
Abstract
Triple-negative breast cancer (TNBC) is a subtype of human breast cancer with one of the worst prognoses, with no targeted therapeutic strategies currently available. Regulated cell death (RCD), also known as programmed cell death (PCD), has been widely reported to have numerous links to the progression and therapy of many types of human cancer. Of note, RCD can be divided into numerous different subroutines, including autophagy-dependent cell death, apoptosis, mitotic catastrophe, necroptosis, ferroptosis, pyroptosis and anoikis. More recently, targeting the subroutines of RCD with small-molecule compounds has been emerging as a promising therapeutic strategy, which has rapidly progressed in the treatment of TNBC. Therefore, in this review, we focus on summarizing the molecular mechanisms of the above-mentioned seven major RCD subroutines related to TNBC and the latest progress of small-molecule compounds targeting different RCD subroutines. Moreover, we further discuss the combined strategies of one drug (e.g., narciclasine) or more drugs (e.g., torin-1 combined with chloroquine) to achieve the therapeutic potential on TNBC by regulating RCD subroutines. More importantly, we demonstrate several small-molecule compounds (e.g., ONC201 and NCT03733119) by targeting the subroutines of RCD in TNBC clinical trials. Taken together, these findings will provide a clue on illuminating more actionable low-hanging-fruit druggable targets and candidate small-molecule drugs for potential RCD-related TNBC therapies.
Graphical abstract

Similar content being viewed by others

Introduction
Hitherto, breast cancer has attained the highest cancer incidence in the world and is also the leading cause of cancer-related death among women worldwide. Based upon gene expression profiles, breast cancer can be divided into five clinical types. According to the status of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor 2 (HER2), the subtypes can be defined as normal-like breast cancer, luminal A breast cancer, luminal B breast cancer, HER2-enriched breast cancer and triple-negative/basal-like breast cancer [1]. Triple-negative breast cancer (TNBC) is the breast cancer subtype with the worst prognosis, and it has a strong invasive and metastatic capacity and easily invaded into blood vessels [2], increasing the recurrence rate. Due to the lack of expressions of ER, PR and HER2, endocrine and targeted therapies achieve comparatively poor outcomes. Therapeutic methods for TNBC are much more limited compared with other breast cancers. Based upon the gene expression profiles of TNBC cases in breast cancer datasets, seven TNBC subtypes were identified: basal-like 1 (BL1), basal-like 2 (BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem-like, luminal androgen receptor, and unclassified (UNS) [3]. The tumors of patients with TNBC show a high genetic diversity ranging from highly proliferative tumors to chemotherapy-resistant tumors with low proliferation and luminal characteristics [4]. These data may be useful for biomarker selection, drug discovery and clinical trial design [5], with the aim of matching patients with TNBC of different subtypes with the appropriate targeted therapies.
In the past decade, the Nomenclature of Cell Death Committee (NCCD) has developed the guidelines for defining and interpreting cell death from the perspectives of morphology, biochemistry and function [6,7,8]. The NCCD divides cell death into accidental death (ACD) and regulated cell death (RCD) [8]. ACD is the instantaneous and catastrophic death of cells under the natural conditions, such as physical stressors (e.g., high pressure, temperature or osmotic force), chemical stressors (e.g., extreme pH change) and mechanical stressors (e.g., shear force). RCD depends on specific molecular mechanisms and is subject to regulation. RCD is also called programmed cell death (PCD) during development or tissue renewal [8], as it plays a key role in these processes. RCD is crucial in the response to injury, infection and inflammation, in this case involving an intracellular suicide pathway [9]. According to its different mechanisms [10], RCD can be divided into three major categories: autophagy, apoptosis and other types of RCD involved in signaling pathways, such as mitotic catastrophe, necroptosis, ferroptosis, pyroptosis and anoikis [11]. RCD can be regulated by pharmacological agents and genetic programs [12] and plays a vital role in tissue homeostasis. Aberrant regulation of this process can be related to a variety of diseases, especially cancer [13, 14].
TNBC has given a rise to the largest proportion in breast cancer-related death, exerting higher recurrence, more aggressive growth and more rapid metastasis. Since TNBC is the absence of hormone receptor and HER2, TNBC patients cannot respond to hormone therapy or any other available targeted agents; therefore, it is imperative to search for innovative therapeutic targets for TNBC. In recent years, a series of small-molecule compounds targeting RCD have made a good progress in the clinical treatment of TNBC. In this review, we summarized the molecular mechanisms of seven major RCD subroutines related to TNBC and the latest progress of small-molecule compounds targeting different RCD subroutines. We also discussed the combined strategies of one drug or more drugs by regulating RCD subroutines in TNBC therapy. Moreover, we demonstrated small-molecule compounds by targeting the subroutines of RCD in TNBC clinical trials.
Targeting apoptotic pathways with small-molecule compounds in TNBC
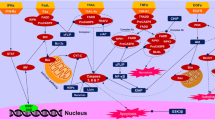
Apoptosis is the main type of programmed cell death (PCD) and has increasingly becoming an essential target for antitumor drug screening [15]. Apoptosis of cancer cells is mainly activated by the following two pathways: extrinsic pathway and intrinsic pathway [16]. The extrinsic pathway is initiated by members of the death receptor family. The binding between death receptors and their ligands leads to the formation of the death-inducing signaling complex (DISC), which eventually activates caspase-8 [17]. The intrinsic (also called mitochondrial) pathway is activated by deoxyribonucleic acid (DNA) damage, growth factor withdrawal, change of osmotic pressure and cytotoxic stimulation of anticancer agents and then regulates the expression of pro-apoptotic and anti-apoptotic proteins, thereby activating caspase-9 and caspase-3 (Fig. 1) [18, 19].

Core apoptotic signaling pathways in triple negative breast cancer (TNBC). In the extrinsic pathway, the interaction between death receptors and their ligands activates caspase 8, which then activates caspase 3, and eventually lead to apoptosis. Death receptors belong to tumor necrosis factor (TNF) receptors superfamily, which is essential for the transmission of intracellular and extracellular signals. The death receptors include FAS, TNFR1, DR4, and DR5. Death receptors bind to the corresponding proapoptotic ligand like FASL, TNF-α, and TNF-related apoptosis-inducing ligand (TRAIL), to trigger extrinsic apoptosis. In mitochondrial dependent apoptotic pathway, when DNA damage occurs, the pro-apoptotic proteins of the Bcl-2 family (such as Bax and Bak) will be upregulated and activated. Anti-apoptotic proteins (such as Bcl-2 and Bcl-xl) inhibits the action of Bax and Bak. A series of apoptogenic factors will be released into the cytosol, including cytochrome c, apaf-1, and procaspase 9, which forms a complex called apoptosome. This complex can activate caspase 9 followed by the transformation of pro-caspase 3 to caspase 3 and thus trigger apoptosis. When cells receive extracellular stimulation, they transmit the signal to inhibitor of kappa-B kinase (IKK), and inhibitor of IκB is separated from the trimer complex formed with NF-κB. The released NF-κB rapidly enters the nucleus and binds to specific sequences on deoxyribonucleic acid (DNA) to participate in physiological processes such as anti-apoptotic effects. Besides, apoptosis can also be induced by regulating the expression of p53 protein. Abbreviations: Apaf-1: Apoptotic protease activating factor 1; Bcl-2: B-cell lymphoma 2; Bcl-xl: B-cell lymphoma-extra large; DR4/5: Death receptor 4/5; FADD: Fas/fas associated via death domain; IκBα: Nuclear factor kappa-B inhibitor α; NF-κB: Nuclear factor kappa-B; RIP: Receptor-interacting protein; STAT3: Signal transducer and activator of transcription 3; TNF-α: Tumor necrosis factor-α; TNFR1: tumor necrosis factor receptor1; TRADD: TNFRSF1A associated via death domain; TRAF2: TNF receptor-associated factor 2; TRAIL: TNF-related apoptosis-inducing ligand
In the extrinsic pathway, the interaction between death receptors and their ligands activates caspase-8, which then activates caspase-3 and eventually leads to apoptosis. Death receptors belong to tumor necrosis factor (TNF) receptors superfamily, which is essential for the transmission of intracellular and extracellular signals. The death receptors include FAS, TNFR1, DR4 and DR5. Death receptors bind to the corresponding proapoptotic ligand like FASL, TNF-α and TNF-related apoptosis-inducing ligand (TRAIL), to trigger extrinsic apoptosis. In mitochondrial-dependent apoptotic pathway, when DNA damage occurs, the pro-apoptotic proteins of the Bcl-2 family (such as Bax and Bak) will be upregulated and activated. Anti-apoptotic proteins (such as Bcl-2 and Bcl-xl) inhibit the action of Bax and Bak. A series of apoptogenic factors will be released into the cytosol, including cytochrome c, apaf-1 and procaspase 9, which forms a complex called apoptosome. This complex can activate caspase-9 followed by the transformation of pro-caspase-3 to caspase-3 and thus trigger apoptosis. When cells receive extracellular stimulation, they transmit the signal to inhibitor of kappa-B kinase (IKK), and inhibitor of IκB is separated from the trimer complex formed with NF-κB. The released NF-κB rapidly enters the nucleus and binds to specific sequences on deoxyribonucleic acid (DNA) to participate in physiological processes such as anti-apoptotic effects. Besides, apoptosis can also be induced by regulating the expression of p53 protein.
Small-molecule drugs play an important role in the treatment of cancer, which combine with specific target molecules in cells to exert their specific functions, such as inducing apoptosis of cancer cells [20, 21]. Targeting the key regulators of apoptosis with the goal of inducing apoptosis in cancer cells was one of the most attractive strategies for drug discovery and development, as well as the hot area of oncology research [22]. Small-molecule drugs had been the focus of research due to their strong specificity, remarkable effect and less damage to normal cells [23, 24]. Small-molecule compounds summarized in this paper include not only drugs that have entered clinical trials, but also synthetic compounds, natural compounds and semisynthetic derivatives of natural compounds. It was estimated that about 60% of marketed drugs were natural compounds or semisynthetic derivatives of natural compounds [25, 26]. Next, we will describe the antitumor activity of small-molecule compounds in TNBC in terms of apoptosis-related signaling pathways and targets, and the main pathways and targets of apoptosis include tumor necrosis factor receptors (TNFR), B-cell lymphoma 2 (Bcl-2) family, apoptotic protease activating factor 1 (Apaf-1) and cytochrome c (Cyt-C), nuclear factor kappa-B (NF-κB) pathway, signal transducer and activator of transcription-3 (STAT3) pathway and p53.
Targeting TNF-related ligands and their receptors
The TNFR family has many members, including Fas, TNFR1, death receptor (DR) 4, and DR5, which bound to their ligands FasL, TNF-α and TNF-related apoptosis-inducing ligand (TRAIL), respectively, triggering a signaling cascade that resulted in the recruitment and activation of caspase-8, ultimately inducing cell apoptosis and causing programmed cell death [27,28,29]. The TNFR had been studied for many years as a potential target for tumor therapy, among which TNF-α and TRAIL had been the most studied [30]. Recently, investigations gradually identified the regulatory effect of small-molecule compounds on TNF-related receptors and their ligands, leading to the induction of apoptosis in TNBC.
V-3–17,18-epoxyeicosatetraenoic acid (C20E), a newly synthesized compound, could stimulate TNFR-1/ASK1/JNK signaling to induce apoptosis of MDA-MB-231 cells, which acted at the intracellular domain of TNFR1, and then activated TNFRSF1A associated via death domain (TRADD), TNF receptor-associated factor 2 (TRAF2) and several downstream signal. Besides, the anti-breast cancer activity of C20E in vivo also relied on its modulatory effect on TNFR-1/ASK1/JNK signaling and apoptosis induction [31]. In addition, CPT211, a novel camptothecin derivative, was found to suppress the proliferation and induce apoptosis of MDA-MB-231 cells effectively, by activating Fas/fas associated via death domain (FADD)/caspase-8 signaling [32].
TRAIL, a proapoptotic molecule, can selectively induce apoptosis in a variety of human tumor cell lines without affecting normal cells [33]. Most small-molecule compounds could be used in combination with TRAIL to promote apoptosis of TNBC cells. For example, pterostilbene (PTER), a natural analogue of resveratrol, was proved to enhance TRAIL-induced apoptosis via reactive oxygen species (ROS)-mediated C/EBP homologous protein (CHOP) activation, leading to the expression of DR4 and DR5 [34]. Like PTER, doscadenamide A could also synergistically interact with TRAIL that induced exogenous apoptosis, and bound to death receptors to induce apoptosis of TNBC cells [35].
Organometallic complexes exhibit a significant anti-breast cancer activity and induced apoptosis against TNBC cells [36]. For an instance, MnIII complex could enhance the activity of caspase-8 and caspase-9, upregulate the expression of Bax/Bcl-2 ratio and promote the binding of TNF-α to its receptor, indicating a simultaneous activation of both internal and external apoptotic pathways in MDA-MB-231 cells. In addition, MnIII complex combined with Adriamycin could synergistically inhibit the growth of TNBC cells [37]. Likewise, numerous small-molecule compounds could trigger apoptosis of TNBC cell lines by influencing TNF-α-related pathway [38, 39] (Table 1).
Targeting Bcl-2 family
Bcl-2 belongs to a growing family of proteins that regulated a unique programmed cell death, namely apoptosis. The Bcl-2 family proteins were central regulators of cell death, which played an important regulatory role in cell apoptosis [40]. The Bcl-2 family proteins regulated the permeability, stability and integrity of mitochondrial outer membrane (MOM), which was especially vital for the release of cytochromic c and the activation of downstream factors [41]. The Bcl-2 family was mainly divided into pro-apoptotic proteins (e.g., Bax, Bak) and anti-apoptotic proteins (e.g., Bcl-2, B-cell lymphoma-extra-large (Bcl-xl)). The balance of proapoptotic protein and anti-apoptotic protein was the key to determine whether cell apoptosis would occur [42,43,44]. Statistically, Bcl-2 antiapoptotic proteins showed abnormal expression in various malignancy, including 70% of breast cancers, 80% of B-cell lymphomas and other forms of cancer [45]. We conclude that several small molecular compounds can induce TNBC cells apoptosis by modulating the biological functions of Bcl-2 family-related proteins.
Novel spirooxindole is reported to have the anticancer effect of them on TNBC cell lines [46]. In the TNBC MDA-MB-231 cells, spirooxindole 6e, with a half maximal inhibitory concentration (IC50) value of 6.40 μM, was identified as the most potent compound among them. It could induce apoptosis in MDA-MB-231 cells through suppression of Bcl-2 protein, upregulation of Bax protein, as well as the activation of caspase-3 [46]. In addition, the concept of molecular hybridization was used and combined the pharmacodynamic elements of isatin and phthalazine or quinazoline in a chemical framework through a hydrazine linker to synthesize a novel 5-chloro-3–(2–(4–(4-chlorophenyl)phthalazin-1-yl)hydrazono)indolin-2-one (compound 10 g) targeting TNBC [47]. Compound 10 g was the most active hybrid with an IC50 value of 12.0 ± 0.13 μM and induced apoptosis through the enhanced expression of Bax, the reduced expression of Bcl-2 and the activated level of caspase-3,9 [47].
Sophoraflavanone G (SG) could inhibit Bcl-2 and Bcl-xl expressions, significantly stimulate Bax expression and inhibit the mitogen-activated protein kinase (MAPK) pathway, resulting in the induction of apoptosis as well as the inhibition of the migration and invasion [48]. IIamycin E, a natural product extracted from deep sea-derived Streptomyces atratus, could effectively suppress the proliferation and arrest G1/S cell cycle of HCC1937 and MDA-MB-468 cells. Meanwhile, IIamycin E induced apoptosis via downregulating Bcl-2 expression [49]. The combination of bioactive compounds exerted an effectively synergistic anticancer activity. Gallic acid (GA) and curcumin (Cur), as naturally plant derivatives, had been reported to have potently exerted anticancer effects through induction of apoptosis [50]. The combination of GA and Cur demonstrated a more significant promotive effect on Bax expression and caspase-3 cleavage than single compound utilized alone. In the meantime, the combination of GA and Cur could also decrease Bcl-2 expression dramatically. These regulatory activities led to a more essential proapoptotic effect of the co-treatment than these reagents used separately [50].
Natural compound derivatives have gradually been becoming a new source for the discovery of anti-TNBC drug candidates. KYZ3 (7-((4-fluorobenzyl)oxy)-2-methyl-2,3-dihydronaphtho[1,2-b].furan-4,5-dione), a cryptotanshinone derivative, exhibited approximately 22–24-fold higher antitumor activity against the MDA-MB-231 cells than its parent compound cryptotanshinone [51]. KYZ3 inhibited signal transducer and activator of transcription 3 (STAT3) phosphorylation, leading to the inhibition of STAT3-transcriptionally activated oncogenes, including Bcl-2. Also, KYZ3 could increase the level of Bax, ultimately leading to the increase in TNBC apoptotic cells [51]. ((1aR,7aS,10aS,10bS,E)-1a-Methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2,3':9,10].cyclodeca[1,2-b].furan-5-yl)methyl (E)-3-(2,6-dimethoxyphenyl)acrylate (Compound 7d) was a parthenolide derivative, which could inhibit proliferation in TNBC cells via apoptosis induction through increasing the level of Bax and Bcl-2 interacting mediator of cell death (Bim) protein and promoting the cleavage of caspase-9. Moreover, Compound 7d could cause G1 phase arrest [52]. Furthermore, a new derivative of SMBA1 (Bax activators), CYD-4-61, was found to enhance the anti-proliferation activity of TNBC cell lines. CYD-4-61 was also reported to activate Bax protein, promote the release of Cyt-C and boost the cleavage of PARP-1 and caspase-3, thus inducing breast cancer cells apoptosis [53].
Combining some chemotherapeutic drugs with natural compound could not only improve the effectiveness of cancer treatment, but also reduced the toxicity and side effects of the chemotherapeutic drugs [54]. Doxorubicin (Dox) was an anthracycline antibiotic and a broad-spectrum anticancer agent; as a cytotoxic anthracycline antibiotic, it is often used as tumor chemotherapy agent for MOA during inhibition of topoisomerase and DNA replication [55]. Although Dox was effective in the treatment of TNBC, its actual use was limited due to side effects including cardiotoxicity [56]. Combination of Dox and small-molecule compounds to reduce the Dox dose could minimize the toxic and side effects [54, 57]. Arctigenin (ATG) exhibited its own anticancer activity, and when combined with Dox, ATG could enhance the cytotoxic effect of Dox on MDA-MB-231 cells. ATG and Dox co-treatment was induced by downregulating expression of Bcl-xl and Bcl-2, through non-major (or off-target) drug effects by promoting the translocalization of Bax to mitochondria, thereby destroying mitochondrial integrity [58]. In addition, coralline, a heterocyclic analog, combined with paclitaxel (PTX) had a synergistic effect on the inhibition of proliferation and migration of TNBC cells without any toxic effect on normal cells. The co-treatment could promote cell apoptosis by suppressing Bcl-2 and increasing Bax [59]. FZU-0038-056, a tetrahydro-β-carboline (THβC) skeleton derivative, was reported to induce TNBC cell lines apoptosis via enhancing the cleavage of caspase-3 and reducing Bcl-2, X-linked inhibitor of apoptosis protein (XIAP), and myeloid cell leukemia-1 (Mcl-1) proteins. Moreover, when it used together with cisplatin, the antitumor activity could be efficiently strengthened [60]. In addition, the study had shown that BCL2 inhibitor ABT199 is generally considered to be effectively only for Bcl-2-dependent cancers, but when combined with cisplatin, it could inhibit TNBC cells viability with less side effects [61]. Combinational drug therapy is a viable and effective strategy for cancer treatment. Apart from the compounds mentioned above, other small-molecule compounds that induce apoptosis of TNBC cells by regulating Bcl-2 family proteins are also summarized in Table 1 [62,63,64,65,66,67,68,69,70].
Targeting Apaf-1 and Cyt-C
Cyt-C is a type of hemoglobin involved in the mitochondrial electron transport chain, which played an important role in REDOX and energy metabolism [71]. Meanwhile, Cyt-C is a crucial material in mitochondria to start the process of apoptosis. In the presence of deoxyadenosine triphosphate (dATP) and adenosine triphosphate (ATP), Cyt-C is released from mitochondria and bound to Apaf-1 to form a poly-complex, which in turn activated the caspase cascade and thus induced apoptosis [72].
Overexpression of macrophage migration inhibitory factor (MIF) has been an important prognostic factor in breast cancer by regulating tumor initiation, aggressiveness and progression. A recent report showed that MIF was overexpressed in TNBC; CPSI-1306 as a MIF inhibitor was found to decrease TNBC tumor growth and metastasis both in vitro and in vivo. It could increase the ROS level in TNBC cells, promote the release of Cyt-C and apoptosis-inducing factor (AIF) from mitochondria and thus induce cell apoptosis [73, 74]. Moreover, a natural isoquinoline alkaloid, berberine (BBR), was reported to induce caspase-9-dependent apoptosis by triggering the release of Cyt-C from mitochondria and downregulating Bcl-2. It also suppressed the tumor growth in TNBC xenograft mice [75] (Table 1).
Targeting NF-κB pathway
NF-κB is a transcription factor of Rel family proteins, which was widely involved in a variety of cellular activities, such as cell cycle, cell proliferation, apoptosis, migration and invasion [76, 77]. In addition, NF-κB was associated with cancer initiation, metastasis and resistance. Particularly in TNBC, the abnormal activation of NF-κB was more frequent, which influenced the expression of its downstream signaling targets [78]. Therefore, selectively targeting NF-κB and its downstream signaling might be a promising therapeutic approach for the treatment of TNBC.
Budlein A methylacrylate (BAM), as an active compound isolated from Helianthus genus plant, showed selective cytotoxicity against TNBC cell lines without any toxic effect on normal cells [79]. BAM inhibited the activity of inhibitor kappa B kinaseβ (IKKβ) and exportin 1 (XPO-1) and then inhibited the NF-κB pathway, leading to TNBC cell apoptosis. Besides, the findings from the in vivo study suggested that it could decrease tumor growth [79]. Icariin was a prenylated flavonol glycoside, which had potent properties in various types of cancers. Icariin could upregulate silent information regulator 6 (SIRT6) expression to inhibit the activation of NF-κB pathways, thereby triggering apoptosis in TNBC cells [80]. Moreover, it also suppressed the tumor growth and pulmonary metastasis in both MDA-MB-231 and 4T1 mouse model [80]. Similarly, crambescidin 800 (C800), a guanidine alkaloid isolated from sponge, could decrease the phosphorylation of Akt, NF-κB and MAPK, resulting in apoptosis in TNBC cells [81]. A novel derivative of sesquiterpene lactone, ACT001, was found to suppress tumor angiogenesis and the accumulation of myeloid-derived suppressor cells (MDSCs) of 4T1 tumor-bearing mice, by inhibiting the activity of NF-κB pathway and successively inducing apoptosis [82]. Cedarone acetate was a compound obtained by acetylation modification of original cedarone molecule; when compared with the original molecule, it could enhance the cytotoxic activity and induce apoptosis through downregulating the level of NF-κB and matrix metallopeptidase 9 (MMP9) [83].
DOX and PTX were the most widely used chemotherapeutic drugs in the treatment of TNBC [84], but their high-dose use would have inherent drug resistance and serious side effects [54]. NF-κB could improve the resistance of chemotherapeutic drugs to TNBC cells by regulating anti-apoptotic pathways. The combination treatment of ginsenoside panaxatriol (GPT) and PTX could inhibit the tumor cells growth and induce apoptosis of TNBC cells resistant to PTX by suppressing interleukin-1 receptor-associated kinase 1 (IRAK1)-mediated NF-κB and ERK pathways [85]. In addition, GPT could increase the sensitivity of TNBC PTX-resistant cells [85]. Meanwhile, it was reported that ginsenoside Rg3 could promote the cytotoxicity and apoptosis of PTX on TNBC by reducing the expression of NF-κB [86]. Oprozomib and carfilzomib were both proteasome inhibitors, which could sensitize TNBC cells to DOX treatment and induce apoptosis by suppressing the activation of NF-κB and JNK/p38 MAPK phosphorylation [87, 88]. DCC-2036 was reported to exert an inhibitory effect on TNBC cells proliferation, migration and invasion, ultimately inducing apoptosis. It targeted Anexelekto (AXL)/MET to inhibit the PI3K/Akt-NF-κB pathway and epithelial–mesenchymal transition (EMT) [89, 90]. In vivo, DCC-2036 could suppress the growth and metastasis of tumor-burden mice. Besides, when DCC-2036 combined with cisplatin or lapatinib, the co-treatment showed a notable synergistic effect on TNBC [89]. In addition to compounds mentioned above, remaining small-molecule compounds targeting NF-κB to trigger apoptosis on TNBC cells are also collected [91,92,93] (Table 1).
Targeting STAT3 pathway
STAT3, which is an essential intracellular signal transduction protein, can participate in the regulation of cancer cell proliferation, differentiation, apoptosis and invasion by acting on downstream related genes [94, 95]. STAT3 was activated in lung, prostate and breast cancer. STAT3 expression level is significantly higher in TNBC than that in other breast cancers and normal tissues [96, 97]. Several small-molecule compounds could induce cancer cells apoptosis to exhibit anticancer properties by suppressing the STAT3 phosphorylation process, which was considered as one of the crucial targets for TNBC treatment [98].
Sorafenib analogue, SC-60, has been reported to reduce TNBC cell viability and induced apoptosis by downregulating phosphorylated STAT3 expression in both a dose- and time-dependent manner. In addition, the combination of SC-60 and docetaxel synergistically enhanced the anticancer effect by inhibiting the SHP-1/STAT3 pathway [99]. Another sorafenib analogue, SC-43, was reported to block STAT3 signaling to increase the sensitivity of cancer cells to chemotherapeutic drugs like docetaxel. SC-43 showed tumor growth inhibition and apoptosis inducing by suppressing the SHP-1-dependent STAT3 expression [100]. SG-1721, a (-)-galiellalactone analogue, was found to inhibit the growth of TNBC cells. It could promote cells apoptosis via suppressing the nuclear translocation and DNA binding of STAT3, as well as reduce the expression of carcinogenic proteins such as Bcl-2, Cyclin D1 and MMP-2. In in vivo experiment, SG-1721 could significantly inhibit the growth of breast xenograft tumor. Moreover, when SG-1721 was combined with radiotherapy, TNBC cells were sensitized to radiation and apoptotic effect was enhanced. It could be a potential agent that targets STAT3 to treat TNBC [101]. Compound 15d as JAK/HDAC dual inhibitor exhibited the antiproliferative and proapoptotic activities by suppressing the activation of LIFR-JAK-STAT signaling and attenuate the drug resistance in tumor cells [102]. Ilamycin C, a new compound isolated from Streptomyces atratus SCSIO ZH16, exerted a strong cytotoxic activity against TNBC cells. It promoted cell apoptosis by inhibiting IL-6-induced STAT3 phosphorylation and suppressed TNBC cells migration and invasion through MMP2/MMP9/vimentin/fascin [103]. A growing body of research suggests that inhibition of STAT3 pathway has been considered as a novel therapeutic strategy to treat TNBC. Other small-molecule compounds target STAT3 to trigger apoptosis on TNBC [104,105,106,107,108,109] (Table 1).
Targeting p53
p53 is an essential tumor suppressor protein, which could regulate diverse cellular processes, including cell apoptosis, DNA repair, cell cycle arrest, etc. [110]. Activating p53 protein could increase the sensitivity of cancer cells to DNA damage factors, so that cells with DNA damage could not enter the replication cycle, and then went to apoptosis [111]. P21, as a target gene of p53 protein, is involved in cell growth arrest and inhibits cell cycle progression by inhibiting cyclin B/Cdc2 through mitosis. Upregulation of p21 acts as the inhibitor of cell cycle dynamics [112]. Furthermore, more than 80% of TNBC patients had p53 mutation, and the mutant p53 protein could effectively promote the malignant transformation of cells, activate other oncogenes and lead to the occurrence of tumors [113, 114]. In recent years, some small-molecule compounds could reactivate the mutant p53 protein and restore it to the wild-type conformation, which made targeting the mutant p53 protein becoming one of the most attractive targets for treating TNBC [115].
APR-246 as a p53 activator could inhibit cell proliferation and migration and induce apoptosis in a p53 mutant-dependent manner of TNBC cells [116]. Recently, the study found that co-treatment of APR-246 and CX-5461, an RNA polymerase I inhibitor, could also significantly inhibit TNBC cells growth and induce apoptosis, which was caused by DNA damage [116, 117]. Furthermore, a third-generation thiosemicarbazone, COTI-2, targeted the p53 protein to upregulate the expression of apoptosis effector genes, such as Bcl-2 binding component 3 (BBC3) and phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1), leading to the induction of apoptosis in BT549 and Hs578t cells. COTI-2 could convert the mutant p53 protein into wild-type p53 to exhibit anticancer activity [118]. In general, the IC50 of COTI-2 was lower than that of APR-246, suggesting that COTI-2 was more active in inhibiting cell proliferation than APR-246 in TNBC cell lines [116, 118]. In addition, PK11007 was a mild mercaptan alkylation agent, which could inhibit cell growth and induce apoptosis in TNBC cells by upregulating of p53 protein. In addition, PK11007 combined with cisplatin could synergistically inhibit the growth of TNBC cell lines [119].
There are some natural and semisynthetic compounds targeting p53 protein to induce apoptosis of TNBC cell lines. In addition, it was found that a series of spirotriazoline oxindoles had been synthesized and 5-bromo-4'-(3-chlorophenyl)-20,50-diphenyl-20,40-dihy-drospiro[indoline-3,3'-[1, 2, 4].triazol].-2-one (compound 5i) was identified as the compound with the most significant inhibitory effect on MDA-MB-231 cell [120]. After compound 5i treated MDA-MB-231 cells, the expression of p53 was upregulated and the expression of murine double minute 2 (MDM2) was downregulated, and then compound 5i could increase the expression of tumor suppressor proteins, arrest cell cycle at G0/G1 phase, inhibit cell proliferation and eventually lead to cell apoptosis [120]. Moreover, it was also found that organometallic complexes, like ruthenium (II)/allopurinol complex, had been shown to have significant cytotoxicity against TNBC cells [121]. In contrast, ruthenium (II)/allopurinol complex binds to tumor cells to cause DNA damage and upregulate p53 protein, then causing the overexpression of bim, beclin-1 and caspase-3, and ultimately induced cell apoptosis [121]. Additional small-molecule compounds target p53 protein to induce apoptosis in TNBC cells [122,123,124,125,126] (Table 1).
Targeting autophagy-dependent cell death pathways with small-molecule compounds in TNBC
Autophagy is a process in which the excess proteins of damaged cellular organelles are degraded by lysosome in order to maintain cell homeostasis under stress [127]. Cells can recover energy and nutrients from autophagy degradation products, so that cells can maintain their own metabolism and enhance their tolerance to adverse stimulation [128]. Thus, autophagy can also be regarded as a self-protection mechanism of cells [129]. Autophagy has been becoming a new target of breast cancer treatment, but the role of autophagy in cancer is quite complex, which acts as a double-edged sword in the tumor treatment [130,131,132]. On the one hand, it can increase tumor cell autophagy activity, which contributes to programmed forms of cell death. On the other hand, autophagy may provide energy for tumor cell metabolism of maintaining cells survival (Fig. 2) [133, 134].

Core autophagy-dependent cell death signaling pathways in triple negative breast cancer (TNBC). Autophagy is a complex regulation process involving many upstream signaling pathways. Mammalian target of rapamycin (mTOR) is a negative regulator of autophagy, which is composed of mammalian target of rapamycin complex (mTORC) 1 and mTORC2. Among them, mTORC1 is the main autophagy regulator and phosphatidylinositol 3 kinase complex 1 (PI3KC1)-protein kinase B (Akt)-mTORC1 pathway inhibits the occurrence of autophagy. P53 pathway negatively regulates mTOR pathways to promote autophagy. When mTORC1 is inhibited, it can indirectly activate unc-51-like kinase 1 (ULK1) complex (including ULK1, autophagy associated protein (ATG) 101, ATG13, and focal adhesion kinase interacting protein of 200 kDa (FIP200)). ULK1 complex is closely related to Beclin1, and ULK1 can phosphorylate ATG14, which promotes the binding of Beclin1 to vacuolar protein sorting 34 (VPS34) and ultimately participates in the regulation of autophagy. Forkhead box O (FoxO) had been shown to regulate autophagy by transcriptional dependent mechanism. P62 can bind light chain 3 (LC3)-labeled autophagosomes to substrates, promote the combination of substrates and autophagosomes, and promote the occurrence of autophagy. Additionally, the inhibiting of Ras-Raf-MAPK pathway and NF-κB pathway could also regulate autophagy. Abbreviations: AKT: Protein kinase B; ATG: Autophagy associated protein; ERK: Extracellular signal-regulated kinase; FIP200: Focal adhesion kinase interacting protein of 200 kDa; FoxO: Forkhead box O; LC3: Light chain 3; MEK: Mitogen-activated protein kinase kinase; mTORC1: Mammalian target of rapamycin complex 1; PI3KC1: Phosphatidylinositol 3 kinase complex 1; ULK1: Unc-51-like kinase 1; Vps34: Vacuolar protein sorting 34
Autophagy is a complex regulation process involving many upstream signaling pathways. Mammalian target of rapamycin (mTOR) is a negative regulator of autophagy, which is composed of mammalian target of rapamycin complex (mTORC) 1 and mTORC2. Among them, mTORC1 is the main autophagy regulator and phosphatidylinositol 3 kinase complex 1 (PI3KC1)-protein kinase B (Akt)-mTORC1 pathway inhibits the occurrence of autophagy. p53 pathway negatively regulates mTOR pathways to promote autophagy. When mTORC1 is inhibited, it can indirectly activate unc-51-like kinase 1 (ULK1) complex (including ULK1, autophagy-associated protein (ATG101, mATG13 and focal adhesion kinase interacting protein of 200 kDa (FIP200)). ULK1 complex is closely related to Beclin1, and ULK1 can phosphorylate ATG14, which promotes the binding of Beclin1 to vacuolar protein sorting 34 (VPS34) and ultimately participates in the regulation of autophagy. Forkhead box O (FoxO) had been shown to regulate autophagy by transcriptional-dependent mechanism. P62 can bind light chain 3 (LC3)-labeled autophagosomes to substrates, promote the combination of substrates and autophagosomes, and promote the occurrence of autophagy. Additionally, the inhibiting of Ras-Raf-MAPK pathway and NF-κB pathway could also regulate autophagy.
Autophagy modulation can be served as a promising target for the development of anti-TNBC drugs, which had profound implications on breast cancer investigation [135, 136]. Several small-molecule compounds could alleviate or treat TNBC by changing the level of autophagy in tumor cells [137]. Moreover, autophagy had great potential in improving the therapeutic efficiency, overcoming chemotherapy resistance of breast cancer [138]. A variety of signaling pathways and target proteins had been implicated in autophagy regulation, including unc-51-like kinase 1 (ULK1) complex, PI3KCI-Akt-mTORC1, Ras-Raf-MAPKs, p53, p62, fork-head box O (FoxO), NF-κB, Beclin-1, etc. [139]. Then, we summarized the research progress of autophagy-related signaling pathways and their corresponding small-molecule compounds in the treatment of TNBC.
Targeting the ULK1 complex
ULK1, a homologous protein of the yeast autophagy-associated protein 1 (ATG1) in mammals, is a serine/threonine protein kinase and a major regulator of autophagy initiation [140]. It interacted with focal adhesion kinase interacting protein of 200 kDa (FIP200), a protein in the adhesion spot kinase family, autophagy-related proteins mammalian autophagy-associated protein 13 (mAtg13) and Atg101 to form the ULK complex, which further activated the downstream autophagy signaling pathway and promoted the formation of autophagosomes [141]. ULK1, as a promoter of autophagy, also played different roles in different tumors. Downregulation of ULK1 expression was closely related to the progression of breast cancer and was accompanied by the decrease in autophagy level [142, 143]. Therefore, activation of ULK1-regulated autophagy cell death is a potential strategy for the treatment of TNBC [143]. There are some small-molecular compounds that directly or indirectly target ULK1 for TNBC therapy by regulating autophagy.
A small-molecular agonist, LYN-1604, was found to target ULK1 with a median effective concentration (EC50) value of 18.94 nm [144]. Based on site-directed mutagenesis and biochemical detection, three amino acid residues (LYS50, LEU53 and TYR89) were identified to be important for LYN-1604 binding and activation of ULK1. LYN-1604 was proved to induce TNBC cell death through autophagy via activating ULK1 and its complex. LYN-1604 could also activate ULK1 interactors including activating transcription factor 3 (ATF3), RAD21 and caspase-3, to induce cell death [144]. FL-411, a bromodomain 4 (BRD4) inhibitor, induced autophagy-associated cell death, which was dependent on the activation of the adenosine 5′-monophosphate-activated protein kinase (AMPK)–mammalian target of rapamycin (mTOR)–ULK1-modulated autophagic pathway [145]. In contrast, SBP-7455 as a ULK1/2 inhibitor could inhibit TNBC cells survival and proliferation by inhibiting starvation-induced autophagic flux. Moreover, SBP-7455, in combination with PARP inhibitor Olaparib, showed an enhanced effect on apoptosis in TNBC cells [146].
Autophagy had also been shown to play a key role in the chemotherapy resistance of TNBC [147]. Phloretin (PH), a dihydrochalcone flavonoid compound, could effectively inhibit the growth of TNBC cell by suppressing of autophagy through the downregulation of mTOR/ULK1 signaling pathway. Besides, PH overcomes resistance to chemotherapy drugs (4OH-Tamoxifen and DOX) by regulating autophagy in TNBC cells [148] (Table 2).
Targeting PI3KC1-Akt-mTORC1
The PI3KC1-Akt-mTORC1 signal transduction pathway was one of the central pathways regulating autophagy and was also involved in the regulation of tumor cell growth, proliferation and metabolism [149]. PI3K was a complex family which can be divided into PI3KC1, PI3KC2 and PI3KC3. PI3KC1 was involved in cell proliferation, insulin signal transduction, immune function and inflammatory response. Akt (serine/threonine kinase) was a protein kinase downstream of PI3K, which could activate and regulate multiple downstream targets [150, 151]. mTOR is a member of the PI3K family of protein kinases and existed in cells in the form of two complexes, mTORC1 and mTORC2. mTORC1 had an essential role in maintaining cellular homeostasis as a sensor of energy and nutrition in the cell [152, 153]. PI3KC1 activated Akt, which then directly phosphorylated and activated mTORC1 or indirectly activated mTORC1 by inhibiting tuberous sclerosis complex 1/2 (TSC1/2) and GTP-active Rheb, thereby inhibiting autophagy [154].
An increasing number of small-molecule compounds could induce TNBC cell death by targeting PI3KC1-Akt-mTORC1. A novel small molecule, SLLN-15, was shown to exhibit anticancer activity and inhibit the growth and proliferation of TNBC cells. These effects were associated with autophagy induction via decreasing the expression of aurora kinase A (AURKA) and blockade of AKT-mTOR signaling pathway [155]. Besides, 2-(4-(9-(6-aminopyridin-3-yl)-2-oxopyrazino[2,3-c].-quinolin-1(2H)-yl)piperidin-1-yl)acetonitrile (compound 9 m), a novel mTOR inhibitor, could induce the G0/G1 phase arrest of cell cycle and autophagy by inhibiting the phosphorylation of Akt, 4E-BP1 and S6. Besides, compound 9 m could significantly induce tumor regression in vivo experiment (Table 2) [156].
Targeting Ras-Raf-MAPKs
MAPK is a signaling pathway that converted extracellular signals into intracellular signals in the form of a tertiary kinase cascade (MAPKKK-MAPKK-MAPK), among which Ras-Raf-MEK-ERK pathway had been studied most deeply [157]. More and more data indicated that MAPK (Ras-Raf-MEK-ERK) pathway was a vital target for TNBC treatment. Ras was a GTP binding protein, which activated Ras by binding to GTP [158], then phosphorylated Raf, causing downstream MEK/ERK activation, and then caused a series of physiological responses. Ras-Raf-MEK-ERK signaling pathway was not only widely involved in the regulation of multiple physiological processes such as cell growth, proliferation and apoptosis, but also involved in the regulation of autophagy and the promotion of autophagy-dependent death of tumor cells [159, 160]. This signaling pathway could also directly induce autophagy by upregulating the expression of autophagy-related proteins such as light chain 3 (LC3) and p62 [161]. Consequently, small-molecular compounds targeting Ras-Raf-MAPK pathway to induce breast cancer cells autophagy death was a promising approach for the treatment of TNBC.
Sometimes, the inhibition of autophagy could restrain the energy of cell metabolism and survival in cancer. HCQ as an autophagy inhibitor could suppress the cell proliferation and invasion and increase the sensitivity of breast cancer cells to 5-FU. HCQ could inhibit autophagy, and its inhibitory effect on SUM190 cells can be achieved by downregulating Ras-Raf-ERK pathway [162, 163]. Y29, a synthetic pyridine derivative, has markedly antiproliferative activity against TNBC cells. Y29 could induce autophagy by targeting platelet-derived growth factor receptor β (PDGFR-β), and it could enhance autophagy-related cell death through regulating AKT-MAPK pathway in MDA-MB-231 cells [164] (Table 2).
Targeting p53
p53 protein not only induces apoptosis, but participates in autophagy as a critical regulator [165]. Recent reports showed that p53 played dual roles in the regulation of autophagy. The regulatory effect of p53 protein on autophagy was dependent on its intracellular localization [166]. In the nucleus, p53 protein induced autophagy and promoted cell death, while in the cytoplasm p53 inhibited autophagy, which could promote the survival of cancer cells in the occurrence and development of tumors [167]. During starvation or hypoxia, p53 activation is induced, which activates AMPK, further phosphorylates TSC1/TSC2, upregulates mTOR inhibitor phosphatase and tensin homologue deleted on chromosome ten (PTEN), and inhibits mTOR to induce autophagy [168].
High expression of mutant p53 protein was found in most TNBC, which is one of the causes of malignant cell proliferation [114]. Small-molecule compounds could target p53 mutant protein, induce cell autophagy and promote cancer cell death. For example, it was reported that small-molecule compound, CP-31398, could reactivate wild-type p53 protein to induce autophagy, thereby enhancing natural killer cell (NK) lysis of p53-mutated MB-MDA-231 cells [169, 170]. This finding suggested a new approach for future combination immunotherapy [171].
Targeting p62, FoxO and NF-κB
p62, also called SQSTM1, is a receptor that could bind to ubiquitin and LC3 proteins, thus targeting autophagosomes and promoting the degradation of ubiquitin proteins [172]. During autophagy, p62 protein was continuously degraded in the cytoplasm, and p62 protein was continuously accumulated in the cytoplasm when autophagy was inhibited, so p62 could be used as a marker of autophagy activation [172, 173]. PC3-15, as a Schisandraceae triterpenoid compound, bound to the ubiquitination enzyme UbcH5b and suppressed the ubiquitination of p62, thereby inhibiting lapatinib-induced autophagy and increasing the sensitivity of TNBC cells to lapatinib therapy both in vitro and in vivo. These findings proved that UbcH5b-p62 axis was a potential therapeutic target for TNBC cell resistance [174].
FoxO is an essential autophagy regulator. FoxO in mammals included FoxO1, FoxO3, FoxO4 and FoxO6 subtypes, among which FoxO1 and FoxO3 had the most extensive effects [175, 176]. FoxO was involved in the regulation of cell proliferation, metabolism, survival and other processes by activating autophagy. FoxO regulated autophagy-associated with multiple signaling pathways, including AMPK, PI3K-Akt, etc. [175, 177]. Therefore, targeted therapy of autophagy genes could be a potential therapeutic strategy in TNBC breast cancer. WX20120108 was a novel IAP inhibitor with strong antitumor and pro-autophagic activity [178]. It also found that WX20120108 promoted the release of ROS to activate FoxO3, thereby inducing autophagy in MDA-MB-231 cell and HeLa cell [178].
In addition to regulating cell survival, apoptosis and inflammatory activation, NF-κB transcription factor was also involved in the regulation of autophagy [179, 180]. The regulatory relationship between NF-κB pathway and autophagy was complex. According to the different external environment, NF-κB could activate or inhibit autophagy, induce the body to produce protective or damage effect. Several small-molecule compounds could play a good therapeutic effect by acting on NF-κB signaling pathway to mediate autophagy [181]. Alisol A, a natural active compound of Alismatis rhizoma, could significantly suppress the proliferation and migration of TNBC cells when autophagy exerted an oncogenic effect. Alisol A induced MDA-MB-231 cells autophagy death by inhibiting both NF-κB and PI3K/AKT/mTOR pathways [182,183,184].
Targeting Beclin-1
Beclin-1 (a homolog of yeast autophagy gene ATG6/Vps30, also known as BECN1) is one of the key autophagy regulatory proteins, which is involved in autophagosome membrane formation. Beclin-1, as an important tumor suppressor gene, was missing in most breast cancer patients, especially in TNBC [185, 186]. When the expression level of Beclin-1 was reduced, the autophagy activity of cells would also be inhibited, so that tumor cells become more aggressive. While Beclin-1, as a key gene for the formation of autophagosome initiation complex, was highly expressed in breast cancer, the proliferation and drug resistance of TNBC were dramatically restrained [163, 187,188,189]. Therefore, Beclin-1 could not only reduce the risk of breast cancer, but also affected the development of breast cancer by regulating autophagy.
Thymoquinone (TQ), a phenolic compound of Nigella sativa, was found to suppress the cell proliferation and invasion by inhibiting LC3, Beclin-1 and related oncogenic signaling (VEGF, MMPs) in TNBC cells, leading to the inhibition of oncogenic autophagy [190]. Rhein is a natural anthraquinone compound with significant antiproliferative and antimetastatic effects. However, due to poor bioavailability and lack of specific targets of this compound [191], Rhein was modified to obtain more efficient Rhein derivative 4F. Rhein 4F was reported to induce autophagy-dependent death of MDA-MB-231 cells through the upregulation of Beclin-1 and LC3-II/I and the downregulation of p62. Since the expression of apoptosis marker-caspase-3 was not affected by Rhein 4F, Rhein 4F-induced MDA-MB-231 cell death was mainly dependent on autophagy rather than apoptosis [192].
Targeting other RCD subroutines with small-molecule compounds in TNBC
Besides the classical apoptosis and autophagy-dependent cell death pathways, there are some other cell death measures belonging to RCD. According to the latest progress, RCD also includes mitotic catastrophe, necroptosis, ferroptosis, pyroptosis and anoikis, with their regulatory signaling pathways in TNBC (Table 3) (Figs. 3, 4).

The key mitotic catastrophe, necroptosis and ferroptosis pathways in triple-negative breast cancer (TNBC). a DNA damage inhibits checkpoint kinase 1 (chk1) and cyclin-dependent kinase (CDK) 2 targets and then inhibits the recovery of cell cycle checkpoints, resulting in mitotic catastrophe. The rad3-related protein (ATR)-chk1 signaling pathway is activated in the absence of G2 checkpoints, restores S/G2 and G2/M cell cycle checkpoints and avoids the production of mitotic catastrophe. PI3K-like kinase (PIKK)/mammalian target of rapamycin (mTOR) inhibitors cause the accumulation of single-stranded deoxyribonucleic acid (ssDNA), replication catastrophe and mitotic failure, and ultimately lead to mitotic catastrophe. Polo-like kinase 1 (Plk1)-interacting checkpoint helicase (PICH) depletion can also lead to mitotic catastrophe. Bromodomain and extraterminal protein (BET) inhibitors eventually cause mitotic catastrophe by inhibiting B-cell lymphoma-extra-large (Bcl-xL). The production of the above mitotic catastrophe will eventually cause the death of TNBC cells; b Aquaporin1 (AQP1) can inhibit receptor-interacting protein (RIPK) 1/RIPK3/mixed lineage kinase domain-like (MLKL)-mediated necroptosis by binding to the D324 site of RIPK1. The fas associated via death domain (FADD)/TNFRSF1A associated via death domain (TRADD) complex depends on both RIPK1/caspase-8-mediated apoptosis and RIPK1/RIPK3/MLKL-mediated necroptosis. The production of the above necroptosis will eventually cause the death of TNBC cells; c Zn protoporphyrin IX (Znpp) inhibits the accumulation of unstable iron pools by inhibiting HO-1, reduces reactive oxygen species (ROS) levels and reduces ferroptosis caused by lipid peroxidation. Cystine enters and exits the cell membrane through solute carrier family 7 member 11 (SLC7A11)/solute carrier family 3 member 2 (SLC3A2), converts to cysteine, causes glutathione (GSH) levels to rise, activates Glutathione peroxidase 4 (GPX4) and inhibits ferroptosis caused by lipid peroxidation. The production of the above ferroptosis will eventually cause the death of TNBC cells

The key pyroptosis and anoikis pathways in triple-negative breast cancer (TNBC). a Maternally expressed gene 3 (MEG3) activates the NLR family, pyrin domain containing 3 (NLRP3)/procaspase-1/apoptosis-associated speck-like protein containing a CRAD (ASC) complex, procaspase-1 is converted to caspase-1, and pyroptosis is induced via gasdermin D (GSDMD). In addition, caspase-11 and caspase-4/5 can also induce pyroptosis via GSDMD. Inhibition of mitochondrial signal transducer and activator of transcription 3 (STAT3) phosphorylation can increase reactive oxygen species (ROS) levels, activate bak and B-cell lymphoma 2 (Bcl-2) targets, activate caspase-9 and caspase-3 in the presence of cytochrome c (Cyt-c), and ultimately promote the cleavage of gasdermin E (GSDME), to transform apoptosis into pyroptosis. In addition, procaspase-8 can also activate caspase-3 to induce pyroptosis via GSDME. The above pyroptosis will eventually cause the death of TNBC cells. b cFLIP inhibits the production of anoikis by inhibiting the conversion of procaspase-8/fas associated via death domain (FADD)/TNFRSF1A associated via death domain (TRADD) complex to caspase-8. After epidermal growth factor receptor (EGFR) is activated, it inhibits the phosphorylation of Tyr705 on STAT3 and resists anoikis. EGFR and SRC/FAK activate the PI3K/Akt pathway, mediate late Bcl-2 interacting mediator of cell death (BIM) degradation, activate myeloid cell leukemia-1 (Mcl-1)/B-cell lymphoma-extra large (Bcl-xL)/Bcl-2, reduce Bax/Bak activity, and inhibit the production of anoikis. SRC/FAK also activates the Ras/Raf/MEK/extracellular signal-regulated kinase (ERK) pathway, relieves the inhibitory effect of proteasome on Mcl-1/Bcl-xL/BCL-2, reduces the activity of Bax/Bak, and inhibits anoikis. In addition, caveolin-1 (cav-1) can also restore the activity of Mcl-1/Bcl-xL/BCL-2 and ultimately inhibit anoikis. After adenosine 5′-monophosphate-activated protein kinase (AMPK) is activated, it can reduce glucose-6-phosphate dehydrogenase (G6PD) and increase phospho-acetyl-CoA carboxylase (p-ACC) and finally activate anoikis. In addition, the overexpression of protein kinase c theta (PRKCQ)/protein kinase C theta (PKCθ), C-X-C motif chemokine receptor 4 (CXCR4), C–C motif chemokine receptor 7 (CCR7) and integrin-β1 will inhibit anoikis. The production of the above anoikis will eventually cause the death of TNBC cells
Targeting mitotic catastrophe
Mitotic catastrophe is a type of cell death during mitosis due to abnormal chromosome separation, such as mitotic checkpoint defects and damage [193, 194]. The treatment of cancer with anticancer drugs such as DNA damage drugs and microtubule targeting could induce mitotic catastrophe [195], while inhibition of mitotic catastrophe would promote the development of chemoresistance and tumor occurrence [196,197,198]. PTX, as a microtubule inhibitor, interfered with microtubule dynamics by stabilizing microtubule polymerization, resulting in abnormal chromosome condensation, and finally formed mitotic catastrophe [199]. Due to the poor water solubility of PTX, currently commonly used solvents of PTX tend to result in many adverse reactions [199]. Nanodiamond (ND), as a drug delivery carbon-based nanomaterial with good biocompatibility, PTX carrying ND reduced the cell viability of TNBC and induced mitotic catastrophe in a concentration-dependent manner. Interestingly, the use of ND alone would not induce cell death [199, 200]. As a common drug for standard chemotherapy in patients with TNBC, these findings about PTX suggested that the study of mitotic catastrophe mechanism of RCD might bring a new vision and direction for the treatment of TNBC. Unfortunately, TNBC tumors were prone to only respond to traditional chemotherapy well at the beginning [201] and often relapsed within 3 years. In recent years, in a variety of cancer mice models, bromodomain and extraterminal protein inhibitors (BETi) prevented the recruitment of bromodomain and extraterminal protein (BET) protein to chromatin and inhibit BET transcriptional activity by selectively targeting the epigenetic reader of BET family [202,203,204]. In a variety of TNBC models, the classic BETi JQ1 could induce polynuclear, which was a characteristic early index of mitotic catastrophes [193], and followed by apoptosis and aging, leading to mitotic catastrophe [205]. Inhibition of anti-apoptotic protein Bcl-xL could promote mitotic catastrophe and apoptosis induced by BETi [206]. High Bcl-xL level may be a potential effective biomarker for BETi in the treatment of TNBC [207]. In addition to the above hot targets, more studies in recent years had focused on new targets for the treatment of TNBC. A study through the breast cancer dataset and Gene Ontology (GO) database found that checkpoint kinase 1 (chk1) gene is significantly overexpressed in TNBC patients, and chk1 as a key target is involved in the DNA repair pathway to treat TNBC. The results show that chk1 inhibitor SB218078 can reduce the cell viability and survival rate of TNBC cells by inducing mitotic catastrophe [208]. In addition, the analysis of human clinical database TCGA shows that NOTCH1 is highly expressed in TNBC. The results show that NOTCH1 restores S/G2 and G2/M cell cycle checkpoints by activating rad3-related protein (ATR)-chk1 signaling pathway [209], so as to inhibit mitotic catastrophes caused by BRCA1 deficiency [210, 211]. Interestingly, TNBC had a unique cell cycle progression mechanism due to its inherent genetic complexity and was sensitive to drugs leading to mitotic catastrophes [212, 213]. Polo-like kinase 1 (Plk1)-interacting checkpoint helicase (PICH), the binding substrate of plk1 which was a key enzyme in M-phase progression, was significantly overexpressed in TNBC through clinical sample analysis, ensuring reliable chromosome separation and promoting the growth of TNBC cells. The results further showed that the depletion of PICH in TNBC cells would lead to the formation of chromatin bridge and late chromosome lag, the formation of binuclear, and finally resulted in mitotic catastrophe and apoptosis. The adenosine triphosphatase (ATPase) activity of PICH is necessary for the proliferation and survival of TNBC cells, suggesting that PICH is a potential new therapeutic target of TNBC [212]. In addition to the above single target inhibition treatment of TNBC, small-molecule drugs that exert a higher level of therapeutic effect by inhibiting a variety of targets are becoming more and more popular. Torin2 and its chemical analogues lead to the accumulation and replication disaster of single-stranded DNA by simultaneously inhibiting mTOR and other PI3K-like kinases (PIKKs), which eventually leads to mitotic catastrophes and the death of TNBC tumor cells [214]. By further developing the combination of existing mTOR and PIKKs inhibitors or torin2 analogues, it may become a potentially effective strategy for the treatment of TNBC. KX-01 downregulated the expression of phosphorylated SRC and proliferation signal molecules by inhibiting SRC and tubulin at the same time, induced G2/M cell cycle arrest, increased aneuploid cell population and multinucleated cells, and finally induced mitotic catastrophe, which effectively delayed the tumor growth of TNBC mouse xenotransplantation model [215]. The inhibition of tubulin overcame the therapeutic limitation that the current SRC inhibitors failed to show clinical benefits in the treatment of TNBC (Table 3) (Fig. 3a) [216, 217].
Targeting necroptosis
Necroptosis is a kind of RCD, resembling necrosis-dying cells cluster, releasing intracellular components and recruiting a large quantity of inflammasomes [8]. Necroptosis shows the morphological type of necrosis. Its function in cancer was mainly to mediate the adaptive function when the stress response failed. Necrosis was mediated by three major kinases: receptor-interacting protein (RIPK) 1, RIPK3 and mixed lineage kinase domain-like (MLKL) [8]. Sometimes, TNBC had no obvious response to chemotherapy [218]. It was urgent to explore TNBC-specific signal pathway and sensitive biomarkers [219]. Aquaporin1 (AQP1), a water transport membrane protein related to tumor development and progression [220, 221], as a carcinogenic biomarker of a variety of cancers [222,223,224], could inhibit RIPK1/MLKL/RIPK3-mediated necroptosis and RIPK1/caspase-8/caspase-3-mediated apoptosis by binding to d324 site of ripk1, driving the progression of TNBC [225]. As a negative mediator, the ectopic expression of RIPK1 could significantly weaken the signal transduction of AQP1 [225], suggesting that RIPK1 could be used as an effective potential target for the treatment of TNBC by affecting necroptosis (Table 3) (Fig. 3b).
Targeting ferroptosis
Ferroptosis is a subroutine of RCD caused by oxidative disturbance of intracellular microenvironment, which was related to the accumulation of toxic lipid peroxides [8]. With acquired resistance of cancer cells, ferroptosis-inducing therapy shifted its importance in recent years [226]. The escape of cancer cells from other types of RCD might be still sensitive to ferroptosis [227]. TNBC cells were sensitive to ferroptosis inducers [228, 229]. Glutathione peroxidase 4 (GPX4) [230], as the main inhibitor of ferroptosis [231], is antioxidant defense enzyme. Its deletion can specifically activate ferroptosis [232]. Gefitinib is a classical epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor for the treatment of TNBC, and some TNBC subtypes are resistant to it [233, 234]. A recent study found that the expression of GPX4 increased in gefitinib-resistant cells. By constructing gefitinib-resistant TNBC cells, it was found that silencing the expression of GPX4 increased the production of MDA and ROS, reduced the level of glutathione (GSH) and finally promoted ferroptosis, resulting in the inhibition of TNBC cell viability and colony forming ability, and the improvement in sensitivity of TNBC cells to gefitinb [235]. TNBC cells are very sensitive to iron poisoning induced by erastin [236], a low molecular weight chemotherapeutic drug. Unfortunately, its application is hindered by the nephrotoxicity caused by side effects and limited water solubility. In recent years, the more advanced development of nanomaterials has made a lot of contributions to the drug delivery system of small-molecule targeted drugs [237,238,239,240]. Exosomes, as micro-membrane vesicles, have attracted extensive attention because they can be used as drug delivery carriers to load low molecular weight chemotherapeutic drugs in cancer [241,242,243,244,245,246]. Researchers had developed a formula for exosomes loaded with erastin which labeled with folate (FA) (erastin@FA-exo) [247]. By inhibiting the GPX4 expression and upregulating the dioxygenase (CDO1) expression, erastin@FA-exo could reduce the level of GSH, increase the level of ROS and promote ferroptosis in TNBC cells with FA receptor overexpression. Compared with free erastin, erastin@FA-exo strongly inhibited the proliferation and migration of TNBC cells [248]. It is suggested that exosome-based drug delivery system may provide a new choice and direction for the treatment of TNBC. Ferroptosis is often accompanied by the accumulation of lethal ROS [249]. As a pleiotropic protein, lactoferrin (Lf) was often used to study the efficacy in cancer recently [250]. As an iron-saturated Lf, hollo lactate (Holo-Lf) induced ferroptosis in TNBC tumors by increasing total iron content and promoting the production of ROS, which showed better anticancer properties than low iron-saturated Lf (Apo-Lf) (Table 3) (Fig. 3c) [251].
Targeting pyroptosis
Pyroptosis, also known as inflammatory cell necrosis, is a subroutine of RCD mediated by gasdermin (GSDM), which is characterized by the pore formation in the plasma membrane, cell swelling and rupture of the membrane, resulting in the release of cell contents and then activating a strong inflammatory response [252,253,254,255]. On the one hand, chemotherapeutic drugs could promote the cleavage of gasdermin E (GSDME) by activating Caspase-3, to transform apoptosis into pyroptosis and promote tumor cell death [256, 257]. Likewise, tetraarsenic hexoxide (As4O6) could inhibit the phosphorylation of mitochondrial STAT3 and activate mitochondrial ROS-mediated GSDME pathway, to induce pyroptotic cell death in TNBC cells, and finally inhibit tumor growth and metastasis of TNBC [258, 259]. On the other hand, chemotherapeutic drugs also played an anti-TNBC role by inducing pyroptosis [260]. Analogously, cisplatin is a classic chemotherapy drug for main mechanism of actions (MOA), is DNA damage, and induced pyroptosis through non-major (or off-target) drug effects by upregulating the long noncoding RNA (lncRNA) maternally expressed gene 3 (MEG3) and activating the NLR family, pyrin domain containing 3 (NLRP3)/caspase-1/gasdermin D (GSDMD) pathway, to treat TNBC [260, 261]. Interestingly, polyI: C, a synthetic double-stranded RNA (dsRNA) analogue traditionally used to activate retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), promoted tumor cell death by inhibiting the anti-pyroptotic function of TGF after transfection into TNBC cells (Table 3) (Fig. 4a) [262].
Targeting anoikis
Anoikis is a special form of programmed cell death induced by the loss of contact between cells and extracellular matrix (ECM) and other cells (Table 3) (Fig. 4b) [8]. The growth of most cancer cells depends on anchoring. Without attachment to ECM in vivo, cancer cells would experience anoikis [263, 264], which played an important role in tumor metastasis [265]. Activating anoikis was the key factor to resist the occurrence and development of tumor. Protein kinase c theta (PRKCQ)/protein kinase C theta (PKCθ), as a member of the novel protein kinase C (PKC) family [266], was a regulatory factor that does not depend on adherence to survival in breast cancer cells [267], and was preferentially expressed in TNBC [268]. The results proved that PRKCQ/PKCθ promoted the phosphorylation of retinoblastoma (Rb), caused growth factor-independent cell cycle arrest, promoted the formation of tumor phenotypes [269, 270] and enhanced anchorage-independent survival, proliferation and migration [268]. Downregulation of the expression level of PRKCQ/PKCθ promoted anoikis of TNBC cells and inhibited the growth of TNBC xenografts [268]. The results showed that AEB071 as a PKCθ inhibitor also inhibited the growth of TNBC cells [268]. Research on PRKCQ/PKCθ promoting the growth of TNBC in vitro and in vivo supported its use as a potential and effective target in the treatment of TNBC. Moreover, a study manifested that synthesized flavor-derived GL-V9 could reduce glucose-6-phosphate dehydrogenase (G6PD) and increase phospho-acetyl-CoA carboxylase (p-ACC) by activating the activity and expression level of AMPK, and finally activate anoikis to inhibit the tumor metastasis of MDA-MB-231 TNBC cell line and TNBC xenograft nude mice [271]. Tubeimoside V (TBMS-V) activated EGFR and ITGB1-FAK by regulating caveolin-1 (cav-1)-related signal pathway [272] and finally made TNBC cancer cells sensitive to anoikis and inhibited TNBC cell growth and metastasis [273]. Disulfiram/copper (DSF/Cu) induced anoikis and significantly inhibited TNBC cell migration and invasion by activating calpain and decomposing vimentin in a Cu-dependent manner [274]. In TNBC xenograft tumor model, DSF also inhibited lung nodule growth and tumor growth by activating calpain [274]. Unfortunately, not all cancer cells could be affected by anoikis. Gaining resistance to anoikis had been identified as a feature of the treatment of advanced cancer cells and a key step in the process of tumor metastasis [275, 276]. CD44+/CD24− stem cell-like population in TNBC tended to be a more aggressive phenotype [277,278,279,280,281], and cancer stem cell (CSC) was resistant to anoikis by allowing replication independent of anchoring [282, 283]. STAT3 could regulate stem cell self-renewal and differentiation and resist anoikis [284, 285]. It was also overexpressed and structurally activated in TNBC cells. During anchoring independent growth, salinomycin could reduce CD44+/CD24− stem cell-like population and inhibit the formation of mammary gland ball. In the meantime, salinomycin exerted a significant inhibitory effect on TNBC cell migration and invasion. Mechanically, salinomycin could not only downregulate MMP-9 and MMP-2 messenger ribonucleic acid (mRNA) levels, but also activate caspase-3 and caspase-8, cleave PARP and inhibit STAT3 phosphorylation tyr705, finally significantly inducing anoikis sensitivity [286]. CSC can also overexpress the chemokine receptor C-X-C motif chemokine receptor 4 (CXCR4) in many cancer types by using the typical pathway of hematopoietic stem cells (HSCs) [287,288,289,290,291,292,293]. POL5551, as a peptidic CXCR4 antagonist, enhanced the susceptibility of tumor cells to anoikis by mobilizing tumor cells into surrounding blood and significantly reduced distant metastasis in TNBC in situ model [294]. The interaction between chemokine and its homologous receptors is very important in tumor metastasis. In addition to the above studies on chemokine receptor CXCR4, chemokine receptor C–C motif chemokine receptor 7 (CCR7) was also found to be a sialylated protein and highly expressed in human breast cancer cells [295]. The homologous ligand chemokine (C–C motif) ligand 19 (CCL19) of CCR7 prevented anoikis and increased invasion by upregulating the survival promoting proteins Bcl-2 and Bcl-xL [296]. Sialyltransferase inhibitor AL10 restrained the proliferation and invasion of TNBC cells by suppressing the abnormal sialylation of CCR7 and then triggering anoikis [296]. Some studies had established a more aggressive anti-anoikis TNBC cell. BBR could promote the growth inhibition of anoikis-resistant TNBC cells by inducing cell cycle arrest at G0/G1 phase, which was more effective than traditional Adriamycin treatment for breast cancer [276]. In addition, HPW-RX40 restored the anoikis sensitivity of TNBC cells resistant to anoikis and induced cell death by reducing the activation and expression of β1 integrin and inhibiting the FAK pathway [297]. Interestingly, in addition to regulating the classic anoikis pathway as described above, certain small-molecule drugs could induce anti-anoikis mechanisms [298]. For instance, vacuolar ATPase (V-ATPase) is a proton pump located on the membrane of acidic organelles, which affected anoikis by regulating receptor recirculation through acidification of endosomes and lysosomes [299,300,301]. A V-ATPase inhibitor archazolid A induced reactive oxygen species and resists anoikis by promoting late BIM degradation mediated by ERK, Src and Akt kinase [298]. The results showed that archazolid A treatment inhibited the metastasis of TNBC cancer cells and reduced the lung metastasis of mouse breast cancer cells in vitro [298]. Inhibition of V-ATPase provides us with a unique perspective to inhibit TNBC cancer metastasis and study anoikis resistance [302].
Combination therapies of RCD subroutines with small-molecule compounds in TNBC
The interrelationships between different RCD subroutines
Autophagy and apoptosis are the main types of RCD of eukaryotic cells. Many strategies for treating TNBC concentrated on regulating apoptosis and autophagy to inhibit cancer initiation and development. Interestingly, autophagy, as a double-edged sword of cancer cells, achieved the purpose of treating cancer by promoting autophagy or inhibiting autophagy in different tumor microenvironments [303]. The induction of cytoprotective autophagy could promote the survival of TNBC cells [304]. Narciclasine could promote autophagy-dependent apoptosis in a dose-dependent manner by upregulating AMPK-ULK1 signal axis [305]. The latest study adopted the structure simplification strategy and obtained N-(1H-benzo[d].imidazole-2-yl)-4-(1-(2-(3-bromobenzoyl)hydrazono)ethyl)benzamide (compound 7C) with excellent mTOR enzyme inhibitory activity through virtual screening and bioactivity determination based on pharmacophore [306]. Compound 7C could induce autophagy cell death and apoptosis in TNBC cell line and exhibited the most effective inhibitory activity on TNBC cells among the analogous synthetics [306]. Interestingly, the inhibition of autophagy flux could also treat TNBC. Cantharidin, a terpenoid compound, could inhibit the transformation from LC3 I to LC3 II and the formation of autophagy with a significant inhibitory activity to cancer in TNBC cells and TNBC nude mouse models. Mechanically, Cantharidin suppressed the expression of beclin-1, finally inhibiting autophagy and inducing apoptosis [307]. Similarly, a new SL active component F1012-2, which consisted of three compounds, namely Eupalinolide G, Eupalinolide I and Eupalinolide J, isolated from Eupatorium lindleyanum DC could induce apoptosis in a caspase-dependent manner through endogenous and exogenous pathways, and the induced apoptosis could be enhanced by inhibiting autophagy and finally suppressing the growth of TNBC cells [308]. Clinical trials for TNBC had a high failure rate, and targeted therapeutic drugs for TNBC were rarely approved by Food and Drug Administration (FDA). Therefore, search for novel approaches of approved drugs might be a very promising and potential strategy for the treatment of TNBC [309]. Flubendazole, a broad-spectrum antibody drug, had been repositioned as a promising anticancer drug. Flubendazole regulated autophagy and apoptosis by targeting the key site thr113 of EVA1A and finally inhibited the proliferation and migration of TNBC [310].
Autophagy can usually prolong the survival time of cancer cells by removing damaged organelles and providing nutrients for cancer cells. Autophagy can also maintain the homeostasis of genome and internal environment, prevent inflammation or oxidative stress, and inhibit the occurrence, proliferation and metastasis of tumor cells [311, 312]. A series of small-molecule compounds targeting autophagy-related proteins (or autophagy process) have shown good anticancer effects in cancer cells. Therefore, targeted autophagy has great potential for the treatment of TNBC patients. However, the relationship between autophagy and apoptosis is not clear. The rationale for using autophagy inhibitors in combination with chemotherapeutic drugs is a better way to improve the efficacy of anticancer treatments and counteract TNBC resistance. Tamoxifen is a selective estrogen receptor modulator (SERMs). Due to the negative expression of ER receptor in TNBC, tamoxifen has a poor prognosis and even drug resistance against TNBC. A study found that tamoxifen induces autophagy in TNBC. Combined treatment with csc-3436 enhanced the tumor growth inhibitory effect of tamoxifen on TNBC compared with tamoxifen alone in vivo. The molecular mechanism may be that CSC-3436 converts tamoxifen-induced autophagy into apoptosis through AMPK/mTOR pathway and cleavage of ATG-5 [313]. Jatamanvaltrate P enhances the cleavage of PARP and caspase, while reducing the expression levels of cell cycle-related cyclin B1, cyclin D1 and cdc-2. It plays its cytotoxic and antitumor role in TNBC cell lines (MDA-MB-231, MDA-MB-453 and MDA-MB-468) and MDA-MB-231 xenografts by inducing apoptosis, and autophagy-dependent cell death [314].
Recently, a new antitumor drug 1,4,5-oxathiazinane-4,4-dioxide (OTD) was designed and synthesized [315]. The results showed that OTD induced necroptosis and apoptosis of TNBC cells, resulting in cell death and inhibition of proliferation in the dose-dependent manner [315]. Clinical studies showed that the poor prognosis of TNBC was related to the activation of PI3K/AKT pathway [316, 317]. A recent study exhibited that PI3K/AKT inhibitor AEZS 126 caused cell death by inducing apoptosis and necroptosis in TNBC cells [318]. Among the basal-like subtypes of TNBC, chemotherapy combined AEZS 126 with good toxicity characteristics and antitumor activity might be a potential strategy for TNBC clinical trials. GPX4 is an important ferroptotic cell death regulator [319], and its expression in TNBC is higher than that in other subtypes of breast cancer [320]. The decrease in GPX4 expression can induce apoptosis [321]. As a derivative of natural product parthenolide, DMOCPTL significantly inhibited the proliferation of TNBC cells by directly binding to GPX4 protein and induced GPX4 ubiquitination to promote ferroptosis. Also, DMOCPTL could upregulate EGR1, resulting in the activation of mitochondrial-mediated apoptosis [320]. The metabolic characteristics of cancer cells are at least partly attributed to the imbalance of specific amino acids and abnormal metabolism of amino acids [322,323,324]. A present study found that TNBC was highly sensitive to cystine starvation. Cystine starvation in TNBC cells increased the phosphorylation at eIF2α serine 51 and the protein expression levels of ATF4 and CHAC1 by activating GCN2, induced mitochondrial rupture, dysfunction, and ROS production, reduced the level of GSH, and induced necroptosis and ferroptosis, resulting in cell death [236, 322, 325].
Chemotherapy is a common scheme for the treatment of TNBC; however, the non-specific distribution and non-targeted side effects of chemotherapy limit its long-term application for patients [326]. The increasing development of nanomedicine was expected to solve these issues by specifically activating drug efficacy [327,328,329,330]. A photo-switchable microtubule inhibitor azobenzene combretastatin A4 (Azo-CA4) was loaded into up-conversion nanocarriers to promote microtubule decomposition and cell cycle arrest in G2/M phase by inducing Azo-CA4 photoisomerization, resulting in cell apoptosis. The reduction of Fe3+ to Fe2+ induced by ultraviolet light caused ferroptosis, significantly reduced the activity of TNBC cells and inhibited the tumor size of xenograft mouse model [331]. Although chemotherapy is widely used during TNBC treatment, the side effect was extremely serious according to the evaluation based on Common Terminology Criteria for Adverse Events. Moreover, chemotherapy could threaten the health of patients with high dosage. Even combining chemotherapy with radiation could not reduce the risk of recurrence and metastasis, or abate the threat of adverse effects [332]. In recent years, an increasing number of researchers focused on unconventional methods, such as bacterial toxins. LT-IIc, a member of the bacterial type II subfamily of heat-labile enterotoxin, which could promote TNBC cell death by inducing apoptosis and necroptosis [333].
Combination therapies of small-molecule compounds
Pharmacological regulation by inducing or inhibiting autophagy [334, 335], combined application of inducing apoptosis to produce effective innovative strategies, could enhance therapeutic activity. The combined application of torin-1 (TOR) and chloroquine (CQ) could show a half-lethal effect by inducing autophagic cell death and apoptotic cell death [336]. The combined application of CQ and DOX induced AVO accumulation through non-major (or off-target) drug effects by inhibiting apoptotic cell death and ultimately increased growth inhibition [336]. Copper (I) nicotinate complex (CNC) combined with DOX could inhibit autophagy by inducing cell cycle arrest and reduced the dose of DOX applied to TNBC cells [336]. The expression level of EGFR was higher in 40% of TNBC patients [337, 338]. However, EGFR inhibitors are mostly ineffective in the treatment of TNBC patients, and increasing evidence supports that autophagy is currently related to the drug resistance of EGFR inhibitors in the treatment of TNBC. Inhibition of autophagy by 3-methyladenine or bafilomycin A1 increased the sensitivity of gefitinib to TNBC cell lines and more strongly inhibited cell viability and colony formation by inducing G0/G1 arrest and DNA damage and activating mitochondrial-dependent apoptosis pathway. The combined application of autophagy inhibitor and gefitinib also improved the antitumor effect in TNBC nude mouse model. These results suggest that targeted autophagy should be considered as an effective therapeutic strategy to enhance the sensitivity of EGFR inhibitors to TNBC [339]. ND, a carbon-based nanomaterial, combined PTX and cetuximab (CET) for the targeted treatment of EGFR-positive TNBC cells. The results showed that ND-PTX-CET enhanced the mitotic catastrophe and apoptosis of TNBC cells by targeting EGFR, which provided a feasible strategy for the treatment of TNBC based on RCD synergistic mechanism [200]. Cetuximab was a classic monoclonal antibody against EGFR [340,341,342,343]. However, with the advancement of the course of treatment, cancer cells rapidly gained resistance to cetuximab. Hence, it is urgent to develop novel combination therapy [344]. MicroRNA (miRNA)-155-5p was found to be upregulated in TNBC cells as a new biomarker [345]. By upregulating gasdermin E-N-terminal subunit (GSDME-N) and cleaving caspase-1 [346], miR-155-5p antagonist combined with cetuximab could promote apoptosis and pyroptosis and inhibit the proliferation and migration of EGFR overexpressing TNBC cells. Meanwhile, miR-155-5p antagonist also enhanced the antitumor effect of cetuximab in TNBC xenotransplantation model [347]. Cancer cells with anoikis resistance are prone to metastasis [348], which is reported to be related to integrin and growth factor receptor [349,350,351,352,353,354]. HPW-RX40, which inhibited integrin, combined with EGFR inhibitor AG1478, could inhibit FAK/paxillin phosphorylation and significantly induce TNBC cell death [297]. Blocking both integrin and growth factor-dependent survival pathways may be a potential strategy to overcome the resistance of TNBC cancer cells to EGFR inhibitors [355]. Chk1 inhibitor could develop as a single drug [208]; in order to improve its anti-TNBC tumor effect, it is worthwhile to explore the potential synergistic effect of chk1 inhibitors and chemotherapy. A recent study found that the long-term treatment of DNA damaging agent carboplatin, as a cycle nonspecific anticancer drug for main mechanism of actions (MOA), is DNA damage and inhibits tumor growth and could induce the mitotic arrest through non-major (or off-target) drug effects of TNBC. The combined application of carboplatin and chk1 inhibitor AZD7762 inhibited the G2-M conversion by inhibiting chk1 pathway, resulting in the accumulation of DNA damage, a significant increase in the incidence of spindle multipolarity and cytokinesis failure. The co-treatment promoted the polynuclear and polyploidization of TNBC tumor cells, eventually leading to mitotic catastrophe and apoptosis. The results showed that the combination of DNA damaging agent and chk1 inhibitor could synergistically inhibit the growth and proliferation of multiple TNBC cell lines in vitro [356, 357]. In addition, ganetespib could also promote mitotic catastrophe and apoptosis when used in combination with docetaxel in vitro [358]. Similarly, their combination exerted significantly synergistic activity in vivo [358]. It is revealed that the synergistic induction of mitotic catastrophe and apoptosis can be used as an alternative strategy for the treatment of TNBC. Interestingly, recent studies achieved effective results in the treatment of TNBC through combination therapy alone or in parallel with chemotherapy. ATR inhibitor AZD6738 and WEE1 inhibitor AZD1775 inactivated RAD51-mediated homologous recombination [359], activated cyclin-dependent kinase 1 (CDK1) activity, forced DNA-damaged cells into mitosis, and induced serious mitotic abnormalities and mitotic catastrophes, eventually resulting in TNBC cell death, and improving the sensitivity of TNBC to cisplatin and PARP inhibitors [360, 361]. BMS-754807, as a dual IGF-1R/InsR inhibitor, combined with the chemotherapeutic drug docetaxel could increase cell apoptosis and induce mitotic catastrophe, inhibit the growth of TNBC primary human tumor transplantation MC1, and regress tumors [362]. Additionally, the combination of IGF-1R inhibitor NVP-AEW541 and autophagy inhibitor 3-mA could improve the therapeutic effect of IGF-1R inhibitors in TNBC cells, which provided a direction for the combined treatment strategy based on IGF-1R inhibitors [363]. Multiple schemes in parallel revealed an innovative and effective targeted therapy in TNBC. Functional studies, clinical dataset analysis and breast cancer specimens showed that TNBC had unique vulnerability to ferroptosis inducers [364, 365], and BRD4 transcripts as well as protein levels were significantly enriched in TNBC [366,367,368]. The results showed that classical bet inhibitor JQ1 and proteasome inhibitor bortezomib (BTZ) showed effective synthetic lethality to major TNBC subtypes by inducing ferroptosis [369]. Likewise, BET inhibitor JQ1 and CXCR2 inhibitor SB225002 had high efficacy in CO-inhibition of M/MSL TNBC subtypes by inducing apoptosis in vivo and in vitro [369]. These effective combination therapies revealed the inherent susceptibility of TNBC to ferroptosis and highlighted the potential strategy of ferroptosis as a drug target of TNBC. The expression level of mucin 1 (MUC1) transmembrane glycoprotein increased in most TNBC [370, 371]. In this case, erastin is ineffective in inducing ferroptosis [372]. Application of xCT inhibitor sulfasalazine could inhibit MUC1 gene transcription by enhancing histone and DNA methylation on MUC1 promoter [372]. The amplification of MUC1 gene led to over-expression of the MUC1 C-terminal subunit (MUC1-C) complex, which could mediate the TNBC cell self-renewal ability and tumor origin [373]. The results showed that xCT inhibitor reduced the expression level of MUC1-C by inhibiting MUC1-C/xCT pathway, induced ferroptosis and finally inhibited the survival rate of TNBC cells [372]. These findings suggested the prospect of the combination of xCT inhibitor and erastin in the treatment of TNBC.
In addition to the conventional combination therapy, virotherapy destroys malignant tumors without damaging normal tissues, making oncolytic virus a promising antitumor drug. A recent study found that Dox could specifically enhance the oncolytic effect of M1 virus, promote virus replication in tumor, further trigger apoptosis and necroptosis by non-major (or off-target) drug effects and significantly inhibit the tumor growth of TNBC in vivo [374]. These data indicated the combination of molecular diagnosis and viral therapy as a promising approach for the development of anti-TNBC strategies. In addition, the combination of radiotherapy and ferroptosis induced exerted coefficient effect on TNBC by affecting ROS [375]. Ferroptosis could make cancer cells more sensitive to radiotherapy [376]. Recently, Holo-Lf was found to promote ROS generation and improved the hypoxic microenvironment by reducing the expression of HIF-1α in TNBC cells. Concurrently, Holo-Lf induced ferroptosis and ultimately promoted radiation-induced DNA damage [375]. The combined application of Holo-Lf and radiotherapy enhanced the sensitivity of TNBC to radiotherapy (Table 4).
Targeted small-molecule compounds in TNBC clinical trials
In preclinical and clinical trials, a series of small-molecule compounds are used to explore their therapeutic effects against TNBC, and some of them have displayed remarkable results. For example, as a dual inhibitor of PI3K and HDAC, CUDC-907 has shown significant anticancer effects on TNBC cell lines in preclinical studies. When it combined with TRAIL, the cleavage of caspase-8, caspase-9 and PARP increased and induced breast cancer cell apoptosis. Besides, CUDC-907 could upregulate the expression of DR5 and downregulate the level of XIAP, Bcl-2 and Bcl-xl to promote apoptosis mediated by TRAIL. A phase 1 clinical trial (NCT02307240) evaluated the safety, tolerability and validity of CUDC-907 in advanced/relapsed solid tumors, including TNBC, high-grade serous ovarian cancer (HGSOC) and NUT midline carcinoma (NMC) [377]. In addition, ONC201, also as a TRAIL-inducing compound, could trigger apoptosis by targeting DR5. In a phase 2 clinical trial (NCT03733119), Akt/ERK inhibitor ONC201 and a methionine-restricted (MR) diet were studied in the treatment of metastatic TNBC. ONC201 could target tumor cells and cleared them without affecting normal cells, and MR diet could enhance the activity of ONC201. In another phase 2 clinical trial (NCT03394027), ONC201 was proved to kill breast cancer and endometrial cancer cells, but was not sure if ONC201 help shrink tumors of TNBC or endometrial cancers [378]. Etoposide (ET) could stimulate apoptosis with TRAIL and when ET, in combination with Dox, could markedly upregulate DR5 expression by non-major (or off-target) drug effects in TNBC cells. A phase 2 clinical trial (NCT04452370) evaluated the effect of the oral topoisomerase-II inhibitor etoposide combined with the anlotinib in the treatment of recurrent or metastatic TNBC [379].
Additionally, ENMD-2076 was a small-molecule inhibitor that was cytotoxic to p53-mutated TNBC cell lines. Significant antitumor, antiproliferative and proapoptotic activities of ENMD-2076 were observed in TNBC cells. The increased expression of p53 and p73 protein could enhance the sensitivity of cancer cells to treatment. A phase 2 clinical trial (NCT01639248) showed that ENMD-2076 could lead to partial response or clinical benefit lasting more than 6 months in patients with previously treated locally advanced or metastatic TNBC [380, 381]. NVP-BEZ235 as a PI3K/mTOR inhibitor was found to induce autophagy by degrading mutant p53 protein. Meanwhile, autophagy promoted by BEZ235 was also related to the downregulation of Akt/mTOR pathway. NVP-BEZ235, the combination of MEK1/2 inhibitor and MEK162 in phase 2 clinical trial (NCT01337765), evaluated the safety and preliminary antitumor activity in the treatment of TNBC, pancreatic cancer and other advanced solid tumors [382]. It was worth noting that more and more preclinical and clinical activities had verified the promising strategy of small molecular compounds to intervene TNBC by activating mitotic catastrophe. Clinical evidence of tumor regression and preclinical activity profile (NCT01677455) showed that ganetespib, as a selective HSP90 inhibitor, could inhibit cell growth in TNBC cell line and inhibit lung metastasis in xenotransplantation model by enhancing DNA damage and mitotic catastrophe and inactivating a variety of carcinogenic pathways. Metastatic lung and lymphatic lesions were also suppressed significantly in patients with ganetespib monotherapy [358] (Table 5).
Concluding remarks and future perspectives
Cell death occurs in many forms to cope with different environmental challenges, and it has been gradually recognized that RCD can involve much more than the classical apoptosis pathway. Particularly, autophagy, necrosis and ferroptosis, belonging to RCD, follow their specific mechanistic procedures and appear in the corresponding conditions to decide cell fate. Abnormal cell growth without typical RCD can lead to many types of disease, including TNBC, which is known as the “hidden killer of women” due to its aggressive properties and limited treatment options. More recently, numerous factors of RCD have been found to be associated with the occurrence and progression TNBC. For instance, TNBC is one of the earliest genetic diseases found to be related to autophagy dysfunction. Autophagy dysfunction can help to inhibit the attack of T-lymphocytes on TNBC tumors, aiding immune escape for these tumors. Under some circumstances, autophagy can degrade tumor cells and inhibit tumor growth and metastasis through its own actions [383], suggesting that targeting autophagy will be an effective therapeutic strategy.
Hitherto, a promising area for small-molecule drug discovery has been taking an advantage of the concept that precisely targeting different molecular characteristics of TNBC subtypes. For instance, the LAR tumor characterized by AR expression is a subtype of TNBC, which is sensitive to the endocrine regulation of AR antagonists, such as enzalutamide [384]. The classifications of TNBC can be utilized as a prognostic or predictive means for better individualized therapies in TNBC patients theoretically. The online tool “TNBCtype-4” (also known as “Lehmann Classifier”) can be used for Lehmann typing, to make more accurate diagnosis, biomarker selection, drug discovery and more appropriate therapeutic strategies in TNBC [385, 386]. With the deepening of the biological behavior and molecular typing of TNBC, the development of more clinical studies and the optimization of different approaches, the diagnosis and treatment of TNBC will be more accurate and individualized.
Although small-molecule drugs have achieved encouraging results for TNBC patients and brought a new hope, they still face many challenges. Firstly, the efficacy of small-molecule drugs is based on the joint standard scheme. If they are not used according to the drug standards, the side effects of drugs will increase, the tolerance of patients will decrease, and ultimately the therapeutic efficacy will be poor. Secondly, small-molecule drug monotherapy can obtain an ideal TNBC tumor cell inhibition, but it is prone to drug-resistant mutations. Multidrug resistance sites are easy to appear after small-molecule drugs used. With the continuous development of such small-molecule drugs with low resistance, high curative effect and few side effects, as well as the research of new drug combination schemes, it is believed that it will bring a new development for the treatment of TNBC.
In summary, it is valuable for development of targeted therapies to conduct more in-depth investigations of RCD on the purpose of more accurately determining the intricate molecular mechanisms of each subroutine of RCD, and further exploring the relationships between them. With a better understanding of the complex regulatory mechanisms of RCD, we can anticipate a breakthrough for discovery of more targeted small-molecule drugs for fighting TNBC in the future.
Availability of data and materials
Not applicable.
Abbreviations
- ACD:
-
Accidental death
- AIF:
-
Apoptosis-inducing factor
- AKT:
-
Protein kinase B
- AMPK:
-
Adenosine 5′-monophosphate-activated protein kinase
- Apaf-1:
-
Apoptotic protease activating factor 1
- AQP1:
-
Aquaporin1
- As4O6:
-
Tetraarsenic hexoxide
- ASC:
-
Apoptosis-associated speck-like protein containing a CRAD
- ATF3:
-
Activating transcription factor 3
- ATG:
-
Arctigenin
- ATG:
-
Autophagy-associated protein
- ATP:
-
Adenosine triphosphate
- ATPase:
-
Adenosine triphosphatase
- ATR:
-
Rad3-related protein
- AURKA:
-
Aurora kinase A
- AXL:
-
Anexelekto
- Azo-CA4:
-
Azobenzene combretastatin A4
- BAM:
-
Budlein A methylacrylate
- BBC3:
-
Bcl-2 binding component 3
- BBR:
-
Berberine
- Bcl-2:
-
B-cell lymphoma 2
- Bcl-xL:
-
B-cell lymphoma extra-large
- BET:
-
Bromodomain and extraterminal protein
- BETi:
-
Bromodomain and extraterminal protein inhibitors
- Bim:
-
Bcl-2 interacting mediator of cell death
- BL1/2:
-
Basal-like 1/2
- BRD4:
-
Bromodomain 4
- BTZ:
-
Bortezomib
- CA:
-
Cinnamic acid
- Cav-1:
-
Caveolin-1
- CCL19:
-
Chemokine (C–C motif) ligand 19
- CCR7:
-
C–C motif chemokine receptor 7
- CET:
-
Cetuximab
- C-FLIP:
-
Cellular FLICE-inhibitory protein
- CDK:
-
Cyclin-dependent kinase
- Chk1:
-
Checkpoint kinase 1
- CHOP:
-
C/EBP homologous protein
- CNC:
-
Copper (I) nicotinate complex
- CSC:
-
Cancer stem cell
- Cu:
-
Copper
- Cur:
-
Curcumin
- CXCR4:
-
C-X-C motif chemokine receptor 4
- Cyt-c:
-
Cytochrome c
- dATP:
-
Deoxyadenosine triphosphate
- DIABLO:
-
Direct IAP-binding protein with low pI
- DISC:
-
Death-inducing signaling complex
- DNA:
-
Deoxyribonucleic acid
- Dox:
-
Doxorubicin
- DR3/4/5:
-
Death receptor 3/4/5
- DSF:
-
Disulfiram
- dsRNA:
-
Double-stranded RNA
- e.g.:
-
Exempli gratia
- EC50:
-
Median effective concentration
- ECM:
-
Extracellular matrix
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial–mesenchymal transition
- ER:
-
Estrogen receptor
- ERK:
-
Extracellular signal-regulated kinase
- ET:
-
Etoposide
- FA:
-
Folate
- FADD:
-
Fas associated via death domain
- FDA:
-
Food and Drug Administration
- FIP200:
-
Focal adhesion kinase interacting protein of 200 kD
- FoxO:
-
Forkhead box O
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- GA:
-
Gallic acid
- GO:
-
Gene ontology
- GPT:
-
Ginsenoside panaxatriol
- GPX4:
-
Glutathione peroxidase 4
- GSDM:
-
Gasdermin
- GSDMD:
-
Gasdermin D
- GSDME:
-
Gasdermin E
- GSDME-N:
-
Gasdermin E-N-terminal subunit
- GSH:
-
Glutathione
- HER2:
-
Human epidermal growth factor 2
- HGSOC:
-
High-grade serous ovarian cancer
- Holo-Lf:
-
Hollo lactate
- HSCs:
-
Hematopoietic stem cells
- HSP70:
-
Heat shock protein 70
- i.e.:
-
Id est
- IC50:
-
Half maximal inhibitory concentration
- IKB:
-
Inhibitor of nuclear factor kappa-B
- IKK:
-
Inhibitor of nuclear factor kappa-B kinase
- IKKβ:
-
Inhibitor kappa-B kinase-β
- IL-6:
-
Interleukin-6
- IM:
-
Immunomodulatory
- IRAK1:
-
Interleukin-1 receptor-associated kinase 1
- KRT15:
-
Keratin 15
- LAR:
-
Luminal androgen receptor
- LC3:
-
Light chain 3
- Lf:
-
Lactoferrin
- lncRNA:
-
Long noncoding RNA
- M:
-
Mesenchymal
- MAPK:
-
Mitogen-activated protein kinase
- mAtg13:
-
Mammalian autophagy-associated protein 13
- Mcl-1:
-
Myeloid cell leukemia-1
- MDM2:
-
Murine double minute 2
- MDSCs:
-
Myeloid-derived suppressor cells
- MEG3:
-
Maternally expressed gene 3
- MEK:
-
Mitogen-activated protein kinase kinase
- MIF:
-
Migration inhibitory factor
- miRNA:
-
MicroRNA
- MLKL:
-
Mixed lineage kinase domain-like
- MMP:
-
Matrix metallopeptidase
- MOM:
-
Mitochondrial outer membrane
- MR:
-
Methionine-restricted
- mRNA:
-
Messenger ribonucleic acid
- MSL:
-
Mesenchymal stem-like
- mTOR:
-
Mammalian target of rapamycin
- mTORC:
-
Mammalian target of rapamycin complex
- MUC1:
-
Mucin 1
- MUC1-C:
-
MUC1 C-terminal subunit
- NCCD:
-
Nomenclature of Cell Death Committee
- ND:
-
Nanodiamond
- NF-κB:
-
Nuclear factor kappa-B
- NLRP3:
-
NLR family, pyrin domain containing 3
- NMC:
-
NUT midline carcinoma
- OL:
-
Oleuropein
- OTD:
-
1,4,5-Oxathiazinane-4,4-dioxide
- p-ACC:
-
Phospho-acetyl-CoA carboxylase
- PAD:
-
Peptidylarginine deiminase
- PARP1:
-
Poly-ADP-ribose polymerase 1
- PCB 104:
-
Polychlorinated biphenyl 104
- PDGFR-β:
-
Platelet-derived growth factor receptor β
- PD-L1:
-
Programmed cell death ligand 1
- PH:
-
Phloretin
- PI3K:
-
Phosphatidylinositol 3 kinase
- PI3KC:
-
Phosphatidylinositol 3 kinase complex
- PICH:
-
Plk1-interacting checkpoint helicase
- PIKK:
-
PI3K-like kinase
- PKC:
-
Protein kinase C
- PKCθ:
-
Protein kinase C theta
- Plk1:
-
Polo-like kinase 1
- PMAIP1:
-
Phorbol-12-myristate-13-acetate-induced protein 1
- POLD1:
-
Polymerase (DNA) delta 1
- PR:
-
Progesterone receptor
- PRKCQ:
-
Protein kinase c theta
- PTEN:
-
Phosphatase and tensin homologue deleted on chromosome ten
- PTER:
-
Pterostilbene
- Rb:
-
Retinoblastoma
- RCD:
-
Regulated cell death
- RIG-I:
-
Retinoic acid-inducible gene-I
- RIPK:
-
Receptor-interacting protein
- RLRs:
-
Retinoic acid-inducible gene-I (RIG-I)-like receptors
- ROS:
-
Reactive oxygen species
- SAPK:
-
Stress-activated protein kinase
- SG:
-
Sophoraflavanone G
- SGNI:
-
Shuganning injection
- SHP-1:
-
Src homology region 2 domain containing phosphatase 1
- SIRT6:
-
Silent information regulator 6
- SJW:
-
St. John's wort
- SJWE:
-
SJW extract
- SLC3A2:
-
Solute carrier family 3 member 2
- SLC7A11:
-
Solute carrier family 7 member 11
- SMAC:
-
Second mitochondria-derived activator of caspase
- ssDNA:
-
Single-stranded deoxyribonucleic acid
- STAT3:
-
Signal transducer and activator of transcription 3
- TBMS-V:
-
Tubeimoside V
- TNBC:
-
Triple-negative breast cancer
- TNFR:
-
Tumor necrosis factor receptors
- TOR:
-
Torin-1
- TRADD:
-
TNFRSF1A associated via death domain
- TRAF2:
-
TNF receptor-associated factor 2
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- TSC1/2:
-
Tuberous sclerosis complex 1/2
- ULK1:
-
Unc-51-like autophagy activating kinase 1
- UNS:
-
Unclassified
- V-ATPase:
-
Vacuolar-ATPase
- Vps34:
-
Vacuolar protein sorting 34
- XIAP:
-
X-linked inhibitor of apoptosis protein
- XPO-1:
-
Exportin 1
- ZnPP:
-
Zn protoporphyrin IX
References
Johansson A, Trewin C, Hjerkind K, Ellingjord-Dale M, Johannesen T, Ursin G. Breast cancer-specific survival by clinical subtype after 7 years follow-up of young and elderly women in a nationwide cohort. Int J Cancer. 2019;144(6):1251–61.
Sharma M, Turaga R, Yuan Y, Satyanarayana G, Mishra F, Bian Z, et al. Simultaneously targeting cancer-associated fibroblasts and angiogenic vessel as a treatment for TNBC. J Exp Med. 2021;218(4):e20200712.
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67.
Islam S, Dasgupta H, Basu M, Roy A, Alam N, Roychoudhury S, et al. Downregulation of beta-catenin in chemo-tolerant TNBC through changes in receptor and antagonist profiles of the WNT pathway: clinical and prognostic implications. Cell Oncol (Dordrecht). 2020;43(4):725–41.
Wang L, Hu X, Wang P, Shao Z. Integrative 3’ untranslated region-based model to identify patients with low risk of axillary lymph node metastasis in operable triple-negative breast cancer. Oncologist. 2019;24(1):22–30.
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri E, Baehrecke E, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3–11.
Galluzzi L, Vitale I, Abrams J, Alnemri E, Baehrecke E, Blagosklonny M, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19(1):107–20.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541.
Daley-Bauer LP, Roback L, Crosby LN, McCormick AL, Feng Y, Kaiser WJ, et al. Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc Natl Acad Sci U S A. 2017;114(13):E2786–95.
Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. 2015;16(6):329–44.
Ke B, Tian M, Li J, Liu B, He G. Targeting programmed cell death using small-molecule compounds to improve potential cancer therapy. Med Res Rev. 2016;36(6):983–1035.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–85.
Zeng C, Matsuda K, Jia WH, Chang J, Kweon SS, Xiang YB, et al. Identification of susceptibility loci and genes for colorectal cancer risk. Gastroenterology. 2016;150(7):1633–45.
Wang G, Zhao Y, Liu Y, Sun D, Zhen Y, Liu J, et al. Discovery of a novel dual-target inhibitor of ERK1 and ERK5 that induces regulated cell death to overcome compensatory mechanism in specific tumor types. J Med Chem. 2020;63(8):3976–95.
Pfeffer CM, Singh ATK. Apoptosis: a target for anticancer therapy. Int J Mol Sci. 2018;19(2):448.
Kiraz Y, Adan A, Yandim MK, Baran Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumor Biol. 2016;37(7):8471–86.
Pfeffer CM, Singh ATK. Apoptosis: a target for anticancer therapy. Int J Mol Sci. 2018;19(2):10.
Derakhshan A, Chen Z, Van Waes C. Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin Cancer Res. 2017;23(6):1379–87.
Croce CM, Reed JC. Finally, an apoptosis-targeting therapeutic for cancer. Cancer Res. 2016;76(20):5914–20.
Guo Y, Zhao Y, Wang G, Chen Y, Jiang Y, Ouyang L, et al. Design, synthesis and structure-activity relationship of a focused library of β-phenylalanine derivatives as novel eEF2K inhibitors with apoptosis-inducing mechanisms in breast cancer. Eur J Med Chem. 2018;143:402–18.
Liu Y, Zhen Y, Wang G, Yang G, Fu L, Liu B, et al. Designing an eEF2K-Targeting PROTAC small molecule that induces apoptosis in MDA-MB-231 cells. Eur J Med Chem. 2020;204:112505.
Bedard PL, Hyman DM, Davids MS, Siu LL. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet. 2020;395(10229):1078–88.
Liao M, Zhang J, Wang G, Wang L, Liu J, Ouyang L, et al. Small-molecule drug discovery in triple negative breast cancer: current situation and future directions. J Med Chem. 2021;64(5):2382–418.
Lee YT, Tan YJ, Oon CE. Molecular targeted therapy: treating cancer with specificity. Eur J Pharmacol. 2018;834:188–96.
Varghese E, Samuel SM, Abotaleb M, Cheema S, Mamtani R, Busselberg D. The, “Yin and Yang” of natural compounds in anticancer therapy of triple-negative breast cancers. Cancers. 2018;10(10):346.
Wang Y, Zhong J, Bai JJ, Tong RS, An FF, Jiao PC, et al. The application of natural products in cancer therapy by targeting apoptosis pathways. Curr Drug Metab. 2018;19(9):739–49.
Varfolomeev E, Vucic D. Intracellular regulation of TNF activity in health and disease. Cytokine. 2018;101:26–32.
Cruceriu D, Baldasici O, Balacescu O, Berindan-Neagoe I. The dual role of tumor necrosis factor-alpha (TNF-alpha) in breast cancer: molecular insights and therapeutic approaches. Cell Oncol. 2020;43(1):1–18.
Rossin A, Miloro G, Hueber AO. TRAIL and FasL functions in cancer and autoimmune diseases: towards an increasing complexity. Cancers. 2019;11(5):18.
Yuan X, Gajan A, Chu Q, Xiong H, Wu KM, Wu GS. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev. 2018;37(4):733–48.
Dyari HRE, Rawling T, Chen YJ, Sudarmana W, Bourget K, Dwyer JM, et al. A novel synthetic analogue of omega-3 17,18-epoxyeicosatetraenoic acid activates TNF receptor-1/ASK1/JNK signaling to promote apoptosis in human breast cancer cells. Faseb J. 2017;31(12):5246–57.
Chiu CF, Lin YQ, Park JM, Chen YC, Hung SW, Chiu CC, et al. The novel camptothecin derivative, CPT211, induces cell cycle arrest and apoptosis in models of human breast cancer. Biomed Pharmacother. 2020;128:110309.
von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer. 2017;17(6):352–66.
Wu YC, Wang HC, Chen CJ, Liu LC, Way TD. Pterostilbene enhances TRAIL-induced apoptosis in TRAIL-resistant triple negative breast cancer cells. Cancer Res. 2017;65(51):11179–91.
Liang X, Chen QY, Seabra GM, Matthew S, Kwan JC, Li CL, et al. Bifunctional doscadenamides activate quorum sensing in gram-negative bacteria and synergize with TRAIL to induce apoptosis in cancer cells. J Nat Prod. 2021;84(3):779–89.
Farghadani R, Rajarajeswaran J, Mohd Hashim NB, Abdulla MA, Muniandy S. A novel β-diiminato manganeseIII complex as the promising anticancer agent induces G0/G1 cell cycle arrest and triggers apoptosis via mitochondrial-dependent pathways in MCF-7 and MDA-MB-231 human breast cancer cells. RSC Adv. 2017;7(39):24387–98.
Farghadani R, Seifaddinipour M, Rajarajeswaran J, Abdulla MA, Hashim NBM, Khaing SL, et al. In vivo acute toxicity evaluation and in vitro molecular mechanism study of antiproliferative activity of a novel indole Schiff base beta-diiminato manganese(III) complex in hormone-dependent and triple negative breast cancer cells. PeerJ. 2019;7:e7686.
Wu J, Ding Y, Chen CH, Zhou ZM, Ding CY, Chen HY, et al. A new oridonin analog suppresses triple-negative breast cancer cells and tumor growth via the induction of death receptor 5. Cancer Lett. 2016;380(2):393–402.
Greer YE, Gilbert SF, Gril B, Narwal R, Brooks DLP, Tice DA, et al. MEDI3039, a novel highly potent tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor 2 agonist, causes regression of orthotopic tumors and inhibits outgrowth of metastatic triple-negative breast cancer. Breast Cancer Res. 2019;21(1):27.
Campbell KJ, Tait SWG. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018;8(5):180002.
Shahar N, Larisch S. Inhibiting the inhibitors: targeting anti-apoptotic proteins in cancer and therapy resistance. Drug Resist Updat. 2020;52:100712.
Knight T, Luedtke D, Edwards H, Taub JW, Ge Y. A delicate balance - The BCL-2 family and its role in apoptosis, oncogenesis, and cancer therapeutics. Biochem Pharmacol. 2019;162:250–61.
Campbell KJ, Tait SWG. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018;8(5):11.
Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017;16(4):273–84.
Hafezi S, Rahmani M. Targeting BCL-2 in cancer: advances, challenges, and perspectives. Cancers (Basel). 2021;13(6):1292.
Eldehna WM, El-Naggar DH, Hamed AR, Ibrahim HS, Ghabbour HA, Abdel-Aziz HA. One-pot three-component synthesis of novel spirooxindoles with potential cytotoxic activity against triple-negative breast cancer MDA-MB-231 cells. J Enzyme Inhibit Med Chem. 2017;33(1):309–18.
Eldehn WM, Almahli H, Al-Ansary GH, Ghabbour HA, Aly MH, Ismael OE, et al. Synthesis and in vitro anti-proliferative activity of some novel isatins conjugated with quinazoline/phthalazine hydrazines against triple-negative breast cancer MDA-MB-231 cells as apoptosis-inducing agents. J Enzyme Inhibit Med Chem. 2017;32(1):600–13.
Huang WC, Gu PY, Fang LW, Huang YL, Lin CF, Liou CJ. Sophoraflavanone G from Sophora flavescens induces apoptosis in triple-negative breast cancer cells. Phytomedicine. 2019;61:152852.
Zhou WH, Fang H, Wu QJ, Wang XY, Liu R, Li FB, et al. Ilamycin E, a natural product of marine actinomycete, inhibits triple-negative breast cancer partially through ER stress-CHOP-Bcl-2. Int J Biol Sci. 2019;15(8):1723–32.
Moghtaderi H, Sepehri H, Delphi L, Attari F. Gallic acid and curcumin induce cytotoxicity and apoptosis in human breast cancer cell MDA-MB-231. Bioimpacts. 2018;8(3):185–94.
Zhang WD, Yu WY, Cai GP, Zhu JW, Zhang C, Li SS, et al. A new synthetic derivative of cryptotanshinone KYZ3 as STAT3 inhibitor for triple-negative breast cancer therapy. Cell Death Dis. 2021;12(6):522.
Ge W, Hao X, Han F, Liu Z, Wang T, Wang M, et al. Synthesis and structure-activity relationship studies of parthenolide derivatives as potential anti-triple negative breast cancer agents. Eur J Med Chem. 2019;166:445–69.
Liu G, Yin T, Kim Y, Ding CY, Yu Z, Wang H, et al. Structure-activity relationship studies on Bax activator SMBA1 for the treatment of ER-positive and triple-negative breast cancer. Eur J Med Chem. 2019;178:589–605.
Nedeljkovic M, Damjanovic A. Mechanisms of chemotherapy resistance in triple-negative breast cancer-how we can rise to the challenge. Cells. 2019;8(9):957.
Cui Y, Huang Y, Wu X, Zheng M, Xia Y, Fu Z, et al. Hypoxia-induced tRNA-derived fragments, novel regulatory factor for doxorubicin resistance in triple-negative breast cancer. J Cell Physiol. 2019;234(6):8740–51.
Ciocan-Cartita CA, Jurj A, Zanoaga O, Cojocneanu R, Pop LA, Moldovan A, et al. New insights in gene expression alteration as effect of doxorubicin drug resistance in triple negative breast cancer cells. J Exp Clin Cancer Res. 2020;39(1):241.
Tang D, Ma J, Chu Z, Wang X, Zhao W, Zhang Q. Apatinib-induced NF-κB inactivation sensitizes triple-negative breast cancer cells to doxorubicin. Am J Transl Res. 2020;12(7):3741–53.
Lee KS, Lee MG, Kwon YS, Nam KS. Arctigenin enhances the cytotoxic effect of doxorubicin in MDA-MB-231 breast cancer cells. Int J Mol Sci. 2020;21(8):2997.
Kumari S, Badana AK, Mohan GM, Naik GS, Malla R. Synergistic effects of coralyne and paclitaxel on cell migration and proliferation of breast cancer cells lines. Biomed Pharmacother. 2017;91:436–45.
Wang DP, Nie Z, Jiang XY, Ye JX, Wei ZM, Cheng DT, et al. Pyrrolo 3,4-b -quinolin-9-amine compound FZU-0038-056 suppresses triple-negative breast cancer partially through inhibiting the expression of Bcl-2. Aging-Us. 2020;12(10):9621–32.
Lucantoni F, Lindner AU, O’Donovan N, Düssmann H, Prehn JHM. Systems modeling accurately predicts responses to genotoxic agents and their synergism with BCL-2 inhibitors in triple negative breast cancer cells. Cell Death Dis. 2018;9(2):42.
Li XX, Wang DQ, Sui CG, Meng FD, Sun SL, Zheng J, et al. Oleandrin induces apoptosis via activating endoplasmic reticulum stress in breast cancer cells. Biomed Pharmacother. 2020;124:109852.
Peng F, Xiong L, Peng C. (-)-Sativan inhibits tumor development and regulates miR-200c/PD-L1 in triple negative breast cancer cells. Front Pharmacol. 2020;11:251.
Shrivastava S, Jeengar MK, Thummuri D, Koval A, Katanaev VL, Marepally S, et al. Cardamonin, a chalcone, inhibits human triple negative breast cancer cell invasiveness by downregulation of Wnt/-beta-catenin signaling cascades and reversal of epithelial-mesenchymal transition. BioFactors. 2017;43(2):152–69.
Pirali M, Taheri M, Zarei S, Majidi M, Ghafouri H. Artesunate, as a HSP70 ATPase activity inhibitor, induces apoptosis in breast cancer cells. Int J Biol Macromol. 2020;164:3369–75.
Peng F, Xiong L, Xie XF, Tang HL, Huang RZ, Peng C. Isoliquiritigenin derivative regulates miR-374a/BAX axis to suppress triple-negative breast cancer tumorigenesis and development. Front Pharmacol. 2020;11:378.
Li J, Liu J, Wang RQ, Chen H, Li C, Zhao MG, et al. Trifluridine selectively inhibits cell growth and induces cell apoptosis of triple-negative breast cancer. Am J Cancer Res. 2020;10(2):507–22.
Li XL, Yu XP, Zhou DF, Chen B, Li WJ, Zheng XR, et al. CCT020312 inhibits triple-negative breast cancer through PERK pathway-mediated G1 phase cell cycle arrest and apoptosis. Front Pharmacol. 2020;11:737.
Ono H, Horinaka M, Sukeno M, Morita M, Yasuda S, Nishimoto E, et al. Novel RAF/MEK inhibitor CH5126766/VS-6766 has efficacy in combination with eribulin for the treatment of triple-negative breast cancer. Cancer Sci. 2021;112(10):4166–75.
Taş İ, Varlı M, Son Y, Han J, Kwak D, Yang Y, et al. Physciosporin suppresses mitochondrial respiration, aerobic glycolysis, and tumorigenesis in breast cancer. Phytomedicine. 2021;91:153674.
Zhang M, Zheng J, Nussinov R, Ma B. Release of cytochrome C from Bax pores at the mitochondrial membrane. Sci Rep. 2017;7(1):2635.
Santucci R, Sinibaldi F, Cozza P, Polticelli F, Fiorucci L. Cytochrome c: an extreme multifunctional protein with a key role in cell fate. Int J Biol Macromol. 2019;136:1237–46.
Charan M, Das S, Mishra S, Chatterjee N, Varikuti S, Kaul K, et al. Macrophage migration inhibitory factor inhibition as a novel therapeutic approach against triple-negative breast cancer. Cell Death Dis. 2020;11(9):774.
Bao X, Shi R, Zhao T, Wang Y, Anastasov N, Rosemann M, et al. Integrated analysis of single-cell RNA-seq and bulk RNA-seq unravels tumour heterogeneity plus M2-like tumour-associated macrophage infiltration and aggressiveness in TNBC. Cancer Immunol Immunother CII. 2021;70(1):189–202.
Zhao YW, Jing ZL, Lv J, Zhang ZW, Lin JT, Cao XJ, et al. Berberine activates caspase-9/cytochrome c-mediated apoptosis to suppress triple-negative breast cancer cells in vitro and in vivo. Biomed Pharmacother. 2017;95:18–24.
Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell. 2017;168(1–2):37–57.
Yang J, Li G, Zhang K. Pro-survival effects by NF-kappaB, Akt and ERK(1/2) and anti-apoptosis actions by Six1 disrupt apoptotic functions of TRAIL-Dr4/5 pathway in ovarian cancer. Biomed Pharmacother. 2016;84:1078–87.
Huang Y, Chen R, Zhou J. E2F1 and NF-κB: Key mediators of inflammation-associated cancers and potential therapeutic targets. Curr Cancer Drug Targets. 2016;16(9):765–72.
Wang XZ, Feng Y, Han YF, Bian Y, Liang J, Wen HM, et al. Budlein A methylacrylate demonstrates potent activity against triple-negative breast cancer by targeting I kappa B alpha kinase and exportin-1. Toxicol Appl Pharm. 2020;408:115263.
Song LJ, Chen X, Mi L, Liu C, Zhu SM, Yang TL, et al. Icariin-induced inhibition of SIRT6/NF-kappa B triggers redox mediated apoptosis and enhances anti-tumor immunity in triple-negative breast cancer. Cancer Sci. 2020;111(11):4242–56.
Shrestha S, Sorolla A, Fromont J, Blancafort P, Flematti GR. Crambescidin 800, isolated from the marine sponge Monanchora viridis, induces cell cycle arrest and apoptosis in triple-negative breast cancer cells. Mar Drugs. 2018;16(2):53.
Liu YY, Wang L, Liu JW, Xie XX, Hu HY, Luo F. Anticancer effects of ACT001 via NF-kappa B suppression in murine triple-negative breast cancer cell line 4T1. Cancer Manag Res. 2020;12:5131–9.
Becceneri AB, Fuzer AM, Popolin CP, Cazal CD, Domingues VD, Fernandes JB, et al. Acetylation of cedrelone increases its cytotoxic activity and reverts the malignant phenotype of breast cancer cells in 3D culture. Chemico-Biol Interact. 2020;316:108920.
Narayan P, Wahby S, Gao J, Amiri-Kordestani L, Ibrahim A, Bloomquist E, et al. FDA approval summary: atezolizumab plus paclitaxel protein-bound for the treatment of patients with advanced or metastatic TNBC whose tumors express PD-L1. Clin Cancer Res. 2020;26(10):2284–9.
Wang PP, Song D, Wan DH, Li LY, Mei WH, Li XY, et al. Ginsenoside panaxatriol reverses TNBC paclitaxel resistance by inhibiting the IRAK1/NF-kappa B and ERK pathways. PeerJ. 2020;8:e9281.
Yuan ZG, Jiang H, Zhu XH, Liu XG, Li JH. Ginsenoside Rg3 promotes cytotoxicity of Paclitaxel through inhibiting NF-kappa B signaling and regulating Bax/Bcl-2 expression on triple-negative breast cancer. Biomed Pharmacother. 2017;89:227–32.
Shi YH, Bieerkehazhi S, Ma H. Next-generation proteasome inhibitor oprozomib enhances sensitivity to doxorubicin in triple-negative breast cancer cells. Int J Clin Exp Pathol. 2018;11(5):2347–55.
Vyas D, Lopez-Hisijos N, Shah P, Deshpande KS, Basson MD, Vyas A, et al. A second-generation proteasome inhibitor and doxorubicin modulates IL-6, pSTAT-3 and NF-kappa B activity in MDA-MB-231 breast cancer cells. J Nanosci Nanotechnol. 2017;17(1):175–85.
Shen YY, Zhang W, Liu JH, He J, Cao RX, Chen XG, et al. Therapeutic activity of DCC-2036, a novel tyrosine kinase inhibitor, against triple-negative breast cancer patient-derived xenografts by targeting AXL/MET. Int J Cancer. 2019;144(3):651–64.
Wang Z, Wang X. miR-122-5p promotes aggression and epithelial-mesenchymal transition in triple-negative breast cancer by suppressing charged multivesicular body protein 3 through mitogen-activated protein kinase signaling. J Cell Physiol. 2020;235(3):2825–35.
Shrestha S, Sorolla A, Fromont J, Blancafort P, Flematti GR. Aurantoside C targets and induces apoptosis in triple negative breast cancer cells. Mar Drugs. 2018;16(10):361.
Kong YJ, Li FB, Nian Y, Zhou ZM, Yang RX, Qiu MH, et al. KHF16 is a leading structure from Cimicifuga foetida that suppresses breast cancer partially by inhibiting the NF-kappa B signaling pathway. Theranostics. 2016;6(6):875–86.
Quisbert-Valenzuela EO, Calaf GM. Apoptotic effect of noscapine in breast cancer cell lines. Int J Oncol. 2016;48(6):2666–74.
Laudisi F, Cherubini F, Monteleone G, Stolfi C. STAT3 interactors as potential therapeutic targets for cancer treatment. Int J Mol Sci. 2018;19(6):1787.
Chai EZ, Shanmugam MK, Arfuso F, Dharmarajan A, Wang C, Kumar AP, et al. Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol Ther. 2016;162:86–97.
Chun J, Song K, Kim YS. Sesquiterpene lactones-enriched fraction of Inula helenium L. induces apoptosis through inhibition of signal transducers and activators of transcription 3 signaling pathway in MDA-MB-231 breast cancer cells. Phytother Res. 2018;32(12):2501–9.
Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: a review. Int J Cancer. 2016;138(11):2570–8.
Qin JJ, Yan L, Zhang J, Zhang WD. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res. 2019;38(1):195.
Liu CY, Su JC, Huang TT, Chu PY, Huang CT, Wang WL, et al. Sorafenib analogue SC-60 induces apoptosis through the SHP-1/STAT3 pathway and enhances docetaxel cytotoxicity in triple-negative breast cancer cells. Mol Oncol. 2017;11(3):266–79.
Liu CY, Chen KF, Chao TI, Chu PY, Huang CT, Huang TT, et al. Sequential combination of docetaxel with a SHP-1 agonist enhanced suppression of p-STAT3 signaling and apoptosis in triple negative breast cancer cells. J Mol Med (Berl). 2017;95(9):965–75.
Ko H, Lee JH, Kim HS, Kim T, Han YT, Suh YG, et al. Novel galiellalactone analogues can target STAT3 phosphorylation and cause apoptosis in triple-negative breast cancer. Biomolecules. 2019;9(5):170.
Liang X, Tang S, Liu X, Liu Y, Xu Q, Wang X, et al. Discovery of novel pyrrolo[2,3-d]pyrimidine-based derivatives as potent JAK/HDAC dual inhibitors for the treatment of refractory solid tumors. J Med Chem. 2022;65(2):1243–64.
Xie Q, Yang Z, Huang X, Zhang Z, Li J, Ju J, et al. Ilamycin C induces apoptosis and inhibits migration and invasion in triple-negative breast cancer by suppressing IL-6/STAT3 pathway. J Hematol Oncol. 2019;12(1):60.
Dai X, Yin C, Zhang Y, Guo G, Zhao C, Wang O, et al. Osthole inhibits triple negative breast cancer cells by suppressing STAT3. J Exp Clin Cancer Res. 2018;37(1):322.
Chen Y, Ji M, Zhang S, Xue N, Xu H, Lin S, et al. Bt354 as a new STAT3 signaling pathway inhibitor against triple negative breast cancer. J Drug Target. 2018;26(10):920–30.
Kim JH, Choi HS, Lee DS. Primaquine inhibits the endosomal trafficking and nuclear localization of EGFR and induces the apoptosis of breast cancer cells by nuclear EGFR/Stat3-mediated c-Myc downregulation. Int J Mol Sci. 2021;22(23):12961.
Lou C, Chen Y, Zhang J, Yang B, Zhao H. Eupalinolide J suppresses the growth of triple-negative breast cancer cells via targeting STAT3 signaling pathway. Front Pharmacol. 2019;10:1071.
Pan L, Chen X, Fu S, Yu W, Li C, Wang T, et al. LLY17, a novel small molecule STAT3 inhibitor induces apoptosis and suppresses cell migration and tumor growth in triple-negative breast cancer. Breast Cancer Res Treat. 2020;181(1):31–41.
Byun WS, Bae ES, Cui J, Park HJ, Oh DC, Lee SK. Antitumor activity of pulvomycin via targeting activated-STAT3 signaling in docetaxel-resistant triple-negative breast cancer cells. Biomedicines. 2021;9(4):436.
Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25(1):104–13.
Kanapathipillai M. Treating p53 mutant aggregation-associated cancer. Cancers (Basel). 2018;10(6):154.
Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6(3):a026104.
Duffy MJ, Synnott NC, Crown J. Mutant p53 in breast cancer: potential as a therapeutic target and biomarker. Breast Cancer Res Tr. 2018;170(2):213–9.
Duffy MJ, Synnott NC, Crown J. Mutant p53 as a target for cancer treatment. Eur J Cancer. 2017;83:258–65.
dos Santos MB, Anselmo DB, de Oliveira JG, Jardim-Perassi BV, Monteiro DA, Silva G, et al. Antiproliferative activity and p53 upregulation effects of chalcones on human breast cancer cells. J Enzyme Inhibit Med Chem. 2019;34(1):1093–9.
Synnott NC, Murray A, McGowan PM, Kiely M, Kiely PA, O’Donovan N, et al. Mutant p53: a novel target for the treatment of patients with triple-negative breast cancer? Int J Cancer. 2017;140(1):234–46.
Makhale A, Nanayakkara D, Raninga P, Khanna KK, Kalimutho M. CX-5461 enhances the efficacy of APR-246 via induction of DNA damage and replication stress in triple-negative breast cancer. Int J Mol Sci. 2021;22(11):5782.
Synnott NC, O’Connell D, Crown J, Duffy MJ. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res Treat. 2020;179(1):47–56.
Synnott NC, Bauer MR, Madden S, Murray A, Klinger R, O’Donovan N, et al. Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: Preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett. 2018;414:99–106.
Ribeiro CJA, Nunes RC, Amaral JD, Goncalves LM, Rodrigues CMP, Moreira R, et al. Spirotriazoline oxindoles: a novel chemical scaffold with in vitro anticancer properties. Eur J Med Chem. 2017;140:494–509.
Travassos IO, Mello-Andrade F, Caldeira RP, Pires WC, da Silva PFF, Correa RS, et al. Ruthenium(II)/allopurinol complex inhibits breast cancer progression via multiple targets. J Biol Inorg Chem. 2021;26(4):385–401.
Liang ZJ, Wan Y, Zhu DD, Wang MX, Jiang HM, Huang DL, et al. Resveratrol mediates the apoptosis of triple negative breast cancer cells by reducing POLD1 expression. Front Oncol. 2021;11:569295.
Zhu X, Wang K, Zhang K, Zhang T, Yin YX, Xu F. Ziyuglycoside I inhibits the proliferation of MDA-MB-231 breast carcinoma cells through inducing p53-mediated G2/M cell cycle arrest and intrinsic/extrinsic apoptosis. Int J Mol Sci. 2016;17(11):1903.
Hafezi K, Hemmati AA, Abbaszadeh H, Valizadeh A, Makvandi M. Anticancer activity and molecular mechanisms of α-conidendrin, a polyphenolic compound present in Taxus yunnanensis, on human breast cancer cell lines. Phytother Res. 2020;34(6):1397–408.
Yang M, Dang XF, Tan Y, Wang MX, Li XJ, Li G. I-7ab inhibited the growth of TNBC cells via targeting HDAC3 and promoting the acetylation of p53. Biomed Pharmacother. 2018;99:220–6.
Beberok A, Wrzesniok D, Rok J, Rzepka Z, Respondek M, Buszman E. Ciprofloxacin triggers the apoptosis of human triple-negative breast cancer MDA-MB-231 cells via the p53/Bax/Bcl-2 signaling pathway. Int J Oncol. 2018;52(5):1727–37.
Yun CW, Jeon J, Go G, Lee JH, Lee SH. The dual role of autophagy in cancer development and a therapeutic strategy for cancer by targeting autophagy. Int J Mol Sci. 2021;22(1):22.
Marinkovic M, Sprung M, Buljubasic M, Novak I. Autophagy modulation in cancer: current knowledge on action and therapy. Oxid Med Cell Longev. 2018;2018:8023821.
Li Y, Yang G, Yang C, Tang P, Chen J, Zhang J, et al. Targeting autophagy-related epigenetic regulators for cancer drug discovery. J Med Chem. 2021;64(16):11798–815.
Hurley JH, Young LN. Mechanisms of autophagy initiation. Annu Rev Biochem. 2017;86:225–44.
Kim D, Hwang HY, Kwon HJ. Targeting autophagy in disease: recent advances in drug discovery. Expert Opin Drug Discov. 2020;15(9):1045–64.
Limpert AS, Lambert LJ, Bakas NA, Bata N, Brun SN, Shaw RJ, et al. Autophagy in cancer: regulation by small molecules. Trends Pharmacol Sci. 2018;39(12):1021–32.
Xiang H, Zhang J, Lin C, Zhang L, Liu B, Ouyang L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm Sin B. 2020;10(4):569–81.
Han YY, Fan SJ, Qin T, Yang JF, Sun Y, Lu Y, et al. Role of autophagy in breast cancer and breast cancer stem cells (review). Int J Oncol. 2018;52(4):1057–70.
Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913–30.
Zhang J, Zou L, Shi D, Liu J, Zhang J, Zhao R, et al. Structure-guided design of a small-molecule activator of sirtuin-3 that modulates autophagy in triple negative breast cancer. J Med Chem. 2021;64(19):14192–216.
Cocco S, Leone A, Piezzo M, Caputo R, Di Lauro V, Di Rella F, et al. Targeting autophagy in breast cancer. Int J Mol Sci. 2020;21(21):7836.
Chang H, Zou Z. Targeting autophagy to overcome drug resistance: further developments. J Hematol Oncol. 2020;13(1):159.
Zhang L, Du Y, Xu S, Jiang Y, Yuan C, Zhou L, et al. DEPDC1, negatively regulated by miR-26b, facilitates cell proliferation via the up-regulation of FOXM1 expression in TNBC. Cancer Lett. 2019;442:242–51.
Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. In: Lane JD, Korolchuk VI, Murray JT, editors. Signalling Mechanisms in Autophagy, vol. 61. London: Portland Press Ltd; 2017. pp. 585–96.
Turco E, Fracchiolla D, Martens S. Recruitment and activation of the ULK1/Atg1 kinase complex in selective autophagy. J Mol Biol. 2020;432(1):123–34.
Papinski D, Kraft C. Regulation of autophagy by signaling through the Atg1/ULK1 complex. J Mol Biol. 2016;428(9):1725–41.
Ouyang L, Zhang L, Fu L, Liu B. A small-molecule activator induces ULK1-modulating autophagy-associated cell death in triple negative breast cancer. Autophagy. 2017;13(4):777–8.
Zhang L, Fu L, Zhang S, Zhang J, Zhao Y, Zheng Y, et al. Discovery of a small molecule targeting ULK1-modulated cell death of triple negative breast cancer in vitro and in vivo. Chem Sci. 2017;8(4):2687–701.
Ouyang L, Zhang L, Liu J, Fu L, Yao D, Zhao Y, et al. Discovery of a small-molecule bromodomain-containing protein 4 (BRD4) inhibitor that induces AMP-activated protein kinase-modulated autophagy-associated cell death in breast cancer. J Med Chem. 2017;60(24):9990–10012.
Ren HY, Bakas NA, Vamos M, Chaikuad A, Limpert AS, Wimer CD, et al. Design, synthesis, and characterization of an orally active dual-specific ULK1/2 autophagy inhibitor that synergizes with the PARP inhibitor olaparib for the treatment of triple-negative breast cancer. J Med Chem. 2020;63(23):14609–25.
Mele L, del Vecchio V, Liccardo D, Prisco C, Schwerdtfeger M, Robinson N, et al. The role of autophagy in resistance to targeted therapies. Cancer Treat Rev. 2020;88:102043.
Chen M, Gowd V, Wang MF, Chen F, Cheng KW. The apple dihydrochalcone phloretin suppresses growth and improves chemosensitivity of breast cancer cells via inhibition of cytoprotective autophagy. Food Funct. 2021;12(1):177–90.
Popova NV, Jücker M. The role of mTOR signaling as a therapeutic target in cancer. Int J Mol Sci. 2021;22(4):1743.
Nascimbeni AC, Codogno P, Morel E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017;284(9):1267–78.
Backer JM. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem J. 2016;473(15):2251–71.
Whitmarsh-Everiss T, Laraia L. Small molecule probes for targeting autophagy. Nat Chem Biol. 2021;17(6):653–64.
Xu ZR, Han X, Ou DM, Liu T, Li ZX, Jiang GM, et al. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol. 2020;104(2):575–87.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169(2):361–71.
Chang CH, Bijian K, Wernic D, Su J, da Silva SD, Yu H, et al. A novel orally available seleno-purine molecule suppresses triple-negative breast cancer cell proliferation and progression to metastasis by inducing cytostatic autophagy. Autophagy. 2019;15(8):1376–90.
Guo Q, Yu C, Zhang C, Li Y, Wang T, Huang Z, et al. Highly selective, potent, and oral mTOR inhibitor for treatment of cancer as autophagy inducer. J Med Chem. 2018;61(3):881–904.
Yuan JM, Dong XD, Yap JJ, Hu JC. The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J Hematol Oncol. 2020;13(1):19.
Ryu W, Lee J, Park J, Cha P, Cho Y, Kim J, et al. Destabilization of β-catenin and RAS by targeting the Wnt/β-catenin pathway as a potential treatment for triple-negative breast cancer. Exp Mol Med. 2020;52(5):832–42.
Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death—apoptosis, autophagy and senescence. Febs J. 2010;277(1):2–21.
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19(3):1997–2007.
Ho CJ, Gorski SM. Molecular mechanisms underlying autophagy-mediated treatment resistance in cancer. Cancers. 2019;11(11):1775.
Wang P, Du Y, Wang J. Indentification of breast cancer subtypes sensitive to HCQ-induced autophagy inhibition. Pathol Res Pract. 2019;215(10):152609.
Wang J, Dang M, Day E. Inhibition of Wnt signaling by Frizzled7 antibody-coated nanoshells sensitizes triple-negative breast cancer cells to the autophagy regulator chloroquine. Nano Res. 2020;13(6):1693–703.
Yao D, Zhou Y, Zhu L, Ouyang L, Zhang J, Jiang Y, et al. Design, synthesis and structure-activity relationship studies of a focused library of pyrimidine moiety with anti-proliferative and anti metastasis activities in triple negative breast cancer. Eur J Med Chem. 2017;140:155–71.
Sui X, Jin L, Huang X, Geng S, He C, Hu X. p53 signaling and autophagy in cancer: a revolutionary strategy could be developed for cancer treatment. Autophagy. 2011;7(6):565–71.
Xu J, Patel NH, Gewirtz DA. Triangular relationship between p53, autophagy, and chemotherapy resistance. Int J Mol Sci. 2020;21(23):8991.
Mrakovcic M, Frohlich LF. p53-mediated molecular control of autophagy in tumor cells. Biomolecules. 2018;8(2):14.
Cordani M, Butera G, Pacchiana R, Donadelli M. Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Bba-Rev Cancer. 2017;1867(1):19–28.
Liu Y, Zhou Y, Huang K, Fang X, Li Y, Wang F, et al. Targeting epidermal growth factor-overexpressing triple-negative breast cancer by natural killer cells expressing a specific chimeric antigen receptor. Cell Prolif. 2020;53(8):e12858.
Sommaggio R, Cappuzzello E, Dalla Pietà A, Tosi A, Palmerini P, Carpanese D, et al. Adoptive cell therapy of triple negative breast cancer with redirected cytokine-induced killer cells. Oncoimmunology. 2020;9(1):1777046.
Chollat-Namy M, Ben Safta-Saadoun T, Haferssas D, Meurice G, Chouaib S, Thiery J. The pharmalogical reactivation of p53 function improves breast tumor cell lysis by granzyme B and NK cells through induction of autophagy. Cell Death Dis. 2019;10(10):695.
Islam MA, Sooro MA, Zhang PH. Autophagic regulation of p62 is critical for cancer therapy. Int J Mol Sci. 2018;19(5):1405.
Moscat J, Karin M, Diaz-Meco MT. p62 in cancer: signaling adaptor beyond autophagy. Cell. 2016;167(3):606–9.
Huang MB, Zhou YF, Duan DZ, Yang CY, Zhou ZM, Li FB, et al. Targeting ubiquitin conjugating enzyme UbcH5b by a triterpenoid PC3-15 from Schisandra plants sensitizes triple-negative breast cancer cells to lapatinib. Cancer Lett. 2021;504:125–36.
Liao R, Yan F, Zeng Z, Farhan M, Little P, Quirion R, et al. Amiodarone-induced retinal neuronal cell apoptosis attenuated by IGF-1 via counter regulation of the PI3k/Akt/FoxO3a pathway. Mol Neurobiol. 2017;54(9):6931–43.
Hou T, Li Z, Zhao Y, Zhu WG. Mechanisms controlling the anti-neoplastic functions of FoxO proteins. Semin Cancer Biol. 2018;50:101–14.
Farhan M, Silva M, Li S, Yan F, Fang J, Peng T, et al. The role of FOXOs and autophagy in cancer and metastasis-Implications in therapeutic development. Med Res Rev. 2020;40(6):2089–113.
Ding R, Wang X, Chen W, Li Z, Wei AL, Wang QB, et al. WX20120108, a novel IAP antagonist, induces tumor cell autophagy via activating ROS-FOXO pathway. Acta Pharmacol Sin. 2019;40(11):1466–79.
Verzella D, Fischietti M, Capece D, Vecchiotti D, Del Vecchio F, Cicciarelli G, et al. Targeting the NF-kappaB pathway in prostate cancer: a promising therapeutic approach? Curr Drug Targets. 2016;17(3):311–20.
Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18(5):309–24.
Verzella D, Pescatore A, Capece D, Vecchiotti D, Ursini MV, Franzoso G, et al. Life, death, and autophagy in cancer: NF-kappa B turns up everywhere. Cell Death Dis. 2020;11(3):210.
Lou C, Xu X, Chen Y, Zhao H. Alisol A suppresses proliferation, migration, and invasion in human breast cancer MDA-MB-231 Cells. Molecules. 2019;24(20):3651.
Li Y, Xiao Y, Lin H, Reichel D, Bae Y, Lee E, et al. In vivo β-catenin attenuation by the integrin α5-targeting nano-delivery strategy suppresses triple negative breast cancer stemness and metastasis. Biomaterials. 2019;188:160–72.
Hu C, Li G, Mu Y, Wu W, Cao B, Wang Z, et al. In vitro discovery of anti-TNBC agents targeting PTP1B: total synthesis, structure-activity relationship, and investigations of jamunones. J Med Chem. 2021;64(9):6008–20.
Liu L, He J, Wei X, Wan G, Lao Y, Xu W, et al. MicroRNA-20a-mediated loss of autophagy contributes to breast tumorigenesis by promoting genomic damage and instability. Oncogene. 2017;36(42):5874–84.
Vega-Rubin-de-Celis S. The role of beclin 1-dependent autophagy in cancer. Biology-Basel. 2020;9(1):4.
Jung YY, Lee YK, Koo JS. The potential of Beclin 1 as a therapeutic target for the treatment of breast cancer. Expert Opin Ther Targets. 2016;20(2):167–78.
Menon MB, Dhamija S. Beclin 1 phosphorylation—at the center of autophagy regulation. Front Cell Dev Biol. 2018;6:137.
Kim S, Ju J, Kang M, Eun J, Kim Y, Raninga P, et al. RNA-binding protein NONO contributes to cancer cell growth and confers drug resistance as a theranostic target in TNBC. Theranostics. 2020;10(18):7974–92.
Unal TD, Hamurcu Z, Delibasi N, Cinar V, Guler A, Gokce S, et al. Thymoquinone inhibits proliferation and migration of MDA-MB-231 triple negative breast cancer cells by suppressing autophagy, Beclin-1 and LC3. Anti-Cancer Agents Med Chem. 2021;21(3):355–64.
Liu X, Jiang J, Jin X, Liu Y, Xu C, Zhang J, et al. Simultaneous determination of YZG-331 and its metabolites in monkey blood by liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal. 2021;193:113720.
Liu YF, Zhong YP, Tian W, Lan F, Kang JK, Pang HF, et al. An autophagy-dependent cell death of MDA-MB-231 cells triggered by a novel Rhein derivative 4F. Anticancer Drugs. 2019;30(10):1038–47.
Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12(6):385–92.
Portugal J, Mansilla S, Bataller M. Mechanisms of drug-induced mitotic catastrophe in cancer cells. Curr Pharm Des. 2010;16(1):69–78.
Dolgin E. Atezolizumab combo approved for PD-L1-positive TNBC. Cancer Discov. 2019;9(5):OF2.
Jiang L, Siu M, Wong O, Tam K, Lu X, Lam E, et al. iASPP and chemoresistance in ovarian cancers: effects on paclitaxel-mediated mitotic catastrophe. Clin Cancer Res. 2011;17(21):6924–33.
Nitta M, Kobayashi O, Honda S, Hirota T, Kuninaka S, Marumoto T, et al. Spindle checkpoint function is required for mitotic catastrophe induced by DNA-damaging agents. Oncogene. 2004;23(39):6548–58.
Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15(7):1153–62.
Jurj A, Pop L, Zanoaga O, Ciocan-Cârtiţă C, Cojocneanu R, Moldovan C, et al. New insights in gene expression alteration as effect of paclitaxel drug resistance in triple negative breast cancer cells. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2020;54(4):648–64.
Liao W, Ho Y, Lin Y, Naveen Raj E, Liu K, Chen C, et al. Targeting EGFR of triple-negative breast cancer enhances the therapeutic efficacy of paclitaxel- and cetuximab-conjugated nanodiamond nanocomposite. Acta Biomater. 2019;86:395–405.
Massa C, Karn T, Denkert C, Schneeweiss A, Hanusch C, Blohmer J, et al. Differential effect on different immune subsets of neoadjuvant chemotherapy in patients with TNBC. J Immunother Cancer. 2020;8(2):e001261.
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith W, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–73.
Shi J, Vakoc C. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54(5):728–36.
Wang Y, Xie Q, Tan H, Liao M, Zhu S, Zheng L, et al. Targeting cancer epigenetic pathways with small-molecule compounds: therapeutic efficacy and combination therapies. Pharmacol Res. 2021;173:105702.
Sahni J, Gayle S, Webb B, Weber-Bonk K, Seachrist D, Singh S, et al. Mitotic vulnerability in triple-negative breast cancer associated with LIN9 is targetable with BET inhibitors. Cancer Res. 2017;77(19):5395–408.
Gayle S, Sahni J, Webb B, Weber-Bonk K, Shively M, Spina R, et al. Targeting BCL-xL improves the efficacy of bromodomain and extra-terminal protein inhibitors in triple-negative breast cancer by eliciting the death of senescent cells. J Biol Chem. 2019;294(3):875–86.
Serrano-Oviedo L, Nuncia-Cantarero M, Morcillo-Garcia S, Nieto-Jimenez C, Burgos M, Corrales-Sanchez V, et al. Identification of a stemness-related gene panel associated with BET inhibition in triple negative breast cancer. Cell Oncol (Dordrecht). 2020;43(3):431–44.
Albiges L, Goubar A, Scott V, Vicier C, Lefèbvre C, Alsafadi S, et al. Chk1 as a new therapeutic target in triple-negative breast cancer. Breast (Edinburgh, Scotland). 2014;23(3):250–8.
Karn T, Denkert C, Weber K, Holtrich U, Hanusch C, Sinn B, et al. Tumor mutational burden and immune infiltration as independent predictors of response to neoadjuvant immune checkpoint inhibition in early TNBC in GeparNuevo. Ann Oncol. 2020;31(9):1216–22.
Miao K, Lei J, Valecha M, Zhang A, Xu J, Wang L, et al. NOTCH1 activation compensates BRCA1 deficiency and promotes triple-negative breast cancer formation. Nat Commun. 2020;11(1):3256.
Sami E, Paul B, Koziol J, ElShamy W. The immunosuppressive microenvironment in BRCA1-IRIS-overexpressing TNBC tumors is induced by bidirectional interaction with tumor-associated macrophages. Cancer Res. 2020;80(5):1102–17.
Huang Y, Li W, Yan W, Wu J, Chen L, Yao X, et al. Loss of PICH promotes chromosome instability and cell death in triple-negative breast cancer. Cell Death Dis. 2019;10(6):428.
Killock D. Pembrolizumab can delay progression of TNBC. Nat Rev Clin Oncol. 2021;18(2):64.
Chopra S, Jenney A, Palmer A, Niepel M, Chung M, Mills C, et al. Torin2 exploits replication and checkpoint vulnerabilities to cause death of PI3K-activated triple-negative breast cancer cells. Cell Syst. 2020;10(1):66-81.e11.
Kim S, Min A, Lee K, Yang Y, Kim T, Lim J, et al. Antitumor effect of KX-01 through inhibiting Src family kinases and mitosis. Cancer Res Treat. 2017;49(3):643–55.
Finn R, Bengala C, Ibrahim N, Roché H, Sparano J, Strauss L, et al. Dasatinib as a single agent in triple-negative breast cancer: results of an open-label phase 2 study. Clin Cancer Res. 2011;17(21):6905–13.
Deng S, Krutilina R, Wang Q, Lin Z, Parke D, Playa H, et al. An orally available tubulin inhibitor, VERU-111, suppresses triple-negative breast cancer tumor growth and metastasis and bypasses taxane resistance. Mol Cancer Ther. 2020;19(2):348–63.
Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell. 2018;173(4):879-93.e13.
Brockwell N, Rautela J, Owen K, Gearing L, Deb S, Harvey K, et al. Tumor inherent interferon regulators as biomarkers of long-term chemotherapeutic response in TNBC. NPJ Precis Oncol. 2019;3:21.
Otterbach F, Callies R, Adamzik M, Kimmig R, Siffert W, Schmid K, et al. Aquaporin 1 (AQP1) expression is a novel characteristic feature of a particularly aggressive subgroup of basal-like breast carcinomas. Breast Cancer Res Treat. 2010;120(1):67–76.
Esteva-Font C, Jin B, Verkman A. Aquaporin-1 gene deletion reduces breast tumor growth and lung metastasis in tumor-producing MMTV-PyVT mice. FASEB J. 2014;28(3):1446–53.
Kao S, Armstrong N, Condon B, Griggs K, McCaughan B, Maltby S, et al. Aquaporin 1 is an independent prognostic factor in pleural malignant mesothelioma. Cancer. 2012;118(11):2952–61.
Morrissey J, Mellnick V, Luo J, Siegel M, Figenshau R, Bhayani S, et al. Evaluation of urine aquaporin-1 and perilipin-2 concentrations as biomarkers to screen for renal cell carcinoma: a prospective cohort study. JAMA Oncol. 2015;1(2):204–12.
Verkman A. Aquaporins in clinical medicine. Annu Rev Med. 2012;63:303–16.
Yin Z, Chen W, Yin J, Sun J, Xie Q, Wu M, et al. RIPK1 is a negative mediator in Aquaporin 1-driven triple-negative breast carcinoma progression and metastasis. NPJ Breast Cancer. 2021;7(1):53.
Bebber C, Müller F, Prieto Clemente L, Weber J, von Karstedt S. Ferroptosis in cancer cell biology. Cancers. 2020;12(1):164.
Hassannia B, Vandenabeele P, Vanden BT. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35(6):830–49.
Doll S, Proneth B, Tyurina Y, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–8.
Timmerman L, Holton T, Yuneva M, Louie R, Padró M, Daemen A, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24(4):450–65.
Beatty A, Fink L, Singh T, Strigun A, Peter E, Ferrer C, et al. Metabolite profiling reveals the glutathione biosynthetic pathway as a therapeutic target in triple-negative breast cancer. Mol Cancer Ther. 2018;17(1):264–75.
Chen L, Hambright W, Na R, Ran Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. 2015;290(47):28097–106.
Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal. 2018;29(1):61–74.
McGovern U, Francis R, Peck B, Guest S, Wang J, Myatt S, et al. Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol Cancer Ther. 2009;8(3):582–91.
McLaughlin R, He J, van der Noord V, Redel J, Foekens J, Martens J, et al. A kinase inhibitor screen identifies a dual cdc7/CDK9 inhibitor to sensitise triple-negative breast cancer to EGFR-targeted therapy. Breast Cancer Res BCR. 2019;21(1):77.
Song X, Wang X, Liu Z, Yu Z. Role of GPX4-mediated ferroptosis in the sensitivity of triple negative breast cancer cells to gefitinib. Front Oncol. 2020;10:597434.
Dixon S, Lemberg K, Lamprecht M, Skouta R, Zaitsev E, Gleason C, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Peng Q, Zhang S, Yang Q, Zhang T, Wei X, Jiang L, et al. Preformed albumin corona, a protective coating for nanoparticles based drug delivery system. Biomaterials. 2013;34(33):8521–30.
Zhang C, Zhang X, Zhao W, Zeng C, Li W, Li B, et al. Chemotherapy drugs derived nanoparticles encapsulating mRNA encoding tumor suppressor proteins to treat triple-negative breast cancer. Nano Res. 2019;12(4):855–61.
Zhao Q, Hai B, Zhang X, Xu J, Koehler B, Liu F. Biomimetic nanovesicles made from iPS cell-derived mesenchymal stem cells for targeted therapy of triple-negative breast cancer. Nanomed Nanotechnol Biol Med. 2020;24:102146.
Fan J, Liu B, Long Y, Wang Z, Tong C, Wang W, et al. Sequentially-targeted biomimetic nano drug system for triple-negative breast cancer ablation and lung metastasis inhibition. Acta Biomater. 2020;113:554–69.
Cocucci E, Meldolesi J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015;25(6):364–72.
Yu D, Wu Y, Shen H, Lv M, Chen W, Zhang X, et al. Exosomes in development, metastasis and drug resistance of breast cancer. Cancer Sci. 2015;106(8):959–64.
Kalani A, Kamat P, Chaturvedi P, Tyagi S, Tyagi N. Curcumin-primed exosomes mitigate endothelial cell dysfunction during hyperhomocysteinemia. Life Sci. 2014;107:1–7.
Tian Y, Li S, Song J, Ji T, Zhu M, Anderson G, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35(7):2383–90.
Kanchanapally R, Deshmukh S, Chavva S, Tyagi N, Srivastava S, Patel G, et al. Drug-loaded exosomal preparations from different cell types exhibit distinctive loading capability, yield, and antitumor efficacies: a comparative analysis. Int J Nanomed. 2019;14:531–41.
Yang S, Wang D, Zhong S, Chen W, Wang F, Zhang J, et al. Tumor-derived exosomal circPSMA1 facilitates the tumorigenesis, metastasis, and migration in triple-negative breast cancer (TNBC) through miR-637/Akt1/β-catenin (cyclin D1) axis. Cell Death Dis. 2021;12(5):420.
Yang C, Zhang J, Liao M, Yang Y, Wang Y, Yuan Y, et al. Folate-mediated one-carbon metabolism: a targeting strategy in cancer therapy. Drug Discov Today. 2021;26(3):817–25.
Yu M, Gai C, Li Z, Ding D, Zheng J, Zhang W, et al. Targeted exosome-encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer Sci. 2019;110(10):3173–82.
Richards C, Vellanki S, Smith Y, Hopkins A. Diterpenoid natural compound C4 (Crassin) exerts cytostatic effects on triple-negative breast cancer cells via a pathway involving reactive oxygen species. Cell Oncol (Dordrecht). 2018;41(1):35–46.
Zhang Y, Lima CF, Rodrigues LR. Anticancer effects of lactoferrin: underlying mechanisms and future trends in cancer therapy. Nutr Rev. 2014;72(12):763–73.
Cutone A, Rosa L, Ianiro G, Lepanto M, Bonaccorsi di Patti M, Valenti P, et al. Lactoferrin’s anti-cancer properties: safety, selectivity, and wide range of action. Biomolecules. 2020;10(3):456.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–5.
Kayagaki N, Stowe I, Lee B, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–71.
He W, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25(12):1285–98.
Trotter T, Shuptrine C, Tsao L, Marek R, Acharya C, Wei J, et al. IL26, a noncanonical mediator of DNA inflammatory stimulation, promotes TNBC engraftment and progression in association with neutrophils. Cancer Res. 2020;80(15):3088–100.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri E. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103.
An H, Heo J, Kim P, Lian Z, Lee S, Park J, et al. Tetraarsenic hexoxide enhances generation of mitochondrial ROS to promote pyroptosis by inducing the activation of caspase-3/GSDME in triple-negative breast cancer cells. Cell Death Dis. 2021;12(2):159.
Zhu Z, Yuan J, Xu X, Wei Y, Yang B, Zhao H. Eucannabinolide, a novel sesquiterpene lactone, suppresses the growth, metastasis and BCSCS-like traits of TNBC via inactivation of STAT3. Neoplasia. 2021;23(1):36–48.
Yan H, Luo B, Wu X, Guan F, Yu X, Zhao L, et al. Cisplatin induces pyroptosis via activation of MEG3/NLRP3/caspase-1/GSDMD pathway in triple-negative breast cancer. Int J Biol Sci. 2021;17(10):2606–21.
Wang L, Liu D, Wu X, Zeng Y, Li L, Hou Y, et al. Long non-coding RNA (LncRNA) RMST in triple-negative breast cancer (TNBC): Expression analysis and biological roles research. J Cell Physiol. 2018;233(10):6603–12.
Tamura Y, Morikawa M, Tanabe R, Miyazono K, Koinuma D. Anti-pyroptotic function of TGF-β is suppressed by a synthetic dsRNA analogue in triple negative breast cancer cells. Mol Oncol. 2021;15(5):1289–307.
Gilmore AP. Anoikis. Cell Death Differ. 2005;12(2):1473–7.
Monteiro H, Silva E, Stern A. Nitric oxide: a potential inducer of adhesion-related apoptosis–anoikis. Nitric Oxide Biol Chem. 2004;10(1):1–10.
D’Amato N, Rogers T, Gordon M, Greene L, Cochrane D, Spoelstra N, et al. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015;75(21):4651–64.
Zhang E, Kong K, Altman A. The yin and yang of protein kinase C-theta (PKCθ): a novel drug target for selective immunosuppression. Adv Pharmacol. 2013;66:267–312.
Irie HY, Shrestha Y, Selfors LM, Frye F, Iida N, Wang Z, et al. PTK6 regulates IGF-1-induced anchorage-independent survival. PLoS ONE. 2010;5(7):e11729.
Byerly J, Halstead-Nussloch G, Ito K, Katsyv I, Irie HY. PRKCQ promotes oncogenic growth and anoikis resistance of a subset of triple-negative breast cancer cells. Breast Cancer Res. 2016;18(1):95.
Elkhalifa D, Siddique A, Qusa M, Cyprian F, El Sayed K, Alali F, et al. Design, synthesis, and validation of novel nitrogen-based chalcone analogs against triple negative breast cancer. Eur J Med Chem. 2020;187:111954.
Guo G, Wang J, Han M, Zhang L, Li L. microRNA-761 induces aggressive phenotypes in triple-negative breast cancer cells by repressing TRIM29 expression. Cellular Oncol (Dordrecht). 2017;40(2):157–66.
Yang L, He Z, Yao J, Tan R, Zhu Y, Li Z, et al. Regulation of AMPK-related glycolipid metabolism imbalances redox homeostasis and inhibits anchorage independent growth in human breast cancer cells. Redox Biol. 2018;17:180–91.
Rigiracciolo D, Santolla M, Lappano R, Vivacqua A, Cirillo F, Galli G, et al. Focal adhesion kinase (FAK) activation by estrogens involves GPER in triple-negative breast cancer cells. J Exp Clin Cancer Res CR. 2019;38(1):58.
Wang K, Zhu X, Chen Y, Yin Y, Ma T. Tubeimoside V sensitizes human triple negative breast cancer MDA-MB-231 cells to anoikis via regulating caveolin-1-related signaling pathways. Arch Biochem Biophys. 2018;646:10–5.
Kim J, Lee N, Kim Y, Cho Y, An H, Oh E, et al. Disulfiram induces anoikis and suppresses lung colonization in triple-negative breast cancer via calpain activation. Cancer Lett. 2017;386:151–60.
Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13(5):555–62.
Shen X, Kramer R. Adhesion-mediated squamous cell carcinoma survival through ligand-independent activation of epidermal growth factor receptor. Am J Pathol. 2004;165(4):1315–29.
Marotta L, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker S, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121(7):2723–35.
Idowu M, Kmieciak M, Dumur C, Burton R, Grimes M, Powers C, et al. CD44(+)/CD24(-/low) cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Human Pathol. 2012;43(3):364–73.
Ma F, Li H, Wang H, Shi X, Fan Y, Ding X, et al. Enriched CD44(+)/CD24(-) population drives the aggressive phenotypes presented in triple-negative breast cancer (TNBC). Cancer Lett. 2014;353(2):153–9.
Teslow E, Mitrea C, Bao B, Mohammad R, Polin L, Dyson G, et al. Obesity-induced MBD2_v2 expression promotes tumor-initiating triple-negative breast cancer stem cells. Mol Oncol. 2019;13(4):894–908.
Tu Z, Schmöllerl J, Cuiffo B, Karnoub A. Microenvironmental regulation of long noncoding RNA LINC01133 promotes cancer stem cell-like phenotypic traits in triple-negative breast cancers. Stem Cells. 2019;37(10):1281–92.
Kruyt F, Schuringa J. Apoptosis and cancer stem cells: implications for apoptosis targeted therapy. Biochem Pharmacol. 2010;80(4):423–30.
He J, Lee H, Saha S, Ruan D, Guo H, Chan C. Inhibition of USP2 eliminates cancer stem cells and enhances TNBC responsiveness to chemotherapy. Cell Death Dis. 2019;10(4):285.
Dontu G, Abdallah W, Foley J, Jackson K, Clarke M, Kawamura M, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17(10):1253–70.
Song H, Luo Q, Deng X, Ji C, Li D, Munankarmy A, et al. VGLL4 interacts with STAT3 to function as a tumor suppressor in triple-negative breast cancer. Exp Mol Med. 2019;51(11):1–13.
An H, Kim JY, Oh E, Lee N, Cho Y, Seo JH. Salinomycin promotes anoikis and decreases the CD44+/CD24-stem-like population via inhibition of STAT3 activation in MDA-MB-231 cells. PLoS ONE. 2015;10(11):e0141919.
Li L, Neaves W. Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 2006;66(9):4553–7.
Shiozawa Y, Pedersen E, Havens A, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121(4):1298–312.
Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31–82.
Mondal T, Shivange G, Tihagam R, Lyerly E, Battista M, Talwar D, et al. Unexpected PD-L1 immune evasion mechanism in TNBC, ovarian, and other solid tumors by DR5 agonist antibodies. EMBO Mol Med. 2021;13(3):e12716.
Bei Y, Cheng N, Chen T, Shu Y, Yang Y, Yang N, et al. CDK5 inhibition abrogates TNBC stem-cell property and enhances anti-PD-1 therapy. Adv Sci. 2020;7(22):2001417.
Sidaway P. Setting dictates efficacy of pembrolizumab in TNBC. Nat Rev Clin Oncol. 2019;16(2):66.
Romero D. Benefit in patients with PD-L1-positive TNBC. Nat Rev Clin Oncol. 2019;16(1):6.
Xiang J, Hurchla MA, Fontana F, Su X, Amend SR, Esser AK, et al. CXCR4 Protein epitope mimetic antagonist POL5551 disrupts metastasis and enhances chemotherapy effect in triple-negative breast cancer. Mol Cancer Ther. 2015;14(11):2473–85.
Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan M, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50–6.
Su M-L, Chang T-M, Chiang C-H, Chang H-C, Hou M-F, Li W-S, et al. Inhibition of chemokine (C-C motif) receptor 7 sialylation suppresses CCL19-stimulated proliferation, invasion and anti-anoikis. PLoS ONE. 2014;9(6):e98823.
Chen IH, Shih H-C, Hsieh P-W, Chang F-R, Wu Y-C, Wu C-C. HPW-RX40 restores anoikis sensitivity of human breast cancer cells by inhibiting integrin/FAK signaling. Toxicol Appl Pharmacol. 2015;289(2):330–40.
Schempp CM, von Schwarzenberg K, Schreiner L, Kubisch R, Müller R, Wagner E, et al. V-ATPase inhibition regulates anoikis resistance and metastasis of cancer cells. Mol Cancer Ther. 2014;13(4):926.
Hinton A, Bond S, Forgac M. V-ATPase functions in normal and disease processes. Pflugers Arch Eur J Physiol. 2009;457(3):589–98.
Kozik P, Hodson N, Sahlender D, Simecek N, Soromani C, Wu J, et al. A human genome-wide screen for regulators of clathrin-coated vesicle formation reveals an unexpected role for the V-ATPase. Nat Cell Biol. 2013;15(1):50–60.
Arjonen A, Alanko J, Veltel S, Ivaska J. Distinct recycling of active and inactive β1 integrins. Traffic. 2012;13(4):610–25.
Kwak T, Lee E. Rapid multilayer microfabrication for modeling organotropic metastasis in breast cancer. Biofabrication. 2020. https://doi.org/10.1088/1758-5090/abbd28.
Chiu H, Yeh Y, Ho S, Wu Y, Wang B, Huang W, et al. A new histone deacetylase inhibitor enhances radiation sensitivity through the induction of misfolded protein aggregation and autophagy in triple-negative breast cancer. Cancers. 2019;11(11):1703.
Lin H, Kuei C, Lee H, Lin C, Chen Y, Chen C, et al. TNFSF13 upregulation confers chemotherapeutic resistance via triggering autophagy initiation in triple-negative breast cancer. J Mol Med. 2020;98(9):1255–67.
Cao C, Huang W, Zhang N, Wu F, Xu T, Pan X, et al. Narciclasine induces autophagy-dependent apoptosis in triple-negative breast cancer cells by regulating the AMPK-ULK1 axis. Cell Prolif. 2018;51(6):e12518.
Xu T, Zhang J, Yang C, Pluta R, Wang G, Ye T, et al. Identification and optimization of 3-bromo-N’-(4-hydroxybenzylidene)-4-methylbenzohydrazide derivatives as mTOR inhibitors that induce autophagic cell death and apoptosis in triple-negative breast cancer. Eur J Med Chem. 2021;219:113424.
Li H, Xia Z, Chen Y, Yang F, Feng W, Cai H, et al. Cantharidin inhibits the growth of triple-negative breast cancer cells by suppressing autophagy and inducing apoptosis in vitro and in vivo. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2017;43(5):1829–40.
Tian S, Chen Y, Yang B, Lou C, Zhu R, Zhao Y, et al. F1012–2 inhibits the growth of triple negative breast cancer through induction of cell cycle arrest, apoptosis, and autophagy. Phytother Res PTR. 2018;32(5):908–22.
Shen L, Jiang X, Li Z, Li J, Wang M, Jia G, et al. Cepharanthine sensitizes human triple negative breast cancer cells to chemotherapeutic agent epirubicin via inducing cofilin oxidation-mediated mitochondrial fission and apoptosis. Acta Pharmacol Sin. 2021;43(1):177–93.
Zhen Y, Zhao R, Wang M, Jiang X, Gao F, Fu L, et al. Flubendazole elicits anti-cancer effects via targeting EVA1A-modulated autophagy and apoptosis in triple-negative breast cancer. Theranostics. 2020;10(18):8080–97.
Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25(5):1037–43.
Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–42.
Wu S, Sun G, Cha T, Kao C, Chang S, Kuo S, et al. CSC-3436 switched tamoxifen-induced autophagy to apoptosis through the inhibition of AMPK/mTOR pathway. J Biomed Sci. 2016;23(1):60.
Yang B, Zhu R, Tian S, Wang Y, Lou S, Zhao H. Jatamanvaltrate P induces cell cycle arrest, apoptosis and autophagy in human breast cancer cells in vitro and in vivo. Biomed Pharmacother Biomed Pharmacother. 2017;89:1027–36.
Jinih M, Wang J, Pfirrmann R, O’Leary D, Corrigan M, Redmond H. Evaluation of the cytotoxic effects of the novel antineoplastic agent 1,4,5-oxathiazinane-4,4-dioxide on triple negative breast cancer cells. Anticancer Res. 2021;41(5):2247–56.
Khan M, Jain V, Rizwanullah M, Ahmad J, Jain K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: a review on drug discovery and future challenges. Drug Discov Today. 2019;24(11):2181–91.
Basho R, Gilcrease M, Murthy R, Helgason T, Karp D, Meric-Bernstam F, et al. Targeting the PI3K/AKT/mTOR pathway for the treatment of mesenchymal triple-negative breast cancer: evidence from a phase 1 trial of mTOR inhibition in combination with liposomal doxorubicin and bevacizumab. JAMA Oncol. 2017;3(4):509–15.
Hahne J, Schmidt H, Meyer S, Engel J, Dietl J, Honig A. Anti-tumour activity of phosphoinositide-3-kinase antagonist AEZS 126 in models of triple-negative breast cancer. J Cancer Res Clin Oncol. 2013;139(6):905–14.
Yang W, SriRamaratnam R, Welsch M, Shimada K, Skouta R, Viswanathan V, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Ding Y, Chen X, Liu C, Ge W, Wang Q, Hao X, et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol. 2021;14(1):19.
Imai H. Biological significance of lipid hydroperoxide and its reducing enzyme, phospholipid hydroperoxide glutathione peroxidase, in mammalian cells. Yakugaku zasshi J Pharm Soc Jpn. 2004;124(12):937–57.
Tang X, Wu J, Ding C, Lu M, Keenan M, Lin C, et al. Cystine deprivation triggers programmed necrosis in VHL-deficient renal cell carcinomas. Cancer Res. 2016;76(7):1892–903.
Galluzzi L, Kepp O, Heiden MGV, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12(11):829–46.
Qiu F, Chen Y, Liu X, Chu C, Shen L, Xu J, et al. Arginine starvation impairs mitochondrial respiratory function in ASS1-deficient breast cancer cells. Sci Signal. 2014;7(319):ra31.
Chen M, Wang S, Hsu C, Yin P, Yeh T, Lee H, et al. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget. 2017;8(70):114588–602.
Lee A, Djamgoz M. Triple negative breast cancer: emerging therapeutic modalities and novel combination therapies. Cancer Treat Rev. 2018;62:110–22.
van der Meel R, Sulheim E, Shi Y, Kiessling F, Mulder WJM, Lammers T. Smart cancer nanomedicine. Nat Nanotechnol. 2019;14(11):1007–17.
Adir O, Poley M, Chen G, Froim S, Krinsky N, Shklover J, et al. Integrating artificial intelligence and nanotechnology for precision cancer medicine. Adv Mater. 2020;32(13):e1901989.
Rosenblum D, Joshi N, Tao W, Karp J, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9(1):1410.
Gao Y, Yang C, Liu X, Ma R, Kong D, Shi L. A multifunctional nanocarrier based on nanogated mesoporous silica for enhanced tumor-specific uptake and intracellular delivery. Macromol Biosci. 2012;12(2):251–9.
Zhu J, Dai P, Liu F, Li Y, Qin Y, Yang Q, et al. Upconverting nanocarriers enable triggered microtubule inhibition and concurrent ferroptosis induction for selective treatment of triple-negative breast cancer. Nano Lett. 2020;20(9):6235–45.
Kalimutho M, Parsons K, Mittal D, López J, Srihari S, Khanna K. Targeted therapies for triple-negative breast cancer: combating a stubborn disease. Trends Pharmacol Sci. 2015;36(12):822–46.
Masso-Welch P, Girald Berlingeri S, King-Lyons N, Mandell L, Hu J, Greene C, et al. LT-IIc, A bacterial type II Heat-labile enterotoxin, induces specific lethality in triple negative breast cancer cells by modulation of autophagy and induction of apoptosis and necroptosis. Int J Mol Sci. 2018;20(1):85.
El-Ashmawy N, Al-Ashmawy G, Amr E, Khedr E. Inhibition of lovastatin- and docosahexaenoic acid-initiated autophagy in triple negative breast cancer reverted resistance and enhanced cytotoxicity. Life Sci. 2020;259:118212.
Meng L, Liu S, Ding P, Chang S, Sang M. Circular RNA ciRS-7 inhibits autophagy of ESCC cells by functioning as miR-1299 sponge to target EGFR signaling. J Cell Biochem. 2020;121(2):1039–49.
Abdel-Mohsen M, Abdel Malak C, El-Shafey E. Influence of copper(I) nicotinate complex and autophagy modulation on doxorubicin-induced cytotoxicity in HCC1806 breast cancer cells. Adv Med Sci. 2019;64(1):202–9.
Nielsen T, Hsu F, Jensen K, Cheang M, Karaca G, Hu Z, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10(16):5367–74.
Fu W, Sun H, Zhao Y, Chen M, Yang X, Liu Y, et al. BCAP31 drives TNBC development by modulating ligand-independent EGFR trafficking and spontaneous EGFR phosphorylation. Theranostics. 2019;9(22):6468–84.
Liu Z, He K, Ma Q, Yu Q, Liu C, Ndege I, et al. Autophagy inhibitor facilitates gefitinib sensitivity in vitro and in vivo by activating mitochondrial apoptosis in triple negative breast cancer. PLoS ONE. 2017;12(5):e0177694.
Xu M, Johnson D, Grandis J. EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev. 2017;36(3):463–73.
Lu Y, Zhao X, Liu Q, Li C, Graves-Deal R, Cao Z, et al. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/β-catenin signaling. Nat Med. 2017;23(11):1331–41.
Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526(7572):263–7.
Islam M, Dasgupta H, Basu M, Roy A, Alam N, Roychoudhury S, et al. Reduction of nuclear Y654-p-β-catenin expression through SH3GL2-meditated downregulation of EGFR in chemotolerance TNBC: clinical and prognostic importance. J Cell Physiol. 2020;235(11):8114–28.
Foidart P, Yip C, Radermacher J, Blacher S, Lienard M, Montero-Ruiz L, et al. Expression of MT4-MMP, EGFR, and RB in triple-negative breast cancer strongly sensitizes tumors to erlotinib and palbociclib combination therapy. Clin Cancer Res. 2019;25(6):1838–50.
Menbari M, Rahimi K, Ahmadi A, Mohammadi-Yeganeh S, Elyasi A, Darvishi N, et al. miR-483-3p suppresses the proliferation and progression of human triple negative breast cancer cells by targeting the HDAC8>oncogene. J Cell Physiol. 2020;235(3):2631–42.
Naorem L, Muthaiyan M, Venkatesan A. Identification of dysregulated miRNAs in triple negative breast cancer: a meta-analysis approach. J Cell Physiol. 2019;234(7):11768–79.
Xu W, Song C, Wang X, Li Y, Bai X, Liang X, et al. Downregulation of miR-155-5p enhances the anti-tumor effect of cetuximab on triple-negative breast cancer cells via inducing cell apoptosis and pyroptosis. Aging. 2021;13(1):228–40.
Berezovskaya O, Schimmer AD, Glinskii AB, Pinilla C, Hoffman RM, Reed JC, et al. Increased expression of apoptosis inhibitor protein XIAP contributes to anoikis resistance of circulating human prostate cancer metastasis precursor cells. Cancer Res. 2005;65(6):2378.
Joseph R, Yazer E, Hanakawa Y, Stadnyk A. Prostaglandins and activation of AC/cAMP prevents anoikis in IEC-18. Apoptosis Int J Program Cell Death. 2005;10(6):1221–33.
Guadamillas MC, Cerezo A, del Pozo MA. Overcoming anoikis—pathways to anchorage-independent growth in cancer. J Cell Sci. 2011;124(19):3189–97.
Lahlou H, Muller W. β1-integrins signaling and mammary tumor progression in transgenic mouse models: implications for human breast cancer. Breast Cancer Res BCR. 2011;13(6):229.
Casey RC, Burleson KM, Skubitz KM, Pambuccian SE, Oegema TR, Ruff LE, et al. β1-integrins regulate the formation and adhesion of ovarian carcinoma multicellular spheroids. Am J Pathol. 2001;159(6):2071–80.
Schwartz SM. Smooth muscle migration in vascular development and pathogenesis. Transpl Immunol. 1997;5(4):255–60.
Mirando A, Patil A, Rafie C, Christmas B, Pandey N, Stearns V, et al. Regulation of the tumor immune microenvironment and vascular normalization in TNBC murine models by a novel peptide. Oncoimmunology. 2020;9(1):1760685.
Shome R, Ghosh S. Tweaking EMT and MDR dynamics to constrain triple-negative breast cancer invasiveness by EGFR and Wnt/β-catenin signaling regulation. Cell Oncol (Dordrecht). 2021;44(2):405–22.
Zhu H, Rao Z, Yuan S, You J, Hong C, He Q, et al. One therapeutic approach for triple-negative breast cancer: checkpoint kinase 1 inhibitor AZD7762 combination with neoadjuvant carboplatin. Eur J Pharmacol. 2021;908:174366.
Park J, Jonas S, Bataillon G, Criscitiello C, Salgado R, Loi S, et al. Prognostic value of tumor-infiltrating lymphocytes in patients with early-stage triple-negative breast cancers (TNBC) who did not receive adjuvant chemotherapy. Ann Oncol. 2019;30(12):1941–9.
Proia D, Zhang C, Sequeira M, Jimenez J, He S, Spector N, et al. Preclinical activity profile and therapeutic efficacy of the HSP90 inhibitor ganetespib in triple-negative breast cancer. Clin Cancer Res. 2014;20(2):413–24.
Llop-Guevara A, Loibl S, Villacampa G, Vladimirova V, Schneeweiss A, Karn T, et al. Association of RAD51 with homologous recombination deficiency (HRD) and clinical outcomes in untreated triple-negative breast cancer (TNBC): analysis of the GeparSixto randomized clinical trial. Ann Oncol. 2021;32(12):1590–6.
Jin J, Fang H, Yang F, Ji W, Guan N, Sun Z, et al. Combined inhibition of ATR and WEE1 as a novel therapeutic strategy in triple-negative breast cancer. Neoplasia (New York, NY). 2018;20(5):478–88.
Shi Y, Jin J, Wang X, Ji W, Guan X. DAXX, as a tumor suppressor, impacts DNA damage repair and sensitizes BRCA-proficient TNBC cells to PARP inhibitors. Neoplasia. 2019;21(6):533–44.
Litzenburger B, Creighton C, Tsimelzon A, Chan B, Hilsenbeck S, Wang T, et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin Cancer Res. 2011;17(8):2314–27.
Wu W, Ma J, Shao N, Shi Y, Liu R, Li W, et al. Co-Targeting IGF-1R and autophagy enhances the effects of cell growth suppression and apoptosis induced by the IGF-1R inhibitor NVP-AEW541 in triple-negative breast cancer cells. PLoS ONE. 2017;12(1):e0169229.
Wang W, Green M, Choi J, Gijón M, Kennedy P, Johnson J, et al. CD8 T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–4.
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W, et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med. 2019;23(8):4900–12.
Shu S, Lin C, He H, Witwicki R, Tabassum D, Roberts J, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529(7586):413–7.
Zhang Y, Xu B, Shi J, Li J, Lu X, Xu L, et al. BRD4 modulates vulnerability of triple-negative breast cancer to targeting of integrin-dependent signaling pathways. Cell Oncol. 2020;43(6):1049–66.
Ren C, Zhang G, Han F, Fu S, Cao Y, Zhang F, et al. Spatially constrained tandem bromodomain inhibition bolsters sustained repression of BRD4 transcriptional activity for TNBC cell growth. Proc Natl Acad Sci U S A. 2018;115(31):7949–54.
Verma N, Vinik Y, Saroha A, Nair NU, Ruppin E, Mills G, et al. Synthetic lethal combination targeting BET uncovered intrinsic susceptibility of TNBC to ferroptosis. Sci Adv. 2020;6(34):eaba8968.
Kufe D. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene. 2013;32(9):1073–81.
Siroy A, Abdul-Karim F, Miedler J, Fong N, Fu P, Gilmore H, et al. MUC1 is expressed at high frequency in early-stage basal-like triple-negative breast cancer. Hum Pathol. 2013;44(10):2159–66.
Hasegawa M, Takahashi H, Rajabi H, Alam M, Suzuki Y, Yin L, et al. Functional interactions of the cystine/glutamate antiporter, CD44v and MUC1-C oncoprotein in triple-negative breast cancer cells. Oncotarget. 2016;7(11):11756–69.
Lacunza E, Baudis M, Colussi A, Segal-Eiras A, Croce M, Abba M. MUC1 oncogene amplification correlates with protein overexpression in invasive breast carcinoma cells. Cancer Genet Cytogenet. 2010;201(2):102–10.
Zhang J, Liu Y, Tan J, Zhang Y, Wong C, Lin Z, et al. Necroptotic virotherapy of oncolytic alphavirus M1 cooperated with Doxorubicin displays promising therapeutic efficacy in TNBC. Oncogene. 2021;40(29):4783–95.
Zhang Z, Lu M, Chen C, Tong X, Li Y, Yang K, et al. Holo-lactoferrin: the link between ferroptosis and radiotherapy in triple-negative breast cancer. Theranostics. 2021;11(7):3167–82.
Sleire L, Skeie B, Netland I, Førde H, Dodoo E, Selheim F, et al. Drug repurposing: sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene. 2015;34(49):5951–9.
Li ZJ, Hou YJ, Hao GP, Pan XX, Fei HR, Wang FZ. CUDC-907 enhances TRAIL-induced apoptosis through upregulation of DR5 in breast cancer cells. J Cell Commun Signal. 2020;14(4):377–87.
Ralff MD, Kline CLB, Kucukkase OC, Wagner J, Lim B, Dicker DT, et al. ONC201 demonstrates antitumor effects in both triple-negative and non-triple-negative breast cancers through TRAIL-dependent and TRAIL-independent mechanisms. Mol Cancer Ther. 2017;16(7):1290–8.
Das S, Tripathi N, Siddharth S, Nayak A, Nayak D, Sethy C, et al. Etoposide and doxorubicin enhance the sensitivity of triple negative breast cancers through modulation of TRAIL-DR5 axis. Apoptosis. 2017;22(10):1205–24.
Diamond JR, Eckhardt SG, Pitts TM, van Bokhoven A, Aisner D, Gustafson DL, et al. A phase II clinical trial of the Aurora and angiogenic kinase inhibitor ENMD-2076 for previously treated, advanced, or metastatic triple-negative breast cancer. Breast Cancer Res. 2018;20(1):82.
Ionkina AA, Tentler JJ, Kim J, Capasso A, Pitts TM, Ryall KA, et al. Efficacy and molecular mechanisms of differentiated response to the aurora and angiogenic kinase inhibitor ENMD-2076 in preclinical models of p53-mutated triple-negative breast cancer. Front Oncol. 2017;7:94.
Cai JJ, Xia JR, Zou J, Wang Q, Ma Q, Sun R, et al. The PI3K/mTOR dual inhibitor NVP-BEZ235 stimulates mutant p53 degradation to exert anti-tumor effects on triple-negative breast cancer cells. FEBS Open Bio. 2020;10(4):535–45.
Nguyen K, Yan Y, Yuan B, Dasgupta A, Sun J, Mu H, et al. ST8SIA1 regulates tumor growth and metastasis in TNBC by activating the FAK-AKT-mTOR signaling pathway. Mol Cancer Ther. 2018;17(12):2689–701.
Traina TA, Miller K, Yardley DA, Eakle J, Schwartzberg LS, O’Shaughnessy J, et al. Enzalutamide for the treatment of androgen receptor-expressing triple-negative breast cancer. J Clin Oncol. 2018;36(9):884–90.
Lehmann BD, Jovanovic B, Chen X, Estrada MV, Johnson KN, Shyr Y, et al. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS ONE. 2016;11(6):e0157368.
Chen X, Li J, Gray WH, Lehmann BD, Bauer JA, Shyr Y, et al. TNBCtype: a subtyping tool for triple-negative breast cancer. Cancer Inform. 2012;11:147–56.
Acknowledgements
We are grateful to Prof. Canhua Huang and Prof. Heng Xu (Sichuan University) for their critical reviews on this manuscript.
Funding
This work was supported by grants from National Natural Science Foundation of China (Grant Nos. 82172649, 81773889, and 82073998), the Science & Technology Department of Sichuan Province (Grant No. 22CXRC0077), Youth Talent Promotion Project of China Association for Science and Technology (Grant No. CACM-2020-QNRC1-01) and Key R&D Program of Sichuan Province (Grant No. 2021YFS0046).
Author information
Authors and Affiliations
Contributions
BL, FP, and BH designed this study. ML, RQ, WH, and HZ drafted the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liao, M., Qin, R., Huang, W. et al. Targeting regulated cell death (RCD) with small-molecule compounds in triple-negative breast cancer: a revisited perspective from molecular mechanisms to targeted therapies. J Hematol Oncol 15, 44 (2022). https://doi.org/10.1186/s13045-022-01260-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01260-0