
Abstract
To commemorate the auspicious occasion of the 30th anniversary of IPC, leading pioneers in the field of cardioprotection gathered in Barcelona in May 2016 to review and discuss the history of IPC, its evolution to IPost and RIC, myocardial reperfusion injury as a therapeutic target, and future targets and strategies for cardioprotection. This article provides an overview of the major topics discussed at this special meeting and underscores the huge importance and impact, the discovery of IPC has made in the field of cardiovascular research.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The year 2016 marks the 30th anniversary since Murry, Jennings and Reimer first discovered the phenomenon of ischaemic preconditioning (IPC) [180]. The seminal discovery in 1986, that brief episodes of ischaemia and reperfusion could dramatically reduce myocardial infarct (MI) size, gave rise to the field of cardioprotection, and has resulted in over 10,000 publications in the research literature. Over the last 30 years enormous efforts have been made to understand the mechanisms underlying IPC and have provided huge insights into the mechanisms of cardiomyocyte death during acute ischaemia/reperfusion injury (IRI), and the complex signalling pathways underlying cytoprotection within the cardiomyocyte and beyond. In addition, the last 30 years have witnessed enormous efforts to translate this endogenous cardioprotective strategy into the clinical setting for patient benefit. In this regard, the evolution of IPC to an intervention which could be applied at the time of reperfusion [ischaemic postconditioning (IPost)] [276] and to a remote organ or tissue [remote ischaemic conditioning (RIC)] [200] has facilitated the translation of IPC into the clinical setting.
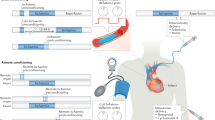
To commemorate the auspicious occasion of the 30th anniversary of IPC, leading pioneers in the field of cardioprotection gathered in Barcelona in May 2016 to review and discuss the history of IPC (Fig. 1), its evolution to IPost and RIC, myocardial reperfusion injury as a therapeutic target, and future targets and strategies for cardioprotection. This article provides an overview of the major topics discussed at this special meeting and underscores the huge importance and impact, the discovery of IPC has made in the field of cardiovascular research.

Faculty photo at the 30 year anniversary celebration of IPC in Barcelona May 2016: “Ischaemic conditioning and targeting reperfusion injury: a 30 year voyage of discovery”. Back row, left to right Michael Rahbek Schmidt, Peter Ferdinandy, Hans Erik Bøtker, Rajesh Kharbanda, Michael Marber, Pasquale Pagliaro, Thomas Engstrom, Karin Przyklenk, Tetsuji Miura, Hector A. Carbrera-Fuentes, Sandine Lecour, Derek Hausenloy, Derek Yellon, Borja Ibanez, Rainer Schulz, Gerd Heusch, Hans Michael Piper, Efstathios Iliodromitis, Miguel A Perez-Pinzon, Gemma Vilahur, Marisol Ruiz-Meana. Front row, left to right David Garcia-Dorado, Javier Inserte, Jose Barrabes, Robert Jennings, Jakob Vinten-Johansen, Andrew Redington, Michel Ovize, Fabio Di Lisa, James Downey
Ischaemic preconditioning
In IPC, several minutes of acute coronary occlusion followed by reperfusion delay the onset of MI from a subsequent period of prolonged lethal ischaemia and reperfusion. The description of IPC 30 years ago in 1986 by Murry et al. [180] was a landmark discovery. It proved once and for all that the final size of a MI was not only a function of the area-at-risk (AAR), ischaemic time and collateral flow, but could indeed be reduced, as had been originally proposed by Braunwald and colleagues years before [165]. The Jennings laboratory was pursuing the observation that a brief ischaemic episode slowed the rate of ATP consumption when the heart was subjected to subsequent episodes of ischaemia. Since virtually no ATP is present in dead cardiomyocytes, they hypothesised that delaying ATP depletion would attenuate the development of cardiomyocyte death [181].
Considering the huge number of papers eventually published on IPC since 1986, it is amazing that it took 4 years before the first confirmatory paper by another laboratory appeared on the subject [149]. However, after that virtually everyone who tried to replicate IPC was able to observe protection that lasted for several hours [258]. In 1991, Liu et al. [153] showed that the preconditioned state resulted from protective signal transduction. Infusing adenosine or an adenosine A1 receptor-selective agonist into the coronary arteries for 5 min prior to occluding a coronary branch put the heart into a protected state identical to IPC. Conversely, an adenosine receptor antagonist completely blocked the IPC protection but had no effect on a non-IPC heart. A1 receptors are Gi-coupled and act to slow the heart rate as opposed to the Gs-coupled adenosine A2 receptors which act to dilate the coronary arteries. In fact it was shown that many of the Gi-coupled receptors in the heart can mimic IPC [40]. A brief coronary occlusion has been found to release ligands for only four of these receptors: adenosine, bradykinin, opioid, and sphingosine. These four receptors act in an additive fashion. Blocking a single receptor subtype only raises the ischaemic threshold for protection rather than abolishing the IPC response. Subsequent studies quickly showed that protein kinase C [155] and ATP-sensitive potassium channels (KATP) [5], which later turned out to be in the mitochondria [154] and could be stimulated by diazoxide (pharmacological preconditioning), were also in the IPC signalling pathway.
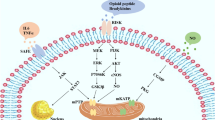
The overall signalling pathway is still not completely understood but extensive research in many laboratories has revealed much of it (Fig. 2) [29, 74, 101, 273]. In 2002, Yellon’s group [86, 94, 218] proposed the Reperfusion Injury Salvage Kinase or RISK Pathway to describe a group of pro-survival kinases that must be activated at the time of reperfusion for IPC to protect against MI. Since protection could be aborted by blocking the RISK pathway at reperfusion, IPC must, therefore, protect against a reperfusion injury. They also went on to demonstrate the importance of this pathway in all forms of the conditioning process, i.e. pre-, post-, remote and pharmacological conditioning [90]. It now appears that much of the cell death in the heart is due to the formation of permeability transition pores (PTPs) in the mitochondrial membranes in the first minutes of reperfusion, and IPC through the RISK signalling protects by suppressing these PTPs [97, 103]. Lecour et al. [146] subsequently identified the Salvage Activating Factor Enhancement (SAFE) pathway which is activated in parallel to the RISK pathway and appears to play a more important role in larger mammals [78, 108, 227, 229].

Figure modified from that appearing in [38]
A proposed map of some of the major signalling pathways involved in ischaemic pre- and postconditioning. The pink coloured boxes indicate pharmacological interventions that have been reported to reduce MI size when administered just prior to reperfusion. They are positioned near their proposed site of action.
The key to understanding IPC is to appreciate why the brief period of reperfusion after the preconditioning ischaemia is so important. The Gi receptor activation leads to opening of mitochondrial KATP channels during ischaemia and potassium entry into the mitochondria. When oxygen is reintroduced during the reperfusion phase of the IPC protocol, elevated mitochondrial potassium stimulates the mitochondria to produce reactive oxygen species (ROS). These ROS cause redox signalling which ultimately results in PKC activation and completion of the IPC signalling pathway [52]. In a non-conditioned heart this pathway is blocked at the redox signalling step during the prolonged ischaemic period as potassium has entered the mitochondria but there is no oxygen available. If the heart is reperfused after a prolonged ischaemic period PTPs will always open before redox signalling can activate the downstream pathway to inhibit them [37]. A large series of recent studies have demonstrated that connexin 43, the protein forming gap junction channels between cardiomyocytes, is also located at the inner mitochondrial membrane [15], where it can form hemi-channels that allow the passage of potassium [175], and that its absence at that location prevents ROS generation during the IPC stimulus [98], and abolishes cardioprotection [205]. There is recent evidence that IPC can protect mitochondria against respiratory inhibition induced by prolonged IRI independently of cytosolic signalling [215].
Although IPC is clearly protective, the need for its application before ischaemia makes it impractical for treating acute myocardial infarction (AMI). However, if the protective pathway can be rapidly activated by a drug administered at reperfusion (known as “pharmacological postconditioning”) then this can “win the race” and protect the heart against reperfusion injury. Because much of conditioning’s signal transduction pathway is now known [101], it has been possible to identify agents effective at inducing pharmacological postconditioning. Figure 2 shows a tentative map of the signalling pathways underlying IPC, and some known interventions that, at least in animal models, reportedly put the heart into a conditioned state.
Second window of protection
The second window of protection (SWOP) describes the increased resistance to myocardial injury that re-appears 12–24 h after the short durations of ischaemia/reperfusion that trigger classic or early preconditioning. This phenomenon was first described in 1993 by independent research groups based in London [164] and Osaka [140].
Yellon’s group in London had been interested in the cardiac protection that followed whole body heart stress—this was known to be associated with the induction of stress proteins and catalase within the myocardium [44]. However, whole body heat stress was associated with a multitude of changes both within and outside the heart. Using an experimental method that involved a support animal providing oxygenated blood to perfuse a donor animal’s isolated heart, evidence had been obtained that whilst whole body heat stress protected the heart, it also triggered extra-cardiac adaptations that aggravated myocardial injury [252]. This observation provided the impetus to find a way of spatially restricting the stress response to the heart, thereby avoiding the deleterious systemic adaptations associated with whole body heart stress. In 1991, Knowlton had observed cardiac stress protein induction beginning 6–8 h after short episodes of myocardial ischaemia [136]. Based on this observation, it was reasoned that sublethal myocardial ischaemia would induce stress proteins without causing the detrimental systemic response associated with whole body heat stress. It was on this basis that Marber et al. designed and performed the experiments that laid the foundation for the SWOP [164].
Following the original observations by Marber [164] and Kuzuya [139], there followed a number of basic science studies that indicated the SWOP had a duration of 72–96 h, and whilst the magnitude of protection may be less robust than that of the first window of protection, there were likely to be important clinical correlates [272].
Studies in patients have broadly fallen into two groups; observational studies, where symptoms and circumstance are related to outcome after spontaneous plaque rupture (type I MI) and interventional studies using controlled, iatrogenic myocardial ischaemia. In the observational studies, it was found that patients who experience repeated episodes of pre-infarction angina more than 24 h before the index event, may develop smaller final MI size than those without pre-infarction angina [102, 184]. Interpretation of these studies is complicated by the variation of each ischaemic episode in terms of its duration, intensity and exact timing before spontaneous coronary artery occlusion. Furthermore, most patients have co-morbidities and medications that have been shown to both facilitate and prevent the manifestation of protection. These uncontrollable variables may explain the discrepancies that appear in the literature regarding the benefit of pre-infarction angina. Consequently, the benefit of the SWOP is more easily demonstrated in interventional studies that use exercise treadmill tests, isotope scintigraphy or coronary angioplasty to cause and document myocardial ischaemia [14, 121, 143, 190].
In conclusion, the SWOP is clinically apparent under pre-specified controlled conditions but is impossible to identify with certainty in observational retrospective studies relevant to everyday clinical practice. Furthermore, the systematic changes in primary percutaneous coronary intervention (PPCI) services ensure early intervention with the surety of reperfusion further secured by advances in antiplatelet drugs and interventional devices [262]. On this background it is very difficult to demonstrate sufficient room for novel cardiac protection [28, 76, 99, 110, 262]. Furthermore, any benefit of SWOP may be subsumed by ischaemic pre- and/or post-conditioning. Nonetheless, one aspect of SWOP which may yet prove important is its potential to reveal novel protective proteins that may form the basis of future therapies.
Remote ischaemic conditioning
Remote ischaemic conditioning is the intriguing phenomenon, first reported by Przyklenk, Whittaker and colleagues [200, 260], that brief periods of ischaemia applied in a distant tissue can render the heart resistant to IRI and reduce MI size. Although first viewed as a specious finding [201], the concept of RIC-induced cardioprotection has, during the past two decades, been corroborated in multiple, diverse models (reviewed in [29, 93, 104, 193, 226]). A recent meta-analysis of experimental studies in RIC found that RIPC reduced MI size as a percentage of AAR by 22.8 %, when compared to untreated controls, and RIPerC/RIPostC reduced MI size by 22.2 % [26]. Moreover, two priorities have emerged: (1) identification of the mechanisms responsible for the infarct-sparing effect of RIC; and (2) translation of RIC to patient cohorts.
The molecular mechanisms contributing to RIC are, without question, complex and remain incompletely resolved [30–32, 93, 101, 104, 193, 226]. In brief, the current hypothesis is that RIC induces a neuro-humoral response which, in turn, induces a cascade of downstream effects. Evidence for a neural component of RIC comes from early observations that pretreatment of animals with the ganglion blocker hexamethonium abolishes the cardioprotective effect of transient mesenteric ischaemia [68], and subsequent studies showing that transection of the ipsilateral femoral nerve [152, 235], or bilateral cervical vagotomy [50], abolishes cardioprotection by RIC induced by limb ischaemia. Conversely, direct stimulation of the femoral nerve [204] or sensory nerves [169] within the limb have been shown to induce cardioprotection. However, there are also controversial data on neuronal involvement in RIC, as hexamethonium [257] or nerve transection [203] did not abrogate cardioprotection. The consequence of any neural stimulus, whether local to the limb [221] or of the cardiac ganglia [194], is the release of dialysable cardioprotective substances into the blood [129]. These include the chemokine SDF-1α [47], Ribonuclease-1 [30], leukotrienes [224], and microRNA 144 [150]. The exact mechanism by which any of these putative effectors are released, and their relative importance remains to be fully understood. Yet, the ultimate effect at the cardiomyocyte level is to induce a protective kinase response [151, 227], and modification of PTP opening [247], similar to that observed with local preconditioning and postconditioning. Unlike local preconditioning and postconditioning, RIC appears to have additional pleiotropic effects that modify pathways involved in the acute and chronic responses to IRI and may contribute to its benefits, including improved vascular endothelial function [158], decreased platelet aggregation [10, 191], and a significant anti-inflammatory effect manifest early by decreased neutrophil adhesion [220], and later by reduced inflammatory cell infiltration, reduced local inflammation [30] and reduced remodelling in the weeks after experimental MI [256].
In the clinical setting, remote ischaemic preconditioning (RIPC) has been administered prior to IRI as three or four cycles of 5 min ischaemia followed by 5 min reperfusion of the upper, or less frequently, lower limb in cardiac and vascular surgery, and elective and emergency angioplasty. The majority of studies in coronary artery bypass graft (CABG) surgery patients have shown reduction of post-operative cardiac biomarker release [2, 33, 35, 84, 138, 239, 240, 250] while others did not [69, 131, 159, 167, 202, 275]. One randomized study of 329 CABG patients demonstrated simultaneous reduction of troponin I release and reduction of all-cause mortality up to 4 years following the operation [91]. In contrast, a randomised study of 1280 patients undergoing off-pump coronary artery bypass graft surgery showed no effect of RIPC before and after the surgery on a comprehensive composite endpoint [113]. Two more recent studies, the ERRICA and the RIPHeart studies, also failed to demonstrate any beneficial effect on major adverse cardiac and cerebral events (MACCE) after 12 months and event free survival after 3 months, respectively [77, 171]. Studies that failed to demonstrate a beneficial effect of RIPC used propofol as an anaesthetic regimen. Similar experiences have been obtained in major non-cardiac surgery [3, 254]. A specific effect of propofol that interacts with neuronal transfer of the protective RIPC signal may interfere with the inherent cardioprotective effect of propofol and further protection by RC [137, 138].
Invasive coronary procedures circumvent any influence from anaesthetics. In this setting, RIPC attenuated the release of ischaemic markers in the majority of studies including patients undergoing elective percutaneous coronary intervention (PCI) [114, 162, 198, 265, 277], and translated into a prognostic benefit in terms of MACCE at follow-up period of up to 6 years [49]. Whilst RIPC can be used in predictable ischaemia, another temporal variant is necessary in unpredictable ischaemia such as ST-segment elevation myocardial infarction (STEMI). Remote ischaemic perconditioning (RIPerC) [217], in which the RIC intervention is applied during evolving MI prior to PPCI, has consistently yielded cardioprotection in proof-of-concept studies using a variety of outcome measures including myocardial salvage, ST-segment resolution and biomarker release (Table 1) [25, 42, 54, 179, 199, 205, 259, 271]. The reduction of MI size translated into a reduction of MACCE [230] and was cost-effective [231] over a 4-year period following the index infarct. This study included 333 patients and was not powered for clinical outcomes. The ongoing CONDI-2/ERIC-PPCI study including 4300 patients will determine the clinical benefit of RIPerC as an adjunct to PPCI in patients with STEMI [79].
Ischaemic postconditioning
Emergence of the concept of ischaemic postconditioning (IPost) was based on four points: (a) myocardial reperfusion injury was not a laboratory curiosity but an pathophysiological entity that exacerbated tissue injury (whether de novo or extending pre-existing injury) after onset of reflow; (b) lethal myocardial reperfusion injury was initiated quickly after the onset of reperfusion; (c) tissue destined to die in the path of the reperfusion injury “wave front” after onset of reflow could be salvaged; (d) reperfusion injury pathology could be avoided or prevented by altering how the ischaemic tissue was reperfused. The latter point was expanded to include modifying the conditions and composition of the reperfusate, including the inclusion of drugs during early reperfusion. Despite considerable controversy over the very existence and clinical importance of myocardial reperfusion injury, there is now compelling evidence that reperfusion contributes to the extent of transient as well as permanent (lethal) post-ischaemic injury to the myocardium [27, 65, 73, 251, 274], and that this injury was initiated within moments after onset of reflow [246]. Early reports of the protective effects of gradual or gentle reperfusion (modified conditions of reperfusion) in the early moments of reperfusion [22, 115, 216] did not capture the attention of the scientific or clinical communities. Although initial trials on IPost were performed in 1992, results were negative due to (a) excessively long durations of the reperfusion-re-occlusion cycles (5 min emulating preconditioning cycles), and (b) a single cycle rather than multiple cycles; studies resumed eight years later using shorter cycle durations in a large animal model which successfully reduced MI size, coronary artery endothelial dysfunction, oedema in the AAR, and apoptosis [276].
Studies confirming and extending the original results were published quickly by independent laboratories [63, 245, 270] as well as by Vinten-Johansen’s laboratory [71, 135]. Kin et al. [135] showed that the cardioprotective effects of IPost were not observed if the manoeuver was delayed by 60 s, confirming that a IPost window opened in the first few minutes of reperfusion which was critical to protection. This IPost window was confirmed by Yang et al. [270] and implied that reperfusion injury interventions should be implemented immediately at or before the onset of reperfusion. There is scant evidence that delayed postconditioning is effective in reducing post-ischaemic injury [209].
IPost has been shown to reduce abnormal alterations in a multitude of end points associated with post-ischaemic injury. These include reduction of (1) MI size and possibly the no-reflow area, (2) apoptosis, (3) interstitial and intracellular oedema, (4) early post-ischaemic arrhythmias, (5) the pro-inflammatory response to reperfusion, (6) explosive (injurious) ROS generation by multiple cell types, and (7) the incidence of heart failure. Whether IPost attenuates transient (stunning) or permanent contractile dysfunction globally or regionally is controversial. In addition, the cardioprotection of IPost may be lost in the presence of comorbidities (diabetes, hypertension, and hypercholesterolemia) or co-medications (such as P2Y12 inhibitors), in older individuals [19, 56, 225]. Although these data may imply limited efficacy in patients that present with isolated or the constellation of comorbidities in the metabolic syndrome, it must be said that efficacy has been shown in patients that present for PCI with these demographics [192].
The mechanisms by which these physiological responses to reperfusion are attenuated include (1) inhibiting PTP opening [4, 85, 103], (2) delaying rapid myocardial re-alkalinisation that, in part, contributes to PTP opening [39], (3) reducing intracellular and intra-mitochondrial calcium accumulation [117], (4) reduced oxidative damage of eNOS and preserved cGMP signalling [125], (5) attenuating endothelial dysfunction (expression of adhesion molecules [276], production of NOˑ and other vasoactive and cardioprotective autacoids such as adenosine) that otherwise trigger the vascular inflammatory response to reperfusion injury, and (6) reducing pro-inflammatory cell activation and expression of cytokines in blood that contribute to the inflammatory response to reperfusion injury [31, 32]. A year after its introduction, Tsang et al. [245] reported that IPost activated the reperfusion injury salvage kinase (RISK) pathway pro-survival kinases PI3K-Akt and downstream targets eNOS and p70S6K (see later section).
Unlike preconditioning whose clinical application is limited by the unpredictability of AMI, IPost immediately caught the attention of interventional cardiologists. In 2005, Staat et al. [234] reported that four episodes of 1-min inflation–deflation cycles of the angioplasty balloon performed immediately after coronary artery re-opening were able to significantly reduce MI size in STEMI patients. This was the first report demonstrating that reperfusion injury exists in man, is of pathophysiological importance, and can be attenuated by a timely intervention. Most [133, 238, 241], but not all [132], clinical studies in patients with these conditions undergoing PCI or cardiac surgery have shown positive outcomes with IPost (reviewed in Heusch [99, 101, 110]). Reasons for such discrepancy are unclear but might include a different use of thrombus aspiration, direct stenting, in-stent balloon inflation–deflation for inducing IPost, as well as the confounding role of new adjunct therapies like P2Y12 inhibitors. The recent phase 3 DANAMI-3 IPOST study (NCT01435408) [111] reported that 4 cycles of 30 s IPost failed to improve clinical outcomes in STEMI patients, but this study used a sub-optimal IPost algorithm and was probably underpowered. Additional studies are awaited to clarify whether or not MI size reduction observed in phase 2 IPost trials can actually provide any clinical benefit to STEMI patients.
Whether IPost is cardioprotective in the presence of comorbidities such as diabetes or hypercholesterolaemia, or in the presence of co-medications, or wanes with age [19] is still controversial [56, 192]. In addition, whether the efficacy of IPost is masked when other forms of cardioprotection are used, such as IPC, P2Y12 inhibitors, or hypothermia and cardioplegia in cardiac surgery, is still unresolved.
Studies should continue to unravel the numerous and interacting mechanisms involved in IPost, and how they relate to other types of conditioning (preconditioning, perconditioning). In addition to clarifying the mechanisms of IPost, these studies may lead to the development of broad spectrum drugs with multiple therapeutic targets emulating IPost’s broad spectrum therapeutic profile. Potential loss of cardioprotection in comorbid circumstances should be further investigated in large animal models with genetic predispositions to the comorbidity spectrum, such as the Ossabaw pig with genetic metabolic syndrome. Similar studies need to define whether efficacy of IPost is lost with advancing age. Studies in large animal models should re-examine the “ischaemic wavefront” to separate the temporal progression of myocardial injury after ischaemia only (without reperfusion) and after ischaemia plus reperfusion to redefine the extent of injury in patients arriving at the catheter laboratory with unresolved occlusions. More clinical studies need to be performed that embrace the design features of randomisation and adequate statistical power that avoids Type II errors, and allows stratification of patients into various subgroups to differentiate responders from non-responders. A combination of different protective interventions, including remote, per- and postconditioning as well as cocktails of drugs may be tested in the future.
Myocardial reperfusion injury
Reperfusion Injury has many facets. Apart from reversible forms of reperfusion injury, including reperfusion arrhythmias and stunning, there is also lethal reperfusion injury, or cell death, occurring at the time of reperfusion, and thus preventable by treatments applied at the time of restoration of blood flow [196]. There is extremely solid evidence of the existence of lethal reperfusion injury in experimental MI models. There is also solid evidence of the occurrence of reperfusion injury in patients with STEMI, although several interventions to reduce lethal reperfusion injury in patients have failed or provided inconsistent results in this clinical setting [76, 106, 110]. The reasons for these failures are more likely dependent on the particular treatments applied or on associated circumstances (age, comorbidities, treatment received) than to inter-species differences. Over the past 30 years, the importance and mechanisms of cardiomyocyte cell death in myocardial reperfusion injury have been elucidated in part. Altered Ca2+ handling and PTP opening have been identified to be complementary pathways of reperfusion-induced cell death, but important questions remain unsolved [118].
Cardiomyocyte death is the main cause of heart failure, arrhythmias and death in patients with STEMI, and depends largely on phenomena occurring within cardiomyocytes themselves, as shown by the fact that it can be recapitulated in isolated cardiomyocytes submitted to transient ischaemia [118], but other cells can contribute, in particular, platelets [8, 9, 174]. Endothelial cells, in which metabolism is largely independent of mitochondrial respiration [163] are more tolerant of ischaemia than cardiomyocytes.
A substantial component of reperfusion-induced cell death occurs during the initial minutes of reperfusion [91]. Apoptosis plays little, if any, role in reperfusion-induced cardiomyocyte cell death [168] and selective lack of expression of executioner caspases 3 or 7, does not modify MI size or post-MI remodeling in mice [122]. Severe ischaemia stops mitochondrial respiration, progressively dissipates mitochondrial membrane potential, and ATP concentration reaches very low levels and triggers rigor contracture [188]. ATP hydrolysis secondary to reversal of respiratory Complex V (ATP synthase) plays an important role [59]. The inactivity of the Na+ ATPase pump leads to Na+ and Ca2+ overload through reverse Na+/Ca2+ exchange [196]. Anaerobic metabolism in combination with reduced catabolite washout causes intracellular acidosis, reaching pH 6.4 within a few minutes. Reperfusion results in the rapid restoration of energy availability [59] and intracellular pH [126], and generation of large amounts of reactive oxygen species (ROS) and additional Ca2+ influx [64].
Altered Ca2+ handling is a key factor in reperfusion injury-cardiomyocyte cell death. Na+ concentration may increase in reperfused cardiomyocytes due to Na+ influx associated with pH normalization and passage of Na+ from adjacent cells via gap junctions favoured by impaired Na+ pump function [213], and Na+ influx favours Ca2+ influx through the Na+/Ca2+ exchanger [164]. Increased Ca2+ and pH normalisation causes calpain activation [124] resulting in damage of the subsarcolemmal cytoskeleton leading to Na+ pump dysfunction. Restoration of ATP availability during initial reperfusion leads to Ca2+ uptake into the sarcoplasmic reticulum (SR) followed, when Ca2+ capacity is exceeded, by Ca2+ release through the Ryanodine receptor channel (RyR2) resulting in oscillations of Ca2+ concentration that propagates across the cell favouring arrhythmias, hypercontracture and mitochondrial Ca2+ overload [210]. Hypercontracture can cause cell death, and transient contractile inhibition during the initial minutes of reperfusion prevents cardiomyocyte death in isolated cardiomyocytes, isolated hearts and intact large animals [66, 222]. Reperfusion-generated ROS cause nitric oxide synthase (NOS) oxidation and reduced NO-cGMP-PKG signaling. PKG modulates phospholamban (PLB) phosphorylation, SR Ca2+ uptake, and Ca2+ oscillations, and treatments normalizing PKG signaling in reperfused myocardium limit MI size in a number of pre-clinical and clinical studies [123].
Resumption of respiration is associated with increased ROS generation, due in part to re-oxidation of succinate accumulated during ischaemia and reverse electron transport between complex II and complex I of the respiratory chain [36] and to restoration of mitochondrial potential favoring mitochondrial Ca2+ uptake through the Ca2+ uniporter and Ca2+ overload [64]. Studies in mitochondrial preparations and cells show that mitochondrial Ca2+ and ROS may trigger an abrupt increase in the permeability of the inner mitochondrial membrane resulting in release of molecules from the mitochondrial matrix into the cytosol, mitochondrial depolarisation and swelling [85, 185, 186]. Mitochondrial permeability transition is supposed to be due to the opening of the PTP, a proposed large conductance cannel in the inner mitochondrial whose molecular structure is not really clear [103], except for the involvement of cyclophilin D and more recently ATP synthase. PTP opening is inhibited by low pH and favored by ROS and low ATP concentration, conditions occurring during myocardial reperfusion [72].
Mitochondrial permeability transition and Ca2+ oscillations/hypercontracture are closely related cell death pathways. This is partly due to the tight physical connection between SR and mitochondria allowing preferential Ca2+ exchange between both organelles [212]. Ca2+ release from mitochondria secondary to PTP opening may cause hypercontracture in Ca2+ overloaded cardiomyocytes [211] while SR-driven Ca2+ oscillations may cause PTP opening [210]. The relative importance of these two pathways may depend on conditions such as the severity of the ischaemic insult [214].
Opening of the PTP has been well documented in mitochondrial preparations exposed to very high Ca2+ concentrations and isolated cardiomyocytes subjected to simulated IRI [72]. The most important evidence for the role of PTP in reperfusion injury is the reduced MI size associated with genetic ablation of cyclophilin D [6, 75, 81, 183]. Inhibition of cyclophilin D with cyclosporine-A (CsA) prevents PTP opening in mitochondrial preparations but reductions in MI size with this agent have not been consistent, particularly when applied exclusively at the time of reperfusion or in large animals [130, 228], and a positive proof-of-concept trial in patients with STEMI [195] was not confirmed in a larger phase III trial [43]. Furthermore, all other drugs aimed at PTP inhibition in STEMI patients have so far failed. Although disappointing, these results are more likely explained by ineffective PTP inhibition by CsA, rather than the PTP not being important in human reperfusion injury.
Reperfusion signalling via the RISK and SAFE pathway
It is now well established that ischaemic conditioning protects the heart from acute IRI through the activation of signal transduction pathways recruited at the onset of reperfusion. These signalling cascades mediate the cardioprotective signal elicited by ischaemic conditioning from the sarcolemma to the mitochondria and include, amongst others, the reperfusion injury salvage kinase (RISK), and the survivor activating factor enhancement (SAFE) pathways (reviewed in [80, 92, 94, 145, 146]).
The RISK pathway refers to the pro-survival kinases, Akt and Erk1/2, the activation of which at the onset of reperfusion reduces MI size [92, 94]. It was first described by Yellon and colleagues in 2002 while studying the signalling mechanisms underlying the cardioprotective effect induced by the growth factor, urocortin [218]. In that study the administration of urocortin specifically at the time of myocardial reperfusion reduced MI size and increased the phosphorylation of myocardial Erk1/2, the effects of which were abrogated by the co-administration of the pharmacological MEK1/2-Erk1/2 inhibitor, PD98059, at the time of reperfusion [218]. A large number of experimental studies have linked the activation of the RISK pathway to the cardioprotection induced by a diverse variety of pharmacological agents including growth factors, cytokines, and other agents such as metformin and statins [92, 94]. The RISK pathway has also been shown to mediate the cardioprotection induced by IPC and IPost, suggesting that it may be a common pathway for cardioprotection [86, 87, 245]. Most of the experimental studies implicating the RISK pathway as a cardioprotective pathway have been performed in small rodent models of AMI, whereas recent studies suggest that the RISK pathway does not appear to mediate the cardioprotection induced by IPost [132, 229], gentle reperfusion [182] or RIC [1, 227] in large animal models, suggesting species differences in the reperfusion signalling pathways underlying ischaemic conditioning.
In 2005, Lecour and colleagues made the unexpected observation that the MI-limiting effects of TNF-α at the onset of reperfusion were mediated independently of the RISK pathway [147, 148]. They subsequently discovered, that TNF-α administered at the onset of myocardial reperfusion recruited an alternative signalling cascade, termed the SAFE pathway [142, 145, 146, 148], by binding to TNF receptor type 2 and activating Janus Kinase (JAK) and Signal transducer and activator of transcription 3 (STAT3) via mechanisms which are still unclear but may involve sphingosine kinase [61]. A number of experimental studies have demonstrated that pharmacological conditioning mimetics which limit myocardial reperfusion injury do so via the activation of the SAFE pathway, including high density lipoproteins [62], melatonin [144], glyceryltrinitrate, and cariporide [140]. In IPC studies, the activation of the SAFE pathway was demonstrated to occur at two time-points, following the IPC protocol and at the onset of reperfusion [148, 236]. The activation of the SAFE pathway by ischaemic conditioning has been confirmed both in small and large animals of AMI [227], whereas in humans, the STAT-5 isoform appears to be preferentially activated [109].
It has been demonstrated that there exists crosstalk between the Akt and Erk1/2 components of the RISK pathway such that the pharmacological inhibition of one kinase activated the other kinase to ensure maximal protection against myocardial reperfusion injury [83]. Interestingly, a crosstalk also exists between the RISK and the SAFE pathways [232]. Both signaling pathways converge on mitochondria where they appear to mediate their cardioprotective effect by inhibiting PTP opening [16, 46]. The mechanism for this is unclear for the RISK pathway, but for the SAFE pathway, STAT-3 has been shown to be present in mitochondria, where it modulates mitochondrial respiration and targets the PTP [16, 108]. TNF-α itself can also directly target mitochondrial function [141].
Whether targeting the RISK and SAFE pathway can benefit patients subjected to acute myocardial IRI has not been directly tested, although pharmacological agents such as atrial natriuretic peptide, erythropoietin and statins, which are known to activate components of these two signaling cascades, have been investigated in the clinical setting with mixed results [160, 161]. Further studies are required to test whether using combination therapy to simultaneous target the RISK, SAFE and other pathways is a more effective cardioprotective strategy than focusing on one single signaling cascade.
Mitochondria as targets of cardioprotection
The apparent paradox of IPC, whereby a short period of ischaemia protects from an otherwise lethal ischaemic episode, is reflected and likely contributed to, by paradoxical actions of Ca2+ and ROS in mitochondria. Undoubtedly, a large and prolonged elevation in Ca2+ and ROS levels causes cell death mainly by favoring PTP opening [13]. However, antioxidants abolish IPC protection [189], that is mimicked by a mild elevation in ROS or Ca2+ levels [264, 266]. Several processes have been proposed to explain the paradoxical involvement of ROS and Ca2+ in both survival and death of cardiomyocytes. First, intra-mitochondrial Ca2+ is necessary to stimulate oxidative phosphorylation by activating key dehydrogenase steps, while sub-lethal levels of ROS activate signaling pathways promoting cell survival [128]. A mild ROS formation has been proposed also to explain the protection related to the opening of mitochondrial KATP channels downstream of PKC-ɛ activation and upstream of PTP inhibition [41].
Besides the elucidation of the molecular nature of PTP, KATP channels and other potential targets of IPC protection, a major challenge in the field is to determine the threshold separating physiological from pathological levels of ROS and Ca2+. This issue can be addressed by exploiting technological advances in Ca2+ and ROS imaging that have largely contributed to our understanding of IRI and IPC protection. Early studies by Michael Piper using the Ca2+-sensitive, fluorescent dye Fura-2 demonstrated that the recovery of ATP production in isolated cardiomyocytes with Ca2+ overload during reoxygenation leads to Ca2+ oscillations and hypercontracture [223]. It was proposed that Ca2+ microdomains in the vicinity of the SR could raise local [Ca2+] to levels sufficient to drive mitochondrial Ca2+ entry. Subsequent experiments using the mitochondria-targeted, Ca2+-sensitive photoprotein, aequorin, helped to validate the concept of these Ca2+ “hot spots”. Importantly, mitochondrial Ca2+ uptake contributes to the buffering of cytoplasmic Ca2+ peaks in cardiomyocytes [53]. However, during reperfusion massive cytosolic Ca2+ oscillations can lead to mitochondrial Ca2+ overload and PTP opening [64]. Oxidative stress during reperfusion can accentuate SR Ca2+ release [45]. These cytosolic and mitochondrial Ca2+ changes also occur in perfused hearts during IRI, as was initially demonstrated by imaging Ca2+ using fluorescent dyes and microscopy [248], and more recently with genetically encoded reporters and multiphoton microscopy [48]. IPC was shown to attenuate ischaemic SR Ca2+ overload in the isolated rabbit heart [34].
The elucidation of signalling pathways related to IPC-induced protection commenced with the seminal discovery of the involvement of the adenosine receptor in IPC [153]. Several of the described pathways converge on cytosolic Akt and/or ERK, which lead to the activation of mitochondrial PKC-ɛ [177]. Activated mitochondrial PKC-ɛ induces not only opening of the mKATP channel but also activation of Akt-GSK3β signalling in mitochondria, both of which contribute to inhibition of PTP opening [176]. Despite the redundancy of IPC-induced signal pathways in the cytosol and mitochondria, several diseases have been shown to significantly impair IPC-induced signalling in the myocardium. Interestingly, diabetes mellitus attenuates activation of Akt in response to upstream signals [116, 173, 244, 245] and also lowers the threshold for PTP opening by enhanced mitochondrial recruitment of non-phosphorylated GSK3β [172, 237] and increased ER stress [127]. Recently, progress has been made in our understanding of PTP and mitochondrial Ca2+ uniporter. However, the intra-mitochondrial localisation of protein kinases and phosphatases and their relationships with PTP, ROS and Ca2+ regulating machineries remain unclear and warrant further investigation.
Beyond its participation in signal transduction during IPC, mitochondria are effectors of cardioprotection. IPC attenuates IRI-induced mitochondrial respiratory failure and oxidative damage independently of PTP opening, even in the absence of cytosolic components [178]. Importantly, Cx43 translocates to and is predominantly present at subsarcolemmal mitochondria [20] and subsarcolemmal mitochondria but not interfibrillar mitochondria are the main targets of IRI damage and IPC protection [20, 206]. Indeed, mitochondria with genetic ablation of Cx43 are resistant to preconditioning [20, 206, 212]. Mitochondrial Cx43 regulates complex 1-mediated respiration [18, 175], ROS production [98] and K+ permeability [175], although its participation in IRI pathophysiology remains unclear in detail. The role of subsarcolemmal mitochondria in cardioprotection is necessarily linked to the function of interfibrillar mitochondria, as the latter are involved in cytosolic calcium buffering, energy demand–supply matching and antioxidant regeneration through privileged communication with SR [51, 212]. Indeed, partial disruption of mitochondria-SR interplay appears to aggravate IRI-induced cytosolic calcium, hypercontracture, PTP opening and cell death in aged mice [59, 60]. Variations in mitochondrial Cx43 contents might explain the resistance against IPC-mediated cardioprotection observed in aged animals [17, 19, 219].
Pharmacological targeting of myocardial IRI
Elucidation of the signaling pathways underlying ischaemic conditioning cardioprotection has identified a large variety of therapeutic targets for pharmacological cardioprotection. In this section, we review some of the more recent pharmacological therapies which have been investigated in the clinical setting to target myocardial IRI in reperfused STEMI patients.
GLP-1
Glucagon-like peptide-1 (GLP-1) is an incretin hormone that regulates plasma glucose, and within the latest 10 years GLP-1 analogues have been introduced for treatment of type-2 diabetes [89, 112]. In addition, receptors for GLP-1 have been found in the heart [7]. In experimental studies GLP-1 or its analogues protect against reperfusion injury-induced cell death [23, 24, 88, 89, 243]. These cardioprotective analogues include exendin-4, a peptide derived from the saliva of the Gila lizard showing a GLP-1-like potency and efficacy at GLP-1 receptors [95, 255]. Exendin-4 was found to be cardioprotective during reperfusion in isolated rat hearts [233], a finding that has been confirmed in several species, e.g. pigs [242].
Intravenous (IV) exenatide was found to increase myocardial salvage by 15 % if administered as a 6 h infusion initiated 10 min before reperfusion in STEMI patients [157]. When examining only patients with short ischaemic times (<132 min) MI size was reduced by 30 % [156]. This effect of exenatide was confirmed in an Asian population, since Woo et al. observed an almost 50 % reduction in MI size when administered subcutaneously [263]. A recent clinical study has failed to demonstrate a cardioprotective effect with exenatide in STEMI patients—it is not clear why this study was neutral, but it may have been related to the dose used [208]. To date no trials have sought to challenge the results from the proof-of-concept studies on a clinical end point.
Cyclosporin-A
Opening of the PTP is a critical signalling hub in the cascade of myocardial reperfusion injury and preventing it from opening has been suggested to be an obvious pharmacological target [82, 85, 185, 186]. CsA is a compound that preserves PTP closure and in addition, it has been reported to affect remodelling following MI [170].
In a small proof-of-concept study Piot et al. demonstrated that CsA can reduce enzyme leakage by 40 % and MI size by 20 % when administered as an IV bolus prior to PPCI [96, 195]. Both infarcts located in RCA and LAD were included but only patients with TIMI 0 were eligible. The more recent CYCLE trial recruited 410 STEMI patients within 6 h of symptom onset (TIMI flow grade 0–1) and randomized them to CsA (2.5 mg/kg) or control [187]. The primary endpoint (ST-segment resolution at 60 min) and secondary endpoints (high-sensitivity cardiac troponin T (hs-cTnT) on day 4, left ventricular (LV) remodelling, and clinical events at 6-months follow-up) were not reduced by CsA [187]. Finally, in the definite hard endpoints-powered CIRCUS trial, 970 anterior STEMI patients (TIMI flow 0–1 in the LAD) were randomised to CsA or placebo. The trial failed to show any effect of IV CsA on a composite endpoint of death, hospitalization for heart failure and adverse LV remodelling [195]. The reasons for CsA to improve clinical outcomes in STEMI patients are not known and have been discussed in several recent articles [76, 100].
Metoprolol
Early beta-blocker therapy in reperfused STEMI patients is controversial and had largely been investigated in the pre-reperfusion era. However, recently it has been shown that IV metoprolol administered prior to reperfusion in a porcine model reduced MI size [120]. This experimental work was followed by a clinical trial (METOCARD-CNIC trial) demonstrating that IV metoprolol administered in the ambulance prior to PPCI reduced MI size and improved clinical outcomes (as a secondary endpoint) in anterior STEMI patients presenting early (<6 h) [119, 197]. More importantly, in the METOCARD-CNIC trial, patients receiving pre-reperfusion IV metoprolol had not only CMR-evaluated smaller infarctions [166], and better long-term left ventricular ejection fraction (LVEF) [197], but also the incidence of LV severe systolic dysfunction was significantly reduced [197]. Very recently, the results of the EARLY BAMI trial have been reported. This trial recruited 600 STEMI patients (any location) presenting within 12 h from symptoms onset. Patients were randomized to IV metoprolol (10 mg) or placebo [207]. Primary endpoint was MI size assessed by CMR one month after infarction. The trial was neutral and MI size was not smaller in patients allocated to IV metoprolol. There were no signs of adverse effects in patients receiving IV metoprolol, and the incidence of ventricular fibrillation was significantly lower in metoprolol-treated patients. These data support the safety of this strategy in Killip I–II STEMI patients. There are important differences between the METOCARD-CNIC and EARLY BAMI trials. Dose and timing of IV metoprolol administration were different between trials. In contrast to the METOCARD-CNIC trial, in the EARLY BAMI study, patients received only one 5 mg dose at recruitment, and per protocol the second dose was given in the catheter laboratory immediately before PCI. In fact, the first dose of metoprolol did not have any effect on blood pressure or heart rate, suggesting an underdosing effect. In this regard, a recent subanalysis from the METOCARD-CNIC trial demonstrated that the longer the “onboard” metoprolol time at the time of reperfusion, the higher the infarct-reduction effect [67]. In fact, patients receiving IV metoprolol close to reperfusion had a very mild protective effect, while those with a longer time from metoprolol 15 mg bolus to reperfusion were those with the largest reduction in MI size and improvement in long-term LVEF. These differences in dose and timing of metoprolol administration might explain the different conclusions from both trials. Given the clear safety profile and the low cost of this therapy, it is worth to continue the clinical research and perform a definite large hard endpoint-powered trial. In the near future, the MOVE ON! Trial will be initiated and more than 1200 anterior STEMI patients will be recruited and randomized to IV metoprolol (15 mg immediately after diagnosis is made in the out of hospital setting) or placebo. The primary endpoint will be the composite of cardiovascular death, heart failure, ICD insertion, or severe LV dysfunction.
P2Y12 inhibitors
State of the art anti-thrombotic therapy in STEMI patients includes the early administration of P2Y12 inhibitors. Ticagrelor, a potent P2Y12 inhibitor was associated with reduced mortality in ACS patients when compared to another P2Y12 inhibitor (clopidogrel) [253]. These benefits may not be fully explained by a pure antiplatelet effect. In this regard, ticagrelor has been shown, to increase the levels of extracellular adenosine [21], a mediator known to exert a wide range of benefits including vasodilation, inhibition of platelet aggregation and leukocyte adherence to the vessel wall. In line with this, cangrelor, a potent and fast acting IV P2Y12 inhibitor, has been shown to reduce MI size in mouse [12], rat [267], rabbit [268], and primates [269]. Interestingly, the protection conferred by cangrelor is dependent upon the presence of platelets with no evidence of protection ex vivo in crystalloid-perfused Langendorff heart [267, 268]. This protection is mediated through pathways typically recruited by ischaemic conditioning, suggesting that P2Y12 inhibition, via a blood component, leads to conditioning-like protection [267, 268]. Therefore, IV P2Y12 inhibition may thus have the dual advantage of optimising both platelet inhibition and offering cardioprotection.
Combination reperfusion therapy—a novel therapeutic strategy
As can be seen above, most attempts to reduce MI size in STEMI patients have relied on using a single agent to target one single component of myocardial IRI. However, myocardial IRI is the result of several mechanisms and thus targeting on individual phenomena will unlikely reduce the MI size. The possibility of targeting several mechanisms simultaneously (either with one agent targeting different pathways or by several agents administered simultaneously) is attractive although not widely undertaken. A recent large animal study [1] showed that the combination of RIC with glucose-insulin-potassium and exenatide had an additive benefit in terms of MI size reduction. The COMBAT-MI trial (NCT02404376) will test the potential benefits of using RIC with exenatide on MI size reduction in STEMI patient.
Problems in translation to the clinic and confounding factors
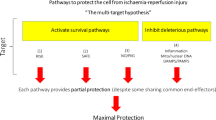
Although much effort has been taken to translate cardioprotection into clinical practice, so far translation has not been successful, as still no drugs are on the market and no therapeutic interventions are available for routine clinical practice that may protect the heart after IRI and thereby prevent the development of post-ischaemic heart failure [11, 28, 76, 99, 106, 107, 110, 118]. There are two major problems of clinical translation to overcome in the future: (1) target discovery and validation taking into consideration the known confounding factors of cardioprotection; and (2) better design of clinical development studies.
The putative mechanisms of cardioprotection explored in the past three decades have so far led to potential drug targets that were not robust enough for their pharmacological use as clinical trials targeting them largely showed no efficacy. Although it has been known from preclinical studies already in the mid-1990s that major cardiovascular co-morbidities and risk factors including aging, hyperlipidemia, diabetes (see for the first extensive review from 1998 on the effect of risk factors on cardioprotection [58], as well as some later specific reviews on aging [19], hyperlipidaemia [55], and diabetes [178, 261]) and also their medications (see for the first extensive review: [57] and its updated version: [56]) largely modify the response of the ischaemic heart to cardioprotective therapies, target discovery and validation were performed and still continue to be performed in young and healthy animals in the vast majority of studies. In the future, at least the major known co-morbidities and their major classes of pharmacological treatments should be used to validate the potential drug target before entering into clinical trials. The second reason may be a simplified and biased way of target selection so far. It is already known from transcriptomics data from the early 2000 years that IRI and cardioprotection trigger multifactorial mechanisms, moreover, co-morbidities and co-medications also significantly modify the cardiac gene expression profile (see for extensive reviews: [249]). Therefore, targeting a single pathway to protect all IHD patients is obviously not an approach that may lead to success. In the future, maybe a multi-omics approach including transcriptomics, proteomics, and metabolomics followed by systems biological network analysis may provide novel targets using this unbiased global approach (see for extensive reviews: [249]).
Although clinical trials of RIC may show some patient benefit in acute MI patients with multiple co-morbidities [104], two recent large clinical studies in patients undergoing cardiovascular surgery (ERICCA, RIPHeart) [77, 171] revealed no evidence for protection. It must be mentioned, however, that in both of these trials propofol was used for anesthesia, although propofol has been shown before to interfere with the efficacy of conditioning [105, 106]. Some larger clinical studies of IPost so far showed no acute cardioprotection [70, 134] nor long-term benefit up to 1 year follow up [71]. Nevertheless, the results of these clinical studies showed that both RIC and IPost have a favorable safety profile [110], so further studies are encouraged using better design and enough power to find out the importance of confounding factors of ischaemic conditioning in clinical reality.
As to the clinical development of cardioprotective drugs, the results so far have been disappointing. Targeting mitochondria by PTP inhibitors and other mitochondrial protective compounds (CIRCUS, CYCLE, Bendavia, Mitocare, EMBRACE STEMI studies) or replacing NO by inhaled NO (NOMI trial) or nitrite administration were ineffective in clinical trials (see for a recent review [76]). It should be noted that the molecular targets of these drugs were not validated properly before entering into clinical trials, i.e. targets were selected by the traditional biased way and no validations have been attempted in any of the animal models with the presence of the confounding factors. In the future, based on careful preclinical validation, phase 2 studies with careful patient selection based on the relevant confounding factors for the specific molecular target(s) may open new perspectives for successful translation of cardioprotection. Also, given the complexity of the cardioprotective signal transduction [101], combined treatment of several targets maybe needed.
Conclusions
In this article we have provided an overview of the major topics discussed at this special meeting to celebrate 30 years of research in the field of IPC and cardioprotection. The huge research literature, which has arisen from the seminal discovery of IPC, has provided important insights into the mechanisms and elucidation of the signalling pathways underlying cytoprotection in the heart and other organs. The evolution of IPC to both IPost and RIC has helped facilitate the translation of this endogenous cardioprotective strategy from the laboratory to the clinical setting. We hope this article provides a worthy account of the huge importance and impact the discovery of IPC has made in the field of cardiovascular research over the last 30 years.
References
Alburquerque-Bejar JJ, Barba I, Inserte J, Miro-Casas E, Ruiz-Meana M, Poncelas M, Vilardosa U, Valls-Lacalle L, Rodriguez-Sinovas A, Garcia-Dorado D (2015) Combination therapy with remote ischaemic conditioning and insulin or exenatide enhances infarct size limitation in pigs. Cardiovasc Res 107:246–254. doi:10.1093/cvr/cvv171
Ali N, Rizwi F, Iqbal A, Rashid A (2010) Induced remote ischemic pre-conditioning on ischemia–reperfusion injury in patients undergoing coronary artery bypass. J Coll Physicians Surg Pak 20:427–431. doi:07.2010/JCPSP.427431
Ali ZA, Callaghan CJ, Lim E, Ali AA, Nouraei SA, Akthar AM, Boyle JR, Varty K, Kharbanda RK, Dutka DP, Gaunt ME (2007) Remote ischemic preconditioning reduces myocardial and renal injury after elective abdominal aortic aneurysm repair: a randomized controlled trial. Circulation 116:I98–105. doi:10.1161/circulationaha.106.679167
Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M (2005) Postconditioning inhibits mitochondrial permeability transition. Circulation 111:194–197. doi:10.1161/01.CIR.0000151290.04952.3B
Auchampach JA, Grover GJ, Gross GJ (1992) Blockade of ischaemic preconditioning in dogs by the novel ATP dependent potassium channel antagonist sodium 5-hydroxydecanoate. Cardiovasc Res 26:1054–1062. doi:10.1093/cvr/26.11.1054
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434:658–662. doi:10.1038/nature03434
Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M (2008) Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 117:2340–2350. doi:10.1161/CIRCULATIONAHA.107.739938
Barrabes JA, Garcia-Dorado D, Mirabet M, Inserte J, Agullo L, Soriano B, Massaguer A, Padilla F, Lidon RM, Soler-Soler J (2005) Antagonism of selectin function attenuates microvascular platelet deposition and platelet-mediated myocardial injury after transient ischemia. J Am Coll Cardiol 45:293–299. doi:10.1016/j.jacc.2004.09.068
Barrabes JA, Inserte J, Mirabet M, Quiroga A, Hernando V, Figueras J, Garcia-Dorado D (2010) Antagonism of P2Y12 or GPIIb/IIIa receptors reduces platelet-mediated myocardial injury after ischaemia and reperfusion in isolated rat hearts. Thromb Haemost 104:128–135. doi:10.1160/TH09-07-0440
Battipaglia I, Scalone G, Milo M, Di Franco A, Lanza GA, Crea F (2011) Upper arm intermittent ischaemia reduces exercise-related increase of platelet reactivity in patients with obstructive coronary artery disease. Heart 97:1298–1303. doi:10.1136/hrt.2011.226415
Bell RM, Botker HE, Carr RD, Davidson SM, Downey JM, Dutka DP, Heusch G, Ibanez B, Macallister R, Stoppe C, Ovize M, Redington A, Walker JM, Yellon DM (2016) 9th Hatter Biannual Meeting: position document on ischaemia/reperfusion injury, conditioning and the ten commandments of cardioprotection. Basic Res Cardiol 111:41. doi:10.1007/s00395-016-0558-1
Bell RM, Sivaraman V, Kunuthur SP, Cohen MV, Downey JM, Yellon DM (2015) Cardioprotective properties of the platelet P2Y12 receptor inhibitor, cangrelor: protective in diabetics and reliant upon the presence of blood. Cardiovasc Drugs Ther 29:415–418. doi:10.1007/s10557-015-6609-2
Bernardi P, Di Lisa F (2015) The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 78:100–106. doi:10.1016/j.yjmcc.2014.09.023
Bilinska M, Rudnicki S, Beresewicz A (2000) Delayed attenuation of myocardial ischemia with repeated exercise in subjects with stable angina: a possible model for the second window of protection? Basic Res Cardiol 95:418–423. doi:10.1007/s003950070042
Boengler K, Dodoni G, Rodriguez-Sinovas A, Cabestrero A, Ruiz-Meana M, Gres P, Konietzka I, Lopez-Iglesias C, Garcia-Dorado D, Di Lisa F, Heusch G, Schulz R (2005) Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc Res 67:234–244. doi:10.1016/j.cardiores.2005.04.014
Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R (2010) Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol 105:771–785. doi:10.1007/s00395-010-0124-1
Boengler K, Konietzka I, Buechert A, Heinen Y, Garcia-Dorado D, Heusch G, Schulz R (2007) Loss of ischemic preconditioning’s cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. Am J Physiol Heart Circ Physiol 292:H1764–H1769. doi:10.1152/ajpheart.01071.2006
Boengler K, Ruiz-Meana M, Gent S, Ungefug E, Soetkamp D, Miro-Casas E, Cabestrero A, Fernandez-Sanz C, Semenzato M, Di Lisa F, Rohrbach S, Garcia-Dorado D, Heusch G, Schulz R (2012) Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J Cell Mol Med 16:1649–1655. doi:10.1111/j.1582-4934.2011.01516.x
Boengler K, Schulz R, Heusch G (2009) Loss of cardioprotection with ageing. Cardiovasc Res 83:247–261. doi:10.1093/cvr/cvp033
Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz-Meana M, Garcia-Dorado D, Heusch G, Schulz R (2009) Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol 104:141–147. doi:10.1007/s00395-009-0007-5
Bonello L, Laine M, Kipson N, Mancini J, Helal O, Fromonot J, Gariboldi V, Condo J, Thuny F, Frere C, Camoin-Jau L, Paganelli F, Dignat-George F, Guieu R (2014) Ticagrelor increases adenosine plasma concentration in patients with an acute coronary syndrome. J Am Coll Cardiol 63:872–877. doi:10.1016/j.jacc.2013.09.067
Bopassa JC, Michel P, Gateau-Roesch O, Ovize M, Ferrera R (2005) Low-pressure reperfusion alters mitochondrial permeability transition. Am J Physiol Heart Circ Physiol 288:H2750–H2755. doi:10.1152/ajpheart.01081.2004
Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM (2005) Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes 54:146–151. doi:10.2337/diabetes.54.1.146
Bose AK, Mocanu MM, Carr RD, Yellon DM (2005) Glucagon like peptide-1 is protective against myocardial ischemia/reperfusion injury when given either as a preconditioning mimetic or at reperfusion in an isolated rat heart model. Cardiovasc Drugs Ther 19:9–11. doi:10.1007/s10557-005-6892-4
Botker HE, Kharbanda R, Schmidt MR, Bottcher M, Kaltoft AK, Terkelsen CJ, Munk K, Andersen NH, Hansen TM, Trautner S, Lassen JF, Christiansen EH, Krusell LR, Kristensen SD, Thuesen L, Nielsen SS, Rehling M, Sorensen HT, Redington AN, Nielsen TT (2010) Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet 375:727–734. doi:10.1016/S0140-6736(09)62001-8
Bromage DI, Pickard JM, Rosello X, Ziff OJ, Burke N, Yellon DM, Davidson SM (2016) Remote ischaemic conditioning reduces infarct size in animal in vivo models of ischaemia-reperfusion injury: a systematic review and meta-analysis. Cardiovasc Res. doi:10.1093/cvr/cvw143
Buckberg GD (1987) Strategies and logic of cardioplegic delivery to prevent, avoid, and reverse ischemic and reperfusion damage. J Thorac Cardiovasc Surg 93:127–139
Bulluck H, Yellon DM, Hausenloy DJ (2016) Reducing myocardial infarct size: challenges and future opportunities. Heart 102:341–348. doi:10.1136/heartjnl-2015-307855
Cabrera-Fuentes HA, Alba-Alba C, Aragones J, Bernhagen J, Boisvert WA, Botker HE, Cesarman-Maus G, Fleming I, Garcia-Dorado D, Lecour S, Liehn E, Marber MS, Marina N, Mayr M, Perez-Mendez O, Miura T, Ruiz-Meana M, Salinas-Estefanon EM, Ong SB, Schnittler HJ, Sanchez-Vega JT, Sumoza-Toledo A, Vogel CW, Yarullina D, Yellon DM, Preissner KT, Hausenloy DJ (2016) Meeting report from the 2nd international symposium on new frontiers in cardiovascular research. Protecting the cardiovascular system from ischemia: between bench and bedside. Basic Res Cardiol 111:7. doi:10.1007/s00395-015-0527-0
Cabrera-Fuentes HA, Niemann B, Grieshaber P, Wollbrueck M, Gehron J, Preissner KT, Boning A (2015) RNase1 as a potential mediator of remote ischaemic preconditioning for cardioprotectiondagger. Eur J Cardiothorac Surg 48(5):732–737. doi:10.1093/ejcts/ezu519
Cabrera-Fuentes HA, Preissner KT (2014) Abstract 20396: induction of ischemia–reperfusion injury by extracellular RNA: a case for tumor necrosis factor (TNF-α)—shedding. Circulation 130:A20396
Cabrera-Fuentes HA, Ruiz-Meana M, Simsekyilmaz S, Kostin S, Inserte J, Saffarzadeh M, Galuska SP, Vijayan V, Barba I, Barreto G, Fischer S, Lochnit G, Ilinskaya ON, Baumgart-Vogt E, Boning A, Lecour S, Hausenloy DJ, Liehn EA, Garcia-Dorado D, Schluter KD, Preissner KT (2014) RNase1 prevents the damaging interplay between extracellular RNA and tumour necrosis factor-alpha in cardiac ischaemia/reperfusion injury. Thromb Haemost 112:1110–1119. doi:10.1160/TH14-08-0703
Candilio L, Malik A, Ariti C, Barnard M, Di Salvo C, Lawrence D, Hayward M, Yap J, Roberts N, Sheikh A, Kolvekar S, Hausenloy DJ, Yellon DM (2015) Effect of remote ischaemic preconditioning on clinical outcomes in patients undergoing cardiac bypass surgery: a randomised controlled clinical trial. Heart 101:185–192. doi:10.1136/heartjnl-2014-306178
Chen W, London R, Murphy E, Steenbergen C (1998) Regulation of the Ca2+ gradient across the sarcoplasmic reticulum in perfused rabbit heart. A 19F nuclear magnetic resonance study. Circ Res 83:898–907. doi:10.1161/01.RES.83.9.898
Cheung MM, Kharbanda RK, Konstantinov IE, Shimizu M, Frndova H, Li J, Holtby HM, Cox PN, Smallhorn JF, Van Arsdell GS, Redington AN (2006) Randomized controlled trial of the effects of remote ischemic preconditioning on children undergoing cardiac surgery: first clinical application in humans. J Am Coll Cardiol 47:2277–2282. doi:10.1016/j.jacc.2006.01.066
Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa AS, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515:431–435. doi:10.1038/nature13909
Cohen MV, Downey JM (2011) Ischemic postconditioning: from receptor to end-effector. Antioxid Redox Signal 14:821–831. doi:10.1089/ars.2010.3318
Cohen MV, Downey JM (2015) Signalling pathways and mechanisms of protection in pre- and postconditioning: historical perspective and lessons for the future. Br J Pharmacol 172:1913–1932. doi:10.1111/bph.12903
Cohen MV, Yang XM, Downey JM (2007) The pH hypothesis of postconditioning: staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation 115:1895–1903. doi:10.1161/CIRCULATIONAHA.106.675710
Cohen MV, Yang XM, Liu GS, Heusch G, Downey JM (2001) Acetylcholine, bradykinin, opioids, and phenylephrine, but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial K(ATP) channels. Circ Res 89:273–278. doi:10.1161/hh1501.094266
Costa AD, Jakob R, Costa CL, Andrukhiv K, West IC, Garlid KD (2006) The mechanism by which the mitochondrial ATP-sensitive K+ channel opening and H2O2 inhibit the mitochondrial permeability transition. J Biol Chem 281:20801–20808. doi:10.1074/jbc.M600959200
Crimi G, Pica S, Raineri C, Bramucci E, De Ferrari GM, Klersy C, Ferlini M, Marinoni B, Repetto A, Romeo M, Rosti V, Massa M, Raisaro A, Leonardi S, Rubartelli P, Oltrona Visconti L, Ferrario M (2013) Remote ischemic post-conditioning of the lower limb during primary percutaneous coronary intervention safely reduces enzymatic infarct size in anterior myocardial infarction: a randomized controlled trial. JACC Cardiovasc Interv 6:1055–1063. doi:10.1016/j.jcin.2013.05.011
Cung TT, Morel O, Cayla G, Rioufol G, Garcia-Dorado D, Angoulvant D, Bonnefoy-Cudraz E, Guerin P, Elbaz M, Delarche N, Coste P, Vanzetto G, Metge M, Aupetit JF, Jouve B, Motreff P, Tron C, Labeque JN, Steg PG, Cottin Y, Range G, Clerc J, Claeys MJ, Coussement P, Prunier F, Moulin F, Roth O, Belle L, Dubois P, Barragan P, Gilard M, Piot C, Colin P, De Poli F, Morice MC, Ider O, Dubois-Rande JL, Unterseeh T, Le Breton H, Beard T, Blanchard D, Grollier G, Malquarti V, Staat P, Sudre A, Elmer E, Hansson MJ, Bergerot C, Boussaha I, Jossan C, Derumeaux G, Mewton N, Ovize M (2015) Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med 373:1021–1031. doi:10.1056/NEJMoa1505489
Currie RW, Karmazyn M, Kloc M, Mailer K (1988) Heat-shock response is associated with enhanced postischemic ventricular recovery. Circ Res 63:543–549. doi:10.1161/01.RES.63.3.543
Davidson SM, Duchen MR (2006) Calcium microdomains and oxidative stress. Cell Calcium 40:561–574. doi:10.1016/j.ceca.2006.08.017
Davidson SM, Hausenloy D, Duchen MR, Yellon DM (2006) Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell Biol 38:414–419. doi:10.1016/j.biocel.2005.09.017
Davidson SM, Selvaraj P, He D, Boi-Doku C, Yellon RL, Vicencio JM, Yellon DM (2013) Remote ischaemic preconditioning involves signalling through the SDF-1alpha/CXCR4 signalling axis. Basic Res Cardiol 108:377. doi:10.1007/s00395-013-0377-6
Davidson SM, Yellon DM, Murphy MP, Duchen MR (2012) Slow calcium waves and redox changes precede mitochondrial permeability transition pore opening in the intact heart during hypoxia and reoxygenation. Cardiovasc Res 93:445–453. doi:10.1093/cvr/cvr349
Davies WR, Brown AJ, Watson W, McCormick LM, West NE, Dutka DP, Hoole SP (2013) Remote ischemic preconditioning improves outcome at 6 years after elective percutaneous coronary intervention: the CRISP stent trial long-term follow-up. Circ Cardiovasc Interv 6:246–251. doi:10.1161/CIRCINTERVENTIONS.112.000184
Donato M, Buchholz B, Rodriguez M, Perez V, Inserte J, Garcia-Dorado D, Gelpi RJ (2013) Role of the parasympathetic nervous system in cardioprotection by remote hindlimb ischaemic preconditioning. Exp Physiol 98:425–434. doi:10.1113/expphysiol.2012.066217
Dorn GW 2nd, Maack C (2013) SR and mitochondria: calcium cross-talk between kissing cousins. J Mol Cell Cardiol 55:42–49. doi:10.1016/j.yjmcc.2012.07.015
Dost T, Cohen MV, Downey JM (2008) Redox signaling triggers protection during the reperfusion rather than the ischemic phase of preconditioning. Basic Res Cardiol 103:378–384. doi:10.1007/s00395-008-0718-z
Drago I, De Stefani D, Rizzuto R, Pozzan T (2012) Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci USA 109:12986–12991. doi:10.1073/pnas.1210718109
Eitel I, Stiermaier T, Rommel KP, Fuernau G, Sandri M, Mangner N, Linke A, Erbs S, Lurz P, Boudriot E, Mende M, Desch S, Schuler G, Thiele H (2015) Cardioprotection by combined intrahospital remote ischaemic perconditioning and postconditioning in ST-elevation myocardial infarction: the randomized LIPSIA CONDITIONING trial. Eur Heart J 36:3049–3057. doi:10.1093/eurheartj/ehv463
Ferdinandy P (2003) Myocardial ischaemia/reperfusion injury and preconditioning: effects of hypercholesterolaemia/hyperlipidaemia. Br J Pharmacol 138:283–285. doi:10.1038/sj.bjp.0705097
Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R (2014) Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev 66:1142–1174. doi:10.1124/pr.113.008300
Ferdinandy P, Schulz R, Baxter GF (2007) Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev 59:418–458. doi:10.1124/pr.107.06002
Ferdinandy P, Szilvassy Z, Baxter GF (1998) Adaptation to myocardial stress in disease states: is preconditioning a healthy heart phenomenon? Trends Pharmacol Sci 19:223–229. doi:10.1016/S0165-6147(98)01212-7
Fernandez-Sanz C, Ruiz-Meana M, Castellano J, Miro-Casas E, Nunez E, Inserte J, Vazquez J, Garcia-Dorado D (2015) Altered FoF1 ATP synthase and susceptibility to mitochondrial permeability transition pore during ischaemia and reperfusion in aging cardiomyocytes. Thromb Haemost 113:441–451. doi:10.1160/TH14-10-0901
Fernandez-Sanz C, Ruiz-Meana M, Miro-Casas E, Nunez E, Castellano J, Loureiro M, Barba I, Poncelas M, Rodriguez-Sinovas A, Vazquez J, Garcia-Dorado D (2014) Defective sarcoplasmic reticulum-mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis 5:e1573. doi:10.1038/cddis.2014.526
Frias MA, Lecour S, James RW, Pedretti S (2012) High density lipoprotein/sphingosine-1-phosphate-induced cardioprotection: role of STAT3 as part of the SAFE pathway. JAKSTAT 1:92–100. doi:10.4161/jkst.19754
Frias MA, Pedretti S, Hacking D, Somers S, Lacerda L, Opie LH, James RW, Lecour S (2013) HDL protects against ischemia reperfusion injury by preserving mitochondrial integrity. Atherosclerosis 228:110–116. doi:10.1016/j.atherosclerosis.2013.02.003
Galagudza M, Kurapeev D, Minasian S, Valen G, Vaage J (2004) Ischemic postconditioning: brief ischemia during reperfusion converts persistent ventricular fibrillation into regular rhythm. Eur J Cardiothorac Surg 25:1006–1010. doi:10.1016/j.ejcts.2004.02.003
Garcia-Dorado D, Ruiz-Meana M, Inserte J, Rodriguez-Sinovas A, Piper HM (2012) Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res 94:168–180. doi:10.1093/cvr/cvs116
Garcia-Dorado D, Ruiz-Meana M, Piper HM (2009) Lethal reperfusion injury in acute myocardial infarction: facts and unresolved issues. Cardiovasc Res 83:165–168. doi:10.1093/cvr/cvp185
Garcia-Dorado D, Theroux P, Duran JM, Solares J, Alonso J, Sanz E, Munoz R, Elizaga J, Botas J, Fernandez-Aviles F et al (1992) Selective inhibition of the contractile apparatus. A new approach to modification of infarct size, infarct composition, and infarct geometry during coronary artery occlusion and reperfusion. Circulation 85:1160–1174. doi:10.1161/01.CIR.85.3.1160
Garcia-Ruiz JM, Fernandez-Jimenez R, Garcia-Alvarez A, Pizarro G, Galan-Arriola C, Fernandez-Friera L, Mateos A, Nuno-Ayala M, Aguero J, Sanchez-Gonzalez J, Garcia-Prieto J, Lopez-Melgar B, Martinez-Tenorio P, Lopez-Martin GJ, Macias A, Perez-Asenjo B, Cabrera JA, Fernandez-Ortiz A, Fuster V, Ibanez B (2016) Impact of the timing of metoprolol administration during STEMI on infarct size and ventricular function. J Am Coll Cardiol 67:2093–2104. doi:10.1016/j.jacc.2016.02.050
Gho BC, Schoemaker RG, van den Doel MA, Duncker DJ, Verdouw PD (1996) Myocardial protection by brief ischemia in noncardiac tissue. Circulation 94:2193–2200. doi:10.1161/01.CIR.94.9.2193
Gunaydin B, Cakici I, Soncul H, Kalaycioglu S, Cevik C, Sancak B, Kanzik I, Karadenizli Y (2000) Does remote organ ischaemia trigger cardiac preconditioning during coronary artery surgery? Pharmacol Res 41:493–496. doi:10.1006/phrs.1999.0611
Hahn JY, Song YB, Kim EK, Yu CW, Bae JW, Chung WY, Choi SH, Choi JH, Bae JH, An KJ, Park JS, Oh JH, Kim SW, Hwang JY, Ryu JK, Park HS, Lim DS, Gwon HC (2013) Ischemic postconditioning during primary percutaneous coronary intervention: the effects of postconditioning on myocardial reperfusion in patients with ST-segment elevation myocardial infarction (POST) randomized trial. Circulation 128:1889–1896. doi:10.1161/CIRCULATIONAHA.113.001690
Hahn JY, Yu CW, Park HS, Song YB, Kim EK, Lee HJ, Bae JW, Chung WY, Choi SH, Choi JH, Bae JH, An KJ, Park JS, Oh JH, Kim SW, Hwang JY, Ryu JK, Lim DS, Gwon HC (2015) Long-term effects of ischemic postconditioning on clinical outcomes: 1-year follow-up of the POST randomized trial. Am Heart J 169:639–646. doi:10.1016/j.ahj.2015.01.015
Halestrap AP, Richardson AP (2015) The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol 78:129–141. doi:10.1016/j.yjmcc.2014.08.018
Halkos ME, Kerendi F, Corvera JS, Wang NP, Kin H, Payne CS, Sun HY, Guyton RA, Vinten-Johansen J, Zhao ZQ (2004) Myocardial protection with postconditioning is not enhanced by ischemic preconditioning. Ann Thorac Surg 78:961–969. doi:10.1016/j.athoracsur.2004.03.033 (discussion 969)
Hausenloy DJ (2013) Cardioprotection techniques: preconditioning, postconditioning and remote conditioning (basic science). Curr Pharm Des 19:4544–4563. doi:10.2174/1381612811319250004
Hausenloy DJ, Boston-Griffiths EA, Yellon DM (2012) Cyclosporin A and cardioprotection: from investigative tool to therapeutic agent. Br J Pharmacol 165:1235–1245. doi:10.1111/j.1476-5381.2011.01700.x
Hausenloy DJ, Botker HE, Engstrom T, Erlinge D, Heusch G, Ibanez B, Kloner RA, Ovize M, Yellon DM, Garcia-Dorado D (2016) Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: trials and tribulations. Eur Heart J. doi:10.1093/eurheartj/ehw145
Hausenloy DJ, Candilio L, Evans R, Ariti C, Jenkins DP, Kolvekar S, Knight R, Kunst G, Laing C, Nicholas J, Pepper J, Robertson S, Xenou M, Clayton T, Yellon DM, Investigators ET (2015) Remote ischemic preconditioning and outcomes of cardiac surgery. N Engl J Med 373:1408–1417. doi:10.1056/NEJMoa1413534
Hausenloy DJ, Iliodromitis EK, Andreadou I, Papalois A, Gritsopoulos G, Anastasiou-Nana M, Kremastinos DT, Yellon DM (2012) Investigating the signal transduction pathways underlying remote ischemic conditioning in the porcine heart. Cardiovasc Drugs Ther 26:87–93. doi:10.1007/s10557-011-6364-y
Hausenloy DJ, Kharbanda R, Rahbek Schmidt M, Moller UK, Ravkilde J, Okkels Jensen L, Engstrom T, Garcia Ruiz JM, Radovanovic N, Christensen EF, Sorensen HT, Ramlall M, Bulluck H, Evans R, Nicholas J, Knight R, Clayton T, Yellon DM, Botker HE (2015) Effect of remote ischaemic conditioning on clinical outcomes in patients presenting with an ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Eur Heart J 36:1846–1848. doi:10.1093/eurheartj/ehv249
Hausenloy DJ, Lecour S, Yellon DM (2011) Reperfusion injury salvage kinase and survivor activating factor enhancement prosurvival signaling pathways in ischemic postconditioning: two sides of the same coin. Antioxid Redox Signal 14:893–907. doi:10.1089/ars.2010.3360
Hausenloy DJ, Lim SY, Ong SG, Davidson SM, Yellon DM (2010) Mitochondrial cyclophilin-D as a critical mediator of ischaemic preconditioning. Cardiovasc Res 88:67–74. doi:10.1093/cvr/cvq113
Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM (2002) Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res 55:534–543. doi:10.1016/S0008-6363(02)00455-8
Hausenloy DJ, Mocanu MM, Yellon DM (2004) Cross-talk between the survival kinases during early reperfusion: its contribution to ischemic preconditioning. Cardiovasc Res 63:305–312. doi:10.1016/j.cardiores.2004.04.011
Hausenloy DJ, Mwamure PK, Venugopal V, Harris J, Barnard M, Grundy E, Ashley E, Vichare S, Di Salvo C, Kolvekar S, Hayward M, Keogh B, MacAllister RJ, Yellon DM (2007) Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: a randomised controlled trial. Lancet 370:575–579. doi:10.1016/S0140-6736(07)61296-3
Hausenloy DJ, Ong SB, Yellon DM (2009) The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol 104:189–202. doi:10.1007/s00395-009-0010-x
Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM (2005) Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol Heart Circ Physiol 288:H971–H976. doi:10.1152/ajpheart.00374.2004
Hausenloy DJ, Tsang A, Yellon DM (2005) The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med 15:69–75. doi:10.1016/j.tcm.2005.03.001
Hausenloy DJ, Whittington HJ, Wynne AM, Begum SS, Theodorou L, Riksen N, Mocanu MM, Yellon DM (2013) Dipeptidyl peptidase-4 inhibitors and GLP-1 reduce myocardial infarct size in a glucose-dependent manner. Cardiovasc Diabetol 12:154. doi:10.1186/1475-2840-12-154
Hausenloy DJ, Yellon DM (2008) GLP-1 therapy: beyond glucose control. Circ Heart Fail 1:147–149. doi:10.1161/CIRCHEARTFAILURE.108.810887
Hausenloy DJ, Yellon DM (2016) Ischaemic conditioning and reperfusion injury. Nat Rev Cardiol 13:193–209. doi:10.1038/nrcardio.2016.5
Hausenloy DJ, Yellon DM (2013) Myocardial ischemia–reperfusion injury: a neglected therapeutic target. J Clin Invest 123:92–100. doi:10.1172/JCI62874
Hausenloy DJ, Yellon DM (2004) New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res 61:448–460. doi:10.1016/j.cardiores.2003.09.024
Hausenloy DJ, Yellon DM (2008) Remote ischaemic preconditioning: underlying mechanisms and clinical application. Cardiovasc Res 79:377–386. doi:10.1093/cvr/cvn114
Hausenloy DJ, Yellon DM (2007) Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev 12:217–234. doi:10.1007/s10741-007-9026-1
Hausenloy DJ, Yellon DM (2012) Taking lizard saliva to heart. Eur Heart J 33:1426–1430. doi:10.1093/eurheartj/ehr382
Hausenloy DJ, Yellon DM (2008) Time to take myocardial reperfusion injury seriously. N Engl J Med 359:518–520. doi:10.1056/NEJMe0803746
Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR (2004) Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol 287:H841–H849. doi:10.1152/ajpheart.00678.2003
Heinzel FR, Luo Y, Li X, Boengler K, Buechert A, Garcia-Dorado D, Di Lisa F, Schulz R, Heusch G (2005) Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ Res 97:583–586. doi:10.1161/01.RES.0000181171.65293.65
Heusch G (2013) Cardioprotection: chances and challenges of its translation to the clinic. Lancet 381:166–175. doi:10.1016/S0140-6736(12)60916-7
Heusch G (2015) CIRCUS: a kiss of death for cardioprotection? Cardiovasc Res 108:215–216. doi:10.1093/cvr/cvv225
Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116:674–699. doi:10.1161/CIRCRESAHA.116.305348
Heusch G (2001) Nitroglycerin and delayed preconditioning in humans: yet another new mechanism for an old drug? Circulation 103:2876–2878. doi:10.1161/01.CIR.103.24.2876
Heusch G, Boengler K, Schulz R (2010) Inhibition of mitochondrial permeability transition pore opening: the Holy Grail of cardioprotection. Basic Res Cardiol 105:151–154. doi:10.1007/s00395-009-0080-9
Heusch G, Botker HE, Przyklenk K, Redington A, Yellon D (2015) Remote ischemic conditioning. J Am Coll Cardiol 65:177–195. doi:10.1016/j.jacc.2014.10.031
Heusch G, Gersh BJ (2016) ERICCA and RIPHeart: two nails in the coffin for cardioprotection by remote ischemic conditioning? Probably not! Eur Heart J 37:200–202. doi:10.1093/eurheartj/ehv606
Heusch G, Gersh BJ (2016) The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J. doi:10.1093/eurheartj/ehw224
Heusch G, Libby P, Gersh B, Yellon D, Bohm M, Lopaschuk G, Opie L (2014) Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 383:1933–1943. doi:10.1016/S0140-6736(14)60107-0
Heusch G, Musiolik J, Gedik N, Skyschally A (2011) Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res 109:1302–1308. doi:10.1161/CIRCRESAHA.111.255604
Heusch G, Musiolik J, Kottenberg E, Peters J, Jakob H, Thielmann M (2012) STAT5 activation and cardioprotection by remote ischemic preconditioning in humans: short communication. Circ Res 110:111–115. doi:10.1161/CIRCRESAHA.111.259556
Heusch G, Rassaf T (2016) Time to give up on cardioprotection? A critical appraisal of clinical studies on ischemic pre-, post-, and remote conditioning. Circ Res 119:676–695u. doi:10.1161/CIRCRESAHA.116.308736
Hofsten DE, Kelbaek H, Helqvist S, Klovgaard L, Holmvang L, Clemmensen P, Torp-Pedersen C, Tilsted HH, Botker HE, Jensen LO, Kober L, Engstrom T, Investigators D (2015) The Third DANish Study of Optimal Acute Treatment of Patients with ST-segment Elevation Myocardial Infarction: ischemic postconditioning or deferred stent implantation versus conventional primary angioplasty and complete revascularization versus treatment of culprit lesion only: Rationale and design of the DANAMI 3 trial program. Am Heart J 169:613–621. doi:10.1016/j.ahj.2015.02.004
Holst JJ (2007) The physiology of glucagon-like peptide 1. Physiol Rev 87:1409–1439. doi:10.1152/physrev.00034.2006
Hong DM, Jeon Y, Lee CS, Kim HJ, Lee JM, Bahk JH, Kim KB, Hwang HY (2012) Effects of remote ischemic preconditioning with postconditioning in patients undergoing off-pump coronary artery bypass surgery–randomized controlled trial. Circ J 76:884–890. doi:10.1253/circj.CJ-11-1068
Hoole SP, Heck PM, Sharples L, Khan SN, Duehmke R, Densem CG, Clarke SC, Shapiro LM, Schofield PM, O’Sullivan M, Dutka DP (2009) Cardiac Remote Ischemic Preconditioning in Coronary Stenting (CRISP Stent) Study: a prospective, randomized control trial. Circulation 119:820–827. doi:10.1161/CIRCULATIONAHA.108.809723
Hori M, Kitakaze M, Sato H, Takashima S, Iwakura K, Inoue M, Kitabatake A, Kamada T (1991) Staged reperfusion attenuates myocardial stunning in dogs. Role of transient acidosis during early reperfusion. Circulation 84:2135–2145. doi:10.1161/01.CIR.84.5.2135
Hotta H, Miura T, Miki T, Togashi N, Maeda T, Kim SJ, Tanno M, Yano T, Kuno A, Itoh T, Satoh T, Terashima Y, Ishikawa S, Shimamoto K (2010) Angiotensin II type 1 receptor-mediated upregulation of calcineurin activity underlies impairment of cardioprotective signaling in diabetic hearts. Circ Res 106:129–132. doi:10.1161/CIRCRESAHA.109.205385
Huang CH, Chiang CY, Pen RH, Tsai MS, Chen HW, Hsu CY, Wang TD, Ma MH, Chen SC, Chen WJ (2015) Hypothermia treatment preserves mitochondrial integrity and viability of cardiomyocytes after ischaemic reperfusion injury. Injury 46:233–239. doi:10.1016/j.injury.2014.10.055
Ibanez B, Heusch G, Ovize M, Van de Werf F (2015) Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol 65:1454–1471. doi:10.1016/j.jacc.2015.02.032
Ibanez B, Macaya C, Sanchez-Brunete V, Pizarro G, Fernandez-Friera L, Mateos A, Fernandez-Ortiz A, Garcia-Ruiz JM, Garcia-Alvarez A, Iniguez A, Jimenez-Borreguero J, Lopez-Romero P, Fernandez-Jimenez R, Goicolea J, Ruiz-Mateos B, Bastante T, Arias M, Iglesias-Vazquez JA, Rodriguez MD, Escalera N, Acebal C, Cabrera JA, Valenciano J, Perez de Prado A, Fernandez-Campos MJ, Casado I, Garcia-Rubira JC, Garcia-Prieto J, Sanz-Rosa D, Cuellas C, Hernandez-Antolin R, Albarran A, Fernandez-Vazquez F, de la Torre-Hernandez JM, Pocock S, Sanz G, Fuster V (2013) Effect of early metoprolol on infarct size in ST-segment-elevation myocardial infarction patients undergoing primary percutaneous coronary intervention: the Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction (METOCARD-CNIC) trial. Circulation 128:1495–1503. doi:10.1161/CIRCULATIONAHA.113.003653
Ibanez B, Prat-Gonzalez S, Speidl WS, Vilahur G, Pinero A, Cimmino G, Garcia MJ, Fuster V, Sanz J, Badimon JJ (2007) Early metoprolol administration before coronary reperfusion results in increased myocardial salvage: analysis of ischemic myocardium at risk using cardiac magnetic resonance. Circulation 115:2909–2916. doi:10.1161/CIRCULATIONAHA.106.679639
Iliodromitis EK, Koutelou M, Paraskevaidis IA, Theodorakos A, Farmakis D, Tsoutsanis J, Kremastinos DT (2008) Treadmill exercise test with dual isotope scintigraphy documents the second window of preconditioning in humans. Atherosclerosis 198:122–128. doi:10.1016/j.atherosclerosis.2007.10.036
Inserte J, Cardona M, Poncelas-Nozal M, Hernando V, Vilardosa U, Aluja D, Parra VM, Sanchis D, Garcia-Dorado D (2016) Studies on the role of apoptosis after transient myocardial ischemia: genetic deletion of the executioner caspases-3 and -7 does not limit infarct size and ventricular remodeling. Basic Res Cardiol 111:18. doi:10.1007/s00395-016-0537-6
Inserte J, Garcia-Dorado D (2015) The cGMP/PKG pathway as a common mediator of cardioprotection: translatability and mechanism. Br J Pharmacol 172:1996–2009. doi:10.1111/bph.12959
Inserte J, Hernando V, Garcia-Dorado D (2012) Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc Res 96:23–31. doi:10.1093/cvr/cvs232
Inserte J, Hernando V, Vilardosa U, Abad E, Poncelas-Nozal M, Garcia-Dorado D (2013) Activation of cGMP/protein kinase G pathway in postconditioned myocardium depends on reduced oxidative stress and preserved endothelial nitric oxide synthase coupling. J Am Heart Assoc 2:e005975. doi:10.1161/JAHA.112.005975
Inserte J, Ruiz-Meana M, Rodriguez-Sinovas A, Barba I, Garcia-Dorado D (2011) Contribution of delayed intracellular pH recovery to ischemic postconditioning protection. Antioxid Redox Signal 14:923–939. doi:10.1089/ars.2010.3312
Itoh T, Kouzu H, Miki T, Tanno M, Kuno A, Sato T, Sunaga D, Murase H, Miura T (2012) Cytoprotective regulation of the mitochondrial permeability transition pore is impaired in type 2 diabetic Goto–Kakizaki rat hearts. J Mol Cell Cardiol 53:870–879. doi:10.1016/j.yjmcc.2012.10.001
Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A (2008) Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med 45:1–17. doi:10.1016/j.freeradbiomed.2008.03.011
Jensen RV, Stottrup NB, Kristiansen SB, Botker HE (2012) Release of a humoral circulating cardioprotective factor by remote ischemic preconditioning is dependent on preserved neural pathways in diabetic patients. Basic Res Cardiol 107:285. doi:10.1007/s00395-012-0285-1
Karlsson LO, Zhou AX, Larsson E, Astrom-Olsson K, Mansson C, Akyurek LM, Grip L (2010) Cyclosporine does not reduce myocardial infarct size in a porcine ischemia–reperfusion model. J Cardiovasc Pharmacol Ther 15:182–189. doi:10.1177/1074248410362074
Karuppasamy P, Chaubey S, Dew T, Musto R, Sherwood R, Desai J, John L, Shah AM, Marber MS, Kunst G (2011) Remote intermittent ischemia before coronary artery bypass graft surgery: a strategy to reduce injury and inflammation? Basic Res Cardiol 106:511–519. doi:10.1007/s00395-011-0185-9
Khalili H, Patel VG, Mayo HG, de Lemos JA, Brilakis ES, Banerjee S, Bavry AA, Bhatt DL, Kumbhani DJ (2014) Surrogate and clinical outcomes following ischemic postconditioning during primary percutaneous coronary intervention of ST—segment elevation myocardial infarction: a meta-analysis of 15 randomized trials. Catheter Cardiovasc Interv 84:978–986. doi:10.1002/ccd.25581
Khan AR, Binabdulhak AA, Alastal Y, Khan S, Faricy-Beredo BM, Luni FK, Lee WM, Khuder S, Tinkel J (2014) Cardioprotective role of ischemic postconditioning in acute myocardial infarction: a systematic review and meta-analysis. Am Heart J 168(512–521):e514. doi:10.1016/j.ahj.2014.06.021
Kim EK, Hahn JY, Song YB, Lee SC, Choi JH, Choi SH, Lee SH, Choe YH, Gwon HC (2015) Effect of ischemic postconditioning on myocardial salvage in patients undergoing primary percutaneous coronary intervention for ST-segment elevation myocardial infarction: cardiac magnetic resonance substudy of the POST randomized trial. Int J Cardiovasc Imaging 31:629–637. doi:10.1007/s10554-015-0589-y
Kin H, Zhao ZQ, Sun HY, Wang NP, Corvera JS, Halkos ME, Kerendi F, Guyton RA, Vinten-Johansen J (2004) Postconditioning attenuates myocardial ischemia–reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res 62:74–85. doi:10.1016/j.cardiores.2004.01.006
Knowlton AA, Brecher P, Apstein CS (1991) Rapid expression of heat shock protein in the rabbit after brief cardiac ischemia. J Clin Invest 87:139–147. doi:10.1172/JCI114963
Kottenberg E, Musiolik J, Thielmann M, Jakob H, Peters J, Heusch G (2014) Interference of propofol with signal transducer and activator of transcription 5 activation and cardioprotection by remote ischemic preconditioning during coronary artery bypass grafting. J Thorac Cardiovasc Surg 147:376–382. doi:10.1016/j.jtcvs.2013.01.005
Kottenberg E, Thielmann M, Bergmann L, Heine T, Jakob H, Heusch G, Peters J (2012) Protection by remote ischemic preconditioning during coronary artery bypass graft surgery with isoflurane but not propofol—a clinical trial. Acta Anaesthesiol Scand 56:30–38. doi:10.1111/j.1399-6576.2011.02585.x
Kuzuya T, Hoshida S, Yamashita N, Fuji H, Oe H, Hori M, Kamada T, Tada M (1993) Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circ Res 72:1293–1299. doi:10.1161/01.RES.72.6.1293
Kwan JC, Gao L, Macdonald PS, Hicks M (2015) Cardio-protective signalling by glyceryl trinitrate and cariporide in a model of donor heart preservation. Heart Lung Circ 24:306–318. doi:10.1016/j.hlc.2014.10.001
Lacerda L, McCarthy J, Mungly SF, Lynn EG, Sack MN, Opie LH, Lecour S (2010) TNFalpha protects cardiac mitochondria independently of its cell surface receptors. Basic Res Cardiol 105:751–762. doi:10.1007/s00395-010-0113-4
Lacerda L, Somers S, Opie LH, Lecour S (2009) Ischaemic postconditioning protects against reperfusion injury via the SAFE pathway. Cardiovasc Res 84:201–208. doi:10.1093/cvr/cvp274
Lambiase PD, Edwards RJ, Cusack MR, Bucknall CA, Redwood SR, Marber MS (2003) Exercise-induced ischemia initiates the second window of protection in humans independent of collateral recruitment. J Am Coll Cardiol 41:1174–1182. doi:10.1016/S0735-1097(03)00055-X
Lamont KT, Somers S, Lacerda L, Opie LH, Lecour S (2011) Is red wine a SAFE sip away from cardioprotection? Mechanisms involved in resveratrol- and melatonin-induced cardioprotection. J Pineal Res 50:374–380. doi:10.1111/j.1600-079X.2010.00853.x
Lecour S (2009) Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: does it go beyond the RISK pathway? J Mol Cell Cardiol 47:32–40. doi:10.1016/j.yjmcc.2009.03.019
Lecour S (2009) Multiple protective pathways against reperfusion injury: a SAFE path without Aktion? J Mol Cell Cardiol 46:607–609. doi:10.1016/j.yjmcc.2009.01.003
Lecour S, Smith RM, Woodward B, Opie LH, Rochette L, Sack MN (2002) Identification of a novel role for sphingolipid signaling in TNF alpha and ischemic preconditioning mediated cardioprotection. J Mol Cell Cardiol 34:509–518. doi:10.1006/jmcc.2002.1533
Lecour S, Suleman N, Deuchar GA, Somers S, Lacerda L, Huisamen B, Opie LH (2005) Pharmacological preconditioning with tumor necrosis factor-alpha activates signal transducer and activator of transcription-3 at reperfusion without involving classic prosurvival kinases (Akt and extracellular signal-regulated kinase). Circulation 112:3911–3918. doi:10.1161/CIRCULATIONAHA.105.581058
Li GC, Vasquez JA, Gallagher KP, Lucchesi BR (1990) Myocardial protection with preconditioning. Circulation 82:609–619. doi:10.1161/01.CIR.82.2.609
Li J, Rohailla S, Gelber N, Rutka J, Sabah N, Gladstone RA, Wei C, Hu P, Kharbanda RK, Redington AN (2014) MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res Cardiol 109:423. doi:10.1007/s00395-014-0423-z
Li J, Xuan W, Yan R, Tropak MB, Jean-St-Michel E, Liang W, Gladstone R, Backx PH, Kharbanda RK, Redington AN (2011) Remote preconditioning provides potent cardioprotection via PI3K/Akt activation and is associated with nuclear accumulation of beta-catenin. Clin Sci (Lond) 120:451–462. doi:10.1042/CS20100466
Lim SY, Yellon DM, Hausenloy DJ (2010) The neural and humoral pathways in remote limb ischemic preconditioning. Basic Res Cardiol 105:651–655. doi:10.1007/s00395-010-0099-y
Liu GS, Thornton J, Van Winkle DM, Stanley AW, Olsson RA, Downey JM (1991) Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation 84:350–356. doi:10.1161/01.CIR.84.1.350
Liu Y, Gao WD, O’Rourke B, Marban E (1996) Synergistic modulation of ATP-sensitive K+ currents by protein kinase C and adenosine. Implications for ischemic preconditioning. Circ Res 78:443–454. doi:10.1161/01.RES.78.3.443
Liu Y, Ytrehus K, Downey JM (1994) Evidence that translocation of protein kinase C is a key event during ischemic preconditioning of rabbit myocardium. J Mol Cell Cardiol 26:661–668. doi:10.1006/jmcc.1994.1078
Lonborg J, Kelbaek H, Vejlstrup N, Botker HE, Kim WY, Holmvang L, Jorgensen E, Helqvist S, Saunamaki K, Terkelsen CJ, Schoos MM, Kober L, Clemmensen P, Treiman M, Engstrom T (2012) Exenatide reduces final infarct size in patients with ST-segment-elevation myocardial infarction and short-duration of ischemia. Circ Cardiovasc Interv 5:288–295. doi:10.1161/CIRCINTERVENTIONS.112.968388
Lonborg J, Vejlstrup N, Kelbaek H, Botker HE, Kim WY, Mathiasen AB, Jorgensen E, Helqvist S, Saunamaki K, Clemmensen P, Holmvang L, Thuesen L, Krusell LR, Jensen JS, Kober L, Treiman M, Holst JJ, Engstrom T (2012) Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. Eur Heart J 33:1491–1499. doi:10.1093/eurheartj/ehr309
Loukogeorgakis SP, Panagiotidou AT, Broadhead MW, Donald A, Deanfield JE, MacAllister RJ (2005) Remote ischemic preconditioning provides early and late protection against endothelial ischemia–reperfusion injury in humans: role of the autonomic nervous system. J Am Coll Cardiol 46:450–456. doi:10.1016/j.jacc.2005.04.044
Lucchinetti E, Bestmann L, Feng J, Freidank H, Clanachan AS, Finegan BA, Zaugg M (2012) Remote ischemic preconditioning applied during isoflurane inhalation provides no benefit to the myocardium of patients undergoing on-pump coronary artery bypass graft surgery: lack of synergy or evidence of antagonism in cardioprotection? Anesthesiology 116:296–310. doi:10.1097/ALN.0b013e318242349a
Ludman AJ, Hausenloy DJ, Babu G, Hasleton J, Venugopal V, Boston-Griffiths E, Yap J, Lawrence D, Hayward M, Kolvekar S, Bognolo G, Rees P, Yellon DM (2011) Failure to recapture cardioprotection with high-dose atorvastatin in coronary artery bypass surgery: a randomised controlled trial. Basic Res Cardiol 106:1387–1395. doi:10.1007/s00395-011-0209-5
Ludman AJ, Yellon DM, Hasleton J, Ariti C, Babu GG, Boston-Griffiths E, Venugopal V, Walker M, Holdright D, Swanton H, Crake T, Brull D, Moon JC, Puranik R, Muthurangu V, Taylor A, Hausenloy DJ (2011) Effect of erythropoietin as an adjunct to primary percutaneous coronary intervention: a randomised controlled clinical trial. Heart 97:1560–1565. doi:10.1136/hrt.2011.223867
Luo SJ, Zhou YJ, Shi DM, Ge HL, Wang JL, Liu RF (2013) Remote ischemic preconditioning reduces myocardial injury in patients undergoing coronary stent implantation. Can J Cardiol 29:1084–1089. doi:10.1016/j.cjca.2012.11.022
Mankad PS, Amrani M, Rothery S, Severs NJ, Yacoub MH (1997) Relative susceptibility of endothelium and myocardial cells to ischaemia-reperfusion injury. Acta Physiol Scand 161:103–112. doi:10.1046/j.1365-201X.1997.00195.x
Marber MS, Latchman DS, Walker JM, Yellon DM (1993) Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation 88:1264–1272. doi:10.1161/01.CIR.88.3.1264
Maroko PR, Libby P, Covell JW, Sobel BE, Ross J Jr, Braunwald E (1972) Precordial S-T segment elevation mapping: an atraumatic method for assessing alterations in the extent of myocardial ischemic injury. The effects of pharmacologic and hemodynamic interventions. Am J Cardiol 29:223–230. doi:10.1016/0002-9149(72)90633-9
Mateos A, Garcia-Lunar I, Garcia-Ruiz JM, Pizarro G, Fernandez-Jimenez R, Huertas P, Garcia-Alvarez A, Fernandez-Friera L, Bravo J, Flores-Arias J, Barreiro MV, Chayan-Zas L, Corral E, Fuster V, Sanchez-Brunete V, Ibanez B, Investigators M-C (2015) Efficacy and safety of out-of-hospital intravenous metoprolol administration in anterior ST-segment elevation acute myocardial infarction: insights from the METOCARD-CNIC trial. Ann Emerg Med 65:318–324. doi:10.1016/j.annemergmed.2014.07.010
McCrindle BW, Clarizia NA, Khaikin S, Holtby HM, Manlhiot C, Schwartz SM, Caldarone CA, Coles JG, Van Arsdell GS, Scherer SW, Redington AN (2014) Remote ischemic preconditioning in children undergoing cardiac surgery with cardiopulmonary bypass: a single-center double-blinded randomized trial. J Am Heart Assoc. doi:10.1161/JAHA.114.000964
McCully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S (2004) Differential contribution of necrosis and apoptosis in myocardial ischemia–reperfusion injury. Am J Physiol Heart Circ Physiol 286:H1923–H1935. doi:10.1152/ajpheart.00935.2003
Merlocco AC, Redington KL, Disenhouse T, Strantzas SC, Gladstone R, Wei C, Tropak MB, Manlhiot C, Li J, Redington AN (2014) Transcutaneous electrical nerve stimulation as a novel method of remote preconditioning: in vitro validation in an animal model and first human observations. Basic Res Cardiol 109:406. doi:10.1007/s00395-014-0406-0
Mewton N, Croisille P, Gahide G, Rioufol G, Bonnefoy E, Sanchez I, Cung TT, Sportouch C, Angoulvant D, Finet G, Andre-Fouet X, Derumeaux G, Piot C, Vernhet H, Revel D, Ovize M (2010) Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. J Am Coll Cardiol 55:1200–1205. doi:10.1016/j.jacc.2009.10.052
Meybohm P, Bein B, Brosteanu O, Cremer J, Gruenewald M, Stoppe C, Coburn M, Schaelte G, Boning A, Niemann B, Roesner J, Kletzin F, Strouhal U, Reyher C, Laufenberg-Feldmann R, Ferner M, Brandes IF, Bauer M, Stehr SN, Kortgen A, Wittmann M, Baumgarten G, Meyer-Treschan T, Kienbaum P, Heringlake M, Schon J, Sander M, Treskatsch S, Smul T, Wolwender E, Schilling T, Fuernau G, Hasenclever D, Zacharowski K, Collaborators RIS (2015) A multicenter trial of remote ischemic preconditioning for heart surgery. N Engl J Med 373:1397–1407. doi:10.1056/NEJMoa1413579
Miki T, Itoh T, Sunaga D, Miura T (2012) Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovasc Diabetol 11:67. doi:10.1186/1475-2840-11-67
Miki T, Miura T, Hotta H, Tanno M, Yano T, Sato T, Terashima Y, Takada A, Ishikawa S, Shimamoto K (2009) Endoplasmic reticulum stress in diabetic hearts abolishes erythropoietin-induced myocardial protection by impairment of phospho-glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeability transition. Diabetes 58:2863–2872. doi:10.2337/db09-0158
Mirabet M, Garcia-Dorado D, Inserte J, Barrabes JA, Lidon RM, Soriano B, Azevedo M, Padilla F, Agullo L, Ruiz-Meana M, Massaguer A, Pizcueta P, Soler-Soler J (2002) Platelets activated by transient coronary occlusion exacerbate ischemia–reperfusion injury in rat hearts. Am J Physiol Heart Circ Physiol 283:H1134–H1141. doi:10.1152/ajpheart.00065.2002
Miro-Casas E, Ruiz-Meana M, Agullo E, Stahlhofen S, Rodriguez-Sinovas A, Cabestrero A, Jorge I, Torre I, Vazquez J, Boengler K, Schulz R, Heusch G, Garcia-Dorado D (2009) Connexin43 in cardiomyocyte mitochondria contributes to mitochondrial potassium uptake. Cardiovasc Res 83:747–756. doi:10.1093/cvr/cvp157
Miura T, Tanno M (2012) The mPTP and its regulatory proteins: final common targets of signalling pathways for protection against necrosis. Cardiovasc Res 94:181–189. doi:10.1093/cvr/cvr302
Miura T, Tanno M, Sato T (2010) Mitochondrial kinase signalling pathways in myocardial protection from ischaemia/reperfusion-induced necrosis. Cardiovasc Res 88:7–15. doi:10.1093/cvr/cvq206
Monteiro P, Goncalves L, Providencia LA (2005) Diabetes and cardiovascular disease: the road to cardioprotection. Heart 91:1621–1625. doi:10.1136/hrt.2005.063008
Munk K, Andersen NH, Schmidt MR, Nielsen SS, Terkelsen CJ, Sloth E, Botker HE, Nielsen TT, Poulsen SH (2010) Remote ischemic conditioning in patients with myocardial infarction treated with primary angioplasty: impact on left ventricular function assessed by comprehensive echocardiography and gated single-photon emission CT. Circ Cardiovasc Imaging 3:656–662. doi:10.1161/CIRCIMAGING.110.957340
Murry CE, Jennings RB, Reimer KA (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74:1124–1136. doi:10.1161/01.CIR.74.5.1124
Murry CE, Richard VJ, Reimer KA, Jennings RB (1990) Ischemic preconditioning slows energy metabolism and delays ultrastructural damage during a sustained ischemic episode. Circ Res 66:913–931. doi:10.1161/01.RES.66.4.913
Musiolik J, van Caster P, Skyschally A, Boengler K, Gres P, Schulz R, Heusch G (2010) Reduction of infarct size by gentle reperfusion without activation of reperfusion injury salvage kinases in pigs. Cardiovasc Res 85:110–117. doi:10.1093/cvr/cvp271
Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y (2005) Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434:652–658. doi:10.1038/nature03317
Noda T, Minatoguchi S, Fujii K, Hori M, Ito T, Kanmatsuse K, Matsuzaki M, Miura T, Nonogi H, Tada M, Tanaka M, Fujiwara H (1999) Evidence for the delayed effect in human ischemic preconditioning: prospective multicenter study for preconditioning in acute myocardial infarction. J Am Coll Cardiol 34:1966–1974. doi:10.1016/S0735-1097(99)00462-3
Ong SB, Dongworth RK, Cabrera-Fuentes HA, Hausenloy DJ (2015) Role of the MPTP in conditioning the heart—translatability and mechanism. Br J Pharmacol 172:2074–2084. doi:10.1111/bph.13013
Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ (2015) The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol 78:23–34. doi:10.1016/j.yjmcc.2014.11.005
Ottani F, Latini R, Staszewsky L, La Vecchia L, Locuratolo N, Sicuro M, Masson S, Barlera S, Milani V, Lombardi M, Costalunga A, Mollichelli N, Santarelli A, De Cesare N, Sganzerla P, Boi A, Maggioni AP, Limbruno U, Investigators C (2016) Cyclosporine A in reperfused myocardial infarction: the multicenter, controlled, open-label CYCLE trial. J Am Coll Cardiol 67:365–374. doi:10.1016/j.jacc.2015.10.081
Padilla F, Garcia-Dorado D, Rodriguez-Sinovas A, Ruiz-Meana M, Inserte J, Soler-Soler J (2003) Protection afforded by ischemic preconditioning is not mediated by effects on cell-to-cell electrical coupling during myocardial ischemia–reperfusion. Am J Physiol Heart Circ Physiol 285:H1909–H1916. doi:10.1152/ajpheart.00438.2003
Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Liu GS, Heusch G, Cohen MV, Downey JM (2000) Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ Res 87:460–466. doi:10.1161/01.RES.87.6.460
Paraskevaidis IA, Iliodromitis EK, Mavrogeni S, Karavolias GK, Theodorakis GN, Georgiadis M, Kremastinos DT (2005) Repeated exercise stress testing identifies early and late preconditioning. Int J Cardiol 98:221–226. doi:10.1016/j.ijcard.2003.10.040
Pedersen CM, Cruden NL, Schmidt MR, Lau C, Botker HE, Kharbanda RK, Newby DE (2011) Remote ischemic preconditioning prevents systemic platelet activation associated with ischemia–reperfusion injury in humans. J Thromb Haemost 9:404–407. doi:10.1111/j.1538-7836.2010.04142.x
Pichot S, Mewton N, Bejan-Angoulvant T, Roubille F, Rioufol G, Giraud C, Boussaha I, Lairez O, Elbaz M, Piot C, Angoulvant D, Ovize M (2015) Influence of cardiovascular risk factors on infarct size and interaction with mechanical ischaemic postconditioning in ST-elevation myocardial infarction. Open Heart 2:e000175. doi:10.1136/openhrt-2014-000175
Pickard JM, Botker HE, Crimi G, Davidson B, Davidson SM, Dutka D, Ferdinandy P, Ganske R, Garcia-Dorado D, Giricz Z, Gourine AV, Heusch G, Kharbanda R, Kleinbongard P, MacAllister R, McIntyre C, Meybohm P, Prunier F, Redington A, Robertson NJ, Suleiman MS, Vanezis A, Walsh S, Yellon DM, Hausenloy DJ (2015) Remote ischemic conditioning: from experimental observation to clinical application: report from the 8th Biennial Hatter Cardiovascular Institute Workshop. Basic Res Cardiol 110:453. doi:10.1007/s00395-014-0453-6
Pickard JM, Davidson SM, Hausenloy DJ, Yellon DM (2016) Co-dependence of the neural and humoral pathways in the mechanism of remote ischemic conditioning. Basic Res Cardiol 111:50. doi:10.1007/s00395-016-0568-z
Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, Andre-Fouet X, Revel D, Kirkorian G, Monassier JP, Derumeaux G, Ovize M (2008) Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 359:473–481. doi:10.1056/NEJMoa071142
Piper HM, Garcia-Dorado D, Ovize M (1998) A fresh look at reperfusion injury. Cardiovasc Res 38:291–300. doi:10.1016/S0008-6363(98)00033-9
Pizarro G, Fernandez-Friera L, Fuster V, Fernandez-Jimenez R, Garcia-Ruiz JM, Garcia-Alvarez A, Mateos A, Barreiro MV, Escalera N, Rodriguez MD, de Miguel A, Garcia-Lunar I, Parra-Fuertes JJ, Sanchez-Gonzalez J, Pardillos L, Nieto B, Jimenez A, Abejon R, Bastante T, Martinez de Vega V, Cabrera JA, Lopez-Melgar B, Guzman G, Garcia-Prieto J, Mirelis JG, Zamorano JL, Albarran A, Goicolea J, Escaned J, Pocock S, Iniguez A, Fernandez-Ortiz A, Sanchez-Brunete V, Macaya C, Ibanez B (2014) Long-term benefit of early pre-reperfusion metoprolol administration in patients with acute myocardial infarction: results from the METOCARD-CNIC trial (Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction). J Am Coll Cardiol 63:2356–2362. doi:10.1016/j.jacc.2014.03.014
Prasad A, Gossl M, Hoyt J, Lennon RJ, Polk L, Simari R, Holmes DR Jr, Rihal CS, Lerman A (2013) Remote ischemic preconditioning immediately before percutaneous coronary intervention does not impact myocardial necrosis, inflammatory response, and circulating endothelial progenitor cell counts: a single center randomized sham controlled trial. Catheter Cardiovasc Interv 81:930–936. doi:10.1002/ccd.24443
Prunier F, Angoulvant D, Saint Etienne C, Vermes E, Gilard M, Piot C, Roubille F, Elbaz M, Ovize M, Biere L, Jeanneteau J, Delepine S, Benard T, Abi-Khalil W, Furber A (2014) The RIPOST-MI study, assessing remote ischemic perconditioning alone or in combination with local ischemic postconditioning in ST-segment elevation myocardial infarction. Basic Res Cardiol 109:400. doi:10.1007/s00395-013-0400-y
Przyklenk K, Bauer B, Ovize M, Kloner RA, Whittaker P (1993) Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 87:893–899. doi:10.1161/01.CIR.87.3.893
Przyklenk K, Whittaker P (2013) Genesis of remote conditioning: action at a distance–’hypotheses non fingo’? J Cardiovasc Med (Hagerstown) 14:180–186. doi:10.2459/JCM.0b013e328358c8eb
Rahman IA, Mascaro JG, Steeds RP, Frenneaux MP, Nightingale P, Gosling P, Townsend P, Townend JN, Green D, Bonser RS (2010) Remote ischemic preconditioning in human coronary artery bypass surgery: from promise to disappointment? Circulation 122:S53–S59. doi:10.1161/CIRCULATIONAHA.109.926667
Rassaf T, Totzeck M, Hendgen-Cotta UB, Shiva S, Heusch G, Kelm M (2014) Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res 114:1601–1610. doi:10.1161/CIRCRESAHA.114.303822
Redington KL, Disenhouse T, Strantzas SC, Gladstone R, Wei C, Tropak MB, Dai X, Manlhiot C, Li J, Redington AN (2012) Remote cardioprotection by direct peripheral nerve stimulation and topical capsaicin is mediated by circulating humoral factors. Basic Res Cardiol 107:241. doi:10.1007/s00395-011-0241-5
Rentoukas I, Giannopoulos G, Kaoukis A, Kossyvakis C, Raisakis K, Driva M, Panagopoulou V, Tsarouchas K, Vavetsi S, Pyrgakis V, Deftereos S (2010) Cardioprotective role of remote ischemic periconditioning in primary percutaneous coronary intervention: enhancement by opioid action. JACC Cardiovasc Interv 3:49–55. doi:10.1016/j.jcin.2009.10.015
Rodriguez-Sinovas A, Boengler K, Cabestrero A, Gres P, Morente M, Ruiz-Meana M, Konietzka I, Miro E, Totzeck A, Heusch G, Schulz R, Garcia-Dorado D (2006) Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ Res 99:93–101. doi:10.1161/01.RES.0000230315.56904.de
Roolvink V, Ibanez B, Ottervanger JP, Pizarro G, van Royen N, Mateos A, Dambrink JH, Escalera N, Lipsic E, Albarran A, Fernandez-Ortiz A, Fernandez-Aviles F, Goicolea J, Botas J, Remkes W, Hernandez-Jaras V, Kedhi E, Zamorano JL, Navarro F, Alfonso F, Garcia-Lledo A, Alonso J, van Leeuwen M, Nijveldt R, Postma S, Kolkman E, Gosselink M, de Smet B, Rasoul S, Piek JJ, Fuster V, van ‘t Hof AW, Investigators E-B (2016) Early intravenous beta-blockers in patients with ST-segment elevation myocardial infarction before primary percutaneous coronary intervention. J Am Coll Cardiol 67:2705–2715. doi:10.1016/j.jacc.2016.03.522
Roos ST, Timmers L, Biesbroek PS, Nijveldt R, Kamp O, van Rossum AC, van Hout GP, Stella PR, Doevendans PA, Knaapen P, Velthuis BK, van Royen N, Voskuil M, Nap A, Appelman Y (2016) No benefit of additional treatment with exenatide in patients with an acute myocardial infarction. Int J Cardiol 220:809–814. doi:10.1016/j.ijcard.2016.06.283
Roubille F, Franck-Miclo A, Covinhes A, Lafont C, Cransac F, Combes S, Vincent A, Fontanaud P, Sportouch-Dukhan C, Redt-Clouet C, Nargeot J, Piot C, Barrere-Lemaire S (2011) Delayed postconditioning in the mouse heart in vivo. Circulation 124:1330–1336. doi:10.1161/CIRCULATIONAHA.111.031864
Ruiz-Meana M, Abellan A, Miro-Casas E, Agullo E, Garcia-Dorado D (2009) Role of sarcoplasmic reticulum in mitochondrial permeability transition and cardiomyocyte death during reperfusion. Am J Physiol Heart Circ Physiol 297:H1281–H1289. doi:10.1152/ajpheart.00435.2009
Ruiz-Meana M, Abellan A, Miro-Casas E, Garcia-Dorado D (2007) Opening of mitochondrial permeability transition pore induces hypercontracture in Ca2+ overloaded cardiac myocytes. Basic Res Cardiol 102:542–552. doi:10.1007/s00395-007-0675-y
Ruiz-Meana M, Fernandez-Sanz C, Garcia-Dorado D (2010) The SR-mitochondria interaction: a new player in cardiac pathophysiology. Cardiovasc Res 88:30–39. doi:10.1093/cvr/cvq225
Ruiz-Meana M, Garcia-Dorado D, Hofstaetter B, Piper HM, Soler-Soler J (1999) Propagation of cardiomyocyte hypercontracture by passage of Na(+) through gap junctions. Circ Res 85:280–287. doi:10.1161/01.RES.85.3.280
Ruiz-Meana M, Inserte J, Fernandez-Sanz C, Hernando V, Miro-Casas E, Barba I, Garcia-Dorado D (2011) The role of mitochondrial permeability transition in reperfusion-induced cardiomyocyte death depends on the duration of ischemia. Basic Res Cardiol 106:1259–1268. doi:10.1007/s00395-011-0225-5
Ruiz-Meana M, Nunez E, Miro-Casas E, Martinez-Acedo P, Barba I, Rodriguez-Sinovas A, Inserte J, Fernandez-Sanz C, Hernando V, Vazquez J, Garcia-Dorado D (2014) Ischemic preconditioning protects cardiomyocyte mitochondria through mechanisms independent of cytosol. J Mol Cell Cardiol 68:79–88. doi:10.1016/j.yjmcc.2014.01.001
Sato H, Jordan JE, Zhao ZQ, Sarvotham SS, Vinten-Johansen J (1997) Gradual reperfusion reduces infarct size and endothelial injury but augments neutrophil accumulation. Ann Thorac Surg 64:1099–1107. doi:10.1016/S0003-4975(97)00734-0
Schmidt MR, Smerup M, Konstantinov IE, Shimizu M, Li J, Cheung M, White PA, Kristiansen SB, Sorensen K, Dzavik V, Redington AN, Kharbanda RK (2007) Intermittent peripheral tissue ischemia during coronary ischemia reduces myocardial infarction through a KATP-dependent mechanism: first demonstration of remote ischemic perconditioning. Am J Physiol Heart Circ Physiol 292:H1883–H1890. doi:10.1152/ajpheart.00617.2006
Schulman D, Latchman DS, Yellon DM (2002) Urocortin protects the heart from reperfusion injury via upregulation of p42/p44 MAPK signaling pathway. Am J Physiol Heart Circ Physiol 283:H1481–H1488. doi:10.1152/ajpheart.01089.2001
Schulz R, Boengler K, Totzeck A, Luo Y, Garcia-Dorado D, Heusch G (2007) Connexin 43 in ischemic pre- and postconditioning. Heart Fail Rev 12:261–266. doi:10.1007/s10741-007-9032-3
Shimizu M, Saxena P, Konstantinov IE, Cherepanov V, Cheung MM, Wearden P, Zhangdong H, Schmidt M, Downey GP, Redington AN (2010) Remote ischemic preconditioning decreases adhesion and selectively modifies functional responses of human neutrophils. J Surg Res 158:155–161. doi:10.1016/j.jss.2008.08.010
Shimizu M, Tropak M, Diaz RJ, Suto F, Surendra H, Kuzmin E, Li J, Gross G, Wilson GJ, Callahan J, Redington AN (2009) Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: evidence suggesting cross-species protection. Clin Sci (Lond) 117:191–200. doi:10.1042/CS20080523
Siegmund B, Klietz T, Schwartz P, Piper HM (1991) Temporary contractile blockade prevents hypercontracture in anoxic-reoxygenated cardiomyocytes. Am J Physiol 260:H426–H435
Siegmund B, Zude R, Piper HM (1992) Recovery of anoxic-reoxygenated cardiomyocytes from severe Ca2+ overload. Am J Physiol 263:H1262–H1269
Singh B, Randhawa PK, Singh N, Jaggi AS (2016) Investigations on the role of leukotrienes in remote hind limb preconditioning-induced cardioprotection in rats. Life Sci 152:238–243. doi:10.1016/j.lfs.2016.04.005
Sivaraman V, Hausenloy DJ, Wynne AM, Yellon DM (2010) Preconditioning the diabetic human myocardium. J Cell Mol Med 14:1740–1746. doi:10.1111/j.1582-4934.2009.00796.x
Sivaraman V, Pickard JM, Hausenloy DJ (2015) Remote ischaemic conditioning: cardiac protection from afar. Anaesthesia 70:732–748. doi:10.1111/anae.12973
Skyschally A, Gent S, Amanakis G, Schulte C, Kleinbongard P, Heusch G (2015) Across-species transfer of protection by remote ischemic preconditioning with species-specific myocardial signal transduction by reperfusion injury salvage kinase and survival activating factor enhancement pathways. Circ Res 117:279–288. doi:10.1161/CIRCRESAHA.117.306878
Skyschally A, Schulz R, Heusch G (2010) Cyclosporine A at reperfusion reduces infarct size in pigs. Cardiovasc Drugs Ther 24:85–87. doi:10.1007/s10557-010-6219-y
Skyschally A, van Caster P, Boengler K, Gres P, Musiolik J, Schilawa D, Schulz R, Heusch G (2009) Ischemic postconditioning in pigs: no causal role for RISK activation. Circ Res 104:15–18. doi:10.1161/CIRCRESAHA.108.186429
Sloth AD, Schmidt MR, Munk K, Kharbanda RK, Redington AN, Schmidt M, Pedersen L, Sorensen HT, Botker HE, Investigators C (2014) Improved long-term clinical outcomes in patients with ST-elevation myocardial infarction undergoing remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention. Eur Heart J 35:168–175. doi:10.1093/eurheartj/eht369
Sloth AD, Schmidt MR, Munk K, Schmidt M, Pedersen L, Sorensen HT, Enemark U, Parner ET, Botker HE, Investigators C (2016) Cost-effectiveness of remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention in patients with ST-elevation myocardial infarction. Eur Heart J Acute Cardiovasc Care. doi:10.1177/2048872615626657
Somers SJ, Frias M, Lacerda L, Opie LH, Lecour S (2012) Interplay between SAFE and RISK pathways in sphingosine-1-phosphate-induced cardioprotection. Cardiovasc Drugs Ther 26:227–237. doi:10.1007/s10557-012-6376-2
Sonne DP, Engstrom T, Treiman M (2008) Protective effects of GLP-1 analogues exendin-4 and GLP-1(9-36) amide against ischemia–reperfusion injury in rat heart. Regul Pept 146:243–249. doi:10.1016/j.regpep.2007.10.001
Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L’Huillier I, Aupetit JF, Bonnefoy E, Finet G, Andre-Fouet X, Ovize M (2005) Postconditioning the human heart. Circulation 112:2143–2148. doi:10.1161/CIRCULATIONAHA.105.558122
Steensrud T, Li J, Dai X, Manlhiot C, Kharbanda RK, Tropak M, Redington A (2010) Pretreatment with the nitric oxide donor SNAP or nerve transection blocks humoral preconditioning by remote limb ischemia or intra-arterial adenosine. Am J Physiol Heart Circ Physiol 299:H1598–H1603. doi:10.1152/ajpheart.00396.2010
Suleman N, Somers S, Smith R, Opie LH, Lecour SC (2008) Dual activation of STAT-3 and Akt is required during the trigger phase of ischaemic preconditioning. Cardiovasc Res 79:127–133. doi:10.1093/cvr/cvn067
Tanno M, Kuno A, Ishikawa S, Miki T, Kouzu H, Yano T, Murase H, Tobisawa T, Ogasawara M, Horio Y, Miura T (2014) Translocation of glycogen synthase kinase-3beta (GSK-3beta), a trigger of permeability transition, is kinase activity-dependent and mediated by interaction with voltage-dependent anion channel 2 (VDAC2). J Biol Chem 289:29285–29296. doi:10.1074/jbc.M114.563924
Thibault H, Piot C, Staat P, Bontemps L, Sportouch C, Rioufol G, Cung TT, Bonnefoy E, Angoulvant D, Aupetit JF, Finet G, Andre-Fouet X, Macia JC, Raczka F, Rossi R, Itti R, Kirkorian G, Derumeaux G, Ovize M (2008) Long-term benefit of postconditioning. Circulation 117:1037–1044. doi:10.1161/CIRCULATIONAHA.107.729780
Thielmann M, Kottenberg E, Boengler K, Raffelsieper C, Neuhaeuser M, Peters J, Jakob H, Heusch G (2010) Remote ischemic preconditioning reduces myocardial injury after coronary artery bypass surgery with crystalloid cardioplegic arrest. Basic Res Cardiol 105:657–664. doi:10.1007/s00395-010-0104-5
Thielmann M, Kottenberg E, Kleinbongard P, Wendt D, Gedik N, Pasa S, Price V, Tsagakis K, Neuhauser M, Peters J, Jakob H, Heusch G (2013) Cardioprotective and prognostic effects of remote ischaemic preconditioning in patients undergoing coronary artery bypass surgery: a single-centre randomised, double-blind, controlled trial. Lancet 382:597–604. doi:10.1016/S0140-6736(13)61450-6
Thuny F, Lairez O, Roubille F, Mewton N, Rioufol G, Sportouch C, Sanchez I, Bergerot C, Thibault H, Cung TT, Finet G, Argaud L, Revel D, Derumeaux G, Bonnefoy-Cudraz E, Elbaz M, Piot C, Ovize M, Croisille P (2012) Post-conditioning reduces infarct size and edema in patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol 59:2175–2181. doi:10.1016/j.jacc.2012.03.026
Timmers L, Henriques JP, de Kleijn DP, Devries JH, Kemperman H, Steendijk P, Verlaan CW, Kerver M, Piek JJ, Doevendans PA, Pasterkamp G, Hoefer IE (2009) Exenatide reduces infarct size and improves cardiac function in a porcine model of ischemia and reperfusion injury. J Am Coll Cardiol 53:501–510. doi:10.1016/j.jacc.2008.10.033
Treiman M, Elvekjaer M, Engstrom T, Jensen JS (2010) Glucagon-like peptide 1—a cardiologic dimension. Trends Cardiovasc Med 20:8–12. doi:10.1016/j.tcm.2010.02.012
Tsang A, Hausenloy DJ, Mocanu MM, Carr RD, Yellon DM (2005) Preconditioning the diabetic heart: the importance of Akt phosphorylation. Diabetes 54:2360–2364. doi:10.2337/diabetes.54.8.2360
Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM (2004) Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ Res 95:230–232. doi:10.1161/01.RES.0000138303.76488.fe
Tsao PS, Aoki N, Lefer DJ, Johnson G 3rd, Lefer AM (1990) Time course of endothelial dysfunction and myocardial injury during myocardial ischemia and reperfusion in the cat. Circulation 82:1402–1412. doi:10.1161/01.CIR.82.4.1402
Turrell HE, Thaitirarot C, Crumbie H, Rodrigo G (2014) Remote ischemic preconditioning of cardiomyocytes inhibits the mitochondrial permeability transition pore independently of reduced calcium-loading or sarcKATP channel activation. Physiol Rep. doi:10.14814/phy2.12231
Varadarajan SG, An J, Novalija E, Smart SC, Stowe DF (2001) Changes in [Na(+)](i), compartmental [Ca(2+)], and NADH with dysfunction after global ischemia in intact hearts. Am J Physiol Heart Circ Physiol 280:H280–H293
Varga ZV, Giricz Z, Bencsik P, Madonna R, Gyongyosi M, Schulz R, Mayr M, Thum T, Puskas LG, Ferdinandy P (2015) Functional genomics of cardioprotection by ischemic conditioning and the influence of comorbid conditions: implications in target identification. Curr Drug Targets 16:904–911. doi:10.2174/1389450116666150427154203
Venugopal V, Hausenloy DJ, Ludman A, Di Salvo C, Kolvekar S, Yap J, Lawrence D, Bognolo J, Yellon DM (2009) Remote ischaemic preconditioning reduces myocardial injury in patients undergoing cardiac surgery with cold-blood cardioplegia: a randomised controlled trial. Heart 95:1567–1571. doi:10.1136/hrt.2008.155770
Vinten-Johansen J, Johnston WE, Mills SA, Faust KB, Geisinger KR, DeMasi RJ, Cordell AR (1988) Reperfusion injury after temporary coronary occlusion. J Thorac Cardiovasc Surg 95:960–968
Walker DM, Pasini E, Kucukoglu S, Marber MS, Iliodromitis E, Ferrari R, Yellon DM (1993) Heat stress limits infarct size in the isolated perfused rabbit heart. Cardiovasc Res 27:962–967. doi:10.1093/cvr/27.6.962
Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, Horrow J, Husted S, James S, Katus H, Mahaffey KW, Scirica BM, Skene A, Steg PG, Storey RF, Harrington RA, Investigators P, Freij A, Thorsen M (2009) Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 361:1045–1057. doi:10.1056/NEJMoa0904327
Walsh SR, Boyle JR, Tang TY, Sadat U, Cooper DG, Lapsley M, Norden AG, Varty K, Hayes PD, Gaunt ME (2009) Remote ischemic preconditioning for renal and cardiac protection during endovascular aneurysm repair: a randomized controlled trial. J Endovasc Ther 16:680–689. doi:10.1583/09-2817.1
Webb IG, Williams R, Marber MS (2009) Lizard spit and reperfusion injury. J Am Coll Cardiol 53:511–513. doi:10.1016/j.jacc.2008.11.006
Wei M, Xin P, Li S, Tao J, Li Y, Li J, Liu M, Li J, Zhu W, Redington AN (2011) Repeated remote ischemic postconditioning protects against adverse left ventricular remodeling and improves survival in a rat model of myocardial infarction. Circ Res 108:1220–1225. doi:10.1161/CIRCRESAHA.110.236190
Weinbrenner C, Nelles M, Herzog N, Sarvary L, Strasser RH (2002) Remote preconditioning by infrarenal occlusion of the aorta protects the heart from infarction: a newly identified non-neuronal but PKC-dependent pathway. Cardiovasc Res 55:590–601. doi:10.1016/S0008-6363(02)00446-7
Wever KE, Hooijmans CR, Riksen NP, Sterenborg TB, Sena ES, Ritskes-Hoitinga M, Warle MC (2015) Determinants of the efficacy of cardiac ischemic preconditioning: a systematic review and meta-analysis of animal studies. PLoS One 10:e0142021. doi:10.1371/journal.pone.0142021
White SK, Frohlich GM, Sado DM, Maestrini V, Fontana M, Treibel TA, Tehrani S, Flett AS, Meier P, Ariti C, Davies JR, Moon JC, Yellon DM, Hausenloy DJ (2015) Remote ischemic conditioning reduces myocardial infarct size and edema in patients with ST-segment elevation myocardial infarction. JACC Cardiovasc Interv 8:178–188. doi:10.1016/j.jcin.2014.05.015
Whittaker P, Przyklenk K (1994) Reduction of infarct size in vivo with ischemic preconditioning: mathematical evidence for protection via non-ischemic tissue. Basic Res Cardiol 89:6–15. doi:10.1007/BF00788673
Whittington HJ, Babu GG, Mocanu MM, Yellon DM, Hausenloy DJ (2012) The diabetic heart: too sweet for its own good? Cardiol Res Pract 2012:845698. doi:10.1155/2012/845698
Windecker S, Bax JJ, Myat A, Stone GW, Marber MS (2013) Future treatment strategies in ST-segment elevation myocardial infarction. Lancet 382:644–657. doi:10.1016/S0140-6736(13)61452-X
Woo JS, Kim W, Ha SJ, Kim JB, Kim SJ, Kim WS, Seon HJ, Kim KS (2013) Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arterioscler Thromb Vasc Biol 33:2252–2260. doi:10.1161/ATVBAHA.113.301586
Xu M, Wang Y, Hirai K, Ayub A, Ashraf M (2001) Calcium preconditioning inhibits mitochondrial permeability transition and apoptosis. Am J Physiol Heart Circ Physiol 280:H899–H908
Xu X, Zhou Y, Luo S, Zhang W, Zhao Y, Yu M, Ma Q, Gao F, Shen H, Zhang J (2014) Effect of remote ischemic preconditioning in the elderly patients with coronary artery disease with diabetes mellitus undergoing elective drug-eluting stent implantation. Angiology 65:660–666. doi:10.1177/0003319713507332
Yaguchi Y, Satoh H, Wakahara N, Katoh H, Uehara A, Terada H, Fujise Y, Hayashi H (2003) Protective effects of hydrogen peroxide against ischemia/reperfusion injury in perfused rat hearts. Circ J 67:253–258. doi:10.1253/circj.67.253
Yang XM, Cui L, Alhammouri A, Downey JM, Cohen MV (2013) Triple therapy greatly increases myocardial salvage during ischemia/reperfusion in the in situ rat heart. Cardiovasc Drugs Ther 27:403–412. doi:10.1007/s10557-013-6474-9
Yang XM, Liu Y, Cui L, Yang X, Liu Y, Tandon N, Kambayashi J, Downey JM, Cohen MV (2013) Platelet P2Y(1)(2) blockers confer direct postconditioning-like protection in reperfused rabbit hearts. J Cardiovasc Pharmacol Ther 18:251–262. doi:10.1177/1074248412467692
Yang XM, Liu Y, Cui L, Yang X, Liu Y, Tandon N, Kambayashi J, Downey JM, Cohen MV (2013) Two classes of anti-platelet drugs reduce anatomical infarct size in monkey hearts. Cardiovasc Drugs Ther 27:109–115. doi:10.1007/s10557-012-6436-7
Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV (2004) Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol 44:1103–1110. doi:10.1016/j.jacc.2004.05.060
Yellon DM, Ackbarkhan AK, Balgobin V, Bulluck H, Deelchand A, Dhuny MR, Domah N, Gaoneadry D, Jagessur RK, Joonas N, Kowlessur S, Lutchoo J, Nicholas JM, Pauvaday K, Shamloll O, Walker JM, Hausenloy DJ (2015) Remote ischemic conditioning reduces myocardial infarct size in STEMI patients treated by thrombolysis. J Am Coll Cardiol 65:2764–2765. doi:10.1016/j.jacc.2015.02.082
Yellon DM, Baxter GF, Marber MS (1996) Angina reassessed: pain or protector? Lancet 347:1159–1162. doi:10.1016/S0140-6736(96)90613-3
Yellon DM, Downey JM (2003) Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev 83:1113–1151. doi:10.1152/physrev.00009.2003
Yellon DM, Hausenloy DJ (2007) Myocardial reperfusion injury. N Engl J Med 357:1121–1135. doi:10.1056/NEJMra071667
Young PJ, Dalley P, Garden A, Horrocks C, La Flamme A, Mahon B, Miller J, Pilcher J, Weatherall M, Williams J, Young W, Beasley R (2012) A pilot study investigating the effects of remote ischemic preconditioning in high-risk cardiac surgery using a randomised controlled double-blind protocol. Basic Res Cardiol 107:256. doi:10.1007/s00395-012-0256-6
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J (2003) Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol 285:H579–H588. doi:10.1152/ajpheart.01064.2002
Zografos TA, Katritsis GD, Tsiafoutis I, Bourboulis N, Katsivas A, Katritsis DG (2014) Effect of one-cycle remote ischemic preconditioning to reduce myocardial injury during percutaneous coronary intervention. Am J Cardiol 113:2013–2017. doi:10.1016/j.amjcard.2014.03.043
Acknowledgments
DJH was funded by the British Heart Foundation (Grant Number FS/10/039/28270), the Rosetrees Trust, and is supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. HEB was funded by the Danish Council for Independent Research (11-108354), the Danish Council for Strategic Research (11-115818), the Novo-Nordisk Foundation and Trygfonden. PF was funded by the Hungarian Scientific Research Fund (OTKA K 109737 and ANN 107803). HACF is funded by a Startup Grant of the “Excellence Cluster Cardio-Pulmonary System” (ECCPS) from the German Research Foundation (DFG, Bonn, Germany) and the “Peter und Traudl Engelhorn-Stiftung” (Weilheim, Germany) and in part by the Russian Government Program for competitive growth of Kazan Federal University, Kazan (Russian Federation). GH was supported by the German Research Foundation (He 1320/18-3 and SFB 1116/B8). TM was funded by the Japan Society for the Promotion of Science (Grant-in-Aid for Scientific Research #23591086, #2646113). MRS was funded by Novo-Nordisk Foundation. DG-D is supported by the Spanish Institute of Health, Instituto de Salud Carlos III (Grants PIE 13/00027, RETICS-RIC, RD12/0042/0021, and PI14/01431). RS and PF were funded by the European Foundation for the Study of Diabetes (EFSD) New Horizons Collaborative Research Initiative from the European Association for the Study of Diabetes (EASD) and by the European Cooperation in Science and Technology (COST EU-ROS). PF was funded by the Hungarian Scientific Research Fund (OTKA K 109737 and ANN 107803).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
HEB is shareholder of CellAegis Inc. PF is a founder and CEO of Pharmahungary, a group of R&D companies. GH served as a consultant to Servier. MO was a consultant for Neurovive Pharmaceuticals. DGD served as consultant to Neurovive Pharmaceuticals. All other authors have no relevant disclosures.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hausenloy, D.J., Barrabes, J.A., Bøtker, H.E. et al. Ischaemic conditioning and targeting reperfusion injury: a 30 year voyage of discovery. Basic Res Cardiol 111, 70 (2016). https://doi.org/10.1007/s00395-016-0588-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-016-0588-8