
Abstract
The high-temperature corrosion behaviors of the equimolar CrCoNi medium-entropy alloy and CrMnFeCoNi high-entropy alloy were studied in a gas atmosphere consisting of a volumetric mixture of 10% H2O, 2% O2, 0.5% SO2, and 87.5% Ar at 800 °C for up to 96 h. Both alloys were initially single-phase fcc with a mean grain size of ~ 50 μm and a homogeneous chemical composition. The oxide layer thickness of CrMnFeCoNi increased linearly with exposure time while it remained constant at ~ 1 μm for CrCoNi. A Cr2O3 layer and minor amounts of (Co,Ni)Cr2O4 developed on the latter while three oxide layers were detected on the former, i.e., a thin and continuous chromium rich oxide layer at the oxide/alloy interface, a dense (Mn,Cr)3O4 layer in the center and a thick and porous layer of Mn3O4 and MnSO4 at the gas/oxide interface. Additionally, a few metal sulfides were observed in the CrMnFeCoNi matrix. These results were found to be in reasonable agreement with thermodynamic calculations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In high-temperature applications like jet engines, gas turbines, or heat exchange tubes, alloys are exposed to reactive gases such as H2O, O2, or SO2, which can mix and harm the integrity of the material even further than a single gas would. Although oxidation studies of high entropy alloys (HEAs) have increased over time, there is still a fundamental need for understanding their behavior under mixed gas atmospheres. Several oxidation studies on HEAs and medium entropy alloys (MEAs) reported so far used synthetic air, laboratory air, CO/CO2, O2, and H2O [1,2,3]. In an early study Laplanche et al. [1] exposed the equiatomic CrMnFeCoNi HEA to laboratory air at different temperatures (600–900 °C) and observed the formation of thick Mn rich oxides (Mn2O3 at 600–800 °C; Mn3O4 at 900 °C). Adomako et al. [2] carried out oxidation tests in dry air (80% N2/20% O2) from 800 to 1000 °C on the CrCoNi, CrMnCoNi, and CrMnFeCoNi alloys. During exposure, a continuous protective Cr2O3 layer grows on the CrCoNi MEA, which makes it more oxidation resistant than CrMnFeCoNi and CrMnCoNi. The authors concluded that the addition of Mn dramatically enhances the oxidation kinetics while Cr reduces them.
Kai et al. [3] investigated the corrosion resistance of CrMnFeCoNi at 950 °C under different O2 partial pressures (pO2 = 10 to 105 Pa) up to 48 h. The thickness of the scales was found to systematically increase with increasing pO2. Growth kinetics were parabolic and, in all atmospheres, the formed scales were trifold: Mn3O4 at the oxide/gas interface, a (Cr,Mn)3O4-layer at the center of the scale, and Cr2O3 at the oxide/alloy interface. In one of our previous works we investigated the corrosion behavior of CrMnFeCoNi and CrCoNi at 800 °C under the following single gas atmospheres: 2% O2 + 98% Ar and 10% H2O + 90% Ar [4]. CrCoNi was found to exhibit a better corrosion resistance due to the formation of a Cr2O3 layer compared to CrMnFeCoNi, which formed Mn rich oxide layers.
Even when the influence of one single corrosive gas is known it is expected that a mixed environment acts as more than the sum of its components. One objective of the present work is thus to evaluate the influence of mixed H2O–O2–SO2 gas atmosphere on the corrosion behavior of CrCoNi at 800 °C, using the well-known Cantor alloy CrMnFeCoNi as a reference. SO2 and water vapor are known to substantially contribute to fast corrosion rates and potential failure and there is a need to identify new materials that can withstand such harsh environments. Previous studies on single elements (Fe [5,6,7], Cr [8, 9], Mn [10], Co [11], and Ni [12]) exposed to a pure SO2 atmosphere revealed the formation of a duplex layer consisting of oxides MO and sulfides MS, M being the metal [13]. Aging of chromium-containing steels in SO2 lead to a duplex layer consisting of an external Fe2O3 layer and an internal iron sulfide. A Cr rich sulfide layer was formed on the steel for a Cr content lower than 10 at%. Steels with higher Cr contents (> 15 at%) formed a protective Cr2O3 layer on the alloy surface [14]. Between 10 and 15% Cr content in the steels, the formation of mixed (Fe,Cr)3O4 oxides and Cr2O3 is observed rather than a single phase, closed, dense Cr2O3 layer. The formation of sulfides could not be observed in these steels due to the low diffusivity of sulfur-containing species through Cr2O3 [8, 15]. Several studies for materials behavior under SO2, H2O, and mixed gas environments were reported for ferritic-austenitic, ferritic-martensitic, and nickel-based materials [16,17,18,19,20,21]. One conclusion of these studies was that oxide scale spallation, breakaway oxidation, and accelerated corrosion could be correlated to even small amounts of sulfur in the gas.
It is expected for the equimolar CrCoNi MEA that a Cr2O3 layer should form during exposure under the mixed oxidizing/sulfurizing atmosphere since it contains enough Cr. Whether or not this layer is dense and fine-grained and thus suppresses fast oxidation and sulfidation, under the given conditions needs to be addressed. When Fe-Cr alloys with more than 10 at% Cr are exposed to high temperatures in 0.5% SO2, Cr2O3 forms widely, Cr-sulfides are frequently found at the alloy grain boundaries and often lead to grain boundary embrittlement [22, 23]. The second objective of the present work is thus an evaluation of how the presence of sulfur affects the oxide layer formation on CrCoNi.
Materials and Methods
Alloy Preparation
Two Cr33.3Co33.3Ni33.3 and Cr20Mn20Fe20Co20Ni20 alloys (nominal compositions in at%) were melted in a vacuum induction furnace starting with high-purity metals (≥ 99.9 wt%). After casting, the cylindrical ingots (diameter Ø = 40 mm) were sealed in evacuated fused silica tubes (p = 3 × 10−5 mbar), homogenized for 48 h at 1200 °C, rotary swaged to reduce their diameter to ~ 17 mm at room temperature (RT), and annealed for 1 h at 1060 °C (CrCoNi) and 1020 °C (CrMnFeCoNi), resulting in similar grain sizes of ~ 50 μm for both alloys. More details about the thermomechanical processing route can be found elsewhere [24, 25]. After metallographic preparation, the compositions of the alloys, measured by electron microprobe analysis (EMPA), were found to be close to the targeted ones, see Table 1.
Gas Exposure Experiments
Penny-shaped specimens (Ø ~17 mm and thickness ~ 3 mm) were cut from the recrystallized rods and their surfaces and edges ground with abrasive paper (grit size: P1200). Before gas exposure, the samples were cleaned with acetone and ethanol, dried, and their dimensions measured with a digital vernier caliper. To avoid contamination by different dissolved metallic ions or corrosion products, two freshly prepared penny-shaped specimens of each alloy were placed vertically on their edge on a custom-made alumina holder and inserted in a Carbolite Gero© horizontal tubular furnace, see Fig. 1. The furnace was flushed with pure argon (Ar 6.0, purity ≥ 99.9999%), and heated to 800 °C at 10 °C/min. The temperature was controlled by a type K thermocouple which was in the vicinity of the specimens. A mixture of corrosive gases including 10% H2O, 2% O2, and 0.5% SO2 (vol%) with Ar as carrier gas was then allowed to stream into the furnace.

Schematic illustration of the experimental setup used to expose CrCoNi and CrMnFeCoNi to a corrosive gas atmosphere (10% H2O, 2% O2, and 0.5% SO2 in 87.5% Ar)
The flow rates of the O2 and SO2 gases were individually controlled using digital mass flow controllers (Bronkhorst©). To adjust the flow rate of water vapor, Ar was injected into a water bottle heated to 63 °C, and the wet gas was transported into the furnace via a frit. Further information about the experimental setup can be found in Ref. [26]. After exposures for 24 h, 48 h, and 96 h, the chamber was flushed with Ar at 800 °C and the specimens furnace cooled down to RT under Ar.
Post-exposure Analyses
After exposure, macro scale investigations of the surfaces were conducted using optical microscopy and micro scale investigations by scanning electron microscopy (SEM) using a TESCAN Vega 3 operating at 20 kV and equipped with an X-Max80 detector (Oxford Instruments). The phase constituents were identified by X-ray diffraction (XRD, Seifert PTS 3003) with Co-Kα radiation. After the collection of diffraction pattern, an X-ray phase analysis was carried out to identify the reaction products at the alloy surface. For this purpose, theoretical diffraction pattern were compared with the measurement, focusing on peak position and intensities. Additionally, to examine the formed oxide layers, the penny-shaped samples were embedded in epoxy resin to avoid oxide scale spallation, cut in two halves, embedded again to prepare a cross-section, ground, and polished to a mirror-finish. Scale thicknesses were determined by optical microscopy (OM) using the software Layers [27, 28]. Ten images were taken along the entire sample’ cross-section using the optical microscope and evaluated by the software “Layers”. A more detailed explanation on how the software Layers works can be found in Weber et al. [29]. The corrosion layer thickness was determined as a function of the exposure time. Note that the sample is not weighed in this method. The oxidation rate constant is calculated directly from the measured layer thickness.
Further microstructural characterizations were carried out in a Zeiss Leo Gemini 1530 VP SEM, operating at 20 kV and equipped with a Bruker energy-dispersive X ray spectrometer (EDS).
Thermodynamic Calculations
Thermodynamic equilibrium calculations of the systems Cr–Co–Ni–O2–H2O–SO2 and Cr–Mn–Fe–Co–Ni–O2–H2O–SO2 were carried out with the software Factsage 7.0 using the FactPS and Factoxid databases. ‘Equilibrium’, ‘phase diagram’ and ‘reaction’ mode were operated. For the ternary equilibrium phase diagrams at T = 800 °C and p = 1 atm of the Cr–Co–Ni and the Cr–Mn–Fe–Co–Ni systems, constant fugacities of p(O2) = 0.0197 atm, p(S2) = 10−28.9 atm and p(H2) = 10−9.3 atm were set, representing the partial pressure of these gases at the alloy-gas interface. These values were calculated before for a volumetric mixture of 10% H2O, 2% O2, 0.5% SO2. In the ternary phase diagrams of the HEA mixture within the Cr–Mn–Fe–Co–Ni system, a fixed molar ratio of x(Co) = x(Ni) = 0.2 was employed, as these metallic elements exhibited relatively minor involvement in the formation of corrosion layers in comparison to the remaining constituents. For the binary phase diagrams of p(O2) vs. p(S2) in the Cr–Co–Ni and Cr–Mn–Fe–Co–Ni system at T = 800 °C and p = 1 atm, a constant partial pressure of hydrogen of p(H2) = 10−9.3 atm was assumed. More details of the thermodynamic calculations can be found in in [30].
Results
Comparison of the Morphologies and Thicknesses of the Oxide Layers
The secondary electron (SE) images in Fig. 2 enable a comparison of the grown oxide layers on the CrCoNi MEA (left column) and the CrMnFeCoNi HEA (right column) after corrosion in the H2O–O2–SO2 atmosphere after 24 h, 48 h, and 96 h at 800 °C. The oxide scale growing on CrCoNi was found to be much thinner than that of CrMnFeCoNi, indicating that the former is more corrosion resistant. For this reason, a higher magnification was used to characterize the oxide scale of CrCoNi (compare the magnifications in the left and right columns of Fig. 2). Oxide inclusions of various sizes and shapes were observed in the matrix of CrMnFeCoNi directly below the oxide/alloy interface and their number density appeared to grow with exposure time. Oxide inclusions were also observed in CrCoNi but much more rarely. After 48 h, the oxide layer on the HEA consisted of three layers numbered from 1 to 3, which differ in gray tone and morphology. Close to the oxide/gas interface lies the porous layer 1. At the oxide/alloy interface, a very thin and continuous oxide layer has formed (layer 2). A few elongated and compact islands are fully embedded in layer 1 (layer 3, Fig. 2f).

BSE images of cross-sections showing the oxide layers grown on CrCoNi (left column) and CrMnFeCoNi (right column) at different magnifications. Exposure times of (a, b) 24 h, (c, d) 48 h, and (e, f) 96 h
The oxidation kinetics were investigated by measuring how the layer thickness evolved with time, Fig. 3. The oxide layer growing on CrCoNi is very thin (< 1 μm). After 24 h, the scale thickness is 0.43 ± 0.08 μm and its value increases to 0.80 ± 0.20 μm after 48 h (Table 2). After 96 h the thickness of the oxide scale decreased to 0.68 ± 0.03 μm because it partially flaked off before it was removed from the corrosion chamber (see sub-Sect. 4.2 CrCoNi MEA). The oxide layers grown on the CrMnFeCoNi HEA were significantly thicker than those on CrCoNi after all durations. The layer thickness remained similar between 24 h (8 ± 1 μm) and 48 h (7 ± 3 μm) and grows significantly to 28 ± 6 μm after 96 h.

Oxide scale thickness on CrCoNi and CrMnFeCoNi as a function of exposure times at 800 °C in Ar–H2O–SO2–O2
The temporal evolution of the thickness, d, of the corrosion layers can be described with the following equation:
where t is the time, k a rate constant of oxidation, and n a parameter related to the corrosion mechanism. The parameters of the oxidation kinetics are summarized in Table 2 for the two alloys. The n-value of CrMnFeCoNi is close to unity and the oxidation rate of 9.8 × 10−2 μm·h−1 is high. In other words, the oxide scale grew at a linear rate and is non-protective, probably due to its porous nature. In contrast, the growth kinetics of the oxide layer on CrCoNi proceeded as a power-law with a time exponent of 1/3 and k = 3.7 × 10−11 μm h−1. This value for CrCoNi is more than nine orders of magnitude lower than that of CrMnFeCoNi, reflecting the better corrosion resistance of CrCoNi in the multi-oxidant atmosphere. However, the exact value of k has to be considered with care since spallation was found to affect the corrosion behaviour of CrCoNi.
CrCoNi MEA
Surface Composition and Morphology
The CrCoNi MEA exhibited greenish surfaces after high-temperature corrosion, see photos of the penny-shaped specimens shown in the left column of Fig. 4. For all corrosion durations, chromia (Cr2O3-space group \(R\stackrel{-}{3}c\)) was identified as the dominant oxide layer by XRD and minor reflections at 2θ ~ 35, 36, and 54° indicated the presence of a spinel-type phase with (Ni, Co)Cr2O4 composition (space group \(Fd\stackrel{-}{3}m\)), Fig. 4a–c. The fcc reflections of the alloy substrate at 2θ ~ 51° and 60° have the strongest intensity at all exposure times, indicating that the oxide layer remains thin. Figure 4d shows a large bright region where the oxide scale spalled off after 24 h. After 48 h clear evidence of oxide scale spallation is visible, i.e., thick spalled-off oxide plates appear dark, freshly re-grown oxide areas are imaged in gray while unprotected regions are depicted bright in Fig. 4e. After 96 h, the oxide layer appears dense and continuous, but some grinding grooves and cracks are visible (see BSE image in Fig. 4f). At this stage, it is unclear whether spallation occurred continuously during high-temperature corrosion, or whether it occurred upon cooling.

Phase analysis of the oxide scale of the CrCoNi MEA after different times. a–c XRD patterns with optical micrographs of the oxidized penny-shaped specimen and d–f BSE micrographs of the corroded surfaces (top-down view) after 24 h, 48 h, and 96 h at 800 °C in Ar–H2O–SO2–O2
SEM Elemental Distribution Maps
Figure 5 shows SEM-EDS elemental distribution maps of Cr, Co, Ni, O, and S, collected on a cross-section through the oxide scale formed on CrCoNi after corrosion in H2O–O2–SO2 at 800 h for (a) 24 h, (b) 48 h, and (c) 96 h. In each case, the corrosion layer is O and Cr rich and there is a corresponding Cr depleted zone below the oxide/alloy interface. Sulfur could not be detected, neither in the corrosion layer nor within the MEA matrix.

SE images of cross-sections showing the oxide layers on CrCoNi and corresponding EDS elemental maps of Cr, Co, and Ni as well as O and S after corrosion in Ar–H2O–O2–SO2 at 800 °C for a 24 h, b 48 h, and c 96 h
CrMnFeCoNi HEA
Surface Composition and Morphology
The photos of the CrMnFeCoNi samples after corrosion at 800 °C show a yellow to greenish surface with greyish borders, see left column of Fig. 6. After 24 h exposure time, the corrosion layer is continuous, but after 48 and 96 h, the corrosion layer is slightly disrupted with signs of spallation at the sample edges (Fig. 6a–c). Hausmannite (Mn3O4-tetragonal spinel-space group I41/amd) is identified by XRD as the dominant oxide. Additionally, distinct reflections at 2θ ~ 28°, 30°, and 39° reveal the presence of sulfates (MnSO4-orthorhombic-space group Cmcm). With increasing exposure time, the intensity of the fcc reflections from the alloy progressively decreases and vanishes after 96 h due to the thickening of the corrosion layers (see Fig. 6). After 48 and 96 h, two porous nodules are observed in Fig. 6e, f, indicating a nucleation and growth process.

Color, constituting phases, and morphology of the oxide scales forming on the CrMnFeCoNi HEA at 800 °C in H2O–SO2–O2 after different times. (From left to right) Optical micrographs of the penny-shaped specimen, XRD patterns, and top-down views of the oxide scales observed with BSE contrast imaging. (From top to bottom) Exposure times of 24 h, 48 h, and 96 h
EDS Elemental maps
In Fig. 7, BSE micrographs of cross-sections through the corrosion layers that formed on CrMnFeCoNi are presented in combination with the corresponding EDS elemental maps of the five alloying elements (Cr, Mn, Fe, Co, and Ni) as well as the gas components O and S after corrosion at 800 h for (a) 24 h, (b) 48 h, and (c) 96 h. The surface of the corrosion products has a rough appearance, especially after 96 h (see Fig. 7c). The porous corrosion layer 1 is enriched in Mn, O, and to a lesser extent, S, and corresponds to hausmannite (Mn3O4, in which S can be dissolved, see XRD patterns in Fig. 6). After 96 h (see Fig. 7c) small areas enriched in S and O are randomly distributed in layer 1. Based on this observation and the XRD pattern in Fig. 6a, these particles are likely sulfates (MnSO4). In contrast to the porous and S rich layer 1, corrosion layers 2 and 3 are compact and depleted in S. Oxide layer 2 formed at the oxide/alloy interface and is mainly enriched in Cr, O, and to a lesser degree, Mn, and likely corresponds to chromia or a spinel (note that these phases were not detected by XRD). Layer 3 is either right above layer 2 or embedded into layer 1. As the corrosion layers are mainly enriched in Mn, the alloy below the oxide/alloy interface is strongly depleted in this element. The thickness of the Mn-depleted region increased from 18 ± 9 μm after 24 h to 20 ± 8 μm after 48 h and finally to 33 ± 10 μm after 96 h. A few regions in the alloy are strongly enriched in S and Mn and most likely correspond to Mn sulfides. These appear in the vicinity of grain boundaries and pores.

BSE images of the oxide layers on the CrMnFeCoNi HEA and corresponding EDS elemental maps of the alloying elements (Cr, Mn, Fe, Co, and Ni) as well as O and S as the corrosive gas components after corrosion in H2O–O2–SO2 at 800 °C for a 24 h, b 48 h, and c 96 h
Discussion
General Remarks
The main findings of the previous section are summarized and illustrated in Table 3; Fig. 8. After exposure to the corrosive atmosphere (10% H2O, 2% O2, 0.5% SO2, and 87.5% Ar) at 800 °C, the CrMnFeCoNi HEA formed a thick (up to 30 μm after 96 h), discontinuous scale consisting of three corrosion layers, which are from top to bottom: outward growing Mn3O4, outward growing MnSO4, and a third phase, which was not identified by XRD but likely corresponds to Cr2O3 or a (Mn,Cr)3O4 spinel (see lower rows of Table 3; Fig. 8). Considering the shape of the bulk surface below this layer it can be considered that the oxide has grown inwards. Below these, narrow Cr and large Mn depleted zones were detected, accompanied by Mn rich sulfides. In contrast, the CrCoNi MEA developed very thin (< 1 μm), continuous oxide layers, consisting of Cr2O3 (also considered an inwards growing oxide, see [31]), below which Cr depleted zones were found in the alloy. These results clearly show that CrCoNi exhibit a superior corrosion resistance than CrMnFeCoNi.

Schematic illustration of the oxide layer formation on CrCoNi (upper row) and CrMnFeCoNi (lower row) during exposure to H2O–SO2–O2 for 24, 48 and 96 h
Impact of Mixed Gas Atmosphere on Oxide Layer Growth on CrCoNi
Thermodynamic Boundary Conditions
Thermodynamic boundary conditions were considered using FactSage 7.0 [32]. Stability diagrams (see Fig. 9a and b) were calculated based on the Fact PS database for oxides, sulfates, and metals as well as the FactMisc database for the sulfides.

a log(pO2) vs. log(pS2) plot for the equimolar CrCoNi MEA at pH2 = 10−9 atm and 800 °C. The filled red circle represents the pO2 and pS2 at the gas/oxide interface. b Stability diagram for the formation of oxides in the Cr–Co–Ni system with a constant partial pressure of O2, S2 (directly linked to SO2) and H2 (directly related to H2O) at 800 °C (pO2 = 0.0197 atm and pS2 = 10–29 atm). The orange area shows the compositional range in which the phases detected by XRD are stable from a thermodynamic point of view. The blue arrow shows the shift from nominal to local composition. Both diagrams in a and b were calculated with the FactSage ‘phase diagram’ tool
The filled circle (red) in Fig. 9a corresponds to the experimental starting conditions and the conditions assumed at the gas/oxide interface. The oxygen partial pressure decreases from the gas/oxide interface towards the oxide/alloy interface. Accordingly, the local O2 or S2 partial pressure required within the oxide layer for the formation of certain corrosion products can be determined from the diagrams log(O2)–log(S2). Neither H2O nor indirectly H2 form hydroxides at 800 °C and the latter are known to remain volatile at this temperature. Therefore, hydrogen is not considered neither in the calculation nor in the representation and consequently remains constant. The ternary diagrams are representative for corrosion products near the oxide/gas interface, since the partial pressures of the reactive species (O2, SO2) are the same as in the gas.
The binary diagram for the CrCoNi alloy shows the sulfur partial pressure pS2 versus oxygen partial pressure pO2 at a constant pH2 = 10−9 atm and 800 °C (Fig. 9a). With increasing either pO2 or pS2, Cr is the first reacting element, which oxidizes to chromia (Cr2O3) or sulfidizes to pyrrhotite (Cr,Co, Ni)1−xS, respectively. At high pO2 and pS2, Cr-sulfate (Cr2(SO4)3) can form on the CrCoNi MEA in an oxidation atmosphere containing O2, SO2 and H2O. The red filled circle in the binary diagram (Fig. 9a) represents the conditions (pO2 = 0.0197 atm pS2 = 10−29 atm) at the gas/oxide interface at 800 °C in which the formation of the spinel CoCr2O4 and the monoxide (Co,Ni)O on the MEA CrCoNi is possible. Neither sulfates nor sulfides were identified in the SEM-EDS elemental distribution maps (see Fig. 5), leading to the assumption that the necessary high sulfur partial pressure was not reached, neither locally nor globally. This indicates that the oxide layer formed was not allowing a sulfur transport through it towards the oxide/alloy interface.
Figure 9b presents the ternary diagram of the Cr–Co–Ni system at 800 °C and constant partial pressures of pH2 = 10−9 atm, pS = 10−29 atm and pO2 = 0.0197 atm, i.e., equal to atmospheric conditions. The blue triangle in the diagram corresponds to the nominal composition of the equimolar CrCoNi alloy, which serves as a base for this consideration. Because of the chromium’s significant diffusion and affinity to oxygen the local composition at the alloy’s surface shifts over time towards a Cr rich corner [33]. This is confirmed by the experimental results: the calculated monoxide (Ni,Co)O was not detected by XRD on the MEA after 96 h of gas exposure, but chromia (Cr2O3) and the mixed ((Co,Ni)Cr2O4 were systematically observed (see Figs. 4 and 5).
Oxide Layer Characteristics in Different Atmospheres
The chromia layer grown on CrCoNi alloy suffered from buckling in some parts. Both buckling and spallation could have be induced by tensile and interfacial cracks due to compressive stresses in the oxide layer and a weak alloy/oxide interface [34]. It will not be discussed further here.
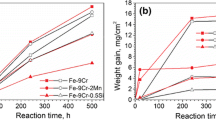
The oxidation kinetics are compared for single (98% Ar-2% O2 and 90% Ar-10% H2O) and mixed gas (87.5% Ar-10% H2O-2% O2-0.5% SO2) atmospheres at 800 °C (see Fig. 10a). The values for single gas atmospheres were taken from a previous work [26], in which the same CrMnFeCoNi and CrCoNi were exposed to 98% Ar-2% O2 and 90% Ar-10% H2O at 800 °C for equal aging times. From the layer thicknesses observed for the 2% O2 and 10% H2O condition, the oxidation rate constants k were determined and compared to the values of the mixed atmosphere (see Table 4).

Evolution with time (24, 48 and 96 h) of the oxide scale thickness at 800 °C in different atmospheres for a CrCoNi and b CrMnFeCoNi. Data on the O2 and H2O atmospheres taken from [26]
The CrCoNi alloy under oxygen exposure shows oxide thicknesses comprised between 1.3 ± 0.5 μm and 2.3 ± 0.4 μm, resulting in an n value of 2.45 ± 0.7 with an oxidation rate constant k = 1.3 × 10−7 μm h−1. In H2O atmosphere (layer thicknesses ranging from 0.9 ± 0.3 μm to 2.3 ± 0.8 μm), the oxidation rate constant of CrCoNi is k = 3.9 × 10−4 μm h−1. In the mixed atmosphere studied here, the oxide layer on the MEA is the thinnest after all exposure times and grows slowly (layer thickness of 0.43 ± 0.05 μm at 24 h and 0.68 ± 0.03 μm at 96 h, bearing in mind that the absolute values are questionable within the order of magnitude due to the spallation). The oxidation rate constant was determined to be k = 3.7 × 10−11 μm·h−1, possibly higher due to the spallation. Still, k was the highest in the 10% H2O atmosphere and the lowest in the mixed gas atmosphere: kH2O > kO2 > kH2O–O2–SO2. The chromia layer on CrCoNi seems to provide protection against fast oxidation and internal sulfidation in the mixed gas atmosphere (H2O–SO2–O2) at 800 °C. Neither sulfides nor sulfates were detected, neither in the oxide layer nor in the bulk material.
Thus, even if pO2 is similar in the 98% Ar-2%O2 and mixed gas atmospheres, the chromia layer seems to have different growth and protection characteristics. A gas corrosion study on a 9 wt% Cr steel at 650 °C in O2–H2O mixed atmosphere demonstrated that the transition from protective Cr rich oxide formation into non-protective Fe rich mixed oxide is governed by the H2O/O2 ratio [35]. The authors assumed the layer growth to be governed by competitive adsorption of oxygen and water vapor molecules on external and internal interfaces of the oxide scale [35]. Note that Cr2O3 grain boundaries were reported to play an important role in chromia growth, since lattice diffusion was found to be very slow [36,37,38,39]. In the mixed gas atmosphere applied here it is suggested that different competitive adsorption and absorption processes with contributions of O2, H2O and SO2 take place on external and internal interfaces of the chromia layer. This led to a different thickness growth of Cr2O3 in the 98% Ar-2% O2, 90% Ar-10% H2O and O2–H2O–SO2 atmospheres. A study of chromia grown on pure Cr in multi-oxidant environments (N2–CO–H2–CO2–H2S with varying gas composition) at 871 °C showed that both inward and outward growth of chromia were inferred at mixed gas conditions [40]. Additionally, the Cr2O3 growth was faster in mixed gases containing water than in those with dry air. Additional works on pure Cr in the temperature range of 800–900 °C have shown that regardless of the pO2, the chromia layer has a duplex morphology with inwards and outwards growing sub-layers [41,42,43,44,45]. It is expected to have the same formation in this study, but this assumption needs to be clarified via more high-resolution observations in the future.
Impact of Mixed Gas Atmosphere on Oxide Layer Growth on CrMnFeCoNi
Thermodynamic Boundary Conditions
The binary phase diagram (log(pO2) − log(pS2)) of CrMnFeCoNi is presented in Fig. 11a for a constant pH2 = 10−9 atm and 800 °C. The (red) filled circle in the binary diagram represents the conditions (pO2 = 0.0197 atm and pS2 = 10−29 atm) at the gas/oxide interface at 800 °C for which the stable reaction products are a mixed (Co,Fe3+)(Co,Fe2+,Ni)2O4 spinel, (Mn,Co)O and Mn3O4 (hausmannite – tetragonal spinel). At higher pO2 (right side of the diagram, pO2 ≈ 10−1 atm), Mn2O3 (bixbyite) is stable.

a log(pO2) vs. log(pS2) plot for the equimolar CrMnFeCoNi HEA at pH2 = 10−9 atm and 800 °C. The red dot in a corresponds to the pO2 and pS2 at the gas/oxide interface. The black dashed lines on the right side of a highlight how the sulfate boundary (see upper right corner) shifts with decreasing temperature to 700 °C and 600 °C. The dashed red circles mark the pS2 values at 700 °C and 600 °C. b Stability diagram for the formation of oxides in the CrMnFe system with a constant (Co+Ni) content (= 0.2) and constant partial pressures of O2, S2 (directly related to SO2) and H2 (directly linked to H2O) at 800 °C (pO2 = 0.0197 atm and pS2 = 10–29 atm). In b, the orange region shows the compositional range in which the phases detected by XRD are stable. The blue arrow shows the shift from nominal to local composition. Both phase diagrams in a and b were calculated with the FactSage ‘phase diagram’ tool
Sulfide and sulfate formation in the corrosion layers of CrMnFeCoNi require a local sulfur partial pressure of at least pS2 ≥ 10−20 atm (sulfides) and pS2 ≥ 10−30 atm (sulfates), see Fig. 11a. Sulfides are formed at low pO2 and high pS2. In the case where the oxide layer on top of an alloy does not provide protection against sulfur diffusion and transport towards the oxide/alloy interface, high local pS2 at simultaneously low pO2 can be present there. This is case here, as metal-sulfides are observed within the alloy by SEM-EDS (see Fig. 7).
Under the given conditions sulfate formation requires high pO2 and pS2. This condition is met during layer growth at the gas/oxide interface, since sulfates were identified in the outward grown oxide layer 1 (see especially Fig. 7c, S and O elemental distribution map after 96 h of exposure). After 96 h of exposure at 800 °C in Ar–H2O–O2–SO2, a porous outer Mn3O4–MnSO4 layer (layer 1) formed, followed by a compact yet interrupted Mn3O4 layer (intermediate layer 3, named layer 3b when slightly enriched in Mn and located further inside layer 1), and a continuous Cr enriched mixed (Mn,Cr)2O3 layer (layer 2) at the oxide/alloy interface. An indication for a (Co,Ni)-spinel was observed by XRD on the alloy after exposure for 24 h.
Unsurprisingly no bixbyite Mn2O3 was observed on the CrMnFeCoNi, as it would only be stable under local and global high pO2, which cannot be achieved in the present conditions.
Figure 11b displays the ternary stability diagram Cr–Mn–Fe with a constant Co+Ni content. Under the conditions studied here, no Mn2O3 could be detected but Mn3O4 was formed. This means that the entire oxide formation sequence over time shifts to higher Mn contents to stabilize Mn3O4 (Fig. 11b), which is highlighted in the diagram by a blue arrow—the local composition is no longer equiatomic CrMnFe(CoNi) but shifted to the Mn rich corner. A (Mn,Cr)2O3 layer (layer 3) was observed near the oxide/alloy interface (see Fig. 7). For Cr2O3 formation, low pO2 is required and obviously achieved locally—otherwise (Co,Fe3+) (Co,Fe2+,Ni)2O4 would form.
The preferential formation of Mn containing reaction products (Mn3O4, MnSO4 and (Mn,Cr)2O3 ± (Co,Ni)Cr2O4) instead of equimolar mixed oxides requires a strong enrichment in Mn and, to a lesser extend, Cr (and therefore a depletion in Co, Ni and Fe). Their formation products is supported by kinetic aspects, i.e. the fast diffusion of Mn compared to the other alloying components (DMn > DCr > DFe > > DCo > DNi) [46, 47] resulting in the formation of Mn oxides and Cr oxide at low pO2. These elements are thus the main components of the corrosion products.
Oxide Layer Characteristics in Different Atmospheres
No matter the atmosphere (air [1, 2, 48]; 2% O2 or 10% H2O [26]), when CrMnFeCoNi is exposed to temperatures larger than 600 °C, it develops non protective, thick Mn oxide containing layers. In direct comparison with single gas exposure (2% O2 or 10% H2O) the layers formed under mixed-gas (10%H2O-2% O2-0.5%SO2) atmosphere are the thickest and strongly porous (see Fig. 10b). The SO2 proportion in the gas causes, additionally to the Mn oxides, a MnSO4 formation in the outward grown layers. The high amount of Mn, which is known to have an adverse effect on oxidation resistance [49] and the fast Mn outward diffusion dominantly influence the formation of the respective layers and lead to disastrous layer characteristics (porous, thick, fast growing) in terms of oxidation protection.
Summary and Conclusion
High-temperature gas corrosion was studied on CrMnFeCoNi and CrCoNi alloys by isothermal exposure experiments in a multiple gas atmosphere consisting of 2% O2, 10% H2O and 0.5% SO2 (volumetric mixture, rest argon) for 24 h < t < 96 h at 800 °C. The outward and inward grown layers were characterized for their thickness, structure, composition and morphology.
CrCoNi developed thin oxide layers (Cr2O3 layer with minor amounts of (Co,Ni)Cr2O4 ; layer thickness mostly < 1 μm) after 96 h. The chromia layer on CrCoNi grew slowly and seemed to prevent the alloy from sulfidation and further oxidation. No sulfides were detected, neither by XRD, nor by SEM-EDS. Thermodynamic equilibrium calculations with FactSage were in good agreement with the observations and confirmed the results.
As expected, CrMnFeCoNi demonstrated a low corrosion resistance due to the fast growth of Mn oxides and Mn sulfates. The thick (up to 30 μm), non-protective oxide layer after 96 h of exposure consisted of an inner continuous (Mn,Cr)2O3, an intermediate dense Mn3O4, and an outer, porous, discontinuous Mn3O4 layer, heterogeneously intermixed with MnSO4 particles. Mn rich sulfides were observed within the bulk material of the HEA after all exposure times, indicating constant sulfur transport towards the alloy through the oxide/sulfate layer. The results were evaluated considering thermodynamic and kinetic boundary conditions. As for our previous experiments in more simple atmospheres (air, 98% Ar-2% O2, 90% Ar-10% H2O,), Mn increased the reaction kinetics in the corrosion process due its fast diffusion in the alloy and through previously formed Cr2O3 or Mn3O4 oxides. The high amount of Mn and the fast Mn outward diffusion are responsible for the fast layer growth.
This study of relatively short time behavior (up to 96 h) proves that CrCoNi is a suitable candidate for high-temperature applications in hot and aggressive sulfur-containing gas atmospheres while CrMnFeCoNi is not.
Data Availability
Data will be shared upon request.
References
G. Laplanche, U.F. Volkert, G. Eggeler, E.P. George, Oxidation behavior of the CrMnFeCoNi high-entropy alloy. Oxid. Met. 85(5), 629–645 (2016). https://doi.org/10.1007/s11085-016-9616-1
N.K. Adomako, J.H. Kim, Y.T. Hyun, High-temperature oxidation behaviour of low-entropy alloy to medium- and high-entropy alloys. J. Therm. Anal. Calorim. 133(1), 13–26 (2018). https://doi.org/10.1007/s10973-018-6963-y
W. Kai, C.C. Li, F.P. Cheng, K.P. Chu, R.T. Huang, L.W. Tsay, J.J. Kai, The oxidation behavior of an equimolar FeCoNiCrMn high-entropy alloy at 950 °C in various oxygen-containing atmospheres. Corros. Sci. 108, 209–214 (2016). https://doi.org/10.1016/j.corsci.2016.03.020
W. Kai, W.L. Jang, R.T. Huang, C.C. Lee, H.H. Hsieh, C.F. Du, Air oxidation of FeCoNi-base Equi-molar alloys at 800–1000 °C. Oxid. Met. 63(3), 169–192 (2005). https://doi.org/10.1007/s11085-004-3198-z
B. Chatterjee, A.J. Dowell, High temperature reaction of Fe in SO2. Corros. Sci. 15(6), 639–648 (1975). https://doi.org/10.1016/0010-938X(75)90029-3
K. Ota, H. Sugimoto, K. Fueki, The high-temperature reaction of Iron with sulfur dioxide. Bull. Chem. Soc. Jpn 62(3), 653–656 (1989). https://doi.org/10.1246/bcsj.62.653
T. Flatley, N. Birks, Oxidation of iron in atmospheres containing sulphur dioxide. J. Iron Steel Inst. (London) 209(7), 523–532 (1971)
C. De Asmundis, F. Gesmundo, C. Bottino, High-temperature corrosion of pure chromium in SO2 (700–1000 °C). Oxid. Met. 14(4), 351–361 (1980). https://doi.org/10.1007/BF00603790
A.G. Andersen, P. Kofstad, Reactions of chromium in SO2-containing atmospheres. Oxid. Met. 43(3), 301–315 (1995). https://doi.org/10.1007/BF01047032
F. Gesmundo, C. De Asmundis, C. Bottino, High temperature corrosion of manganese in pure SO2. Oxid. Met. 14(1), 15–29 (1980). https://doi.org/10.1007/bf00604101
K. Holthe, P. Kofstad, High temperature corrosion of cobalt in SO2. Corros. Sci. 20(7), 919–936 (1980). https://doi.org/10.1016/0010-938X(80)90124-9
F. Gesmundo, C. de Asmundis, P. Nanni, The corrosion of nickel in 1 atm of pure SO2 at 600–1000 °C and the mechanism of formation of the two-layered scales. Oxid. Met. 20(5), 217–240 (1983). https://doi.org/10.1007/BF00656842
F. Gesmundo, D.J. Young, S.K. Roy, The high temperature corrosion of metals in sulfidizing-oxidizing environments: a critical review, high temp. Mater. Proc. (London) 8(3), 149–190 (1989). https://doi.org/10.1515/HTMP.1989.8.3.149
C.S. Giggins, F.S. Pettit, Corrosion of metals and alloys in mixed gas environments at elevated temperatures. Oxid. Met. 14(5), 363–413 (1980). https://doi.org/10.1007/BF00603609
T.T. Huang, B. Peterson, D.A. Shores, E. Pfender, XPS and AES studies of the high temperature corrosion mechanism of Fe-30Cr alloy. Corros. Sci. 24(3), 167–173 (1984). https://doi.org/10.1016/0010-938X(84)90048-9
P. Huczkowski, T. Olszewski, M. Schiek, B. Lutz, G.R. Holcomb, V. Shemet, W. Nowak, G.H. Meier, L. Singheiser, W.J. Quadakkers, Effect of SO2 on oxidation of metallic materials in CO2/H2O-rich gases relevant to oxyfuel environments. Mater. Corros. 65(2), 121–131 (2014). https://doi.org/10.1002/maco.201307306
P. Huczkowski, D.J. Young, T. Olszewski, A. Chyrkin, W.J. Quadakkers, Effect of sulphur on the oxidation behaviour of possible construction materials for heat exchangers in oxyfuel plants in the temperature range 550–700 °C. Oxid. Met. 89(5), 651–681 (2018). https://doi.org/10.1007/s11085-017-9809-2
K. Chandra, A. Kranzmann, R. Saliwan Neumann, G. Oder, F. Rizzo, High temperature oxidation behavior of 9–12% Cr ferritic/martensitic steels in a simulated dry oxyfuel environment. Oxid. Met. 83(3), 291–316 (2015). https://doi.org/10.1007/s11085-014-9521-4
B.A. Pint, Invited review paper in commemoration of over 50 years of oxidation of metals: addressing the role of water vapor on long-term stainless steel oxidation behavior. Oxid. Met. 95(5), 335–357 (2021). https://doi.org/10.1007/s11085-020-10012-9
W.J. Quadakkers, J. Żurek, M. Hänsel, Effect of water vapor on high-temperature oxidation of FeCr alloys. JOM. 61(7), 44–50 (2009). https://doi.org/10.1007/s11837-009-0102-y
J. Żurek, M. Michalik, F. Schmitz, T.U. Kern, L. Singheiser, W.J. Quadakkers, The effect of water-vapor content and gas flow rate on the oxidation mechanism of a 10%Cr-ferritic steel in Ar-H2O mixtures. Oxid. Met. 63(5), 401–422 (2005). https://doi.org/10.1007/s11085-005-4394-1
K. Nützmann, N. Wollschläger, C. Rockenhäuser, A. Kranzmann, C. Stephan-Scherb, Identification and 3D reconstruction of Cr5S6 precipitates along grain boundaries in Fe13Cr. JOM 70(8), 1478–1483 (2018). https://doi.org/10.1007/s11837-018-2940-y
F. Falk, O. Sobol, C. Stephan-Scherb, The impact of the microstructure of Fe-16Cr-0.2 C on high-temperature oxidation – sulphidation in SO2. Corros. Sci. 190, 109618 (2021). https://doi.org/10.1016/j.corsci.2021.109618
G. Laplanche, O. Horst, F. Otto, G. Eggeler, E.P. George, Microstructural evolution of a CoCrFeMnNi high-entropy alloy after swaging and annealing. J. Alloys Compd. 647, 548–557 (2015). https://doi.org/10.1016/j.jallcom.2015.05.129
M. Schneider, E.P. George, T.J. Manescau, T. Záležák, J. Hunfeld, A. Dlouhý, G. Eggeler, G. Laplanche, Analysis of strengthening due to grain boundaries and annealing twin boundaries in the CrCoNi medium-entropy alloy. Int. J. Plast. 124, 155–169 (2020). https://doi.org/10.1016/j.ijplas.2019.08.009
C. Stephan-Scherb, W. Schulz, M. Schneider, S. Karafiludis, G. Laplanche, High-temperature oxidation in dry and humid atmospheres of the Equiatomic CrMnFeCoNi and CrCoNi High- and medium-entropy alloys. Oxid. Met. 95(1), 105–133 (2021). https://doi.org/10.1007/s11085-020-10014-7
Gesellschaft zur Förderung angewandter Informatik [Society for the Promotion of Applied Computer Science], Layers (2012)
K. Nützmann, A. Kranzmann, C. Stephan-Scherb, The influence of chromium on early high temperature corrosion of ferritic alloys under SO2 atmosphere. Mater. High. Temp. 35(6), 558–568 (2018). https://doi.org/10.1080/09603409.2018.1446705
K. Weber, Schädigungsmechanismen von Eisen-Chrom-Legierungen in sulfidierend-oxidierender Atmosphäre, Geowissenschaften (Freie Universität Berlin, Berlin, 2019). https://doi.org/10.17169/refubium-2217
S. Karafiludis, The impact of exhaust gas atmospheres on the high temperature gas corrosion of high and medium entropy alloys (Freie Universität Berlin, Berlin, 2020)
M.G.C. Cox, B. McEnaney, V.D. Scott, A chemical diffusion model for partitioning of transition elements in oxide scales on alloys. Philos. Mag.: J. Theor. Exp. Appl. Phys. 26(4), 839–851 (1972). https://doi.org/10.1080/14786437208226960
C.W. Bale, E. Bélisle, P. Chartrand, S.A. Decterov, G. Eriksson, A.E. Gheribi, K. Hack, I.H. Jung, Y.B. Kang, J. Melançon, A.D. Pelton, S. Petersen, C. Robelin, J. Sangster, P. Spencer, M.A. Van Ende, FactSage thermochemical software and databases, 2010–2016. Calphad 54, 35–53 (2016). https://doi.org/10.1016/j.calphad.2016.05.002
A. Durand, L. Peng, G. Laplanche, J.R. Morris, E.P. George, G. Eggeler, Interdiffusion in Cr–Fe–Co–Ni medium-entropy alloys. Intermetallics. 122, 106789 (2020). https://doi.org/10.1016/j.intermet.2020.106789
H.E. Evans, Cracking and spalling of protective oxide layers. Mater. Sci. Eng.: A (1989). https://doi.org/10.1016/0921-5093(89)90731-4
J. Ehlers, D.J. Young, E.J. Smaardijk, A.K. Tyagi, H.J. Penkalla, L. Singheiser, W.J. Quadakkers, Enhanced oxidation of the 9%Cr steel P91 in water vapour containing environments. Corros. Sci. 48(11), 3428–3454 (2006). https://doi.org/10.1016/j.corsci.2006.02.002
A.C.S. Sabioni, A.M. Huntz, F. Millot, C. Monty, Self-diffusion in Cr2O3 III. Chromium and oxygen grain-boundary diffusion in polycrystals. Philos. Mag A 66(3), 361–374 (1992). https://doi.org/10.1080/01418619208201562
A.C.S. Sabioni, A.M. Huntz, F. Millot, C. Monty, Self-diffusion in Cr2O3 II. Oxygen diffusion in single crystals. Philos. Mag A 66(3), 351–360 (1992). https://doi.org/10.1080/01418619208201561
A.C.S. Sabioni, A.M. Huntz, J. Philibert, B. Lesage, C. Monty, Relation between the oxidation growth rate of chromia scales and self-diffusion in Cr2O3. J. Mater. Sci. 27(17), 4782–4790 (1992). https://doi.org/10.1007/BF01166020
A.C.S. Sabioni, B. Lesage, A.M. Huntz, J.C. Pivin, C. Monty, Self-diffusion in Cr2O3 I. Chromium diffusion in single crystals. Philos. Mag A 66(3), 333–350 (1992). https://doi.org/10.1080/01418619208201560
S. Soltanattar, P. Nowakowski, C.S. Bonifacio, P. Fischione, B. Gleeson, Use of Microanalysis to Better understand the high-temperature corrosion behavior of Chromium exposed to Multi-oxidant environments. Oxid. Met. 91(1), 11–31 (2019). https://doi.org/10.1007/s11085-018-9882-1
L. Latu-Romain, Y. Parsa, S. Mathieu, M. Vilasi, A. Galerie, Y. Wouters, Towards the growth of stoichiometric chromia on pure chromium by the control of temperature and oxygen partial pressure. Corros. Sci. 126, 238–246 (2017). https://doi.org/10.1016/j.corsci.2017.07.005
L. Latu-Romain, Y. Parsa, S. Mathieu, M. Vilasi, M. Ollivier, A. Galerie, Y. Wouters, Duplex n- and p-Type Chromia grown on pure chromium: a photoelectrochemical and microscopic study. Oxid. Met. 86(5), 497–509 (2016). https://doi.org/10.1007/s11085-016-9648-6
L. Latu-Romain, Y. Parsa, S. Mathieu, M. Vilasi, Y. Wouters, Chromia Scale thermally grown on pure chromium under controlled p(O2) atmosphere: I. Spallation Investigation using Photoelectrochemical techniques at a Mesoscale. Oxid. Met. 90(3), 255–266 (2018). https://doi.org/10.1007/s11085-018-9844-7
L. Latu-Romain, Y. Parsa, M. Ollivier, S. Mathieu, M. Vilasi, G. Renou, F. Robaut, Y. Wouters, About the control of semiconducting properties of chromia: investigation using photoelectrochemistry and orientation mapping in a TEM. Mater. High. Temp. 35(1–3), 159–167 (2018). https://doi.org/10.1080/09603409.2017.1389113
L. Latu-Romain, Y. Parsa, Y. Wouters, Spallation study of chromia scales thermally grown on pure chromium in synthetic air. Mater. Char. 152, 58–66 (2019). https://doi.org/10.1016/j.matchar.2019.04.011
M. Vaidya, K.G. Pradeep, B.S. Murty, G. Wilde, S.V. Divinski, Bulk tracer diffusion in CoCrFeNi and CoCrFeMnNi high entropy alloys. Acta Mater. 146, 211–224 (2018). https://doi.org/10.1016/j.actamat.2017.12.052
K.Y. Tsai, M.H. Tsai, J.W. Yeh, Sluggish diffusion in Co–Cr–Fe–Mn–Ni high-entropy alloys. Acta Mater. 61(13), 4887–4897 (2013). https://doi.org/10.1016/j.actamat.2013.04.058
G.R. Holcomb, J. Tylczak, C. Carney, Oxidation of CoCrFeMnNi High Entropy alloys. JOM. 67(10), 2326–2339 (2015). https://doi.org/10.1007/s11837-015-1517-2
A.L. Marasco, D.J. Young, The oxidation of Iron-chromium-manganese alloys at 900 °C. Oxid. Met. 36(1), 157–174 (1991). https://doi.org/10.1007/BF00938460
Acknowledgements
The authors gratefully acknowledge Heike Nitschke and Romeo Saliwan Neumann for the metallographic work and the extensive SEM–EDS analysis. Mike Schneider acknowledges funding from the Alexander von Humboldt Foundation through a Feodor Lynen Research Fellowship. Guillaume Laplanche acknowledges funding from the German Research Foundation (Deutsche Forschungsgemeinschaft DFG) through the Priority Program SPP 2006 “Compositionally Complex Alloys—High Entropy Alloys” Project LA 3607/3-2.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
WM: conceptualization, investigation, writing—original draft, project management. SK: conceptualization, investigation, data analysis. AMM: writing—review and editing. MS: sample preparation. GL: writing—review and editing. CS-S: conceptualization, writing—original draft, project management.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohring, W., Karafiludis, S., Manzoni, A.M. et al. High-Temperature Corrosion of High- and Medium-Entropy Alloys CrMnFeCoNi and CrCoNi Exposed to a Multi-Oxidant Atmosphere H2O–O2–SO2. High Entropy Alloys & Materials 2, 16–32 (2024). https://doi.org/10.1007/s44210-023-00026-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s44210-023-00026-8