
Abstract
The notion that the central nervous system is an immunologically immune-exempt organ has changed over the past two decades, with increasing evidence of strong links and interactions between the central nervous system and the peripheral immune system, both in the healthy state and after ischemic and hemorrhagic stroke. Although primary injury after stroke is certainly important, the limited therapeutic efficacy, poor neurological prognosis and high mortality have led researchers to realize that secondary injury and damage may also play important roles in influencing long-term neurological prognosis and mortality and that the neuroinflammatory process in secondary injury is one of the most important influences on disease progression. Here, we summarize the interactions of the central nervous system with the peripheral immune system after ischemic and hemorrhagic stroke, in particular, how the central nervous system activates and recruits peripheral immune components, and we review recent advances in corresponding therapeutic approaches and clinical studies, emphasizing the importance of the role of the peripheral immune system in ischemic and hemorrhagic stroke.
Similar content being viewed by others

Introduction
Because of the tolerance of the central nervous system (CNS) to antigen-induced immune responses, coupled with the presence of specific physical barriers, such as the blood‒brain barrier (BBB) and the blood‒cerebrospinal fluid barrier (BCSFB), which isolate the CNS from the peripheral immune system, the CNS has always been regarded as an immune-privileged system, and the presence of a peripheral immune component in the CNS in a healthy state has been considered a manifestation of pathology. However, over the past 20 years, this perception has changed more objectively [1]. Increasing evidence suggests that the CNS and the peripheral immune system are not two completely isolated systems but rather represent a complex network of interactions [2, 3]. Indeed, the peripheral immune system maintains surveillance of the CNS, and it not only recognizes external pathogens but also plays an important role in aseptic CNS injury, such as ischemic/hemorrhagic stroke and traumatic brain injury (TBI) [4,5,6]. In the past, researchers have focused on exploring the pathophysiological mechanisms underlying these primary sterile CNS injury lesions, such as the sudden cessation of blood supply to vascular regions in ischemic stroke leading to the death of nerve cells to form ischemic cores, the sudden overflow of vascular blood components in cerebral hemorrhage, and, in more complex subarachnoid hemorrhage (SAH), hypoxia following hemorrhage [7, 8]. However, while targeted treatment partially controls further injury, the poor long-term prognosis and lower survival rates have led a growing number of investigators realizing that secondary injury following primary injury may play an important role in long-term prognosis [9].
Thus, the role of the peripheral immune system in secondary damage following CNS injury has important implications for the long-term prognosis of the injury. Indeed, a more accurate description of the whole process is that the CNS already activates and recruits components of the peripheral immune system after the onset of injury through a variety of pathways, including the autonomic nervous system, the neuroendocrine system, and the meningeal lymphatics; these components enter the CNS through a variety of modalities, such as the BBB and the BCSFB; and these components exert both deleterious and beneficial effects through interactions with the intrinsic cells of the CNS. Therefore, targeted monitoring and intervention of the immune system not only are important for the prediction of disease progression but also hold promise for improving long-term neurological prognosis and reducing mortality.
Brain–peripheral crosstalk in inflammatory infiltration after stroke
As a central regulator of organisms, the brain is responsible for maintaining the homeostasis of organisms, while the advantages and significance of CNS regulation of the peripheral immune system are mainly reflected in the following aspects. First, the brain constantly monitors the internal and external environment and integrates this information to regulate the immune system in synchronization with the cardiovascular system and digestive system to play a beneficial role under physiological and pathological conditions. Second, an important role of the brain is to predict threats and coordinate the body’s response after damage occurs, which requires the brain to play a strong regulatory role in numerous systems that protect and repair the body, including the immune system. Finally, a significant advantage of the nervous system over the immune system is its speed of response, even within milliseconds of injury. In response to injury, the CNS responds quickly and powerfully to modulate immune responses in an effective manner [2]. We next explored the interaction between the CNS and the peripheral immune system after injury.
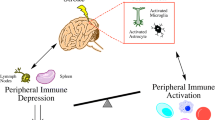
Peripheral immune responses induced by stroke (Fig. 1)

Schematic diagram of peripheral immune responses induced by hemorrhagic stroke
Autonomic nervous system (ANS)
The most direct influence is that of the neural signaling output network, in which the sympathetic nervous system (SNS) network and the parasympathetic nervous system (PSNS) network play major roles [10]. The most obvious role of sympathetic nerves is to help the body produce a stress response, and these nerves are the main source of epinephrine and norepinephrine after CNS injury. Most importantly, immune cells express both α-adrenergic and β-adrenergic receptors, and the level of receptor expression correlates with cellular status and whether the cells are mature or activated [11, 12]. Through the activation of these receptors, the CNS can influence immune cell migration and cytokine production [13,14,15], but these effects are likely to be double-edged swords because recent research has shown contradictions in terms of whether this effect is beneficial [16]. For example, β-adrenergic receptor activation can play a role in inducing CD4+ T-cell proliferation and cytokine production [17, 18] while inhibiting macrophage responses to lipopolysaccharide (LPS) [19]. In addition, it has been demonstrated that the norepinephrine produced by sympathetic nerves in the spleen can partially activate memory T cells and that the acetylcholine produced by activated T cells can in turn inhibit cytokine production in macrophages [3].
An important component of the parasympathetic nervous system is the vagus nerve. The afferent arc of the vagus nerve senses the stimulation of inflammatory cells and secretes ACH in the efferent arc in the periphery, thereby suppressing the inflammatory response [20]. In autoimmune diseases, it has also been shown that suppressing the immune response by stimulating the vagus nerve can be therapeutic in rheumatoid arthritis [21]. Specifically, macrophages are the first peripheral immune cells shown to be affected by the vagus nerve, which inhibits the release of TNF-α from macrophages [22,23,24]. In addition, the vagus nerve reduces the level of CD11b on the surface of neutrophils, thus attenuating tissue damage during inflammation [25]. However, the interactions between the nervous system and the immune system are extremely complex. The most direct evidence is that immune cells themselves can secrete different neuronal factors [26] and that many functions of the vagus nerve are mediated through sympathetic nerves [27]. To further complicate matters, sympathetic influences on peripheral inflammation can even differ on different sides of the body [28], and in arthritic patients with stroke, the antigen-specific response of T cells is significantly stronger in the limb on the side affected by the stroke [29], suggesting that there may be differences in the regulation of the peripheral immune system in each of the different cerebral hemispheres [30,31,32,33].
Neuroendocrine system
Cutting-edge research has confirmed that the neuroendocrine system is also an important way in which the nervous system achieves central-to-peripheral regulation. The neuroendocrine system consists of the hypothalamic neuropituitary system, which primarily secretes oxytocin and arginine vasopressin (AVP), and the hypothalamic-pituitary portal system, which secretes adrenocorticotropic hormone (ACTH), thyroid stimulating hormone (TSH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), and growth hormone (GH) in an endocrine manner [34,35,36]. Oxytocin and AVP in the hypothalamic-pituitary system have been shown to correlate with immunoreactivity after injury; specifically, oxytocin inhibits proinflammatory cytokines [37, 38], and the anti-inflammatory effects of AVP are mediated through the activation of AVP-producing neurons by inflammatory factors [39, 40]. In the hypothalamic-pituitary portal system, sex steroids are involved in the programming of the immune system [41], and testosterone is immunosuppressive, whereas estrogen is, in contrast, immunopotentiating [42]. The hypothalamic-pituitary-thyroid axis has also been shown to induce lymphocyte proliferation and to be involved in immune activation [43, 44], which explains the diminished immune response after thyroidectomy [45]. However, the specific mechanisms and factors influencing these neuroendocrine effects need to be further investigated.
Meningeal lymphatic vessels
Recent findings have shown the presence of a traditional functional lymphatic system in the meninges that allows the drainage of fluid, macromolecules and immune cells from the CNS into the deep cervical lymph nodes [46,47,48]. Anatomically, lymphatic vessels composed of lymphatic endothelial cells line up along the dural sinuses and leave the CNS from the base of the skull, and more importantly, these cells express traditional lymphatic vessel cell markers, such as Prox1 and CD31 [1, 49]. Under normal conditions, immune cells (T cells, B cells and dendritic cells) are present in the meningeal lymphatics, suggesting that in the steady state, the meningeal lymphatics are involved in the transport of immune cells from the CNS meninges and cerebrospinal fluid [46]. In disease states, the brain can regulate the peripheral immune system by introducing brain tissue in situ products and immune cells, such as brain-specific antigens, into the peripheral immune system through meningeal lymphatics [50]. It has been demonstrated in a mouse model of multiple sclerosis that suppression of the T-cell inflammatory response and attenuation of CNS damage can be achieved through ablation of meningeal lymphatics [51]. It has also been demonstrated in Alzheimer’s disease that meningeal lymphatics can promote amyloid-B clearance [52].
For brain-specific antigens, immunohistochemistry revealed that neuron-derived antigens, such as microtubule-associated protein 2 (MAP2) and myelin basic protein (MBP), were found in the deep cervical lymph nodes of acute stroke patients [53]. These antigens can be presented by antigen-presenting cells (APCs) to the T-cell compartment of the lymph node and induce an autoreactive T-cell response [53, 54]. In addition, noninfectious immune responses against injured tissues are initiated after CNS injury; these patterns, known as danger-associated molecular patterns (DAMPs) [3], are a group of molecular determinants derived from cellular debris, intracellular proteins/enzymes, or nuclear DNA/RNA that are released from the cell after CNS injury [55]. Proteins with a greater impact on the peripheral immune system include high-mobility group box 1 (HMGB1), S100 and ATP [56]. HMGB1 is a nuclear protein that binds to DNA and regulates gene transcription [57]; it can be produced by damaged neurons after injury and survives activation of the immune system by binding to RAGE on microglia/macrophages, linking nerve injury to microglia/macrophages [58, 59]. Like HMGB1, S100B binds to RAGE on microglia/macrophages and stimulates the upregulation of nitric oxide (NO), IL-1β, NF-κB, tumor necrosis factor (TNF)-a, several chemokines (CCL3, CCL5 and CXCL12) and chemokine receptors (CCR1 and CCR5) [60,61,62]. The source and role of ATP are relatively widespread, as ATP can be produced by damaged vascular cells and blood cells in addition to neuronal cells [63], and ATP has been demonstrated to lead to worsened outcomes in numerous models of disease (ischemic brain injury, spinal cord injury, and autoimmune diseases, among others) [56]. For example, ATP can act on purinergic receptors such as P2 × 7 or P2 × 4, which can activate microglia and astrocytes, in turn stimulating the activation of inflammatory cells and the release of a range of inflammatory substances, including inflammasome vesicles [64, 65].
Stroke-induced immune depression syndrome (SIDS)
An important phenomenon after stroke is the systemic immunosuppressive effect, which usually manifests as splenic atrophy accompanied by increased apoptosis or cellular dysfunction [66], along with the suppression of numerous inflammatory factors, including IL10, IL-1b, TNF-α, and IL-6. This state of immunosuppression has been referred to as SIDS [67]. Although this may be intended to attenuate the inflammatory response in the CNS, it has become an important cause of poststroke infections [68, 69]. The benefits of immunosuppression have been demonstrated in numerous studies; for example, the suppression of the immune response by splenectomy before middle cerebral artery occlusion (MCAO) or neonatal hypoxia-ischemia reduces cerebral infarction [70] and is accompanied by a significant reduction in the number of infiltrating neutrophils and activated microglia in the injured brain [71]. Poststroke splenic irradiation has also been shown to significantly reduce cerebral infarction in rats [72]. The mechanisms of injury-induced immunosuppression are largely unknown, and cutting-edge literature suggests that this suppressive effect is likely related in several ways, as described previously [73,74,75]. It has been shown that sympathetic nerves suppress the inflammatory response by inhibiting natural killer T-cell activity through the action of norepinephrine in the liver [76]. In addition, studies in the SAH model have shown that the hypothalamic‒pituitary‒adrenal axis after hemorrhage can lead to splenic contraction [77], which is a very important feature of immunosuppression [78]. Specifically, SNS-induced splenic crumpling may be achieved through the activation of a1 adrenergic receptors on smooth muscle cells [79]. Further studies have shown that cerebral hemorrhage leads to a significant reduction in splenic-derived leukocytes and lymphocytes in the peripheral blood [80], and the greater the hemorrhage is, the more severe the splenic constriction, the greater the risk of late infection, and the worse the prognosis [81, 82]. In addition to its immunosuppressive effects, CNS injury affects many systems and organs. For example, in the brain-gut axis, the neuroinflammatory response during stroke can lead to dysbiosis of the gut flora [83], reduced motility and increased permeability [84, 85]. In turn, changes in gut microbiology can affect the inflammatory response in the CNS, e.g., leading to altered T-cell homeostasis, thereby exacerbating the inflammatory response and poor prognosis [86]. Studies in the renal field have shown that hemorrhagic stroke-mediated production of inflammatory mediators is a central mechanism contributing to renal insufficiency [87], but concomitant activation of cholinergic anti-inflammatory pathways in splenocytes can achieve renal protection [88]. In addition, activation of the hypothalamic‒pituitary‒adrenal (HPA) axis, sympathetic and parasympathetic neuromodulation, and dysregulation of the intestinal microbiota after hemorrhagic stroke may also contribute to cardiac damage, such as heart attack, congestive heart failure, cardiac arrest, and atrial fibrillation [89,90,91].
Pathway by which peripheral immune cells enter the central nervous system (Fig. 2)

Pathways through which peripheral immune cells enter the central nervous system
Crossing the blood‒brain barrier
The most common route to cross the BBB is through small postcapillary veins, which has been confirmed by many studies. This process involves multiple steps: (1) After CNS injury, many peripheral immune cells are recruited to the cerebral vasculature due to the various effects described previously. (2) Depending on the action of P-, E-, and L-selectins, leukocytes initiate contact with the endothelium, reduce their velocity, and roll on the side of endothelial cells. (3) This reduction in velocity allows immune cells to pass through G protein-coupled receptors (GPCRs) and to recognize endothelial cell chemokines, which in turn activate adhesion molecules of the integrin family, resulting in the cell being firmly anchored to the luminal surface of the endothelial cell. (4) Finally, immune cells cross the BBB (which, strictly speaking, also requires the crossing of glial cell boundaries or glial limitans (GLs), the contingency barriers formed by astrocytes after injury) into the brain parenchyma through sites such as endothelial cell junctions [92,93,94,95].
Specifically, the BBB becomes permeable 3 min after injury [96], and even on the fourth day after CNS injury, the aggregation of neutrophils in the vasculature and the release of inflammatory factors are observed [97]; these changes are due to global vascular inflammation and are present in numerous models of stroke, including cerebral hemorrhage [98,99,100]. Vascular endothelial cells are rapidly activated after injury and upregulate numerous cytokines, including vascular cell adhesion molecule (VCAM-1), which promotes peripheral inflammatory cell adhesion and migration [101, 102]. Using molecular imaging techniques, researchers have found that this endothelial cell activation persists until at least 5 days postinjury in an ischemic infarction model, and this activation is global, as in acute ischemic cerebral infarction, as confirmed by positron emission computed tomography imaging of MMP9, which promotes BBB injury [103]. A similar effect has been demonstrated in the SAH model, where MMP9 released by neutrophils in the subarachnoid space damages the BBB [104], which leads to more peripheral immune cells infiltrating the brain parenchyma, which in turn leads to the production of more MMP9 and thus a vicious cycle [105, 106]. Numerous studies have demonstrated that attenuating the adhesion of neutrophils to endothelial cells via cell adhesion molecules (CAMs), such as VLA4 (a protein closely related to cell adhesion and migration), can significantly ameliorate the activation of microglia and neuronal damage in the brain parenchyma [97, 107]. In addition, IL-6 produced by neutrophils can exacerbate the disruption of tight junctions between endothelial cells in cerebral blood vessels, leading to an increase in BBB permeability and correlating with vasospasm [108, 109], suggesting that there is an outwardly activated immune mechanism from outside-in at the vascular/cerebral interface after SAH [110].
Core points of peripheral-central interactions in the choroid plexus
In addition to crossing the classic pathway of the BBB, there is a second important pathway through which peripheral immune cells enter the brain parenchyma. The choroidal plexus serves as a communication hub between the central nervous system and periphery, and under physiological conditions, there is a large circulation of immune cells in the cerebrospinal fluid (CSF), mainly in the choroid plexus, that generates CSF, which includes neutrophils, monocytes, T cells, B cells, and dendritic cells; this concept is supported by the recent discovery of meningeal lymphatic vessels in the CNS [111, 112]. This finding suggested that after primary CNS injury, the peripheral immune system contributes to secondary CNS injury by interacting with the CNS [113]. Indeed, after the onset of injury, immune cells cross the choroid plexus epithelium into the ventricles and then pass through the ventricular cell layer into the periventricular region [114]. The barrier of the choroid plexus epithelium is also often referred to as the BCSFB because of the tight network of connections that exist in the choroid plexus epithelium, thereby limiting the entry of cells and molecules from the stroma into the cerebrospinal fluid [115]. It has been demonstrated in experimental autoimmune cerebrospinal (EAE) inflammation models that early inflammatory cells originate from the choroidal stroma [116]. In addition, a marked increase in the number of macrophages, granulocytes and CD8+ T cells in the choroid plexus was found in multiple sclerosis patients [117, 118], and further studies have shown that this change is most likely due to the absence of the key tight junction protein claudin-3 [119]. In chronic diseases such as amyotrophic lateral sclerosis, Alzheimer’s disease and Parkinson’s disease [120], prolonged exposure to inflammatory factors such as reactive oxygen species (ROS) and dysregulated signaling between barrier cell types leads to increased BCSFB permeability, permitting the invasion of other peripheral immune cells, such as monocytes, into the CNS [121, 122]. Thus, the choroid plexus is a hub for peripheral-central nervous system interactions and plays an important role both in the surveillance and storage of inflammatory cells in the healthy state and in proinflammatory and anti-inflammatory effects after CNS injury.
Direct migration of skull bone marrow cells
Recent cutting-edge studies labeling local cells with spectrally resolved membrane dyes in models of ischemia and chemically induced acute brain inflammation have revealed the occurrence of a more rapid mode of neutrophil entry into the brain parenchyma: medullary-like cells (especially those of cranial bone marrow origin) migrate toward the inflamed brain through microscopic channels through the cranial endothelium, thereby directly connecting the cranial bone marrow cavity to the dura mater [123], and the relaxation of the extracellular matrix by proteases released by activated mast cells and intradural macrophages may also help in this process [124,125,126]. Anatomically, most of the blood vessels within the skull are located at the parietal bone, and the average diameter of these vessels is approximately 1.5 mm, which can be observed by corrosion casting of the human body. Corrosion casting shows that these blood vessels have many branches toward the bone marrow cavity [127, 128] and connecting to the meningeal vascular system [129], which is precisely the site of the early inflammatory immune response in many chronic diseases, such as Alzheimer’s disease [130]. Following CNS injury, these vessels can act as bidirectional conduits, and injury signals may preferentially reach the cranial bone marrow and directly cause local alterations in the proliferation and differentiation of hematopoietic stem cells, which further results in bone marrow-derived leukocytes being stronger than other sources of leukocytes in terms of both their inflammatory properties and their interactions with other cells [131,132,133]. There is also recent evidence that immune cells, such as neutrophils, can enter the subarachnoid space through head microvessels or meningeal vessels near the site of brain injury [134]; these cells in turn enter the brain parenchyma through GLs, although recent studies have been conducted mainly on T cells and disease models [135, 136]. Notably, recent studies have shown that cerebrospinal fluid can enter deep cervical lymph nodes not only from meningeal lymphatics but also from the nasopharyngeal lymphatic plexus through sieve lymphatics and that lymphatic endothelial cells show a variety of aberrant features with age, with exponential increases in the number of phosphorylated tau and apoptotic cells and a marked decrease in the amount of cerebrospinal fluid discharged. In conclusion, in-depth research is needed to determine how to target peripheral immune cells to support a more rapid, direct, and effective pathway to enter and thus influence the CNS.
Effect of aging on brain–peripheral crosstalk
Stroke is largely a disease of aging. In fact, the incidence of stroke doubles every decade after age 55 or older, and 80% of strokes occur in people over 65 years of age [137, 138]. Moreover, stroke disease in the older age group (which often represents a greater biological age) also exhibits more severe damage, slower recovery, and higher mortality rates [139]. Studies have shown that aging leads to significant and complex changes in the immune system [140] and that low-grade chronic inflammation (inflammation) associated with aging increases the risk of stroke [141]. This alteration of the immune system plays a key role in secondary brain damage after stroke and leads to a poorer prognosis in older patients [142].
Aging has been shown to affect the interaction between the central nervous system and the peripheral immune system after stroke. Aging is a state of irreversible replication arrest and leads to changes in gene expression and phenotype [143]. Researchers have proposed an important concept called the “senescence-associated secretory phenotype”, whereby aging brain cells release more proinflammatory signals, such as IL, IL-1α, IL-1β, IL-6, and IL-8, which amplify the inflammatory response after stroke in elderly patients [144]. In addition, the intrinsic immune cells in the CNS are also affected by senescence, thereby affecting the immune microenvironment. For example, microglia are in dynamic equilibrium under normal conditions, where activation and inhibitory signals regulate each other, but with increasing age, the interaction of inhibitory signals, such as CD200, CXCL1, and CD47, with ligands is disrupted, which leads to the overactivation of microglia [145]. It has been shown that the expression level of transforming growth factor β (TGF-β) further increases with age, and TGF-β not only attenuates the level of anti-inflammatory cytokines secreted by microglia [146] but also inhibits the conversion of microglia from a proinflammatory phenotype to an anti-inflammatory phenotype by decreasing interferon-regulating factor 7 [147]. Further studies have confirmed that amyloid beta protein (Aβ) accumulates in the CNS during aging, which also leads to increased levels of proinflammatory factor secretion by microglia [148, 149]. In addition to directly affecting the inflammatory response in the CNS, aging also affects the way peripheral immune cells enter the CNS. The blood‒brain barrier, an important barrier protecting the brain parenchyma, contains vascular endothelial cells as its main constituent cells, and as endothelial cells are transformed to a senescent phenotype, endothelial cells are coupled to neurovascular contacts by attenuating the regulation of vascular endothelial growth factor (VEGF) and through the ROS/NO axis [150]. More importantly, endothelial cells of the senescent phenotype can also promote peripheral immune cell infiltration by upregulating proinflammatory factors (e.g., IL-6 and IL-1β) and vascular adhesion molecules (e.g., VCAM1), and this infiltration further impairs the barrier function of the BBB [151, 152]. In the blood cerebrospinal fluid barrier, tumor necrosis factor (INF) signaling in senescent choroidal cells impairs brain function, and at the same time, more monocyte-derived macrophages enter the brain parenchyma through the blood cerebrospinal fluid barrier [153]. Moreover, it has been demonstrated in aged mice that the meningeal lymphatic system also shows impairments such as reduced area coverage, smaller diameter and reduced cerebrospinal fluid drainage to the deep cervical lymph nodes with age, which may be associated with the downregulation of lymphangiogenic factor signaling in aging meningeal lymphatic vessel endothelial cells [52]. In conclusion, many studies have confirmed that stroke affects not only the signal secretion pattern of the central nervous system after stroke but also the way and pathway through which peripheral immune cells enter the central nervous system, and future studies on the effects of aging on the interaction between the central nervous system and the peripheral immune system after stroke are of great clinical significance.
Infiltration of peripheral inflammatory cells and their crosstalk with the central nervous system
The CNS has long been viewed as an immune-privileged system, but this perceived paradigm has slowly changed over the past two decades [154], and it is clear from the previous section that the peripheral immune system plays a critically important role through its interaction with the CNS after CNS injury [155, 156]. A large body of evidence suggests that the immune system maintains a constant state of immune surveillance, looking for signals not only from external pathogens but also from damaged tissues, especially in the case of aseptic injuries such as traumatic brain injury (TBI), spinal cord injury, and stroke [1]. Indeed, the initial inflammatory response to CNS injury may be a mechanism of the innate immune response characterized by the initial production of DAMPs, the production of inflammatory cytokines/chemokines by resident innate immune cells (microglia and astrocytes) and the subsequent recruitment of infiltrating innate immune cells. These infiltrating innate immune cells include granulocytes (basophils, eosinophils, and neutrophils), monocytes (which subsequently differentiate into macrophages), T cells, B cells, and others [9]. Both CNS resident immune cells and infiltrating immune cells play a beneficial/harmful role in the immune response after injury, depending on multiple influences, such as the type of injury, the time of day, and even individual differences, such as age, which is corroborated by the effects observed in multiple sclerosis, Alzheimer’s disease, and other diseases through the blocking of immune or inflammatory responses [157, 158].
Neutrophils
Neutrophils, one of the most widespread and common types of immune cells in the peripheral immune system, are the first cells to mobilize to the CNS after CNS injury, and they play important roles in the inflammatory response [159]. Neutrophils are the first to respond in the first few hours after injury, with a marked increase in lifespan and number that lasts up to 48 h [160, 161], and the infiltration of neutrophils can be observed as early as 30 min postinjury and peaks at 2–3 days [162]. However, there is also evidence that during this period (especially within 24 h), neutrophils in the human brain mainly accumulate in blood vessels and perivascular spaces and start to infiltrate into the brain parenchyma only 3–5 days after injury [163, 164]. This finding was further supported by a recent flow cytometry study in which only 40% of leukocytes in the brain parenchyma were derived from blood at 24 h after cerebral hemorrhage, and almost all of them were derived on day five [165]. Notably, the evidence that neutrophils accumulate in blood vessels rather than in parenchymal tissues after stroke is controversial, and more in-depth studies are needed at this time. Over time, neutrophils are gradually phagocytized by macrophages and microglia; thus, they gradually decrease in number [166]; however, the number of neutrophils in the brain parenchyma does not return to normal until the seventh day [167], and even up to 32 days after stroke, neutrophils can still be detected [168]. This late detection of neutrophils raises the possibility that neutrophils may be confined to the subarachnoid space due to changes in BBB reconstruction and cerebrospinal fluid flow, which leads to the degranulation of neutrophils and the release of inflammatory factors throughout the CNS, possibly leading to delayed brain injury [169].
The role of neutrophils in phagocytosis and inflammatory factor release has been extensively studied, and many factors, including interleukins (IL-6, IL-1α, IL-1β, and IL-8), TNF-α, LFA-1, leukotrienes, arachidonic acid, vWF, matrix metalloproteinase (MMP-9) and VEGF [170,171,172], have been identified as DAMPs associated with neutrophils, suggesting that neutrophils play numerous roles in the CNS. In addition, recent research has shown that neutrophil-released neutrophil extracellular traps (NETs) play important roles in both thrombosis and trapping invading microorganisms [173, 174]. In response to the stimulus of injury, a large meshwork of structures containing histones, DNA, and granule proteins, among others [175], is formed, and this meshwork may be a central crossroads of platelet aggregation, microthrombosis, and microglia-mediated inflammation [176, 177] and thus play an important role in irreversible impairment of the microcirculation [178].
Neutrophils also play an important role in interactions with other cellular components. In the TBI model, neutrophil depletion attenuates brain edema and microglial activation [179], and similar results have been found in CCL2 and CXCR2 gene deficiency models [180, 181]. On the other hand, microglia can be activated by neutrophils and blood catabolic products, increasing CXCL2 expression and interacting with CXCR2 on the surface of neutrophils [182], thereby recruiting more inflammatory cells into the subarachnoid space through chemoattraction and facilitating the clearance of blood substances, which is beneficial for neuronal recovery [169, 183, 184]. Moreover, although neutrophils act as phagocytes, the release of ROS (which are necessary for immune defense) via NADPH oxidase and myeloperoxidase [185] leads to the activation of microglia, the amplification of inflammatory processes, and a poor prognosis [186,187,188]; therefore, the elimination of free radicals in cerebral hemorrhage has demonstrated potential therapeutic benefits [189]. With the intensive study of microglia–neutrophil interactions, Neumann et al. provided the first in vivo evidence of physical interaction between microglia and infiltrating neutrophils for the first time in 2014, in which infiltrating neutrophils could induce microglial morphology alterations directly by physical contact [107, 190], thus enabling the capture of infiltrating neutrophils.
Mononuclear/macrophages
The functions of monocytes/macrophages during injury include killing pathogens by phagocytosis, expressing inflammatory cytokines and enzymes, and subsequently producing ROS and reactive nitrogen species (RNS) [191]; monocytes/macrophages can also differentiate into inflammatory dendritic cells (DCs) or macrophages during inflammation [192, 193]. In rodents, two circulating monocyte subpopulations are known to exist in the peripheral immune system: “inflammatory monocytes” and “patrol” monocytes. “Inflammatory” monocytes are characterized by high levels of lymphocyte antigen 6 complex site C (LY6C), low to moderate levels of CX [3]-C motif chemokine receptor 1 (CX3CR1), and C-C chemokine receptor type 2 (CCR2) [194, 195]. These cells are actively recruited to the site of injury after CNS injury, where they promote an inflammatory response that increases significantly at 3 days postinjury [196, 197]. “Patrolling” monocytes, characterized by low levels of Ly6C, high levels of CXC3CR1, and no CCR2 [198, 199], have a relatively long lifespan, “patrol” the vascular lumen and contribute to the maintenance of vascular homeostasis, with peak numbers occurring at 6 days postinjury; additionally, these cells are actively recruited to the site of injury and contribute to the inflammatory response, with a significant increase occurring at 3 days postinjury. The number of invading cells peaks at 6 days after injury [200, 201].
Numerous studies have also confirmed the strong association between monocytes and other immune cells. Previous studies have demonstrated that CD16+ monocytes can lead to a threefold increase in APC activity to the original level in peripheral blood [202] and can promote T-cell proliferation [203]; specifically, monocytes can act as APCs along with dendritic cells, thereby activating T cells [204]. After TBI, myelin basic proteins, glial fibrillary acidic proteins, and other proteins produced as a result of brain cell injury are recognized by APCs and enter the lymphatic system through meningeal lymphatic vessels to activate T and B cells through antigen presentation [46].
In the CNS, microglial populations are renewed through local expansion and replenishment of circulating monocytes [205]. The activation of microglia after cerebral ischemia precedes and can be facilitated by blood-derived macrophages [206, 207], which are not recruited to the site of injury until 3–4 h after stroke [208], but their numbers remain low until the next day, with peak macrophage aggregation occurring on the seventh day after stroke [209, 210]. Conversely, monocyte/macrophage infiltration also further activates microglia, and Bhalala et al. demonstrated that it is possible to suppress peripheral immune cells by splenectomy, thereby attenuating the inflammatory response of microglia in the brain parenchyma [211]. In an in-depth study utilizing the MCAO mouse model, the differences in gene expression between blood-derived macrophages and microglia at 7 days poststroke were analyzed. Blood-derived macrophages preferred the M2 phenotype, and the expression of genes primarily involved in migration, proliferation, and calcium signaling was upregulated [212]. Using flow cytometry, Li distinguished between peripherally derived monocytes/macrophages and microglia and reported that NADPH-2, IL-1b and CD68 levels were greater in macrophages, whereas TGF-β1, IL-6 and TNF-a levels were greater in microglia [213]. It can be hypothesized that although microglia are rapidly activated and function in the acute phase, macrophages may be key factors exacerbating the inflammatory response. A recent study revealed the beneficial effects of the combination of macrophage colony-stimulating factor (M-CSF), IL-6, and transforming growth factor-beta (TGF-β), particularly in terms of cosecretion by microglia, macrophages, and endothelial cells [214].
Moreover, monocytes entering the CNS can be activated by DAMPs such as ATP, ADP and clone-stimulating factors, which results in activated monocytes/macrophages recruiting more neutrophils and monocytes to the site of injury via neurotrophic factors (e.g., BDNF, NGF) and chemotactic factors [215]. In turn, neutrophil depletion can reduce monocyte/macrophage infiltration [216]. Moreover, in a spinal cord injury model, peripheral macrophages can instead inhibit microglial activation [217], which once again demonstrates the dual-edged sword of characterizing inflammatory cells. However, the effects of the monocyte/macrophage subpopulation and age have yet to be thoroughly investigated [218]. For example, it has been demonstrated that, compared with adults, children under 10 years of age have less monocyte chemotaxis, which may reduce monocyte damage after traumatic brain injury [219, 220].
T cells
Several recent studies have clearly demonstrated that T cells can act as patrolling lymphocytes, even in a healthy state, and that T cells can continuously monitor the brain and migrate toward the healthy brain, whereas subsequent interactions with brain-resident cells [135] are important for the maintenance of physiological function and plasticity [221,222,223]. Specifically, T cells have been shown to influence neural cell genesis [224]. T-cell entry into the subarachnoid space and perivascular space has been shown to be achieved by transendothelial cell migration through small postcapillary veins [95]. In the healthy state, however, patrolling T cells eventually terminate their patrols and enter deep cervical lymph nodes rather than the brain parenchyma due to a lack of antigen recognition [225]. This concept is supported by the activation of the immune response in the deep cervical lymph nodes by antigens derived from brain tissues [217, 226], although this immune response is more biased toward a B-cell-mediated humoral immune response, probably to inhibit the cytotoxicity of T cells [227, 228]. Through this mechanism, lymphocytes in the brain can be replaced in a healthy state in only approximately 12 h. Researchers have found that there are also many T cells, mainly CD4+ T cells, in the choroid plexus of the healthy brain [229]; these T cells may provide a cellular pool for T-cell responses to injury during brain injury. There is growing evidence of the important role of adaptive immunity, particularly T lymphocytes, in brain injury secondary to ischemia [230,231,232]. After cerebral hemorrhage, flow cytometry showed that CD4+ T-cell infiltration was significant and peaked on day 5 [233], whereas in cerebral ischemia, T-cell infiltration, especially that of CD8+ T cells, was lower than that in cerebral hemorrhage [234].
T cells are important for interacting with other immune cells. T cells may act on glial cells such as microglia and astrocytes to alter the brain microenvironment [235]. Different cytokines produced by different T-cell subsets may not have the same activating effect on microglia [236]. Upon activation, microglia can promote neural precursor cell (NPC) proliferation by secreting IGF-1 [237] and promoting oligodendrocyte formation [238]. Studies of T cells on astrocytes have shown that inflammatory factors produced by T cells can induce astrocytes to produce neutrophil chemokines, thereby promoting neutrophil infiltration [239]. In turn, the production of IL-15 by astrocytes and microglia after ischemic stroke has been shown to activate CD8+ T cells and NK cells and to accumulate in the brain parenchyma [240]. Findings from studies in humans support that IL-15 significantly enhances the cytotoxic effect of CD8+ T cells on target organs in the target tissues of patients with rheumatoid arthritis [241, 242], autoimmune myositis [243], obesity [244], celiac disease [245], and multiple sclerosis [246, 247], which is thought to be one of the mechanisms that leads to tissue destruction. Moreover, as another T-cell subtype, Treg cells have been shown to play a primarily protective role after injury. Specifically, in addition to directly suppressing the increase in inflammatory factor levels, Treg cells can also act directly on astrocytes and microglia to convert them to an anti-inflammatory phenotype [248]. In addition, Treg cells directly signal oligodendrocyte progenitors to differentiate into myelin-producing oligodendrocytes, and mature oligodendrocytes bind to demyelinated axons and rewrap these axons with myelin to restore nerve function and neuroprotection [249]. In addition to their ability to recruit cells [250], T cells can also cause microvascular dysfunction by binding to endothelial cells and platelets [251], which in turn impairs neuronal function. More in-depth studies have shown that the interaction of specific regulatory T cells with endothelial cells may be achieved through LFA-1/ICAM-1 signaling [252]. Therefore, it is more logical to put forth more effort into preventing thrombosis by blocking T cells and thus thrombosis than working on means of thrombolysis after thrombosis. Studies on T cells and monocytes/macrophages have shown a significant increase in ipsilateral thalamic CD4+ and CD8+ T cells by flow cytometry 14 days after stroke, and the sites of T-cell aggregation overlap with the areas of activated macrophage aggregation, which suggests that there may be interactions between T cells and macrophages [253]; however, there is also evidence that T-cell-produced IL-4 can promote the conversion of macrophages to the M2 phenotype, thereby suppressing inflammation [254, 255].
B cells
There is a relative paucity of studies on the role of B cells after CNS injury, which may be related to their low infiltration rate after injury [165]. Up to 8 weeks after MCAO in mice, B cells migrate to distal brain regions (e.g., the dentate gyrus, hypothalamus, olfactory regions, and cerebellum), which is associated with neurogenesis and functional recovery and has specific positive effects on motor recovery [256]. This migration may explain and support the results of another study in which motor recovery improved despite continued thalamic and hippocampal degeneration and associated cognitive dysfunction [257], which has important implications for neurological recovery. Although the infiltration rate is low, the role of humoral immunity induced by B cells cannot be ignored [217], and B cells may be involved in brain injury by producing autoantibodies against antigens such as MBP and GFAP [217, 258]. For example, in a study of children (7–16 years old) with chronic posttraumatic headache secondary to mild craniocerebral injury, B cells were shown to produce autoantibodies against AMPA and NMDA receptor subunits up to one year after injury, thus leading to a poor prognosis [259]. Although B cells play a minimal role in the formation of focal brain injury in the acute phase [260, 261], analysis of CSF composition has confirmed that B cells can mediate the activation of humoral immunity during stroke [217] and cocluster with the immunoglobulins they produce in the thalamus, which in turn leads to cognitive dysfunction [262].
Peripheral inflammatory cells participate in the pathophysiological process after stroke
Ischemic stroke
In ischemic stroke, the primary injury is the sudden cessation of blood supply to the vascular region, leading to neuronal cell death and the formation of an ischemic core surrounded by a hypoperfused area known as the hemidiaphragm, and peripheral immune cells play an important role in the secondary injury caused by the primary injury [263, 264]. Studies have confirmed that neutrophils are the predominant cells in the ischemic hemisphere 3 days after MCAO [265]. Increased neutrophils are positively correlated with infarct volume and functional defects [162]. More importantly, they can contribute to tissue damage by releasing elastase, which destroys thin-walled tissue, and by producing reactive oxygen species that lead to BBB destruction [266]. The elevation of neutrophils may be due to increased release from the bone marrow and spleen along with decreased apoptosis of neutrophils [267]. In contrast to patients with neutrophils, patients with ischemic stroke have a reduced number of lymphocytes and therefore an elevated neutrophil ratio. This ratio is strongly correlated with infarct size and mortality [268]. However, despite their association with tissue damage, there is no clear correlation between the number of circulating neutrophils and the extent of ischemic injury [269], suggesting a possible beneficial effect of neutrophils. This idea was first proposed in the field of oncology research, where by analogy to the M1M2 phenotype of macrophages, researchers found that neutrophils also exist in different proinflammatory or anti-inflammatory subtypes, where the proinflammatory type was named N1 and the anti-inflammatory type was named N2 [270] and that TRL4 of the TLR family promotes the conversion of neutrophils to the N1 phenotype [271], while rosiglitazone promotes the conversion of neutrophil granulocytes to the N2 phenotype [272]. Furthermore, in the MCAO model, investigators found that type N2 neutrophils are more easily and efficiently cleared by microglia than other subtypes, which provides new ideas for future treatment [273, 274].
Monocytes also play an important role in cerebral ischemia. Sensory-motor functional CCR + monocytes/macrophages are not only associated with the maintenance of neurovascular unit integrity but also help to induce the migration of neuroblasts from areas of neurogenesis to the site of injury [275]. This may explain why CCR + monocytes can promote the recovery of sensorimotor nerve function in the first week after cerebral ischemia [276]. In the MCAO model, the phenotypic transformation of CCR2 + monocytes to a more activated state after infiltration into ischemic tissues was found at 14 days postinjury, suggesting that monocytes/macrophages play a pleiotropic role in mediating the injury and repair process after cerebral ischemia [277]. Because blood-derived monocytes/macrophages are morphologically and functionally very similar to resident microglia in the brain parenchyma, finding specific labeling methods to differentiate between the two cell types is a difficult challenge [278]. The excellent work of Tanaka et al. using chimeric mice with enhanced green fluorescent protein (GFP) bone marrow provides a useful tool to distinguish between resident macrophages and blood-derived macrophages in ischemic brain injury [206], and further studies have shown that it is also possible to explore differences in CD45 expression between microglia and monocytes/macrophages using flow cytometry to differentiate [279, 280]. These studies once again confirmed that blood-derived macrophages infiltrate later than microglia, but this, on the other hand, suggested a possible role for blood-derived macrophages in tissue remodeling [281]. This approach has important clinical and therapeutic applications because circulating monocytes are more receptive to drug manipulation than microglia located behind the BBB.
T cells, in contrast to neutrophils and monocytes/macrophages, have rarely been reported to be present in the vasculature or brain parenchyma within 24 h of injury and are recruited into the brain parenchyma mainly in the later stages of injury [261]. Experimental results in experimental cerebral ischemic injury models (including the MCAO model and the photochemically induced focal ischemia model) have shown that T-cell infiltration does not appear in large numbers until the third day and is mainly concentrated in the marginal zone of the lesion and that the number of T cells gradually increases between the third and the seventh day [282, 283]; T-cell infiltration is still detected in the brain parenchyma even for at least 30 days after cerebral infarction [284]. It has been shown that T-cell deletion has a neuroprotective effect in a focal cerebral ischemia model and that this effect disappears after T cells are retransplanted into immunodeficient mice [285, 286]. Many intensive studies have investigated different T-cell types, including CD4 + T cells, CD8 + T cells and Tregs [287], and especially in the delayed brain injury phase, an increasing number of studies have attempted to intervene in Treg cells because Treg cell infiltration occurs significantly later than that of other T-cell subsets and because of the neuroprotective function of Tregs [288]. During neuroregeneration from ischemic injury, the number of Tregs in the lateral ventricle of the ischemic hemisphere is increased and correlates with the proliferation of NPCs [289], which suggests that T cells may play a role in the migration of NPCs to the site of injury to repair damage [290, 291]. This is corroborated by the fact that in the healthy state, T cells can come into direct contact with NPCs (neural precursor cells) and interact with them functionally [292]. It has been shown that the removal of Tregs using CD25 on the seventh day after MCAO-induced cerebral ischemia in mice leads to increased neuronal damage [293, 294], and clinical studies have confirmed that increasing the number or function of Tregs can have a neuroprotective effect [287]. In terms of neuroprotective effects, researchers have demonstrated in stroke models that the mTOR inhibitor rapamycin protects neurons through the anti-inflammatory effects of Tregs [295, 296]. However, different methods for inhibiting Tregs have led to controversial results [297].
Hemorrhagic stroke
Hemorrhagic stroke can include two major types of cerebral hemorrhage and subarachnoid space hemorrhage, in which the primary brain damage caused by cerebral hemorrhage is caused mainly by the rupture of blood vessels, leading to the extravasation of blood components directly into the brain and the formation of hematomas, which cause structural damage. However, whether cerebral ischemia occurs after hemorrhage is controversial, as is whether this change is correlated with the severity of hemorrhage, etc [9]. The most immediate damage after SAH consists of two main events: (1) sudden blood invasion in the subarachnoid space and (2) hypoxia due to cerebral circulatory disturbances and increased intracranial pressure [178]. However, a growing number of studies have confirmed the importance of secondary injury caused by inflammatory infiltration in terms of mortality and neurological prognosis. The development of secondary brain injury involves numerous factors, such as the induction of neuroinflammatory factors produced by hemolytic products [298] and the effects of metabolites produced in situ by brain tissue [299, 300], all of which are inextricably linked to inflammatory cell infiltration [301].
Vasospasm is considered one of the strongest risk factors for secondary brain injury after hemorrhagic stroke and is significantly associated with delayed cerebral ischemia (DCI) [172]. In patients with vasospasm, the number of neutrophils and their enzymes myeloperoxidase and NADPH oxidase are greater [170, 302], and vasospasm is significantly improved if neutrophils are depleted or the CD11b/CD18 complex is blocked [303]. In addition, neutrophil surface Toll-like receptor 4 (TLR-4), TLR-related interferon activator (TRIF) and myeloid differentiation primary response gene (MyD88) are important mediators of neuronal apoptosis and cerebral vasospasm, respectively [304]. Although 70% of angiograms after aneurysmal SAH show vasospasm, the incidence of DCI is in fact only 30% [305], and more importantly, even when the incidence of vasospasm is reduced by pharmacological treatment, clinical outcomes are not improved [306]. This finding suggested that factors such as neuroinflammation, BBB disruption, and microthrombosis should be considered in addition to vasospasm [307] and may lead to secondary delayed cerebral ischemia or secondary brain injury [308, 309]. As a means of detection, neutrophil counts are correlated with the occurrence and prognosis of DCI, both in the early stages of injury [310] and three days after the onset of aneurysmal SAH [311]. In addition to these findings [312], there is evidence that NLRs are correlated with neurological symptoms such as amnesia and the presence of CT positivity in lesions [313]. However, due to the complexity of the etiology and progression of DCI, there is still a long way to go in predicting or diagnosing DCI or delayed brain injury by using inflammation as an entry point, and as yet, there is no method that can be repeated many times [172]. Another important factor in DCI is thrombosis, and neutrophils are incredibly perceptive of thrombi; even in reverse blood flow, the movement of neutrophils can be achieved in vessels without thrombi, which may explain why the number of neutrophils is ten times greater than that of other lymphocytes [231]. This strong ability to cope with shear stress may be achieved by sticky membrane slings [314]; unfortunately, the resolution of in vivo imaging is not sufficient to visualize these structures. Future studies could focus on identifying inflammatory cells with microthrombi, delayed axonal degeneration, etc [315].
Within the secondary injury phase after hemorrhagic stroke, although many of the physiological changes in the acute phase, such as cerebral hemorrhage and increased intracranial pressure, gradually subside and the subarachnoid space is progressively cleared of clots, a second focus of neuronal damage occurs between 14 and 28 days after hemorrhage [316]. This second focus may be related to a second vasospasm accompanied by thickening of the vascular wall that occurs at the end of the acute phase [303, 317]; however, ischemia alone is not sufficient to cause neutrophils to cross the BBB [318]. Although the current literature includes a series of studies on the mechanisms that may lead to delayed neurological deficits (DNDs) after hemorrhagic stroke, the main focus has been on cytokines [308], including the inflammatory cytokine profile in the brain and periphery and their impact on vascular dysregulation [110], and on the role of inflammatory cells in DNDs revolving around the association with vasospasm only; however, there is a lack of relevant evidence for the direct role of inflammatory cells in DNDs [319]. Studies have demonstrated that neutrophils play a key role in the progression of DNDs; that neutrophils modulate the expression of NMDAR subunits in the hippocampus, thereby leading to spatial memory deficits; and that neutrophil depletion can eliminate DNDs [320].
BBB disruption after hemorrhagic stroke leads to the infiltration of inflammatory cells [321, 322], among which monocytes play an important role and are the second peripheral immune cells to enter the CNS after neutrophils [161]. After neutrophil invasion, monocytes adhere to the vessel wall and move toward the ischemic area. Blood-derived monocyte/macrophage counts reach a peak at 1–2 days after injury [323], reach maximal activity at 3–7 days [162] and persist for several weeks [324]. Blood degradation products after hemorrhagic stroke are sufficient to activate [184] the release of ET-1 [325, 326]. In addition, monocyte infiltration has been associated with elevated levels of many inflammatory factors, such as CXCL-1, CXCL-9 and CXCL-10, which have been shown to be associated with DCI after aneurysmal SAH [327]. However, a change in cytokine concentration is positively correlated with a change in blood flow velocity [328]. This process results in a vicious cycle in which monocytes produce cytokines, leading to vasoconstriction; blood flow slows, and cytokine concentrations rise, leading to further vasoconstriction. Monocytes infiltrate the brain parenchyma after CNS injury, activate macrophages and initiate the phagocytosis and degradation of cellular products [184]. The current view is that there are two phenotypes of macrophages, M1 proinflammatory and M2 inhibitory inflammatory types, and that the M1 type is associated with increased production of proinflammatory cytokines (e.g., TNF-α, IL-1b, and NADPH), which ultimately leads to inflammation in the nervous system [329, 330]. The macrophage phenotype is regulated by a variety of factors; for example, blood metabolites such as hemoglobin and iron promote macrophage conversion to the M1 type [331]; a variety of factors such as metabolite clearance promote macrophage conversion to the M2 type [332, 333]; and numerous efforts have been made to promote monocyte/macrophage phenotypic conversion to the M2 type. For example, studies have shown that atorvastatin and cordycepin inhibit microglial/macrophage proinflammatory polarization and promote anti-inflammatory polarization after TBI. This effect may further inhibit BBB disruption and neutrophil infiltration [334, 335].
After hemorrhagic stroke, activated T cells infiltrate the brain parenchyma and contribute to neuronal damage and poor prognosis by releasing ROS and cytokines [336]. In addition, cell-based CD28 expression in the adaptive immune system is a coactivation signal for T cells, leading not only to increased CD4 + and CD8 + T-cell activation [337] but also to increased cytokine production by T cells [338]. T-cell accumulation can be observed in the thalamus distal to the primary injury site, which has been shown to undergo secondary neuronal degeneration, and it has been found that the infiltration of T cells continues at least two weeks after stroke, with more CD4 + T cells than CD8 + T cells [253], which coincides with the severe period of poststroke secondary neurodegeneration (SND) [339]. In the middle to late stages of DCI, CD3 + Tregs are significantly elevated compared to those in the EBI stage and are significantly associated with infection after SAH [340]. This may be because Tregs inhibit T-cell proliferation and reduce the secretion of cytokines (e.g., IL-10 and TNF-α), thereby attenuating the inflammatory response [341, 342]. In addition, Tregs also protect against BBB damage, which is mainly achieved by inhibiting peripheral MMP-9 production [343]. These findings show that the role of T cells in the development of inflammation is bidirectional, as cytokines can both eliminate damaged cells and clear microorganisms, leading to a severe inflammatory cascade [344, 345]. Angiogenesis is an important repair mechanism after stroke, and the effect of T cells on angiogenesis has been demonstrated in numerous organs. In tumors and lung ischemia, proinflammatory Th1 cells are thought to inhibit vascular growth, whereas anti-inflammatory Treg cells are associated with increased angiogenesis [346]; moreover, these various facilitating effects are reflected not only by the fact that Treg cells can inhibit effector T cells but also by their active secretion of chemokines, vascular endothelial growth factor, and transforming growth factor to promote neoangiogenesis [347, 348]. In rat corneal tissue, T-cell-derived IL-17 has also been shown to promote neoangiogenesis [349]. Although the role of T cells in promoting angiogenesis has not been studied in stroke, this is not a valuable research direction.
Potential treatment strategies and status evaluation by targeting brain–peripheral immune responses after stroke
The important role of inflammatory injury in CNS disease is receiving increasing attention, and treatments targeting inflammation have numerous advantages over other approaches (Table 1). First, anti-inflammatory treatments have an advantage in terms of time window limitations; in ischemic stroke, the window for thrombolysis is only 4.5 [350], but treatments targeting inflammation are effective 12–24 h after stroke [351]. In addition, inflammatory treatments are beneficial even for hemorrhagic strokes, as they are more broadly available and less demanding in terms of diagnosing the type of injury [352]. Finally, regardless of the type of CNS injury, the beneficial role of treatments targeting inflammation in ischemia‒reperfusion can help smooth the recovery period in patients treated for primary injury [353].
Ischemic stroke
There has been some progress in research targeting peripheral inflammatory infiltrating cells. First, a series of attempts have been made to treat ischemic stroke. In acute ischemic stroke, neurological recovery can be improved, and the volume of brain tissue damage can be reduced by oral administration of fingolimod [354, 355], which is attributed to the fact that fingolimod can promote the polarization of microglia to the M2 type [356, 357]. Another clinical study revealed that ischemic stroke patients could achieve better MRS scores at 90 days after taking minocycline orally and that minocycline is safe when combined with tissue plasminogen activator [358]. Splenic tyrosine kinase (SYK) is a nonreceptor tyrosine kinase found in organs such as the spleen and thymus and is closely associated with inflammation. SYK inhibitors protect against thrombosis and attenuate cerebral infarction injury [359], and more importantly, this effect does not affect the hemostatic effect of platelets. Recombinant T-cell receptors (RTLs) are a type of major histocompatibility complex (MCH) II that are predominantly present in the MCAO model in association with mouse brain tissue and the spleen. Treatment with its ligand recombinant T-cell receptor ligand (RTL) resulted in a 50% reduction in the infarct area and a reduction in the number of immune cells recruited to the brain [360, 361]. Moreover, researchers found that bexarotene, a retinoid X receptor (RXR) agonist, increased the number of N2 neutrophils in MCAO mice, which alleviated inflammatory damage and produced neuroprotection [362]. Bone marrow mononuclear cells (BMNCs), which include hematopoietic spectrum cells, stem cells, and progenitor cells, have been shown to promote neural stem cell proliferation [363, 364]; therefore, investigators explored the role of BMNC transplantation in ischemic stroke and confirmed that BMNCs are associated with improved neurological function [365, 366]. The two main types of grafts are arterial and venous grafts, and researchers have found that although arterial grafts are relatively risky, the grafts increase growth factor levels and decrease inflammatory factor levels. In contrast, vein grafts are safe but have no beneficial effects [367, 368]. Granulocyte colony-stimulating factor (G-CSF) can stimulate the infiltration of BMNCs to the site of injury after ischemic stroke [369], in addition to promoting axonal growth and the polarization of microglia toward the M2 phenotype [370, 371]; however, more studies are needed to confirm the exact efficacy of G-CSF in clinical translation. In a phase IIa clinical study of acute-phase ischemic stroke, investigators demonstrated by recombinant human IL-1 receptor antagonists that IL-1 receptor antagonists not only significantly improved patients’ NIH stroke scores three months after injury but also led to better performance on the modified Rankin scale (mRS) score [372]. In a study of natalizumab, investigators found that in acute ischemic stroke, natalizumab prevents T cells from entering the CNS by blocking α4-integrin, which is beneficial for reducing the volume of cerebral infarction [373].
Hemorrhagic stroke
In hemorrhagic stroke, numerous strategies have also been adopted for the infiltration of peripheral immune cells. Clinical studies have shown that fingolimod not only works in treating ischemic stroke, as mentioned earlier but also has a therapeutic effect on hemorrhagic stroke. Fingolimod blocks the release of lymphocytes from lymph nodes, which results in fewer T cells and NK cells infiltrating the CNS [374]; moreover, fingolimod attenuates neutrophil adherence to blood vessels after SAH, thereby improving neurological function after SAH [375]. Minocycline also plays a therapeutic role in intracerebral hemorrhage (ICH), and studies have shown that on the fifth day after ICH, minocycline reduces the number of microglia and macrophages around the hematoma, attenuates cerebral edema and decreases the expression levels of TNFα and MMP12 [376]. Neutrophil recruitment to and activation in the CNS are associated with formyl peptide receptor 1 (FPR1) expression on microglia and are in fact mediated through IL-1β. The activation of neutrophils by microglia is attenuated by the FPR1 inhibitor T-0080, which reduces cerebral edema and improves neurological function after ICH [377, 378]. Similarly, myeloperoxidase (MPO) can mediate the activation of microglia against neutrophils, and MPO knockout can play a beneficial role in the recovery of neurological function after SAH; however, further investigation is needed to determine how these findings can be translated into clinical application [379]. The beneficial role of Treg cells in hemorrhagic stroke has been previously described. During the recovery phase of hemorrhagic stroke, rapamycin inhibits mTOR activation and increases the number of Treg cells in brain tissue; more importantly, these effects lead to increased levels of IL-10 and transforming growth factor-b, which are important for improving neurological function after ICH [380]. Moreover, multicenter clinical trial findings have confirmed that selective inhibition of cyclooxygenase 2 (COX2) using celecoxib after cerebral hemorrhage reduces inflammatory cell infiltration, cerebral edema, and perihematomal cell death [381]. Rosiglitazone has been shown to have a therapeutic effect not only on diabetes mellitus but also on ICH, after which it may act as an agonist of PPAR-γ and upregulate the expression of CD36, which has been shown to regulate the phagocytosis of microglia and phagocytes in ICH, suggesting that rosiglitazone can promote hematoma clearance after ICH [382]. In addition, it has been experimentally confirmed that low doses of IL-2 can attenuate neuronal damage after SAH, which is achieved by inhibiting proinflammatory factors and peripheral neutrophils [340].
Perspectives and conclusion
Over the past two decades, there has been increasing interest in the interactions between the CNS and the peripheral immune system after CNS injury. Here, we summarize the effects of CNS injury on the peripheral immune system. CNS injury is signaled to the peripheral immune system through the autonomic nervous system, neuroendocrine system, and meningeal lymphatic vascular system, the activation of which allows peripheral immune cells to enter the CNS through the ruptured BBB and BCSFB and direct infiltration into the bone marrow of the skull to play a role in various acute CNS injuries. We conclude by summarizing the monitoring methods, clinical findings, and corresponding therapeutic advances for this interaction. A more complete description of the effects on the peripheral immune system after acute CNS injury and the effects of the corresponding inflammatory cell infiltration in secondary injury after primary injury is presented. Regarding the existing studies and future research priorities, we make the following points: (1) The inflammatory response is a complex network with a complex interplay of effects, both in terms of the relationship between the CNS and the peripheral immune system and in terms of the relationships between the different inflammatory components. Future studies of a particular inflammatory factor should not only be more in-depth but also explore the overall effects at the level of the network of inflammatory relationships. (2) The role of inflammatory cells in CNS injury is often a double-edged sword, and it is clear that simply promoting or inhibiting a factor in response to a certain factor does not have an absolute positive effect; for example, although the immunosuppressive effect or artificial immunosuppression after injury reduces the inflammatory response at the site of injury to a certain extent, it also leads to more infections, and ultimately, there is no clear evidence of a beneficial effect on prognosis or survival. Therefore, a combination of multifactorial therapies or interventions targeting beneficial effects should be considered for the treatment of inflammatory injuries in the future. (3) Although the inflammatory patterns induced by different CNS injuries are very similar, there are many factors affecting the inflammatory response, including not only different types of injuries but also different periods of disease, severity of disease, and even age; therefore, future research should be conducted to investigate the inflammatory response and treatment modalities. Therefore, the generalizability of the inflammatory response and treatment modalities should be further investigated in the future.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- CNS:
-
Central nervous system
- BBB:
-
Blood‒brain barrier
- BCSFB:
-
Blood–cerebrospinal fluid barrier
- SAH:
-
Subarachnoid hemorrhage
- ANS:
-
Autonomic nervous system
- SNS:
-
Sympathetic nervous system
- PSNS:
-
Parasympathetic nervous system
- LPS:
-
Lipopolysaccharide
- AVP:
-
Arginine vasopressin
- ACTH:
-
Adrenocorticotropic hormone
- TSH:
-
Thyroid stimulating hormone
- FSH:
-
Follicle-stimulating hormone
- LH:
-
Luteinizing hormone
- GH:
-
Growth hormone
- MAP2:
-
Microtubule-associated protein 2
- MBP:
-
Myelin basic protein
- APCs:
-
Antigen-presenting cells
- DAMPs:
-
Danger-associated molecular patterns
- HMGB1:
-
High-mobility group box 1
- NO:
-
Nitric oxide
- TNF:
-
Tumor necrosis factor
- SIDS:
-
Stroke-induced immune depression syndrome
- MCAO:
-
Middle cerebral artery occlusion
- HPA:
-
Hypothalamic‒pituitary‒adrenal
- GPCRs:
-
G protein-coupled receptors
- GLs:
-
Glial limitans
- VCAM-1:
-
Vascular cell adhesion molecule
- CAMs:
-
Cell adhesion molecules
- VLA4:
-
Very late antigen-4
- CSF:
-
Cerebrospinal fluid
- EAE:
-
Experimental autoimmune cerebrospinal
- ROS:
-
Reactive oxygen species
- TBI:
-
Traumatic brain injury
- MMP-9:
-
Matrix metalloproteinase
- NETs:
-
Neutrophil extracellular traps
- LY6C:
-
Lymphocyte antigen 6 complex site C
- CX3CR1:
-
CX(3)-C motif chemokine receptor 1
- CCR2:
-
C-C chemokine receptor type 2
- M-CSF:
-
Macrophage colony-stimulating factor
- TGF-β:
-
Transforming growth factor-beta
- NPC:
-
Neural precursor cell
- DCI:
-
Delayed cerebral ischemia
- TLR-4:
-
Toll-like receptor 4
- TRIF:
-
TLR-related interferon activator
- MyD88:
-
Myeloid differentiation primary response gene
- DNDs:
-
Delayed neurological deficits
- SND:
-
Poststroke secondary neurodegeneration
- ICH:
-
Intracerebral hemorrhage
- FPR1:
-
Formyl peptide receptor 1
- MPO:
-
Myeloperoxidase
- COX2:
-
Cyclooxygenase 2
References
Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS Immune Privilege. Trends Immunol. 2015;36(10):569–77.
Schiller M, Ben-Shaanan TL, Rolls A. Neuronal regulation of immunity: why, how and where? Nat Rev Immunol. 2021;21(1):20–36.
An C, Shi Y, Li P, Hu X, Gan Y, Stetler RA, et al. Molecular dialogs between the ischemic brain and the peripheral immune system: dualistic roles in injury and repair. Prog Neurobiol. 2014;115:6–24.
Claassen J, Park S. Spontaneous subarachnoid haemorrhage. Lancet. 2022;400(10355):846–62.
Cordonnier C, Demchuk A, Ziai W, Anderson CS. Intracerebral haemorrhage: current approaches to acute management. Lancet. 2018;392(10154):1257–68.
Russo MV, McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science. 2016;353(6301):783–5.
Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5(1):53–63.
Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808.
Shi K, Tian DC, Li ZG, Ducruet AF, Lawton MT, Shi FD. Global brain inflammation in stroke. Lancet Neurol. 2019;18(11):1058–66.
Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav Immun. 2007;21(6):736–45.
Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve–an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52(4):595–638.
Scanzano A, Cosentino M. Adrenergic regulation of innate immunity: a review. Front Pharmacol. 2015;6:171.
Maestroni GJ. Dendritic cell migration controlled by alpha 1b-adrenergic receptors. J Immunol. 2000;165(12):6743–7.
Kohm AP, Sanders VM. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4 + T and B lymphocyte function in vitro and in vivo. Pharmacol Rev. 2001;53(4):487–525.
Szelenyi J, Kiss JP, Vizi ES. Differential involvement of sympathetic nervous system and immune system in the modulation of TNF-alpha production by alpha2- and beta-adrenoceptors in mice. J Neuroimmunol. 2000;103(1):34–40.
Bellinger DL, Millar BA, Perez S, Carter J, Wood C, ThyagaRajan S, et al. Sympathetic modulation of immunity: relevance to disease. Cell Immunol. 2008;252(1–2):27–56.
Loza MJ, Peters SP, Foster S, Khan IU, Penn RB. beta-agonist enhances type 2 T-cell survival and accumulation. J Allergy Clin Immunol. 2007;119(1):235–44.
Swanson MA, Lee WT, Sanders VM. IFN-gamma production by Th1 cells generated from naive CD4 + T cells exposed to norepinephrine. J Immunol. 2001;166(1):232–40.
Chelmicka-Schorr E, Kwasniewski MN, Czlonkowska A. Sympathetic nervous system modulates macrophage function. Int J Immunopharmacol. 1992;14(5):841–6.
Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9(6):418–28.
Koopman FA, Chavan SS, Miljko S, Grazio S, Sokolovic S, Schuurman PR, et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2016;113(29):8284–9.
Bernik TR, Friedman SG, Ochani M, DiRaimo R, Susarla S, Czura CJ, Tracey KJ. Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion. J Vasc Surg. 2002;36(6):1231–6.
Guarini S, Altavilla D, Cainazzo MM, Giuliani D, Bigiani A, Marini H, et al. Efferent vagal fibre stimulation blunts nuclear factor-kappab activation and protects against hypovolemic hemorrhagic shock. Circulation. 2003;107(8):1189–94.
Pavlov VA, Ochani M, Yang LH, Gallowitsch-Puerta M, Ochani K, Lin X, et al. Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med. 2007;35(4):1139–44.
Saeed RW, Varma S, Peng-Nemeroff T, Sherry B, Balakhaneh D, Huston J, et al. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med. 2005;201(7):1113–23.
Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334(6052):98–101.
Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405(6785):458–62.
Tarkowski E, Naver H, Wallin BG, Blomstrand C, Tarkowski A. Lateralization of T-lymphocyte responses in patients with stroke. Effect Sympathetic Dysfunction? Stroke. 1995;26(1):57–62.
Tarkowski E, Ekelund P, Tarkowski A. Enhancement of antigen-specific T-cell reactivity on the affected side in stroke patients. J Neuroimmunol. 1991;34(1):61–7.
Moshel YA, Durkin HG, Amassian VE. Lateralized neocortical control of T lymphocyte export from the thymus I. increased export after left cortical stimulation in behaviorally active rats, mediated by sympathetic pathways in the upper spinal cord. J Neuroimmunol. 2005;158(1–2):3–13.
Neveu PJ, Barneoud P, Vitiello S, Betancur C, Le Moal M. Brain modulation of the immune system: association between lymphocyte responsiveness and paw preference in mice. Brain Res. 1988;457(2):392–4.
Betancur C, Neveu PJ, Vitiello S, Le Moal M. Natural killer cell activity is associated with brain asymmetry in male mice. Brain Behav Immun. 1991;5(2):162–9.
Neveu PJ. Asymmetrical brain modulation of the immune response. Brain Res Brain Res Rev. 1992;17(2):101–7.
Churchland PS, Winkielman P. Modulating social behavior with oxytocin: how does it work? What does it mean? Horm Behav. 2012;61(3):392–9.
Neumann ID. Brain oxytocin: a key regulator of emotional and social behaviours in both females and males. J Neuroendocrinol. 2008;20(6):858–65.
Boone M, Deen PM. Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflugers Arch. 2008;456(6):1005–24.
Jankowski M, Bissonauth V, Gao L, Gangal M, Wang D, Danalache B, et al. Anti-inflammatory effect of oxytocin in rat myocardial infarction. Basic Res Cardiol. 2010;105(2):205–18.
Oliveira-Pelegrin GR, Saia RS, Carnio EC, Rocha MJ. Oxytocin affects nitric oxide and cytokine production by sepsis-sensitized macrophages. Neuroimmunomodulation. 2013;20(2):65–71.
Boyd JH, Holmes CL, Wang Y, Roberts H, Walley KR. Vasopressin decreases sepsis-induced pulmonary inflammation through the V2R. Resuscitation. 2008;79(2):325–31.
Palin K, Moreau ML, Sauvant J, Orcel H, Nadjar A, Duvoid-Guillou A, et al. Interleukin-6 activates arginine vasopressin neurons in the supraoptic nucleus during immune challenge in rats. Am J Physiol Endocrinol Metab. 2009;296(6):E1289–99.
Tanriverdi F, Silveira LF, MacColl GS, Bouloux PM. The hypothalamic-pituitary-gonadal axis: immune function and autoimmunity. J Endocrinol. 2003;176(3):293–304.
Taneja V. Sex hormones determine Immune Response. Front Immunol. 2018;9:1931.
Pawlikowski M, Stepien H, Komorowski J. Hypothalamic-pituitary-thyroid axis and the immune system. Neuroimmunomodulation. 1994;1(3):149–52.
Barreiro Arcos ML, Gorelik G, Klecha A, Genaro AM, Cremaschi GA. Thyroid hormones increase inducible nitric oxide synthase gene expression downstream from PKC-zeta in murine tumor T lymphocytes. Am J Physiol Cell Physiol. 2006;291(2):C327–36.
Klecha AJ, Genaro AM, Gorelik G, Barreiro Arcos ML, Silberman DM, Schuman M, et al. Integrative study of hypothalamus-pituitary-thyroid-immune system interaction: thyroid hormone-mediated modulation of lymphocyte activity through the protein kinase C signaling pathway. J Endocrinol. 2006;189(1):45–55.
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–41.
Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212(7):991–9.
Andres KH, von During M, Muszynski K, Schmidt RF. Nerve fibres and their terminals of the dura mater encephali of the rat. Anat Embryol (Berl). 1987;175(3):289–301.
Truman LA, Bentley KL, Smith EC, Massaro SA, Gonzalez DG, Haberman AM, et al. ProxTom lymphatic vessel reporter mice reveal Prox1 expression in the adrenal medulla, megakaryocytes, and platelets. Am J Pathol. 2012;180(4):1715–25.
Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Science. 2016;353(6301):766–71.
Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci. 2018;21(10):1380–91.
Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. 2018;560(7717):185–91.
Planas AM, Gomez-Choco M, Urra X, Gorina R, Caballero M, Chamorro A. Brain-derived antigens in lymphoid tissue of patients with acute stroke. J Immunol. 2012;188(5):2156–63.
Tsuchida T, Parker KC, Turner RV, McFarland HF, Coligan JE, Biddison WE. Autoreactive CD8 + T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci U S A. 1994;91(23):10859–63.
Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4(6):469–78.
Zhang M, Li W, Niu G, Leak RK, Chen J, Zhang F. ATP induces mild hypothermia in rats but has a strikingly detrimental impact on focal cerebral ischemia. J Cereb Blood Flow Metab. 2013;33(1):e1–10.
Kim JB, Lim CM, Yu YM, Lee JK. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J Neurosci Res. 2008;86(5):1125–31.
Yang QW, Lu FL, Zhou Y, Wang L, Zhong Q, Lin S, et al. HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent toll-like receptor 4 signaling. J Cereb Blood Flow Metab. 2011;31(2):593–605.
Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, et al. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci. 2006;26(24):6413–21.
Adami C, Bianchi R, Pula G, Donato R. S100B-stimulated NO production by BV-2 microglia is independent of RAGE transducing activity but dependent on RAGE extracellular domain. Biochim Biophys Acta. 2004;1742(1–3):169–77.
Bianchi R, Giambanco I, Donato R. S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol Aging. 2010;31(4):665–77.
Bianchi R, Kastrisianaki E, Giambanco I, Donato R. S100B protein stimulates microglia migration via RAGE-dependent up-regulation of chemokine expression and release. J Biol Chem. 2011;286(9):7214–26.
Bune LT, Thaning P, Johansson PI, Bochsen L, Rosenmeier JB. Effects of nucleotides and nucleosides on coagulation. Blood Coagul Fibrinolysis. 2010;21(5):436–41.
Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, et al. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J Biol Chem. 2009;284(27):18143–51.
Chu K, Yin B, Wang J, Peng G, Liang H, Xu Z, et al. Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J Neuroinflammation. 2012;9:69.
Hug A, Dalpke A, Wieczorek N, Giese T, Lorenz A, Auffarth G, et al. Infarct volume is a major determiner of post-stroke immune cell function and susceptibility to infection. Stroke. 2009;40(10):3226–32.
Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6(10):775–86.
Shi K, Wood K, Shi FD, Wang X, Liu Q. Stroke-induced immunosuppression and poststroke infection. Stroke Vasc Neurol. 2018;3(1):34–41.
Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T, et al. Stroke-induced immunodepression: experimental evidence and clinical relevance. Stroke. 2007;38(2 Suppl):770–3.
Fathali N, Ostrowski RP, Hasegawa Y, Lekic T, Tang J, Zhang JH. Splenic immune cells in experimental neonatal hypoxia-ischemia. Transl Stroke Res. 2013;4(2):208–19.
Ajmo CT Jr., Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, Pennypacker KR. The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res. 2008;86(10):2227–34.
Ostrowski RP, Schulte RW, Nie Y, Ling T, Lee T, Manaenko A, et al. Acute splenic irradiation reduces brain injury in the rat focal ischemic stroke model. Transl Stroke Res. 2012;3(4):473–81.
Winklewski PJ, Radkowski M, Demkow U. Cross-talk between the inflammatory response, sympathetic activation and pulmonary infection in the ischemic stroke. J Neuroinflammation. 2014;11:213.
Dorrance AM, Fink G. Effects of Stroke on the autonomic nervous system. Compr Physiol. 2015;5(3):1241–63.
Radak D, Resanovic I, Isenovic ER. Changes in hypothalamus-pituitary-adrenal axis following transient ischemic attack. Angiology. 2014;65(8):723–32.
Wong CH, Jenne CN, Lee WY, Leger C, Kubes P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science. 2011;334(6052):101–5.
Soto-Tinoco E, Guerrero-Vargas NN, Buijs RM. Interaction between the hypothalamus and the immune system. Exp Physiol. 2016;101(12):1463–71.
Jin R, Zhu X, Liu L, Nanda A, Granger DN, Li G. Simvastatin attenuates stroke-induced splenic atrophy and lung susceptibility to spontaneous bacterial infection in mice. Stroke. 2013;44(4):1135–43.
Ajmo CT Jr., Collier LA, Leonardo CC, Hall AA, Green SM, Womble TA, et al. Blockade of adrenoreceptors inhibits the splenic response to stroke. Exp Neurol. 2009;218(1):47–55.
Chiu NL, Kaiser B, Nguyen YV, Welbourne S, Lall C, Cramer SC. The volume of the spleen and its correlates after Acute Stroke. J Stroke Cerebrovasc Dis. 2016;25(12):2958–61.
Illanes S, Liesz A, Sun L, Dalpke A, Zorn M, Veltkamp R. Hematoma size as major modulator of the cellular immune system after experimental intracerebral hemorrhage. Neurosci Lett. 2011;490(3):170–4.
Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117(2):289–96.
Hanscom M, Loane DJ, Shea-Donohue T. Brain-gut axis dysfunction in the pathogenesis of traumatic brain injury. J Clin Invest. 2021;131(12).
Mangalam AK, Taneja V, David CS. HLA class II molecules influence susceptibility versus protection in inflammatory diseases by determining the cytokine profile. J Immunol. 2013;190(2):513–8.
Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol. 2014;35(3):347–69.
Yu X, Zhou G, Shao B, Zhou H, Xu C, Yan F, et al. Gut Microbiota Dysbiosis Induced by Intracerebral Hemorrhage aggravates Neuroinflammation in mice. Front Microbiol. 2021;12:647304.
Zhao Q, Yan T, Chopp M, Venkat P, Chen J. Brain-kidney interaction: renal dysfunction following ischemic stroke. J Cereb Blood Flow Metab. 2020;40(2):246–62.
Tanaka S, Okusa MD. Crosstalk between the nervous system and the kidney. Kidney Int. 2020;97(3):466–76.
Chen Z, Venkat P, Seyfried D, Chopp M, Yan T, Chen J. Brain-Heart Interaction: Cardiac complications after Stroke. Circ Res. 2017;121(4):451–68.
Battaglini D, Robba C, Lopes da Silva A, Dos Santos Samary C, Leme Silva P, Dal Pizzol F, et al. Brain-heart interaction after acute ischemic stroke. Crit Care. 2020;24(1):163.
Samuels MA. The brain-heart connection. Circulation. 2007;116(1):77–84.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8.
Vink R, Gabrielian L, Thornton E. The role of substance P in secondary pathophysiology after traumatic brain Injury. Front Neurol. 2017;8:304.
Kubes P, Ward PA. Leukocyte recruitment and the acute inflammatory response. Brain Pathol. 2000;10(1):127–35.
Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;26(9):485–95.
Povlishock JT, Becker DP, Sullivan HG, Miller JD. Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain Res. 1978;153(2):223–39.
Atangana E, Schneider UC, Blecharz K, Magrini S, Wagner J, Nieminen-Kelha M, et al. Intravascular inflammation triggers Intracerebral activated Microglia and contributes to secondary brain Injury after experimental subarachnoid hemorrhage (eSAH). Transl Stroke Res. 2017;8(2):144–56.
Gauberti M, Montagne A, Marcos-Contreras OA, Le Behot A, Maubert E, Vivien D. Ultra-sensitive molecular MRI of vascular cell adhesion molecule-1 reveals a dynamic inflammatory penumbra after strokes. Stroke. 2013;44(7):1988–96.
Quenault A, Martinez de Lizarrondo S, Etard O, Gauberti M, Orset C, Haelewyn B, et al. Molecular magnetic resonance imaging discloses endothelial activation after transient ischaemic attack. Brain. 2017;140(1):146–57.
Siler DA, Berlow YA, Kukino A, Davis CM, Nelson JW, Grafe MR, et al. Soluble Epoxide Hydrolase in Hydrocephalus, cerebral edema, and vascular inflammation after subarachnoid hemorrhage. Stroke. 2015;46(7):1916–22.
Gob E, Reymann S, Langhauser F, Schuhmann MK, Kraft P, Thielmann I, et al. Blocking of plasma kallikrein ameliorates stroke by reducing thromboinflammation. Ann Neurol. 2015;77(5):784–803.
De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F, Kleinschnitz C. Thromboinflammation in stroke brain damage. Stroke. 2016;47(4):1165–72.
Villringer K, Sanz Cuesta BE, Ostwaldt AC, Grittner U, Brunecker P, Khalil AA, et al. DCE-MRI blood-brain barrier assessment in acute ischemic stroke. Neurology. 2017;88(5):433–40.
Mehta V, Russin J, Spirtos A, He S, Adamczyk P, Amar AP, Mack WJ. Matrix metalloproteinases in Cerebral Vasospasm following Aneurysmal Subarachnoid Hemorrhage. Neurol Res Int. 2013;2013:943761.
Guo Z, Sun X, He Z, Jiang Y, Zhang X, Zhang JH. Matrix metalloproteinase-9 potentiates early brain injury after subarachnoid hemorrhage. Neurol Res. 2010;32(7):715–20.
Peeyush Kumar T, McBride DW, Dash PK, Matsumura K, Rubi A, Blackburn SL. Endothelial cell dysfunction and Injury in Subarachnoid Hemorrhage. Mol Neurobiol. 2019;56(3):1992–2006.
Neumann J, Riek-Burchardt M, Herz J, Doeppner TR, Konig R, Hutten H, et al. Very-late-antigen-4 (VLA-4)-mediated brain invasion by neutrophils leads to interactions with microglia, increased ischemic injury and impaired behavior in experimental stroke. Acta Neuropathol. 2015;129(2):259–77.
Fassbender K, Hodapp B, Rossol S, Bertsch T, Schmeck J, Schutt S, et al. Inflammatory cytokines in subarachnoid haemorrhage: association with abnormal blood flow velocities in basal cerebral arteries. J Neurol Neurosurg Psychiatry. 2001;70(4):534–7.
Blecharz-Lang KG, Wagner J, Fries A, Nieminen-Kelha M, Rosner J, Schneider UC, Vajkoczy P. Interleukin 6-Mediated endothelial barrier disturbances can be attenuated by blockade of the IL6 receptor expressed in Brain Microvascular endothelial cells. Transl Stroke Res. 2018;9(6):631–42.
Schneider UC, Xu R, Vajkoczy P. Inflammatory events following subarachnoid hemorrhage (SAH). Curr Neuropharmacol. 2018;16(9):1385–95.
Ma Q, Ineichen BV, Detmar M, Proulx ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun. 2017;8(1):1434.
Absinta M, Ha SK, Nair G, Sati P, Luciano NJ, Palisoc M et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife. 2017;6.
Saghazadeh A, Rezaei N. The role of timing in the treatment of spinal cord injury. Biomed Pharmacother. 2017;92:128–39.
Robert SM, Reeves BC, Kiziltug E, Duy PQ, Karimy JK, Mansuri MS, et al. The choroid plexus links innate immunity to CSF dysregulation in hydrocephalus. Cell. 2023;186(4):764–85.e21.
Dando SJ, Mackay-Sim A, Norton R, Currie BJ, St John JA, Ekberg JA, et al. Pathogens penetrating the central nervous system: infection pathways and the cellular and molecular mechanisms of invasion. Clin Microbiol Rev. 2014;27(4):691–726.
Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10(5):514–23.
Vercellino M, Votta B, Condello C, Piacentino C, Romagnolo A, Merola A, et al. Involvement of the choroid plexus in multiple sclerosis autoimmune inflammation: a neuropathological study. J Neuroimmunol. 2008;199(1–2):133–41.
Rodriguez-Lorenzo S, Konings J, van der Pol S, Kamermans A, Amor S, van Horssen J, et al. Inflammation of the choroid plexus in progressive multiple sclerosis: accumulation of granulocytes and T cells. Acta Neuropathol Commun. 2020;8(1):9.
Kooij G, Kopplin K, Blasig R, Stuiver M, Koning N, Goverse G, et al. Disturbed function of the blood-cerebrospinal fluid barrier aggravates neuro-inflammation. Acta Neuropathol. 2014;128(2):267–77.
Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci U S A. 2014;111(11):E1035–42.
Witt KA, Mark KS, Hom S, Davis TP. Effects of hypoxia-reoxygenation on rat blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. 2003;285(6):H2820–31.
Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125(1):111–20.
Herisson F, Frodermann V, Courties G, Rohde D, Sun Y, Vandoorne K, et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci. 2018;21(9):1209–17.
McKittrick CM, Lawrence CE, Carswell HV. Mast cells promote blood brain barrier breakdown and neutrophil infiltration in a mouse model of focal cerebral ischemia. J Cereb Blood Flow Metab. 2015;35(4):638–47.
Arac A, Grimbaldeston MA, Nepomuceno AR, Olayiwola O, Pereira MP, Nishiyama Y, et al. Evidence that meningeal mast cells can worsen stroke pathology in mice. Am J Pathol. 2014;184(9):2493–504.
Sellner J, Leib SL. In bacterial meningitis cortical brain damage is associated with changes in parenchymal MMP-9/TIMP-1 ratio and increased collagen type IV degradation. Neurobiol Dis. 2006;21(3):647–56.
Zenker W, Kubik S. Brain cooling in humans–anatomical considerations. Anat Embryol (Berl). 1996;193(1):1–13.
Rangel de Lazaro G, de la Cuetara JM, Pisova H, Lorenzo C, Bruner E. Diploic vessels and computed tomography: segmentation and comparison in modern humans and fossil hominids. Am J Phys Anthropol. 2016;159(2):313–24.
Adeeb N, Mortazavi MM, Tubbs RS, Cohen-Gadol AA. The cranial dura mater: a review of its history, embryology, and anatomy. Childs Nerv Syst. 2012;28(6):827–37.
Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. 2011;365(23):2188–97.
Park L, Uekawa K, Garcia-Bonilla L, Koizumi K, Murphy M, Pistik R, et al. Brain perivascular macrophages initiate the neurovascular dysfunction of Alzheimer abeta peptides. Circ Res. 2017;121(3):258–69.
Faraco G, Park L, Anrather J, Iadecola C. Brain perivascular macrophages: characterization and functional roles in health and disease. J Mol Med (Berl). 2017;95(11):1143–52.
Abtin A, Jain R, Mitchell AJ, Roediger B, Brzoska AJ, Tikoo S, et al. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat Immunol. 2014;15(1):45–53.
Szmydynger-Chodobska J, Shan R, Thomasian N, Chodobski A. The involvement of Pial Microvessels in Leukocyte Invasion after mild traumatic brain Injury. PLoS ONE. 2016;11(12):e0167677.
Bartholomaus I, Kawakami N, Odoardi F, Schlager C, Miljkovic D, Ellwart JW, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. 2009;462(7269):94–8.
Schlager C, Korner H, Krueger M, Vidoli S, Haberl M, Mielke D, et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature. 2016;530(7590):349–53.
Finger CE, Moreno-Gonzalez I, Gutierrez A, Moruno-Manchon JF, McCullough LD. Age-related immune alterations and cerebrovascular inflammation. Mol Psychiatry. 2022;27(2):803–18.
Chen Q, Wu M, Tang Q, Yan P, Zhu L. Age-related alterations in Immune function and inflammation: focus on ischemic stroke. Aging Dis. 2023.
Androvic P, Kirdajova D, Tureckova J, Zucha D, Rohlova E, Abaffy P, et al. Decoding the Transcriptional response to ischemic stroke in young and aged mouse brain. Cell Rep. 2020;31(11):107777.
Malaguarnera L, Ferlito L, Imbesi RM, Gulizia GS, Di Mauro S, Maugeri D, et al. Immunosenescence: a review. Arch Gerontol Geriatr. 2001;32(1):1–14.
Liberale L, Bonetti NR, Puspitasari YM, Vukolic A, Akhmedov A, Diaz-Canestro C, et al. TNF-alpha antagonism rescues the effect of ageing on stroke: perspectives for targeting inflamm-ageing. Eur J Clin Invest. 2021;51(11):e13600.
Iadecola C, Buckwalter MS, Anrather J. Immune responses to stroke: mechanisms, modulation, and therapeutic potential. J Clin Invest. 2020;130(6):2777–88.
Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–72.
Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21(12):1424–35.
Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10(4):217–24.
Doyle KP, Cekanaviciute E, Mamer LE, Buckwalter MS. TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflammation. 2010;7:62.
Cohen M, Matcovitch O, David E, Barnett-Itzhaki Z, Keren-Shaul H, Blecher-Gonen R, et al. Chronic exposure to TGFbeta1 regulates myeloid cell inflammatory response in an IRF7-dependent manner. EMBO J. 2014;33(24):2906–21.
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405.
Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 2007;10(1):61–74.
Graves SI, Baker DJ. Implicating endothelial cell senescence to dysfunction in the ageing and diseased brain. Basic Clin Pharmacol Toxicol. 2020;127(2):102–10.
Han Y, Kim SY. Endothelial senescence in vascular diseases: current understanding and future opportunities in senotherapeutics. Exp Mol Med. 2023;55(1):1–12.
Yousef H, Czupalla CJ, Lee D, Chen MB, Burke AN, Zera KA, et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat Med. 2019;25(6):988–1000.
Ge R, Tornero D, Hirota M, Monni E, Laterza C, Lindvall O, Kokaia Z. Choroid plexus-cerebrospinal fluid route for monocyte-derived macrophages after stroke. J Neuroinflammation. 2017;14(1):153.
Castellani G, Croese T, Peralta Ramos JM, Schwartz M. Transforming the understanding of brain immunity. Science. 2023;380(6640):eabo7649.
Negi N, Das BK. CNS: not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int Rev Immunol. 2018;37(1):57–68.
Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017;18(2):123–31.
Khalid SI, Ampie L, Kelly R, Ladha SS, Dardis C. Immune Modulation in the treatment of amyotrophic lateral sclerosis: a review of clinical trials. Front Neurol. 2017;8:486.
Spencer B, Masliah E. Immunotherapy for Alzheimer’s disease: past, present and future. Front Aging Neurosci. 2014;6:114.
Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J. Neuroimmunology of Traumatic Brain Injury: time for a paradigm shift. Neuron. 2017;95(6):1246–65.
Rhind SG, Crnko NT, Baker AJ, Morrison LJ, Shek PN, Scarpelini S, Rizoli SB. Prehospital resuscitation with hypertonic saline-dextran modulates inflammatory, coagulation and endothelial activation marker profiles in severe traumatic brain injured patients. J Neuroinflammation. 2010;7:5.
Ceulemans AG, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J Neuroinflammation. 2010;7:74.
Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab. 2015;35(6):888–901.
Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505(7482):223–8.
Holmin S, Soderlund J, Biberfeld P, Mathiesen T. Intracerebral inflammation after human brain contusion. Neurosurgery. 1998;42(2):291–8. discussion 8–9.
Mracsko E, Javidi E, Na SY, Kahn A, Liesz A, Veltkamp R. Leukocyte invasion of the brain after experimental intracerebral hemorrhage in mice. Stroke. 2014;45(7):2107–14.
Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–75.
Gris T, Laplante P, Thebault P, Cayrol R, Najjar A, Joannette-Pilon B, et al. Innate immunity activation in the early brain injury period following subarachnoid hemorrhage. J Neuroinflammation. 2019;16(1):253.
Weston RM, Jones NM, Jarrott B, Callaway JK. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. J Cereb Blood Flow Metab. 2007;27(1):100–14.
de Oliveira Manoel AL, Macdonald RL. Neuroinflammation as a target for intervention in Subarachnoid Hemorrhage. Front Neurol. 2018;9:292.
Provencio JJ. Inflammation in subarachnoid hemorrhage and delayed deterioration associated with vasospasm: a review. Acta Neurochir Suppl. 2013;115:233–8.
Sarrafzadeh A, Schlenk F, Gericke C, Vajkoczy P. Relevance of cerebral interleukin-6 after aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2010;13(3):339–46.
Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol. 2014;10(1):44–58.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5.
Hao X, Zeng Z, Liang L, Feng Z, Li W, Xiong B, et al. The role of Neutrophil Extracellular traps in Early Microthrombosis and Brain Injury after Subarachnoid Hemorrhage in mice. Transl Stroke Res. 2023;14(5):752–65.
Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014;8(3):883–96.
Zeng H, Fu X, Cai J, Sun C, Yu M, Peng Y, et al. Neutrophil Extracellular traps may be a potential target for treating early Brain Injury in Subarachnoid Hemorrhage. Transl Stroke Res. 2022;13(1):112–31.
Zhou J, Guo P, Hao X, Sun X, Feng H, Chen Z. Neutrophil Extracellular traps (NETs): a New Therapeutic Target for Neuroinflammation and Microthrombosis after Subarachnoid Hemorrhage? Transl Stroke Res. 2023;14(4):443–5.
Chen Y, Galea I, Macdonald RL, Wong GKC, Zhang JH. Rethinking the initial changes in subarachnoid haemorrhage: focusing on real-time metabolism during early brain injury. EBioMedicine. 2022;83:104223.
Tian F, Yuan C, Hu L, Shan S. MicroRNA-93 inhibits inflammatory responses and cell apoptosis after cerebral ischemia reperfusion by targeting interleukin-1 receptor-associated kinase 4. Exp Ther Med. 2017;14(4):2903–10.
Semple BD, Bye N, Ziebell JM, Morganti-Kossmann MC. Deficiency of the chemokine receptor CXCR2 attenuates neutrophil infiltration and cortical damage following closed head injury. Neurobiol Dis. 2010;40(2):394–403.
Xie X, Peng L, Zhu J, Zhou Y, Li L, Chen Y, et al. miR-145-5p/Nurr1/TNF-alpha Signaling-Induced Microglia activation regulates Neuron Injury of Acute Cerebral Ischemic/Reperfusion in rats. Front Mol Neurosci. 2017;10:383.
Zarbock A, Ley K. Neutrophil adhesion and activation under flow. Microcirculation. 2009;16(1):31–42.
Gallia GL, Tamargo RJ. Leukocyte-endothelial cell interactions in chronic vasospasm after subarachnoid hemorrhage. Neurol Res. 2006;28(7):750–8.
Pradilla G, Chaichana KL, Hoang S, Huang J, Tamargo RJ. Inflammation and cerebral vasospasm after subarachnoid hemorrhage. Neurosurg Clin N Am. 2010;21(2):365–79.
Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92(9):3007–17.
Nguyen HX, O’Barr TJ, Anderson AJ. Polymorphonuclear leukocytes promote neurotoxicity through release of matrix metalloproteinases, reactive oxygen species, and TNF-alpha. J Neurochem. 2007;102(3):900–12.
Han N, Ding SJ, Wu T, Zhu YL. Correlation of free radical level and apoptosis after intracerebral hemorrhage in rats. Neurosci Bull. 2008;24(6):351–8.
Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10(6):427–39.
Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27(5):894–908.
Neumann J, Sauerzweig S, Ronicke R, Gunzer F, Dinkel K, Ullrich O, et al. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci. 2008;28(23):5965–75.
Saha P, Geissmann F. Toward a functional characterization of blood monocytes. Immunol Cell Biol. 2011;89(1):2–4.
Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15(10):929–37.
Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, et al. Adult langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med. 2012;209(6):1167–81.
Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV Jr., Broxmeyer HE, Charo IF. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest. 1997;100(10):2552–61.
Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci U S A. 1997;94(22):12053–8.
Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327(5966):656–61.
Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. 2013;153(2):362–75.
Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, et al. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187(4):601–8.
Chu HX, Arumugam TV, Gelderblom M, Magnus T, Drummond GR, Sobey CG. Role of CCR2 in inflammatory conditions of the central nervous system. J Cereb Blood Flow Metab. 2014;34(9):1425–9.
Naert G, Rivest S. A deficiency in CCR2 + monocytes: the hidden side of Alzheimer’s disease. J Mol Cell Biol. 2013;5(5):284–93.
Gliem M, Mausberg AK, Lee JI, Simiantonakis I, van Rooijen N, Hartung HP, Jander S. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol. 2012;71(6):743–52.
Grage-Griebenow E, Zawatzky R, Kahlert H, Brade L, Flad H, Ernst M. Identification of a novel dendritic cell-like subset of CD64(+) / CD16(+) blood monocytes. Eur J Immunol. 2001;31(1):48–56.
Balboa L, Romero MM, Basile JI, Sabio y Garcia CA, Schierloh P, Yokobori N, et al. Paradoxical role of CD16 + CCR2 + CCR5 + monocytes in tuberculosis: efficient APC in pleural effusion but also mark disease severity in blood. J Leukoc Biol. 2011;90(1):69–75.
Fraunberger E, Esser MJ. Neuro-inflammation in Pediatric Traumatic Brain Injury-from mechanisms to Inflammatory Networks. Brain Sci. 2019;9(11).
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10(12):1538–43.
Tanaka R, Komine-Kobayashi M, Mochizuki H, Yamada M, Furuya T, Migita M, et al. Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience. 2003;117(3):531–9.
Kokovay E, Li L, Cunningham LA. Angiogenic recruitment of pericytes from bone marrow after stroke. J Cereb Blood Flow Metab. 2006;26(4):545–55.
Breckwoldt MO, Chen JW, Stangenberg L, Aikawa E, Rodriguez E, Qiu S, et al. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci U S A. 2008;105(47):18584–9.
Schilling M, Besselmann M, Muller M, Strecker JK, Ringelstein EB, Kiefer R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: an investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2005;196(2):290–7.
Schilling M, Strecker JK, Schabitz WR, Ringelstein EB, Kiefer R. Effects of monocyte chemoattractant protein 1 on blood-borne cell recruitment after transient focal cerebral ischemia in mice. Neuroscience. 2009;161(3):806–12.
Bhalala US, Koehler RC, Kannan S. Neuroinflammation and neuroimmune dysregulation after acute hypoxic-ischemic injury of developing brain. Front Pediatr. 2014;2:144.
Kronenberg G, Uhlemann R, Richter N, Klempin F, Wegner S, Staerck L, et al. Distinguishing features of microglia- and monocyte-derived macrophages after stroke. Acta Neuropathol. 2018;135(4):551–68.
Li G, Xiao L, Qin H, Zhuang Q, Zhang W, Liu L, et al. Exosomes-carried microRNA-26b-5p regulates microglia M1 polarization after cerebral ischemia/reperfusion. Cell Cycle. 2020;19(9):1022–35.
Cai G, Cai G, Zhou H, Zhuang Z, Liu K, Pei S, et al. Mesenchymal stem cell-derived exosome mir-542-3p suppresses inflammation and prevents cerebral infarction. Stem Cell Res Ther. 2021;12(1):2.
Beschorner R, Nguyen TD, Gozalan F, Pedal I, Mattern R, Schluesener HJ, et al. CD14 expression by activated parenchymal microglia/macrophages and infiltrating monocytes following human traumatic brain injury. Acta Neuropathol. 2002;103(6):541–9.
Sansing LH, Harris TH, Kasner SE, Hunter CA, Kariko K. Neutrophil depletion diminishes monocyte infiltration and improves functional outcome after experimental intracerebral hemorrhage. Acta Neurochir Suppl. 2011;111:173–8.
Greenhalgh AD, Zarruk JG, Healy LM, Baskar Jesudasan SJ, Jhelum P, Salmon CK, et al. Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PLoS Biol. 2018;16(10):e2005264.
Makinde HM, Cuda CM, Just TB, Perlman HR, Schwulst SJ. Nonclassical monocytes mediate secondary Injury, Neurocognitive Outcome, and Neutrophil Infiltration after Traumatic Brain Injury. J Immunol. 2017;199(10):3583–91.
Yegin O. Chemotaxis in childhood. Pediatr Res. 1983;17(3):183–7.
Klein RB, Fischer TJ, Gard SE, Biberstein M, Rich KC, Stiehm ER. Decreased mononuclear and polymorphonuclear chemotaxis in human newborns, infants, and young children. Pediatrics. 1977;60(4):467–72.
Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, et al. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9(2):268–75.
Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci U S A. 2004;101(21):8180–5.
Lewitus GM, Wilf-Yarkoni A, Ziv Y, Shabat-Simon M, Gersner R, Zangen A, Schwartz M. Vaccination as a novel approach for treating depressive behavior. Biol Psychiatry. 2009;65(4):283–8.
Schwartz M, Shechter R. Protective autoimmunity functions by intracranial immunosurveillance to support the mind: the missing link between health and disease. Mol Psychiatry. 2010;15(4):342–54.
Goldmann J, Kwidzinski E, Brandt C, Mahlo J, Richter D, Bechmann I. T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J Leukoc Biol. 2006;80(4):797–801.
Schiefenhovel F, Immig K, Prodinger C, Bechmann I. Indications for cellular migration from the central nervous system to its draining lymph nodes in CD11c-GFP(+) bone-marrow chimeras following EAE. Exp Brain Res. 2017;235(7):2151–66.
Hatterer E, Davoust N, Didier-Bazes M, Vuaillat C, Malcus C, Belin MF, Nataf S. How to drain without lymphatics? Dendritic cells migrate from the cerebrospinal fluid to the B-cell follicles of cervical lymph nodes. Blood. 2006;107(2):806–12.
Louveau A, Plog BA, Antila S, Alitalo K, Nedergaard M, Kipnis J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J Clin Invest. 2017;127(9):3210–9.
Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, et al. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc Natl Acad Sci U S A. 2013;110(6):2264–9.
Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113(17):2105–12.
Liesz A, Zhou W, Mracsko E, Karcher S, Bauer H, Schwarting S, et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134(Pt 3):704–20.
Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8(7):401–10.
Mracsko E, Veltkamp R. Neuroinflammation after intracerebral hemorrhage. Front Cell Neurosci. 2014;8:388.
Chaitanya GV, Schwaninger M, Alexander JS, Babu PP. Granzyme-b is involved in mediating post-ischemic neuronal death during focal cerebral ischemia in rat model. Neuroscience. 2010;165(4):1203–16.
Cramer JV, Benakis C, Liesz A. T cells in the post-ischemic brain: troopers or paramedics? J Neuroimmunol. 2019;326:33–7.
Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M. Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol Cell Neurosci. 2005;29(3):381–93.
Gudi V, Skuljec J, Yildiz O, Frichert K, Skripuletz T, Moharregh-Khiabani D, et al. Spatial and temporal profiles of growth factor expression during CNS demyelination reveal the dynamics of repair priming. PLoS ONE. 2011;6(7):e22623.
Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, et al. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31(1):149–60.
Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120(18):3793–802.
Li M, Li Z, Yao Y, Jin WN, Wood K, Liu Q, et al. Astrocyte-derived interleukin-15 exacerbates ischemic brain injury via propagation of cellular immunity. Proc Natl Acad Sci U S A. 2017;114(3):E396–405.
Ferrari-Lacraz S, Zanelli E, Neuberg M, Donskoy E, Kim YS, Zheng XX, et al. Targeting IL-15 receptor-bearing cells with an antagonist mutant IL-15/Fc protein prevents disease development and progression in murine collagen-induced arthritis. J Immunol. 2004;173(9):5818–26.
Yoshida S, Ikari K, Yano K, Toyama Y, Taniguchi A, Yamanaka H, Momohara S. Lack of association between IL-15 genetic variants and progression of joint destruction in Japanese patients with rheumatoid arthritis. Ann Rheum Dis. 2014;73(4):784–5.
Huang PL, Hou MS, Wang SW, Chang CL, Liou YH, Liao NS. Skeletal muscle interleukin 15 promotes CD8(+) T-cell function and autoimmune myositis. Skelet Muscle. 2015;5:33.
Quinn LS, Anderson BG, Conner JD, Pistilli EE, Wolden-Hanson T. Overexpression of interleukin-15 in mice promotes resistance to diet-induced obesity, increased insulin sensitivity, and markers of oxidative skeletal muscle metabolism. Int J Interferon Cytokine Mediat Res. 2011;3:29–42.
van Bergen J, Mulder CJ, Mearin ML, Koning F. Local communication among mucosal immune cells in patients with celiac disease. Gastroenterology. 2015;148(6):1187–94.
Saikali P, Antel JP, Pittet CL, Newcombe J, Arbour N. Contribution of astrocyte-derived IL-15 to CD8 T cell effector functions in multiple sclerosis. J Immunol. 2010;185(10):5693–703.
Broux B, Mizee MR, Vanheusden M, van der Pol S, van Horssen J, Van Wijmeersch B, et al. IL-15 amplifies the pathogenic properties of CD4 + CD28- T cells in multiple sclerosis. J Immunol. 2015;194(5):2099–109.
Liston A, Pasciuto E, Fitzgerald DC, Yshii L. Brain regulatory T cells. Nat Rev Immunol. 2023.
Machhi J, Kevadiya BD, Muhammad IK, Herskovitz J, Olson KE, Mosley RL, Gendelman HE. Harnessing regulatory T cell neuroprotective activities for treatment of neurodegenerative disorders. Mol Neurodegener. 2020;15(1):32.
Clausen F, Lorant T, Lewen A, Hillered L. T lymphocyte trafficking: a novel target for neuroprotection in traumatic brain injury. J Neurotrauma. 2007;24(8):1295–307.
Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurol Res. 2008;30(8):783–93.
Kleinschnitz C, Kraft P, Dreykluft A, Hagedorn I, Gobel K, Schuhmann MK, et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood. 2013;121(4):679–91.
Jones KA, Maltby S, Plank MW, Kluge M, Nilsson M, Foster PS, Walker FR. Peripheral immune cells infiltrate into sites of secondary neurodegeneration after ischemic stroke. Brain Behav Immun. 2018;67:299–307.
Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604.
Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098.
Ortega SB, Torres VO, Latchney SE, Whoolery CW, Noorbhai IZ, Poinsatte K, et al. B cells migrate into remote brain areas and support neurogenesis and functional recovery after focal stroke in mice. Proc Natl Acad Sci U S A. 2020;117(9):4983–93.
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43(11):3063–70.
Ankeny DP, Popovich PG. B cells and autoantibodies: complex roles in CNS injury. Trends Immunol. 2010;31(9):332–8.
Goryunova AV, Bazarnaya NA, Sorokina EG, Semenova NY, Globa OV, Semenova Zh B, et al. Glutamate receptor autoantibody concentrations in children with chronic post-traumatic headache. Neurosci Behav Physiol. 2007;37(8):761–4.
Fu Y, Liu Q, Anrather J, Shi FD. Immune interventions in stroke. Nat Rev Neurol. 2015;11(9):524–35.
Feng Y, Liao S, Wei C, Jia D, Wood K, Liu Q, et al. Infiltration and persistence of lymphocytes during late-stage cerebral ischemia in middle cerebral artery occlusion and photothrombotic stroke models. J Neuroinflammation. 2017;14(1):248.
Doyle KP, Quach LN, Sole M, Axtell RC, Nguyen TV, Soler-Llavina GJ, et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci. 2015;35(5):2133–45.
Urday S, Kimberly WT, Beslow LA, Vortmeyer AO, Selim MH, Rosand J, et al. Targeting secondary injury in intracerebral haemorrhage–perihaematomal oedema. Nat Rev Neurol. 2015;11(2):111–22.
Xi G, Strahle J, Hua Y, Keep RF. Progress in translational research on intracerebral hemorrhage: is there an end in sight? Prog Neurobiol. 2014;115:45–63.
Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40(5):1849–57.
Ikegame Y, Yamashita K, Hayashi S, Yoshimura S, Nakashima S, Iwama T. Neutrophil elastase inhibitor prevents ischemic brain damage via reduction of vasogenic edema. Hypertens Res. 2010;33(7):703–7.
Tsai NW, Chang WN, Shaw CF, Jan CR, Lu CH. Leucocyte apoptosis in patients with acute ischaemic stroke. Clin Exp Pharmacol Physiol. 2010;37(9):884–8.
Gokhan S, Ozhasenekler A, Mansur Durgun H, Akil E, Ustundag M, Orak M. Neutrophil lymphocyte ratios in stroke subtypes and transient ischemic attack. Eur Rev Med Pharmacol Sci. 2013;17(5):653–7.
Strecker JK, Sevimli S, Schilling M, Klocke R, Nikol S, Schneider A, Schabitz WR. Effects of G-CSF treatment on neutrophil mobilization and neurological outcome after transient focal ischemia. Exp Neurol. 2010;222(1):108–13.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: N1 versus N2 TAN. Cancer Cell. 2009;16(3):183–94.
Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115(12):1599–608.
Cuartero MI, Ballesteros I, Moraga A, Nombela F, Vivancos J, Hamilton JA, et al. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARgamma agonist rosiglitazone. Stroke. 2013;44(12):3498–508.
Adrover JM, Nicolas-Avila JA, Hidalgo A. Aging: a temporal dimension for neutrophils. Trends Immunol. 2016;37(5):334–45.
Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chevre R, N AG, et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell. 2013;153(5):1025–35.
Yan YP, Sailor KA, Lang BT, Park SW, Vemuganti R, Dempsey RJ. Monocyte chemoattractant protein-1 plays a critical role in neuroblast migration after focal cerebral ischemia. J Cereb Blood Flow Metab. 2007;27(6):1213–24.
Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J, et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci. 2016;36(15):4182–95.
Garcia-Bonilla L, Faraco G, Moore J, Murphy M, Racchumi G, Srinivasan J, et al. Spatio-temporal profile, phenotypic diversity, and fate of recruited monocytes into the post-ischemic brain. J Neuroinflammation. 2016;13(1):285.
Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2003;183(1):25–33.
Sansing LH, Harris TH, Welsh FA, Kasner SE, Hunter CA, Kariko K. Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann Neurol. 2011;70(4):646–56.
Hammond MD, Ai Y, Sansing LH. Gr1 + macrophages and dendritic cells dominate the inflammatory infiltrate 12 hours after experimental intracerebral hemorrhage. Transl Stroke Res. 2012;3(1):s125–31.
Kleinschnitz C, Bendszus M, Frank M, Solymosi L, Toyka KV, Stoll G. In vivo monitoring of macrophage infiltration in experimental ischemic brain lesions by magnetic resonance imaging. J Cereb Blood Flow Metab. 2003;23(11):1356–61.
Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27(11):1798–805.
Jander S, Kraemer M, Schroeter M, Witte OW, Stoll G. Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J Cereb Blood Flow Metab. 1995;15(1):42–51.
Stubbe T, Ebner F, Richter D, Engel O, Klehmet J, Royl G, et al. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab. 2013;33(1):37–47.
Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15(8):946–50.
Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, et al. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115(18):3835–42.
Xie L, Yang SH. Interaction of astrocytes and T cells in physiological and pathological conditions. Brain Res. 2015;1623:63–73.
Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 2019;16(1):142.
Wang J, Xie L, Yang C, Ren C, Zhou K, Wang B, et al. Activated regulatory T cell regulates neural stem cell proliferation in the subventricular zone of normal and ischemic mouse brain through interleukin 10. Front Cell Neurosci. 2015;9:361.
Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol. 2002;52(6):802–13.
Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8(9):963–70.
Kokaia Z, Martino G, Schwartz M, Lindvall O. Cross-talk between neural stem cells and immune cells: the key to better brain repair? Nat Neurosci. 2012;15(8):1078–87.
Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15(2):192–9.
Saino O, Taguchi A, Nakagomi T, Nakano-Doi A, Kashiwamura S, Doe N, et al. Immunodeficiency reduces neural stem/progenitor cell apoptosis and enhances neurogenesis in the cerebral cortex after stroke. J Neurosci Res. 2010;88(11):2385–97.
Xie L, Sun F, Wang J, Mao X, Xie L, Yang SH, et al. mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J Immunol. 2014;192(12):6009–19.
Yang Z, Yu A, Liu Y, Shen H, Lin C, Lin L, et al. Regulatory T cells inhibit microglia activation and protect against inflammatory injury in intracerebral hemorrhage. Int Immunopharmacol. 2014;22(2):522–5.
Ren X, Akiyoshi K, Vandenbark AA, Hurn PD, Offner H. CD4 + FoxP3 + regulatory T-cells in cerebral ischemic stroke. Metab Brain Dis. 2011;26(1):87–90.
Akeret K, Buzzi RM, Schaer CA, Thomson BR, Vallelian F, Wang S, et al. Cerebrospinal fluid hemoglobin drives subarachnoid hemorrhage-related secondary brain injury. J Cereb Blood Flow Metab. 2021;41(11):3000–15.
Ho WM, Gorke AS, Glodny B, Oberacher H, Helbok R, Thome C, Petr O. Time Course of Metabolomic Alterations in Cerebrospinal Fluid after Aneurysmal Subarachnoid Hemorrhage. Front Neurol. 2020;11:589.
Wang KC, Tang SC, Lee JE, Li YI, Huang YS, Yang WS, et al. Cerebrospinal fluid high mobility group box 1 is associated with neuronal death in subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2017;37(2):435–43.
Andersen CR, Presseau J, Saigle V, Etminan N, Vergouwen MDI, English SW. Outcomes in Subarachnoid Haemorrhage Working G. Core outcomes for subarachnoid haemorrhage. Lancet Neurol. 2019;18(12):1075–6.
Provencio JJ, Fu X, Siu A, Rasmussen PA, Hazen SL, Ransohoff RM. CSF neutrophils are implicated in the development of vasospasm in subarachnoid hemorrhage. Neurocrit Care. 2010;12(2):244–51.
Provencio JJ, Altay T, Smithason S, Moore SK, Ransohoff RM. Depletion of Ly6G/C(+) cells ameliorates delayed cerebral vasospasm in subarachnoid hemorrhage. J Neuroimmunol. 2011;232(1–2):94–100.
Hanafy KA. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J Neuroinflammation. 2013;10:83.
Rowland MJ, Hadjipavlou G, Kelly M, Westbrook J, Pattinson KT. Delayed cerebral ischaemia after subarachnoid haemorrhage: looking beyond vasospasm. Br J Anaesth. 2012;109(3):315–29.
Etminan N, Vergouwen MD, Ilodigwe D, Macdonald RL. Effect of pharmaceutical treatment on vasospasm, delayed cerebral ischemia, and clinical outcome in patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. J Cereb Blood Flow Metab. 2011;31(6):1443–51.
Geraghty JR, Testai FD. Delayed cerebral ischemia after subarachnoid hemorrhage: beyond vasospasm and towards a multifactorial pathophysiology. Curr Atheroscler Rep. 2017;19(12):50.
Geraghty JR, Davis JL, Testai FD. Neuroinflammation and Microvascular Dysfunction after experimental subarachnoid hemorrhage: Emerging Components of Early Brain Injury related to Outcome. Neurocrit Care. 2019;31(2):373–89.
McBride DW, Blackburn SL, Peeyush KT, Matsumura K, Zhang JH. The role of thromboinflammation in delayed cerebral ischemia after subarachnoid hemorrhage. Front Neurol. 2017;8:555.
Chamling B, Gross S, Stoffel-Wagner B, Schubert GA, Clusmann H, Coburn M, Hollig A. Early diagnosis of delayed cerebral ischemia: possible relevance for inflammatory biomarkers in routine clinical practice? World Neurosurg. 2017;104:152–7.
Da Silva IRF, Gomes JA, Wachsman A, de Freitas GR, Provencio JJ. Hematologic counts as predictors of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J Crit Care. 2017;37:126–9.
Al-Mufti F, Amuluru K, Damodara N, Dodson V, Roh D, Agarwal S, et al. Admission neutrophil-lymphocyte ratio predicts delayed cerebral ischemia following aneurysmal subarachnoid hemorrhage. J Neurointerv Surg. 2019;11(11):1135–40.
Marchese P, Lardone C, Canepele A, Biondi S, Roggi C, Massart F, et al. Pediatric traumatic brain injury: a new relation between outcome and neutrophil-to-lymphocite ratio. Acta Biomed. 2022;92(S4):e2021417.
Sundd P, Gutierrez E, Koltsova EK, Kuwano Y, Fukuda S, Pospieszalska MK, et al. Slings’ enable neutrophil rolling at high shear. Nature. 2012;488(7411):399–403.
Legg LA, Rudberg AS, Hua X, Wu S, Hackett ML, Tilney R, et al. Selective serotonin reuptake inhibitors (SSRIs) for stroke recovery. Cochrane Database Syst Rev. 2021;11(11):CD009286.
Schneider UC, Davids AM, Brandenburg S, Muller A, Elke A, Magrini S, et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. 2015;130(2):215–31.
Cahill J, Calvert JW, Solaroglu I, Zhang JH. Vasospasm and p53-induced apoptosis in an experimental model of subarachnoid hemorrhage. Stroke. 2006;37(7):1868–74.
Enzmann G, Mysiorek C, Gorina R, Cheng YJ, Ghavampour S, Hannocks MJ, et al. The neurovascular unit as a selective barrier to polymorphonuclear granulocyte (PMN) infiltration into the brain after ischemic injury. Acta Neuropathol. 2013;125(3):395–412.
Coulibaly AP, Provencio JJ. Aneurysmal Subarachnoid Hemorrhage: an overview of inflammation-Induced Cellular Changes. Neurotherapeutics. 2020;17(2):436–45.
Provencio JJ, Swank V, Lu H, Brunet S, Baltan S, Khapre RV, et al. Neutrophil depletion after subarachnoid hemorrhage improves memory via NMDA receptors. Brain Behav Immun. 2016;54:233–42.
Doczi T, Joo F, Adam G, Bozoky B, Szerdahelyi P. Blood-brain barrier damage during the acute stage of subarachnoid hemorrhage, as exemplified by a new animal model. Neurosurgery. 1986;18(6):733–9.
Doczi T, Joo F, Sonkodi S, Adam G. Increased vulnerability of the blood-brain barrier to experimental subarachnoid hemorrhage in spontaneously hypertensive rats. Stroke. 1986;17(3):498–501.
Soares HD, Hicks RR, Smith D, McIntosh TK. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J Neurosci. 1995;15(12):8223–33.
Mao M, Xu Y, Zhang XY, Yang L, An XB, Qu Y, et al. MicroRNA-195 prevents hippocampal microglial/macrophage polarization towards the M1 phenotype induced by chronic brain hypoperfusion through regulating CX3CL1/CX3CR1 signaling. J Neuroinflammation. 2020;17(1):244.
Fassbender K, Hodapp B, Rossol S, Bertsch T, Schmeck J, Schutt S, et al. Endothelin-1 in subarachnoid hemorrhage: an acute-phase reactant produced by cerebrospinal fluid leukocytes. Stroke. 2000;31(12):2971–5.
Zheng VZ, Wong GKC. Neuroinflammation responses after subarachnoid hemorrhage: a review. J Clin Neurosci. 2017;42:7–11.
Mohme M, Sauvigny T, Mader MM, Schweingruber N, Maire CL, Runger A, et al. Immune characterization in Aneurysmal Subarachnoid Hemorrhage reveals distinct monocytic activation and chemokine patterns. Transl Stroke Res. 2020;11(6):1348–61.
Johnston SC, Selvin S, Gress DR. The burden, trends, and demographics of mortality from subarachnoid hemorrhage. Neurology. 1998;50(5):1413–8.
Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44(3):450–62.
Minogue AM. Role of infiltrating monocytes/macrophages in acute and chronic neuroinflammation: effects on cognition, learning and affective behaviour. Prog Neuropsychopharmacol Biol Psychiatry. 2017;79(Pt A):15–8.
Ganz T. Macrophages and Iron Metabolism. Microbiol Spectr. 2016;4(5).
Saber M, Kokiko-Cochran O, Puntambekar SS, Lathia JD, Lamb BT. Triggering receptor expressed on myeloid cells 2 Deficiency alters Acute Macrophage distribution and improves recovery after traumatic brain Injury. J Neurotrauma. 2017;34(2):423–35.
Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, et al. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood. 2016;127(4):473–86.
Wei P, Wang K, Luo C, Huang Y, Misilimu D, Wen H, et al. Cordycepin confers long-term neuroprotection via inhibiting neutrophil infiltration and neuroinflammation after traumatic brain injury. J Neuroinflammation. 2021;18(1):137.
Xu X, Gao W, Cheng S, Yin D, Li F, Wu Y, et al. Anti-inflammatory and immunomodulatory mechanisms of atorvastatin in a murine model of traumatic brain injury. J Neuroinflammation. 2017;14(1):167.
Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab. 2012;32(4):598–611.
Moraes L, Grille S, Morelli P, Mila R, Trias N, Brugnini A, et al. Immune cells subpopulations in cerebrospinal fluid and peripheral blood of patients with Aneurysmal Subarachnoid Hemorrhage. Springerplus. 2015;4:195.
Lim HS, Cordoba SP, Dushek O, Goyette J, Taylor A, Rudd CE, van der Merwe PA. Costimulation of IL-2 production through CD28 is dependent on the size of its ligand. J Immunol. 2015;195(11):5432–9.
Jones KA, Zouikr I, Patience M, Clarkson AN, Isgaard J, Johnson SJ, et al. Chronic stress exacerbates neuronal loss associated with secondary neurodegeneration and suppresses microglial-like cells following focal motor cortex ischemia in the mouse. Brain Behav Immun. 2015;48:57–67.
Dong G, Li C, Hu Q, Wang Y, Sun J, Gao F, et al. Low-dose IL-2 treatment affords Protection against Subarachnoid Hemorrhage Injury by Expanding Peripheral Regulatory T Cells. ACS Chem Neurosci. 2021;12(3):430–40.
Mirlekar B, Patil S, Bopanna R, Chattopadhyay S. MAR binding protein SMAR1 favors IL-10 mediated regulatory T cell function in acute colitis. Biochem Biophys Res Commun. 2015;464(2):647–53.
Saand AR, Yu F, Chen J, Chou SH. Systemic inflammation in hemorrhagic strokes - a novel neurological sign and therapeutic target? J Cereb Blood Flow Metab. 2019;39(6):959–88.
Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y, et al. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol. 2013;74(3):458–71.
Sarrafzadeh A, Schlenk F, Meisel A, Dreier J, Vajkoczy P, Meisel C. Immunodepression after aneurysmal subarachnoid hemorrhage. Stroke. 2011;42(1):53–8.
Jin J, Duan J, Du L, Xing W, Peng X, Zhao Q. Inflammation and immune cell abnormalities in intracranial aneurysm subarachnoid hemorrhage (SAH): relevant signaling pathways and therapeutic strategies. Front Immunol. 2022;13:1027756.
Zhong Q, Jenkins J, Moldobaeva A, D’Alessio F, Wagner EM. Effector T cells and Ischemia-Induced systemic angiogenesis in the lung. Am J Respir Cell Mol Biol. 2016;54(3):394–401.
Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475(7355):226–30.
D’Alessio FR, Zhong Q, Jenkins J, Moldobaeva A, Wagner EM. Lung angiogenesis requires CD4(+) forkhead homeobox Protein-3(+) Regulatory T cells. Am J Respir Cell Mol Biol. 2015;52(5):603–10.
Wang H, Yan FL, Cunningham M, Deng QW, Zuo L, Xing FL, et al. Potential specific immunological indicators for stroke associated infection are partly modulated by sympathetic pathway activation. Oncotarget. 2016;7(32):52404–15.
Fonarow GC, Smith EE, Saver JL, Reeves MJ, Bhatt DL, Grau-Sepulveda MV, et al. Timeliness of tissue-type plasminogen activator therapy in acute ischemic stroke: patient characteristics, hospital factors, and outcomes associated with door-to-needle times within 60 minutes. Circulation. 2011;123(7):750–8.
Gesuete R, Storini C, Fantin A, Stravalaci M, Zanier ER, Orsini F, et al. Recombinant C1 inhibitor in brain ischemic injury. Ann Neurol. 2009;66(3):332–42.
Prestigiacomo CJ, Kim SC, Connolly ES Jr., Liao H, Yan SF, Pinsky DJ. CD18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke. 1999;30(5):1110–7.
Stowe AM, Adair-Kirk TL, Gonzales ER, Perez RS, Shah AR, Park TS, Gidday JM. Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol Dis. 2009;35(1):82–90.
Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A. 2014;111(51):18315–20.
Zhu Z, Fu Y, Tian D, Sun N, Han W, Chang G, et al. Combination of the Immune Modulator Fingolimod with Alteplase in Acute ischemic stroke: a pilot trial. Circulation. 2015;132(12):1104–12.
Nazari M, Keshavarz S, Rafati A, Namavar MR, Haghani M. Fingolimod (FTY720) improves hippocampal synaptic plasticity and memory deficit in rats following focal cerebral ischemia. Brain Res Bull. 2016;124:95–102.
Qin C, Fan WH, Liu Q, Shang K, Murugan M, Wu LJ, et al. Fingolimod protects against ischemic white matter damage by modulating Microglia toward M2 polarization via STAT3 pathway. Stroke. 2017;48(12):3336–46.
Fagan SC, Waller JL, Nichols FT, Edwards DJ, Pettigrew LC, Clark WM, et al. Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke. 2010;41(10):2283–7.
van Eeuwijk JM, Stegner D, Lamb DJ, Kraft P, Beck S, Thielmann I, et al. The novel oral syk inhibitor, Bl1002494, protects mice from arterial thrombosis and thromboinflammatory brain infarction. Arterioscler Thromb Vasc Biol. 2016;36(6):1247–53.
Zhu W, Dotson AL, Libal NL, Lapato AS, Bodhankar S, Offner H, Alkayed NJ. Recombinant T-cell receptor ligand RTL1000 limits inflammation and decreases infarct size after experimental ischemic stroke in middle-aged mice. Neuroscience. 2015;288:112–9.
Dotson AL, Chen Y, Zhu W, Libal N, Alkayed NJ, Offner H. Partial MHC constructs treat thromboembolic ischemic stroke characterized by early Immune expansion. Transl Stroke Res. 2016;7(1):70–8.
Certo M, Endo Y, Ohta K, Sakurada S, Bagetta G, Amantea D. Activation of RXR/PPARgamma underlies neuroprotection by bexarotene in ischemic stroke. Pharmacol Res. 2015;102:298–307.
Sharma S, Yang B, Strong R, Xi X, Brenneman M, Grotta JC, et al. Bone marrow mononuclear cells protect neurons and modulate microglia in cell culture models of ischemic stroke. J Neurosci Res. 2010;88(13):2869–76.
Nakano-Doi A, Nakagomi T, Fujikawa M, Nakagomi N, Kubo S, Lu S, et al. Bone marrow mononuclear cells promote proliferation of endogenous neural stem cells through vascular niches after cerebral infarction. Stem Cells. 2010;28(7):1292–302.
Moniche F, Montaner J, Gonzalez-Marcos JR, Carmona M, Pinero P, Espigado I, et al. Intra-arterial bone marrow mononuclear cell transplantation correlates with GM-CSF, PDGF-BB, and MMP-2 serum levels in stroke patients: results from a clinical trial. Cell Transpl. 2014;23(Suppl 1):S57–64.
Steinberg GK, Kondziolka D, Wechsler LR, Lunsford LD, Coburn ML, Billigen JB, et al. Clinical outcomes of transplanted modified bone marrow-derived mesenchymal stem cells in stroke: a phase 1/2a study. Stroke. 2016;47(7):1817–24.
Prasad K, Sharma A, Garg A, Mohanty S, Bhatnagar S, Johri S, et al. Intravenous autologous bone marrow mononuclear stem cell therapy for ischemic stroke: a multicentric, randomized trial. Stroke. 2014;45(12):3618–24.
Hess DC, Wechsler LR, Clark WM, Savitz SI, Ford GA, Chiu D, et al. Safety and efficacy of multipotent adult progenitor cells in acute ischaemic stroke (MASTERS): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2017;16(5):360–8.
England TJ, Abaei M, Auer DP, Lowe J, Jones DR, Sare G, et al. Granulocyte-colony stimulating factor for mobilizing bone marrow stem cells in subacute stroke: the stem cell trial of recovery enhancement after stroke 2 randomized controlled trial. Stroke. 2012;43(2):405–11.
Cui L, Duchamp NS, Boston DJ, Ren X, Zhang X, Hu H, Zhao LR. NF-kappaB is involved in brain repair by stem cell factor and granulocyte-colony stimulating factor in chronic stroke. Exp Neurol. 2015;263:17–27.
Guo Y, Zhang H, Yang J, Liu S, Bing L, Gao J, Hao A. Granulocyte colony-stimulating factor improves alternative activation of microglia under microenvironment of spinal cord injury. Neuroscience. 2013;238:1–10.
Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, et al. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76(10):1366–72.
Schabitz WR, Dirnagl U. Are we ready to translate T-cell transmigration in stroke? Stroke. 2014;45(6):1610–1.
Li YJ, Chang GQ, Liu Y, Gong Y, Yang C, Wood K, et al. Fingolimod alters inflammatory mediators and vascular permeability in intracerebral hemorrhage. Neurosci Bull. 2015;31(6):755–62.
Xu HL, Pelligrino DA, Paisansathan C, Testai FD. Protective role of fingolimod (FTY720) in rats subjected to subarachnoid hemorrhage. J Neuroinflammation. 2015;12:16.
Wu J, Yang S, Hua Y, Liu W, Keep RF, Xi G. Minocycline attenuates brain edema, brain atrophy and neurological deficits after intracerebral hemorrhage. Acta Neurochir Suppl. 2010;106:147–50.
Han D, Liu H, Gao Y, Feng J. Targeting brain-spleen crosstalk after stroke: New insights into Stroke Pathology and Treatment. Curr Neuropharmacol. 2021;19(9):1590–605.
Li X, Chen G. CNS-peripheral immune interactions in hemorrhagic stroke. J Cereb Blood Flow Metab. 2023;43(2):185–97.
Coulibaly AP, Pezuk P, Varghese P, Gartman W, Triebwasser D, Kulas JA, et al. Neutrophil enzyme myeloperoxidase modulates neuronal response in a model of subarachnoid hemorrhage by venous Injury. Stroke. 2021;52(10):3374–84.
Lu Q, Gao L, Huang L, Ruan L, Yang J, Huang W, et al. Inhibition of mammalian target of rapamycin improves neurobehavioral deficit and modulates immune response after intracerebral hemorrhage in rat. J Neuroinflammation. 2014;11:44.
Lee SH, Park HK, Ryu WS, Lee JS, Bae HJ, Han MK, et al. Effects of celecoxib on hematoma and edema volumes in primary intracerebral hemorrhage: a multicenter randomized controlled trial. Eur J Neurol. 2013;20(8):1161–9.
Graham DI, McIntosh TK, Maxwell WL, Nicoll JA. Recent advances in neurotrauma. J Neuropathol Exp Neurol. 2000;59(8):641–51.
Acknowledgements
Not applicable.
Funding
This work was supported by National Natural Science Foundation of China (No. 82371333 to Yujie Chen, No. 82030036 to Hua Feng) and Chongqing Medical Scientific Research Project (Joint Project of Chongqing Health Commission and Science & Technology Bureau, No. 2023MSXM138 to Yujie Chen).
Author information
Authors and Affiliations
Contributions
YC and HF conceived and designed this review. MD, YX, and YL drafted a substantial portion of the manuscript. MD and YC revised the manuscript. All authors read and approved the present version of the manuscript to be published.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Duan, M., Xu, Y., Li, Y. et al. Targeting brain-peripheral immune responses for secondary brain injury after ischemic and hemorrhagic stroke. J Neuroinflammation 21, 102 (2024). https://doi.org/10.1186/s12974-024-03101-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-024-03101-y