
Abstract
Cancer is characterized as a complex disease caused by coordinated alterations of multiple signaling pathways. The Ras/RAF/MEK/ERK (MAPK) signaling is one of the best-defined pathways in cancer biology, and its hyperactivation is responsible for over 40% human cancer cases. To drive carcinogenesis, this signaling promotes cellular overgrowth by turning on proliferative genes, and simultaneously enables cells to overcome metabolic stress by inhibiting AMPK signaling, a key singular node of cellular metabolism. Recent studies have shown that AMPK signaling can also reversibly regulate hyperactive MAPK signaling in cancer cells by phosphorylating its key components, RAF/KSR family kinases, which affects not only carcinogenesis but also the outcomes of targeted cancer therapies against the MAPK signaling. In this review, we will summarize the current proceedings of how MAPK-AMPK signalings interplay with each other in cancer biology, as well as its implications in clinic cancer treatment with MAPK inhibition and AMPK modulators, and discuss the exploitation of combinatory therapies targeting both MAPK and AMPK as a novel therapeutic intervention.
Similar content being viewed by others

Introduction
The Ras/RAF/MEK/ERK (MAPK) signaling is a fundamental pathway in cell biology, and its alteration causes human cancers or developmental disorders. Given its crucial roles in physiology and pathology, this pathway has been extensively studied for over two decades. Unfortunately, the regulation of MAPK signaling remains ambiguous till now by virtue of its intrinsic complexity and diverse crosstalks with other signalings. Here, we focus on the complicated interplays between the MAPK and the AMPK signalings in cellular carcinogenesis and their implications in current targeted cancer therapies. We hope this review would provide a conceptual framework for developing more effective therapeutic approaches against hyperactive MAPK signaling-driven cancers.
The Ras/RAF/MEK/ERK (MAPK) signaling and its aberrant activation in cancers
The Ras/RAF/MEK/ERK (MAPK) signaling
The Ras/RAF/MEK/ERK (MAPK, mitogen-activated protein kinase) signaling is a central pathway that regulates cellular proliferation, differentiation, and survival. This signaling pathway was discovered in the 1970s–1980s, when Ras small GTPases were identified as first oncogenes from sarcoma viruses [1,2,3,4,5,6]. Later, studies on viral oncogenes had also led to the discovery of a N-terminal truncated version of RAF Ser/Thr kinase (RAF1 or CRAF) [1,2,3,4,5]. In contrast, the other two components of this signaling pathway, MEK (mitogen-activated protein kinase kinase) and ERK (mitogen-activated protein kinase) were identified as cytoplasmic protein kinases activated by mitogens in the 1990s [7,8,9,10,11]. Following these discoveries, RAF was identified as the upstream kinase of MEK in 1992 and the first direct effector of Ras in 1993 [12, 13], resulting in the delineation of the whole MAPK signaling pathway, which is considered as a milestone in our understanding of how cell senses external stimuli.
The first component of MAPK signaling, Ras small GTPases, have three gene isoforms: H-ras, K-ras, and N-ras, that encode four proteins with splicing isoforms of K-ras giving rise to K-ras4A and K-ras4B. Although all Ras proteins possess highly homologous sequences, they have quite different activities, tissue expression patterns, and effector preferences, which lead to their differential physiological and pathological functions [14,15,16,17].
The downstream of Ras small GTPases is the RAF/MEK/ERK kinase cascade [18]. The first kinases in this cascade, RAF/KSR (kinase suppressor of Ras) family kinases, include three RAF isoforms, i.e., CRAF, BRAF, and ARAF, and two close pseudokinases, i.e., KSR1 and KSR2. All RAF isoforms have highly homologous sequences and similar structures with three conserved regions: conserved region 1 (CR1) contains RAS-binding domain (RBD) and a Cys-rich domain [19, 20]; conserved region 2 (CR2) is characterized by a Ser/Thr-rich sequence; conserved region 3 (CR3) comprises of a putative kinase domain with a N-terminal acidic motif (NTA) [21,22,23] and a C-terminal regulatory tail [24,25,26]. Nevertheless, RAF isoforms have variable kinase activities with an order as BRAF>CRAF>ARAF likely by virtue of their distinct NTA motifs and APE motifs that contribute to the dimerization-driven transactivation of RAFs [27,28,29,30]. In contrast to RAF isoforms, KSR proteins replace the RBD at the N-terminus with a coiled-coil fused sterile α-motif and Pro-rich stretch that are responsible for recruiting proteins to the plasma membrane upon stimulation, and lack the catalytic lysine in VAIK motif of kinase domain which impairs their catalytic activity [31, 32]. Given their associations with MEK and ERK as well as low kinase activity, KSR proteins have been thought as scaffold proteins in a long term. However, recent studies have indicated that KSR proteins can also function as allosteric activators to stimulate the catalytic activity of RAF proteins through dimerization [27, 32,33,34,35,36,37]. The side-to-side dimerization of RAF/KSR family kinases is critical not only for their activation but also for their catalytic activity towards downstream kinases [25, 38,39,40,41,42]. MEKs (MEK1 and MEK2) are the second kinases of the RAF/MEK/ERK kinase cascade, which have both redundant and non-redundant functions [43, 44]. These two dual-specific kinases comprise a short regulatory N-terminus and a canonic kinase domain. The N-terminal regulatory region of MEK1/2 contains a docking site for substrate ERKs, a nuclear export sequence that controls the cytoplasmic-nuclear shuttling of proteins, and a negative regulatory sequence that forms a helix and locks kinase in an inactive conformation [11, 43, 44]. Further, through its kinase domain, MEK1/2 forms a face-to-face heterodimer with RAF/KSR, or a homodimer/heterodimer with itself, which is indispensable for its activation stimulated by RAF and for its activity towards ERKs [28, 45, 46]. Like MEKs, the terminal kinases of MAPK signaling, ERKs, also include two highly homologous members, ERK1 and ERK2, which have a central kinase domain flanked by short N- and C-terminal tails. These two isoforms also have redundant functions albeit different expression patterns [7,8,9,10]. However, unlike RAFs and MEKs that have very limited substrates, ERKs recognize and phosphorylate numerous substrates that include transcription factors, protein kinases and phosphatases, and other functional proteins [47,48,49,50,51].
It should be noted that active Ras also turns on other signaling pathways such as PI3K/AKT/mTORC, which regulate different cellular functions [52]. In this review, we focus only on the MAPK signaling given its dominant role in cancer biology.
Hyperactive Ras/RAF/MEK/ERK (MAPK) signaling in cancers
The MAPK signaling plays a crucial role in cell biology and is tightly regulated in normal cells. Upon engagement of receptor tyrosine kinases (RTKs) or other stimulations, Ras small GTPases are activated by GTP/GDP exchange factors (GEFs), which in turn recruit RAF/MEK complexes to the plasma membrane and trigger the RAF/MEK/ERK kinase cascade through facilitating RAF/RAF (or KSR), RAF/MEK, and MEK/MEK interactions as well as subsequent phosphorylations [53]. Active ERKs are further translocated into the nuclei or stay in the cytoplasm, where they phosphorylate a number of substrates that regulate cell functions [49,50,51, 54, 55]. On the other hand, active MAPK signaling also turns on some negative feedback loops, which help cells return to quiescent status [56,57,58]. An aberrant activation of MAPK signaling frequently induces human cancers or developmental disorders, though an extremely high MAPK signaling may induce cell death or senescence under some conditions [59,60,61,62,63].
Hyperactive MAPK signaling exists in over 85% of cancers, which is caused directly by genetic alterations of its upstream activators or components, including RTKs, Ras, and BRAF, or indirectly by those independent of Ras or RAF [64,65,66], and significantly promotes disease progression [67]. Since genetic alterations of RTKs in cancers have been extensively reviewed in recent years [68,69,70,71,72,73], here we focus on oncogenic mutations of Ras and BRAF. As a small GTPase, Ras cycles between active GTP-bound status and inactive GDP-bound status, which is regulated by GEFs and GTPase-activating proteins (GAPs). Oncogenic Ras mutations can be mainly classified into two groups: (1) mutations on glycine 12 or 13 (G12/13) that impair GAP associations and (2) mutations on glutamine 61 (Q61) that diminish the intrinsic GTPase activity of Ras [74], both of which lead to an extended half-life of GTP-loaded Ras. Oncogenic Ras mutations have both isoform and cancer-type preferences. K-ras is mostly mutated in all cancers (85%), followed by N-ras (12%) and H-ras (3%), and its mutations prevail in pancreatic cancers, while those of N-ras in myeloma and melanomas, and H-ras in adrenal gland cancers [75, 76]. This phenomenon may reflect underlying fundamental signaling landscapes, and RAS mutants interplay with these landscapes. As the downstream effector of Ras, RAF is another dominant target of oncogenic mutations in the MAPK signaling pathway. Similarly, RAF mutations have isoform preference in cancers as Ras mutations with BRAF >> CRAF > ARAF, which may arise from their different basal activities. Overall, a single point mutation that converts Val 600 into Glu in the activation loop of BRAF accounts for > 90% cases [77]. Although BRAF (V600E) exists only in ~ 7% of all cancers, it is highly prevalent in some tissue-specific cancers such as melanoma (50~60%), thyroid cancer (40~50%), and histiocytosis (~50%) [78,79,80,81], albeit the underlying molecular mechanism(s) remains unknown. In contrast to Ras and RAF, MEK and ERK have rare mutations in cancers though their mutations have been shown to be responsible for some RAF inhibitor (RAFi)-resistant cases in current cancer therapies [82,83,84,85].
Targeting the Ras/RAF/MEK/ERK (MAPK) signaling pathway for cancer therapy: promising but challenging
Given their high prevalence in cancers, great efforts have been made to develop specific inhibitors against oncogenic Ras and RAF mutants in the last decades. These inhibitors that have been approved for clinic treatment of Ras/RAF-mutated cancers or under clinical trials are listed in Table 1. However, none of these inhibitors can effectively target the large portion of Ras mutants in cancers. Since having no attractive docking sites suitable for designing high-affinity and selective small molecule inhibitors, Ras mutants have been thought as “undruggable” cancer drivers in a long term. Until recently, a group of covalent small inhibitors that are docked into a previously unknown pocket of GDP-bound Ras and are linked to the adventive cysteine of Ras(G12C) have been developed and achieved encouraging outcomes for treating Ras(G12C)-driven cancers as a single agent in clinical trials [86,87,88,89,90,91] (Fig. 1). To further enhance their efficacy, these Ras(G12C) inhibitors are also undergoing clinical evaluation when combined with SHP2 (Src homology region 2 domain-containing phosphatase-2) inhibitors that block the pathway reactivation caused by the relief of negative feedback loops [92, 93] (Clinical Trial: NCT04330664). In addition, these inhibitors have also been further developed into Ras(G12C) degraders by conjugating with ligands of ubiquitin E3 ligases, which effectively deplete Ras mutant proteins in cancer cells [94, 95] though their efficacy in vivo remains unknown. Unlike Ras(G12C), the majority of Ras mutants remain “undruggable” at present [96].

Target hyperactive Ras/RAF/MEK/ERK (MAPK) signaling for cancer therapy. The Ras/RAF/MEK/ERK (MAPK) signaling functions downstream of receptor tyrosine kinases (RTKs). Upon engagement by their ligands, RTKs activates guanine exchange factors, Sos proteins, which load GTP to Ras GTPases. Then, GTP-bound Ras GTPases recruit RAF/MEK heterodimers in cytosol to plasma membrane where they form transient tetramers through the side-to-side dimerization of RAFs. The RAF dimerization not only turns on RAFs but also loosens RAF/MEK heterodimerization and facilitates MEK homodimerization on RAF dimer surface, which leads to the activation of MEKs by RAFs. Once MEKs are activated, they phosphorylate ERKs, and then active ERKs phosphorylate a number of downstream effectors. In cancer cells, hyperactive Ras/RAF/MEK/ERK (MAPK) signaling arising from genetic mutations of Ras GTPases and BRAF can be targeted by small molecular inhibitors of Ras G12C, BRAF(V600E), MEK, and ERK
It has been shown that Ras activates downstream effectors through direct interactions. Therefore, disrupting Ras/effector interactions might be an alternative approach that can effectively block cancer growth driven by Ras mutations. Such a type of small molecule blockers include rigosertib, sulindac, and MCP110, and among which, the therapeutic efficacy of rigosertib combined with nivolumab for Ras-mutated cancers is being determined by phase I/II clinical trials currently [97] (Clinical Trial: NCT04263090). However, it has to be noted that these inhibitors impair the MAPK signaling in both Ras-mutated cancers and normal tissues and thereby their therapeutic index may not be high.
Genetic studies have revealed that the ablation of the RAF/MEK/ERK kinase cascade but not other effector pathways is a most efficient approach to inhibit the growth of Ras-mutated cancers [98], which leads to extensive developments of specific inhibitors against this kinase cascade for treating Ras-mutated cancers. Moreover, these inhibitors should be also effective for treating RAF-mutated cancers. Indeed, a number of RAF/MEK/ERK inhibitors have been developed and applied to clinical trials for treating Ras/RAF-mutated cancers [67, 99,100,101,102,103,104,105,106,107]. At present, three RAF inhibitors and three MEK inhibitors have been approved to treat late-stage BRAF(V600E)-harboring cancers as a single agent or in combination with other chemotherapeutics and exhibited excellent efficacies [101, 108,109,110,111,112,113,114,115,116] (Fig. 1). However, Ras-mutated cancers possess intrinsic resistance to both RAF and MEK inhibitors [98], and even BRAF (V600E)-harboring cancers develop acquired resistance after 6–10 months treatment [111, 117]. Mechanistic studies have shown that active Ras facilitates the RAF dimerization on plasma membrane, which leads to both intrinsic and acquired resistance to RAF inhibitors [118,119,120]. To overcome the drug resistance arising from enhanced RAF dimerization, the second-generation RAF inhibitors such as PLX8394, BGB283, TAK-580, and CCT3833 have been developed and are undergoing clinical evaluations (Clinical Trials: NCT02428712, NCT02610361, NCT03905148, NCT02327169, NCT02437227). These novel RAF inhibitors reduce the RAF dimerization-driven resistance through distinct mechanisms: (1) PLX8394 and BGB283 impair RAF dimerization upon loading on RAF proteins [121,122,123]; (2) TAK-580 binds to and inhibits both protomers in RAF dimers [124]; (3) CCT3833 inhibits both RAF and upstream kinases of Ras and thereby prevents the activation of Ras by the relief of negative feedback loops [125, 126]. Besides these second-generation RAF inhibitors, a unique RAF/MEK dual inhibitor, RO5126766, has been developed and exhibited a strong potential against both Ras- and RAF-mutated cancers in phase I clinical trials [127,128,129,130]. This allosteric inhibitor docks on MEK and prevents the release of MEK from RAF as well as the subsequent phosphorylation of MEK by RAF [128], which gives it much more advantages than all other known RAF inhibitors according to the regulatory mechanism of the RAF/MEK/ERK kinase cascade [46]. As to small molecule inhibitors that target the terminal kinase, ERK, although a number of them have been developed and are undergoing clinical trials [131, 132], their therapeutic values for treating Ras/RAF-mutated cancers remain unknown. Like MEK inhibitors, these ERK inhibitors may not achieve a good therapeutic index as single agents by virtue of their inhibitory role in both malignant and normal tissues. However, they may contribute to anti-Ras/RAF cancer therapy as synergetic agents combined with Ras/RAF inhibitors.
Overall, targeting hyperactive MAPK signaling has achieved exciting outcomes for treating Ras/RAF-mutated cancers. However, although some effective small molecule inhibitors have been developed and applied to clinical treatment, drug resistance and side effects remain remarkable challenges and there is still a long way to develop a long-effective approach with manageable side effects for treating Ras/RAF-mutated cancers.
Although hyperactive MAPK signaling has a dominant role in cancer biology, it is fine-tuned by other signalings such as PI3K/AKT/mTORC and AMPK during disease progression [133]. These signaling interplays have important impacts on both cancer progression and clinical treatment based on MAPK inhibition. In this review, we will focus on the crosstalk between MAPK and AMPK signalings.
AMPK signaling and its roles in cancer biology
AMPK signaling and cellular metabolism
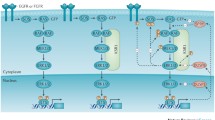
AMPK (AMP-activated protein kinase) is an energy sensor that monitors the AMP:ADP:ATP ratio in eukaryotic cells. This atypical protein kinase was firstly discovered as a contaminant during the purification of acetyl-CoA carboxylase (ACC), a well-studied substrate of AMPK for fatty acid (FA) synthesis nowadays [134,135,136] (Fig. 2). However, the phosphorylation of ACC by AMPK in response to the high AMP/ATP ratio had not been revealed until a decade later [137], and the enzyme was thus named as AMPK thereafter [138] (Fig. 2). Biochemical studies have shown that AMPK consists of three subunits including the catalytic α subunit and the regulatory β and γ subunits [139,140,141,142,143,144,145,146,147,148] (Fig. 2). In mammals, AMPK subunits are encoded as several isoforms (α1, α2; β1, β2; γ1, γ2, γ3), which are preferentially expressed in specific tissues or organisms [145, 149, 150]. For instance, the α2 subunit associates only with β1 in type I muscle fibers, while it binds to both β1 and β2 in type II muscle fibers [150, 151]. Also, the liver formulation of AMPK subunits differs among species as that α1β2γ1 is dominant in human whereas α1β1γ1 and α2β1γ1 in dog and rat, respectively [152]. Although an isoform replacement of AMPK subunits may not extensively affect the basal activity of AMPK as adaptive responses such as exercise do [153], it alters AMPK’s subcellular locations and sensitivity as well as interactions with other signaling pathways [147]. The organism/tissue/stage-specific selectivity of subunit isoforms complicates AMPK’s regulation.

AMPK signaling and its downstream effectors. AMPK is activated by liver kinase B1 (LKB1) or calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2/β) through phosphorylation on Thr172 of α subunit and is inactivated through dephosphorylation of this site by protein phosphatases in response to changes of cellular AMP:ADP:ATP ratio. Downstream effectors activated by AMPK are indicated as arrows, and those inhibited by AMPK are shown as bar-headed lines
As a key sensor of cellular energy stress, the activity of AMPK is predominantly regulated by cellular AMP/ADP/ATP that competitively binds to the γ subunit of AMPK and thus promotes or inhibits the phosphorylation of Thr172 on α subunit by the tumor suppressor liver kinase B1 (LKB1) or the dephosphorylation of this site by phosphatases [154, 155] (Fig. 2). Besides adenine nucleotides, intracellular calcium ions activate AMPK through calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2, also called CAMKKβ) [156,157,158] (Fig. 2), which acts downstream of the hormone-activated receptors such as muscarinic receptors and ghrelin receptor on endothelial cells or neuron cells [159,160,161,162]. On the other hand, AMPK can be inhibited by a metabolite of glucose, fructose 1,6-bisphosphate (FBP), which binds to the aldolase and prevents the interaction of AMPK with LKB1 in glucose-rich environments [163] (Fig. 2). Active AMPK has more than 100 downstream substrates that regulate the metabolism of lipids, cholesterol, carbohydrates, and amino acids.
Active AMPK promotes the oxidation of fatty acids and inhibits the synthesis of fatty acids and cholesterol, which involves largely in acetyl-CoA. AMPK phosphorylates and inhibits HMG-CoA reductase (HMGR) that requires acetyl-CoA in its reduction reaction [138, 164, 165] (Fig. 2). Also, AMPK phosphorylates ACC that converts acetyl-CoA to malonyl-CoA and therefore slows down the de novo fatty acid (FA) synthesis and increases the FA oxidation [166] (Fig. 2). Alternatively, AMPK regulates the lipid metabolism through altering the mitochondria structure and function. In the mitochondria, AMPK phosphorylates A-kinase anchoring protein 1 (AKAP1), a key scaffold protein for protein kinase A (PKA), and facilitates the phosphorylation of a mitochondria fusion factor, dynamin-related protein 1 (DRP1) by PKA, which promotes mitochondrial fusion and oxidative phosphorylation [167]. Moreover, AMPK accelerates the mitochondria biogenesis likely through phosphorylating and activating the transcriptional activator, proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) [168, 169] (Fig. 2). However, upon energy stress, AMPK plays an opposite role in mitochondria biology. Under this condition, AMPK is essential for the fragmentation of mitochondria. AMPK phosphorylates mitochondrial fission factor (MFF) on Ser129 and thereby facilitates the translocation of DRP1 from cytosol to mitochondria membrane in energy stress-driven mitochondria fission [170, 171]. Then, AMPK promotes the clearance of damaged mitochondria through autophagy. In this process, AMPK binds directly to and phosphorylates the unc-51-like autophagy activating kinase 1 (ULK1), Autophagy-related gene 9 (ATG9), and Beclin 1, which triggers the autophagosome formation [172,173,174,175] (Fig. 2).
Active AMPK directly regulates the carbohydrate metabolism or indirectly through altering the fatty acid metabolism as described above. Activation of AMPK stimulates the expression and plasma membrane translocation of solute carrier family member (GLUT) proteins and thereby facilitates glucose import [152, 176,177,178,179,180,181] (Fig. 2). Intracellularly, AMPK phosphorylates and activates 6-phosphofructo-2-kinase (PFK2) that is responsible for the synthesis of fructose 2,6-bisphosphate, a potent stimulator of glycolysis, and thus accelerates glycolysis [182] (Fig. 2). Furthermore, AMPK appears to phosphorylate and inhibit glycogen synthase in the liver, which dampens glycogen synthesis and thus indirectly enhances glycolysis [183].
Active AMPK maintains cellular amino acid homeostasis mainly by controlling the activity of mammalian target of rapamycin complex 1 (mTORC1). The mTORC1 is a central sensor of cellular amino acids that samples amino acids in both cytosol and lysosome [184, 185]. Upon activation by amino acids, mTORC1 stimulates protein synthesis by phosphorylating ribosomal protein S6 kinase B1 (S6K) and eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), which enhances the consumption of cellular amino acids. Moreover, active mTORC1 blocks cellular autophagy by phosphorylating ULK1 and impairs the recycling of amino acids [186]. Both effects of mTORC1 lead to a remarkable drop of cellular amino acid reservoir. Active AMPK has been shown to inhibit the activity of mTORC1 direct and indirectly upon energy stress, which limits the expenditure of amino acids. Alternatively, active AMPK can restrict protein synthesis by phosphorylating and thereby inhibiting eukaryotic translation elongation factor 2 (eEF2) kinase, a key regulator of protein synthesis [187]. To restore cellular amino acid reservoir, active AMPK stimulates cellular autophagy as discussed above, which degrades surplus or dysfunctional proteins into amino acids [186]. In addition, it is worth noted that cellular amino acids can affect the activity of AMPK reversely. Dependent on conditions/contexts, either amino acids may inhibit or stimulate the activity of AMPK though underlying molecular mechanisms remain ambiguous [188,189,190].
AMPK signaling in cancer biology
It is well known that AMPK is a putative substrate of tumor suppressor, LKB1 [154, 155, 191] (Fig. 2). Therefore, AMPK has been generally considered as a key effector that mediates the tumor-suppressive function of LKB1. Indeed, a genetic ablation of the AMPK α subunit in mice accelerates Myc-driven lymphomagenesis through facilitating a metabolic shift to aerobic glycolysis [192]. Simultaneously, AMPK inhibitors (AMPKi) promote epithelial-to-mesenchymal transition (EMT) in breast and prostate cancers [193]. These studies validate AMPK as a tumor suppressor under certain circumstances. Further mechanistic studies have demonstrated that AMPK prevents cancers through phosphorylating multiple targets that play indispensable roles on different layers of disease progression. AMPK phosphorylates angiomotin like 1 (AMOTL1), an adaptor protein in the Hippo-Yap pathway, and thus blocks Yes1 associated transcriptional regulator (YAP) activity, which impairs cancer cells’ proliferation and survival [194]. AMPK also phosphorylates TSC complex subunit 2 (TSC2) and regulatory associated protein of MTOR complex 1 (Raptor) and thereby inactivates mTORC1 [195, 196], which in turn elevates cellular autophagy activity and inhibits cancer initiation. To bypass this inhibitory effect, cancer cells can activate the MAGE family member A 3/6 (MAGEA3/6)-tripartite motif containing 28 (TRIM28) ubiquitin ligase complex that targets the AMPK α subunit for degradation and thus re-activates mTORC1 to restrict cellular autophagy [197]. Moreover, AMPK is able to phosphorylate enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) and thereby disrupts the polycomb repressive complex 2 (PRC2), which relieves the epigenetic silence of tumor suppressors in cancers [198]. Alternatively, AMPK phosphorylates and stabilizes another epigenetic master regulator, Tet methylcytosine dioxygenase 2 (TET2), which functions as a putative tumor suppressor to prevent tumorigenesis [199]. Altogether, these findings indicate that AMPK has a pronounced anti-tumor activity as its upstream kinase, LKB1 does.
Although significant studies have shown that AMPK dampens the pathogenesis of cancers, some emerging findings indicate that it may promote disease progression under other circumstances. In T cell acute lymphoblastic leukemia (T-ALL), oncogenic Notch signaling induces a high level of aerobic glycolysis which needs to be restrained by AMPK, and loss of AMPK results in energy stress-driven apoptosis of leukemic cells and slows down disease progression [200]. Similarly, in acute myeloid leukemia (AML), metabolic stress elevates the ROS level and induces DNA damage in leukemia-initiating cells (LICs), and AMPK confers metabolic stress resistance to LICs [201]. AMPK knockout or pharmaceutical inhibition under metabolic stress kills LICs and inhibits leukemogenesis. Moreover, AMPK plays a determinant role in maintaining the NADPH homeostasis in cancer cells upon energy stress, which is critical for cancer cell survival [202]. Depletion of the AMPK α subunit or its upstream kinase, LKB1 makes cancer cells susceptible to death upon energy stress, such as glucose limitations, anchorage-independent growth, and solid tumor formation in vivo. In KrasG12D-driven non-small cell lung cancer, the failure of AMPK activation by virtue of LKB1 mutation sensitizes cancer cells for phenformin-induced metabolic stress, further supporting that AMPK adapts cancer cells for metabolic stress [203]. Alternatively, a synthetic lethal screening has revealed that AMPK activation by AMPK-related kinase 5 (ARK5) is essential for Myc-driven cancer progression [204]. Consistent with this finding, AMPK has been shown to promote survival of Myc-positive melanoma cells with N-Ras mutation by restraining oxidative stress [205]. In addition, AMPK sustains the activation of oncogenic protein kinase B (AKT) signaling upon stress or epidermal growth factor receptor (EGFR) engagement in breast cancers [206]. Besides these direct effects on cancer cells, AMPK may promote cancer progression by altering the cancer microenvironment. AMPK signaling has been shown to intrinsically promote the immunoregulatory activity of myeloid-derived suppressor cells (MDSC), which dysfunctions T cells in cancer tissue [207]. All these findings indicate that AMPK can significantly contribute to the disease progression of variable cancers via distinct manners.
Unlike LKB1, which is frequently mutated or deleted in cancer genomes [208,209,210], AMPK has nearly no mutations, and on the contrary, is upregulated in some types of cancers such as glioblastoma [211], suggesting that it may play a paradoxical role in carcinogenesis. Dependent on origins of cancers, driver mutations, developmental stages, and external conditions, AMPK may dampen or promote the disease progression of cancers, and uncovering underlying mechanisms would propel cancer therapy development by targeting this signaling pathway.
The crosstalk between MAPK and AMPK signalings
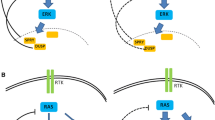
As described above, the MAPK signaling controls cellular proliferation, differentiation, and survival, whereas the AMPK signaling regulates cellular metabolism. However, many studies have indicated that these two distinct signalings have profound and complicated interplays in both physiological and pathological processes. In quiescent cells, the AMPK signaling maintains energy homeostasis by switching on catabolic pathways that generate ATP, while switching off anabolic pathways that are required for cell growth [142, 146, 176,177,178,179,180,181,182,183, 204, 212,213,214,215]. Upon mitogen stimulation, the MAPK signaling is turned on and drives cellular proliferation/differentiation, which needs cells shifting their metabolic program from catabolic to anabolic for biomass synthesis [216, 217]. To achieve this, the MAPK signaling activates transcription factors such as Myc and Hypoxia inducible factor 1 subunit alpha (HIF-1α), which control the expression of glycolytic enzymes and promote aerobic glycolysis [218,219,220,221]. Furthermore, the MAPK signaling directly regulates AMPK signaling and thus constrains the AMPK signaling-driven oxidative phosphorylation of biomaterials [167, 222]. These interplays frequently occur with marginal coordination when cells response to different stimuli such as oncogenesis and cell stress. Recent studies have revealed that the MAPK signaling regulates AMPK signaling on different layers under distinct circumstances. Firstly, ERK and ribosomal protein S6 kinase A (RSK), two downstream kinases of MAPK signaling, have been shown to phosphorylate and inhibit the upstream activator of AMPK, LKB1, and thereby block the activation of AMPK by LKB1 in BRAF(V600E)-driven melanoma [223] (Fig. 3a). Secondly, ERK likely phosphorylates the α subunit of AMPK directly on negative regulatory sites Ser485/491 and impairs its catalytic activity, which is essential for C-C motif chemokine receptor 7 (CCR7)-dependent survival of mature dendritic cells [224]. Thirdly, KSR, one of the key components of MAPK module, has been shown to interact with all AMPK subunits and regulate the AMPK-dependent energy expenditure [225, 226] (Fig. 3b). In addition, the MAPK signaling controls the subcellular localization of AMPK and thus alters its function under cell stress [227]. All these findings suggest that AMPK could function as a downstream effector of MAPK signaling.

AMPK signaling is inhibited by hyperactive Ras/RAF/MEK/ERK (MAPK) signaling in cancers. a In BRAF(V600E)-harboring cancers, hyperactive ERKs and downstream RSKs phosphorylate LKB1 on Ser325 and Ser428 sites, which inactivates LKB1 and thus blocks the activation of AMPK by LKB1. b In Ras-mutated cancers, the activity of AMPK is partially inhibited likely by hyperactive MAPK signaling, though the underlying molecular mechanism remains ambiguous. However, this moderate AMPK activity is indispensable for disease progression in Ras-mutated cancers
The interplays between MAPK and AMPK signalings are binary, and the AMPK signaling can regulate MAPK signaling reversely. Conclusive evidence shows that AMPK can directly phosphorylate the RAF/KSR family kinases, the pivotal components of MAPK module, and alter their activities under variable conditions. It is well established that the hetero-/homo-dimerization of RAF/KSR family kinases plays a determinant role in the activation of MAPK signaling, which requires the association of 14-3-3, a dimeric scaffold protein with their carboxyl-terminus [228, 229]. Mechanistic studies have revealed that a 14-3-3 dimer associates with the C-terminus of two individual RAF/KSR molecules and facilitates their dimerization and subsequent activation [25, 230, 231] (Fig. 4a). Since RAF/KSR family kinases have the other conserved 14-3-3 binding site at the N-terminus, however, if a 14-3-3 dimer binds to the N- and C-terminus of a single RAF/KSR intramolecularly, it will stabilize RAF/KSR in an autoinhibitory conformation and thus prevent the dimerization-driven activation of kinases [38, 77, 232] (Fig. 4b). AMPK has been shown to phosphorylate the C-terminal 14-3-3 binding site of RAF/KSR family kinases and promote the intra- or inter-molecular 14-3-3 associations with these kinases respectively [233, 234] (Fig. 4). Among RAF/KSR family kinases, CRAF is the first member that has been shown being phosphorylated by AMPK on its C-terminal 14-3-3 binding site [234]. AMPKi by pharmaceutical inhibitors abolishes the dimer-dependent paradoxical activation of MAPK signaling driven by the RAF inhibitors in Ras-mutated cancers, suggesting that AMPK-mediated phosphorylation of the C-terminal 14-3-3 binding site on CRAF promotes the intermolecular association of 14-3-3 dimers with CRAF homo- or hetero-dimers [231] (Fig. 4a). This molecular mechanism may also be responsible for the hyperactive MAPK signaling induced by metabolic stress in Ras-mutated melanoma. Upon metabolic perturbations, AMPK is activated in this type of melanoma cells and promotes KSR/CRAF heterodimerization likely through altering 14-3-3 binding manners, which leads to a highly activated MAPK signaling [230]. Besides CRAF and KSR, the association of BRAF with 14-3-3 is also regulated by AMPK-mediated phosphorylation. In BRAF(V600E)-harboring melanoma, metabolic stress-activated AMPK phosphorylates the C-terminal 14-3-3 binding site of BRAF and promotes the intramolecular association of a single BRAF molecule with a 14-3-3 dimer [233], which breaks the BRAF/KSR heterodimer and thus inhibits MAPK signaling [230], although whether active AMPK phosphorylates the N-terminal 14-3-3 binding site of BRAF under this condition needs further investigation (Fig. 4b). Consistent with these findings, AMPK activators have been shown to inhibit the proliferation of BRAF(V600E)-harboring melanoma and enhance the therapeutic efficacy of BRAF inhibitors on this type of melanoma [235, 236]. Over all, the distinct regulations of RAF/KSR family kinases by AMPK lead to completely different outputs of MAPK signaling, which determine cell fates under variable conditions.

AMPK regulates differentially hyperactive Ras/RAF/MEK/ERK (MAPK) signaling in Ras- versus BRAF(V600E)-mutated cancers. a In Ras-mutated cancers, the C-terminal 14-3-3 binding site of CRAF is phosphorylated by AMPK, which facilitates CRAF dimerization through improving the association of CRAF dimer with 14-3-3 dimer and thus elevates the activity of CRAF, particularly upon RAF inhibitor treatment or metabolic stress. Under these conditions, CRAF forms homodimers with itself or heterodimers with KSR or BRAF. b In BRAF(V600E)-harboring cancers, AMPK phosphorylates the C-terminal 14-3-3 binding site of BRAF(V600E), which prevents BRAF(V600E) dimerization with KSR through enhancing the association of a single BRAF(V600E) molecule with 14-3-3 dimer and thus blocks the activity of BRAF(V600E) upon metabolic stress
The interplays between MAPK and AMPK signalings also alter cellular autophagy, particularly that of cancer cells. Cancer cells with Ras/RAF mutations have much higher activity of autophagy [237,238,239,240], which significantly contributes to disease progression [238, 240,241,242,243,244,245,246], although how autophagy is upregulated in these cancer cells remains unknown. Elevated autophagy in Ras/RAF-mutated cancer cells preserves mitochondrial and glycolytic functions by recycling dysfunctional mitochondria [247, 248]. Disruption of autophagy by depleting Atg7 or Atg5 induces cellular senescence and reduces cancer burden in these diseases [238, 240,241,242,243,244,245,246]. The critical role of autophagy in K-ras-driven cancers is further confirmed by a synthetic lethal screening for factors that support K-ras addiction, which identified Atg7 and RAF kinases as a minimal oncoeffector combination that best discriminates K-ras cancer cells from normal cells [249]. It is well known that AMPK is a prominent regulator of autophagy in spite of its key role as an energy sensor, which drives cellular autophagy machinery via the LKB1/AMPK/ULK1 axis [250,251,252]. Since LKB1 is inhibited by hyperactive MAPK signaling, this signal axis should not be responsible for elevated activity of autophagy in Ras/RAF-driven cancers. However, it provides cancer cells a protective strategy for adapting themselves to MAPK inhibition [248, 253, 254]. Indeed, MAPK inhibition (MAPKi) by RAF/MEK/ERK inhibitors in Ras/RAF-mutated cancer cells further elevates autophagic flux through AMPK, which restores cellular metabolic hemostasis and leads to tolerance towards MAPKi.
Combinatorial targeting of MAPK and AMPK signalings to treat Ras/RAF-mutated cancers
Hyperactive MAPK signaling is responsible for a large portion of cancers, and genetic alterations that aberrantly activate this pathway mainly occur on receptor tyrosine kinases (RTKs), Ras small GTPases, and BRAF [63]. In current cancer therapies, hyperactive RTKs can be effectively targeted by tyrosine kinase inhibitors (TKIs) or neutralizing antibodies [255,256,257,258,259,260,261,262,263], while there are no drugs that are able to specifically target most Ras mutants [264]. To treat Ras/BRAF-mutated cancers, RAF/MEK inhibitors such as vemurafenib, dabrafenib, encorafenib, trametinib, cobimetinib, and binimetinib have been developed and applied to disease management [99,100,101, 265]. These inhibitors have exhibited a promising efficacy towards most BRAF-mutated cancers [101, 108,109,110,111,112,113,114,115,116] (Fig. 5). In contrast, Ras-mutated cancers are intrinsically resistant to these drugs, which do not inhibit but paradoxically activate the MAPK signaling through promoting RAF family kinases’ dimerization [266]. Furthermore, even BRAF-mutated cancers develop adaptive resistance to these drugs after 6~10 months treatment by either activating Ras or alternatively splicing BRAF mutant [266]. Therefore, for most cases, once cancer cells possess high Ras activity, these drugs lose their efficacy as a monotherapy. To improve the efficacy of MAPKi against Ras/RAF-mutated cancers, emerging evidence indicates that disruption of MAPK signaling complex, particularly dimerization of RAF family kinases, and/or synergistic targeting of synthetic lethality of MAPK signaling should be two feasible strategies [267,268,269,270,271,272], both of which are involved in AMPK signaling.

Combinatorial targeting of hyperactive Ras/RAF/MEK/ERK (MAPK) signaling and AMPK-mediated autophagy to treat Ras/RAF-mutated cancers. Blocking hyperactive Ras/RAF/MEK/ERK (MAPK) signaling by MAPK inhibitors in Ras/RAF-mutated cancer cells elevates autophagic flux through relieving LKB1/AMPK/ULK1 axis and inhibiting glycolysis and mitochondrial functions, which leads to drug tolerance and/or acquired resistance. Combinatorial inhibition of both hyperactive MAPK signaling and autophagy remarkably improves therapeutic efficacy of drugs against these cancers
It has been shown that the components of MAPK signaling form a super complex in cancer cells with active Ras [273, 274], which leads to resistance towards MAPKi. Assembly of this complex involves in RAF/RAF (or KSR), RAF (or KSR)/MEK, MEK/MEK, as well as RAF (or KSR)/14-3-3 interactions, and disruption of these interactions contributes to an effective inhibition of MAPK signaling. As discussed above, AMPK directly regulates RAF (or KSR)/14-3-3 interaction by phosphorylating the 14-3-3 binding sites on RAF (or KSR) and thus facilitates or impairs RAF/RAF (or KSR) dimerization. In Ras-mutated cancer cells or RAFi-resistant cancer cells with active Ras, CRAF is the key isoform of RAF family kinases responsible for disease progression and drug resistance [27, 32, 275,276,277,278], whose phosphorylation on the C-terminal 14-3-3 binding site by AMPK plays a determinant role in the paradoxical effect of RAF inhibitors, and AMPKi sensitizes these cancer cells to RAF inhibitors both in vitro and in vivo [231] (Wang & Hu, unpublished data) (Fig. 4b). Although the components of MAPK signaling do not assemble a super complex in BRAF-mutated cancer cells, constitutively active BRAF mutant still functions as homo- or hetero-dimers (BRAF/BRAF or BRAF/KSR) that can be disrupted by AMPK-driven phosphorylation of both N- and C-terminal 14-3-3 binding sites [230, 231, 233]. In this type of cancers, AMPK activators have been shown to significantly enhance the therapeutic efficacy of RAF inhibitors [235, 236]. Taken together, altering RAF/KSR dimerization by using either AMPK inhibitors or activators may remarkably improve the targeted therapies of Ras/RAF-mutated cancers with RAF inhibitors.
Since most oncogenic Ras mutants are undruggable, efforts for developing effective approaches against Ras-mutated cancers have been switched to identify and target synthetic lethal vulnerabilities of Ras mutants over decades, which led to the discovery of some putative factors essential for in vitro growth of Ras-mutated cancer cells [267, 279]. Unfortunately, most factors except those regulating cellular autophagy exhibit little-to-no therapeutic values for treating Ras-mutated cancers in vivo so far. As we know, Ras/RAF-mutated cancer cells have a high basal activity of autophagy though hyperactive MAPK signaling inhibits the LKB1-AMPK-ULK1 signaling axis, which is critical for maintaining cellular metabolic homeostasis. MAPKi relives the LKB1-AMPK-ULK1 axis and thus further elevates autophagic flux in Ras/RAF-mutated cancer cells, which adapts Ras-mutated cancer cells to MAPKi [25, 254], or promotes drug tolerance and subsequent resistance of RAF-mutated cancer cells [253] (Fig. 5). Pharmaceutical blocking of AMPK by compound C has been shown to remarkably reduce the RAFi-resistant clones arising from BRAF-mutated melanoma [231]. Furthermore, combinations of autophagy inhibitors with RAF/MEK/ERK inhibitors (chloroquine plus vemurafenib, hydroxychloroquine plus trametinib, or chloroquine plus SCH772984) can effectively block the growth of K-ras-mutated pancreatic ductal adenocarcinoma, N-ras-mutated melamona, as well as BRAF-mutated colorectal cancer and melanoma in vivo [248, 253, 254, 280] (Fig. 5). However, it has to be noted that although both AMPK inhibitors and activators may synergistically enhance the therapeutic efficacy of RAF inhibitors against BRAF-mutated cancers, molecular mechanisms underlying these phenomena are completely different.
Combinatorial inhibition of both MAPK and AMPK signalings has shown promising potentials for treating Ras/RAF-mutated cancers. To target MAPK signaling, the first-generation RAF/MEK/ERK inhibitors have been developed and applied to clinic treatment, and the second-generation drugs that can inhibit RAF mutants with elevated dimer affinity or have much less paradoxical effect have also undergone clinical trials [102, 121, 281, 282]. In contrast, the drug development of AMPK-specific activators and inhibitors lags far behind the needs for treating cancers, though some AMPK activators such as O304 are undergoing clinical trials for other diseases [148]. Currently, only two non-specific AMPK activators, phenformin and metformin, that have been approved for treating type II diabetes are undergoing clinic evaluations as combinatorial agents for treating BRAF-mutated melanoma together with vemurafenib or dabrafenib/trametinib. As to AMPK-specific inhibitors, only compound C has been tested in animal models at present [283]. Although a combination of compound C with RAF inhibitor, vemurafenib can effectively inhibit the growth of Ras-mutated cancer cells in vitro [231], and its therapeutic efficacy/benefit needs further investigations by using preclinical animal models and through clinical trials. Overall, these unmet needs for AMPK-specific activators and inhibitors in targeted cancer therapies appeal to accelerate their pharmaceutical development.
Conclusions and perspectives
Recent studies have provided compelling evidence that interplays between MAPK and AMPK signalings play a critical role in cell physiology and have important implications in disease treatment, particularly for cancer. Combinatory targeting both MAPK and AMPK signalings represents for a promising therapeutic intervention. However, although the framework by which these two signalings interact with each other has been illustrated, the precise molecular basis and their impacts on cancer therapies remain largely unresolved. For instance, how the AMPK signaling differentially regulates the dimerization of different RAF isoforms (BRAF versus CRAF) and thus distinctly alters the outputs of MAPK signaling in Ras- versus RAF-mutated cancers is unclear. Besides elevating autophagic flux, does the AMPK signaling plays other roles in the MAPKi-resistance of Ras/RAF-mutated cancers? Addressing these questions would deepen our understanding of MAPK/AMPK interplays and help us develop better combinatorial therapies for cancers and other diseases. In addition, developing AMPK-specific activators/inhibitors would be an attractive research topic for both academy and pharmaceutical industry in the next years given their absence and unmet needs in clinic treatment.
Availability of data and materials
Not applicable.
Abbreviations
- MAPK:
-
Mitogen-activated protein kinase
- AMPK:
-
AMP-activated protein kinase
- Ras:
-
Ras proto-oncogene
- GTPase; RAF:
-
Raf proto-oncogene, serine/threonine kinase
- MEK:
-
Mitogen-activated protein kinase kinase
- ERK:
-
Mitogen-activated protein kinase
- KSR:
-
Kinase suppressor of Ras
- CR:
-
Conserved region
- RBD:
-
RAS-binding domain
- NTA:
-
N-terminal acidic motif
- PI3K:
-
Phosphoinositide 3-kinase
- AKT:
-
Protein kinase B
- mTORC:
-
Mammalian target of rapamycin complex
- RTKs:
-
Receptor tyrosine kinases
- GEFs:
-
GTP/GDP exchange factors
- GAPs:
-
GTPase activating proteins
- SHP2:
-
Src homology region 2 domain-containing phosphatase-2
- ACC:
-
Acetyl-CoA carboxylase
- FA:
-
Fatty acid
- LKB1:
-
Liver kinase B1
- CAMKK2:
-
Calcium/calmodulin-dependent protein kinase kinase 2
- FBP:
-
Fructose 1,6-bisphosphate
- HMGR:
-
HMG-CoA reductase
- AKAP1:
-
A-kinase anchoring protein 1
- PKA:
-
Protein kinase A
- DRP1:
-
Dynamin-related protein 1
- PGC1α:
-
Proliferator-activated receptor gamma coactivator 1-alpha
- MFF:
-
Mitochondrial fission factor
- ULK1:
-
Unc-51-like autophagy activating kinase 1
- ATG:
-
Autophagy-related gene
- GLUT:
-
Solute carrier family 2 member
- PFK2:
-
6-Phosphofructo-2-kinase
- S6K:
-
Ribosomal protein S6 kinase B1
- 4E-BP1:
-
Eukaryotic translation initiation factor 4E binding protein 1
- eEF2:
-
Eukaryotic translation elongation factor 2
- EMT:
-
Epithelial-to-mesenchymal transition
- AMOTL1:
-
Angiomotin like 1
- YAP:
-
Yes1 associated transcriptional regulator
- TSC2:
-
TSC complex subunit 2
- Raptor:
-
Regulatory associated protein of MTOR complex 1
- MAGEA:
-
MAGE family member A
- TRIM28:
-
Tripartite motif containing 28
- EZH2:
-
Enhancer of zeste 2 polycomb repressive complex 2 subunit
- PRC2:
-
Polycomb repressive complex 2
- TET2:
-
Tet methylcytosine dioxygenase 2
- T-ALL:
-
T cell acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- LICs:
-
Leukemia-initiating cells
- ARK5:
-
AMPK-related kinase 5
- EGFR:
-
Epidermal growth factor receptor
- MDSC:
-
Myeloid-derived suppressor cells
- HIF-1α:
-
Hypoxia inducible factor 1 subunit alpha
- RSK:
-
Ribosomal protein S6 kinase A
- CCR7:
-
C-C motif chemokine receptor 7
References
Rapp UR, Todaro C. Generation of new mouse sarcoma viruses in cell culture. Science. 1978;201(4358):821–4.
Jansen HW, Lurz R, Bister K, Bonner TI, Mark GE, Rapp UR. Homologous cell-derived oncogenes in avian carcinoma virus MH2 and murine sarcoma virus 3611. Nature. 1984;307(5948):281–4.
Jansen HW, Ruckert B, Lurz R, Bister K. Two unrelated cell-derived sequences in the genome of avian leukemia and carcinoma inducing retrovirus MH2. EMBO J. 1983;2(11):1969–75.
Bonner T, O'Brien SJ, Nash WG, Rapp UR, Morton CC, Leder P. The human homologs of the raf (mil) oncogene are located on human chromosomes 3 and 4. Science. 1984;223(4631):71–4.
Kozak C, Gunnell MA, Rapp UR. A new oncogene, c-raf, is located on mouse chromosome 6. J Virol. 1984;49(1):297–9.
Bonner TI, Kerby SB, Sutrave P, Gunnell MA, Mark G, Rapp UR. Structure and biological activity of human homologs of the raf/mil oncogene. Mol Cell Biol. 1985;5(6):1400–7.
Ahn NG, Weiel JE, Chan CP, Krebs EG. Identification of multiple epidermal growth factor-stimulated protein serine/threonine kinases from Swiss 3T3 cells. J Biol Chem. 1990;265(20):11487–94.
Ray LB, Sturgill TW. Characterization of insulin-stimulated microtubule-associated protein kinase. Rapid isolation and stabilization of a novel serine/threonine kinase from 3T3-L1 cells. J Biol Chem. 1988;263(25):12721–7.
Rossomando AJ, Payne DM, Weber MJ, Sturgill TW. Evidence that pp42, a major tyrosine kinase target protein, is a mitogen-activated serine/threonine protein kinase. Proc Natl Acad Sci U S A. 1989;86(18):6940–3.
Boulton TG, Nye SH, Robbins DJ, Ip NY, Radzlejewska E, Morgenbesser SD, et al. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65(4):663–75.
Crews CM, Erikson RL. Purification of a murine protein-tyrosine/threonine kinase that phosphorylates and activates the Erk-1 gene product: relationship to the fission yeast byr1 gene product. Proc Natl Acad Sci U S A. 1992;89(17):8205–9.
Rapp UR, Goldsborough MD, Mark GE, Bonner TI, Groffen J, Reynolds FH Jr, et al. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc Natl Acad Sci U S A. 1983;80(14):4218–22.
Moelling K, Heimann B, Beimling P, Rapp UR, Sander T. Serine- and threonine-specific protein kinase activities of purified gag-mil and gag-raf proteins. Nature. 1984;312(5994):558–61.
Cox AD, Der CJ. Ras history: the saga continues. Small GTPases. 2010;1(1):2–27.
Ehrenreiter K, Piazzolla D, Velamoor V, Sobczak I, Small JV, Takeda J, et al. Raf-1 regulates Rho signaling and cell migration. J Cell Biol. 2005;168(6):955–64.
Rubiolo C, Piazzolla D, Meissl K, Beug H, Huber JC, Kolbus A, et al. A balance between Raf-1 and Fas expression sets the pace of erythroid differentiation. Blood. 2006;108(1):152–9.
Kolbus A, Pilat S, Husak Z, Deiner EM, Stengl G, Beug H, et al. Raf-1 antagonizes erythroid differentiation by restraining caspase activation. J Exp Med. 2002;196(10):1347–53.
Kolch W, Heidecker G, Lloyd P, Rapp UR. Raf-1 protein kinase is required for growth of induced NIH/3T3 cells. Nature. 1991;349(6308):426–8.
Daum G, Eisenmann-Tappe I, Fries HW, Troppmair J, Rapp UR. The ins and outs of Raf kinases. Trends Biochem Sci. 1994;19(11):474–80.
Fischer A, Hekman M, Kuhlmann J, Rubio I, Wiese S, Rapp UR. B- and C-RAF display essential differences in their binding to Ras: the isotype-specific N terminus of B-RAF facilitates Ras binding. J Biol Chem. 2007;282(36):26503–16.
Diaz B, Barnard D, Filson A, MacDonald S, King A, Marshall M. Phosphorylation of Raf-1 serine 338-serine 339 is an essential regulatory event for Ras-dependent activation and biological signaling. Mol Cell Biol. 1997;17(8):4509–16.
Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999;18(8):2137–48.
Kohler M, Brummer T. B-Raf activation loop phosphorylation revisited. Cell Cycle. 2016;15(9):1171–3.
Dhillon AS, Yip YY, Grindlay GJ, Pakay JL, Dangers M, Hillmann M, et al. The C-terminus of Raf-1 acts as a 14-3-3-dependent activation switch. Cell Signal. 2009;21(11):1645–51.
Kondo Y, Ognjenovic J, Banerjee S, Karandur D, Merk A, Kulhanek K, et al. Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science. 2019;366(6461):109–15.
Hobbs GA, Der CJ. RAS mutations are not created equal. Cancer Discov. 2019;9(6):696–8.
Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJS, et al. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154(5):1036–46.
Yuan J, Ng WH, Lam PYP, Wang Y, Xia H, Yap J, et al. The dimer-dependent catalytic activity of RAF family kinases is revealed through characterizing their oncogenic mutants. Oncogene. 2018;37(43):5719–34.
Huleihel M, Goldsborough M, Cleveland J, Gunnell M, Bonner T, Rapp UR. Characterization of murine A-raf, a new oncogene related to the v-raf oncogene. Mol Cell Biol. 1986;6(7):2655–62.
Beck TW, Huleihel M, Gunnell M, Bonner TI, Rapp UR. The complete coding sequence of the human A-raf-1 oncogene and transforming activity of a human A-raf carrying retrovirus. Nucleic Acids Res. 1987;15(2):595–609.
Therrien M, Chang HC, Solomon NM, Karim FD, Wassarman DA, Rubin GM. KSR, a novel protein kinase required for RAS signal transduction. Cell. 1995;83(6):879–88.
Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009;461(7263):542–5.
Hu J, Yu H, Kornev AP, Zhao J, Filbert EL, Taylor SS, et al. Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc Natl Acad Sci U S A. 2011;108(15):6067–72.
Hu J, Ahuja LG, Meharena HS, Kannan N, Kornev AP, Taylor SS, et al. Kinase regulation by hydrophobic spine assembly in cancer. Mol Cell Biol. 2015;35(1):264–76.
Shaw AS, Kornev AP, Hu J, Ahuja LG, Taylor SS. Kinases and pseudokinases: lessons from RAF. Mol Cell Biol. 2014;34(9):1538–46.
Taylor SS, Shaw A, Hu J, Meharena HS, Kornev A. Pseudokinases from a structural perspective. Biochem Soc Trans. 2013;41(4):981–6.
Yap J, Yuan J, Tee ZH, Huang X, Ng WH, Hu J. Characterize disease-related mutants of RAF family kinases by using a set of practical and feasible methods. J Vis Exp. 2019;(149). https://doi.org/10.3791/59795.
Park E, Rawson S, Li K, Kim BW, Ficarro SB, Pino GG, et al. Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature. 2019;575(7783):545–50.
Varga A, Ehrenreiter K, Aschenbrenner B, Kocieniewski P, Kochanczyk M, Lipniacki T, et al. RAF1/BRAF dimerization integrates the signal from RAS to ERK and ROKalpha. Sci Signal. 2017;10(469):eaai8482.
Weber CK, Slupsky JR, Kalmes HA, Rapp UR. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001;61(9):3595–8.
Rushworth LK, Hindley AD, O'Neill E, Kolch W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol. 2006;26(6):2262–72.
Baljuls A, Mahr R, Schwarzenau I, Muller T, Polzien L, Hekman M, et al. Single substitution within the RKTR motif impairs kinase activity but promotes dimerization of RAF kinase. J Biol Chem. 2011;286(18):16491–503.
Kyriakis JM, App H, Zhang XF, Banerjee P, Brautigan DL, Rapp UR, et al. Raf-1 activates MAP kinase-kinase. Nature. 1992;358(6385):417–21.
Alessi DR, Saito Y, Campbell DG, Cohen P, Sithanandam G, Rapp U, et al. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994;13(7):1610–9.
Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11(12):1192–7.
Yuan J, Ng WH, Tian Z, Yap J, Baccarini M, Chen Z, et al. Activating mutations in MEK1 enhance homodimerization and promote tumorigenesis. Sci Signal. 2018;11(554):eaar6795.
Wang HG, Rapp UR, Reed JC. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87(4):629–38.
Panka DJ, Atkins MB, Mier JW. Targeting the mitogen-activated protein kinase pathway in the treatment of malignant melanoma. Clin Cancer Res. 2006;12(7 Pt 2):2371s–5s.
Baumann B, Weber CK, Troppmair J, Whiteside S, Israel A, Rapp UR, et al. Raf induces NF-kappaB by membrane shuttle kinase MEKK1, a signaling pathway critical for transformation. Proc Natl Acad Sci U S A. 2000;97(9):4615–20.
Troppmair J, Hartkamp J, Rapp UR. Activation of NF-kappa B by oncogenic Raf in HEK 293 cells occurs through autocrine recruitment of the stress kinase cascade. Oncogene. 1998;17(6):685–90.
Norris JL, Baldwin AS Jr. Oncogenic Ras enhances NF-kappaB transcriptional activity through Raf-dependent and Raf-independent mitogen-activated protein kinase signaling pathways. J Biol Chem. 1999;274(20):13841–6.
Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22.
Wojnowski L, Stancato LF, Larner AC, Rapp UR, Zimmer A. Overlapping and specific functions of Braf and Craf-1 proto-oncogenes during mouse embryogenesis. Mech Dev. 2000;91(1-2):97–104.
Zhang Z, Kobayashi S, Borczuk AC, Leidner RS, Laframboise T, Levine AD, et al. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis. 2010;31(4):577–86.
Ekerot M, Stavridis MP, Delavaine L, Mitchell MP, Staples C, Owens DM, et al. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem J. 2008;412(2):287–98.
Brummer T, Naegele H, Reth M, Misawa Y. Identification of novel ERK-mediated feedback phosphorylation sites at the C-terminus of B-Raf. Oncogene. 2003;22(55):8823–34.
Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17(2):215–24.
Ritt DA, Monson DM, Specht SI, Morrison DK. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol. 2010;30(3):806–19.
Valis K, Novak P. Targeting ERK-Hippo interplay in cancer therapy. Int J Mol Sci. 2020;21(9):3236.
Ohm AM, Affandi T, Reyland ME. EGF receptor and PKCdelta kinase activate DNA damage-induced pro-survival and pro-apoptotic signaling via biphasic activation of ERK and MSK1 kinases. J Biol Chem. 2019;294(12):4488–97.
Unni AM, Harbourne B, Oh MH, Wild S, Ferrarone JR, Lockwood WW, et al. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. Elife. 2018;7:e33718.
Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death—apoptosis, autophagy and senescence. FEBS J. 2010;277(1):2–21.
Degirmenci U, Wang M, Hu J. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells. 2020;9(1):198.
Jun S, Lee S, Kim HC, Ng C, Schneider AM, Ji H, et al. PAF-mediated MAPK signaling hyperactivation via LAMTOR3 induces pancreatic tumorigenesis. Cell Rep. 2013;5(2):314–22.
Drosten M, Sum EY, Lechuga CG, Simon-Carrasco L, Jacob HK, Garcia-Medina R, et al. Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway. Proc Natl Acad Sci U S A. 2014;111(42):15155–60.
Xu S, Khoo S, Dang A, Witt S, Do V, Zhen E, et al. Differential regulation of mitogen-activated protein/ERK kinase (MEK)1 and MEK2 and activation by a Ras-independent mechanism. Mol Endocrinol. 1997;11(11):1618–25.
Maik-Rachline G, Seger R. The ERK cascade inhibitors: towards overcoming resistance. Drug Resist Updat. 2016;25:1–12.
Aoki Y, Niihori T, Inoue S, Matsubara Y. Recent advances in RASopathies. J Hum Genet. 2016;61(1):33–9.
Quilliam LA, Castro AF, Rogers-Graham KS, Martin CB, Der CJ, Bi C. M-Ras/R-Ras3, a transforming ras protein regulated by Sos1, GRF1, and p120 Ras GTPase-activating protein, interacts with the putative Ras effector AF6. J Biol Chem. 1999;274(34):23850–7.
Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19(9):495–509.
Martinez P, Mak RH, Oxnard GR. Targeted therapy as an alternative to whole-brain radiotherapy in EGFR-mutant or ALK-positive non-small-cell lung cancer with brain metastases. JAMA Oncol. 2017;3(9):1274–5.
Loibl S, Gianni L. HER2-positive breast cancer. Lancet. 2017;389(10087):2415–29.
Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019;16(2):105–22.
Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129(7):1287–92.
Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10):2457–67.
Tessoulin B, Moreau-Aubry A, Descamps G, Gomez-Bougie P, Maiga S, Gaignard A, et al. Whole-exon sequencing of human myeloma cell lines shows mutations related to myeloma patients at relapse with major hits in the DNA regulation and repair pathways. J Hematol Oncol. 2018;11(1):137.
Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16(5):281–98.
Tiacci E, Pettirossi V, Schiavoni G, Falini B. Genomics of Hairy Cell Leukemia. J Clin Oncol. 2017;35(9):1002–10.
Punt CJ, Koopman M, Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol. 2017;14(4):235–46.
Bible KC, Ryder M. Evolving molecularly targeted therapies for advanced-stage thyroid cancers. Nat Rev Clin Oncol. 2016;13(7):403–16.
Schadendorf D, Fisher DE, Garbe C, Gershenwald JE, Grob JJ, Halpern A, et al. Melanoma. Nat Rev Dis Primers. 2015;1:15003.
Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106(48):20411–6.
Gao Y, Maria A, Na N, da Cruz Paula A, Gorelick AN, Hechtman JF, et al. V211D mutation in MEK1 causes resistance to MEK inhibitors in colon cancer. Cancer Discov. 2019;9(9):1182–91.
Kakadia S, Yarlagadda N, Awad R, Kundranda M, Niu J, Naraev B, et al. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther. 2018;11:7095–107.
Goetz EM, Ghandi M, Treacy DJ, Wagle N, Garraway LA. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014;74(23):7079–89.
Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13(11):828–51.
Westover KD, Janne PA, Gray NS. Progress on covalent inhibition of KRAS(G12C). Cancer Discov. 2016;6(3):233–4.
Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351(6273):604–8.
Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant cancers with a covalent G12C-specific inhibitor. Cell. 2018;172(3):578–89 e17.
Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10(1):54–71.
Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–23.
Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol. 2018;20(9):1064–73.
Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRAS(G12C) inhibition. Clin Cancer Res. 2020;26(7):1633–43.
Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM. Targeted degradation of oncogenic KRASG12C by VHL-recruiting PROTACs. ACS Central Sci. 2020. https://doi.org/10.1021/acscentsci.0c00411.
Zeng M, Xiong Y, Safaee N, Nowak RP, Donovan KA, Yuan CJ, et al. Exploring targeted degradation strategy for oncogenic KRAS(G12C). Cell Chem Biol. 2020;27(1):19–31 e6.
Stalnecker CA, Der CJ. RAS, wanted dead or alive: advances in targeting RAS mutant cancers. Sci Signal. 2020;13(624):eaay6013.
Athuluri-Divakar SK, Vasquez-Del Carpio R, Dutta K, Baker SJ, Cosenza SC, Basu I, et al. A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signaling. Cell. 2016;165(3):643–55.
Hymowitz SG, Malek S. Targeting the MAPK pathway in RAS mutant cancers. Cold Spring Harb Perspect Med. 2018;8(11):a031492.
Flaherty K, Puzanov I, Sosman J, Kim K, Ribas A, McArthur G, et al. Phase I study of PLX4032: proof of concept for V600E BRAF mutation as a therapeutic target in human cancer. J Clin Oncol. 2009;27(15_suppl):9000.
Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105(8):3041–6.
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65.
Yao Z, Gao Y, Su W, Yaeger R, Tao J, Na N, et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat Med. 2019;25(2):284–91.
Herrero A, Pinto A, Colon-Bolea P, Casar B, Jones M, Agudo-Ibanez L, et al. Small molecule inhibition of ERK dimerization prevents tumorigenesis by RAS-ERK pathway oncogenes. Cancer Cell. 2015;28(2):170–82.
Gunderwala AY, Nimbvikar AA, Cope NJ, Li Z, Wang Z. Development of allosteric BRAF peptide inhibitors targeting the dimer interface of BRAF. ACS Chem Biol. 2019;14(7):1471–80.
Fremin C, Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol. 2010;3:8.
Huang T, Karsy M, Zhuge J, Zhong M, Liu D. B-Raf and the inhibitors: from bench to bedside. J Hematol Oncol. 2013;6:30.
Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D. MEK and the inhibitors: from bench to bedside. J Hematol Oncol. 2013;6:27.
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16.
Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–14.
Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76.
Long GV, Weber JS, Infante JR, Kim KB, Daud A, Gonzalez R, et al. Overall survival and durable responses in patients with BRAF V600-mutant metastatic melanoma receiving dabrafenib combined with trametinib. J Clin Oncol. 2016;34(8):871–8.
Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–9.
Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland A, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–16.
Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol. 2018;36(1):7–13.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703.
Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877–88.
Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–7.
Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–9.
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–30.
Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526(7574):583–6.
Yuan X, Tang Z, Du R, Yao Z, Cheung SH, Zhang X, et al. RAF dimer inhibition enhances the antitumor activity of MEK inhibitors in K-RAS mutant tumors. Mol Oncol. 2020;14(8):1833–49.
Desai J, Gan H, Barrow C, Jameson MB, Solomon B, Atkinson V, et al. Abstract CT002: a phase IB study of RAF dimer inhibitor BGB-283 in patients with B-RAF or K-RAS/N-RAS mutated solid tumors. Cancer Res. 2017;77(13 Supplement):CT002–CT.
Sun Y, Alberta JA, Pilarz C, Calligaris D, Chadwick EJ, Ramkissoon SH, et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro Oncol. 2017;19(6):774–85.
Tang Z, Yuan X, Du R, Cheung SH, Zhang G, Wei J, et al. BGB-283, a novel RAF kinase and EGFR inhibitor, displays potent antitumor activity in BRAF-mutated colorectal cancers. Mol Cancer Ther. 2015;14(10):2187–97.
Saturno G, Lopes F, Girotti MR, Niculescu-Duvaz I, Niculescu-Duvaz D, Zambon A, et al. Abstract LB-212: therapeutic efficacy of the paradox-breaking panRAF and SRC drug CCT3833/BAL3833 in KRAS-driven cancer models. Cancer Res. 2016;76(14 Supplement):LB-212–LB.
Martinez-Garcia M, Banerji U, Albanell J, Bahleda R, Dolly S, Kraeber-Bodere F, et al. First-in-human, phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of RO5126766, a first-in-class dual MEK/RAF inhibitor in patients with solid tumors. Clin Cancer Res. 2012;18(17):4806–19.
Ishii N, Harada N, Joseph EW, Ohara K, Miura T, Sakamoto H, et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res. 2013;73(13):4050–60.
Wada M, Horinaka M, Yamazaki T, Katoh N, Sakai T. The dual RAF/MEK inhibitor CH5126766/RO5126766 may be a potential therapy for RAS-mutated tumor cells. PLoS One. 2014;9(11):e113217.
Knauf JA, Luckett KA, Chen KY, Voza F, Socci ND, Ghossein R, et al. Hgf/Met activation mediates resistance to BRAF inhibition in murine anaplastic thyroid cancers. J Clin Invest. 2018;128(9):4086–97.
Sullivan RJ, Infante JR, Janku F, Wong DJL, Sosman JA, Keedy V, et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: results of a phase I dose-escalation and expansion study. Cancer Discov. 2018;8(2):184–95.
Moschos SJ, Sullivan RJ, Hwu WJ, Ramanathan RK, Adjei AA, Fong PC, et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight. 2018;3(4):e92352.
Coussy F, El Botty R, Lavigne M, Gu C, Fuhrmann L, Briaux A, et al. Combination of PI3K and MEK inhibitors yields durable remission in PDX models of PIK3CA-mutated metaplastic breast cancers. J Hematol Oncol. 2020;13(1):13.
Carlson CA, Kim KH. Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. J Biol Chem. 1973;248(1):378–80.
Yeh LA, Lee KH, Kim KH. Regulation of rat liver acetyl-CoA carboxylase. Regulation of phosphorylation and inactivation of acetyl-CoA carboxylase by the adenylate energy charge. J Biol Chem. 1980;255(6):2308–14.
Ly S, Kim KH. Inactivation of hepatic acetyl-CoA carboxylase by catecholamine and its agonists through the alpha-adrenergic receptors. J Biol Chem. 1981;256(22):11585–90.
Lent B, Kim KH. Purification and properties of a kinase which phosphorylates and inactivates acetyl-CoA carboxylase. J Biol Chem. 1982;257(4):1897–901.
Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223(2):217–22.
Hardie DG. AMPK—sensing energy while talking to other signaling pathways. Cell Metab. 2014;20(6):939–52.
Oakhill JS, Scott JW, Kemp BE. AMPK functions as an adenylate charge-regulated protein kinase. Trends Endocrinol Metab. 2012;23(3):125–32.
Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445(1):11–27.
Hardie DG, Schaffer BE, Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016;26(3):190–201.
Davies SP, Hawley SA, Woods A, Carling D, Haystead TA, Hardie DG. Purification of the AMP-activated protein kinase on ATP-gamma-sepharose and analysis of its subunit structure. Eur J Biochem. 1994;223(2):351–7.
Mitchelhill KI, Stapleton D, Gao G, House C, Michell B, Katsis F, et al. Mammalian AMP-activated protein kinase shares structural and functional homology with the catalytic domain of yeast Snf1 protein kinase. J Biol Chem. 1994;269(4):2361–4.
Dasgupta B, Chhipa RR. Evolving lessons on the complex role of AMPK in normal physiology and cancer. Trends Pharmacol Sci. 2016;37(3):192–206.
Vara-Ciruelos D, Russell FM, Hardie DG. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? (dagger). Open Biol. 2019;9(7):190099.
Ross FA, MacKintosh C, Hardie DG. AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. FEBS J. 2016;283(16):2987–3001.
Steinberg GR, Carling D. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov. 2019;18(7):527–51.
Quentin T, Kitz J, Steinmetz M, Poppe A, Bar K, Kratzner R. Different expression of the catalytic alpha subunits of the AMP activated protein kinase—an immunohistochemical study in human tissue. Histol Histopathol. 2011;26(5):589–96.
Dasgupta B, Ju JS, Sasaki Y, Liu X, Jung SR, Higashida K, et al. The AMPK beta2 subunit is required for energy homeostasis during metabolic stress. Mol Cell Biol. 2012;32(14):2837–48.
Chen Z, Heierhorst J, Mann RJ, Mitchelhill KI, Michell BJ, Witters LA, et al. Expression of the AMP-activated protein kinase beta1 and beta2 subunits in skeletal muscle. FEBS Lett. 1999;460(2):343–8.
Wu J, Puppala D, Feng X, Monetti M, Lapworth AL, Geoghegan KF. Chemoproteomic analysis of intertissue and interspecies isoform diversity of AMP-activated protein kinase (AMPK). J Biol Chem. 2013;288(50):35904–12.
Jorgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, et al. Effects of alpha-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J. 2005;19(9):1146–8.
Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101(10):3329–35.
Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13(22):2004–8.
Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2(1):9–19.
Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280(32):29060–6.
Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2(1):21–33.
Anderson KA, Ribar TJ, Lin F, Noeldner PK, Green MF, Muehlbauer MJ, et al. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008;7(5):377–88.
Stahmann N, Woods A, Carling D, Heller R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Mol Cell Biol. 2006;26(16):5933–45.
Thornton C, Sardini A, Carling D. Muscarinic receptor activation of AMP-activated protein kinase inhibits orexigenic neuropeptide mRNA expression. J Biol Chem. 2008;283(25):17116–22.
Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem. 2004;279(13):12005–8.
Zhang CS, Hawley SA, Zong Y, Li M, Wang Z, Gray A, et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature. 2017;548(7665):112–6.
Ingebritsen TS, Geelen MJ, Parker RA, Evenson KJ, Gibson DM. Modulation of hydroxymethylglutaryl-CoA reductase activity, reductase kinase activity, and cholesterol synthesis in rat hepatocytes in response to insulin and glucagon. J Biol Chem. 1979;254(20):9986–9.
Loh K, Tam S, Murray-Segal L, Huynh K, Meikle PJ, Scott JW, et al. Inhibition of adenosine monophosphate-activated protein kinase-3-hydroxy-3-methylglutaryl coenzyme A reductase signaling leads to hypercholesterolemia and promotes hepatic steatosis and insulin resistance. Hepatol Commun. 2019;3(1):84–98.
Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–77.
Hoffman NJ, Parker BL, Chaudhuri R, Fisher-Wellman KH, Kleinert M, Humphrey SJ, et al. Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. 2015;22(5):922–35.
Wan Z, Root-McCaig J, Castellani L, Kemp BE, Steinberg GR, Wright DC. Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity (Silver Spring). 2014;22(3):730–8.
Zhang H, Guan M, Townsend KL, Huang TL, An D, Yan X, et al. MicroRNA-455 regulates brown adipogenesis via a novel HIF1an-AMPK-PGC1alpha signaling network. EMBO Rep. 2015;16(10):1378–93.
Ducommun S, Deak M, Sumpton D, Ford RJ, Nunez Galindo A, Kussmann M, et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal. 2015;27(5):978–88.
Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Loson OC, Hellberg K, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351(6270):275–81.
Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5(11):e15394.
Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466(7302):68–76.
Weerasekara VK, Panek DJ, Broadbent DG, Mortenson JB, Mathis AD, Logan GN, et al. Metabolic-stress-induced rearrangement of the 14-3-3zeta interactome promotes autophagy via a ULK1- and AMPK-regulated 14-3-3zeta interaction with phosphorylated Atg9. Mol Cell Biol. 2014;34(24):4379–88.
Zhang D, Wang W, Sun X, Xu D, Wang C, Zhang Q, et al. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy. 2016;12(9):1447–59.
Taylor EB, An D, Kramer HF, Yu H, Fujii NL, Roeckl KS, et al. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J Biol Chem. 2008;283(15):9787–96.
Treebak JT, Glund S, Deshmukh A, Klein DK, Long YC, Jensen TE, et al. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes. 2006;55(7):2051–8.
Liu Y, Lai YC, Hill EV, Tyteca D, Carpentier S, Ingvaldsen A, et al. Phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) is an AMPK target participating in contraction-stimulated glucose uptake in skeletal muscle. Biochem J. 2013;455(2):195–206.
Kim JH, Park JM, Yea K, Kim HW, Suh PG, Ryu SH. Phospholipase D1 mediates AMP-activated protein kinase signaling for glucose uptake. PLoS One. 2010;5(3):e9600.
Abbud W, Habinowski S, Zhang JZ, Kendrew J, Elkairi FS, Kemp BE, et al. Stimulation of AMP-activated protein kinase (AMPK) is associated with enhancement of Glut1-mediated glucose transport. Arch Biochem Biophys. 2000;380(2):347–52.
Fryer LG, Foufelle F, Barnes K, Baldwin SA, Woods A, Carling D. Characterization of the role of the AMP-activated protein kinase in the stimulation of glucose transport in skeletal muscle cells. Biochem J. 2002;363(Pt 1):167–74.
Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10(20):1247–55.
Bultot L, Guigas B, Von Wilamowitz-Moellendorff A, Maisin L, Vertommen D, Hussain N, et al. AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase. Biochem J. 2012;443(1):193–203.
Broer S, Broer A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem J. 2017;474(12):1935–63.
Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med. 2012;18(9):524–33.
Hu CA, Wu Z, Wang J. Amino acids and autophagy: their crosstalk, interplay and interlock. Amino Acids. 2015;47(10):2035–6.
Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004;279(31):32771–9.
Saha AK, Xu XJ, Lawson E, Deoliveira R, Brandon AE, Kraegen EW, et al. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes. 2010;59(10):2426–34.
Gleason CE, Lu D, Witters LA, Newgard CB, Birnbaum MJ. The role of AMPK and mTOR in nutrient sensing in pancreatic beta-cells. J Biol Chem. 2007;282(14):10341–51.
Dalle Pezze P, Ruf S, Sonntag AG, Langelaar-Makkinje M, Hall P, Heberle AM, et al. A systems study reveals concurrent activation of AMPK and mTOR by amino acids. Nat Commun. 2016;7:13254.
Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6(1):91–9.
Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17(1):113–24.
Chou CC, Lee KH, Lai IL, Wang D, Mo X, Kulp SK, et al. AMPK reverses the mesenchymal phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a signaling axis. Cancer Res. 2014;74(17):4783–95.
DeRan M, Yang J, Shen CH, Peters EC, Fitamant J, Chan P, et al. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 2014;9(2):495–503.
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26.
Choo AY, Kim SG, Vander Heiden MG, Mahoney SJ, Vu H, Yoon SO, et al. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol Cell. 2010;38(4):487–99.
Pineda CT, Ramanathan S, Fon Tacer K, Weon JL, Potts MB, Ou YH, et al. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell. 2015;160(4):715–28.
Wan L, Xu K, Wei Y, Zhang J, Han T, Fry C, et al. Phosphorylation of EZH2 by AMPK suppresses PRC2 methyltransferase activity and oncogenic function. Mol Cell. 2018;69(2):279–91 e5.
Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature. 2018;559(7715):637–41.
Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 2016;23(4):649–62.
Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK Protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell. 2015;17(5):585–96.
Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485(7400):661–5.
Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23(2):143–58.
Liu L, Ulbrich J, Muller J, Wustefeld T, Aeberhard L, Kress TR, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012;483(7391):608–12.
Kfoury A, Armaro M, Collodet C, Sordet-Dessimoz J, Giner MP, Christen S, et al. AMPK promotes survival of c-Myc-positive melanoma cells by suppressing oxidative stress. EMBO J. 2018;37(5):e97673.
Han F, Li CF, Cai Z, Zhang X, Jin G, Zhang WN, et al. The critical role of AMPK in driving Akt activation under stress, tumorigenesis and drug resistance. Nat Commun. 2018;9(1):4728.
Trillo-Tinoco J, Sierra RA, Mohamed E, Cao Y, de Mingo-Pulido A, Gilvary DL, et al. AMPK alpha-1 intrinsically regulates the function and differentiation of tumor myeloid-derived suppressor cells. Cancer Res. 2019;79(19):5034–47.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4.
Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50.
Chhipa RR, Fan Q, Anderson J, Muraleedharan R, Huang Y, Ciraolo G, et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat Cell Biol. 2018;20(7):823–35.
Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49(6):1167–75.
Creancier L, Morello D, Mercier P, Prats AC. Fibroblast growth factor 2 internal ribosome entry site (IRES) activity ex vivo and in transgenic mice reveals a stringent tissue-specific regulation. J Cell Biol. 2000;150(1):275–81.
Dobson T, Chen J, Krushel LA. Dysregulating IRES-dependent translation contributes to overexpression of oncogenic Aurora A Kinase. Mol Cancer Res. 2013;11(8):887–900.
Pantovic A, Krstic A, Janjetovic K, Kocic J, Harhaji-Trajkovic L, Bugarski D, et al. Coordinated time-dependent modulation of AMPK/Akt/mTOR signaling and autophagy controls osteogenic differentiation of human mesenchymal stem cells. Bone. 2013;52(1):524–31.
Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14(12):1295–304.
Papa S, Choy PM, Bubici C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene. 2019;38(13):2223–40.
Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem. 2003;278(16):14013–9.
Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999;3(2):169–79.
Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14(19):2501–14.
Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6(4):308–18.
Pelletier A, Joly E, Prentki M, Coderre L. Adenosine 5′-monophosphate-activated protein kinase and p38 mitogen-activated protein kinase participate in the stimulation of glucose uptake by dinitrophenol in adult cardiomyocytes. Endocrinology. 2005;146(5):2285–94.
Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, et al. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell. 2009;33(2):237–47.
Lopez-Cotarelo P, Escribano-Diaz C, Gonzalez-Bethencourt IL, Gomez-Moreira C, Deguiz ML, Torres-Bacete J, et al. A novel MEK-ERK-AMPK signaling axis controls chemokine receptor CCR7-dependent survival in human mature dendritic cells. J Biol Chem. 2015;290(2):827–40.
Costanzo-Garvey DL, Pfluger PT, Dougherty MK, Stock JL, Boehm M, Chaika O, et al. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab. 2009;10(5):366–78.
Fernandez MR, Henry MD, Lewis RE. Kinase suppressor of Ras 2 (KSR2) regulates tumor cell transformation via AMPK. Mol Cell Biol. 2012;32(18):3718–31.
Kodiha M, Rassi JG, Brown CM, Stochaj U. Localization of AMP kinase is regulated by stress, cell density, and signaling through the MEK-->ERK1/2 pathway. Am J Physiol Cell Physiol. 2007;293(5):C1427–36.
Freed E, Symons M, Macdonald SG, McCormick F, Ruggieri R. Binding of 14-3-3 proteins to the protein kinase Raf and effects on its activation. Science. 1994;265(5179):1713–6.
Fu H, Xia K, Pallas DC, Cui C, Conroy K, Narsimhan RP, et al. Interaction of the protein kinase Raf-1 with 14-3-3 proteins. Science. 1994;266(5182):126–9.
Verlande A, Krafcikova M, Potesil D, Trantirek L, Zdrahal Z, Elkalaf M, et al. Metabolic stress regulates ERK activity by controlling KSR-RAF heterodimerization. EMBO Rep. 2018;19(2):320–36.
Yuan J, Ng WH, Yap J, Chia B, Huang X, Wang M, et al. The AMPK inhibitor overcomes the paradoxical effect of RAF inhibitors through blocking phospho-Ser-621 in the C terminus of CRAF. J Biol Chem. 2018;293(37):14276–84.
Mischak H, Seitz T, Janosch P, Eulitz M, Steen H, Schellerer M, et al. Negative regulation of Raf-1 by phosphorylation of serine 621. Mol Cell Biol. 1996;16(10):5409–18.
Shen CH, Yuan P, Perez-Lorenzo R, Zhang Y, Lee SX, Ou Y, et al. Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol Cell. 2013;52(2):161–72.
Sprenkle AB, Davies SP, Carling D, Hardie DG, Sturgill TW. Identification of Raf-1 Ser621 kinase activity from NIH 3T3 cells as AMP-activated protein kinase. FEBS Lett. 1997;403(3):254–8.
Petti C, Vegetti C, Molla A, Bersani I, Cleris L, Mustard KJ, et al. AMPK activators inhibit the proliferation of human melanomas bearing the activated MAPK pathway. Melanoma Res. 2012;22(5):341–50.
Yuan P, Ito K, Perez-Lorenzo R, Del Guzzo C, Lee JH, Shen CH, et al. Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proc Natl Acad Sci U S A. 2013;110(45):18226–31.
Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25(5):460–70.
Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3(11):1272–85.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25(7):717–29.
Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4(8):905–13.
Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27(13):1447–61.
Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5(1):3056.
Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300.
Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov. 2018;8(3):276–87.
Xie X, Koh JY, Price S, White E, Mehnert JM. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. 2015;5(4):410–23.
Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913–30.
Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016;30(15):1704–17.
Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25(4):628–40.
Lee CS, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci U S A. 2019;116(10):4508–17.
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–61.
Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41.
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–23.
Sanduja S, Feng Y, Mathis RA, Sokol ES, Reinhardt F, Halaban R, et al. AMPK promotes tolerance to Ras pathway inhibition by activating autophagy. Oncogene. 2016;35(40):5295–303.
Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019;25(4):620–7.
Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med. 2007;357(1):39–51.
Messersmith WA, Ahnen DJ. Targeting EGFR in colorectal cancer. N Engl J Med. 2008;359(17):1834–6.
Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023–34.
Johnston S, Pippen J Jr, Pivot X, Lichinitser M, Sadeghi S, Dieras V, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2009;27(33):5538–46.
Tripathy D, Slamon DJ, Cobleigh M, Arnold A, Saleh M, Mortimer JE, et al. Safety of treatment of metastatic breast cancer with trastuzumab beyond disease progression. J Clin Oncol. 2004;22(6):1063–70.
Montemurro F, Donadio M, Clavarezza M, Redana S, Jacomuzzi ME, Valabrega G, et al. Outcome of patients with HER2-positive advanced breast cancer progressing during trastuzumab-based therapy. Oncologist. 2006;11(4):318–24.
Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–11.
Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343(2):342–50.
Schiering N, Knapp S, Marconi M, Flocco MM, Cui J, Perego R, et al. Crystal structure of the tyrosine kinase domain of the hepatocyte growth factor receptor c-Met and its complex with the microbial alkaloid K-252a. Proc Natl Acad Sci U S A. 2003;100(22):12654–9.
Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2(3):359–72.
Li Z, Jiang K, Zhu X, Lin G, Song F, Zhao Y, et al. Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016;370(2):332–44.
Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4(1):94–109.
Downward J. RAS synthetic lethal screens revisited: still seeking the elusive prize? Clin Cancer Res. 2015;21(8):1802–9.
Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137(5):821–34.
Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, et al. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell. 2017;168(5):890–903 e15.
Anderson GR, Winter PS, Lin KH, Nussbaum DP, Cakir M, Stein EM, et al. A landscape of therapeutic cooperativity in KRAS mutant cancers reveals principles for controlling tumor evolution. Cell Rep. 2017;20(4):999–1015.
Martin TD, Cook DR, Choi MY, Li MZ, Haigis KM, Elledge SJ. A role for mitochondrial translation in promotion of viability in K-Ras mutant cells. Cell Rep. 2017;20(2):427–38.
Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23(1):121–8.
Sos ML, Levin RS, Gordan JD, Oses-Prieto JA, Webber JT, Salt M, et al. Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Cell Rep. 2014;8(4):1037–48.
Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, et al. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell. 2014;26(3):402–13.
Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, Curtin JA, et al. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 2006;66(19):9483–91.
Cox AD, Der CJ. The raf inhibitor paradox: unexpected consequences of targeted drugs. Cancer Cell. 2010;17(3):221–3.
Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140(2):209–21.
Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431–5.
Aguirre AJ, Hahn WC. Synthetic lethal vulnerabilities in KRAS-mutant cancers. Cold Spring Harb Perspect Med. 2018;8(8):a031518.
Sueda T, Sakai D, Kawamoto K, Konno M, Nishida N, Koseki J, et al. BRAF V600E inhibition stimulates AMP-activated protein kinase-mediated autophagy in colorectal cancer cells. Sci Rep. 2016;6:18949.
Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell. 2015;28(3):384–98.
Tutuka CSA, Andrews MC, Mariadason JM, Ioannidis P, Hudson C, Cebon J, et al. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol Cancer. 2017;16(1):112.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–74.
Acknowledgments
We thank all the members of the Laboratory of Cancer Signaling (NCCS) for the helpful comments on this manuscript.
Funding
This study was funded by Asia Fund for Cancer Research (AFCR-2017/2019-JH), SingHealth Foundation (AM/TP011/2018), and NMRC (OFIRG18nov-0078) to Jiancheng Hu.
Author information
Authors and Affiliations
Contributions
Conceptualization, J.Y. and J.H.; original draft writing, J.Y., X.D., J.Y., and J.H.; review and editing, J.H.; visualization, J.Y.; supervision, J.H.; funding acquisition, J.H. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yuan, J., Dong, X., Yap, J. et al. The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J Hematol Oncol 13, 113 (2020). https://doi.org/10.1186/s13045-020-00949-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00949-4