
Abstract
Subarachnoid hemorrhage (SAH) is a prevalent cerebrovascular disease with significant global mortality and morbidity rates. Despite advancements in pharmacological and surgical approaches, the quality of life for SAH survivors has not shown substantial improvement. Traditionally, vasospasm has been considered a primary contributor to death and disability following SAH, but anti-vasospastic therapies have not demonstrated significant benefits for SAH patients' prognosis. Emerging studies suggest that early brain injury (EBI) may play a crucial role in influencing SAH prognosis. Sirtuins (SIRTs), a group of NAD + -dependent deacylases comprising seven mammalian family members (SIRT1 to SIRT7), have been found to be involved in neural tissue development, plasticity, and aging. They also exhibit vital functions in various central nervous system (CNS) processes, including cognition, pain perception, mood, behavior, sleep, and circadian rhythms. Extensive research has uncovered the multifaceted roles of SIRTs in CNS disorders, offering insights into potential markers for pathological processes and promising therapeutic targets (such as SIRT1 activators and SIRT2 inhibitors). In this article, we provide an overview of recent research progress on the application of SIRTs in subarachnoid hemorrhage and explore their underlying mechanisms of action.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Subarachnoid hemorrhage (SAH) is a prevalent cerebrovascular disease with varying incidence rates across different countries and regions. Globally, the overall incidence is estimated to be around 6 per 100,000 individuals annually [1]. The occurrence of subarachnoid hemorrhage tends to increase with age. Despite notable advancements in surgical interventions such as aneurysm clamping and endovascular occlusion, the long-term prognosis for SAH patients remains suboptimal. Delayed cerebral ischemia (DCI), which typically manifests 3–14 days after SAH, has long been recognized as a significant factor contributing to the poor prognosis of SAH patients [2].
Historically, cerebral vasospasm (CVS) has been regarded as the primary cause of DCI following SAH. Consequently, many studies over the past decades have focused on reducing episodes of vasospasm to improve the prognosis of SAH patients [3,4,5,6,7]. However, recent large-scale clinical trials have revealed that treating vasospasm does not yield substantial improvements in patient outcomes [8,9,10]. This suggests that other mechanisms of injury may play a role in determining the prognosis of SAH patients. An increasing body of evidence highlights the importance of early brain injury (EBI), which refers to brain damage occurring within 72 h after subarachnoid hemorrhage [11].
Recent findings indicate that EBI after SAH may lay the foundation for the subsequent development of DCI, characterized by complex pathophysiological changes such as increased intracranial pressure, reduced cerebral blood flow, and direct hematoma toxicity in brain tissue. These processes are followed by blood–brain barrier (BBB) disruption, oxidative stress injury, cell death, inflammatory response, microcirculatory dysfunction, and mitochondrial dysfunction, all contributing to neurological damage and poor prognosis after SAH [12,13,14,15,16].
In recent years, there has been a growing interest in the early diagnosis and treatment of SAH, focusing on initiating neuroprotective measures and early prevention of complications during the initial bleeding stages. This approach aims to mitigate the impact of EBI and improve patient outcomes.
Sirtuins (SIRTs) are a class of NAD + -dependent protein lysine deacylases and ADP ribosylases present in both prokaryotes and eukaryotes. Within the family of histone deacetylases (HDACs), SIRTs are classified as class III HDACs, acting in opposition to the activity of histone acetyltransferases (HATs) [17, 18, 19]. Currently, there are seven known isoforms of sirtuins (SIRT1-7) that exhibit distinct subcellular localizations and substrate specificities. SIRT1, SIRT6, and SIRT7 are primarily located in the nucleus, although a fraction of SIRT1 is also found in the cytoplasm. Conversely, SIRT2 is predominantly cytoplasmic, but certain splice isoforms can be present in the nucleus under specific conditions. SIRT3-5 are primarily localized within mitochondria [20].
Sirtuins play crucial roles in various biological processes, including cell differentiation, transcriptional regulation, cell cycle progression, apoptosis, inflammation, metabolism, as well as neurological and cardiovascular physiology. Over the past two decades, there has been a growing interest in exploring the functions of sirtuins, and they have garnered significant attention [21]. In recent years, the role and mechanisms of sirtuins in SAH have been extensively investigated. The findings suggest that sirtuins are involved in multiple mechanisms that alleviate brain injury following SAH, thereby reducing neurological impairment and improving prognosis. Consequently, modulating sirtuin activity has emerged as a promising therapeutic approach. This review aims to summarize and discuss the existing literature on the involvement of sirtuins in EBI after SAH. The goal is to elucidate the underlying mechanisms responsible for inducing neuroprotection and provide a theoretical basis for understanding the mechanisms and treatment options for EBI following SAH.
The Role and Mechanism of Sirtuins in Subarachnoid Hemorrhage
Sirtuins and Mitochondrial Dysfunction
Mitochondria, double-membrane organelles, are pivotal in energy production within eukaryotic cells. They play crucial roles in cell growth, differentiation, function, signal transduction, cell cycle regulation, apoptosis, and survival [22,23,24]. Impaired mitochondrial function can have detrimental effects, including the loss of mitochondrial membrane potential (ΔΨm), excessive reactive oxygen species (ROS) production, release of apoptogenic proteins, compromised mitochondrial dynamics, and activation of inflammation associated with mitochondria [25, 26]. Recent evidence highlights mitochondrial dysfunction as a novel mechanism and target for EBI in relation to delayed cerebral ischemia (DCI) and SAH outcomes [27]. Additionally, mitochondrial autophagy, known as mitophagy, is activated following SAH and may act as a neuroprotective mechanism [28].
Mitochondrial dynamics, which involve fusion and division processes, have gained considerable attention due to their involvement in various biological phenomena such as apoptosis, senescence, and mitochondrial autophagy [29, 30]. Mitochondrial fusion relies on three guanosine triphosphate (GTP) hydrolases: mitochondrial fusion proteins (Mfn) 1 and 2 facilitate outer membrane fusion, while optic atrophy-associated protein 1 (OPA-1) mediates inner membrane fusion [31]. Under normal circumstances, mitochondrial fusion maintains organelle integrity through the diffusion and sharing of components [32]. Animal studies have demonstrated a significant decrease in the expression of Mfn1 and Mfn2 in the central nervous system (CNS) 24 h after SAH, while OPA-1 expression decreased starting from 3 h post-SAH and continued to decline for up to 72 h [33, 34]. Interestingly, both studies revealed that increased expression of Mfn1/2 and OPA-1 facilitated the mitigation of EBI following SAH [33, 34]. Mitochondrial autophagy, a selective process responsible for removing damaged mitochondria, promotes mitochondrial homeostasis. Accumulation of impaired mitochondria can increase oxygen consumption and ROS production, leading to cellular degeneration and activation of cell death pathways [35]. Abundant evidence supports mitochondrial dysfunction after SAH and its association with EBI and neurological outcomes [36,37,38,39]. Therefore, further investigations are warranted to elucidate the underlying mechanisms of mitochondrial dysfunction in the development of EBI after SAH (Table 1).
Sirtuins have been implicated in the regulation of mitochondrial function in SAH. Activation of the MC1R by BMS-470539 has been shown to attenuate EBI after SAH by promoting mitochondrial biogenesis and controlling mitochondrial metabolism through the AMP-activated protein kinase (AMPK)/SIRT1/PGC-1α pathway [40]. Additionally, fucoxanthin (FX), a derivative of luteolin, restores mitochondrial morphology by activating SIRT1 and preventing cytochrome c release from mitochondria [41].
SIRT3 is predominantly localized in the mitochondrial matrix and plays a crucial role in regulating mitochondrial metabolism, the tricarboxylic acid (TCA) cycle, urea cycle, amino acid metabolism, fatty acid oxidation, electron transport chain (ETC)/oxidative phosphorylation (OXPHOS), detoxification of reactive oxygen species, mitochondrial dynamics, and mitochondrial unfolded protein response (UPR) [42, 43]. SIRT3 improves EBI after SAH by promoting mitochondrial fusion in an AMPK-dependent manner. Activation of SIRT3 by Honikiol (HKL) increases the levels of mitochondrial fusion proteins Mfn1 and Mfn2, thereby maintaining mitochondrial morphology, protecting mitochondrial function, and promoting neuronal cell survival [33].
Moreover, IIPKC is an isoform of protein kinase C (PKC) that interacts with Mfn1 to mediate mitochondrial dysfunction and neuronal damage after SAH, both in vitro and in vivo. The interaction between Mfn1 and IIPKC can be blocked by activating the SIRT3 pathway, as observed in experiments where Sirt3 was knocked down with small interfering RNA (siRNA) [44]. Additionally, protein lysine succinylation serves as a biochemical marker of metabolic crisis after SAH. In animal studies of SAH, resveratrol (RVS) activation of SIRT5-mediated blockade of lysine desuccinylation protected mitochondrial metabolism after SAH, ameliorating neuronal cell death and neurological deficits [45].
Therapies targeting mitochondria in the context of SAH have shown potential in animal studies. The regulation of mitochondrial function by SIRTs offers new perspectives for future studies on the mechanisms and treatment of EBI after SAH.
Sirtuins and Ferroptosis
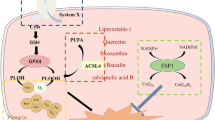
Ferroptosis, a novel form of cell death first defined in 2012, is characterized by non-apoptotic cell demise, iron dependency, and accumulation of reactive lipid substances (RLS) [46]. Ferroptosis exhibits distinct morphological and bioenergetic features that set it apart from other established forms of regulated cell death. Morphologically, it primarily affects mitochondria, leading to smaller mitochondria, increased mitochondrial membrane density, disrupted or absent mitochondrial cristae, and damaged outer mitochondrial membranes, while the nuclear morphology remains unaffected. Bioenergetically, ferroptosis is characterized by iron accumulation and lipid peroxidation [47].
The regulatory mechanism of ferroptosis is complex and still being extensively studied. Current research suggests that the regulation of iron, lipid peroxidation, and various antioxidant systems, such as the GSH/GPX4 pathway, CoQ10/FSP1 pathway, and others, play crucial roles in the regulation of ferroptosis [48]. Iron's involvement in ferroptosis primarily occurs through the Fenton reaction, which catalyzes the generation of ROS and ultimately leads to cell death [49]. Lipid peroxidation and subsequent accumulation of oxidized products are hallmark features of ferroptosis [50]. However, it should be noted that only the esterification of phospholipids into the membrane can induce ferroptosis, and lipid peroxidation can affect various cellular membranes, including the lipid bilayer and subcellular membranes of mitochondria, endoplasmic reticulum, and lysosomes [51].
Iron-induced apoptosis has been implicated in the pathology of EBI after SAH. Notably, ferroptosis is not an isolated pathological process but rather shares commonalities with other forms of cell death, including apoptosis, necroptosis, necrosis, and autophagy [52,53,54]. Furthermore, ferroptosis exhibits regulatory interactions with pathological processes such as inflammation and oxidative stress [55, 56]. Although ferroptosis is a relatively new and recently discovered type of cell death, it shows promising potential for attenuating EBI after SAH and may serve as a novel and effective target for SAH treatment.
Ferroptosis is a recently discovered form of cell death, and there is limited clinical research and translation focused on targeting iron-induced cell death for the treatment of SAH). Studies investigating the role of sirtuins in this context are even scarcer. Only one study conducted in the past two decades, utilizing a mouse model of SAH with crossed anterior pool injection, has demonstrated that activation of SIRT1 can inhibit iron-induced cell death by increasing the expression levels of GPX4 and FSP1 after SAH. This study elucidated, for the first time, the involvement of FSP1/CoQ10-mediated iron depletion in the development of EBI after SAH. Furthermore, the study revealed multiple mechanisms through which ferroptosis contributes to the pathogenesis of EBI after SAH, including upregulation of ASCL4 expression, increased expression of iron uptake proteins (e.g., TFR and DMT1) due to iron overload, reduced GPX4 expression, and inactivation of the FSP1-mediated antioxidant pathway [57]. Therefore, SIRT1 may represent a novel therapeutic target for inhibiting ferroptosis following SAH.
Sirtuins and Neuroinflammation
During the early stages of subarachnoid hemorrhage (SAH), degradation products from erythrocytes rapidly accumulate in the subarachnoid space, triggering an inflammatory response that contributes to the progression of brain injury [58, 59]. Increasing evidence suggests that the neuroinflammatory response is a pervasive factor in both EBI and delayed brain injury (DBI) following SAH, playing a crucial role in the pathogenesis of EBI in particular [60]. The inflammatory cascade is primarily initiated by molecules released from extravasated blood and damaged brain tissue, accompanied by activation and infiltration of immune cells at the site of injury [61]. In addition to the classical neuroinflammation, SAH also leads to aseptic neurogenic inflammation, characterized by the release of potent vasoactive neuropeptides. This aseptic neurogenic inflammatory response is an independent predictor of mortality in SAH patients and can exacerbate classical neuroinflammation through a positive feedback loop involving inflammatory mediators [62,63,64].
The central nervous system (CNS), protected by the blood–brain barrier (BBB), has its own innate immune system, primarily composed of microglia, along with astrocytes and oligodendrocytes [65]. Following SAH, damage-associated molecular patterns (DAMPs) are released by neurons, astrocytes, microglia, and endothelial cells due to tissue injury. DAMPs activate local and peripheral immune cells, leading to the release of inflammation-associated proteins and cytokines that fuel the inflammatory response [66]. Microglial activation occurs within minutes of SAH onset, resulting in rapid morphological changes characterized by enlarged cell bodies, shortened and thickened axons, and an amoeboid appearance [67, 68]. Activated microglia release various factors such as IL-1, IL-6, iNOS, TNF-α, NO, and MMP-9, which contribute to detrimental consequences including cerebral vasospasm, microthrombosis, blood–brain barrier disruption, and neuroapoptosis. This has a significant impact on disease regression and progression in patients [69]. Additionally, the transition of microglia from an anti-inflammatory phenotype (M2) to a pro-inflammatory phenotype (M1) plays a crucial role in microglial activation and the subsequent neuroinflammatory response. The M1 phenotype facilitates the release of pro-inflammatory cytokines that worsen nerve injury, while the M2 phenotype promotes the release of neurotrophic factors that contribute to nerve repair [70]. Similar to microglia, astrocytes have the ability to synthesize and secrete inflammatory factors, including cytokines and chemokines, which participate in the inflammatory process of SAH [71].
Apart from inflammatory cells, inflammation-associated proteins such as nuclear factor-kB (NF-kB), intercellular adhesion molecule-1 (ICAM-1), high mobility group box 1 (HMGB1), NLRP3 inflammasomes, toll-like receptors (TLRs), and mitogen-activated protein kinase (MAPK) also play critical roles in post-SAH inflammation [72]. Furthermore, pro-inflammatory cytokines like interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) upregulate specific cell adhesion molecules on brain capillary endothelial cells, directly damage peripheral nerve cells, increase leukocyte recruitment, induce neuroinflammation, degrade the brain capillary endothelial basement membrane, leading to BBB disruption, and contribute to various apoptotic cell death processes, further exacerbating brain injury after SAH [73].
Therefore, modulating the neuroinflammatory response may alleviate early EBI following SAH and potentially improve patient prognosis.
Sirtuins have a regulatory role in the inflammatory response of the CNS. Numerous studies have reported a close association between SIRT1 and neuroinflammation, with NF-kB, p53, Nrf2, FOXO, and HIF identified as substrates for SIRT1 deacetylation, both in histone and non-histone proteins [74]. Interestingly, levels of SIRT1 protein were found to be significantly elevated in the early stages of SAH, peaking at 24 h after the event. The endogenous SIRT1-regulated p53 pathway reduces p53 acetylation by inhibiting FOXO1 and NF-кB. Activation of the SIRT1 pathway after SAH leads to a significant reduction in levels of IL-1b, IL-6, and TNF-a, decreased activation of microglia, reduced levels of Bax and cleaved-caspase-3, increased expression of Bcl-2, and improved neuroinflammation following SAH [75,76,77]. SIRT1 has also been found to be closely associated with neuroinflammation in multiple studies. TLR4, highly expressed in microglia, can induce the production of various pro-inflammatory cytokines and chemokines, including IL-1b, IL-6, TNF-a, and ICAM-1. Astaxanthin (ATX) effectively alleviates brain inflammation by activating SIRT1 and inhibiting the TLR4 signaling pathway. However, this effect is ineffective in mice lacking the TLR4 gene [78]. Pinocembrin upregulates SIRT1 expression, suppresses NF-кB and microglia activation, and ameliorates inflammation following EBI [79]. In EBI after SAH, resveratrol (RSV) inhibits pro-inflammatory cytokines (IL-1b, IL-6, and TNF-a) through the AMPK/SIRT1 cascade [80]. Furthermore, melatonin has been shown to downregulate Ac-NF-kB and Bax expression while upregulating SIRT1 expression, indicating that melatonin attenuates neuroinflammation after SAH partly through the melatonin receptor (MR)/SIRT1/NF-kB signaling pathway [81]. Phosphodiesterase-4 (PDE-4) plays a crucial role in various CNS injuries. As a PDE4 inhibitor, rolipram significantly increases the expression of SIRT1, leading to the inhibition of NF-kB activation in EBI after SAH. Rolipram ameliorates inflammation by upregulating the expression of the protective cytokine IL-10, inhibiting the expression of pro-inflammatory cytokines TNFa, IL-1ß, and IL-6, and suppressing microglial activation [82].
Additionally, intense brain inflammation following SAH is associated with the substantial activation of the HMGB1/NF-kB pathway [83, 84]. Growing evidence suggests that SIRT1 regulates HMGB1 hyperacetylation and inhibits HMGB1 translocation release. Increased expression of SIRT1 inhibits HMGB1/NF-KB activation-mediated inflammatory responses after SAH. The selective SIRT1 inhibitor EX527 reverses the SIRT1 activation induced by cycloastragenol (CAG) and attenuates the anti-inflammatory and neuroprotective effects of CAG on SAH, as evidenced by the upregulation of TNF-a, IL-1b, IL-6, ICAM-1 release, and microglial activation [76].
Oleanolic acid (OA) reduces the acetylation level of HMGB1 by increasing the expression of SIRT1, rather than inhibiting the JAK/STAT3 pathway. This promotion of HMGB1 deacetylation inhibits its translocation from the nucleus to the cytoplasm, thus reducing the serum level of HMGB1. OA exerts its anti-inflammatory effects through the SIRT1 signaling pathway, downregulating the expression of TLR4, TNF-a, IL-1b, and NF-kB, thereby exerting anti-inflammatory effects. Furthermore, it has been observed that HMGB1 is predominantly expressed in neurons and associated with apoptosis in EBI after SAH, while in DBI after SAH, it is primarily expressed in microglia and associated with immune activation. This finding remains controversial but represents a valuable issue that merits further investigation [85].
Furthermore, NLRP3 inflammasomes, functioning as multiprotein oligomers, play a pivotal role in activating the inflammatory response, promoting IL-1b maturation, and inducing IL-1b release, ultimately resulting in inflammation and tissue damage [86]. Activation of SIRT1 has been found to reduce the acetylation of FoxO1 and P53 and inhibit NLRP3 inflammasome activation [87]. Another study demonstrated that SIRT1 could alleviate EBI after SAH by modulating NLRP3 inflammasome signaling to shift microglial phenotype from M1 to M2 phenotype [88]. Recently, in an estrogen-deficient aneurysm model, the deficiency of estrogen receptor alpha (ERα) and SIRT1 may contribute to inflammation and tissue damage, promoting the activation of the NLRP3/IL-1b/MMP-9 pathway, thereby increasing the risk of intracranial aneurysm rupture and SAH [89]. Experimental evidence also suggests the involvement of other members of the SIRT family in neuroinflammation after SAH. For instance, bexarotene activates retinoid X receptors (RXR) to reduce neuroinflammation after SAH through the PPARγ/SIRT6/FoxO3a pathway [90].
Neuroinflammation is a common cause of brain injury in SAH, and while some studies suggest that targeting neuroinflammation could be a therapeutic option, other clinical studies have found that modulating inflammation after SAH does not yield beneficial effects [91]. Acute neuroinflammation is often considered a protective response, whereas chronic neuroinflammation is deemed harmful and damaging to neural tissue. It is important to recognize that inflammatory pathways and mediators may have both protective and detrimental roles at different stages [92]. Therefore, the management of neuroinflammation after SAH should not solely focus on suppressing inflammation; it should also consider avoiding the blockade of neuroprotective immune responses. In the future, individualized treatments tailored to the timing and intensity of the inflammatory response in each patient should be developed. Modulation of SIRTs presents a promising avenue for achieving this goal.
Sirtuins and Oxidative Stress
Oxidative stress (OS) is a crucial factor contributing to early brain injury (EBI) after subarachnoid hemorrhage (SAH). The brain is highly vulnerable to oxidative damage due to its abundance of polyunsaturated fatty acids in the tissue. Following SAH, an excessive generation of free radicals occurs, depleting the endogenous antioxidant system and leading to the downregulation of key antioxidant enzymes such as superoxide dismutase (SOD), glutathione peroxidase, and catalase in brain tissue, thereby disrupting redox homeostasis. Additionally, SAH-induced CVS and cerebral edema result in cerebral ischemia, which leads to the production of substantial amounts of reactive oxygen species (ROS) such as oxygen ions (O2-) and hydrogen peroxide (H2O2) [93, 94]. The release of oxyhemoglobin or hemoglobin from erythrocytes following erythrocyte lysis after SAH further exacerbates the situation as these molecules readily undergo oxidation to form methemoglobin [95]. High levels of Fe2 + and Fe3 + can react with H2O2 and O2- through the Fenton reaction, generating hydroxyl radicals, the most toxic ROS. Hydroxyl radicals directly damage neurovascular units, leading to neurological impairment, and the remaining ROS contribute to mitochondrial dysfunction, perpetuating a detrimental cycle of ROS production. Moreover, oxidative stress induced by free radicals results in lipid peroxidation, protein degradation, and DNA damage, ultimately leading to neuronal apoptosis, endothelial cell damage, and BBB disruption [96, 97]. Importantly, a growing body of evidence suggests that the prevention of excessive ROS can alleviate EBI after SAH [98, 99]. Therefore, targeting the prevention of oxidative damage may represent a promising therapeutic approach to improve the prognosis following subarachnoid hemorrhage.
Despite the substantial evidence of oxidative stress in SAH, the use of antioxidants as a treatment modality for EBI in SAH is not recommended in the definitive guidelines [100, 101]. Nevertheless, numerous studies have highlighted the regulatory role of SIRTs in various biological processes, with SIRT1 particularly known for its antioxidant properties. SIRT1 deacetylation has been shown to inhibit oxidative pathways mediated by FoxOs, NF-кB, and to some extent, P53, offering neuroprotection against EBI in animal models. Additionally, a decrease in SIRT1 levels was associated with exacerbated cortical oxidative damage, microglial activation, and the release of pro-inflammatory cytokines [75, 76]. Another antioxidant, ATX, exerts its protective effects against SAH-induced oxidative stress by increasing SIRT1 expression and inhibiting the TLR4 signaling pathway [78]. Fx, derived from seaweed, attenuates SAH-induced reactive oxygen species (ROS) production and lipid peroxidation through a SIRT1-dependent pathway. Fx inhibits the acetylation of downstream substrates FoxO1 and p53, restores the activity of endogenous antioxidant enzymes, blocks mitochondrial cytochrome c release, and restores mitochondrial morphology following SAH [41].
Activation of the melanocortin 1 receptor by BMS-470539 inhibits oxidative stress and mitochondrial division after EBI through the AMPK/SIRT1/PGC-1α signaling pathway [40]. Nrf2, a basic leucine zipper protein (bZIP), plays a crucial role in maintaining cellular redox homeostasis. Activation of Nrf2 induces the expression of several antioxidant genes, including glutathione peroxidase (GSH-Px), superoxide dismutase (SOD), and heme oxygenase-1 (HO-1) [102]. The Keap1/Nrf2/ARE pathway has been identified as an antioxidant target in models of the oxidative stress response after SAH [103]. Under the regulation of SIRT1 activation, salvianolic acid B ameliorates oxidative damage by promoting Nrf2 nuclear translocation. Knockdown of Nrf2 significantly reverses the antioxidant effect of salvianolic acid B, indicating a positive feedback loop between SIRT1 and Nrf2 signaling [104]. Two animal studies have shown that isoglycyrrhizin (ISL) and the resveratrol (RVS) analog pterostilbene (PTE) can mitigate severe oxidative damage by activating the SIRT1/Nrf2 pathway and enhancing the activity of endogenous antioxidant enzymes [105, 106]. Recently, other members of the SIRT family, particularly SIRT3, have gained attention for their antioxidant mechanisms in various neurological disorders, including ischemic stroke, Huntington's disease, and Alzheimer's disease [107]. SIRT3 is significantly activated in animal models of SAH, both in vivo and in vitro. The transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) is involved in regulating the antioxidant activity of SIRT3 after SAH, thereby enhancing the endogenous antioxidant response [108]. In a time-dependent manner, the expression of SIRT3 in cortical neurons was found to decrease, and both mRNA and protein expression of SOD2 showed a positive correlation with SIRT3 expression [109]. Furthermore, melatonin was shown to reduce ROS levels, inhibit the expression of SOD2 and the lipid peroxidation marker malondialdehyde (MDA), and regulate SIRT3 expression [110]. SIRT6 has demonstrated protective effects against cardiac I/R injury by upregulating antioxidants and suppressing OS. Expanding on this success, activation of the RXR could potentially ameliorate certain neurological deficits after SAH by modulating the PPARγ/SIRT6/FOXO3a pathway [90]. Currently, four free radical scavengers, including edaravone, tirazadex mesylate, nicardipine, and ebselenolide, are undergoing clinical trials, but unfortunately, no neuroprotective effects have been reported [93]. The modulation of SIRTs to reduce oxidative stress after SAH holds promise as a new avenue of investigation that merits further exploration.
Sirtuins and Apoptosis Versus Autophagy
Apoptosis is a prominent pathological process in EBI, and experimental studies have demonstrated its close association with brain injury following SAH [111]. Apoptosis is characterized by distinct morphological changes such as cell shrinkage, nuclear fragmentation, chromatin condensation, and chromosomal DNA fragmentation [112]. Neuronal apoptosis has been observed within 10 min of SAH onset and continues for up to 24 h [113]. Due to the limited regenerative capacity of the human brain, EBI-induced neuronal injury can lead to permanent damage and long-term neurological impairment [114]. Apoptosis can occur through three different pathways: the extrinsic pathway, the intrinsic pathway, and the endoplasmic reticulum stress-induced pathway, depending on the site of apoptosis. Molecular mechanisms such as p53 and oxidative stress pathways are also implicated in SAH-induced apoptosis [115]. The extrinsic pathway, also known as the death receptor pathway, involves pro-apoptotic death receptors such as TNFR1, TNFR2, Fas, and TRAIL-R1 (DR 4) and TRAIL-R2 (DR 5) [116]. The upregulation of Fas and TNF ligands following SAH binds to death receptors, activating the caspase cascade [117, 118]. The intrinsic pathway, known as the mitochondrial pathway, is primarily regulated by the B-cell lymphoma-2 (Bcl-2) family of proteins. Upon apoptotic stimulation during SAH, mitochondrial outer membrane permeability increases, and cytochrome C is released from the mitochondria into the cytoplasm, where it forms apoptotic complexes with apoptotic protease activator 1 (Apaf-1), leading to the activation of caspase-9. Caspase-3 is subsequently activated, triggering apoptosis [119, 120]. Endoplasmic reticulum stress (ERS) occurs due to imbalanced ion levels and the accumulation of misfolded or unfolded proteins in the endoplasmic reticulum [121]. ERS disrupts calcium homeostasis and affects mitochondrial and Bcl-2 family protein activities, resulting in apoptosis. It also activates the cysteine protease cascade, further influencing apoptosis [122, 123]. Recent studies have revealed morphological changes in the endoplasmic reticulum within 6 h of SAH, with cortical neuron swelling peaking at 24 h and subsiding within 24–48 h [124]. These pathways interact with each other and contribute to the initiation and regulation of apoptosis after SAH. By targeting these pathways, it may be possible to effectively alleviate apoptosis, thereby mitigating nerve injury and promoting neurological recovery after SAH.
Autophagy is a cellular repair process that maintains intracellular homeostasis by selectively degrading and recycling cytoplasmic components and eliminating unwanted cellular entities [125]. Following SAH, the autophagic pathway is activated and reaches its peak at 24 h [22]. Activation of autophagy has been shown to exert neuroprotective effects [126]. In a rat model of intravascular perforation, autophagy activation reduced the translocation of Bax from the cytosol to the mitochondrial membrane, thereby counteracting apoptotic effects [127]. The mitochondrial pathway is believed to be involved in autophagy-mediated regulation of apoptosis in EBI after SAH [128]. Studies have consistently demonstrated that proper regulation of autophagic mechanisms has a pro-survival effect and reduces apoptotic cell death after SAH. However, when SAH surpasses a certain stress threshold, autophagic mechanisms can contribute to increased apoptotic cell death [129,130,131,132]. Besides autophagy and apoptosis, necrosis may also occur simultaneously in neurons after SAH, resulting in a mixed pattern of cell death morphology. Moreover, extensive crosstalk exists between autophagic and apoptotic pathways [133]. Recent research has highlighted the importance of reducing neuronal death to ameliorate neurological dysfunction and improve patient prognosis [134, 135]. Therefore, future studies should focus on understanding the interplay between autophagy, apoptosis, and necrosis rather than studying them in isolation. A comprehensive understanding of the relationship among these pathways holds promise for advancing SAH treatment options.
Several studies have demonstrated the neuroprotective effect of sirtuins on apoptosis in EBI cells after subarachnoid hemorrhage and their involvement in autophagy regulation. Numerous studies focusing on SIRT1 have revealed its ability to ameliorate apoptosis after SAH through multiple pathways. For instance, wogonoside and resveratrol can activate SIRT1, deacetylate p53, inhibit p53-mediated transcriptional activity, increase cleaved caspase-3 and Bax levels, and increase Bcl-2 levels, thereby blocking apoptosis [76, 136, 137]. Pterostilbene (PTE) effectively upregulates SIRT1 expression, promotes nuclear Nrf2 accumulation, inhibits microglial activation and pro-inflammatory mediators after SAH, and attenuates oxidative damage and neuroinflammation, directly inhibiting apoptosis [106]. Magnesium lithospermate B (MLB) and melatonin (Mel), extracted from Salvia miltiorrhiza, induce upregulation of Bcl-2 expression and downregulation of Bax expression through the SIRT1/NF-κB pathway, exerting anti-apoptotic effects and inhibiting apoptosis after SAH [77, 81]. Moreover, inhibition of phosphodiesterase-4 (PDE4), an enzyme that hydrolyzes cAMP, significantly increases SIRT1 expression after SAH, upregulates Akt phosphorylation, and decreases the expression of apoptotic proteins, thereby inhibiting neuronal apoptosis after SAH [138]. Other members of the sirtuin family, such as SIRT3, have also been implicated in the regulation of apoptosis after SAH in animal studies. Puerarin (Pur) reduces cortical neuronal degeneration and apoptosis by downregulating the Bcl-2/Bax/cleaved caspase-3 apoptotic pathway and upregulating the SIRT3/SOD2 antiapoptotic pathway [139]. Tauroursodeoxycholic acid (TUDCA) attenuates apoptosis after SAH by activating the TGR5/SIRT3 pathway, leading to the inhibition of apoptotic protein expression. Conversely, knockdown of TGR5 by siRNA abolishes the beneficial effects of TUDCA [140]. Unfortunately, the role of SIRTs in autophagy after SAH remains poorly studied. However, resveratrol has been shown to mediate autophagy and apoptosis in SAH through the regulation of the Akt/mTOR pathway. Resveratrol stimulates autophagy through the AMPK/SIRT1 signaling pathway, inhibiting the release of pro-inflammatory cytokines and neuronal apoptosis, thereby uncovering a novel molecular mechanism for its protective effect in subarachnoid hemorrhage [80]. Autophagy plays a crucial role in maintaining intracellular homeostasis in the brain following SAH, exerting a pro-survival effect and reducing apoptotic cell death. Thus, modulating autophagy and the crosstalk with apoptosis may hold therapeutic benefits in the context of SAH. Additionally, sirtuins can regulate both apoptosis and autophagy. The relationship between apoptosis and autophagy in SAH presents an intriguing target for further research and therapeutic interventions.
Sirtuins and Blood–Brain Barrier Disruption
The blood–brain barrier primarily consists of capillary endothelial cells, pericytes, astrocytes, and the vascular basement membrane. Capillary endothelial cells are closely connected, resulting in minimal cell gaps. In normal physiological conditions, the BBB prevents most substances, except for a few lipid-soluble molecules and gases, from crossing into the brain, including plasma components and red blood cells [141, 142]. However, under pathological conditions, the BBB becomes compromised, allowing harmful blood components such as thrombin and fibrinogen to enter the brain parenchyma and directly expose the brain tissue to these toxic substances [143]. Increased BBB permeability also facilitates the infiltration and restricted migration of immune cells, like leukocytes, into the brain parenchyma, leading to the release of various cytokines, chemokines, reactive oxygen species, and proteases. This further exacerbates brain tissue damage, raises intracranial pressure, triggers neuronal apoptosis, and can contribute to epilepsy. Additionally, BBB permeability disruption can cause dysfunction in endothelial cells and the breakdown of tight junctions, resulting in the formation of cerebral edema [144,145,146].
Brain edema following SAH is primarily categorized as vasogenic and cytotoxic. Vasogenic edema occurs due to the leakage of plasma proteins and fluid accumulation in the brain interstitium [147]. On the other hand, cytotoxic edema is a consequence of reduced cerebral blood flow (CBF) resulting from increased intracranial pressure (ICP) after SAH. This reduction in blood flow leads to whole-brain ischemia, ATP depletion, and loss of energy from key "pumps" like Na + -K + -ATPase and Ca2 + -ATPase, eventually causing cellular swelling [148, 149]. Studies have shown that BBB disruption occurs soon after SAH onset, even before observable changes on MRI. Researchers have provided evidence that BBB damage begins as early as 30 min after SAH, peaks at 3 h, and can be assessed at 72 h by evaluating tight junction proteins such as occludin and ZO-1. Clinical data also indicate that approximately 8% of patients present with whole-brain edema upon admission using cranial CT, and an additional 12% develop significant brain edema within 6 days of SAH [12, 150]. Severe cerebral edema often leads to elevated ICP, acute cerebral ischemia, brain herniation, and potentially fatal outcomes for patients. Therefore, it is crucial to safeguard the integrity of the blood–brain barrier, reduce the occurrence of cerebral edema, and improve the prognosis of individuals with SAH [151].
Hypertonic saline, hyperosmolar agents, therapeutic hypothermia, barbiturates, non-peptide antidiuretic hormone receptor antagonists, calcium channel blockers, and decompression with debridement are commonly utilized in clinical practice to mitigate cerebral edema and manage intracranial pressure. However, the precise efficacy of these interventions has not been adequately evaluated or retrospectively analyzed [152,153,154,155]. Cerebral edema, which results from blood–brain barrier dysfunction, is a significant independent risk factor for the high morbidity and mortality associated with subarachnoid hemorrhage [156]. Maintaining the integrity of the blood–brain barrier relies on tight junction proteins, including claudin-5, occludin, and ZO-1, which are crucial components of the blood–brain barrier structure and regulation [157,158,159]. Recent experimental studies have demonstrated that astragaloside, resveratrol, and MEL, as SIRT1 activators, enhance SIRT1 expression and suppress p53 activation through deacetylation. Additionally, they downregulate the activity and expression of matrix metalloproteinase-9 (MMP-9) while increasing the expression of ZO-1, claudin-5, and occludin. These actions contribute to protecting the blood–brain barrier and ameliorating brain edema. Notably, the administration of the potent SIRT1 inhibitor sirtinol (SIR) hampers SIRT1 activation, reverses the aforementioned protein expression and outcomes, and exacerbates brain edema following experimental subarachnoid hemorrhage [136, 137, 160]. Modulating SIRT1 may therefore represent a promising therapeutic target for effectively reducing brain edema subsequent to subarachnoid hemorrhage.
Conclusion and Outlook
SAH represents a fatal type of stroke, and even those patients who survive often face a grim prognosis, including conditions like hemiplegia, aphasia, cognitive dysfunction, or even death. These outcomes significantly impact patients' quality of life and pose a substantial socioeconomic burden [1, 161, 162]. Experimental studies have highlighted the intricate nature of early EBI after SAH, which arises from a combination of mechanisms rather than a singular cause. These mechanisms can exhibit synergistic effects and temporal variability. However, most animal model studies exploring the role and mechanisms of sirtuins in regulating SAH have focused solely on one aspect, neglecting the investigation of other potential mechanisms underlying EBI. Additionally, there remains controversy regarding the expression of SIRT1 in astrocytes. Despite extensive research into the mechanisms of EBI, the exact molecular pathway remains unclear, impeding the development of effective and targeted therapeutic interventions. Sirtuins, as epigenetic regulators, participate in numerous physiological activities such as genome stabilization, cancer, stress response, apoptosis, metabolism, aging, proliferation, and inflammation [163,164,165]. Generally, sirtuins confer neuroprotective effects on CNS cells, with a possible synergistic enhancement of intracerebral homeostasis through distinct cellular regulatory pathways involving SIRT1-SIRT7 proteins. Among the sirtuins, SIRT1 was the first to be identified and extensively studied for its involvement in various disease-modulating mechanisms [166]. Presently, studies examining sirtuins in the context of SAH are primarily limited to animal and cellular models, with a scarcity of clinical evidence. Although animal models, including endovascular perforation and autologous blood injection, are widely utilized to mimic SAH [167], more comprehensive investigations are required to establish a solid understanding of the role of sirtuins in SAH pathophysiology.
Regrettably, the intricate physiological conditions associated with human SAH are difficult to fully replicate in animal models. Consequently, it is crucial to identify a more suitable SAH model for future investigations. Moreover, extensive clinical studies involving humans are necessary to confirm the mechanisms of action, efficacy, and safety of potential therapies. Sirtuins have garnered significant attention as promising therapeutic targets, leading to the development of specific sirtuin activators and inhibitors. These compounds serve as valuable tools to study sirtuin activity and hold potential as therapies for associated diseases. Clinical trials utilizing sirtuin activators have been conducted to prevent age-related conditions [168, 178]. Furthermore, many sirtuin modulators, including substances like resveratrol, melatonin, and tannic acid, are derived from natural sources, boasting advantages such as environmental friendliness, accessibility, and minimal side effects [169, 170]. Additionally, approximately one-third of SAH patients experience DCI occurring between days 4 and 14 or even later following EBI [171, 172]. Recently, a new perspective has emerged challenging the conventional classification of SAH stages, suggesting that DCI is a continuation of EBI rather than a distinct phase [173]. Intriguingly, a significant body of research has demonstrated the protective effects of enhanced SIRT1 expression in DCI after SAH [174,175,176,177]. However, translating the findings of experimental studies on sirtuins into clinical trials poses several challenges. These include the complex and bidirectional nature of sirtuin activity, genetic and biochemical differences between human diseases and animal models, and the difficulty of identifying suitable candidate molecules with favorable pharmacodynamic profiles and bioavailability [178]. Nevertheless, the field of sirtuin modulation has emerged as a promising avenue for drug development. Its potential in treating brain injury and improving prognosis following SAH presents an exciting direction for future research, warranting further exploration despite the associated risks and challenges.
References
Etminan N, Chang HS, Hackenberg K, de Rooij NK, Vergouwen MDI, Rinkel GJE, Algra A. Worldwide incidence of aneurysmal subarachnoid hemorrhage according to region, time period, blood pressure, and smoking prevalence in the population: a systematic review and meta-analysis. JAMA Neurol. 2019;76(5):588–97. https://doi.org/10.1001/jamaneurol.2019.0006.
Claassen J, Park S. Spontaneous subarachnoid haemorrhage. Lancet. 2022;400(10355):846–62. https://doi.org/10.1016/S0140-6736(22)00938-2.
Kassell NF, Sasaki T, Colohan AR, Nazar G. Cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Stroke. 1985;16(4):562–72. https://doi.org/10.1161/01.str.16.4.562.
Macdonald RL, Weir BK. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke. 1991;22(8):971–82. https://doi.org/10.1161/01.str.22.8.971.
Cook DA. Mechanisms of cerebral vasospasm in subarachnoid haemorrhage. Pharmacol Ther. 1995;66(2):259–84. https://doi.org/10.1016/0163-7258(94)00080-m.
Crowley RW, Medel R, Kassell NF, Dumont AS. New insights into the causes and therapy of cerebral vasospasm following subarachnoid hemorrhage. Drug Discov Today. 2008;13(5–6):254–60. https://doi.org/10.1016/j.drudis.2007.11.010.
Etminan N, Macdonald RL. Neurovascular disease, diagnosis, and therapy: subarachnoid hemorrhage and cerebral vasospasm. Handb Clin Neurol. 2021;176:135–69. https://doi.org/10.1016/B978-0-444-64034-5.00009-2.
Macdonald RL, Kassell NF, Mayer S, Ruefenacht D, Schmiedek P, Weidauer S, Frey A, Roux S, Pasqualin A, CONSCIOUS-1 Investigators. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke. 2008;39(11):3015–21. https://doi.org/10.1161/STROKEAHA.108.519942.
Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I, Bach D, Frey A, Marr A, Roux S, Kassell N. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol. 2011;10(7):618–25. https://doi.org/10.1016/S1474-4422(11)70108-9.
Shen J, Pan JW, Fan ZX, Xiong XX, Zhan RY. Dissociation of vasospasm-related morbidity and outcomes in patients with aneurysmal subarachnoid hemorrhage treated with clazosentan: a meta-analysis of randomized controlled trials. J Neurosurg. 2013;119(1):180–9. https://doi.org/10.3171/2013.3.JNS121436.
Cahill J, Zhang JH. Subarachnoid hemorrhage: is it time for a new direction? Stroke. 2009;40(3 Suppl):S86-7. https://doi.org/10.1161/STROKEAHA.108.533315.
Cahill J, Calvert JW, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26(11):1341–53. https://doi.org/10.1038/sj.jcbfm.9600283. (Erratum in: J Cereb Blood Flow Metab. 2006 Nov;26(11):1463. Cahill, W Julian [corrected to Cahill, Julian]; Calvert, John H [corrected to Calvert, John W]).
Ostrowski RP, Colohan AR, Zhang JH. Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28(4):399–414. https://doi.org/10.1179/016164106X115008.
Sehba FA, Hou J, Pluta RM, Zhang JH. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol. 2012;97(1):14–37. https://doi.org/10.1016/j.pneurobio.2012.02.003.
Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, Zhang JH. Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Transl Stroke Res. 2013;4(4):432–46. https://doi.org/10.1007/s12975-013-0257-2.
Ji C, Chen G. Signaling pathway in early brain injury after subarachnoid hemorrhage: news update. Acta Neurochir Suppl. 2016;121:123–6. https://doi.org/10.1007/978-3-319-18497-5_21.
Ho TCS, Chan AHY, Ganesan A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J Med Chem. 2020;63(21):12460–84. https://doi.org/10.1021/acs.jmedchem.0c00830.
Fiorentino F, Mai A, Rotili D. Lysine acetyltransferase inhibitors from natural sources. Front Pharmacol. 2020;12(11):1243. https://doi.org/10.3389/fphar.2020.01243.
Mautone N, Zwergel C, Mai A, Rotili D. Sirtuin modulators: where are we now? A review of patents from 2015 to 2019. Expert Opin Ther Pat. 2020;30(6):389–407. https://doi.org/10.1080/13543776.2020.1749264.
Dang W. The controversial world of sirtuins. Drug Discov Today Technol. 2014;12:e9–17. https://doi.org/10.1016/j.ddtec.2012.08.003.
Fiorentino F, Mautone N, Menna M, D’Acunzo F, Mai A, Rotili D. Sirtuin modulators: past, present, and future perspectives. Future Med Chem. 2022;14(12):915–39. https://doi.org/10.4155/fmc-2022-0031.
Chen S, Wu H, Tang J, Zhang J, Zhang JH. Neurovascular events after subarachnoid hemorrhage: focusing on subcellular organelles. Acta Neurochir Suppl. 2015;120:39–46. https://doi.org/10.1007/978-3-319-04981-6_7.
Yang LQ, Chen M, Ren DL, Hu B. Dual oxidase mutant retards mauthner-cell axon regeneration at an early stage via modulating mitochondrial dynamics in Zebrafish. Neurosci Bull. 2020;36(12):1500–12. https://doi.org/10.1007/s12264-020-00600-9.
Elfawy HA, Das B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: etiologies and therapeutic strategies. Life Sci. 2019;1(218):165–84. https://doi.org/10.1016/j.lfs.2018.12.029.
Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125(7):1241–52. https://doi.org/10.1016/j.cell.2006.06.010.
Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619–42. https://doi.org/10.1146/annurev.physiol.60.1.619.
Zhang Z, Zhang A, Liu Y, Hu X, Fang Y, Wang X, Luo Y, Lenahan C, Chen S. New mechanisms and targets of subarachnoid hemorrhage: a focus on mitochondria. Curr Neuropharmacol. 2022;20(7):1278–96. https://doi.org/10.2174/1570159X19666211101103646.
Huang L, Wan J, Chen Y, Wang Z, Hui L, Li Y, Xu D, Zhou W. Inhibitory effects of p38 inhibitor against mitochondrial dysfunction in the early brain injury after subarachnoid hemorrhage in mice. Brain Res. 2013;23(1517):133–40. https://doi.org/10.1016/j.brainres.2013.04.010.
van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013;5(6):a011072. https://doi.org/10.1101/cshperspect.a011072.
Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11(12):872–84. https://doi.org/10.1038/nrm3013.
Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. 2020;24(15):235–59. https://doi.org/10.1146/annurev-pathmechdis-012419-032711.
Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280(28):26185–92. https://doi.org/10.1074/jbc.M503062200.
Wu X, Luo J, Liu H, Cui W, Feng D, Qu Y. SIRT3 protects against early brain injury following subarachnoid hemorrhage via promoting mitochondrial fusion in an AMPK dependent manner. Chin Neurosurg J. 2020;3(6):1. https://doi.org/10.1186/s41016-019-0182-7.
Zhang T, Xu S, Wu P, Zhou K, Wu L, Xie Z, Xu W, Luo X, Li P, Ocak U, Ocak PE, Travis ZD, Tang J, Shi H, Zhang JH. Mitoquinone attenuates blood-brain barrier disruption through Nrf2/PHB2/OPA1 pathway after subarachnoid hemorrhage in rats. Exp Neurol. 2019;317:1–9. https://doi.org/10.1016/j.expneurol.2019.02.009.
Palikaras K, Tavernarakis N. Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp Gerontol. 2014;56:182–8. https://doi.org/10.1016/j.exger.2014.01.021.
Jacobsen A, Nielsen TH, Nilsson O, Schalén W, Nordström CH. Bedside diagnosis of mitochondrial dysfunction in aneurysmal subarachnoid hemorrhage. Acta Neurol Scand. 2014;130(3):156–63. https://doi.org/10.1111/ane.12258.
Chou SH, Lan J, Esposito E, Ning M, Balaj L, Ji X, Lo EH, Hayakawa K. Extracellular mitochondria in cerebrospinal fluid and neurological recovery after subarachnoid hemorrhage. Stroke. 2017;48(8):2231–7. https://doi.org/10.1161/STROKEAHA.117.017758.
Chen Y, Chen J, Sun X, Shi X, Wang L, Huang L, Zhou W. Evaluation of the neuroprotective effect of EGCG: a potential mechanism of mitochondrial dysfunction and mitochondrial dynamics after subarachnoid hemorrhage. Food Funct. 2018;9(12):6349–59. https://doi.org/10.1039/c8fo01497c.
Shi G, Cui L, Chen R, Liang S, Wang C, Wu P. TT01001 attenuates oxidative stress and neuronal apoptosis by preventing mitoNEET-mediated mitochondrial dysfunction after subarachnoid hemorrhage in rats. NeuroReport. 2020;31(11):845–50. https://doi.org/10.1097/WNR.0000000000001492.
Xu W, Yan J, Ocak U, Lenahan C, Shao A, Tang J, Zhang J, Zhang JH. Melanocortin 1 receptor attenuates early brain injury following subarachnoid hemorrhage by controlling mitochondrial metabolism via AMPK/SIRT1/PGC-1α pathway in rats. Theranostics. 2021;11(2):522–39. https://doi.org/10.7150/thno.49426.
Zhang XS, Lu Y, Tao T, Wang H, Liu GJ, Liu XZ, Liu C, Xia DY, Hang CH, Li W. Fucoxanthin Mitigates subarachnoid hemorrhage-induced oxidative damage via sirtuin 1-dependent pathway. Mol Neurobiol. 2020;57(12):5286–98. https://doi.org/10.1007/s12035-020-02095-x.
Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, Archer SL, Chan DC, Gupta MP. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. 2014;34(5):807–19. https://doi.org/10.1128/MCB.01483-13.
Papa L, Germain D. Correction for Papa and Germain, “SirT3 Regulates a Novel Arm of the Mitochondrial Unfolded Protein Response.” Mol Cell Biol. 2017;37(13):e00191-e217. https://doi.org/10.1128/MCB.00191-17. (Erratum for: Mol Cell Biol. 2014 Feb;34(4):699-710).
Chen T, Wang Y, Wang YH, Hang CH. The Mfn1-βIIPKC interaction regulates mitochondrial dysfunction via Sirt3 following experimental subarachnoid hemorrhage. Transl Stroke Res. 2022;13(5):845–57. https://doi.org/10.1007/s12975-022-00999-5.
Xiao ZP, Lv T, Hou PP, Manaenko A, Liu Y, Jin Y, Gao L, Jia F, Tian Y, Li P, Zhang JH, Hu Q, Zhang X. Sirtuin 5-mediated lysine desuccinylation protects mitochondrial metabolism following subarachnoid hemorrhage in mice. Stroke. 2021;52(12):4043–53. https://doi.org/10.1161/STROKEAHA.121.034850.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72. https://doi.org/10.1016/j.cell.2012.03.042.
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, Sun B, Wang G. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88. https://doi.org/10.1038/s41419-020-2298-2.
Xu Y, Li K, Zhao Y, Zhou L, Liu Y, Zhao J. Role of ferroptosis in stroke. Cell Mol Neurobiol. 2023;43(1):205–22. https://doi.org/10.1007/s10571-022-01196-6.
Stoyanovsky DA, Tyurina YY, Shrivastava I, Bahar I, Tyurin VA, Protchenko O, Jadhav S, Bolevich SB, Kozlov AV, Vladimirov YA, Shvedova AA, Philpott CC, Bayir H, Kagan VE. Iron catalysis of lipid peroxidation in ferroptosis: regulated enzymatic or random free radical reaction? Free Radic Biol Med. 2019;133:153–61. https://doi.org/10.1016/j.freeradbiomed.2018.09.008.
D’Herde K, Krysko DV. Ferroptosis: oxidized PEs trigger death. Nat Chem Biol. 2017;13(1):4–5. https://doi.org/10.1038/nchembio.2261.
Capelletti MM, Manceau H, Puy H, Peoc’h K. Ferroptosis in liver diseases: an overview. Int J Mol Sci. 2020;21(14):4908. https://doi.org/10.3390/ijms21144908.
Lee YS, Lee DH, Choudry HA, Bartlett DL, Lee YJ. Ferroptosis-induced endoplasmic reticulum stress: cross-talk between ferroptosis and apoptosis. Mol Cancer Res. 2018;16(7):1073–6. https://doi.org/10.1158/1541-7786.MCR-18-0055.
Liu J, Guo ZN, Yan XL, Huang S, Ren JX, Luo Y, Yang Y. Crosstalk between autophagy and ferroptosis and its putative role in ischemic stroke. Front Cell Neurosci. 2020;14:577403. https://doi.org/10.3389/fncel.2020.577403.
Zhou Y, Liao J, Mei Z, Liu X, Ge J. Insight into crosstalk between ferroptosis and necroptosis: novel therapeutics in ischemic stroke. Oxid Med Cell Longev. 2021;25(2021):9991001. https://doi.org/10.1155/2021/9991001.
Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, Xu S, Gao Y, Chen X, Sui X, Li G. The emerging role of ferroptosis in inflammation. Biomed Pharmacother. 2020;127:110108. https://doi.org/10.1016/j.biopha.2020.110108.
Liu J, He H, Wang J, Guo X, Lin H, Chen H, Jiang C, Chen L, Yao P, Tang Y. Oxidative stress-dependent frataxin inhibition mediated alcoholic hepatocytotoxicity through ferroptosis. Toxicology. 2020;445:152584. https://doi.org/10.1016/j.tox.2020.152584.
Yuan B, Zhao XD, Shen JD, Chen SJ, Huang HY, Zhou XM, Han YL, Zhou LJ, Lu XJ, Wu Q. Activation of SIRT1 alleviates ferroptosis in the early brain injury after subarachnoid hemorrhage. Oxid Med Cell Longev. 2022;9(2022):9069825. https://doi.org/10.1155/2022/9069825.
Schallner N, Pandit R, LeBlanc R 3rd, Thomas AJ, Ogilvy CS, Zuckerbraun BS, Gallo D, Otterbein LE, Hanafy KA. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J Clin Invest. 2015;125(7):2609–25. https://doi.org/10.1172/JCI78443.
Zhang ZH, Han YL, Wang CX, Zhou CH, Wu LY, Zhang HS, Chen Q, Fan JM, Zhou ML, Hang CH. The effect of subarachnoid erythrocyte lysate on brain injury: a preliminary study. Biosci Rep. 2016;36(4):e00359. https://doi.org/10.1042/BSR20160100.
Wang L, Geng G, Zhu T, Chen W, Li X, Gu J, Jiang E. Progress in research on TLR4-mediated inflammatory response mechanisms in brain injury after subarachnoid hemorrhage. Cells. 2022;11(23):3781. https://doi.org/10.3390/cells11233781.
Schneider UC, Xu R, Vajkoczy P. Inflammatory events following subarachnoid hemorrhage (SAH). Curr Neuropharmacol. 2018;16(9):1385–95. https://doi.org/10.2174/1570159X16666180412110919.
Dagistan Y, Kilinc E, Balci CN. Cervical sympathectomy modulates the neurogenic inflammatory neuropeptides following experimental subarachnoid hemorrhage in rats. Brain Res. 2019;1722:146366. https://doi.org/10.1016/j.brainres.2019.146366.
Luo F, Li Y, Zhao Y, Sun M, He Q, Wen R, Xie Z. Systemic immune-inflammation index predicts the outcome after aneurysmal subarachnoid hemorrhage. Neurosurg Rev. 2022;45(2):1607–15. https://doi.org/10.1007/s10143-021-01681-4.
Devlin P, Ishrat T, Stanfill AG. A systematic review of inflammatory cytokine changes following aneurysmal subarachnoid hemorrhage in animal models and humans. Transl Stroke Res. 2022;13(6):881–97. https://doi.org/10.1007/s12975-022-01001-y.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553. https://doi.org/10.1152/physrev.00011.2010.
Balança B, Desmurs L, Grelier J, Perret-Liaudet A, Lukaszewicz AC. DAMPs and RAGE pathophysiology at the acute phase of brain injury: an overview. Int J Mol Sci. 2021;22(5):2439. https://doi.org/10.3390/ijms22052439.
Heinz R, Brandenburg S, Nieminen-Kelhä M, Kremenetskaia I, Boehm-Sturm P, Vajkoczy P, Schneider UC. Microglia as target for anti-inflammatory approaches to prevent secondary brain injury after subarachnoid hemorrhage (SAH). J Neuroinflammation. 2021;18(1):36. https://doi.org/10.1186/s12974-021-02085-3.
Chen J, Zheng ZV, Lu G, Chan WY, Zhang Y, Wong GKC. Microglia activation, classification and microglia-mediated neuroinflammatory modulators in subarachnoid hemorrhage. Neural Regen Res. 2022;17(7):1404–11. https://doi.org/10.4103/1673-5374.330589.
Jiang CT, Wu WF, Deng YH, Ge JW. Modulators of microglia activation and polarization in ischemic stroke (Review). Mol Med Rep. 2020;21(5):2006–18. https://doi.org/10.3892/mmr.2020.11003.
Kumar A, Stoica BA, Loane DJ, Yang M, Abulwerdi G, Khan N, Kumar A, Thom SR, Faden AI. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J Neuroinflammation. 2017;14(1):47. https://doi.org/10.1186/s12974-017-0819-4.
Zhang L, Guo K, Zhou J, Zhang X, Yin S, Peng J, Liao Y, Jiang Y. Ponesimod protects against neuronal death by suppressing the activation of A1 astrocytes in early brain injury after experimental subarachnoid hemorrhage. J Neurochem. 2021;158(4):880–97. https://doi.org/10.1111/jnc.15457.
Jin J, Duan J, Du L, Xing W, Peng X, Zhao Q. Inflammation and immune cell abnormalities in intracranial aneurysm subarachnoid hemorrhage (SAH): relevant signaling pathways and therapeutic strategies. Front Immunol. 2022;23(13):1027756. https://doi.org/10.3389/fimmu.2022.1027756.
Okada T, Suzuki H. Mechanisms of neuroinflammation and inflammatory mediators involved in brain injury following subarachnoid hemorrhage. Histol Histopathol. 2020;35(7):623–36. https://doi.org/10.14670/HH-18-208.
Zhang F, Wang S, Gan L, Vosler PS, Gao Y, Zigmond MJ, Chen J. Protective effects and mechanisms of sirtuins in the nervous system. Prog Neurobiol. 2011;95(3):373–95. https://doi.org/10.1016/j.pneurobio.2011.09.001.
Zhang XS, Wu Q, Wu LY, Ye ZN, Jiang TW, Li W, Zhuang Z, Zhou ML, Zhang X, Hang CH. Sirtuin 1 activation protects against early brain injury after experimental subarachnoid hemorrhage in rats. Cell Death Dis. 2016;7(10):e2416. https://doi.org/10.1038/cddis.2016.292. (Erratum in: Cell Death Dis. 2022 Nov 17;13(11):964).
Lin W, Yao H, Lai J, Zeng Y, Guo X, Lin S, Hu W, Chen J, Chen X. Cycloastragenol confers cerebral protection after subarachnoid hemorrhage by suppressing oxidative insults and neuroinflammation via the SIRT1 signaling pathway. Oxid Med Cell Longev. 2022;2(2022):3099409. https://doi.org/10.1155/2022/3099409.
Peng Y, He P, Fan L, Xu H, Li J, Chen T, Ruan W, Dou Z, Chen G. Neuroprotective effects of magnesium Lithospermate B against subarachnoid hemorrhage in rats. Am J Chin Med. 2018;46(6):1225–41. https://doi.org/10.1142/S0192415X18500647.
Zhang X, Lu Y, Wu Q, Dai H, Li W, Lv S, Zhou X, Zhang X, Hang C, Wang J. Astaxanthin mitigates subarachnoid hemorrhage injury primarily by increasing sirtuin 1 and inhibiting the Toll-like receptor 4 signaling pathway. FASEB J. 2019;33(1):722–37. https://doi.org/10.1096/fj.201800642RR.
Zeng Y, Fang Z, Lai J, Wu Z, Lin W, Yao H, Hu W, Chen J, Guo X, Chen X. Activation of sirtuin-1 by pinocembrin treatment contributes to reduced early brain injury after subarachnoid hemorrhage. Oxid Med Cell Longev. 2022;16(2022):2242833. https://doi.org/10.1155/2022/2242833.
Li Z, Han X. Resveratrol alleviates early brain injury following subarachnoid hemorrhage: possible involvement of the AMPK/SIRT1/autophagy signaling pathway. Biol Chem. 2018;399(11):1339–50. https://doi.org/10.1515/hsz-2018-0269.
Zhao L, Liu H, Yue L, Zhang J, Li X, Wang B, Lin Y, Qu Y. Melatonin attenuates early brain injury via the melatonin Receptor/Sirt1/NF-κB signaling pathway following subarachnoid hemorrhage in mice. Mol Neurobiol. 2017;54(3):1612–21. https://doi.org/10.1007/s12035-016-9776-7.
Peng Y, Jin J, Fan L, Xu H, He P, Li J, Chen T, Ruan W, Chen G. Rolipram attenuates early brain injury following experimental subarachnoid hemorrhage in rats: possibly via regulating the SIRT1/NF-κB pathway. Neurochem Res. 2018;43(4):785–95. https://doi.org/10.1007/s11064-018-2480-4.
Sun Q, Wu W, Hu YC, Li H, Zhang D, Li S, Li W, Li WD, Ma B, Zhu JH, Zhou ML, Hang CH. Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J Neuroinflammation. 2014;12(11):106. https://doi.org/10.1186/1742-2094-11-106.
Zhang XH, Peng L, Zhang J, Dong YP, Wang CJ, Liu C, Xia DY, Zhang XS. Berberine ameliorates subarachnoid hemorrhage injury via induction of sirtuin 1 and inhibiting HMGB1/Nf-κB pathway. Front Pharmacol. 2020;10(11):1073. https://doi.org/10.3389/fphar.2020.01073.
Han Y, Tong Z, Wang C, Li X, Liang G. Oleanolic acid exerts neuroprotective effects in subarachnoid hemorrhage rats through SIRT1-mediated HMGB1 deacetylation. Eur J Pharmacol. 2021;893:173811. https://doi.org/10.1016/j.ejphar.2020.173811.
Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, Yoshimura K, Aoki H, Tsutsui H, Noda T, Sagara J, Taniguchi S, Takahashi M. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35(1):127–36. https://doi.org/10.1161/ATVBAHA.114.303763.
Zhang XS, Lu Y, Li W, Tao T, Wang WH, Gao S, Zhou Y, Guo YT, Liu C, Zhuang Z, Hang CH, Li W. Cerebroprotection by dioscin after experimental subarachnoid haemorrhage via inhibiting NLRP3 inflammasome through SIRT1-dependent pathway. Br J Pharmacol. 2021;178(18):3648–66. https://doi.org/10.1111/bph.15507.
Xia DY, Yuan JL, Jiang XC, Qi M, Lai NS, Wu LY, Zhang XS. SIRT1 promotes M2 microglia polarization via reducing ROS-mediated NLRP3 inflammasome signaling after subarachnoid hemorrhage. Front Immunol. 2021;12:770744. https://doi.org/10.3389/fimmu.2021.770744.
Yamaguchi T, Miyamoto T, Shikata E, Yamaguchi I, Shimada K, Yagi K, Tada Y, Korai M, Kitazato KT, Kanematsu Y, Takagi Y. Activation of the NLRP3/IL-1β/MMP-9 pathway and intracranial aneurysm rupture associated with the depletion of ERα and Sirt1 in oophorectomized rats. J Neurosurg. 2022;138(1):191–8. https://doi.org/10.3171/2022.4.JNS212945.
Zuo Y, Huang L, Enkhjargal B, Xu W, Umut O, Travis ZD, Zhang G, Tang J, Liu F, Zhang JH. Activation of retinoid X receptor by bexarotene attenuates neuroinflammation via PPARγ/SIRT6/FoxO3a pathway after subarachnoid hemorrhage in rats. J Neuroinflammation. 2019;16(1):47. https://doi.org/10.1186/s12974-019-1432-5.
Lucke-Wold BP, Logsdon AF, Manoranjan B, Turner RC, McConnell E, Vates GE, Huber JD, Rosen CL, Simard JM. Aneurysmal subarachnoid hemorrhage and neuroinflammation: a comprehensive review. Int J Mol Sci. 2016;17(4):497. https://doi.org/10.3390/ijms17040497.
Corwin C, Nikolopoulou A, Pan AL, Nunez-Santos M, Vallabhajosula S, Serrano P, Babich J, Figueiredo-Pereira ME. Prostaglandin D2/J2 signaling pathway in a rat model of neuroinflammation displaying progressive parkinsonian-like pathology: potential novel therapeutic targets. J Neuroinflammation. 2018;15(1):272. https://doi.org/10.1186/s12974-018-1305-3.
Yang Y, Chen S, Zhang JM. The updated role of oxidative stress in subarachnoid hemorrhage. Curr Drug Deliv. 2017;14(6):832–42. https://doi.org/10.2174/1567201813666161025115531.
Shao A, Lin D, Wang L, Tu S, Lenahan C, Zhang J. Oxidative stress at the crossroads of aging, stroke and depression. Aging Dis. 2020;11(6):1537–66. https://doi.org/10.14336/AD.2020.0225.
Kwon MS, Woo SK, Kurland DB, Yoon SH, Palmer AF, Banerjee U, Iqbal S, Ivanova S, Gerzanich V, Simard JM. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int J Mol Sci. 2015;16(3):5028–46. https://doi.org/10.3390/ijms16035028.
Lu Y, Zhang XS, Zhou XM, Gao YY, Chen CL, Liu JP, Ye ZN, Zhang ZH, Wu LY, Li W, Hang CH. Peroxiredoxin 1/2 protects brain against H2O2-induced apoptosis after subarachnoid hemorrhage. FASEB J. 2019;33(2):3051–62. https://doi.org/10.1096/fj.201801150R.
Wu F, Liu Z, Li G, Zhou L, Huang K, Wu Z, Zhan R, Shen J. Inflammation and oxidative stress: potential targets for improving prognosis after subarachnoid hemorrhage. Front Cell Neurosci. 2021;15:739506. https://doi.org/10.3389/fncel.2021.739506.
Liu H, Guo W, Guo H, Zhao L, Yue L, Li X, Feng D, Luo J, Wu X, Cui W, Qu Y. Bakuchiol attenuates oxidative stress and neuron damage by regulating Trx1/TXNIP and the phosphorylation of AMPK after subarachnoid hemorrhage in mice. Front Pharmacol. 2020;15(11):712. https://doi.org/10.3389/fphar.2020.00712.
Zhang T, Wu P, Budbazar E, Zhu Q, Sun C, Mo J, Peng J, Gospodarev V, Tang J, Shi H, Zhang JH. Mitophagy reduces oxidative stress via Keap1 (Kelch-Like Epichlorohydrin-Associated Protein 1)/Nrf2 (Nuclear Factor-E2-Related Factor 2)/PHB2 (Prohibitin 2) pathway after subarachnoid hemorrhage in rats. Stroke. 2019;50(4):978–88. https://doi.org/10.1161/STROKEAHA.118.021590. (Erratum in: Stroke. 2020 Mar;51(3):e57).
Bederson JB, Connolly ES Jr, Batjer HH, Dacey RG, Dion JE, Diringer MN, Duldner JE Jr, Harbaugh RE, Patel AB, Rosenwasser RH, American Heart Association. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke. 2009;40(3):994–1025. https://doi.org/10.1161/STROKEAHA.108.191395. (Erratum in: Stroke. 2009 Jul;40(7):e518).
Connolly ES Jr, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, Hoh BL, Kirkness CJ, Naidech AM, Ogilvy CS, Patel AB, Thompson BG, Vespa P, American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/american Stroke Association. Stroke. 2012;43(6):1711–37. https://doi.org/10.1161/STR.0b013e3182587839.
Huang Mei-Zhou, et al. Aspirin eugenol ester attenuates oxidative injury of vascular endothelial cells by regulating NOS and Nrf2 signalling pathways. Br J Pharmacol. 2019;176(7):906–18. https://doi.org/10.1111/bph.14592.
Liu Y, Qiu J, Wang Z, You W, Wu L, Ji C, Chen G. Dimethylfumarate alleviates early brain injury and secondary cognitive deficits after experimental subarachnoid hemorrhage via activation of Keap1-Nrf2-ARE system. J Neurosurg. 2015;123(4):915–23. https://doi.org/10.3171/2014.11.JNS132348.
Zhang X, Wu Q, Lu Y, Wan J, Dai H, Zhou X, Lv S, Chen X, Zhang X, Hang C, Wang J. Cerebroprotection by salvianolic acid B after experimental subarachnoid hemorrhage occurs via Nrf2- and SIRT1-dependent pathways. Free Radic Biol Med. 2018;124:504–16. https://doi.org/10.1016/j.freeradbiomed.2018.06.035.
Liu JQ, Zhao XT, Qin FY, Zhou JW, Ding F, Zhou G, Zhang XS, Zhang ZH, Li ZB. Isoliquiritigenin mitigates oxidative damage after subarachnoid hemorrhage in vivo and in vitro by regulating Nrf2-dependent Signaling pathway via targeting of SIRT1. Phytomedicine. 2022;105:154262. https://doi.org/10.1016/j.phymed.2022.154262.
Zhang Z, Fang J, Zhou J, Ding F, Zhou G, Zhao X, Zhuang Z, Lu Y. Pterostilbene attenuates subarachnoid hemorrhage-induced brain injury through the SIRT1-dependent Nrf2 signaling pathway. Oxid Med Cell Longev. 2022;30(2022):3550204. https://doi.org/10.1155/2022/3550204.
Dai SH, Chen T, Wang YH, Zhu J, Luo P, Rao W, Yang YF, Fei Z, Jiang XF. Sirt3 protects cortical neurons against oxidative stress via regulating mitochondrial Ca2+ and mitochondrial biogenesis. Int J Mol Sci. 2014;15(8):14591–609. https://doi.org/10.3390/ijms150814591.
Zhang K, Cheng H, Song L, Wei W. Inhibition of the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α)/Sirtuin 3 (SIRT3) pathway aggravates oxidative stress after experimental subarachnoid hemorrhage. Med Sci Monit. 2020;26:e923688. https://doi.org/10.12659/MSM.923688.
Huang W, Huang Y, Huang RQ, Huang CG, Wang WH, Gu JM, Dong Y. SIRT3 expression decreases with reactive oxygen species generation in rat cortical neurons during early brain injury induced by experimental subarachnoid hemorrhage. Biomed Res Int. 2016;2016:8263926. https://doi.org/10.1155/2016/8263926.
Yang S, Chen X, Li S, Sun B, Hang C. Melatonin treatment regulates SIRT3 Expression in Early Brain Injury (EBI) due to Reactive Oxygen Species (ROS) in a mouse model of Subarachnoid Hemorrhage (SAH). Med Sci Monit. 2018;24:3804–14. https://doi.org/10.12659/MSM.907734.
Chen J, Wang L, Wu C, Hu Q, Gu C, Yan F, Li J, Yan W, Chen G. Melatonin-enhanced autophagy protects against neural apoptosis via a mitochondrial pathway in early brain injury following a subarachnoid hemorrhage. J Pineal Res. 2014;56(1):12–9. https://doi.org/10.1111/jpi.12086.
Wu LY, Enkhjargal B, Xie ZY, Travis ZD, Sun CM, Zhou KR, Zhang TY, Zhu QQ, Hang CH, Zhang JH. Recombinant OX40 attenuates neuronal apoptosis through OX40-OX40L/PI3K/AKT signaling pathway following subarachnoid hemorrhage in rats. Exp Neurol. 2020;326:113179. https://doi.org/10.1016/j.expneurol.2020.113179.
Friedrich V, Flores R, Sehba FA. Cell death starts early after subarachnoid hemorrhage. Neurosci Lett. 2012;512(1):6–11. https://doi.org/10.1016/j.neulet.2012.01.036.
Rass V, Helbok R. Early brain injury after poor-grade subarachnoid hemorrhage. Curr Neurol Neurosci Rep. 2019;19(10):78. https://doi.org/10.1007/s11910-019-0990-3.
Hasegawa Y, Suzuki H, Sozen T, Altay O, Zhang JH. Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochir Suppl. 2011;110(Pt 1):43–8. https://doi.org/10.1007/978-3-7091-0353-1_8.
Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104(4):487–501. https://doi.org/10.1016/s0092-8674(01)00237-9.
Gorojod RM, Alaimo A, Porte Alcon S, Saravia F, Kotler ML. Interplay between lysosomal, mitochondrial and death receptor pathways during manganese-induced apoptosis in glial cells. Arch Toxicol. 2017;91(9):3065–78. https://doi.org/10.1007/s00204-017-1936-7.
Cha Z, Cheng J, Xiang H, Qin J, He Y, Peng Z, Jia J, Yu H. Celastrol enhances TRAIL-induced apoptosis in human glioblastoma via the death receptor pathway. Cancer Chemother Pharmacol. 2019;84(4):719–28. https://doi.org/10.1007/s00280-019-03900-8.
Ceccatelli S, Tamm C, Sleeper E, Orrenius S. Neural stem cells and cell death. Toxicol Lett. 2004;149(1–3):59–66. https://doi.org/10.1016/j.toxlet.2003.12.060.
Bornstein R, Gonzalez B, Johnson SC. Mitochondrial pathways in human health and aging. Mitochondrion. 2020;54:72–84. https://doi.org/10.1016/j.mito.2020.07.007.
Zeeshan HM, Lee GH, Kim HR, Chae HJ. Endoplasmic reticulum stress and associated ROS. Int J Mol Sci. 2016;17(3):327. https://doi.org/10.3390/ijms17030327.
Burton GJ, Yung HW, Murray AJ. Mitochondrial - endoplasmic reticulum interactions in the trophoblast: stress and senescence. Placenta. 2017;52:146–55. https://doi.org/10.1016/j.placenta.2016.04.001.
Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, Giorgi C, Pinton P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62–72. https://doi.org/10.1016/j.ceca.2017.05.003.
Tian XS, Xu H, He XJ, Li Y, He B, Zhao D. Endoplasmic reticulum stress mediates cortical neuron apoptosis after experimental subarachnoid hemorrhage in rats. Int J Clin Exp Pathol. 2020;13(7):1569–77.
Liu J, Fan L, Wang H, Sun G. Autophagy, a double-edged sword in anti-angiogenesis therapy. Med Oncol. 2016;33(1):10. https://doi.org/10.1007/s12032-015-0721-9.
Zheng Y, Zhou Z, Han F, Chen Z. Special issue: Neuroinflammatory pathways as treatment targets in brain disorders autophagic regulation of neuroinflammation in ischemic stroke. Neurochem Int. 2021;148:105114. https://doi.org/10.1016/j.neuint.2021.105114.
Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G. Autophagy in acute brain injury. Nat Rev Neurosci. 2016;17(8):467–84. https://doi.org/10.1038/nrn.2016.51.
Wang Z, Shi XY, Yin J, Zuo G, Zhang J, Chen G. Role of autophagy in early brain injury after experimental subarachnoid hemorrhage. J Mol Neurosci. 2012;46(1):192–202. https://doi.org/10.1007/s12031-011-9575-6.
Jing CH, Wang L, Liu PP, Wu C, Ruan D, Chen G. Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience. 2012;28(213):144–53. https://doi.org/10.1016/j.neuroscience.2012.03.055.
Shao A, Wang Z, Wu H, Dong X, Li Y, Tu S, Tang J, Zhao M, Zhang J, Hong Y. Enhancement of autophagy by histone deacetylase inhibitor trichostatin A ameliorates neuronal apoptosis after subarachnoid hemorrhage in rats. Mol Neurobiol. 2016;53(1):18–27. https://doi.org/10.1007/s12035-014-8986-0.
Wang J, Wang Y, Zuo Y, Duan J, Pan A, Li JM, Yan XX, Liu F. MFGE8 mitigates brain injury in a rat model of SAH by maintaining vascular endothelial integrity via TIGβ5/PI3K/CXCL12 signaling. Exp Brain Res. 2021;239(7):2193–205. https://doi.org/10.1007/s00221-021-06111-x.
Huang W, Li N, Zhang Y, Wang X, Yin M, Lei QY. AHCYL1 senses SAH to inhibit autophagy through interaction with PIK3C3 in an MTORC1-independent manner. Autophagy. 2022;18(2):309–19. https://doi.org/10.1080/15548627.2021.1924038.
Wu HJ, Pu JL, Krafft PR, Zhang JM, Chen S. The molecular mechanisms between autophagy and apoptosis: potential role in central nervous system disorders. Cell Mol Neurobiol. 2015;35(1):85–99. https://doi.org/10.1007/s10571-014-0116-z.
Dong Y, Fan C, Hu W, Jiang S, Ma Z, Yan X, Deng C, Di S, Xin Z, Wu G, Yang Y, Reiter RJ, Liang G. Melatonin attenuated early brain injury induced by subarachnoid hemorrhage via regulating NLRP3 inflammasome and apoptosis signaling. J Pineal Res. 2016;60(3):253–62. https://doi.org/10.1111/jpi.12300.
Ding PF, Zhu Q, Sheng B, Yang H, Xu HJ, Tao T, Peng Z, Chen XX, Li XJ, Zhou Y, Zhang HS, Gao YY, Zhuang Z, Hang CH, Li W. Alpha-ketoglutarate alleviates neuronal apoptosis induced by central insulin resistance through inhibiting S6K1 phosphorylation after subarachnoid hemorrhage. Oxid Med Cell Longev. 2022;25(2022):9148257. https://doi.org/10.1155/2022/9148257.
Cheng Y, Li S, Liu Y, Li J, Chen Y, Zhao H. Treatment of brain edema by wogonoside is associated with inhibition of neuronal apoptosis and SIRT1 activation in rats. Med Sci Monit. 2020;26:e921250. https://doi.org/10.12659/MSM.921250.
Qian C, Jin J, Chen J, Li J, Yu X, Mo H, Chen G. SIRT1 activation by resveratrol reduces brain edema and neuronal apoptosis in an experimental rat subarachnoid hemorrhage model. Mol Med Rep. 2017;16(6):9627–35. https://doi.org/10.3892/mmr.2017.7773.
Li Q, Peng Y, Fan L, Xu H, He P, Cao S, Li J, Chen T, Ruan W, Chen G. Phosphodiesterase-4 inhibition confers a neuroprotective efficacy against early brain injury following experimental subarachnoid hemorrhage in rats by attenuating neuronal apoptosis through the SIRT1/Akt pathway. Biomed Pharmacother. 2018;99:947–55. https://doi.org/10.1016/j.biopha.2018.01.093.
Zhang Y, Yang X, Ge X, Zhang F. Puerarin attenuates neurological deficits via Bcl-2/Bax/cleaved caspase-3 and Sirt3/SOD2 apoptotic pathways in subarachnoid hemorrhage mice. Biomed Pharmacother. 2019;109:726–33. https://doi.org/10.1016/j.biopha.2018.10.161.
Wu H, Yu N, Wang X, Yang Y, Liang H. Tauroursodeoxycholic acid attenuates neuronal apoptosis via the TGR5/ SIRT3 pathway after subarachnoid hemorrhage in rats. Biol Res. 2020;53(1):56. https://doi.org/10.1186/s40659-020-00323-1.
Daneman R, Barres BA. The blood-brain barrier–lessons from moody flies. Cell. 2005;123(1):9–12. https://doi.org/10.1016/j.cell.2005.09.017.
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell. 2015;163(5):1064–78. https://doi.org/10.1016/j.cell.2015.10.067.
Keep RF, Zhou N, Xiang J, Andjelkovic AV, Hua Y, Xi G. Vascular disruption and blood-brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS. 2014;10(11):18. https://doi.org/10.1186/2045-8118-11-18.
Chen S, Feng H, Sherchan P, Klebe D, Zhao G, Sun X, Zhang J, Tang J, Zhang JH. Controversies and evolving new mechanisms in subarachnoid hemorrhage. Prog Neurobiol. 2014;115:64–91. https://doi.org/10.1016/j.pneurobio.2013.09.002.
Fujimoto M, Shiba M, Kawakita F, Liu L, Shimojo N, Imanaka-Yoshida K, Yoshida T, Suzuki H. Effects of Tenascin-C knockout on cerebral vasospasm after experimental subarachnoid hemorrhage in mice. Mol Neurobiol. 2018;55(3):1951–8. https://doi.org/10.1007/s12035-017-0466-x.
Peeyush Kumar T, McBride DW, Dash PK, Matsumura K, Rubi A, Blackburn SL. Endothelial cell dysfunction and injury in subarachnoid hemorrhage. Mol Neurobiol. 2019;56(3):1992–2006. https://doi.org/10.1007/s12035-018-1213-7.
Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab. 2016;36(3):513–38. https://doi.org/10.1177/0271678X15617172.
Okada T, Suzuki H, Travis ZD, Zhang JH. The stroke-induced blood-brain barrier disruption: current progress of inspection technique, mechanism, and therapeutic target. Curr Neuropharmacol. 2020;18(12):1187–212. https://doi.org/10.2174/1570159X18666200528143301.
Chen S, Shao L, Ma L. Cerebral edema formation after stroke: emphasis on blood-brain barrier and the lymphatic drainage system of the brain. Front Cell Neurosci. 2021;15:716825. https://doi.org/10.3389/fncel.2021.716825.
Wang KC, Tang SC, Lee JE, Li YI, Huang YS, Yang WS, Jeng JS, Arumugam TV, Tu YK. Cerebrospinal fluid high mobility group box 1 is associated with neuronal death in subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2017;37(2):435–43. https://doi.org/10.1177/0271678X16629484.
Michinaga S, Koyama Y. Pathogenesis of brain edema and investigation into anti-edema drugs. Int J Mol Sci. 2015;16(5):9949–75. https://doi.org/10.3390/ijms16059949.
de Oliveira Manoel AL, Goffi A, Zampieri FG, Turkel-Parrella D, Duggal A, Marotta TR, Macdonald RL, Abrahamson S. The critical care management of spontaneous intracranial hemorrhage: a contemporary review. Crit Care. 2016;18(20):272. https://doi.org/10.1186/s13054-016-1432-0.
Rowland MJ, Ezra M, Winkler A, Garry P, Lamb C, Kelly M, Okell TW, Westbrook J, Wise RG, Douaud G, Pattinson KT. Calcium channel blockade with nimodipine reverses MRI evidence of cerebral oedema following acute hypoxia. J Cereb Blood Flow Metab. 2019;39(2):285–301. https://doi.org/10.1177/0271678X17726624.
Corry JJ, Asaithambi G, Shaik AM, Lassig JP, Marino EH, Ho BM, Castle AL, Banerji N, Tipps ME. Conivaptan for the reduction of cerebral edema in intracerebral hemorrhage: a safety and tolerability study. Clin Drug Investig. 2020;40(5):503–9. https://doi.org/10.1007/s40261-020-00911-9.
Hinson HE, Sun E, Molyneaux BJ, von Kummer R, Demchuk A, Romero J, Taylor Kimberly W, Sheth KN. Osmotherapy for malignant cerebral edema in a phase 2 prospective, double blind, randomized, placebo-controlled study of IV glibenclamide. J Stroke Cerebrovasc Dis. 2020;29(7):104916. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.104916.
Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke. 2002;33(5):1225–32. https://doi.org/10.1161/01.str.0000015624.29071.1f.
Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275(12):8945–51. https://doi.org/10.1074/jbc.275.12.8945.
Liebner S, Kniesel U, Kalbacher H, Wolburg H. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur J Cell Biol. 2000;79(10):707–17. https://doi.org/10.1078/0171-9335-00101.
Kniesel U, Wolburg H. Tight junctions of the blood-brain barrier. Cell Mol Neurobiol. 2000;20(1):57–76. https://doi.org/10.1023/a:1006995910836.
Zhou XM, Zhang X, Zhang XS, Zhuang Z, Li W, Sun Q, Li T, Wang CX, Zhu L, Shi JX, Zhou ML. SIRT1 inhibition by sirtinol aggravates brain edema after experimental subarachnoid hemorrhage. J Neurosci Res. 2014;92(6):714–22. https://doi.org/10.1002/jnr.23359.
van Dijk BJ, Vergouwen MD, Kelfkens MM, Rinkel GJ, Hol EM. Glial cell response after aneurysmal subarachnoid hemorrhage - Functional consequences and clinical implications. Biochim Biophys Acta. 2016;1862(3):492–505. https://doi.org/10.1016/j.bbadis.2015.10.013.
Osgood ML. Aneurysmal subarachnoid hemorrhage: review of the pathophysiology and management strategies. Curr Neurol Neurosci Rep. 2021;21(9):50. https://doi.org/10.1007/s11910-021-01136-9.
Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460(7255):587–91. https://doi.org/10.1038/nature08197.
Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS, Jarho E, Lahtela-Kakkonen M, Mai A, Altucci L. Sirtuin functions and modulation: from chemistry to the clinic. Clin Epigenetics. 2016;25(8):61. https://doi.org/10.1186/s13148-016-0224-3.
Mendes KL, Lelis DF, Santos SHS. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017;38:98–105. https://doi.org/10.1016/j.cytogfr.2017.11.001.
Donmez G. The neurobiology of sirtuins and their role in neurodegeneration. Trends Pharmacol Sci. 2012;33(9):494–501. https://doi.org/10.1016/j.tips.2012.05.007.
Grüter BE, Croci D, Schöpf S, Nevzati E, d’Allonzo D, Lattmann J, Roth T, Bircher B, Muroi C, Dutilh G, Widmer HR, Plesnila N, Fandino J, Marbacher S. Systematic review and meta-analysis of methodological quality in in vivo animal studies of subarachnoid hemorrhage. Transl Stroke Res. 2020;11(6):1175–84. https://doi.org/10.1007/s12975-020-00801-4.
Grabowska W, Sikora E, Bielak-Zmijewska A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology. 2017;18(4):447–76. https://doi.org/10.1007/s10522-017-9685-9.
Hong JY, Lin H. Sirtuin modulators in cellular and animal models of human diseases. Front Pharmacol. 2021;12:735044. https://doi.org/10.3389/fphar.2021.735044.
Fang C, Xu H, Yuan L, Zhu Z, Wang X, Liu Y, Zhang A, Shao A, Lou M. Natural compounds for SIRT1-mediated oxidative stress and neuroinflammation in stroke: a potential therapeutic target in the future. Oxid Med Cell Longev. 2022;5(2022):1949718. https://doi.org/10.1155/2022/1949718.
Budohoski KP, Guilfoyle M, Helmy A, Huuskonen T, Czosnyka M, Kirollos R, Menon DK, Pickard JD, Kirkpatrick PJ. The pathophysiology and treatment of delayed cerebral ischaemia following subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry. 2014;85(12):1343–53. https://doi.org/10.1136/jnnp-2014-307711.
Suzuki H, Nakano F. To improve translational research in subarachnoid hemorrhage. Transl Stroke Res. 2018;9(1):1–3. https://doi.org/10.1007/s12975-017-0546-2.
Solár P, Zamani A, Lakatosová K, Joukal M. The blood-brain barrier and the neurovascular unit in subarachnoid hemorrhage: molecular events and potential treatments. Fluids Barriers CNS. 2022;19(1):29. https://doi.org/10.1186/s12987-022-00312-4.
Wan W, Ding Y, Xie Z, Li Q, Yan F, Budbazar E, Pearce WJ, Hartman R, Obenaus A, Zhang JH, Jiang Y, Tang J. PDGFR-β modulates vascular smooth muscle cell phenotype via IRF-9/SIRT-1/NF-κB pathway in subarachnoid hemorrhage rats. J Cereb Blood Flow Metab. 2019;39(7):1369–80. https://doi.org/10.1177/0271678X18760954.
Diwan D, Vellimana AK, Aum DJ, Clarke J, Nelson JW, Lawrence M, Han BH, Gidday JM, Zipfel GJ. Sirtuin 1 mediates protection against delayed cerebral ischemia in subarachnoid hemorrhage in response to hypoxic postconditioning. J Am Heart Assoc. 2021;10(20):e021113. https://doi.org/10.1161/JAHA.121.021113.
Liu M, Jayaraman K, Giri T, Zipfel GJ, Athiraman U. Role of SIRT1 in isoflurane conditioning-induced neurovascular protection against delayed cerebral ischemia secondary to subarachnoid hemorrhage. Int J Mol Sci. 2021;22(8):4291. https://doi.org/10.3390/ijms22084291.
Clarke JV, Brier LM, Rahn RM, Diwan D, Yuan JY, Bice AR, Imai SI, Vellimana AK, Culver JP, Zipfel GJ. SIRT1 mediates hypoxic postconditioning- and resveratrol-induced protection against functional connectivity deficits after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2022;42(7):1210–23. https://doi.org/10.1177/0271678X221079902.
Curry AM, White DS, Donu D, Cen Y. Human sirtuin regulators: the “Success” Stories. Front Physiol. 2021;12:752117. https://doi.org/10.3389/fphys.2021.752117.
Funding
This work was supported by grants from the National Natural Science Foundation of China (NO. 81960229); Guizhou Provincial Department of Science and Technology (Qiankeheji [2020] 1Y319); Doctoral Research Initiation Fund of the Affiliated Hospital of Zunyi Medical University (Hospital Zi (2019) 01); and Zunyi Honghuagang District (Zunhongkeheji [2021] 16).
Author information
Authors and Affiliations
Contributions
The idea and design were made by RF and LG. Drafting the article was done by KL, RW, JW, XL, EZ. Critically revising the article was made by all authors. Reviewed submitted version of manuscript was made by all authors. RF and LG approved the final version of the manuscript on behalf of all the authors.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lei, K., Wu, R., Wang, J. et al. Sirtuins as Potential Targets for Neuroprotection: Mechanisms of Early Brain Injury Induced by Subarachnoid Hemorrhage. Transl. Stroke Res. (2023). https://doi.org/10.1007/s12975-023-01191-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12975-023-01191-z