
Abstract
It is now established that crystalline cellulose is held together not just by hydrogen bonding, but also by dispersion forces and by electrostatic attraction modulated by stereoelectronic factors such as the exo-anomeric effect. The surface chains of native cellulose microfibrils differ in C6 conformation from crystalline cellulose and therefore form different hydrogen bonds, both outward and inward. Dispersion and electrostatic forces, influenced by cellulose conformation, also operate at the microfibril surface. The surface conformation depends on whether cellulose interacts with water, with the surfaces of other microfibrils or with non-cellulosic polymers. Cellulose-water binding competes with other binding interactions, so that diverse surface interactions are finely balanced in free energy, difficult to simulate, and dependent on local details of water structuring about which little is known, especially in the presence of dispersed chains of hemicellulosic or pectic polymers. An example is the influence of hydration on the aggregation of microfibrils as measured by neutron scattering, which is large for primary-wall cellulose and small for hardwood microfibrils. There are many consequent uncertainties about the surface interactions of hydrated cellulose microfibrils, for example how pectins associate with cellulose or why cellulose-xylan interfaces resist hydration. Evidence from a range of experimental technologies, alongside simulations, will be needed to resolve these uncertainties. The practical implications are wide-ranging, from the mechanism of plant growth and the mechanical resilience of wood to the development of novel, wood-based building materials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The surfaces of cellulose microfibrils are often described as either hydrophilic, exposing the hydroxyl-rich edge of each sheet of chains; or hydrophobic, where the face of a sheet of chains is exposed (Medronho et al. 2012; Glasser et al. 2012). Hydrophilic means, literally, having an affinity for water (Pingali et al. 2020; Chandler 2005). Both the hydrophilicity of cellulose microfibrils and their interactions with other molecules have been routinely ascribed to hydrogen bonding (Nissan 1976), with the cellulosic hydroxyl groups as key hydrogen-bond donors.
An insightful recent review (Wohlert et al. 2022) questioned the assumption that hydrogen bonding was the dominant factor in the internal cohesion of cellulose microfibrils, the sorption of non-cellulosic polymers to their surfaces and their aggregation with one another. They pointed out that there are substantial contributions to the binding energy from dispersion forces (Nishiyama 2018), and electrostatic interactions other than OH-O hydrogen bonding (Chen et al. 2014), particularly between C and O atoms with partial atomic charges modulated by the anomeric and related stereoelectronic effects (Loerbroks et al. 2013; Cardamone and Popelier 2015) (Fig. 1) The relative importance of hydrogen bonding in fixing the chain conformation in cellulose has also been questioned (Ling et al. 2020; Wohlert et al. 2022).
Given accurate structures, these non-covalent bonding contributions can be estimated by molecular simulation, with limitations specific to each interaction type. We have the necessary structural information for crystalline cellulose such as the Iβ form (Nishiyama 2009). We have much less structural detail for less ordered, native cell-wall microfibrils or for matrix polymers. This detail is emerging from solid-state NMR studies, especially on cellulose microfibrils (Zhao et al. 2020; Kirui et al. 2019; Cresswell et al. 2021; Phyo and Hong 2019), but is still incomplete.
An alternative approach is to use molecular dynamics simulations starting from a known crystalline structure that is allowed to change in response to environmental factors such as the presence of water or interactions with other cell-wall polymers (Wohlert et al. 2022; Cresswell et al. 2021). However, in native, hydrated cell walls there is so much scope for alternative structures with very similar free energy (Ling et al. 2020) that making predictions from such simulations is rather uncertain. With the growing availability of structural information from spectroscopic and scattering methods (Wang et al. 2016a; Zhao et al. 2021; Thomas et al. 2021; Cresswell et al. 2021; Shekar et al. 2022), it would seem advisable to carry out a reality check on simulated structures against the available experimental data before attempting to calculate the prevalence of any kind of non-covalent bonding. Such checks have been made, (e.g. Bergenstrahle-Wohlert et al. 2016; Chen et al. 2019b) but not always in this context.
Direct experimental estimation of the relative contributions of different non-covalent bonding mechanisms is generally difficult. The O–H stretching frequencies in vibrational (FTIR, Raman, NIR or SFG) spectra (Akerholm et al. 2004; Lindh et al. 2016; Gierlinger 2018; Guo and Altaner 2018) are guides to hydrogen-bond direction and enthalpy, provided that the O–H stretching bands are correctly assigned (Altaner et al. 2014a; Altaner et al. 2014b; Marechal and Chanzy 2000; Nishiyama 2018). Because chemical shifts from 13C NMR depend on partial atomic charges i.e. local electron density, and hence magnetic shielding, they can give some indirect indication of stereoelectronic effects such as the anomeric and gamma-gauche effects. For example, the large difference in the 13C chemical shift of the C4 signal between interior (C6 tg, 89 ppm) and surface cellulose (C6 gt, 84 ppm) implies, in principle, reduced electron density and increased partial positive charge on C4, in the interior chains, contributing to electrostatic interactions and influenced by the C6 conformation (Phyo et al. 2018; Newman and Davidson 2004). More detailed information on electron density distribution could be obtained from 13C chemical shift anisotropy experiments (Ghosh et al. 2019) or from high-resolution 1H chemical shifts, which have recently become accessible for cellulose (Phyo et al. 2018; Phyo and Hong 2019). Electron density around oxygen atoms would in theory be still more informative but 17O NMR is not amenable to high-resolution chemical shift measurement (Ni et al. 2013).
Dispersion effects are not directly measurable by any experimental method, although broadly the dispersion contribution will favour binding more when cellulose chains sit closer together, as expressed in the a and b dimensions of the crystallographic unit cell; or will lead to an increase in energy when the mean separation of water molecules is increased in low-density water domains. Dispersion effects in crystalline cellulose can be calculated if the full three-dimensional structure is known, by performing simulations with and without the dispersion term left out (Chen et al. 2021). This approach assumes that different forms of non-covalent bonding are independent and additive, which is not necessarily the case (Chen et al. 2022). For example, removing the dispersion correction in a DFT simulation of crystalline cellulose under tension (Chen et al. 2021) led to an expansion of the lattice that would presumably, to varying extents, reduce the attractive stereoelectronic and hydrogen bonding terms. The converse would also be expected, e.g. interchain hydrogen bonds would be expected to reduce interchain distances and thus augment the contribution of dispersion forces.
It seems safest to say that none of the contributions to non-covalent cohesion should be discounted. Their relative importance may not be the same when resisting dissolution or mechanical disruption, nor between different modes of mechanical disruption (reversible or irreversible tension, compression or shear in any or the three dimensions, or peeling of single chains).
Wohlert et al. (2022) also point out that hydrogen bonds between cellulose chains, or between cellulose and another polymer, are usually in competition with hydrogen bonding between cellulose and water (Nissan 1976). Vibrational spectroscopy shows that cellulose-cellulose, cellulose-water and water-water hydrogen bonds lie within similar, rather broad ranges of bond enthalpies (Igarashi et al. 2020; Salmen et al. 2021; Auer and Skinner 2008) so the net enthalpy change when water inserts into a cellulose-cellulose hydrogen bond is not large and can potentially be either positive or negative. The entropy contribution to the free energy change (Kishani et al. 2021) depends on the freedom of movement of the water molecule both before and after insertion. Even before insertion, the water may be more confined and less mobile than in the bulk liquid state (Amann-Winkel et al. 2016). Water mobility within cellulosic materials can be estimated by neutron inelastic scattering (Amann-Winkel et al. 2016) or 1H broadline NMR relaxation (Cox et al. 2010; Furman et al. 2021), but it is not straightforward to calculate entropy levels from these data.
Hydrogen bonds in the core of a cellulose microfibril are not, of course, subject to competition from water (Wang et al. 2021). The poorly understood process by which the free energy of displaced water is compensated when parallel, newly synthesised cellulose chains coalesce into a microfibril (Zhang et al. 2014), may be regarded as a remarkable instance of irreversible thermodynamics in action (Wohlert et al. 2022).
Each molecule of liquid water is transiently both a donor and an acceptor of hydrogen bonds, while within each sheet of chains in a cellulose microfibril the interchain hydrogen bonds obviously have both donor and acceptor oxygens (Fig. 1A). The acceptors include the ring oxygen, and occasionally the glycosidic oxygen, which have no hydrogen atom to donate (Nishiyama 2009). When we think of the hydrophilicity of cellulose we tend to focus on the hydrogen-bond donor function of its equatorial hydroxyl groups, but hydrogen-bond acceptors are also relevant (Ling et al. 2020).
These considerations suggest that a more detailed understanding of non-covalent bonding at cellulose surfaces might be useful. In this review the emphasis is on native cellulose, direct experimental evidence and unanswered questions.
Structure and non-covalent bonding in the interior of cellulose microfibrils
To understand what forms of non-covalent bonding may be expected at cellulose surfaces, it is instructive to look first at the microfibril interior. In crystalline native celluloses, the tg conformation at C6 allows O6 to lie alongside the glycosidic linkage (Nishiyama 2009). It can then accept a hydrogen bond from O2 of the preceding glucosyl unit in the same chain and donate an intermolecular hydrogen bond to O3 of the adjacent chain, which in turn is the donor for a hydrogen bond to the ring oxygen on the preceding residue in the same chain (Fig. 1A). There is some disorder in the O6 and O2 hydrogen bonding (Nishiyama 2009) and some hydrogen bonds have two simultaneous acceptor oxygens. In cellulose II, with antiparallel rather than parallel chains, the C6 conformation is gt (Wohlert et al. 2022) and O6 and O2 are therefore too far apart for hydrogen bonding between them. However, the O3’H–O5 hydrogen bond is an almost universal structural motif in dry cellulose (and hemicelluloses) in their characteristic flat-ribbon 21 helical conformation.
It would be natural to assume that the flat-ribbon chain conformation of cellulose is simply the result of the inter-residue hydrogen bonding, but this has been questioned (Ling et al. 2020). The simulated conformations of the glycosidic linkage in xylobiose, and in analogues that lack hydroxyl groups, suggest that the favoured conformation results to a considerable extent from steric and stereoelectronic factors, involving C6 and modulated by solvation, with the inter-residue hydrogen bonding being a consequence as much as a cause (Ling et al. 2020).
In cellulose Iβ each chain lies above the interface between two chains in the sheet below, with a slight sideways displacement expressed in the monoclinic angle of the unit cell (Nishiyama et al. 2002). Intersheet OH–O hydrogen bonding is absent but weak intersheet CH–O hydrogen bonds have been suggested based on the crystal geometry (Nishiyama et al. 2003) and supported by Atoms-In-Molecules simulations (Parthasarathi et al. 2011; French et al. 2014). Wohlert et al. (2022) focused on attractive electrostatic and dispersion forces between the sheets. Both electrostatic and dispersion forces increase as the distance between the sheets decreases. The electrostatic interaction is augmented by several short intersheet distances between an oxygen atom with a partial negative charge and a carbon atom with a partial positive charge (Parthasarathi et al. 2011; Chen et al. 2014) (Fig. 1B). The resulting electrostatic effect pulls the sheets closer together, decreasing the a dimension of the unit cell and enhancing the attractive dispersion force (Fig. 1C). The components of the electrostatic attraction and the proposed intersheet CH–O hydrogen bonds, projected in the b and c directions, will then influence the monoclinic angle (Chen et al. 2014) (Fig. 1B) and the axial stagger, respectively. Similar forces contribute to the very different packing arrangement of the antiparallel chains in cellulose II (Chen et al. 2014).

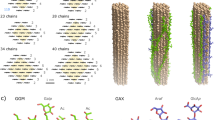
Non-covalent binding interactions in crystalline cellulose Iβ.
A. Hydrogen bonding. Hydrogen bonds between glucosyl monomer residues of each chain (Nishiyama et al. 2002) (blue arrows) and between adjacent chains within the same sheet (green arrows).
B. Electrostatic attraction between sheets. One centre chain is shown slightly offset above the junction between two origin chains in the cellulose Iβ lattice, with the shortest-distance intersheet interactions (< 0.35 nm) between oxygen and carbon atoms shown as red, solid lines (Chen et al. 2014). The oxygens carry negative partial atomic charges and the carbons carry positive partial atomic charges. The intersheet CH–O hydrogen bonds proposed by Parthasarathi et al. (2011) and French et al. (2014) are shown as blue dashed lines. The lateral offset corresponds to the monoclinic angle of 96.5° maximising the net intersheet electrostatic attraction (including weaker attractions between more distant C/O pairs and repulsive C/C and O/O interactions). The lateral components of the electrostatic forces and CH–O hydrogen bonds are balanced, influencing the monoclinic angle (Chen et al.2014).
C. Dispersion interaction between sheets. The strength of the long-range attractive dispersion component decreases steeply with the spacing a/2 between the origin and centre sheets. Although predominantly between sheets, these dispersion interactions also operate in the other dimensions.
The electrostatic character of hydrogen bonds means that an oxygen functioning as a hydrogen-bond donor acquires a greater negative charge, and thereby becomes a stronger acceptor, if it receives a hydrogen bond from another donor. Thus chains of hydrogen-bonded hydroxyls can form, and there may be some co-operative stabilisation (Araujo et al. 2018; Jarvis 2003). Since charge can be redistributed by conformation-dependent stereoelectronic effects, as well as by hydrogen bonding, it is to be expected that these effects add to and interact with hydrogen-bonding patterns (Newman and Davidson 2004) and C–O electrostatic interactions (Chen et al. 2014). Such interactions are included in molecular simulations, in so far as they are captured by the force fields used (Wohlert et al. 2022), but the components are not readily separated either in simulations or experimentally.
Surface structures of cellulose microfibrils
In the large, highly crystalline microfibrils from algae or tunicates the cellulose Iα or Iβ lattice extends to the surface [110] and [1–10] planes, diagonal to the ring plane (Baker et al. 2000; Malm et al. 2010). The thinner, less ordered microfibrils of higher plants have more than half of their chains located at the surface (Jarvis 2018). Modelling studies (Kubicki et al. 2018) suggest that a large part of the surface area is occupied by the [110] and [1–10] planes, and that the chains at these surfaces are placed roughly in the positions specified by the cellulose I lattice. It has therefore become conventional (Jarvis 2018; Kubicki et al. 2018) to use the lattice indexing for cellulose Iβ (Nishiyama et al. 2002) to denote the crystal planes and the exposed surfaces of the microfibrils of higher-plant celluloses, even though none of their chains is identical with either the Iα or the Iβ crystalline allomorph (Wang et al. 2016b). That nomenclature is followed here, in the current absence of a detailed structure for the 3 nm microfibrils of higher plants.
These thin microfibrils contain about seven kinds of glucosyl monomers identifiable by NMR. (Dupree et al. 2015; Cresswell et al. 2021; Wang et al. 2014, 2016b). Their locations within the microfibril structure are incompletely known. In particular, it is not wholly clear if conformationally distinguishable monomers alternate along any one glucan chain, which might be expected if the environments of the two edges of the chain differ as at the hydrophilic surfaces (Oehme et al. 2015; Funahashi et al. 2017). However the maximum abundance of the C6 gt conformer, observed where cellulose microfibrils are in contact with water (Dupree et al. 2015; Sturcova et al. 2004; Newman and Davidson 2004) is consistent with all monomers in the surface chains having that conformation as commonly assumed (Fig. 2). If so the reason is perhaps stereoelectronic. An alternative suggestion (Oehme et al. 2015) is that outward-facing C6 hydroxyls on each surface chain and on the chain below have the gt conformation, with the rest tg. Being less rigidly crowded, the surface chains of thin microfibrils are more mobile than the interior chains, especially in contact with water (Chen et al. 2019b). Whether the tg or the gt C6 conformation predominates depends on what is in contact with the surface: water, a non-cellulosic polymer or another microfibril, as discussed in the next sections.

Section of a higher-plant microfibril, across a 4-chain sheet with the surface chains (top and bottom) having the C6 gt conformation at all glucosyl residues rather than the C6 tg conformation found in the interior chains. The surface chains have outward-pointing hydroxyl groups due to the gt conformation. These hydroxyls are well placed to act as donors for hydrogen bonds to water or other microfibrils. Other arrangements of gt and tg conformations are possible (Oehme et al. 2015). With the arrangement shown there are gaps (blue ellipses) between the surface and interior chains, in which there might be enough space for confined water
It is usually assumed that the surface chains are not axially displaced with respect to the chains inside them (Fig. 2), an assumption consistent with the retention of the 004 as the principal axial reflection in the diffraction patterns from thin microfibrils. Oehme et al. (2015) simulated a small axial displacement. Minor axial reflections are also present and differ in intensity between celluloses in cell walls from different taxa (Thomas et al. 2013b, 2014, 2021), but may reflect their hemicellulose composition rather than cellulose structure. As not all the C6 conformations at the junction between the surface chain and the underlying chain are tg, the hydrogen bonding pattern there cannot be the same as in crystalline cellulose (Oehme et al. 2015). There might be room for trapped water between the interior and surface chains (Fig. 2), although spin-diffusion NMR experiments do not confirm close proximity of water to C6 tg cellulose (Gelenter et al. 2017; Wang et al. 2015; White et al. 2014). Essentially all cellulose hydroxyl groups function as hydrogen-bond donors: hydroxyls that are not hydrogen-bonded to any acceptor would have a distinctive O–H stretching signal in the vibrational spectra. This signal is weak (Makarem et al. 2020) or unobserved (Marechal and Chanzy 2000; Hofstetter et al. 2006).
A surface chain on a slanting [110] or [1–10] face of the microfibril is in contact with only one cellulose chain in the sheet above, so that less intersheet electrostatic attraction is expected (Chen et al. 2014). The a dimension of the unit cell is therefore greater in thin microfibrils, with a greater percentage of surface chains, than in crystalline cellulose Iβ (Fernandes et al. 2011; Thomas et al. 2013a, 2013b). The wider intersheet spacing presumably reduces the dispersion forces (Chen et al. 2014). The monoclinic angle is often reduced in thin microfibrils (Thomas et al. 2013b), but is sensitive to hydration (Zabler et al. 2010). Further steric and stereoelectronic effects of the C6 conformation may be assumed (Newman and Davidson 2004), but the details are unclear.
In most models of the shape of cellulose microfibrils from higher plants (Kubicki et al. 2018) the ‘hydrophobic’ faces, corresponding to the [100] and [200] lattice planes, are quite narrow, often only two chains wide. If wider, their hydroxyl groups would be tied up in intra- or interchain hydrogen bonds. However, there are exposed oxygens suitably sited to act as hydrogen-bond acceptors, particularly between chains and at glycosidic linkages (Miyamoto et al. 2014), and hydroxyls project at the corners. These surfaces are not, therefore, uniformly hydrophobic.
Cellulose-cellulose interactions and microfibril aggregation
In wood, microfibrils are laterally aggregated, giving rise to a distinctive small-angle scattering neutron scattering (SANS) feature on deuteration, from which the centre-to-centre ‘characteristic’ spacing of microfibrils within a macrofibril can be calculated. The ‘characteristic’ spacing reflects the distribution of spacings across hydrophilic junctions, but is not an arithmetic mean: it is dominated by the larger spacings where there is more interposed deuterium to provide contrast (Fernandes et al. 2011). The intensity of this SANS feature diminishes to near zero on drying as scattering contrast is lost, but when extrapolated to the dry state it corresponds to a 2.4–4 nm centre-to-centre spacing, varying with cell-wall type (Fernandes et al. 2011; Thomas et al. 2013b, 2014, 2015; Nishiyama et al. 2014; Zitting et al. 2021; Penttila et al. 2021a). Interestingly, there are similar observations for regenerated celluloses (Sawada et al. 2021). The centre-to-centre spacing in the dry state has been taken as a measure of the microfibril diameter, assuming that the microfbrils are then in contact (Fernandes et al. 2011). This is an approximation. For example, a hypothetical 18-chain (2,3,4,4,3,2) microfibril ((Kubicki et al. 2018) with the lattice spacings observed for dry wood (Zabler et al. 2010) has diameters varying from 2.4 to 3.0 nm across the different crystal faces.
It should be noted that the same SANS experiments can also yield an independent, normally smaller, estimate of the microfibril diameter through the form factor (Penttila et al. 2019; Kennedy et al. 2007a), rather than the structure factor as described above. The form factor depends on local radii, which are small at corners. The approximation that microfibrils are hard, smooth and cylindrical biases both of these approaches, probably in opposite directions. However, the characteristic centre-to-centre SANS spacings of up to about 3 nm, observed in softwoods (Penttila et al. 2021a; Thomas et al. 2020; Fernandes et al. 2011) and grasses (Thomas et al. 2015), have been interpreted in terms of 18-chain or slightly larger microfibrils aggregated in cellulose-cellulose contact (Jarvis 2018). In hardwoods it has been suggested that a glucuronoarabinoxylan coating on one side leads to the observed centre-to-centre spacings of nearer 4 nm (Thomas et al. 2014; Nishiyama et al. 2014). These ̴4 nm spacings are detectable at lower intensity in softwoods (Thomas et al. 2020) as well as the ̴3 nm spacings assumed to represent cellulose-cellulose contact. The irregularity of microfibril shapes, especially when hemicellulose-coated, means that such contacts are also likely to be irregular.
In primary cell walls, microfibril aggregation appears to be rather limited (Zhang et al. 2016; Ye et al. 2018) although possibly important for growth. An exception is the celery collenchyma system (Chen et al. 2019a), where aggregation of well-aligned microfibrils is much more extensive and leads to a strong SAXS or SANS peak (Thomas et al. 2013b; Kennedy et al. 2007a), converging towards a 3 nm centre-to-centre microfibril spacing in the dry state.
From the C4 and C6 chemical shifts in 13C NMR (Phyo et al. 2018), the C6 gt conformer predominates in the surface chains of celery collenchyma microfibrils (Thomas et al. 2013b; Kennedy et al. 2007a) and other primary-wall systems (Newman and Davidson 2004; Phyo et al. 2018) and in softwoods (Fernandes et al. 2011) and grasses (Duan et al. 2021; Thomas et al. 2015). Many of the NMR measurements on woody cell walls were made using hydrated samples, because spectral resolution tends to be reduced in the dry state. Dried pine wood showed a higher proportion of the tg form than hydrated, perhaps due to condensation of xylan chains onto cellulose surfaces, but a substantial proportion of gt remained (Cresswell et al. 2021). Repeated drying of regenerated cellulose reduced the ratio of surface to interior residues (Idström et al. 2016).
The implication is that where aggregation occurs directly between uncoated cellulose surfaces, many of the surface chains have the C6 gt conformation. The outward direction of the O6 and O2 hydroxyls with the C6 gt conformation would be consistent with both of these hydroxyls acting as hydrogen-bond donors to the adjacent microfibril (Oehme et al. 2015).
A puzzling question is: why does crystallographic fusion not occur when microfibrils are aggregated in this way? The intervention of water might be a sufficient explanation in vivo, but not in dried cellulosic materials. Twisting of the microfibrils was suggested to prevent their fusion (Jarvis 2018) but other explanations are possible. It is not known for certain if aggregated microfibrils lie parallel or antiparallel (Li et al. 2016). An antiparallel configuration would certainly prevent fusion into a single cellulose I lattice, but might be expected to lead to some of the spectral signatures of cellulose II, which are not observed in native cellulosic materials (Marechal and Chanzy 2000).
Supporting the above model for microfibril aggregation, simulations (Zhang et al. 2021) showed stick–slip behaviour of the interface between dry, parallel microfibrils under shear. The transient stability of the stick phase correlated with the number of hydrogen bonds formed. There were two unequal stick points per cellobiosyl repeat displacement (1.04 nm) but neither of these corresponded to the in-register lateral alignment of the two chains as found in crystalline cellulose: they were axially displaced by about half a monomer unit (Zhang et al. 2021), which would inhibit crystalline fusion.
These experiments do not imply that only hydrogen bonding was involved. At the stick points, increased proximity of the chains, whether due to hydrogen bonding or reduced steric interference, would be expected to augment dispersion and perhaps electrostatic interactions. Similar simulations in the presence of water showed more irregular stick–slip behaviour averaging about (1.04/4) nm apart (Zhang et al. 2021), weaker than in the dry state as would be expected from the intervention of at least one ‘lubricating’ layer of hydrogen-bonded water molecules between the cellulose surfaces (Oehme et al. 2015). Intervening water, as explained below, would be expected in in vivo situations such as elongation of primary cell walls or plastic deformation of softwoods, bamboo or straw.
Several imaging methods appear to show discrete bundles of aggregated microfibrils, sometimes called ‘macrofibrils’. Macrofibrils can be distinguished with difficulty in ESEM and cryo-TEM images of wood cell walls, close to their native state (Donaldson 2007; Reza et al. 2014). They are more evident when their boundaries appear as lines of weakness along the fracture plane in cryo-SEM (Sell and Zimmerman 1993; Donaldson 2007; Cresswell et al. 2021) or AFM images (Guo et al. 2020), or when the boundary domains between them are delignified (Adobes-Vidal 2020) or swollen with polyethylene glycol (Fahlen and Salmen 2005; Penttila et al. 2020), which also allows SANS signals to be recorded (Penttila et al. 2020). The measured width of the macrofibrils is typically some tens of nm, but varies between imaging techniques, probably because a variable proportion of the boundaries are distinguished (Fahlen and Salmen 2005; Donaldson 2007; Penttila et al. 2020; Cresswell et al. 2021). Lignin and associated glucomannans appear to be enriched between macrofibrils (Altaner et al. 2006), but if so these polymers do not provide enough contrast for clear TEM imaging in the native state (Reza et al. 2014). The lignin-rich domains may be axially discontinuous (Tershima et al. 2009).
Cellulose interactions with water
Water being the classic hydrogen-bonded liquid, it is common to think of cellulose-water interaction in terms of hydrogen bonding and to consider the hydrophilicity of cellulose surfaces in terms of hydrogen-bond free energy (Willems 2018), but other electrostatic and dispersion contributions to water binding are probably involved as well, as in cellulose-cellulose interactions (Chen et al. 2021). The thermodynamics of hydration in wood and other cellulosic materials have been extensively reviewed (Thybring et al. 2021; Arzola-Villegas et al. 2019), but it was recently concluded that none of the physical models on which thermodynamic descriptions were based is a good match for what happens at the cellulose-water interface (Thybring et al. 2021).
Binding of water molecules to cellulose depends on the nature of the water. The process of hydration is commonly understood as the interaction of a rigid, plane, solid surface with an infinite volume of bulk water (Chandler 2005). That model has been widely adopted in discussions and simulations of cellulose hydration, but for wood cellulose it is misleading. Much of the water does not bind to existing bare surfaces but inserts itself between microfibrils, swelling the wood laterally (Arzola-Villegas et al. 2019). The thermodynamics of water binding then includes a contribution from detachment of the two microfibril surfaces, in addition to the enthalpy and entropy of the bound water (Bertinetti et al. 2016). From the neutral temperature dependence of hydration, enthalpy and entropy contributions seem to be balanced (Willems 2014).
Isotherms like Fig. 3 express the equilibria between water and cellulosic materials like wood (Willems 2016; Nopens et al. 2019; Lovikka et al. 2018), demonstrating the presence of binding sites that vary in free energy (Arzola-Villegas et al. 2019). It is only in very dry wood that any space appears to be available for ingress of water without swelling (Nopens et al. 2019). While microfibrils of the shapes assumed (Kubicki et al. 2018) could in principle be close-packed, it seems that in wood the packing is irregular enough to leave some gaps at interstices. Strongly bound water, in gaps that would exist in its absence, may therefore lead to the downturn in the isotherms at the lower limit of the RH scale, which corresponds typically to about 5–10% of non-swelling water in wood (Arzola-Villegas et al. 2019; Nopens et al. 2019), depending on whether the wood has ever previously been dried (Thybring et al. 2020). The percentage is less in other cellulosic materials (Lovikka et al. 2018) and approximately zero in hydrolysed nanocellulose fibres (Guo and Wu 2018). The net binding energy of water in such locations is high because no polymer–polymer hydrogen bonding need be broken to make way for it, and because an unfilled gap implies unfavourable dispersion energy. A somewhat different explanation for the low-RH region of the softwood isotherms, in terms of hydration free energy at interfaces, is suggested by Bertinetti et al. (2016). There is also strongly bound water in gaps between bound xylan and cellulose (Jarvis 2018; Cresswell et al. 2021).

Typical water sorption isotherm for a softwood, showing suggested locations for the water molecules bound, more or less successively, at each stage in the forward sorption direction (Nopens et al. 2019). There is considerable uncertainty concerning the strength of binding of water that is associated with hemicelluloses and lignin and located between aggregates of microfibrils
Above the inflection at the low-RH end of the isotherms, swelling and water uptake by wood are positively correlated (Nopens et al. 2019) and are roughly equal if the wood is locally free to swell, as in pillars dissected from the S2 layer (Rafsanjani et al. 2014). In intact wood cells the volume expansion is constrained and directed inward into the cell lumina (Plaza 2019; Guo et al. 2020) by the S1 and S3 layers, which resist hoop stress. It may not be coincidence that the biexponential kinetics of wood hydration, swelling and plastic deformation are all similar (Salmen and Olsson 2016; Hill and Xie 2011; Thomas et al. 2021; Nopens et al. 2019).
The increase in the characteristic centre-to-centre SANS spacing (Fernandes et al. 2011; Zitting et al. 2021; Arzola-Villegas et al. 2019) is a reasonable starting point for observing how much the microfibrils move apart on hydration. In celery collenchyma, a model for primary cell walls (but with much more aligned and aggregated microfibrils than primary walls usually contain) the characteristic spacing increased from 3 to 5–6 nm on hydration (Kennedy et al. 2007b; Thomas et al. 2013b). In woody cell walls, expansion is much more constrained. In softwoods (conifers), the characteristic spacing increases from 3 or less, to 4 nm at saturation (Fernandes et al. 2011; Thomas et al. 2020; Zitting et al. 2021; Arzola-Villegas et al. 2019). Estimates of pore size in hydrated pine cell walls by size exclusion (Thybring et al. 2018) were similarly of the order of 1 nm. The dynamics and chemical reactivity of water in these confined spaces are anomalous (Beaumont et al. 2021).
In the grass bamboo, the characteristic spacing increased less, from 3.0 in the dry state to 3.2 nm when hydrated (Thomas et al. 2015). In hardwoods there was little or no increase in the 4 nm microfibril spacing in the presence of water (Thomas et al. 2014; Nishiyama et al. 2014), and it was suggested that the outer face of the arabinoglucuronoxylans coating one microfibril bound to the uncoated hydrophilic surface of the adjacent microfibril in a way that resisted water penetration (Thomas et al. 2014). Since hardwoods swell as much as softwoods (Arzola-Villegas et al. 2019; Nopens et al. 2019), there may be additional ways of packing hardwood microfibrils that are too irregular to give a coherent SANS signal.
Calculating the number of layers of water implied by increases in spacing on hydration is imprecise due to the irregularity of the microfibril surfaces and the uncertain density of the associated water. The volume of a molecule of bulk water, density 1000 kg/m3, is 18/No = 0.030 nm3. The partial volume of a molecule of bound water, averaged across much variation between hydrated solid biomolecules, is 0.024 nm3 (Gerstein and Chothia 1996; Sirotkin et al. 2012). On a cubic basis these figures give a mean monolayer thickness of about 0.3 nm, suggesting that within the macrofibrils of softwood cell walls at saturation, more than half of the water molecules are associated with one or more hydrophilic cellulose surfaces, while in grasses the increased SANS spacings on hydration amount to no more than a single water monolayer, and in hardwoods little increase in spacing is detectable by SANS.
In addition to water between aggregated microfibrils, some water must penetrate between microfibril aggregates. Water molecules in these wider spaces (Arzola-Villegas et al. 2019), and between the widely spaced microfibrils in hydrated primary walls (Kennedy et al. 2007b) are more likely to be associated with one another (Willems 2018) as in the structure of bulk liquid water (Amann-Winkel et al. 2016), but the water structure may be influenced by solute-like xylan (Zhang et al. 2020), glucomannan (Cresswell et al. 2021) or pectin chains (Ha et al. 2005).
When wood or wood pulp is hydrated there are minor, but distinct changes in the structure of cellulose. The preponderance of the C6 gt conformation in the hydrophilic surface chains becomes more marked, perhaps due to release of xylans (Cresswell et al. 2021). In the C6 gt conformation there is also a possibility of hydrogen bonding to a surface chain in the next sheet of the same microfibril (Terrett et al. 2019; Cresswell et al. 2021). The microfibrils become stretched in the axial dimension (Zabler et al. 2010; Salmen et al. 2021), either due to swelling pressure in the hydrated matrix (Salmen et al. 2021) or through straightening of the surface chains as inter-residue hydrogen bonding between O6’ and O2 is disrupted (Altaner et al. 2014b). The intersheet dimension of the microfibrils contracts (Salmen et al. 2021; Zabler et al. 2010) perhaps because the conformational changes on stretching (Altaner et al. 2014b; Djahedi et al. 2016) allow a closer fit under dispersion and electrostatic forces (Chen et al. 2021).
Details of the hydrogen bonding to water can be inferred from vibrational spectroscopy (Araujo et al. 2018; Guo et al. 2018; Cichosz and Masek 2020; Salmen et al. 2021). The O–H stretching regions of the polarised FTIR and NIR spectra show that the hydrogen bonds newly formed on hydration are predominantly transversely oriented (Driemeier et al. 2015; Lindh et al. 2016; Guo and Altaner 2019). This spectral region is difficult to disentangle because of overlaps and vibrational coupling, but O2 and O6 are potential hydrogen-bond donors to water, with some uncertainty as to which is preferred (Araujo et al. 2018; Hofstetter et al. 2006).
Lindh (2016) concluded that the (O3H–O5) hydrogen bond remained intact, resisting 2H exchange during hydration with 2H2O in contrast with its behaviour in β-chitin (Sawada et al. 2012). This conclusion of Lindh et al. (2016) was based on deconvolution of the (1H–2H) FTIR difference spectra from microcrystalline cellulose, and on deconvolution of a 2H T1 NMR relaxation experiment. In each case the data could be fitted with only two components, assignable to O22H and O62H. The deconvolution step is challenging (Altaner et al. 2014b; Lindh et al. 2017), being subject to baseline and saturation errors as well as coupling problems. Deconvoluting the multiexponential NMR data is more challenging still. Qualitatively similar FTIR difference spectra have been recorded from wood pulp cellulose (Hofstetter et al. 2006), cotton (Lindh et al. 2017; Maréchal and Chanzy 2000), Avicel (Driemeier et al. 2015) and spruce wood (Fernandes et al. 2011; Altaner et al. 2014b; Penttila et al. 2021b). When hydrogen-bonded to O5, O3H does appear to be more resistant to cleavage by water than the other hydroxyl groups of surface cellulose chains (Lindh et al. 2016; Lindh and Salmen 2017), but it is not clear if it is fully resistant in all materials at full hydration. Deuterium exchange kinetics (Lindh and Salmen 2017; Penttila et al. 2021b) show that accessibility of sites at microfibril interfaces is somewhat restricted, especially at low moisture content (Driemeier et al. 2015).
The FTIR difference spectra seem to indicate that the O–H stretching frequencies of the bound water itself are bimodally centred around the upper and lower boundaries of the frequency range of cellulose or of bulk water (Igarashi et al. 2020; Guo et al. 2018). In principle this bimodal frequency distribution could arise from saturation artefacts, but FTIR-AFM experiments (Igarashi et al. 2020) imply that it is genuine. Also, the FTIR band assigned to H–O–H deformation of water has been deconvoluted into two components (Cichosz and Masek 2020); a narrow band and a broader band that increases in intensity more steeply with hydration, shifting simultaneously to lower frequency. The FTIR data are consistent with the insertion of water molecules, with a mix of higher- and lower-enthalpy hydrogen bonds, between cellulose surfaces that would otherwise be in direct contact.
The simulations by Trentin et al (2021) predicted that nanoscale (but still multilayer) water droplets behave differently on different cellulose surfaces. The [1–10] surfaces were fully wetted with simulated contact angles < 10°, and C6 was largely in the gt conformation permitting both O6 and O2 to act as outward-facing hydrogen-bond donors. Surprisingly, the [110] surfaces had much larger simulated contact angles. However, in the simulations the [110] surface chains retained more than half of the C6 groups in the tg conformation (Trentin et al. 2021), which is inconsistent with the experimental data from NMR experiments on pure celluloses (Sturcova et al. 2004), so the force fields used may not have precisely captured the balance of intermolecular forces at the interface. At wide [100] and [200] surfaces, equilibrated contact angles of > 30° (Trentin et al. 2021) were consistent with properties more hydrophobic than at [1–10] surfaces, but not strongly hydrophobic in absolute terms.
The water domains in hydrated softwoods are highly elongated in the direction of the microfibrils (Furman et al. 2021). The water comprises two mobility classes, distinguishable by broadline 1H NMR (Cox et al. 2010), which may correspond to the multiple domains detectable by FTIR (Igarashi et al. 2020). The less mobile fraction is filled first during hydration (Topgaard and Soderman 2002). Its presence increases the mobility of the surface cellulose chains (Garvey et al. 2019; Phyo et al. 2017). This fraction does not freeze (Cox et al. 2010; Igarashi et al. 2020). The presence of non-freezing water in pores between fibrils (Igarashi et al. 2020) might be attributed to capillarity and described by variants of the Kelvin Equation (Liu et al. 2003), but that approach assumes wrongly that the material is rigid and that all pores emptied of water fill with air.
It might be supposed that two hydrophobic cellulose surfaces would associate through hydrophobic interactions. In the simulations of Oehme et al. (2015), however, a monolayer of water was quite strongly retained between two [100] or [200] microfibril faces. This is the opposite of what would be expected for hydrophobic association driven by the expulsion of water into a domain where its free energy is lower, such as a bulk liquid phase. For any interaction that displaces water from a cellulose surface, the free energy change depends on the nature of the water removed and its destination. When the destination of water is within the cell wall, its free energy there may be higher than that of liquid water.
On current understanding of hydrophobic interactions (Rego and Patel 2022; Monroe et al. 2020), the free energy change when water is displaced is dominated by the enthalpic term when the dimensions of the surface exceed about 1 nm, and by the entropic term for narrower surfaces or solutes, with a reduction in the associated water density. The width of a two-chain hydrophobic surface is about 1.5 nm, or somewhat less if measured between the hydroxyl-rich edges, and is thus around the crossover dimension between enthalpic and entropic dominance. The [100] and [200] surfaces simulated by Trentin et al. (2021) were wider. However the simulated behaviour of water over extended hydrophobic cellulose surfaces (Miyamoto et al. 2014) was not uniform: lines of low-density water overlay the glucose rings, separated by denser water over the hydrogen-bonded interfaces between the chains. Heterogeneous, patterned surfaces are known to show non-intuitive anomalies in the overall density and free energy of the associated water (Monroe et al. 2021). Whether [100] and [200] surfaces should really be called hydrophobic is an open question, although the term has a long history.
Low-density water domains with limited mobility and structural resemblances to ice might reasonably be implicated in restricting shear between microfibrils. It has recently emerged that microcrystalline and nanocrystalline celluloses can nucleate ice crystallisation from supercooled bulk water (Hou et al. 2021). Cellulosic materials in atmospheric aerosols may therefore contribute to cloud formation (Hiranuma et al. 2015). A more potent, slow-acting, property of nanocelluloses is to inhibit ice recrystallisation (Li et al. 2022, 2019).
These properties resemble some of the diverse activities of mineral ice-binding surfaces and ice-binding proteins (Cui et al. 2022). Bacterial ice-nucleating proteins promote freezing (Qiu et al. 2019), whereas antifreeze proteins from animals and plants block the growth of ice crystals by binding to their surfaces (Voets 2017). By related mechanisms, polymers useful in frozen food products inhibit recrystallisation and the consequent coarsening of ice crystal texture (Bachtiger et al. 2021). Comparison with cellulose provides clues about the nature of water in contact with cellulose surfaces.
In some ice-binding proteins and minerals, adhesion to ice depends on a specific surface pattern of hydrogen-bond donor, hydrogen-bond acceptor and hydrophobic sites, complementary to the ice surface (Hudait et al. 2018) and matching the inter-oxygen spacings on the basal (0.451 nm) or prismatic (0.277, 0.451, 0.735 nm) faces of ice (Soni and Patey 2021). However, the exposed lattice d-spacings (Nishiyama et al. 2002) of native cellulose microfibrils ([110] = 0.548 nm, [1–10] = 0.593 nm, [001] = 1.038 nm) show a complete mismatch with ice. There are other ice-binding proteins and minerals that lack lattice matching (Qiu et al. 2017). It has been suggested that these have plane surfaces which can bind the first monolayer of ice irregularly with moderate strength and enough flexibility to let the next and more distant monolayers settle into the ice lattice (Qiu et al. 2017). That model would seem appropriate for the observed binding of ice by cellulose surfaces in contact with bulk-like water, as in nanocellulose preparations (Li et al. 2019) and perhaps in primary cell walls, although solute-like pectic chains (Ha et al. 2005) might also contribute to the structuring, mobility and freezing behaviour of primary-wall water (Takahashi et al. 2021; Phyo et al. 2019). In bacterial cellulose, with thicker microfibrils separated by wider pores, layers of non-freezing water were associated with the exposed cellulose surfaces and freezing water was considered to lie between (O'Neill et al. 2017). A liquid-like, non-freezing structure for the monolayers of water between microfibrils in grasses or wood could facilitate shear at these interfaces and assist energy absorption, fracture diversion (Guo et al. 2020) and freezing tolerance (Takahashi et al. 2021).
Binding of hemicelluloses
The hemicelluloses (xylans, glucomannans and xyloglucans) all resemble cellulose in structure and can adopt its flat-ribbon 21 helical chain conformation (Salmen 2022), retaining the O3H–O5 hydrogen bond and, at least in the case of xylans, other stabilising factors (Ling et al. 2020). All lack inter-residue hydrogen bonding between O6 and O2, for different reasons. In xylans O6 is lacking, while in most residues of xyloglucans it is substituted. In the mannose residues of glucomannans O2 points the wrong way.
In principle a hemicellulose chain in the 21 helical conformation could take the place of a surface cellulose chain in a microfibril, if its monomer sequence allowed the substituents to face outward. To what extent this happens in practice is not fully established, and depends on the hemicellulose. The best understood example is the binding of alternately substituted dicot glucuronoarabinoxylans to hydrophilic faces of cellulose microfibrils, inducing the underlying cellulose chain to adopt the C6 tg conformation characteristic of the microfibril interior (Simmons et al. 2016; Martinez-Abad et al. 2017). Binding is reduced by hydration. Arabinoxylans can also bind to hydrophobic cellulose faces (Martinez-Abad et al. 2017; Duan et al. 2021), possibly through electrostatic and dispersion forces resembling those between cellulose sheets, although the arabinoxylan geometry is unclear.
Some, but not all, softwood glucomannan chain segments bind to cellulose in a similar 21 helical conformation (Terrett et al. 2019; Martinez-Abad et al. 2020) in proximity to cellulose monomers with both tg and gt conformations at C6, with gt predominating in hydrated pine wood (Cresswell et al. 2021). Xyloglucans, although apparently as capable of adopting cellulose-like conformations and binding to microfibril surfaces, are not so extensively found in close association with the microfibrils of dicot primary cell walls (Wang et al. 2013; Kishani et al. 2021). Xyloglucan-cellulose contacts at low abundance may however be key participants in the cell-wall loosening that permits growth (Phyo et al. 2017; Zhang et al. 2019; Wang et al. 2013).
The change on binding to a conformation that gives a disaccharide repeat distance matching cellulose suggests that glucuronoarabinoxylan chains, at least, co-align with the cellulose axis, as supported by simulations (Falcoz-Vigne et al. 2017; Cresswell et al. 2021). However, it is not clear if bound hemicelluloses lie parallel or antiparallel to the underlying cellulose chains. The spin-diffusion experiments on which this model is based (e.g. Simmons et al. 2016) do not distinguish directly between these arrangements. The secretion of hemicelluloses in soluble form suggests a random parallel/antiparallel mixture. A parallel arrangement would be needed for the non-covalent binding to resemble that between surface and interior cellulose chains (Gupta et al. 2021), but even then, the binding arrangement cannot be identical. For example, the absence of C6 and O6 in xylose probably reduces the potential for interchain hydrogen bonding and interlayer electrostatic attraction, and creates a gap that may be filled by water (Jarvis 2018; Cresswell et al. 2021). The substituents on the outer edge of the xylan chain – arabinosyl, acetyl and 4-O-methyl glucuronosyl—can in some cases form associations with cellulose chains in the next sheet exposed on the same hydrophilic microfibril surface (Cresswell et al. 2021; Simmons et al. 2016). Dispersion and C–O electrostatic interactions may contribute to these associations. In mannosyl residues O2 points out of the ring plane and therefore cannot participate in hydrogen bonding with an adjacent cellulose chain, its place being taken by a water molecule in the simulations of Cresswell et al. (2021).
If bound hemicelluloses are considered as part of the microfibril, the substituents ranged along their outer edges will modulate the microfibril’s surface properties. An extreme example is dispersal of the microfibrils of quince mucilage by charge repulsion between the unusually abundant 4-O-methylgucuronosyl substituents on the bound xylan (Vignon and Gey 1998). Otherwise, outward-facing carboxyl groups on 4-O-methylgucuronosyl residues can function as strong hydrogen-bond acceptors or interact electrostatically with the cellulosic surfaces of adjacent microfibrils (Jarvis 2018; Thomas et al. 2020). Neutral carbohydrates (arabinosyl residues on xylans, galactosyl residues on glucomannans) increase the number of exposed hydroxyls that can potentially act as both hydrogen-bond donors and acceptors. Acetyl carbonyls are also potential hydrogen-bond acceptors, but block a hydroxyl on the main hemicellulose chain. The surface roughness of hemicellulose-coated microfibrils may affect the potential for close-fitting interactions with favourable electrostatic or dispersion binding.
Binding of pectins and lignin
There is evidence that some of the lignin in wood cell walls forms discrete clusters between macrofibrils (Altaner et al. 2006; Terashima et al. 2009), associated with and covalently bound to hemicelluloses (Terrett and Dupree 2019; Kirui et al. 2022). However other lignin domains are non-covalently associated with cellulose (Kirui et al. 2022; Dupree et al. 2015). The monomethoxyl substituents of a minor fraction of the guaiacyl lignins associate with acetyls on bound xylans (Kirui et al. 2022). In dicots, linear chains of syringyl lignin appear to stack onto the hydrophobic faces of microfibrils (Kang et al. 2019) like the tyrosine residues in Type A cellulose-binding domains (Sprenger et al. 2021), but these interactions need not be wholly hydrophobic in character. They could, for example, involve specific stacking interactions between the aromatic and pyranose rings.
Intimate spatial association of pectic galacturonan and rhamnogalacturonan I chains has been demonstrated by NMR spin-diffusion experiments (Wang et al. 2012, 2015; Phyo and Hong 2019). The nature of this association remains unexplained. It appears to include an electrostatic contribution, because suppression of the negative charge on galacturonic acid by acidification reduced pectin-cellulose association and increased cellulose-water, pectin-water and pectin-pectin associations (Phyo et al. 2019). These observations imply existing or induced partial positive charges on cellulose, which could be on carbon atoms as implicated in the internal electrostatic interactions within microfibrils; and/or on hydrogen, implying hydrogen bonding from cellulose with pectic carboxyls as acceptors. From their C4 13C chemical shifts (Wang et al. 2015) the galacturonan chains are in an approximate 31 helical conformation with a trisaccharide repeat distance that does not match the cellulose axial repeat (Jarvis and Apperley 1995). Nor are known rhamnogalacturonan conformations axially matched with cellulose, although they do permit asymmetry with carboxyls on one side and mobile, hydrated arabinan and galactan side-chains on the other (Makshakova et al. 2017). The mismatched repeat distances argue against axially repetitive binding if the pectic chains are aligned along the microfibril axis. Electrostatic binding might be delocalised, as in the counterion condensation model (Manning and Ray 1998; Manning 2007). Pectin-cellulose binding does not seem to require divalent counterions like Ca2+ (nor galacturonoyl ester linkages) as it survives sequential extraction with CDTA (Goldberg et al. 1996) and Na2CO3 (White et al. 2014; Wang et al. 2015). The counterions present in vivo (Goldberg et al. 1996) may modulate the water activity and dielectric constant around the microfibrils, and might influence both cross-linking and transverse electrostatic swelling as the cell-wall pH falls during growth (Phyo et al. 2019; Cosgrove 2022). Speculatively, nanophase separation might occur within the pectic component (MacDougall et al. 1997) for example with galacturonan nanofilaments (Haas et al. 2020) as one of the phases.
In principle, attached pectic polymers would be expected to separate microfibrils by charge repulsion and osmotic water uptake, assuming that water is available to let the cell wall swell (Kennedy et al. 2007b). An extreme case is the fully dispersed microfibrils of Arabidopsis seed mucilage where the non-cellulosic polysaccharides are pectic in nature (Griffiths and North 2017). However, in primary cell walls cation binding (Goldberg et al. 1996) might lead to effects on microfibril aggregation that are difficult to predict (Ray and Manning 1994), and there is a need for more data on these phenomena before the implications for growth are known. It should be noted that although microfibril separation can be measured by small-angle scattering (Kennedy et al. 2007b) this technique is biased towards the larger microfibril spacings and is not well suited to detect the localised points of contact that have been implicated in growth (Cosgrove, 2022).
Discussion
It is now apparent that a cellulose microfibril is held together not just by hydrogen bonding, but also by dispersion forces and by electrostatic, mainly C–O, attraction, modulated by stereoelectronic factors like the exo-anomeric effect (Wohlert et al. 2022; Ling et al. 2020). As detailed above, there is evidence for a similar range of binding types at the microfibril surface where cellulose interacts with water molecules, with the surfaces of other microfibrils or with non-cellulosic polymers. The relative contributions of these interactions to the binding enthalpy are not well understood. Cellulose-water binding competes with binding to other sites (Wohlert et al. 2022), but is dependent on details of local water structuring about which little is known, especially when the water structure is perturbed by dispersed chains of hemicelluloses (Zhang et al. 2020) or pectins (Ha et al. 2005). The entropy contribution to water binding (Bertinetti et al. 2016) is influenced by the variable freedom of movement of the bound water molecules (Cox et al. 2010), and of water wherever it goes when displaced.
There remain many uncertainties, therefore, about the surface interactions of hydrated cellulose microfibrils. Evidence from a range of experimental methods, alongside simulations, will be needed to resolve these and other uncertainties.
As an example, the role of surface chains and their outer environment in the response of microfibrils to tensile stress is not well understood. FTIR and X-ray scattering experiments demonstrated that cellulose chains in wood straighten out under tension, resisted by synergy between hydrogen and covalent bonding. But this mechanism requires the C6 tg conformation, and therefore excludes many of the surface chains. Simulations of the interior chains have shown that additionally, the glycosidic linkages and the glucopyranosyl rings stretched (Djahedi et al. 2016), in ways that can now be traced by bandshift analysis in the fingerprint region of the FTIR spectra (Thomas et al. 2021). Some preliminary distinctions can be made between the contributions of surface cellulose chains and hemicelluloses to the FTIR spectra under tension (Thomas et al. 2021), but the detail falls far short of what can be discerned by multidimensional NMR, e.g. (Cresswell et al. 2021; Kirui et al. 2019)—in which, of course, tensile experiments are not possible. The expulsion of water under tension (Guo et al. 2020) suggests that its structural involvement cannot be ignored. The initial microfibril structure does not simply elongate, but changes in complex ways that depend on the surface environment and must influence both electrostatic and dispersion contributions to the free energy of stretching. The substantial dispersion term simulated by subtraction (Chen et al. 2021) is non-intuitive in view of the large contraction in the a dimension of the unit cell of wood microfibrils in tension (Thomas et al. 2021; Salmen et al. 2021), which would be expected to lower the free energy of the stretched structure. In principle these structural changes under tensile stress should now be accessible by crystallographic methods, since displacements of reflections can be separated from their intensities by correlative shift mapping (Thomas et al. 2021). However suitably high-resolution diffraction patterns have not yet been recorded under stress.
The implications of the surface interactions of cellulose are wide-ranging. For example, the controlled, cellulose-scaffolded coherence of primary cell walls is central to the mechanism of plant growth (Zhang et al. 2019; Cosgrove 2022). The primary-wall skeleton of a herbaceous plant gives it shape and rigidity when inflated elastically by turgor, yet somehow a controlled transition to non-elastic deformation permits and directs growth. How, we do not yet really understand; but the key controlling events seem to be located between hydrated microfibrils (Phyo et al. 2017; Cosgrove 2022). Wood cell walls do not grow, but wood close to its breaking stress has a similar capability for energy-absorbing deformation (Guo et al. 2020). Cell walls under stress deform on several length scales, and there remains much to be learned about how cell-scale forces decompose into local, nm-scale, shear and tensile stresses within each layer of a primary or secondary cell wall. Nevertheless, the central issue is how the interfaces between microfibrils or macrofibrils respond to these local stresses.
More practically, many of the limiting engineering properties of cellulose-based products–shrinkage, warping, fungal decay and the disintegration of wet chipboard–can be traced to the ingress of water. In response to climate change we need, urgently, new wood- and bamboo-derived products to replace steel, concrete and other unsustainable building materials. The greatest challenge in the design of such materials is stability against water penetration between microfibrils.
Availability of data and materials
Not applicable.
References
Adobes-Vidal M, Frey M, Keplinger T (2020) Atomic force microscopy imaging of delignified secondary cell walls in liquid conditions facilitates interpretation of wood ultrastructure. J Struct Biol. https://doi.org/10.1016/j.jsb.2020.107532
Akerholm M, Hinterstoisser B, Salmen L (2004) Characterization of the crystalline structure of cellulose using static and dynamic FT-IR spectroscopy. Carbohyd Res 339:569–578. https://doi.org/10.1016/j.carres.2003.11.012
Altaner C, Apperley DC, Jarvis MC (2006) Spatial relationships between polymers in Sitka spruce: proton spin-diffusion studies. Holzforschung 60:665–673. https://doi.org/10.1515/hf2006.112
Altaner CM, Horikawa Y, Sugiyama J, Jarvis MC (2014a) Cellulose I beta investigated by IR-spectroscopy at low temperatures. Cellulose 21:3171–3179. https://doi.org/10.1007/s10570-014-0360-x
Altaner CM, Thomas LH, Fernandes AN, Jarvis MC (2014b) How cellulose stretches: synergism between covalent and hydrogen bonding. Biomacromol 15:791–798. https://doi.org/10.1021/bm401616n
Amann-Winkel K, Bellissent-Funel M-C, Bove LE, Loerting T, Nilsson A, Paciaroni A, Schlesinger D, Skinner L (2016) X-ray and neutron scattering of water. Chem Rev 116:7570–7589. https://doi.org/10.1021/acs.chemrev.5b00663
Araujo C, Freire CSR, Nolasco MM, Ribeiro-Claro PJA, Rudic S, Silvestre AJD, Vaz PD (2018) Hydrogen bond dynamics of cellulose through inelastic neutron scattering spectroscopy. Biomacromol 19:1305–1313. https://doi.org/10.1021/acs.biomac.8b00110
Arzola-Villegas X, Lakes R, Plaza NZ, Jakes JE (2019) Wood moisture-induced swelling at the cellular scale-ab intra. Forests 10:996. https://doi.org/10.3390/f10110996
Auer BM, Skinner JL (2008) IR and Raman spectra of liquid water: Theory and interpretation. J Chem Phys 128:224511. https://doi.org/10.1063/1.2925258
Bachtiger F, Congdon TR, Stubbs C, Gibson MI, Sosso GC (2021) The atomistic details of the ice recrystallisation inhibition activity of PVA. Nat Commun 12:1323. https://doi.org/10.1038/s41467-021-21717-z
Baker AA, Helbert W, Sugiyama J, Miles MJ (2000) New insight into cellulose structure by atomic force microscopy shows the I-alpha crystal phase at near-atomic resolution. Biophys J 79:1139–1145
Beaumont M, Jusner P, Gierlinger N, King AWT, Potthast A, Rojas OJ, Rosenau T (2021) Unique reactivity of nanoporous cellulosic materials mediated by surface-confined water. Nat Commun 12:2513. https://doi.org/10.1038/s41467-021-22682-3
Bergenstrahle-Wohlert M, d’Ortoli TA, Sjoberg NA, Widmalm G, Wohlert J (2016) On the anomalous temperature dependence of cellulose aqueous solubility. Cellulose 23:2375–2387. https://doi.org/10.1007/s10570-016-0991-1
Bertinetti L, Fratzl P, Zemb T (2016) Chemical, colloidal and mechanical contributions to the state of water in wood cell walls. New J Phys. https://doi.org/10.1088/1367-2630/18/8/083048
Cardamone S, Popelier PLA (2015) Prediction of conformationally dependent atomic multipole moments in carbohydrates. J Comput Chem 36:2361–2373. https://doi.org/10.1002/jcc.24215
Chandler D (2005) Interfaces and the driving force of hydrophobic assembly. Nature 437:640–647. https://doi.org/10.1038/nature04162
Chen P, Nishiyama Y, Mazeau K (2014) Atomic partial charges and one Lennard-Jones parameter crucial to model cellulose allomorphs. Cellulose 21:2207–2217. https://doi.org/10.1007/s10570-014-0279-2
Chen D, Melton LD, McGillivray DJ, Ryan TM, Harris PJ (2019a) Changes in the orientations of cellulose microfibrils during the development of collenchyma cell walls of celery (Apium graveolens L.). Planta 250:1819–1832. https://doi.org/10.1007/s00425-019-03262-8
Chen P, Terenzi C, Furo I, Berglund LA, Wohlert J (2019b) Quantifying localized macromolecular dynamics within hydrated cellulose fibril aggregates. Macromolecules 52:7278–7288. https://doi.org/10.1021/acs.macromol.9b00472
Chen P, Nishiyama Y, Wohlert J (2021) Quantifying the influence of dispersion interactions on the elastic properties of crystalline cellulose. Cellulose 28:10777–10786. https://doi.org/10.1007/s10570-021-04210-0
Chen P, Zhao C, Wang H, Li Y, Tan G, Shao Z, Nishiyama Y, Hu T, Wohlert J (2022) Quantifying the contribution of the dispersion interaction and hydrogen bonding to the anisotropic elastic properties of chitin and chitosan. Biomacromol 23:1633–1642. https://doi.org/10.1021/acs.biomac.1c01488
Cichosz S, Masek A (2020) IR study on cellulose with the varied moisture contents: insight into the supramolecular structure. Materials 13:4573. https://doi.org/10.3390/ma13204573
Cosgrove DJ (2022) Building an extensible cell wall. Plant Physiol 189:1246–1277. https://doi.org/10.1093/plphys/kiac184
Cox J, McDonald PJ, Gardiner BA (2010) A study of water exchange in wood by means of 2D NMR relaxation correlation and exchange. Holzforschung 64:259–266. https://doi.org/10.1515/hf.2010.036
Cresswell R, Dupree R, Brown SP, Pereira CS, Skaf MS, Sorieul M, Dupree P, Hill S (2021) Importance of water in maintaining softwood secondary cell wall nanostructure. Biomacromol 22:4669–4680. https://doi.org/10.1021/acs.biomac.1c00937
Cui S, Zhang W, Shao X, Cai W (2022) Do antifreeze proteins generally possess the potential to promote ice growth? Phys Chem Chem Phys 24:7901–7908. https://doi.org/10.1039/d1cp05431g
Djahedi C, Bergenstrahle-Wohlert M, Berglund LA, Wohlert J (2016) Role of hydrogen bonding in cellulose deformation: the leverage effect analyzed by molecular modeling. Cellulose 23:2315–2323. https://doi.org/10.1007/s10570-016-0968-0
Donaldson L (2007) Cellulose microfibril aggregates and their size variation with cell wall type. Wood Sci Technol 41(5):443. https://doi.org/10.1007/s00226-006-0121-6
Driemeier C, Mendes FM, Ling LY (2015) Hydrated fractions of cellulosics probed by infrared spectroscopy coupled with dynamics of deuterium exchange. Carbohyd Polym 127:152–159. https://doi.org/10.1016/j.carbpol.2015.03.068
Duan P, Kaser SJ, Lyczakowski JJ, Phyo P, Tryfona T, Dupree P, Hong M (2021) Xylan structure and dynamics in native Brachypodium grass cell walls investigated by solid-state NMR spectroscopy. ACS Omega 6:15460–15471. https://doi.org/10.1021/acsomega.1c01978
Dupree R, Simmons TJ, Mortimer JC, Patel D, Iuga D, Brown SP, Dupree P (2015) Probing the molecular architecture of Arabidopsis thaliana secondary cell walls using two- and three-dimensional C-13 solid state nuclear magnetic resonance spectroscopy. Biochemistry 54:2335–2345. https://doi.org/10.1021/bi501552k
Fahlen J, Salmen L (2005) Pore and matrix distribution in the fiber wall revealed by atomic force microscopy and image analysis. Biomacromol 6:433–438. https://doi.org/10.1021/bm040068x
Falcoz-Vigne L, Ogawa Y, Molina-Boisseau S, Nishiyama Y, Meyer V, Petit-Conil M, Mazeau K, Heux L (2017) Quantification of a tightly adsorbed monolayer of xylan on cellulose surface. Cellulose 24:3725–3739. https://doi.org/10.1007/s10570-017-1401-z
Fernandes AN, Thomas LH, Altaner CM, Callow P, Forsyth VT, Apperley DC, Kennedy CJ, Jarvis MC (2011) Nanostructure of cellulose microfibrils in spruce wood. Proc Natl Acad Sci USA 108:E1195–E1203. https://doi.org/10.1073/pnas.1108942108
French AD, Concha M, Dowd MK, Stevens ED (2014) Electron (charge) density studies of cellulose models. Cellulose 21:1051–1063. https://doi.org/10.1007/s10570-013-0042-0
Funahashi R, Okita Y, Hondo H, Zhao M, Saito T, Isogai A (2017) Different conformations of surface cellulose molecules in native cellulose microfibrils revealed by layer-by-layer peeling. Biomacromol 18:3687–3694. https://doi.org/10.1021/acs.biomac.7b01173
Furman G, Goren S, Meerovich V, Panich A, Sokolovsky V, Xia Y (2021) Anisotropy of transverse and longitudinal relaxations in liquids entrapped in nano- and micro-cavities of a plant stem. J Magn Reson 331:107051. https://doi.org/10.1016/j.jmr.2021.107051
Garvey CJ, Simon GP, Whittaker AK, Parker IH (2019) Moisture-activated dynamics on crystallite surfaces in cellulose. Colloid Polym Sci 297:521–527. https://doi.org/10.1007/s00396-018-04464-4
Gelenter MD, Wang T, Liao S-Y, O’Neill H, Hong M (2017) H-2-C-13 correlation solid-state NMR for investigating dynamics and water accessibilities of proteins and carbohydrates. J Biomol NMR 68:257–270. https://doi.org/10.1007/s10858-017-0124-7
Gerstein M, Chothia C (1996) Packing at the protein-water interface. Proc Natl Acad Sci USA 93:10167–10172. https://doi.org/10.1073/pnas.93.19.10167
Ghosh M, Kango N, Dey KK (2019) Investigation of the internal structure and dynamics of cellulose by C-13-NMR relaxometry and 2DPASS-MAS-NMR measurements. J Biomol NMR 73:601–616. https://doi.org/10.1007/s10858-019-00272-2
Gierlinger N (2018) New insights into plant cell walls by vibrational microspectroscopy. Appl Spectrosc Rev 53:517–551. https://doi.org/10.1080/05704928.2017.1363052
Glasser WG, Atalla RH, Blackwell J, Brown RM Jr, Burchard W, French AD, Klemm DO, Nishiyama Y (2012) About the structure of cellulose: debating the Lindman hypothesis. Cellulose 19:589–598. https://doi.org/10.1007/s10570-012-9691-7
Goldberg R, Morvan C, Jauneau A, Jarvis MC (1996) Methyl-esterification, de-esterification and gelation of pectins in the primary cell wall. In: International Symposium on Pectins and Pectinases, Wageningen, Netherlands, Dec 03–07 1995. Progress in Biotechnology. pp 151–172. https://doi.org/10.1016/s0921-0423(96)80253-x
Griffiths JS, North HM (2017) Sticking to cellulose: exploiting Arabidopsis seed coat mucilage to understand cellulose biosynthesis and cell wall polysaccharide interactions. New Phytol 214:959–966. https://doi.org/10.1111/nph.14468
Guo F, Altaner CM (2018) Molecular deformation of wood and cellulose studied by near infrared spectroscopy. Carbohyd Polym 197:1–8. https://doi.org/10.1016/j.carbpol.2018.05.064
Guo F, Altaner CM (2019) Effects of mechanical stretching, desorption and isotope exchange on deuterated eucalypt wood studied by near infrared spectroscopy. Spectrochimica Acta Part A–Molecular Biomol Spectrosc 211:254–259. https://doi.org/10.1016/j.saa.2018.12.012
Guo X, Wu Y (2018) IN SITU visualization of water adsorption in cellulose nanofiber film with micrometer spatial resolution using micro-FTIR imaging. J Wood Chem Technol 38:361–370. https://doi.org/10.1080/02773813.2018.1488869
Guo X, Liu L, Wu J, Fan J, Wu Y (2018) Qualitatively and quantitatively characterizing water adsorption of a cellulose nanofiber film using micro-FTIR spectroscopy. RSC Adv 8:4214–4220. https://doi.org/10.1039/c7ra09894d
Guo F, Altaner CM, Jarvis MC (2020) Thickness-dependent stiffness of wood: potential mechanisms and implications. Holzforschung 74:1079–1087. https://doi.org/10.1515/hf-2019-0311
Gupta M, Rawal TB, Dupree P, Smith JC, Petridis L (2021) Spontaneous rearrangement of acetylated xylan on hydrophilic cellulose surfaces. Cellulose 28:3327–3345. https://doi.org/10.1007/s10570-021-03706-z
Ha MA, Vietor RJ, Jardine GD, Apperley DC, Jarvis MC (2005) Conformation and mobility of the arabinan and galactan side-chains of pectin. Phytochemistry 66:1817–1824. https://doi.org/10.1016/j.phytochem.2005.06.001
Haas KT, Wightman R, Meyerowitz EM, Peaucelle A (2020) Pectin homogalacturonan nanofilament expansion drives morphogenesis in plant epidermal cells. Science 367:1003–1007. https://doi.org/10.1126/science.aaz5103
Hill CAS, Xie Y (2011) The dynamic water vapour sorption properties of natural fibres and viscoelastic behaviour of the cell wall: is there a link between sorption kinetics and hysteresis? J Mater Sci 46:3738–3748. https://doi.org/10.1007/s10853-011-5286-1
Hiranuma N, Moehler O, Yamashita K, Tajiri T, Saito A, Kiselev A, Hoffmann N, Hoose C, Jantsch E, Koop T, Murakami M (2015) Ice nucleation by cellulose and its potential contribution to ice formation in clouds. Nat Geosci 8:273–277. https://doi.org/10.1038/ngeo2374
Hofstetter K, Hinterstoisser B, Salmen L (2006) Moisture uptake in native cellulose–the roles of different hydrogen bonds: a dynamic FT-IR study using Deuterium exchange. Cellulose 13:131–145. https://doi.org/10.1007/s10570-006-9055-2
Hou Y, Sun X, Dou M, Lu C, Liu J, Rao W (2021) Cellulose nanocrystals facilitate needle-like ice crystal growth and modulate molecular targeted ice crystal nucleation. Nano Lett 21:4868–4877. https://doi.org/10.1021/acs.nanolett.1c00514
Hudait A, Odendahl N, Qiu Y, Paesani F, Molinero V (2018) Ice-nucleating and antifreeze proteins recognize ice through a diversity of anchored clathrate and ice-like motifs. J Am Chem Soc 140:4905–4912. https://doi.org/10.1021/jacs.8b01246
Idström A, Schantz S, Sundberg J, Chmelka BF, Gatenholm P, Nordstierna L (2016) C-13 NMR assignments of regenerated cellulose from solid-state 2D NMR spectroscopy. Carbohyd Polym 151:480–487. https://doi.org/10.1016/j.carbpol.2016.05.107
Igarashi T, Hoshi M, Nakamura K, Kaharu T, Murata K-I (2020) Direct observation of bound water on cotton surfaces by atomic force microscopy and atomic force microscopy-infrared spectroscopy. J Phys Chem C 124:4196–4201. https://doi.org/10.1021/acs.jpcc.0c00423
Jarvis M (2003) Chemistry–cellulose stacks up. Nature 426(6967):611–612. https://doi.org/10.1038/426611a
Jarvis MC (2018) Structure of native cellulose microfibrils, the starting point for nanocellulose manufacture. Philos Transact Royal Soc A-Math Phys Eng Sci 376:2112. https://doi.org/10.1098/rsta.2017.0045
Jarvis MC, Apperley DC (1995) Chain conformation in concentrated pectic gels–evidence from C-13 NMR. Carbohyd Res 275:131–145. https://doi.org/10.1016/0008-6215(95)00033-p
Kang X, Kirui A, Widanage MCD, Mentink-Vigier F, Cosgrove DJ, Wang T (2019) Lignin-polysaccharide interactions in plant secondary cell walls revealed by solid-state NMR. Nat Commun 10:347. https://doi.org/10.1038/s41467-018-08252-0
Kennedy CJ, Cameron GJ, Sturcova A, Apperley DC, Altaner C, Wess TJ, Jarvis MC (2007a) Microfibril diameter in celery collenchyma cellulose: X-ray scattering and NMR evidence. Cellulose 14:235–246. https://doi.org/10.1007/s10570-007-9116-1
Kennedy CJ, Sturcova A, Jarvis MC, Wess TJ (2007b) Hydration effects on spacing of primary-wall cellulose microfibrils: a small angle X-ray scattering study. Cellulose 14:401–408. https://doi.org/10.1007/s10570-007-9129-9
Kirui A, Ling Z, Kang X, Widanage MCD, Mentink-Vigier F, French AD, Wang T (2019) Atomic resolution of cotton cellulose structure enabled by dynamic nuclear polarization solid-state NMR. Cellulose 26:329–339. https://doi.org/10.1007/s10570-018-2095-6
Kirui A, Zhao W, Deligey F, Yang H, Kang X, Mentink-Vigier F, Wang T (2022) Carbohydrate-aromatic interface and molecular architecture of lignocellulose. Nat Commun 13:538. https://doi.org/10.1038/s41467-022-28165-3
Kishani S, Benselfelt T, Wagberg L, Wohlert J (2021) Entropy drives the adsorption of xyloglucan to cellulose surfaces–a molecular dynamics study. J Colloid Interface Sci 588:485–493. https://doi.org/10.1016/j.jcis.2020.12.113
Kubicki JD, Yang H, Sawada D, O’Neill H, Oehme D, Cosgrove D (2018) The shape of native plant cellulose microfibrils. Sci Rep 8:13983. https://doi.org/10.1038/s41598-018-32211-w
Li S, Bashline L, Zheng Y, Xin X, Huang S, Kong Z, Kim SH, Cosgrove DJ, Gu Y (2016) Cellulose synthase complexes act in a concerted fashion to synthesize highly aggregated cellulose in secondary cell walls of plants. Proc Natl Acad Sci USA 113:11348–11353. https://doi.org/10.1073/pnas.1613273113
Li T, Zhao Y, Zhong Q, Wu T (2019) Inhibiting ice recrystallization by nanocelluloses. Biomacromol 20:1667–1674. https://doi.org/10.1021/acs.biomac.9b00027
Li M, Luckett CR, Wu T (2022) Potent time-dependent ice recrystallization inhibition activity of cellulose nanocrystals in sucrose solutions. Biomacromol 23:497–504. https://doi.org/10.1021/acs.biomac.1c01201
Lindh EL, Salmen L (2017) Surface accessibility of cellulose fibrils studied by hydrogen-deuterium exchange with water. Cellulose 24:21–33. https://doi.org/10.1007/s10570-016-1122-8
Lindh EL, Bergenstrahle-Wohlert M, Terenzi C, Salmen L, Furo I (2016) Non-exchanging hydroxyl groups on the surface of cellulose fibrils: the role of interaction with water. Carbohyd Res 434:136–142. https://doi.org/10.1016/j.carres.2016.09.006
Ling Z, Edwards JV, Nam S, Xu F, French AD (2020) Conformational analysis of xylobiose by DFT quantum mechanics. Cellulose 27:1207–1224. https://doi.org/10.1007/s10570-019-02874-3
Liu ZH, Muldrew K, Wan RG, Elliott JAW (2003) Measurement of freezing point depression of water in glass capillaries and the associated ice front shape. Phys Rev E 67:061602. https://doi.org/10.1103/PhysRevE.67.061602
Loerbroks C, Rinaldi R, Thiel W (2013) The electronic nature of the 1,4-beta-glycosidic bond and its chemical environment: DFT insights into cellulose chemistry. Chemistry-a European Journal 19:16282–16294. https://doi.org/10.1002/chem.201301366
Lovikka VA, Rautkari L, Maloney TC (2018) Changes in the hygroscopic behavior of cellulose due to variations in relative humidity. Cellulose 25:87–104. https://doi.org/10.1007/s10570-017-1570-9
MacDougall AJ, Rigby NM, Ring SG (1997) Phase separation of plant cell wall polysaccharides and its implications for cell wall assembly. Plant Physiol 114:353–362. https://doi.org/10.1104/pp.114.1.353
Makarem M, Kim H, Emami P, Melendez J, Steinbach A, Lipkie T, Deleris I, Desmet C, Wallecan J, Kim SH (2020) Impact of drying on meso- and nanoscale structures of citrus fiber: a study by SFG, ATR-IR, XRD, and DLS. Ind Eng Chem Res 59:2718–2724. https://doi.org/10.1021/acs.iecr.9b06194
Makshakova ON, Gorshkova TA, Mikshina PV, Zuev YF, Perez S (2017) Metrics of rhamnogalacturonan I with beta-(1 -> 4)-galactan side chains and structural basis for its self-aggregation. Carbohyd Polym 158:93–101. https://doi.org/10.1016/j.carbpol.2016.11.082
Malm E, Bulone V, Wickholm K, Larsson PT, Iversen T (2010) The surface structure of well-ordered native cellulose fibrils in contact with water. Carbohyd Res 345:97–100. https://doi.org/10.1016/j.carres.2009.10.020
Manning GS (2007) Counterion condensation on charged spheres, cylinders, and planes. J Phys Chem B 111:8554–8559. https://doi.org/10.1021/jp0670844
Manning GS, Ray J (1998) Counterion condensation revisited. J Biomol Struct Dyn 16:461–476. https://doi.org/10.1080/07391102.1998.10508261
Maréchal Y, Chanzy H (2000) The hydrogen bond network in I-beta cellulose as observed by infrared spectrometry. J Mol Struct 523:183–196. https://doi.org/10.1016/s0022-2860(99)00389-0
Martinez-Abad A, Berglund J, Toriz G, Gatenholm P, Henriksson G, Lindstrom M, Wohlert J, Vilaplana F (2017) Regular motifs in xylan modulate molecular flexibility and interactions with cellulose surfaces. Plant Physiol 175:1579–1592. https://doi.org/10.1104/pp.17.01184
Martinez-Abad A, Jimenez-Quero A, Wohlert J, Vilaplana F (2020) Influence of the molecular motifs of mannan and xylan populations on their recalcitrance and organization in spruce softwoods. Green Chem 22:3956–3970. https://doi.org/10.1039/d0gc01207f
Medronho B, Romano A, Miguel MG, Stigsson L, Lindman B (2012) Rationalizing cellulose (in)solubility: reviewing basic physicochemical aspects and role of hydrophobic interactions. Cellulose 19:581–587. https://doi.org/10.1007/s10570-011-9644-6
Miyamoto H, Schnupf U, Brady JW (2014) Water structuring over the hydrophobic surface of cellulose. J Agric Food Chem 62:11017–11023. https://doi.org/10.1021/jf501763r
Monroe JI, Jiao S, Davis RJ, Brown DR, Katz LE, Shell MS (2021) Affinity of small-molecule solutes to hydrophobic, hydrophilic, and chemically patterned interfaces in aqueous solution. Proc Natl Acad Sci USA 118:e2020205118. https://doi.org/10.1073/pnas.2020205118
Monroe J, Barry M, DeStefano A, Gokturk PA, Jiao S, Robinson-Brown D, Webber T, Crumlin EJ, Han S, Shell MS (2020) Water structure and properties at hydrophilic and hydrophobic surfaces. In: Doherty MF, Segalman RA (eds) Annual Review of Chemical and Biomolecular Engineering, Vol 11, vol 11. Annual Review of Chemical and Biomolecular Engineering. pp 523–557. https://doi.org/10.1146/annurev-chembioeng-120919-114657
Newman RH, Davidson TC (2004) Molecular conformations at the cellulose-water interface. Cellulose 11:23–32. https://doi.org/10.1023/B:CELL.0000014778.49291.c6
Ni QZ, Daviso E, Can TV, Markhasin E, Jawla SK, Swager TM, Temkin RJ, Herzfeld J, Griffin RG (2013) High frequency dynamic nuclear polarization. Acc Chem Res 46:1933–1941. https://doi.org/10.1021/ar300348n
Nishiyama Y (2009) Structure and properties of the cellulose microfibril. J Wood Sci 55:241–249. https://doi.org/10.1007/s10086-009-1029-1
Nishiyama Y (2018) Molecular interactions in nanocellulose assembly. Philos Transact Royal Soc A-Math Phys Eng Sci 376 (2112). https://doi.org/10.1098/rsta.2017.0047
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose 1 beta from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124:9074–9082. https://doi.org/10.1021/ja0257319
Nishiyama Y, Sugiyama J, Chanzy H, Langan P (2003) Crystal structure and hydrogen bonding system in cellulose 1(alpha), from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 125:14300–14306. https://doi.org/10.1021/ja037055w
Nishiyama Y, Langan P, O’Neill H, Pingali SV, Harton S (2014) Structural coarsening of aspen wood by hydrothermal pretreatment monitored by small- and wide-angle scattering of X-rays and neutrons on oriented specimens. Cellulose 21:1015–1024. https://doi.org/10.1007/s10570-013-0069-2
Nissan AH (1976) H-Bond dissociation in hydrogen-bond dominated solids. Macromolecules 9(840):850. https://doi.org/10.1021/ma60053a026
Nopens M, Riegler M, Hansmann C, Krause A (2019) Simultaneous change of wood mass and dimension caused by moisture dynamics. Sci Rep 9:10309. https://doi.org/10.1038/s41598-019-46381-8
Oehme DP, Doblin MS, Wagner J, Bacic A, Downton MT, Gidley MJ (2015) Gaining insight into cell wall cellulose macrofibril organisation by simulating microfibril adsorption. Cellulose 22:3501–3520. https://doi.org/10.1007/s10570-015-0778-9
O’Neill H, Pingali SV, Petridis L, He J, Mamontov E, Hong L, Urban V, Evans B, Langan P, Smith JC, Davison BH (2017) Dynamics of water bound to crystalline cellulose. Sci Rep 7:11840. https://doi.org/10.1038/s41598-017-12035-w
Parthasarathi R, Bellesia G, Chundawat SPS, Dale BE, Langan P, Gnanakaran S (2011) Insights into hydrogen bonding and stacking interactions in cellulose. J Phys Chem A 115:14191–14202. https://doi.org/10.1021/jp203620x
Penttila PA, Altgen M, Carl N, van der Linden P, Morfin I, Osterberg M, Schweins R, Rautkari L (2019) Moisture-related changes in the nanostructure of woods studied with X-ray and neutron scattering. Cellulose 27:71–87. https://doi.org/10.1007/s10570-019-02781-7
Penttila PA, Altgen M, Awais M, Osterberg M, Rautkari L, Schweins R (2020) Bundling of cellulose microfibrils in native and polyethylene glycol-containing wood cell walls revealed by small-angle neutron scattering. Sci Rep. https://doi.org/10.1038/s41598-020-77755-y
Penttila PA, Paajanen A, Ketoja JA (2021a) Combining scattering analysis and atomistic simulation of wood-water interactions. Carbohyd Polym 251:117064. https://doi.org/10.1016/j.carbpol.2020.117064
Penttila PA, Zitting A, Lourencon T, Altgen M, Schweins R, Rautkari L (2021b) Water-accessibility of interfibrillar spaces in spruce wood cell walls. Cellulose 28:11231–11245. https://doi.org/10.1007/s10570-021-04253-3
Phyo P, Hong M (2019) Fast MAS H-1-C-13 correlation NMR for structural investigations of plant cell walls. J Biomol NMR 73:661–674. https://doi.org/10.1007/s10858-019-00277-x
Phyo P, Wang T, Kiemle SN, O’Neill H, Pingali SV, Hong M, Cosgrove DJ (2017) Gradients in wall mechanics and polysaccharides along growing inflorescence stems. Plant Physiol 175:1593–1607. https://doi.org/10.1104/pp.17.01270
Phyo P, Wang T, Yang Y, O’Neil H, Hong M (2018) Direct determination of hydroxymethyl conformations of plant cell wall cellulose using H-1 polarization transfer solid-state NMR. Biomacromol 19:1485–1497. https://doi.org/10.1021/acs.biomac.8b00039
Phyo P, Gu Y, Hong M (2019) Impact of acidic pH on plant cell wall polysaccharide structure and dynamics: insights into the mechanism of acid growth in plants from solid-state NMR. Cellulose 26:291–304. https://doi.org/10.1007/s10570-018-2094-7
Pingali SV, Smith MD, Liu S-H, Rawal TB, Pu Y, Shah R, Evans BR, Urban VS, Davison BH, Cai CM, Ragauskas AJ, O’Neill HM, Smith JC, Petridis L (2020) Deconstruction of biomass enabled by local demixing of cosolvents at cellulose and lignin surfaces. Proc Natl Acad Sci USA 117:16776–16781. https://doi.org/10.1073/pnas.1922883117
Plaza NZ (2019) On the experimental assessment of the molecular-scale interactions between wood and water. Forests 10:616. https://doi.org/10.3390/f10080616
Qiu Y, Odendahl N, Hudait A, Mason R, Bertram AK, Paesani F, DeMott PJ, Molinero V (2017) Ice nucleation efficiency of hydroxylated organic surfaces is controlled by their structural fluctuations and mismatch to ice. J Am Chem Soc 139:3052–3064. https://doi.org/10.1021/jacs.6b12210
Qiu Y, Hudait A, Molinero V (2019) How size and aggregation of ice-binding proteins control their ice nucleation efficiency. J Am Chem Soc 141:7439–7452. https://doi.org/10.1021/jacs.9b01854
Rafsanjani A, Stiefel M, Jefimovs K, Mokso R, Derome D, Carmeliet J (2014) Hygroscopic swelling and shrinkage of latewood cell wall micropillars reveal ultrastructural anisotropy. J R Soc Interface. https://doi.org/10.1098/rsif.2014.0126
Ray J, Manning GS (1994) An attractive force between two rodlike polyions mediated by the sharing of condensed counterions. Langmuir 10:2450–2461. https://doi.org/10.1021/la00019a071
Rego NB, Patel AJ (2022) Understanding hydrophobic effects: insights from water density fluctuations. Ann Rev Condens Matter Phys 13:303–324. https://doi.org/10.1146/annurev-conmatphys-040220-045516
Reza M, Ruokolainen J, Vuorinen T (2014) Out-of-plane orientation of cellulose elementary fibrils on spruce tracheid wall based on imaging with high-resolution transmission electron microscopy. Planta 240:565–573. https://doi.org/10.1007/s00425-014-2107-1
Salmen L (2022) On the organization of hemicelluloses in the wood cell wall. Cellulose 29:1349–1355. https://doi.org/10.1007/s10570-022-04425-9
Salmen L, Olsson A-M (2016) Physical properties of cellulosic materials related to moisture changes. Wood Sci Technol 50:81–89. https://doi.org/10.1007/s00226-015-0777-x
Salmen L, Stevanic JS, Holmqvist C, Yu S (2021) Moisture induced straining of the cellulosic microfibril. Cellulose 28:3347–3357. https://doi.org/10.1007/s10570-021-03712-1
Sawada D, Nishiyama Y, Langan P, Forsyth VT, Kimura S, Wada M (2012) Water in crystalline fibers of dihydrate beta-chitin results in unexpected absence of intramolecular hydrogen bonding. PLoS ONE. https://doi.org/10.1371/journal.pone.0039376
Sawada D, Nishiyama Y, Roder T, Porcar L, Zahra H, Trogen M, Sixta H, Hummel M (2021) Process-dependent nanostructures of regenerated cellulose fibres revealed by small angle neutron scattering. Polymer. https://doi.org/10.1016/j.polymer.2021.123510
Sell J, Zimmermann T (1993) Radial fibril agglomerations of the S2 on transverse fracture surfaces of the tracheid of tension-loaded spruce and white fir. Holz Als Roh-Und Werkstoff 51:384–384. https://doi.org/10.1007/bf02628234
Shekar SC, Zhao W, Fernando LD, Hung I, Wang T (2022) A 13C three-dimensional DQ-SQ-SQ correlation experiment for high-resolution analysis of complex carbohydrates using solid-state NMR. J Magn Reson 336:107148–107148. https://doi.org/10.1016/j.jmr.2022.107148
Simmons TJ, Mortimer JC, Bernardinelli OD, Poppler A-C, Brown SP, DeAzevedo ER, Dupree R, Dupree P (2016) Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nat Commun 7:13902. https://doi.org/10.1038/ncomms13902
Sirotkin VA, Komissarov IA, Khadiullina AV (2012) Hydration of proteins: excess partial volumes of water and proteins. J Phys Chem B 116:4098–4105. https://doi.org/10.1021/jp300726p
Soni A, Patey GN (2021) How microscopic features of mineral surfaces critically influence heterogeneous ice nucleation. J Phys Chem C 125:10723–10737. https://doi.org/10.1021/acs.jpcc.1c01740
Sprenger K, Roeters SJ, Mauri S, Mertig R, Nishiyama Y, Pfaendtner J, Weidner T (2021) Direct evidence for aligned binding of cellulase enzymes to cellulose surfaces. J Phys Chem Lett 12:10684–10688. https://doi.org/10.1021/acs.jpclett.1c02757
Sturcova A, His I, Apperley DC, Sugiyama J, Jarvis MC (2004) Structural details of crystalline cellulose from higher plants. Biomacromol 5:1333–1339. https://doi.org/10.1021/bm034517p
Takahashi D, Willick IR, Kasuga J, Livingston DP (2021) Responses of the plant cell wall to sub-zero temperatures: a brief update. Plant Cell Physiol 62:1858–1866. https://doi.org/10.1093/pcp/pcab103
Terashima N, Kitano K, Kojima M, Yoshida M, Yamamoto H, Westermark U (2009) Nanostructural assembly of cellulose, hemicellulose, and lignin in the middle layer of secondary wall of Ginkgo tracheid. J Wood Sci 55:409–416. https://doi.org/10.1007/s10086-009-1049-x
Terrett OM, Dupree P (2019) Covalent interactions between lignin and hemicelluloses in plant secondary cell walls. Curr Opin Biotechnol 56:97–104. https://doi.org/10.1016/j.copbio.2018.10.010
Terrett OM, Lyczakowski JJ, Yu L, Iuga D, Franks WT, Brown SP, Dupree R, Dupree P (2019) Molecular architecture of softwood revealed by solid-state NMR. Nat Commun 10:4978–4978. https://doi.org/10.1038/s41467-019-12979-9
Thomas LH, Altaner CM, Jarvis MC (2013a) Identifying multiple forms of lateral disorder in cellulose fibres. J Appl Crystallogr 46:972–979. https://doi.org/10.1107/s002188981301056x
Thomas LH, Forsyth VT, Sturcova A, Kennedy CJ, May RP, Altaner CM, Apperley DC, Wess TJ, Jarvis MC (2013b) Structure of cellulose microfibrils in primary cell walls from collenchyma. Plant Physiol 161:465–476. https://doi.org/10.1104/pp.112.206359
Thomas LH, Forsyth VT, Martel A, Grillo I, Altaner CM, Jarvis MC (2014) Structure and spacing of cellulose microfibrils in woody cell walls of dicots. Cellulose 21:3887–3895. https://doi.org/10.1007/s10570-014-0431-z
Thomas LH, Forsyth VT, Martel A, Grillo I, Altaner CM, Jarvis MC (2015) Diffraction evidence for the structure of cellulose microfibrils in bamboo, a model for grass and cereal celluloses. BMC Plant Biol. https://doi.org/10.1186/s12870-015-0538-x
Thomas LH, Martel A, Grillo I, Jarvis MC (2020) Hemicellulose binding and the spacing of cellulose microfibrils in spruce wood. Cellulose 27:4249–4254. https://doi.org/10.1007/s10570-020-03091-z
Thomas LH, Altaner CM, Forsyth VT, Mossou E, Kennedy CJ, Martel A, Jarvis MC (2021) Nanostructural deformation of high-stiffness spruce wood under tension. Sci Rep 11:453. https://doi.org/10.1038/s41598-020-79676-2
Thybring EE, Kymalainen M, Rautkari L (2018) Experimental techniques for characterising water in wood covering the range from dry to fully water-saturated. Wood Sci Technol 52:297–329. https://doi.org/10.1007/s00226-017-0977-7
Thybring EE, Piqueras S, Tarmian A, Burgert I (2020) Water accessibility to hydroxyls confined in solid wood cell walls. Cellulose 27:5617–5627. https://doi.org/10.1007/s10570-020-03182-x
Thybring EE, Boardman CR, Zelinka SL, Glass SV (2021) Common sorption isotherm models are not physically valid for water in wood. Colloids Surf A-Physicochem Eng Asp 627:127214. https://doi.org/10.1016/j.colsurfa.2021.127214
Topgaard D, Soderman O (2002) Changes of cellulose fiber wall structure during drying investigated using NMR self-diffusion and relaxation experiments. Cellulose 9:139–147. https://doi.org/10.1023/a:1020158524621
Trentin LN, Pereira CS, Silveira RL, Hill S, Sorieul M, Skaf MS (2021) Nanoscale wetting of crystalline cellulose. Biomacromol 22:4251–4261. https://doi.org/10.1021/acs.biomac.1c00801
Vignon MR, Gey C (1998) Isolation, H-1 and C-13 NMR studies of (4-O-methyl-D-glucurono)-D-xylans from luffa fruit fibres, jute bast fibres and mucilage of quince tree seeds. Carbohyd Res 307:107–111. https://doi.org/10.1016/s0008-6215(98)00002-0
Voets IK (2017) From ice-binding proteins to bio-inspired antifreeze materials. Soft Matter 13:4808–4823. https://doi.org/10.1039/c6sm02867e
Wang T, Zabotina O, Hong M (2012) Pectin-cellulose interactions in the Arabidopsis primary cell wall from two-dimensional magic-angle-spinning solid-state nuclear magnetic resonance. Biochemistry 51:9846–9856. https://doi.org/10.1021/bi3015532
Wang T, Park YB, Caporini MA, Rosay M, Zhong L, Cosgrove DJ, Hong M (2013) Sensitivity-enhanced solid-state NMR detection of expansin’s target in plant cell walls. Proc Natl Acad Sci USA 110:16444–16449. https://doi.org/10.1073/pnas.1316290110
Wang T, Salazar A, Zabotina OA, Hong M (2014) Structure and dynamics of Brachypodium primary cell wall polysaccharides from two-dimensional C-13 solid-state nuclear magnetic resonance spectroscopy. Biochemistry 53:2840–2854. https://doi.org/10.1021/bi500231b
Wang T, Park YB, Cosgrove DJ, Hong M (2015) Cellulose-pectin spatial contacts are inherent to never-dried Arabidopsis primary cell walls: evidence from solid-state nuclear magnetic resonance. Plant Physiol 168:871–884. https://doi.org/10.1104/pp.15.00665
Wang T, Phyo P, Hong M (2016a) Multidimensional solid-state NMR spectroscopy of plant cell walls. Solid State Nucl Magn Reson 78:56–63. https://doi.org/10.1016/j.ssnmr.2016.08.001
Wang T, Yang H, Kubicki JD, Hong M (2016b) Cellulose structural polymorphism in plant primary cell walls investigated by high-field 2D solid-state NMR spectroscopy and density functional theory calculations. Biomacromol 17:2210–2222. https://doi.org/10.1021/acs.biomac.6b00441
Wang Y, Kiziltas A, Drews AR, Tamrakar S, Blanchard P, Walsh TR (2021) Dynamical water ingress and dissolution at the amorphous-crystalline cellulose interface. Biomacromol 22:3884–3891. https://doi.org/10.1021/acs.biomac.1c00690
White PB, Wang T, Park YB, Cosgrove DJ, Hong M (2014) Water-polysaccharide interactions in the primary cell wall of Arabidopsis thaliana from polarization transfer solid-state NMR. J Am Chem Soc 136:10399–10409. https://doi.org/10.1021/ja504108h
Willems W (2014) Hydrostatic pressure and temperature dependence of wood moisture sorption isotherms. Wood Sci Technol 48:483–498. https://doi.org/10.1007/s00226-014-0616-5
Willems W (2016) Equilibrium thermodynamics of wood moisture revisited: presentation of a simplified theory. Holzforschung 70:963–970. https://doi.org/10.1515/hf-2015-0251
Willems W (2018) Hygroscopic wood moisture: single and dimerized water molecules at hydroxyl-pair sites? Wood Sci Technol 52:777–791. https://doi.org/10.1007/s00226-018-0998-x
Wohlert M, Benselfelt T, Wagberg L, Furo I, Berglund LA, Wohlert J (2022) Cellulose and the role of hydrogen bonds: not in charge of everything. Cellulose 29:1–23. https://doi.org/10.1007/s10570-021-04325-4
Ye D, Kiemle SN, Rongpipi S, Wang X, Wang C, Cosgrove DJ, Gomez EW, Gomez ED (2018) Resonant soft X-ray scattering reveals cellulose microfibril spacing in plant primary cell walls. Sci Rep 8:12449. https://doi.org/10.1038/s41598-018-31024-1
Zabler S, Paris O, Burgert I, Fratzl P (2010) Moisture changes in the plant cell wall force cellulose crystallites to deform. J Struct Biol 171:133–141. https://doi.org/10.1016/j.jsb.2010.04.013
Zhang T, Mahgsoudy-Louyeh S, Tittmann B, Cosgrove DJ (2014) Visualization of the nanoscale pattern of recently-deposited cellulose microfibrils and matrix materials in never-dried primary walls of the onion epidermis. Cellulose 21:853–862. https://doi.org/10.1007/s10570-013-9996-1
Zhang T, Zheng YZ, Cosgrove DJ (2016) Spatial organization of cellulose microfibrils and matrix polysaccharides in primary plant cell walls as imaged by multichannel atomic force microscopy. Plant J 85:179–192. https://doi.org/10.1111/tpj.13102
Zhang T, Tang H, Vavylonis D, Cosgrove DJ (2019) Disentangling loosening from softening: insights into primary cell wall structure. Plant J 100:1101–1117. https://doi.org/10.1111/tpj.14519
Zhang C, Coasne B, Guyer R, Derome D, Carmeliet J (2020) Moisture-induced crossover in the thermodynamic and mechanical response of hydrophilic biopolymer. Cellulose 27:89–99. https://doi.org/10.1007/s10570-019-02808-z
Zhang C, Keten S, Derome D, Carmeliet J (2021) Hydrogen bonds dominated frictional stick-slip of cellulose nanocrystals. Carbohyd Polym 258:117682. https://doi.org/10.1016/j.carbpol.2021.117682
Zhao W, Fernando LD, Kirui A, Deligey F, Wang T (2020) Solid-state NMR of plant and fungal cell walls: A critical review. Solid State Nucl Magn Reson 107:101660. https://doi.org/10.1016/j.ssnmr.2020.101660
Zhao W, Kirui A, Deligey F, Mentink-Vigier F, Zhou Y, Zhang B, Wang T (2021) Solid-state NMR of unlabeled plant cell walls: high-resolution structural analysis without isotopic enrichment. Biotechnol Biofuels 14:14. https://doi.org/10.1186/s13068-020-01858-x
Zitting A, Paajanen A, Rautkari L, Penttila PA (2021) Deswelling of microfibril bundles in drying wood studied by small-angle neutron scattering and molecular dynamics. Cellulose 28:10765–10776. https://doi.org/10.1007/s10570-021-04204-y
Acknowledgments
The author thanks many colleagues, especially C. Altaner, for informative discussions.
Funding
No grants or other financial support were received during the preparation of this review.
Author information
Authors and Affiliations
Contributions
Not applicable.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The author has no competing interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jarvis, M.C. Hydrogen bonding and other non-covalent interactions at the surfaces of cellulose microfibrils. Cellulose 30, 667–687 (2023). https://doi.org/10.1007/s10570-022-04954-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-022-04954-3