632, B73, Mo17) and their hybrids (A619 x A632 and B73 x Mo17). Maize parental inbred lines and their hybrids: 1—A632 (PI587140); 2—hybrid A619 x A632 (Ames23710); 3—A619 (PI587139); 4—Mo17 (PI558532); 5—hybrid B73 x Mo17 (Ames19097); 6—B73 (PI550473). M—Thermo Scientific GeneRuler DNA Ladder Mix (100–10,000 bp)
The IRAP banding profiles, which displayed from 21 to 95% polymorphism, were used to identify genetic similarity in the tested maize hybrid lines (Table 5). The highest similarity value (95%) was observed between the two white triple-cross hybrids (H-11 and H-12), which possess a common ancestor and share two parents, as seen in their pedigree (Table 1). In contrast, the lowest genetic similarity value (21%) was detected between the single-cross hybrid (H-4) and the triple-cross white maize (H-11), indicating that these two hybrids were the most divergent genotypes. This dissimilarity can be attributed to the two genotypes inheriting their genetic makeup from different ancestors (Table 1). Genotypes that have low genetic similarity are of great interest for maize breeders. Weising et al. [65] mentioned that it is mandatory that genetically divergent parents be chosen that exhibit sufficient numbers of polymorphisms, but are not so distant as to cause sterility of the progeny. Estimation of genetic similarity based on molecular data is dependent on several factors, such as the number of markers analyzed, their distribution throughout the genome, and the quality of marker scoring. It is difficult to compare genetic distance between different studies, due to the difference in materials, number of genotypes analyzed, the number of alleles detected per marker, and the genetic diversity of the markers.
The genetic similarity coefficient determined for the maize hybrids was employed to develop a dendrogram based on IRAP data, as shown in Fig. 3. Cluster analysis resolved the 16 maize hybrids into two main clusters (A and B). The first cluster (A) was divided into two subclusters (C and D). The first subcluster (C) contained hybrid H-6 as a separate group, while the other subcluster (D) contained two groups; the first of which included H-4 and H-5, while the other contained H-9 (white single-cross) as a separate group. All yellow single-cross maize hybrids were clearly grouped in the main cluster A. In contrast, cluster B was divided into two subclusters (E and F). Subcluster E contained all white triple-cross maize hybrids (H-11, H-12, H-10, H-13, H-14, and H-15), whereas the yellow triple-cross (H-16) was separated into a different group. Subcluster F contained all white single-cross maize hybrids in a separate group, while H-7 branched out into a different group.

Dendrogram of maize genotypes generated by the IRAP primers
Conclusion
Here, we developed and applied a high-throughput IRAP technique for five LTR retrotransposon families to detect genetic polymorphisms among maize germplasm. These RLX families included Opie, Ji, Cinful, Huck, and Grande, which together comprise a large fraction of the maize genome. The RLX polymorphism captures the record of integration events, which are driven by retrotransposon activation and replication, that have been fixed in the germ line and inherited, and their subsequent fate in plant populations. The main goal of this study was to find efficient and high-throughput LTR retrotransposon markers for diversity analyses and to assess the polymorphism of these markers among maize genotypes originating from differing ecogeographical origins.
We successfully characterized the maize genotypes in worldwide and Egyptian collections, using high-throughput IRAP fingerprinting DNA markers. The DNA analysis of the lines of maize germplasm showed that even single LTR primers can be successfully used in the assessment of genetic differences at the line level and display several advantages, such as robustness, informativeness, and efficiency in breeding selection. We demonstrated here that the IRAP marker system provides a useful and simple electrophoretic technique for studying genetic diversity in maize, as they have in other plant species. The LTR primers used yielded multilocus fingerprints, displaying sufficiently high levels of polymorphism to differentiate between maize accessions and grouping them according to their cross level and kernel color. The markers are informative, reliable, and inexpensive for maize breeders and researchers. The number of differences between maize lines was sufficient to easily identify them as separate genotypes, correlated with their phenotypic differences.
Abbreviations
- IRAP:
-
Inter-retrotransposon-amplified polymorphism
- TE:
-
Transposable element
- LTR:
-
Long terminal repeat
- RLX:
-
LTR retrotransposon
References
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982. https://doi.org/10.1038/nrg2165
Schulman AH (2013) Retrotransposon replication in plants. Curr Opin Virol 3:604–614. https://doi.org/10.1016/j.coviro.2013.08.009
Belyayev A, Kalendar R, Brodsky L, Nevo E, Schulman AH, Raskina O (2010) Transposable elements in a marginal plant population: temporal fluctuations provide new insights into genome evolution of wild diploid wheat. Mob DNA. https://doi.org/10.1186/1759-8753-1-6
Sanchez DH, Gaubert H, Drost HG, Zabet NR, Paszkowski J (2017) High-frequency recombination between members of an LTR retrotransposon family during transposition bursts. Nat Commun 8:1283. https://doi.org/10.1038/s41467-017-01374-x
Arkhipova IR, Yushenova IA (2019) Giant transposons in eukaryotes: is bigger better? Genome Biol Evol 11:906–918. https://doi.org/10.1093/gbe/evz041
Wolfgruber TK, Nakashima MM, Schneider KL, Sharma A, Xie Z, Albert PS, Xu R, Bilinski P, Dawe RK, Ross-Ibarra J, Birchler JA, Presting GG (2016) High quality maize centromere 10 sequence reveals evidence of frequent recombination events. Front Plant Sci 7:308. https://doi.org/10.3389/fpls.2016.00308
Michael TP (2014) Plant genome size variation: bloating and purging DNA. Brief Funct Genomics 13:308–317. https://doi.org/10.1093/bfgp/elu005
Vicient CM, Casacuberta JM (2017) Impact of transposable elements on polyploid plant genomes. Ann Bot 120:195–207. https://doi.org/10.1093/aob/mcx078
Underwood CJ, Henderson IR, Martienssen RA (2017) Genetic and epigenetic variation of transposable elements in Arabidopsis. Curr Opin Plant Biol 36:135–141. https://doi.org/10.1016/j.pbi.2017.03.002
Shang Y, Yang F, Schulman AH, Zhu J, Jia Y, Wang J, Zhang XQ, Jia Q, Hua W, Yang J, Li C (2017) Gene deletion in barley mediated by LTR-retrotransposon BARE. Sci Rep 7:43766. https://doi.org/10.1038/srep43766
Kalendar R, Vicient CM, Peleg O, Anamthawat-Jonsson K, Bolshoy A, Schulman AH (2004) Large retrotransposon derivatives: abundant, conserved but nonautonomous retroelements of barley and related genomes. Genetics 166:1437–1450. https://doi.org/10.1534/genetics.166.3.1437
Kalendar R, Tanskanen J, Chang W, Antonius K, Sela H, Peleg O, Schulman AH (2008) Cassandra retrotransposons carry independently transcribed 5S RNA. Proc Natl Acad Sci USA 105:5833–5838. https://doi.org/10.1073/pnas.0709698105
Mascagni F, Giordani T, Ceccarelli M, Cavallini A, Natali L (2017) Genome-wide analysis of LTR-retrotransposon diversity and its impact on the evolution of the genus Helianthus (L). BMC Genomics 18:634. https://doi.org/10.1186/s12864-017-4050-6
Vicient CM, Jaaskelainen MJ, Kalendar R, Schulman AH (2001) Active retrotransposons are a common feature of grass genomes. Plant Physiol 125:1283–1292. https://doi.org/10.1104/pp.125.3.1283
Ramallo E, Kalendar R, Schulman AH, Martínez-Izquierdo JA (2008) Reme1, a Copia retrotransposon in melon, is transcriptionally induced by UV light. Plant Mol Biol 66:137–150. https://doi.org/10.1007/s11103-007-9258-4
Goodier JL (2016) Restricting retrotransposons: a review. Mob DNA 7:16. https://doi.org/10.1186/s13100-016-0070-z
Kalendar R, Amenov A, Daniyarov A (2019) Use of retrotransposon-derived genetic markers to analyse genomic variability in plants. Funct Plant Biol 46:15–29. https://doi.org/10.1071/fp18098
Kalendar R, Schulman AH (2014) Transposon-based tagging: IRAP, REMAP, and iPBS. Methods Mol Biol 1115:233–255. https://doi.org/10.1007/978-1-62703-767-9_12
Kalendar R, Schulman AH (2006) IRAP and REMAP for retrotransposon-based genotyping and fingerprinting. Nat Protoc 1:2478–2484. https://doi.org/10.1038/nprot.2006.377
Baumel A, Ainouche M, Kalendar R, Schulman AH (2002) Retrotransposons and genomic stability in populations of the young allopolyploid species Spartina anglica CE Hubbard (Poaceae). Mol Biol Evol 19:1218–1227
Flavell AJ, Knox MR, Pearce SR, Ellis THN (1998) Retrotransposon-based insertion polymorphisms (RBIP) for high throughput marker analysis. Plant J 16:643–650
Waugh R, McLean K, Flavell AJ, Pearce SR, Kumar A, Thomas BB, Powell W (1997) Genetic distribution of Bare-1-like retrotransposable elements in the barley genome revealed by sequence-specific amplification polymorphisms (S-SAP). Mol Gen Genet 253:687–694
Kalendar R, Flavell AJ, Ellis THN, Sjakste T, Moisy C, Schulman AH (2011) Analysis of plant diversity with retrotransposon-based molecular markers. Heredity 106:520–530. https://doi.org/10.1038/hdy.2010.93
Kalendar RN, Aizharkyn KS, Khapilina ON, Amenov AA, Tagimanova DS (2017) Plant diversity and transcriptional variability assessed by retrotransposon-based molecular markers. Vavilovskii Zhurnal Genetiki i Selektsii 21:128–134. https://doi.org/10.18699/vj17.231
Kalendar R, Antonius K, Smykal P, Schulman AH (2010) iPBS: a universal method for DNA fingerprinting and retrotransposon isolation. Theor Appl Genet 121:1419–1430. https://doi.org/10.1007/s00122-010-1398-2
Kalendar R, Grob T, Regina M, Suoniemi A, Schulman A (1999) IRAP and REMAP: two new retrotransposon-based DNA fingerprinting techniques. Theor Appl Genet 98:704–711. https://doi.org/10.1007/s001220051124
Leigh F, Kalendar R, Lea V, Lee D, Donini P, Schulman AH (2003) Comparison of the utility of barley retrotransposon families for genetic analysis by molecular marker techniques. Mol Genet Genomics 269:464–474. https://doi.org/10.1007/s00438-003-0850-2
Chang RY, O'Donoughue LS, Bureau TE (2001) Inter-MITE polymorphisms (IMP): a high throughput transposon-based genome mapping and fingerprinting approach. Theor Appl Genet 102:773–781. https://doi.org/10.1007/s001220051709
Boyko E, Kalendar R, Korzun V, Fellers J, Korol A, Schulman AH, Gill BS (2002) A high-density cytogenetic map of the Aegilops tauschii genome incorporating retrotransposons and defense-related genes: Insights into cereal chromosome structure and function. Plant Mol Biol 48:767–789. https://doi.org/10.1023/a:1014831511810
Tanhuanpaa P, Kalendar R, Schulman AH, Kiviharju E (2007) A major gene for grain cadmium accumulation in oat (Avena sativa L.). Genome 50:588–594. https://doi.org/10.1139/G07-036
Tanhuanpaa P, Kalendar R, Laurila J, Schulman AH, Manninen O, Kiviharju E (2006) Generation of SNP markers for short straw in oat (Avena sativa L.). Genome 49:282–287. https://doi.org/10.1139/g05-100
Manninen OM, Jalli M, Kalendar R, Schulman A, Afanasenko O, Robinson J (2006) Mapping of major spot-type and net-type netblotch resistance genes in the Ethiopian barley line Cl 9819. Genome 49:1564–1571. https://doi.org/10.1139/G06-119
Milovanov A, Zvyagin A, Daniyarov A, Kalendar R, Troshin L (2019) Genetic analysis of the grapevine genotypes of the Russian Vitis ampelographic collection using iPBS markers. Genetica 147:91–101. https://doi.org/10.1007/s10709-019-00055-5
Doungous O, Kalendar R, Filippova N, Ngane BK (2019) Utility of iPBS retrotransposons markers for molecular characterization of African Gnetum species. Plant Biosyst 154:1–10. https://doi.org/10.1080/11263504.2019.1651782
Vuorinen A, Kalendar R, Fahima T, Korpelainen H, Nevo E, Schulman A (2018) Retrotransposon-based genetic diversity assessment in wild emmer wheat (Triticum turgidum ssp. dicoccoides). Agronomy 8:107. https://doi.org/10.3390/agronomy8070107
Abdollahi Mandoulakani B, Yaniv E, Kalendar R, Raats D, Bariana HS, Bihamta MR, Schulman AH (2015) Development of IRAP- and REMAP-derived SCAR markers for marker-assisted selection of the stripe rust resistance gene Yr15 derived from wild emmer wheat. Theor Appl Genet 128:211–219. https://doi.org/10.1007/s00122-014-2422-8
Doungous O, Kalendar R, Adiobo A, Schulman AH (2015) Retrotransposon molecular markers resolve cocoyam (Xanthosoma sagittifolium) and taro (Colocasia esculenta) by type and variety. Euphytica 206:541–554. https://doi.org/10.1007/s10681-015-1537-6
Vukich M, Schulman AH, Giordani T, Natali L, Kalendar R, Cavallini A (2009) Genetic variability in sunflower (Helianthus annuus L.) and in the Helianthus genus as assessed by retrotransposon-based molecular markers. Theor Appl Genet 119:1027–1038. https://doi.org/10.1007/s00122-009-1106-2
Hosid E, Brodsky L, Kalendar R, Raskina O, Belyayev A (2012) Diversity of Long Terminal repeat retrotransposon genome distribution in natural populations of the wild diploid wheat Aegilops speltoides. Genetics 190:263–U412. https://doi.org/10.1534/genetics.111.134643
Grandbastien MA, Audeon C, Bonnivard E, Casacuberta JM, Chalhoub B, Costa AP, Le QH, Melayah D, Petit M, Poncet C, Tam SM, Van Sluys MA, Mhiri C (2005) Stress activation and genomic impact of Tnt1 retrotransposons in Solanaceae. Cytogenet Genome Res 110:229–241. https://doi.org/10.1159/000084957
Teo CH, Tan SH, Ho CL, Faridah QZ, Othman YR, Heslop-Harrison JS, Kalendar R, Schulman AH (2005) Genome constitution and classification using retrotransposon-based markers in the orphan crop banana. J Plant Biol 48:96–105. https://doi.org/10.1007/BF03030568
Smykal P, Bacova-Kerteszova N, Kalendar R, Corander J, Schulman AH, Pavelek M (2011) Genetic diversity of cultivated flax (Linum usitatissimum L.) germplasm assessed by retrotransposon-based markers. Theor Appl Genet 122:1385–1397. https://doi.org/10.1007/s00122-011-1539-2
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, Minx P, Reily AD, Courtney L, Kruchowski SS, Tomlinson C, Strong C, Delehaunty K, Fronick C, Courtney B, Rock SM, Belter E, Du F, Kim K, Abbott RM, Cotton M, Levy A, Marchetto P, Ochoa K, Jackson SM, Gillam B, Chen W, Yan L, Higginbotham J, Cardenas M, Waligorski J, Applebaum E, Phelps L, Falcone J, Kanchi K, Thane T, Scimone A, Thane N, Henke J, Wang T, Ruppert J, Shah N, Rotter K, Hodges J, Ingenthron E, Cordes M, Kohlberg S, Sgro J, Delgado B, Mead K, Chinwalla A, Leonard S, Crouse K, Collura K, Kudrna D, Currie J, He R, Angelova A, Rajasekar S, Mueller T, Lomeli R, Scara G, Ko A, Delaney K, Wissotski M, Lopez G, Campos D, Braidotti M, Ashley E, Golser W, Kim H, Lee S, Lin J, Dujmic Z, Kim W, Talag J, Zuccolo A, Fan C, Sebastian A, Kramer M, Spiegel L, Nascimento L, Zutavern T, Miller B, Ambroise C, Muller S, Spooner W, Narechania A, Ren L, Wei S, Kumari S, Faga B, Levy MJ, McMahan L, Van Buren P, Vaughn MW, Ying K, Yeh CT, Emrich SJ, Jia Y, Kalyanaraman A, Hsia AP, Barbazuk WB, Baucom RS, Brutnell TP, Carpita NC, Chaparro C, Chia JM, Deragon JM, Estill JC, Fu Y, Jeddeloh JA, Han Y, Lee H, Li P, Lisch DR, Liu S, Liu Z, Nagel DH, McCann MC, SanMiguel P, Myers AM, Nettleton D, Nguyen J, Penning BW, Ponnala L, Schneider KL, Schwartz DC, Sharma A, Soderlund C, Springer NM, Sun Q, Wang H, Waterman M, Westerman R, Wolfgruber TK, Yang L, Yu Y, Zhang L, Zhou S, Zhu Q, Bennetzen JL, Dawe RK, Jiang J, Jiang N, Presting GG, Wessler SR, Aluru S, Martienssen RA, Clifton SW, McCombie WR, Wing RA, Wilson RK (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326:1112–1115. https://doi.org/10.1126/science.1178534
Döring H-P, Starlinger P (1984) Barbara McClintock's controlling elements: now at the DNA level. Cell 39:253–259
SanMiguel P, Tikhonov A, Jin YK, Motchoulskaia N, Zakharov D, Melake-Berhan A, Springer PS, Edwards KJ, Lee M, Avramova Z, Bennetzen JL (1996) Nested retrotransposons in the intergenic regions of the maize genome. Science 274:765–768
Forestan C, Farinati S, Aiese Cigliano R, Lunardon A, Sanseverino W, Varotto S (2017) Maize RNA PolIV affects the expression of genes with nearby TE insertions and has a genome-wide repressive impact on transcription. BMC Plant Biol 17:161. https://doi.org/10.1186/s12870-017-1108-1
Diez CM, Meca E, Tenaillon MI, Gaut BS (2014) Three groups of transposable elements with contrasting copy number dynamics and host responses in the maize (Zea mays ssp. mays) genome. PLoS Genet 10:1004298. https://doi.org/10.1371/journal.pgen.1004298
Estep MC, DeBarry JD, Bennetzen JL (2013) The dynamics of LTR retrotransposon accumulation across 25 million years of panicoid grass evolution. Heredity (Edinb) 110(2):194–204. https://doi.org/10.1038/hdy.2012.99
Chuck G, Robbins T, Nijjar C, Ralston E, Courtney-Gutterson N, Dooner HK (1993) Tagging and cloning of a petunia flower color gene with the maize transposable element activator. Plant Cell 5:371–378. https://doi.org/10.1105/tpc.5.4.371
García-Martínez J, Martínez-Izquierdo JA (2003) Study on the evolution of the Grande retrotransposon in the Zea genus. Mol Biol Evol 20:831–841. https://doi.org/10.1093/molbev/msg095
Dotto BR, Carvalho EL, Silva AF, Duarte Silva LF, Pinto PM, Ortiz MF, Wallau GL (2015) HTT-DB: horizontally transferred transposable elements database. Bioinformatics 31(17):2915–2917. https://doi.org/10.1093/bioinformatics/btv281
Pietzenuk B, Markus C, Gaubert H, Bagwan N, Merotto A, Bucher E, Pecinka A (2016) Recurrent evolution of heat-responsiveness in Brassicaceae COPIA elements. Genome Biol 17:209. https://doi.org/10.1186/s13059-016-1072-3
Aledo R, Raz R, Monfort A, Vincient CM, Puigdomèbnech P, Martínez-Izquierdo JA (1995) Chromosomal localization and characterization of a family of long interspersed repetitive DNA elements from the genus Zea. Theor Appl Genet 90:1094–1100. https://doi.org/10.1007/BF00222927
Kuhn BC, Lopez-Ribera I, da Silva Machado MDF, Vicient CM (2014) Genetic diversity of maize germplasm assessed by retrotransposon-based markers. Electrophoresis 35(12–13):1921–1927. https://doi.org/10.1002/elps.201400038
Kalendar R (2019) Universal DNA isolation protocol. protocolsio. https://doi.org/10.17504/protocols.io.z2jf8cn
Corpet F (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16:10881–10890
Kalendar R, Khassenov B, Ramanculov E, Samuilova O, Ivanov KI (2017) FastPCR: an in silico tool for fast primer and probe design and advanced sequence analysis. Genomics 109(3–4):312–319. https://doi.org/10.1016/j.ygeno.2017.05.005
Kalendar R, Tselykh TV, Khassenov B, Ramanculov EM (2017) Introduction on using the FastPCR Software and the related Java web tools for PCR and oligonucleotide assembly and analysis. Methods Mol Biol 1620:33–64. https://doi.org/10.1007/978-1-4939-7060-5_2
Kalendar R, Muterko A, Shamekova M, Zhambakin K (2017) In Silico PCR tools for a fast primer, probe, and advanced searching. Methods Mol Biol 1620:1–31. https://doi.org/10.1007/978-1-4939-7060-5_1
Excoffier L, Laval G, Schneider S (2007) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Swofford DL (1998) PAUP* Phylogenetic analysis using parsimony (*and other methods), 4th edn. Sinauer Associates, Sunderland
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Hollingsworth M (2006) DNA fingerprinting in plants: principles, methods, and applications. Ann Bot 97(3):476–477. https://doi.org/10.1093/aob/mcj057
Acknowledgements
Open access funding provided by University of Helsinki including Helsinki University Central Hospital. This work was supported under "Enhancing Development of Water Use Efficient Crops & Production Methods to Dry and Saline Conditions" (Grant No. EGY/IKY MTT/FCRI 14208006) funded by the Ministry of Foreign Affairs of Finland, and partially by the Science Committee of the Ministry of Education and Science of the Republic of Kazakhstan in the framework of program funding for research (Grant Nos. BR05236334, BR06349586). The authors wish to thank Dr. James Thompson (University of Helsinki) for outstanding editing and proofreading of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest. All authors read and approved the final manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary file1 (XLSX 23 kb)
Supporting Information 1 List of the maize lines used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ghonaim, M., Kalendar, R., Barakat, H. et al. High-throughput retrotransposon-based genetic diversity of maize germplasm assessment and analysis. Mol Biol Rep 47, 1589–1603 (2020). https://doi.org/10.1007/s11033-020-05246-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05246-4