
Abstract
Neuroinflammation is a key element in the ischemic cascade after cerebral ischemia that results in cell damage and death in the subacute phase. However, anti-inflammatory drugs do not improve outcome in clinical settings suggesting that the neuroinflammatory response after an ischemic stroke is not entirely detrimental. This review describes the different key players in neuroinflammation and their possible detrimental and protective effects in stroke. Because of its inhibitory influence on several pathways of the ischemic cascade, hypothermia has been introduced as a promising neuroprotective strategy. This review also discusses the influence of hypothermia on the neuroinflammatory response. We conclude that hypothermia exerts both stimulating and inhibiting effects on different aspects of neuroinflammation and hypothesize that these effects are key to neuroprotection.
Similar content being viewed by others


Introduction
Inflammation is an essential tool to defend oneself against infectious organisms. However, it becomes detrimental when it is prolonged or attacks self antigens [1]. Stroke and neurodegenerative diseases such as Alzheimer's disease, multiple sclerosis and Parkinson's disease are associated with a chronic inflammatory response [2, 3]. After ischemic stroke, the death of ischemic neurons and especially the release of necrotic cell debris triggers inflammation resulting in strong activation of phagocytic cells [1, 4]. Over the past two decades, our understanding of the inflammatory response after stroke and in other diseases has increased due to extensive research. Previously, it was thought that the inflammatory response in brain was beneficial and necessary for repair. Later, it became clear that this "neuro" inflammatory response could be detrimental too, and that even peripheral immune responses can be regulated by the brain [5]. Furthermore, injury to the brain can make the body more vulnerable to systemic infections. For example, a central nervous system injury-induced immunodepression syndrome has been identified in experimental stroke models leading to spontaneous systemic bacterial infections within 3 days after stroke [6–8].
Stroke
Stroke is a broad term that includes conditions caused by occlusion of or hemorrhage from a blood vessel supplying the brain [7, 9]. Its incidence remains high and the number of approved therapies low. As our society ages, the number of stroke patients continues to increase, and will become an important socio-economic burden, as 80% of patients who survive their stroke remain permanently disabled. Ischemic strokes represent more than 80% of all cases and are characterized by the occlusion of a blood vessel due to a thrombus or embolus [10, 11]. The location and the size of the ischemic area of the brain varies, depending on which artery is occluded, thereby causing metabolic and functional dysregulations [12, 13]. Furthermore, the occlusion can be permanent or transient, the latter meaning that reperfusion will occur. After ischemic stroke, two main regions of damage can be defined according to the remaining blood supply. The area in the brain where complete abolishment of blood supply occurred (blood flow reduced to less than 12 ml/100g/min), is called the core of the insult. Complete, or almost complete, energetic failure resulting in necrosis, defines this region. The penumbra is the area surrounding the core which is hypoperfused during the occlusion period. Collateral blood supply from surrounding arteries ensures that a flow of approximately 30 ml/100g/min is maintained. Some energetic metabolism persists in this region and if reperfusion can be restored quickly, this tissue can be salvaged [14–17]. Consequently, the penumbra is an attractive target to rescue brain tissue as this region can remain potentially viable for 16 to 48 hours, enabling clinicians to intervene and reduce post-stroke disability [7, 15].
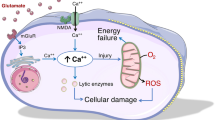
There are excellent reviews elaborating extensively on the complex ischemic cascade after stroke [5, 7, 14, 17, 18]. Briefly and much simplified, three phases can be characterized in infarct progression. The acute phase starts within minutes to a few hours after stroke onset in which the decrease in cerebral blood flow perturbs the ionic homeostasis. This leads to increased intracellular calcium concentrations and stimulation of glutamate release, causing excitotoxicity and a spreading depression throughout the ischemic region [14, 18, 19]. Water shifts to the intracellular space due to osmotic gradients and cells swell. The resulting vasogenic edema can influence reperfusion negatively and causes intracranial pressure, vascular compression and herniation [18]. Furthermore, generation of reactive oxygen species (ROS), especially if reperfusion takes place, can damage membranes, mitochondria and DNA, leading to misfolding of proteins and enzyme dysfunctions. In the second, subacute phase (a few hours to a few days after ischemia), an apoptotic and neuroinflammatory response develops as a result of the stimulatory influences of the acute phase [14, 17, 18]. The high intracellular calcium concentrations built up during the acute phase lead to an overactivation of several proteolytic enzyme systems. Finally, in the chronic phase, which can last up to some months after the actual ischemic stroke, repair and regeneration will determine the ultimate extent of damage [19]. As contradictory as it may appear, reperfusion following an occlusion may exacerbate neuronal injury through increased production of ROS and a stronger inflammatory response [16, 20]. On the other hand, quickly re-establishing the blood supply is vital as it may allow salvation of neurons in the penumbra from irreversible damage, as energy metabolites and cellular membrane ionic gradients would be restored to normal levels [10]. Recanalization is the only approved and effective therapy for stroke to date. Recombinant tissue plasminogen-activator (rt-PA) must be administered within 4.5 hours after stroke onset to restore reperfusion. Unfortunately, due to the short time window and several risk factors, this treatment can only be administered in 5-10% of cases [11, 15]. This emphasizes the need for other neuroprotective therapies not influencing revascularisation, but a strategy that antagonizes, interrupts or slows down injurious biochemical and molecular events in cases treatment with rt-PA is not indicated [21]. Such neuroprotective strategies would primarily focus on reducing the extent of damage in the penumbral region and thus the outcome after stroke [15]. Combining quick reperfusion with the right neuroprotective strategy to address the downstream damaging cascades induced by the occlusion could lead to a better outcome. Nowadays, hypothermia seems to be the most promising neuroprotective therapy that has been translated to clinical practice [22, 23]. Many other proven neuroprotectants in experimental models have not improved outcome after ischemic stroke in clinical settings. Several reasons may explain this discrepancy. Firstly, research is performed in many different stroke models ranging from permanent occlusion models to transient ones. Furthermore, the duration of transient occlusion can range between 30 minutes to 3 hours. Therefore, as functional and structural damage after ischemia depends on its severity and duration, it is important to stress which experimental model was used [9]. Secondly, experiments are generally performed in healthy young rodents. But in reality, most patients who suffer stroke have several risk factors (e.g. age, diabetes and hypertension) which negatively interfere with the ischemic cascade. Thirdly, the adaptation mechanism of high phylogenic species such as human beings is expected to be different from lower phylogenic ones. Most experimental approaches involve occlusion of the middle cerebral artery (MCA), the same artery that is most commonly occluded in clinical stroke [11]. In order to improve the relevance of preclinical studies to clinical practice, STAIR (Stroke Therapy Academic Industry Roundtable) criteria were set up [10, 22]. Neuroprotective agents that target the acute effects of the ischemic cascade (e.g. glutamate release) do not produce any significant improvement in outcome in a clinical setting, partly due to the (too) short time window to start such therapies. Modulation of later events, like inflammation, could lead to successful neuroprotective therapy [18]. However, in clinical trials, anti-inflammatory drugs have not yet shown the expected results [20, 21]. It is hypothesized that such discrepancies indicate that the neuroinflammatory response after stroke is not entirely detrimental and that, without this response, the damage would be even greater. Inflammation causes cell death acutely, but in a later stage, it may help restore body balance [24, 25]. Or, vice versa, chronic inflammation can cause tissue damage as well. Therefore, it is necessary to understand the detrimental and neuroprotective effects of each component of the neuroinflammatory response at different time points after ischemic stroke. This will allow a more specific approach to develop therapies targeting different aspects of neuroinflammation (see Table 1).
The neuroinflammatory response after ischemic stroke
General
Cerebral ischemia leads to the activation of microglia and astrocytes and subsequent production of inflammatory mediators. Such molecules will increase the vulnerability of neurons, cause blood-brain barrier (BBB) disruption and further stimulate gliosis. Consequently, more cell damage and death will occur [7, 26, 27]. Furthermore, cytokines stimulate the expression of adhesion molecules, mediating the adherence and extravasation of neutrophils and monocytes into the ischemic tissue [26]. Local production of chemokines attracts (via chemotaxis) extravasated leukocytes to the ischemic tissue (Figure 1) [26, 28, 29].

Schematic overview of the neuroinflammatory response after ischemic stroke. Microglia become activated after ischemia (grey area) and release pro- and anti-inflammatory mediators. Astrocytes are activated as well and will neglect the maintenance of the neurons, which are most vulnerable to ischemia, and produce neurotoxic and neurotrophic factors. In the ischemic core, neurons die due to necrosis and release necrotic debris into the ischemic tissue, thereby stimulating further activation of glial cells. Astrocytes, together with the attachment of astrocyte endfeet to endothelium and connection with neurons define the neurovascular unit. Neutrophils roll onto the endothelial surface (which is primed by pro-inflammatory cytokines (blue and purple)) until they have slowed down to such a degree that they stick to the endothelium. After binding of selectins to sialyl-Lewisx and CAMs to integrins, the neutrophils undergo conformational changes and flatten. Subsequently, the neutrophils crawl on the endothelium to find an intercellular junction between the endothelial cells for extravasation to the abluminal side and transmigration to the ischemic tissue under the influence of chemokines (red and yellow). Adapted from [26].
Inflammatory mediators
Cytokines
Cytokines represent a family of pleiotropic polypeptides (8-26 kDa) that regulate cell activation, proliferation and differentiation [30, 31]. Normally, cytokines in the brain are hardly detectable. Their receptors are constitutively expressed, albeit at very low levels. After brain injury, pro- and anti-inflammatory cytokines are quickly and extensively upregulated [26, 27, 32]. However, the spatial and temporal upregulation of cytokines and their receptors depends on the ischemic model used [33]. Pro-inflammatory cytokines stimulate and aggravate the inflammatory response. The most prominent ones after stroke are interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α) and IL-6 [17, 27, 33]. On the other hand, anti-inflammatory cytokines inhibit the expression of pro-inflammatory cytokines and reduce inflammation. Transforming growth factor-β (TGF-β) and IL-10 belong to this category and are most studied after ischemic stroke [27, 34]. Other cytokines contribute to tissue damage or repair, but with less pronounced effects than those mentioned. However, cytokines cannot unequivocally be divided into pro- or anti-inflammatory cytokines and may exert neurotoxic as well as neuroprotective effects [12, 30, 34]. The balance between deleterious and beneficial effects of cytokines will depend on the physiological and biochemical context in the brain [34].
IL-1β
A biphasic release pattern has been established for this cytokine in transient as well as permanent MCA occlusion (MCAo) models with a first peak 1 hour after reperfusion and a second peak after 6-24 hours [27, 32, 35]. Activated microglia account for the larger part of the early production of IL-1β, followed by astrocytes, neurons and endothelial cells [36]. Late expression is due to an influx of inflammatory cells into the central nervous system [35, 37]. IL-1β is an endogenous pyrogen which centrally will contribute to an exacerbation of neuronal loss [1]. In the central nervous system, IL-1β stimulates its own production and the expression of other pro-inflammatory mediators such as cytokines and adhesion molecules. Secondly, IL-1β will contribute to the activation and proliferation of microglia and astrocytes. Such a reaction will lead to the upregulation of genes encoding for more neurotoxic mediators, creating a spellbound damaging cycle. Thirdly, IL-1β will stimulate an influx of calcium into neurons, thereby increasing their vulnerability to ischemia. Finally, IL-1β induces edema formation and primes the endothelium for leukocyte adherence [1, 12, 37–39]. But besides all these neurotoxic effects, even IL-1β can contribute to tissue salvation. IL-1β stimulates the activation of astrocytes, which in their turn will produce survival-promoting factors [1, 39]. Moreover, increases in IL-1β will lead to increased levels of IL-1ra, an antagonist of IL-1β. The temporal induction profile of IL-1ra after ischemia virtually parallels that of IL-1β which may suggest that the balance between IL-1β and IL-1ra is more important than the levels of IL-1β itself [18, 39]. In permanent and transient MCAo models, administration of IL-1ra reduces tissue damage significantly [40–42]. Even if the administration is delayed up to 3 hours after a 1-hour MCAo, rats show a reduction of 60% in cortical lesion volume [43]. A clinical phase II safety trial performed in acute stroke patients with the recombinant methionylated form of human Il-1ra (Anakinra or Kineret®) proved the product to be safe and improved functional outcome [39, 44]. Recently, a dose-ranging study has been performed in stroke patients to assess if Anakinra can easily cross the BBB and reach effective concentrations when administered intravenously. The results were favorable and showed that IL-1ra can enter the CSF and that the rate of entry can be modulated by altering the administration regime [45]. If the right therapeutic window can now be determined in patients, this agent might be a promising clinical effective neuroprotective agent after stroke.
TNF-α
Contrary to IL-1β, TNF-α is a more pleiotropic cytokine with neurotoxic and neuroprotective effects [46]. TNF-α also shows a biphasic release pattern with a first peak 1-3 hours and a second peak 24-36 hours after an ischemic insult [27, 32, 47]. However, it is important to make a distinction between soluble and membrane-bound TNF-α [48]. Activated microglia and macrophages are major producers of soluble TNF-α within the first 6 hours after cerebral ischemia [49–51]. Although IL-1β and TNF-α often work synergistically, they are produced by non-overlapping subsets of microglia/macrophages. A maximum of 1.2% of microglia and 4.5% of macrophages co-express IL-1β and TNF-α within the first day after a permanent MCAo [52]. Furthermore, TNF-α may show higher production rates in certain regions (e.g. striatum) and, depending on the region, TNF-α can either be detrimental or beneficial. For example, TNF-α released in the striatum is considered to cause neurodegeneration, while release in the hippocampus could promote neuroprotection [30]. TNF-α can potentiate excitotoxicity in vitro by inhibiting glutamate uptake [30, 53, 54]. It also activates glia, thereby promoting its own production and that of other neurotoxic mediators, leading to more oxidative outburst [50, 53]. Such activation also boosts the production of neurotrophic factors such as BDNF, GDNF and VEGF. Furthermore, TNF-α can increase neuronal vulnerability by stimulating intraneuronal calcium signaling in vitro [55]. TNF-α also controls the (down-) regulation of extracellular calcium in astrocytes, thereby reducing damage [30, 56]. TNF-α stimulates apoptosis of endothelial cells and contributes to vasogenic edema and, due to this BBB breakdown, infiltration of circulatory inflammatory cells are stimulated [57]. On the other hand, TNF-α activates repair processes of the cerebral microvasculature and mediates neuronal plasticity [30]. Perhaps most importantly, TNF-α activates the NF-κB pathway that is involved in signaling cell death (apoptosis) as well as cell survival. NF-κB will stimulate the production of pro-inflammatory cytokines [30, 53, 58]. Ultimately, the balance between the two signals will determine the toxic degree of TNF-α [12, 30, 54]. Several hypotheses exist. One suggests that the detrimental effects occur in the early phase of the inflammatory response and the more beneficial effects in a later stage [24]. Another hypothesis relates to the receptors to which TNF-α binds. Soluble TNF-α (which binds to TNF receptor 1) would cause primarily detrimental effects whereas membrane bound TNF-α (which binds to TNF receptor 2) would signal for neuroprotection [33, 59]. Other studies suggest that TNF-α can also be neuroprotective by acting through TNF receptor 1 [48, 50, 60]. Whatever the case may be, research shows that inhibition of TNF-α, using antibodies given at the onset of reperfusion, reduces infarct volume after a 2-hour MCAo, but only by 16% [41, 47]. In contrast, mice lacking both TNF receptors show increased infarct volume after ischemic stroke [61], and administration of TNF-α 48 hours prior to induction of MCAo results in a neuroprotective effect. Thus, TNF-α can cause ischemic tolerance and may stimulate tissue repair [41]. In conclusion, there is no consensus on the effect of TNF-α after ischemic stroke. Neurotoxic or neuroprotective effects will depend on several factors such as the extent of microglial activation in specific brain regions, timing and threshold of TNF-α expression and of its receptors and on the conditions that stimulate TNF-α signaling [12, 30]. It is important to know in which form TNF-α is induced, in which cells, and on which receptor it will act. For instance, TNF-α in neutrophils and endothelial cells stimulate injury whereas in neurons it is neuroprotective [62].
IL-6
IL-6 is detected 4 hours after stroke onset, with peak concentrations after a day, and remains detectable for up to 14 days [24, 32, 63]. Activated microglia, followed by astrocytes, neurons and invading cells of the immune system, comprise the main source for IL-6 [24, 35]. Like IL-1β, IL-6 is an endogenous pyrogen. It helps to attract T-lymphocytes to the brain and will in general contribute to an exacerbation of the inflammatory response. However, IL-6 can upregulate IL-1ra, and IL-6-deficient mice do not show improved outcome after stroke [12, 26, 64]. In clinical studies, IL-6 is often suggested as a good marker to predict the severity of an insult as high plasma levels correlate with the severity of the stroke [65, 66].
Anti-inflammatory cytokines
Only two of the five different TGF-β cytokines are prominent after stroke; these peak 6 hours after the onset of a lesion. TGF-β1 is primarily produced by activated microglia and macrophages, but some production is also visible in neurons and T-cells [34, 67]. TGF-β2 on the other hand is produced by astrocytes and neurons [68]. Generally, TGF-β controls cellular processes like proliferation, differentiation, apoptosis and migration [63, 69]. More specifically after stroke, it reduces glial activation, decreases the expression and efficacy of other cytokines, suppresses the release of harmful oxygen and nitrogen derived products, promotes angiogenesis in the penumbral area, causes less brain edema with less neutrophil adherence to endothelial cells, and stimulates the release of IL-1ra [41, 70]. However, as TGF-β1 can inhibit apoptosis of neurons, but not necrosis, its possible protective influence is consequently limited to the penumbra. On the other hand, TGF-β1 stimulates glial scar formation and production of beta amyloid precursor, which can lead to a higher risk of cognitive deficit. Although TGF-β works on several inflammatory pathways simultaneously to protect the brain, the endogenous concentration is too low to inhibit serious damage after stroke. Administration of TGF-β before the induction of an ischemic insult rescues neurons from cell death [33, 69, 71].
IL-10 is another constitutively expressed anti-inflammatory cytokine with peak levels 3 days after stroke onset. The primary source is activated microglia and astrocytes. IL-10 inhibits cytokine production and the expression of their receptors and attenuates astrocytic activation. IL-10 administration can reduce damage after MCAo [12, 27, 34].
High-mobility group box (HMGB) protein family
Neuroinflammation can also be triggered by members of the HMGB protein family, which are related to cytokines. They function as sentinels of cellular stress (e.g. necrosis). HMGB proteins are ubiquitously expressed in many cell types. Specific structures mediate their direct binding to DNA and intracellular proteins (e.g. histones and transcription factors) to regulate, among others, DNA structure, repair and transcription [72, 73]. HMGB1 is released in response to pathogen-associated molecular patterns or cellular stress (e.g. stroke) and functions as a pro-inflammatory cytokine [74, 75]. It translocates from the nucleus to the cytoplasm or even completely disappears from the cells within 1 hour after induction of cerebral ischemia in mice [76, 77]. HMGB1 is secreted by neurons and cells of the immune system and passive release also occurs from necrotic cells [77]. Although apoptotic cells release only small amounts of HMGB1, the release of this compound by phagocytosis of apoptotic cells cannot be underestimated [78]. The effects of recombinant HMGB1 have been addressed in vitro as well as in vivo. In glial cell cultures, HMGB1 induces the expression of iNOS, IL-1β and TNF-α; augments excitotoxicity-induced neuronal cell death and induces microglial activation [79]. Microinjection of HMGB1 into the cortex of mice results in increased expression of iNOS and IL-1β [77]. Three cell surface receptors can bind extracellular HMGB1 and activate NF-κB subsequently: Toll-like receptors (TLR) 2 and 4 and the receptor for advanced glycated end products (RAGE). Binding of HMGB1 to RAGE also stimulates chemotaxis [80–82]. A deficiency in these receptors was observed in an MCAo model for cerebral stroke, and this leads to increased infarct size [82]. A role for HMGB1 in cerebral ischemia is indicated by the observation that downregulation of HMGB1 by RNA interference or by using neutralizing antibodies leads to reduced infarct volume and suppression of microglial activation and neuroinflammation in rat and mouse MCAo models [79, 83]. In patients who suffer cerebral ischemia, HMGB1 levels in the serum are increased [84].
Chemokines
Chemokines are small (8-10 kDa), inducible molecules, structurally and functionally related to cytokines, that stand out because of their chemotactic activity for leukocytes [85–87]. There are 40 different known chemokines thus far, which all share a common structural pattern with 4 cysteine residues, which leads to their classification into 4 subfamilies of which 2 are important after stroke: the C-X-C and C-C family [29, 41, 88]. The C-X-C family attracts neutrophils and the C-C family monocytes/macrophages [41, 85]. Constitutively, chemokines and their receptors are expressed in very low concentrations [29, 86]. Cytokines (especially IL-1β and TNF-α) stimulate the production and release of specific chemokines after cerebral ischemia, such as CINC (cytokine induced neutrophil chemoattractant), MCP-1 (monocyte chemoattractant 1), fractalkine, MRF-1 (microglial response factor-1) and MIP-1 (macrophage inflammatory protein 1). These are upregulated in the first 3 hours after ischemic stroke and remain high for at least 6 hours [29]. The earliest part of the production is ascribed to activated microglia, followed by astrocytes and injured neurons [27, 28]. However, depending on cell type, other chemokines are upregulated. While neurons express fractalkine and MCP-1 after cerebral ischemia, microglia show increased expression of MIP-1-α, MIP-2 and MRF-1 [33]. Furthermore, when reperfusion is established, other chemokines, like IL-8, are upregulated as well [26, 89]. Under normal conditions, chemokines control the positioning of cells in tissues and recruit leukocytes to site of inflammation [87]. After brain injury, chemokines serve as signals released into the extracellular fluid and cerebrospinal fluid to recruit microglia, neutrophils and monocytes (chemotaxis) [7, 29, 33]. To achieve leukocyte recruitment, chemokines co-operate with adhesion molecules and affect BBB permeability to ensure diapedesis through the vessel wall [27–29]. Furthermore, chemokines stimulate apoptosis and phagocytosis [28].
IL-8 is a major chemotactic cytokine in humans, but has not been identified in rodents, only its equivalent CINC. Inhibition of CINC, using antibodies, reduces infarct size in a transient MCAo model when given before (1 day) and upon reperfusion [89]. Blocking the activity of MCP-1 with antibodies or MCP-1 deficiency in mice reduces infarct size in transient and permanent MCAo models [90, 91]. Inhibiting the activity of MIP-3-α reduces infarct size after transient MCAo [92]. Fractalkine-deficient mice provide neuroprotection in transient (30 minutes to 2 hours) MCAo models [33, 93, 94]. In general, transgenic mice overexpressing chemokines show chronic neutrophil infiltration, continuous glial activation and BBB disruption. They ultimately die by a terminal wasting syndrome. Chemokine knock-out mice, on the other hand, show deficiency in leukocyte recruitment [29]. Therapy with a broad spectrum pan-chemokine inhibitor, given at the onset of reperfusion, can reduce infarct volume by 50% after a 1-hour MCAo in rats [95]. These animals showed less macrophage accumulation in the peri-infarct area, but not in the core of the insult. This supports the hypothesis that inflammatory cells contribute to extended damage to the penumbra [95].
Free oxygen radicals
Oxidative stress can damage the organism if the physiological balance between oxidants and anti-oxidants is disrupted in favor of the former. Oxidative stress is induced after cerebral ischemia by various pathways of the ischemic cascade; especially inflammation and reperfusion increase the production of ROS [7]. The key radical after stroke is superoxide anion, produced by xanthine oxidase and NADPH oxidase. OH• radicals are increased 2 hours after stroke onset [96]. L-arginin is transformed into nitric oxide (NO) via 3 types of NO synthases: neuronal, endothelial and inducible (n-, e-, iNOS respectively). These NOS are increased after brain ischemia [7]. In rodents, eNOS is upregulated directly whereas nNOS is upregulated a day after ischemia onset and iNOS even later. In the core of the insult, production is seen in neutrophils and macrophages, while at the margins of the infarct it is seen in microglia, blood vessels and astrocytes [38, 60, 96]. NO has actually a dual role. On the one hand, NO can reduce damage after stroke by helping to restore blood supply to the ischemic area; on the other hand, NO can form radicals by reacting, primarily with superoxide anion radical or other molecules with free electrons, to form peroxynitrite [33]. These radicals contribute to lipid peroxidation and protein biochemistry disruption by causing an imbalance between signal transduction mechanisms and cellular toxicity [33]. Furthermore, NO can stimulate the expression of inflammatory mediators and adhesion molecules, induce iron loss from cells, and inhibit enzymes for DNA replication (causing DNA damage) [97]. Which of these contradictory effects will prevail depends on the concentration of NO, the sensitivity of the cell type to NO and the difference between the acute or chronic phase of inflammation [98]. Knock-out mice for nNOS show reduced infarct volumes whereas in knock-out mice for eNOS the injury after ischemic stroke is increased [62, 97]. This suggests that the early production of NO from nNOS leads to tissue damage while NO from eNOS has a protective function by dilating the vessels and thereby regulating blood flow to the penumbra [99]. Depletion of iNOS has no effect on infarct volume after 1 day, but it should be noted that the increase in iNOS occurs at later time points [62, 96, 97].
Matrix metalloproteinases (MMPs)
MMPs are a group of at least 28 zinc-dependent endopeptidases that can proteolytically degrade a variety of molecules that constitute the extracellular matrix components [100–102]. Physiologically, these proteases are involved in tissue development, wound healing, bone growth, ovulation and angiogenesis. After brain trauma, MMPs are upregulated and activated after cleavage of their pro-forms under influence of inflammatory mediators [103, 104]. After a transient MCAo, MMP-2 (gelatinase A) and MMP-9 (gelatinase-B) appear within 3 hours in the core and stay detectable up to 7 days [16, 100, 101]. However, there are studies that report no obvious increase of MMP-2 after ischemic stroke compared to the observed constitutive expression [24, 38, 62, 104, 105]. If MMP-2 is upregulated, it peaks later. MMP-9 is first produced by endothelial cells and neutrophils and later predominantly by macrophages (after 5 days) [106]. Indeed, neutrophils contain and degranulate gelatinase granules resulting in release of pro-MMP-9 and MMP-9 within a day after stroke onset [107]. Although complete neutropenia can reduce the increased levels of MMP-9 after transient MCAo, it cannot entirely abolish the upregulation after ischemic stroke [108]. MMP-9 expression by endothelial cells within and at the periphery of the ischemic lesion is said to be the key mechanism by which the endothelial wall is compromised [106]. The fact that MMP-2 and -9 degrade type IV collagen, laminin and fibronectin, which are major components of the basal lamina around cerebral blood vessels and the extracellular matrix, will certainly underpin this theory [10, 38, 101, 106]. This digestion starts as early as 2 hours after ischemia which correlates with BBB breakdown 3 hours after ischemia [16]. In patients, a correlation could be made between the biphasic opening pattern of the BBB and the involvement of MMPs. In the early reversible opening (after 3 hours), there is an increase in plasma MMP-2. One to 2 days after ischemia, the severe and late opening of the BBB correlates with increased MMP-9 [109]. However, even such destructive molecules have some good qualities. It has been noticed that 7 days post-ischemia, MMPs are involved in plasticity, recovery and repair [109]. The late increase may aid in the migration of macrophages into the ischemic lesion and contribute to clearing of cellular debris [106, 109]. Broad-spectrum inhibitors of MMPs, such as BB-94 and KB-R7785, administered after stroke onset, reduce damage after permanent MCAo in mice by 26% [100, 101]. This is also combined with reduced BBB opening [110]. Such treatments however, cause serious side effects due to their low specificity and explain why these inhibitors have not been used in clinical practice [47, 103]. On that account, more selective inhibitors or knock-out mice for MMP-2 or -9 have been explored. MMP-2 knock-out mice subjected to 2 hours occlusion show massive upregulation of MMP-9 and obviously no improvement could be observed [111]. MMP-9 knock-out mice, on the other hand, do show better outcome after ischemic stroke [107, 112, 113], and inhibition of MMP-9 as late as 4 hours after ischemia onset decreases infarct size [16, 106]. A selective inhibitor for MMP-2 and -9 (SB-3CT) in mice subjected to a 2-hour MCAo reduces lesion size up to 6 hours after ischemia onset, and this inhibitor is well tolerated in animals [103]. Additionally, MMP levels in plasma could be a good biomarker to predict the severity of stroke. In rats, levels of pro-MMP-9 and MMP-9 correlate with infarct volume [105].
Adhesion molecules
Adhesion molecules comprise three groups of molecules that all serve one purpose: adherence and extravasation of neutrophils and monocytes out of the blood vessel, through the BBB into the ischemic tissue. Therefore, some necessary bindings must occur: first selectins bind to neutrophils and integrin receptors on neutrophils bind to cellular adhesion molecules (CAMs) on the endothelial vessel wall [7, 26, 27].
Selectins
Selectins are membrane-bound glycoproteins [26]. There are three selectins, characterized by a common sequence and structural features, E- (endothelial), P- (platelet) and L- (leukocyte) selectin [114]. Selectins bind to carbohydrate residues (sialyl-Lewisx) on neutrophils and monocytes, thereby promoting a first binding to the endothelium which is necessary to slow down neutrophils and monocytes and promote rolling over the endothelium during the early stage of activation [26]. E-selectin is produced by stimulated endothelium and leukocytes and P-selectin also by platelets, hence the name. Stimulation occurs via cytokines and other inflammatory mediators. Four hours after the onset of stroke and up to 70 hours after reperfusion, increased levels of E-selectin are noticed [26]. There is a constitutive expression of P-selectin, although basal levels almost escape routine detection. Due to preformed pools of P-selectin in endothelial cells and platelets, P-selectin can be upregulated quickly and faster than E-selectin after stroke [26, 115]. Looking at the temporal profile of P-selectin, a biphasic release pattern can be distinguished with a first peak 15 minutes after the insult. This then falls back to basal levels within 1 hour after the insult. The second peak, at 6 hours after reperfusion in transient MCAo models, is ascribed to new synthesis of P-selectin protein [116, 117]. Plasma levels of P-selectin are often determined to assess the severity of an insult. This is possible as there are actually 2 isoforms, one with and one without a transmembrane region. The former is expressed on the cell surface and will only be shed after proteolysis while the latter is secreted directly into the circulation from the preformed pools [115]. Inhibiting the activity of P-selectin alone by treatment with monoclonal antibodies (ARP 2-4, RMP-1) after the onset of the insult, does not reduce the infarct volume significantly [26, 118–120]. This suggests that the involvement of P-selectin in the inflammatory response after ischemic injury starts early. The exact function of L-selectin is not known. It is believed that it guides unstimulated leukocytes to the endothelium to facilitate transmigration [26, 27]. However, post-treatment with inhibitors of L-selectin using monoclonal antibodies shows no improvement of lesion size in rabbits subjected to 2 hours occlusion [121]. Inhibition of the binding of selectins to their ligands by administration of a synthetic analogue of sialyl-Lewisx reduced infarct volume after a transient MCAo, even if it was given as late as 2 hours after onset of the insult [119, 122].
Cellular adhesion molecules (CAMs)
The CAMs belong to the immunoglobulin superfamily. After stroke, intracellular adhesion molecule (ICAM) -1 and -2, vascular adhesion molecule (VCAM)-1 and platelet endothelial cell adhesion molecule (PECAM)-1 will contribute to the inflammatory response by attaching neutrophils and monocytes more tightly to the endothelial wall for facilitating and even stimulating diapedesis through the vessel wall to the site of injury [26, 27, 123]. These CAMs are constitutively expressed on cell membrane of endothelial cells, leukocytes, epithelial cells and fibroblasts. Furthermore, higher levels are observed in hypertensive than in normotensive rats [41]. After trauma, inducible ICAM-1 is quickly upregulated, especially in the core of the insult [62]. The increases in ICAM-1 and VCAM-1 after stroke are influenced by IL-1β and TNF-α [26, 27, 124]. ICAM-1 levels peak 12 hours after stroke onset and are maintained during 24 hours whereas VCAM-1 only peaks after a day in a 1-hour MCAo model [24, 27, 123, 124]. Soluble isoforms of these adhesion molecules can be quantified in peripheral blood after they have been shed from the surface of activated cells. In patients, VCAM-1 was increased up to 5 days after symptom onset while ICAM-2 and PECAM-1 were not increased after injury [123, 124]. As these polypeptides contribute to aggravation of damage after stroke, antibodies for these CAMs were evaluated. Administration of a murine anti-ICAM-1 antibody (enlimomab) after a 2-hour MCAo resulted in a decrease in infarct volume of 44% [125, 126]. However, in permanent occlusion models there was no such positive influence of this treatment. Similar results were seen with antisense oligonucleotides for ICAM-1 [127]. These data suggest that there is a need for reperfusion for activation of the inflammatory response [126]. An anti-VCAM-1 monoclonal antibody was tested too, but showed no reduction in infarct size after transient cerebral ischemia in rodents [128]. In a phase III clinical trial, enlimomab was given within 6 hours after symptom onset in cerebral ischemia and maintained over 4 days. Unfortunately, the treated group showed a 43% higher mortality, possibly due to the murine origin of the antibody [20, 124, 129]. Surprisingly, for PECAM-1, there is discussion about possible neuroprotective properties aside from the neurotoxic ones. PECAM-1 knock-out mice show facilitated leukocyte transendothelial migration after histamine treatment which contradicts the current hypothesis that PECAM-1 stimulates migration [123].
Integrins
CAMs bind to integrins on neutrophils and monocytes. After stroke, CD18 or β2 integrins are activated; these can be further divided into 3 different receptors with the same β-chain, but 3 distinct different α-chains, CD11a/CD18 (or LFA-1), CD11b/CD18 (or Mac-1) and CD11c/CD18. The first one primarily binds to ICAM-1 and -2, the second to ICAM-1 and the third to complement fragments [20, 124, 130]. LFA-1 or lymphocyte function-associated antigen-1 is expressed on peripheral blood lymphocytes [131]. Mac-1 or the leukocyte adhesion receptor macrophage-1 antigen is constitutively expressed on leukocytes and is transformed into an activated conformation as well as quantitatively upregulated on the endothelial cell surface after ischemia [130]. Physiologically, integrins connect endothelial cells to components of the underlying basal lamina [41, 132]. CD18 integrin production can be influenced by cytokines (especially TNF-α), chemokines and the adhesion of neutrophils to E-selectin [26, 124, 133]. Due to upregulation of LFA-1 and Mac-1 after stroke, more neutrophils will adhere to the vessel wall and undergo the necessary conformational changes to cross the BBB [133]. Therefore, research has focused on these integrins as potential targets for stroke therapy. Mice with null mutations for LFA-1, subjected to 1-hour occlusion, showed less leukocyte and platelet recruitment compared to their wild types [129]. Mac-1-deficient mice or treatment with monoclonal antibodies showed reduced infarct size after a transient occlusion of 3 hours in mice, but this coincided with serious side effects including peripheral white blood cell depletion [20, 130]. A humanized anti-Mac-1 antibody (called Leukarrest) was evaluated in a phase III clinical trial in which the agent was administered up to 12 hours after symptom onset. The trial showed no improvement in treated subjects, possibly due to the large time-window for treatment onset [20]. Research was also performed with a humanized monoclonal antibody to all CD11/CD18 integrins. In rabbits, post-treatment with this antibody after a 2-hour occlusion attenuates infarct size after the insult [20, 134]. However, no protection was observed in permanent occlusion models [20, 135].
Cellular inflammatory response
There is a hierarchy of vulnerability to ischemia. Unfortunately, neurons are the most vulnerable, followed by astrocytes, microglia and endothelial cells [38, 136]. Not only are other cells less vulnerable, they will also participate in and aggravate the inflammatory response, thereby inducing mostly deleterious but also some beneficial effects [7, 26, 27].
Glia
Glial cells (microglia and astrocytes) are necessary to support the central nervous system.
Microglia represent 5-20% of the total glial population and are key modulators of the immune response in the brain [137, 138]. Their main characteristic is their sensitivity to changes. Quiescent resting microglia show a ramified state characterized by a small cell soma and extensive branches projecting out of the cell body [2, 33, 139]. When a foreign aggressor occurs, microglia change from this monitoring state to one of protection and repair, characterized by an amoeboid form very similar to macrophages and are therefore called "resident" macrophages. Using phagocytosis, microglia will clear foreign organisms [27, 33, 137–140]. However, after ischemia, this immune response turns on its own cells, becomes destructive and will lead to cell damage. How microglia become activated after ischemic stroke is still not clear. A possible mechanism is rupture of necrotic neurons in the core of the insult, leading to release of their contents into the extracellular space and scavenging of these contents by microglia [4, 141]. ROS play a major part in the activation and proliferation of microglia [27]. The penumbra in particular is vulnerable to microglial activation where this causes considerable secondary cell death [33]. Resident macrophages are detected as soon as 2 hours after ischemia onset, whereas blood-born macrophages do not enter the brain before 10 hours. At 22-46 hours, blood-born as well as resident macrophages are distributed over the entire lesion and stay detectable up to 1 week after the insult [13, 14, 32, 52, 137]. Microglia in the core of the insult survive the period of reduced blood flow if it does not exceed 90 minutes of occlusion [142]. Nevertheless, microglia/macrophages surrounding the ischemic tissue will migrate toward the ischemic lesion and engage in close contact with neurons ("capping"). As these neurons die later on, this capping ensures early recognition and fast phagocytic removal of dying/dead neurons [139, 142]. Surprisingly, defective expression of LFA-1 eliminates the capacity of microglial cells to migrate toward injured neurons [131]. In an activated state, microglia will produce inflammatory and cytotoxic mediators contributing to cell damage and cell death. On the other hand, microglia are a major producer of TGF-β1 which supports the hypothesis that microglial activation is also neuroprotective [33]. There are some possible mechanisms supporting this theory. First of all, microglia produce neurotrophic factors which stimulate neurogenesis and plasticity. Secondly, phagocytosis of neutrophils by activated microglia in the lesion prevents the release of extra toxic mediators [2, 143]. Finally, resident macrophages scavenge and remove necrotic debris and harmful components. Indeed, these data suggest that early activation is detrimental and later activation beneficial [2]. Different subsets of microglia may have different roles after ischemic stroke and thus improve or reduce the chances of survival of ischemic neurons [144]. Perhaps an ideal therapy should modulate the microglial response in order to stimulate neurogenesis [145]. Transgenic mice in which microglial proliferation can be inhibited (eliminated) show increased infarct volume (by 13%) after a 1-hour occlusion, which suggests that proliferating resident microglial cells exert a neuroprotective role after ischemia [146].
Astrocytes are essential for the maintenance of the central nervous system. They too will proliferate and differentiate (astrogliosis) after stroke, which coincides with increased production of glial fibrillary acidic protein (GFAP). Astrogliosis can be potentially destructive after an insult [12, 27]. A massive astroglial response, starting in the core of the lesion 4 hours after the trauma, is observed up to 28 days after stroke onset in the photothrombosis model [147]. However, 10 minutes of occlusion results in astrogliosis only after 1 day with peak activity 4 days after the insult [69]. Astrocytes supply energy to neurons, produce precursors of neurotransmitters, form an anti-oxidative defense and secrete neuroprotective and neurogenic factors. Their end-feet form, together with the endothelial cells, the BBB and thereby they contribute to the maintenance of the ionic homeostasis [38, 147, 148]. Astrocytic activation leads to the production of inflammatory mediators and cytotoxic molecules (ROS, NOS, proteases, etc.) [138, 147]. They express high levels of chemokines and chemokine receptors, but it is not proven that astrocytes can migrate toward these factors [86]. Glial scar tissue will replace damaged tissue which has neurotoxic as well as neuroprotective effects. On the one hand, this barrier will prevent axonal ingrowth and reinnervation, thereby impeding recovery. On the other hand, this barrier will isolate the damaged tissue from viable tissue [147].
Endothelial cells and the blood-brain barrier (BBB)
The BBB comprises a ternary structure of endothelial cells with tight junctions distinguishable from the more fenestrated peripheral endothelium, followed by a basal lamina, which is an extension of the extracellular matrix, and finally the end-foot processes of astrocytes [116, 149]. The interconnection is mediated by integrins [41]. This BBB is needed to maintain the fragile extracellular microenvironment via a twofold function. On the one hand, it protects sensitive neurons from contact with potentially toxic activated plasma proteases [148, 150]. On the other hand, the BBB ensures the supply of nutrients by specific transport systems [148]. To improve understanding of the BBB and its effect on neurons, the hypothesis of a neurovascular unit was suggested. This consists of microvessels (with the above mentioned ternary structure) that communicate with astrocytes which in turn are connected to neurons and their axons. Microglia contribute to the integrity of the neurovascular unit. Adjacent neurovascular units communicate with each other [38, 102]. After ischemia, subtle or dynamical changes in BBB permeability occur and can be transient or permanent depending on the severity of the insult. The latter is characterized by endothelial swelling, astrocyte detachment and blood vessel rupture in the ischemic area while transient BBB disruption shows endothelial hyperpermeability to macromolecules in the peri-infarct area [151]. Transient BBB disruption shows a biphasic pattern with an initial opening 2-3 hours after the onset of the insult, while 24-48 hours after reperfusion a second opening occurs, leading to vasogenic edema and increased intracranial pressure [10, 151]. Furthermore, production of pro-inflammatory cytokines and adhesion molecules will be stimulated. Such a disruption results in rapid but significant changes in the molecular relationship between astrocytes and the microvascular extracellular matrix, which has a feedback effect on the neurons they supply and protect [132].
Leukocytes
Neutrophils invade ischemic tissue first, followed by monocytes.
Five hours after reperfusion, neutrophils are already present in damaged tissue [27, 32]. In a permanent MCAo model, neutrophils peak 12 hours after insult onset [63]. Due to increased adhesion molecules after stroke, neutrophils undergo conformational changes (actually modifications in the cellular cytoskeleton) and migrate through the endothelial vessel wall (diapedesis) [26]. After this crossing, chemokines will attract neutrophils to ischemic tissue via a concentration gradient (chemotaxis) [18, 26, 27, 152]. Once there, neutrophils contribute to secondary injury of potentially viable tissue by releasing pro-inflammatory cytokines and other cytotoxic products (proteases, ROS, MMPs) [26, 27, 89]. The release of proteolytic enzymes damage endothelial cell membranes and basal lamina, increases BBB permeability and contributes to post-ischemic edema. Neutrophil activation can alter cerebral artery vasoreactivity [41]. Moreover, due to their adherence onto the vessel wall, they create a secondary obstruction in the cerebral microvasculature, called the no-reflow phenomenon [18, 26, 153]. Inhibiting neutrophil infiltration does improve outcome, but only in transient experimental stroke models [27, 95]. Four to 6 hours after invasion of neutrophils, monocytes adhere to vessel walls as well and migrate into ischemic tissue with peak activity 3-7 days after the onset of the insult [63, 154]. These monocytes will replace the neutrophils [20, 153]. When activated, they transform into blood-borne macrophages. On the one hand, macrophages generate superoxide anions, secrete pro-inflammatory cytokines and create a pro-thrombotic environment, while on the other hand, phagocytosis will remove necrotic cell debris and neutrophils [20].
Hypothermia
As stroke is a multi-faceted phenomenon, a neuroprotective approach of choice should act on several levels of the ischemic cascade. With respect to inflammation, toxic effects should be inhibited while the associated repair mechanisms should be stimulated. Moderate (30-32°C) to mild (32-34°C) hypothermia could be a good option, as it acts on several pathways of the ischemic cascade and exerts differential effects on the inflammatory response at different time points [23]. Indeed, data from the literature indicate that stimulating and inhibiting effects may be involved in the neuroprotective effects of hypothermia. In cardiac arrest, hypothermia has already proven to be neuroprotective in a clinical setting [155]. Furthermore, in experimental models of transient cerebral ischemia, moderate-to-mild hypothermia can reduce infarct volume by more than 50% [156, 157]. However, in permanent MCAo models the effect is less clear.
Fever occurs often after ischemic stroke and increases cell damage and cell death [158]. If hypothermia is induced, more neurons survive the ischemic period [159]. However, the neuroprotective effect of hypothermia is not yet fully elucidated. Hypothermia has no effect on the cerebral blood flow in the penumbra during ischemia, but can block hyperperfusion after ischemic insult [23, 160]. Furthermore, the neuroprotective effect of hypothermia cannot be explained by solely a reduction in cerebral metabolic demand or glutamate release as hypothermia still reduces infarct volume when initiated in the subacute phase of the ischemic cascade, when the initial increase in glutamate has already passed [156, 161, 162]. Consequently, hypothermia affects several pathways.
Hypothermia has effects on the neuroinflammatory response. Unlike anti-inflammatory drugs, hypothermia exerts both stimulating and inhibiting effects on the inflammatory response (see table 1). Hypothermia reduces the increase of IL-1β after stroke [153, 162–164]. It would be interesting to know what a combination of hypothermia with IL-1ra could accomplish. While there is no consensus on the role of TNF-α after stroke, there is also as of yet no consensus on the effect of hypothermia on TNF-α. Most studies show a reduction of TNF-α after hypothermic treatment. More interesting is that mild hypothermia can also influence the expression of TNF-α receptors and the activation of NF-κB [23, 165]. Hypothermia can reduce the increased expression of TNF receptor 1 as early as 15 minutes after traumatic brain injury [165]. This receptor could be associated with detrimental effects after brain injury. It would be interesting to know if hypothermia also decreases the expression of TNF receptor 2. A recent study using a transient MCAo model showed that pre- as well as post-ischemic hypothermia is able to attenuate HMGB1 protein levels in brain and plasma [166]. However, little literature exists regarding the influence of hypothermia on the expression of the main receptors of HMGB1. TNF receptors and TLRs are known to primarily mediate activation of NF-κB. Indeed, hypothermia has anti-apoptotic effects that could also be mediated by affecting NF-κB [96, 153, 157]. In normal conditions, NF-κB is present in the cytoplasm, but is bound to a family of inhibiting proteins. To become activated, IκB kinase must phosphorylate these inhibiting proteins to liberate NF-κB and let it enter the nucleus and induce gene expression [153, 167]. In a 2-hour MCAo model, increased NF-κB binding was observed as early as 2 hours after ischemia. Intra- and post-ischemic hypothermia was able to reduce IκB kinase expression, resulting in less NFκB translocation which could be the result of two pathways [153, 167, 168]: (1) hypothermia may directly influence the expression of IκB kinase, or (2) cells that are protected by hypothermia may influence surrounding cells by releasing fewer inflammatory stimuli [167]. Inhibition of NF-κB activation could be key to the neuroprotective effects of hypothermia, but it should be taken into account that NF-κB also exerts anti-apoptotic effects. Indeed, there are conflicting reports on this subject as both increases and decreases in ischemic injury have been observed when inhibiting the activity of NF-κB [167–169]. Ultimately, it will be the balance between type I and type II TNF-α receptor expression and the signaling pathways involved that will determine the outcome [170]. Chemokines are reduced by hypothermic treatment. Recently, a study showed downregulation of MCP-1 after application of mild hypothermia in vitro [92]. However, these results need to be confirmed in vivo. Hypothermia can suppress oxidative stress after an insult. Superoxide anion radical production is attenuated by a post-ischemic moderate hypothermic treatment [166]. These results also confirm earlier findings that hypothermia reduces the production of hydroxyl radicals after a transient MCAo in rats [96]. Further research has shown that post- but not intra-ischemic hypothermia reduces the release of superoxide anions, as most ROS are only produced in the reperfusion phase [171]. Concerning NOS, it has been shown that hypothermia decreases nNOS by 50% in the penumbra, while no effect is seen on iNOS [96]. Intra-ischemic mild hypothermia reduces expression of MMP-9 in rats subjected to 2 hours of MCAo [104]. There is no effect on MMP-2, neither after stroke nor after hypothermic treatment up to 24 hours after the insult [104]. Also, MMP-9 expression is low in patients after hypothermia, which could suggest a temperature sensitivity of MMPs [172]. In vitro studies show that hypothermia can reversibly inhibit E-selectin, without affecting L-selectin [173, 174]. Furthermore, 2 hours of mild post-ischemic hypothermia reduces ICAM-1 levels in brain and plasma for up to 7 days after 2 hours of MCAo [162, 166, 175]. In vitro, as well as in patients with cardiopulmonary bypass, hypothermia influences integrin expression. Hypothermia (27°C) delays upregulation of Mac-1 and CD11c, but has no effect on LFA-1 [174, 176]. Hypothermia can inhibit but possibly only delay glial activation [177, 178]. Hypothermia appears to decrease tissue density of microglia/macrophages and to inhibit microglial activation. This inhibition leads to a 54% reduction in microglial activation after 3 days if intra- or early post-ischemic hypothermic treatment in rats (subjected to a 2-hour MCAo) is maintained for 2 hours [153, 162, 168, 179]. However, as the phagocytic activity of activated microglia and macrophages exerts beneficial effects in the long term, such parameters should be studied at later time points after the induction of hypothermia. Hypothermia also reduces BBB disruption occurring after stroke, which especially has implications for the passage of large molecules. Permeability to small molecules remains for at least 4 days post-injury [180]. Infiltration of neutrophils and monocytes can be reduced after transient MCAo with post-ischemic mild hypothermic treatment at 3 and 7 days post-injury [162]. This reduction can increase to approximately 75% if the hypothermic treatment is started 1-2 hours after a MCAo of 2 hours [157]. Similar results have been observed in other traumatic injury models [181].
Mild-to-moderate intra-ischemic hypothermia has a pan-inhibiting effect [23, 182]. It reduces ATP depletion, anoxic depolarization, glutamate release, and apoptosis; maintains BBB integrity, inhibits white matter injury and blocks necrosis (if started during ischemia itself) [23, 160, 183]. The effects of hypothermia on inflammation show a more modulating response. However, hypothermia has been suggested to only delay neuronal damage rather than to provide permanent protection. Seven hours after a hypothermic treatment, a hyperthermic episode has been described which may revive or re-initiate pathophysiological mediators which had been quiet [159]. The challenge in hypothermic treatment is thus to know when to start cooling, how long to maintain it and how deep to cool. Deep cooling has no advantage over mild or moderate hypothermia, and actually causes more side effects [157]. Furthermore, a distinction has to be made between short and long cooling periods. It has been suggested that the disadvantages of delayed cooling could be overcome by performing a prolonged hypothermic protocol [22, 182]. However, the modulating influence of hypothermia on neuroinflammation could also differ depending on these parameters and should be further investigated [184]. Depth, start of cooling and duration all influence outcome in both experimental and clinical settings, and make translation to the clinic difficult [185].
Conclusions
The neuroinflammatory response after ischemic stroke involves several parameters, all connected with each other and connected to other pathways of the ischemic cascade. This makes it difficult to draw conclusions from research observations and to extrapolate them to clinical settings, because a limited number of differences between man and animal models may affect multiple levels of the inflammatory response. Many inflammatory components have neurotoxic as well as neuroprotective effects that may either stimulate or reduce cell damage after ischemic stroke. Inhibition of only one part of the neuroinflammatory response after ischemic stroke does not induce sufficient protection to improve recovery in patients. There is sufficient data that hypothermia acts at multiple levels of the ischemic cascade and of the neuroinflammatory response, and does not simply inhibit all processes induced by cerebral ischemia. It should be noted that hypothermia influences the expression of several inflammatory parameters at certain time points, but more research is required to discern which positive or negative effects contribute to neuroprotection. The ability of hypothermia to modulate many aspects of the inflammatory response may render translation to the clinic feasible. Another advantage of hypothermia could be the creation of a larger therapeutic time window to administer other neuroprotective agents and thus improve outcome after transient focal cerebral ischemia.
References
Simi A, Tsakiri N, Wang P, Rothwell NJ: Interleukin-1 and inflammatory neurodegeneration. Biochem Soc Trans. 2007, 35: 1122-1126. 10.1042/BST0351122.
Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG: Does neuroinflammation fan the flame in neurodegenerative diseases?. Mol Neurodegener. 2009, 4: 47-10.1186/1750-1326-4-47.
Lucas SM, Rothwell NJ, Gibson RM: The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006, 147 (Suppl 1): S232-240.
Lelekov-Boissard T, Chapuisat G, Boissel JP, Grenier E, Dronne MA: Exploration of beneficial and deleterious effects of inflammation in stroke: dynamics of inflammation cells. Philos Transact A Math Phys Eng Sci. 2009, 367: 4699-4716. 10.1098/rsta.2009.0184.
Arvin B, Neville LF, Barone FC, Feuerstein GZ: The role of inflammation and cytokines in brain injury. Neurosci Biobehav Rev. 1996, 20: 445-452. 10.1016/0149-7634(95)00026-7.
Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T, Prass K, Meisel A: Stroke-induced immunodepression: experimental evidence and clinical relevance. Stroke. 2007, 38: 770-773. 10.1161/01.STR.0000251441.89665.bc.
Lakhan SE, Kirchgessner A, Hofer M: Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009, 7: 97-10.1186/1479-5876-7-97.
Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U: Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005, 6: 775-786. 10.1038/nrn1765.
Kriz J, Lalancette-Hebert M: Inflammation, plasticity and real-time imaging after cerebral ischemia. Acta Neuropathol. 2009, 117: 497-509. 10.1007/s00401-009-0496-1.
Candelario-Jalil E: Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr Opin Investig Drugs. 2009, 10: 644-654.
Durukan A, Tatlisumak T: Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav. 2007, 87: 179-197. 10.1016/j.pbb.2007.04.015.
Kadhim HJ, Duchateau J, Sebire G: Cytokines and brain injury: invited review. J Intensive Care Med. 2008, 23: 236-249. 10.1177/0885066608318458.
Stoll G, Jander S, Schroeter M: Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol. 1998, 56: 149-171. 10.1016/S0301-0082(98)00034-3.
Dirnagl U, Iadecola C, Moskowitz MA: Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999, 22: 391-397. 10.1016/S0166-2236(99)01401-0.
De Keyser J, Uyttenboogaart M, Koch MW, Elting JW, Sulter G, Vroomen PC, Luijckx GJ: Neuroprotection in acute ischemic stroke. Acta Neurol Belg. 2005, 105: 144-148.
Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH: Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999, 842: 92-100. 10.1016/S0006-8993(99)01843-0.
Hossmann KA: Pathophysiology and therapy of experimental stroke. Cell Mol Neurobiol. 2006, 26: 1057-1083. 10.1007/s10571-006-9008-1.
Barone FC, Feuerstein GZ: Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999, 19: 819-834. 10.1097/00004647-199908000-00001.
Chu LS, Wei EQ, Yu GL, Fang SH, Zhou Y, Wang ML, Zhang WP: Pranlukast reduces neutrophil but not macrophage/microglial accumulation in brain after focal cerebral ischemia in mice. Acta Pharmacol Sin. 2006, 27: 282-288. 10.1111/j.1745-7254.2006.00290.x.
Sughrue ME, Mehra A, Connolly ES, D'Ambrosio AL: Anti-adhesion molecule strategies as potential neuroprotective agents in cerebral ischemia: a critical review of the literature. Inflamm Res. 2004, 53: 497-508. 10.1007/s00011-004-1282-0.
Ginsberg MD: Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008, 55: 363-389. 10.1016/j.neuropharm.2007.12.007.
Kollmar R, Schwab S: Hypothermia in focal ischemia: implications of experiments and experience. J Neurotrauma. 2009, 26: 377-386. 10.1089/neu.2008.0564.
Zhao H, Steinberg GK, Sapolsky RM: General versus specific actions of mild-moderate hypothermia in attenuating cerebral ischemic damage. J Cereb Blood Flow Metab. 2007, 27: 1879-1894. 10.1038/sj.jcbfm.9600540.
Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT: Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. Febs J. 2009, 276: 13-26. 10.1111/j.1742-4658.2008.06766.x.
Somera-Molina KC, Nair S, Van Eldik LJ, Watterson DM, Wainwright MS: Enhanced microglial activation and proinflammatory cytokine upregulation are linked to increased susceptibility to seizures and neurologic injury in a 'two-hit' seizure model. Brain Res. 2009, 1282: 162-172. 10.1016/j.brainres.2009.05.073.
Huang J, Upadhyay UM, Tamargo RJ: Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006, 66: 232-245. 10.1016/j.surneu.2005.12.028.
Wang Q, Tang XN, Yenari MA: The inflammatory response in stroke. J Neuroimmunol. 2007, 184: 53-68. 10.1016/j.jneuroim.2006.11.014.
Bajetto A, Bonavia R, Barbero S, Florio T, Schettini G: Chemokines and their receptors in the central nervous system. Front Neuroendocrinol. 2001, 22: 147-184. 10.1006/frne.2001.0214.
Mennicken F, Maki R, de Souza EB, Quirion R: Chemokines and chemokine receptors in the CNS: a possible role in neuroinflammation and patterning. Trends Pharmacol Sci. 1999, 20: 73-78. 10.1016/S0165-6147(99)01308-5.
Sriram K, O'Callaghan JP: Divergent roles for tumor necrosis factor-alpha in the brain. J Neuroimmune Pharmacol. 2007, 2: 140-153. 10.1007/s11481-007-9070-6.
Zhao B, Schwartz JP: Involvement of cytokines in normal CNS development and neurological diseases: recent progress and perspectives. J Neurosci Res. 1998, 52: 7-16. 10.1002/(SICI)1097-4547(19980401)52:1<7::AID-JNR2>3.0.CO;2-I.
Nilupul Perera M, Ma HK, Arakawa S, Howells DW, Markus R, Rowe CC, Donnan GA: Inflammation following stroke. J Clin Neurosci. 2006, 13: 1-8. 10.1016/j.jocn.2005.07.005.
Lai AY, Todd KG: Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006, 84: 49-59. 10.1139/Y05-143.
Vitkovic L, Maeda S, Sternberg E: Anti-inflammatory cytokines: expression and action in the brain. Neuroimmunomodulation. 2001, 9: 295-312. 10.1159/000059387.
Legos JJ, Whitmore RG, Erhardt JA, Parsons AA, Tuma RF, Barone FC: Quantitative changes in interleukin proteins following focal stroke in the rat. Neurosci Lett. 2000, 282: 189-192. 10.1016/S0304-3940(00)00907-1.
Utagawa A, Truettner JS, Dietrich WD, Bramlett HM: Systemic inflammation exacerbates behavioral and histopathological consequences of isolated traumatic brain injury in rats. Exp Neurol. 2008, 211: 283-291. 10.1016/j.expneurol.2008.02.001.
Rothwell NJ, Luheshi GN: Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci. 2000, 23: 618-625. 10.1016/S0166-2236(00)01661-1.
del Zoppo GJ: Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience. 2009, 158: 972-982. 10.1016/j.neuroscience.2008.08.028.
Rothwell N: Interleukin-1 and neuronal injury: mechanisms, modification, and therapeutic potential. Brain Behav Immun. 2003, 17: 152-157. 10.1016/S0889-1591(02)00098-3.
Clark SR, McMahon CJ, Gueorguieva I, Rowland M, Scarth S, Georgiou R, Tyrrell PJ, Hopkins SJ, Rothwell NJ: Interleukin-1 receptor antagonist penetrates human brain at experimentally therapeutic concentrations. J Cereb Blood Flow Metab. 2008, 28: 387-394. 10.1038/sj.jcbfm.9600537.
Pantoni L, Sarti C, Inzitari D: Cytokines and cell adhesion molecules in cerebral ischemia: experimental bases and therapeutic perspectives. Arterioscler Thromb Vasc Biol. 1998, 18: 503-513.
Banwell V, Sena ES, Macleod MR: Systematic review and stratified meta-analysis of the efficacy of interleukin-1 receptor antagonist in animal models of stroke. J Stroke Cerebrovasc Dis. 2009, 18: 269-276. 10.1016/j.jstrokecerebrovasdis.2008.11.009.
Mulcahy NJ, Ross J, Rothwell NJ, Loddick SA: Delayed administration of interleukin-1 receptor antagonist protects against transient cerebral ischaemia in the rat. Br J Pharmacol. 2003, 140: 471-476. 10.1038/sj.bjp.0705462.
Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, Tyrrell PJ: A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005, 76: 1366-1372. 10.1136/jnnp.2004.054882.
Galea J, Ogungbenro K, Hulme S, Greenhalgh A, Aarons L, Scarth S, Hutchinson P, Grainger S, King A, Hopkins SJ, Rothwell N, Tyrrell P: Intravenous anakinra can achieve experimentally effective concentrations in the central nervous system within a therapeutic time window: results of a dose-ranging study. J Cereb Blood Flow Metab. 2010.
Pan W, Kastin AJ: Tumor necrosis factor and stroke: role of the blood-brain barrier. Prog Neurobiol. 2007, 83: 363-374. 10.1016/j.pneurobio.2007.07.008.
Hosomi N, Ban CR, Naya T, Takahashi T, Guo P, Song XY, Kohno M: Tumor necrosis factor-alpha neutralization reduced cerebral edema through inhibition of matrix metalloproteinase production after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2005, 25: 959-967. 10.1038/sj.jcbfm.9600086.
McCoy MK, Tansey MG: TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008, 5: 45-10.1186/1742-2094-5-45.
Gregersen R, Lambertsen K, Finsen B: Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2000, 20: 53-65. 10.1097/00004647-200001000-00009.
Lambertsen KL, Clausen BH, Babcock AA, Gregersen R, Fenger C, Nielsen HH, Haugaard LS, Wirenfeldt M, Nielsen M, Dagnaes-Hansen F, Bluethmann H, Faergeman N, Meldgaard M, Deierborg T, Finsen B: Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci. 2009, 29: 1319-1330. 10.1523/JNEUROSCI.5505-08.2009.
Pettigrew LC, Kindy MS, Scheff S, Springer JE, Kryscio RJ, Li Y, Grass DS: Focal cerebral ischemia in the TNFalpha-transgenic rat. J Neuroinflammation. 2008, 5: 47-10.1186/1742-2094-5-47.
Clausen BH, Lambertsen KL, Babcock AA, Holm TH, Dagnaes-Hansen F, Finsen B: Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflammation. 2008, 5: 46-10.1186/1742-2094-5-46.
Brabers NA, Nottet HS: Role of the pro-inflammatory cytokines TNF-alpha and IL-1beta in HIV-associated dementia. Eur J Clin Invest. 2006, 36: 447-458. 10.1111/j.1365-2362.2006.01657.x.
Zou JY, Crews FT: TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res. 2005, 1034: 11-24. 10.1016/j.brainres.2004.11.014.
Park KM, Yule DI, Bowers WJ: Tumor necrosis factor-alpha potentiates intraneuronal Ca2+ signaling via regulation of the inositol 1,4,5-triphosphate receptor. J Biol Chem. 2008, 283: 33069-33079. 10.1074/jbc.M802209200.
Köller H, Trimborn M, von Giesen H, Schroeter M, Arendt G: TNFalpha reduces glutamate induced intracellular Ca(2+) increase in cultured cortical astrocytes. Brain Res. 2001, 893: 237-243. 10.1016/S0006-8993(00)03318-7.
Christov A, Ottman JT, Grammas P: Vascular inflammatory, oxidative and protease-based processes: implications for neuronal cell death in Alzheimer's disease. Neurol Res. 2004, 26: 540-546. 10.1179/016164104225016218.
Wilde GJ, Pringle AK, Sundstrom LE, Mann DA, Iannotti F: Attenuation and augmentation of ischaemia-related neuronal death by tumour necrosis factor-alpha in vitro. Eur J Neurosci. 2000, 12: 3863-3870. 10.1046/j.1460-9568.2000.00273.x.
Fontaine L, Meynial-Salles I, Girbal L, Yang X, Croux C, Soucaille P: Molecular characterization and transcriptional analysis of adhE2, the gene encoding the NADH-dependent aldehyde/alcohol dehydrogenase responsible for butanol production in alcohologenic cultures of Clostridium acetobutylicum ATCC 824. J Bacteriol. 2002, 184: 821-830. 10.1128/JB.184.3.821-830.2002.
Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP: Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab. 1998, 18: 1283-1287. 10.1097/00004647-199812000-00001.
Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP: Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996, 2: 788-794. 10.1038/nm0796-788.
Sharp FR, Lu A, Tang Y, Millhorn DE: Multiple molecular penumbras after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000, 20: 1011-1032. 10.1097/00004647-200007000-00001.
Kim JS: Cytokines and adhesion molecules in stroke and related diseases. J Neurol Sci. 1996, 137: 69-78. 10.1016/0022-510X(95)00338-3.
Clark WM, Rinker LG, Lessov NS, Hazel K, Hill JK, Stenzel-Poore M, Eckenstein F: Lack of interleukin-6 expression is not protective against focal central nervous system ischemia. Stroke. 2000, 31: 1715-1720.
Orion D, Schwammenthal Y, Reshef T, Schwartz R, Tsabari R, Merzeliak O, Chapman J, Mekori YA, Tanne D: Interleukin-6 and soluble intercellular adhesion molecule-1 in acute brain ischaemia. Eur J Neurol. 2008, 15: 323-328. 10.1111/j.1468-1331.2008.02066.x.
Tuttolomondo A, Di Sciacca R, Di Raimondo D, Serio A, D'Aguanno G, La Placa S, Pecoraro R, Arnao V, Marino L, Monaco S, Natalè E, Licata G, Pinto A: Plasma levels of inflammatory and thrombotic/fibrinolytic markers in acute ischemic strokes: relationship with TOAST subtype, outcome and infarct site. J Neuroimmunol. 2009, 215: 84-89. 10.1016/j.jneuroim.2009.06.019.
Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R: Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009, 15: 192-199. 10.1038/nm.1927.
Stoll G, Schroeter M, Jander S, Siebert H, Wollrath A, Kleinschnitz C, Bruck W: Lesion-associated expression of transforming growth factor-beta-2 in the rat nervous system: evidence for down-regulating the phagocytic activity of microglia and macrophages. Brain Pathol. 2004, 14: 51-58. 10.1111/j.1750-3639.2004.tb00497.x.
Zhu Y, Roth-Eichhorn S, Braun N, Culmsee C, Rami A, Krieglstein J: The expression of transforming growth factor-beta1 (TGF-beta1) in hippocampal neurons: a temporary upregulated protein level after transient forebrain ischemia in the rat. Brain Res. 2000, 866: 286-298. 10.1016/S0006-8993(00)02240-X.
Pang L, Ye W, Che XM, Roessler BJ, Betz AL, Yang GY: Reduction of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-ss1 expression. Stroke. 2001, 32: 544-552.
Ma M, Ma Y, Yi X, Guo R, Zhu W, Fan X, Xu G, Frey WH, Liu X: Intranasal delivery of transforming growth factor-beta1 in mice after stroke reduces infarct volume and increases neurogenesis in the subventricular zone. BMC Neurosci. 2008, 9: 117-10.1186/1471-2202-9-117.
Stros M: HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. 1799: 101-113.
Ueda T, Yoshida M: HMGB proteins and transcriptional regulation. Biochim Biophys Acta. 1799: 114-118.
Yang H, Tracey KJ: Targeting HMGB1 in inflammation. Biochim Biophys Acta. 1799: 149-156.
Yang H, Wang H, Czura CJ, Tracey KJ: The cytokine activity of HMGB1. J Leukoc Biol. 2005, 78: 1-8. 10.1189/jlb.1104648.
Qiu J, Nishimura M, Wang Y, Sims JR, Qiu S, Savitz SI, Salomone S, Moskowitz MA: Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab. 2008, 28: 927-938. 10.1038/sj.jcbfm.9600582.
Faraco G, Fossati S, Bianchi ME, Patrone M, Pedrazzi M, Sparatore B, Moroni F, Chiarugi A: High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem. 2007, 103: 590-603. 10.1111/j.1471-4159.2007.04788.x.
Qin S, Wang H, Yuan R, Li H, Ochani M, Ochani K, Rosas-Ballina M, Czura CJ, Huston JM, Miller E, Lin X, Sherry B, Kumar A, LaRosa G, Newman W, Tracey KJ, Yang H: Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006, 203: 1637-1642. 10.1084/jem.20052203.
Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL, Park JS, Lee JK: HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci. 2006, 26: 6413-6421. 10.1523/JNEUROSCI.3815-05.2006.
Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP: Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005, 83: 876-886. 10.1007/s00109-005-0688-7.
Palumbo R, Galvez BG, Pusterla T, De Marchis F, Cossu G, Marcu KB, Bianchi ME: Cells migrating to sites of tissue damage in response to the danger signal HMGB1 require NF-kappaB activation. J Cell Biol. 2007, 179: 33-40. 10.1083/jcb.200704015.
Vicente Miranda H, Outeiro TF: The sour side of neurodegenerative disorders: the effects of protein glycation. J Pathol. 221: 13-25. 10.1002/path.2682.
Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G, Nawroth PP, Bierhaus A, Schwaninger M: The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008, 28: 12023-12031. 10.1523/JNEUROSCI.2435-08.2008.
Goldstein RS, Gallowitsch-Puerta M, Yang L, Rosas-Ballina M, Huston JM, Czura CJ, Lee DC, Ward MF, Bruchfeld AN, Wang H, Lesser ML, Church AL, Litroff AH, Sama AE, Tracey KJ: Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock. 2006, 25: 571-574. 10.1097/01.shk.0000209540.99176.72.
Yamagami S, Tamura M, Hayashi M, Endo N, Tanabe H, Katsuura Y, Komoriya K: Differential production of MCP-1 and cytokine-induced neutrophil chemoattractant in the ischemic brain after transient focal ischemia in rats. J Leukoc Biol. 1999, 65: 744-749.
Bacon KB, Harrison JK: Chemokines and their receptors in neurobiology: perspectives in physiology and homeostasis. J Neuroimmunol. 2000, 104: 92-97. 10.1016/S0165-5728(99)00266-0.
Chen Y, Hallenbeck JM, Ruetzler C, Bol D, Thomas K, Berman NE, Vogel SN: Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab. 2003, 23: 748-755. 10.1097/01.WCB.0000071885.63724.20.
Minami M, Satoh M: Chemokines and their receptors in the brain: pathophysiological roles in ischemic brain injury. Life Sci. 2003, 74: 321-327. 10.1016/j.lfs.2003.09.019.
Yamasaki Y, Matsuo Y, Zagorski J, Matsuura N, Onodera H, Itoyama Y, Kogure K: New therapeutic possibility of blocking cytokine-induced neutrophil chemoattractant on transient ischemic brain damage in rats. Brain Res. 1997, 759: 103-111. 10.1016/S0006-8993(97)00251-5.
Che X, Ye W, Panga L, Wu DC, Yang GY: Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 2001, 902: 171-177. 10.1016/S0006-8993(01)02328-9.
Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C: Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab. 2002, 22: 308-317. 10.1097/00004647-200203000-00008.
Terao Y, Ohta H, Oda A, Nakagaito Y, Kiyota Y, Shintani Y: Macrophage inflammatory protein-3alpha plays a key role in the inflammatory cascade in rat focal cerebral ischemia. Neurosci Res. 2009, 64: 75-82. 10.1016/j.neures.2009.01.017.
Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV: Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke. 2007, 38: 1345-1353. 10.1161/01.STR.0000259709.16654.8f.
Soriano SG, Amaravadi LS, Wang YF, Zhou H, Yu GX, Tonra JR, Fairchild-Huntress V, Fang Q, Dunmore JH, Huszar D, Pan Y: Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J Neuroimmunol. 2002, 125: 59-65. 10.1016/S0165-5728(02)00033-4.
Beech JS, Reckless J, Mosedale DE, Grainger DJ, Williams SC, Menon DK: Neuroprotection in ischemia-reperfusion injury: an antiinflammatory approach using a novel broad-spectrum chemokine inhibitor. J Cereb Blood Flow Metab. 2001, 21: 683-689. 10.1097/00004647-200106000-00006.
Van Hemelrijck A, Hachimi-Idrissi S, Sarre S, Ebinger G, Michotte Y: Post-ischaemic mild hypothermia inhibits apoptosis in the penumbral region by reducing neuronal nitric oxide synthase activity and thereby preventing endothelin-1-induced hydroxyl radical formation. Eur J Neurosci. 2005, 22: 1327-1337. 10.1111/j.1460-9568.2005.04331.x.
Loihl AK, Asensio V, Campbell IL, Murphy S: Expression of nitric oxide synthase (NOS)-2 following permanent focal ischemia and the role of nitric oxide in infarct generation in male, female and NOS-2 gene-deficient mice. Brain Res. 1999, 830: 155-164. 10.1016/S0006-8993(99)01388-8.
Liu P, Xu B, Hock CE, Nagele R, Sun FF, Wong PY: NO modulates P-selectin and ICAM-1 mRNA expression and hemodynamic alterations in hepatic I/R. Am J Physiol. 1998, 275: H2191-2198.
Ambrosini A, Louin G, Croci N, Plotkine M, Jafarian-Tehrani M: Characterization of a rat model to study acute neuroinflammation on histopathological, biochemical and functional outcomes. J Neurosci Methods. 2005, 144: 183-191.
Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH: Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000, 20: 1681-1689. 10.1097/00004647-200012000-00007.
Jiang X, Namura S, Nagata I: Matrix metalloproteinase inhibitor KB-R7785 attenuates brain damage resulting from permanent focal cerebral ischemia in mice. Neurosci Lett. 2001, 305: 41-44. 10.1016/S0304-3940(01)01800-6.
del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Berg GI, Koziol JA: Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke. 2007, 38: 646-651. 10.1161/01.STR.0000254477.34231.cb.
Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA: A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005, 25: 6401-6408. 10.1523/JNEUROSCI.1563-05.2005.
Lee JE, Yoon YJ, Moseley ME, Yenari MA: Reduction in levels of matrix metalloproteinases and increased expression of tissue inhibitor of metalloproteinase-2 in response to mild hypothermia therapy in experimental stroke. J Neurosurg. 2005, 103: 289-297. 10.3171/jns.2005.103.2.0289.
Park KP, Rosell A, Foerch C, Xing C, Kim WJ, Lee S, Opdenakker G, Furie KL, Lo EH: Plasma and brain matrix metalloproteinase-9 after acute focal cerebral ischemia in rats. Stroke. 2009, 40: 2836-2842. 10.1161/STROKEAHA.109.554824.
Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC: Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998, 29: 1020-1030.
Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS: Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005, 289: H558-568. 10.1152/ajpheart.01275.2004.
Justicia C, Panes J, Sole S, Cervera A, Deulofeu R, Chamorro A, Planas AM: Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab. 2003, 23: 1430-1440. 10.1097/01.WCB.0000090680.07515.C8.
Lucivero V, Prontera M, Mezzapesa DM, Petruzzellis M, Sancilio M, Tinelli A, Di Noia D, Ruggieri M, Federico F: Different roles of matrix metalloproteinases-2 and -9 after human ischaemic stroke. Neurol Sci. 2007, 28: 165-170. 10.1007/s10072-007-0814-0.
Gasche Y, Fujimura M, Morita-Fujimura Y, Copin JC, Kawase M, Massengale J, Chan PH: Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 1999, 19: 1020-1028. 10.1097/00004647-199909000-00010.
Asahi M, Sumii T, Fini ME, Itohara S, Lo EH: Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. Neuroreport. 2001, 12: 3003-3007. 10.1097/00001756-200109170-00050.
Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH: Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001, 21: 7724-7732.
Svedin P, Hagberg H, Savman K, Zhu C, Mallard C: Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. J Neurosci. 2007, 27: 1511-1518. 10.1523/JNEUROSCI.4391-06.2007.
Haring HP, Berg EL, Tsurushita N, Tagaya M, del Zoppo GJ: E-selectin appears in nonischemic tissue during experimental focal cerebral ischemia. Stroke. 1996, 27: 1386-1391. discussion 1391-1382
Frijns CJ, Kappelle LJ, van Gijn J, Nieuwenhuis HK, Sixma JJ, Fijnheer R: Soluble adhesion molecules reflect endothelial cell activation in ischemic stroke and in carotid atherosclerosis. Stroke. 1997, 28: 2214-2218.
del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ: Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000, 10: 95-112. 10.1111/j.1750-3639.2000.tb00247.x.
Zhang R, Chopp M, Zhang Z, Jiang N, Powers C: The expression of P- and E-selectins in three models of middle cerebral artery occlusion. Brain Res. 1998, 785: 207-214. 10.1016/S0006-8993(97)01343-7.
Connolly ES, Winfree CJ, Prestigiacomo CJ, Kim SC, Choudhri TF, Hoh BL, Naka Y, Solomon RA, Pinsky DJ: Exacerbation of cerebral injury in mice that express the P-selectin gene: identification of P-selectin blockade as a new target for the treatment of stroke. Circ Res. 1997, 81: 304-310.
Goussev AV, Zhang Z, Anderson DC, Chopp M: P-selectin antibody reduces hemorrhage and infarct volume resulting from MCA occlusion in the rat. J Neurol Sci. 1998, 161: 16-22. 10.1016/S0022-510X(98)00262-7.
Suzuki H, Hayashi T, Tojo SJ, Kitagawa H, Kimura K, Mizugaki M, Itoyama Y, Abe K: Anti-P-selectin antibody attenuates rat brain ischemic injury. Neurosci Lett. 1999, 265: 163-166. 10.1016/S0304-3940(99)00229-3.
Yenari MA, Sun GH, Kunis DM, Onley D, Vexler V: L-selectin inhibition does not reduce injury in a rabbit model of transient focal cerebral ischemia. Neurol Res. 2001, 23: 72-78. 10.1179/016164101101198154.
Zhang RL, Chopp M, Zhang ZG, Phillips ML, Rosenbloom CL, Cruz R, Manning A: E-selectin in focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab. 1996, 16: 1126-1136. 10.1097/00004647-199611000-00006.
Kalinowska A, Losy J: PECAM-1, a key player in neuroinflammation. Eur J Neurol. 2006, 13: 1284-1290. 10.1111/j.1468-1331.2006.01640.x.
Frijns CJ, Kappelle LJ: Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke. 2002, 33: 2115-2122. 10.1161/01.STR.0000021902.33129.69.
Shyu KG, Chang H, Lin CC: Serum levels of intercellular adhesion molecule-1 and E-selectin in patients with acute ischaemic stroke. J Neurol. 1997, 244: 90-93. 10.1007/s004150050055.
Zhang RL, Chopp M, Jiang N, Tang WX, Prostak J, Manning AM, Anderson DC: Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke. 1995, 26: 1438-1442. discussion 1443
Vemuganti R, Dempsey RJ, Bowen KK: Inhibition of intercellular adhesion molecule-1 protein expression by antisense oligonucleotides is neuroprotective after transient middle cerebral artery occlusion in rat. Stroke. 2004, 35: 179-184. 10.1161/01.STR.0000106479.53235.3E.
Justicia C, Martin A, Rojas S, Gironella M, Cervera A, Panes J, Chamorro A, Planas AM: Anti-VCAM-1 antibodies did not protect against ischemic damage either in rats or in mice. J Cereb Blood Flow Metab. 2006, 26: 421-432. 10.1038/sj.jcbfm.9600198.
Kusterer K, Bojunga J, Enghofer M, Heidenthal E, Usadel KH, Kolb H, Martin S: Soluble ICAM-1 reduces leukocyte adhesion to vascular endothelium in ischemia-reperfusion injury in mice. Am J Physiol. 1998, 275: G377-380.
Soriano SG, Coxon A, Wang YF, Frosch MP, Lipton SA, Hickey PR, Mayadas TN: Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999, 30: 134-139.
Arumugam TV, Salter JW, Chidlow JH, Ballantyne CM, Kevil CG, Granger DN: Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. Am J Physiol Heart Circ Physiol. 2004, 287: H2555-2560. 10.1152/ajpheart.00588.2004.
Wagner S, Tagaya M, Koziol JA, Quaranta V, del Zoppo GJ: Rapid disruption of an astrocyte interaction with the extracellular matrix mediated by integrin alpha 6 beta 4 during focal cerebral ischemia/reperfusion. Stroke. 1997, 28: 858-865.
Tsai NW, Chang WN, Shaw CF, Jan CR, Huang CR, Chen SD, Chuang YC, Lee LH, Lu CH: The value of leukocyte adhesion molecules in patients after ischemic stroke. J Neurol. 2009, 256: 1296-1302. 10.1007/s00415-009-5117-3.
Yenari MA, Kunis D, Sun GH, Onley D, Watson L, Turner S, Whitaker S, Steinberg GK: Hu23F2G, an antibody recognizing the leukocyte CD11/CD18 integrin, reduces injury in a rabbit model of transient focal cerebral ischemia. Exp Neurol. 1998, 153: 223-233. 10.1006/exnr.1998.6876.
Zhang L, Zhang ZG, Zhang RL, Lu M, Krams M, Chopp M: Effects of a selective CD11b/CD18 antagonist and recombinant human tissue plasminogen activator treatment alone and in combination in a rat embolic model of stroke. Stroke. 2003, 34: 1790-1795. 10.1161/01.STR.0000077016.55891.2E.
Gurer G, Gursoy-Ozdemir Y, Erdemli E, Can A, Dalkara T: Astrocytes are more resistant to focal cerebral ischemia than neurons and die by a delayed necrosis. Brain Pathol. 2009, 19: 630-641. 10.1111/j.1750-3639.2008.00226.x.
Zhang Z, Chopp M, Powers C: Temporal profile of microglial response following transient (2 h) middle cerebral artery occlusion. Brain Res. 1997, 744: 189-198. 10.1016/S0006-8993(96)01085-2.
Schubert P, Morino T, Miyazaki H, Ogata T, Nakamura Y, Marchini C, Ferroni S: Cascading glia reactions: a common pathomechanism and its differentiated control by cyclic nucleotide signaling. Ann N Y Acad Sci. 2000, 903: 24-33. 10.1111/j.1749-6632.2000.tb06346.x.
Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K: Microglia provide neuroprotection after ischemia. Faseb J. 2006, 20: 714-716.
Basu A, Krady JK, Enterline JR, Levison SW: Transforming growth factor beta1 prevents IL-1beta-induced microglial activation, whereas TNFalpha- and IL-6-stimulated activation are not antagonized. Glia. 2002, 40: 109-120. 10.1002/glia.10118.
Kaushal V, Schlichter LC: Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008, 28: 2221-2230. 10.1523/JNEUROSCI.5643-07.2008.
Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, Allan SM: Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab. 2007, 27: 1941-1953. 10.1038/sj.jcbfm.9600495.
Weston RM, Jones NM, Jarrott B, Callaway JK: Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. J Cereb Blood Flow Metab. 2007, 27: 100-114. 10.1038/sj.jcbfm.9600324.
Denes A, Thornton P, Rothwell NJ, Allan SM: Inflammation and brain injury: Acute cerebral ischaemia, peripheral and central inflammation. Brain Behav Immun. 2009
Ekdahl CT, Kokaia Z, Lindvall O: Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience. 2009, 158: 1021-1029. 10.1016/j.neuroscience.2008.06.052.
Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J: Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007, 27: 2596-2605. 10.1523/JNEUROSCI.5360-06.2007.
Nowicka D, Rogozinska K, Aleksy M, Witte OW, Skangiel-Kramska J: Spatiotemporal dynamics of astroglial and microglial responses after photothrombotic stroke in the rat brain. Acta Neurobiol Exp (Wars). 2008, 68: 155-168.
Petty MA, Lo EH: Junctional complexes of the blood-brain barrier: permeability changes in neuroinflammation. Prog Neurobiol. 2002, 68: 311-323. 10.1016/S0301-0082(02)00128-4.
Dietrich PY, Walker PR, Saas P: Death receptors on reactive astrocytes: a key role in the fine tuning of brain inflammation?. Neurology. 2003, 60: 548-554.
Milner R, Hung S, Wang X, Berg GI, Spatz M, del Zoppo GJ: Responses of endothelial cell and astrocyte matrix-integrin receptors to ischemia mimi those observed in the neurovascular unit. Stroke. 2008, 39: 191-197. 10.1161/STROKEAHA.107.486134.
Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV: Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2006, 26: 797-810. 10.1038/sj.jcbfm.9600229.
Yamasaki Y, Matsuo Y, Matsuura N, Onodera H, Itoyama Y, Kogure K: Transient increase of cytokine-induced neutrophil chemoattractant, a member of the interleukin-8 family, in ischemic brain areas after focal ischemia in rats. Stroke. 1995, 26: 318-322. discussion 322-313
Yenari MA, Han HS: Influence of hypothermia on post-ischemic inflammation: role of nuclear factor kappa B (NFkappaB). Neurochem Int. 2006, 49: 164-169. 10.1016/j.neuint.2006.03.016.
Grau AJ, Reis A, Buggle F, Al-Khalaf A, Werle E, Valois N, Bertram M, Becher H, Grond-Ginsbach C: Monocyte function and plasma levels of interleukin-8 in acute ischemic stroke. J Neurol Sci. 2001, 192: 41-47. 10.1016/S0022-510X(01)00590-1.
Hachimi-Idrissi S, Zizi M, Nguyen DN, Schiettecate J, Ebinger G, Michotte Y, Huyghens L: The evolution of serum astroglial S-100 beta protein in patients with cardiac arrest treated with mild hypothermia. Resuscitation. 2005, 64: 187-192. 10.1016/j.resuscitation.2004.08.008.
Van Hemelrijck A, Vermijlen D, Hachimi-Idrissi S, Sarre S, Ebinger G, Michotte Y: Effect of resuscitative mild hypothermia on glutamate and dopamine releae, apoptosis and ischaemic brain damage in the endothelin-1 rat model for focal cerebral ischaemia. J Neurochem. 2003, 87: 66-75. 10.1046/j.1471-4159.2003.01977.x.
Maier CM, Ahern K, Cheng ML, Lee JE, Yenari MA, Steinberg GK: Optimal depth and duration of mild hypothermia in a focal model of transient cerebral ischemia: effects on neurologic outcome, infarct size, apoptosis, and inflammation. Stroke. 1998, 29: 2171-2180.
Gordon CJ: The therapeutic potential of regulated hypothermia. Emerg Med J. 2001, 18: 81-89. 10.1136/emj.18.2.81.
Coimbra C, Drake M, Boris-Moller F, Wieloch T: Long-lasting neuroprotective effect of postischemic hypothermia and treatment with an anti-inflammatory/antipyretic drug. Evidence for chronic encephalopathic processes following ischemia. Stroke. 1996, 27: 1578-1585.
Liu L, Yenari MA: Therapeutic hypothermia: neuroprotective mechanisms. Front Biosci. 2007, 12: 816-825. 10.2741/2104.
Sakoh M, Gjedde A: Neuroprotection in hypothermia linked to redistribution of oxygen in brain. Am J Physiol Heart Circ Physiol. 2003, 285: H17-25.
Wang GJ, Deg HY, Maier CM, Sun GH, Yenari MA: Mild hypothermia reduces ICAM-1 expression, neutrophil infiltration and microglia/monocyte accumulation following experimental stroke. Neuroscience. 2002, 114: 1081-1090.
Truettner JS, Suzuki T, Dietrich WD: The effect of therapeutic hypothermia on the expression of inflammatory response genes following moderate traumatic brain injury in the rat. Brain Res Mol Brain Res. 2005, 138: 124-134. 10.1016/j.molbrainres.2005.04.006.
Zhang H, Zhou M, Zhang J, Mei Y, Sun S, Tong E: Therapeutic effect of post-ischemic hypothermia duration on cerebral ischemic injury. Neurol Res. 2008, 30: 332-336. 10.1179/174313208X300279.
Lotocki G, de Rivero Vaccari JP, Perez ER, Alonso OF, Curbelo K, Keane RW, Dietrich WD: Therapeutic hypothermia modulates TNFR1 signaling in the traumatized brain via early transient activation of the JNK pathway and suppression of XIAP cleavage. Eur J Neurosci. 2006, 24: 2283-2290. 10.1111/j.1460-9568.2006.05123.x.
Koda Y, Tsuruta R, Fujita M, Miyauchi T, Kaneda K, Todani M, Aoki T, Shitara M, Izumi T, Kasaoka S, Yuasa M, Maekawa T: Moderate hypothermia suppresses jugular venous superoxide anion radical, oxidative stress, early inflammation, and endothelial injury in forebrain ischemia/reperfusion rats. Brain Res. 1311: 197-205. 10.1016/j.brainres.2009.11.028.
Han HS, Karabiyikoglu M, Kelly S, Sobel RA, Yenari MA: Mild hypothermia inhibits nuclear factor-kappaB translocation in experimental stroke. J Cereb Blood Flow Metab. 2003, 23: 589-598. 10.1097/01.WCB.0000059566.39780.8D.
Webster CM, Kelly S, Koike MA, Chock VY, Giffard RG, Yenari MA: Inflammation and NFkappaB activation is decreased by hypothermia following global cerebral ischemia. Neurobiol Dis. 2009, 33: 301-312. 10.1016/j.nbd.2008.11.001.
Hill WD, Hess DC, Carroll JE, Wakade CG, Howard EF, Chen Q, Cheng C, Martin-Studdard A, Waller JL, Beswick RA: The NF-kappaB inhibitor diethyldithiocarbamate (DDTC) increases brain cell death in a transient middle cerebral artery occlusion model of ischemia. Brain Res Bull. 2001, 55: 375-386. 10.1016/S0361-9230(01)00503-2.
Baud V, Karin M: Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11: 372-377. 10.1016/S0962-8924(01)02064-5.
Maier CM, Sun GH, Cheng D, Yenari MA, Chan PH, Steinberg GK: Effects of mild hypothermia on superoxide anion production, superoxide dismutase expression, and activity following transient focal cerebral ischemia. Neurobiol Dis. 2002, 11: 28-42. 10.1006/nbdi.2002.0513.
Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S: Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke. 2003, 34: 2165-2170. 10.1161/01.STR.0000088062.86084.F2.
Haddix TL, Pohlman TH, Noel RF, Sato TT, Boyle EM, Verrier ED: Hypothermia inhibits human E-selectin transcription. J Surg Res. 1996, 64: 176-183. 10.1006/jsre.1996.0325.
Le Deist F, Menasche P, Kucharski C, Bel A, Piwnica A, Bloch G: Hypothermia during cardiopulmonary bypass delays but does not prevent neutrophil-endothelial cell adhesion. A clinical study. Circulation. 1995, 92: II354-358.
Deng H, Han HS, Cheng D, Sun GH, Yenari MA: Mild hypothermia inhibits inflammation after experimental stroke and brain inflammation. Stroke. 2003, 34: 2495-2501. 10.1161/01.STR.0000091269.67384.E7.
Rowin ME, Xue V, Irazuzta J: Hypothermia attenuates beta1 integrin expression on extravasated neutrophils in an animal model of meningitis. Inflammation. 2001, 25: 137-144. 10.1023/A:1011044312536.
Han HS, Qiao Y, Karabiyikoglu M, Giffard RG, Yenari MA: Influence of mild hypothermia on inducible nitric oxide synthase expression and reactive nitrogen production in experimental stroke and inflammation. J Neurosci. 2002, 22: 3921-3928.
Inamasu J, Suga S, Sato S, Horiguhi T, Akai K, Mayanagi K, Kawase T: Post-ischemic hypothermia delayed neutrophil accumulation and microglial activation following transient focal ischemia in rats. J Neuroimmunol. 2000, 109: 66-74. 10.1016/S0165-5728(00)00211-3.
Fukui O, Kinugasa Y, Fukuda A, Fukuda H, Tskitishvili E, Hayashi S, Song M, Kanagawa T, Hosono T, Shimoya K, Murata Y: Post-ischemic hypothermia reduced IL-18 expression and suppressed microglial activation in the immature brain. Brain Res. 2006, 1121: 35-45. 10.1016/j.brainres.2006.08.121.
Lotocki G, de Rivero Vaccari JP, Perez ER, Sanchez-Molano J, Furones-Alonso O, Bramlett HM, Dietrich WD: Alterations in blood-brain barrier permeability to large and small molecules and leukocyte accumulation after traumatic brain injury: effects of post-traumatic hypothermia. J Neurotrauma. 2009, 26: 1123-1134. 10.1089/neu.2008.0802.
Chatzipanteli K, Yanagawa Y, Marcillo AE, Kraydieh S, Yezierski RP, Dietrich WD: Posttraumatic hypothermia reduces polymorphonuclear leukocyte accumulation following spinal cord injury in rats. J Neurotrauma. 2000, 17: 321-332. 10.1089/neu.2000.17.321.
Lyden PD, Krieger D, Yenari M, Dietrich WD: Therapeutic hypothermia for acute stroke. Int J Stroke. 2006, 1: 9-19. 10.1111/j.1747-4949.2005.00011.x.
Dietrich WD, Atkins CM, Bramlett HM: Protection in animal models of brain and spinal cord injury with mild to moderate hypothermia. J Neurotrauma. 2009, 26: 301-312. 10.1089/neu.2008.0806.
Tang XN, Liu L, Yenari MA: Combination therapy with hypothermia for treatment of cerebral ischemia. J Neurotrauma. 2009, 26: 325-331. 10.1089/neu.2008.0594.
Ohta H, Terao Y, Shintani Y, Kiyota Y: Therapeutic time window of post-ischemic mild hypothermia and the gene expression associated with the neuroprotection in rat focal cerebral ischemia. Neurosci Res. 2007, 57: 424-433. 10.1016/j.neures.2006.12.002.
Acknowledgements
This work was supported by the Research Fund from the Vrije Universiteit Brussel (OZR-VUB) and from the Instituut voor de Aanmoediging van Innovatie door Wetenschap en Technologie in Vlaanderen (IWT-TBM) and the Fonds voor Wetenschappelijk Onderzoek (FWO-Vlaanderen) (G.0215.08). AGC is a research fellow of the FWO-Vlaanderen.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
The manuscript was written by AGC as part of her doctoral thesis. All authors provided editorial assistance and have read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ceulemans, AG., Zgavc, T., Kooijman, R. et al. The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J Neuroinflammation 7, 74 (2010). https://doi.org/10.1186/1742-2094-7-74
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-7-74