
Abstract
Background
Saraca asoca (Roxb.) De Wilde is a valuable tree used in traditional medicine against a variety of ailments. Almost all parts of the tree are used for various commercial herbal preparations. Due to overexploitation, the species is rapidly disappearing from Western Ghats where it is native and grew extensively until recently. Conservation of this important medicinal plant is therefore an urgent need. To plan effective conservation strategies, a scientific assessment of the genetic diversity and distribution is needed.
Methods
Random Amplified Polymorphic DNA fingerprinting was employed and the population genetic structure of seven wild and three cultivated populations totalling 160 individuals of S. asoca in the Western Ghats region of Karnataka, Maharashtra and Goa for analysis.
Results
Variation of 89% was observed within populations while 11% was observed among the populations of S. asoca. Gene flow (Nm) of 2.01 was observed, and 0.19 was the degree of genetic differentiation recorded. Unweighted Pair Group Method with Arithmetic average (UPGMA) cluster analysis generated a dendrogram that showed an admixture of all genotypes but with two major clusters, which was also supported by a STRUCTURE-based Bayesian model. One wild population was well, but inexplicably, differentiated from the rest.
Conclusions
The study shows that there is still considerable genetic diversity existing in natural populations of S. asoca, suggesting good natural cross-pollination, giving encouraging indications that the gene pool is under no immediate threat. Any conservation strategy should utilise the observed genetic variation in the choice of planting stock for programmes of conservation, propagation and reforestation.
Similar content being viewed by others

Background
Trees constitute the backbone of natural forest habitats. However, most forests are stressed from overexploitation due to increased demand for timber and other natural products coupled with anthropogenic activities such as forest clearance for farming. Excessive commercial exploitation rapidly depletes populations, resulting in loss of valuable biodiversity, ecological imbalance, and often outright species loss. This process occurs much faster with trees than with most, smaller plants because regeneration of trees is slower. Some species have medicinal value but unethical collection practices (including collection of immature/vital parts) can cause extensive damage to trees and further reduce reproduction/regeneration. Lack of sufficient knowledge and training (not only among collectors but also among forest officials) and unethical use of various plant resources in a booming and under-regulated crude drug market make the situation even more serious. Therefore, protection of not only the number but also the diversity of trees in a forest is a priority.
Saraca asoca (Roxb.) De Wilde, (Family: Caesalpiniaceae), a slow-growing climax forest tree species, is immensely valued for its medicinal properties. Saraca asoca is being depleted rapidly from its natural habitat in the Western Ghats range of India and has been ‘red listed’ and categorised as ‘vulnerable’ by the International Union for Conservation of Nature (IUCN) (Senapati et al. 2012; Mohan et al. 2017). Saraca asoca, commonly known as the ‘Ashoka’ is distributed in evergreen forests of India up to an elevation of about 750 m and is also cultivated (Warrier and Nambiar 1993). Its bark, leaves, flowers and seeds have well-proven medicinal properties, in both modern and traditional systems. The bark of this tree is one of the most important and widely used ingredients of several commercial Ayurvedic preparations like ‘Ashokrishtam’ and ‘Ashokaghritham’, which are prescribed as pharmaceuticals for several gynaecological disorders (Tiwari 1979; Hegde et al. 2017a). The bark is also used as an astringent, anthelminthic, styptic, stomachic, antipyretic, and demulcent and to treat menorrhagia, bleeding haemorrhoids, haemorrhagic dysentery and disorders associated with the menstrual cycle (Bhandary et al. 1995; Singh et al. 2015). As a consequence of its medicinal importance, the plant is widely exploited in the legitimate and black-market herbal drug trades (Hegde et al. 2017b). With increasing demand, there has been an increase in harvesting from the wild (which is the main source of raw material), which has led to the tree becoming endangered in a very short space of time (Hegde et al. 2018b).
A clear understanding of population structure in a forest is essential to understand genetic diversity and to regularly assess the impact of protection strategies (Deshpande et al. 2001; Ezzat et al. 2016; Das et al. 2017). Wild germplasm characterisation, particularly in an endangered plant species, provides baseline data to develop management strategies not only for protection and conservation but also for sustainable utilisation (Dawson and Powell 1999; Hegde et al. 2018a). Conservation and preservation of rare plants require accurate assessment of their genetic diversity through objective indices and parameters (Nongrum et al. 2012; Iranjo et al. 2016). For such purposes, molecular genetic markers (which are heritable characteristics of a plant) are used for identification and assessment of population genetic parameters (Petit et al. 1998; Joshi et al. 2004). Therefore, the use of molecular genetic tools is often crucial for both management and conservation programmes. These markers also find applications in plant genetics and breeding programmes (Hilfiker et al. 2004; Arslam and Okumus 2006; Li et al. 2007). Among the plethora of molecular fingerprinting techniques available, one of the simplest is Random Amplified Polymorphic DNA (RAPD) (Williams et al. 1990; Ginwal et al. 2011; Mohan et al. 2017). This technique serves the purpose of differentiating species, types and varieties. Most importantly, it provides much valuable information on the population genetic parameters required by foresters and conservationists, despite certain drawbacks like problems with production of reproducible fingerprints. This technique is widely used to study plant populations at species and cultivar levels because it is simple to perform, rapid, cost-effective and does not require prior knowledge of DNA sequences (Lamine and Mliki 2015; Mucciarelli et al. 2015; Pendkar et al. 2016).
In the present study, the RAPD fingerprinting technique was employed to understand the population genetic structure of Indian S. asoca populations in the Western Ghats region of Karnataka, Maharashtra and Goa, with the aim of helping stakeholders design strategies for its effective conservation.
Methods
Plant material
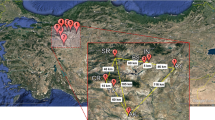
Fresh leaves were collected from 160 S. asoca trees in seven different forest areas as well as from three different cultivated sites (≥ 20 m radius separation within each population) located in central and north-central Western Ghats (Table 1; Fig. 1). All individual samples were assigned a laboratory identification code and kept separately at − 80 °C for genetic analyses. The sample size varied among sites depending on the number of S. asoca trees available in each forest area, and the voucher sample deposited at ICMR – NITM herbarium (voucher sample number: RMRC 1156).

Map of Western Ghats of India showing S. asoca collection sites. Sampled locations are labelled according to population code (see Table 1)
DNA extraction
Total genomic DNA was extracted from the collected leaf samples of all the individual plants using the modified CTAB (cetyltrimethylammonium bromide/hexadecyltrimethylammonium bromide) method (Doyle and Doyle 1990; Richards et al. 1994; Padmalatha and Prasad 2006). The quality and quantity of isolated DNA were determined using Nanodrop Spectrophotometer (JH BIO, USA) at 260/280 nm as well as visually by horizontal electrophoresis on 1% w/v agarose gels stained with GelRed (Biotium Inc., USA). Each sample was diluted to 30 ng/μL with TE buffer (10 mM Tris HCI, pH 8.0 and 0.1 mM EDTA, pH 8.0) and stored at − 20 °C.
RAPD fingerprinting assay
Amplification of RAPD fragments was performed in 25 μL volumes containing 30 ng genomic DNA, 10 pmol primer (Merck, India; Table 2), 200 μM of each dNTP and 1.5 units of Taq DNA polymerase (Merck, India) in PCR buffer supplied (TrisHCl, pH 9.0; 15 mM MgCl2). The amplification reaction consisted of an initial denaturation at 94 °C for 3 min followed by 30 cycles of denaturation at 94 °C for 45 s, annealing at 37 °C for 60 s, and extension at 72 °C for 1 min with final extension at 72 °C for 7 min.
RAPD amplifications were performed in iCycler (BioRad Inc., USA) thermal cycler. The amplified PCR products were visualised through gel electrophoresis (BioRad Inc., USA) on 2% w/v agarose gels in 1X TAE buffer (40 mmol/L Tris, 20 mmol/L acetic acid, 1 mmol/L EDTA) using GelRed (Biotium Inc., USA) as the staining dye, for 2.5 h at a constant voltage of 100 V. The agarose gels were visualised and documented under UV light using a gel documentation system with Alpha Imager software (Alpha Innotech Corporation, USA). Amplification with each primer was repeated at least twice to confirm the reproducibility of the bands and only the consistent and reproducible bands were scored for data analysis. A negative control reaction containing ultra-pure water in place of genomic DNA was included for each PCR run.
Data analysis
Each band obtained, regardless of primer, was treated as an independent locus. Bands were scored only if they were prominently stained and reproduced each time PCR was carried out. Each individual plant was scored for the presence or absence of a particular amplified band. Presence was marked as 1 (one) and absence as 0 (zero). The genetic diversity within and among the populations was estimated in terms of HT (total heterozygosity), h (Nei’s genetic diversity; Nei 1973), HS (heterozygosity within population), DST (heterozygosity among populations; Nei 1973), GST (genetic differentiation) and Nm (number of migrants per generation or gene flow; Slatkin and Barton 1989), using POPGENE ver. 1.31 (Yeh et al. 1997). Analysis of molecular variance (AMOVA) was carried out to partition the variance among geographic regions, among different populations and among individuals within the populations using GenAlEx 6.4. (Peakall and Smouse 2012). The Polymorphism Information Content (PIC) was calculated using the formula 1 − p2 − q2, where q is the frequency of no bands and p is the frequency of present bands (Rajwade et al. 2010; Bhagwat et al. 2014). The primer index (SPI) was then calculated using loci amplified by the same primer with summing up the PIC values (Bhagwat et al. 2014). The Marker Index (MI) for each primer was calculated as a product of PIC and effective multiplex ratio (EMR) (Zhang et al. 2018); MI = EMR × PIC (Pecina-Quintero et al. 2011). The genetic distance data obtained from the analysis of molecular variance was used to perform principal coordinate analysis (PCoA) (Peakall and Smouse 2012; Abraham et al. 2018), which showed the percentage variation explained by the first three axes and provided graphical representation of the genetic relationships among populations. The PCoA was executed with the pairwise genetic distance matrix in GenAlEx 6.5 (Peakall and Smouse 2006; Peakall and Smouse 2012).
The population structure of S. asoca was also determined using a Bayesian approach by employing the software STRUCTURE v. 2.3.4 (Pritchard et al. 2000). This approach assumes a model with admixture, and the number of populations (K) was estimated from 1 to 10 for each value with 5000 burn-in iterations, followed by 50,000 Markov Chain Monte Carlo (MCMC) replications after burn-in. The population structure analysis was run with five independent iterations. The ΔK of the Evanno method was performed to estimate the value of K that best fitted the data (Evanno et al. 2005), using the Structure Harvester v. 0.6.94 (Earl and vonHoldt 2012). To visualise the genetic structure, Discriminant Analysis of Principal Components (DAPC), which does not rely on a particular model, was performed using Adegenet version 2.1.1 and Poppr R Packages version 2.8.0 (Jombart 2008). Two DAPC runs were performed initially to understand the optimal K according to the Bayesian information criterion (BIC). Based on the BIC value, this defined the clusters, which was further applied to the dataset. With the dataset of 10 populations, an analysis was carried out including all individuals in one run, while another analysis was carried out, where the three cultivated small populations (SAM, SAR, SADU) were excluded.
A dendrogram was constructed by using Unweighted Pair Group Method with Arithmetic average (UPGMA) cluster analysis based on the matrix of Nei’s genetic distances with TFPGA (Tools For Population Genetic Analyses) ver.1.3 (Miller 1997) to show a representation of genetic relationships among the ten S. asoca populations and also done only for wild populations.
Results
Polymorphism
RAPD fingerprinting assays were carried out with 10 random primers (Table 2). Eight primers showed consistent and clear bands and were selected for the study. In all, 722 bands were produced by RAPD fingerprinting assays from a total of 160 samples. A total of 75 amplification products where all the bands were polymorphic were scored. An average number of around nine loci was scored in RAPD, with a maximum of 11 obtained with RPi - C6 and RPi - C8. The minimum amplification product was eight, with primers RPi - C3 and RPi - C9 (Table 2). For the eight primers used for the study, the PIC values ranged from 0.368 to 0.429. RPi - C2 showed minimum PIC while RPi - C10 showed maximum, with an average value of 0.40. The MI showed a maximum of 2.80 (RPi - C8), with a minimum of 1.81 (RPi - C2) and an average value of 2.21 (Table 2).
Genetic diversity
Genetic variations in the 10 populations were estimated using various methods of analysis (Table 3). The mean observed number of alleles (Na) ranged from 1.42 (SADU) to 2.0 (SAD), with the average being 1.85 (standard deviation (SD) ± 0.245). The mean effective number of alleles (Ne) per locus ranged from 1.34 (SADU) to 1.71 (SAD) with average of 1.60 (SD ± 0.302). The mean Nei’s gene diversity (h) ranged from 0.18 (SADU) to 0.40 (SAD) with average of 0.34 (SD ± 0.151), and the mean Shannon’s Information index (I) was 0.27 (SADU) to 0.58 (SAD) with average of 0.49 (SD ± 0.214). The expected heterozygosity under Hardy–Weinberg equilibrium averaged 0.42 (SD ± 0.006) and 0.34 (SD ± 0.006) across all populations (HT) and pooled within other populations (HS) respectively. The mean genetic differentiation (GST) was 0.19 and showed an estimated gene flow (Nm) of 2.01 (Table 4). AMOVA showed 89% variance within the populations and 11% among the populations. In general, the results revealed that genetic variability within the populations was quite high. The remaining genetic variation was due to differences among populations of S. asoca.
The relationships among all the populations of S. asoca were also depicted by cluster analysis through Nei’s genetic distance based on UPGMA dendrogram using TFPGA software (Fig. 2a, b). The populations clustered in two distinct main groups and the distribution of populations were not according to the geographical areas of the collection. The first group contained all the populations (SANAG, SAM, SAB, SAGJ, SAR, SAD, SABG, SAT, SAKT), except SADU which showed distinct separation into a second group. Within the first group, the SAD, SAM and SAR populations clustered separately and three minor clusters of other populations (SAKT, SAT; SAGJ, SAB; SABG, SANAG) appeared (Fig. 2a). The populations showed admixture, and no particular assortment was noticed. Interestingly, the populations of SAM, SAD and SAR appeared distinct and SADU separated as another group (Fig. 2a). In the second run, only wild populations were considered in which the SAD population stood out from other three groups (viz, SAT, SAKT, SAB, SAGJ and SANAG, SABG) (Fig. 2b). Graphical representation of the relationships between populations obtained by PCoA analysis is shown in Fig. 3. The first axis accounted for 48.3%, second 36.8% and third 14.8% of the total variation observed. While the first principal coordinate distinguished SAD and SAR, the second coordinate showed that the populations were broadly scattered and often overlapped (Fig. 3).

UPGMA dendrograms based on Nei’s genetic distances showing clustering patterns of a ten S. asoca accessions and b seven wild S. asoca accessions

Principal coordinate analysis (PCoA) plot of RAPD profiles of the ten S. asoca populations
The Bayesian clustering was carried out by employing STRUCTURE version 2.3.1 (Pritchard et al. 2000) and setting the number of probable clusters from 1 to 10. The results of this analysis indicated that K = 2 clusters signify the most informative illustration representing maximum log likelihood (Fig. 4). Numerous subpopulations were allocated at the higher values of K with specific clusters (Additional file 1: Figure S1). It was found that the individuals from the populations SAM, SAD, SAR and SADU were distinctly grouped in the cluster analysis, forming two main clusters whereas the populations SADU and SAD separated out from the others. The population structure analysis performed by DAPC involved combining all individuals (both cultivated and wild) in a single dataset without any a prior group assignment. The results from this analysis identified six groups although members of at least two groups overlapped with each other while a group of from the SAD population was positioned away from the other groups (Fig. 5a). When only wild populations (i.e. SAD, SANAG, SABG, SAT, SAKT, SAB, SAGJ) were analysed, five clusters were found with two of them comprising overlapping populations (Fig. 5b). The SAD population formed a distinct group with no overlap with any other population (Fig. 5a, b).

a Maximum likelihoods based on the Bayesian model for alternative values of K and b population structure analysis of the ten populations of S. asoca based on the Bayesian approach (K = 2) (population labelling, 1-27:SAD; 28-43:SANAG; 44-59:SABG; 60-72:SAT; 73-82:SAKT; 83-85:SADU; 86-91:SAM; 92-125:SAB; 126-157:SAGJ; 158-160:SAR)

Scatterplot showing S. asoca DAPC analysis for the first two principal components. Inset shows discriminant analysis eigenvalues in histogram. a All 160 accessions. b Seven wild populations
Discussion
Using AMOVA, most of the variance was found between individuals within the populations (89%) while a small amount of variation was found among populations (11%). However, Senapati et al. (2012) reported 36.12% variation among populations and the remaining 63.88% resided among individuals within population of S. asoca collected from Orissa state of India. In a recent study using fluorescent-labelled RAPD markers, six accessions of S. asoca revealed high degree (92.7%) of polymorphism (Mohan et al. 2017). In another study, AMOVA revealed a 43% variation within the populations and 57% variation among the populations of S. asoca using inter simple sequence repeat (ISSR) markers (Hegde et al. 2018a). The results of the PCoA undertaken in the current study showed that the three populations SAD, SAR and SADU formed a distinct group and were genetically closer than the other populations. Within this group, SADU and SAR were cultivated populations and, therefore, their divergence is perhaps due to the difference in the cultivar used. In contrast, individuals within the SAM population were genetically diverse, which indicates that genetic segregation occurred even within cultivated populations. Among the natural populations, out of seven, six (SANAG, SAGJ, SABG, SAB, SAT, SAKT) grouped together albeit with overlaps, whereas one natural population (SAD) separated from the rest indicating perhaps long-standing genetic differentiation. However, it is to be noted that the size of some populations was very small due to presence of very few trees at those sites, so any conclusions must be drawn with caution. The variation observed by the PCoA, i.e. 48.3%, 36.8% and 14.8% by the first, second and third axis, respectively, revealed a similar pattern of variation as indicated by the cluster analysis.
Cluster analysis revealed that the cultivated populations (SAR, SADU, SAM) formed distinct clusters compared with the natural populations. Even among the natural populations, one (SAD) formed a separate cluster depicting more genetic distance from all the other (six) natural populations. The cultivated group (SADU) and the wild group (SAD) ordinate separately and deviated from the rest for no clear reason. Bayesian clustering showing admixture of genotypes indicated two major clusters when K = 2, which were strongly supported by the UPGMA dendrogram (Fig. 4). The results were in agreement with those obtained by Senapati et al. (2012), where fragmentation, isolation and particularly anthropogenic activities have been suggested to have caused rapid extirpation of several species and may lead to low genetic diversity in S. asoca, if such processes continue unabated.
Population genetic parameters, such as Nei’s genetic diversity, Shannon’s diversity index and AMOVA, detected a similar genetic variation pattern for the ten populations of S. asoca. The effective number of alleles was 0.7, and a relatively high number of polymorphic loci indicate the high genetic differentiation using RAPD primers. This result was also supported by AMOVA where a high level of intra-specific variation was observed within the individual populations. Despite limited numbers, six accessions of S. asoca revealed high polymorphism using fluorescent-labelled RAPD primers in earlier studies (Mohan et al. 2017). Estimated gene flow (Nm) was higher (2.0) than that reported earlier (0.513) (Senapati et al. 2012), which might be due to the differences in the study populations and geographic locations. Use of DAPC (Jombart et al. 2010) is an approach that does not rely on a particular population genetics model and can also identify and describe clusters of genetically related individuals. Genetic structure analysed using DAPC summarises the situation, maximising the variability among groups and minimising the within-group variance (Jombart et al. 2010). Analysis of the entire molecular binary dataset (without prior information on group assignments) indicated that the populations split into six overlapped clusters along with one differentiated cluster corresponded to most individuals of SAD. Further, analysis of only wild populations also revealed SAD to be clearly distinguished from other clusters. The reason(s) behind the distinctiveness of the SAD cluster was not determined within the scope of this study, although it corroborated the results of UPGMA analysis of wild populations.
It is well recognised that genetic variation is essential for a species to evolve and adapt to changing environmental conditions. The sustained ability of forest trees to provide goods and services thus depend on the maintenance and management of forest genetic resources (Katwal et al. 2003). The high genetic variation of 89% found between individuals within the natural populations, the small degree of variation (11%) found among populations and the lack of significant variation among regions are ecologically very encouraging signs that indicate genetic mixing is occurring naturally via pollination. Earlier studies on the breeding system and floral biology of this species indicate the strong predominance of outcrossing, which helps to maintain the heterozygosity in the various populations (Smitha and Thondaiman 2016). Therefore, the gene pool is not likely to be threatened immediately. However, lack of tolerance of S. asoca seeds to drying to reduce seed moisture content (recalcitrant nature of seeds; Smitha and Das 2016) and poor seed viability (Smitha and Thondaiman 2016) might be reasons for the ‘vulnerable’ status of S. asoca. Nonetheless, it has been suggested that the difficulty of using seeds to obtain plants might be overcome through micropropagation of selected accessions of S. asoca (Mohan et al. 2017).
In conservation biology, differentiating between closely related species with the use of molecular markers is considered important for biodiversity studies (Agarwal et al. 2008; Sheeja et al. 2013). The null alleles, which are present only in heterozygous genotypes, cannot be detected from dominant RAPD markers, and RAPD variation cannot be identified as adaptive genes (Bekessy et al. 2003). Marker diversity is far from a completely reliable guide to functional genetic diversity but, with limitations of time and expense, diversity of various markers is used as an indirect measure. Therefore, despite known differences in robustness of various rapid fingerprinting techniques, the use of RAPD fingerprinting technique has proven to be a very useful tool for rapid assessment of the genetic diversity, population structure and gene flow. Although the adoption of Next-Generation Sequencing (NGS) holds promise, it is still impractical to sequence all individuals of different populations of any plant species. Therefore, the use of RAPD markers in prediction of genetic variation and in correlation with respective phenotypes is appropriate (Agarwal et al. 2008). Genomic limitations of the RAPD technique are the issue of reproducibility and the fact that it cannot pinpoint the exact genomic sites and extent of polymorphisms. However, the technique served its purpose in the current study.
Conclusions
This study has attempted to generate genetic information on S. asoca, which is a very important tree species widely traded as a crude drug source. This species is rapidly depleting and needs immediate implementation of conservation measures. This study revealed that considerable genetic diversity still exists in natural populations of S. asoca. These results suggest good natural cross-pollination and indicate that the gene pool is under no immediate threat. This study also makes a strong case for further research on understanding of the relationship between the observed variation and the adaptive potential of various tree populations.
Abbreviations
- AMOVA:
-
Analysis of Molecular Variance
- BIC:
-
Bayesian information criterion
- CTAB:
-
Cetyltrimethylammonium bromide/hexadecyltrimethylammonium bromide
- DAPC:
-
Discriminant Analysis of Principal Components
- D ST :
-
Heterozygosity among populations
- EMR:
-
Effective Multiplex Ratio
- G ST :
-
Genetic differentiation
- h :
-
Nei’s genetic diversity
- H S :
-
Heterozygosity within population
- H T :
-
Total heterozygosity
- ISSR:
-
Inter Simple Sequence Repeat
- IUCN:
-
International Union for Conservation of Nature
- MCMC:
-
Markov Chain Monte Carlo
- MI:
-
Marker index
- NGS:
-
Next-Generation Sequencing
- Nm:
-
Gene flow
- PCoA:
-
Principal coordinate analysis
- PIC:
-
Polymorphism Information Content
- RAPD:
-
Random Amplified Polymorphic DNA
- TFPGA:
-
Tools for Population Genetic Analysis
- UPGMA:
-
Unweighted Pair Group Method with Arithmetic average
References
Abraham, E. M., Aftzalanidou, A., Ganopoulos, I., Osathanunkul, M., Xanthopoulou, A., Avramidou, E., Sarrou, E., Aravanopoulos, F., & Madesis, P. (2018). Genetic diversity of Thymus sibthorpii Bentham in mountainous natural grasslands of Northern Greece as related to local factors and plant community structure. Industrial Crops and Products, 111, 651–659.
Agarwal, M., Shrivastava, N., & Padh, H. (2008). Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Reports, 27(4), 617–631.
Arslam, B., & Okumus, A. (2006). Genetic and geographic polymorphism of cultivated tobaccos (Nicotiana tabacum) in Turkey. Russian Journal of Genetics, 42(6), 667–671.
Bekessy, S. A., Ennos, R. A., Burgman, M. A., Newton, A. C., & Ades, P. K. (2003). Neutral DNA markers fail to detect genetic divergence in an ecologically important trait. Biological Conservation, 110(2), 267–275.
Bhagwat, R. M., Banu, S., Dholakia, B. B., Kadoo, N. Y., Lagu, M. D., & Gupta, V. S. (2014). Evaluation of genetic variability in Symplocos laurina Wall. from two biodiversity hotspots of India. Plant Systematics and Evolution, 300(10), 2239–2247.
Bhandary, M. J., Chandrashekar, K. R., & Kaveriappa, K. M. (1995). Medical ethnobotany of the Siddis of Uttara Kannada district, Karnataka, India. Journal of Ethnopharmacology, 47(3), 149–158.
Das, S., Singh, Y. P., Negi, Y. K., & Shrivastav, P. C. (2017). Genetic variability in different growth forms of Dendrocalamus strictus: Deogun revisited. New Zealand Journal of Forestry Science, 47(1), 23.
Dawson, I. K., & Powell, W. (1999). Genetic variation in the Afromontane tree Prunus africana, an endangered medicinal species. Molecular Ecology, 8(1), 151–156.
Deshpande, A. U., Apte, G. S., Bahulikar, R. A., Lagu, M. D., Kulkarni, B. G., Suresh, H. S., Singh, N. P., Rao, M. K. V., Gupta, V. S., Pant, A., & Ranjekar, P. K. (2001). Genetic diversity across natural populations of three montane plant species from the Western Ghats, India revealed by inter simple sequence repeats. Molecular Ecology, 10(10), 2397–2408.
Doyle, J. J., & Doyle, J. L. (1990). Isolation of plant DNA from fresh tissue. Focus, 12, 3–15.
Earl, D. A., & vonHoldt, B. M. (2012). Structure Harvester: a website and program for visualizing structure output and implementing the Evanno method. Conservation Genetics Resources, 4(2), 359–361.
Evanno, G., Regnaut, S., & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology, 14(8), 2611–2620.
Ezzat, S. M., El Sayed, A. M., & Salama, M. M. (2016). Use of Random Amplified Polymorphic DNA (RAPD) technique to study the genetic diversity of eight aloe species. Planta Medica, 82(15), 1381–1386.
Ginwal, H. S., Maurya, S. S., & Chauhan, P. (2011). Genetic diversity and relationship between cultivated clones of Dalbergia sissoo of wide geographical origin using RAPD markers. Journal of Forestry Research, 22(4), 507–517.
Hegde, S., Hegde, H. V., Jalalpure, S. S., Peram, M. R., Pai, S. R., & Roy, S. (2017a). Resolving identification issues of Saraca asoca from its adulterant and commercial samples using phytochemical markers. Pharmacognosy Magazine, 13(Suppl. 2), S266.
Hegde, S., Pai, S. R., Bhagwat, R. M., Saini, A., Rathore, P. K., Jalalpure, S. S., Hegde, H. V., Sugunan, A. P., Gupta, V. S., Kholkute, S. D., & Roy, S. (2018a). Genetic and phytochemical investigations for understanding population variability of the medicinally important tree Saraca asoca to help develop conservation strategies. Phytochemistry, 156, 43–54.
Hegde, S., Pai, S. R., & Roy, S. (2017b). Combination of DNA isolation and RP-HPLC analysis method for bark samples of Saraca asoca and its adulterant. 3Biotech, 7(3), 208.
Hegde, S., Saini, A., Hegde, H. V., Kholkute, S. D., & Roy, S. (2018b). Molecular identification of Saraca asoca from its substituents and adulterants. 3Biotech, 8(3), 161.
Hilfiker, K., Gugerli, F., Schutz, J., Rotach, P., & Holderegger, R. (2004). Low RAPD variation and female-biased sex ratio indicate genetic drift in small populations of the dioecious conifer Taxus baccata in Switzerland. Conservation Genetics, 5(3), 357–365.
Iranjo, P., NabatiAhmadi, D., Sorkheh, K., Memeari, H. R., & Ercisli, S. (2016). Genetic diversity and phylogenetic relationships between and within wild Pistacia species populations and implications for its conservation. Journal of Forestry Research, 27(3), 685–697.
Jombart, T. (2008). Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics, 24(11), 1403–1405.
Jombart, T., Devillard, S., & Balloux, F. (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics, 11(1), 94.
Joshi, K., Chavan, P., Warude, D., & Patwardhan, B. (2004). Molecular markers in herbal drug technology. Current Science, 87: 159–65
Katwal, R. P. S., Srivastava, R. K., Kumar, S., & Jeeva, V. (2003). Status of forest genetic resources conservation and management in India. In Asia Pacific Forest Genetic Resources Programme (APFORGEN). Rome: Forest Resources Development Service Working Paper FGR/65E, Forest Resources Division FAO.
Lamine, M., & Mliki, A. (2015). Elucidating genetic diversity among sour orange rootstocks: A comparative study of the efficiency of RAPD and SSR markers. Applied Biochemistry and Biotechnology, 175(6), 2996–3013.
Li, L., Li, J., Zou, L., Bai, S. Y., Niu, L. M., & Ma, Y. K. (2007). RAPD analysis of genetic diversity of nine strains of Auricularia auricular cultivated in Heilongjiang Province. Journal of Forestry Research, 18(2), 136–138.
Miller, M. P. (1997). Tools for population genetic analysis (TFPGA) Version 1.3, Department of Biological Science, Northern Arizona University, Arizona, USA.
Mohan, C., Reddy, M. S., Naresh, B., Kumar, S. M., Fatima, S., Manzelat, B., & Cherku, P. D. (2017). RAPD studies of Saraca asoca by fluorescent-labeled primers and development of micropropagation protocol for its conservation. International Journal of Applied Agricultural Research, 12(2), 137–151.
Mucciarelli, M., Ferrazzini, D., & Belletti, P. (2015). Genetic variability and population divergence in the rare Fritillaria tubiformis subsp. moggridgeirix (Liliaceae) as revealed by RAPD analysis. PLoS One, 9(7), e101967.
Nei, M. (1973). Analysis of gene diversity in subdivided populations. Proceedings of the National Academy of Sciences of the United States of America, 70(12), 3321–3323.
Nongrum, I., Kumar, S., Kumaria, S., & Tandon, P. (2012). Genetic variation and gene flow estimation of Nepenthes khasiana Hook. F – a threatened insectivorous plant of India as revealed by RAPD markers. Journal of Crop Science and Biotechnology, 15(2), 101–105.
Padmalatha, K., & Prasad, M. N. V. (2006). Optimization of DNA isolation and PCR protocol for RAPD analysis of selected medicinal and aromatic plants of conservation concern from Peninsular India. African Journal of Biotechnology, 5(3), 230–234.
Peakall, R., & Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics, 28, 2537–2539.
Peakall, R. O. D., & Smouse, P. E. (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Resources, 6(1), 288–295.
Pecina-Quintero, V., Anaya-López, J. L., Colmenero, A. Z., García, N. M., Colín, C. A. N., Bonilla, J. L. S., Aguilar-Rangel, M. R., Langarica, H. R. G., & Bustamante, D. J. M. (2011). Molecular characterisation of Jatropha curcas L. genetic resources from Chiapas, México through AFLP markers. Biomass and Bioenergy, 35(5), 1897–1905.
Pendkar, S. K., Hegde, S., Nayak, S. U., Hegde, H., Kholkute, S. D., & Roy, S. (2016). Detection of adulteration by Wedelia calendulacea in Eclipta alba through ISSR and RAPD markers. Planta Medica International Open, 3(02), e43–e46.
Petit, R. J., El Mousadik, A., & Pons, O. (1998). Identifying populations for conservation on the basis of genetic markers. Conservation Biology, 12(4), 844–855.
Pritchard, J. K., Stephens, M., & Donnelly, P. J. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959.
Rajwade, A. V., Arora, R. S., Kadoo, N. Y., Harsulkar, A. M., Ghorpade, P. B., & Gupta, V. S. (2010). Relatedness of Indian flax genotypes (Linum usitatissimum L.): an inter-simple sequence repeat (ISSR) primer assay. Molecular Biotechnology, 45(2), 161–170.
Richards, E., Reichardt, M., & Rogers, S. (1994). Preparation of genomic DNA from plant tissue. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, & K. Struhl (Eds.). Current Protocols in Molecular Biology, 27. New York: Wiley.
Senapati, S. K., Das, G. K., Aparajita, S., & Rout, G. R. (2012). Assessment of genetic variability in the Asoka Tree of India. Biodiversity, 13(1), 16–23.
Sheeja, T. E., Anju, P. R., Shalini, R. S., Siju, S., Dhanya, K., & Krishnamoorthy, B. (2013). RAPD, SCAR and conserved 18S rDNA markers for a red listed and endemic medicinal plant species, Knemaand amanica (Myristicaceae). Physiology and Molecular Biology of Plants, 19(2), 245–250.
Singh, S., Krishna, A. T., Kamalraj, S., Kuriakose, G. C., Valayil, J. M., & Jayabaskaran, C. (2015). Phytomedicinal importance of Saraca asoca (Ashoka): an exciting past, an emerging present and a promising future. Current Science, 109(10), 1790–1801.
Slatkin, M., & Barton, N. H. (1989). A comparison of three indirect methods for estimating average levels of gene flow. Evolution, 43(7), 1349–1368.
Smitha, G. R., & Das, M. (2016). Effect of seed moisture content, temperature and storage period on seed germination of Saraca asoca - an endangered medicinal plant. Medicinal Plants, 8(1), 60–64.
Smitha, G. R., & Thondaiman, V. (2016). Reproductive biology and breeding system of Saraca asoca (Roxb.) De Wilde: a vulnerable medicinal plant. Springer Plus, 5(1), 2025.
Tiwari, S. D. N. (1979). The phytogeography of legumes of Madhya Pradesh (Central India). Dehradun: Bishan Singh, Mahendrapal Singh.
Warrier, P. K., & Nambiar, V. P. K. (1993). Indian medicinal plants: a compendium of 500 species (Vol. 5). Hyderabad: Orient Blackswan.
Williams, J. G., Kubelik, A. R., Livak, K. J., Rafalski, J. A., & Tingey, S. V. (1990). DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Research, 18(22), 6531–6535.
Yeh, F. C., Yang, R. C., Boyle, T. B., Ye, Z. H., & Mao, J. X. (1997). POPGENE, the user-friendly shareware for population genetic analysis. Edmonton: Molecular Biology and Biotechnology Centre, University of Alberta, Canada. https://sites.ualberta.ca/~fyeh/popgene_info.html.
Zhang, Y., Zhang, X., Chen, X., Sun, W., & Li, J. (2018). Genetic diversity and structure of tea plant in Qinba area in China by three types of molecular markers. Hereditas, 155(1), 22. https://doi.org/10.1186/s41065-018-0058-4.
Acknowledgements
The authors are indebted to the members of the Scientific Advisory Committee of ICMR-NITM (Formerly Regional Medical Research Centre), Belagavi, for providing necessary inputs and support. The authors are grateful to Dr. Divakar Mesta for the help in sample collection and Mr. Sanjay Deshpande for the bioinformatics support. AS is grateful to ICMR, New Delhi, for providing financial support (ICMR-Post Doctoral Fellowship).
Funding
Indian Council of Medical Research, New Delhi, India
Availability of data and materials
Please contact the author for data requests.
Author information
Authors and Affiliations
Contributions
AS and SH carried out the molecular studies. SH, AS and SR performed interpretation, analysis of results and drafted the manuscript. SR, HVH and SDK designed the study, participated in the interpretation and correction of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Population structure analysis of the ten populations of S. asoca based on the Bayesian approach (K = 3; K = 4; K = 5; K = 6; K = 7) (Population labelling, 1-27:SAD; 28-43:SANAG; 44-59:SABG; 60-72:SAT; 73-82:SAKT; 83-85:SADU; 86-91:SAM; 92-125:SAB; 126-157:SAGJ; 158-160:SAR). (DOCX 789 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Saini, A., Hegde, S., Hegde, H.V. et al. Assessment of genetic diversity of Saraca asoca (Roxb.) De Wilde: a commercially important, but endangered, forest tree species in Western Ghats, India. N.Z. j. of For. Sci. 48, 17 (2018). https://doi.org/10.1186/s40490-018-0122-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40490-018-0122-x