
Abstract
Laves phases with their comparably simple crystal structure are very common intermetallic phases and can be formed from element combinations all over the periodic table resulting in a huge number of known examples. Even though this type of phases is known for almost 100 years, and although a lot of information on stability, structure, and properties has accumulated especially during the last about 20 years, systematic evaluation and rationalization of this information in particular as a function of the involved elements is often lacking. It is one of the two main goals of this review to summarize the knowledge for some selected respective topics with a certain focus on non-stoichiometric, i.e., non-ideal Laves phases. The second, central goal of the review is to give a systematic overview about the role of Laves phases in all kinds of materials for functional and structural applications. There is a surprisingly broad range of successful utilization of Laves phases in functional applications comprising Laves phases as hydrogen storage material (Hydraloy), as magneto-mechanical sensors and actuators (Terfenol), or for wear- and corrosion-resistant coatings in corrosive atmospheres and at high temperatures (Tribaloy), to name but a few. Regarding structural applications, there is a renewed interest in using Laves phases for creep-strengthening of high-temperature steels and new respective alloy design concepts were developed and successfully tested. Apart from steels, Laves phases also occur in various other kinds of structural materials sometimes effectively improving properties, but often also acting in a detrimental way.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
It was during the 1930s when the German mineralogist and crystallographer Fritz Laves (1906–1978) published his crucial findings about the crystallographic similarity and relationship between various intermetallic compounds of the cubic MgCu2 and the hexagonal MgZn2 and MgNi2 structure types [1, 2]. In 1939, the designation ‘Laves phases’ for this class of AB2 intermetallic compounds was suggested by Gustav E.R. Schulze [3], whose pioneering work contributed a lot to the fundamental understanding of these intermetallic phases [4, 5].
Since then and continuing until today, there was and is a tremendous interest in these phases for several reasons. Laves phases form a huge class of intermetallic compounds; already in ‘Pearson’s Handbook of Crystallographic Data for Intermetallic Phases’ from 1991 [6] more than 1400 different Laves phases were listed [7], and this number has further increased during the last 30 years. Metal combinations from all over the periodic system can form Laves phases, and especially in multi-component materials, there is a good chance to encounter a Laves phase, the presence of which sometimes is wanted but in many cases also is detrimental. Excellent examples for that may be high-temperature steels and superalloys, where frequently Laves phase precipitates occur which were regarded for a long time as detrimental phases that have to be avoided. However, more recently it was recognized that by a proper control of the precipitation process, Laves phase can well serve as strengthening particles in the matrix. Moreover, based on such findings completely new alloying concepts have been developed for designing improved ferritic as well as austenitic high-temperature steels (as will be described in Sects. 5.2 and 5.3). Furthermore, in the new materials class of so-called high-entropy alloys (HEAs) (which actually are compositionally complex alloys consisting of five or more principal elements), Laves phases were not only identified as the most frequent intermetallic phase in a recent statistical analysis [8], but also various single-phase ‘high-entropy’ Laves phases were found, see, for example, [9] (cf. also Sect. 5.6.5). The use of Laves phases in various functional as well as structural applications is already discussed for a long time. Some Laves phases based on transition metals feature very high melting temperatures and their resulting excellent high-temperature strength encouraged J. D. Livingston to entitle his 1992 review paper “Laves phase superalloys?” [10].
Besides their relevance for structural applications and their utilization in various functional applications, Laves phases are also well-suited candidates to study fundamental properties of intermetallic phases in general. Compared to most other intermetallic compounds, their crystal structure is still simple enough to allow detailed investigations of basic phenomena, while on the other hand, their behavior is already characteristic and representative for intermetallic phases. Therefore, the structure of this review article is as follows: After some introductory remarks about the structure of Laves phases and some peculiarities of this very common structure type (Sect. 2), this review is divided into three main parts:
Section 3 deals with a discussion of some selected topics related to more recent insights into the understanding of the behavior of Laves phases from a more fundamental point of view. This includes discussions about the stability of and site preference in the different polytypes (Sect. 3.1), the occurrence of point defects and their relation to the frequently observed extended homogeneity ranges (Sect. 3.2), and the polytypism of Laves phases with insights on the mechanism and kinetics of transformations between the polytypes (Sect. 3.3). In materials development, theoretical prediction of phase stability, phase equilibria, and kinetics of their formation or transformation plays a central role. A helpful and nowadays routinely used tool is modeling by the CALPHAD approach (see, for example, [11]) the success of which crucially depends on the employed models for the involved phases. The description of Laves phases in CALPHAD modeling is discussed in Sect. 3.4. Section 3.5 treats different structural variants of Laves phase, which, for example, can result from slight distortions either due to changes in electronic structure or due to atomic ordering. Some more complex structural variants are also introduced here. Atomically resolved scanning transmission electron microscopy (STEM) allows interesting insights into the structure and chemistry of planar defects in Laves phases. Such planar defects as well as intergrowth and intergrowth related faulting are the topics of Sect. 3.6. Section 3.7 of this part deals with an improved understanding of the plasticity of Laves phases and with the softening effect of mechanical properties, which was observed in some Laves phases with extended homogeneity ranges as a result of deviations from the stoichiometric composition. While the pronounced brittleness of Laves phases impedes their investigation by classical mechanical testing, more recently introduced micromechanical testing methods have proven to be an efficient tool for getting an improved understanding of the mechanical behavior of Laves phases.
Section 4 of this review summarizes the status of knowledge regarding Laves phases in functional materials. It is especially remarkable that Laves phases not only show many interesting physical properties, but have progressed to the next step forward being in use today in various real functional applications. Topics discussed here comprise the very recent developments related to hydrogen storage in Laves phases (‘Hydraloy’, see Sect. 4.1), the well-established application of Laves phases as wear- and corrosion-resistant materials (‘Tribaloy’, Sect. 4.2), and the utilization of their special magnetic properties, for example as magneto-mechanical sensors and actuators (‘Terfenol’, Sect. 4.3). Many Laves phases also show superconducting behavior (Sect. 4.4), and finally, there are also some more exotic possibilities for functional applications such as for the adjustment of the color of coins via the content of Laves phase (Sect. 4.5).
Section 5 focuses on the role of Laves phases in structural materials. While single-phase or Laves phase-dominated materials appear not to be suited for structural applications due to their pronounced brittleness and, hence, insufficient fracture toughness (Sect. 5.1), it is especially their role in ferritic and austenitic steels, which is of extraordinary importance and is discussed very comprehensively and controversially in the literature (Sects. 5.2 and 5.3). Similarly, Laves phases often occur in Ni- and Co-based superalloys (Sect. 5.4) as well as in other structural materials based on metal M solid solutions (M = Cr, Cu, Mg, Nb, Ti, or Zr; Sect. 5.5) or based on intermetallic phases (mostly aluminides) or HEAs (Sect. 5.6). Adding Laves phase to usually comparably soft metal solid solutions helps to significantly strengthen the material, but simultaneously to this beneficial effect there is an increase in brittleness. Therefore, only a few alloys of this kind have managed the step to real applications even though a huge number of studies were performed to design improved, Laves phase-strengthened structural materials.
Although Laves phases were already frequently reviewed in various textbooks and survey papers including detailed descriptions of their structures and properties (see, for example, [4, 5, 12,13,14,15,16,17,18]), there are still a lot of open questions and discussion about these phases, and a huge amount of new results related to Laves phases was published especially in recent years. The present article tries to focus on some of these more recently discussed aspects and is intended to give an overview especially of the more recent literature.
Some remarks about the Laves phase structures and their particular role and uniqueness among intermetallic phases
The important finding of Fritz Laves was that all phases of the MgCu2, MgZn2, and MgNi2 structure types are homeotect meaning that they are constructed by a common geometrical principle leading to an optimal space filling and maximum coordination number [2, 3, 19,20,21]. They belong to the group of tetrahedrally close-packed (t.c.p.) structures [22,23,24,25], the interstices are exclusively tetrahedral and the coordination polyhedra in these AB2 phases are Frank–Kasper polyhedra with coordination numbers (CN) of 12 (for the smaller B atoms) and 16 (for the larger A atoms). The highest packing density is achieved for an ideal atomic radius ratio of rA/rB = (3/2)1/2 ≈ 1.225 resulting in a space filling of 71% (for more detailed discussions of such general aspects of the crystal structure, see also the aforementioned textbooks and reviews about Laves phases). The coordination polyhedra of the three Laves phase structure types are shown in Fig. 1, and the crystallographic information is summarized in Table 1.

Coordination polyhedra of the three Laves phases structure types cubic C15, hexagonal C14 and hexagonal C36. Symmetry of the positions as well as type and number of nearest-neighbor atoms are listed in Table 1
The close relationship between the three structure types becomes most obvious when describing them as layer stacking of alternating A atom and B atom sheets that are packed perpendicular to the [0001] direction in C14 or [111] direction in C15. A stack of four of such layers forms the fundamental unit for all types of Laves phases. This stack consists of one B atom layer ordered in a Kagomé net of regular triangles and hexagons followed by a triple layer A-B-A with triangularly ordered atoms in each layer. As this triple layer can be placed in two ways on top of the single B layer, there are two versions of the four-layered structural unit, which might be called X and X′. Now—similar as in simple, dense-packed fcc and hcp metals—a dense stacking of these units results in the cubic C15 structure for XYZXYZ…, stacking, hexagonal C14 for XY′XY′…, stacking, and hexagonal C36 for XY′X′ZXY′X′Z…, stacking. This idea of describing the crystal structure was already introduced by Laves himself [2] and later on in a more detailed way by Komura [27]. The above three stacking variants C14, C15, and C36 are the most simple and most common ones. More complex, long-periodic stacking sequences are possible but were only very rarely observed in real systems [28, 29]. In ternary systems, fully ordered derivatives of the C14 and C15 Laves phase structures are known to exist. This topic will be discussed in Sect. 3.5.
Among the numerous structure types of intermetallic phases, the Laves phase structures have an exceptional position. Their uniqueness and particular role is demonstrated here by three examples related to (i) the similarity of the atom arrangements to liquids and quasicrystalline phases, (ii) their occurrence as defect clusters in bcc-metals, and (iii) their tendency to form as solid phases in gas mixtures under high pressures, and, related to that, the self-assembly of non-reacting particle mixtures resulting in Laves phase ordering:
-
(i)
The icosahedral atom arrangements that are characteristic for liquids, metallic glasses, and quasicrystalline phases have similarity to the coordination polyhedra occurring in t.c.p. phases such as the Laves phases (see, for example, [30,31,32]). Therefore, Laves phase structures can be regarded as a bridge between liquids/metallic glass structures and classical close-packed metal structures [33,34,35]. Ghosh et al. [35] studied the glass forming ability of Fe-early transition metal binary and ternary alloys in various systems containing Laves phases. They found that the composition at which the glassy state will be most stable (compared to the solid solution phase) coincides with that of the Laves phase [35]. In an aqueous suspension of spherical polystyrene particles of two diameters (ratio 1.4 to 1.7) with sizes of some hundred nanometers, an amorphous state appeared first after stirring until after a few hours the nucleation of Laves phase ordering was observed [36] (for this aspect of the self-assembly of particles, see also (iii)).
-
(ii)
Irradiation of bcc-Fe with high-energetic ions or neutrons can result in the formation of nano-sized self-interstitial Fe atom clusters with C15 Laves-type structures as was predicted by density functional theory (DFT) calculations [37,38,39] and molecular dynamics simulations [40, 41]. Such irradiation-induced defect clusters can be highly stable and immobile and exhibit large antiferromagnetic moments [37]. When growing to diameters exceeding 1.5 nm, the C15 clusters start dissolving and dislocation loops become the more stable defect configuration [41]. Experimental evidence for the existence of such kind of Laves phase-ordered clusters, however, still appears to be lacking.
-
(iii)
A quite exotic type of phase are so-called van der Waals compounds, which are solid compounds obtained from gas mixtures (rare gases or molecular gases as, for example, H2, N2, O2, or CH4) under high pressures. A very frequently found structure type of such van der Waals compounds are Laves phases. This was experimentally proven, for example, for the rare gas binary systems He-Ne, Ar-Ne, and Ar-Xe, where the only observed room-temperature, high-pressure compounds are hexagonal C14 NeHe2 (above 12.8 GPa) [42], C14 ArNe2 (above 4.6 GPa) [43], and cubic C15 XeAr2 (above 1.1 GPa) [44]. Similarly, high-pressure Laves phases were also found to crystallize in molecular gas systems with A and/or B components being molecules. Examples are C14 Ar(H2)2 (> 4.3 GPa) [45], C15 Xe(O2)2 (> 3.1 GPa) [46], C15 Xe(N2)2 (> 4.9 GPa) [47]. In a 2H2 + CH4 gas mixture, the van der Waals compound CH4(H2)2 crystallizes with C14 Laves phase structure at pressures above 5.4 GPa [48]. First principles calculations of the stability of high-pressure phases in the systems He-Ne and Ar-He confirm the existence of the NeHe2 Laves phase with C14 structure and indicate for very high pressures above 120 GPa a transformation to the cubic C15 structure [49]. For the Ar-He system, the same authors predict the crystallization of a cubic C15 Laves phase, which should transform to an AlB2-type structure above 13.8 GPa [49].
The frequent observation of the preferred crystallization of Laves-type structures at such high-pressure in interaction-free systems fits well to the results of Monte Carlo simulations on the stability of various dense-packed crystal structures in binary mixtures of large and small hard spheres. By calculating composition versus pressure diagrams, Hynninen et al. [50] could show that for radius ratios rA/rB in the range 1.19 to 1.32, Laves phases are the most stable configuration and the only occurring solid compound in the entire composition range. Moreover, when comparing the stability of the three Laves structures C14, C15, and C36, they find that—even though the energy differences are very small—hexagonal C14 should be the most stable variant. Experiments with colloidal suspensions of uncharged hard spheres confirm the preferential formation of C14 Laves phase [51]. Figure 2 shows the calculated phase diagram of binary A + B hard spheres with a radius ratio of rA/rB = 1.22 (which is very near to the ideal radius ratio of the Laves phases, see above) [50].

Calculated composition-versus-reduced pressure phase diagram of binary hard-sphere mixtures of large A and small B particles for a rA/rB size ratio of 1.22 (x and p are the dimensionless composition x = NB/(NA + NB) and pressure p = Pσ 3/kBT with P: pressure, σ: diameter of the A spheres). “fccA” and “fccB” denote the fcc crystals of pure A and pure B spheres, respectively. The light-blue marked regions indicate the pressure-composition range of existence of Laves phase (adapted with permission from [50])
Simulations of crystallization in polydispersive hard-sphere fluids reveal that such mixtures tend to fractionate based on particle size and that in large regions bimodal subpopulations form C14 and C15 Laves phase. While the size ratio—as expected—is a critical parameter for the formation of Laves phase-type ordering, the mixing ratio of small versus large particles interestingly does not play a role and the unused particles remain as coexisting disordered phase [52]. Interestingly, it was also shown by computer simulations that equilibrium Laves phase in binary hard-sphere mixtures of ideal size ratio can contain an extraordinarily high concentration of antisite defects. Stable regions were found where up to 2% of the large-particle lattice sites are occupied by a small particle. Moreover, the calculations indicate that a hard-sphere Laves phase should never be thermodynamically stable at its ideal composition [53].
The effect of the self-assembly of non-reacting particle mixtures with formation of Laves phase ordering was not only predicted by several computational simulations [50, 52, 54,55,56,57,58], but was also observed experimentally in colloidal or copolymeric particle mixtures [51, 59,60,61,62,63,64], in mixtures of DNA-coated colloidal spheres [65] or of monodisperse, hydrophobically coated Au nanoparticles [34], and in aqueous dispersions of soft spherical, oil-swollen micelles [66].
The self-assembly of colloidal C15 Laves phase was also suggested from computer simulations as a route to produce photonic crystals as the tetrahedral sublattice of the small particles can exhibit a bandgap in the visible region [54,55,56,57,58].
Regarding the stability of hard-sphere Laves phases compared to alternative structures, detailed investigations and interesting conclusions were reported by Filion and Dijkstra [67]. For the ideal radius ratio of Laves phases of rA/rB = 1.225, there are several other crystal structure types such as α-IrV (orthorhombic, oS8, Cmmm), γ-CuTi (tetragonal, tP4, P4/nmm), AuTe2 (monoclinic, mS6, C2/m), and Ag2Se (orthorhombic, oP12, P222) offering a higher packing density than Laves phases. Nevertheless, full free-energy calculations (which were also reported by the same group in Ref. [50]) show that the only stable structures at this size ratio are the Laves phases. Obviously the binary hard-sphere system seems to favor the more symmetric crystal structure over the most dense-packed structure, meaning that optimal space filling alone does not decide about the stable structure type even for hard spheres [67].
These findings on atomic arrangements in non-interacting binary systems also confirm the importance and central role of geometrical factors for the stability of Laves phases.
Fundamental aspects
This section reviews selected topics related to the crystal and defect structure of Laves phases as well as the related thermodynamic and mechanical phenomena. Thus it can be regarded as a kind of continuation of the reviews [16, 17], which are about structure and stability of Laves phases. Here we focus on more recent results in the literature and aspects less emphasized in these two earlier reviews rather than try to cover the entire field of fundamental studies of Laves phases.
Stability and site preference
If a Laves phase AB2 with a given elemental composition (binary, ternary, multinary) is stated to exist, it is usually implied that this Laves phase is thermodynamically stable under certain conditions or that it can be produced under thermodynamic or kinetic control and retained for observation. Using this type of definition, it has to be kept in mind that formation of a phase is not only influenced by its own thermodynamic stability with respect to the elements, but also by the fact that its formation competes, on a thermodynamic and kinetic level, with formation of other phases of the same or of different composition. So, even if formation of a Laves phase AB2 from the two elements is thermodynamically favorable, as, maybe, revealed by first-principles calculations, this Laves phase may never be observed if other phases are even more stable and/or form more rapidly. The fact that ‘stability of a phase’ in the described sense cannot be predicted without consideration of alternative states of the system puts limitations on approaches, which try to predict stability of a Laves phase (or of other phases) simply on the basis of its composition. Nevertheless, such approaches (e.g., based on the atomic radius ratio rA/rB, see Sect. 2) have some reasonable but limited success for Laves phases, as reviewed in 2004 [16]. A newer approach allows rationalizing existence of Laves phases in comparison to other Frank–Kasper phases using structure maps constructed based on energies from first-principles calculations [68, 69].
Some quite peculiar systems exhibiting Laves phases or Laves-phase-like structure elements such as van der Waals compounds, irradiated metals, polymer particles and pressurized van der Waals compounds were already mentioned in Sect. 2. In a couple of more ‘ordinary’ binary metallic systems, which do not contain Laves phases at ambient pressure, they may form at high pressure as summarized in a review [70]. Notable examples of such Laves phases, which are only stable at elevated pressures, but which can be retained at ambient pressure, are AAl2 with alkaline earth metals A = Sr or Ba, and AZn2 with A = Ca or Sr [70,71,72,73]. Except for BaAl2, a CeCu2/KHg2-type phase is formed at ambient pressure as a stable phase instead of the Laves phase. On replacing A by lighter alkaline earth metal elements, the corresponding Laves phases CaAl2 and MgZn2 are obtained, both of which are stable at ambient pressure. A low-pressure CeCu2-type and a high-pressure C14 polymorph also appear to exist for YbAg2 [74]. Similarly, a pressure-induced CeCu2 → C15 transition was predicted for YCu2, but it could not be confirmed experimentally [75]. Pressure also appears to allow or at least ease preparation of a series of C15 and C14 AFe2 phases with A being rare-earth metal elements [76, 77], in particular for A metals with large atomic radius. Also preparation of CaCo2 Laves phase was achieved at elevated pressure [78]. Another notable Laves phase requiring high pressure for preparation is C14 KAg2 [79], whereas KAu2 is also accessible at ambient pressure [80]. All these examples have in common that the A atom appears to be too large for the Laves phase to form at ambient pressure, i.e., the atomic radius ratio rA/rB is very large. This is compensated for by a higher compressibility of the A atoms (as compared to the B atoms) at elevated pressures.
In the case of Laves phases with three or more elements, the question arises how the elements will be distributed on the A and B sites. This question of so-called site preferenceFootnote 1 was discussed, for example, in Refs. [16, 17, 82,83,84,85,86,87]. Upon adding a third element C to a Laves AB2, i.e., moving into the ternary system A-B-C, C may substitute either A or B to form a solid solution of the general formula (A1-xCx)(B1-yCy)2, if we, for a moment neglect the possibility of A and B occupying the opposite sublattices.
In most cases there will be some more or less pronounced site preference, i.e., C will substitute preferentially either A (y = 0) or B (x = 0). The most obvious cases are those, where either a Laves phase CB2 exists in the B-C system or AC2 in the A-C system, implying, in most (but not all) cases, complete solid solubility between the two respective Laves phases, see Fig. 3a and b (neglecting possible two-phase regions occurring upon change of the Laves phase polytype; see Sect. 3.3 for this aspect). If the homogeneity range is only of limited extension (e.g., in case of non-existence of CB2 or AC2), preferential site occupation can be determined by refining the site occupancies on the basis of Bragg diffraction intensity data or by using the ALCHEMI (Atom Location by Channeling Enhanced Microanalysis) technique [88]. Note that experimental investigations on site preference are complemented more and more frequently by calculations predicting site preference by means of first-principles methods.

Schematic extensions of homogeneity ranges of Laves phases in ternary systems shown in blue (as arrows: length may or may not extend to the respective endmembers), partially following ideas from Ref. [87]. Preferential substitution in a binary Laves phase AB2 of a A by C and b B by C, especially if corresponding Laves phase CB2 and AC2 exist. c Existence of a true ternary Laves phase Nb(Ni1-yAly)2 in the Al-Nb-Ni system [108, 109]. Case when C can either substitute A or B, as encountered for d the Fe-Mo-Zr systems [111] and for e the Hf-Nb-V system [114, 115]. f Corresponds to the situation in the Al-Ni-Ti system [116] being a variant of (c) where Ti is able to additionally occupy the B sites
NbCr2 Laves phase is an example for which various experimental methods were applied to reveal site preferences for a considerable series of third elements. The mere shape of the experimentally determined homogeneity range allowed to conclude that Ti substitutes Nb on the A sites (extending to the TiCr2 Laves phase, see below) [89] and Al [90,91,92,93] and Si [94] substitute Cr on the B sites. In the case of Al, also X-ray diffraction-based site occupancies indicate the same [95]. ALCHEMI, X-ray diffraction and the shape of the homogeneity range also located V on the B sites [96, 97]. Moreover, ALCHEMI revealed preference of Ti for the A sites and V, Mo, W, Ti for the B sites [98]. Phase compositions of Laves phases in further ternary Nb-Cr-C systems indicate that C = Fe [99], Co [100, 101] and Ni [102] substitute Cr on the B sites, where the former two systems also form Laves phases NbC2. Information about many other elements can be obtained for elements from the shape of the homogeneity ranges. Such studies were complemented by many types of first-principles calculations, referring either strictly to T = 0 K or also to elevated temperatures, and handling a wide series of elements [103,104,105,106,107]. In most instances, the site preferences agree with experimental findings where available. However, V, Mo, W [106] and Mo, W, Pd, Au [107] are predicted to show only moderate site preference, which may depend substantially on factors such as temperature or even on the presence of other elements.
Certain ternary systems containing no Laves phases in the binary subsystems form true ternary Laves phases with homogeneity ranges of different shapes, as also reviewed previously [16]. Figure 3c sketches the homogeneity range as found in the Al-Nb-Ni system [108,109,110] extending along the line NbAl2-NbNi2, but lacking Laves phases in the Al-Nb and Nb-Ni binary subsystems. Obviously, Nb occupies the A sites, whereas Ni and Al take over the role of the small atoms in the sense of A(B1-yCy)2.
Composition-dependent site preference can occur for an element C with an intermediate size between A and B so that both CB2 and AC2 may exist or are only potentially existing Laves phases. As a result, quite peculiar homogeneity ranges can arise. A ternary system showing three binary Laves phases and extended homogeneity ranges is the Fe-Mo-Zr system [111], with Mo being the atom of intermediate size, which can occupy both A and B sites. Starting from the binary ZrFe2 Laves phase, quite distinct homogeneity ranges extend toward MoFe2 and ZrMo2, which merge around the ZrFe2 composition, see Fig. 3d. Similar homogeneity ranges appear to exist in the Fe-Nb-Zr system [87, 112, 113] with Nb as the intermediate-size element. The homogeneity range, however, does not reach the composition of a binary ZrNb2 phase, which appears not to exist. In the Hf-Nb-V system, HfV2 is the only binary Laves phase with the intermediate-sized element being Nb. Nb is able to substitute both Hf and V [114, 115], leading to a homogeneity range as schematically indicated in Fig. 3e. Composition-dependent site preference leading to a strongly non-linear homogeneity range is also known for the truly ternary C14 Laves phase in the Al-Ni-Ti system. The A sites are exclusively occupied by Ti, but all three elements may occupy the B sites of the Laves phase in the sense of Ti(Ni1-y-zAlyTiz)2 [116], see Fig. 3f. In this context it may be mentioned that a recent study [117] refuted earlier results, which had implied that C14 Mn(Cu,Si)2 and C14 Mn(Ni,Si)2 Laves phase exist at very Mn-rich compositions requiring substantial occupation of the B sublattice by Mn.
Point defects—the binary case
Point defects were already discussed above in terms of site preference in ternary Laves phases. In the present section, typically less abundant point defects in binary Laves phases will be discussed.
The contribution of configurational entropy to the thermodynamics of crystals dictates that in single-element crystals point defects must occur to some extent in equilibrium at T > 0 (thermal point defects). In ordinary metals, these thermal equilibrium point defects are practically only vacancies. In intermetallic compounds with more than one single (crystallographically distinct) sublattice, naturally more different types of (thermal) point defects can exist. At the same time, the third law of thermodynamics predicts complete order at T = 0 K, requiring for binary and multinary intermetallic compounds complete ordering over the sublattices and, hence, fixed composition at 0 K. At T > 0 K, homogeneity ranges have to exist due to generation of configurational entropy by constitutional point defects at T > 0 K.
Comprehensive models for the defect thermodynamics are able to describe the point defect thermodynamics as a function of temperature and composition [118,119,120]. Such thermodynamic models also predict that the highest point defect densities are present at off-stoichiometric compositions in the form of what is called constitutional point defects. The substitution of elements A and B by a third element C in Laves phases in ternary and higher-order systems was already dealt with in Sect. 3.1. The changes in the site occupancies due to constitutional point defects in binary Laves phases are, in most cases, much smaller than the changes, which can occur in ternary and higher-order systems.
The composition dependence of the lattice parameters and of the volume per unit cell as measured by diffraction techniques already may give insights about, e.g., changes in the point defect mechanism. It is usually expected that the average atomic volume of an intermetallic phase increases monotonously with an increasing molar fraction of the larger element, i.e., with xA in the case of binary AB2 Laves phases. However, changes in the metallic bonding and magnetism with composition of the Laves phase but also the occurrence of constitutional vacancies may lead to behavior, which is difficult to predict. Hence, a combination of composition-dependent measurements of the mass density and of lattice-parameters is expected to give more reliable insight into the (predominant) type of constitutional point defects. However, mass-density measurements require high-quality alloys, e.g., without porosity, the presence of which would incorrectly suggest the presence of vacancies. Furthermore, the refinement of site occupancies from diffraction-based intensity data already mentioned in Sect. 3.1 can also give valuable information. This, however, requires a critical comparison of the quality of refinement for different types of point defect models. Thereby, it should be kept in mind that, based on the Bragg reflection intensities alone, mutual presence of vacancies on all sublattices cannot be assessed due to complete correlation of the occupancies with the scale factor of the refinement model. The ALCHEMI method is likely not sufficiently sensitive to study the usually low point defect densities in binary Laves phases [88]. Also in the field of predicting the type of constitutional point defects, first-principles calculations play an increasingly important role.
These introductory comments on the different methods to determine point defect characteristics in Laves should be kept in mind when assessing reports in the literature.
Constitutional point defects
The most commonly considered types of constitutional point defects are
-
constitutional antisite atoms being the analogues of the ternary substitutional atoms predominating in ternary Laves phases (see Sect. 3.1). These are denoted, for example, as AB meaning an A atom on a B site, but also
-
constitutional vacancies, denoted, for example, as VaA for vacancies on an A site.
Note, however, that more types of point defects were considered in the past, for example substitution of B4 tetrahedra by a single A atom [5, 121]. It appears, however, that only antisite atoms and vacancies seem relevant in reality in Laves phases.
In order to appropriately consider the effect of antisite atoms and vacancies on the results of mass density or diffraction measurements one can first consider the general chemical formula
where the different y(X) are (or contribute to) the indices in the chemical formula Eq. (1) and simultaneously correspond to the site fractions of the indicated species X on the A or B sites. The molar fraction xB of the component B of such a Laves phase is:
Assuming that only one single type of defect is realized at given side of xB = 2/3 (xB < 2/3 or xB > 2/3), one obtains site fractions of antisite atoms \( y\left( {B_{A} } \right) \) and \( y\left( {A_{B} } \right) \) as a function of the composition of the Laves phase given in terms of the molar fraction xB:
as well as
Similar expressions can be derived for occurrence of only constitutional vacancies.
As detailed in the following, the majority of literature implies predominance of constitutional antisite atoms, A on B sites AB for a molar fraction xB < 2/3 as well as antisite B atoms on A sites, BA for xB > 2/3. The latter is in contrast to the repeatedly supposed unlikeliness that large A atoms can occupy the sites of the smaller B atoms [122,123,124,125,126]. This in turn leads to the assumption of occurrence of VB vacancies for xB > 2/3, which, however, could not be demonstrated to be present. Only in specific cases clear evidence for vacancies VA exist for xB < 2/3; see below.
Many Laves phases AB2 with A being a main-group metal have relatively narrow homogeneity ranges [127]. The ‘classical’ Mg-based Laves phases MgCu2 [128, 129], MgZn2 [5] (and citations therein), and MgNi2 [129], however, were stated to exhibit significant homogeneity ranges. The best experimental evidence appears to exist for MgZn2 quenched from elevated temperatures. For this Laves phase, antisite atoms exist on both sides of xB = 2/3 as was shown by combined lattice parameter and mass-density measurements, see Fig. 4a [5].

a Composition dependence of observed mass-density of C14 MgZn2 (data points) as compared to calculated ones (lines) predicted from experimentally determined lattice parameters and from different models for constitutional point defects (S: substitutional, V: constitutional vacancies, T and DT: substitution models involving substitution of Zn4 tetrahedra by Mg atoms and vice versa). Adapted with permission from [5]. b Formation enthalpies of C14 MgZn2 and different supercell structures involving different types of antisite atoms and vacancies, implying preference of antisite atoms on both sides of xZn = 1 − xMg = 2/3 (adapted with permission from [131])
For MgZn2 first-principles calculations on different kinds of point defects were reported, considering the energetics of the static structures applying supercell approaches for the defects [130, 131] and also phonon calculations to account for T > 0 K effects. These investigations confirm that generation of antisite atoms is more favorable than generation of constitutional vacancies (see Fig. 4b). Although clear experimental evidence is lacking, first-principles calculations on MgCu2 also suggest predominance of constitutional antisite atoms as compared to constitutional vacancies [132]. Among Laves phases with A being a main-group metal, comparable computational investigations exist for CaAl2 [133] and CaMg2 [134], but again with little experimental evidence.
Convincing experimental evidence for the presence of constitutional vacancies exists for C15 ANi2 phases with A being rare earth metals Y and La-Lu [135]. Preferentially at low temperatures and ambient pressure, also vacancy-ordered superstructure variants exist for this type of Laves phase [136, 137] (see also Sect. 3.5). Disordering was reported to occur at elevated temperatures and upon applying elevated pressures [138,139,140,141]. The observed maximum fraction of vacancies seems to be largest for La with the largest atomic radius [141], see Fig. 5. In fact, the stoichiometric composition LaNi2 is not stable at ambient pressure [136], which was also confirmed by first-principles calculations [142,143,144], see Fig. 6.

Maximum content of constitutional A vacancies in C15 ANi2 Laves phases (formula (A1-xVax)Ni2) with A = Y, La-Lu, indicating a clear correlation between this content and the atomic radius rA; redrawn from [141]

Formation energies of various real and hypothetical La-Ni phases (red squares and black points) as obtained by first-principles calculations. The convex hull is shown as red continuous line (the dashed black line is unrelated to the present considerations). Note that the energies of the hypothetical stoichiometric C15/C36/C14 Laves phases are located well above the convex hull (adapted with permission from [143])
Under equilibrium conditions, C15 YAl2 appears to have a quite narrow homogeneity range. Rapid solidification of Y-Al melts and subsequent diffraction analysis indicated, however, that under such conditions the Laves phase might form over a wide range of composition xAl = 0.58–0.82 [123]. Based on the xAl dependence of the cubic lattice parameter, it was concluded that constitutional vacancies VaAl dominate for xAl > 2/3 and constitutional antisite atoms AlY prevail for xAl < 2/3. For this, it was assumed that the larger Y atoms are unlikely to occupy the sites of the smaller Al atoms. In contrast to this, first-principles calculations on defect formation in YAl2 predict preference of constitutional antisite atoms prior to constitutional vacancies on both sides of xAl = 2/3 [145]. This casts some doubt on the interpretation of the xAl dependence of the lattice parameters, which was not paired with mass-density measurements (see begin of Sect. 3.2). Likewise, Ir-rich YIr2 seems to be realized by antisite IrY atoms as deduced from powder-diffraction data evaluated by Rietveld refinement based on critically comparing vacancy and antisite atom models [146].
There are numerous binary Laves phases in systems with A and B being elements from the 3d, 4d and 5d transition metal series with often wide homogeneity ranges. Accordingly, there are many investigations on the kind of point defects, both employing density measurements and comparison with lattice parameter evolutions or diffraction analysis leading to refined occupancies. Table 2 summarizes investigations giving experimental and theoretical evidence, without explicit reference to the Laves phase polytypes considered. The very large majority of the results indicate predominance of antisite atoms on both sides of the stoichiometric composition, while there is apparently no compelling evidence for constitutional vacancies.
In the Co-Nb system the C14, C15 and C36 polytypes occur with increasing Co content in the sequence C14, C15 and C36, where C14 and C36 are high temperature phases but can be retained by quenching, see Fig. 7 [161]. Two quite general phenomena relevant for off-stoichiometric binary Laves phases were investigated in quite some detail in this system: preferential site occupation and static atomic displacements.

Phase diagram Co-Nb featuring three different Laves phase polytypes C15 (around the ideal composition corresponding to the molar fraction xCo = 2/3) as well as C14 (Co-poor) and C36 (Co-rich) (adapted with permission from [161])
For the Co-poor C14 polytype (xCo < 2/3), it was shown [155, 162] based on a single-crystal diffraction-based analysis (assigned crystal composition Nb1.07Co1.93) that Nb antisite atoms NbCo are located only on the Co2a sites, while the Co6h sites are only occupied by Co (compare Fig. 1 and Table 1). At the same time, it was noted that the atomic displacement parameters are larger than those obtained from a similarly performed structure analysis on a stoichiometric C15 NbCo2 single crystal [155]. These atomic displacement parameters are also found to be larger for a Co-rich C15 NbCo2 single crystal (Nb0.88Co1.12), for which Co antisite CoNb atoms are encountered. Such a composition-dependent evolution of refined atomic displacement parameters (pertaining to ambient temperature) implies a static contribution to the atomic displacements due to the point-defect-induced disorder. These static displacements add up to the thermal contributions, which should be the only contribution for the stoichiometric composition.
A C36 polytype develops at xCo > 2/3 with quite high Co contents [159, 161]. Due to the high content of CoNb antisite atoms, a more detailed evaluation of the diffraction data was possible based on a single crystal with assigned composition Nb0.735Co1.265 (“NbCo3”). As in the case of C14 NbCo2 (Nb1.07Co1.93), the antisite atoms are not evenly distributed over the available A sites (now: Nb4f and Nb4e, see Fig. 1 and Table 1) [155, 160, 162]. The static atomic displacements caused by the point defects lead to a pronounced non-Gaussian distribution of the electron density (see Fig. 8), which was modeled upon structure refinement in terms of off-center displacements of the Co antisite atoms, which are ‘too small’ for the Nb sites. Detailed reasons for the observed displacements and for the differences in the occupation of the two different Nb sites (supported by first-principles calculations) are discussed in Refs. [155, 160, 162].

Electron density difference maps originating from structure refinements on single-crystal-based X-ray diffraction data of Co-rich C36 NbCo2 Laves phase. A pronounced, peculiar-shaped electron density difference in the vicinity of the two crystallographically distinct A sites (4e and 4f Wyckoff sites) was obtained after refinement using mixed occupation of these sites by both Nb and Co (with refined anisotropic displacement parameters). This justified a more advanced refinement model involving split positions for the final refinements (adapted with permission from [155])
The preferential occupation of the Co2a sites in Nb-rich C14 NbCo2 Laves phase (see Fig. 1 and Table 1) appears to have similarities to the preferred occupation of such sites in Ti-rich C14 TiMn2 [153]. Nb-rich NbMn2, however, seems to be a counterexample with the antisite Nb atoms preferentially occupying the 6f sites [155]; see also Sect. 3.3 for the role of similar site preferences in ternary C14 and C36 Laves phases.
Thermal point defects
At T > 0 K, point defects in Laves phases can also be generated at the stoichiometric composition xB = 2/3. They can, however, only occur in combination to retain composition as resulting from Eqs. (1-2). Table 3 lists the relation between the amounts of defects, if two types of defects balance each other at the stoichiometric composition.
There are only a few experimental studies determining and quantifying the kind of thermal point defects in Laves phases. For GdPt2, GdIr2, GdRh2, GdAl2 with typically narrow homogeneity ranges, the occurrence of (non-equilibrium) quadruple defects (i.e., combinations of BA + 3VaB; see Table 3) was concluded from an observed ball-milling-induced reduction of the lattice parameters and changes in the magnetic behavior [163,164,165,166]. The same group concluded GdMg2 to exhibit pairs of antisite atoms (BA + AB; see Table 3) [166]. Thereby, it was proposed that the point defects generated upon mechanical activation should be the same as formed thermally.
Comparison of composition-dependent mass-density with lattice parameters of quenched NbCr2 [151, 152], NbFe2 and NbCo2 [151] as well as ZrCo2 [157] revealed dominance of constitutional antisite atoms in all cases (see Sect. 3.2.1). In the case of NbCr2, NbCo2 and ZrCo2, some measurable contents of thermal vacancies were shown to form, which increase with the equilibration temperature of the respective alloys. Only NbFe2 did not show any measurable evidence of thermal vacancies [151]. Note however, that these density measurements do not reveal the presence of thermal antisite atoms.
More recently, first-principles calculations on point defects in Laves phases paired with modeling of defect thermodynamics was used to predict thermal defect contents at T > 0 [103, 131,132,133,134, 145, 150, 154]. While in all these calculations the constitutional point defects are found to be antisite atoms (see Sect. 3.2.1), pairs of antisite atoms also constitute the predominant thermal point defects at the stoichiometric composition (see, for example, Fig. 9). An exception is YAl2 [145]. While the calculations for this Laves phase predict predominance of antisite defects in both Al-poor and Al-rich YAl2 in line with the other studies, considerable importance of thermal vacancies is predicted at the stoichiometric composition (combination of triple and quadruple defects according to Table 3 [145]). This agrees with findings for the electronically similar rare earth-Al compound GdAl2 [163]. Nevertheless, all the mentioned first-principles-based calculations indicate the formation of vacancies to a strongly temperature-dependent extent at T > 0, showing no obvious systematics as function of the A and B elements. These amounts of vacancies are, of course, quite relevant for diffusion of the A and B elements in Laves phases [133, 167,168,169,170].

Composition dependence of the site fractions of antisite atoms and vacancies on the Zr and Co sublattices (see Eq. (1)) predicted for ZrCo2 Laves phase at 1000 K (adapted with permission from [145]). The data indicate dominance of antisite atoms as constitutional vacancies in both Co-poor and Co-rich ZrCo2 as well as in the form of pairs of antisite atoms at the stoichiometric composition of xCo = 66.7%. Note the non-linear abscissa. Moreover, the content variables (site fractions) were adapted to comply with the quantities used in the present work
The use of CALPHAD-type thermodynamic descriptions to estimate disordering by formation of antisite atoms at the stoichiometric composition of NbCr2 is described in Sect. 3.4.
Polytypism
As indicated in Sect. 2, there is an infinite number of Laves phase polytypes differing by their stacking sequences. The simplest polytypes are C15 (MgCu2 type), C14 (MgZn2 type) and C36 (MgNi2 type), but many others were reported in special systems, see, for example, [27, 29, 171]. As indicated in a review on polytypic structures [172], there can be several reasons for occurrence of distinct polytypes:
-
(i)
There are polytypic phases, which are stable under different thermodynamic conditions (temperature, pressure, composition).
-
(ii)
Non-equilibrium polytypes may develop during growth from the melt, from the gas phase or from a structurally unrelated solid because growth of the non-equilibrium polytype is more rapid than growth of the equilibrium polytype.
-
(iii)
Non-equilibrium polytypes may develop from an already existing polytype in the course of a phase transition between two polytypes (polytypic transition). Thereby, a non-equilibrium polytype is formed because it develops more rapidly than the equilibrium polytype.
The possibility to form non-equilibrium polytypes according to (ii) and (iii) should always be taken into account upon interpreting experimental data. Moreover, it should be noted that the observation of some irregular stacking sequence does not automatically mean the discovery of a new polytype as is stated occasionally in the literature. Instead, a given stacking sequence should be repeated several times, although no specific limit exists defining how often some stacking sequence must be repeated to call it a new polytype.
As reviewed [16], Laves and Witte [173] had already pointed out the obviously polytype-influencing role of the valence electron concentration VEC (or, more precisely, the number of valence electrons per atom) in A = Mg-based Laves phases. Similar schemes were reported for transition metal Laves phases [174]. Studies revealing sequences of characteristically changing polytypes are typically performed on alloy series A(B,C)2 with B, C providing different numbers of valence electrons. Recent investigations of this type, which are often supplemented by electronic structure calculations, deal with x-/VEC-dependent changes of the stacking sequences in Ca(Al1-xMgx)2 [175, 176], Mg(Zn1-xPdx)2 [177], Mg(Ni1-xGex)2 [178], and Gd(Co1-xGax)2 [179]. It was, however, emphasized in Ref. [16], that a pure VEC dependence cannot explain all trends in polytype formation, and observations from different systems are contradictory. An early reported additional influencing factor modifying the trends implied by a VEC dependence of the polytype in ternary transition metal Laves phases A(B,C)2 was proposed to be the difference of the group number of two elements B and C [180].
Much more recently, the following experimentally observed trends on polytype stability in Laves phases with the A atoms being group-IV/V transition metals were rationalized [181]:
-
(i)
If in a binary system several Laves phase polytypes exist with a composition-dependent polytype stability, the C15 phase is found around the stoichiometric composition given by xB = 2/3, whereas at B-poor compositions the C14 polytype is encountered and at more B-rich compositions the C36 may be found (not in all cases all three polytypes are encountered). The best investigated system showing this systematic is the Co-Nb system (see Sect. 3.2.1 and, in particular, Fig. 7). As already mentioned, NbCo antisite atoms are located very preferably on the Co2a sites of the C14 Laves phase in the Co-poor regime, and a less pronounced site preference occurs for CoNb antisite atoms in the Co-rich C36 Laves phase. Consistent with this, if a binary Laves phase assumes the C14 structure at xB = 2/3, no polytypism is encountered upon deviating from the stoichiometric composition.
-
(ii)
Alloying of a binary C15 Laves phase AB2 by a third element C, which substitutes the B element, frequently leads to a change to a C14 polytype. In several cases, it was demonstrated that also in this situation, a preferred occupation of the 2a site by C occurs at low C contents. This, for example, is known for the extended solid solution series with C = Al, Si [17]. Several of such systems were explored more recently in some detail, as Al-Cr-Nb [93, 95] and Al-Co-Nb [182]. In systems ranging from C15 Laves phase to C15 Laves phase such as NbCr2-NbCo2 [155, 183, 184] and ZrV2-ZrCo2 [184], C14 Laves phase occurs at intermediate compositions with enrichment of the respective minority element on the 2a site; see Fig. 10.Footnote 2 Energetics of such preferred site occupation was also supplemented by first-principles calculations [183]. In contrast to the effects upon alloying binary C15 Laves phases with a third element, alloying of binary C14 binary Laves phases (e.g., C14 TiFe2 with Si) does not lead to a change of the polytype.
Figure 10 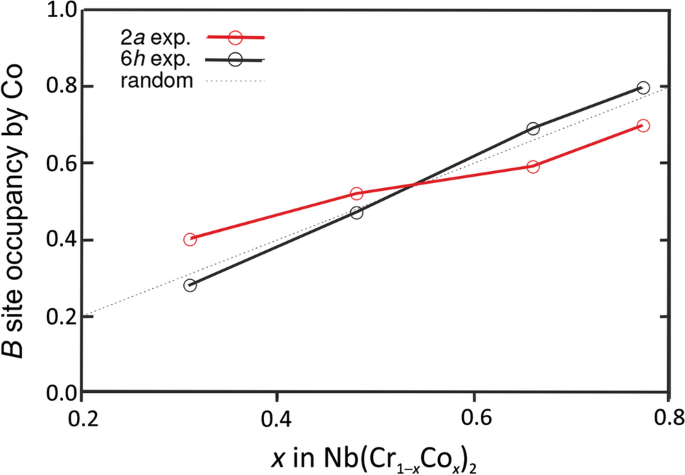
Occupancies of the B2a and B6h sites by Co in C14 Nb(Cr1-xCox)2 Laves phase [183, 184] as determined on the basis of single-crystal-based X-ray diffraction data, revealing preferred occupation of the 2a sites by the minority B element being accompanied with an inversion of the occupancies at x ≈ 0.5 (adapted with permission from [184])

Both these observations imply that if a binary C15 Laves phase exists for some stoichiometric binary composition, introduction of substitutional point defects (antisite atoms in the binary system or C atoms on B sites upon making the system ternary) promotes occurrence of hexagonal polytypes. The C36 polytype occurs when different types of species occupy the A sites, i.e., in B-rich binary Laves phases, whereas the C14 polytype occurs when different types of species occupy the B sites (B-poor binary and A(B,C)2 ternary Laves phases). It was made likely [181] that the larger number of structural degrees of freedom of the hexagonal polytypes and, in particular, the crystallographically independent B (C14, C36) and A (C36) sites (see Fig. 1 and Table 1), which allow for preferential site substitution on the B or on the A sites, stabilize the C14/C36 polytypes with respect to the original C15 polytype. Energies from first-principles calculations confirm this stabilization for C14 with respect to C15 [183, 184]. Assuming that only the simplest polytype is formed, which is necessary to allow for some energetically favorable preferred site occupation, explains why in case of mixed occupation of the B sites the C14 polytype develops whereas for mixed occupation of the A sites the more complicated C36 polytype forms.
Different Laves phase polytypes must essentially be regarded as different phases in the sense of the phase rule, with transitions between different equilibrium polytypes occurring in a first-order fashion. If, as discussed above, polytypes occur in a binary (or ternary) system with varying composition at constant temperature, two-phase regions should be located between these polytypes. These two-phase regions typically appear to be quite narrow in binary and ternary systems. Such two-phase regions C14 + C15 and C15 + C36 are shown in Fig. 7. The widths of such two-phase regions are typically determined from heat-treated, macroscopically homogeneous alloys, where it is, however, difficult to hit the composition of a narrow two-phase field. An alternative method is preparation of diffusion couples, which were used in Ref. [161] leading to composition profiles as shown in Fig. 11 (see also [160]). Whereas the presence of different phases was established by electron backscatter diffraction, expected composition steps indicating the width of the two-phase region are not always visible, see Fig. 11. Other examples for non-detectability of a two-phase region can be found in investigations on the Co-Ta [185] and the Co-Ti systems [186]. As briefly indicated in Ref. [186], the similar crystal structures of the various polytypes likely have quite similar energies but also allow for formation of coherent interfaces upon introducing moderate coherency stresses. As a consequence, the two-phase regions expected due to the continuum thermodynamics of the systems can become narrowed or even absent in diffusion couples due to coherency effects [187,188,189].

Composition profiles measured by electron probe microanalysis on diffusion couples Co-Nb (left) and NbCo2-Nb (right) after heat treatments at different temperatures [161]. All phases expected from the equilibrium phase diagram (compare Fig. 7) were formed. Whereas a composition step is visible between the regions of the C15 and C14 Laves phase, such a step is not discernible between C36 and C15 (adapted with permission from [161])
There are a couple of binary systems containing Laves phases showing temperature-dependent equilibrium polytypism. It appears that, if several polytypes occur as equilibrium phases, C15 appears to be the lowest-temperature phase and C14 is highest-temperature phase, with possibly intermediate C36 [17]. This temperature-dependent equilibrium polytypism was explored in quite some detail in the Cr-Ti [190,191,192,193,194] and Hf-Cr systems [195,196,197] dealing with transformation kinetics [192, 193, 196] and with the fault structure as a consequence of the transformation [190, 191, 195, 197].
Temperature-dependent equilibrium polytypism with a C14 high-temperature phase had also previously been concluded from various types of experimental evidence for NbCr2 (see [198, 199] for a review of such evidence): (i) It is possible to obtain C14 NbCr2 (which can turn out to be actually C36) in as-cast alloys, whereas subsequent annealing produces C15. (ii) Thermal analysis data often show an endothermic thermal signal upon heating just prior to melting, which was attributed to the C15 → C14 transition. A correspondingly exothermic signal is also observed upon cooling. (iii) Extrapolation from the ternary Al-Cr-Nb system to pure NbCr2 suggests presence of a C14 phase in a small range of temperature.
Quenching experiments on alloys equilibrated in the apparent C14 field were unsuccessful and because C14 was unusually distributed in as-solidified arc-melted ingots [200], doubts arose about the existence of an equilibrium C14 NbCr2 high-temperature phase. By use of in situ high-temperature neutron diffraction it was shown that C15 NbCr2 melts without formation of a C14 phase [198]. Moreover, it was shown that the signal in thermal analysis arose due to melting of an η-carbide-type impurity phase NbCrXz [198, 199]; see also below. Hence, it was concluded that the C14 phase occasionally observed in as-cast alloy is only metastable at all temperatures.
More detailed high-resolution electron microscopy and powder X-ray diffraction investigations of the apparent C14 NbCr2 phase formed in as-cast alloy indicated that, at least in the investigated specimens, the majority of the hexagonal Laves phase was actually faulted C36 NbCr2 [191] which is, however, also only metastable at all temperatures. Remnants of true C14 suggested formation of metastable C14 upon solidification, which quite easily transforms to C36. This rapid transformation suggests an important role of (synchro) Shockley partial dislocation dipoles [201]. Such dipoles were indeed observed in C36 NbCr2 [197], and the irregularities in the stacking sequence of C36 NbCr2 (and of C36 TiCr2 having formed from equilibrium C14 TiCr2) [191] imply that the transformation C14 → C36 is indeed carried out by Shockley partial dislocation dipoles on adjacent lattice planes. This kind of transformation is not possible for the stable C15 phase, which evidently forms much more sluggishly [196].
Beyond the above-mentioned investigations dealing experimentally and theoretically with polytypism, there are further first-principles-based calculations demonstrating the power of these methods to reveal the thermodynamics of polytypism of the Laves phases. For example, the relative stability of polytypes of stoichiometric binary Laves phases at T = 0 K can reliably be predicted for HfV2 [202], (Ti,Zr,Hf)Cr2 [150, 203, 204], ZrMn2 [154, 205], TaV2 [206], NbCr2 [202, 207, 208] and TaCr2 [208]. Dealing with non-stoichiometry, the stability of an intermediate C36 polytype on the CaAl2-CaMg2 section [209, 210] was confirmed. Use of the quasiharmonic approach to predict T > 0 thermodynamics of TiCr2 polytypes [211] gave at least a qualitative agreement with experimental evidence for stability of C15 TiCr2 at 0 K and increasing stability of C36 and C14 with increasing temperature [194], see also paragraph above. The contribution of magnetic ordering to the binding energy is substantial, so that this contribution can even change the relative stability of polytypes as compared to the nonmagnetic state, as, for example, detailed in Ref. [205] for the case of ZrMn2.
Two aspects remain to be mentioned in the context of diffraction analysis of Laves phase-containing alloys, which accompany virtually all experimental studies dealing with polytypism. Firstly, care has to be taken upon identifying the polytype present based on diffraction evidence, e.g., powder X-ray diffraction, selected area electron diffraction or by electron backscatter diffraction. As outlined in detail in [195] for the case of diffraction analysis, there are fundamental reflections (or Kikuchi bands), which are common to all Laves phase polytypes, and polytype reflections (or Kikuchi bands), which are specific for the polytype. Hence, it is impossible to assign the type of polytype present simply on the basis of observation of only the (usually strong) fundamental reflections. Moreover, faulting of the stacking sequences can considerably broaden the polytype reflections, possibly hampering polytype identification, whereas the fundamental reflections remain unaffected; see, for example, [191, 195].
Secondly, high-temperature corrosion of Laves phases with A being a group-IV/V transition can lead to the formation of peculiar intermetallic compounds stabilized by small amounts of X = O, N or C. These intermetallic phases can obstruct interpretation of, in particular, powder-diffraction patterns. One example is the η-carbide-type NbCrXz phase [198, 199] already mentioned above. Such η-carbide type (or Ti2Ni-type; differing only by the distribution of the metal atoms) phases were also reported in Laves phase-containing alloys in the systems Co-Nb [155], Fe-Nb [155, 212], Mn-Zr [213], Fe-Ta-V [184, 214], and Co-Cr-Nb [183, 184]. In the Fe-Nb system also an M23C6-type phase was reported [215], whereas impurity-stabilized Th6Fe23-type phases were observed in Fe-Zr-based alloys [216, 217]. Moreover, a metastable face-centered cubic phase with a lattice parameter of a = 8.17 Å and unknown atomic structure incompatible with the previously mentioned structures was reported in Cr-36 at.% Zr alloy [218], which might also be an impurity phase. Note that this list is definitely incomplete since such impurity phases are often only mentioned in passing. Furthermore, it should be mentioned that due to the low quantity of O/N/C needed to stabilize such phases, uptake of small amounts of such impurities during handling at elevated temperatures can lead to formation of appreciable amounts of the impurity phase.
The frequent occurrence of such phases in turn suggests very low or even negligible solubility of O/N/C in Laves phases, most likely because the tetrahedral interstices are simply too small to accommodate O/N/C. There is, however, some atom-probe field ion microscopy evidence for incorporation of C into (Mo,W)(Fe,Cr)2 Laves phase in some high-Cr ferritic steels [219, 220]. DFT calculations were performed to estimate the energetics of N and C incorporation into C14 NbFe2 Laves phase [221]. This study indicates that the dissolution energy of N into the most favorable tetrahedral sites of NbFe2 is similar to that for N dissolution into α-Fe (showing only low N solubility), whereas C incorporation into NbFe2 appears to be even less favorable. As discussed in Sect. 4.1, these interstices can, however, well accommodate appreciable amounts of hydrogen.
CALPHAD modeling
Description of the thermodynamics of multicomponent systems using the CALPHAD (CALculation of PHAse Diagrams) method [11, 86] is nowadays routine. This method involves development of a composition-dependent description of the temperature-dependent Gibbs energy for each phase of the system. The simplest way to model the Gibbs energy of Laves phases is in the form of stoichiometric compounds. This was done for Laves phases especially in early publications (but also in later studies, if non-stoichiometry is not an issue) as, for example, in the systems Fe-Ti [222], Cu-Mg [223], and Mg-Zn [224]. Gibbs energies of potentially non-stoichiometric, multicomponent crystalline phases require more complex functional descriptions. In early times, descriptions for non-ideal, simple substitutional solid solutions were employed when describing non-stoichiometric Laves phase in the Co-Cr-Zr system [225], at that time not considering polytypism. As, however, also discussed above, Laves phase polytypes are distinct phases. Hence, they should be (and indeed usually are nowadays) treated as such within the CALPHAD approach.
In current research, modeling of non-stoichiometry is predominantly realized using the compound-energy formalism (CEF) [226, 227] as it allows handling appropriately at least ideal configurational entropy in crystalline phases with several, crystallographically distinct sublattices. Depending on the type of constitutional point defects, which are considered to be responsible for deviations from ideal AB2 composition, different sublattice models can be used, with or without Redlich–Kister-type description of the excess Gibbs energy. Initially, various types of sublattice models were used for Laves phases, for example in the Cr-Ti [228], Cr-Zr [229], Fe-Ti [125, 230], Mn-Zr [126], and Cr-Ta [231] systems, partially also considering predominance of constitutional vacancies in contrast with later obtained experimental evidence (see Sect. 3.2). The variability of the models arises also from the fact that the C14 and C36 polytypes contain more than two sublattices; see Table 1.
There is a strong desire to develop CALPHAD descriptions of certain systems by using thermodynamic models for phases, which allow the extension into higher-order systems while employing a minimum number of independent parameters. In view of this desire, it was recommended to work, for a given Laves phase polytype, with only one single A and one single B sublattice [7]. Each sublattice can be occupied by both types of atoms, neglecting the possibility of occupation by vacancies.Footnote 3 In some cases this procedure is exactly followed. Sometimes the crystallographically distinct A and B sublattices (according to Table 1) are considered separately in a formal sense, whereas the relevant parameters are constrained such that the model behaves as a model with only two sublattices.
Within the CEF formalism, consideration of two sublattices for a Laves phase in a binary system implies the assessment of the Gibbs energies of the endmember compounds AB2 (the stoichiometric, ideally ordered Laves phase), AA2 and BB2 (pure A and B in the Laves phase structure), and BA2 (Laves phase structure with inverse occupation of the sublattice). Note that in all known cases of Laves phases, the AA2, BB2 and BA2 compounds are not accessible experimentally so that corresponding experimental Gibbs energies are not available. The only experimental data available to assess estimated values is the energetics of the Laves phase in the relatively narrow observed homogeneity regions. In view of this, a series of strategies to arrive at thermodynamic descriptions of binary and higher-order systems were employed in the literature [7]. Nowadays, typically direct experimental data are used to assess the Gibbs energy of stoichiometric AB2, while estimated values for the Gibbs energies are used for AA2 and BB2 ensuring that the Laves phase is sufficiently unstable with respect to pure elemental A and B. The Gibbs energy of the BA2 endmember is typically expressed in terms of the energies of the remaining three endmembers AB2, AA2 and BB2, yielding effectively a Wagner-Schottky model, which should be valid for dilute point defect contents [7, 227]. The available information about the homogeneity range of the actual Laves phase is then typically employed to optimize the Redlich–Kister parameters. Such type of strategy was applied, e.g., for the systems Al-Ca-Mg [232], (Al-)Cu-Mg-Zn [233,234,235], Ca-Mg-Zn [236], Cr-Ti(-Al) [237], Fe-Ti [238], Co-Ti [239], Al-Ni-Ti [240], Fe-Zr [241], Fe-Mo-Zr [111], Mn-Zr [213], Cr-Nb [242], (Al-)Cr-Nb [92], Co-Nb(-Al) [243] and Co-Ta [244].
An increasing number of studies makes use of energy values obtained from first-principles calculations. In particular, formation energy values are calculated for the experimentally not accessible endmembers BA2, AA2 and BB2Footnote 4 of the CEF and the corresponding values are used in the thermodynamic descriptions. Examples for such modeling works are descriptions of the systems Cu-Mg [247], Ca-Mg-Zn [248], Al-Ca-Mg [249,250,251], Cr-Ti [203], Cr-Zr [252], Fe(-Sn)-Zr [253], Cr-Hf [203], Hf-W [254], Cr-Nb [208, 252, 255, 256], Cr-Nb-Fe [257], Cr-Ta [208], Fe-Ta [258], Cr-Ta [259], Co-Ta [260], Ta-V [206], Cr-Nb-Zr [252]. It should be noted that first-principles calculations can yield sublattice-specific Gibbs energies (see, for example, [203, 206]) allowing to arrive at energies of the endmembers of more complex sublattice models for the C14 and C36 polytypes. Recent publications follow the approach of CALPHAD modeling with respect to 0 K [256, 259] (third generation CALPHAD databases).
Thermodynamic descriptions can also be used to predict the difficult-to-measure thermal point defect content, see Sect. 3.2.2. This was done in Fig. 12 for the amount of antisite atoms developing in stoichiometric NbCr2 (using a thermodynamic description from Ref. [255]).

Fraction of Cr sites occupied by Nb atoms y(NbCr) in stoichiometric C15 NbCr2 as a function of temperature calculated from a thermodynamic description of the Cr-Nb system [255]: a linear fashion and b in an Arrhenius-type fashion
Going beyond the CEF, recently the cluster expansion approach based on quasi-random structures of Laves phases was used to investigate the thermodynamics in the homogeneity ranges of various C15 Laves phases [261].
Structure variants
Structure variants of the Laves phases are, in the sense of the current section, crystal structures, which result from breaking the space group symmetry of any of the Laves phase polytypes. The cases discussed in the following can be roughly subdivided into (i) simple, largely homogeneous distortions due to changes in the electronic structure, for example by the onset of magnetic ordering or changes in the bonding pattern; see Sect. 3.5.1. Other types of distortions can occur due to (ii) orderly occupation of the A or B sites by different types of species, i.e., different types of atoms or also by vacancies; see Sect. 3.5.2. More complex structures occur by (iii) combination of Laves phase structures with fragments from other crystal structures, typically breaking up the tetrahedral network of the B atoms; see Sect. 3.5.3.
Distortions due to changes in the electronic structure
Ferro-, antiferro-, or ferrimagnetic ordering below some magnetic order–disorder temperature can result in magnetostriction associated with largely homogeneous strain of the crystal structure. In the simplest case, non-symmetry-breaking volume effects are encountered, possibly accompanied by non-symmetry-breaking effects on the axial ratio in the case of hexagonal polytypes. These effects can be pronounced over a narrow temperature range, if first-order magnetic phase transitions can be brought about, for example in the form of a transition from high-moment/high-volume ferromagnetic to low-moment/low-volume antiferromagnetic, which was studied in a series of publications on (Hf,Ta)Fe2 Laves phases [262,263,264,265].
Generally, in the case of uniaxial (e.g., ferro)magnetic ordering, magnetostriction conforming with the magnetization direction is always expected to break the cubic (nuclear) symmetry of the C15 polytype, and, if the axis of magnetization does not agree with the stacking direction, also the hexagonal symmetry of other polytypes. These effects may be undetectable by conventional diffraction techniques in many systems, but they can become considerable for rare-earth element-based materials, as in (usually C15) Laves phase AB2 with A a rare-earth element and B typically Mn, Fe or Co. While the magnetic ordering is frequently more complicated (e.g., ferrimagnetic and sometimes antiferromagnetic), the resulting distorted nuclear structures derived from diffraction experiments were described as tetragonal (I41/amd, moment direction 〈100〉C15, NdCo2 [266, 267], HoCo2 [268], YMn2 [269]), rhombohedral (\( R\bar{3}m \), moment direction 〈111〉C15, TbFe2 [270], SmFe2 [271]), or orthorhombic (Imma, moment direction 〈110〉C15, NdCo2 [267], SmFe2 [271]) distortion variants, largely resulting from homogeneous strain. Please note however, that in many studies on functional properties, crystal structure is not described explicitly in conventional crystallographic terms (e.g., indicating the space group symmetry) but more in terms of the observed direction of magnetostriction.
In line with considerations on the local symmetry of the rare-earth metal atoms [272], the largest distortions are encountered for the rhombohedral structures with moment direction along 〈111〉C15. To make the materials magnetically sufficiently soft and in order to make polycrystalline materials suitable for such applications, magnetic anisotropy is reduced by alloying with rare-earth elements favoring the 〈100〉C15 moment direction. This was also the alloying strategy to arrive at Tb0.3Dy0.7Fe2, Terfenol-D, see Fig. 13 [273] and Sect. 4.3.1.

Pseudobinary phase diagram TbFe2-DyFe2 redrawn from [273] with the binary endmembers forming different distortion variants due to magnetostriction of the C15 Laves phase at low temperatures. The varying magnetic anisotropy leads, under the constraint of constant composition, to the morphotropic phase boundary (curved line). Compositions on the rhombohedral side close to this boundary show the best properties for polycrystalline magnetic actuator materials (see also Sect. 4.3.1)
Furthermore, some symmetry-breaking distortive phase transitions, which do not appear to be related to magnetism, are known to occur in some Laves phases either with decreasing temperature or with increasing pressure. Examples are C15 UMn2 [274, 275], C14 URe2 [276], C15 ZrV2 [277, 278], C15 HfV2 [278,279,280,281,282], C15/C14 CaLi2 [283] and C15 PbAu2 [284]. The resulting space groups are the same as for magnetostrictively distorted magnetic Laves phases. Such transitions are often found to be related to special quantum phenomena such as superconductivity, for example in the cases of ZrV2 and HfV2; see also Sect. 4.4.
Internal structural distortions are responsible for a cubic-cubic transition in KBi2-xPbx (x = 0…0.8), as characterized by experimental and theoretical methods [285]. While KBi2 and the compounds with up to x = 0.6 have ordinary C15 structure with \( Fd\bar{3}m \) symmetry, for higher Pb contents a superstructure with cubic \( F\bar{4}3m \) symmetry was found. The symmetry reduction is accomplished by an alternate contraction and expansion of the B atom Bi4-2xPb2x tetrahedra, see Fig. 14. For the experimentally unattainable value of x = 1, the small tetrahedra can be imagined to correspond to electron-precise Zintl anions [Bi2Pb2]2− [285]. Hence, although atomic substitution occurs, the symmetry reduction is obviously due to changes in the chemical bonding induced by the change in the number of electrons in the system, although occurrence of atomic ordering cannot be excluded to accompany the symmetry reduction as a secondary process.

Intermetallic distances in a pair of tetrahedra within the crystal structure of KBi2-xPbx [285] sharing a common corner: (left) at x = 0, with an atomic structure and symmetry of an ordinary C15 KBi2 Laves phase. The “i” implies a center of inversion. (right) x = 0.8 with the upper tetrahedron expanded and the lower one contracted. Assuming electron transfer from K, the lower, small tetrahedra corresponds to an electron-precise [Bi2Pb2]2− Zintl anion for the idealized case of x = 1 (redrawn with slight modifications from [285])
Distortions due to atomic ordering
While in the case of the KBi2-xPbx, no obvious occupational ordering occurred on the A or B sites, symmetry-broken Laves phase variants are sometimes formed to accommodate different types of atoms in an ordered fashion on the A or on the B sites. It was noted already in Sect. 3.3 that such a symmetry reduction is necessary if two different atoms have to orderly occupy the B sites in the C15-type structure, whereas this is not the case in the C14 structure, which already contains two different sites B2a and B6h (see Table 1). Table 4 lists a couple of cases of ordering-induced symmetry reductions for the C15 and C14 polytypes, including also vacancy ordering in rare-earth nickelides (see also Sect. 3.2.1). Some examples for resulting structures are shown in Fig. 15.

Example structures showing symmetry reduction by ordering in C15-based Laves phase variants as depicted in (pseudo)cubic unit cells: a Mg2Ni3Si in \( R\bar{3}m \) symmetry [286] after transformation into a face-centered pseudo-cubic structure, unique axis corresponds to [111] direction of the shown unit cell, b Mg2Cu3Si (real composition poorer in Si) in P4332 symmetry [293]. a and b show Cu/Ni versus Si ordering on the B sites, respectively. c Y15(Y0.24Va0.76)Ni32 with \( F\bar{4}3m \) and 2 × 2 × 2 superstructure [317] showing ordering of vacancies (partially occupied by Y) against Y on the A sites
It is striking that all substitutionally ordered cases involving symmetry reduction listed in Table 4 are cases with A = alkali, alkaline earth or rare earth metal. Examples with A = group-IV/V transition metals lack and only occur for the indicated case of the non-symmetry-breaking ordering on the 2a and 6f sites already inequivalent in the C14 structure. This indicates, as already implied by the examples in Sect. 3.3, that the valence electron concentration is a more important polytype-determining criterion in Laves phases with A = alkali metal, alkaline earth than in the case of A = group-IV/V transition metal.
More complex Laves phase variants
Further crystal structures can be considered showing ‘similarity’ with the crystal structures of Laves phases, lacking, however, a one-to-one correspondence of the atomic or vacancy sites with the sites of a particular Laves phase polytype. These structures are as diverse as the diffuse term ‘similar’ may imply. One group of such Laves structure variants results from the fact that the Laves phases belong to the larger group of Frank–Kasper phases, which can be regarded as tetrahedrally close-packed structures of differently sized atoms. Each of these atoms is surrounded by a triangulated polyhedron with the coordination numbers 12, 14, 15 or 16 [22, 23]. All Frank–Kasper phases can be described as layered structures consisting of regular stackings of a particular combination of certain structure fragments (or structural motifs), which can be combined, in principle, in an infinite number of ways [25, 318]. Therefore, Frank–Kasper phases containing fragments of the Laves phase structures might in a sense be regarded as variants of the Laves phases. One example is the μ phase, which contains structure fragments of the Laves phase and the Zr4Al3 phase [319], and which exists in many Laves phase-containing binary transition metal systems. The already discussed Co-Nb system is one example containing the Nb6Co7 (µ) phase [320]; see Fig. 7. Laves and Zr4Al3 fragments also build the crystal structure of the monoclinic Mg4Zn7 phase [321, 322]. Combinations of fragments or layers from Laves phase structures with layers containing non-Frank–Kasper structure elements such as the CaCu5 structure can lead to layered structures of compositions such as A2B7 or AB3 [323, 324]. Other complex types of structures obtained from stackings of Laves phase and non-Frank–Kasper structure fragments occur in a series of Sr-Al and Ba-Al intermetallic phases [176, 325,326,327,328]. Interestingly, Laves phase itself actually is only stable at elevated pressure in both systems (see Sect. 3.1).
Finally, the alkali metal-Au Laves phases, which are known to exist in the systems Au-Na (C15 NaAu2) [329, 330] and Au-K (C14 KAu2) [331], appear to be the basis for a wide range of structure variants. To begin with the binary systems, a RbAu2 Laves does not appear to exist, likely due to the large size of the Rb atoms, whereas a Rb3Au7 (RbAu2.33) phase [332, 333] exists with a structure that can be derived from the Laves phase (see Fig. 16a). This compound contains chains of partially corner-sharing Au4 tetrahedra as structure motif, within which the large Rb atoms are embedded and in which isolated corners of the Au4 tetrahedra, which can be regarded as fragments of the B network of a C15 Laves phase, form contacts to another type of Au atoms showing square-planar coordination. Crystal structures with somehow fragmented C15 Laves phase-related networks of partially corner-sharing Au4 tetrahedra were also reported for a couple of A-Au-B′ compounds with A = K, Rb and B′ = Ga, In, Tl, Ge, Sn, Pb (heavy main-group III and IV metals). All Au atoms of the C15-type tetrahedral network either are common corners of two Au4 tetrahedra, or they form some apparently directional bond to the additional B′ atoms. Examples are A4Au7B′2 (AAu1.75B′0.5) with (A, B′) = (K, Ge) or (K/Rb/Cs, Sn) or (Rb/Cs, Pb) [334,335,336], K3Au5B′ (KAu1.67B′0.33) with B′ = In, Tl, Pb [337, 338], Rb2Au3Tl (RbAu1.5Tl0.5) [338], K4Au8Ga (KAu2Ga0.125) [339], and K12Au21Sn4 (KAu1.75Sn0.33) [340]; see Fig. 16b and c for example. The main-group III and IV metal atoms appear to be tetrahedrally bonded to themselves or to the surrounding Au atoms; see also a recent review on polar gold intermetallic compounds [341].

Crystal structures derived from non-existing C15 RbAu2 Laves phase still featuring the Au4 tetrahedra (yellow) partially sharing common additional bonds toward a single Au in the middle of the unit cell depicted in Rb3Au7 [332, 333], b toward Tl atoms of an infinite zigzag chain of Tl atoms in Rb2Au3Tl [338], and c toward Sn dumbbells in Rb4Au7Sn2 [334]
Planar defects, intergrowth, and intergrowth-related faulting
Peculiar planar defects distinct from faults associated with a variable Laves phase stacking sequence [342] were reported in a series of studies mainly based on observations by transmission electron microscopy (TEM) techniques. The apparently earliest investigation of this type was performed on C14 MgZn2. By conventional TEM, some features parallel to \( \left\{ {10\bar{1}0} \right\}_{{{\text{C}}14}} \) were observed that were interpreted as Mg2Zn3 precipitates [343]. A later high-resolution TEM study on the relation of C14 MgZn2 and Mg4Zn7 (which was previously described as Mg2Zn3) rationalized such an intergrowth [344, 345].
In an early high-resolution TEM investigation of an Fe-rich, multicomponent (Mo,Ti,W)(Fe,Cr,Ni)2 C14 Laves phase, which had been extracted from an Fe-based superalloy [319], planar faults were observed on basal (\( \left\{ {0001} \right\}_{{{\text{C}}14}} \)), prismatic (\( \left\{ {10\bar{1}0} \right\}_{{{\text{C}}14}} \)), and pyramidal (\( \left\{ {10\bar{1}1} \right\}_{{{\text{C}}14}} \)) planes [346]. Analysis of the images was based on comparison with an ideal C14 phase and a µ phase (consisting itself of structural motifs of the Laves phase and Zr4Al3 stacked along [0001]C14) using a tiling scheme worked out in Ref. [319] (similar to the scheme used in [344, 345]). It was concluded that all three types of planar defects can be perceived as layers out of the µ phase [346].
More recent atomically resolved scanning TEM (STEM) investigations on single-phase Nb-rich C14 NbFe2 Laves phase gave direct experimental evidence confirming that the observed planar defects on basal \( \left\{ {0001} \right\}_{{{\text{C}}14}} \) (see Fig. 17) and pyramidal \( \left\{ {10\bar{1}1} \right\}_{{{\text{C}}14}} \) planes consist of structural motifs from the µ phase [347]. STEM energy dispersive X-ray spectroscopy (EDS) proved these layers to be rich in Nb as expected from the ideal Nb6Fe7 composition of the µ phase. Indeed, such kind of planar defects were only observed in Nb-rich NbFe2 while they were not found in stoichiometric material. Interestingly, none of the observed variants of extended basal planar faults shows a perfect μ phase stacking sequence, even though all of them are composed of the characteristic structural motifs of the µ phase. First-principles calculations showed that such extended defects in off-stoichiometric Laves phase can be thermodynamically more stable than formation of a basal synchroshear-formed stacking fault (see Sect. 3.7) defect structure with a simultaneous accommodation of the excess Nb in correctly stacked, true μ phase precipitates with a constrained basal lattice parameter. Besides the occurrence of antisite atoms, the formation of such kind of coherent planar faults obviously is a way for the accommodation of excess Nb [347].

a High angle annular dark field STEM micrograph of Fe-poor (Nb-rich) C14 NbFe2 Laves phase with a coherent planar defect consisting of structural motifs from the µ phase (reproduced with permission from [347]). b Atomic structure model in higher magnification (small atoms: Fe, large atoms: Nb) revealing the typical layer arrangements of Nb atoms in the planar defect which is characteristic for the µ phase (note that the thickness of the planar fault in the sketch is reduced compared to the experimental micrograph). For a more detailed description of this kind of defects, see [347]
Whereas the above examples concern B-poor (mainly C14) Laves phase, planar defects in B-rich C15 YIr2 on \( \left\{ {111} \right\}_{{{\text{C}}15}} \) planes appear to be associated with the intergrowth of PuNi3-type YIr3 [146]. As already indicated in the previous Sect. 3.5.3, the periodic combination of fragments of different crystal structures can lead to new crystal structure types. The same scheme can, as shown here, also be used to construct lower dimensional features in crystal structures such as planar faults.
Plastic deformation
The deformation mechanism of Laves phases was widely studied beginning in the 1960s and 1970s with detailed investigations on dislocation density and mobility and observation of twinning as the dominating deformation mode, see, for example, [190, 348,349,350]. Most of this early, fundamental work was performed by the group of Paufler and Schulze in Dresden, Germany. A summary of the main results of their partially German-written papers related to mechanical properties of MgZn2 and other Laves phases is given in two more recent reviews [4, 5]. Today it is generally accepted that plastic deformation in Laves phases occurs via a dislocation-mediated process by (basal) slip or twinning, see, for example, [351,352,353]. The underlying mechanism is the so-called synchroshear process, which is schematically explained in Fig. 18. Synchroshear in Laves phases occurs by the cooperative motion of two coupled Shockley partial dislocations on the adjacent planes of a triple layer of the Laves phase structure [354,355,356,357,358]. Deformation by undulating slip was proposed as an alternative mechanism to synchroshear [359], but from a theoretical comparison of the two alternatives it was concluded later that both models finally describe the same mechanism and only differ in the way of describing the deformation process [358]. Synchroshear as the most likely mechanism for dislocation-mediated plastic deformation in Laves phases had been already suggested by Krämer and Schulze in 1968 from theoretical considerations [360]. Their geometrical analysis of possible slip systems and types of dislocations revealed that this type of synchronous shearing of adjacent planes is the only possibility that results in a stacking fault without lattice expansion. Prismatic and pyramidal slip modes were also identified as energetically possible but their activation was found to be much less favorable [360].

Schematic illustration of the synchroshear mechanism: a Initial arrangement of the big A (orange) and small B (dark blue) atoms in the three-layer stack with the surrounding B single-atom Kagomé layers above and below the three-layer stack indicated by dashed circles. The bottom gray block is considered stationary and the top block responding to a shear stress by moving to the left. b Atomic arrangement after synchroshear. c Illustration of the atomic motions during the synchroshear process. The upper gray block including all the big A atoms in the top layer of the three-layer stack moves in the plane of the paper (y–z plane) to the left by a distance b as shown by the red arrow, while simultaneously the small B atoms move on the vertical plane marked in blue (x–y plane) at 60° to the –y direction as shown by the blue arrow, producing the final configuration shown in (b) (figure and text adapted with permission from [356])
Successful direct observation of synchroshear-induced stacking faults in Laves phases by high-resolution scanning transmission electron microscopy (STEM) was reported for the first time by Chiswick et al. [356]. Since then synchroshear-induced stacking faults in different deformed C14 and C15 Laves phases were observed by various authors [361,362,363,364]. Figure 19 shows a synchro-Shockley dislocation in C14 HfCr2 Laves phase with the dislocation core in the center of the image as explained in the figure caption [356].

Atomic resolution STEM Z-contrast image of a [\( 11\bar{2}0 \)] projection of a synchro-Shockley dislocation bounding a stacking fault in the C14 variant of the HfCr2 Laves phase. The fault comes in from the right (indicated by the arrows) and terminates at the dislocation core in the center of the image. The Burgers circuit (indicated by the white line) made from Hf atoms failed to close. The schematic below the image, obtained directly from the STEM image, shows a proposed core structure (adapted with permission from [356])
Laves phases (similar to most other intermetallic phases) are brittle at room temperature and exhibit a transition from brittle-to-ductile behavior at high homologous temperatures T/Tm (Tm is the melting temperature) in the range 0.6–0.75 [127, 365,366,367]. Therefore, many transition metal Laves phases remain brittle up to temperatures above 1000 °C. For example, in case of the intensively studied C15 Laves phase NbCr2, this BDTT (brittle-to-ductile transition temperature) is about 1200 °C and plastic deformation of macroscopic bulk samples is only observable in experiments performed above this temperature [367,368,369]. A chance to study plastic deformation of such brittle phases at room temperature came up with the establishment of micromechanical testing methods in materials science, see, for example, [370]. Micropillar compression tests of single-crystal cubic C15 and hexagonal C14 and C36 Laves phases revealed the occurrence of plastic deformation by basal and, in case of the hexagonal crystals, also non-basal, prismatic and pyramidal slip [364, 371,372,373,374]. An example for a plastically deformed C14 NbCo2 micropillar is shown in Fig. 20 [373].

SEM image (side view) of a room-temperature-deformed, single-crystal Laves phase (C14 NbCo2) micropillar showing slip traces. The micropillar was compressed vertically, the arrows indicate the activated slip plane and direction (adapted with permission from [373])
As already discussed in Sect. 3.2 (‘point defects’), many Laves phases exhibit extended homogeneity ranges. Regarding the effect of compositional deviations from the ideal, stoichiometric AB2 value, the mechanical properties of Laves phases reveal a very interesting behavior. The first detailed investigations on the composition dependence of mechanical properties of C14 MgZn2 were performed by Paufler et al. during the 1970s [5, 375,376,377,378,379]. Both room-temperature microhardness [375, 376, 378] and yield stress at 350 °C [379] reveal softening at off-stoichiometric compositions (Fig. 21a). Also the stationary creep rate increases with deviating from stoichiometry (Fig. 21b) [378]. This is attributed to a strong increase of the density of mobile dislocations, which overcompensates the simultaneous decrease of dislocation velocity (Fig. 21b) [5, 378].

Composition dependence of mechanical properties of single-phase C14 MgZn2 Laves phase: a Microhardness HV (room temperature) [378] and upper yield stress σu (at 350 °C) [379] showing macroscopic softening for off-stoichiometric compositions. b Stationary creep rate \( \dot{\varepsilon } \) (350 °C, shear stress 10 MPa) [378], dislocation density d [378], and dislocation velocity v (399 °C) [377] also exhibit extrema at the stoichiometric composition (adapted with permission from [5])
Transition metal Laves phases, which are well known for frequently possessing extended homogeneity ranges (cf. Sect. 3.2.1), are perfectly suited to examine if this particular behavior is specific for C14 MgZn2 or is a more general characteristic of Laves phases. Regarding the effect of deviation from stoichiometry on the hardness, contradicting results were reported in the literature. While hardening by off-stoichiometry was reported in two studies (for C15 ZrCr2 [380], C15 NbCr2, C15 NbCo2, and C14 NbFe2 [151]), other experimental investigations confirm the aforementioned softening behavior, for example for C15 TiCr2 [381], C15 ZrCr2 [382], C15 NbCo2 [383, 384], C14 NbFe2 [383, 385], and C15 HfCo2 [386].
As already discussed in Sect. 3.2, the point defects needed for stabilization of off-stoichiometric compositions in MgZn2 and in the majority of the transition metal Laves phases are antisite atoms on both sides of the stoichiometric composition. In general, such kind of substitutional defects are expected to result in hardening at low homologous temperatures as is well known for metals (e.g., [387]) and for intermetallics [388,389,390,391]. Solid solution hardening is generally explained as originating from elastic interactions of the solid atom or defect with dislocations. Obviously, the presence of defects in Laves phases instead tends to soften the material in many systems, meaning that the defects can promote the deformation process by facilitating the movement of partial dislocations in the synchroshear process [386]. Another idea discussed by Chen et al. [386] is related to the changes in chemical bonding resulting from wrong site occupations. The strength of Laves phases originates from the strong bonding of dissimilar atoms in the ideal structure. Replacing an A atom by a B atom (or vice versa) will locally increase the number of B-B (or A-A) bonds, i.e., more metallic bonding is created locally, by which deformability might be improved. However, if this argument is correct, the effect should be even more pronounced in B2-type intermetallic compounds, because in this bcc-based AB structure the replacement of, e.g., an A by a B atom changes the number of dissimilar next neighbors from eight to zero. Against this expectation, it was clearly shown that for B2 compounds constitutional defects result in solid solution hardening (e.g., [388, 389]) indicating that this simple chemical bonding model is not suited to explain the observed softening behavior.
In order to study in a systematic way the dependence of mechanical properties of single-crystalline Laves phases on composition, crystal orientation, and crystal structure, Luo et al. [364, 384, 392,393,394] focused on the binary system Co-Nb, in which the Laves phase NbCo2 exists as stable phase with C36, C15, and C14 structure in adjacent phase fields (from approximately 24.0 to 25.0, 25.0 to 34.3, and 35.5 to 37.0 at.% Nb, respectively). By producing diffusion couples, they obtained several hundred µm thick layers of Laves phase covering the entire homogeneity ranges of the three polytypes. Electron channeling contrast imaging revealed low dislocation densities d of the order of 10−11 m−2 in the C15 composition range around the stoichiometric composition, while d increases by one order of magnitude close to the boundaries to the hexagonal variants C36 (near 25 at.% Nb) and C14 (near 35 at.% Nb) [384]. The occurrence of widely extended stacking faults in these areas close to the transitions C15-C36 and C15-C14 indicates a significant decrease of the stacking fault energy (SFE). Hardness and elastic modulus, which were obtained from nanoindentation experiments along the concentration gradient, show a similar compositional behavior with continuously decreasing values at off-stoichiometric compositions near the boundaries of the homogeneity range. This implies reduction of the Peierls stress [384] as had already been suggested as explanation for the above-described behavior of MgZn2 [5]. Strength (by micropillar compression tests [364]) and toughness (by microcantilever bending tests [393]) of NbCo2 were also studied as a function of composition using the same diffusion couples. The critical resolved shear stress behaves very similar to the hardness with highest values in the central region of the C15 homogeneity range and a decrease when approaching the off-stoichiometric boundaries, which is explained by the reduction of the shear modulus and stacking fault energy [364]. The microcantilever bending tests revealed a linear elastic fracture behavior for all compositions. Within the experimental accuracy, the fracture toughness was found to be independent of chemical composition and crystal structure type of the NbCo2 Laves phase [393].
The crystal orientation of single-crystalline samples was found to have only a negligible effect on strength and hardness in micromechanical testing. Although the pillar orientation can influence the activated slip systems, the energy barriers of dislocation nucleation and motion, which determine the strength of the NbCo2 micropillars, are independent of orientation [364]. Nanohardness values measured in differently oriented grains of polycrystalline cubic C15 as well as hexagonal C14 and C36 NbCo2 alloys are shown in Fig. 22. The hardness values of the C15 alloys with 25.6 at.% (b) and 33 at.% Nb (c) as well as those of the C36 alloy (a) are independent of orientation within the given accuracy. For the C14 NbCo2 Laves phase (d), the hardness measured perpendicular to the basal plane was found to be slightly higher (in the order of 5%) than in the other orientations [384]. A similar, slight anisotropy in the hardness of hexagonal C14 Laves phase with highest values for [0001] orientation was also reported by Paufler [5] for MgZn2 and CaMg2 Laves phase.

Inverse pole figures showing nanohardness values of cubic C15 (b, c) and hexagonal C14 (d) and C36 (a) NbCo2 Laves phase for various crystal orientations (adapted with permission from [384])
The role of the crystal structure type of a Laves phase for the mechanical properties can only be studied in an indirect way, because a change of the stable structure type is always connected with a change in composition (or temperature). Nevertheless, the above-described investigations of cubic C15 and hexagonal C14 and C36 NbCo2 Laves phase indicate that there is no effect of crystal structure. In all cases, a continuous, smooth change of properties is observed when crossing the boundaries from the C15 to C36 or C15 to C14 homogeneity ranges. This is true for the continuously decreasing values of hardness, elastic modulus [384], and critical resolved shear stress [364] as well as for the composition-independent fracture toughness values [393].
Functional applications
Hydrogen storage materials
Intermetallic phases as effective hydrogen storage materials are discussed since the late 1960s, for example, as fuel tanks in automotive applications [395, 396]. Since then and until today, metal hydrides and their potential application for hydrogen storage are a topic of great interest as described in a wealth of review articles, see, for example, [397,398,399,400] to name but a few. Besides offering a safe and efficient method to store hydrogen, metal hydrides are also materials for the negative electrode of nickel–metal hydride (Ni/MH) batteries. A recent compilation of articles related to the topic ‘Nickel Metal Hydride Batteries’ [401] gives an excellent overview about the status of development of this type of material and also contains a comprehensive review of Laves phase metal hydrides for battery applications [402].
Besides AB5 compounds and bcc-metal (mostly Ti and Zr)-based alloys, the AB2 Laves phases are the most important interstitial hydride-forming materials [402, 403]. It was recognized already in very early studies that Laves phases can absorb relatively high amounts of hydrogen (up to 2 wt%) and possess high hydrogenation/dehydrogenation cycle stability [404,405,406]. Compared to the AB5 compounds, where especially LaNi5 is widely used in commercial applications as the negative electrode in Ni/MH batteries, most of the Laves phases show faster kinetics, longer life times and have relatively low costs. However, a major problem for application of binary Laves phase alloys is the very high stability of their hydrides at room temperature leading to poor electrochemical properties in alkaline electrolytes. As the hydriding properties of Laves phases very sensitively depend on composition, there were many attempts in the literature to overcome the stability problem and improve their properties by diverse alloying concepts [397,398,399].
As Laves phases belong to the class of tetrahedrally close-packed (t.c.p.) phases, all possible interstitial sites have four nearest neighbors resulting in either A2B2, AB3, or B4 tetrahedral interstices, see Fig. 23 [407]. The preferential occupation of particular interstitial sites of the C14 and C15 Laves phase lattices by hydrogen atoms and the relative stability of atomic hydrogen at various interstitial sites as defined by the bonding energies is the topic of several investigations, see, for example, [407,408,409,410]. According to some simple criteria formulated by Westlake [411], interstices in stable hydrides must have minimum radii of 40 pm and the hydrogen–hydrogen distances must be a minimum of 210 pm. Shoemaker and Shoemaker [408] performed a detailed theoretical analysis of the Laves phase crystal lattice with respect to the different interstitial sites and found a maximum possible occupancy of about six hydrogen atoms per AB2 formula unit. Already before, Jacob et al. [412] had studied Zr-based binary Laves phases and developed a phenomenological model based on short-range ordering effects. Their results indicated somewhat lower hydrogen solubilities as was later confirmed by neutron diffraction experiments [413,414,415,416,417] and more recent ab initio total energy calculations [410]. As only particular interstitial sites are occupied, ordering of hydrogen on such sites may occur resulting in crystallographic symmetries of the hydride different from that of the parent Laves phase. An overview of the crystallographic aspects of hydrogen ordering in hydrides of Laves phases is given in a recent review by Kohlmann [407].

The three types of tetrahedral interstices in the crystal structure of a cubic AB2 Laves phase (C15 type) (A: large, blue spheres; B: small, red spheres) offering possible positions for hydrogen: A2B2 (light gray), AB3 (middle gray), and B4 (dark gray) tetrahedral interstices. The number of such interstices per unit cell are 96, 32, and 8, respectively. The 8 B4 tetrahedra are marked with green faces (with the exception of the one that is dark grey). For reasons of clarity, only one interstice of each kind is drawn (reproduced with permission from [407])
Regarding the effect of the type of crystal structure (hexagonal C14 vs. cubic C15) of the Laves phase on the hydrogen storage properties, partially contradicting observations were reported [418,419,420,421,422,423]. The stable structure type cannot be changed without changing chemical composition (or temperature). As it was found that such variations of composition (type and concentration of elements) can have a pronounced effect on the hydrogen storage performance, it is difficult to draw a general conclusion on the effect of crystal structure. Type and number of interstitial sites in cubic and hexagonal Laves phase are identical and it can be assumed that the type of crystal structure does not or only marginally affect the hydrogen storage behavior.
The first detailed investigations on Laves phases as potential hydrogen storage materials date back to the 1970s, when the hydriding properties of ZrFe2- and ZrCo2-based cubic C15 Laves phases with small ternary additions of either Al, V, Cr, or Mn were tested [405, 412, 424]. Studies on these Laves phase systems were continued for more than twenty years [425,426,427], but problems with an either too low hydrogen storage capability or a too high stability of the hydrides could not be overcome.
After these first, at least partially promising studies on hydrogen storage properties of this class of compounds, a huge amount of projects started on various other AB2 Laves phases. Some of these investigations focused on light-weight Laves phases such as (Mg,Ca)Ni2 [428, 429], CaLi2 [430], or CaMg2 [431,432,433], but their hydrogenation properties were found to be insufficient from the viewpoint of applications. Until today clearly the most promising and by far most intensively investigated Laves phases are based on Zr and/or Ti as the large A metal, while the B lattice sites are occupied by one or—in most cases—more of the 3d metals Co, Cr, Fe, Mn, Ni, V. The compositions of both the alloys with mainly Zr on the A sites (e.g., [434,435,436,437,438,439,440,441]) and with mainly Ti on the A sites (e.g., [442,443,444,445,446,447,448,449]) became more and more complex. Many of the currently discussed alloys for applications contain six to nine components, where in some cases small amounts of main group elements such as Al or Sn (e.g., [421, 450, 451]), or lanthanides [452] were added. More recently, also some multi-component, equiatomic or nearly equiatomic alloys (frequently called ‘high-entropy alloys’ (HEAs)) such as CoFeMnTixVyZrz (0.5 ≤ x ≤ 2.5, 0.4 ≤ y ≤ 3.0, and 0.4 ≤ z ≤ 3.0) [453], CrFeNiTiVZr [454] and CrFeMnNiTiZr [455] were studied with respect to their hydrogen storage properties (cf. Sect. 5.6.5). These alloys consist nearly completely of C14 Laves phase, but do not show any especially remarkable behavior compared to other Zr/Ti-based alloys [453,454,455]. According to Ovshinsky’s concept of compositional disorder [456], the variety of elements in multicomponent alloys can offer a multitude of hydrogen bonding possibilities increasing the hydrogen storage capacity and improving the catalytic activity.
Moreover, it was found that deviations from the stoichiometric composition strongly affect the hydrogen storage properties [439, 457,458,459]. Young et al. [460] studied the effect of vanadium excess in C14 Laves phase alloys for Ni/MH battery applications. Vanadium atoms usually occupy B atom sites in Zr- and Ti-based Laves phase, but due to their relatively large atomic radius, they can partially move from B to A sites in case their number exceeds the available B sites [461]. Such a substitution of vanadium on A sites was reported not only to increase the degree of disorder but also to provide more hydrogen interstitial sites for reversible hydrogen storage [460, 462]. More recently, this strategy of partial substitution on the A site was adopted to C15 Y(Fe,Al)2 alloys by replacing 10% of Y by Zr, Ti, or V resulting in a remarkable increase of the hydrogen desorption capacity [463].
Besides compositional disorder and off-stoichiometry of a Laves phase, a multiphase microstructure was found to be another important characteristic of Laves phase-based hydrogen storage alloys needed to qualify them for application [456]. The so-called ‘Laves-phase-related bcc solid solution’ concept is an early example for multiphase hydrogen storage alloys [464, 465]. Such alloys are composed of a bcc solid solution based on Ti, V, or Zr coexisting with a Laves phase. The bcc solid solutions can reach high hydrogen storage capabilities, but their activation and the very low plateau pressures are a problem. However, in combination with an easily to activate Laves phase, good hydriding properties can be achieved as was especially demonstrated for bcc-V solid solutions in combination with Laves phase [466,467,468,469,470]. It should be mentioned that in addition to the AB2 Laves phases also alloys based on the structurally closely related phases A2B7 (Ce2Ni7 type) and AB3 (PuNi3 type), which contain fragments of the Laves phase structure (cf. Sect. 3.5.3), were discussed as electrode materials for batteries, see, for example, [471,472,473].
Regarding industrial applications, a lot of effort was put into the development of alloys for the negative electrode in rechargeable Ni/MH batteries for electrical vehicles. A class of (Ti,Zr)(V,Cr,Mn,Ni)2 alloys consisting mainly of a C14 and a C15 Laves phase was found to have excellent properties in tests under application conditions [456, 474,475,476,477].
The only commercially available Laves phase-based hydride-forming class of alloys, which is already successfully in application, is so-called Hydralloy [450, 478,479,480,481,482,483]. Hydralloy is always based on a C14 Laves phase containing Ti and Mn in a 1:1.5 ratio as main constituents. Commercially available alloys are Hydralloy C2 (Ti0.98Zr0.02Mn1.46V0.41Cr0.05Fe0.08), C51 (Ti0.95Zr0.05Mn1.48V0.43Fe0.08Al0.01) and C52 (Ti0.955Zr0.045Mn1.52V0.43Fe0.12Al0.03). As for several battery applications a high-dynamic tank operation is required, storage materials with high heat conductivity are needed. For this reason, pelletized composites of Hydralloy C5 and expanded natural graphite (ENG) were developed, see Fig. 24 [481, 483,484,485].

Hydraloy-ENG pellet consisting of a mixture of Hydraloy C52 powder and 5 wt% expanded natural graphite (ENG). A comparison of the as-compacted state to that after 85 hydrogenation cycles reveals that shape and dimensions are kept and that the pellets preserve their mechanical integrity throughout cycling. Graphite is added to ensure high thermal conductivity. The diagram in the middle shows the uptake and release of hydrogen during a typical hydrogenation-dehydrogenation cycle (adapted with permission from [481])
Hydralloy has only a medium gravimetric hydrogen storage capacity (about 1.5 wt% H2), but a comparably high volumetric hydrogen storage capacity of about 80 g-H2 l−1. Moreover, it can be easily hydrogenated at moderate hydrogen pressures in the temperature range −20 °C to +100 °C. Therefore, even though it is not well suited, for example, for automotive applications, Hydralloy is in operation in some niche applications, where the high weight of a potential hydrogen tank does not pose a problem [481, 482]. An example for that is the successful use of Hydralloy as hydride tank material for maritime fuel cells in the German Navy submarines of the U212A type (since 2003) and its export version U214 (since 2004) [400, 482]. Another application, where the high weight is even an advantage, are electric forklifts. In this case, heavy counterweights are essential. Series of performance tests with a 3-ton electric forklift were carried out yielding promising results [486,487,488]. Another successful demonstration of the applicability of Hydralloy C5 was its usage as hydrogen tank material on a fuel cell-powered, 4-ton mining locomotive [489]. Hydralloy C5 was also tested as hydrogen tank for fuel cell city cars. Promising intermediate results were reported, but an application is not yet to be expected [490]. More recently, a metal hydride air-conditioning system for fuel cell vehicles consisting of two plate reactors filled with Hydralloy C2 and coupled to a polymer electrolyte membrane fuel cell was developed and suggested as technology to reutilize the compression work in the hydrogen pressure tank [491, 492].
Wear- and corrosion-resistant materials
Tribaloy
The very high hardness of high-melting transition-metal-based Laves phases can be effectively used not only to strengthen high-temperature structural materials, but also for applications requiring a good wear and friction behavior. Tribaloy is the registered trade name (Deloro Stellite Holdings Corporation) for a family of wear-resistant Co- (or Ni-) based Co/Ni-Mo-Cr–Si alloys with very high Mo contents (up to 35 wt%). These alloys were introduced in 1974/5 [493,494,495,496] and today are used extensively especially as coating materials in corrosive environments. Tribaloy alloys contain high volume fractions of up to 65 vol.% C14 Mo(Co,Cr,Si)2 Laves phase embedded in a softer matrix of Co solid solution or eutectic (which is Co solid solution + Laves phase). A typical microstructure is shown in Fig. 25 (taken from [497]). Due to the high content of hard Laves phase and with Co as main constituent of the material, the Tribaloy alloys show a combination of excellent resistance to high-temperature wear, galling, and corrosion, i.e., the material is especially used for situations where strong wear occurs in combination with high temperatures and corrosive media. The high Mo contents result in good dry-running properties making these alloys especially suitable for use where lubrication is a problem. Maximum application temperatures are in the range 800 to 1000 °C [493,494,495,496, 498,499,500].

Microstructure of T-800 Tribaloy coating after laser deposition on a Ni-based superalloy substrate. The Tribaloy microstructure consists of large, primary Laves phase particles in a matrix of Co solid solution and Co + Laves phase eutectic (reproduced with permission from [497])
At room temperature, the fracture toughness of Tribaloy alloys is low. The matrix of as-cast Tribaloy contains both hcp- and fcc-Co, and this can be strongly varied by the cooling conditions and subsequent heat treatments. It was shown that the faster the cooling rate the greater the volume fraction of primary Laves phase and the larger the proportion of the fcc Co solid solution. With respect to the hardness and fracture behavior, the best combination of room-temperature properties was obtained with the highest volume contents of fcc solid solution, which can also be stabilized by additions of 5–15 wt% Fe [501,502,503].
Until today, only very little work related to further alloy development of this class of wear-resistant alloys was performed. Table 5 shows a complete list of the commercially available Tribaloy alloys. Out of the first-generation Tribaloy alloys (Co-based T-100, T-400, T-800, and the Ni-based counterpart T-700), especially T-400 and T-800 entered into industrial application and were later on starting materials for development of improved alloys. About 30 years after its introduction in the literature, the first-generation Tribaloy T-400 was modified to improve its toughness and corrosion behavior. Tribaloy T-400C contains an increased amount of Cr (14 instead of 8.5 wt%). Immersion corrosion tests in various oxidizing and reducing acids demonstrated an excellent corrosion resistance superior to the T-400 alloy [504, 505]. The newly developed Tribaloy T-401 has a lower Mo and Si content combined with an increased Cr content. Due to the significantly reduced Mo content, the primary phase is no longer the Laves phase but the Co solid solution. This results in a higher ductility but lower hardness and wear resistance compared to T-400 [505, 506]. In tests on the corrosion resistance in molten Zn-Al baths, Tribaloy T-401 performs better than T-400 and T-800 [507]. The increased Cr contents in T-400C and T-401 lead to the formation of Cr-oxide layers, which are not only beneficial for the oxidation behavior, but also for the high-temperature wear resistance [508]. While very thin oxide films on T-400C specimens were found to be removed from the surface during wear exposure, thicker oxide layers resisted this attack and were observed to be squeezed and embedded into the specimen surface, thereby enhancing the hardness and wear resistance of the surface [509].
Tribaloy T-800 has a lower Co content compared to T-400 and, therefore, contains a higher amount of Mo(Co,Cr,Si)2 Laves phase. This results in a significant hardening and further improvement of the wear behavior in dry conditions as was, for example, demonstrated by hardness measurements and sliding wear tests (ball-on-disk and block-on-ring configurations) of Tribaloy T-800 coatings deposited by laser cladding on a stainless steel (AISI 304) substrate without using lubrication [510]. However, the brittle nature of Laves phases promotes the formation and propagation of cracks in T-800, and this increased cracking susceptibility complicates the development of coatings [511]. Tribaloy T-900 is a modification of the T-800 alloy with a reduced volume fraction of hard Laves phase by replacing some of the Laves phase forming alloying elements (Co, Mo, Si) with Ni. This reduction in Laves phase content results in a higher ductility and fracture toughness and improved crack resistance [511].
Besides mechanical properties, the oxidation behavior of Tribaloy alloys at high temperatures is of central importance for their applicability. A comparison of the oxidation behavior of Tribaloy T-400 and T-800 during long-time tests (up to 1000 h) at 900 °C in synthetic air revealed that T-800 provides a much better oxidation resistance than T-400. Due to the higher Cr content in T-800, dense Cr2O3 layers formed after long times, while the scale on T-400 additionally contains the Co-rich spinel Co2CrO4 and a much smaller fraction of Cr2O3. Moreover, the oxides were observed to protrude deeper into the Laves phase [512]. Short-time (< 60 h) isothermal oxidation tests of T-800 confirmed a good oxidation behavior at 800 °C indicated by a parabolic mass gain with formation of a complex oxide scale. However, a very poor oxidation behavior with a linearly increasing mass gain was observed in similar tests at 1000 °C resulting from the formation of voids and some loose internal oxide SiO2 at the interface between Cr2O3 and the alloy [513]. An application temperature of 1000 °C, therefore, seems to be too high for the T-800 alloy. Cyclic oxidation tests comparing the T-900 and T-401 alloys revealed a much better performance of T-900. This was attributed to the formation of dense continuous Cr2O3 layers (with an upper thinner continuous layer of CoCr2O4 and NiCr2O4 oxides). For the T-401 specimen, severe spallation after 3 cycles (45 h) between room temperature and 1000 °C was observed [514]. Modification of the Tribaloy T-700 [515], T-800 [516], and T-900 [517] alloy compositions by additions of Al (5 wt%) and Y (0.5 wt%) resulted in a significant improvement of the cyclic oxidation resistance due to the formation of protective, compact and continuous external layers of Al2O3 at 800 and 1000 °C [515,516,517]. Moreover, the added Y was reported to enhance the adherence of oxide scales during sliding wear [517].
Tribaloy coatings (nearly exclusively T-800, and today often less precisely termed CoMoCrSi coatings) are deposited either by laser cladding techniques [497, 510, 511, 518] or by spraying methods such as the high velocity oxygen fuel (HVOF) thermal spray technique [519,520,521,522,523,524,525,526] or atmospheric plasma spraying (APS) [527]. Tobar et al. [511] report that, when compared to the spraying methods, laser cladding gives coatings of higher quality on steel substrates as it produces dense and pore-free layers with finer microstructure and better adherence with the substrate. HVOF-sprayed layers of T-800 alloy were found to be mostly amorphous in the as-deposited state resulting especially in a worsened wear behavior. However, after short-time heat treatment for 1 h at 600 °C, the Laves phase and Co solid solution crystallized again and the coating showed the expected beneficial protecting properties [523, 524].
Besides pure Tribaloy coatings, also composite coatings consisting of a tin-bronze matrix reinforced by T-401 alloy particles were successfully deposited by HVOF spraying [528, 529]. Bushings of a journal bearing coated with the composite were found to exhibit superior tribological properties to the leaded tin/bronze bushings [528]. Likewise, polytetrafluoroethylene (PTFE)-based composite coatings containing T-401 Tribaloy particles were developed for sliding bearing applications [530].
In general, Laves phase-dominated Tribaloy coatings have proven to be suitable for applications in various industrial fields such as aerospace, turbine, oil, pump, energy, and mining industry [525]. They are candidates for hard-facings of valve spindles and seat rings in natural gas engines [531, 532], they were successfully tested for the repair of gas turbine shafts [519] and low Cr-Mo steel components used in steam circuits in thermal power stations [518], and they are currently in use for the protection of fuel nozzles against fretting wear in high-temperature gas turbine engines [521].
Other Laves phase-based materials
Besides Tribaloy, also other Laves phase-based materials were suggested as wear- or corrosion-resistant coatings. Very similar to the Tribaloy composite concept of combining a very hard, high-melting Si-stabilized C14 Laves phase with a more ductile metal or intermetallic phase, a class of wear-resistant ternary alloys was developed which are composed of an A2B3Si-type (i.e., A(B0.75Si0.25)2) C14 Laves phase with A = Ti, Mo or W and B = Co or Ni in combination with varying second phases for toughening. This group of alloys comprises Ti(Ni0.75Si0.25)2 Laves phase composites with cubic B2 NiTi [533,534,535,536,537] or hexagonal Ni3Ti [538], Mo(Ni0.75Si0.25)2 Laves phase with bcc Mo solid solution [539] or with orthorhombic NiSi [540,541,542], W(Ni0.75Si0.25)2 Laves phase with bcc W solid solution [543, 544] or tetragonal W5Si3 [535, 545], and Mo(Co0.75Si0.25)2 Laves phase with fcc Co solid solution [546,547,548,549]. The combinations of hard but brittle Laves phase with a toughening second phase such as the metal solid solutions allow the adjustment of properties via variation of the phase contents. Laser-clad coatings were produced on various substrates and wear and corrosion tests revealed very good properties. However, to the best of the authors’ knowledge these alloys did not enter into industrial applications.
To protect oxidation and corrosion-sensitive Ti- and TiAl-alloys, it was suggested to coat them with Ti(Cr,Al)2 Laves phase which is known to form a dense alumina scale on the surface, see Sect. 5.6.3.
High-entropy alloys consisting of a tough disordered solid solution and a hard Laves phase (see Sect. 5.6.5) were tested as coatings on steels [550,551,552,553,554,555,556] and pure Ti [557]. Because of the high Laves phase contents, the coatings were reported in all cases to show excellent wear and oxidation behavior.
Magnetic materials
Magnetostrictive applications
Laves phase rare earth–iron compounds AFe2 (A = rare earth metal) have attracted a lot of attention due to their extraordinary magnetic properties. The large anisotropic 4f charge density of the rare earth A3+ ions and the strong Fe-Fe and A-Fe exchange interactions result in large magnetic anisotropy and magnetostriction up to room temperature [558,559,560], see also Sect. 3.5.1. The field dependence of the magnetostriction for the AFe2 compounds (A = Sm, Tb, Dy, Er, Tm) is shown in Fig. 26. The C15 Laves phases TbFe2 and SmFe2 reach giant room-temperature magnetostriction coefficients exceeding those of traditional magnetostrictive materials by one order of magnitude [271, 561,562,563].

Room temperature magnetostriction of rare earth-Fe2 Laves phase polycrystals (redrawn from [558]). λ||–λ⊥ denotes the fractional change in length as an applied field is rotated from perpendicular to parallel to the measurement direction
A maximum magnetostriction/anisotropy ratio, λ/K1, was found for the pseudobinary compound TbxDyl-xFe2 with x = 0.3 [558, 559, 561]. This Laves phase compound with an exact composition Tb0.3Dy0.7Fe1.93 was developed by the Naval Ordnance Laboratory (NOL) and registered under the trade name Terfenol-D. It offers the highest strain of any magnetostrictive material, which makes it an excellent choice for applications in magnetomechanical sensors and actuators, for example, as acoustic and ultrasonic transducers [558, 559, 564,565,566,567,568,569,570,571,572,573,574]. In a more recent study, further improvements of its magnetostrictive properties by partial substitution of Dy by Nd were reported [575]. In order to reduce the brittleness of the material, low-melting Dy-Cu alloy was added to cover the grain boundary with a ductile (Dy,Tb)Cu phase. While the magnetostriction remained at a high value, the bending strength was found to be significantly improved [576]. An overview of the manifold applications of Terfenol-D is given by Olabi and Grunwald [565], for reviews on the magnetostrictive properties, see [577,578,579]. The spectrum of applications ranges from magnetostrictive underwater sound transducers in naval sonar systems [564] to more recent developments of magnetostrictive–piezoelectric composites of Terfenol-D and PZT (Pb(Zr,Ti)O3) for energy harvesting to supply low-power electronics [573, 574].
The giant magnetostrictive effects in TbFe2 leads to a distortion of the cubic C15 structure resulting in a symmetry reduction to rhombohedral \( R\bar{3}m \) (hR18) symmetry [270]. The same is true for other strongly magnetostrictive C15 rare earth Laves phases AFe2 (A = Sm, Tb, Dy, Ho, Er, Tm) [580]. This kind of symmetry reduction in C15 Laves phases is also discussed above in Sect. 3.5.1. In case of SmFe2, spin reorientation and a change of the direction of easy magnetization from [111]cubic to [110]cubic leads to a transformation from the rhombohedral \( R\bar{3}m \) room-temperature structure to an orthorhombic Imma low-temperature structure at 200 K [271].
Magnetocaloric applications
Rare-earth-containing Laves phases AB2 (with A = rare earth metal, B = Al, Co and Ni) belong to the magnetic materials which exhibit a large or unusual magnetocaloric effect (MCE) [581]. With respect to applications, there is a lot of discussion about utilizing materials with a large MCE for magnetic refrigerators that could replace conventional refrigerators which are based on vapor–gas cycles [582, 583]. ACo2 and AAl2 (A = Er, Ho, Dy) Laves phase compounds show MCEs at very low transition temperatures and were suggested as magnetocaloric materials for hydrogen liquefaction [584,585,586,587]. Magnetization and heat capacity measurements of the C15 Laves phase TbMn1.6Fe0.4 indicate that this phase could be a promising candidate for magnetic refrigeration in the temperature range 70 to 162 K [588]. Studies on the MCE and magnetostrictive behavior of a series of single-phase Laves phase alloys Tb0.2Dy0.8-xGdxCo2-yAly (x = 0.3, 0.4, and 0.5; y = 0 and 0.1) did not indicate an improvement of properties by the compositional variations [589]. Magnetocaloric Laves phases with transitions near room temperature that could be suitable for application as magnetic refrigerators were not reported.
Hard magnetic applications
Pure Laves phases are unsuitable as permanent magnets, because they tend to have low magnetization, low Curie temperature, and in the cubic structure low anisotropy [590]. Among the few Laves phases that might be an exception to this rule, C14 FeBe2 was identified as an interesting candidate. Powders of this ferromagnetic phase were investigated revealing a comparably high Curie temperature (823 K) and magnetic moment (1.95 μB/Fe), but—most likely because of the toxicity of Be—no further investigations were reported [591, 592]. More recently, hard magnetic properties of off-stoichiometric Zr27Fe73-xSix (0 ≤ x ≤ 15) and Zr33−yFe52+ySi15 (0 ≤ y ≤ 11) C14 Laves phases [593], nanocrystalline C14 Ti0.75Zr0.25Fe2+x (x = 0–0.4) and Ti0.75−yByZr0.25Fe2.4 (y = 0–0.35) [594], and melt-spun nanocomposites based on Fe-enriched out-of-equilibrium C14 NbFe2+x and TaFe2+x Laves phases [595] were studied. However, in all cases these materials were found not to be suitable for application as permanent magnets.
Laves phases are not only unsuitable as permanent magnets, but their presence in magnets may even be detrimental. An example is the role of C15 Laves phase CeFe2 in Ce-based hard magnetic materials. As an alternative to the well-established but expensive, rare-earth-based (Nd,Dy)-Fe-B permanent magnets, the hard magnetic phase CeFe11Ti is currently under discussion. The main challenge for production of this new type of hard magnetic material indeed is the formation of the more stable C15 CeFe2 Laves phase, which tends to dominate the microstructure and retards the crystallization of the desired hard magnetic phase. In experimental investigations, the CeFe11Ti phase was only observed at high temperatures above 1000 K. Below this temperature, only the phases C15 CeFe2 and Ce2Fe17 were detected. This is confirmed by ab initio free-energy calculations, which reveal that the presence of the CeFe2 Laves phase suppresses the formation of CeFe11Ti up to 700 K [596, 597]. In Ce-based Ce-Fe-B alloys containing hard-magnetic Ce2Fe14B, partial substitution of Ce by La was found to suppress the Laves phase CeFe2 and to promote the formation of (Ce,La)2Fe14B with increased saturation magnetization and Curie temperature [598].
Superconducting materials
Together with the A15 (Cr3Si-type) and B1 (NaCl-type) crystal structures, the C15 and C14 Laves phases form the group of the most favorable compound structures for the occurrence of superconductivity [599, 600]. The observation of superconductivity in some Laves phases ARu2 (A = Sc, Y, La, Ce) was already described as early as in 1958 [601]. Since then, a very large number of other superconducting Laves phases were detected. However, the superconducting transition temperatures Tc in most cases are below 5 K and critical fields Hc are low, which means that these compounds are not suited for applications. The by far highest transition temperatures among the Laves phases were found for the C15 compounds ZrV2 (8.8 K [600]) and HfV2 (9.6 K [602]). Interestingly, both of them show a structural transition below about 120 K [277, 279, 603], where the C15 lattice is distorted in a way that it becomes orthorhombic in case of HfV2 [279] and rhombohedral in case of ZrV2 [277], see also Sect. 3.5.1. Such structural instabilities are known to occur especially in high-Tc superconducting phases, but the reason for this relation is not well understood [603, 604]. Experimental results for the pseudobinary system HfxZr1−xV2, indicate an inverse relation between superconducting transition temperature Tc and the structural transformation temperature Ts, see Fig. 27 [604]. Mixing Hf and Zr on the A lattice site results in an increase of Tc and the record holder is Hf0.5Zr0.5V2 with a Tc of 10.1 K [605].

Superconducting transition temperature Tc and structural transformation temperature Ts as a function of Zr/Hf ratio in the C15 (Hf,Zr)V2 Laves phase (adapted with permission from [604])
Because of its very good superconducting properties (high Tc, upper critical fields of µ0Hc2 = 21 T and critical current densities of about Jc = 1 × 105 A/cm2 at 13 T and 4.2 K) and as it is less brittle than the high-Tc A15 compounds (e.g., Nb3Al) and shows high tolerance to neutron irradiation, (Hf,Zr)V2 Laves phase attracted a lot of interest for applications as high-field superconductor in fusion reactors [606,607,608,609]. To further improve the superconducting properties and especially the workability, replacement of Zr by Nb and addition of Ti were also tested [610, 611]. Various fabrication methods for the production of (Hf,Zr)V2 tapes and wires were applied, all of them are multi-step composite processes starting, for example, from Hf-Zr binary alloy rods and V-based tubes or from elemental Hf, Zr, and V metal powders. Figure 28 shows an example of material in an intermediate production step. The final steps were either cold-drawing to wires or cold-rolling to tapes always followed by high-temperature heat treatments in the range of 1000 °C to transform the phase mixtures to single-phase Laves phase material [606, 612,613,614]. To avoid the high-temperature annealing step, which often resulted in an only incomplete transformation leaving over unwanted phases, an alternative method was developed applying a rapidly heating/quenching process [607,608,609].

Intermediate step in the production of superconducting (Hf,Zr)V2 wires showing a cross section of a 1 mm thick, 1425-core Hf-Zr-V wire with V-1at.%Hf alloy matrix and Zr-45at.%Hf cores. To obtain this structure, Hf-Zr rods had been inserted into drilled holes in a V-1at.%Hf alloy matrix, followed by cold-drawing into wires, cutting into short pieces and packing into a V-1at.%Hf alloy tube (reproduced with permission from [606])
Colored materials
Intermetallic phases often have characteristic colors that cannot be produced by pure metals or alloys, which is why they are frequently used in jewelry. Recently the Laves phase MgCu2 was tested as an addition to Cu to adjust the color of coins. By changing the Mg content of the alloys, the ratio of Cu-to-MgCu2 phase was systematically changed resulting in color variations from the reddish orange of Cu to the pale yellow color of MgCu2, see Fig. 29 [615].

Compositional tuning of the color of Cu-Mg coins containing 0, 10, 13, and 15 at.% Mg corresponding to increasing amounts of C15 MgCu2 Laves phase from 0 to about 40 vol.%. While the reflectivity of Cu shows a drastic increase to values above 90% in the orange and red region of the wavelengths of visible light (explaining its reddish orange color), MgCu2 instead shows a more continuous increase resulting in its less red and more brownish yellow color (reproduced with permission from [615])
Structural applications
Laves phase-based alloys
Single-phase: Laves phases, especially those based on transition metals, often have very high melting temperatures and can keep high strength up to temperature ranges even higher than that of superalloys. Figure 30 shows high-temperature proof stresses of some ternary C14 Laves phases in comparison to the oxide dispersion strengthened (ODS) Ni-base superalloy MA6000 [616]. However, even though this seems to make Laves phases natural candidates for high-temperature structural applications, single-phase Laves phase alloys are not suited as structural materials, especially due to their usually extreme brittleness at ambient temperatures, see, for example, [10, 15, 367, 617,618,619].

A multitude of studies were undertaken to find if and how the mechanical behavior of single-phase Laves phases, especially the room-temperature fracture toughness, changes by compositional deviations from the ideal AB2 stoichiometry (see, for example, [151, 364, 368, 380,381,382, 384, 386, 393, 394, 621]) or by substitutional replacement of the A or B metal by alloying additions (see, for example, [89, 102, 148, 361, 621,622,623,624,625]). There was also a theoretical attempt to predict which element addition should be most effective in enhancing the fracture toughness. Taking NbCr2 as an example and assuming that the elements which can increase the atomic free volume are likely to improve the fracture toughness, rhenium was predicted to be the best choice to improve the fracture toughness of NbCr2 [626]. However, even though partially very strong compositional effects on the high-temperature strength were reported [621], the room-temperature fracture toughness of single-phase Laves phase alloys always stays on a very low level and in many cases no visible effect on the fracture toughness was observed. Therefore, it must be concluded that—as already stated in Liu et al.’s review article [15]—unfortunately the room-temperature fracture toughness of single-phase Laves phase cannot be effectively improved by the introduction of defects such as antisite atoms or solid solution of alloying additions. Likewise, the introduction of thermal vacancies does not affect the fracture toughness at room temperature as was found for stoichiometric ZrCo2 containing a thermal vacancy concentration of 1% [157]. For a discussion about off-stoichiometry of Laves phases and about its role on mechanical properties from a more fundamental point of view, see Sects. 3.2 and 3.7, respectively.
Addition of a second phase: Besides the above-described attempts to improve the room-temperature fracture toughness of single-phase Laves phase materials, dual-phase alloys consisting of a Laves phase toughened by the addition of ductile precipitates such as soft metals is another concept. For example, toughening of C15 NbCr2 Laves phase by introducing fine and uniformly distributed, ductile bcc-Nb or bcc-Cr particles was already suggested in 1990/1991 by Anton and Shah [627] and Takeyama and Liu [628], respectively, and this concept was pursued for the same system for a long time, see, for example, [629,630,631,632,633]. In NbCr2 Laves phase fabricated by spark plasma sintering (SPS), trace amounts of Nb and Cr solid solutions are observed to distribute among the grain boundaries. This kind of dual-phase toughening also resulted in an increase of fracture toughness, which, however, is still far from being sufficient for applications [634,635,636]. After a 500 h heat treatment at 1000 °C, a (Ti,Mo)Fe2-based Laves phase alloy, which was slightly poor in Fe (nominal composition Fe-10Mo-30Ti in at.%), contained a volume fraction of about 12% of fine, ~ 250 nm sized, bcc-(Mo,Ti) precipitates. The fracture toughness was found to be increased from 1.1 to 2.2 MPa m1/2, which still corresponds to an extreme brittleness [637].
Also toughening of Laves phase alloys by addition of larger amounts of a ductile metal phase was tried out in many studies (see also Sects. 5.2.5 and 5.5), but such efforts likewise were not sufficient to qualify the material for real structural applications.
It must be concluded that Laves phase-based alloys, neither in a single-phase state toughened by off-stoichiometry or alloying additions nor as dual-phase materials containing soft second-phase precipitates, are suited as structural materials, which mainly is because of their insufficient room-temperature fracture toughness.
Nevertheless, Laves phases can find application in structural materials when introduced as minor phase added to strengthen ductile metallic matrices as is discussed in the following sections.
Ferritic(-martensitic) steels and alloys
Introduction
Laves phases play an important role in ferritic steels for high-temperature structural applications. Especially the need to increase the effectiveness of fossil fuel-fired power plants by increasing the working temperature has led to intensive research for the development of improved, high-temperature creep- and corrosion-resistant ferritic steels for boiler, pipework and steam turbine parts.
To qualify ferritic steels for high-temperature application, usually various metallic and non-metallic elements are added to the α-Fe (bcc) matrix. While the corrosion resistance is especially improved by Cr additions of at least ~ 9 wt% (leading to the formation of protecting Cr-oxide scales), the creep resistance is enhanced by solid-solution- and/or precipitation-strengthening of the material. Besides several types of carbides (and, less frequently, carbonitrides or borides), these strengthening particles can also be intermetallic phases such as a Laves phase, see, for example, [638,639,640].
It is not possible to draw a general conclusion stating that Laves phase particles have either a beneficial or a detrimental effect on the mechanical high-temperature behavior of ferritic steels. As will become obvious in this section below, the effect of Laves phase particles strongly depends on factors such as the composition of the steel, the mechanical load, and the temperature and time of its application. Already in 1956, Gemmill et al. [641] wrote in a study on the effect of Ti additions to a creep-resistant ferritic 8Cr-3Mo steel (numbers are wt%) that it would be desirable to avoid the precipitation of TiFe2 Laves phase because the Ti consumed in the Laves phase is no longer available as solid solution strengthener for the ferritic matrix. On the other hand, improving the strength of iron by Laves phase particles instead of carbides is also a rather old idea [642,643,644,645,646]. Bhandarkar et al. [645, 646] reported that a dispersion of spherical TaFe2 Laves phase particles within the grains of a ferritic Fe-Cr alloy produced good high-temperature strength up to 600 °C without low-temperature embrittlement. Nevertheless, Laves phases still have a reputation as an embrittling phase in steels and other structural alloys. Even though in various studies it was shown that finely dispersed Laves phase precipitates can strongly improve the creep behavior, the effect of Laves phase particles on the high-temperature mechanical properties of ferritic(-martensitic) steels is a topic of controversial discussions until today.
In the following subsections, the vast (and, therefore, only selectively cited) amount of literature dealing with Laves phases in ferritic steels and alloys is roughly divided into four groups which are (i) the so-called ‘9-12Cr’ (9 to 12 wt% Cr) ferritic-martensitic steels, (ii) stainless steels with about 15 to 22 wt% Cr and C contents below ~ 0.1 wt%, (iii) superferritic steels with more than ~ 25 wt% Cr and very low C contents (below ~ 0.02 wt%, often termed ‘ultra-low C steels’), and (iv) C-free ferritic α-Fe alloys containing some Laves phase. As in the entire literature about steels, it is customary to express compositions in wt%, we will follow this convention here.
The ‘9-12Cr’ steels
Ferritic-martensitic steels containing 9 to 12 wt% Cr and 0.1 wt% C are currently applied materials for the hot parts of fossil fuel power plants, where steam temperatures are exceeding 600 °C. Due to the urgent need to reduce the enormous emission of harmful greenhouse gases, the energy efficiency of power plants must be increased, which is most effectively done by further increasing the steam temperature. Therefore, for many years an impressive amount of work is going on world-wide aiming at the development of modified 9-12Cr ferritic(-martensitic) steels with improved temperature capability. The most critical property is the long-term creep resistance at application temperature, which could be improved by controlled, fine dispersion of long-term stable strengthening particles. This is where Laves phases can play an important role.
Critical point and reason why there are so many studies about this kind of high-temperature materials is the fact that these steels are not in an equilibrium state during application. Prior to being built into the power plant, the cast material experiences some heat treatments for normalizing/austenitization and subsequent formation of a martensitic matrix with or without certain precipitates resulting in a metastable material. During application at high temperature, this metastable state moves in the direction of thermodynamic equilibrium with a composition-, temperature-, and also mechanical-load-dependent kinetics resulting in a very complex interplay of growing or dissolving particles, formation of new phases, shifting of the matrix composition, and changes of the microstructure in general. As in parallel with the microstructure also the mechanical properties such as the creep resistance will change, it is an important goal to slow down the kinetics (especially that of particle coarsening) as far as possible. Investigations of long-time aged steels and modeling of phase evolution in such steels show that even after more than 10 years, equilibrium may not yet be achieved, and new phases such as Laves phases or so-called Z phase (a complex (Cr,Fe)(Nb,V)N nitride [647,648,649]) may nucleate and grow only after long times (see, for example, [639, 650,651,652]). Because such aging times of 10 or more years of course by far exceed available times in experimental studies, the kinetics of phase precipitation and coarsening was often investigated by theoretical simulations [650, 651, 653,654,655,656,657,658,659,660]. An example of a simulation of the time-dependent change of phase fractions in two different steels is shown in Fig. 31 [651], details are discussed later in this section.

Time dependence of phase fractions of precipitates in P91 (a) and P92 (b) steels at 600 °C as obtained by numerical simulations (adapted with permission from [651])
The two most important alloying additions to increase high-temperature strength are Mo and W, and both form C14 Laves phases with Fe. Mo-containing ferritic steels are already in use since the 1950s [641, 661, 662] and Mo contents usually are in the range 0.5 to 2 wt%. Most prominent representative of this group of ferritic-martensitic steels is so-called P91 steel (9Cr-1Mo + various minor additions), see, for example, [639, 652, 656, 663,664,665,666,667,668]. While in 9Cr-2Mo steels, a significant loss of toughness after only 1000 h at 600 °C was observed that was attributed to precipitation of MoFe2 Laves phase [669,670,671], P91 (1 wt% Mo) steels were successfully creep-tested for more than 100000 h at 550 °C [672, 673] and 600 °C [652] without fracturing but finally also showing creep damage that at least partially was attributed to strongly coarsened MoFe2 Laves phase precipitates.
With respect to strengthening, W is an even more effective alloying addition than Mo. W diffuses more slowly than Mo and WFe2 Laves phase precipitates coarsen less rapidly than MoFe2 particles (see, for example, [674, 675]) and are also stable to higher temperatures compared to MoFe2. While the Mo-modified 9Cr steel P91 was introduced as steam pipe steel in fossil fuel power plants in the late 1980s, the improved W-modified steels P92 (9Cr-2W-0.5Mo + minor additions) and E911 (9Cr-1Mo-1W + minor additions) came into application in 2001 and 2002, respectively [639]. The Laves phase-containing P92 steel until today is a kind of benchmark for high-temperature steels and its properties and behavior especially with respect to the Laves phase were discussed in many publications, see, for example, [639, 660, 676,677,678,679,680,681,682,683].
Figure 31 shows a comparison of the variation of phase fractions of the diverse precipitates in P91 (left) and P92 (right) steel at 600 °C as a function of time as obtained from numerical simulations [651]. In both steels, Laves phase only starts to precipitate after 100–1000 h from the Mo/W supersaturated matrix and it needs in the range of 10000 h until the complete amount of Laves phase has precipitated. In the following time, where the material is still far from equilibrium, the precipitates will start to coarsen, and finally after times in the range of 100000 h (i.e., more than 10 years) Z phase will start to form in parallel to dissolution of V nitride.
There is general agreement in the literature that the precipitation of fine (Mo,W)Fe2 Laves phase particles increases the strength of the steels as long as the coarsening process has not yet started. Simultaneously with the formation of Laves phase, the effect of solid solution strengthening decreases as Mo and W are consumed from the supersaturated matrix. However, from a rough estimation of the solid solution strengthening effect by Mo and W and comparing it with the precipitation hardening effect of the Laves phase, Hald [639] concluded that solid solution strengthening from Mo and W actually has no significant effect on the long-term microstructure stability of 9-12Cr steels and that Laves phase precipitation is much more effective for strengthening. As, for example, shown by Abe [674, 675], precipitation of Laves phases leads to a decrease of the creep rate, but with the onset of rapid coarsening the creep rate reaches a minimum value and starts to strongly increase again. The nucleation of Laves phase precipitates was found to start at grain boundaries near M23C6 carbides (see Fig. 32) [684,685,686], followed by incoherent (and, therefore, comparatively rapid) growth into the matrix. This can result in void formation and de-cohesion effects initiating crack formation, which finally may lead to the failure of the steel. An especially critical situation regarding unwanted precipitation and coarsening of Laves phase particles exists in weld joints (for example in similar welds of P92 [687] or dissimilar welds of 9Cr steel with austenitic steels [688,689,690,691,692]), where microstructural changes during melting and subsequent cooling are difficult to control.

STEM images of a Laves phase precipitate connecting two M23C6 particles on a grain boundary of the matrix of a 12Cr-1Mo steel after 2400 h creep at 550 °C and 120 MPa: a HAADF image, b bright field image, and c corresponding EDX map (reproduced with permission from [685])
Therefore, a central goal of developing optimized ferritic-martensitic high-temperature steels is to minimize the coarsening rate of Laves phase precipitates, which as fine particles are very beneficial to increase the creep resistance. The risk of void formation at the interface between a Laves phase particle and the matrix does not only depend on the particle size, but also on the level of strain energy at the interface, which in turn will depend on the composition of matrix and particles. By systematically studying the role of the amount and ratio of Mo/W additions and of the Cr content, it was shown that compositional changes of the Laves phase particles may strongly vary their embrittling effect independent of their size [693]. Regarding changes of the Cr content, various types of (10-)12Cr-xW steels were developed including the P122 (12Cr-2W) steels [220, 694,695,696,697] and a group of steels containing higher amount of W (3–5%) and further alloying additions [650, 657, 698,699,700,701,702]. With respect to the Mo and W contents, various modifications of the 9Cr steels P91 and P92 were studied such as the 1Mo-1W steels with 9% Cr (designated as E911 steels [639, 662, 667, 678]) or 10% Cr [703,704,705,706], or model steels with varying Mo/W ratios [658, 671, 674, 675, 707].
Moreover, quite a lot of studies dealing with the effect of further alloying additions to 9-12Cr steels were performed always including effects on Laves phase precipitation. For example, alloying 3(−5) % Co to 9-12Cr steels (e.g., [708,709,710,711,712,713,714,715,716,717,718,719]) was found to promote the precipitation of Laves phase (due to the fact that Co reduces the solubility of Mo/W in the Fe matrix). In addition, Co was predicted (by a computational design approach coupling thermodynamic and kinetic data involving a genetic algorithm optimization routine) to drastically reduce the coarsening rate of Laves and M23C6 particles, thereby yielding a much improved long-time creep resistance compared to the Co-free counterpart steel [659]. Cu is another frequent alloying addition aiming at positively affecting the precipitation processes [657, 699,700,701]. Cu should be beneficial for the creep resistance as it affects a finer distribution of the precipitates [640]. In order to study the precipitation of (Mo,W)Fe2 Laves phase particles and the impact on creep behavior as a separate effect (i.e., unaffected by carbides), a series of C- and N-free (< 50 ppm) Fe-9Cr-X alloys with varying amounts of X = W, Mo, Co and further typical steel additions were creep-tested at 650 °C [720]. For up to about 3% W + Mo, Laves phase was found to precipitate only on grain boundaries and, hence, contributed little to precipitation strengthening. For 4.5% W + Mo, Laves phase precipitates were observed both at the grain boundary and homogeneously within the grains with a precipitation-free zone on both sides of the grain boundaries. The creep rupture time was mainly influenced by the size of Laves phase particles precipitated at grain boundaries [720]. However, it has to be kept in mind that the precipitation process in real steels is different due to the simultaneous presence of carbide/carbonitride particles.
Finally, it should be mentioned that creep-resistant ferritic-martensitic 9-12Cr high-temperature steels are not only applied in fossil fuel power plants, but they are also candidates for application in nuclear power plants both for fission as well as fusion reactors [640, 721,722,723,724,725,726]. High-energy neutron irradiation leads to significant changes in the microstructure and the formation of Laves phase precipitation can be completely suppressed by irradiation [640]. In case Laves phase is already present in a steel, larger particles can become completely amorphous by ion irradiation and smaller Laves phase particles may completely disintegrate, while instead some new, non-equilibrium phases may nucleate and grow [722, 726]. These partially complex processes are not yet well understood.
Ferritic 15–22% Cr steels
Ferritic steels with increased Cr contents in the range 15 to 22 wt% were developed as oxidation-resistant structural materials for the temperature range of about 600 to 900 °C. Intended fields of application are a) improved high-temperature power plant steels for temperatures above 620 °C, b) automotive parts for the hot regions of the engines such as exhaust manifolds, which see temperatures of up to 900 °C or even higher, and c) interconnects for solid oxide fuel cells (SOFCs), where temperatures in the same range of about 650 to 900 °C can be reached. In all cases, Laves phases play an important role as is briefly discussed in the following.
A reason for the limitation of the 9-12Cr steels to maximum temperatures of 600–620 °C is the insufficient steam oxidation resistance at higher temperatures. This could be significantly improved by higher Cr contents. However, at the same time higher Cr contents lead to strongly increased formation of large Z-phase particles replacing fine, strengthening MX (M = Nb,V and X = C,N) carbonitrides, which causes a breakdown of creep strength [647, 727]. Starting from typical 9-12Cr steel compositions, WFe2 Laves phase containing 15Cr steels with fully ferritic matrix were developed by a Japanese group and continuously improved over the last twenty years for application at 650 °C [728,729,730,731,732,733,734,735,736,737,738,739]. The loss in creep strength due to the increased Cr content is compensated by increased W contents up to 6 wt% and the addition of 3 wt% Co [728, 729] leading to precipitation of not only WFe2 Laves phase but also of intermetallic µ (Fe7W6)- and χ (Fe36Cr12W10)-phase particles as well as M23C6 carbides [730]. As a decrease in long-term creep strength was attributed to the large amount of coarse carbide particles, the C content was reduced from the initial amount of 0.1 wt% to 0.05 wt% and even C-free alloys were tested [733]. Addition of Ni up to 2 wt% led to formation of a two-phase matrix consisting of ferrite and some martensite both containing Laves phase precipitates [732, 736,737,738]. Testing the creep properties of these 15Cr steels at temperatures of 650 to 750 °C gave partially promising results, but also indicated that further optimization is needed before they can be applied in power plants [735, 737,738,739].
NbFe2-containing ferritic stainless steels with Cr contents in the range 15 to 19 wt% and low C contents of 0.01 wt% or less were developed for application in automotive exhaust systems, which currently are working at temperatures in the range of up to 900 °C [740,741,742,743,744,745,746,747,748,749,750,751,752,753,754,755,756]. Typical Nb additions are in the range 0.4 to 0.8 wt% and lead to significant solid solution strengthening of the ferrite matrix with precipitation of NbFe2 Laves phase and small amounts of Nb(C,N) and Fe3Nb3C during aging. Owing to the low C and high Cr contents, the Laves phase can—compared to 9–12Cr steels—exist as stable phase up to much higher temperatures (> 1000 °C depending on composition [750, 754]). While the incongruently growing Laves phase was reported to be detrimental for the long-term strength at 700 °C [742], its formation along grain boundaries was found to effectively prevent grain growth at higher temperatures, thus preserving the high-temperature strength over a long time period [750, 755]. To reduce emissions of conventional car engines, higher discharge temperatures are needed and the operating temperatures of exhaust manifolds will approach 1000 °C or more [747, 752]. A very critical issue at such high temperatures is the corrosion resistance of the material. Therefore, the oxidation behavior of a 19Cr-0.5Nb ferritic stainless steel containing additions Mo, W, and Ce was studied at 950 to 1100 °C. It was found that the Laves phase precipitates can effectively hinder the inward growth of the oxide film. However, with a W content increased to 1 wt%, a reduction of the adhesion of the oxide film was observed [752]. Effective retardation of oxidation by Laves phase precipitates was also reported for a Nb-containing 20Cr steel tested at 800 and 900 °C [757].
Interconnects for SOFC stacks are another application of high-Cr stainless ferritic steels containing Laves phase [758,759,760,761,762,763,764,765,766,767,768,769,770,771]. Alloying elements and concentrations are similar to those in steels for automotive exhaust systems, C contents are < 0.01 wt% and Cr contents are in the range 18 to 22 wt% to ensure formation of protecting chromia scales. Interconnect materials must not only have good high-temperature mechanical properties, but also a low electrical resistivity, high thermal conductivity, and very good corrosion resistance and chemical stability in both anode and cathode gases, i.e., in reducing and oxidizing atmosphere [762]. The role of Laves phase is mainly to provide good mechanical properties [759, 763, 764], but there is also a complex interplay between Laves phase precipitation and the formation of different types of surface oxides [771].
One of the alloys originally developed for the application in SOFCs is so-called Crofer 22 H, which is a commercially available 22Cr ferritic steel containing some (Nb,W)(Fe,Cr)2 Laves phase, see, for example, [759, 762, 763, 772, 773]. Due to its promising high-temperature mechanical properties, the idea came up to modify and optimize this Laves phase forming, fully ferritic steel for structural applications at 600 to 650 °C in power plants [774,775,776,777]. Very good properties were obtained for a series of modifications with 17 wt% Cr and varying amounts of Nb and W, which were patented as HiperFer (High Performance Ferritic) steels [778,779,780,781,782]. The Cr content was reduced from 22 to 17 wt% in order to minimize FeCr (σ) phase formation at temperatures of 600 to 650 °C [774]. Detailed tests of the high-temperature creep performance and cyclic fatigue behavior indicated that this kind of high-strength, Laves phase-forming, fully ferritic steels has a potential to replace currently applied martensitic 9–12 Cr steels in power plants enabling working temperatures > 620 °C [778,779,780,781]. Promising results were also obtained in first tests performed at the linear plasma generator PSI-2 at Forschungszentrum Jülich to study possible applications of HiperFer as a reduced activation ferritic steel for nuclear fusion applications [783].
Superferritic steels (≥ 25% Cr)
Following a definition given by Azevedo and Padilha [784], superferritic steels are stainless ferritic alloys containing less than 0.02 wt% C, more than 25 wt% Cr, between 2 and 4 wt% Mo as well as Ni, and sometimes additions of Nb or Ti. They are especially suited as structural materials for applications in harsh, corrosive environments at ambient temperatures up to a maximum of about 300 °C. At higher temperatures, detrimental embrittling intermetallic phases such as σ (FeCr), χ (Fe36Cr12M10), and Laves phase (Mo,Nb,Ti)Fe2 can precipitate. Due to the high Cr contents, the Cr-rich σ phase is the most frequently observed precipitating phase, while the occurrence of Laves phase has only rarely been described [784,785,786,787,788,789,790,791,792,793]. In a typical superferritic alloy of composition 25Cr-(2-3)Mo-4Ni containing 0.3 wt% Nb, Laves phase was found to nucleate as first intermetallic phase during aging at 850 °C. It precipitates as fine particles along the ferrite grain boundaries and does not significantly grow due to the start of precipitation of σ phase and the exhaustion of the available Nb [785, 787]. Similar observations were made in 27Cr-4Mo-2Ni superferritic steel after hot rolling, solution treatment at 1050 °C and/or aging between 600 and 800 °C [789,790,791,792]. Increased Cr contents were found to promote the precipitation of σ and Laves phase and to widen the precipitation temperature region of the Laves phase, but annealing at temperatures above 1000 °C can result in complete dissolution of the detrimental phases [788]. A review about the role of the deleterious effect of σ, χ, and Laves phases on the processing and service life of superferritic steel components is given by Azevedo and Padilha [784].
Other ferritic C-free alloys
Precipitation of AFe2 Laves phase from α-Fe solid solution is known for many systems (e.g., A = Hf, Mo, Nb, Ta, Ti, W, Zr) and was already studied long time ago as a measure to improve the strength of iron (see, e.g., the aforementioned (Sect. 5.2.1) early studies on Fe-Nb [642, 643], Fe–Ta [644,645,646], and Fe-Ti [643] α-Fe + AFe2 alloys). Due to the poor oxidation behavior of Fe, usually Cr (as in the above discussed steels) and/or Al are added to produce protective chromia or alumina scales on the alloy surfaces. Some examples for Laves phase containing α-Fe alloys suggested for high-temperature applications are described in the following. It should be noted that particular care must be taken when comparing alloy compositions in different publications in this research field as both ‘wt%’ as well as ‘at.%’ are frequently used. To allow comparison with the above discussed ferritic steels, we stick to the wt%-description in this section. In cases where original compositions were given in at.%, we also include this information here.
(i) A = Nb, Mo, W: NbFe2-strengthened Fe-7.6Al-8.7Nb alloys (Fe-15Al-5Nb in at.%) with or without some minor additions of Zr and B were hot-forged at 1150 °C resulting in a uniform dispersion of coarse Laves particles arranged along the triple junctions of a fine-grained α-Fe matrix. The forged alloy showed a good creep resistance at 700 °C [794, 795]. Heat-resisting Fe-Cr-Nb(-Ni) alloys with about 9–14 wt% Cr, 1–8 wt% Nb, and 0–5 wt% Ni (Fe-(10–15)Cr-(0.5–5)Nb-(0–5)Ni in at.%) with varying amounts of NbFe2 Laves phase adjusted by the Nb content were tensile-tested at 600 and 700 °C as well as at room temperature. A good balance between room-temperature ductility and high-temperature strength was obtained for about 1.5–2.5 wt% Nb (1–1.5 at.%) [796, 797]. Replacing Nb by 4.5 wt% W (1.4 at.%) and adding further elements such as Co and Si resulted in more complex precipitation processes as besides Laves phase, a rhombohedral R phase and a quasicrystalline phase I also appear [32, 798].
Nb- and/or Mo-containing Fe-Cr-Al alloys with small additions of Y (Fe-13Cr-(4.5–6)Al-0.15Y-(0-2)Mo-(0-2)Nb) consisting of a ferritic matrix + fine (Mo,Nb)Fe2 precipitates were developed as promising accident-tolerant fuel cladding for light water reactors [799, 800]. By applying a thermomechanical treatment, a fine dispersion of Laves phase particles especially along grain/subgrain boundaries in the α-Fe matrix was obtained, which effectively stabilized the recovered and recrystallized microstructures [800].
High-Cr (30Cr-3Al) ferritic Fe-Cr-Al alloys with strengthening Nb- or W-based Laves phase (1 or 2 wt% Nb and 0 or 6 wt% W + minor amounts of Zr, Mn, and Si) were developed aiming at application in future fossil fuel power plants operating at 700 °C. The very high Cr content was chosen to improve the ash-corrosion resistance in fire-side corrosive environments encountered in fossil fuel-fired power plants. A good combination of corrosion resistance and creep strength was achieved comparable or superior to commercially available austenitic high-temperature stainless steels [801,802,803].
By addition of 3 wt% Ni and 3 wt% W (+ 1 wt% Zr) to Fe-12Cr-3Al, dual precipitation of coherent B2 NiAl and incoherent (W,Zr)Fe2 Laves phase was achieved. Creep properties at 650 °C were tested for this alloy, which is intended for application in nuclear power plants. Creep fracture was primarily ductile, but strongly coarsened Laves particles locally led to partially brittle fracture [726, 804].
(ii) A = Ti: Ti is another transition metal leading to the formation of strongly hardening Laves phase when added to α-Fe, see, for example, [643, 805]. Investigations of the temperature dependence of mechanical properties of Fe-based two-phase ferrite + Ti(Fe,Si)2 alloys in the ternary system Fe-Si-Ti were performed by hardness, compression and bending tests. Si tends to dissolve in the Laves phase, and increasing Ti as well as Si content lead to significant strengthening and loss of ductility by Laves phase particles precipitated inside the ferrite grains and on the grain boundaries. Alloys with additions of about 5 at.% Si and 4 at.% Ti were suggested for further alloy development [806].
(iii) A = Zr: Yield strength and flexural fracture strain of two-phase α-Fe + ZrFe2 Laves phase alloys were studied as a function of temperature up to 1200 °C for Fe-based Al-Fe-Zr with 0 or 5 wt% Al (10 at.%) and varying Zr contents increasing from 0 to 43 wt% Zr (0 to 30 at.%, corresponding to phase fractions of the Laves from 0 to 100%). Figure 33 shows the strong increase of the yield strength with increasing content of Laves phase, which however goes in parallel to an increase of brittleness. The brittle-to-ductile transition temperatures (BDTT) obtained from the temperature dependence of the measured flexural fracture strains strongly increases from below room temperature for the Laves phase-free material to, e.g., about 600 °C for alloys with 1:1 volume content of α-Fe and Laves phase. With respect to the oxidation behavior, Zr additions were found to have a very negative effect resulting in strongly increasing oxidation rates [807].

0.2% yield stress as function of temperature for different amounts of Zr(Fe,Al)2 Laves phase in Fe-based two-phase α-Fe + Laves phase Al-Fe-Zr alloys with a fixed Al content of about 5 wt% (10 at.%) (adapted with permission from [807])
A new class of ZrFe2-strengthened Fe-(9-12)Cr-(3-10)Zr-based alloys (with some quaternary additions of Ni or W) was reported to be promising for applications as cladding materials in nuclear reactors because of the enhanced high-temperature strength and resistance to creep and radiation hardening compared to traditional P91 ferritic-martensitic steels [808,809,810].
(iv) A = Hf: An alternative way to produce homogeneously distributed, fine Laves phase particles for strengthening of a ferritic matrix was recently introduced by Kobayashi et al. [811,812,813,814]. In Fe-Cr-Hf alloys with about 9 wt% Cr and low Hf contents ≤ 0.5 wt% (0.15 at.%), regularly arranged rows of fine HfFe2 particles with sizes of about 20 nm were produced through so-called interphase precipitation (see [815, 816]) along the reaction path δ-ferrite → γ-austenite + HfFe2 with a subsequent phase transformation of the γ phase into the α-ferrite phase [811,812,813,814], see Fig. 34.

BSE (back-scattered electron) SEM image of regularly precipitated, nanometer-sized HfFe2 Laves phase particles in a ferritic Fe-9Cr matrix obtained by interphase precipitation (reproduced with permission from [811])
Austenitic steels and alloys
Heat-resistant austenitic steels contain high amounts of Cr (for oxidation resistance, usually in the range 15 to 25 wt%) and Ni (as austenite stabilizer, strongly varying between about 8 and 30 wt%). Compared to the ferritic 9–12Cr steels, they can be applied at higher temperatures up to 675 °C, but due to the high Cr + Ni contents they are much costlier. Very similar to their ferritic counterparts, both solid solution strengthening and precipitation strengthening by alloying additions contribute to their high-temperature strength and creep resistance. Most used alloying metals for this purpose, which can also lead to precipitation of Laves phases, are Mo, Nb, and, to a much lesser extent, Ti and W.
Similar as in the case of ferritic steels, for a long time the occurrence of precipitation of Laves phases in the austenitic matrix was regarded as a deleterious effect leading to embrittlement and creep fracture, see, for example, [817,818,819,820,821,822,823,824]. The common opinion was that in development of new austenitic steels it is most important to suppress the formation of Laves phase [820], even though in a review about precipitation in creep-resistant austenitic stainless steels, it had been stated that “Whether or not Laves phase has a detrimental effect on creep properties is still discussed” [825].
A concept to benefit from controlled precipitation of finely dispersed Laves phase particles in the austenite (γ-Fe) grains and on the grain boundaries was introduced by Takeyama et al. [826]. Based on their earlier phase diagram studies [827, 828], a C-free alloy with a composition of Fe-20Cr-30Ni-2Nb (at.%, corresponding to Fe-18.4Cr-33.1Ni-3.3Nb in wt%) was designed [826]. While at 1200 °C the alloy is single-phase fcc-Fe, it is two-phase with Nb(Fe,Cr,Ni)2 Laves phase precipitates at 800 °C, and becomes three-phase by additional precipitation of γ” (Ni3Nb) in the fcc-matrix during cooling to 700 °C [829, 830]. In a series of investigations, the properties of this alloy were later on further improved by modifications of composition and heat treatments [829,830,831,832,833,834,835,836,837,838]. These studies showed that strengthening in this alloy is especially determined by the grain boundary precipitation of Laves phase and that the coverage of the grain boundaries by precipitates and their morphology can be controlled by small additions of B. The authors showed that the creep strength directly depends on the grain boundary fraction ρ covered by Laves phase precipitates [829, 830]. Figure 35 shows a comparison of microstructure and creep behavior of the alloy without and with B-doping (0.03 at.%). A higher coverage ρ of the grain boundaries results in an increased creep life and reduced creep rate, which was found to decrease linearly with ρ [829]. A more detailed study regarding the effect of B-doping in this alloy showed that B segregates to the grain boundaries and promotes the precipitation of Laves phase effecting not only a higher number but also a refinement of the Laves phase precipitates [834]. From room temperature tensile and Charpy impact tests, it was concluded that the Laves phase on the grain boundaries has no negative effect on the room temperature ductility. There was no indication of crack propagation within the Laves phase precipitates or along the interface between γ-Fe matrix and Laves phase, and fracture was found to be always transgranular [836].

Microstructures of Fe-20Cr-30Ni-2Nb (at.%) without (a) and with 0.03 at.% B doping (b) after aging for 1200 h at 800 °C, and (c) creep-rates at 700 °C as a function of time of the two aged alloys revealing a strong improvement in creep life by increasing the grain boundary coverage with Laves phase from 52 to 89% (adapted with permission from [829])
Thermokinetic calculations combined with experimental observations on the precipitation process of the NbFe2 Laves phase and the γ” (Ni3Nb) phase in Fe-20Cr-30Ni-2Nb (at.%) revealed that above 700 °C, the Laves phase first nucleates on grain boundaries and precipitation within the grain interior starts later. The nose of the time–temperature-transformation curve for the Laves phase is located at about 1000 °C [837]. The effect of partial or complete replacement of Nb by Ta on the alloy microstructure and kinetics of the precipitation processes of the Laves and γ” phases was found to be complex. For Ta/(Nb + Ta) ratios above 0.6, Ta was found to stabilize the Laves phase against γ” [838].
On the basis of the Fe-20Cr-30Ni-2Nb (at.%) alloy composition suggested by Takeyama [826, 827], a series of Laves phase strengthened austenitic alloys was designed and tested at Oak Ridge National Laboratory [802, 831, 839,840,841,842,843,844,845,846,847,848]. The most important difference is the addition of 2.5 to 4 wt% Al (about 5 to 8 at.%) to the base composition resulting in the formation of a dense, protective alumina scale on the alloy surface. Such beneficial, external alumina scales exhibit one to two orders of magnitude slower oxide growth kinetics compared to chromia scales, and are far more stable in water vapor containing environments at elevated temperatures [849, 850]. This is why they were introduced as a new class of stainless steels termed alumina-forming austenitic steels or ‘AFA steels’ [842, 843]. Overviews of the development of this type of austenitic steels, which besides Laves phase also contain Al-stabilized B2-NiAl and, depending on composition, γ′(L12) Ni3Al precipitates, are given by Yamamoto et al. [802, 847].
The growth of Laves phase and B2 precipitates at 800 °C in the ‘base’ alloy Fe-20Cr-30Ni-2Nb-5Al (at.%) was studied with samples heat-treated up to 1325 h. Within the austenitic matrix, Laves phase was found to grow only very slowly compared to B2 precipitates. However, the growth of both phases was much faster on the grain boundaries, and the coverage of the grain boundaries by precipitates increased from an initial value of 56% to 93% after 1325 h. This also resulted in an increased yield strength, while interestingly the elongation at room temperature still reached a high value of 19% [851]. As the authors tentatively assigned the surprisingly high room-temperature ductility to the low-misfits of the precipitates with the austenite matrix, they additionally analyzed the orientation relationships of both phases with the matrix in another study [852]. For the interfaces between Laves phase (Lp) and matrix (γ), they confirmed an orientation relationship (111)γ//(0001)Lp, [\( \bar{1}10 \)]γ//[\( 10\bar{1}0 \)]Lp, which had already been reported before by Denham and Silcock [853] for hexagonal C14 NbFe2 Laves phase in an fcc austenitic matrix. For the B2 precipitates, they found a Kurdjumov–Sachs orientation relationship (111)γ//(011)B2, [\( \bar{1}01 \)]γ//[\( \overline{11} 1 \)]B2 [852].
Trotter and Baker also studied the effect of severe plastic deformation on the microstructure and precipitation process of this alloy [854]. By cold working of the solution-treated (1250 °C) alloy, the thickness was reduced by 90% resulting in a high density of dislocations that served as nucleation sites for precipitation during subsequent aging at 700 and 800 °C. The introduced defects not only caused an accelerated precipitation in the matrix but also an increase in the total volume fraction of precipitates as compared to material that had been aged at the same temperatures without prior cold working [854]. The same observations were also made for a cold-worked AFA steel of wt%-composition Fe-14Cr-32Ni-3Nb-3Al-2Ti-0.1C (corresponding in at.% to Fe-15Cr-30Ni-2Nb-6Al-2Ti-0.45C) [855]. By introducing 2 wt% W in a Laves phase-containing 14Cr-20Ni-3Mo-2.5Al-2Mn-1Nb-0.1C (wt%) AFA steel (and reducing Mo to 1.5%), a significantly increased creep lifetime was obtained in tests at 700 °C. This improved behavior was traced back to an increased fraction and reduced size of Laves phase precipitates due to an increased content of W in the Laves phase [856]. The same positive effect of W addition was reported for an AFA stainless steel Fe-15Cr-25Ni-3Al-NbWCu (wt%) [857].
The Laves phase ZrFe2 plays an especially important role in nuclear waste management, where austenitic stainless steels alloyed with 15 wt% Zr alloys were developed as nuclear waste form materials [858,859,860,861,862,863]. Neutron diffraction results showed that the addition of U does not result in the formation of discrete U-rich phases. The lattice parameters of the ZrFe2 Laves phase are larger in U-containing 15Zr stainless steel alloys and are consistent with the substitution of U at Zr sites of the ZrFe2 lattice indicating that U (and also other actinides) dissolves in the Laves phase [863].
Superalloys
Ni- and Co-based superalloys are developed for structural applications at very high temperatures as, for example, in the hot parts of gas turbines, where steels can no longer be used. High-temperature strengthening of the fcc Ni- or Co-matrix (‘γ phase’) is usually achieved by coherent intermetallic precipitates (such as fcc-ordered γ′ or bct-ordered γ″ phases) and by solid solution hardening. This is why superalloys are usually highly alloyed materials. An often observed, unwanted effect of these alloying additions is the precipitation of incoherent intermetallic phases, which can negatively affect the mechanical behavior [864]. Very frequently these unwanted intermetallic phases are Laves phases, which were found in both Ni-based and Co-based superalloys, see, for example, [865, 866].
Laves phases in superalloys are considered as very detrimental for the mechanical behavior. The best investigated example is the Ni-based superalloy Inconel 718 (IN718, typical composition in wt% Ni-19Cr-18Fe-5Nb-3Mo-1Ti and minor other additions). IN718 is by far the most important superalloy for applications, and it is used in gas turbine components since the early 1960s and until today [867]. Laves phase forms due to the high Nb content and can impair the mechanical properties of IN718 through several mechanisms, which were summarized in an overview by Schirra et al. [868]. According to these authors, the most dominant mechanism probably is brittle fracture of the Laves phase. In addition, similar to steels, the formation of Laves phase consumes large amounts of Nb depleting the matrix of the principal hardening element. Finally, during welding processes the Laves phase may melt, and cooling of the partially melted material can lead to the initiation and propagation of microcracks in the re-solidified material [868]. Especially in superalloy welds [869,870,871,872,873,874,875] and dissimilar welds of superalloys with steels [876,877,878,879,880,881], Laves phases were consistently considered as highly detrimental to the mechanical properties. The effect of liquation cracking also plays an important role in the production of superalloys by additive manufacturing (AM) methods (e.g., [882, 883]). Parts as well as coatings made of Ni-based superalloys (mostly IN718) are often produced by electron beam melting or laser AM, and a large number of investigations were performed to understand and control the formation and distribution of Laves phase during the AM process, see, for example, [884,885,886,887,888,889,890,891] to name but a few.
Amount, size and morphology of Laves phase precipitates sensitively depend on the cooling process. Therefore, several detailed studies (see, for example, [892,893,894,895,896,897,898]) were performed to precisely understand the solidification process of superalloys. The most common method to reduce the amount of Laves phase precipitates is their decomposition at high temperatures by heat treatments above the eutectic temperature, see, for example, [897, 899,900,901]. However, while it is possible to dissolve most of the interdendritic Laves phase in IN718 superalloy, at the same time needle-shaped δ phase precipitates and the matrix grains may coarsen substantially [899]. Minor alloying additions may also play an important role for stabilizing the Laves phase. Doping with elements such as P and B was reported to promote the formation of Laves phase, i.e., both should be avoided [902,903,904]. From DFT-based ab initio calculations on the effect of different solute elements on the interface energies between Laves phases and matrix grains, it was concluded that Ru additions should notably increase the interfacial energy and, thereby, increase the difficulty of nucleation of Laves phase [905]. Another interesting approach to reduce the amount of Laves phase was presented by Chen et al., who were able to significantly suppress the formation of Laves phase by introducing carbon nanotubes in a laser melting process of IN718 [906].
It should be noted that the occurrence of Laves phases in superalloys is not always regarded as detrimental. Sui et al. [888] studied differently heat-treated IN718 samples with varying sizes and morphologies of Laves phase precipitates. They conclude that the size and the morphology are the key factors in determining whether Laves phase precipitates are good for the mechanical properties of IN718 superalloy or not. A certain amount of small and granular Laves phase is found to be beneficial for the room temperature tensile properties [888].
There is also a class of superalloys neither containing γ′ nor γ″ phase, but being purely strengthened by Laves phase. Superalloys such as the W-rich Ni-based alloys HR6W (Ni-23Cr-25Fe-7W-Ti-Nb) and HR35 (Ni-30Cr-15Fe-6W-Ti) (which is additionally strengthened by α-Cr) were developed for boiler tubes and pipes in advanced ultra-supercritical (USC) power plants with 700 °C steam temperature. Even after long-term aging of WFe2-strengthened HR6W superalloy for 3000 h at 750 °C, the microstructure exclusively contained fine Laves phase precipitates and M23C6 carbides, and no σ phase precipitation was observed [907]. Creep tests conducted at 800 °C (with 70 MPa load) and 700 °C (100 MPa load) revealed that the amount and size of Laves phase precipitates remained constant over time [908].
Cr-, Cu-, Mg-, Nb-, Ti-, or Zr-based alloys
Strengthening of soft metal solid solutions by strong Laves phase particles is an old concept for designing new structural materials, which is not only applied to Fe-, Co-, and Ni-based alloys (such as the above discussed steels and superalloys), but was also considered and tested for several other metals. Here, some examples of Laves phase-forming alloy systems are briefly summarized.
Cr-based alloys
Alloys based on Cr are of longstanding interest as high-temperature structural materials especially due to their good high-temperature oxidation and corrosion resistance in combination with a high melting point and moderate density. Laves phase in equilibrium with bcc-Cr solid solution exists in the Cr-X systems with X = Ti, Zr, Nb, Hf, or Ta (and interestingly in all these binary systems, Laves phase is the only intermetallic phase). The microstructures and mechanical behavior of two-phase alloys from all these five systems have been intensively explored, see, for example, [381, 909, 910] for Cr + TiCr2 alloys, [911, 912] for Cr + ZrCr2, [15, 628, 630, 631, 911,912,913,914,915,916,917,918] for Cr + NbCr2, [15, 919,920,921] for Cr + HfCr2, and [15, 922,923,924,925,926] for Cr + TaCr2 alloys. Increased high-temperature strength in parallel to decreased room-temperature fracture toughness are the two opposing effects resulting from addition of Laves phase to the bcc solid solution. Various attempts were made to get improved property combinations, for example, by producing directionally solidified eutectic material [913, 914] or by various alloying additions such as Ni, Co, Fe, Al and Re at levels up to 16 at.% to Cr + NbCr2 alloys [927], Si [926, 928,929,930,931], Al, Mo [929], Fe [932], or Ru [933] to Cr + TaCr2 alloys. For a fully eutectic quinary Cr-12Nb-5Ti-5Mo-5Si alloy consisting of Cr solid solution and about 55 vol.% NbCr2-based Laves phase, a room-temperature yield strength of nearly 2000 MPa and a fracture toughness in the range of 14.0–14.8 MPa m1/2 was reported [633]. To increase the strength of the ductile bcc-matrix, a three-phase Cr-Mo-Nb ternary alloy with 30 at.% Mo and 20 at.% Nb was designed containing two bcc solid solution phases, one rich in Cr and the other rich in Mo, in combination with strengthening NbCr2 Laves phase. During aging at 1200 °C, the alternating bcc-Cr/bcc-Mo morphology remained stable, but the growing Laves phase on the grain boundary consumed more and more of the Cr-rich bcc phase [934].
Cu-based alloys
Because of their excellent electrical and thermal conductivity properties, Cu-based alloys are extensively applied especially in electronic materials. As the low mechanical strength of Cu often is an issue in applications of such materials, strengthening by alloying is an important topic. A very good combination of high strength and good conductivity was achieved by alloying Cu with Be. However, the problem in production and handling of such alloys is the enormous toxicity of Be. Two-phase Cu + MgCu2 alloys were suggested as an alternative reaching nearly the same strength and conductivity values as Cu-Be alloys, see Fig. 36 [935]. Early well documented investigations of mechanical properties of Cu-based alloys strengthened by Laves phase had already been reported in a study on directionally solidified Cu + MgCu2 alloyed with up to 4 at.% Ni [936].

Yield strength versus electrical conductivity for different Cu-based alloys (logarithmic plot) revealing a very good property combination for Cu + MgCu2 Laves phase alloys (adapted with permission from [935])
At the NASA Lewis Research Center (renamed to Glenn Research Center in 1999), Cu alloys strengthened by NbCr2 Laves phase were developed in the early 1990s as liners in rocket engine main combustion chambers [937,938,939,940,941,942,943,944]. Best properties were obtained for a Cu alloy containing 8 at.% Cr and 4 at.% Nb, which consists of a Cu matrix with about 14 vol.% of NbCr2 Laves phase. This alloy received the brand name GRCop-84 (for Glenn Research Center Copper-8Cr-4Nb) [944]. A comparison of mechanical properties between GRCop-84 and various other Cu-based alloys with high thermal conductivity proved it to be superior to the other alloys due to its high strength and creep resistance at temperatures above 500 °C and very good ductility at room temperature [945]. Parts of this material are usually processed by consolidation of powders followed by warm-rolling or extrusion (see, for example, [946, 947]), more recently material of this alloy was also produced by additive manufacturing techniques such as selective laser melting (SLM) [948, 949]. Figure 37a shows a micrograph of GRCop-84 powder particles revealing fine NbCr2 Laves precipitated in the pure copper matrix, and the right-hand photograph presents some parts obtained by vacuum plasma spraying of such powder [944].

a GRCop-84 powder obtained by gas atomization consisting of NbCr2 Laves phase particles embedded in a pure copper matrix. NbCr2 immediately precipitates from the molten metal when the melt temperature starts to drop. The volume fraction of NbCr2 particles is about 14%. b In addition to extrusion and HIPing, GRCop-84 can be vacuum plasma sprayed (VPS) into complex shapes. The photograph shows a full-scale space shuttle main engine (SSME) main combustion chamber (MCC) liner and some other parts as produced by NASA Marshall Space Flight Center (pictures reproduced from [944])
A Cu-Cr-Nb alloy containing about 0.6 at.% Cr and 0.1 at.% Nb (actual composition given in wt% in the reference: Cu-0.47Cr-0.16Nb) was cold rolled to 80% reduction after homogenization at 950 °C, and aged at 450 °C for 30 min. Besides NbCr2 Laves phase precipitates, the authors observed the formation and growth of Cr-rich precipitates during aging, resulting in a good strength of the alloy at the elevated temperature [950].
Cu-based Cu-Cr-Nb alloys containing Laves phase were also suggested as components in fusion reactors. As besides high-temperature strength and creep resistance combined with good thermal and electrical conductivity, also a suitable neutron irradiation resistance at 300–450 °C is needed, the effect of small additions of Zr (0.1 at.%) and variation of the Cr/Nb ratio was systematically investigated [951, 952].
Mg-based alloys
As a consequence of their excellent strength to weight ratio, Mg-based alloys are used in various ambient-temperature structural applications such as automotive, railway and aerospace; see, e.g., the review of Nie [953]. Thermodynamically stable binary Laves phases containing Mg are the AMg2 phases with A = Ca, Sr, Y, Ba and the MgB2 phases with B = Co, Ni, Cu, Zn [954]. For Mg-based materials, the ones of highest relevance are the hexagonal C14 Laves phases CaMg2 and MgZn2 as well as the cubic C15 Laves phase CaAl2 [953]. The system studied in most detail is Mg-Al-Ca. Besides the two Laves phases C14 CaMg2 and C15 CaAl2, as-cast and high-temperature heat-treated alloys of this system also contain ternary C36 Laves phase (approximate composition Ca(Al0.67Mg0.33)2), which decomposes into C14 + C15 Laves phase at lower temperatures [175, 176, 209, 955,956,957].
Three-phase Mg-Al-Ca alloys with a Mg matrix and a strengthening combination of C14 CaMg2 and C15 CaAl2 Laves phases were intensively discussed and characterized in detail as mechanical properties can be adjusted by the Ca/Al ratio, which controls the ratio and amount of the two Laves phase components; see, for example, [958,959,960,961,962] and references therein. According to Khorasani et al. [960, 961], CaAl2 Laves phase is harder than CaMg2 Laves, which explains the observation of decreasing tensile strength with increasing Ca/Al ratio in extruded material. A more complex behavior was reported by Zhang et al. [959], who performed room-temperature tensile tests and found decreasing ultimate tensile strength, elongation, and strain hardening rate with increasing Ca/Al ratio, while the yield strength showed an inverse tendency. Finally, Zubair et al. [962] reported that a higher Ca/Al ratio improves the yield strength and creep resistance, which mainly was explained by an increased volume fraction of Laves phase.
MgZn2-containing alloys were studied to a lesser extent. This is because Mg-based alloys with Zn can contain the Laves phase only in a metastable state since the Mg-richer phase MgZn should be in equilibrium with Mg. Examples for studies on the mechanical properties of such alloys are investigations on the deformation behavior of Mg95.9Zn3.5Gd0.6 and Mg94.4Zn3.5Gd0.6Cu1.5 alloys reinforced with MgZn2 Laves phase and the icosahedral quasicrystalline I phase [963, 964]. Quaternary Mg-Al-Zn-Ca alloys with 2 wt% Ca and 8 wt% Al + Zn contained a C15- and a C36-type Laves phase. Increasing the Al content increased the amount of C36 Laves phase, and the alloy containing the majority of networked C36 phase was found to show optimal mechanical properties at both room and elevated temperature [965].
A Mg-base alloy strengthened by Laves phase was also tested for application as biodegradable medical implants. Applying the metal injection molding (MIM) technique, orthopedic implants for biomedical applications were produced from pure Mg containing 0.9% Ca, which exhibited a Mg matrix with small C14 CaMg2 Laves phase precipitates [966].
Nb-based alloys
Because of the very high melting temperature of Nb of more than 2400 °C, Nb-based alloys are of interest for the development of materials for very high temperatures, not only to replace the currently applied Ni-based superalloys but also to go beyond. Nb forms a high-melting eutectic with the C15 Laves phase NbCr2 (1681 °C [967]) and microstructure and mechanical properties of respective Nb + NbCr2 alloys were studied by several authors [618, 627, 629, 913, 914, 968, 969]. To improve the fracture behavior, ternary additions such as Ti and Hf were shown to be beneficial [89, 623, 970,971,972,973].
Three-phase alloys consisting of Nb solid solution, NbCr2 Laves phase and Nb9(Si,Cr)5 silicide form a ternary eutectic and respective alloys were found to have a very high creep resistance [974]. High-temperature mechanical properties of Nb-silicide containing in situ composites consisting of (Nb), Nb5Si3, and NbCr2 Laves phase were reviewed by Bewlay et al. [975] and various further alloying additions such as Ti, Hf, Ge [976] or Ti, Hf, Mo, and Al [84, 977] were tested. However, fracture toughness remained as a very critical issue for these types of alloy impeding their application as structural materials.
Ti-based alloys
Laves phase containing, Ti-based alloys are especially of interest for hydrogen storage applications, see Sect. 4.1. Much less work was done regarding structural applications. Microstructure and mechanical properties of binary β-Ti (bcc) + C15-TiCr2 alloys with high volume fractions of Laves phase were studied by Chen et al. [381, 909, 910], but this concept was not further explored later on.
A very important Ti-based alloy being in use for various structural and biomedical applications is so-called Ti64 (Ti-6Al-4 V in wt%), which does not contain Laves phase. However, in an attempt to further improve the properties, an alloy of composition Ti-6.0Al-4.5Cr-1.5Mn (wt%) consisting of α-Ti (hcp) solid solution and TiCr2 Laves phase was developed and tested, but was found to show poor plasticity compared to other Ti-base alloys [978].
The mechanical properties of β-Ti-based Ti-Zr-Fe-X alloys (33–35 wt% Zr, 3–7 wt% Fe) with either C15 Laves phase (X = 2–4 wt%Cr [979,980,981]) or C14 Laves phase (X = 0–8 wt% Mn [981, 982] or 1–2 wt% Sn [983]) were investigated at room temperature by compression and Vickers hardness tests. From comparing the results, the authors conclude that addition of the C14 Laves phase increases the strength of the β-Ti-based alloys while the C15 Laves phase improves plastic deformability [981].
As a potential material for graded medical implants in spinal fixation surgeries, a Ti-xCr (9 ≤ x ≤ 28 at.%) graded alloy containing TiCr2 Laves phase was additively manufactured via laser-engineered net shaping. The as-deposited alloy consisted of α-Ti and Laves phase particles in a metastable β-Ti matrix with volume fractions changing along the concentration gradient. Heat treatments of the metastable material were applied to initiate partial transformation of the β-Ti matrix allowing to adjust the Young’s modulus [984].
Zr-based alloys
Zr is widely used in the nuclear industry because of its excellent corrosion resistance, low thermal neutron cross section, and good heat resistance. Since the construction of the first nuclear reactors, Zr-based alloys are applied as cladding material for uranium fuel rods and for other nuclear applications. Well-established alloys are Zircaloy-2 (approximate composition Zr-1.5Sn-0.2Fe-0.1Cr-0.05Ni, in wt%), Zircaloy-4 (same composition but no Ni and reduced Fe content to reduce hydrogen up-take) [985], and ZIRLO™ (Zr-1Nb-1Sn-0.1Fe) [986]. Owing to the alloyed Fe, Cr and Nb, these Zr alloys contain ZrFe2, Zr(Fe,Cr)2 or Zr(Fe,Nb)2 Laves phase precipitates, the presence and behavior of which strongly affect the materials properties. Therefore, detailed studies have been performed on structure and precipitation of the Laves phases [987,988,989,990,991,992,993] and on the irradiation behavior [994,995,996,997,998,999,1000,1001,1002]. It has long been known that irradiation by neutrons and bombardment by protons or various types of ions can result in amorphization of the Laves phase particles as was summarized in a review by Yan et al. [1002] (see Fig. 38). The authors assume that the irradiation initially produces single defects in the Laves phase structure and their accumulation finally results in amorphization. According to investigations by Shishov et al. [990, 999], neutron irradiation results in a transfer of Fe atoms from the Laves phase precipitates to the matrix leading to transformation and dissolution of Laves phase particles, while they did not observe any amorphization.

Zr-based, Laves phase containing alloys were also studied with respect to their behavior in nuclear waste management. Zr alloyed with 8 wt% stainless steel (Zr-8SS) was developed as a baseline waste form for Zr-based and Zircaloy-clad spent nuclear fuels [858, 861, 862, 1003]. The ZrFe2 Laves phase was found to incorporate and immobilize highly radioactive and long-lived constituents that are present in the waste forms [858].
Intermetallic-phase-based and HEA-based materials
Fe3Al-/FeAl-based alloys
Fe-Al alloys based on the aluminides Fe3Al and FeAl (which are the D03-ordered and B2-ordered variants of the disordered bcc-Fe(Al) solid solution) have long been considered as excellent candidates to replace heat-resistant steels or possibly even superalloys in high-temperature applications up to 800 °C especially because of their excellent oxidation- and corrosion resistance even in aggressive environments [1004,1005,1006,1007]. Compared to steels and superalloys they have a lower density and are a relatively cheap material, while they suffer from their poor high-temperature strength. Strengthening by intermetallic phases such as Laves phases is one of the concepts tested to improve the properties of iron aluminide-based alloys [1005, 1007,1008,1009]. Systems in which an iron aluminide matrix can exist in thermodynamic equilibrium with a Laves phase and which have been tested with respect to the mechanical properties are those with addition of one of the metals A = Nb [1010,1011,1012,1013,1014,1015,1016], Ta [1017,1018,1019,1020], Ti [805, 1021], or Zr [807, 1013, 1022,1023,1024,1025]. The resulting C14 A(Fe,Al)2 Laves phases either form as fine particles through precipitation from the supersaturated aluminide matrix, or—depending on the alloying amount—exist in a eutectic mixture with iron aluminide. A major problem for high temperature application of such alloys is the strong coarsening tendency of the incoherently growing Laves phase particles. In attempts to better control the precipitation and growth of Laves phase particles, small amounts of non-metallic elements such as B and C have been added resulting in formation of borides [1024, 1026] or carbides [1027,1028,1029]. In Fe-Al-Nb-B alloys containing 26–33 at.% Al, 1–2 at.% Nb, and 0.07 at.% B, the B-doping was found to lead to preferential precipitation of the Laves phase along grain boundaries and—with higher supersaturation of Nb in the Fe-Al matrix—to an even distribution of additional precipitates within the grains. However, no improvement of the high-temperature creep properties was reported [1026].
NiAl-based alloys
The cubic B2-ordered intermetallic phase NiAl was considered for gas turbine applications to replace Ni-base superalloy owing to its very high melting temperature, very good oxidation resistance, comparably low density, and a good thermal conductivity [1030, 1031]. To improve its insufficient high-temperature creep resistance, various alloying additions were tested for strengthening. This especially included the C14 Laves phase forming metals Nb [616, 1032,1033,1034] and Ta [616, 1034,1035,1036,1037,1038,1039]. The solubility of Ta in NiAl is very low and additions up to 3 at.% Ta result in Ta(Ni,Al)2 Laves phase precipitates primarily on the grain boundaries of the B2 NiAl grains. At higher Ta contents, the Laves phase can cover the grain boundaries completely to form a continuous skeleton [1036]. Nb and Ta can completely replace each other. In a systematic study, the role of the Nb/Ta ratio as well as the additional effect of other alloying additions such as Cr, Fe, or Si were investigated [1036, 1037, 1040]. Optimum properties for application at high temperatures above 1000 °C were achieved for an alloy with a composition NiAl-2.5Ta-7.5Cr (in at.%). This alloy, which was termed IP75 and later on also patented [1041], was found to show a good combination of high-temperature strength and creep resistance, tolerable brittleness at room temperature and excellent corrosion and thermo-shock resistance at temperatures up to 1350 °C [1034, 1038, 1039, 1042]. Prototype pins for application in diesel-engine pre-chambers were produced by powder injection molding, and cast IP 75 plates were brazed to a commercial brake disk for cars, which was tested successfully under service conditions [1039]. In addition, panels were produced by investment casting and by hot isostatic pressing [1043,1044,1045,1046], and tested as liners for the combustion chamber of stationary gas turbines [1044].
Properties and further possible applications of IP75 were discussed in several more recent investigations [1047,1048,1049,1050,1051,1052]. NiAl-Ta-Cr IP75 coatings were produced by thermal spraying applying the HVOF (high-velocity oxygen fuel) technique [1047], and fiber composites consisting of single-crystal α-Al2O3 fibers that were PVD-coated with IP75 and hot-pressed in vacuum were tested with respect to their mechanical properties [1048, 1049]. However, a thick amorphous layer formed at the interfaces where debonding occurred making this kind of composite material unsuitable for high-temperature applications [1048]. A Cr-free variant containing additions of Nb and consisting of NiAl grains with (Nb,Ta)(Ni,Al)2 Laves phase covering the grain boundaries was patented in 2015 for gas turbine applications [1053], but no further results about the materials’ properties were published.
TiAl-based alloys
γ-TiAl-based alloys are one of the most successful intermetallics-based structural materials, they found industrial application, e.g., as energy-saving turbine blades in the low-pressure section of jet engines. However, in contrast to FeAl and NiAl, they have a poor high-temperature oxidation resistance. Their Al content is too low to allow the formation of dense, oxidation- and corrosion-resistant Al2O3 surface layers, and instead fast growing TiO2 is formed. Interestingly, the substitution of 8 or more at.% Cr for Ti in Ti-Al was found to reduce the level of Al needed for protective Al2O3 scale formation [1054, 1055]. This so-called “Cr effect” results from formation of Ti(Cr,Al)2 Laves phase, which has a low oxygen permeability and is capable of alumina scale formation despite its relatively low Al content of 37–42 at.% Al [1054]. The beneficial effect of the formation of continuous Laves phase layers was later on confirmed in several studies, see, for example, [1056,1057,1058,1059]. Oxidation tests at 800 and 900 °C in air were performed on Ti-48Al-2Ag (at.%) alloys with Cr additions varying from 0 to 7 at.%. The best oxidation resistance was observed for the highest Cr content, as in this alloy a continuous Ti(Cr,Al)2 Laves phase layer existed beneath the external Al2O3 scale [1056]. Smaller amounts of Cr are not sufficient to result in continuous layers, but nevertheless Ti(Cr,Al)2 Laves phase forms as an equilibrium phase. This was shown for Ti-46.5Al-4(Cr,Nb,Ta,B)-0.22O (at.%), so-called γ-MET alloy, by thermodynamic calculations and TEM investigations revealing the formation of C14 Ti(Cr,Al)2 precipitates on α2/γ interfaces [1060, 1061]. Some efforts were also put into the application of Laves phase containing Ti-Al-Cr alloys as coatings for Ti-Al alloys [1058, 1059, 1062, 1063]. However, strong interdiffusion between substrate and coating at high temperatures can lead to depletion of that phase and results in poor oxidation resistance for long-term use [1059, 1063].
FeTi-based alloys
The observation of deformation-induced precipitation of C14 Laves phase in the B2 FeTi phase of a sintered Co-Fe-Ti alloy was reported in Ref. [1064]. An alloy with composition Ti-26.5Fe-10Co (at.%) was produced by spark plasma sintering and subsequently deformed under compression to a total strain of 5%. The sintered material consisted of B2 FeTi, bcc β-Ti, and cubic C15 TiCo2 Laves phase. After deformation to 5%, additional C14 TiFe2 Laves phase precipitates were found inside the B2 FeTi grains. TEM analysis revealed that these 30–100 nm sized Laves phase particles have coherently precipitated in dislocation walls within the deformed FeTi parent phase. Subsequent compression tests show a markedly increased strength and ductility compared to the sintered material. The authors conclude that the fine-scaled, deformation-induced Laves phase particles significantly contribute to this improved mechanical behavior [1064].
HEA-based alloys
The designation ‘high-entropy alloys’ (HEAs) was originally introduced for a group of multicomponent, equiatomic alloys forming single-phase bcc or fcc solid solutions [1065, 1066]. It was later on shown that high configurational entropy is not the critical factor for the formation of a single-phase multicomponent alloy [1067] and it actually would be better to use a more general term such as ‘multi-principal element alloys’ [1068]. Nevertheless, the notation HEA is kept until today and used for all kind of multicomponent, near-equiatomic alloys, which in many cases also contain secondary phases besides the bcc or fcc solid solution. Interestingly, it was found that these secondary phases very often are close-packed Frank–Kasper phases such as σ, µ, or Laves phases (see, for example, [1069]), and a more recent statistical analysis even proved Laves phases to be the most frequent intermetallic phase in HEAs [8]. As discussed in Ref. [181] (cf. also Sect. 3.3), the hexagonal C14 Laves phase crystal structure possesses a higher flexibility to accommodate atoms of different sizes when compared to the cubic C15 version. This might explain why the structure type of Laves phases in HEAs nearly always is of the C14 type. An exception to this rule (and the only one known to the present authors) are CrNbTiZr and CrNbTiVZr, which are reported to consist of bcc solid solution and C15 Laves phase [1070, 1071].
Discussions about criteria for the formation of Laves phases in HEAs revealed that atomic size difference, valence electron concentration, and enthalpy of mixing are the main factors defining their occurrence [9, 1072, 1073]. Gorban’ et al. [9] studied a large number of equiatomic HEAs regarding especially the role of the number of valence electrons for the occurrence of phases. Results for some HEAs with very high content of C14 Laves phase are listed in Table 6 (including some additional data from the literature), and Fig. 39 shows plots of Laves phase content as well as alloy hardness and modulus of elasticity as a function of the average valence electron concentration (VEC). The left diagram indicates a clear dependence of Laves phase content on the VEC. In a certain range between about 6 and 7 electrons/atom, single-phase C14 HEAs (sort of ‘high-entropy Laves phases’) can exist as was also confirmed in other studies [453, 1074, 1075]. Another interesting observation is that also the type of the disordered cubic solid solution coexisting with the C14 Laves phase depends on the VEC, i.e., for VECs below about 6 this is always bcc, while for VEC > 7 it is fcc. This behavior qualitatively agrees with a rule formulated by Guo et al. [1076] for single-phase, cubic solid solution HEAs saying that for VEC < 6.87 bcc is the stable structure while single-phase fcc solid solutions exist for VEC > 8.

Numerous examples for bcc + Laves phase (see, for example, [554, 557, 1071, 1077,1078,1079,1080]) as well as fcc + Laves phase (see, for example, [553, 1081,1082,1083,1084,1085,1086,1087,1088,1089,1090]) two-phase HEAs were reported in the literature. Sometimes ordering of the solid solution phase is observed as, for example, B2-ordering of the bcc solid solution in B2 + C14 Al-Cr-Nb-Ti-V-Zr [1091] or L12-ordering in the superalloy-type three-phase fcc + L12 + C14 HEA Al-Co-Cr-Fe-Nb-Ni [1092]. There are also several reports about three-phase HEAs consisting of C14 Laves phase and two different solid solutions such as bcc + fcc + C14 [1093, 1094], bcc + hcp + C14 [1095], and bcc1 + bcc2 + C14 [1078, 1096,1097,1098], some authors also added carbon for further strengthening by carbides [555, 556, 1099].
Regarding high-temperature structural applications, one focus is on two-phase fcc + C14 HEAs based on CoCrFeNi with additions of Nb [553, 1081,1082,1083,1084,1085, 1087, 1089, 1100], Hf [1090], Ta [1088, 1089], Mn + Nb [551, 1086], or Cu + Nb [550, 1101]. All of these alloys contain a fine eutectic mixture of a tough disordered fcc solid solution and a hard Laves phase the amount of which can be controlled by the added elements. Similar as for steels and other applied alloys, the occurrence of hard and brittle Laves phase precipitates was regarded as detrimental for a long time, see, for example, [1078]. However, today HEAs are intensively discussed as wear- and oxidation-resistant coatings for structural materials [1102], and the high hardness and excellent wear resistance of Laves phase have been recognized as very beneficial to improve the properties of such coatings [550,551,552,553,554,555,556,557].
Finally, it should be mentioned that HEAs are also under investigation as hydrogen storage material. As already discussed in Sect. 4.1, multi-element Laves phases—especially those based on Ti and/or Zr—can provide very good properties for hydrogen storage. Therefore, HEAs based on Laves phases might be natural candidates, and, e.g., a CrFeMnNiTiZr HEA containing 95 wt% C14 Laves phase can absorb and desorb 1.7 wt% of hydrogen at room temperature with a fast kinetics and without activation treatment [455]. However, in general HEAs were found to offer no especially remarkable hydrogen storage behavior compared to other Zr/Ti-based Laves phase alloys.
Conclusions and outlook
With the present review, we have tried to contribute to a better understanding of Laves phases in general and their potential for applications. Out of the huge number of possible crystal structures that can be formed from two or more metallic elements, the comparably simple and very frequently found Laves phase structures appear to play a very special role. The large number of representatives, their polytypism and their frequently extended homogeneity ranges along with their not too complicated crystal structure make Laves phases ideal candidates to study fundamentals of intermetallic phases.
Their structure, stability, and properties in case of deviation from the ideal composition were discussed here in some detail. Defects, which in most cases could be proven to be antisite atoms, may stabilize Laves phases to strongly off-stoichiometric compositions resulting in wide homogeneity ranges. The different number of available sublattices (i.e., of crystallographically different symmetry sites) of the three Laves phase structures C15 (1 A and 1 B site), C14 (1 A and 2 B sites), and C36 (2 A and 3 B sites) and the occurrence of site preference, i.e., preferred occupation of specific sublattices, result in an unequal stability of the three structure types in case of off-stoichiometry. This kind of higher structural flexibility of the hexagonal structure types can, at least qualitatively, explain the observation that A(B,C)2 Laves phases in ternary systems in most cases have the C14 structure and those of the (A,B)C2 type tend to adopt the C36 structure. For the same reason, Laves phases stabilized at off-stoichiometric compositions in binary systems as a second (or third) stable polytype are always C14-type Laves phases on the A-rich side, and C36 Laves phase on the B-rich side, while the cubic C15 variant is only stable at and around the stoichiometric composition.
Besides point defects, excess atoms resulting from off-stoichiometry can also be accommodated by planar defects. High-resolution TEM investigations of different kind allowed detailed investigations of structure and composition of such kind of faults revealing that they contain structural fragments of the neighboring intermetallic phases.
Another possible effect resulting from defects is related to atomic ordering or changes in the electronic structure. In such cases slight, regular distortions can result which lead to various new structure variants of the original Laves phase structure.
A very interesting and for some time controversially discussed point is the effect of off-stoichiometry on the mechanical behavior. It is now clearly proven that deviations from the ideal composition can result in softening in Laves phases. In case of the classical solid solution hardening, defects serve as obstacles for the movement of dislocations, while in Laves phases the presence of defects obviously assists the more complex plastic deformation process. Detailed investigations on plastic behavior and its composition dependence were reported in the literature, but still the reasons for the softening behavior are not completely clear.
The second focus of the review was on the role of Laves phases in functional and structural materials for real applications. The main goal was to provide a systematic overview for both classes of materials. On the one hand, this gives an idea about the manifold of possible applications, and on the other hand, it was also intended to show ‘the dark side of Laves phases’ as especially in many metal- or intermetallic phase-based structural materials, the presence of Laves phases can have a detrimental effect on the materials’ properties.
In summary, it can be stated that the fundamentals of Laves phases regarding their stability, structure and properties are still far from being completely understood, even though during recent years important new findings were reported, which led to a significant increase in knowledge about this kind of intermetallic phase. In particular, it was attempted to review and systematize this knowledge in view of the constituting elements by comparison of the data and observations for a multitude of Laves phases. Following the literature about applications of Laves phases, one finds a very diverse situation. In some cases, Laves phase materials are well established in application, as, for example, Tribaloy for wear- and corrosion-resistant coatings or Terfenol in magnetomechanical sensors and actuators. Then there is a second group of examples, in which the first step to applications has been done and material was successfully tested or is in use in some niche applications. Some examples for this group are Hydraloy and other Laves phase compounds for hydrogen storage, grain boundary precipitation-strengthened austenitic or HiperFer ferritic high-temperature steels, or IP75 as a liner for gas turbine combustion chambers. Finally, in many cases the potential of a Laves phase for a certain application has been well recognized, but the first steps into application are still missing. Due to the strong current and future need of improved or completely new materials, for example, for saving energy or reduction of greenhouse gases, one might expect and hope that the interesting, often still unexplored properties of Laves phases can help in future materials development for respective applications.
Notes
Already the preferred occupation of the two different sites in a C15 Laves phase by A and B atoms can be conceived as site preference or as ordering as it reduces configurational entropy. In terms of a notion used especially in [81], such a non-symmetry-breaking ordering (the space group of C15 Laves phase with randomly distributed A and B or by only one kind of atom would still be \( Fd\bar{3}m \)) is sometimes called non-convergent ordering. In contrast, convergent ordering involves change (reduction) of space group symmetry, because certain sites become inequivalent by symmetry due to unequal (i.e., ordered) average occupation by different types of atoms. Here we will use the terms site preference and ordering as synonyms. However, it will always be emphasized, whether this involves a breaking of symmetry with respect to some parent structure or not.
A broader view on site preferences in Frank–Kasper phases in general, showing similar phenomena also in σ phases was reported in a recent review [86].
It appears that the two-sublattice CEF approach according to the recommendations in Ref. [7] is sufficient to describe the thermodynamics of most systems. It must, however, be noted that the site preference described in Sect. 3.3 for binary and ternary C14 and C36 Laves phases cannot be described by such simplified models, and a simplified two-sublattice CEF description will lead to an incorrect description of the configurational entropy.
References
Laves F, Löhberg K (1934) Die Kristallstruktur von intermetallischen Verbindungen der Formel AB2 (The crystal structure of intermetallic compounds with formula AB2; in German). Nachr Ges Wiss Göttingen, Math-Phys Kl IV, Neue Folge 1(6):59–66
Laves F, Witte H (1935) Die Kristallstruktur des MgNi2 und seine Beziehungen zu den Typen des MgCu2 und MgZn2 (The crystal structure of MgNi2 and its relations to the MgCu2 and MgZn2 types; in German). Metallwirtsch Metallwiss Metalltech 14:645–649
Schulze GER (1939) Zur Kristallchemie der intermetallischen AB2-Verbindungen (Laves-Phasen) (About the crystal chemistry of the intermetallic AB2 compounds (Laves phases); in German). Z Elektrochem 45:849–865. https://doi.org/10.1002/bbpc.19390451202
Paufler P, Gustav ER (2006) Schulze’s pioneering work on Laves phases. Z Kristallogr 221:311–318. https://doi.org/10.1524/zkri.2006.221.5-7.311
Paufler P (2011) Early work on Laves phases in East Germany. Intermetallics 19:599–612. https://doi.org/10.1016/j.intermet.2010.11.032
Villars P, Calvert LD (1991) Pearson’s handbook of crystallographic data for intermetallic phases, 2nd edn. ASM International, Materials Park
Ansara I, Chart TG, Fernández Guillermet A, Hayes FH, Kattner UR, Pettifor DG, Saunders N, Zeng K (1997) Thermodynamic modelling of selected topologically close-packed intermetallic compounds. Calphad 21:171–218. https://doi.org/10.1016/S0364-5916(97)00021-7
Tsai M-H, Tsai R-C, Chang T, Huang W-F (2019) Intermetallic phases in high-entropy alloys: statistical analysis of their prevalence and structural inheritance. Metals 9:247. https://doi.org/10.3390/met9020247
Gorban’ VF, Krapivka NA, Firstov SA (2017) High-entropy alloys: interrelations between electron concentration, phase composition, lattice parameter, and properties. Phys Met Metallogr 118:970–981. https://doi.org/10.1134/S0031918X17080051
Livingston JD (1992) Laves-phase superalloys? Phys Status Solidi A 131:415–423. https://doi.org/10.1002/pssa.2211310215
Lukas HL, Fries SG, Sundman B (2007) Computational thermodynamics: the calphad method. Cambridge University Press, Cambridge ISBN 978-0521868112
Thoma DJ (2001) Intermetallics: Laves phases. In: Buschow KHJ, Cahn RW, Flemmings MC, Ilschner B, Kramer EJ, Mahajan S (eds) Encyclopedia of materials: science and technology. Elsevier, Amsterdam, pp 4205–4213. https://doi.org/10.1016/B0-08-043152-6/00739-7
Westbrook JH, Fleischer RL (2002) Intermetallic compounds, Vol. 1 - Principles (1995), Vol. 2 - Practice (1995), Vol. 3 - Progress. Wiley, Chichester
Kumar KS, Hazzledine PM (2004) Polytypic transformations in Laves phases. Intermetallics 12:763–770. https://doi.org/10.1016/j.intermet.2004.02.017
Liu CT, Zhu JH, Brady MP, McKamey CG, Pike LM (2000) Physical metallurgy and mechanical properties of transition-metal Laves phase alloys. Intermetallics 8:1119–1129. https://doi.org/10.1016/S0966-9795(00)00109-6
Stein F, Palm M, Sauthoff G (2004) Structure and stability of Laves phases Part I—Critical assessment of factors controlling Laves phase stability. Intermetallics 12:713–720. https://doi.org/10.1016/j.intermet.2004.02.010
Stein F, Palm M, Sauthoff G (2005) Structure and stability of Laves phases Part II—Structure type variations in binary and ternary systems. Intermetallics 13:1056–1074. https://doi.org/10.1016/j.intermet.2004.11.001
Ferro R, Saccone A (2008) Intermetallic chemistry. Elsevier, Amsterdam
Laves F (1942) Über den Einfluß geometrischer Faktoren auf die stöchiometrische Formel metallischer Verbindungen, gezeigt an der Kristallstruktur des KNa2 (About the effect of geometrical factors on the stoichiometric formula of metallic compounds, shown for the crystal structure of KNa2; in German). Z Anorg Allg Chem 250:110–120. https://doi.org/10.1002/zaac.19422500110
Schulze GER (1961) Dichte und Raumerfüllung bei intermetallischen Verbindungen, insbesondere Laves-Phasen (Density and space filling in intermetallic compounds, especially Laves phases; in German). Z Kristallogr 115:261–268. https://doi.org/10.1524/zkri.1961.115.3-4.261
Parthé E (1961) Space filling of crystal structures—a contribution to the graphical presentation of geometrical relationships in simple crystal structures. Z Kristallogr 115:52–79. https://doi.org/10.1524/zkri.1961.115.1-2.52
Frank FC, Kasper JS (1958) Complex alloy structures regarded as sphere packings. I. Definitions and basic principles. Acta Crystallogr 11:184–190. https://doi.org/10.1107/S0365110X58000487
Frank FC, Kasper JS (1959) Complex alloy structures regarded as sphere packings. II. Analysis and classification of representative structures. Acta Crystallogr 12:483–499. https://doi.org/10.1107/S0365110X59001499
Yarmolyuk YP, Kripyakevich PI (1974) Mean weighted coordination numbers and the origin of close-packed structures with atoms of unequal size but normal coordination polyhedra. Sov Phys Crystallogr 19:334–337
Shoemaker DP, Shoemaker CB (1986) Concerning the relative numbers of atomic coordination types in tetrahedrally close packed metal structures. Acta Crystallogr B 42:3–11. https://doi.org/10.1107/S0108768186098671
Aroyo MI (ed) (2016) International tables for crystallography, volume A: space-group symmetry, 6th edn. Wiley, New York. https://doi.org/10.1107/97809553602060000114
Komura Y (1962) Stacking faults and two new modifications of the Laves phase in Mg-Cu-Al system. Acta Crystallogr 15:770–778. https://doi.org/10.1107/S0365110X62002017
Komura Y, Tokunaga K (1980) Structural studies of stacking variants in Mg-base Friauf-Laves phases. Acta Crystallogr B 36:1548–1554. https://doi.org/10.1107/S0567740880006565
Komura Y, Kitano Y (1977) Long-period stacking variants and electron-concentration dependence in the Mg-base Friauf-Laves phases. Acta Crystallogr B 33:2496–2501. https://doi.org/10.1107/S0567740877008784
Zemp J, Celino M, Schönfeld B, Löffler JF (2015) Crystal-like rearrangements of icosahedra in simulated copper-zirconium metallic glasses and their effect on mechanical properties. Phys Rev Lett 115:165501. https://doi.org/10.1103/PhysRevLett.115.165501
Zemp J, Celino M, Schönfeld B, Löffler JF (2014) Icosahedral superclusters in Cu64Zr36 metallic glass. Phys Rev B 90:144108. https://doi.org/10.1103/PhysRevB.90.144108
Yamamoto K, Kimura Y, Mishima Y (2004) Precipitation of the icosahedral quasicrystalline phase, R-phase and Laves phase in ferritic alloys. Mater Trans 45:357–360. https://doi.org/10.2320/matertrans.45.357
Natarajan AR, Van der Ven A (2018) Connecting the simpler structures to topologically close-packed phases. Phys Rev Lett 121:255701. https://doi.org/10.1103/PhysRevLett.121.255701
Hajiw S, Pansu B, Sadoc J-F (2015) Evidence for a C14 Frank–Kasper phase in one-size gold nanoparticle superlattices. ACS Nano 9:8116–8121. https://doi.org/10.1021/acsnano.5b02216
Ghosh S, Basu J, Ramachandran D, Mohandas E, Vijayalakshmi M (2012) A unified approach to phase and microstructural stability for Fe-ETM alloys through Miedema’s model. Intermetallics 23:148–157. https://doi.org/10.1016/j.intermet.2011.11.016
Hasaka M, Nakashima H, Oki K (1984) Structure of the Laves phase observed in polystyrene latexes. Trans Jpn Inst Met 25:65–72. https://doi.org/10.2320/matertrans1960.25.65
Marinica MC, Willaime F, Crocombette JP (2012) Irradiation-induced formation of nanocrystallites with C15 Laves phase structure in bcc iron. Phys Rev Lett 108:025501. https://doi.org/10.1103/PhysRevLett.108.025501
Dézerald L, Marinica M-C, Ventelon L, Rodney D, Willaime F (2014) Stability of self-interstitial clusters with C15 Laves phase structure in iron. J Nucl Mater 449:219–224. https://doi.org/10.1016/j.jnucmat.2014.02.012
Alexander R, Marinica MC, Proville L, Willaime F, Arakawa K, Gilbert MR, Dudarev SL (2016) Ab initio scaling laws for the formation energy of nanosized interstitial defect clusters in iron, tungsten, and vanadium. Phys Rev B 94:024103. https://doi.org/10.1103/PhysRevB.94.024103
Zhang Y, Bai X-M, Tonks MR, Biner SB (2015) Formation of prismatic loops from C15 Laves phase interstitial clusters in body-centered cubic iron. Scr Mater 98:5–8. https://doi.org/10.1016/j.scriptamat.2014.10.033
Chartier A, Marinica MC (2019) Rearrangement of interstitial defects in alpha-Fe under extreme condition. Acta Mater 180:141–148. https://doi.org/10.1016/j.actamat.2019.09.007
Loubeyre P, Jean-Louis M, LeToullec R, Charon-Gérard L (1993) High pressure measurements of the He-Ne binary phase diagram at 296 K: evidence for the stability of a stoichiometric Ne(He)2 solid. Phys Rev Lett 70:178–181. https://doi.org/10.1103/PhysRevLett.70.178
Dewaele A, Rosa AD, Guignot N (2019) Argon-neon binary diagram and ArNe2 Laves phase. J Chem Phys 151:124708. https://doi.org/10.1063/1.5119419
Yan XZ, Chen YM, Geng HY (2019) Prediction of the reactivity of argon with xenon under high pressures. ACS Omega 4:13640–13644. https://doi.org/10.1021/acsomega.9b00638
Loubeyre P, Letoullec R, Pinceaux J-P (1994) Compression of Ar(H2)2 up to 175 GPa: a new path for the dissociation of molecular hydrogen? Phys Rev Lett 72:1360–1363. https://doi.org/10.1103/PhysRevLett.72.1360
Weck G, Dewaele A, Loubeyre P (2010) Oxygen/noble gas binary phase diagrams at 296 K and high pressures. Phys Rev B 82:014112. https://doi.org/10.1103/PhysRevB.82.014112
Laniel D, Weck G, Loubeyre P (2016) Xe(N2)2 compound to 150 GPa: reluctance to the formation of a xenon nitride. Phys Rev B 94:174109. https://doi.org/10.1103/PhysRevB.94.174109
Somayazulu MS, Finger LW, Hemley RJ, Mao HK (1996) High-pressure compounds in methane-hydrogen mixtures. Science 271:1400–1402. https://doi.org/10.1126/science.271.5254.1400
Cazorla C, Errandonea D, Sola E (2009) High-pressure phases, vibrational properties, and electronic structure of Ne(He)2 and Ar(He)2: a first-principles study. Phys Rev B 80:064105. https://doi.org/10.1103/PhysRevB.80.064105
Hynninen A-P, Filion L, Dijkstra M (2009) Stability of LS and LS2 crystal structures in binary mixtures of hard and charged spheres. J Chem Phys 131:064902. https://doi.org/10.1063/1.3182724
Schaertl N, Botin D, Palberg T, Bartsch E (2018) Formation of Laves phases in buoyancy matched hard sphere suspensions. Soft Matter 14:5130–5139. https://doi.org/10.1039/C7SM02348K
Lindquist BA, Jadrich RB, Truskett TM (2018) Communication: from close-packed to topologically close-packed: formation of Laves phases in moderately polydisperse hard-sphere mixtures. J Chem Phys 148:191101. https://doi.org/10.1063/1.5028279
van der Meer B, Smallenburg F, Dijkstra M, Filion L (2020) High antisite defect concentrations in hard-sphere colloidal Laves phases. Soft Matter 16:4155–4161. https://doi.org/10.1039/D0SM00335B
Hynninen A-P, Thijssen JHJ, Vermolen ECM, Dijkstra M, van Blaaderen A (2007) Self-assembly route for photonic crystals with a bandgap in the visible region. Nat Mater 6:202. https://doi.org/10.1038/nmat1841
Vermolen ECM, Thijssen JHJ, Moroz A, Megens M, Av Blaaderen (2009) Comparing photonic band structure calculation methods for diamond and pyrochlore crystals. Opt Express 17:6952–6961. https://doi.org/10.1364/OE.17.006952
Avvisati G, Dijkstra M (2016) Colloidal Laves phases as precursors of photonic crystals. arXiv:160307591v1 1–5
Avvisati G, Dasgupta T, Dijkstra M (2017) Fabrication of colloidal Laves phases via hard tetramers and hard spheres: bulk phase diagram and sedimentation behavior. ACS Nano 11:7702–7709. https://doi.org/10.1021/acsnano.7b00505
Dasgupta T, Dijkstra M (2018) Towards the colloidal Laves phase from binary hard-sphere mixtures via sedimentation. Soft Matter 14:2465–2475. https://doi.org/10.1039/C8SM00237A
Yoshimura S, Hachisu S (1983) Order formation in binary mixtures of monodisperse latices. Prog Colloid Polym Sci 68:59–70
Ma GH, Fukutomi T, Morone N (1994) Preparation and analysis of ordered structure of binary mixtures composed of poly(4-vinylpyridine) and polystyrene microgels. J Colloid Interface Sci 168:393–401. https://doi.org/10.1006/jcis.1994.1435
Cabane B, Li J, Artzner F, Botet R, Labbez C, Bareigts G, Sztucki M, Goehring L (2016) Hiding in plain view: colloidal self-assembly from polydisperse populations. Phys Rev Lett 116:208001. https://doi.org/10.1103/PhysRevLett.116.208001
Kim K, Schulze MW, Arora A, Lewis RM, Hillmyer MA, Dorfman KD, Bates FS (2017) Thermal processing of diblock copolymer melts mimics metallurgy. Science 356:520–523. https://doi.org/10.1126/science.aam7212
Kim K, Arora A, Lewis RM, Liu M, Li W, Shi A-C, Dorfman KD, Bates FS (2018) Origins of low-symmetry phases in asymmetric diblock copolymer melts. Proc Natl Acad Sci 115:847–854. https://doi.org/10.1073/pnas.1717850115
Mueller AJ, Lindsay AP, Jayaraman A, Lodge TP, Mahanthappa MK, Bates FS (2020) Emergence of a C15 Laves phase in diblock polymer/homopolymer blends. ACS Macro Lett 9:576–582. https://doi.org/10.1021/acsmacrolett.0c00124
Ducrot É, He M, Yi G-R, Pine DJ (2017) Colloidal alloys with preassembled clusters and spheres. Nature Mater 16:652–657. https://doi.org/10.1038/nmat4869
Baez-Cotto CM, Mahanthappa MK (2018) Micellar mimicry of intermetallic C14 and C15 Laves phases by aqueous lyotropic self-assembly. ACS Nano 12:3226–3234. https://doi.org/10.1021/acsnano.7b07475
Filion L, Dijkstra M (2009) Prediction of binary hard-sphere crystal structures. Phys Rev E 79:046714. https://doi.org/10.1103/PhysRevE.79.046714
Seiser B, Drautz R, Pettifor DG (2011) TCP phase predictions in Ni-based superalloys: structure maps revisited. Acta Mater 59:749–763. https://doi.org/10.1016/j.actamat.2010.10.013
Seiser B, Hammerschmidt T, Kolmogorov AN, Drautz R, Pettifor DG (2011) Theory of structural trends within 4d and 5d transition metal topologically close-packed phases. Phys Rev B 83:224116. https://doi.org/10.1103/PhysRevB.83.224116
Demchyna R, Leoni S, Rosner H, Schwarz U (2006) High-pressure crystal chemistry of binary intermetallic compounds. Z Kristallogr 221:420–434. https://doi.org/10.1524/zkri.2006.221.5-7.420
Kal S, Stoyanov E, Belieres JP, Groy TL, Norrestam R, Häussermann U (2008) High-pressure modifications of CaZn2, SrZn2, SrAl2, and BaAl2: implications for Laves phase structural trends. J Solid State Chem 181:3016–3023. https://doi.org/10.1016/j.jssc.2008.07.036
Cordier G, Czech E, Schäfer H (1982) Über eine Hochdruckmodifikation des SrAl2 (About a high pressure modification of SrAl2; in German). Z Naturforsch B 37:1442–1445. https://doi.org/10.1515/znb-1982-1115
Cordier G, Czech E, Schäfer H (1984) Zur Kenntnis der Hochdruckphase BaAl2 (On BaAl2, a high pressure phase; in German). Z Naturforsch B 39:421–423. https://doi.org/10.1515/znb-1984-0403
Tsvyashchenko AV, Menushenkov AP, Sidorov VA, Petrova AE, Fomicheva LN, Chernysheva OV, Lebed YB, Axenov SN, Bud’ko SL, Sun L, Zhao Z (2015) High pressure synthesis of a new phase of YbAg2: structure, valence of Yb and properties. J Alloys Compd 650:295–298. https://doi.org/10.1016/j.jallcom.2015.06.203
Lindbaum A, Hafner J, Gratz E, Heathman S (1998) Structural stability of YM2 compounds (M = Al, Ni, Cu) studied by ab initio total-energy calculations and high-pressure X-ray diffraction. J Phys: Condens Matter 10:2933–2945. https://doi.org/10.1088/0953-8984/10/13/011
Cannon JF, Robertson DL, Hall HT (1972) Synthesis of lanthanide-iron Laves phases at high pressures and temperatures. Mater Res Bull 7:5–12. https://doi.org/10.1016/0025-5408(72)90063-3
Tsvyashchenko AV, Popova SV (1985) New phases melt quenched under high pressure in R-Fe systems (R ≡ Pr, Sm, Dy, Tb, Ho, Er, Tm, Yb, Lu). J Less-Common Met 108:115–121. https://doi.org/10.1016/0022-5088(85)90436-9
Tsvyashchenko AV, Fomicheva LN, Magnitskaya MV, Sidorov VA, Kuznetsov AV, Eremenko DV, Trofimov VN (1998) New ferromagnetic compound CaCo2 (C15) synthesized at high pressure. J Exp Theor Phys Lett 68:908–914. https://doi.org/10.1134/1.567954
Hasegawa M, Atou T, Badding JV (1997) High-pressure synthesis of an alkali metal–transition metal Laves phase: KAg2. J Solid State Chem 130:311–315. https://doi.org/10.1006/jssc.1997.7391
Kienast G, Verma J, Klemm W (1961) Das Verhalten der Alkalimetalle zu Kupfer, Silber und Gold (The behavior of alkali metals towards Cu, Ag, and Au; in German). Z Anorg Allg Chem 310:143–169. https://doi.org/10.1002/zaac.19613100304
Thompson JB Jr (1969) Chemical reactions in crystals. Am Mineral 54:341–375
Watson RE, Bennett LH (1984) Transition-metal alloy formation. The occurrence of topologically close packed phases-I. Acta Metall 32:477–489. https://doi.org/10.1016/0001-6160(84)90058-0
Watson RE, Bennett LH (1984) Model predictions of volume contractions in transition-metal alloys and implications for Laves phase formation–II. Acta Metall 32:491–502. https://doi.org/10.1016/0001-6160(84)90059-2
Tsakiropoulos P (2018) Alloying and properties of C14–NbCr2 and A15–Nb3X (X = Al, Ge, Si, Sn) in Nb–silicide-based alloys. Materials 11:395. https://doi.org/10.3390/ma11030395
Yamanouchi T, Miura S (2018) Influence of atomic size factors on the phase stability of Laves phase in Nb-Cr-Ni-Al and Nb-V-Ni-Al phase diagrams. Mater Trans 59:546–555. https://doi.org/10.2320/matertrans.MJ201604
Joubert J-M, Crivello J-C (2012) Non-stoichiometry and Calphad modeling of Frank–Kasper phases. Appl Sci 2:669–681. https://doi.org/10.3390/app2030669
Joubert J-M, Andasmas M, Crivello J-C (2015) Irregular homogeneity domains in ternary intermetallic systems. Appl Sci 5:1570. https://doi.org/10.3390/app5041570
Spence JCH, Taftø J (1983) ALCHEMI: a new technique for locating atoms in small crystals. J Microsc 130:147–154. https://doi.org/10.1111/j.1365-2818.1983.tb04213.x
Thoma DJ, Nibur KA, Chen KC, Cooley JC, Dauelsberg LB, Hults WL, Kotula PG (2002) The effect of alloying on the properties of (Nb, Ti)Cr2 C15 Laves phases. Mater Sci Eng A 329–331:408–415. https://doi.org/10.1016/S0921-5093(01)01614-8
Mahdouk K, Gachon JC (2001) A thermodynamic study of the Al-Cr-Nb ternary system. J Alloys Compd 321:232–236. https://doi.org/10.1016/S0925-8388(01)00961-6
Zhao J-C, Jackson MR, Peluso LA (2004) Evaluation of phase relations in the Nb-Cr-Al system at 1000°C using a diffusion-multiple approach. J Phase Equilib 25:152–159. https://doi.org/10.1007/s11669-004-0008-x
He C, Stein F (2010) Thermodynamic assessment of the Cr-Al-Nb system. Int J Mater Res 101:1369–1375. https://doi.org/10.3139/146.110420
Prymak O, Stein F (2012) The ternary Cr-Al-Nb phase diagram: experimental investigations of isothermal sections at 1150, 1300 and 1450°C. J Alloys Compd 513:378–386. https://doi.org/10.1016/j.jallcom.2011.10.053
Zhao J-C, Jackson MR, Peluso LA (2003) Determination of the Nb-Cr-Si phase diagram using diffusion multiples. Acta Mater 51:6395–6405. https://doi.org/10.1016/j.actamat.2003.08.007
Prymak O, Stein F, Kerkau A, Ormeci A, Kreiner G, Frommeyer G, Raabe D (2009) Phase equilibria in the ternary Nb-Cr-Al system and site occupation in the hexagonal C14 Laves phase Nb(AlxCr1-x)2. Mater Res Soc Symp Proc 1128:499–504. https://doi.org/10.1557/PROC-1128-U08-11
Chu F, Thoma DJ, Kotula PG, Gerstl S, Mitchell TE, Anderson IM, Bentley J (1998) Phase stability and defect structure of the C15 Laves phase Nb(Cr, V)2. Acta Mater 46:1759–1769. https://doi.org/10.1016/S1359-6454(97)00291-7
Chu F, Zhu Q, Thoma DJ, Mitchell TE (1998) Structural and defect analysis of V-alloyed C15 NbCr2 from high resolution synchrotron X-ray powder diffraction. Philos Mag A 78:551–565. https://doi.org/10.1080/01418619808241922
Okaniwa H, Shindo D, Yoshida M, Takasugi T (1999) Determination of site occupancy of additives-X (X = V, Mo, W and Ti) in the Nb-Cr-X Laves phase by ALCHEMI. Acta Mater 47:1987–1992. https://doi.org/10.1016/S1359-6454(99)00065-8
Jacob A, Schmetterer C, Grüner D, Wessel E, Hallstedt B, Singheiser L (2015) The Cr–Fe–Nb ternary system: experimental isothermal sections at 700 °C, 1050 °C and 1350 °C. J Alloys Compd 648:168–177. https://doi.org/10.1016/j.jallcom.2015.06.137
Putland F, Chart T, Dinsdale A (1980) Thermodynamically calculated phase diagrams for the Co-Cr-Ta and Co-Cr-Nb systems. Calphad 4:133–141. https://doi.org/10.1016/0364-5916(80)90030-9
Zhang X, Yang S, Zhao CH, Lu Y, Liu X, Wang C (2013) Experimental Investigation of Phase Equilibria in the Co-Cr-Nb System at 1000, 1100, and 1200°C. J Phase Equilibr Diffus 34:313–321. https://doi.org/10.1007/s11669-013-0237-y
Nie XW, Lu SQ, Wang KL, Chen TC, Niu CL (2009) Fabrication and toughening of NbCr2 matrix composites alloyed with Ni obtained by powder metallurgy. Mater Sci Eng A 502:85–90. https://doi.org/10.1016/j.msea.2008.10.004
Jiang C (2007) Site preference of early transition metal elements in C15 NbCr2. Acta Mater 55:1599–1605. https://doi.org/10.1016/j.actamat.2006.10.020
Yao Q, Sun J, Zhang Y, Jiang B (2006) First-principles studies of ternary site occupancy in the C15 NbCr2 Laves phase. Acta Mater 54:3585–3591. https://doi.org/10.1016/j.actamat.2006.03.039
Yao Q, Sun J, Lin D, Liu S, Jiang B (2007) First-principles studies of defects, mechanical properties and electronic structure of Cr-based Laves phases. Intermetallics 15:694–699. https://doi.org/10.1016/j.intermet.2006.10.022
Wei Z, Yang Y, Huang J, Wu B, Sa B, Huang Y, Wang S, Lin M, Tsai C-Ty, Bai K (2018) Prediction of site occupancy of C15 Laves phase at finite temperature based on quasi-harmonic approximation model. Intermetallics 96:33–40. https://doi.org/10.1016/j.intermet.2018.02.007
Long Q, Wang J, Du Y, Nie X, Jin Z (2017) Site occupancy of transition elements in C15 NbCr2 Laves phase: a first-principles study. J Min Metall B 53:13–18. https://doi.org/10.2298/JMMB151115020L
Du Y, Chang YA, Gong W, Huang B, Xu H, Jin Z, Zhang F, Chen S-L (2003) Thermodynamic properties of the Al-Nb-Ni system. Intermetallics 11:995–1013. https://doi.org/10.1016/S0966-9795(03)00123-7
Yan X, Grytsiv A, Rogl P, Schmidt H, Giester G (2009) The ternary Laves phase Nb(Ni1-xAlx)2 with MgZn2-type. Calphad 33:11–16. https://doi.org/10.1016/j.calphad.2008.07.006
Lopes ESN, Dal Bó MR, Opini VC, de Mello MG, Rios CT, Fraser HL, Caram R (2019) Crystallographic features of the Al3Nb, Nb2Al and Nb(Ni1−XAlX)2 phases in a directionally solidified ternary eutectic microstructure. Mater Charact 147:303–310. https://doi.org/10.1016/j.matchar.2018.10.030
Zinkevich M, Mattern N (2002) Thermodynamic modeling of the Fe-Mo-Zr system. Acta Mater 50:3373–3383. https://doi.org/10.1016/S1359-6454(02)00140-4
Granovsky MS, Canay M, Lena E, Arias D (2002) Experimental investigation of the Zr corner of the ternary Zr-Nb-Fe phase diagram. J Nucl Mater 302:1–8. https://doi.org/10.1016/S0022-3115(02)00718-3
Ramos C, Saragovi C, Granovsky MS (2007) Some new experimental results on the Zr-Nb-Fe system. J Nucl Mater 366:198–205. https://doi.org/10.1016/j.jnucmat.2007.01.216
Chu F, Pope DP (1992) The Laves phase field in the Hf-V-Nb system. Scr Metall Mater 26:399–404. https://doi.org/10.1016/0956-716X(92)90619-P
Chu F, Lu Y-C, Kotula PG, Mitchell TE, Thoma DJ (1998) Phase stability and defect structure of the C15 phase in the Hf-V-Nb system. Philos Mag A 77:941–956. https://doi.org/10.1080/01418619808221221
Grytsiv A, Chen XQ, Witusiewicz VT, Rogl P, Podloucky R, Pomjakushin V, Maccio D, Saccone A, Giester G, Sommer F (2006) Atom order and thermodynamic properties of the ternary Laves phase Ti(TiyNixAl1-x-y)2. Z Kristallogr 221:334–348. https://doi.org/10.1524/zkri.2006.221.5-7.334
Yan X, Pieper MW, Michor H, Hilscher G, Reissner M, Grytsiv A, Rogl P, Pomjakushin V, Giester G, Bauer E, Paschen S (2013) Phase relations, crystal chemistry, and physical properties of MgZn2-type Laves phases in the Mn-Cu-Si and Mn-Ni-Si systems. Phys Rev B 88:174416. https://doi.org/10.1103/PhysRevB.88.174416
Krachler R, Ipser H, Komarek KL (1989) Thermodynamics of intermetallic B2-phases. A generalized model. J Phys Chem Solids 50:1127–1135. https://doi.org/10.1016/0022-3697(89)90022-X
de Novion C (1995) Point defects. In: Westbrook JH, Fleischer RL (eds) Intermetallic Compounds: principles and Practice, vol 1. Wiley, Chichester, pp 559–583
Cai W, Nix WD (2016) Imperfections in crystalline solids. Cambridge University Press, Cambridge ISBN 978-1107123137
Wieting J (1962) Geometrische Beziehungen zwischen einigen Strukturtypen intermetallischer AB2-Verbindungen (Geometric relations between some structure types of intermetallic AB2 compounds; in German). Monatsber Dt Akad Wissensch Berlin 4:603–606
Thoma DJ, Perepezko JH (1992) An experimental evaluation of the phase relationships and solubilities in the Nb-Cr system. Mater Sci Eng A 156:97–108. https://doi.org/10.1016/0921-5093(92)90420-6
Foley JC, Thoma DJ, Perepezko JH (1994) Supersaturation of the AI2Y laves phase by rapid solidification. Metall Mater Trans A 25:230–233. https://doi.org/10.1007/BF02646693
Thoma DJ, Perepezko JH (1995) A geometric analysis of solubility ranges in Laves phases. J Alloys Compd 224:330–341. https://doi.org/10.1016/0925-8388(95)01557-4
Hari Kumar KC, Wollants P, Delaey L (1994) Thermodynamic reassessment and calculation of Fe-Ti phase diagram. Calphad 18:223–234. https://doi.org/10.1016/0364-5916(94)90028-0
Flandorfer H, Rogl P, Groebner J, Stamou A, Hassiotis N, Saccone A, Maccio D, Ferro R, Wouters R, Delaey L, Seifert H, Effenberg G, Haidemenopoulos G (1997) Experimental investigation and thermodynamic calculation of the ternary system Mn-Y-Zr. Z Metallkd 88:529–538
Paufler P, Schulze GER (1971) Über einige physikalische Eigenschaften von Laves-Phasen, insbesondere MgZn2 (About some physical properties of Laves phases, especially MgZn2; in German). Z Kristallogr 133:7–17. https://doi.org/10.1524/zkri.1971.133.16.7
Nayeb-Hashemi AA, Clark JB (1984) The Cu-Mg (copper-magnesium) system. Bull Alloy Phase Diagrams 5:36–43. https://doi.org/10.1007/BF02868722
Bagnoud P, Feschotte P (1978) Les systemes binaires magnesium-cuivre et magnesium-nickel en particulier non-stoechiométrie des phases de Laves MgCu2 et MgNi2 (The binary magnesium-copper and magnesium-nickel systems, especially the non-stoichiometric Laves phases MgCu2 and MgNi2; in French). Z Metallkd 69:114–120
Andersson M, de Boissieu M, Brühne S, Drescher C, Assmus W, Ohahshi S, Tsai AP, Mihalkovič M, Krajčí M, Rapp Ö (2010) Electronic and structural properties of Laves-phase MgZn2 of varying chemical disorder. Phys Rev B 82:024202. https://doi.org/10.1103/PhysRevB.82.024202
Shao L, Shi T-T, Zheng J, Wang H-C, Pan X-Z, Tang B-Y (2016) First-principles study of point defects in C14 MgZn2 Laves phase. J Alloys Compd 654:475–481. https://doi.org/10.1016/j.jallcom.2015.09.142
Zheng J, Tian X, Shao L, Pan X-Z, Tang P-Y, Tang B-Y (2016) Point defects and Zn-doping in defective Laves phase C15 MgCu2: a first-principles study. Comput Mater Sci 122:159–166. https://doi.org/10.1016/j.commatsci.2016.05.026
Tian X, Wang J-N, Wang Y-P, Shi X-F, Tang B-Y (2017) First-principles investigation of point defect and atomic diffusion in Al2Ca. J Phys Chem Solids 103:6–12. https://doi.org/10.1016/j.jpcs.2016.11.017
Shao L, Shi T-T, Zheng J, Pan X-Z, Tang B-Y (2015) The native point defects in C14 Mg2Ca Laves phase: a first-principles study. Intermetallics 65:29–34. https://doi.org/10.1016/j.intermet.2015.05.010
Gschneidner KA Jr, Pecharsky VA (2006) Binary rare earth Laves phases—an overview. Z Kristallogr 221:375–381. https://doi.org/10.1524/zkri.2006.221.5-7.375
Klimyenko AV, Seuntjens J, Miller LL, Beaudry BJ, Jacobson RA, Gschneidner KA (1988) Structure of LaNi2.286 and the La-Ni system from LaNi1.75 to LaNi2.50. J Less-Common Met 144:133–141. https://doi.org/10.1016/0022-5088(88)90357-8
Latroche M, Paul-Boncour V, Percheron-Guegan A, Achard JC (1990) Structure determination of Y0.95Ni2 by X-ray powder diffraction. J Less-Common Met 161:L27–L31. https://doi.org/10.1016/0022-5088(90)90334-G
Gratz E, Kottar A, Lindbaum A, Mantler M, Latroche M, Paul-Boncour V, Acet M, Barner C, Holzapfel WB, Pacheco V, Yvon K (1996) Temperature- and pressure-induced structural transitions in rare-earth-deficient (R = Y, Sm, Gd, Tb) Laves phases. J Phys: Condens Matter 8:8351–8361. https://doi.org/10.1088/0953-8984/8/43/026
Lindbaum A, Gratz E, Heathman S (2002) Pressure-induced order-disorder transitions in RNi2 compounds. Phys Rev B 65:134114. https://doi.org/10.1103/PhysRevB.65.134114
Lindbaum A, Heathman S, Gratz E (2002) Pressure induced disordering of vacancies in RNi2 compounds. Int J High Press Res 22:411–414. https://doi.org/10.1080/08957950212805
Paul-Boncour V, Lindbaum A, Latroche M, Heathman S (2006) Homogeneity range and order-disorder transitions in R1−xNi2 Laves phase compounds. Intermetallics 14:483–490. https://doi.org/10.1016/j.intermet.2005.08.003
Paul-Boncour V, Lindbaum A (2002) Ab initio calculations on the formation of La1−xNi2 compounds. J Phys: Condens Matter 14:3921–3929. https://doi.org/10.1088/0953-8984/14/15/306
Crivello J-C, Zhang J, Latroche M (2011) Structural stability of ABy phases in the (La, Mg)–Ni system obtained by density functional theory calculations. J Phys Chem C 115:25470–25478. https://doi.org/10.1021/jp204835z
Lindbaum A, Hafner J, Gratz E (1999) Ab initio studies of the formation of a Y1−xNi2 superstructure with ordered Y vacancies. J Phys: Condens Matter 11:1177–1187. https://doi.org/10.1088/0953-8984/11/5/006
Krčmar M, Fu CL (2007) Point defect structures of YAl2 and ZrCo2 Laves phase compounds by first-principles calculations. Intermetallics 15:15–20. https://doi.org/10.1016/j.intermet.2006.02.004
Sekido N, Yamabe-Mitarai Y (2010) Growth behavior and interfacial character of Ir3Y precipitates in the Ir2Y Laves phase matrix. Mater Trans 51:447–454. https://doi.org/10.2320/matertrans.MBW200901
Baumann W, Leineweber A (2010) Solid solubility by anti-site atoms in the C36-TiCr2 Laves phase revealed by single-crystal X-ray diffractometry. J Alloys Compd 505:492–496. https://doi.org/10.1016/j.jallcom.2010.06.117
Fleischer RL (1992) Substitutional solutes in the Laves phase intermetallic compounds Cr2Zr and Cu2Mg—solution hardening and defect structures. Scr Metall Mater 27:799–804. https://doi.org/10.1016/0956-716X(92)90395-U
Krčmar M, Fu CL (2003) First-principles study of point-defect structures in C15 ZrCo2 and ZrCr2 and B2 ZrCo. Phys Rev B 68:134110–134118. https://doi.org/10.1103/PhysRevB.68.134110
Chen XQ, Wolf W, Podloucky R, Rogl P (2005) Ab initio study of ground-state properties of the Laves phase compounds TiCr2, ZrCr2, and HfCr2. Phys Rev B 71:174101. https://doi.org/10.1103/PhysRevB.71.174101
Zhu JH, Pike LM, Liu CT, Liaw PK (1999) Point-defects in binary Laves phase alloys. Acta Mater 47:2003–2018. https://doi.org/10.1016/S1359-6454(99)00090-7
Zhu JH, Pike LM, Liu CT, Liaw PK (1998) Point defects in binary NbCr2 Laves-phase alloys. Scr Mater 39:833–838. https://doi.org/10.1016/S1359-6462(98)00263-2
Fruchart D, Soubeyroux JL, Hempelmann R (1984) Neutron diffraction in Ti1.2Mn1.8 deuteride: structural and magnetic aspects. J Less-Common Met 99:307–319. https://doi.org/10.1016/0022-5088(84)90229-7
Chen XQ, Wolf W, Podloucky R, Rogl P, Marsman M (2005) Ab initio study of ground-state properties of the Laves-phase compound ZrMn2. Phys Rev B 72:054411–054440
Grüner D (2007) Untersuchungen zur Natur der Laves-Phasen in Systemen der Übergangsmetalle (Investigations on the nature of Laves phases in transition metal systems; in German). Doct Thesis, TU Dresden, Germany. https://nbn-resolving.de/urn:nbn:de:swb:14-1172078219643-48967:1-276
Brückner W, Kleinstück K, Schulze GER (1967) Atomic arrangement in the homogeneity range of the Laves phases ZrFe2 and TiFe2. Phys Status Solidi 23:475–480. https://doi.org/10.1002/pssb.19670230206
Zhu JH, Liu CT (2000) Defect structures in ZrCo2 Laves phase. Acta Mater 48:2339–2347. https://doi.org/10.1016/S1359-6454(00)00014-8
Chen KC, Peterson EJ, Thoma DJ (2001) HfCo2 Laves phase intermetallics—Part I: solubility limits and defect mechanisms. Intermetallics 9:771–783. https://doi.org/10.1016/S0966-9795(01)00068-1
Saito S, Beck PA (1960) Co-rich intermediate phases in the Cb-Co system. Trans Metall Soc AIME 218:670–674
Grüner D, Stein F, Palm M, Konrad J, Ormeci A, Schnelle W, Grin Y, Kreiner G (2006) Preparation, phase stability, and structure of the C36 Laves phase Nb1−xCo2+x. Z Kristallogr 221:319–333. https://doi.org/10.1524/zkri.2006.221.5-7.319
Stein F, Jiang D, Palm M, Sauthoff G, Grüner D, Kreiner G (2008) Experimental reinvestigation of the Co-Nb phase diagram. Intermetallics 16:785–792. https://doi.org/10.1016/j.intermet.2008.02.017
Kreiner G, Grüner D, Grin Y, Stein F, Palm M, Ormeci A (2009) Structure and disorder of the Laves phases in the Co-Nb system. Mater Res Soc Symp Proc 1128:487–492. https://doi.org/10.1557/PROC-1128-U08-08
Zhou GF, Bakker H (1995) Mechanically induced structural and magnetic changes in the GdAl2 Laves phase. Phys Rev B 52:9437–9445. https://doi.org/10.1103/PhysRevB.52.9437
Modder IW, Bakker H (1997) On quadruple-defect formation in C15 Laves phases. Phys Status Solidi B 199:369–378. https://doi.org/10.1002/1521-3951(199702)199:2%3C369:AID-PSSB369%3E3.0.CO;2-E
Modder IW, Kuin MJ, Bakker H (1998) Estimates of the formation enthalpies of point defects in intermetallic compounds with the C15 structure and comparison with experiments. Intermetallics 6:537–546. https://doi.org/10.1016/S0966-9795(97)00101-5
Modder IW, Bakker H, Zhou GF (1999) Defects and magnetism in cubic GdX2 Laves phase compounds. Phys B (Amsterdam, Neth) 262:141–158. https://doi.org/10.1016/S0921-4526(98)00657-7
Mehrer H (2014) Self-diffusion, solute-diffusion and interdiffusion in binary intermetallics. Diffus Found 2:1–72. https://doi.org/10.4028/www.scientific.net/DF.2.1
Mehrer H, Sprengel W (2013) Reaction diffusion and interdiffusion in some binary metallic systems. Philos Mag 93:1971–1986. https://doi.org/10.1080/14786435.2012.734637
Baheti VA, Roy S, Ravi R, Paul A (2013) Interdiffusion and the phase boundary compositions in the Co–Ta system. Intermetallics 33:87–91. https://doi.org/10.1016/j.intermet.2012.09.020
Balam SSK, Paul A (2011) Study of interdiffusion and growth of topologically closed packed phases in the Co–Nb system. J Mater Sci 46:889–895. https://doi.org/10.1007/s10853-010-4831-7
Kitano Y, Akitoh S, Mitarai M, Ohnishi K, Noguchi K, Numata M (1998) New stacking variant of Laves phase found in (Ti0.95V0.05)Co2 alloy. Microsc Res Tech 40:277–283. https://doi.org/10.1002/(SICI)1097-0029(19980215)40:4%3C277:AID-JEMT4%3E3.0.CO;2-P
Trigunayat GC (1991) A survey of the phenomenon of polytypism in crystals. Solid State Ionics 48:3–70. https://doi.org/10.1016/0167-2738(91)90200-U
Laves F, Witte H (1936) Der Einfluß von Valenzelektronen auf die Kristallstruktur ternärer Magnesiumlegierungen (The effect of valence electrons on the crystal structure of magnesium alloys; in German). Metallwirtsch, Metallwiss, Metalltech 15:840–842
Zhu JH, Liu CT, Liaw PK (1999) Phase stability and mechanical behavior of NbCr2-based Laves phases. Intermetallics 7:1011–1016. https://doi.org/10.1016/S0966-9795(99)00010-2
Amerioun S, Simak SI, Häussermann U (2003) Laves phase structural changes in the system CaAl2−xMgx. Inorg Chem 42:1467–1474. https://doi.org/10.1021/ic020596m
Amerioun S, Yokosawa T, Lidin S, Häussermann U (2004) Phase stability in the systems AeAl2−xMgx (Ae = Ca, Sr, Ba): electron concentration and size controlled variations on the Laves phase structural theme. Inorg Chem 43:4751–4760. https://doi.org/10.1021/ic0400235
Thimmaiah S, Miller GJ (2015) Influence of valence electron concentration on Laves phases: structures and phase stability of pseudo-binary MgZn2−xPdx. Z Anorg Allg Chem 641:1486–1494. https://doi.org/10.1002/zaac.201500197
Siggelkow L, Hlukhyy V, Fässler TF (2017) The influence of the valence electron concentration on the structural variation of the Laves phases MgNi2−xGex. Z Anorg Allg Chem 643:1424–1430. https://doi.org/10.1002/zaac.201700180
Yuan F, Toombs A, Miller GJ, Mozharivskyj Y (2018) Gd(Co1−xGax)2: synthesis, crystal structures, and investigation of structural transformations and magnetic properties. J Solid State Chem 264:68–76. https://doi.org/10.1016/j.jssc.2018.05.009
Johannes RL, Haydock R, Heine V (1976) Phase stability in transition-metal Laves phases. Phys Rev Lett 36:372–376. https://doi.org/10.1103/PhysRevLett.36.372
Stein F (2011) Consequences of crystal structure differences between C14, C15, and C36 Laves phase polytypes for their coexistence in transition-metal-based systems. Mater Res Soc Symp Proc 1295:299–310. https://doi.org/10.1557/opl.2011.211
Dovbenko O, Stein F, Palm M, Prymak O (2010) Experimental determination of the ternary Co-Al-Nb phase diagram. Intermetallics 18:2191–2207. https://doi.org/10.1016/j.intermet.2010.07.004
Kerkau A, Grüner D, Ormeci A, Prots Y, Borrmann H, Schnelle W, Bischoff E, Grin Y, Kreiner G (2009) Site occupation reversal in the C14 Laves phase Nb(Cr1−xCox)2. Z Anorg Allg Chem 635:637–648. https://doi.org/10.1002/zaac.200801411
Kerkau A (2012) Disorder in Laves phases. Doct Thesis, TU Dresden, Germany. https://nbn-resolving.org/urn:nbn:de:bsz:14-qucosa-109917:1-227
Shinagawa K, Chinen H, Omori T, Oikawa K, Ohnuma I, Ishida K, Kainuma R (2014) Phase equilibria and thermodynamic calculation of the Co–Ta binary system. Intermetallics 49:87–97. https://doi.org/10.1016/j.intermet.2014.01.015
Stein F, Merali M, Watermeyer P (2019) The Co–Ti system revisited: about the cubic-to-hexagonal Laves phase transformation and other controversial features of the phase diagram. Calphad 67:101681. https://doi.org/10.1016/j.calphad.2019.101681
Williams RO (1984) The calculation of coherent phase equilibria. Calphad 8:1–14. https://doi.org/10.1016/0364-5916(84)90024-5
Cahn JW, Larché F (1984) A simple model for coherent equilibrium. Acta Metall 32:1915–1923. https://doi.org/10.1016/0001-6160(84)90173-1
Liu Z-K, Ågren J (1990) On two-phase coherent equilibrium in binary alloys. Acta Metall Mater 38:561–572. https://doi.org/10.1016/0956-7151(90)90210-8
Allen CW, Delavignette P, Amelinckx S (1972) Electron microscopic study of the Laves phases TiCr2 and TiCo2. Phys Status Solidi A 9:237–246. https://doi.org/10.1002/pssa.2210090127
Aufrecht J, Baumann W, Leineweber A, Duppel V, Mittemeijer EJ (2010) Layer-stacking irregularities in C36-type Nb-Cr and Ti-Cr Laves phases and their relation with polytypic phase transformations. Philos Mag 90:3149–3175. https://doi.org/10.1080/14786435.2010.482068
Baumann W, Leineweber A, Mittemeijer EJ (2010) The influence of plastic deformation on polytypic phase transformations in TiCr2 Laves phases. Scr Mater 63:1041–1044. https://doi.org/10.1016/j.scriptamat.2010.07.020
Baumann W, Leineweber A, Mittemeijer EJ (2011) The kinetics of a polytypic Laves phase transformation in TiCr2. Intermetallics 19:526–535. https://doi.org/10.1016/j.intermet.2010.11.027
Murray JL (1981) The Cr-Ti (chromium-titanium) system. Bull Alloy Phase Diagrams 2:174–181. https://doi.org/10.1007/BF02881474
Aufrecht J, Leineweber A, Duppel V, Mittemeijer EJ (2011) Polytypic transformations of the HfCr2 Laves phase—part I: structural evolution as a function of temperature, time and composition. Intermetallics 19:1428–1441. https://doi.org/10.1016/j.intermet.2011.05.011
Aufrecht J, Leineweber A, Mittemeijer EJ (2011) Polytypic transformations of the HfCr2 Laves phase—part II: kinetics of the polymorphic C14 - > C15 transformation. Intermetallics 19:1442–1447. https://doi.org/10.1016/j.intermet.2011.05.010
Aufrecht J, Leineweber A, Duppel V, Mittemeijer EJ (2010) Transformation–dislocation dipoles in Laves phases: a high-resolution transmission electron microscopy analysis. J Mater Res 25:1983–1991. https://doi.org/10.1557/JMR.2010.0265
Aufrecht J, Leineweber A, Senyshyn A, Mittemeijer EJ (2010) The absence of a stable hexagonal Laves phase modification (NbCr2) in the Nb-Cr system. Scr Mater 62:227–230. https://doi.org/10.1016/j.scriptamat.2009.11.002
Leineweber A, Aufrecht J, Senyshyn A, Mittemeijer EJ (2011) Reply to comments on “The absence of a stable hexagonal Laves phase modification (NbCr2) in the Nb-Cr system”. Scr Mater 64:994–997. https://doi.org/10.1016/j.scriptamat.2011.01.035
Aufrecht J, Leineweber A, Mittemeijer EJ (2009) Metastable hexagonal modifications of the NbCr2 Laves phase as function of cooling rate. Mater Res Soc Symp Proc 1128:481–486. https://doi.org/10.1557/PROC-1128-U08-07
Allen CW, Liao KC (1982) Dislocation models for shear transformations. Phys Status Solidi A 74:673–681. https://doi.org/10.1002/pssa.2210740236
Ormeci A, Chu F, Wills JM, Mitchell TE, Albers RC, Thoma DJ, Chen SP (1996) Total-energy study of electronic structure and mechanical behavior of C15 Laves phase compounds: NbCr2 and HfV2. Phys Rev B 54:12753–12762. https://doi.org/10.1103/PhysRevB.54.12753
Pavlů J, Vřešťál J, Šob M (2010) Thermodynamic modeling of Laves phases in the Cr-Hf and Cr-Ti systems: reassessment using first-principles results. Calphad 34:215–221. https://doi.org/10.1016/j.calphad.2010.03.003
Sun J, Jiang B (2004) Ab initio calculation of the phase stability, mechanical properties and electronic structure of ZrCr2 Laves phase compounds. Philos Mag 84:3133–3144. https://doi.org/10.1080/14786430410001720345
Chen XQ, Wolf W, Podloucky R, Rogl P, Marsman M (2004) A new polymorphic material? Structural degeneracy of ZrMn2. Europhys Lett 67:807–813. https://doi.org/10.1209/epl/i2004-10119-4
Pavlů J, Vřešťál J, Chen XQ, Rogl P (2011) Thermodynamic modeling of Laves phases in the Ta-V system: reassessment using first-principles results. Calphad 35:103–108. https://doi.org/10.1016/j.calphad.2010.12.002
Hong S, Fu CL (1999) Phase stability and elastic moduli of Cr2Nb by first-principles calculations. Intermetallics 7:5–9. https://doi.org/10.1016/S0966-9795(98)00005-3
Pavlů J, Vřešťál J, Šob M (2009) Re-modeling of Laves phases in the Cr-Nb and Cr-Ta systems using first-principles results. Calphad 33:179–186. https://doi.org/10.1016/j.calphad.2008.04.006
Zhong Y, Liu J, Witt RA, Sohn YH, Liu ZK (2006) Al2(Mg, Ca) phases in Mg-Al-Ca ternary system: first-principles prediction and experimental identification. Scr Mater 55:573–576. https://doi.org/10.1016/j.scriptamat.2006.03.068
Zhong Y, Luo AA, Sofo JO, Liu Z-K (2005) First-principles investigation of Laves phases in Mg-Al-Ca system. Mater Sci Forum 488–489:169–176. https://doi.org/10.4028/www.scientific.net/MSF.488-489.169
He L-Z, Zhu J, Zhang L (2018) First-principles study of structural phase transition, electronic, elastic and thermodynamic properties of C15-type Laves phase TiCr2 under pressure. Phys B (Amsterdam, Neth) 531:79–84. https://doi.org/10.1016/j.physb.2017.11.051
Schön CG, Tenório JAS (1996) The chemistry of the iron-niobium intermetallics. Intermetallics 4:211–216. https://doi.org/10.1016/0966-9795(95)00035-6
Kuprava A, Saenko I, Fabrichnaya O (2020) Heat capacity measurement of C14–ZrMn2 and thermodynamic re-assessment of the Mn–Zr system. Calphad 68:101745. https://doi.org/10.1016/j.calphad.2020.101745
Brando M, Kerkau A, Todorova A, Yamada Y, Khuntia P, Förster T, Burkhard U, Baenitz M, Kreiner G (2016) Quantum phase transitions and multicriticality in Ta(Fe1−xVx)2. J Phys Soc Jpn 85:084707. https://doi.org/10.7566/JPSJ.85.084707
Voß S, Stein F, Palm M, Raabe D (2010) Synthesis of defect-free single-phase bars of high-melting Laves phases through modified cold crucible levitation melting. Mater Sci Eng A 527:7848–7853. https://doi.org/10.1016/j.msea.2010.08.066
Stein F, Sauthoff G, Palm M (2002) Experimental determination of intermetallic phases, phase equilibria, and invariant reaction temperatures in the Fe-Zr system. J Phase Equilib 23:480–494. https://doi.org/10.1361/105497102770331172
Stein F, Sauthoff G, Palm M (2004) Phases and phase equilibria in the Fe-Al-Zr system. Z Metallkd 95:469–485
Bodega J, Garcés G, Leardini F, Ares JR, Fernández JF, Adeva P, Sánchez C (2010) A new metastable crystalline phase in the Cr–Zr system. Intermetallics 18:1099–1101. https://doi.org/10.1016/j.intermet.2009.12.002
Lundin LM (1996) Direct measurement of carbon solubility in the intermetallic (Fe,Cr)2(Mo,W) Laves phase using atom-probe field-ion microscopy. Scr Mater 34:741–747. https://doi.org/10.1016/1359-6462(95)00569-2
Hättestrand M, Schwind M, Andrén H-O (1998) Microanalysis of two creep resistant 9–12% chromium steels. Mater Sci Eng A 250:27–36. https://doi.org/10.1016/S0921-5093(98)00532-2
Ladines AN, Drautz R, Hammerschmidt T (2017) Ab-initio study of C and N point defects in the C14-Fe2Nb phase. J Alloys Compd 693:1315–1322. https://doi.org/10.1016/j.jallcom.2016.10.030
Kaufman L, Nesor H (1978) Coupled phase diagrams and thermochemical data for transition metal binary systems-I. Calphad. https://doi.org/10.1016/0364-5916(78)90005-6
Coughanowr CA, Ansara I, Luoma R, Hämäläinen M, Lukas HL (1991) Assessment of the Cu-Mg system. Z Metallkd 82:574–581
Agarwal R, Fries SG, Lukas HL, Petzow G, Sommer F, Chart TG, Effenberg G (1992) Assessment of the Mg-Zn system. Z Metallkd 83:216–223
Chart T, Putland F (1979) A thermodynamically calculated phase diagram for the Co-Cr-Zr system. Calphad 3:9–18. https://doi.org/10.1016/0364-5916(79)90017-8
Hillert M (2001) The compound energy formalism. J Alloys Compd 320:161–176. https://doi.org/10.1016/S0925-8388(00)01481-X
Sundman B, Chen Q, Du Y (2018) A review of Calphad modeling of ordered phases. J Phase Equilib Diffus 39:678–693. https://doi.org/10.1007/s11669-018-0671-y
Zhuang W, Shen J, Liu Y, Ling L, Shang S, Du Y, Schuster JC (2000) Thermodynamic optimization of the Cr-Ti system. Z Metallkd 91:121–127
Zeng K, Hämäläinen M, Lilius K (1993) Thermodynamic modeling of the Laves phases in the Cr-Zr system. Calphad 17:101–107. https://doi.org/10.1016/0364-5916(93)90038-D
Jonsson S (1998) Assessment of the Fe-Ti system. Metall Mater Trans B 29:361–370. https://doi.org/10.1007/s11663-998-0113-z
Dupin N, Ansara I (1993) Thermodynamic assessment of the Cr-Ta system. J Phase Equilibr 14:451–456. https://doi.org/10.1007/BF02671963
Janz A, Gröbner J, Cao H, Zhu J, Chang YA, Schmid-Fetzer R (2009) Thermodynamic modeling of the Mg–Al–Ca system. Acta Mater 57:682–694. https://doi.org/10.1016/j.actamat.2008.10.037
Liang P, Tarfa T, Robinson JA, Wagner S, Ochin P, Harmelin MG, Seifert HJ, Lukas HL, Aldinger F (1998) Experimental investigation and thermodynamic calculation of the Al–Mg–Zn system. Thermochim Acta 314:87–110. https://doi.org/10.1016/S0040-6031(97)00458-9
Buhler T, Fries SG, Spencer PJ, Lukas HL (1997) Modelling non-stoichiometric phases in the system Al-Cu-Mg-Zn. J Chim Phys 94:1043–1048. https://doi.org/10.1051/jcp/1997941043
Buhler T, Fries SG, Spencer PJ, Lukas HL (1998) A thermodynamic assessment of the Al-Cu-Mg ternary system. J Phase Equilibr 19:317–333. https://doi.org/10.1361/105497198770342058
Wasiur-Rahman S, Medraj M (2009) Critical assessment and thermodynamic modeling of the binary Mg–Zn, Ca–Zn and ternary Mg–Ca–Zn systems. Intermetallics 17:847–864. https://doi.org/10.1016/j.intermet.2009.03.014
Cupid DM, Kriegel MJ, Fabrichnaya O, Ebrahimi F, Seifert HJ (2011) Thermodynamic assessment of the Cr-Ti and first assessment of the Al-Cr-Ti systems. Intermetallics 19:1222–1235. https://doi.org/10.1016/j.intermet.2011.03.031
Dumitrescu LFS, Hillert M, Saunders N (1998) Comparison of Fe-Ti assessments. J Phase Equilibr 19:441–448. https://doi.org/10.1361/105497198770341923
Cacciamani G, Ferro R, Ansara I, Dupin N (2000) Thermodynamic modelling of the Co-Ti system. Intermetallics 8:213–222. https://doi.org/10.1016/S0966-9795(99)00098-9
Zeng K, Schmid-Fetzer R, Huneau B, Rogl P, Bauer J (1999) The ternary system Al-Ni-Ti Part II: thermodynamic assessment and experimental investigation of polythermal phase equilibria. Intermetallics 7:1347–1359. https://doi.org/10.1016/S0966-9795(99)00055-2
Saenko I, Kuprava A, Udovsky A, Fabrichnaya O (2019) Heat capacity measurement of Zr2Fe and thermodynamic re-assessment of the Fe–Zr system. Calphad 66:101625. https://doi.org/10.1016/j.calphad.2019.05.002
Costa Neto JG, Fries SG, Lukas HL, Gama S, Effenberg G (1993) Thermodynamic optimisation of the Nb-Cr system. Calphad 17:219–228. https://doi.org/10.1016/0364-5916(93)90001-R
He C, Stein F, Palm M (2015) Thermodynamic description of the systems Co–Nb, Al–Nb and Co–Al–Nb. J Alloys Compd 637:361–375. https://doi.org/10.1016/j.jallcom.2015.02.182
Liu ZK, Chang YA (1999) Thermodynamic assessment of the Co-Ta system. Calphad 23:339–356. https://doi.org/10.1016/S0364-5916(00)00005-5
Sluiter MHF (2006) Ab initio lattice stabilities of some elemental complex structures. Calphad 30:357–366. https://doi.org/10.1016/j.calphad.2006.09.002
Sluiter MHF (2007) Lattice stability prediction of elemental tetrahedrally close-packed structures. Acta Mater 55:3707–3718. https://doi.org/10.1016/j.actamat.2007.02.016
Zhou S, Wang Y, Shi FG, Sommer F, Chen L-Q, Liu Z-K, Napolitano RE (2007) Modeling of thermodynamic properties and phase equilibria for the Cu-Mg binary system. J Phase Equilibr Diffus 28:158–166. https://doi.org/10.1007/s11669-007-9022-0
Brubaker CO, Liu Z-K (2004) A computational thermodynamic model of the Ca–Mg–Zn system. J Alloys Compd 370:114–122. https://doi.org/10.1016/j.jallcom.2003.08.097
Ozturk K, Zhong Y, Liu Z-K, Luo AA (2003) Creep resistant Mg-Al-Ca alloys: computational thermodynamics and experimental investigation. JOM 55:40–44. https://doi.org/10.1007/s11837-003-0208-6
Zhong Y, Luo AA, Sofo JO, Liu Z-K (2004) Laves phases in Mg-Al-Ca alloys. In: Luo AA (ed) Magnesium Technology 2004, Charlotte, NC, US. TMS, Warrendale, pp 317–323
Zhang H, Shang S-L, Wang Y, Chen L-Q, Liu Z-K (2012) Thermodynamic properties of Laves phases in the Mg–Al–Ca system at finite temperature from first-principles. Intermetallics 22:17–23. https://doi.org/10.1016/j.intermet.2011.08.019
Lu H-J, Wang W-B, Zou N, Shen J-Y, Lu X-G, He Y-L (2015) Thermodynamic modeling of Cr–Nb and Zr–Cr with extension to the ternary Zr–Nb–Cr system. Calphad 50:134–143. https://doi.org/10.1016/j.calphad.2015.06.002
Lafaye P, Toffolon-Masclet C, Crivello J-C, Joubert J-M (2020) Thermodynamic modelling of the Fe–Sn–Zr system based on new experiments and first-principles calculations. J Alloys Compd 821:153200. https://doi.org/10.1016/j.jallcom.2019.153200
Lieser AC, Zacherl CL, Saengdeejing A, Liu Z-K, Kecskes LJ (2012) First-principles calculations and thermodynamic re-modeling of the Hf–W system. Calphad 38:92–99. https://doi.org/10.1016/j.calphad.2012.04.005
Schmetterer C, Khvan A, Jacob A, Hallstedt B, Markus T (2014) A new theoretical study of the Cr-Nb system. J Phase Equilib Diffus 35:434–444. https://doi.org/10.1007/s11669-014-0313-y
Jiang Y, Zomorodpoosh S, Roslyakova I, Zhang L (2018) Thermodynamic re-assessment of binary Cr-Nb system down to 0 K. Calphad 62:109–118. https://doi.org/10.1016/j.calphad.2018.06.001
Jacob A, Schmetterer C, Khvan A, Kondratiev A, Ivanov D, Hallstedt B (2016) Liquidus projection and thermodynamic modeling of the Cr-Fe-Nb ternary system. Calphad 54:1–15. https://doi.org/10.1016/j.calphad.2016.04.013
Rajkumar VB, Hari Kumar KC (2015) Gibbs energy modeling of Fe–Ta system by Calphad method assisted by experiments and ab initio calculations. Calphad 48:157–165. https://doi.org/10.1016/j.calphad.2014.12.006
Jiang Y, Zomorodpoosh S, Roslyakova I, Zhang L (2019) Thermodynamic re-assessment of the binary Cr–Ta system down to 0 K. Int J Mater Res 110:797–807. https://doi.org/10.3139/146.111812
Wang P, Koßmann J, Kattner UR, Palumbo M, Hammerschmidt T, Olson GB (2019) Thermodynamic assessment of the Co-Ta system. Calphad 64:205–212. https://doi.org/10.1016/j.calphad.2018.12.002
Crivello J-C, Joubert J-M, Mohri T (2019) Atomic interactions in C15 Laves phases. J Mater Sci 54:4742–4753. https://doi.org/10.1007/s10853-018-3169-4
Nishihara Y, Yamaguchi Y (1983) Magnetic phase transitions in itinerant electron magnets Hf1−xTaxFe2. J Phys Soc Jpn 52:3630–3636. https://doi.org/10.1143/JPSJ.52.3630
Diop LVB, Amara M, Isnard O (2013) Large magnetovolume effects due to transition from the ferromagnetic to antiferromagnetic state in Hf0.825Ta0.175Fe2 intermetallic compound. J Phys: Condens Matter 25:416007. https://doi.org/10.1088/0953-8984/25/41/416007
Li B, Luo XH, Wang H, Ren WJ, Yano S, Wang CW, Gardner JS, Liss KD, Miao P, Lee SH, Kamiyama T, Wu RQ, Kawakita Y, Zhang ZD (2016) Colossal negative thermal expansion induced by magnetic phase competition on frustrated lattices in Laves phase compound (Hf, Ta)Fe2. Phys Rev B 93:224405. https://doi.org/10.1103/PhysRevB.93.224405
Diop LVB, Isnard O, Amara M, Gay F, Itié JP (2020) Giant negative thermal expansion across the first-order magnetoelastic transition in Hf0.86Ta0.14Fe2. J Alloys Compd 845:156310. https://doi.org/10.1016/j.jallcom.2020.156310
Ouyang ZW, Wang FW, Huang Q, Liu WF, Xiao YG, Lynn JW, Liang JK, Rao GH (2005) Magnetic structure, magnetostriction, and magnetic transitions of the Laves-phase compound NdCo2. Phys Rev B 71:064405. https://doi.org/10.1103/PhysRevB.71.064405
Xiao YG, Huang Q, Ouyang ZW, Lynn JW, Liang JK, Rao GH (2006) Crystal and magnetic structures of Laves phase compound NdCo2 in the temperature range between 9 and 300K. J Alloys Compd 420:29–33. https://doi.org/10.1016/j.jallcom.2005.10.073
Mudryk Y, Paudyal D, Pathak AK, Pecharsky VK, Gschneidner KA (2016) Balancing structural distortions via competing 4f and itinerant interactions: a case of polymorphism in magnetocaloric HoCo2. J Mater Chem C 4:4521–4531. https://doi.org/10.1039/C6TC00867D
Ritter C, Cywinski R, Kilcoyne SH (1997) Structural distortions in doped YMn2 compounds. Phys B (Amsterdam, Neth) 234–236:596–598. https://doi.org/10.1016/S0921-4526(96)01204-5
Dwight AE, Kimball CW (1974) TbFe2, a rhombohedral Laves phase. Acta Crystallogr B 30:2791–2793. https://doi.org/10.1107/S0567740874008156
Liu X, Lin K, Gao Q, Zhu H, Li Q, Cao Y, Liu Z, You L, Chen J, Ren Y, Huang R, Lapidus SH, Xing X (2018) Structure and phase transformation in the giant magnetostriction Laves-phase SmFe2. Inorg Chem 57:689–694. https://doi.org/10.1021/acs.inorgchem.7b02525
Cullen JR, Clark AE (1977) Magnetostriction and structural distortion in rare-earth intermetallics. Phys Rev B 15:4510–4515. https://doi.org/10.1103/PhysRevB.15.4510
Newnham RE (1998) Phase transformations in smart materials. Acta Crystallogr A 54:729–737. https://doi.org/10.1107/S010876739800912X
Lindbaum A, Heathman S, Le Bihan T, Rogl P (2000) Pressure-induced orthorhombic distortion of UMn2. J Alloys Compd 298:177–180. https://doi.org/10.1016/S0925-8388(99)00627-1
Lawson AC, Smith JL, Willis JO, O’Rourke JA, Faber J, Hitterman RL (1985) Orthorhombic structure of UMn2 at low temperatures. J Less-Common Met 107:243–248. https://doi.org/10.1016/0022-5088(85)90083-9
Hatt BA (1961) The crystal structure of URe2. Acta Crystallogr 14:119–123. https://doi.org/10.1107/S0365110X61000516
Moncton DE (1973) Lattice transformation in the superconductivity ZrV2 by neutron diffraction. Solid State Commun 13:1779–1782. https://doi.org/10.1016/0038-1098(73)90728-X
Chu F, Thoma DJ, Mitchell TE, Lin CL, Šob M (1998) Phase stability of C15 MV2 (M = Zr, Hf or Ta): an electronic structure investigation. Philos Mag B 77:121–136. https://doi.org/10.1080/13642819808206387
Lawson AC, Zachariasen WH (1972) Low temperature lattice transformation of HfV2. Phys Lett A 38:1. https://doi.org/10.1016/0375-9601(72)90948-6
Zhao Y, Chu F, Von Dreele RB, Zhu Q (2000) Structural phase transitions of HfV2 at low temperatures. Acta Crystallogr B 56:601–606. https://doi.org/10.1107/s0108768100003633
Krcmar M, Fu CL (2013) Effect of lattice anharmonicity in the structural phase transformation of Laves phase HfV2 alloy: a first-principles investigation. Acta Mater 61:7473–7480. https://doi.org/10.1016/j.actamat.2013.08.059
Keuter P, Music D, Stuer M, Schneider JM (2019) Temperature and impurity induced stabilization of cubic HfV2 Laves phase. Condens Matter 4:63. https://doi.org/10.3390/condmat4030063
Feng J, Ashcroft NW, Hoffmann R (2007) Theoretical indications of singular structural and electronic features of Laves-phase CaLi2 under pressure. Phys Rev Lett 98:247002. https://doi.org/10.1103/PhysRevLett.98.247002
Schoop LM, Xie LS, Chen R, Gibson QD, Lapidus SH, Kimchi I, Hirschberger M, Haldolaarachchige N, Ali MN, Belvin CA, Liang T, Neaton JB, Ong NP, Vishwanath A, Cava RJ (2015) Dirac metal to topological metal transition at a structural phase change in Au2Pb and prediction of Z2 topology for the superconductor. Phys Rev B 91:214517. https://doi.org/10.1103/PhysRevB.91.214517
Ponou S, Müller N, Fässler TF, Häussermann U (2005) KBi2−xPbx (0 < x < 1): a Zintl phase evolving from a distortion of the cubic Laves-phase structure. Inorg Chem 44:7423–7430. https://doi.org/10.1021/ic050603h
Noréus D, Eriksson L, Göthe L, Werner PE (1985) Structure determination of Mg2SiNi3. J Less-Common Met 107:345–349. https://doi.org/10.1016/0022-5088(85)90093-1
Keimes V, Mewis A (1992) Strukturvarianten des MgCu2-Typs: Die Verbindungen Mg2Ni3P und Mg2Ni3As/Variants of the MgCu2 type: The compounds Mg2Ni3P and Mg2Ni3As. Z Naturforsch B 47:1351–1354. https://doi.org/10.1515/znb-1992-1002
Doverbratt I, Ponou S, Lidin S (2013) Ca2Pd3Ge, a new fully ordered ternary Laves phase structure. J Solid State Chem 197:312–316. https://doi.org/10.1016/j.jssc.2012.09.003
Toombs A, Miller GJ (2018) Rhombohedral distortion of the cubic MgCu2-type structure in Ca2Pt3Ga and Ca2Pd3Ga. Crystals 8:186. https://doi.org/10.3390/cryst8050186
Cenzual K, Chabot B, Parthé E (1987) Y2Rh3Ge, a rhombohedral substitution variant of the MgCu2 type. J Solid State Chem 70:229–234. https://doi.org/10.1016/0022-4596(87)90061-2
Seidel S, Janka O, Benndorf C, Mausolf B, Haarmann F, Eckert H, Heletta L, Pöttgen R (2017) Ternary rhombohedral Laves phases RE2Rh3Ga (RE = Y, La–Nd, Sm, Gd–Er). Z Naturforsch B 72:289. https://doi.org/10.1515/znb-2016-0265
Vernière A, Lejay P, Bordet P, Chenavas J, Brison JP, Haen P, Boucherle JX (1994) Crystal structures and physical properties of some new ternary compounds U2T3X (T = Ru, Os; X = Si, Ge). J Alloys Compd 209:251–255. https://doi.org/10.1016/0925-8388(94)91108-8
Matsunaga T, Kodera E, Komura Y (1984) A new ordered structure of the C15-type Laves phase, Mg28.4Cu57.9Si13.7. Acta Crystallogr C 40:1668–1670. https://doi.org/10.1107/S0108270184009100
Stegemann F, Benndorf C, Zhang Y, Bartsch M, Zacharias H, Fokwa BPT, Eckert H, Janka O (2017) Network formation by condensed tetrahedral [Au3Al] units in Na2Au3Al: crystal and electronic structure, spectroscopic investigations, and physical properties of an ordered ternary auride. Inorg Chem 56:1919–1931. https://doi.org/10.1021/acs.inorgchem.6b02480
Osters O, Nilges T, Schöneich M, Schmidt P, Rothballer J, Pielnhofer F, Weihrich R (2012) Cd4Cu7As, the first representative of a fully ordered, orthorhombically distorted MgCu2 Laves phase. Inorg Chem 51:8119–8127. https://doi.org/10.1021/ic3005213
Witte H (1939) Untersuchungen im System Magnesium-Kupfer-Silicium mit besonderer Berücksichtigung des Schnittes MgCu2–MgSi2 (Investigations in the magnesium-copper-silicon system with special emphasis on the section MgCu2–MgSi2; in German). Metallwirtsch, Metallwiss, Metalltech 18:459–463
Range K-J, Rau F, Klement U (1990) Na2LiAu3, eine ternäre Laves-Phase vom MgZn2-Typ (Na2LiAu3, a ternary Laves phase with MgZn2-type structure; in German). Z Naturforsch B 45b:1333–1334. https://doi.org/10.1515/znb-1990-0918
Kotur BY, Gratz E, Bauer E, Hilscher G, Kottar A, Michor H, Reichl C, Wiesinger G, Markosyan AS (1998) Magnetic and electrical properties of Sc(M1−xSix)2−y (M = Fe Co, Ni). J Alloys Compd 278:49–59. https://doi.org/10.1016/S0925-8388(98)00642-2
Le Bihan T, Levet JC, Noël H (1996) Crystal structure and magnetic behavior of the Laves-type phases U4Mo5Si3 and U4Cr6Si2. J Solid State Chem 121:479–482. https://doi.org/10.1006/jssc.1996.0066
Melnyk G, Tremel W (2003) The titanium–iron–antimony ternary system and the crystal and electronic structure of the interstitial compound Ti5FeSb2. J Alloys Compd 349:164–171. https://doi.org/10.1016/S0925-8388(02)00921-0
Yan X, Chen XQ, Grytsiv A, Witusiewicz VT, Rogl P, Podloucky R, Pomjakushin V, Giester G (2006) Site preference, thermodynamic, and magnetic properties of the ternary Laves phase Ti(Fe1−xAlx)2 with the crystal structure of the MgZn2-type. Z Metallkd 97:450–460. https://doi.org/10.3139/146.101238
Yan XL, Chen XQ, Grytsiv A, Witusiewicz VT, Rogl P, Podloucky R, Giester G (2007) On the ternary Laves phases {Sc, Ti}2M3Si (M = Cr, Mn, Fe Co, Ni) with MgZn2-type. J Alloys Compd 429:10–18. https://doi.org/10.1016/j.jallcom.2006.03.086
Henriques MS, Tougait O, Noel H, Pereira LCJ, Waerenborgh JC, Gonçalves AP (2008) Evidence of uranium magnetic ordering on U2Fe3Ge. Solid State Commun 148:159–162. https://doi.org/10.1016/j.ssc.2008.07.029
Hoffmann R-D, Pöttgen R, Chevalier B, Gaudin E, Matar SF (2013) The ternary germanides UMnGe and U2Mn3Ge. Solid State Sci 21:73–80. https://doi.org/10.1016/j.solidstatesciences.2013.04.006
Kudo K, Hiiragi H, Honda T, Fujimura K, Idei H, Nohara M (2020) Superconductivity in Mg2Ir3Si: A Fully Ordered Laves Phase. J Phys Soc Jpn 89:013701. https://doi.org/10.7566/JPSJ.89.013701
Eustermann F, Stegemann F, Gausebeck S, Janka O (2018) Structural and magnetic investigations of the pseudo-ternary RE2TAl3 series (RE = Sc, Y, La–Nd, Sm, Gd–Lu; T = Ru, Rh, Ir)—size dependent formation of two different structure types. Z Naturforsch B 73:819–830. https://doi.org/10.1515/znb-2018-0124
Heletta L, Block T, Klenner S, Pöttgen R (2019) Ternary transition metal gallides with TiNiSi, ZrBeSi and MgZn2-type structure. Z Naturforsch B. https://doi.org/10.1515/znb-2019-0002
Pyon S, Kudo K, Matsumura J-I, Ishii H, Matsuo G, Nohara M, Hojo H, Oka K, Azuma M, Garlea VO, Kodama K, Shamoto S-I (2014) Superconductivity in noncentrosymmetric iridium silicide Li2IrSi3. J Phys Soc Jpn 83:093706. https://doi.org/10.7566/JPSJ.83.093706
Seidel S, Pöttgen R (2017) Yb6Ir5Ga7-A MgZn2 superstructure. Z Anorg Allg Chem 643:261–265. https://doi.org/10.1002/zaac.201600422
Eustermann F, Pominov A, Pöttgen R (2018) Rare earth (RE) gallides with closely related compositions: REIrGa and RE6Ir5Ga7. Z Anorg Allg Chem 644:1297–1303. https://doi.org/10.1002/zaac.201800287
Conrad M, Pohling C, Webert H, Harbrecht B (2006) Atomic ordering in the Laves phases L1 V(Co1−xSix)2 (x = 0.43 and 0.56). Z Kristallogr 221:349–356. https://doi.org/10.1524/zkri.2006.221.5-7.349
Misch L (1935) Die Kristallstruktur des AuBe5 und PdBe5 und ihre Beziehung zur kubischen AB2-Struktur (The crystal structure of AuBe5 and PdBe5 and their relation to the cubic AB2-structure; in German). Metallwirtsch, Metallwiss, Metalltech 14:897–899
Baenziger NC, Rundle RE, Snow AI, Wilson AS (1950) Compounds of uranium with the transition metals of the first long period. Acta Crystallogr 3:34–40. https://doi.org/10.1107/S0365110X50000082
Babizhetskyy V, Myakush O, Simon A, Kotur B (2013) X-ray investigation of the Y-Zr-Ni system at 870 K. Intermetallics 38:44–48. https://doi.org/10.1016/j.intermet.2013.02.017
Kadir K, Noréus D, Yamashita I (2002) Structural determination of AMgNi4 (where A = Ca, La, Ce, Pr, Nd and Y) in the AuBe5 type structure. J Alloys Compd 345:140–143. https://doi.org/10.1016/S0925-8388(02)00323-7
Provino A, Paudyal D, Fornasini ML, Dhiman I, Dhar SK, Das A, Mudryk Y, Manfrinetti P, Pecharsky VK (2013) Unexpected crystal and magnetic structures in MnCu4In and MnCu4Sn. Acta Mater 61:2236–2243. https://doi.org/10.1016/j.actamat.2012.12.043
Percheron-Guégan A, Paul-Boncour V, Latroche M, Achard JC, Bouree-Vigneron F (1991) Structure of Y0.95Ni2 and its hydride. J Less-Common Met 172–174:198–205. https://doi.org/10.1016/0022-5088(91)90448-D
Dutour Sikirić M, Delgado-Friedrichs O, Deza M (2010) Space fullerenes: a computer search for new Frank–Kasper structures. Acta Crystallogr A 66:602–615. https://doi.org/10.1107/s0108767310022932
Ye HQ, Li DX, Kuo KH (1985) Domain structures of tetrahedrally close-packed phases with juxtaposed pentagonal antiprisms I. Structure description and HREM images of the C14 Laves and μ phases. Philos Mag A 51:829–837. https://doi.org/10.1080/01418618508237590
Wagner V, Conrad M, Harbrecht B (1995) The μ-phase of Co6.3Nb6.7. Acta Crystallogr C 51:1241–1243. https://doi.org/10.1107/S0108270195000552
Yarmolyuk YP, Kripyakevich PI, Mel’nik EV (1975) Crystal structure of the compound Mg4Zn7. Sov Phys Crystallogr 20:329–331
Singh A, Rosalie JM (2018) Lattice correspondence and growth structures of monoclinic Mg4Zn7 phase growing on an icosahedral quasicrystal. Crystals 8:194. https://doi.org/10.3390/cryst8050194
Parthé E, Lemaire R (1975) Structure block stacking in intermetallic compounds. I. The rhombohedral-hexagonal Mn+1X5n−1 and the monoclinic-hexagonal-trigonal-orthorhombic Mn+1X5n+2 structure series. Acta Crystallogr B 31:1879–1889. https://doi.org/10.1107/S0567740875006413
Gladyshevskii EI, Bodak O (1995) Topologically close-packed structures. In: Westbrook JH, Fleischer RL (eds) Intermetallic compounds: principles and practice, vol 1. Wiley, Chichester, pp 403–418
Fornasini ML, Bruzzone G (1975) The crystal structure of the Ba7Al13 phase. J Less-Common Met 40:335–340. https://doi.org/10.1016/0022-5088(75)90078-8
Manyako NB, Zarechnyuk OS, Yanson TI (1987) Crystal structure of Sr5Al9. Sov Phys Crystallogr 32:196–198
Jehle M, Scherer H, Wendorff M, Röhr C (2009) Barium aluminides BaxAl5 (x = 3,3.5,4). J Solid State Chem 182:1129–1135. https://doi.org/10.1016/j.jssc.2009.02.005
Prots Y, Lange F, Drathen C, Schmidt M, Grin Y (2016) The new barium compound Ba4Al7+x: formation and crystal structure. Z Naturforsch B 71:611–619. https://doi.org/10.1515/znb-2016-0051
Haucke W (1937) Über Gold-Natrium-Legierungen (About gold-sodium alloys; in German). Naturwissenschaften 25:61. https://doi.org/10.1007/bf01492370
Zachwieja U (1993) Einkristallzüchtung und Strukturverfeinerung von NaAu2 — ein neuer Syntheseweg für Alkalimetall-Gold-Verbindungen (Single crystal growth and structure refinement of NaAu2—a new way of synthesis for alkali metall-gold-compounds; in German). J Alloys Compd 196:171–172. https://doi.org/10.1016/0925-8388(93)90590-J
Range K-J, Rau F, Klement U (1988) Potassium digold, KAu2. Acta Cryst C44:1485–1486. https://doi.org/10.1107/S0108270188003890
Zachwieja U (1993) Eine neue Verbindung im System Rubidium-Gold: Synthese und Struktur von Rb3Au7 (A new compound in the system rubidium-gold: synthesis and structure of Rb3Au7; in German). J Alloys Compd 199:115–118. https://doi.org/10.1016/0925-8388(93)90435-P
Range K-J, Grosch HH, Rau F, Klement U (1994) Gold-Alkalimetallsysteme, III. Hochdrucksynthese und Kristallstruktur von Rb3Au7 [1]/Gold-alkali metal-systems, III. High pressure synthesis and crystal structure of Rb3Au7 [1]. Z Naturforsch B 49:27–30. https://doi.org/10.1515/znb-1994-0107
Sinnen H-D, Schuster H-U (1981) Rb4Au7Sn2, eine intermetallische Phase mit siebenatomigen Goldclustern/Rb4Au7Sn2, an intermetallic phase with sevenatomic gold-clusters. Z Naturforsch B 36:833–836. https://doi.org/10.1515/znb-1981-0710
Zachwieja U (1995) K4Au7Ge2: eine Gerüststruktur mit Au7-Doppeltetraedern und Ge2-Hanteln (K4Au7Ge2: a skeleton structure with Au7-double tetrahedra and Ge2-dumbbells; in German). Z Anorg Allg Chem 621:975–978. https://doi.org/10.1002/zaac.19956210613
Zachwieja U, Müller J, Wlodarski J (1998) Zintl-Verbindungen mit Gold: M3AuSn4 mit M = K, Rb, Cs und M3AuPb4 mit M = Rb, Cs (Zintl compounds with gold: M3AuSn4 with M = K, Rb, Cs and M3AuPb4 with M = Rb, Cs; in German). Z Anorg Allg Chem 624:853–858. https://doi.org/10.1002/(sici)1521-3749(199805)624:5%3c853:aid-zaac853%3e3.0.co;2-n
Zachwieja U, Wlodarski J (1998) K3Au5Pb: eine Schichtstruktur mit [AuAu3/2]-Gold-Tetraedern und [Pb2/2]-Blei-Ketten (K3Au5Pb: a layer structure with [AuAu3/2]-gold-tetrahedra and [Pb2/2]-lead-chains; in German). Z Anorg Allg Chem 624:1569–1572. https://doi.org/10.1002/(sici)1521-3749(199810)624:10%3c1569:aid-zaac1569%3e3.0.co;2-v
Li B, Kim S-J, Miller GJ, Corbett JD (2009) Gold tetrahedra as building blocks in K3Au5Tr (Tr = In, Tl) and Rb2Au3Tl and in other compounds: a broad group of electron-poor intermetallic phases. Inorg Chem 48:6573–6583. https://doi.org/10.1021/ic9004856
Müller J, Zachwieja U (2000) K4Au8Ga: eine Auffüllungsvariante des MgCu2-Typs (K4Au8Ga: a filling variant of the MgCu2-type; in German). Z Anorg Allg Chem 626:1867–1870. https://doi.org/10.1002/1521-3749(200009)626:9%3c1867:aid-zaac1867%3e3.0.co;2-z
Li B, Kim S-J, Miller GJ, Corbett JD (2009) Synthesis, structure, and bonding in K12Au21Sn4. A polar intermetallic compound with dense Au20 and open AuSn4 layers. Inorg Chem 48:11108–11113. https://doi.org/10.1021/ic901493j
Smetana V, Rhodehouse M, Meyer G, Mudring A-V (2017) Gold polar intermetallics: structural versatility through exclusive bonding motifs. Acc Chem Res 50:2633–2641. https://doi.org/10.1021/acs.accounts.7b00316
Kitano Y, Komura Y, Kajiwara H, Watanabe E (1980) Two-dimensional lattice image of the Mg-base Friauf-Laves phase and a new type of defect. Acta Crystallogr A 36:16–21. https://doi.org/10.1107/S0567739480000034
Krämer U, Eichler K (1975) Transmissionselektronenmikroskopische Untersuchung der Defektstruktur der intermetallischen Verbindung MgZn2 (Transmission-electron-microscopical investigation of the defect structure of the intermetallic compound MgZn2; in German). Krist Tech 10:813–820. https://doi.org/10.1002/crat.19750100804
Rosalie JM, Somekawa H, Singh A, Mukai T (2010) Structural relationships among MgZn2 and Mg4Zn7 phases and transition structures in Mg-Zn-Y alloys. Philos Mag 90:3355–3374. https://doi.org/10.1080/14786435.2010.484659
Rosalie JM, Somekawa H, Singh A, Mukai T (2011) Orientation relationships between icosahedral clusters in hexagonal MgZn2 and monoclinic Mg4Zn7 phases in Mg-Zn(-Y) alloys. Philos Mag 91:2634–2644. https://doi.org/10.1080/14786435.2010.541168
Ye HQ, Wang DN, Kuo KH (1985) Domain structures of tetrahedrally close-packed phases with juxtaposed pentagonal antiprisms II. Domain boundary structures of the C14 Laves phase. Philos Mag A 51:839–848. https://doi.org/10.1080/01418618508237591
Šlapáková M, Zendegani A, Liebscher CH, Hickel T, Neugebauer J, Hammerschmidt T, Ormeci A, Grin J, Dehm G, Kumar KS, Stein F (2020) Atomic scale configuration of planar defects in the Nb-rich C14 Laves phase NbFe2. Acta Mater 183:362–376. https://doi.org/10.1016/j.actamat.2019.11.004
Paufler P, Schulze GER (1967) Plastic deformation of the intermetallic compound MgZn2. Phys Status Solidi B 24:77–87. https://doi.org/10.1002/pssb.19670240107
Paufler P, Schulze GER (1967) Zur Zwillingsbildung in MgZn2 (About twinning in MgZn2; in German). Krist Tech 2:231–244. https://doi.org/10.1002/crat.19670020209
Paufler P, Marschner J, Schulze GER (1970) The mobility of grown-in dislocations in the intermetallic compound MgZn2 - I. Stress dependence for edge dislocations in prism slip at 390°C. Phys Status Solidi 40:573–579. https://doi.org/10.1002/pssb.19700400216
Livingston JD, Hall EL (1990) Room-temperature deformation in a Laves phase. J Mater Res 5:5–8. https://doi.org/10.1557/JMR.1990.0005
Kimura Y, Luzzi DE, Pope DP (2002) Deformation twinning in a HfV2 + Nb-based Laves phase alloy. Mater Sci Eng A 329–331:241–248. https://doi.org/10.1016/S0921-5093(01)01583-0
Kazantzis AV, Aindow M, Jones IP, Triantafyllidis GK, De Hosson JTM (2007) The mechanical properties and the deformation microstructures of the C15 Laves phase Cr2Nb at high temperatures. Acta Mater 55:1873–1884. https://doi.org/10.1016/j.actamat.2006.10.048
Hazzledine PM, Pirouz P (1993) Synchroshear transformations in Laves phases. Scr Metall Mater 28:1277–1282. https://doi.org/10.1016/0956-716X(93)90468-8
Kumar KS (1997) Laves phase-based materials: microstructure, deformation modes and properties. Mater Res Soc Symp Proc 460:677–688. https://doi.org/10.1557/PROC-460-677
Chisholm MF, Kumar S, Hazzledine P (2005) Dislocations in complex materials. Science 307:701–703. https://doi.org/10.1126/science.1105962
Ma L, Pan R-K, Zhou S-C, Luo T-P, Wu D-H, Fan T-W, Tang B-Y (2014) Ab initio study of stacking faults and deformation mechanism in C15 Laves phases Cr2X (X = Nb, Zr, Hf). Mater Chem Phys 143:702–706. https://doi.org/10.1016/j.matchemphys.2013.09.056
Guénolé J, Mouhib F-Z, Huber L, Grabowski B, Korte-Kerzel S (2019) Basal slip in Laves phases: the synchroshear dislocation. Scr Mater 166:134–138. https://doi.org/10.1016/j.scriptamat.2019.03.016
Zhang W, Yu R, Du K, Cheng Z, Zhu J, Ye HQ (2011) Undulating slip in Laves phase and implications for deformation in brittle materials. Phys Rev Lett 106:165505. https://doi.org/10.1103/PhysRevLett.106.165505
Krämer U, Schulze GER (1968) Gittergeometrische Betrachtung der plastischen Verformung von Lavesphasen (Lattice geometrical examination of the plastic deformation of Laves phases; in German). Krist Tech 3:417–430. https://doi.org/10.1002/crat.19680030308
Takata N, Ghassemi-Armaki H, Takeyama M, Kumar S (2016) Nanoindentation study on solid solution softening of Fe-rich Fe2Nb Laves phase by Ni in Fe–Nb–Ni ternary alloys. Intermetallics 70:7–16. https://doi.org/10.1016/j.intermet.2015.11.003
Takata N, Ishikawa S, Matsuo T, Takeyama M (2009) Transmission electron microscopy of Fe2Nb Laves phase with C14 structure in Fe-Nb-Ni alloys. Mater Res Soc Symp Proc 1128:475–480. https://doi.org/10.1557/proc-1128-u08-06
Zhang Y, Du K, Zhang W, Du B, Qi D, Li W, Song M, Sheng L, Ye HQ (2019) Shear deformation determined by short-range configuration of atoms in topologically close-packed crystal. Acta Mater 179:396–405. https://doi.org/10.1016/j.actamat.2019.08.056
Luo W, Kirchlechner C, Zavašnik J, Lu W, Dehm G, Stein F (2020) Crystal structure and composition dependence of mechanical properties of single-crystalline NbCo2 Laves phase. Acta Mater 184:151–163. https://doi.org/10.1016/j.actamat.2019.11.036
Wetzig K, Wittig H (1972) On the temperature dependence of the microhardness of some high melting Laves phases. Phys Status Solidi A 9:K1–K3. https://doi.org/10.1002/pssa.2210090147
Bewilogua K, Reichelt R, Wetzig K, Wittig H (1972) Emissionselektronenmikroskopische Untersuchungen am Zweistoffsystem Niob-Eisen (Emission-electron-microsopical investigations on the binary system niobium-iron; in German). Krist Tech 7:601–609. https://doi.org/10.1002/crat.19720070512
von Keitz A, Sauthoff G (2002) Laves phases for high temperatures - Part II: stability and mechanical properties. Intermetallics 10:497–510. https://doi.org/10.1016/S0966-9795(02)00025-0
Takasugi T, Hanada S, Yoshida M (1995) High temperature mechanical properties of C15 Laves phase Cr2Nb intermetallics. Mater Sci Eng A 192(193):805–810. https://doi.org/10.1016/0921-5093(94)03319-6
Kazantzis AV, Aindow M, Jones IP (1997) Deformation behavior of the C15 Laves phase Cr2Nb. Mater Sci Eng A 233:44–49. https://doi.org/10.1016/S0921-5093(97)00047-6
Korte-Kerzel S (2017) Microcompression of brittle and anisotropic crystals: recent advances and current challenges in studying plasticity in hard materials. MRS Commun 7:109–120. https://doi.org/10.1557/mrc.2017.15
Korte S, Clegg WJ (2012) Studying plasticity in hard and soft Nb-Co intermetallics. Adv Eng Mater 14:991–997. https://doi.org/10.1002/adem.201200175
Takata N, Ghassemi Armaki H, Terada Y, Takeyama M, Kumar KS (2013) Plastic deformation of the C14 Laves phase (Fe, Ni)2Nb. Scr Mater 68:615–618. https://doi.org/10.1016/j.scriptamat.2012.12.019
Luo W (2019) Mechanical properties of the cubic and hexagonal NbCo2 Laves phases studied by micromechanical testing. Doct Thesis, Ruhr Universität Bochum, Germany, pp 1–125. https://doi.org/10.13154/294-6539
Zehnder C, Czerwinski K, Molodov KD, Sandlöbes-Haut S, Gibson JSKL, Korte-Kerzel S (2019) Plastic deformation of single crystalline C14 Mg2Ca Laves phase at room temperature. Mater Sci Eng A 759:754–761. https://doi.org/10.1016/j.msea.2019.05.092
Paufler P, Eichler K, Schulze GER (1970) Einfluß der Abweichungen von der Stöchiometrie intermetallischer Verbindungen auf deren plastische Verformbarkeit (Effect of deviations from the stoichiometry of intermetallic compounds on their plastic deformability). Monatsber Dt Akad Wissensch Berlin 12:950–958
Eichler K, Siegel S, Kubsch H, Paufler P (1971) Über das Verhalten einiger physikalischer Meßgrößen im Homogenitätsbereich von MgZn2 (About the behavior of some physical properties within the homogeneity range of MgZn2; in German). Wiss Z TU Dresden 20:399–402
Kubsch H, Paufler P, Schulze GER (1973) The mobility of grown-in dislocations in the intermetallic compound MgZn2. III. Dependence of basal slip on chemical composition within the homogeneity range and on temperature. Phys Status Solidi B 56:231–234. https://doi.org/10.1002/pssb.2220560121
Eichler K, Kubsch H, Müller T, Paufler P (1976) Änderung von Verformungseigenschaften der intermetallischen Verbindung MgZn2 im Homogenitätsbereich (Changes of deformation properties of the intermetallic compound MgZn2 within its homogeneity range; in German). Krist Tech 11:1185–1188. https://doi.org/10.1002/crat.19760111110
Müller T, Paufler P (1977) Yield strength of the monocrystalline intermetallic compound MgZn2. Phys Status Solidi A 40:471–477. https://doi.org/10.1002/pssa.2210400213
Ohta Y, Nakagawa Y, Kaneno Y, Inoue H, Takasugi T (2003) Microstructures and mechanical properties of NbCr2 andZrCr2 Laves phase alloys prepared by powder metallurgy. J Mater Sci 38:657–665. https://doi.org/10.1023/A:1021807519728
Chen KC, Allen SM, Livingston JD (1998) Factors affecting the room-temperature mechanical properties of TiCr2-base Laves phase alloys. Mater Sci Eng A 242:162–173. https://doi.org/10.1016/S0921-5093(97)00526-1
Sun X-S, Sun F, Sun J (2005) Defect structure and its softening effect in ZrCr2 Laves phase compound. Chin J Non-Ferrous Met 15:626–630
Voß S, Stein F, Palm M, Grüner D, Kreiner G, Frommeyer G, Raabe D (2009) Composition dependence of the hardness of Laves phases in the Fe-Nb and Co-Nb systems. Mater Res Soc Symp Proc 1128:469–474. https://doi.org/10.1557/proc-1128-u08-05
Luo W, Kirchlechner C, Li J, Dehm G, Stein F (2020) Composition dependence of hardness and elastic modulus of the cubic and hexagonal NbCo2 Laves phase polytypes studied by nanoindentation. J Mater Res 35:185–195. https://doi.org/10.1557/jmr.2019.384
Leitner G (1971) Untersuchungen über das Auftreten, die Verwandtschaft und die Variabilität einiger Gittertypen intermetallischer Verbindungen (Investigations about the occurrence, relationship and variability of some lattice types of intermetallic compounds; in German). Doct Thesis, TU Dresden, Germany, pp 1–205
Chen KC, Chu F, Kotula PG, Thoma D (2001) HfCo2 Laves phase intermetallics - Part II: elastic and mechanical properties as a function of composition. Intermetallics 9:785–798. https://doi.org/10.1016/S0966-9795(01)00067-X
Fleischer RL (1961) Solution hardening. Acta Metall 9:996–1000. https://doi.org/10.1016/0001-6160(61)90242-5
Westbrook JH (1956) Temperature dependence of hardness of the equi-atomic iron group aluminides. J Electrochem Soc 103:54–63. https://doi.org/10.1149/1.2430233
Westbrook JH (1957) Defect structure and the temperature dependence of hardness of an intermetallic compound. J Electrochem Soc 104:369–373. https://doi.org/10.1149/1.2428584
Westbrook JH (1960) Mechanical properties of intermetallic compounds. In: Westbrook JH (ed) Mechanical properties of intermetallic compounds. Wiley, New York
Fleischer RL (1992) Solid solution hardening of intermetallic compounds. Mater Res Soc Symp Proc 288:165–170. https://doi.org/10.1557/PROC-288-165
Luo W, Kirchlechner C, Dehm G, Stein F (2017) Deformation of micropillars of cubic and hexagonal NbCo2 Laves phases under uniaxial compression at room temperature. In: Heilmaier M, Krüger M, Mayer S, Palm M, Stein F (eds) Intermetallics 2017, Bad Staffelstein. Conventus Congressmanagement & Marketing GmbH, Jena, pp 199–200
Luo W, Kirchlechner C, Fang X, Brinckmann S, Dehm G, Stein F (2018) Influence of composition and crystal structure on the fracture toughness of NbCo2 Laves phase studied by micro-cantilever bending tests. Mater Des 145:116–121. https://doi.org/10.1016/j.matdes.2018.02.045
Luo W, Kirchlechner C, Dehm G, Stein F (2019) Micromechanics of Co-Nb Laves phases: Strength, fracture toughness, and hardness as function of composition and crystal structure. In: Shingledecker J, Takeyama M (eds) Proc Joint EPRI-123HiMAT International Conference on Advances in High Temperature Materials, Nagasaki, Japan. ASM International, Materials Park, pp 11–21 ISBN 9781627082716
Hoffman KC, Winsche WE, Wiswall RH, Reilly JJ, Sheehan TV, Waide CH (1969) Metal hydrides as a source of fuel for vehicular propulsion. SAE Trans 78:981–986
Hoffman KC, Reilly JJ, Salzano FJ, Waide CH, Wiswall RH, Winsche WE (1976) Metal hydride storage for mobile and stationary applications. Int J Hydrogen Energy 1:133–151. https://doi.org/10.1016/0360-3199(76)90067-7
Bououdina M, Grant D, Walker G (2006) Review on hydrogen absorbing materials—structure, microstructure, and thermodynamic properties. Int J Hydrogen Energy 31:177–182. https://doi.org/10.1016/j.ijhydene.2005.04.049
Sakintuna B, Lamari-Darkrim F, Hirscher M (2007) Metal hydride materials for solid hydrogen storage: a review. Int J Hydrogen Energy 32:1121–1140. https://doi.org/10.1016/j.ijhydene.2006.11.022
Ouyang L, Huang J, Wang H, Liu J, Zhu M (2017) Progress of hydrogen storage alloys for Ni-MH rechargeable power batteries in electric vehicles: a review. Mater Chem Phys 200:164–178. https://doi.org/10.1016/j.matchemphys.2017.07.002
Bellosta von Colbe J, Ares J-R, Barale J, Baricco M, Buckley C, Capurso G, Gallandat N, Grant DM, Guzik MN, Jacob I, Jensen EH, Jensen T, Jepsen J, Klassen T, Lototskyy MV, Manickam K, Montone A, Puszkiel J, Sartori S, Sheppard DA, Stuart A, Walker G, Webb CJ, Yang H, Yartys V, Züttel A, Dornheim M (2019) Application of hydrides in hydrogen storage and compression: achievements, outlook and perspectives. Int J Hydrogen Energy 44:7780–7808. https://doi.org/10.1016/j.ijhydene.2019.01.104
Young K (2018) Nickel metal hydride batteries 2017. MDPI, Basel ISBN 978-3-03842-828-2
Young K-H, Chang S, Lin X (2017) C14 Laves phase metal hydride alloys for Ni/MH batteries applications. Batteries 3:27. https://doi.org/10.3390/batteries3030027
Felderhoff M (2012) Functional materials for hydrogen storage. In: Kilner JA, Skinner SJ, Irvine SJC, Edwards PP (eds) Functional Materials for Sustainable Energy Applications. Woodhead Publishing, Cambridge, pp 217–246e. https://doi.org/10.1533/9780857096371.2.217
Pebler A, Gulbransen EA (1967) Equilibrium studies on the systems ZrCr2-H2, ZrV2-H2, and ZrMo2-H2 between 0 and 900°C. Trans Metall Soc AIME 239:1593–1600
Shaltiel D, Jacob I, Davidov D (1977) Hydrogen absorption and desorption properties of AB2 Laves-phase pseudobinary compounds. J Less-Common Met 53:117–131. https://doi.org/10.1016/0022-5088(77)90162-X
Ivey DG, Northwood DO (1986) Storing hydrogen in AB2 Laves-type compounds. Z Phys Chem NF 147:191–209. https://doi.org/10.1524/zpch.1986.147.1_2.191
Kohlmann H (2020) Hydrogen order in hydrides of Laves phases. Z Kristallogr Cryst Mater 235:319–332. https://doi.org/10.1515/zkri-2020-0043
Shoemaker DP, Shoemaker CB (1979) Concerning atomic sites and capacities for hydrogen absorption in the AB2 Friauf-Laves phases. J Less-Common Met 68:43–58. https://doi.org/10.1016/0022-5088(79)90271-6
Hong S, Fu CL (2002) Hydrogen in Laves phase ZrX2 (X = V, Cr, Mn, Fe Co, Ni) compounds: binding energies and electronic and magnetic structure. Phys Rev B 66:094109. https://doi.org/10.1103/PhysRevB.66.094109
Gesari SB, Pronsato ME, Visintin A, Juan A (2010) Hydrogen storage in AB2 Laves phase (A = Zr, Ti; B = Ni, Mn, Cr, V): binding energy and electronic structure. J Phys Chem C 114:16832–16836. https://doi.org/10.1021/jp106036v
Westlake DG (1984) Hydrogen sites in A2BHy (A = Ca, Sr, Eu; B = Ir, Rh, Ru). J Solid State Chem 53:130–135. https://doi.org/10.1016/0022-4596(84)90235-4
Jacob I, Shaltiel D, Davidov D, Miloslavski I (1977) A phenomenological model for the hydrogen absorption capacity in pseudobinary Laves phase compounds. Solid State Commun 23:669–672. https://doi.org/10.1016/0038-1098(77)90546-4
Fruchart D, Rouault A, Shoemaker CB, Shoemaker DP (1980) Neutron diffraction studies of the cubic ZrCr2Dx and ZrV2Dx (Hx) phases. J Less-Common Met 73:363–368. https://doi.org/10.1016/0022-5088(80)90330-6
Soubeyroux JL, Bououdina M, Fruchart D, Pontonnier L (1995) Phase stability and neutron diffraction studies of Laves phases Zr(Cr1−xMx)2 with M = Mn, Fe Co, Ni, Cu and 0 < x<0.2 and their hydrides. J Alloys Compd 219:48–54. https://doi.org/10.1016/0925-8388(94)05019-8
Soubeyroux JL, Fruchart D, Biris AS (1999) Structural studies of Laves phases ZrCo(V1−xCrx) with 0 ≤ x≤1 and their hydrides. J Alloys Compd 293–295:88–92. https://doi.org/10.1016/S0925-8388(99)00305-9
Bououdina M, Soubeyroux JL, de Rango P, Fruchart D (2000) Phase stability and neutron diffraction studies of the Laves phase compounds Zr(Cr1−xMox)2 with 0.0 <=x <=0.5 and their hydrides. Int J Hydrogen Energy 25:1059–1068. https://doi.org/10.1016/S0360-3199(99)00087-7
Lushnikov SA, Verbetsky VN, Glazkov VP, Somenkov VA (2006) Structure of deuterided NbVCo. Inorg Mater 42:733–738. https://doi.org/10.1134/s0020168506070089
Huot J, Akiba E, Ogura T, Ishido Y (1995) Crystal structure, phase abundance and electrode performance of Laves phase compounds (Zr, A)V0.5Ni1.1Mn0.2Fe0.2 (A ≡ Ti, Nb or Hf). J Alloys Compd 218:101–109. https://doi.org/10.1016/0925-8388(94)01370-5
Zhang QA, Lei YQ, Yang XG, Ren K, Wang QD (1999) Annealing treatment of AB2-type hydrogen storage alloys: II. Electrochemical properties. J Alloys Compd 292:241–246. https://doi.org/10.1016/S0925-8388(99)00486-7
Song XY, Zhang XB, Lei YQ, Zhang Z, Wang QD (1999) Effect of microstructure on the properties of Zr–Mn–V–Ni AB2 type hydride electrode alloys. Int J Hydrogen Energy 24:455–459. https://doi.org/10.1016/S0360-3199(98)00098-6
Young K, Ouchi T, Huang B, Chao B, Fetcenko MA, Bendersky LA, Wang K, Chiu C (2010) The correlation of C14/C15 phase abundance and electrochemical properties in the AB2 alloys. J Alloys Compd 506:841–848. https://doi.org/10.1016/j.jallcom.2010.07.091
Young K-H, Nei J, Wan C, Denys RV, Yartys VA (2017) Comparison of C14-and C15-predomiated AB2 metal hydride alloys for electrochemical applications. Batteries 3:Article No. 22. https://doi.org/10.3390/batteries3030022
Young K-H, Koch JM, Wan C, Denys RV, Yartys VA (2017) Cell performance comparison between C14- and C15-predomiated AB2 metal hydride alloys. Batteries 3:29. https://doi.org/10.3390/batteries3040029
Jacob I, Shaltiel D (1978) Hydrogen absorption in Zr(AlxFe1−x)2 Laves phase compounds. Solid State Commun 27:175–180. https://doi.org/10.1016/0038-1098(78)90826-8
Israel A, Jacob I (1997) Kinetics and thermodynamics of H absorption in the Zr(Al0.1Fe0.9)2 intermetallic compound. J Alloys Compd 260:131–134. https://doi.org/10.1016/S0925-8388(97)00130-8
Israel A, Jacob I, Soubeyroux JL, Fruchart D, Pinto H, Melamud M (1997) Neutron diffraction study of atomic bonding properties in the hydrogen-absorbing Zr(AlxFe1−x)2 system. J Alloys Compd 253–254:265–267. https://doi.org/10.1016/S0925-8388(96)03029-0
Bereznitsky M, Jacob I, Bloch J, Mintz MH (2003) Thermodynamic and structural aspects of hydrogen absorption in the Zr(AlxFe1−x)2 system. J Alloys Compd 351:180–183. https://doi.org/10.1016/S0925-8388(02)01074-5
Terashita N, Kobayashi K, Sasai T, Akiba E (2001) Structural and hydriding properties of (Mg1−xCax)Ni2 Laves phase alloys. J Alloys Compd 327:275–280. https://doi.org/10.1016/S0925-8388(01)01563-8
Terashita N, Akiba E (2006) Hydriding properties of (Mg1−xMx)Ni2 C15-type Laves phase alloys. Mater Trans 47:1890–1893. https://doi.org/10.2320/matertrans.47.1890
Liu X, Asano K, Terashita N, Akiba E (2009) Hydrogenation of C14 Laves phase alloy: CaLi2. Int J Hydrogen Energy 34:1472–1475. https://doi.org/10.1016/j.ijhydene.2008.12.011
Terashita N, Akiba E (2004) Hydrogenation properties of CaMg2 based alloys. Mater Trans 45:2594–2597. https://doi.org/10.2320/matertrans.45.2594
Ma M, Duan R, Ouyang L, Zhu X, Chen Z, Peng C, Zhu M (2017) Hydrogen storage and hydrogen generation properties of CaMg2-based alloys. J Alloys Compd 691:929–935. https://doi.org/10.1016/j.jallcom.2016.08.307
Ma M, Chen K, Jiang J, Yang X, Wang H, Shao H, Liu J, Ouyang L (2020) Enhanced hydrogen generation performance of CaMg2-based materials by ball milling. Inorg Chem Front 7:918–929. https://doi.org/10.1039/C9QI01299K
Joubert J-M, Sun D, Latroche M, Percheron-Guegan A (1997) Electrochemical performances of ZrM2 (M = V, Cr, Mn, Ni) Laves phases and the relation to microstructures and thermodynamical properties. J Alloys Compd 253–254:564–569. https://doi.org/10.1016/S0925-8388(96)03088-5
Cui XY, Li Q, Chou KC, Chen SL, Lin GW, Xu KD (2008) A comparative study on the hydriding kinetics of Zr-based AB2 hydrogen storage alloys. Intermetallics 16:662–667. https://doi.org/10.1016/j.intermet.2008.02.009
Wu T, Xue XY, Zhang T, Hu R, Kou H, Li J (2015) Microstructures and hydrogenation properties of (ZrTi)(V1−xAlx)2 Laves phase intermetallic compounds. J Alloys Compd 645:358–368. https://doi.org/10.1016/j.jallcom.2015.05.110
Cao Z, Ouyang L, Wang H, Liu J, Sun L, Felderhoff M, Zhu M (2016) Development of ZrFeV alloys for hybrid hydrogen storage system. Int J Hydrogen Energy 41:11242–11253. https://doi.org/10.1016/j.ijhydene.2016.04.083
Watanuki Y, Toyama T, Kojima Y, Nishimiya N (2016) Hydrogen occlusion behaviors of ZrVFe alloys under mild hydrogen pressures and lowering of their hydrogen desorption temperatures. Int J Hydrogen Energy 41:425–430. https://doi.org/10.1016/j.ijhydene.2015.11.051
Wijayanti ID, Denys R, Suwarno Volodin AA, Lototskyy MV, Guzik MN, Nei J, Young K, Roven HJ, Yartys V (2020) Hydrides of Laves type Ti–Zr alloys with enhanced H storage capacity as advanced metal hydride battery anodes. J Alloys Compd 828:154354. https://doi.org/10.1016/j.jallcom.2020.154354
Wan C, Jiang X, Yin X, Ju X (2020) High-capacity Zr-based AB2-type alloys as metal hydride battery anodes. J Alloys Compd 828:154402. https://doi.org/10.1016/j.jallcom.2020.154402
Qin C, Zhou C, Ouyang L, Liu J, Zhu M, Sun T, Wang H (2020) High-pressure hydrogen storage performances of ZrFe2 based alloys with Mn, Ti, and V addition. Int J Hydrogen Energy 45:9836–9844. https://doi.org/10.1016/j.ijhydene.2019.11.242
Kandavel M, Bhat VV, Rougier A, Aymard L, Nazri GA, Tarascon JM (2008) Improvement of hydrogen storage properties of the AB2 Laves phase alloys for automotive application. Int J Hydrogen Energy 33:3754–3761. https://doi.org/10.1016/j.ijhydene.2008.04.042
Guo X, Wang S, Liu X, Li Z, Lü F, Mi J, Hao L, Jiang L (2011) Laves phase hydrogen storage alloys for super-high-pressure metal hydride hydrogen compressors. Rare Met 30:227–231. https://doi.org/10.1007/s12598-011-0373-7
Cao Z, Ouyang L, Wang H, Liu J, Sun L, Zhu M (2015) Composition design of Ti–Cr–Mn–Fe alloys for hybrid high-pressure metal hydride tanks. J Alloys Compd 639:452–457. https://doi.org/10.1016/j.jallcom.2015.03.196
Ulmer U, Dieterich M, Pohl A, Dittmeyer R, Linder M, Fichtner M (2017) Study of the structural, thermodynamic and cyclic effects of vanadium and titanium substitution in Laves-phase AB2 hydrogen storage alloys. Int J Hydrogen Energy 42:20103–20110. https://doi.org/10.1016/j.ijhydene.2017.06.137
Liu P, Xie X, Xu L, Li X, Liu T (2017) Hydrogen storage properties of (Ti0.85Zr0.15)1.05Mn1.2Cr0.6V0.1M0.1 (M = Ni, Fe, Cu) alloys easily activated at room temperature. Prog Nat Sci: Mater Int 27:652–657. https://doi.org/10.1016/j.pnsc.2017.09.007
Pickering L, Lototskyy MV, Wafeeq Davids M, Sita C, Linkov V (2018) Induction melted AB2-type metal hydrides for hydrogen storage and compression applications. Mater Today: Proc 5:10470–10478. https://doi.org/10.1016/j.matpr.2017.12.378
Li J, Jiang X, Xu L, Li X (2019) Model for hydrogen desorption plateau pressure of AB2 type-Tix(CrFeMn)2 alloys. Intermetallics 107:1–5. https://doi.org/10.1016/j.intermet.2018.12.016
Murashkina TL, Syrtanov MS, Laptev RS, Stepanova EN, Lider AM (2019) Structure and defects evolution at temperature and activation treatments of the TiCr2 intermetallic compound of Laves phase C36-type. Int J Hydrogen Energy 44:10732–10743. https://doi.org/10.1016/j.ijhydene.2019.02.154
Skripnyuk VM, Ron M (2003) Hydrogen desorption kinetics in intermetallic compounds C2, C51 and C52 with Laves phase structure. Int J Hydrogen Energy 28:303–309. https://doi.org/10.1016/S0360-3199(02)00081-2
Song C, Klebanoff LE, Johnson TA, Chao BS, Socha AF, Oros JM, Radley CJ, Wingert S, Breit JS (2014) Using metal hydride H2 storage in mobile fuel cell equipment: design and predicted performance of a metal hydride fuel cell mobile light. Int J Hydrogen Energy 39:14896–14911. https://doi.org/10.1016/j.ijhydene.2014.07.069
Volodin AA, Denys RV, Wan CB, Wijayanti ID, Suwarno Tarasov BP, Antonov VE, Yartys VA (2019) Study of hydrogen storage and electrochemical properties of AB2-type Ti0.15Zr0.85La0.03Ni1.2Mn0.7V0.12Fe0.12 alloy. J Alloys Compd 793:564–575. https://doi.org/10.1016/j.jallcom.2019.03.134
Kao Y-F, Chen S-K, Sheu J-H, Lin J-T, Lin W-E, Yeh J-W, Lin S-J, Liou T-H, Wang C-W (2010) Hydrogen storage properties of multi-principal-component CoFeMnTixVyZrz alloys. Int J Hydrogen Energy 35:9046–9059. https://doi.org/10.1016/j.ijhydene.2010.06.012
Kunce I, Polanski M, Bystrzycki J (2013) Structure and hydrogen storage properties of a high entropy ZrTiVCrFeNi alloy synthesized using Laser Engineered Net Shaping (LENS). Int J Hydrogen Energy 38:12180–12189. https://doi.org/10.1016/j.ijhydene.2013.05.071
Edalati P, Floriano R, Mohammadi A, Li Y, Zepon G, Li H-W, Edalati K (2020) Reversible room temperature hydrogen storage in high-entropy alloy TiZrCrMnFeNi. Scr Mater 178:387–390. https://doi.org/10.1016/j.scriptamat.2019.12.009
Ovshinsky SR, Fetcenko MA, Ross J (1993) A nickel metal hydride battery for electric vehicles. Science 260:176–181. https://doi.org/10.1126/science.260.5105.176
Young K, Nei J, Huang B, Fetcenko MA (2011) Studies of off-stoichiometric AB2 metal hydride alloy: Part 2. Hydrogen storage and electrochemical properties. Int J Hydrogen Energy 36:11146–11154. https://doi.org/10.1016/j.ijhydene.2011.05.056
Zhang YL, Li JS, Zhang TB, Hu R, Xue XY (2013) Microstructure and hydrogen storage properties of non-stoichiometric Zr–Ti–V Laves phase alloys. Int J Hydrogen Energy 38:14675–14684. https://doi.org/10.1016/j.ijhydene.2013.09.040
Manickam K, Grant DM, Walker GS (2015) Optimization of AB2 type alloy composition with superior hydrogen storage properties for stationary applications. Int J Hydrogen Energy 40:16288–16296. https://doi.org/10.1016/j.ijhydene.2015.09.157
Young K, Fetcenko MA, Li F, Ouchi T, Koch J (2009) Effect of vanadium substitution in C14 Laves phase alloys for NiMH battery application. J Alloys Compd 468:482–492. https://doi.org/10.1016/j.jallcom.2008.01.033
Züttel A, Chartouni D, Gross K, Bächler M, Schlapbach L (1997) Structural- and hydriding-properties of the Zr(V0.25Ni0.75)α (1≤α≤4) alloy system. J Alloys Compd 253–254:587–589. https://doi.org/10.1016/S0925-8388(96)02989-1
Agostino RG, Liberti G, Formoso V, Colavita E, Züttel A, Nützenadel C, Schlapbach L, Santaniello A, Gauthier C (2000) In situ x-ray absorption study of Zr(V0.29Ni0.71)3 hydride electrodes. Phys Rev B 61:13647–13654. https://doi.org/10.1103/PhysRevB.61.13647
Li Z, Wang H, Ouyang L, Liu J, Zhu M (2019) Achieving superior de-/hydrogenation properties of C15 Laves phase Y-Fe-Al alloys by A-side substitution. J Alloys Compd 787:158–164. https://doi.org/10.1016/j.jallcom.2019.02.074
Akiba E, Iba H (1998) Hydrogen absorption by Laves phase related BCC solid solution. Intermetallics 6:461–470. https://doi.org/10.1016/S0966-9795(97)00088-5
Akiba E (1999) Hydrogen-absorbing alloys. Curr Opin Solid State Mater Sci 4:267–272. https://doi.org/10.1016/S1359-0286(99)00026-1
Guéguen A, Joubert JM, Latroche M (2011) Influence of the C14 Ti35.4V32.3Fe32.3 Laves phase on the hydrogenation properties of the body-centered cubic compound Ti24.5V59.3Fe16.2. J Alloys Compd 509:3013–3018. https://doi.org/10.1016/j.jallcom.2010.10.213
Chen XY, Chen RR, Ding X, Fang HZ, Guo JJ, Ding HS, Su YQ, Fu HZ (2018) Crystal structure and hydrogen storage properties of Ti-V-Mn alloys. Int J Hydrogen Energy 43:6210–6218. https://doi.org/10.1016/j.ijhydene.2018.02.009
Okada M, Kuriiwa T, Kamegawa A, Takamura H (2002) Role of intermetallics in hydrogen storage materials. Mater Sci Eng A 329–331:305–312. https://doi.org/10.1016/S0921-5093(01)01580-5
Zhu Y, Pan H, Gao M, Liu Y, Wang Q (2003) Influence of annealing treatment on Laves phase compound containing a V-based bcc solid solution phase - Part II: electrochemical properties. Int J Hydrogen Energy 28:395–401. https://doi.org/10.1016/S0360-3199(02)00151-9
Wang HB, Wang Q, Dong C, Yuan L, Xu F, Sun LX (2008) Composition design for Laves phase-related BCC-V solid solution alloys with large hydrogen storage capacities. J Phys: Conf Series 98:012018. https://doi.org/10.1088/1742-6596/98/1/012018
Zhang J, Han SM, Li Y, Liu J, Yang S, Zhang L, Wang J (2013) Effects of PuNi3- and Ce2Ni7-type phase abundance on electrochemical characteristics of La–Mg–Ni-based alloys. J Alloys Compd 581:693–698. https://doi.org/10.1016/j.jallcom.2013.07.180
Ouyang LZ, Cao ZJ, Li LL, Wang H, Liu JW, Min D, Chen YW, Xiao FM, Tang RH, Zhu M (2014) Enhanced high-rate discharge properties of La11.3Mg6.0Sm7.4Ni61.0Co7.2Al7.1 with added graphene synthesized by plasma milling. Int J Hydrogen Energy 39:12765–12772. https://doi.org/10.1016/j.ijhydene.2014.06.111
Gao Z, Geng Y, Lin Z, Wei Y, Luo Y, Li H-W (2020) Synergistic effects of Gd and Co on the phase evolution mechanism and electrochemical performances of Ce2Ni7-type La–Mg–Ni-based alloys. Dalton Trans 49:156–163. https://doi.org/10.1039/C9DT03944A
Chao BS, Young RC, Ovshinsky SR, Pawlik DA, Huang B, Im JS, Chakoumakos BC (2000) Effect of alloy composition on the structure of Zr based metal alloys. Mater Res Soc Symp Proc 575:193–198. https://doi.org/10.1557/PROC-575-193
Young RC, Chao B, Li Y, Myasnikov V, Huang B, Ovshinsky SR (2004) A hydrogen ICE vehicle powered by Ovonic metal hydride storage. SAE Technical Paper 2004-01-0699. https://doi.org/10.4271/2004-01-0699
Lupu D, Biris AR, Biris AS, Indrea E, Misan I, Züttel A, Schlapbach L (2003) Cobalt-free over-stoichiometric Laves phase alloys for Ni-MH batteries. J Alloys Compd 350:319–323. https://doi.org/10.1016/S0925-8388(02)00991-X
Chang S, Young KH, Nei J, Fierro C (2016) Reviews on the U.S. patents regarding nickel/metal hydride batteries. Batteries 2:Article No. 10. https://doi.org/10.3390/batteries2020010
Vanhanen JP, Lund PD, Hagström MT (1996) Feasibility study of a metal hydride hydrogen store for a self-sufficient solar hydrogen energy system. Int J Hydrogen Energy 21:213–221. https://doi.org/10.1016/0360-3199(95)00064-X
Hagström MT, Vanhanen JP, Lund PD (1998) AB2 metal hydrides for high-pressure and narrow temperature interval applications. J Alloys Compd 269:288–293. https://doi.org/10.1016/S0925-8388(98)00213-8
Dieterich M, Bürger I, Linder M (2017) Open and closed metal hydride system for high thermal power applications: preheating vehicle components. Int J Hydrogen Energy 42:11469–11481. https://doi.org/10.1016/j.ijhydene.2017.03.024
Pohlmann C, Röntzsch L, Heubner F, Weißgärber T, Kieback B (2013) Solid-state hydrogen storage in Hydralloy–graphite composites. J Power Sources 231:97–105. https://doi.org/10.1016/j.jpowsour.2012.12.044
Eberle U, Felderhoff M, Schüth F (2009) Chemical and physical solutions for hydrogen storage. Ang Chem, Int Ed 48:6608–6630. https://doi.org/10.1002/anie.200806293
Bürger I, Dieterich M, Pohlmann C, Röntzsch L, Linder M (2017) Standardized hydrogen storage module with high utilization factor based on metal hydride-graphite composites. J Power Sources 342:970–979. https://doi.org/10.1016/j.jpowsour.2016.12.108
Herbrig K, Röntzsch L, Pohlmann C, Weißgärber T, Kieback B (2013) Hydrogen storage systems based on hydride–graphite composites: computer simulation and experimental validation. Int J Hydrogen Energy 38:7026–7036. https://doi.org/10.1016/j.ijhydene.2013.03.104
Dieterich M, Pohlmann C, Bürger I, Linder M, Röntzsch L (2015) Long-term cycle stability of metal hydride-graphite composites. Int J Hydrogen Energy 40:16375–16382. https://doi.org/10.1016/j.ijhydene.2015.09.013
Lototskyy MV, Tolj I, Davids MW, Klochko YV, Parsons A, Swanepoel D, Ehlers R, Louw G, van der Westhuizen B, Smith FJ, Pollet BG, Sita C, Linkov V (2016) Metal hydride hydrogen storage and supply systems for electric forklift with low-temperature proton exchange membrane fuel cell power module. Int J Hydrogen Energy 41:13831–13842. https://doi.org/10.1016/j.ijhydene.2016.01.148
Lototskyy MV, Tolj I, Parsons A, Smith FJ, Sita C, Linkov V (2016) Performance of electric forklift with low-temperature polymer exchange membrane fuel cell power module and metal hydride hydrogen storage extension tank. J Power Sources 316:239–250. https://doi.org/10.1016/j.jpowsour.2016.03.058
Lototskyy M, Tolj I, Klochko YV, Davids MW, Swanepoel D, Linkov V (2020) Metal hydride hydrogen storage tank for fuel cell utility vehicles. Int J Hydrogen Energy 45:7958–7967. https://doi.org/10.1016/j.ijhydene.2019.04.124
Miller AR, Barnes DL (2002) Advanced underground vehicle power and control fuel cell mine locomotive. In: Proceedings of the 2002 US DOE Hydrogen Program Review NREL/CP-610-32405, pp 1–7
Capurso G, Schiavo B, Jepsen J, Lozano G, Metz O, Saccone A, De Negri S, Bellosta von Colbe JM, Klassen T, Dornheim M (2016) Development of a modular room-temperature hydride storage system for vehicular applications. Appl Phys A 122:236. https://doi.org/10.1007/s00339-016-9771-x
Weckerle C, Nasri M, Hegner R, Linder M, Bürger I (2019) A metal hydride air-conditioning system for fuel cell vehicles—performance investigations. Appl Energy 256:113957. https://doi.org/10.1016/j.apenergy.2019.113957
Weckerle C, Nasri M, Hegner R, Bürger I, Linder M (2020) A metal hydride air-conditioning system for fuel cell vehicles—functional demonstration. Appl Energy 259:114187. https://doi.org/10.1016/j.apenergy.2019.114187
Cameron CB, Ferriss DP (1974) Tribaloy intermetallic materials: New wear- and corrosion-resistant alloys. Cobalt 1974 No.3:49-53 (reprinted 1975 in Anti-Corros. Methods Mater. 1922 No.1974:1975-1978)
Cameron CB, Hoffman RA, Poskitt RW (1975) Tribaloy intermetallic alloy compositions: new materials or additives for wear resistant applications. Progr Powder Metall 31:41–51
Schmidt RD, Ferriss DP (1975) New materials resistant to wear and corrosion to 1000°C. Wear 32:279–289. https://doi.org/10.1016/0043-1648(75)90316-6
Ferriss DP, Cameron CB (1975) Wear properties of cast vs plasma-sprayed Laves intermetallic alloys. J Vac Sci Technol 12:795–799. https://doi.org/10.1116/1.568674
Durejko T, Łazińska M, Dworecka-Wójcik J, Lipiński S, Varin RA, Czujko T (2019) The Tribaloy T-800 coatings deposited by laser engineered net shaping (LENS™). Materials 12:1366. https://doi.org/10.3390/ma12091366
Price MO, Wolfla TA, Tucker RC Jr (1977) Some comparative properties of Laves- and carbide-strengthened coatings deposited by plasma or detonation gun. Thin Solid Films 45:309–319. https://doi.org/10.1016/0040-6090(77)90264-4
Johnson RN, Farwick DG (1978) Friction, wear and corrosion of Laves-hardened nickel alloy hardsurfacing in sodium. Thin Solid Films 53:365–373. https://doi.org/10.1016/0040-6090(78)90229-8
Deloro Wear Solutions GmbH (2020) Metallurgical Expertise. https://www.delorocom/solutions/metallurgical-expertise. Accessed 19 June 2020
Halstead A, Rawlings RD (1984) Structure and hardness of Co-Mo-Cr-Si wear resistant alloys (Tribaloys). Met Sci 18:491–500. https://doi.org/10.1179/030634584790253146
Halstead A, Rawlings RD (1985) The fracture behaviour of two Co-Mo-Cr-Si wear resistant alloys (“Tribaloys”). J Mater Sci 20:1248–1256. https://doi.org/10.1007/BF01026320
Halstead A, Rawlings RD (1985) The effect of iron additions on the microstructure and properties of the “Tribaloy” Co-Mo-Cr-Si wear resistant alloys. J Mater Sci 20:1693–1704. https://doi.org/10.1007/BF00555273
Yao MX, Wu JBC, Yick S, Xie Y, Liu R (2006) High temperature wear and corrosion resistance of a Laves phase strengthened Co-Mo-Cr-Si alloy. Mater Sci Eng A 435–436:78–83. https://doi.org/10.1016/j.msea.2006.07.054
Xu W, Liu R, Patnaik PC, Yao MX, Wu XJ (2007) Mechanical and tribological properties of newly developed Tribaloy alloys. Mater Sci Eng A 452–453:427–436. https://doi.org/10.1016/j.msea.2006.10.088
Liu R, Xu W, Yao MX, Patnaik PC, Wu XJ (2005) A newly developed Tribaloy alloy with increased ductility. Scr Mater 53:1351–1355. https://doi.org/10.1016/j.scriptamat.2005.08.033
Yao MX, Wu JBC, Liu R (2005) Microstructural characteristics and corrosion resistance in molten Zn–Al bath of Co–Mo–Cr–Si alloys. Mater Sci Eng A 407:299–305. https://doi.org/10.1016/j.msea.2005.07.054
Nsoesie S, Liu R, Jiang K, Liang M (2013) High-temperature hardness and wear resistance of cobalt-based Tribaloy alloys. Int J Mater Mech Eng 2:48–56
Jiang K, Liu R, Chen K, Liang M (2013) Microstructure and tribological properties of solution-treated Tribaloy alloy. Wear 307:22–27. https://doi.org/10.1016/j.wear.2013.08.018
Navas C, Cadenas M, Cuetos JM, de Damborenea J (2006) Microstructure and sliding wear behaviour of Tribaloy T-800 coatings deposited by laser cladding. Wear 260:838–846. https://doi.org/10.1016/j.wear.2005.04.020
Tobar MJ, Amado JM, Álvarez C, García A, Varela A, Yáñez A (2008) Characteristics of Tribaloy T-800 and T-900 coatings on steel substrates by laser cladding. Surf Coat Technol 202:2297–2301. https://doi.org/10.1016/j.surfcoat.2007.11.025
Peng J, Fang X, Marx V, Jasnau U, Palm M (2018) Isothermal oxidation behavior of Tribaloy™ T400 and T800. NPJ Mater Degrad 2:38. https://doi.org/10.1038/s41529-018-0060-3
Zhang YD, Yang ZG, Zhang C, Lan H (2008) Oxidation behavior of Tribaloy T-800 alloy at 800 and 1,000 °C. Oxid Met 70:229–239. https://doi.org/10.1007/s11085-008-9117-y
Rehman A, Liang Y, Bidabadi MHS, Yu Z, Zhang C, Chen H, Yang Z-G (2019) Comparative cyclic oxidation behaviour and effect of oxides on hardness of wear resistance coating alloys T-401 and T-900. J Iron Steel Res Int 26:1069–1079. https://doi.org/10.1007/s42243-019-00258-2
Zhang XH, Zhang C, Zhang YD, Salam S, Wang HF, Yang ZG (2014) Effect of yttrium and aluminum additions on isothermal oxidation behavior of Tribaloy T-700 alloys. Corros Sci 88:405–415. https://doi.org/10.1016/j.corsci.2014.07.053
Zhang YD, Zhang C, Lan H, Hou PY, Yang ZG (2011) Improvement of the oxidation resistance of Tribaloy T-800 alloy by the additions of yttrium and aluminium. Corros Sci 53:1035–1043. https://doi.org/10.1016/j.corsci.2010.11.038
Rehman A, Liang Y, Bidabadi MHS, Yu Z, Zhang C, Chen H, Yang ZG (2020) Enhancement of cyclic oxidation resistance and effect of transition oxides on wear mechanism of yttrium plus aluminium modified tribaloy T-900 alloy. Corros Sci 163:108283. https://doi.org/10.1016/j.corsci.2019.108283
Díaz E, Amado JM, Montero J, Tobar MJ, Yáñez A (2012) Comparative study of Co-based alloys in repairing low Cr-Mo steel components by laser cladding. Phys Procedia 39:368–375. https://doi.org/10.1016/j.phpro.2012.10.050
Sahraoui T, Fenineche N-E, Montavon G, Coddet C (2004) Alternative to chromium: characteristics and wear behavior of HVOF coatings for gas turbine shafts repair (heavy-duty). J Mater Process Technol 152:43–55. https://doi.org/10.1016/j.jmatprotec.2004.02.061
Sahraoui T, Feraoun HI, Fenineche N, Montavon G, Aourag H, Coddet C (2004) HVOF-sprayed Tribaloy©-400: microstructure and first principle calculations. Mater Lett 58:2433–2436. https://doi.org/10.1016/j.matlet.2004.02.025
Koiprasert H, Dumrongrattana S, Niranatlumpong P (2004) Thermally sprayed coatings for protection of fretting wear in land-based gas-turbine engine. Wear 257:1–7. https://doi.org/10.1016/S0043-1648(03)00174-1
Vackel A (2015) In-situ observation of Laves phase precipitation and oxidation of HVOF deposited tribaloyTM T-800 (CoMoCrSi alloy) coatings. In: McDonald A, Agarwal A, Bolelli G, Concustell A, Lau Y-C, Toma F-L, Turunen E, Widener C (eds) Thermal Spray 2015: Proceedings from the International Thermal Spray Conference, Long Beach, CA. ASM International, Long Beach
Bolelli G, Lusvarghi L (2006) Heat treatment effects on the tribological performance of HVOF sprayed Co–Mo–Cr–Si coatings. J Therm Spray Technol 15:802–810. https://doi.org/10.1361/105996306X146721
Bolelli G, Lusvarghi L (2007) Tribological properties of HVOF as-sprayed and heat treated Co–Mo–Cr–Si coatings. Tribol Lett 25:43–54. https://doi.org/10.1007/s11249-006-9136-2
Guo H, Wang Y, Hao E, Li B, An Y, Chen J, Zhou H, Yan P, Wu Z (2019) CoMoCrSi coatings prepared by high-velocity oxygen fuel spraying: microstructure and mechanical properties at elevated temperatures up to 800°C. Mater Res Express 6:1265–1269. https://doi.org/10.1088/2053-1591/ab6374
Yang W-J, Zou L, Cao X-Y, Liu J-H, Li D-J, Cai Z-B (2019) Fretting wear properties of HVOF-sprayed CoMoCrSi coatings with different spraying parameters. Surf Coat Technol 358:994–1005. https://doi.org/10.1016/j.surfcoat.2018.12.039
Wang Y, Liu J, Kang N, Darut G, Poirier T, Stella J, Liao H, Planche M-P (2016) Cavitation erosion of plasma-sprayed CoMoCrSi coatings. Tribol Int 102:429–435. https://doi.org/10.1016/j.triboint.2016.06.014
Gao F, Liu R, Wu XJ (2011) Tribaloy alloy reinforced tin–bronze composite coating for journal bearing applications. Thin Solid Films 519:4809–4817. https://doi.org/10.1016/j.tsf.2011.01.035
Tavakoli A, Liu R, Wu XJ (2008) Improved mechanical and tribological properties of tin–bronze journal bearing materials with newly developed tribaloy alloy additive. Mater Sci Eng A 489:389–402. https://doi.org/10.1016/j.msea.2007.12.030
Khoddamzadeh A, Liu R, Wu X (2009) Novel polytetrafluoroethylene (PTFE) composites with newly developed Tribaloy alloy additive for sliding bearings. Wear 266:646–657. https://doi.org/10.1016/j.wear.2008.08.007
Renz A, Kürten D, Lehmann O (2017) Wear of hardfaced valve spindles in highly loaded stationary lean-burn large bore gas engines. Wear 376–377:1652–1661. https://doi.org/10.1016/j.wear.2016.12.045
Renz A, Prakash B, Hardell J, Lehmann O (2018) High-temperature sliding wear behaviour of Stellite(R)12 and Tribaloy(R) T400. Wear 402–403:148–159. https://doi.org/10.1016/j.wear.2018.02.013
Wang HM, Cao F, Cai LX, Tang HB, Yu RL, Zhang LY (2003) Microstructure and tribological properties of laser clad Ti2Ni3Si/NiTi intermetallic coatings. Acta Mater 51:6319–6327. https://doi.org/10.1016/S1359-6454(03)00465-8
Wang Y, Wang HM (2004) Wear resistance of laser clad Ti2Ni3Si reinforced intermetallic composite coatings on titanium alloy. Appl Surf Sci 229:81–86. https://doi.org/10.1016/j.apsusc.2004.01.045
Wang HM, Cai LX (2006) State of the art and prospects on laser clad multiphase transition metal silicides wear and corrosion resistant coatings for the aerospace and petrochemical industries. Solid State Phenom 118:235–242. https://doi.org/10.4028/www.scientific.net/SSP.118.235
Dong L, Wang H (2008) Microstructure and electrochemical characterization of laser melt-deposited Ti2Ni3Si/NiTi intermetallic alloys. Mater Charact 59:1587–1593. https://doi.org/10.1016/j.matchar.2008.02.003
Dong LX, Wang HM (2008) Microstructure and corrosion properties of laser-melted deposited Ti2Ni3Si/NiTi intermetallic alloy. J Alloys Compd 465:83–89. https://doi.org/10.1016/j.jallcom.2007.10.065
Wang HM, Tang HB, Cai LX, Cao F, Zhang LY, Yu RL (2005) Microstructure and wear properties of laser clad Ti2Ni3Si/Ni3Ti multiphase intermetallic coatings. Appl Phys A 80:1677–1682. https://doi.org/10.1007/s00339-003-2446-4
Gui YL, Wang HM (2007) Microstructure and dry sliding wear resistance of Moss-toughened Mo2Ni3Si metal silicide alloys. Int J Refract Met Hard Mater 25:433–439. https://doi.org/10.1016/j.ijrmhm.2006.11.001
Lu XD, Wang HM (2004) High-temperature phase stability and tribological properties of laser clad Mo2Ni3Si/NiSi metal silicide coatings. Acta Mater 52:5419–5426. https://doi.org/10.1016/j.actamat.2004.08.006
Lu XD, Wang HM (2005) Dry sliding wear behavior of laser clad Mo2Ni3Si/NiSi metal silicide composite coatings. Thin Solid Films 472:297–301. https://doi.org/10.1016/j.tsf.2004.07.006
Lu XD, Wang HM (2005) Corrosive sliding wear behavior of laser clad Mo2Ni3Si/NiSi intermetallic coating. Appl Surf Sci 245:346–352. https://doi.org/10.1016/j.apsusc.2004.10.031
Wang HM, Luan DY, Cai LX (2003) Microstructure and sliding-wear behavior of tungsten-reinforced W-Ni-Si metal-silicide in situ composites. Metall Mater Trans A 34:2005–2015. https://doi.org/10.1007/s11661-003-0165-2
Wang HM, Luan DY, Zhang LY (2003) Microstructure and wear resistance of laser melted W/W2Ni3Si metal silicides matrix in situ composites. Scr Mater 48:1179–1184. https://doi.org/10.1016/S1359-6462(02)00571-7
Cai LX, Wang HM (2004) Microstructure and dry-sliding wear resistance of laser clad tungsten reinforced W5Si3/W2Ni3Si intermetallic coatings. Appl Surf Sci 235:501–506. https://doi.org/10.1016/j.apsusc.2004.03.218
Liu Y, Wang HM (2005) Elevated temperature wear behaviors of a Co–Mo–Si ternary metal silicide alloy. Scr Mater 52:1235–1240. https://doi.org/10.1016/j.scriptamat.2005.03.002
Liu Y, Wang HM (2010) A novel load-insensitive Co3Mo2Si reinforced in situ metal-matrix composite coating for wear resistance application. Mater Lett 64:2494–2497. https://doi.org/10.1016/j.matlet.2010.08.002
Liu Y, Wang HM (2010) Microstructure and wear property of laser-clad Co3Mo2Si/Coss wear resistant coatings. Surf Coat Technol 205:377–382. https://doi.org/10.1016/j.surfcoat.2010.06.064
Liu Y, Wang HM (2013) Toughening and dry sliding wear resistance of Co3Mo2Si alloys. Wear 297:952–957. https://doi.org/10.1016/j.wear.2012.11.021
Cheng JB, Liang XB, Xu BS (2014) Effect of Nb addition on the structure and mechanical behaviors of CoCrCuFeNi high-entropy alloy coatings. Surf Coat Technol 240:184–190. https://doi.org/10.1016/j.surfcoat.2013.12.053
Huo W-Y, Shi H-F, Ren X, Zhang JY (2015) Microstructure and wear behavior of CoCrFeMnNbNi high-entropy alloy coating by TIG cladding. Adv Mater Sci Eng 2015:647351. https://doi.org/10.1155/2015/647351
Jiang L, Wu W, Cao Z, Deng D, Li T (2016) Microstructure evolution and wear behavior of the laser cladded CoFeNi2V0.5Nb0.75 and CoFeNi2V0.5Nb high-entropy alloy coatings. J Therm Spray Technol 25:806–814. https://doi.org/10.1007/s11666-016-0397-5
Ma M, Xiong W, Lian Y, Han D, Zhao CH, Zhang J (2020) Modeling and optimization for laser cladding via multi-objective quantum-behaved particle swarm optimization algorithm. Surf Coat Technol 381:125129. https://doi.org/10.1016/j.surfcoat.2019.125129
Liang H, Gao BY, Li YN, Nie QX, Cao ZQ (2019) Microstructures and wear resistance of Al1.5CrFeNiTi0.5 and Al1.5CrFeNiTi0.5W0.5 high entropy alloy coatings manufactured by laser cladding. Mater Sci Forum 956:154–159. https://doi.org/10.4028/www.scientific.net/MSF.956.154
Wang H, Liu QZ, Guo Y, Lan H (2019) MoFe1.5CrTiWAlNbx refractory high-entropy alloy coating fabricated by laser cladding. Intermetallics 115:106613. https://doi.org/10.1016/j.intermet.2019.106613
Guo Y, Wang H, Liu QZ (2020) Microstructure evolution and strengthening mechanism of laser-cladding MoFexCrTiWAlNby refractory high-entropy alloy coatings. J Alloys Compd 834:155147. https://doi.org/10.1016/j.jallcom.2020.155147
Xiang K, Chen LY, Chai L, Guo N, Wang H (2020) Microstructural characteristics and properties of CoCrFeNiNbx high-entropy alloy coatings on pure titanium substrate by pulsed laser cladding. Appl Surf Sci 517:146214. https://doi.org/10.1016/j.apsusc.2020.146214
Clark AE (1974) Magnetic and magnetoelastic properties of highly magnetostrictive rare earth-iron Laves phase compounds. AIP Conf Proc 18:1015–1029. https://doi.org/10.1063/1.2947192
Clark AE (1979) Magnetostrictive RFe2 intermetallic compounds. In: Gschneidner KA Jr, Eyring L (eds) Handbook on the physics and chemistry of rare earths. North-Holland, Amsterdam, pp 231–258. https://doi.org/10.1016/S0168-1273(79)02005-5
Clark AE (1988) Magnetostrictive rare earth-Fe2 compounds. In: Hamonic B, Decarpigny JN (eds) Power sonic and ultrasonic transducers design. Springer, Berlin, pp 43–99. https://doi.org/10.1007/978-3-642-73263-8_6
Clark AE, Belson HS (1972) Giant room-temperature magnetostrictions in TbFe2 and DyFe2. Phys Rev B 5:3642–3644. https://doi.org/10.1103/PhysRevB.5.3642
Pinkerton FE, Capehart TW, Herbst JF, Brewer EG, Murphy CB (1997) Magnetostrictive SmFe2/metal composites. Appl Phys Lett 70:2601–2603. https://doi.org/10.1063/1.118930
Samata H, Fujiwara N, Nagata Y, Uchida T, Der Lan M (1999) Magnetic anisotropy and magnetostriction of SmFe2 crystal. J Magn Magn Mater 195:376–383. https://doi.org/10.1016/S0304-8853(99)00127-4
Meeks SW, Timme RW (1977) Rare earth iron magnetostrictive underwater sound transducer. J Acoust Soc Am 62:1158–1164. https://doi.org/10.1121/1.381650
Olabi AG, Grunwald A (2008) Design and application of magnetostrictive materials. Mater Des 29:469–483. https://doi.org/10.1016/j.matdes.2006.12.016
Moffett MB, Clark AE, Wun-Fogle M, Linberg J, Teter JP, McLaughlin EA (1991) Characterization of Terfenol-D for magnetostrictive transducers. J Acoust Soc Am 89:1448–1455. https://doi.org/10.1121/1.400678
Savage HT, Abbundi R, Clark AE, McMasters OD (1979) Permeability, magnetomechanical coupling and magnetostriction in grain-oriented rare earth–iron alloys. J Appl Phys 50:1674–1676. https://doi.org/10.1063/1.327234
Sewell JM, Kuhn PM (1988) Opportunities and challenges in the use of Terfenol for sonar transducers. In: Hamonic B, Decarpigny JN (eds) Power sonic and ultrasonic transducers design. Springer, Berlin, pp 134–142. https://doi.org/10.1007/978-3-642-73263-8_9
Houqing Z, Jianguo L, Xiurong W, Yanhong X, Hongping Z (1997) Applications of Terfenol-D in China. J Alloys Compd 258:49–52. https://doi.org/10.1016/S0925-8388(97)00068-6
Snodgrass JD, McMasters OD (1997) Optimized TERFENOL-D manufacturing processes. J Alloys Compd 258:24–29. https://doi.org/10.1016/S0925-8388(97)00067-4
Lim SH, Kim SR, Kang SY, Park JK, Nam JT, Son D (1999) Magnetostrictive properties of polymer-bonded Terfenol-D composites. J Magn Magn Mater 191:113–121. https://doi.org/10.1016/S0304-8853(98)00315-1
Zhang T, Jiang C, Liu X, Xu H (2005) Dynamic magnetostrain properties of giant magnetostrictive alloy actuators for damping. Smart Mater Struct 14:N38–N41. https://doi.org/10.1088/0964-1726/14/4/n06
Lafont T, Gimeno L, Delamare J, Lebedev GA, Zakharov DI, Viala B, Cugat O, Galopin N, Garbuio L, Geoffroy O (2012) Magnetostrictive–piezoelectric composite structures for energy harvesting. J Micromech Microeng 22:094009. https://doi.org/10.1088/0960-1317/22/9/094009
Issindou V, Viala B, Gimeno L, Cugat O, Rado C, Bouat S (2017) Fabrication methods for high-performance miniature disks of Terfenol-D for energy harvesting. Sens Actuators, A 284:1–5. https://doi.org/10.1016/j.sna.2018.09.003
Murtaza A, Li Y, Mi J, Zuo W, Ghani A, Dai Z, Yao K, Hao C, Yaseen M, Saeed A, Yang S, Song X (2020) Spin configuration, magnetic and magnetostrictive properties of Tb0.27Dy0.73−xNdxFe2 compounds. Mater Chem Phys 249:122951. https://doi.org/10.1016/j.matchemphys.2020.122951
Zhou Z, Li J, Bao X, Zhou Y, Gao X (2020) Improvement of bending strength via introduced (Dy, Tb)Cu phase at grain boundary on giant magnetostrictive Tb-Dy-Fe alloy by diffusing Dy–Cu alloys. J Alloys Compd 826:153959. https://doi.org/10.1016/j.jallcom.2020.153959
Liu JH, Jiang CB, Xu HB (2012) Giant magnetostrictive materials. Sci China Technol Sci 55:1319–1326. https://doi.org/10.1007/s11431-012-4810-0
Ren W-J, Zhang Z-D (2013) Progress in bulk MgCu2-type rare-earth iron magnetostrictive compounds. Chin Phys B 22:077507. https://doi.org/10.1088/1674-1056/22/7/077507
Grössinger R, Turtelli RS, Mehmood N (2014) Materials with high magnetostriction. IOP Conf Ser: Mater Sci Eng 60:012002. https://doi.org/10.1088/1757-899x/60/1/012002
Barbara B, Giraud JP, Laforest J, Lemaire R, Siaud E, Schweizer J (1977) Spontaneous magnetoelastic distortion in some rare earth-iron Laves phases. Physica B + C 86–88:155–157. https://doi.org/10.1016/0378-4363(77)90267-4
Gschneidner KA Jr, Pecharsky VK, Tsokol AO (2005) Recent developments in magnetocaloric materials. Rep Prog Phys 68:1479–1539. https://doi.org/10.1088/0034-4885/68/6/R04
Tishin AM, Spichkin YI (2003) The magnetocaloric effect and its applications. Taylor & Francis, Boca Raton
Franco V, Blázquez JS, Ingale B, Conde A (2012) The magnetocaloric effect and magnetic refrigeration near room temperature: materials and models. Annu Rev Mater Res 42:305–342. https://doi.org/10.1146/annurev-matsci-062910-100356
Matsumoto K, Kouen T, Nishida R, Abe S, Kamiya K, Numazawa T (2006) Magnetocaloric effect of RCo2 (R: Er, Ho, Dy) compounds for regenerative magnetic refrigeration. AIP Conf Proc 850:1581–1582. https://doi.org/10.1063/1.2355310
Hashimoto T, Matsumoto K, Kurihara T, Numazawa T, Tomokiyo A, Yayama H, Goto T, Todo S, Sahashi M (1986) Investigations on the possibility of the RAl2 system as a refrigerant in an Ericsson type magnetic refrigerator. In: Reed RP, Clark AF (eds) Advances in cryogenic engineering materials. Springer, Boston, pp 279–286. https://doi.org/10.1007/978-1-4613-9871-4_33
Wikus P, Canavan E, Heine ST, Matsumoto K, Numazawa T (2014) Magnetocaloric materials and the optimization of cooling power density. Cryogenics 62:150–162. https://doi.org/10.1016/j.cryogenics.2014.04.005
Sánchez Llamazares JL, Zamora J, Sánchez-Valdés CF, Álvarez-Alonso P (2020) Design and fabrication of a cryogenic magnetocaloric composite by spark plasma sintering based on the RAl2 Laves phases (R = Ho, Er). J Alloys Compd 831:154779. https://doi.org/10.1016/j.jallcom.2020.154779
Gao T, Qi N, Zhang Y, Zhou T (2014) Magnetic properties and large magnetocaloric effect in Laves phase metallic compound. J Phys: Conf Ser 568:042006. https://doi.org/10.1088/1742-6596/568/4/042006
Politova GA, Pankratov NY, Vanina PY, Filimonov AV, Rudskoy AI, Burkhanov GS, Ilyushin AS, Tereshina IS (2019) Magnetocaloric effect and magnetostrictive deformation in Tb-Dy-Gd-Co-Al with Laves phase structure. J Magn Magn Mater 470:50–54. https://doi.org/10.1016/j.jmmm.2017.11.016
Kumar P, Kashyap A, Balamurugan B, Shield JE, Sellmyer DJ, Skomski R (2014) Permanent magnetism of intermetallic compounds between light and heavy transition-metal elements. J Phys: Condens Matter 26:064209. https://doi.org/10.1088/0953-8984/26/6/064209
Ohta K, Nagata Y (1978) Magnetic properties of iron-beryllium compounds (in Japanese). J Jpn Soc Powder Powder Metall 25:141–144. https://doi.org/10.2497/jjspm.25.141
Samata H, Nagata Y, Morita S, Tanaka G, Mitsuhashi T, Yashiro S, Abe S (2000) Magnetic properties of the Laves-phase compounds Fe1−xCoxBe2. J Alloys Compd 302:29–35. https://doi.org/10.1016/S0925-8388(00)00686-1
Gabay AM, Hadjipanayis GC (2018) Assessment of off-stoichiometric Zr33−xFe52+xSi15 C14 Laves phase compounds as permanent magnet materials. AIP Adv 8:056204. https://doi.org/10.1063/1.5006488
Zhang WY, Li XZ, Valloppilly S, Skomski R, Shield JE, Sellmyer DJ (2014) Development and intrinsic properties of hexagonal ferromagnetic (Zr,Ti)Fe2. J Appl Phys 115:17A769. https://doi.org/10.1063/1.4868696
Gabay AM, Hadjipanayis GC (2019) Semi-hard magnetic nanocomposites based on out-of-equilibrium Fe2+δNb and Fe2+δTa Laves phases. AIP Adv 9:035143. https://doi.org/10.1063/1.5079727
Sözen HI, Ener S, Maccari F, Skokov KP, Gutfleisch O, Körmann F, Neugebauer J, Hickel T (2019) Ab initio phase stabilities of Ce-based hard magnetic materials and comparison with experimental phase diagrams. Phys Rev Mater 3:084407. https://doi.org/10.1103/PhysRevMaterials.3.084407
Sözen HI, Hickel T, Neugebauer J (2020) Impact of magnetism on the phase stability of rare-earth based hard magnetic materials. Calphad 68:101731. https://doi.org/10.1016/j.calphad.2019.101731
Li Z, Yue M, Liu X, Zhang H, Liu W, Li Y, Zhang Z (2020) Tuning the structure and intrinsic magnetic properties of Ce2Fe14B alloys by elimination of CeFe2 with La substitution. J Magn Magn Mater 505:166747. https://doi.org/10.1016/j.jmmm.2020.166747
Matthias BT, Geballe TH, Compton VB (1963) Superconductivity. Rev Mod Phys 35:1–22. https://doi.org/10.1103/RevModPhys.35.1
Matthias BT, Compton VB, Corenzwit E (1961) Some new superconducting compounds. J Phys Chem Solids 19:130–133. https://doi.org/10.1016/0022-3697(61)90066-X
Matthias BT, Suhl H, Corenzwit E (1958) Spin exchange in superconductors. Phys Rev Lett 1:92–94. https://doi.org/10.1103/PhysRevLett.1.92
Sadagopan V, Pollard E, Gatos HC (1965) Superconducting transition temperatures of ZrMo2, HfV2 and PbAu2. Solid State Commun 3:97–98. https://doi.org/10.1016/0038-1098(65)90230-9
Rapp Ö, Benediktsson G (1979) Latent heat of structural transformations in ZrV2 and HfV2. Phys Lett A 74:449–452. https://doi.org/10.1016/0375-9601(79)90250-0
Takashima T, Hayashi H (1974) Lattice instabilities and superconductivity of (Hf, Zr)V2 compounds. Phys Lett A 47:209–210. https://doi.org/10.1016/0375-9601(74)90011-5
Inoue K, Tachikawa K, Iwasa Y (1971) A new high-field superconductor, V2HfxZr1−x. Appl Phys Lett 18:235–237. https://doi.org/10.1063/1.1653644
Inoue K, Kuroda T, Tachikawa K (1985) Superconducting properties of V2(Hf, Zr) Laves phase multifilamentary wires. J Nucl Mater 133–134:815–818. https://doi.org/10.1016/0022-3115(85)90264-8
Hishinuma Y, Kikuchi A, Iijima Y, Yoshida Y, Takeuchi T, Nishimura A, Inoue K (2004) Microstructure and superconductivity of V-based Laves-phase superconductor tape synthesized by a rapidly-heating/quenching process. J Nucl Mater 329–333:1580–1584. https://doi.org/10.1016/j.jnucmat.2004.04.166
Hishinuma Y, Kikuchi A, Iijima Y, Yoshida Y, Takeuchi T, Nishimura A, Inoue K (2005) Fabrication of V-based Laves phase compound multifilamentary wires by applying a rapidly-heating/quenching process to PIT precursors and using a V tube. IEEE Trans Appl Supercond 15:3536–3539. https://doi.org/10.1109/TASC.2005.849353
Hishinuma Y, Kikuchi A, Iijima Y, Yoshida Y, Takeuchi T, Nishimura A, Inoue K (2006) Research and development of low activation V-based superconducting materials based on the requirement for an advanced fusion reactor application. Fusion Eng Des 81:975–980. https://doi.org/10.1016/j.fusengdes.2005.09.028
Inoue K, Kuroda T, Tachikawa K (1979) Some improvements in Laves phase high-field superconductors. IEEE Trans Magn 15:635–638. https://doi.org/10.1109/TMAG.1979.1060070
Olzi E, Matacotta FC, Setina P (1988) The role of niobium in the processing of Laves phase superconducting wires. J Less-Common Met 139:123–132. https://doi.org/10.1016/0022-5088(88)90335-9
Inoue K, Tachikawa K (1974) Superconducting current capacities of composite-processed Laves phase tapes, V/HfxZr1−x. Appl Phys Lett 25:94–96. https://doi.org/10.1063/1.1655295
Kuroda T, Inoue K, Wada H, Tachikawa K (1983) Superconducting properties of V2(Hf, Zr) Laves phase multifilamentary wires. IEEE Trans Magn 19:410–413. https://doi.org/10.1109/TMAG.1983.1062417
Hazelton D (1987) Development of manufacturing techniques to prepare V2(Hf-Zr) Laves phase (C15) superconductor. IEEE Trans Magn 23:1026–1029. https://doi.org/10.1109/TMAG.1987.1065147
Jeong YB, Kim JT, Hong SH, Lee HD, Choi SY, Kim KB (2019) Compositional tuning-induced permanent color adjustment and mechanical properties: binary Cu-Mg colored metallic system. Mater Des 175:107814. https://doi.org/10.1016/j.matdes.2019.107814
Machon L, Sauthoff G (1996) Deformation behaviour of Al-containing C14 Laves phase alloys. Intermetallics 4:469–481. https://doi.org/10.1016/0966-9795(96)00023-4
Anton DL, Shah DM (1992) High temperature evaluation of topologically close packed intermetallics. Mater Sci Eng A 153:410–415. https://doi.org/10.1016/B978-1-85166-822-9.50066-2
Anton DL, Shah DM (1993) Prospects, promises and properties of refractory intermetallics. Mater Res Soc Symp Proc 288:141–150. https://doi.org/10.1557/PROC-288-141
Stoloff NS (2001) Intermetallics: mechanical Properties. In: Buschow KHJ, Cahn RW, Flemings MC, Ilschner B, Kramer EJ, Mahajan S, Veyssiere P (eds) Encyclopedia of materials: science and technology. Elsevier, Oxford, pp 4213–4225
INCO Alloys International (1982) INCO Alloy MA 6000, INCO MAP (Mechanical Alloyed Products) brochure. Huntington WV, USA
Voß S, Stein F, Palm M, Raabe D (2011) Compositional dependence of the compressive yield strength of Fe-Nb(-Al) and Co-Nb Laves phases. Mater Res Soc Symp Proc 1295:311–316. https://doi.org/10.1557/opl.2011.459
Takasugi T, Yoshida M, Hanada S (1996) Deformability improvement in C15 NbCr2 intermetallics by addition of ternary elements. Acta Mater 44:669–674. https://doi.org/10.1016/1359-6454(95)00174-3
Chan KS (2005) Alloying effects on the fracture toughness of Nb-based silicides and Laves phases. Mater Sci Eng A 409:257–269. https://doi.org/10.1016/j.msea.2005.06.077
Lu SQ, Zheng HZ, Fu MW (2009) The fracture toughness of hot-pressed NbCr2 alloys doped by rare earth yttrium. Scr Mater 61:205–207. https://doi.org/10.1016/j.scriptamat.2009.03.049
Lu S, Zheng H, Deng L, Yao J (2013) Effect of silicon on the fracture toughness and oxidation behavior of hot pressed NbCr2 alloys. Mater Des 51:432–437. https://doi.org/10.1016/j.matdes.2013.04.033
Long Q, Wang J, Du Y, Holec D, Nie X, Jin Z (2016) Predicting an alloying strategy for improving fracture toughness of C15 NbCr2 Laves phase: a first-principles study. Comput Mater Sci 123:59–64. https://doi.org/10.1016/j.commatsci.2016.06.009
Anton DL, Shah DM (1990) Ductile phase toughening of brittle intermetallics. Mater Res Soc Symp Proc 194:45–52. https://doi.org/10.1557/PROC-194-45
Takeyama M, Liu CT (1991) Microstructure and mechanical properties of Laves phase alloys based on Cr2Nb. Mater Sci Eng A 132:61–66. https://doi.org/10.1016/0921-5093(91)90361-P
Takasugi T, Hanada S, Miyamoto K (1993) High temperature mechanical properties of Cr2Nb-based intermetallics. J Mater Res 8:3069–3077. https://doi.org/10.1557/JMR.1993.3069
Li KW, Li SM, Xue YL, Fu HZ (2013) Microstructure characterization and mechanical properties of a Laves-phase alloy based on Cr2Nb. Int J Refract Met Hard Mater 36:154–161. https://doi.org/10.1016/j.ijrmhm.2012.08.009
Li KW, Li SM, Zhao SX, Zhong H, Xue YL, Fu HZ (2013) Phase stability of the Laves phase Cr2Nb in a two-phase Cr-Cr2Nb alloy. Acta Metall Sin (Engl Lett) 26:687–692. https://doi.org/10.1007/s40195-013-0140-x
Xue YL, Li SM, Zhong H, Fu HZ (2015) Characterization of fracture toughness and toughening mechanisms in Laves phase Cr2Nb based alloys. Mater Sci Eng A 638:340–347. https://doi.org/10.1016/j.msea.2015.04.077
Xue Y, Li S, Wu Y, Liu H, Li K (2020) The improved strength and toughness in the quinary Cr2Nb/Cr in situ composite. J Alloys Compd 825:153957. https://doi.org/10.1016/j.jallcom.2020.153957
Li KW, Wang T, Di C, Gong DQ (2016) Enhanced mechanical properties of NbCr2 Laves phase by spark plasma sintering. Vacuum 131:28–33. https://doi.org/10.1016/j.vacuum.2016.05.029
Li KW, Gao F, Wang L (2018) Microstructure and phase evolution in spark plasma sintering of the NbCr2 Laves phase matrix composite toughened with ductile Cr phase. J Mater Res 33:4329–4339. https://doi.org/10.1557/jmr.2018.371
Gao F, Li S, Li K (2020) Formation of non-equilibrium ductile solid solutions and textures in NbCr2 bulks produced by mechanical milling and spark plasma sintering. J Alloys Compd 826:154130. https://doi.org/10.1016/j.jallcom.2020.154130
Knowles AJ, Bhowmik A, Purkayastha S, Jones NG, Giuliani F, Clegg WJ, Dye D, Stone HJ (2017) Laves phase intermetallic matrix composite in situ toughened by ductile precipitates. Scr Mater 140:59–62. https://doi.org/10.1016/j.scriptamat.2017.06.043
Abe F (2008) Strengthening mechanisms in steel for creep and creep rupture. In: Abe F, Kern T-U, Viswanathan R (eds) Creep-resistant steels. Woodhead Publishing, Cambridge, pp 279–304. https://doi.org/10.1533/9781845694012.2.279
Hald J (2008) Microstructure and long-term creep properties of 9–12% Cr steels. Int J Press Vessel Pip 85:30–37. https://doi.org/10.1016/j.ijpvp.2007.06.010
Klueh RL (2005) Elevated temperature ferritic and martensitic steels and their application to future nuclear reactors. Int Mater Rev 50:287–310. https://doi.org/10.1179/174328005X41140
Gemmill MG, Hughes H, Murray JD, Pickering FB, Andrews KW (1956) Study of 7% and 8% chromium creep-resisting steels for use in steam power plant. J Iron Steel Inst 184:122–144
Wever F, Peter W (1942) Ausscheidungshärtung und Dauerstandfestigkeit von Eisen-Niob-Legierungen und nioblegierten Stählen (Precipitation hardening and creep strength of iron-niobium alloys and niobium-alloyed steels; in German). Arch Eisenhüttenwes 15:357–363. https://doi.org/10.1002/srin.194200608
Speich GR (1962) Precipitation of Laves phases from iron-niobium (columbium) and iron-titanium solid solutions. Trans Metall Soc AIME 224:850–858
Jones RH, Zackay VF, Parker ER (1972) Laves phase precipitation in Fe-Ta alloys. Metall Trans 3:2835–2842. https://doi.org/10.1007/BF02652850
Bhandarkar MD, Bhat MS, Zackay VF, Parker ER (1975) Structure and elevated temperature properties of carbon-free ferritic alloys strengthened by a Laves phase. Metall Trans A 6:1281–1289. https://doi.org/10.1007/BF02658538
Bhandarkar MD, Bhat MS, Parker ER, Zackay VF (1976) Creep and fracture of a Laves phase strengthened ferritic alloy. Metall Trans A 7:753–760. https://doi.org/10.1007/BF03186808
Danielsen HK, Hald J (2006) Behaviour of Z phase in 9–12%Cr steels. Energy Mater 1:49–57. https://doi.org/10.1179/174892306X99732
Golpayegani A, Andrén H-O, Danielsen H, Hald J (2008) A study on Z-phase nucleation in martensitic chromium steels. Mater Sci Eng A 489:310–318. https://doi.org/10.1016/j.msea.2007.12.022
Cipolla L, Danielsen HK, Venditti D, Di Nunzio PE, Hald J, Somers MAJ (2010) Conversion of MX nitrides to Z-phase in a martensitic 12% Cr steel. Acta Mater 58:669–679. https://doi.org/10.1016/j.actamat.2009.09.045
Schneider A, Inden G (2005) Simulation of the kinetics of precipitation reactions in ferritic steels. Acta Mater 53:519–531. https://doi.org/10.1016/j.actamat.2004.10.008
Srinivas Prasad BS, Rajkumar VB, Hari Kumar KC (2012) Numerical simulation of precipitate evolution in ferritic–martensitic power plant steels. Calphad 36:1–7. https://doi.org/10.1016/j.calphad.2011.10.006
Panait CG, Bendick W, Fuchsmann A, Gourgues-Lorenzon AF, Besson J (2010) Study of the microstructure of the Grade 91 steel after more than 100,000 h of creep exposure at 600°C. Int J Press Vessel Pip 87:326–335. https://doi.org/10.1016/j.ijpvp.2010.03.017
Robson JD, Bhadeshia HKDH (1996) Kinetics of precipitation in power plant steels. Calphad 20:447–460. https://doi.org/10.1016/S0364-5916(97)00007-2
Robson JD, Bhadeshia HKDH (1997) Modelling precipitation sequences in power plant steels Part 1—kinetic theory. Mater Sci Technol 13:631–639. https://doi.org/10.1179/mst.1997.13.8.631
Robson JD, Bhadeshia HKDH (1997) Modelling precipitation sequences in power plant steels Part 2—application of kinetic theory. Mater Sci Technol 13:640–644. https://doi.org/10.1179/mst.1997.13.8.640
Hald J, Korcakova L (2003) Precipitate stability in creep resistant ferritic steels—experimental investigations and modelling. ISIJ Int 43:420–427. https://doi.org/10.2355/isijinternational.43.420
Prat O, Garcia J, Rojas D, Carrasco C, Inden G (2010) Investigations on the growth kinetics of Laves phase precipitates in 12% Cr creep-resistant steels: experimental and DICTRA calculations. Acta Mater 58:6142–6153. https://doi.org/10.1016/j.actamat.2010.07.033
Prat O, Garcia J, Rojas D, Sauthoff G, Inden G (2013) The role of Laves phase on microstructure evolution and creep strength of novel 9%Cr heat resistant steels. Intermetallics 32:362–372. https://doi.org/10.1016/j.intermet.2012.08.016
Lu Q, Xu W, van der Zwaag S (2014) The computational design of W and Co-containing creep-resistant steels with barely coarsening Laves phase and M23C6 as the strengthening precipitates. Metall Mater Trans A 45:6067–6074. https://doi.org/10.1007/s11661-014-2557-x
Xia Z-X, Wang C-Y, Lei C, Lal Y-T, Zhao Y-F, Zhang L (2016) Growth kinetics of Laves phase and its effect on creep rupture behavior in 9Cr heat resistant steel. J Iron Steel Res Int 23:685–691. https://doi.org/10.1016/S1006-706X(16)30106-6
Fujita T (1992) Current progress in advanced high Cr ferritic steels for high-temperature applications. ISIJ Int 32:175–181. https://doi.org/10.2355/isijinternational.32.175
Hald J (1996) Metallurgy and creep properties of new 9–12%Cr steels. Steel Res 67:369–374. https://doi.org/10.1002/srin.199605503
Zhang XZ, Wu XJ, Liu R, Liu J, Yao MX (2017) Influence of Laves phase on creep strength of modified 9Cr-1Mo steel. Mater Sci Eng A 706:279–286. https://doi.org/10.1016/j.msea.2017.08.111
Sarwat SG, Basu J (2019) Understanding Laves phase precipitation induced embrittlement of modified 9Cr–1Mo steel. SN Appl Sci 1:89. https://doi.org/10.1007/s42452-018-0101-0
Sathyanarayanan S, Basu J, Moitra A, Sasikala G, Singh V (2013) Effect of thermal aging on ductile-brittle transition temperature of modified 9Cr-1Mo steel evaluated with reference temperature approach under dynamic loading condition. Metall Mater Trans A 44:2141–2155. https://doi.org/10.1007/s11661-012-1510-0
Thomas Paul V, Saroja S, Vijayalakshmi M (2008) Microstructural stability of modified 9Cr–1Mo steel during long term exposures at elevated temperatures. J Nucl Mater 378:273–281. https://doi.org/10.1016/j.jnucmat.2008.06.033
Ennis PJ, Czyrska-Filemonowicz A (2003) Recent advances in creep-resistant steels for power plant applications. Sadhana 28:709–730. https://doi.org/10.1007/BF02706455
Abe F (2015) Creep behavior, deformation mechanisms, and creep life of Mod. 9Cr-1Mo steel. Metall Mater Trans A 46:5610–5625. https://doi.org/10.1007/s11661-015-3144-5
Hosoi Y, Wade N, Kunimitsu S, Urita T (1986) Precipitation behavior of Laves phase and its effect on toughness of 9Cr-2Mo ferritic-martensitic steel. J Nucl Mater 141–143:461–467. https://doi.org/10.1016/S0022-3115(86)80083-6
Hosoi Y, Wade N, Kunimitsu S, Urita T (1990) A proposal to mechanism of embrittlement of 9Cr-2Mo heat resisting steel during long-term aging. Tetsu-to-Hagané 76:1116–1123. https://doi.org/10.2355/tetsutohagane1955.76.7_1116
Kunimitsu S, You Y, Kasuya N, Sasaki Y, Hosoi Y (1991) Effect of thermo-mechanical treatment on toughness of 9Cr-W ferritic-martensitic steels during aging. J Nucl Mater 179–181:689–692. https://doi.org/10.1016/0022-3115(91)90182-7
Aghajani A, Richter F, Somsen C, Fries SG, Steinbach I, Eggeler G (2009) On the formation and growth of Mo-rich Laves phase particles during long-term creep of a 12% chromium tempered martensite ferritic steel. Scr Mater 61:1068–1071. https://doi.org/10.1016/j.scriptamat.2009.08.031
Aghajani A, Somsen C, Eggeler G (2009) On the effect of long-term creep on the microstructure of a 12% chromium tempered martensite ferritic steel. Acta Mater 57:5093–5106. https://doi.org/10.1016/j.actamat.2009.07.010
Abe F (2001) Creep rates and strengthening mechanisms in tungsten-strengthened 9Cr steels. Mater Sci Eng A 319–321:770–773. https://doi.org/10.1016/S0921-5093(00)02002-5
Abe F (2005) Effect of fine precipitation and subsequent coarsening of Fe2W Laves phase on the creep deformation behavior of tempered martensitic 9Cr-W steels. Metall Mater Trans A 36:321–332. https://doi.org/10.1007/s11661-005-0305-y
Korcakova L, Hald J, Somers MAJ (2001) Quantification of Laves phase particle size in 9CrW steel. Mater Charact 47:111–117. https://doi.org/10.1016/S1044-5803(01)00159-0
Lee JS, Armaki HG, Maruyama K, Muraki T, Asahi H (2006) Causes of breakdown of creep strength in 9Cr–1.8W–0.5Mo–VNb steel. Mater Sci Eng A 428:270–275. https://doi.org/10.1016/j.msea.2006.05.010
Výrostková A, Homolová V, Pecha J, Svoboda M (2008) Phase evolution in P92 and E911 weld metals during ageing. Mater Sci Eng A 480:289–298. https://doi.org/10.1016/j.msea.2007.07.036
Falat L, Výrostková A, Homolová V, Svoboda M (2009) Creep deformation and failure of E911/E911 and P92/P92 similar weld-joints. Eng Failure Anal 16:2114–2120. https://doi.org/10.1016/j.engfailanal.2009.02.004
Wang W, Yan W, Sha W, Shan Y, Yang K (2012) Microstructural evolution and mechanical properties of short-term thermally exposed 9/12Cr heat-resistant steels. Metall Mater Trans A 43:4113–4122. https://doi.org/10.1007/s11661-012-1240-3
Dudko V, Belyakov A, Molodov D, Kaibyshev R (2013) Microstructure evolution and pinning of boundaries by precipitates in a 9 pct Cr heat resistant steel during creep. Metall Mater Trans A 44:162–172. https://doi.org/10.1007/s11661-011-0899-1
Sakthivel T, Vasudevan M, Laha K, Parameswaran P, Chandravathi KS, Panneer Selvi S, Maduraimuthu V, Mathew MD (2014) Creep rupture behavior of 9Cr–1.8W–0.5Mo–VNb (ASME grade 92) ferritic steel weld joint. Mater Sci Eng A 591:111–120. https://doi.org/10.1016/j.msea.2013.10.071
Saini NL, Mulik RS, Mahapatra MM (2018) Study on the effect of ageing on Laves phase evolution and their effect on mechanical properties of P92 steel. Mater Sci Eng A 716:179–188. https://doi.org/10.1016/j.msea.2018.01.035
Isik MI, Kostka A, Eggeler G (2014) On the nucleation of Laves phase particles during high-temperature exposure and creep of tempered martensite ferritic steels. Acta Mater 81:230–240. https://doi.org/10.1016/j.actamat.2014.08.008
Isik MI, Kostka A, Yardley VA, Pradeep KG, Duarte MJ, Choi PP, Raabe D, Eggeler G (2015) The nucleation of Mo-rich Laves phase particles adjacent to M23C6 micrograin boundary carbides in 12% Cr tempered martensite ferritic steels. Acta Mater 90:94–104. https://doi.org/10.1016/j.actamat.2015.01.027
Dimmler G, Weinert P, Kozeschnik E, Cerjak H (2003) Quantification of the Laves phase in advanced 9–12% Cr steels using a standard SEM. Mater Charact 51:341–352. https://doi.org/10.1016/j.matchar.2004.02.003
Wang X, Pan Q-G, Liu ZJ, Zeng H-Q, Tao YS (2011) Creep rupture behaviour of P92 steel weldment. Eng Failure Anal 18:186–191. https://doi.org/10.1016/j.engfailanal.2010.08.020
Cao J, Gong Y, Yang Z-G (2011) Microstructural analysis on creep properties of dissimilar materials joints between T92 martensitic and HR3C austenitic steels. Mater Sci Eng A 528:6103–6111. https://doi.org/10.1016/j.msea.2011.04.057
Cao J, Gong Y, Yang Z-G, Luo X-M, Gu F-M, Hu Z-F (2011) Creep fracture behavior of dissimilar weld joints between T92 martensitic and HR3C austenitic steels. Int J Press Vessel Pip 88:94–98. https://doi.org/10.1016/j.ijpvp.2011.01.003
Chen G, Zhang Q, Liu J, Wang J, Yu X, Hua J, Bai X, Zhang T, Zhang J, Tang W (2013) Microstructures and mechanical properties of T92/Super304H dissimilar steel weld joints after high-temperature ageing. Mater Des 44:469–475. https://doi.org/10.1016/j.matdes.2012.08.022
Kim M-Y, Kwak S-C, Choi I-S, Lee Y-K, Suh J-Y, Fleury E, Jung W-S, Son T-H (2014) High-temperature tensile and creep deformation of cross-weld specimens of weld joint between T92 martensitic and Super304H austenitic steels. Mater Charact 97:161–168. https://doi.org/10.1016/j.matchar.2014.09.011
Zhai X-W, Du J-F, Li L-P, Zhou C, Zhang Z (2019) Creep behavior and damage evolution of T92/Super304H dissimilar weld joints. J Iron Steel Res Int 26:751–760. https://doi.org/10.1007/s42243-019-00249-3
Bae C, Kim R, Heo JH, Kim J (2018) Effect of compositional changes of Laves phase precipitate on grain boundary embrittlement in long-term annealed 9 Pct Cr ferritic steel. Metall Mater Trans A 49:4595–4603. https://doi.org/10.1007/s11661-018-4782-1
Miyata K, Sawaragi Y, Okada H, Masuyama F, Yokoyama T, Komai N (2000) Microstructural evolution of a 12Cr-2W-Cu-V-Nb steel during three-year service exposure. ISIJ Int 40:1156–1163. https://doi.org/10.2355/isijinternational.40.1156
Hättestrand M, Andrén H-O (2001) Evaluation of particle size distributions of precipitates in a 9% chromium steel using energy filtered transmission electron microscopy. Micron 32:789–797. https://doi.org/10.1016/S0968-4328(00)00086-X
Li Q (2006) Precipitation of Fe2W Laves phase and modeling of its direct influence on the strength of a 12Cr-2W steel. Metall Mater Trans A 37:89–97. https://doi.org/10.1007/s11661-006-0155-2
Zhang ZB, Fan ZD, Ma YC, Niu K, Liu CX (2019) Microstructural evolution and mechanical properties of T122 steel tube in a 1000 MW ultra supercritical unit after long-term service. In: Shingledecker J, Takeyama M (eds) Proc Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 265–272
Murata Y, Morinaga M, Hashizume R, Takami K, Azuma T, Tanaka Y, Ishiguro T (2000) Effect of carbon content on the mechanical properties of 10Cr–5W ferritic steels. Mater Sci Eng A 282:251–261. https://doi.org/10.1016/S0921-5093(99)00780-7
Knežević V, Sauthoff G, Vilk J, Inden G, Schneider A, Agamemnone R, Blum W, Wang Y, Scholz A, Berger C (2002) Martensitic/ferritic super heat-resistant 650°C steels—design and testing of model alloys. ISIJ Int 42:1505–1514. https://doi.org/10.2355/isijinternational.42.1505
Knežević V, Balun J, Sauthoff G, Inden G, Schneider A (2008) Design of martensitic/ferritic heat-resistant steels for application at 650°C with supporting thermodynamic modelling. Mater Sci Eng A 477:334–343. https://doi.org/10.1016/j.msea.2007.05.047
Rojas D, Garcia J, Prat O, Carrasco C, Sauthoff G, Kaysser-Pyzalla AR (2010) Design and characterization of microstructure evolution during creep of 12% Cr heat resistant steels. Mater Sci Eng A 527:3864–3876. https://doi.org/10.1016/j.msea.2010.02.056
Sanhueza JP, Rojas D, Prat O, Garcia J, Melendrez M (2019) Laves phase in a 12% Cr martensitic/ferritic steel: evolution and characterization of nanoparticles at 650°C. J Nanosci Nanotechnol 19:2971–2976. https://doi.org/10.1166/jnn.2019.15836
Foldyna V, Kubon Z, Filip M, Mayer K-H, Berger C (1996) Evaluation of structural stability and creep resistance of 9–12% Cr steels. Steel Res 67:375–381. https://doi.org/10.1002/srin.199605504
Cui H, Sun F, Chen K, Zhang L, Wan R, Shan A, Wu J (2010) Precipitation behavior of Laves phase in 10%Cr steel X12CrMoWVNbN10-1-1 during short-term creep exposure. Mater Sci Eng A 527:7505–7509. https://doi.org/10.1016/j.msea.2010.08.013
Xu Y, Wang M, Wang Y, Gu T, Chen L, Zhou X, Ma Q, Liu Y, Huang J (2015) Study on the nucleation and growth of Laves phase in a 10% Cr martensite ferritic steel after long-term aging. J Alloys Compd 621:93–98. https://doi.org/10.1016/j.jallcom.2014.09.204
Xu Y, Nie Y, Wang M, Li W, Jin X (2017) The effect of microstructure evolution on the mechanical properties of martensite ferritic steel during long-term aging. Acta Mater 131:110–122. https://doi.org/10.1016/j.actamat.2017.03.045
Abe F, Ohba T, Miyazaki H, Toda Y, Tabuchi M (2019) Effect of boron nitrides and aluminum nitrides on long-term creep life and rupture ductility of martensitic 9 to 12Cr steels. In: Shingledecker J, Takeyama M (eds) Proc Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 336–347
Abe F (2008) Precipitate design for creep strengthening of 9% Cr tempered martensitic steel for ultra-supercritical power plants. Sci Technol Adv Mater 9:013002. https://doi.org/10.1088/1468-6996/9/1/013002
Hu P, Yan W, Sha W, Wang W, Guo Z-L, Shan Y-Y, Yang K (2009) Study on Laves phase in an advanced heat-resistant steel. Front Mater Sci China 3:434. https://doi.org/10.1007/s11706-009-0063-7
Kipelova A, Kaibyshev R, Belyakov A, Molodov D (2011) Microstructure evolution in a 3%Co modified P911 heat resistant steel under tempering and creep conditions. Mater Sci Eng A 528:1280–1286. https://doi.org/10.1016/j.msea.2010.10.006
Dudova N, Plotnikova A, Molodov D, Belyakov A, Kaibyshev R (2012) Structural changes of tempered martensitic 9%Cr–2%W–3%Co steel during creep at 650°C. Mater Sci Eng A 534:632–639. https://doi.org/10.1016/j.msea.2011.12.020
Fedorova I, Kipelova A, Belyakov A, Kaibyshev R (2013) Microstructure evolution in an advanced 9 pct Cr martensitic steel during creep at 923 K (650°C). Metall Mater Trans A 44:S128–S135. https://doi.org/10.1007/s11661-012-1182-9
Fedoseeva A, Dudova N, Kaibyshev R (2016) Creep strength breakdown and microstructure evolution in a 3%Co modified P92 steel. Mater Sci Eng A 654:1–12. https://doi.org/10.1016/j.msea.2015.12.027
Mishnev R, Dudova N, Kaibyshev R (2018) On the origin of the superior long-term creep resistance of a 10% Cr steel. Mater Sci Eng A 713:161–173. https://doi.org/10.1016/j.msea.2017.12.066
Dudova N, Mishnev R, Kaibyshev R (2019) Creep behavior of a 10%Cr heat-resistant martensitic steel with low nitrogen and high boron contents at 650°C. Mater Sci Eng A 766:138353. https://doi.org/10.1016/j.msea.2019.138353
Nikitin I, Fedoseeva A, Dudova N, Kaibyshev R (2019) On the precipitation of the Laves phase particles in a martensitic 10% Cr-3% Co-3% W-0.2% Re steel during creep at 650°C. In: Shingledecker J, Takeyama M (eds) Proc Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 227–234
Gong XF, Wang TJ, Gao ZH, Zhong J, Miki K, Ohsaki S, Muto Y, Mito Y, Yamane Y, Takasawa K, Takahashi T (2019) The development of the middle scale prototype for the rotor used in the 630°C steam turbine. In: Shingledecker J, Takeyama M (eds) Proc Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 1262–1269
Tkachev E, Belyakov A (2019) Effect of heat treatment on microstructure and mechanical properties of an advanced Ta-alloyed 9%Cr steel. In: Shingledecker J, Takeyama M (eds) Proc Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 116–122
Tkachev E, Belyakov A, Kaibyshev R (2020) Creep strength breakdown and microstructure in a 9%Cr steel with high B and low N contents. Mater Sci Eng A 772:138821. https://doi.org/10.1016/j.msea.2019.138821
Zhu S, Yang M, Song XL, Zhang Z, Wang LB, Tang S, Xiang ZD (2014) A few observations on Laves phase precipitation in relation to its effects on creep rupture strength of ferritic steels based on Fe–9Cr (wt%) alloys at 650°C. Mater Sci Eng A 619:47–56. https://doi.org/10.1016/j.msea.2014.09.059
Klueh RL, Ehrlich K, Abe F (1992) Ferritic/martensitic steels: promises and problems. J Nucl Mater 191–194:116–124. https://doi.org/10.1016/S0022-3115(09)80018-4
Tanigawa H, Sakasegawa H, Ogiwara H, Kishimoto H, Kohyama A (2007) Radiation induced phase instability of precipitates in reduced-activation ferritic/martensitic steels. J Nucl Mater 367–370:132–136. https://doi.org/10.1016/j.jnucmat.2007.03.155
Saroja S, Dasgupta A, Divakar R, Raju S, Mohandas E, Vijayalakshmi M, Bhanu Sankara Rao K, Raj B (2011) Development and characterization of advanced 9Cr ferritic/martensitic steels for fission and fusion reactors. J Nucl Mater 409:131–139. https://doi.org/10.1016/j.jnucmat.2010.09.022
Huang L, Hu X, Yan W, Sha W, Xiao F, Shan Y, Yang K (2014) Laves-phase in the China Low Activation Martensitic steel after long-term creep exposure. Mater Des 63:333–335. https://doi.org/10.1016/j.matdes.2014.06.028
Wang W, Liu S, Xu G, Zhang B, Huang Q (2016) Effect of thermal aging on microstructure and mechanical properties of China Low-Activation Martensitic steel at 550 °C. Nucl Eng Technol 48:518–524. https://doi.org/10.1016/j.net.2015.11.004
He L, Tan L, Yang Y, Sridharan K (2019) Evolution of B2 and Laves phases in a ferritic steel under Fe2+ ion irradiation at 475 °C. J Nucl Mater 525:102–110. https://doi.org/10.1016/j.jnucmat.2019.07.024
Danielsen HK, Hald J (2009) On the nucleation and dissolution process of Z-phase Cr(V, Nb)N in martensitic 12%Cr steels. Mater Sci Eng A 505:169–177. https://doi.org/10.1016/j.msea.2008.11.019
Kimura K, Seki K, Toda Y, Abe F (2001) Development of high strength 15Cr ferritic creep resistant steel with addition of tungsten and cobalt. ISIJ Int 41:S121–S125. https://doi.org/10.2355/isijinternational.41.Suppl_S121
Kimura K, Toda Y, Kushima H, Abe F (2001) Development of high strength 15Cr creep resistant steel for low emission power plant. J Adv Sci 13:294–297. https://doi.org/10.2978/jsas.13.294
Toda Y, Seki K, Kimura K, Abe F (2002) Effects of W and Co on the microstructure and creep strength of the precipitation strengthened 15Cr ferritic steels. JSME Int J Ser A 45:25–29. https://doi.org/10.1299/jsmea.45.25
Toda Y, Seki K, Kimura K, Abe F (2003) Effects of W and Co on long-term creep strength of precipitation strengthened 15Cr ferritic heat resistant steels. ISIJ Int 43:112–118. https://doi.org/10.2355/isijinternational.43.112
Toda Y, Iijima M, Kushima H, Kimura K, Abe F (2005) Effects of Ni and heat treatment on long-term creep strength of precipitation strengthened 15Cr ferritic heat resistant steels. ISIJ Int 45:1747–1753. https://doi.org/10.2355/isijinternational.45.1747
Toda Y, Tohyama H, Kushima H, Kimura K, Abe F (2005) Improvement in creep strength of precipitation strengthened 15Cr ferritic steel by controlling carbon and nitrogen contents. JSME Int J Ser A 48:35–40. https://doi.org/10.1299/jsmea.48.35
Toda Y, Tohyama H, Kushima H, Kimura K, Abe F (2005) Influence of chemical composition and heat treatment condition on impact toughness of 15Cr ferritic creep resistant steel. JSME Int J Ser A 48:125–131. https://doi.org/10.1299/jsmea.48.125
Kimura K, Toda Y, Kushima H, Sawada K (2010) Creep strength of high chromium steel with ferrite matrix. Int J Pressure Vessels Piping 87:282–288. https://doi.org/10.1016/j.ijpvp.2010.03.016
Shibuya M, Toda Y, Sawada K, Kushima H, Kimura K (2011) Effect of nickel and cobalt addition on the precipitation-strength of 15Cr ferritic steels. Mater Sci Eng A 528:5387–5393. https://doi.org/10.1016/j.msea.2011.03.088
Shibuya M, Toda Y, Sawada K, Kushima H, Kimura K (2014) Effect of precipitation behavior on creep strength of 15%Cr ferritic steels at high temperature between 923 and 1023 K. Mater Sci Eng A 592:1–5. https://doi.org/10.1016/j.msea.2013.10.085
Shibuya M, Toda Y, Sawada K, Kushima H, Kimura K (2016) Improving the high-temperature creep strength of 15Cr ferritic creep-resistant steels at temperatures of 923–1023K. Mater Sci Eng A 652:1–6. https://doi.org/10.1016/j.msea.2015.11.068
Toda Y, Yamabe-Mitarai Y, Kaseya A, Umezawa O (2019) Improvement in creep and steam oxidation resistance of precipitation strengthened ferritic steels. In: Shingledecker J, Takeyama M (eds) Proc Joint EPRI–123HiMAT International Conference on Advances in High Temperature Materials. Nagasaki, Japan, ASM International, pp 96–103
Fujita N, Ohmura K, Kikuchi M, Suzuki T, Funaki S, Hiroshige I (1996) Effect of Nb on high-temperature properties for ferritic stainless steel. Scr Mater 35:705–710. https://doi.org/10.1016/1359-6462(96)00214-X
Fujita N, Bhadeshia HKDH, Kikuchi M (2004) Precipitation sequence in niobium-alloyed ferritic stainless steel. Modell Simul Mater Sci Eng 12:273–284. https://doi.org/10.1088/0965-0393/12/2/008
Sim GM, Ahn JC, Hong SC, Lee KJ, Lee KS (2005) Effect of Nb precipitate coarsening on the high temperature strength in Nb containing ferritic stainless steels. Mater Sci Eng A 396:159–165. https://doi.org/10.1016/j.msea.2005.01.030
Ghosh S (2010) Kinetic study on the coarsening behaviour of equilibrium phases in Nb alloyed ferritic stainless steels at 700°C. Mater Chem Phys 124:13–16. https://doi.org/10.1016/j.matchemphys.2010.06.013
Kato Y, Ito M, Kato Y, Furukimi O (2010) Effect of Si on precipitation behavior of Nb-Laves phase and amount of Nb in solid solution at elevated temperature in high purity 17%Cr-0.5%Nb steels. Mater Trans 51:1531–1535. https://doi.org/10.2320/matertrans.M2010112
Sello MP, Stumpf WE (2010) Laves phase embrittlement of the ferritic stainless steel type AISI 441. Mater Sci Eng A 527:5194–5202. https://doi.org/10.1016/j.msea.2010.04.058
Sello MP, Stumpf WE (2011) Laves phase precipitation and its transformation kinetics in the ferritic stainless steel type AISI 441. Mater Sci Eng A 528:1840–1847. https://doi.org/10.1016/j.msea.2010.09.090
Faivre L, Santacreu P-O, Acher A (2013) A new ferritic stainless steel with improved thermo-mechanical fatigue resistance for exhaust parts. Mater High Temp 30:36–42. https://doi.org/10.3184/096034013X13637176323444
Juuti T, Rovatti L, Mäkelä A, Karjalainen LP, Porter D (2014) Influence of long heat treatments on the Laves phase nucleation in a type 444 ferritic stainless steel. J Alloys Compd 616:250–256. https://doi.org/10.1016/j.jallcom.2014.06.201
Ikeda K-I, Yamoah NKG, Reynolds WT Jr, Hamada J-I, Murayama M (2015) Effect of Laves phase on high-temperature deformation and microstructure evolution in an 18Cr-2Mo-0.5Nb ferritic stainless steel. Metall Mater Trans A 46:3460–3469. https://doi.org/10.1007/s11661-015-2936-y
Nabiran N, Klein S, Weber S, Theisen W (2015) Evolution of the Laves phase in ferritic heat-resistant steels during long-term annealing and its influence on the high-temperature strength. Metall Mater Trans A 46:102–114. https://doi.org/10.1007/s11661-014-2505-9
Beck T, Kuhn B, Eckardt T, Singheiser L (2016) Microstructure, creep and thermomechanical fatigue of novel solid solution and Laves phase strengthened Cr2O3 and Al2O3 forming ferrites for car engine exhaust and heat exchanger systems. Trans Indian Inst Met 69:379–385. https://doi.org/10.1007/s12666-015-0747-x
Wei L, Zheng J, Chen L, Misra RDK (2018) High temperature oxidation behavior of ferritic stainless steel containing W and Ce. Corros Sci 142:79–92. https://doi.org/10.1016/j.corsci.2018.07.017
Juuti T, Rovatti L, Porter D, Angella G, Kömi J (2018) Factors controlling ambient and high temperature yield strength of ferritic stainless steel susceptible to intermetallic phase formation. Mater Sci Eng A 726:45–55. https://doi.org/10.1016/j.msea.2018.04.074
Juuti T, Manninen T, Uusikallio S, Kömi J, Porter D (2019) New ferritic stainless steel for service temperatures up to 1050°C utilizing intermetallic phase transformation. Metals 9:Article No 664. https://doi.org/10.3390/met9060664
Hu YK, Mao WM, Jing J, Cheng XM (2019) The high-temperature mechanical properties of EN 1.4509 stainless steel cast using investment casting. J Mater Eng Perform 28:6603–6613. https://doi.org/10.1007/s11665-019-04353-z
Kang Y, Mao WM, Chen YJ, Jing J, Cheng M (2016) Influence of Nb content on grain size and mechanical properties of 18wt% Cr ferritic stainless steel. Mater Sci Eng A 677:453–464. https://doi.org/10.1016/j.msea.2016.09.080
Ide S, Funakawa Y, Kato Y, Furukimi O (2007) Retardation of 20%Cr steel oxidation with Laves phase precipitation. Mater Sci Forum 539–543:4887–4890. https://doi.org/10.4028/www.scientific.net/MSF.539-543.4887
Horita T, Kishimoto H, Yamaji K, Xiong Y, Sakai N, Brito ME, Yokokawa H (2008) Evaluation of Laves-phase forming Fe–Cr alloy for SOFC interconnects in reducing atmosphere. J Power Sources 176:54–61. https://doi.org/10.1016/j.jpowsour.2007.10.041
Froitzheim J, Meier GH, Niewolak L, Ennis PJ, Hattendorf H, Singheiser L, Quadakkers WJ (2008) Development of high strength ferritic steel for interconnect application in SOFCs. J Power Sources 178:163–173. https://doi.org/10.1016/j.jpowsour.2007.12.028
Yang ZG, Xia G-G, Wang C-M, Nie Z, Templeton J, Stevenson JW, Singh P (2008) Investigation of iron–chromium–niobium–titanium ferritic stainless steel for solid oxide fuel cell interconnect applications. J Power Sources 183:660–667. https://doi.org/10.1016/j.jpowsour.2008.05.037
Jablonski PD, Cowen CJ, Sears JS (2010) Exploration of alloy 441 chemistry for solid oxide fuel cell interconnect application. J Power Sources 195:813–820. https://doi.org/10.1016/j.jpowsour.2009.08.023
Palcut M, Mikkelsen L, Neufeld K, Chen M, Knibbe R, Hendriksen PV (2010) Corrosion stability of ferritic stainless steels for solid oxide electrolyser cell interconnects. Corros Sci 52:3309–3320. https://doi.org/10.1016/j.corsci.2010.06.006
Kuhn B, Jimenez CA, Niewolak L, Hüttel T, Beck T, Hattendorf H, Singheiser L, Quadakkers WJ (2011) Effect of Laves phase strengthening on the mechanical properties of high Cr ferritic steels for solid oxide fuel cell interconnect application. Mater Sci Eng A 528:5888–5899. https://doi.org/10.1016/j.msea.2011.03.112
Chiu Y-T, Lin C-K (2012) Effects of Nb and W additions on high-temperature creep properties of ferritic stainless steels for solid oxide fuel cell interconnect. J Power Sources 198:149–157. https://doi.org/10.1016/j.jpowsour.2011.09.056
Ali-Löytty H, Jussila P, Juuti T, Karjalainen LP, Zakharov AA, Valden M (2012) Influence of precipitation on initial high-temperature oxidation of Ti–Nb stabilized ferritic stainless steel SOFC interconnect alloy. Int J Hydrogen Energy 37:14528–14535. https://doi.org/10.1016/j.ijhydene.2012.07.097
Garcia-Fresnillo L, Shemet V, Chyrkin A, de Haart LGJ, Quadakkers WJ (2014) Long-term behaviour of solid oxide fuel cell interconnect materials in contact with Ni-mesh during exposure in simulated anode gas at 700 and 800 °C. J Power Sources 271:213–222. https://doi.org/10.1016/j.jpowsour.2014.07.189
Safikhani A, Esmailian M, Salmani MR, Aminfard M (2014) Effect of Ni–Mo addition on cyclic and isothermal oxidation resistance and electrical behavior of ferritic stainless steel for SOFCs interconnect. Int J Hydrogen Energy 39:11210–11223. https://doi.org/10.1016/j.ijhydene.2014.05.122
Niewolak L, Savenko A, Grüner D, Hattendorf H, Breuer U, Quadakkers WJ (2015) Temperature dependence of Laves phase composition in Nb, W and Si-alloyed high chromium ferritic steels for SOFC interconnect applications. J Phase Equilib Diffus. https://doi.org/10.1007/s11669-015-0403-5
Niewolak L, Zurek J, Wessel E, Hattendorf H, Quadakkers WJ (2017) Temperature dependence of phase composition in W and Si-alloyed high chromium ferritic steels for SOFC interconnect applications. J Alloys Compd 717:240–253. https://doi.org/10.1016/j.jallcom.2017.05.113
Safikhani A, Esmailian M, Tinatiseresht T, Darband GB (2016) High temperature cyclic oxidation behavior of ferritic stainless steel with addition of alloying elements Nb and Ti for use in SOFCs interconnect. Int J Hydrogen Energy 41:6045–6052. https://doi.org/10.1016/j.ijhydene.2016.01.090
Ali-Löytty H, Hannula M, Juuti T, Niu Y, Zakharov AA, Valden M (2018) The role of (FeCrSi)2(MoNb)-type Laves phase on the formation of Mn-rich protective oxide scale on ferritic stainless steel. Corros Sci 132:214–222. https://doi.org/10.1016/j.corsci.2017.12.026
Hsiao Z-W, Kuhn B, Chen D, Singheiser L, Kuo J-C, Lin D-Y (2015) Characterization of Laves phase in Crofer 22 H stainless steel. Micron 74:59–64. https://doi.org/10.1016/j.micron.2015.04.007
Hsiao Z-W, Chen D, Kuo J-C, Lin D-Y (2017) Effect of prior deformation on microstructural development and Laves phase precipitation in high-chromium stainless steel. J Microsc 266:35–47. https://doi.org/10.1111/jmi.12516
Kuhn B, Talik M, Niewolak L, Zurek J, Hattendorf H, Ennis PJ, Quadakkers WJ, Beck T, Singheiser L (2014) Development of high chromium ferritic steels strengthened by intermetallic phases. Mater Sci Eng A 594:372–380. https://doi.org/10.1016/j.msea.2013.11.048
Lopez Barrilao J, Kuhn B, Wessel E (2017) Identification, size classification and evolution of Laves phase precipitates in high chromium, fully ferritic steels. Micron 101:221–231. https://doi.org/10.1016/j.micron.2017.07.010
Lopez Barrilao J, Kuhn B, Wessel E, Talík M (2017) Microstructure of intermetallic particle strengthened high-chromium fully ferritic steels. Mater Sci Technol 33:1056–1064. https://doi.org/10.1080/02670836.2016.1244039
Lopez Barrilao J, Kuhn B, Wessel E (2018) Microstructure evolution and dislocation behaviour in high chromium, fully ferritic steels strengthened by intermetallic Laves phases. Micron 108:11–18. https://doi.org/10.1016/j.micron.2018.02.008
Kuhn B, Talik M, Lopez Barrilao J, Singheiser L, Yamamoto Y (2016) Development status of high performance ferritic (HiperFer) steels. In: Parker J, Shingledecker J, Siefert J (eds) Advances in materials technology for fossil power plants: proceedings from the 8th Int Conf, Albufeira, Portugal. ASM International, Materials Park, pp 1018–1026
Talik M (2016) Influence of initial thermomechanical treatment on high temperature properties of Laves phase strengthened ferritic steels. Schriften des Forschungszentrums Jülich Reihe Energie & Umwelt/Energy & Environment 338:1–163. INIS: http://inis.iaea.org/search/search.aspx?orig_q=RN:48062864
Kuhn B, Yamamoto Y, Talik M, Lopez Barrilao J (2017) High performance ferrite (HiperFer)—a new alloy family. In: Proc 4th Int ECCC Creep & Fracture Conf, Duesseldorf, Germany, pp 1–10. https://www.osti.gov/servlets/purl/1474714
Kuhn B, Lopez Barrilao J, Fischer T (2019) “Reactive” microstructure: The key to cost-effective, fatigue-resistant high-temperature structural materials. In: Shingledecker J, Takeyama M (eds) Proc Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 1–10
Lopez Barrilao J, Kuhn B, Fischer T (2019) Properties and microstructure evolution of advanced high performance ferritic (HiperFer) steels. In: Shingledecker J, Takeyama M (eds) Proc Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 246–252
Möller S, Kuhn B, Rayaprolu R, Heuer S, Rasinski M, Kreter A (2018) HiperFer, a reduced activation ferritic steel tested for nuclear fusion applications. Nucl Mater Energy 17:9–14. https://doi.org/10.1016/j.nme.2018.06.010
Azevedo CRF, Padilha AF (2019) The most frequent failure causes in super ferritic stainless steels: are they really super? Procedia Struct Integr 17:331–338. https://doi.org/10.1016/j.prostr.2019.08.044
Brown EL, Burnett ME, Purtscher PT, Krauss G (1983) Intermetallic phase formation in 25Cr-3Mo-4Ni ferritic stainless steel. Metall Trans A 14:791–800. https://doi.org/10.1007/BF02644282
Ng PG, Clarke E, Khoo CA, Fourlaris G (2006) Microstructural evolution during aging of novel superferritic stainless steel produced by the HIP process. Mater Sci Technol 22:852–858. https://doi.org/10.1179/174328406X91140
de Andrade TF, Kliauga AM, Plaut RL, Padilha AF (2008) Precipitation of Laves phase in a 28%Cr–4%Ni–2%Mo–Nb superferritic stainless steel. Mater Charact 59:503–507. https://doi.org/10.1016/j.matchar.2007.03.006
Ma L, Hu S, Shen J, Han J, Zhu Z (2016) Effects of Cr content on the microstructure and properties of 26Cr–3.5Mo–2Ni and 29Cr–3.5Mo–2Ni super ferritic stainless steels. J Mater Sci Technol 32:552–560. https://doi.org/10.1016/j.jmst.2016.03.022
Lu H-H, Luo Y, Guo H-K, Li W-Q, Li J-C, Liang W (2018) Microstructural evolution and mechanical properties of 27Cr-4Mo-2Ni ferritic stainless steel during isothermal aging. Mater Sci Eng A 735:31–39. https://doi.org/10.1016/j.msea.2018.08.031
Lu H-H, Guo H-K, Luo Y, Liu Z-G, Li W-Q, Li J-C, Liang W (2018) Microstructural evolution, precipitation and mechanical properties of hot rolled 27Cr-4Mo-2Ni ferritic steel during 800 °C aging. Mater Des 160:999–1009. https://doi.org/10.1016/j.matdes.2018.10.039
Lu HH, Lei WW, Luo Y, Li JC, Liu ZG, Liang W (2019) Microstructural evolution, precipitation and mechanical properties of 27Cr-4Mo-2Ni super-ferritic stainless steels. JOM 71:4086–4095. https://doi.org/10.1007/s11837-019-03473-0
Lu H-H, Guo H-K, Du L-Y, Liu Z-G, Li J-C, Liang W (2019) Formation of intermetallics and its effect on microstructure and mechanical properties of 27Cr-4Mo-2Ni super ferritic steels. Mater Charact 151:470–479. https://doi.org/10.1016/j.matchar.2019.03.028
Guo H-K, Lu H-H, Zhang W-G, Meng L-X, Liang W (2019) Cooling effects on microstructure and mechanical properties of 27Cr–4Mo–2Ni ferritic stainless steel. Mater Sci Technol 35:1212–1219. https://doi.org/10.1080/02670836.2019.1618047
Morris DG, Muñoz-Morris MA (2012) Room and high temperature deformation behaviour of a forged Fe–15Al–5Nb alloy with a reinforcing dispersion of equiaxed Laves phase particles. Mater Sci Eng A 552:134–144. https://doi.org/10.1016/j.msea.2012.05.022
Morris DG, Muñoz-Morris MA (2012) Refinement of second phase dispersions in iron aluminide intermetallics by high-temperature severe plastic deformation. Intermetallics 23:169–176. https://doi.org/10.1016/j.intermet.2011.11.020
Yamamoto K, Kimura Y, Wei FG, Mishima Y (2002) Design of Laves phase strengthened ferritic heat resisting steels in the Fe-Cr-Nb(-Ni) system. Mater Sci Eng A 329–331:249–254. https://doi.org/10.1016/S0921-5093(01)01586-6
Yamamoto K, Kimura Y, Mishima Y (2003) Effect of matrix substructures on precipitation of the Laves phase in Fe-Cr-Nb-Ni system. ISIJ Int 43:1253–1259. https://doi.org/10.2355/isijinternational.43.1253
Yamamoto K, Kimura Y, Mishima Y (2006) Effect of matrix microstructure on precipitation of Laves phase in Fe-10Cr-1.4W(-Co) alloys. Intermetallics 14:515–520. https://doi.org/10.1016/j.intermet.2005.09.005
Yamamoto Y, Pint BA, Terrani KA, Field KG, Yang Y, Snead LL (2015) Development and property evaluation of nuclear grade wrought FeCrAl fuel cladding for light water reactors. J Nucl Mater 467:703–716. https://doi.org/10.1016/j.jnucmat.2015.10.019
Sun Z, Edmondson PD, Yamamoto Y (2018) Effects of Laves phase particles on recovery and recrystallization behaviors of Nb-containing FeCrAl alloys. Acta Mater 144:716–727. https://doi.org/10.1016/j.actamat.2017.11.027
Shassere B, Yamamoto Y, Poplawsky J, Guo W, Babu SS (2017) Heterogeneous creep deformations and correlation to microstructures in Fe-30Cr-3Al alloys strengthened by an Fe2Nb Laves phase. Metall Mater Trans A 48:4598–4614. https://doi.org/10.1007/s11661-017-4274-8
Yamamoto Y, Brady MP, Muralidharan G, Pint BA, Maziasz PJ, Shin D, Shassere B, Babu SS, Kuo C-H (2018) Development of creep-resistant, alumina-forming ferrous alloys for high-temperature structural use. ASME 2018 symposium on elevated temperature application of materials for fossil, nuclear, and petrochemical industries, Seattle, WA, US. ASME, Digital Collection: ETAM2018-6727. https://doi.org/10.1115/etam2018-6727
Yamamoto Y, Pint BA, Brady MP, Babu S, Kuo CH, Shassere B (2019) Alloy design and development of high Cr containing FeCrAl ferritic alloys for extreme environments. In: Shingledecker J, Takeyama M (eds) Proc Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 628–639
Chen T, Parish CM, Yang Y, Tan L (2018) High-temperature strengthening mechanisms of Laves and B2 precipitates in a novel ferritic alloy. Mater Sci Eng A 720:110–116. https://doi.org/10.1016/j.msea.2018.02.013
Prakash U, Sauthoff G (2001) Structure and properties of Fe-Al-Ti intermetallic alloys. Intermetallics 9:107–112. https://doi.org/10.1016/S0966-9795(00)00101-1
Löffler F, Palm M, Sauthoff G (2004) Iron-rich iron-titanium-silicon alloys with strengthening intermetallic Laves phase precipitates. Steel Res Int 75:766–772. https://doi.org/10.1002/srin.200405840
Stein F, Palm M, Sauthoff G (2005) Mechanical properties and oxidation behaviour of two-phase iron aluminium alloys with Zr(Fe, Al)2 Laves phase or Zr(Fe, Al)12 τ1 phase. Intermetallics 13:1275–1285. https://doi.org/10.1016/j.intermet.2004.08.013
Tan L, Yang Y (2015) Microstructure and mechanical properties of Laves phase-strengthened Fe-Cr-Zr alloys. Metall Mater Trans A 46:1188–1195. https://doi.org/10.1007/s11661-014-2695-1
Chen T, Yang Y, Tan L (2017) Phase stability in the Fe-rich Fe-Cr-Ni-Zr alloys. Metall Mater Trans A 48:5009–5016. https://doi.org/10.1007/s11661-017-4253-0
Jin X, Chen S, Rong L (2018) Effect of Fe2Zr phase on the mechanical properties and fracture behavior of Fe-Cr-W-Zr ferritic alloy. Mater Sci Eng A 722:173–181. https://doi.org/10.1016/j.msea.2018.03.033
Kobayashi S, Kimura K, Tsuzaki K (2014) Interphase precipitation of Fe2Hf Laves phase in a Fe–9Cr/Fe–9Cr–Hf diffusion couple. Intermetallics 46:80–84. https://doi.org/10.1016/j.intermet.2013.10.017
Kobayashi S (2015) Formation of the Fe2Hf Laves phase through eutectoid type reaction of δ → γ+Fe2Hf in ferritic heat resistant steels. MRS Proc 1760:1708. https://doi.org/10.1557/opl.2015.207
Kobayashi S, Hibaru T (2015) Formation of the Fe2Hf Laves phase along the eutectoid-type reaction path of δ-Fe → γ-Fe + Fe2Hf in an Fe-9Cr based alloy. ISIJ Int 55:293–299. https://doi.org/10.2355/isijinternational.55.293
Yuan ZT, Kobayashi S, Takeyama M (2019) Microstructure control using the formation of Laves phase through interphase precipitation in ferritic heat resistant steels. In: Shingledecker J, Takeyama M (eds) Proc Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 90–95
Davenport AT, Berry FG, Honeycombe RWK (1968) Interphase precipitation in iron alloys. Met Sci J 2:104–106. https://doi.org/10.1179/030634568790443341
Davenport AT, Honeycombe RWK (1971) Precipitation of carbides at γ-α boundaries in alloy steels. Proc R Soc Lond A 322:191–205. https://doi.org/10.1098/rspa.1971.0063
Kestenbach HJ, Bueno LO (1984) Effect of Fe2Nb precipitation on the creep properties of niobium-bearing austenitic stainless steels. Mater Sci Eng 66:L19–L23. https://doi.org/10.1016/0025-5416(84)90157-5
Lee EH, Mansur LK (2000) Fe–15Ni–13Cr austenitic stainless steels for fission and fusion reactor applications. II. Effects of minor elements on precipitate phase stability during thermal aging. J Nucl Mater 278:11–19. https://doi.org/10.1016/S0022-3115(99)00235-4
Padilha AF, Machado IF, Plaut RL (2005) Microstructures and mechanical properties of Fe–15% Cr–15% Ni austenitic stainless steels containing different levels of niobium additions submitted to various processing stages. J Mater Process Technol 170:89–96. https://doi.org/10.1016/j.jmatprotec.2005.05.002
Klotz UE, Solenthaler C, Uggowitzer PJ (2008) Martensitic–austenitic 9–12% Cr steels—alloy design, microstructural stability and mechanical properties. Mater Sci Eng A 476:186–194. https://doi.org/10.1016/j.msea.2007.04.093
Tarasenko LV, Shal’kevich AB (2011) Formation of Laves phase in a refractory austenitic steel due to long-term heating. Met Sci Heat Treat 53:123–126. https://doi.org/10.1007/s11041-011-9353-7
Nikulin I, Kipelova A, Kaibyshev R (2012) Effect of high-temperature exposure on the mechanical properties of 18Cr–8Ni–W–Nb–V–N stainless steel. Mater Sci Eng A 554:61–66. https://doi.org/10.1016/j.msea.2012.06.011
Akita M, Uematsu Y, Kakiuchi T, Nakajima M, Nakamura Y (2014) Effect of Laves phase precipitation on fatigue properties of niobium-containing austenitic stainless steel type 347 in laboratory air and in 3%NaCl solution. Procedia Mater Sci 3:517–523. https://doi.org/10.1016/j.mspro.2014.06.086
Zhou Y, Liu Y, Zhou X, Liu C, Yu J, Huang Y, Li H, Li W (2017) Precipitation and hot deformation behavior of austenitic heat-resistant steels: a review. J Mater Sci Technol 33:1448–1456. https://doi.org/10.1016/j.jmst.2017.01.025
Sourmail T (2001) Precipitation in creep resistant austenitic stainless steels. Mater Sci Technol 17:1–14. https://doi.org/10.1179/026708301101508972
Takeyama M (2007) Novel concept of austenitic heat resistant steels strengthened by intermetallics. Mater Sci Forum 539–543:3012–3017. https://doi.org/10.4028/www.scientific.net/MSF.539-543.3012
Takeyama M, Morita S, Yamauchi A, Yamanaka M, Matsuo T (2001) Phase equilibria among γ, Ni3Nb-δ and Fe2Nb-ε phases in Ni-Nb-Fe and Ni-Nb-Fe-Cr systems at elevated temperatures. In: Loria EA (ed) Superalloys 718, 625, 706 and various derivatives, Pittsburgh, PA, USA. TMS, Warrendale, pp 333–344
Takeyama M, Gomi N, Morita S, Matsuo T (2005) Phase equilibria and lattice parameters of Fe2Nb Laves phase in Fe-Ni-Nb ternary system at elevated temperatures. Mater Res Soc Symp Proc 842:461–466. https://doi.org/10.1557/PROC-842-S5.37
Tarigan I, Kurata K, Takata N, Matsuo T, Takeyama M (2011) Novel concept of creep strengthening mechanism using grain boundary Fe2Nb Laves phase in austenitic heat resistant steel. Mater Res Soc Symp Proc 1295:317–322. https://doi.org/10.1557/opl.2011.558
Tarigan I, Takata N, Takeyama M (2012) Grain boundary precipitation strengthening mechanism by Fe2Nb Laves phase in creep of Fe-20Cr-30Ni-2Nb austenitic heat resistant steel. In: Proceedings of the 12th international conference on creep and fracture of engineering materials and structures, Kyoto, Japan. Japan Institute of Metals, Sendai, Japan, pp 55–58
Yamamoto Y, Takeyama M, Lu ZP, Liu CT, Evans ND, Maziasz PJ, Brady MP (2008) Alloying effects on creep and oxidation resistance of austenitic stainless steel alloys employing intermetallic precipitates. Intermetallics 16:453–462. https://doi.org/10.1016/j.intermet.2007.12.005
Lyta Ueda M, Kawamura K, Takeyama M, Maruyama T (2013) Microstructure development of oxide scale during steam oxidation of the Fe-20Cr-30Ni-2Nb (at%) austenitic steel at 1073 K. Mater Trans 54:2276–2284. https://doi.org/10.2320/matertrans.M2013258
Misosaku Y, Tarigan I, Kimura T, Takata N, Ueda M, Maruyama T, Takeyama M (2014) Effect of grain boundary Fe2Nb Laves phase on creep of austenitic heat resistant steel of Fe-20Cr-30Ni-2Nb in steam atmosphere. Tetsu-to-Hagané 100:1158–1164. https://doi.org/10.2355/tetsutohagane.100.1158
Chen SW, Zhang C, Xia ZX, Ishikawa H, Yang ZG (2014) Precipitation behavior of Fe2Nb Laves phase on grain boundaries in austenitic heat resistant steels. Mater Sci Eng A 616:183–188. https://doi.org/10.1016/j.msea.2014.07.104
Yoshihara M, Takata N, Takeyama M (2015) In-situ tensile observation at 1073 K in Laves phase strengthened Fe-20Cr-30Ni-2Nb steels. In: Murata Y, Ohmori T, Igarashi M, Kimura K, Takeyama M (eds) Proc 123HiMAT-2015 Sapporo, Japan. Japan Society for the Promotion of Science, Japan, pp 153–156
Kanno N, Yoshimura K, Takata N, Tarigan I, Takeyama M (2016) Mechanical properties of austenitic heat-resistant Fe–20Cr–30Ni–2Nb steel at ambient temperature. Mater Sci Eng A 662:551–563. https://doi.org/10.1016/j.msea.2016.03.038
Kikuchi K, Nakashima H, Takeyama M (2019) Experimental and computational study on grain boundary and grain interior precipitation of Fe2Nb Laves phase in Fe-Cr-Ni-Nb austenitic heat resistant steels. In: Shingledecker J, Takeyama M (eds) Proceedings of the Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 1408–1417
Oh S, Nakashima H, Kobayashi S, Takeyama M (2019) Precipitation kinetics of TCP (Fe2Nb) and GCP (Ni3Nb) phases in Fe-Cr-Ni-Nb austenitic heat-resistant steels with partial replacement of Nb with Ta. In: Shingledecker J, Takeyama M (eds) Proceedings of the Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 1418–1428
Yamamoto Y, Brady MP, Lu ZP, Liu CT (2006) Multi-phase high temperature alloys: exploration of Laves-strengthened steels. In: Proceedings of the 20th Ann Conf Fossil Energy Mater, Knoxville, TN, USA. National Energy Technology Laboratory, Pittsburgh, PA, US, pp 20–29. ISBN: 978-1-61567-738-2
Yamamoto Y, Brady MP, Lu ZP, Liu CT, Takeyama M, Maziasz PJ, Pint BA (2007) Alumina-forming austenitic stainless steels strengthened by Laves phase and MC carbide precipitates. Metall Mater Trans A 38:2737–2746. https://doi.org/10.1007/s11661-007-9319-y
Yamamoto Y, Brady MP, Lu ZP, Maziasz PJ, Liu CT, Pint BA, More KL, Meyer HM, Payzant EA (2007) Creep-resistant, Al2O3-forming austenitic stainless steels. Science 316:433–436. https://doi.org/10.1126/science.1137711
Brady MP, Yamamoto Y, Santella ML, Maziasz PJ, Pint BA, Liu CT, Lu ZP, Bei H (2008) The development of alumina-forming austenitic stainless steels for high-temperature structural use. JOM 60:12. https://doi.org/10.1007/s11837-008-0083-2
Brady MP, Yamamoto Y, Lu ZP, Maziasz PJ, Liu CT, Pint BA, Santella ML (2008) Alumina-forming austenitics: a new class of heat-resistant stainless steels. Stainless Steel World Mag March 2008:23–29
Brady MP, Yamamoto Y, Santella ML, Walker LR (2009) Composition, microstructure, and water vapor effects on internal/external oxidation of alumina-forming austenitic stainless steels. Oxid Met 72:311–333. https://doi.org/10.1007/s11085-009-9161-2
Yamamoto Y, Santella ML, Brady MP, Bei H, Maziasz PJ (2009) Effect of alloying additions on phase equilibria and creep resistance of alumina-forming austenitic stainless steels. Metall Mater Trans A 40:1868–1880. https://doi.org/10.1007/s11661-009-9886-1
Bei H, Yamamoto Y, Brady MP, Santella ML (2010) Aging effects on the mechanical properties of alumina-forming austenitic stainless steels. Mater Sci Eng A 527:2079–2086. https://doi.org/10.1016/j.msea.2009.11.052
Yamamoto Y, Brady MP, Santella ML, Bei H, Maziasz PJ, Pint BA (2011) Overview of strategies for high-temperature creep and oxidation resistance of alumina-forming austenitic stainless steels. Metall Mater Trans A 42:922–931. https://doi.org/10.1007/s11661-010-0295-2
Yamamoto Y, Muralidharan G, Brady MP (2013) Development of L12-ordered Ni3(Al, Ti)-strengthened alumina-forming austenitic stainless steel alloys. Scr Mater 69:816–819. https://doi.org/10.1016/j.scriptamat.2013.09.005
Opila EJ, Jacobson NS, Myers DL, Copland EH (2006) Predicting oxide stability in high-temperature water vapor. JOM 58:22–28. https://doi.org/10.1007/s11837-006-0063-3
Opila EJ (2004) Volatility of common protective oxides in high-temperature water vapor: current understanding and unanswered questions. Mater Sci Forum 461–464:765–774. https://doi.org/10.4028/www.scientific.net/MSF.461-464.765
Trotter G, Baker I (2015) The effect of aging on the microstructure and mechanical behavior of the alumina-forming austenitic stainless steel Fe–20Cr–30Ni–2Nb–5Al. Mater Sci Eng A 627:270–276. https://doi.org/10.1016/j.msea.2014.12.072
Trotter G, Baker I (2015) Orientation relationships of Laves phase and NiAl particles in an AFA stainless steel. Philos Mag 95:4078–4094. https://doi.org/10.1080/14786435.2015.1111529
Denham AW, Silcock JM (1969) Precipitation of Fe2Nb in a 16 wt%Ni 16 wt%Cr steel, and the effect of Mn and Si additions. J Iron Steel Inst (London) 207:585–592
Trotter G, Rayner G, Baker I, Munroe PR (2014) Accelerated precipitation in the AFA stainless steel Fe–20Cr–30Ni–2Nb–5Al via cold working. Intermetallics 53:120–128. https://doi.org/10.1016/j.intermet.2014.04.018
Hu B, Trotter G, Baker I, Miller MK, Yao L, Chen S, Cai Z (2015) The effects of cold work on the microstructure and mechanical properties of intermetallic strengthened alumina-forming austenitic stainless steels. Metall Mater Trans A 46:3773–3785. https://doi.org/10.1007/s11661-015-2981-6
Jang M-H, Moon J, Kang J-Y, Ha H-Y, Choi BG, Lee T-H, Lee C (2015) Effect of tungsten addition on high-temperature properties and microstructure of alumina-forming austenitic heat-resistant steels. Mater Sci Eng A 647:163–169. https://doi.org/10.1016/j.msea.2015.09.018
Meng H, Wang J, Wang L, Fang X, Dong N, Zhang C, Han P (2020) The precipitation control in aged alumina-forming austenitic stainless steels Fe-15Cr-25Ni-3Al-NbWCu by W addition and its effect on the mechanical properties. Mater Charact 163:110233. https://doi.org/10.1016/j.matchar.2020.110233
Abraham DP, Dietz N (2002) Role of Laves intermetallics in nuclear waste disposal. Mater Sci Eng A 329–331:610–615. https://doi.org/10.1016/S0921-5093(01)01512-X
Abraham DP, McDeavitt SM, Park J (1996) Microstructure and phase identification in type-304 stainless steel-zirconium alloys. Metall Mater Trans A 27:2151–2159. https://doi.org/10.1007/BF02651870
Abraham DP, Richardson JW Jr, McDeavitt SM (1997) Laves intermetallics in stainless steel-zirconium alloys. Mater Sci Eng A 239–240:658–664. https://doi.org/10.1016/S0921-5093(97)00649-7
McDeavitt SM, Abraham DP, Park JY, Keiser DD Jr (1997) Stainless steel-zirconium waste forms from the treatment of spent nuclear fuel. JOM 49:29–32. https://doi.org/10.1007/BF02914763
McDeavitt SM, Abraham DP, Park JY (1998) Evaluation of stainless steel-zirconium alloys as high-level nuclear waste forms. J Nucl Mater 257:21–34. https://doi.org/10.1016/S0022-3115(98)00433-4
Keiser DD Jr, Abraham DP, Sinkler W, RichardsonJr JW, McDeavitt SM (2000) Actinide distribution in a stainless steel-15 wt% zirconium high-level nuclear waste form. J Nucl Mater 279:234–244. https://doi.org/10.1016/S0022-3115(00)00002-7
Wilson AS (2017) Formation and effect of topologically close-packed phases in nickel-base superalloys. Mater Sci Technol 33:1108–1118. https://doi.org/10.1080/02670836.2016.1187335
Wang L, Song L, Stark A, Liu Y, Oehring M, Lorenz U, Pyczak F (2019) Identification of Laves phases in a Zr or Hf containing γ-γ′ Co-base superalloy. J Alloys Compd 805:880–886. https://doi.org/10.1016/j.jallcom.2019.07.121
Koßmann J, Zenk CH, Lopez-Galilea I, Neumeier S, Kostka A, Huth S, Theisen W, Göken M, Drautz R, Hammerschmidt T (2015) Microsegregation and precipitates of an as-cast Co-based superalloy—microstructural characterization and phase stability modelling. J Mater Sci 50:6329–6338. https://doi.org/10.1007/s10853-015-9177-8
Qi H (2012) Review of INCONEL 718 alloy: its history, properties, processing and developing substitutes. J Mater Eng 2:92–100. https://doi.org/10.3969/j.issn.1001-4381.2013.06.001
Schirra JJ, Caless RH, Hatala RW (1991) The effect of Laves phase on the mechanical properties of wrought and cast + HIP Inconel 718. In: Loria EA (ed) Superalloys 718, 625 and various derivatives, Pittsburgh, PA, USA. TMS, Warrendale, pp 375–388
Vincent R (1985) Precipitation around welds in the nickel-base superalloy, Inconel 718. Acta Metall 33:1205–1216. https://doi.org/10.1016/0001-6160(85)90231-7
Radhakrishna CH, Rao KP (1997) The formation and control of Laves phase in superalloy 718 welds. J Mater Sci 32:1977–1984. https://doi.org/10.1023/a:1018541915113
Ram GDJ, Reddy AV, Rao KP, Reddy GM (2004) Control of Laves phase in Inconel 718 GTA welds with current pulsing. Sci Technol Weld Join 9:390–398. https://doi.org/10.1179/136217104225021788
Reddy GM, Murthy CVS, Rao KS, Rao KP (2009) Improvement of mechanical properties of Inconel 718 electron beam welds—influence of welding techniques and postweld heat treatment. Int J Adv Manuf Technol 43:671–680. https://doi.org/10.1007/s00170-008-1751-7
Manikandan SGK, Sivakumar D, Rao KP, Kamaraj M (2015) Laves phase in alloy 718 fusion zone—microscopic and calorimetric studies. Mater Charact 100:192–206. https://doi.org/10.1016/j.matchar.2014.11.035
Manikandan SGK, Sivakumar D, Kamaraj M (2019) Welding the Inconel 718 superalloy: Reduction of micro-segregation and Laves phases. Elsevier, Amsterdam ISBN 978-0128181834
Ramkumar KD, Abraham WS, Viyash V, Arivazhagan N, Rabel AM (2017) Investigations on the microstructure, tensile strength and high temperature corrosion behaviour of Inconel 625 and Inconel 718 dissimilar joints. J Manuf Process 25:306–322. https://doi.org/10.1016/j.jmapro.2016.12.018
Pang M, Liu CZ, Xu GH, Ma JH (2012) The formation and control of Laves phase in K418 and 42CrMo dissimilar metal laser weld. Appl Mech Mater 166–169:1839–1842. https://doi.org/10.4028/www.scientific.net/AMM.166-169.1839
Li G, Huang J, Wu Y (2015) An investigation on microstructure and properties of dissimilar welded Inconel 625 and SUS 304 using high-power CO2 laser. Int J Adv Manuf Technol 76:1203–1214. https://doi.org/10.1007/s00170-014-6349-7
Hinojos A, Mireles J, Reichardt A, Frigola P, Hosemann P, Murr LE, Wicker RB (2016) Joining of Inconel 718 and 316 stainless steel using electron beam melting additive manufacturing technology. Mater Des 94:17–27. https://doi.org/10.1016/j.matdes.2016.01.041
Ko G, Seo KM, Kim HJ, Hong H (2017) Characteristics of hot cracking in dissimilar joint of A690 overlay and stainless steel clad. Weld World 61:945–953. https://doi.org/10.1007/s40194-017-0477-2
Hejripour F, Aidun DK (2017) Consumable selection for arc welding between Stainless Steel 410 and Inconel 718. J Mater Process Technol 245:287–299. https://doi.org/10.1016/j.jmatprotec.2017.02.013
Dev S, Ramkumar KD, Arivazhagan N, Rajendran R (2018) Investigations on the microstructure and mechanical properties of dissimilar welds of Inconel 718 and sulphur rich martensitic stainless steel, AISI 416. J Manuf Processes 32:685–698. https://doi.org/10.1016/j.jmapro.2018.03.035
Chen Y, Zhang K, Huang J, Hosseini SRE, Li Z (2016) Characterization of heat affected zone liquation cracking in laser additive manufacturing of Inconel 718. Mater Des 90:586–594. https://doi.org/10.1016/j.matdes.2015.10.155
Zhang Y, Yang L, Chen T, Pang S, Zhang W (2017) Sensitivity of liquation cracking to deposition parameters and residual stresses in laser deposited IN718 alloy. J Mater Eng Perform 26:5519–5529. https://doi.org/10.1007/s11665-017-2966-2
Kirka MM, Unocic KA, Raghavan N, Medina F, Dehoff RR, Babu SS (2016) Microstructure development in electron beam-melted Inconel 718 and associated tensile properties. JOM 68:1012–1020. https://doi.org/10.1007/s11837-016-1812-6
Popovich VA, Borisov EV, Popovich AA, Sufiiarov VS, Masaylo DV, Alzina L (2017) Functionally graded Inconel 718 processed by additive manufacturing: crystallographic texture, anisotropy of microstructure and mechanical properties. Mater Des 114:441–449. https://doi.org/10.1016/j.matdes.2016.10.075
Xiao H, Li S, Han X, Mazumder J, Song L (2017) Laves phase control of Inconel 718 alloy using quasi-continuous-wave laser additive manufacturing. Mater Des 122:330–339. https://doi.org/10.1016/j.matdes.2017.03.004
Sui S, Chen JS, Ming X, Zhang S, Lin X, Huang W (2017) The failure mechanism of 50% laser additive manufactured Inconel 718 and the deformation behavior of Laves phases during a tensile process. Int J Adv Manuf Technol 91:2733–2740. https://doi.org/10.1007/s00170-016-9901-9
Sui S, Tan H, Chen JS, Zhong C, Li Z, Fan W, Gasser A, Huang W (2019) The influence of Laves phases on the room temperature tensile properties of Inconel 718 fabricated by powder feeding laser additive manufacturing. Acta Mater 164:413–427. https://doi.org/10.1016/j.actamat.2018.10.032
Wang X, Gong X, Chou K (2017) Review on powder-bed laser additive manufacturing of Inconel 718 parts. Proc Inst Mech Eng Part B J Eng Manuf 231:1890–1903. https://doi.org/10.1177/0954405415619883
Zhang Y, Yang L, Lu W, Wei D, Meng T, Gao S (2020) Microstructure and elevated temperature mechanical properties of IN718 alloy fabricated by laser metal deposition. Mater Sci Eng A 771:138580. https://doi.org/10.1016/j.msea.2019.138580
Xie H, Yang K, Li F, Sun C, Yu Z (2020) Investigation on the Laves phase formation during laser cladding of IN718 alloy by CA-FE. J Manuf Process 52:132–144. https://doi.org/10.1016/j.jmapro.2020.01.050
Antonsson T, Fredriksson H (2005) The effect of cooling rate on the solidification of INCONEL 718. Metall Mater Trans B 36:85–96. https://doi.org/10.1007/s11663-005-0009-0
Formenti A, Eliasson A, Mitchell A, Fredriksson H (2005) Solidification sequence and carbide precipitation in Ni-base superalloys IN718, IN625 and IN939. High Temp Mater Process 24:239–258. https://doi.org/10.1515/HTMP.2005.24.4.239
Nie P, Ojo OA, Li Z (2014) Numerical modeling of microstructure evolution during laser additive manufacturing of a nickel-based superalloy. Acta Mater 77:85–95. https://doi.org/10.1016/j.actamat.2014.05.039
Ling L, Han Y, Zhou W, Gao HY, Shu D, Wang J, Kang M, Sun B (2015) Study of microsegregation and Laves phase in INCONEL718 superalloy regarding cooling rate during solidification. Metall Mater Trans A 46:354–361. https://doi.org/10.1007/s11661-014-2614-5
Keller T, Lindwall G, Ghosh S, Ma L, Lane BM, Zhang F, Kattner UR, Lass EA, Heigel JC, Idell Y, Williams ME, Allen AJ, Guyer JE, Levine LE (2017) Application of finite element, phase-field, and CALPHAD-based methods to additive manufacturing of Ni-based superalloys. Acta Mater 139:244–253. https://doi.org/10.1016/j.actamat.2017.05.003
Sohrabi MJ, Mirzadeh H, Rafiei M (2018) Solidification behavior and Laves phase dissolution during homogenization heat treatment of Inconel 718 superalloy. Vacuum 154:235–243. https://doi.org/10.1016/j.vacuum.2018.05.019
Wang W, Zhu C, Zeng J, Lu C, Qian H, Xu H, Lyu P (2020) Microstructures and Nb-rich precipitation behaviors of Inconel 718 superalloy under sub-rapid solidification process. Metall Mater Trans A 51:2306–2317. https://doi.org/10.1007/s11661-020-05696-2
Qi H, Azer M, Ritter A (2009) Studies of standard heat treatment effects on microstructure and mechanical properties of laser net shape manufactured INCONEL 718. Metall Mater Trans A 40:2410–2422. https://doi.org/10.1007/s11661-009-9949-3
Chlebus E, Gruber K, Kuźnicka B, Kurzac J, Kurzynowski T (2015) Effect of heat treatment on the microstructure and mechanical properties of Inconel 718 processed by selective laser melting. Mater Sci Eng A 639:647–655. https://doi.org/10.1016/j.msea.2015.05.035
Li X, Shi JJ, Wang CH, Cao GH, Russell AM, Zhou ZJ, Li CP, Chen GF (2018) Effect of heat treatment on microstructure evolution of Inconel 718 alloy fabricated by selective laser melting. J Alloys Compd 764:639–649. https://doi.org/10.1016/j.jallcom.2018.06.112
Miao ZJ, Shan AD, Lu J, Song HW (2012) Microstructure and solidification behaviour characterisation of phosphorus and boron doped IN718 superalloy. Mater Sci Technol 28:334–341. https://doi.org/10.1179/1743284711Y.0000000055
Antonov S, Chen W, Lu S, Isheim D, Seidman DN, Feng Q, Sun E, Tin S (2019) The effect of phosphorus on the formation of grain boundary Laves phase in high-refractory content Ni-based superalloys. Scr Mater 161:44–48. https://doi.org/10.1016/j.scriptamat.2018.10.015
Li L, Tin S (2020) Effect of phosphorus content and grain size on the long-term phase stability of Ni-base superalloys. J Alloys Compd 829:154352. https://doi.org/10.1016/j.jallcom.2020.154352
Jin H, Zhang J, Zhang Y, Zhang W, Li P (2020) First-principles investigations of effects of solute elements on stability and electronic structure of Laves phase/matrix interface in Ni-based superalloys. J Phys Chem Solids 136:109166. https://doi.org/10.1016/j.jpcs.2019.109166
Chen Y, Zhang Q, Chen Z, Wang L, Yao J, Kovalenko V (2019) Study on the element segregation and Laves phase formation in the carbon nanotubes reinforced IN718 superalloy by laser cladding. Powder Technol 355:163–171. https://doi.org/10.1016/j.powtec.2019.07.063
Semba H, Okada H, Hamaguchi T, Kurihara S, Tanaka K (2019) Creep strength and microstructure of Ni-base alloys for advanced USC boiler tubes and pipes. In: Shingledecker J, Takeyama M (eds) Proceedings of the Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 513–522
Shioda Y, Nakagawa H, Nomura K, Yamazaki N, Kubushiro K (2019) Creep damage assessment of 47Ni-23Cr-23Fe-7W alloy. In: Shingledecker J, Takeyama M (eds) Proceedings of the Joint EPRI–123HiMAT international conference on advances in high temperature materials, Nagasaki, Japan. ASM International, Materials Park, pp 592–602
Chen KC, Allen SM, Livingston JD (1996) Assessment of the compositional influences on the toughness of TiCr2-base Laves-phase alloys. Mater Res Soc Symp Proc 460:695–700. https://doi.org/10.1557/PROC-460-695
Chen KC, Allen SM, Livingston JD (1997) Microstructures of two-phase Ti-Cr alloys containing the TiCr2 Laves phase intermetallic. J Mater Res 12:1472–1480. https://doi.org/10.1557/JMR.1997.0203
Takasugi T, Kumar KS, Liu CT, Lee EH (1999) Microstructure and mechanical properties of two-phase Cr-Cr2Nb, Cr-Cr2Zr and Cr-Cr2(Nb, Zr) alloys. Mater Sci Eng A 260:108–123. https://doi.org/10.1016/S0921-5093(98)00981-2
Kumar KS, Pang L, Horton JA, Liu CT (2003) Structure and composition of Laves phases in binary Cr-Nb, Cr-Zr and ternary Cr-(Nb, Zr) alloys. Intermetallics 11:677–685. https://doi.org/10.1016/S0966-9795(03)00065-7
Bewlay BP, Sutliff JA, Jackson MR, Lipsitt HA (1994) Microstructural and crystallographic relationships in directionally solidified Nb-Cr2Nb and Cr-Cr2Nb eutectics. Acta Metall Mater 42:2869–2878. https://doi.org/10.1016/0956-7151(94)90228-3
Bewlay BP, Lipsitt HA, Jackson MR, Reeder WJ, Sutliff JA (1995) Solidification processing of high temperature intermetallic eutectic-based alloys. Mater Sci Eng A 192:534–543. https://doi.org/10.1016/0921-5093(95)03299-1
Kumar KS, Liu CT (1997) Precipitation in a Cr-Cr2Nb alloy. Acta Mater 45:3671–3686. https://doi.org/10.1016/S1359-6454(97)00050-5
Aoyama N, Hanada S (1997) Microstructure and strength of NbCr2/Cr in situ composites. Mater Trans JIM 38:155–162. https://doi.org/10.2320/matertrans1989.38.155
Lee S, Liaw PK, Liu CT, Chou YT (1999) Cracking in Cr-Cr2Nb eutectic alloys due to thermal stresses. Mater Sci Eng A 268:184–192. https://doi.org/10.1016/S0921-5093(99)00068-4
Li KW, Wang XB, Li SM, Zhong H, Xue YL, Fu HZ (2016) Microstructure and fracture toughness of Cr/Cr2Nb alloys with trace Y addition. Mater Sci Technol 32:195–199. https://doi.org/10.1179/1743284715Y.0000000085
Kumar KS, Miracle DB (1994) Microstructural evolution and mechanical properties of a Cr-Cr2Hf alloy. Intermetallics 2:257–274. https://doi.org/10.1016/0966-9795(94)90012-4
Ravichandran KS, Miracle DB, Mendiratta MG (1994) Fracture toughness of two Cr2Hf + Cr intermetallic composites as a function temperature. Mater Res Soc Symp Proc 350:249–254. https://doi.org/10.1557/PROC-350-249
Ravichandran KS (1996) Creep of Cr2Hf + Cr in situ intermetallic composites. Scr Mater 34:1819–1824. https://doi.org/10.1016/1359-6462(96)00076-0
Brady MP, Zhu JH, Liu CT, Tortorelli PF, Walker LR (2000) Oxidation resistance and mechanical properties of Laves phase reinforced Cr in situ composites. Intermetallics 8:1111–1118. https://doi.org/10.1016/S0966-9795(00)00046-7
Brady MP, Zhu JH, Liu CT, Tortorelli PF, Walker LR, McKamey CG, Wright JL, Carmichael CA, Larson DJ, Miller MK, Porter WD (1999) Intermetallic reinforced Cr alloys for high-temperature use. Mater High Temp 16:189–193. https://doi.org/10.1179/mht.1999.018
Kumar KS, Pang L, Liu CT, Horton J, Kenik EA (2000) Structural stability of the Laves phase Cr2Ta in a two-phase Cr-Cr2Ta alloy. Acta Mater 48:911–923. https://doi.org/10.1016/S1359-6454(99)00377-8
Bhowmik A, Knowles KM, Stone HJ (2012) Microstructural evolution and interfacial crystallography in Cr–Cr2Ta. Intermetallics 31:34–47. https://doi.org/10.1016/j.intermet.2012.06.001
Xue Y, Li S, Wu Y, Liu C, Liu H, Yuan L (2020) Strengthening and toughening effects in Laves phase Cr2Ta/Cr in situ composites by Si additions. Vacuum 174:109202. https://doi.org/10.1016/j.vacuum.2020.109202
Liu CT, Tortorelli PF, Horton JA, Carmichael CA (1996) Effects of alloy additions on the microstructure and properties of Cr-Cr2Nb alloys. Mater Sci Eng A 214:23–32. https://doi.org/10.1016/0921-5093(96)10197-0
Bhowmik A, Pang HT, Neumeier S, Stone HJ, Edmonds I (2011) Microstructure and oxidation resistance of Cr-Ta-Si alloys. Mater Res Soc Symp Proc 1295:323–328. https://doi.org/10.1557/opl.2011.38
Bhowmik A, Jones CN, Edmonds IM, Stone HJ (2012) Effect of Mo, Al and Si on the microstructure and mechanical properties of Cr–Cr2Ta based alloys. J Alloys Compd 530:169–177. https://doi.org/10.1016/j.jallcom.2012.03.070
Bhowmik A, Neumeier S, Barnard JS, Zenk CH, Göken M, Rae CMF, Stone HJ (2014) Microstructure and mechanical properties of Cr–Ta–Si Laves phase-based alloys at elevated temperatures. Philos Mag 94:3914–3944. https://doi.org/10.1080/14786435.2014.971086
Bhowmik A, Bennett RJ, Monserrat B, Conduit GJ, Connor LD, Parker JE, Thompson RP, Jones CN, Stone HJ (2014) Alloys based on Cr–Cr2Ta containing Si. Intermetallics 48:62–70. https://doi.org/10.1016/j.intermet.2013.11.002
Brady MP, Liu CT, Zhu JH, Tortorelli PF, Walker LR (2005) Effects of Fe additions on the mechanical properties and oxidation behavior of Cr2Ta Laves phase reinforced Cr. Scr Mater 52:815–819. https://doi.org/10.1016/j.scriptamat.2005.01.016
Tien RH, Zhu JH, Liu CT, Walker LR (2005) Effect of Ru additions on microstructure and mechanical properties of Cr-TaCr2 alloys. Intermetallics 13:361–366. https://doi.org/10.1016/j.intermet.2004.07.017
Peng L, Ikeda K-I, Horiuchi T, Miura S (2019) Microstructural evolution and mechanical properties of a three-phase alloy in the Cr–Mo–Nb system. Mater Trans 60:246–253. https://doi.org/10.2320/matertrans.M2018269
Gorsse S, Ouvrard B, Gouné M, Poulon-Quintin A (2015) Microstructural design of new high conductivity—high strength Cu-based alloy. J Alloys Compd 633:42–47. https://doi.org/10.1016/j.jallcom.2015.01.234
Fehrenbach PJ, Kerr HW, Niessen P (1973) Unidirectional solidification of monovariant Cu-Ni-Mg eutectic alloys II. Microstructures and properties. J Crystal Growth 18:151–158. https://doi.org/10.1016/0022-0248(73)90193-0
Ellis DL, Michal GM (1989) Formation of Cr and Cr2Nb precipitates in rapidly solidified Cu-Cr-Nb ribbon. Ultramicroscopy 30:210–216. https://doi.org/10.1016/0304-3991(89)90189-7
Ellis DL, Michal GM, Orth NW (1990) Production and processing of Cu-Cr-Nb alloys. Scr Metall Mater 24:885–890. https://doi.org/10.1016/0956-716X(90)90130-9
Anderson KR, Groza JR, Dreshfield RL, Ellis D (1993) Microstructural evolution and thermal stability of precipitation-strengthened Cu-8Cr-4Nb alloy. Mater Sci Eng A 169:167–175. https://doi.org/10.1016/0921-5093(93)90611-H
Groza JR (1993) Microstructural features of a new precipitation-strengthened Cu-8Cr-4Nb alloy. Mater Charact 31:133–141. https://doi.org/10.1016/1044-5803(93)90054-Y
Anderson KR, Groza JR, Dreshfield RL, Ellis D (1995) High-performance dispersion-strengthened Cu-8 Cr-4 Nb alloy. Metall Mater Trans A 26:2197–2206. https://doi.org/10.1007/BF02671235
Anderson KR (1996) High temperature coarsening of Cr2Nb precipitates in Cu-8 Cr-4 Nb alloy. NASA-CR-198492:1–68
Anderson KR, Groza JR (2001) Microstructural size effects in high-strength high-conductivity Cu-Cr-Nb alloys. Metall Mater Trans A 32:1211–1224. https://doi.org/10.1007/s11661-001-0130-x
Ellis DL (2005) GRCop-84: a high-temperature copper alloy for high-heat-flux applications. NASA/TM-2005-213566
de Groh HC, Ellis DL, Loewenthal WS (2008) Comparison of GRCop-84 to other Cu alloys with high thermal conductivities. J Mater Eng Perform 17:594–606. https://doi.org/10.1007/s11665-007-9175-3
Vettraino LG, Heelan JL, Faconti CA, Walley JL, Garg A, Groza JR, Gibeling JC (2008) Influence of processing on the microstructure of Cu–8Cr–4Nb. J Mater Sci 43:6546–6555. https://doi.org/10.1007/s10853-008-2802-z
Shukla AK, Suresh Kumar R, Narayana Murty SVS, Mondal K (2013) Enhancement of high temperature ductility of hot-pressed Cu–Cr–Nb alloy by hot rolling. Mater Sci Eng A 577:36–42. https://doi.org/10.1016/j.msea.2013.04.043
Gradl PR, Greene SE, Protz CS, Ellis DL, Lerch BA, Locci IE (2017) Development and hot-fire testing of additively manufactured copper combustion chambers for liquid rocket engine applications. In: 53rd AIAA/SAE/ASEE joint propulsion conference, Atlanta, GA, US. American Institute of Aeronautics and Astronautics, AIAA-2017-4670. https://doi.org/10.2514/6.2017-4670
Gradl PR, Protz CS, Ellis DL, Greene SE (2019) Progress in additively manufactured copper-alloy GRCop-84, GRCop-42, and bimetallic combustion chambers for liquid rocket engines. In: 70th international astronautical congress (IAC), Washington DC, US. IAC-19.C14.13.15x52514
Guo X, Xiao Z, Qiu W, Li Z, Zhao Z, Wang X, Jiang Y (2019) Microstructure and properties of Cu-Cr-Nb alloy with high strength, high electrical conductivity and good softening resistance performance at elevated temperature. Mater Sci Eng A 749:281–290. https://doi.org/10.1016/j.msea.2019.02.036
Yang Y, Wang L, Snead LL, Zinkle SJ (2018) Development of novel Cu-Cr-Nb-Zr alloys with the aid of computational thermodynamics. Mater Des 156:370–380. https://doi.org/10.1016/j.matdes.2018.07.003
Wang L, Zheng C, Kombaiah B, Tan L, Sprouster DJ, Snead LL, Zinkle SJ, Yang Y (2020) Contrasting roles of Laves Cr2Nb precipitates on the creep properties of novel CuCrNbZr alloys. Mater Sci Eng A 779:139110. https://doi.org/10.1016/j.msea.2020.139110
Nie J-F (2012) Precipitation and hardening in magnesium alloys. Metall Mater Trans A 43:3891–3939. https://doi.org/10.1007/s11661-012-1217-2
Kawano S, Iikubo S, Ohtani H (2018) Thermodynamic stability of Mg-based Laves phases. Mater Trans 59:890–896. https://doi.org/10.2320/matertrans.M2018079
Suzuki A, Saddock ND, Jones JW, Pollock TM (2005) Solidification paths and eutectic intermetallic phases in Mg–Al–Ca ternary alloys. Acta Mater 53:2823–2834. https://doi.org/10.1016/j.actamat.2005.03.001
Suzuki A, Saddock ND, Jones JW, Pollock TM (2006) Phase equilibria in the Mg-Al-Ca ternary system at 773 and 673 K. Metall Mater Trans A 37:975–983. https://doi.org/10.1007/s11661-006-0070-6
Zhong Y, Luo A, Nie JF, Sofo JO, Liu Z-K (2016) New phases in Mg-Al-Ca system. In: Mathaudhu SN, Luo AA, Neelameggham NR, Nyberg EA, Sillekens WH (eds) Essential readings in magnesium technology. Springer, Cham, pp 427–432. https://doi.org/10.1007/978-3-319-48099-2_68
Suzuki A, Saddock ND, TerBush JR, Powell BR, Jones JW, Pollock TM (2008) Precipitation strengthening of a Mg-Al-Ca–based AXJ530 die-cast alloy. Metall Mater Trans A 39:696–702. https://doi.org/10.1007/s11661-007-9455-4
Zhang L, Deng K-K, Nie K-B, Xu F-J, Su K, Liang W (2015) Microstructures and mechanical properties of Mg–Al–Ca alloys affected by Ca/Al ratio. Mater Sci Eng A 636:279–288. https://doi.org/10.1016/j.msea.2015.03.100
Khorasani F, Pourbahari B, Emamy M, Malekan M, Salehian S (2018) Effects of Ca/Al ratio and extrusion process on Mg–Al–Ca alloys to produce a high toughness in situ composite. Philos Mag 98:2826–2844. https://doi.org/10.1080/14786435.2018.1508898
Khorasani F, Emamy M, Malekan M, Mirzadeh H, Pourbahari B, Krajnák T, Minárik P (2019) Enhancement of the microstructure and elevated temperature mechanical properties of as-cast Mg-Al2Ca-Mg2Ca in situ composite by hot extrusion. Mater Charact 147:155–164. https://doi.org/10.1016/j.matchar.2018.10.018
Zubair M, Sandlöbes S, Wollenweber MA, Kusche CF, Hildebrandt W, Broeckmann C, Korte-Kerzel S (2019) On the role of Laves phases on the mechanical properties of Mg-Al-Ca alloys. Mater Sci Eng A 756:272–283. https://doi.org/10.1016/j.msea.2019.04.048
Liu Y, Yuan G, Ding W, Lu C (2007) Deformation behavior of Mg–Zn–Gd-based alloys reinforced with quasicrystal and Laves phases at elevated temperatures. J Alloys Compd 427:160–165. https://doi.org/10.1016/j.jallcom.2006.03.027
Yuan G, Liu Y, Lu C, Ding W (2008) Effect of quasicrystal and Laves phases on strength and ductility of as-extruded and heat treated Mg-Zn-Gd-based alloys. Mater Sci Eng A 472:75–82. https://doi.org/10.1016/j.msea.2007.03.016
Wang F, Hu T, Zhang Y, Xiao WJ, Ma CA (2017) Effects of Al and Zn contents on the microstructure and mechanical properties of Mg-Al-Zn-Ca magnesium alloys. Mater Sci Eng A 704:57–65. https://doi.org/10.1016/j.msea.2017.07.060
Wolff M, Schaper JG, Suckert MR, Dahms M, Ebel T, Willumeit-Römer R, Klassen T (2016) Magnesium powder injection molding (MIM) of orthopedic implants for biomedical applications. JOM 68:1191–1197. https://doi.org/10.1007/s11837-016-1837-x
Stein F, He C, Wossack I (2014) The liquidus surface of the Cr–Al–Nb system and re-investigation of the Cr–Nb and Al–Cr phase diagrams. J Alloys Compd 598:253–265. https://doi.org/10.1016/j.jallcom.2014.02.045
Thoma DJ (1992) Microstructural development in NbCr2-based alloys. PhD, The University of Wisconsin-Madison, Wisconsin, US
Chen KC, Kotula PG, Cady CM, Mauro ME, Thoma DJ (1998) Microstructures and mechanical properties of two-phase alloys based on NbCr2. Mater Res Soc Symp Proc 552:KK7.4.1–KK7.4.7. https://doi.org/10.1557/PROC-552-KK7.4.1
Davidson DL, Chan KS (1998) The effect of microstructure on the fracture resistance of Nb-Cr-Ti in situ composites. Scr Mater 38:1155–1161. https://doi.org/10.1016/S1359-6462(97)00562-9
Davidson DL, Chan KS (2002) Microstructural and fracture characterization of Nb-Cr-Ti mechanically alloyed materials. Metall Trans A 33:401–416. https://doi.org/10.1007/s11661-002-0101-x
Bewlay BP, Jackson MR (1996) The effect of Hf and Ti additions on microstructure and properties of Cr2Nb–Nb in situ composites. J Mater Res 11:1917–1922. https://doi.org/10.1557/JMR.1996.0242
Chan KS (2005) Linking electronic and dislocation parameters to fracture resistance in Nb-Ti-Cr alloys and Laves phases. Philos Mag 85:239–259. https://doi.org/10.1080/14786430412331315725
Gang F, Kauffmann A, Heilmaier M (2018) Phase evolution in and creep properties of Nb-rich Nb-Si-Cr eutectics. Metall Mater Trans A 49:763–771. https://doi.org/10.1007/s11661-017-4367-4
Bewlay BP, Jackson MR, Zhao J-C, Subramanian PR (2003) A review of very-high-temperature Nb-silicide-based composites. Metall Mater Trans A 34:2043–2052. https://doi.org/10.1007/s11661-003-0269-8
Chan KS, Davidson DL (2003) Improving the fracture toughness of constituent phases and Nb-based in situ composites by a computational alloy design approach. Metall Mater Trans A 34:1833–1849. https://doi.org/10.1007/s11661-003-0149-2
Tsakiropoulos P (2018) On Nb silicide based alloys: alloy design and selection. Materials 11:844. https://doi.org/10.3390/ma11050844
Wang H-B, Wang S-S, Gao P-Y, Jiang T, Lu X-G, Li C-H (2016) Microstructure and mechanical properties of a novel near-α titanium alloy Ti6.0Al4.5Cr1.5Mn. Mater Sci Eng A 672:170–174. https://doi.org/10.1016/j.msea.2016.06.083
Rabadia CD, Liu YJ, Wang L, Sun H, Zhang LC (2018) Laves phase precipitation in Ti-Zr-Fe-Cr alloys with high strength and large plasticity. Mater Des 154:228–238. https://doi.org/10.1016/j.matdes.2018.05.035
Rabadia CD, Liu YJ, Jawed SF, Wang LQ, Sun H, Zhang LC (2020) Deformation and toughness behavior of β -type titanium alloys comprising C15-type Laves phase. Mater Today Sustain 9:100034. https://doi.org/10.1016/j.mtsust.2020.100034
Rabadia CD, Liu YJ, Chen LY, Jawed SF, Wang LQ, Sun H, Zhang LC (2019) Deformation and strength characteristics of Laves phases in titanium alloys. Mater Des 179:107891. https://doi.org/10.1016/j.matdes.2019.107891
Rabadia CD, Liu YJ, Jawed SF, Wang L, Li YH, Zhang XH, Sercombe TB, Sun H, Zhang LC (2018) Improved deformation behavior in Ti-Zr-Fe-Mn alloys comprising the C14 type Laves and β phases. Mater Des 160:1059–1070. https://doi.org/10.1016/j.matdes.2018.10.049
Rabadia CD, Liu YJ, Zhao CH, Wang JC, Jawed SF, Wang LQ, Chen LY, Sun H, Zhang LC (2019) Improved trade-off between strength and plasticity in titanium based metastable beta type Ti-Zr-Fe-Sn alloys. Mater Sci Eng A 766:138340. https://doi.org/10.1016/j.msea.2019.138340
Thomas J, Mogonye JE, Mantri SA, Choudhuri D, Banerjee R, Scharf TW (2020) Additive manufacturing of compositionally graded laser deposited titanium-chromium alloys. Addit Manuf 33:101132. https://doi.org/10.1016/j.addma.2020.101132
Whitmarsh CL (1962) Review of Zircaloy-2 and Zircaloy-4 properties relevant to N.S. Savannah reactor design. Oak Ridge National Laboratory Report No ORNL-3281. https://doi.org/10.2172/4827123
Sabol GP (2005) ZIRLO™—an alloy development success. J ASTM Int 2:JAI12942. https://doi.org/10.1520/JAI12942
Meng XY, Northwood DO (1985) Intermetallic precipitates in zircaloy-4. J Nucl Mater 132:80–87. https://doi.org/10.1016/0022-3115(85)90398-8
Toffolon-Masclet C, Brachet JC, Jago G (2002) Studies of second phase particles in different zirconium alloys using extractive carbon replica and an electrolytic anodic dissolution procedure. J Nucl Mater 305:224–231. https://doi.org/10.1016/S0022-3115(02)00911-X
Toffolon-Masclet C, Brachet JC, Servant C, Joubert JM, Barberis P, Dupin N, Zeller P (2008) Contribution of thermodynamic calculations to metallurgical studies of multi-component zirconium based alloys. J ASTM Int 5:JAI101122. https://doi.org/10.1520/STP48166S
Shishov VN, Peregud MM, Nikulina AV, Konkov VF, Novikov VV, Markelov VA, Khokhunova TN, Kobylyansky GP, Novoselov AE, Ostrovsky ZE, Obukhov AV (2009) Structure-phase state, corrosion and irradiation properties of Zr-Nb-Fe-Sn system alloys. J ASTM Int 5:JAI101127. https://doi.org/10.1520/JAI101127
de Gabory B, Motta AT, Wang K (2015) Transmission electron microscopy characterization of Zircaloy-4 and ZIRLO™ oxide layers. J Nucl Mater 456:272–280. https://doi.org/10.1016/j.jnucmat.2014.09.073
Cao G, Yun Y, Yang L, Yuan G, Yue Q, Shao G, Hu J (2017) The formation and stacking faults of Fe and Cr containing Laves phase in Zircaloy-4 alloy. Mater Lett 191:203–205. https://doi.org/10.1016/j.matlet.2016.12.062
Liu C, Li G, Yuan F, Han F, Zhang Y, Gu H (2018) Stacking faults in Zr(Fe, Cr)2 Laves structured secondary phase particle in Zircaloy-4 alloy. Nanoscale 10:2249–2254. https://doi.org/10.1039/C7NR08963E
Lefebvre F, Lemaignan C (1989) Heavy ion-induced amorphisation of Zr(Fe, Cr)2 precipitates in Zircaloy-4. J Nucl Mater 165:122–127. https://doi.org/10.1016/0022-3115(89)90240-7
Motta AT, Lefebvre F, Lemaignan C (1991) Amorphization of precipitates in Zircaloy under neutron and charged-particle irradiation. In: Eucken C, Garde A (eds) Zirconium in the nuclear industry: ninth international symposium. ASTM International, West Conshohocken, pp 718–739. https://doi.org/10.1520/STP25536S
Taylor DF, Peters HR, Yang WJS (1999) Amorphization of Laves-phase precipitates in Zircaloy-4 by neutron irradiation. In: Bruemmer S, Ford P, Was G (eds) Proceedings of the ninth international symposium on environmental degradation of materials in nuclear power systems - water reactors, Newport Beach, CA, US. TMS, Warrendale, pp 1153–1160. https://doi.org/10.1002/9781118787618.ch121
Taylor DF, Peters HR, Yang WJS (1999) A simple kinetic model of Zircaloy Zr(Fe, Cr)2 precipitate amorphization during neutron irradiation. In: Bruemmer S, Ford P, Was G (eds) Proceedings of the ninth international symposium on environmental degradation of materials in nuclear power systems - water reactors, Newport Beach, CA, US. TMS, Warrendale, pp 1161–1168. https://doi.org/10.1002/9781118787618.ch122
Zu XT, Sun K, Atzmon M, Wang LM, You LP, Wan FR, Busby JT, Was GS, Adamson RB (2005) Effect of proton and Ne irradiation on the microstructure of Zircaloy 4. Philos Mag 85:649–659. https://doi.org/10.1080/14786430412331320017
Shishov V (2010) The evolution of microstructure and deformation stability in Zr-Nb-(Sn, Fe) alloys under neutron irradiation. J ASTM Int 7:JAI103005. https://doi.org/10.1520/JAI103005
Shen HH, Peng SM, Xiang X, Naab FN, Sun K, Zu XT (2014) Proton irradiation effects on the precipitate in a Zr–1.6Sn–0.6Nb–0.2Fe–0.1Cr alloy. J Nucl Mater 452:335–342. https://doi.org/10.1016/j.jnucmat.2014.05.042
Shen HH, Zhang JM, Peng SM, Xiang X, Sun K, Zu XT (2014) In situ TEM investigation of amorphization and recrystallization of Zr(Fe, Cr, Nb)2 precipitates under Ne ion irradiation. Vacuum 110:24–29. https://doi.org/10.1016/j.vacuum.2014.08.002
Yan C, Wang R, Wang Y, Wang X, Bai G (2015) Effects of ion irradiation on microstructure and properties of zirconium alloys—a review. Nucl Eng Technol 47:323–331. https://doi.org/10.1016/j.net.2014.12.015
Abraham DP, Richardson JW, McDeavitt SM (2001) Microscopy and neutron diffraction study of a zirconium-8 wt% stainless steel alloy. J Mater Sci 36:5143–5154. https://doi.org/10.1023/a:1012433526079
McKamey CG (1996) Iron aluminides. In: Stoloff NS, Sikka VK (eds) Physical metallurgy and processing of intermetallic compounds. Springer, Boston, pp 351–391. https://doi.org/10.1007/978-1-4613-1215-4_9
Morris DG, Muñoz-Morris MA (2011) Recent developments toward the application of iron aluminides in fossil fuel technologies. Adv Eng Mater 13:43–47. https://doi.org/10.1002/adem.201000210
Zamanzade M, Barnoush A, Motz C (2016) A review on the properties of iron aluminide intermetallics. Crystals 6:10. https://doi.org/10.3390/cryst6010010
Palm M, Stein F, Dehm G (2019) Iron aluminides. Ann Rev Mater Res 49:297–326. https://doi.org/10.1146/annurev-matsci-070218-125911
Schneider A, Sauthoff G (2004) Iron aluminium alloys with strengthening carbides and intermetallic phases for high-temperature applications. Steel Res Int 75:55–61. https://doi.org/10.1002/srin.200405927
Palm M (2009) Fe-Al materials for structural applications at high temperatures: current research at MPIE. Int J Mater Res 100:277–287. https://doi.org/10.3139/146.110056
Dimiduk DM, Mendiratta MG, Banerjee D, Lipsitt HA (1988) A structural study of ordered precipitates in an ordered matrix within the Fe-Al-Nb system. Acta Metall 36:2947–2958. https://doi.org/10.1016/0001-6160(88)90177-0
Morris DG, Munoz-Morris MA, Requejo LM, Baudin C (2006) Strengthening at high temperatures by precipitates in Fe-Al-Nb alloys. Intermetallics 14:1204–1207. https://doi.org/10.1016/j.intermet.2005.10.015
Morris DG, Munoz-Morris MA (2007) Development of creep-resistant iron aluminides. Mater Sci Eng A 462:45–52. https://doi.org/10.1016/j.msea.2005.10.083
Matsumoto N, Kaneno Y, Takasugi T (2007) Strengthening and ductilization of D03-type Fe3Al intermetallic alloys by dispersion of Laves phases Fe2Zr and Fe2Nb. Mater Sci Forum 561–565:395–398. https://doi.org/10.4028/www.scientific.net/MSF.561-565.395
Milenkovic S, Palm M (2008) Microstructure and mechanical properties of directionally solidified Fe-Al-Nb eutectic. Intermetallics 16:1212–1218. https://doi.org/10.1016/j.intermet.2008.07.007
Yildirim M, Akdeniz MV, Mekhrabov AO (2016) Microstructural evolution and room-temperature mechanical properties of as-cast and heat-treated Fe50Al50−nNbn alloys (n = 1, 3, 5, 7, and 9 at%). Mater Sci Eng A 664:17–25. https://doi.org/10.1016/j.msea.2016.03.128
Kratochvíl P, Švec M, Král R, Veselý J, Lukáč P, Vlasák T (2018) The effect of Nb addition on the microstructure and the high-temperature strength of Fe3Al aluminide. Metall Mater Trans A 49:1598–1603. https://doi.org/10.1007/s11661-018-4524-4
Janschek P, Bauer-Partenheim K, Krein R, Hanus P, Palm M (2009) Forging of steam turbine blades with an Fe3Al-based alloy. Mater Res Soc Symp Proc 1128:47–52. https://doi.org/10.1557/PROC-1128-U02-02
Risanti DD, Sauthoff G (2011) Microstructures and mechanical properties of Fe–Al–Ta alloys with strengthening Laves phase. Intermetallics 19:1727–1736. https://doi.org/10.1016/j.intermet.2011.07.008
Prokopčáková P, Švec M, Palm M (2016) Microstructural evolution and creep of Fe–Al–Ta alloys. Int J Mater Res 107:396–405. https://doi.org/10.3139/146.111357
Emdadi A, Sizova I, Bambach M, Hecht U (2019) Hot deformation behavior of a spark plasma sintered Fe-25Al-1.5Ta alloy with strengthening Laves phase. Intermetallics 109:123–134. https://doi.org/10.1016/j.intermet.2019.03.017
Palm M, Sauthoff G (2004) Deformation behaviour and oxidation resistance of single-phase and two-phase L21-ordered Fe-Al-Ti alloys. Intermetallics 12:1345–1359. https://doi.org/10.1016/j.intermet.2004.03.017
Wasilkowska A, Bartsch M, Stein F, Palm M, Sztwiertnia K, Sauthoff G, Messerschmidt U (2004) Plastic deformation of Fe-Al polycrystals strengthened with Zr-containing Laves phases - I. Microstructure of undeformed materials. Mater Sci Eng A 380:9–19. https://doi.org/10.1016/j.msea.2004.04.005
Wasilkowska A, Bartsch M, Stein F, Palm M, Sauthoff G, Messerschmidt U (2004) Plastic deformation of Fe-Al polycrystals strengthened with Zr-containing Laves phases–II. Mechanical properties. Mater Sci Eng A 381:1–15. https://doi.org/10.1016/j.msea.2004.04.039
Morris DG, Gutierrez-Urrutia I, Munoz-Morris MA (2007) The high-temperature creep behaviour of an Fe-Al-Zr alloy strengthened by intermetallic precipitates. Scr Mater 57:449–452. https://doi.org/10.1016/j.scriptamat.2007.05.038
Kratochvíl P, Kejzlar P, Král R, Vodicková V (2012) The effect of Zr addition on the structure and high temperature strength of Fe-30 at.% Al type alloys. Intermetallics 20:39–46. https://doi.org/10.1016/j.intermet.2011.08.007
Azmi SA, Michalcová A, Senčekova L, Palm M (2017) Microstructure and mechanical properties of Fe–Al–Nb–B alloys. MRS Adv 2:1353–1359. https://doi.org/10.1557/adv.2017.138
Falat L, Schneider A, Sauthoff G, Frommeyer G (2005) Mechanical properties of Fe-Al-M-C (M = Ti, V, Nb, Ta) alloys with strengthening carbides and Laves phase. Intermetallics 13:1256–1262. https://doi.org/10.1016/j.intermet.2004.05.010
Kratochvíl P, Málek P, Cieslar M, Hanus P, Hakl J, Vlasák T (2007) High-temperature mechanical properties of Zr alloyed Fe3Al-type iron aluminide. Intermetallics 15:333–337. https://doi.org/10.1016/j.intermet.2006.07.005
Kratochvíl P, Vodičková V, Král R, Švec M (2016) The effect of Laves phase (Fe, Al)2Zr on the high-temperature strength of carbon-alloyed Fe3Al aluminide. Metall Mater Trans A 47:1128–1131. https://doi.org/10.1007/s11661-015-3309-2
Darolia R (2000) Ductility and fracture toughness issues related to implementation of NiAl for gas turbine applications. Intermetallics 8:1321–1327. https://doi.org/10.1016/S0966-9795(00)00081-9
Sauthoff G (1995) Intermetallics. Wiley-VCH, Weinheim
Sherman M, Vedula K (1986) High temperature dispersion strengthening of NiAl. J Mater Sci 21:1974–1980. https://doi.org/10.1007/BF00547936
Whittenberger JD, Reviere R, Noebe RD, Oliver BF (1992) Compressive strength of directionally solidified NiAl-NiAlNb intermetallics at 1200 and 1300 K. Scr Metall Mater 26:987–992. https://doi.org/10.1016/0956-716X(92)90695-B
Sauthoff G (2000) Multiphase intermetallic alloys for structural applications. Intermetallics 8:1101–1109. https://doi.org/10.1016/S0966-9795(00)00045-5
Johnson DR, Oliver BF, Noebe RD, Whittenberger JD (1995) NiAl-based polyphase in situ composites in the NiAl-Ta-X (X = Cr, Mo, or V) systems. Intermetallics 3:493–503. https://doi.org/10.1016/0966-9795(95)00008-M
Zeumer B, Sauthoff G (1997) Intermetallic NiAl-Ta alloys with strengthening Laves phase for high-temperature applications. I. Basic properties. Intermetallics 5:563–577. https://doi.org/10.1016/S0966-9795(97)00031-9
Zeumer B, Sauthoff G (1997) Deformation behaviour of intermetallic NiAl-Ta alloys with strengthening Laves phase for high-temperature applications. II. Effects of alloying with Nb and other elements. Intermetallics 5:641–649. https://doi.org/10.1016/S0966-9795(97)00046-0
Zeumer B, Sauthoff G (1998) Deformation behaviour of intermetallic NiAl-Ta alloys with strengthening Laves phase for high-temperature applications III. Effects of alloying with Cr. Intermetallics 6:451–460. https://doi.org/10.1016/S0966-9795(97)00094-0
Zeumer B, Sanders W, Sauthoff G (1999) Deformation behaviour of intermetallic NiAl-Ta alloys with strengthening Laves phase for high-temperature applications IV. Effects of processing. Intermetallics 7:889–899. https://doi.org/10.1016/S0966-9795(98)00137-X
Zeumer B, Wunnike-Sanders W, Sauthoff G (1995) Mechanical properties and high-temperature deformation behaviour of particle-strengthened NiAl alloys. Mater Sci Eng A 192–193:817–823. https://doi.org/10.1016/0921-5093(95)03329-7
Sauthoff G, Zeumer B (2001) Nickel-Aluminium Intermetallic Basis Alloy. European Patent EP 0760869 B1:1-8
Palm M, Sauthoff G (2001) Long-term creep and oxidation behavior of a Laves phase-strengthened NiAl-Ta-Cr alloy for gas turbine applications. Mater Res Soc Symp Proc 646:N6.8.1–N6.8.6. https://doi.org/10.1557/PROC-646-N6.8.1
Scheppe F, Sahm PR, Hermann W, Paul U, Preuhs J (2002) Nickel aluminides: a step toward industrial application. Mater Sci Eng A 329–331:596–601. https://doi.org/10.1016/S0921-5093(01)01587-8
Thien V, Hermann W, Paul U, Sahm PR, Scheppe F (2001) Intermetallic NiAl-components for systems of environmentally friendly energy conversion. Partial project: heat shields, made out of intermetallic NiAl-alloys for stationary gas turbines (Intermetallische NiAl-Komponenten fuer Systeme zur umweltfreundlichen Energieumwandlung. Teilvorhaben: Hitzeschilde aus intermetallischen NiAl-Legierungen fuer stationaere Gasturbinen). Final report, BMBF 03N2009A
Palm M, Preuhs J, Sauthoff G (2003) Production-scale processing of a new intermetallic NiAl–Ta–Cr alloy for high-temperature application: Part I. Production of master alloy remelt ingots and investment casting of combustor liner model panels. J Mater Process Technol 136:105–113. https://doi.org/10.1016/S0924-0136(03)00130-4
Palm M, Preuhs J, Sauthoff G (2003) Production scale processing of a new intermetallic NiAl–Ta–Cr alloy for high-temperature application: Part II. Powder metallurgical production of bolts by hot isostatic pressing. J Mater Process Technol 136:114–119. https://doi.org/10.1016/S0924-0136(02)01104-4
Zhao L, He D, Bobzin K, Lugscheider E (2006) Production and characterization of NiAl-Ta-Cr intermetallic coatings sprayed by high velocity oxy-fuel. Rare Met Mater Eng 35:974–977
Zhong Y, Hajas D, Hu W, Chen H, Gottstein G (2007) Microstructure and mechanical properties of continuous Al2O3 fibre reinforced Ni45Al45Cr7.5Ta2.5 alloy (IP75) matrix composites. Philos Mag 87:1019–1032. https://doi.org/10.1080/14786430601019458
Hajas DE, Gebhardt T, Chen H, Schneider JM (2008) Tensile Strength of Ni45Al45Cr7.5Ta2.5-Coated Sapphire Fibers. J Am Ceram Soc 91:4009–4015. https://doi.org/10.1111/j.1551-2916.2008.02754.x
Xinghao D, Jianting G, Baolin W (2007) Compressive properties of high-pressure die cast IP 75 alloy with strengthening Laves phase. Mater Lett 61:288–291. https://doi.org/10.1016/j.matlet.2006.04.052
Wan X, Lin J (2011) Microstructural evolution in multiphase NiAl-2.5Ta-7.5Cr alloy during annealing at different temperatures. Rare Met Mater Eng 40:757–760. https://doi.org/10.1016/S1875-5372(11)60032-5
Wang Z, Zhang M, Yang S, Guo J, Zhou L, Chen Z (2013) Microstructure, mechanical, friction and wear properties of NiAl-2.5Ta-7.5Cr-1B alloy. Acta Metall Sin 49:1325–1332. https://doi.org/10.3724/sp.j.1037.2013.00452
Smarsly W (2015) Lightweight structural NiAl alloy with a high high-temperature strength. US Patent US2015/0315919 A1:1-3
Brady MP, Smialek JL, Smith J, Humphrey DL (1997) The role of Cr in promoting protective alumina scale formation by γ-based Ti-Al-Cr alloys—I. Compatibility with alumina and oxidation behavior in oxygen. Acta Mater 45:2357–2369. https://doi.org/10.1016/S1359-6454(96)00362-X
Brady MP, Smialek JL, Humphrey DL, Smith J (1997) The role of Cr in promoting protective alumina scale formation by γ-based Ti-Al-Cr alloys— II. Oxidation behavior in air. Acta Mater 45:2371–2382. https://doi.org/10.1016/S1359-6454(96)00361-8
Tang Z, Shemet V, Niewolak L, Singheiser L, Quadakkers WJ (2003) Effect of Cr addition on oxidation behavior of Ti–48Al–2Ag alloys. Intermetallics 11:1–8. https://doi.org/10.1016/S0966-9795(02)00062-6
Xi YJ, Liu YJ, Wang ZX, Liu JB (2012) Oxidation behavior of TiAl alloy for various times. Anti-Corros Methods Mater 59:178–181. https://doi.org/10.1108/00035591211242005
Zhou C, Yang Y, Gong S, Xu H (2001) Effect of Ti–Al–Cr coatings on the high temperature oxidation behavior of TiAl alloys. Mater Sci Eng A 307:182–187. https://doi.org/10.1016/S0921-5093(00)01956-0
Fröhlich M, Braun R, Leyens C (2010) Ti-Al-Cr based coatings for high temperature oxidation protection of γ-TiAl. Mater Sci Forum 638–642:1306–1311. https://doi.org/10.4028/www.scientific.net/MSF.638-642.1306
Chatterjee A, Dehm G, Scheu C, Clemens H (2000) Onset of microstructural instability in a fully lamellar Ti-46.5 at.% Al-4 at.% (Cr, Nb, Ta, B) alloy during short-term creep. Z Metallkd 91:755–760
Beschliesser M, Clemens H, Kestler H, Jeglitsch F (2003) Phase stability of a γ-TiAl based alloy upon annealing: comparison between experiment and thermodynamic calculations. Scr Mater 49:279–284. https://doi.org/10.1016/S1359-6462(03)00293-8
Fox-Rabinovich GS, Wilkinson DS, Veldhuis SC, Dosbaeva GK, Weatherly GC (2006) Oxidation resistant Ti-Al-Cr alloy for protective coating applications. Intermetallics 14:189–197. https://doi.org/10.1016/j.intermet.2005.05.011
Leyens C, Braun R, Fröhlich M, Hovsepian PE (2006) Recent progress in the coating protection of gamma titanium-aluminides. JOM 58:17–21. https://doi.org/10.1007/s11837-006-0062-4
Kang LM, Yang C, Wang F, Qu SG, Li XQ, Zhang WW (2019) Deformation induced precipitation of MgZn2-type Laves phase in Ti-Fe-Co alloy. J Alloys Compd 778:795–802. https://doi.org/10.1016/j.jallcom.2018.11.236
Yeh JW, Chen SK, Lin SJ, Gan JY, Chin TS, Shun TT, Tsau CH, Chang SY (2004) Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv Eng Mater 6:299–303. https://doi.org/10.1002/adem.200300567
Cantor B, Chang ITH, Knight P, Vincent AJB (2004) Microstructural development in equiatomic multicomponent alloys. Mater Sci Eng A 375–377:213–218. https://doi.org/10.1016/j.msea.2003.10.257
Otto F, Yang Y, Bei H, George EP (2013) Relative effects of enthalpy and entropy on the phase stability of equiatomic high-entropy alloys. Acta Mater 61:2628–2638. https://doi.org/10.1016/j.actamat.2013.01.042
Miracle DB, Senkov ON (2017) A critical review of high entropy alloys and related concepts. Acta Mater 122:448–511. https://doi.org/10.1016/j.actamat.2016.08.081
Murty BS, Yeh JW, Ranganathan S (2014) Chapter 7-Intermetallics, interstitial compounds and metallic glasses in high-entropy alloys. In: Murty BS, Yeh JW, Ranganathan S (eds) High entropy alloys. Butterworth-Heinemann, Boston, pp 119–131. https://doi.org/10.1016/B978-0-12-800251-3.00007-9
Senkov ON, Senkova SV, Woodward C, Miracle DB (2013) Low-density, refractory multi-principal element alloys of the Cr–Nb–Ti–V–Zr system: microstructure and phase analysis. Acta Mater 61:1545–1557. https://doi.org/10.1016/j.actamat.2012.11.032
Yurchenko NY, Stepanov ND, Shaysultanov DG, Tikhonovsky MA, Salishchev GA (2016) Effect of Al content on structure and mechanical properties of the AlxCrNbTiVZr (x = 0; 0.25; 0.5; 1) high-entropy alloys. Mater Charact 121:125–134. https://doi.org/10.1016/j.matchar.2016.09.039
Yurchenko N, Stepanov N, Salishchev G (2017) Laves-phase formation criterion for high-entropy alloys. Mater Sci Technol 33:17–22. https://doi.org/10.1080/02670836.2016.1153277
Mishra SS, Yadav TP, Srivastava ON, Mukhopadhyay NK, Biswas K (2020) Formation and stability of C14 type Laves phase in multi component high-entropy alloys. J Alloys Compd 832:153764. https://doi.org/10.1016/j.jallcom.2020.153764
Yadav TP, Mukhopadhyay S, Mishra SS, Mukhopadhyay NK, Srivastava ON (2017) Synthesis of a single phase of high-entropy Laves intermetallics in the Ti–Zr–V–Cr–Ni equiatomic alloy. Philos Mag Lett 97:494–503. https://doi.org/10.1080/09500839.2017.1418539
Park HJ, Na YS, Hong SH, Kim JT, Kim YS, Lim KR, Park JM, Kim KB (2016) Phase evolution, microstructure and mechanical properties of equi-atomic substituted TiZrHfNiCu and TiZrHfNiCuM (M = Co, Nb) high-entropy alloys. Met Mater Int 22:551–556. https://doi.org/10.1007/s12540-016-6034-5
Guo S, Ng C, Lu J, Liu CT (2011) Effect of valence electron concentration on stability of fcc or bcc phase in high entropy alloys. J Appl Phys 109:103505. https://doi.org/10.1063/1.3587228
Ma SG, Zhang Y (2012) Effect of Nb addition on the microstructure and properties of AlCoCrFeNi high-entropy alloy. Mater Sci Eng A 532:480–486. https://doi.org/10.1016/j.msea.2011.10.110
Senkov ON, Woodward C, Miracle DB (2014) Microstructure and properties of aluminum-containing refractory high-entropy alloys. JOM 66:2030–2042. https://doi.org/10.1007/s11837-014-1066-0
Fazakas É, Zadorozhnyy V, Varga LK, Inoue A, Louzguine-Luzgin DV, Tian F, Vitos L (2014) Experimental and theoretical study of Ti20Zr20Hf20Nb20X20 (X = V or Cr) refractory high-entropy alloys. Int J Refract Met Hard Mater 47:131–138. https://doi.org/10.1016/j.ijrmhm.2014.07.009
Chen H, Kauffmann A, Gorr B, Schliephake D, Seemüller C, Wagner JN, Christ HJ, Heilmaier M (2016) Microstructure and mechanical properties at elevated temperatures of a new Al-containing refractory high-entropy alloy Nb-Mo-Cr-Ti-Al. J Alloys Compd 661:206–215. https://doi.org/10.1016/j.jallcom.2015.11.050
He F, Wang Z, Cheng P, Wang Q, Li J, Dang Y, Wang J, Liu CT (2016) Designing eutectic high entropy alloys of CoCrFeNiNbx. J Alloys Compd 656:284–289. https://doi.org/10.1016/j.jallcom.2015.09.153
Liu WH, Yang T, Liu CT (2018) Precipitation hardening in CoCrFeNi-based high entropy alloys. Mater Chem Phys 210:2–11. https://doi.org/10.1016/j.matchemphys.2017.07.037
Chanda B, Das J (2018) Composition dependence on the evolution of nanoeutectic in CoCrFeNiNbx (0.45 ≤ x ≤ 0.65) high entropy alloys. Adv Eng Mater 20:1700908. https://doi.org/10.1002/adem.201700908
Chung D, Ding Z, Yang Y (2019) Hierarchical eutectic structure enabling superior fracture toughness and superb strength in CoCrFeNiNb0.5 eutectic high entropy alloy at room temperature. Adv Eng Mater 21:1801060. https://doi.org/10.1002/adem.201801060
Yu Y, He F, Qiao Z, Wang Z, Liu W, Yang J (2019) Effects of temperature and microstructure on the triblogical properties of CoCrFeNiNbx eutectic high entropy alloys. J Alloys Compd 775:1376–1385. https://doi.org/10.1016/j.jallcom.2018.10.138
Qin G, Li Z, Chen R, Zheng H, Fan C, Wang L, Su YQ, Ding HS, Guo J, Fu H (2019) CoCrFeMnNi high-entropy alloys reinforced with Laves phase by adding Nb and Ti elements. J Mater Res 34:1011–1020. https://doi.org/10.1557/jmr.2018.468
Sunkari U, Reddy SR, Rathod BDS, Kumar D, Saha R, Chatterjee S, Bhattacharjee PP (2020) Tuning nanostructure using thermo-mechanical processing for enhancing mechanical properties of complex intermetallic containing CoCrFeNi2.1Nbx high entropy alloys. Mater Sci Eng A 769:138489. https://doi.org/10.1016/j.msea.2019.138489
Ai C, Wang G, Liu L, Guo M, He F, Zhou J, Chen Y, Wang Z, Gan B (2020) Effect of Ta addition on solidification characteristics of CoCrFeNiTax eutectic high entropy alloys. Intermetallics 120:106769. https://doi.org/10.1016/j.intermet.2020.106769
Fan A-C, Li J-H, Tsai M-H (2020) On the phase constituents of three CoCrFeNiX (X = V, Nb, Ta) high-entropy alloys after prolonged annealing. J Alloys Compd 823:153524. https://doi.org/10.1016/j.jallcom.2019.153524
Ma H, Shek CH (2020) Effects of Hf on the microstructure and mechanical properties of CoCrFeNi high entropy alloy. J Alloys Compd 827:154159. https://doi.org/10.1016/j.jallcom.2020.154159
Yurchenko N, Panina E, Tikhonovsky M, Salishchev G, Zherebtsov S, Stepanov N (2020) Structure and mechanical properties of an in situ refractory Al20Cr10Nb15Ti20V25Zr10 high entropy alloy composite. Mater Lett 264:127372. https://doi.org/10.1016/j.matlet.2020.127372
Antonov S, Detrois M, Tin S (2018) Design of novel precipitate-strengthened Al-Co-Cr-Fe-Nb-Ni high-entropy superalloys. Metall Mater Trans A 49:305–320. https://doi.org/10.1007/s11661-017-4399-9
Dong Y, Lu YP, Zhang JJ, Li TJ (2013) Microstructure and properties of multi-component AlxCoCrFeNiTi0.5 high-entropy alloys. Mater Sci Forum 745–746:775–780. https://doi.org/10.4028/www.scientific.net/MSF.745-746.775
Pauzi SSM, Harun MK, Talari M (2016) Effect of Ta addition on microstructure and hardness of FeCrNiMnCoTax and Al0.5FeCrNiMnCoTax high-entropy alloys. Mater Sci Forum 846:13–19. https://doi.org/10.4028/www.scientific.net/MSF.846.13
Pacheco V, Lindwall G, Karlsson D, Cedervall J, Fritze S, Ek G, Berastegui P, Sahlberg M, Jansson U (2019) Thermal stability of the HfNbTiVZr high-entropy alloy. Inorg Chem 58:811–820. https://doi.org/10.1021/acs.inorgchem.8b02957
Senkov ON, Senkova SV, Woodward C (2014) Effect of aluminum on the microstructure and properties of two refractory high-entropy alloys. Acta Mater 68:214–228. https://doi.org/10.1016/j.actamat.2014.01.029
Senkov ON, Woodward CF (2011) Microstructure and properties of a refractory NbCrMo0.5Ta0.5TiZr alloy. Mater Sci Eng A 529:311–320. https://doi.org/10.1016/j.msea.2011.09.033
Yan J, Li K, Wang Y, Qiu J (2019) Microstructure and mechanical properties of WMoNbCrTi HEAs sintered from the powders milled for different durations. JOM 71:2489–2497. https://doi.org/10.1007/s11837-019-03432-9
Lv S, Zu Y, Chen G, Fu X, Zhou W (2019) An ultra-high strength CrMoNbWTi-C high entropy alloy co-strengthened by dispersed refractory IM and UHTC phases. J Alloys Compd 788:1256–1264. https://doi.org/10.1016/j.jallcom.2019.02.318
Liu WH, He JY, Huang HL, Wang H, Lu ZP, Liu CT (2015) Effects of Nb additions on the microstructure and mechanical property of CoCrFeNi high-entropy alloys. Intermetallics 60:1–8. https://doi.org/10.1016/j.intermet.2015.01.004
Qin G, Wang S, Chen R, Gong X, Wang L, Su YQ, Guo J, Fu H (2018) Microstructures and mechanical properties of Nb-alloyed CoCrCuFeNi high-entropy alloys. J Mater Sci Technol 34:365–369. https://doi.org/10.1016/j.jmst.2017.11.007
Li J, Huang Y, Meng XY, Xie Y (2019) A review on high entropy alloys coatings: fabrication processes and property assessment. Adv Eng Mater 21:1900343. https://doi.org/10.1002/adem.201900343
Funding
Open Access funding enabled and organized by Projekt DEAL.. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Handling Editor: P. Nash.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stein, F., Leineweber, A. Laves phases: a review of their functional and structural applications and an improved fundamental understanding of stability and properties. J Mater Sci 56, 5321–5427 (2021). https://doi.org/10.1007/s10853-020-05509-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-020-05509-2