
Abstract
Triple negative breast cancer (TNBC), which is typically lack of expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), represents the most aggressive and mortal subtype of breast cancer. Currently, only a few treatment options are available for TNBC due to the absence of molecular targets, which underscores the need for developing novel therapeutic and preventive approaches for this disease. Recent evidence from clinical trials and preclinical studies has demonstrated a pivotal role of signal transducer and activator of transcription 3 (STAT3) in the initiation, progression, metastasis, and immune evasion of TNBC. STAT3 is overexpressed and constitutively activated in TNBC cells and contributes to cell survival, proliferation, cell cycle progression, anti-apoptosis, migration, invasion, angiogenesis, chemoresistance, immunosuppression, and stem cells self-renewal and differentiation by regulating the expression of its downstream target genes. STAT3 small molecule inhibitors have been developed and shown excellent anticancer activities in in vitro and in vivo models of TNBC. This review discusses the recent advances in the understanding of STAT3, with a focus on STAT3’s oncogenic role in TNBC. The current targeting strategies and representative small molecule inhibitors of STAT3 are highlighted. We also propose potential strategies that can be further examined for developing more specific and effective inhibitors for TNBC prevention and therapy.
Similar content being viewed by others

Background
Triple negative breast cancer (TNBC) is the most aggressive form of breast cancer and accounts for much higher recurrence and metastasis rates [1]. Due to the absence of the expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), TNBC is unresponsive to endocrine and HER2-targeted therapies, which results in the high mortality of patients with this disease [1]. When patients are diagnosed with TNBC at the early stage, combination chemotherapy (anthracyclines, taxanes, platinum salts, etc.) with or without radiotherapy is used as standard non-surgical therapy and has shown some efficacy in patients with both primary and metastatic diseases [2]. Because of the inter- and the intratumoral heterogeneities of TNBC, the intrinsic chemoresistance as well as severe side effects are often observed and lead to limited success in the clinic [3, 4]. Targeted therapies (e.g., poly (ADP-ribose) polymerase (PARP) inhibitors and epidermal growth factor receptor (EGFR) inhibitors) and immunotherapies have also shown some promise in preliminary clinical studies, but further investigations are critically needed [5,6,7]. More recently, many efforts have been made to identify targetable molecules for treating TNBC via genomic profiling and several critical alternations have been discovered, including the overexpression and aberrant activation of signal transducer and activator of transcription 3 (STAT3) [8, 9]. The emerging data suggest that STAT3 may be a potential molecular target and biomarker for TNBC.
The STAT family of transcription factors is comprised of seven members with high structural and functional similarity, including STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6 [10, 11]. All STAT proteins consist of an amino acid domain (NH2), a coiled-coil domain (CCD) for binding with interactive proteins, a DNA binding domain (DBD), a linker domain, a SRC homology 2 (SH2) domain for phosphorylation and dimerization, and a C-terminal transactivation domain (TAD) [11]. Most of these domains are highly conserved among STAT proteins and only TAD is divergent and mainly contributes to their structure diversity [12]. STAT3 was initially discovered to bind to DNA in response to interleukin-6 (IL-6) and epidermal growth factor (EGF) in 1994 [13, 14]. Over the past decades, STAT3 has become one of the most investigated oncogenic transcription factors and is highly associated with cancer initiation, progression, metastasis, chemoresistance, and immune evasion [15, 16]. The recent evidence from both preclinical and clinical studies have demonstrated that STAT3 plays a critical role in TNBC and STAT3 inhibitors have shown efficacy in inhibiting TNBC tumor growth and metastasis.
Considering that there is an unmet medical need for TNBC treatment and innovative therapeutic agents are urgently required, an in-depth understanding of the roles of STAT3 in TNBC will facilitate the development of STAT3-targeted therapeutics and pave the way for a novel TNBC treatment approach. In this review, we focus on the recent findings related to STAT3’s role in TNBC as well as STAT3 inhibitors and current targeting strategies. We also discuss other potential strategies for developing new STAT3 inhibitors for TNBC treatment.
The STAT3 signaling pathway
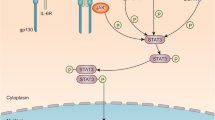
The classical STAT3 signaling pathway that is activated through the binding of cytokines or growth factors to their corresponding cell surface receptors has been extensively reviewed [16,17,18]. Here, we present a brief overview of the STAT3 signaling pathway, nonreceptor tyrosine kinases of STAT3, and its intrinsic inhibitors and coactivators, which are depicted in Fig. 1. Briefly, the overexpressed cytokine receptors, e.g., interleukin-6 receptor (IL-6R) and interleukin-10 receptor (IL-10R) and the hyperactive growth factor receptors, e.g., epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR) and insulin-like growth factor receptor (IGFR) always trigger the tyrosine phosphorylation cascade through the binding of ligands to these receptors, leading to the aberrant activation of STAT3 and the transcription of its downstream target genes [17]. Once the ligands bind to their receptors on the cell surface, these receptors further form dimers and successively recruit glycoprotein 130 (gp130) and Janus kinases (JAKs), thus phosphorylating and activating JAKs [19]. Conversely, the cytoplasmic tyrosine residues of these receptors are phosphorylated by the activated JAKs and then interact with the SH2 domain of STAT3, resulting in STAT3 phosphorylation at Tyr705 by JAKs [16]. In addition, STAT3 can be phosphorylated and activated by several nonreceptor tyrosine kinases, e.g., Src and Abl [20]. The phosphorylated STAT3 (pSTAT3) further forms a homodimer through interaction between their phosphorylated Tyr705 site and SH2 domain, triggering the dissociation of STAT3 dimers from the cell surface receptors and its translocation from cytoplasm to the nucleus [21, 22]. With the help of a variety of coactivator proteins, including NCOA/SRC1a, apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE/Ref-1), and CREB-binding protein (CBP)/p300, the nuclear STAT3 binds to specific DNA sequences and activates the transcription of genes that regulate various phenotypes of cancer cells [17, 18].

The STAT3 signaling pathway in cancer cells. Under normal physiological conditions, STAT3 activation is strictly controlled by the endogenous inhibitors, including the protein inhibitor of activated STAT (PIAS), the suppressor of cytokine signaling (SOCS), and several protein tyrosine phosphatases (PTPs). Once the upstream cytokines (e.g., IL-6) or growth factors (e.g., EGF, FGF, and VEGF) bind to cell surface receptors, STAT3 is phosphorylated and activated by JAK or Src. The nonreceptor tyrosine kinases (e.g., Src and Abl) also phosphorylate STAT3. The phosphorylated STAT3 undergoes dimerization and translocates from cytoplasm into the nucleus. The activated STAT3 further binds to DNA and its coactivators (e.g., NCOA, APE, and CBP) and induces the transcription of its downstream target genes
STAT3 is also highly expressed in some normal tissues and organs, including the bone marrow, peripheral nervous system, and digestive tract and plays a physiological role [23,24,25]. In the normal physiological conditions, STAT3 phosphorylation and activation are tightly controlled by several intrinsic inhibitors, including protein tyrosine phosphatases (PTPs), the suppressors of cytokine signaling (SOCS), and the protein inhibitor of activated STAT (PIAS) [26]. The Src homology domain-containing tyrosine phosphatases 1/2 (SHP-1/2) directly interact and dephosphorylate JAK and STAT3, resulting in their inactivation [27, 28]. The nuclear PTPs, including TC45 and T-cell protein-tyrosine phosphatase (TC-PTP) induce the inactivation of STAT3 through its dephosphorylation and translocation from nucleus to the cytoplasm [29, 30]. Other PTPs, such as PTP1B and PTPeC have also been reported to regulate STAT3 dephosphorylation and inactivation [31]. Moreover, SOCS directly interacts with JAK and STAT3 and inhibits their phosphorylation and activation via forming a negative feedback loop with JAK-STAT3 signaling pathway [32]. PIAS inhibits the binding of nuclear STAT3 to DNA and induces STAT3 dephosphorylation via protein tyrosine phosphatase receptor T (PTPRT), leading to the reduced expression of its downstream target genes [33]. In addition, the stability of STAT3 protein is also regulated by the ubiquitin-proteasome system via the ubiquitin ligase TRAF6 (tumor necrosis factor receptor-associated factor 6) [34]. Recent studies have also reported that miR-544 directly targets the 3′-untranslated region (UTR) on STAT3 mRNA, thus down-regulating STAT3 expression in TNBC cells [35]. Due to the presence of these endogenous inhibitors, STAT3 is strictly governed to exert its physiological functions in normal cells [36]. Herein, both direct inhibition of STAT3 and activation of the endogenous inhibitors may be considered as potential STAT3-inhibiting strategies for developing novel cancer therapeutics.
The STAT3 signaling pathway in triple negative breast cancer
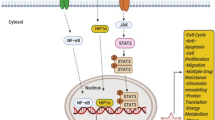
The oncogenic potential of STAT3 has been widely recognized through its involvement in regulating the expression of genes related to cancer cell proliferation, anti-apoptosis, migration, invasion, angiogenesis, chemoresistance, immune suppression, stem cell self-renewal and maintenance, and autophagy (as shown in Fig. 2) [17, 18]. Importantly, STAT3 is overexpressed and constitutively activated in TNBC, which is highly related to TNBC initiation, progression, metastasis, resistance to chemotherapy, and the poor survival outcomes [8]. STAT3 is not only capable of eliciting the expression of cancer-related genes, but also physically interacts and functionally cooperates with other oncogenic transcription factors, e.g., GLI1, promoting the aggressiveness of TNBC [8]. A recent study has also found a reduction of the gene associated with retinoic-interferon-induced mortality 19 (GRIM-19), an intrinsic inhibitor of STAT3 transcription accompanied by STAT3 overexpression in TNBC [37]. In addition, TCPTP, including two splice variants TC45 and TC48 are down-regulated in TNBC cells in vitro and in vivo, which also contributes to the activation of STAT3 signaling [38]. Indeed, STAT3 has also been found to localize in the mitochondria, where it is termed mitoSTAT3 and regulates the mitochondrial functions, including electron transport chain, ATP synthesis, calcium homeostasis, and reactive oxygen species (ROS) accumulation [39, 40]. Moreover, mitoSTAT3 has been shown to promote breast cancer cell growth, in which the phosphorylation of Serine 727 plays a critical role [41].

Activation of STAT3 signaling promotes growth, metastasis, chemoresistance, immune escape, and stemness in TNBC. One the upstream regulators are activated, STAT3 is phosphorylated, dimerized, and translocated into the nucleus, where it activates the transcription of the target genes that regulate cell proliferation, anti-apoptosis, migration, invasion, angiogenesis, chemoresistance, immune escape, stem cell phenotypes, and autophagy
A recent study has shown that acetylated STAT3 is highly elevated in TNBC, causing the methylation and inactivation of tumor-suppressor gene promoters [42]. Importantly, mutation of STAT3 at Lys685 or reducing STAT3 acetylation by resveratrol could induce demethylation and activation of the estrogen receptor-α gene and sensitize TNBC cells to antiestrogens. Considering the emerging data that demonstrate the critical role of STAT3 in TNBC, we herein present a comprehensive overview of its oncogenic functions in this section.
Role of STAT3 in TNBC cell proliferation and anti-apoptosis
Several studies have demonstrated that STAT3 promotes cell proliferation and inhibits apoptosis in TNBC by increasing the expression of target genes, including survivin, c-Myc, cyclin D1, B-cell lymphoma-2 (Bcl-2), and B-cell lymphoma-extra large (Bcl-xL) [21]. In TNBC, STAT3 directly binds to the survivin promoter and promotes its transcription [43, 44], which can be blocked by inhibiting the nuclear export factor, exportin 1 (XPO1) and CBP-mediated STAT3 acetylation [45]. In addition, Galectin-1, a β-galactoside binding protein has also been shown to contribute to TNBC progression through binding to integrin β1 and activating the integrin β1/FAK/c-Src/ERK/STAT3/survivin pathway [46]. Conversely, WW domain-containing oxidoreductase (Wwox) inhibits TNBC cell proliferation by interacting with JAK2 and suppressing JAK2 and STAT3 phosphorylation [47]. Wwox also represses the binding of STAT3 to the IL-6 promoter, therefore decreasing the expression of IL-6 cytokine. A tumor suppressor gene, gametogenetin-binding protein 2 (GGNBP2) has been found to inhibit breast cancer cell proliferation and induce apoptosis, independent of ER expression [48]. A further study has indicated that the inhibition of IL-6/STAT3 signaling by GGNBP2 is mainly responsible for its inhibitory effects on TNBC growth and metastasis [48].
STAT3 also promotes TNBC cell proliferation and inhibits apoptosis through the crosstalk with SET and MYND domain 2 (SMYD2) and nuclear factor-kappa B (NF-κB) [49]. SMYD2 is highly expressed in TNBC cell lines and tissues, which is correlated with increased TNBC cell proliferation and survival. Mechanistically, SMYD2 physically interacts with STAT3 and NF-κB p65 and increases their methylation and phosphorylation, promoting tumor growth and metastasis [49]. STAT3 recruits the acetyltransferase p300 to enhance NF-κB acetylation and prolong its nuclear retention [50]. In addition, STAT3 and NF-κB also contribute to each other’s activation via SMYD2 [49]. Interestingly, a recent study has reported an opposite role of STAT3 in TNBC cells [51]. It was observed that STAT3 knockdown did not inhibit but promoted the growth of MDA-MB-231 cells-derived xenograft tumors, implying that the oncogenic role of STAT3 in TNBC might be context-specific [51].
Role of STAT3 in TNBC cell migration and invasion
The role of STAT3 in promoting cell migration and invasion has been linked to the upregulated expression of matrix metalloproteinase 2 (MMP2), MMP9, TWIST, and Vimentin [52]. As discussed earlier, the STAT3 signaling is frequently activated through the binding of cytokines and growth factors to their corresponding receptors in cancer cells. A newly discovered cytokine termed interleukin-22 (IL-22) was recently reported to promote the migration of TNBC cells and induce their chemoresistance by activating the JAK/STAT3/MAPKs/AKT signaling pathway. The increased levels of the IL-22 producing (Th22) cells were also observed in normal, paratumor, and tumor tissues from patients with TNBC, which confirmed the importance of IL-22/JAK/STAT3/MAPKs/AKT in metastasis of this disease [53].
Recent studies reported that several upstream regulators of STAT3 signaling are involved in TNBC metastasis. Wwox blocks JAK2-STAT3 interaction and inhibits STAT3 phosphorylation, therefore repressing STAT3-driven TNBC metastasis [47]. G protein-coupled estrogen receptor (GPER) has been demonstrated as a TNBC metastasis suppressor. Mechanistically, activation of GPER can inhibit the NF-κB/IL-6/STAT3 signals, cause STAT3 dephosphorylation and inactivation, and then suppress migration and angiogenesis of TNBC [54]. GPER also triggers Y397 phosphorylation of focal adhesion kinase (FAK) in TNBC while the activation of both GPER and FAK promotes the migration of TNBC cells by increasing STAT3 nuclear accumulation and gene expression [55].
Role of STAT3 in angiogenesis of TNBC
The pro-angiogenic role of STAT3 has been partially attributed to the upregulation of vascular endothelial growth factor (VEGF), hypoxia-inducible factor 1-alpha (HIF-1α), hepatocyte growth factor (HGF), and basic fibroblast growth factor (bFGF) via STAT3 transactivation [52]. A recent study demonstrated that lymphatic endothelial cells (LECs) promote angiogenesis and metastasis through pSTAT3-mediated CCL5 expression in TNBC [56]. LECs are an important component of lymphatic vessels (LVs), which are prevailingly considered as the routes for cancer metastasis. Lee et al. have found that IL-6 secretion from TNBC cells causes STAT3 phosphorylation and activation, therefore inducing HIF-1α and VEGF expression. pSTAT3 also forms a ternary complex with phosphorylated c-Jun (pc-Jun) and phosphorylated activating transcription factor 2 (pATF2), which induces CCL5 expression in LECs and accelerates metastasis [56]. It was also observed that estrogen activates G protein-coupled estrogen receptor-1 (GPER-1), inhibits the expression VEGF at both protein and mRNA levels, and suppresses the tumor growth and angiogenesis in TNBC xenograft tumor models, in which STAT3 is involved [57].
Role of STAT3 in chemoresistance of TNBC
It has frequently been observed that blocking STAT3 signaling enhances the anticancer activity of chemotherapies in TNBC cells in vitro and in vivo, which endorses a critical role of STAT3 in chemosensitivity of TNBC [58,59,60,61]. Several recent studies revealed the mechanisms underlying STAT3-mediated chemoresistance in different subsets of TNBC cell lines [62]. NF-κB is highly associated with resistance to cancer therapies, while the overexpression and constitutive activation STAT3-NF-κB signaling pathway have been shown to confer chemoresistance in TNBC cells [63]. Mechanistically, STAT3 upregulates the expression of a target gene TNFRSF1A (tumor necrosis factor receptor superfamily member 1A), which recruits TNFα to the cell surface and triggers the activation of NF-κB signaling pathway [64]. The aberrant activation of STAT3 also increases the expression levels of pluripotency transcription factors octamer-binding transcription factor-4 (Oct-4) and c-Myc, which regulate stemness-mediated doxorubicin resistance in TNBC [65]. The restoration of doxorubicin sensitivity of TNBC cells by a STAT3 inhibitor WP1066 further confirms a pivotal role of this oncogene in chemoresistance.
STAT3-mediated microRNA (miRNA) expression is emerging as a mechanism for regulating chemoresistance in TNBC. Niu et al. found that miR-181a expression is increased in TNBC due to doxorubicin treatment and contributes to acquired resistance and metastasis of this disease through repressing the expression of its target gene Bax (Bcl-2-associated x protein) [66]. Further studies have indicated that pSTAT3 at S727 not only directly binds to MIR181A1 promoter but also recruits MSK1 (mitogen- and stress-activated protein kinase-1) and stabilizes its binding to MIR181A1 promoter, facilitating the transactivation [67]. The effectiveness of targeting STAT3-mediated MIR181A1 transactivation for sensitizing cells to chemotherapy and preventing metastasis has also been validated in a TNBC orthotopic model.
STAT3 is also involved in hypoxia-induced chemoresistance in TNBC [67]. Under hypoxia, the intracellular uptake of chemotherapy, especially cisplatin is dramatically reduced due to the upregulated expression of ATP-binding cassette (ABC) drug transporters. Although the expression level and activity of HIF-1α was increased by hypoxia in TNBC, no significant improvement in chemoresistance was observed in TNBC cells that were treated by HIF-1α siRNA. Intriguingly, STAT3 was found to increase the expression levels of ABC transporters, especially ABCC2 (also known as multidrug resistance protein 2, MRP2) and ABCC6 (also known as MRP6) in hypoxia-treated TNBC cells, therefore conferring chemoresistance to cisplatin [67, 68]. However, another study reported that IL-6-mediated STAT3 activation induces HIF-1α expression in TNBC cells, which consequently attenuates chemotherapy-induced cytotoxicity and cell apoptosis through regulating the expression of apoptosis-related proteins (Bax and Bcl-2) and drug transporters (P-glycoprotein and MRP1) [68]. The transfer RNA-derived fragments (tDRs), particularly tDR-0009 and tDR-7336 are upregulated in TNBC under hypoxia and facilitate the doxorubicin resistance through phosphorylating and activating STAT3 [69]. In addition, the combination treatment with HIF-1α and STAT3 inhibitors significantly enhances the cytotoxicity of cisplatin against TNBC cells and overcomes hypoxia-induced chemoresistance [70]. However, the role of STAT3-induced HIF-1α expression in hypoxia-induced chemoresistance is not clear so far, and further investigation is critically needed.
Role of STAT3 in immune suppression
Recent findings have established STAT3 as a powerful regulator of tumor-mediated immune suppression [21, 71]. STAT3 is not only overexpressed and activated in cancer cells but also in tumor-associated immune cells, inducing the expression of immune-suppression related genes, including IL-6, IL-10, TGF-β and VEGF and driving the escape of cancer cells from immune-mediated elimination [71]. In TNBC, STAT3 and its homolog STAT1 are also involved in regulating the expression of programmed death ligand 1 (PD-L1), a critical immune checkpoint that modulates the magnitude and the functional profile of T cell responses [72]. PD-L1 and PD-L2 are actually also amplified and overexpressed in TNBC cell lines due to JAK-mediated STAT3 phosphorylation and activation [73]. The mechanism studies have shown that pSTAT1 and pSTAT3 form heterodimers in the cytoplasm and translocate into the nucleus, where the pSTAT1-pSTAT3 dimers bind to the PD-L1 promoter and activate its transcription [72]. Another study has shown that syntenin1 is highly expressed in TNBC tissues and increases the expression level of PD-L1 by activating STAT3, consequently attenuates the response of TNBC to anti-PD-L1 treatment [74]. Moreover, direct inhibition of STAT3 overcomes the resistance of TNBC to immunotherapies, which confirms its immunosuppressive activity [72, 74].
Role of STAT3 in TNBC stem cell phenotypes
Early studies on STAT3 signaling disclosed an important role in stem cells self-renewal and differentiation [75]. The increasing evidence has also demonstrated that the constitutive activation of IL-6/STAT3 signaling pathway contributes to the stemness of TNBC stem cells under both normal and hypoxia conditions [76, 77]. In addition, the VEGF-VEGFR-2 binding-induced STAT3 phosphorylation and activation was found to promote the self-renewal of breast cancer cells, especially TNBC cells by upregulating the expression of Myc and Sox2 (SRY-related HMG-box 2) [78]. The crosstalk of STAT3 with NF-κB and Wnt signaling pathways was also observed in TNBC cells and serves as a feed-forward loop for regulating the TNBC stem cell function [79]. Moreover, Syndecan-1 (CD138) is highly expressed in TNBC, especially inflammatory TNBC and contributes to the poor prognosis of this disease [80]. Syndecan-1 was recently reported to promote TNBC stem cells through modulating the STAT3, NF-κB, and Wnt signaling pathways together [76]. Another study by Ibrahim et al. has demonstrated the importance of IL-6/STAT3 signaling pathway in Syndecan-1-modulated cancer stem cell phenotype [81]. Furthermore, Notch and EGFR signaling pathways are also implicated in the modulatory effects of Syndecan-1 on TNBC stem cells [81].
Except for cytokines and growth factors, adipokines, e.g., Leptin are also involved in the constitutive activation of the STAT3 signaling pathway. Leptin and its long form of leptin receptor (LEPRb) are enriched in breast cancer tissues and promote cell proliferation, migration, and angiogenesis [82]. Recently studies have shown that the binding of Leptin to LEPRb initiates the activation of JAK2/STAT3 signaling pathway, which further induces self-renewal and maintains the stem-cell state in TNBC stem cells [83]. Moreover, a new upstream regulator of the LEPR-STAT3 signaling pathway termed hematological and neurological expressed 1-like (HN1L) was also discovered to promote TNBC stem cell properties [84]. HN1L is overexpressed in TNBC tissues and correlates with the shorter survival of patients with this disease. The HN1L silencing experiments further confirmed its regulatory effects on LEPR-STAT3 signaling pathway and on TNBC stem cell population and lung metastasis [84].
Role of STAT3 in autophagy of TNBC cells
Autophagy is capable of regulating STAT3 phosphorylation status in TNBC cells [85]. Maycotte et al. discovered that the autophagy-dependent survival under unstressed conditions is enriched in TNBC, which reduces the response of cancer cells to therapy. Further studies have indicated that autophagy promotes TNBC cell survival by regulating STAT3 phosphorylation and activation [85]. Therefore, pharmacological inhibition of STAT3 may be a promising strategy for treating autophagy-dependent TNBC.
Targeting STAT3 for TNBC prevention and therapy
Abundant evidence has suggested that STAT3 may be a promising molecular target for TNBC therapy [86]. Various STAT3 inhibitors have been developed and shown some efficacy in TNBC models in vitro and in vivo, which have been summarized in Table 1. In this section, we discuss the current STAT3-targeting strategies (as shown in Fig. 3) for treating and preventing TNBC, as well as the challenges in developing more specific and effective STAT3 inhibitors.

Inhibiting STAT3 signaling at multiple levels for cancer therapy. Currently, the majority of STAT3 inhibitors have been developed through (1) targeting the upstream regulators of STAT3, (2) binding to the SH2 domain of STAT3 and inhibiting its activation, (3) inhibiting STAT3 phosphorylation or acetylation, or (4) blocking STAT3-DNA binding. Other potential strategies, such as (5) inhibiting the binding of STAT3 with its co-activators, (6) modulating the binding of STAT3 with other interactive proteins, and (7) promoting STAT3 ubiquitination and proteasomal degradation may also be evaluated for developing novel STAT3 inhibitors
Target upstream regulators of STAT3
The majority of STAT3 inhibitors have been identified to target the upstream regulators of STAT3 signaling. STAT3 activation is often initiated through the binding of cytokines and growth factors to their corresponding cell surface receptors. Therefore, small molecules and natural products that are able to inhibit IL-6 secretion and production, e.g., carfilzomib [87], manuka honey [88], bazedoxifene [89, 90], and Ganoderma lucidum extract [91] or suppress EGFR expression and phosphorylation, e.g., deguelin [92], picrasidine G [93], cantharidin [94], and silibinin [95] have shown significant inhibitory effects on STAT3 signaling as well as the expression of its downstream target genes in TNBC cell lines. In addition, arsenic trioxide (ATO) was reported to inhibit IL-6-mediated STAT3 activation, consequently reducing the expression of VEGF and suppressing angiogenesis [96]. Further studies have demonstrated that ATO blocks the interaction between enhancer of zeste homolog 2 (EZH2) and NF-κB p65, herein suppressing the activity of NF-κB and reducing the expression of IL-6. All these indirect STAT3 inhibitors have exhibited potent in vitro and in vivo anti-TNBC activities (Table 1). However, most of them have also been found to inhibit other signaling pathways that are triggered by ligand-cell surface receptor binding in cancer cells, indicating a low level of specificity in targeting the STAT3 signaling pathway.
As discussed earlier, several protein tyrosine kinases, such as JAK2 contribute to STAT3 phosphorylation and activation in both receptor-dependent and/or receptor-independent manners. JAK2 inhibitors, including silibinin [97] and ganoderic acid A [98] were found to inhibit TNBC cell viability, migration, and invasion and induce apoptosis in vitro through inhibiting the JAK2/STAT3 signaling pathway. However, their in vivo efficacy still needs further investigation. Targeting the intrinsic STAT3 inhibitors, such as PTPs and SOCS have been considered as a potential strategy for repressing STAT3 signaling pathway. Several natural and synthetic compounds were identified to activate one of the STAT3 PTPs, SHP-1. Among them, nintedanib and SC-78 significantly increase SHP-1 activity without affecting its expression [99, 100], while 1,2,3,4,6-penta-O-galloyl-beta-D-glucose (PGG) and SC-2001 largely induce the expression of SHP-1 [101, 102]. All these SHP-1 activators were also shown to inhibit STAT3 phosphorylation and the expression of its downstream target genes, thus suppressing TNBC cell growth and migration and inducing apoptosis in vitro and in vivo [99,100,101,102]. In addition, isolinderalactone was reported to increase SOCS3 expression and then enhance SOCS3-mediated STAT3 dephosphorylation and inactivation [103].
As one of the major client proteins of heat shock protein 90 (HSP90), STAT3 can be degraded through inhibiting HSP90. Two deguelin-derived HSP90 inhibitors, termed compound 57 and L80 have been observed to inhibit STAT3 expression and phosphorylation by interacting with the C-terminal ATP-binding pocket of HSP90 and blocking its function [104, 105]. Both compounds have also exerted their anticancer activities in TNBC models in vitro and in vivo [104, 105]. Moreover, nor-wogonin was found to inhibit the expression of transforming growth factor β-activated kinase 1 (TAK1), therefore dephosphorylating STAT3 without affecting its total expression level [106]. The dopamine receptor D2 (DRD2)-targeting drug thioridazine inhibits TNBC cell self-renewal through reducing DRD2-mediated STAT3 activation [107]. Due to the highly conserved structures among STAT family members, targeting the upstream regulators always results in the wide-spectrum inhibition of all STAT proteins, causing off-target effects. Therefore, directly targeting STAT3 and/or inhibiting its functions may be more promising strategies for developing safe and effective anticancer therapeutics.
Directly bind to STAT3 and inhibit its activation
Due to advances in the understanding of the structural biology of STAT3, small molecule inhibitors have been developed to directly bind to STAT3 and inhibit its activity. Currently, many small molecule inhibitors have been designed to target the SH2 domain and block its phosphorylation, dimerization, and nuclear translocation. Several STAT3-binding small molecule inhibitors that are under preclinical and clinical investigations have shown excellent efficacy in TNBC cells in vitro and in vivo.
Recently, a dual-luciferase assay-based screening of 1563 compounds for STAT3 inhibitors was performed, leading to the identification of Bt354 [108]. Further studies have shown that Bt354 inhibits STAT3 phosphorylation and nuclear translocation, which may be attributed to the binding of this compound to the SH2 domain of STAT3. Bt354 did not cause significant changes in the expression of STAT3 upstream regulators JAK2 and Src, indicating a specific targeting effect on STAT3 [108]. Moreover, this small molecule inhibitor also suppresses the viability of TNBC cells with constitutively activated STAT3, induces the G2/M phase arrest and late apoptosis, and impairs cell migration in vitro and represses the growth of TNBC xenograft tumors in vivo [108]. Additionally, several natural products, including osthole [109], arctigenin [110], and alantolactone [111] have also been shown to directly bind to the SH2 domain of STAT3, inhibit its phosphorylation and activation, and suppress the growth and metastasis of TNBC in vitro and in vivo. Cryptotanshinone is a well-documented natural product inhibitor of STAT3, which also binds to the SH2 domain and inhibits the phosphorylation and dimerization of STAT3 [112]. KYZ3, a synthetic derivative of cryptotanshinone has recently been developed and shown to exert anticancer activity in TNBC cells in vitro and in vivo through binding to and inhibiting STAT3 activation [113]. However, none of these compounds have been evaluated for their binding affinity to STAT3. Their selectivity among STAT3 and other STAT family members is yet to be determined.
Inhibit STAT3 phosphorylation or acetylation
Except for the STAT3-binding small molecule inhibitors that we discussed above, a number of natural products and their derivatives were found to inhibit STAT3 phosphorylation and/or nuclear translocation without affecting the upstream regulators. Sesquiterpene lactones, which are enriched in the hexane fraction from Inula helenium L. have been shown to suppress tumor growth in vitro and in vivo by inhibiting STAT3 phosphorylation and decreasing the expression of the downstream target genes, including cyclin D1, c-Myc, and Bcl-2 [114]. Another crude extract from the fruits of Rhus coriaria was also discovered to inhibit angiogenesis, tumor growth and metastasis in TNBC models in vitro and in vivo by repressing STAT3 phosphorylation and STAT3-mediated VEGF expression [115]. Moreover, several natural compounds and derivatives, including schisandrin B [116], eupalinolide J [117], galiellalactone analogs 16 and 17 [118], and ursolic acid derivative FZU-03,010 [119] have shown in vitro and in vivo efficacy in TNBC models through inhibition of STAT3 phosphorylation and/or nuclear translocation. None of them have been investigated for the binding ability with STAT3. Considering that these compounds did not show any significant effects on STAT3 regulators and interactive proteins, further studies for examining the potential binding between STAT3 and these compounds would provide important information regarding their underlying molecular mechanisms.
Of note, several approved drugs have shown potent inhibitory effects on pSTAT3 and may be repositioned as anticancer drugs. Niclosamide, an FDA-approved anthelmintic drug was identified as a potent STAT3 inhibitor. A recent study demonstrated that niclosamide not only inhibits TNBC cell viability but also sensitizes TNBC cells to ionizing irradiation (IR) by blocking IR-induced STAT3 phosphorylation and activation [120]. Flubendazole, another wildly used anthelmintic agent and disulfiram, a clinical drug for treating chronic alcoholism were found to eradicate TNBC stem cells-like cells that express high levels of pSTAT3 [121, 122]. Further studies showed that both drugs were able to cause TNBC cell growth arrest and apoptosis in vitro and suppress TNBC tumor growth, angiogenesis, and metastasis in vivo by inhibiting STAT3 [121, 122]. Moreover, salinomycin, an antibacterial and coccidiostat ionophore therapeutic drug and metformin, an antidiabetic drug have exhibited potent inhibitory effects on STAT3 phosphorylation and TNBC cell growth in vitro [123, 124]. However, further evaluation of their anti-TNBC efficacy in in vivo models is critically needed.
Recent studies have disclosed that targeting STAT3 acetylation may be a potential therapeutic approach for treating cancer. SH-I-14, a newly synthesized carbazole was shown to inhibit STAT3 phosphorylation through increasing SHP-1 expression [125]. A follow-up study reported that SH-I-14 also inhibited STAT3 acetylation and disrupted DNMT1-STAT3 interaction, resulting in DNA demethylation and re-expression of tumor suppressor genes [126]. Its in vitro and in vivo activity has also been demonstrated in TNBC model, suggesting the effectiveness of inhibiting STAT3 acetylation in TNBC therapy.
Block STAT3-DNA binding
STAT3 induces the expression of its downstream targets through binding to DNA and activating the transcription. Therefore, inhibition of STAT3-DNA binding has been considered as a promising strategy to develop targeted cancer therapies. Several STAT3-DNA binding inhibitors have been developed and shown potent anticancer efficacy in TNBC cells. Methylsulfonyl-methane (MSM), a dietary supplement was found to inhibit TNBC cell viability and induce apoptosis by blocking the DNA binding abilities of STAT3 to VEGF promoter and STAT5 to IGF-1R (IGF-1 receptor) promoter and repressing the expression of VEGF and IGF-1R [127]. Considering the extremely low toxicity of MSM, it could be developed as a preventive agent for cancers harboring overexpressed and aberrantly activated STAT3. Two natural compounds, isoharringtonine and salidroside have also been demonstrated to exert their anti-TNBC activities by blocking the binding of STAT3 to Nanog and MMP2 promoters, respectively [128, 129]. However, their binding affinity to STAT3 and in vivo efficacy are yet to be studied.
As discussed above, several strategies (as shown in Fig. 3) have been developed to inhibit STAT3 signaling, i.e. 1) targeting the upstream regulators, 2) directly binding to STAT3 SH2 domain and inhibiting its activation, 3) inhibiting STAT3 phosphorylation or acetylation, and 4) blocking STAT3-DNA binding. Many small molecules have been developed and shown efficacy in preventing and treating TNBC in preclinical studies (Table 1). Several STAT3 inhibitors also enter clinical trials [130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148], which have been summarized in Table 2. There are other STAT3-targeting strategies (as shown in Fig. 3) that have not been examined, including 1) inhibiting the binding of STAT3 with its co-activators (e.g., NCOA/SRC1a, APE/Ref-1, and CBP/p300) and repressing its transcriptional activity, 2) modulating the binding of STAT3 with other interactive proteins (e.g., SMYD2 and TRAF6) that regulate its activity and stability, and 3) developing STAT3-targeting PROTACs (proteolysis targeting chimeras) for promoting STAT3 ubiquitination and proteasomal degradation. Because most of the small molecule STAT3 inhibitors have been developed to inhibit its phosphorylation and activation but not affect the protein stability, long-term treatment of these inhibitors may result in the compensatory activation of other signaling pathways, finally causing drug resistance. Therefore, small molecules, such as PROTACs that can induce STAT3 protein degradation may be used more efficiently in combination with current inhibitors for cancer therapy.
Conclusions
TNBC is still a treatable but incurable disease with complex genetic heterogeneity. The STAT3 oncogene is overexpressed and constitutively activated in TNBC and is associated with the high metastatic risk and poor survival outcomes. Moreover, STAT3 not only acts as a transcription factor to activate the expression of its downstream target genes but also localizes to mitochondria and regulates its functions, then regulating the various aspects of TNBC cells. Many STAT3-targeted therapies have been successfully developed and shown efficacy in preclinical models of TNBC in vitro and in vivo; several STAT3 inhibitors even enter clinical trials and are currently under investigation in various human cancers, including TNBC. In addition to its role in cancer cells, STAT3 also plays a pivotal role in the immune system. Indeed, STAT3 inhibitors have been found to suppress tumor cells but also boost immune cell responses. Therefore, the STAT3 oncogene is a promising target for TNBC prevention and therapy.
Of note, targeting STAT3 alone has shown excellent anti-TNBC activities in preclinical settings. However, TNBC has been reported to harbor multiple genetic alterations, including STAT3 overexpression and constitutive activation which contribute to the initiation, progression, metastasis, and drug resistance of this disease. Therefore, STAT3 inhibition combined with other targeted therapies may be more effective in treating TNBC. Considering that STAT3 plays a crucial role in chemoresistance, the combination of STAT3 inhibitors with other chemotherapies may exert synergistic effects in treating TNBC. Therefore, further studies are warranted to demonstrate the preventive and therapeutic efficacy of STAT3 inhibitors alone or in combination with chemotherapy and/or other targeted therapies in clinical studies. Moreover, new targeting strategies, i.e. inducing the degradation of STAT3 protein through PROTAC or inhibiting the binding of STAT3 to its co-activators and other interactive proteins can be examined, which may lead to more specific and effective inhibitors for TNBC prevention and therapy.
Abbreviations
- ABC:
-
ATP-binding cassette
- AML:
-
Acute myeloid leukemia
- APE/Ref-1:
-
Apurinic/apyrimidinic endonuclease-1/redox factor-1
- ATO:
-
Arsenic trioxide
- Bax:
-
Bcl-2-associated x protein
- Bcl-2:
-
B-cell lymphoma-2
- Bcl-xL:
-
B-cell lymphoma-extra Large
- bFGF:
-
Basic fibroblast growth factor
- CBP:
-
CREB-binding protein
- CCD:
-
Coiled-coil domain
- CLL:
-
Chronic lymphocytic leukemia
- DBD:
-
DNA binding domain
- DLBCL:
-
Diffuse large B-cell lymphoma
- DRD2:
-
Dopamine receptor D2
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor
- ER:
-
Estrogen receptor
- EZH2:
-
Enhancer of zeste homolog 2
- FAK:
-
Focal adhesion kinase
- FGFR:
-
Fibroblast growth factor receptor
- GGNBP2:
-
Gametogenetin-binding protein 2
- GIC:
-
Gastrointestinal cancer
- gp130:
-
Glycoprotein 130
- GPER:
-
G protein-coupled estrogen receptor
- GPER-1:
-
G protein-coupled estrogen receptor-1
- GRIM-19:
-
Gene associated with retinoic-interferon-induced mortality 19
- HCC:
-
Hepatocellular carcinoma
- HER2:
-
Human epidermal growth factor receptor 2
- HGF:
-
Hepatocyte growth factor
- HIF-1α:
-
Hypoxia-inducible factor 1-alpha
- HN1L:
-
Hematological and neurological expressed 1-like
- HNSCC:
-
Head and neck squamous cell carcinoma
- HSP90:
-
Heat shock protein 90
- IGF-1R:
-
IGF-1 receptor
- IGFR:
-
Insulin-like growth factor receptor
- IL-10R:
-
Interleukin-10 receptor
- IL-22:
-
Interleukin-22
- IL-6:
-
Interleukin-6
- IL-6R:
-
Interleukin-6 receptor
- IR:
-
Ionizing irradiation
- JAKs:
-
Janus kinases
- LECs:
-
Lymphatic endothelial cells
- LEPRb:
-
Long form of leptin receptor
- LVs:
-
Lymphatic vessels
- MCC:
-
Metastatic colorectal cancer
- miRNA:
-
MicroRNA
- MMP:
-
Matrix metalloproteinase
- MRP2:
-
Multidrug resistance protein 2
- MSK1:
-
Mitogen- and stress-activated protein kinase-1
- MSM:
-
Methylsulfonyl-methane
- NF-κB:
-
Nuclear factor-kappa B
- NHL:
-
Non-Hodgkin lymphoma
- NSCLC:
-
Non-small cell lung cancer
- Oct-4:
-
Octamer-binding transcription factor-4
- PARP:
-
Poly (ADP-ribose) polymerase
- pATF2:
-
Phosphorylated activating transcription factor 2
- pc-Jun:
-
Phosphorylated c-Jun
- PD-L1:
-
Programmed death ligand 1
- PGG:
-
1,2,3,4,6-penta-O-galloyl-beta-D-glucose
- PIAS:
-
Protein inhibitor of activated STAT
- PR:
-
Progesterone receptor
- PROTACs:
-
Proteolysis targeting chimeras
- pSTAT3:
-
Phosphorylated STAT3
- PTPRT:
-
Protein tyrosine phosphatase receptor T
- PTPs:
-
Protein tyrosine phosphatases
- ROS:
-
Reactive oxygen species
- SH2:
-
SRC homology 2
- SHP-1/2:
-
Src homology domain-containing tyrosine phosphatases 1/2
- SLL:
-
Small lymphocytic leukemia
- SMYD2:
-
SET and MYND domain 2
- SOCS:
-
Suppressors of cytokine signaling
- Sox2:
-
SRY-related HMG-box 2
- STAT3:
-
Signal transducer and activator of transcription 3
- TAD:
-
Transactivation domain
- TAK1:
-
Transforming growth factor β-activated kinase 1
- TC-PTP:
-
T-cell protein-tyrosine phosphatase
- tDRs:
-
Transfer RNA-derived fragments
- TNBC:
-
Triple negative breast cancer
- TNFRSF1A:
-
Tumor necrosis factor receptor superfamily member 1A
- TRAF6:
-
Tumor necrosis factor receptor-associated factor 6
- UTR:
-
3′-untranslated region
- VEGF:
-
Vascular endothelial growth factor
- Wwox:
-
WW domain-containing oxidoreductase
- XPO1:
-
Exportin 1
References
Waks AG, Winer EP. Breast Cancer treatment: a review. JAMA. 2019;321(3):288–300.
Sharma P. Update on the treatment of early-stage triple-negative breast Cancer. Curr Treat Options in Oncol. 2018;19(5):22.
Walsh EM, Keane MM, Wink DA, Callagy G, Glynn SA. Review of triple negative breast Cancer and the impact of inducible nitric oxide synthase on tumor biology and patient outcomes. Crit Rev Oncog. 2016;21(5–6):333–51.
Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance evolution in triple-negative breast Cancer delineated by single-cell sequencing. Cell. 2018;173(4):879–93 e813.
Robert M, Patsouris A, Frenel JS, Gourmelon C, Augereau P, Campone M. Emerging PARP inhibitors for treating breast cancer. Expert Opin Emerg Drugs. 2018;23(3):211–21.
Costa R, Shah AN, Santa-Maria CA, Cruz MR, Mahalingam D, Carneiro BA, et al. Targeting epidermal growth factor receptor in triple negative breast cancer: new discoveries and practical insights for drug development. Cancer Treat Rev. 2017;53:111–9.
Vikas P, Borcherding N, Zhang W. The clinical promise of immunotherapy in triple-negative breast cancer. Cancer Manag Res. 2018;10:6823–33.
Sirkisoon SR, Carpenter RL, Rimkus T, Anderson A, Harrison A, Lange AM, et al. Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene. 2018;37(19):2502–14.
Gupta I, Sareyeldin RM, Al-Hashimi I, Al-Thawadi HA, Al Farsi H, Vranic S, et al. Triple Negative Breast Cancer Profile, from Gene to microRNA, in Relation to Ethnicity. Cancers (Basel). 2019;11(3):363.
Bousoik E, Montazeri Aliabadi H. “Do we know Jack” about JAK? A closer look at JAK/STAT signaling pathway. Front Oncol. 2018;8:287.
Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and approaches of targeting STAT3 for Cancer treatment. ACS Chem Biol. 2016;11(2):308–18.
Zhuang S. Regulation of STAT signaling by acetylation. Cell Signal. 2013;25(9):1924–31.
Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77(1):63–71.
Zhong Z, Wen Z, Darnell JE Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264(5155):95–8.
Huynh J, Chand A, Gough D, Ernst M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat Rev Cancer. 2019;19(2):82–96.
Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15(4):234–48.
Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–46.
Guanizo AC, Fernando CD, Garama DJ, Gough DJ. STAT3: a multifaceted oncoprotein. Growth Factors. 2018;36(1–2):1–14.
Garbers C, Aparicio-Siegmund S, Rose-John S. The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol. 2015;34:75–82.
Karras JG, Wang Z, Huo L, Howard RG, Frank DA, Rothstein TL. Signal transducer and activator of transcription-3 (STAT3) is constitutively activated in normal, self-renewing B-1 cells but only inducibly expressed in conventional B lymphocytes. J Exp Med. 1997;185(6):1035–42.
Wang Y, Shen Y, Wang S, Shen Q, Zhou X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018;415:117–28.
Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7(1):41–51.
Huang YH, Molavi O, Alshareef A, Haque M, Wang Q, Chu MP, et al. Constitutive activation of STAT3 in myeloma cells cultured in a three-dimensional, reconstructed bone marrow model. Cancers (Basel). 2018;10(6):206.
Quarta S, Baeumer BE, Scherbakov N, Andratsch M, Rose-John S, Dechant G, et al. Peripheral nerve regeneration and NGF-dependent neurite outgrowth of adult sensory neurons converge on STAT3 phosphorylation downstream of neuropoietic cytokine receptor gp130. J Neurosci. 2014;34(39):13222–33.
Hruz P, Dann SM, Eckmann L. STAT3 and its activators in intestinal defense and mucosal homeostasis. Curr Opin Gastroenterol. 2010;26(2):109–15.
Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018;27(12):1984–2009.
Bousquet C, Susini C, Melmed S. Inhibitory roles for SHP-1 and SOCS-3 following pituitary proopiomelanocortin induction by leukemia inhibitory factor. J Clin Invest. 1999;104(9):1277–85.
Kim H, Baumann H. Dual signaling role of the protein tyrosine phosphatase SHP-2 in regulating expression of acute-phase plasma proteins by interleukin-6 cytokine receptors in hepatic cells. Mol Cell Biol. 1999;19(8):5326–38.
Kim DJ, Tremblay ML, Digiovanni J. Protein tyrosine phosphatases, TC-PTP, SHP1, and SHP2, cooperate in rapid dephosphorylation of Stat3 in keratinocytes following UVB irradiation. PLoS One. 2010;5(4):e10290.
Wakahara R, Kunimoto H, Tanino K, Kojima H, Inoue A, Shintaku H, et al. Phospho-Ser727 of STAT3 regulates STAT3 activity by enhancing dephosphorylation of phospho-Tyr705 largely through TC45. Genes Cells. 2012;17(2):132–45.
Martin-Granados C, Prescott AR, Le Sommer S, Klaska IP, Yu T, Muckersie E, et al. A key role for PTP1B in dendritic cell maturation, migration, and T cell activation. J Mol Cell Biol. 2015;7(6):517–28.
Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, et al. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387(6636):924–9.
Yagil Z, Nechushtan H, Kay G, Yang CM, Kemeny DM, Razin E. The enigma of the role of protein inhibitor of activated STAT3 (PIAS3) in the immune response. Trends Immunol. 2010;31(5):199–204.
Wei J, Yuan Y, Jin C, Chen H, Leng L, He F, et al. The ubiquitin ligase TRAF6 negatively regulates the JAK-STAT signaling pathway by binding to STAT3 and mediating its ubiquitination. PLoS One. 2012;7(11):e49567.
Zhu Z, Wang S, Zhu J, Yang Q, Dong H, Huang J. MicroRNA-544 down-regulates both Bcl6 and Stat3 to inhibit tumor growth of human triple negative breast cancer. Biol Chem. 2016;397(10):1087–95.
Zhang W, Qu X, Chen B, Snyder M, Wang M, Li B, et al. Critical roles of STAT3 in beta-adrenergic functions in the heart. Circulation. 2016;133(1):48–61.
Zhou T, Chao L, Rong G, Wang C, Ma R, Wang X. Down-regulation of GRIM-19 is associated with STAT3 overexpression in breast carcinomas. Hum Pathol. 2013;44(9):1773–9.
Shields BJ, Wiede F, Gurzov EN, Wee K, Hauser C, Zhu HJ, et al. TCPTP regulates SFK and STAT3 signaling and is lost in triple-negative breast cancers. Mol Cell Biol. 2013;33(3):557–70.
Yang R, Rincon M. Mitochondrial Stat3, the need for design thinking. Int J Biol Sci. 2016;12(5):532–44.
Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323(5915):793–7.
Zhang Q, Raje V, Yakovlev VA, Yacoub A, Szczepanek K, Meier J, et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J Biol Chem. 2013;288(43):31280–8.
Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc Natl Acad Sci U S A. 2012;109(20):7765–9.
Diaz N, Minton S, Cox C, Bowman T, Gritsko T, Garcia R, et al. Activation of stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin Cancer Res. 2006;12(1):20–8.
Gritsko T, Williams A, Turkson J, Kaneko S, Bowman T, Huang M, et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res. 2006;12(1):11–9.
Cheng Y, Holloway MP, Nguyen K, McCauley D, Landesman Y, Kauffman MG, et al. XPO1 (CRM1) inhibition represses STAT3 activation to drive a survivin-dependent oncogenic switch in triple-negative breast cancer. Mol Cancer Ther. 2014;13(3):675–86.
Nam K, Son SH, Oh S, Jeon D, Kim H, Noh DY, et al. Binding of galectin-1 to integrin beta1 potentiates drug resistance by promoting survivin expression in breast cancer cells. Oncotarget. 2017;8(22):35804–23.
Chang R, Song L, Xu Y, Wu Y, Dai C, Wang X, et al. Loss of Wwox drives metastasis in triple-negative breast cancer by JAK2/STAT3 axis. Nat Commun. 2018;9(1):3486.
Liu J, Liu L, Yague E, Yang Q, Pan T, Zhao H, et al. GGNBP2 suppresses triple-negative breast cancer aggressiveness through inhibition of IL-6/STAT3 signaling activation. Breast Cancer Res Treat. 2019;174(1):65–78.
Li LX, Zhou JX, Calvet JP, Godwin AK, Jensen RA, Li X. Lysine methyltransferase SMYD2 promotes triple negative breast cancer progression. Cell Death Dis. 2018;9(3):326.
Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15(4):283–93.
Banerjee K, Pru C, Pru JK, Resat H. STAT3 knockdown induces tumor formation by MDA-MB-231 cells. Clin Oncol Res. 2018;1(1).
Kamran MZ, Patil P, Gude RP. Role of STAT3 in cancer metastasis and translational advances. Biomed Res Int. 2013;2013:421821.
Wang S, Yao Y, Yao M, Fu P, Wang W. Interleukin-22 promotes triple negative breast cancer cells migration and paclitaxel resistance through JAK-STAT3/MAPKs/AKT signaling pathways. Biochem Biophys Res Commun. 2018;503(3):1605–9.
Liang S, Chen Z, Jiang G, Zhou Y, Liu Q, Su Q, et al. Activation of GPER suppresses migration and angiogenesis of triple negative breast cancer via inhibition of NF-kappaB/IL-6 signals. Cancer Lett. 2017;386:12–23.
Rigiracciolo DC, Santolla MF, Lappano R, Vivacqua A, Cirillo F, Galli GR, et al. Focal adhesion kinase (FAK) activation by estrogens involves GPER in triple-negative breast cancer cells. J Exp Clin Cancer Res. 2019;38(1):58.
Lee E, Fertig EJ, Jin K, Sukumar S, Pandey NB, Popel AS. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat Commun. 2014;5:4715.
Wang C, Li J, Ye S, Zhang Y, Li P, Wang L, et al. Oestrogen inhibits VEGF expression and angiogenesis in triple-negative breast Cancer by activating GPER-1. J Cancer. 2018;9(20):3802–11.
Liu CY, Su JC, Huang TT, Chu PY, Huang CT, Wang WL, et al. Sorafenib analogue SC-60 induces apoptosis through the SHP-1/STAT3 pathway and enhances docetaxel cytotoxicity in triple-negative breast cancer cells. Mol Oncol. 2017;11(3):266–79.
Liu CY, Chen KF, Chao TI, Chu PY, Huang CT, Huang TT, et al. Sequential combination of docetaxel with a SHP-1 agonist enhanced suppression of p-STAT3 signaling and apoptosis in triple negative breast cancer cells. J Mol Med (Berl). 2017;95(9):965–75.
Fatehi D, Soltani A, Ghatrehsamani M. SRT1720, a potential sensitizer for radiotherapy and cytotoxicity effects of NVB-BEZ235 in metastatic breast cancer cells. Pathol Res Pract. 2018;214(6):889–95.
Tzeng YT, Liu PF, Li JY, Liu LF, Kuo SY, Hsieh CW, et al. Kinome-wide siRNA screening identifies Src-enhanced resistance of chemotherapeutic drugs in triple-negative breast Cancer cells. Front Pharmacol. 2018;9:1285.
Moreira MP, da Conceicao Braga L, Cassali GD, Silva LM. STAT3 as a promising chemoresistance biomarker associated with the CD44(+/high)/CD24(−/low)/ALDH(+) BCSCs-like subset of the triple-negative breast cancer (TNBC) cell line. Exp Cell Res. 2018;363(2):283–90.
Kuo WY, Hwu L, Wu CY, Lee JS, Chang CW, Liu RS. STAT3/NF-kappaB-regulated lentiviral TK/GCV suicide gene therapy for cisplatin-resistant triple-negative breast Cancer. Theranostics. 2017;7(3):647–63.
Egusquiaguirre SP, Yeh JE, Walker SR, Liu S, Frank DA. The STAT3 target gene TNFRSF1A modulates the NF-kappaB pathway in breast Cancer cells. Neoplasia. 2018;20(5):489–98.
Cheng CC, Shi LH, Wang XJ, Wang SX, Wan XQ, Liu SR, et al. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int J Oncol. 2018;53(1):339–48.
Niu J, Xue A, Chi Y, Xue J, Wang W, Zhao Z, et al. Induction of miRNA-181a by genotoxic treatments promotes chemotherapeutic resistance and metastasis in breast cancer. Oncogene. 2016;35(10):1302–13.
Soleymani Abyaneh H, Gupta N, Radziwon-Balicka A, Jurasz P, Seubert J, Lai R, et al. STAT3 but not HIF-1alpha is important in mediating hypoxia-induced Chemoresistance in MDA-MB-231, a triple negative breast Cancer cell line. Cancers (Basel). 2017;9(10):137.
Wang K, Zhu X, Zhang K, Yin Y, Chen Y, Zhang T. Interleukin-6 contributes to chemoresistance in MDA-MB-231 cells via targeting HIF-1alpha. J Biochem Mol Toxicol. 2018;32(3):e22039.
Cui Y, Huang Y, Wu X, Zheng M, Xia Y, Fu Z, et al. Hypoxia-induced tRNA-derived fragments, novel regulatory factor for doxorubicin resistance in triple-negative breast cancer. J Cell Physiol. 2019;234(6):8740–51.
Soleymani Abyaneh H, Soleimani AH, Vakili MR, Soudy R, Kaur K, Cuda F, et al. Modulation of hypoxia-induced Chemoresistance to polymeric micellar cisplatin: the effect of ligand modification of micellar carrier versus inhibition of the mediators of drug resistance. Pharmaceutics. 2018;10(4):196.
Kitamura H, Ohno Y, Toyoshima Y, Ohtake J, Homma S, Kawamura H, et al. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017;108(10):1947–52.
Sasidharan Nair V, Toor SM, Ali BR, Elkord E. Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD-L1 in human breast cancer cells. Expert Opin Ther Targets. 2018;22(6):547–57.
Chen M, Pockaj B, Andreozzi M, Barrett MT, Krishna S, Eaton S, et al. JAK2 and PD-L1 amplification enhance the dynamic expression of PD-L1 in triple-negative breast Cancer. Clin Breast Cancer. 2018;18(5):e1205–15.
Liu J, Yang Y, Wang H, Wang B, Zhao K, Jiang W, et al. Syntenin1/MDA-9 (SDCBP) induces immune evasion in triple-negative breast cancer by upregulating PD-L1. Breast Cancer Res Treat. 2018;171(2):345–57.
Galoczova M, Coates P, Vojtesek B. STAT3, stem cells, cancer stem cells and p63. Cell Mol Biol Lett. 2018;23:12.
Ibrahim SA, Hassan H, Vilardo L, Kumar SK, Kumar AV, Kelsch R, et al. Syndecan-1 (CD138) modulates triple-negative breast cancer stem cell properties via regulation of LRP-6 and IL-6-mediated STAT3 signaling. PLoS One. 2013;8(12):e85737.
Soleymani Abyaneh H, Gupta N, Alshareef A, Gopal K, Lavasanifar A, Lai R. Hypoxia induces the Acquisition of Cancer Stem-like Phenotype via Upregulation and Activation of signal transducer and activator of Transcription-3 (STAT3) in MDA-MB-231, a triple negative breast Cancer cell line. Cancer Microenviron. 2018;11(2–3):141–52.
Zhao D, Pan C, Sun J, Gilbert C, Drews-Elger K, Azzam DJ, et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene. 2015;34(24):3107–19.
Jarnicki A, Putoczki T, Ernst M. Stat3: linking inflammation to epithelial cancer - more than a “gut” feeling? Cell Div. 2010;5:14.
Nguyen TL, Grizzle WE, Zhang K, Hameed O, Siegal GP, Wei S. Syndecan-1 overexpression is associated with nonluminal subtypes and poor prognosis in advanced breast cancer. Am J Clin Pathol. 2013;140(4):468–74.
Ibrahim SA, Gadalla R, El-Ghonaimy EA, Samir O, Mohamed HT, Hassan H, et al. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, notch and EGFR signaling pathways. Mol Cancer. 2017;16(1):57.
Zheng Q, Dunlap SM, Zhu J, Downs-Kelly E, Rich J, Hursting SD, et al. Leptin deficiency suppresses MMTV-Wnt-1 mammary tumor growth in obese mice and abrogates tumor initiating cell survival. Endocr Relat Cancer. 2011;18(4):491–503.
Thiagarajan PS, Zheng Q, Bhagrath M, Mulkearns-Hubert EE, Myers MG, Lathia JD, et al. STAT3 activation by leptin receptor is essential for TNBC stem cell maintenance. Endocr Relat Cancer. 2017;24(8):415–26.
Liu Y, Choi DS, Sheng J, Ensor JE, Liang DH, Rodriguez-Aguayo C, et al. HN1L promotes triple-negative breast Cancer stem cells through LEPR-STAT3 pathway. Stem Cell Reports. 2018;10(1):212–27.
Maycotte P, Gearheart CM, Barnard R, Aryal S, Mulcahy Levy JM, Fosmire SP, et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014;74(9):2579–90.
Huynh J, Etemadi N, Hollande F, Ernst M, Buchert M. The JAK/STAT3 axis: a comprehensive drug target for solid malignancies. Semin Cancer Biol. 2017;45:13–22.
Vyas D, Lopez-Hisijos N, Shah P, Deshpande KS, Basson MD, Vyas A, et al. A second-generation proteasome inhibitor and doxorubicin modulates IL-6, pSTAT-3 and NF-kB activity in MDA-MB-231 breast Cancer cells. J Nanosci Nanotechnol. 2017;17(1):175–85.
Aryappalli P, Al-Qubaisi SS, Attoub S, George JA, Arafat K, Ramadi KB, et al. The IL-6/STAT3 signaling pathway is an early target of Manuka honey-induced suppression of human breast Cancer cells. Front Oncol. 2017;7:167.
Tian J, Chen X, Fu S, Zhang R, Pan L, Cao Y, et al. Bazedoxifene is a novel IL-6/GP130 inhibitor for treating triple-negative breast cancer. Breast Cancer Res Treat. 2019.
Fu S, Chen X, Lo HW, Lin J. Combined bazedoxifene and paclitaxel treatments inhibit cell viability, cell migration, colony formation, and tumor growth and induce apoptosis in breast cancer. Cancer Lett. 2019;448:11–9.
Rios-Fuller TJ, Ortiz-Soto G, Lacourt-Ventura M, Maldonado-Martinez G, Cubano LA, Schneider RJ, et al. Ganoderma lucidum extract (GLE) impairs breast cancer stem cells by targeting the STAT3 pathway. Oncotarget. 2018;9(89):35907–21.
Mehta R, Katta H, Alimirah F, Patel R, Murillo G, Peng X, et al. Deguelin action involves c-met and EGFR signaling pathways in triple negative breast cancer cells. PLoS One. 2013;8(6):e65113.
Yamashita N, Kondo M, Zhao S, Li W, Koike K, Nemoto K, et al. Picrasidine G decreases viability of MDA-MB 468 EGFR-overexpressing triple-negative breast cancer cells through inhibition of EGFR/STAT3 signaling pathway. Bioorg Med Chem Lett. 2017;27(11):2608–12.
Chun J, Park MK, Ko H, Lee K, Kim YS. Bioassay-guided isolation of cantharidin from blister beetles and its anticancer activity through inhibition of epidermal growth factor receptor-mediated STAT3 and Akt pathways. J Nat Med. 2018;72(4):937–45.
Kim S, Jeon M, Lee J, Han J, Oh SJ, Jung T, et al. Induction of fibronectin in response to epidermal growth factor is suppressed by silibinin through the inhibition of STAT3 in triple negative breast cancer cells. Oncol Rep. 2014;32(5):2230–6.
Jiang F, Li Y, Si L, Zhang Z, Li Z. Interaction of EZH2 and P65 is involved in the arsenic trioxide-induced anti-angiogenesis in human triple-negative breast cancer cells. Cell Biol Toxicol. 2019.
Byun HJ, Darvin P, Kang DY, Sp N, Joung YH, Park JH, et al. Silibinin downregulates MMP2 expression via Jak2/STAT3 pathway and inhibits the migration and invasive potential in MDA-MB-231 cells. Oncol Rep. 2017;37(6):3270–8.
Yang Y, Zhou H, Liu W, Wu J, Yue X, Wang J, et al. Ganoderic acid a exerts antitumor activity against MDA-MB-231 human breast cancer cells by inhibiting the Janus kinase 2/signal transducer and activator of transcription 3 signaling pathway. Oncol Lett. 2018;16(5):6515–21.
Liu CY, Huang TT, Chu PY, Huang CT, Lee CH, Wang WL, et al. The tyrosine kinase inhibitor nintedanib activates SHP-1 and induces apoptosis in triple-negative breast cancer cells. Exp Mol Med. 2017;49(8):e366.
Su JC, Mar AC, Wu SH, Tai WT, Chu PY, Wu CY, et al. Disrupting VEGF-A paracrine and autocrine loops by targeting SHP-1 suppresses triple negative breast cancer metastasis. Sci Rep. 2016;6:28888.
Lee HJ, Seo NJ, Jeong SJ, Park Y, Jung DB, Koh W, et al. Oral administration of penta-O-galloyl-beta-D-glucose suppresses triple-negative breast cancer xenograft growth and metastasis in strong association with JAK1-STAT3 inhibition. Carcinogenesis. 2011;32(6):804–11.
Liu CY, Su JC, Ni MH, Tseng LM, Chu PY, Wang DS, et al. Obatoclax analog SC-2001 inhibits STAT3 phosphorylation through enhancing SHP-1 expression and induces apoptosis in human breast cancer cells. Breast Cancer Res Treat. 2014;146(1):71–84.
Yen MC, Shih YC, Hsu YL, Lin ES, Lin YS, Tsai EM, et al. Isolinderalactone enhances the inhibition of SOCS3 on STAT3 activity by decreasing miR-30c in breast cancer. Oncol Rep. 2016;35(3):1356–64.
Kim HS, Hoang VH, Hong M, Chul Kim K, Ann J, Nguyen CT, et al. Investigation of B,C-ring truncated deguelin derivatives as heat shock protein 90 (HSP90) inhibitors for use as anti-breast cancer agents. Bioorg Med Chem. 2019;27(7):1370–81.
Cho TM, Kim JY, Kim YJ, Sung D, Oh E, Jang S, et al. C-terminal HSP90 inhibitor L80 elicits anti-metastatic effects in triple-negative breast cancer via STAT3 inhibition. Cancer Lett. 2019;447:141–53.
Abd El-Hafeez AA, Khalifa HO, Mahdy EAM, Sharma V, Hosoi T, Ghosh P, et al. Anticancer effect of nor-wogonin (5, 7, 8-trihydroxyflavone) on human triple-negative breast cancer cells via downregulation of TAK1, NF-kappaB, and STAT3. Pharmacol Rep. 2019;71(2):289–98.
Tegowski M, Fan C, Baldwin AS. Thioridazine inhibits self-renewal in breast cancer cells via DRD2-dependent STAT3 inhibition, but induces a G1 arrest independent of DRD2. J Biol Chem. 2018;293(41):15977–90.
Chen Y, Ji M, Zhang S, Xue N, Xu H, Lin S, et al. Bt354 as a new STAT3 signaling pathway inhibitor against triple negative breast cancer. J Drug Target. 2018;26(10):920–30.
Dai X, Yin C, Zhang Y, Guo G, Zhao C, Wang O, et al. Osthole inhibits triple negative breast cancer cells by suppressing STAT3. J Exp Clin Cancer Res. 2018;37(1):322.
Feng T, Cao W, Shen W, Zhang L, Gu X, Guo Y, et al. Arctigenin inhibits STAT3 and exhibits anticancer potential in human triple-negative breast cancer therapy. Oncotarget. 2017;8(1):329–44.
Chun J, Li RJ, Cheng MS, Kim YS. Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA-MB-231 cells. Cancer Lett. 2015;357(1):393–403.
Shin DS, Kim HN, Shin KD, Yoon YJ, Kim SJ, Han DC, et al. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer Res. 2009;69(1):193–202.
Zhang W, Yu W, Cai G, Zhu J, Zhang C, Li S, et al. A new synthetic derivative of cryptotanshinone KYZ3 as STAT3 inhibitor for triple-negative breast cancer therapy. Cell Death Dis. 2018;9(11):1098.
Chun J, Song K, Kim YS. Sesquiterpene lactones-enriched fraction of Inula helenium L. induces apoptosis through inhibition of signal transducers and activators of transcription 3 signaling pathway in MDA-MB-231 breast cancer cells. Phytother Res. 2018;32(12):2501–9.
El Hasasna H, Saleh A, Al Samri H, Athamneh K, Attoub S, Arafat K, et al. Rhus coriaria suppresses angiogenesis, metastasis and tumor growth of breast cancer through inhibition of STAT3, NFkappaB and nitric oxide pathways. Sci Rep. 2016;6:21144.
Dai X, Yin C, Guo G, Zhang Y, Zhao C, Qian J, et al. Schisandrin B exhibits potent anticancer activity in triple negative breast cancer by inhibiting STAT3. Toxicol Appl Pharmacol. 2018;358:110–9.
Yang B, Shen JW, Zhou DH, Zhao YP, Wang WQ, Zhu Y, et al. Precise discovery of a STAT3 inhibitor from Eupatorium lindleyanum and evaluation of its activity of anti-triple-negative breast cancer. Nat Prod Res. 2019;33:477-85.
Kim HS, Kim T, Ko H, Lee J, Kim YS, Suh YG. Identification of galiellalactone-based novel STAT3-selective inhibitors with cytotoxic activities against triple-negative breast cancer cell lines. Bioorg Med Chem. 2017;25(19):5032–40.
Li W, Zhang H, Nie M, Tian Y, Chen X, Chen C, et al. Ursolic acid derivative FZU-03,010 inhibits STAT3 and induces cell cycle arrest and apoptosis in renal and breast cancer cells. Acta Biochim Biophys Sin Shanghai. 2017;49(4):367–73.
Lu L, Dong J, Wang L, Xia Q, Zhang D, Kim H, et al. Activation of STAT3 and Bcl-2 and reduction of reactive oxygen species (ROS) promote radioresistance in breast cancer and overcome of radioresistance with niclosamide. Oncogene. 2018;37(39):5292–304.
Oh E, Kim YJ, An H, Sung D, Cho TM, Farrand L, et al. Flubendazole elicits anti-metastatic effects in triple-negative breast cancer via STAT3 inhibition. Int J Cancer. 2018;143(8):1978–93.
Kim YJ, Kim JY, Lee N, Oh E, Sung D, Cho TM, et al. Disulfiram suppresses cancer stem-like properties and STAT3 signaling in triple-negative breast cancer cells. Biochem Biophys Res Commun. 2017;486(4):1069–76.
An H, Kim JY, Oh E, Lee N, Cho Y, Seo JH. Salinomycin promotes Anoikis and decreases the CD44+/CD24- stem-like population via inhibition of STAT3 activation in MDA-MB-231 cells. PLoS One. 2015;10(11):e0141919.
Deng XS, Wang S, Deng A, Liu B, Edgerton SM, Lind SE, et al. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012;11(2):367–76.
Hou S, Yi YW, Kang HJ, Zhang L, Kim HJ, Kong Y, et al. Novel carbazole inhibits phospho-STAT3 through induction of protein-tyrosine phosphatase PTPN6. J Med Chem. 2014;57(15):6342–53.
Kang HJ, Yi YW, Hou SJ, Kim HJ, Kong Y, Bae I, et al. Disruption of STAT3-DNMT1 interaction by SH-I-14 induces re-expression of tumor suppressor genes and inhibits growth of triple-negative breast tumor. Oncotarget. 2017;8(48):83457–68.
Lim EJ, Hong DY, Park JH, Joung YH, Darvin P, Kim SY, et al. Methylsulfonylmethane suppresses breast cancer growth by down-regulating STAT3 and STAT5b pathways. PLoS One. 2012;7(4):e33361.
Chen W, Wang H, Cheng M, Ni L, Zou L, Yang Q, et al. Isoharringtonine inhibits breast cancer stem-like properties and STAT3 signaling. Biomed Pharmacother. 2018;103:435–42.
Kang DY, Sp N, Kim DH, Joung YH, Lee HG, Park YM, et al. Salidroside inhibits migration, invasion and angiogenesis of MDAMB 231 TNBC cells by regulating EGFR/Jak2/STAT3 signaling via MMP2. Int J Oncol. 2018;53(2):877–85.
Njatcha C, Farooqui M, Kornberg A, Johnson DE, Grandis JR, Siegfried JM. STAT3 cyclic Decoy demonstrates robust antitumor effects in non-small cell lung Cancer. Mol Cancer Ther. 2018;17(9):1917–26.
Reilley MJ, McCoon P, Cook C, Lyne P, Kurzrock R, Kim Y, et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial. J Immunother Cancer. 2018;6(1):119.
Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015;7(314):314ra185.
Jung KH, Yoo W, Stevenson HL, Deshpande D, Shen H, Gagea M, et al. Multifunctional effects of a small-molecule STAT3 inhibitor on NASH and hepatocellular carcinoma in mice. Clin Cancer Res. 2017;23(18):5537–46.
Wong AL, Soo RA, Tan DS, Lee SC, Lim JS, Marban PC, et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann Oncol. 2015;26(5):998–1005.
Jonker DJ, Nott L, Yoshino T, Gill S, Shapiro J, Ohtsu A, et al. Napabucasin versus placebo in refractory advanced colorectal cancer: a randomised phase 3 trial. Lancet Gastroenterol Hepatol. 2018;3(4):263–70.
Oh DY, Lee SH, Han SW, Kim MJ, Kim TM, Kim TY, et al. Phase I study of OPB-31121, an Oral STAT3 inhibitor, in patients with advanced solid tumors. Cancer Res Treat. 2015;47(4):607–15.
Yoo C, Kang J, Lim HY, Kim JH, Lee MA, Lee KH, et al. Phase I dose-finding study of OPB-111077, a novel STAT3 inhibitor, in patients with advanced hepatocellular carcinoma. Cancer Res Treat. 2019;51(2):510–8.
Khan MW, Saadalla A, Ewida AH, Al-Katranji K, Al-Saoudi G, Giaccone ZT, et al. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol Immunother. 2018;67(1):13–23.
Wang ST, Ho HJ, Lin JT, Shieh JJ, Wu CY. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017;8(2):e2626.
A Study of DSP-0337 in Patients With Advanced Solid Tumors to Determine the Safety and the Pharmacokinetic Profile. https://clinicaltrials.gov/ct2/show/NCT03416816. Accessed 26 Apr 2019.
Venook AP, Niedzwiecki D, Lenz HJ, Innocenti F, Fruth B, Meyerhardt JA, et al. Effect of first-line chemotherapy combined with Cetuximab or bevacizumab on overall survival in patients with KRAS wild-type advanced or metastatic colorectal Cancer: a randomized clinical trial. JAMA. 2017;317(23):2392–401.
Medina PJ, Goodin S. Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther. 2008;30(8):1426–47.
Song L, Morris M, Bagui T, Lee FY, Jove R, Haura EB. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res. 2006;66(11):5542–8.
Chung SY, Chen YH, Lin PR, Chao TC, Su JC, Shiau CW, et al. Two novel SHP-1 agonists, SC-43 and SC-78, are more potent than regorafenib in suppressing the in vitro stemness of human colorectal cancer cells. Cell Death Discov. 2018;4:25.
Bissonnette R, Maari C, Forman S, Bhatia N, Lee M, Fowler J, et al. The Oral JAK/SYK inhibitor ASN002 demonstrates efficacy and improves associated systemic inflammation in patients with moderate-to-severe atopic dermatitis: results from a randomised, double-blind, placebo-controlled study. Br J Dermatol. 2019.
Zhang M, Xu CR, Shamiyeh E, Liu F, Yin JY, von Moltke LL, et al. A randomized, placebo-controlled study of the pharmacokinetics, pharmacodynamics, and tolerability of the oral JAK2 inhibitor fedratinib (SAR302503) in healthy volunteers. J Clin Pharmacol. 2014;54(4):415–21.
Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16(6):487–97.
Verstovsek S, Manshouri T, Quintas-Cardama A, Harris D, Cortes J, Giles FJ, et al. WP1066, a novel JAK2 inhibitor, suppresses proliferation and induces apoptosis in erythroid human cells carrying the JAK2 V617F mutation. Clin Cancer Res. 2008;14(3):788–96.
Acknowledgements
We thank the current and former members of our laboratories and collaborators for their contributions to the publications cited in this review article. The research field in STAT3 is rapidly growing, and we apologize for not being able to cite all the recent publications, due to space limitation.
Funding
JJQ was supported by Zhejiang Chinese Medical University Startup Funding (111100E014). WDZ was supported by Professor of Chang Jiang Scholars Program, NSFC (81520108030, 21472238), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (16DZ2280200), the Scientific Foundation of Shanghai China (13401900103, 13401900101), and the National Key Research and Development Program of China (2017YFC1700200).
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
JJQ and WDZ conceptualized the manuscript. JJQ, LY, and JZ collected the literature, wrote the manuscript and made the figures. JJQ and WDZ edited and made significant revisions to the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Qin, JJ., Yan, L., Zhang, J. et al. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res 38, 195 (2019). https://doi.org/10.1186/s13046-019-1206-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-019-1206-z