
Abstract
In spite of the current optimal therapy, the mortality of patients with ischemic heart disease (IHD) remains high, particularly in cases with diabetes mellitus (DM) as a co-morbidity. Myocardial infarct size is a major determinant of prognosis in IHD patients, and development of a novel strategy to limit infarction is of great clinical importance. Ischemic preconditioning (PC), postconditioning (PostC) and their mimetic agents have been shown to reduce infarct size in experiments using healthy animals. However, a variety of pharmacological agents have failed to demonstrate infarct size limitation in clinical trials. One of the possible reasons for the discrepancy between the results of animal experiments and clinical trials is that co-morbidities, including DM, modified myocardial responses to ischemia/reperfusion and to cardioprotective agents. Here we summarize observations of the effects of DM on myocardial infarct size and ischemic PC and PostC and discuss perspectives for protection of DM hearts.
Similar content being viewed by others

Introduction
Cardiovascular diseases are the leading cause of death, accounting for approximately 30% of all deaths worldwide [1]. Among the various cardiovascular diseases, acute myocardial infarction (AMI) has a high rate of mortality, and infarct size is a primary determinant of prognosis in these patients. The only established and clinically approved method to limit infarct size is restoration of coronary blood flow by percutaneous coronary interventions (PCI), thrombolytic agents or coronary bypass surgery. However, our recent review of clinical infarct size data indicated that infarct size after current reperfusion therapy was larger than 75% of the area at risk and larger than 20% of the left ventricle in one fourth of AMI patients [2]. Lack of substantial myocardial salvage in those patients cannot be simply attributable to long symptom-to-reperfusion time, and involvement of reperfusion injury [3] is also possible. Nevertheless, significant percentages of AMI patients suffer from insufficient cardiac function and have a poor prognosis even after successful restoration of coronary flow [2]. Therefore, the establishment of a novel strategy to limit the extent of infarction during ischemia/reperfusion is of great clinical importance.
As a cardioprotective strategy, ischemic preconditioning (PC) has received much attention for its potent infarct size-limiting effect since its first report by Murry et al. in 1986 [4]. Zhao et al. found that “conditioning” with repetitive ischemia/reperfusion at the time of reperfusion also affords protection and named the phenomenon ischemic postconditioning (PostC) [5]. Laboratory investigations to date have clarified outlines of the intracellular mechanisms of PC and PostC [6–9]. In addition, several agents targeting signaling relevant to PC and PostC have been shown to act as PostC mimetics [6–8]. However, clinical benefits of PC, PostC and their mimetics were not always demonstrated in clinical trials [8, 10]. The discrepancy between results of animal studies and clinical trials may be explained by notable differences between animal models of AMI and AMI in humans [2, 10]. First, there are species differences in regulatory mechanisms of cardiomyocytes such as contribution of sarcolemmal Ca2+ ATPase to overall Ca2+ handling. Second, the timing of administration of the test agent was not exactly the same in the positive animal studies and negative clinical trials. Third, most of the patients with AMI were pretreated with pharmacological agents for their co-morbidities at the onset of AMI, whereas animals were untreated, except for anesthetic agents, before AMI in most of the previous studies. Pharmacological agents used for treatment of hypertension, dyslipidemia and/or diabetes mellitus (DM) potentially modify intracellular signaling relevant to cytoprotection and thus myocardial responses to PC, Post and PC mimetics. Fourth, AMI patients recruited to clinical studies were generally old and had concomitant diseases, whereas relatively young and healthy animals were used in most of the previous animal studies. Aging and co-morbidities have been shown to modify or even abrogate the infarct size-limiting effects of interventions in animal experiments [6, 11] and thus possibly underlie the negative results in clinical trials.
Of morbidities that potentially compromise the protective mechanisms of the heart, DM appears primarily important to study today. The number of patients with DM has been increasing worldwide in the past two decades, and these patients are predisposed to serious cardiovascular morbidity and mortality [12]. Despite recent progress in coronary intervention strategies, DM is associated with higher mortality after AMI due to more extensive atherosclerotic lesions and also hypertrophied and dysfunctional left ventricle [13–15]. Interestingly, Lamblin et al. recently showed that rates of cardiovascular death and heart failure after the first AMI were higher in DM patients than in non-DM patients, though ventricular function and remodeling were comparable in the two groups [16]. The increased mortality after AMI in DM patients is unlikely to be prevented simply by improvement of glycemic control, since intensive glycemic controls failed to reduce cardiovascular events as well as mortality rate in DM patients in recent large-scale clinical trials [17–19]. Hence, we need novel strategies for protecting DM patients with coronary artery diseases from excessive myocardial ischemia-reperfusion injury. To gain an insight into a novel strategy, we thoroughly review reported effects of DM on myocardial infarct size and on protective mechanisms against infarction and discuss future perspectives in this article. Our discussion on pathophysiology and clinical approach to diabetic cardiomyopathy was recently published elsewhere [20].
Changes in myocardial susceptibility to infarction by DM
Clinical studies showed that DM increased the susceptibility of the myocardium to ischemia/reperfusion injury [21, 22]. However, results of infarct size studies in diabetic animal models were contradictory (Tables 1 and 2), and the reason for the inconsistency remains unclear.
Multiple mechanisms are involved in cardiomyocyte necrosis induced by ischemia/reperfusion. Depletion of intracellular ATP, intracellular overloading with Na+ and Ca2+, and increased fragility of the cell membrane are major changes during ischemia relevant to cell necrosis. Accumulating evidence supports the notion that the mitochondrial permeability transition pore (mPTP) is primed during ischemia and is opened by Ca2+ overload and burst of reactive oxygen species at the time of reperfusion, leading to irreversible loss of mitochondrial functions and cell necrosis [23]. Theoretically, modification of ischemia-induced changes or reperfusion-induced opening of the mPTP should underlie the change in myocardial susceptibility to infarction in DM hearts. However, such modification has not been understood well.
Experimental studies
Infarct size in animal models of DM was larger or smaller than or similar to, depending on reports, that in non-diabetic controls (Table 1). As shown in Table 1, there were numerous differences in experimental preparations and protocols, and a single factor cannot entirely explain the discrepancy in the effects of DM on infarct size. Duration of the diabetic state and plasma level of insulin (i.e., type 1 DM vs. type 2 DM) appear to influence myocardial tolerance against infarction. In a study by Ravingerová et al. [24], infarct size after 30-min ischemia was smaller in diabetic rat hearts at 1 week after streptozotocin (STZ) injection than in controls, but this infarct size limitation was not detected 8 weeks later. Two other studies have also shown that resistance of diabetic hearts to ischemia/reperfusion injury was detectable at the early phase of DM and disappeared later [25, 26]. However, enlargement of infarct size was observed as early as 8 days after STZ injection [27–29], indicating that duration of DM is not the only factor responsible for inconsistent effects of DM on infarct size.
Difference in metabolic profiles associated with DM (i.e., presence or absence of hyperinsulinemia and types of dyslipidemia) may also be responsible for different changes in infarct size in DM animals. With a few exceptions [30, 31], DM models with obesity and hyperinsulinemia showed increased myocardial susceptibility to ischemia/reperfusion-induced necrosis [32–41]. In contrast, high glucose level induced simply by glucose or dextrose infusion did not affect infarct size in the majority of studies, including two recent studies [42, 43] (Table 2).
Clinical studies
Increased myocardial susceptibility to infarction in DM patients was indicated by two clinical studies: infarct size determined by SPECT imaging after reperfusion therapy was larger by 30 ~ 70% in DM patients than in non-DM patients [21, 22]. The change in myocardial tolerance to infarction is consistent with worse short- and long-term prognosis after AMI in DM patients [44, 45]. Of particular relevance, Haffner et al. [46] showed that cardiovascular event risk in DM patients without prior myocardial infarction was comparable to the risk in non-diabetic patients with prior myocardial infarction. However, the untoward effects of DM on prognosis after AMI cannot be explained solely by increased myocardial vulnerability to ischemia/reperfusion injury, since the risk of heart failure and death was significantly higher in DM patients than in non-DM patients with comparable infarct sizes and left ventricular ejection fractions [47]. Myopathic changes (“diabetic cardiomyopathy”), impaired collateral recruitment, microcirculatory abnormalities, and underuse of evidence-based therapies are possible explanations, in addition to increased susceptibility to infarction, for detrimental prognosis after AMI in DM patients.
Defects in intracellular protective signaling in DM hearts
DM is one of the co-morbidities that potentially modify myocardial response to protective interventions, including PC and PostC. A number of studies have been carried out to determine the efficacy of ischemic PC and PostC for protecting the myocardium in animal models of DM (Tables 3 and 4) and diabetic patients (Table 5). The majority of those studies showed that DM interferes with protective mechanisms of cardioprotective interventions. Myocardial protection by PC and PostC is achieved by activation of multiple protective signaling pathways that appear to converge to inhibit mPTP opening upon reperfusion via phosphorylation of glycogen synthase kinase-3β (GSK-3β) at Ser9 [6, 8, 9], and DM-induced defects in the protective signaling may be different depending on the model and/or phase of DM.
Experimental studies
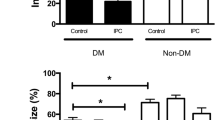
With a few exceptions, previous studies demonstrated that cardioprotection achieved by ischemic PC or PostC is impaired in DM: the infarct size-limiting effect was lost or required extra cycles of ischemia/reperfusion in experimental diabetes (Table 3). Tsang et al. [48] reported that one, two and three cycles of ischemic PC significantly reduced infarct size in normal rats; however, three cycles of ischemic PC were required to limit infarct size in diabetic Goto-Kakizaki rats. Their findings suggested that the threshold for PC protection is increased in the diabetic myocardium. Liu et al. [49] were the first to examine the effects of DM on PC and they reported preservation of PC protection in STZ-induced DM. However, since they used a PC protocol with multiple cycles of ischemia/reperfusion only, the change in the threshold for inducing PC protection might have been missed in their study. In fact, impairment of PC by DM has been observed in multiple species (rats, rabbits and dogs) and different models of DM (STZ-induced type 1 DM and genetic models of type 2 DM) [30, 37, 50–54]. Mimetics of ischemic PC and PostC (diazoxide, erythropoietin, [D-Ala2, D-Leu5-enkephalin acetate [DADLE] and isoflurane) were also ineffective for limitation of infarct size in DM hearts [33, 34, 37, 55–59], confirming that DM impairs intracellular signaling mechanisms relevant to myocardial protection.
Several steps in cytoprotective signaling have been found to be disrupted by DM in the heart. Blocked or attenuated signaling via phosphoinsitide-3 kinase (PI3K)-Akt, extracellular signal-regulated kinase (ERK) and/or signal transducer and activator of transcription-3 (STAT3) pathways was detected in association with loss or reduced efficiency of PC in protection against infarction [31, 33, 34, 36, 48, 57–59]. Our recent studies have shown that phosphorylation of Jak2, being upstream of PI3K-Akt signaling, is inhibited by enhanced calcineurin activity and that phosphorylation of GSK-3β by ERK is lost by an endoplasmic reticulum stress-dependent mechanism in OLETF, a rat model of obese type 2 DM [33, 34]. Furthermore, protein level of active GSK-3β, a pro-necrotic and pro-apoptotic kinase, was increased in mitochondria, leading to reduction in the threshold for mPTP opening in response to Ca2+ overload [34]. On the other hand, a protective mechanism downstream of GSK-3β phosphorylation appears to be intact in diabetic hearts, since direct pharmacological inhibitors of GSK-3β limited infarct size in diabetic hearts as in non-diabetic animals [33, 34, 54, 57, 58].
An important and clinically relevant issue is whether hyperglycemia per se is responsible for disruption of protective signaling in PC and PostC. Acute hyperglycemia induced by dextrose infusion attenuated infarct size limitation by ischemic PC, a mitochondrial ATP-sensitive K+ channel (KATP channel) opener and anesthetic agents (Table 4). These results indicate a primary role of hyperglycemia in impairment of protective signaling. Interestingly, Przyklenk et al. [31] reported that the cardioprotective effect of ischemic PostC was re-established in STZ-induced diabetic mice by pancreas islet cell transplantation. Transplantation of islet cells in diabetic mice normalized both blood glucose level and free fatty acid level and cellular signaling, such as ERK phosphorylation, activated by ischemic PostC. Since dyslipidemia was also shown to attenuate the infarct size-limiting effect of ischemic PC [60, 61], restoration of myocardial response to PostC in the diabetic heart by islet cell transplantation might be achieved by normalization of both plasma glucose level and lipid profile. Nevertheless, circumstantial evidence in animal experiments supports the notion that normalization of the metabolic profile restores protective signaling mechanisms in the DM heart.
Clinical studies on PC
PC protection in human hearts has been demonstrated by results of in vitro experiments using human ventricular myocytes [62] and atrial trabeculae [63] and by detailed analyses of myocardial responses in patients with naturally occurring ischemic syndromes [64, 65]. Since ischemic PC and PC-like phenomena in human hearts have been recently reviewed elsewhere [7, 65], we briefly summarize the results that are relevant to the effects of DM on ischemic PC. As for PC-like effects of preinfarct angina, different groups reported that pre-infarct angina was associated with smaller infarct size determined by creatine kinase (CK) release, improved left ventricular function and reduced mortality [64, 66]. Furthermore, in overall analysis of AMI patients, angina before AMI was associated with better long-term prognosis [67, 68]. However, in patients with DM, beneficial effects of pre-infarct angina were not detected [69]. Ishihara et al. [69] showed that CK release and recovery of cardiac function and in-hospital mortality after AMI were similar in DM patients with and without pre-infarct angina. DM-induced impairment of ischemic PC protection in human hearts has also been indicated by studies in which myocardial injury was assessed during angioplasty [70] and during a treadmill exercise test [71] by use of electrocardiography.
Direct evidence for diabetes-induced loss of ischemic PC protection in the human myocardium was provided by an in vitro experiment using atrial trabeculae obtained at open heart surgery. CK release and contractile dysfunction after hypoxia/reoxygenation in vitro was significantly suppressed by PC in trabeculae from non-diabetic patients but not in trabeculae from diabetic patients [63, 72].
Clinical studies on PostC
In contrast to PC, PostC or administration of its mimetic at the time of reperfusion is clinically feasible. In 2005, Staat et al. [73] translated for the first time the concept of ischemic PostC into the clinical setting. PostC with four cycles of 1-min inflation and 1-min deflation before full reperfusion successfully reduced CK release in a small and selected cohort of patients with AMI. Efficacy of ischemic PostC in patients with AMI was subsequently confirmed in a number of small-scale studies, using as endpoints cardiac biomarkers, ECG ST-segment resolution, and anatomic infarct size (Table 5). However, three recent studies failed to show protection by ischemic PostC in terms of CK and troponin release, LV function or infarct size that was determined by delayed enhancement magnetic resonance imaging (MRI) [74–76]. The reasons for the discrepant results remain uncertain, but there are some possibilities: differences in the protocols of ischemic PostC, presence of co-morbidities and use of different pharmacological agents for the co-morbidities.
In the PostC protocol, duration from reperfusion to the first re-occlusion and number of re-occlusions have been shown to influence the infarct size-limiting effects of PostC in animal models of AMI [77–79]. Retrospective analyses by Darling et al. [80] and Wang et al. [81] suggest that larger numbers of balloon inflations–deflations were associated with greater protection in patients with AMI. However, an appropriate algorithm of PostC stimulus to maximally protect the human myocardium has not been established. In addition, protection by PostC in animal models of AMI is attenuated with aging and presence of co-morbidities, such as DM, hypertension and hypercholesterolemia [6, 11]. Although characteristics of patients, such as age and prevalence of DM, seem to be similar in the human studies (Table 5), difference in durations of the co-morbidities might have been involved in the different outcomes. Nevertheless, the effect of DM on protection afforded by PostC has not been specifically examined in clinical studies.
The effects of pharmacological PostC (i.e., administration of pharmacological agents at reperfusion) on myocardial injury have been examined in a number of clinical trials. Results of the trials are a mixture of positive and negative results, and the reason for the discrepancy remains unknown [82–92]. Adenosine infusion limited infarct size, but neither the death rate during 6-month follow-up nor the rate of re-hospitalization for heart failure were reduced by the adenosine treatment [82, 84]. A post hoc analysis suggested that only patients with early reperfusion therapy received clinical benefits of adenosine, such as a reduction in death or heart failure [85]. In the J-WIND trial [86], the effects of nicorandil and ANP on infarct size in different study arms were examined. Administration of nicorandil failed to significantly limit infarct size, though recombinant ANP infusion was shown to induce a small but significant reduction in infarct size. The results of studies on the effect of erythropoietin administered at reperfusion were discrepant [87–92]. Taken together, there are a few promising agent for infarct size limitation in AMI patients, but none of their effects has been unequivocally demonstrated to be sufficient for improving prognosis.
Effects of anti-diabetic medications on AMI in diabetic patients
Possible adverse effects of sulfonylureas on cardiac events in DM patients have been debated for decades, and recent studies have indicated that cardiovascular risk was significantly higher in DM patients treated with sulfonylureas than in those who received metformin monotherapy [93, 94]. Sulphonylureas have been shown to inhibit protection of PC and PostC by blocking the ATP-sensitive K+ channel (KATP channel) in cardiomyocytes, raising concern that such actions of sulphonylureas may be detrimental at the time of AMI in DM patients. However, the effects on PC and PostC are actually not uniform across sulfonylureas. Glibenclamide, a non-specific blocker of the KATP channel, abrogated the infarct size-limiting effect of PC and PostC in non-diabetic animals [6, 7, 95]. In contrast, glimepiride, a second-generation sulfonylurea, did not block the protective effect of PC in the animal myocardium or human myocardium [96, 97], presumably because this agent has few cardiac actions [98]. In addition, glimepiride was shown to have a PostC-mimetic action by activation of PI3K/Akt in the rabbit myocardium [99]. However, the effects of sulfonylureas on PC in diabetes have been tested in only a few studies [51, 96], since PC and PostC failed to limit infarction in most of the DM models. Interestingly, Nieszner et al. [51] reported that infarct-sparing effect of PC in alloxan-treated diabetic rabbits was restored by pretreatment with a low dose, but not a high dose, of glibenclamide. The mechanism by which glibenclamide restored myocardial response to PC in the DM rabbit is unclear. However, beneficial effects of glycemic control, possibly on PI3K/Akt signaling, might have overwhelmed the effect of partial inhibition of cardiac KATP channels when an appropriate dose of glibenclamide was selected.
Metformin is an insulin-sensitizing agent that reduces hepatic glucose output and increases the uptake of glucose in peripheral tissues, including the skeletal muscle. In non-diabetic animals, metformin reduced infarct size by activation of AMP-activated protein kinase and/or Akt [95]. However, whether metformin is additive to ischemic PC or PostC has not been examined. Infarct size-limiting effect of metformin in diabetic animals was controversial: there are two positive reports [40, 100] and one negative report [101].
Thiazolidinediones (pioglitazone, rosiglitazone) administered before ischemia reduced infarct size in both normal and diabetic animals [39, 41, 95]. However, the impact of the cardioprotective effect of thiazolidinediones on clinical outcome is unclear, since the effects of pioglitazone and rosiglitazone on risk of cardiovascular events appear discrepant [102].
In the past few years, growing evidence has demonstrated cardiovascular effects of incretin-based therapy using glucagon-like peptide-1 (GLP-1) receptor agonists and inhibitors of dipeptidyl peptidase-4 (DPP-4) [95, 103–105]. However, the effects of GLP-1 receptor agonists or DPP-4 inhibitors on ischemia/reperfusion injury have not been examined in diabetic animals except for a study by Huisamen et al. [106] They reported that enlargement of infarct size in obese pre-diabetic rats was reduced by treatment with a DPP-4 inhibitor for 4 weeks [106]. Further investigation is necessary for clarifying the effects of GLP-1 analogues and DPP-4 inhibitors on intracellular protective signaling and its modification by DM, though there are a number of on-going large-scale clinical trials to test the effects of incretin-based therapy on cardiovascular outcomes [107].
Conclusions and perspectives
DM potentially increases myocardial susceptibility to ischemia/reperfusion injury, though it cannot be explained by chronic hyperglycemia alone. DM also modifies myocardial responses to ischemic and pharmacological PC and PostC by disruption of intracellular signaling responsible for enhancement of resistance to cell death. These alterations in the diabetic heart appear to underlie the poor prognosis of DM patients after AMI. In addition, a defect of the protective mechanisms caused by aging, DM and possibly other co-morbidities is one of possible reasons why infarct size-limiting effects of agents in animal studies could not have been translated into AMI patients.
Analyses of modifications of cytoprotective signaling by DM, heart failure, dyslipidemia and hypertension generally showed that defects in the signaling by the pathological factors are upstream of a step of GSK-3β phosphorylation and mPTP regulation [6, 9]. Hence, a reasonable approach to protect the heart in DM patients is manipulation of protective mechanisms at the level of GSK-3β or mPTP. Blockade of mPTP opening by cyclosporine A at the time of reperfusion is one such approach, and its effect was first examined in 58 AMI patients [108]. Infarct size determined by MRI at 5 days after AMI was significantly limited by cyclosporine A and the beneficial effects persisted until 6 month after AMI [109]. The results are very promising and support the feasibility of a strategy for cardioprotection targeting the mPTP. However, how DM modifies the mPTP regulation and how we can suppress opening of the mPTP at the time of reperfusion need to be further investigated for devising a novel therapy for cardioprotection in DM patients.
Abbreviations
- AMI:
-
Acute myocardial infarction
- CK:
-
Creatine kinase
- DADLE:
-
[D-Ala2,D-Leu5]-enkephalin acetate
- DM:
-
Diabetes mellitus
- DPP-4:
-
Dipeptidyl peptidase-4
- ERK:
-
Extracellular signal-regulated kinase
- GLP-1:
-
Glucagon-like peptide-1
- GSK-3β:
-
Glycogen synthase kinase-3β
- IHD:
-
Ischemic heart disease
- KATP channel:
-
ATP-sensitive K+ channel
- MPTP:
-
Mitochondrial permeability transition pore
- MRI:
-
Magnetic resonance imaging
- PC:
-
Preconditioning
- PCI:
-
Percutaneous coronary interventions
- PostC:
-
Postconditioning
- PI3K:
-
Phosphoinositide-3 kinase
- PKC:
-
Protein kinase C
- STZ:
-
Streptozotocin.
References
Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al: Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2012, 125 (1): e2-e220.
Miura T, Miki T: Limitation of myocardial infarct size in the clinical setting: current status and challenges in translating animal experiments into clinical therapy. Basic Res Cardiol. 2008, 103 (6): 501-513. 10.1007/s00395-008-0743-y.
Yellon DM, Hausenloy DJ: Myocardial reperfusion injury. N Eng J Med. 2007, 357 (11): 1121-1135. 10.1056/NEJMra071667.
Murry CE, Jennings RB, Reimer KA: Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986, 74 (5): 1124-1136. 10.1161/01.CIR.74.5.1124.
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J: Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003, 285 (2): H579-H588.
Ferdinandy P, Schulz R, Baxter GF: Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007, 59 (4): 418-458. 10.1124/pr.107.06002.
Yellon DM, Downey JM: Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003, 83 (4): 1113-1151.
Ovize M, Baxter GF, Di Lisa F, Ferdinandy P, Garcia-Dorado D, Hausenloy DJ, Heusch G, Vinten-Johansen J, Yellon DM, Schulz R: Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2010, 87 (3): 406-423. 10.1093/cvr/cvq129.
Sanada S, Komuro I, Kitakaze M: Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011, 301 (5): H1723-H1741. 10.1152/ajpheart.00553.2011.
Bolli R, Becker L, Gross G, Mentzer R, Balshaw D, Lathrop DA: Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res. 2004, 95 (2): 125-134. 10.1161/01.RES.0000137171.97172.d7.
Przyklenk K: Efficacy of cardioprotective 'conditioning' strategies in aging and diabetic cohorts: the co-morbidity conundrum. Drugs Aging. 2011, 28 (5): 331-343. 10.2165/11587190-000000000-00000.
Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Farzadfar JK, Farzadfar F, Khang YH, Stevens GA, et al: National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011, 378 (9785): 31-40. 10.1016/S0140-6736(11)60679-X.
Kirtane AJ, Ellis SG, Dawkins KD, Colombo A, Grube E, Popma JJ, Fahy M, Leon MB, Moses JW, Mehran R, et al: Paclitaxel-eluting coronary stents in patients with diabetes mellitus: pooled analysis from 5 randomized trials. J Am Coll Cardiol. 2008, 51 (7): 708-715. 10.1016/j.jacc.2007.10.035.
Cubbon RM, Wheatcroft SB, Grant PJ, Gale CP, Barth JH, Sapsford RJ, Ajjan R, Kearney MT, Hall AS: Temporal trends in mortality of patients with diabetes mellitus suffering acute myocardial infarction: a comparison of over 3000 patients between 1995 and 2003. Eur Heart J. 2007, 28 (5): 540-545.
Norhammar A, Lindback J, Ryden L, Wallentin L, Stenestrand U: Improved but still high short- and long-term mortality rates after myocardial infarction in patients with diabetes mellitus: a time-trend report from the Swedish Register of Information and Knowledge about Swedish Heart Intensive Care Admission. Heart (British Cardiac Society). 2007, 93 (12): 1577-1583.
Lamblin N, Fertin M, de Groote P, Bauters C: Cardiac remodeling and heart failure after a first anterior myocardial infarction in patients with diabetes mellitus. J Cardiovasc Med (Hagerstown). 2012, 13 (6): 353-359. 10.2459/JCM.0b013e328353694b.
Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, Marre M, Cooper M, Glasziou P, Grobbee D, et al: Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Eng J Med. 2008, 358 (24): 2560-2572.
Gerstein HC, Miller ME, Byington RP, Goff DC, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH, et al: Effects of intensive glucose lowering in type 2 diabetes. N Eng J Med. 2008, 358 (24): 2545-2559.
Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven PD, Zieve FJ, Marks J, Davis SN, Hayward R, et al: Glucose control and vascular complications in veterans with type 2 diabetes. N Eng J Med. 2009, 360 (2): 129-139. 10.1056/NEJMoa0808431.
Miki T, Yuda S, Kouzu H, Miura T: Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Fail Rev. Epub ahead of print
Marso SP, Miller T, Rutherford BD, Gibbons RJ, Qureshi M, Kalynych A, Turco M, Schultheiss HP, Mehran R, Krucoff MW, et al: Comparison of myocardial reperfusion in patients undergoing percutaneous coronary intervention in ST-segment elevation acute myocardial infarction with versus without diabetes mellitus (from the EMERALD Trial). Am J Cardiol. 2007, 100 (2): 206-210. 10.1016/j.amjcard.2007.02.080.
Alegria JR, Miller TD, Gibbons RJ, Yi QL, Yusuf S: Infarct size, ejection fraction, and mortality in diabetic patients with acute myocardial infarction treated with thrombolytic therapy. Am Heart J. 2007, 154 (4): 743-750. 10.1016/j.ahj.2007.06.020.
Halestrap AP: A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans. 2010, 38 (4): 841-860. 10.1042/BST0380841.
Ravingerova T, Neckar J, Kolar F: Ischemic tolerance of rat hearts in acute and chronic phases of experimental diabetes. Mol Cell Biochem. 2003, 249 (1–2): 167-174.
Xu G, Takashi E, Kudo M, Ishiwata T, Naito Z: Contradictory effects of short- and long-term hyperglycemias on ischemic injury of myocardium via intracellular signaling pathway. Exp Mol Pathol. 2004, 76 (1): 57-65. 10.1016/j.yexmp.2003.08.003.
Ma G, Al-Shabrawey M, Johnson JA, Datar R, Tawfik HE, Guo D, Caldwell RB, Caldwell RW: Protection against myocardial ischemia/reperfusion injury by short-term diabetes: enhancement of VEGF formation, capillary density, and activation of cell survival signaling. Naunyn Schmiedebergs Arch Pharmacol. 2006, 373 (6): 415-427. 10.1007/s00210-006-0102-1.
Marfella R, D'Amico M, Di Filippo C, Piegari E, Nappo F, Esposito K, Berrino L, Rossi F, Giugliano D: Myocardial infarction in diabetic rats: role of hyperglycaemia on infarct size and early expression of hypoxia-inducible factor 1. Diabetologia. 2002, 45 (8): 1172-1181. 10.1007/s00125-002-0882-x.
Marfella R, Di Filippo C, Esposito K, Nappo F, Piegari E, Cuzzocrea S, Berrino L, Rossi F, Giugliano D, D'Amico M: Absence of inducible nitric oxide synthase reduces myocardial damage during ischemia reperfusion in streptozotocin-induced hyperglycemic mice. Diabetes. 2004, 53 (2): 454-462. 10.2337/diabetes.53.2.454.
Di Filippo C, Marfella R, Cuzzocrea S, Piegari E, Petronella P, Giugliano D, Rossi F, D'Amico M: Hyperglycemia in streptozotocin-induced diabetic rat increases infarct size associated with low levels of myocardial HO-1 during ischemia/reperfusion. Diabetes. 2005, 54 (3): 803-810. 10.2337/diabetes.54.3.803.
Kristiansen SB, Lofgren B, Nielsen JM, Stottrup NB, Buhl ES, Nielsen-Kudsk JE, Nielsen TT, Rungby J, Flyvbjerg A, Botker HE: Comparison of two sulfonylureas with high and low myocardial K(ATP) channel affinity on myocardial infarct size and metabolism in a rat model of type 2 diabetes. Diabetologia. 2011, 54 (2): 451-458. 10.1007/s00125-010-1970-y.
Przyklenk K, Maynard M, Greiner DL, Whittaker P: Cardioprotection with postconditioning: loss of efficacy in murine models of type-2 and type-1 diabetes. Antioxid Redox Signal. 2011, 14 (5): 781-790. 10.1089/ars.2010.3343.
Jordan JE, Simandle SA, Tulbert CD, Busija DW, Miller AW: Fructose-fed rats are protected against ischemia/reperfusion injury. J Pharmacol Exp Ther. 2003, 307 (3): 1007-1011. 10.1124/jpet.103.055970.
Hotta H, Miura T, Miki T, Togashi N, Maeda T, Kim SJ, Tanno M, Yano T, Kuno A, Itoh T, et al: Angiotensin II type 1 receptor-mediated upregulation of calcineurin activity underlies impairment of cardioprotective signaling in diabetic hearts. Circ Res. 2010, 106 (1): 129-132. 10.1161/CIRCRESAHA.109.205385.
Miki T, Miura T, Hotta H, Tanno M, Yano T, Sato T, Terashima Y, Takada A, Ishikawa S, Shimamoto K: Endoplasmic reticulum stress in diabetic hearts abolishes erythropoietin-induced myocardial protection by impairment of phospho-glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeability transition. Diabetes. 2009, 58 (12): 2863-2872. 10.2337/db09-0158.
La Bonte LR, Davis-Gorman G, Stahl GL, McDonagh PF: Complement inhibition reduces injury in the type 2 diabetic heart following ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2008, 294 (3): H1282-H1290.
Bouhidel O, Pons S, Souktani R, Zini R, Berdeaux A, Ghaleh B: Myocardial ischemic postconditioning against ischemia-reperfusion is impaired in ob/ob mice. Am J Physiol Heart Circ Physiol. 2008, 295 (4): H1580-H1586. 10.1152/ajpheart.00379.2008.
Katakam PV, Jordan JE, Snipes JA, Tulbert CD, Miller AW, Busija DW: Myocardial preconditioning against ischemia-reperfusion injury is abolished in Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol. 2007, 292 (2): R920-R926.
Zhu SG, Xi L, Kukreja RC: Type 2 diabetic Obese db/db mice are refractory to myocardial ischemic postconditioning In Vivo: potential role for Hsp20, F(1) -ATPase delta, and Echs1. J Cell Mol Med. 2012, 16 (4): 950-958. 10.1111/j.1582-4934.2011.01376.x.
Yue TL, Bao W, Gu JL, Cui J, Tao L, Ma XL, Ohlstein EH, Jucker BM: Rosiglitazone treatment in Zucker diabetic Fatty rats is associated with ameliorated cardiac insulin resistance and protection from ischemia/reperfusion-induced myocardial injury. Diabetes. 2005, 54 (2): 554-562.
Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, Lefer DJ: Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008, 57 (3): 696-705. 10.2337/db07-1098.
Honda T, Kaikita K, Tsujita K, Hayasaki T, Matsukawa M, Fuchigami S, Sugiyama S, Sakashita N, Ogawa H, Takeya M: Pioglitazone, a peroxisome proliferator-activated receptor-gamma agonist, attenuates myocardial ischemia-reperfusion injury in mice with metabolic disorders. J Mol Cell Cardiol. 2008, 44 (5): 915-926. 10.1016/j.yjmcc.2008.03.004.
Ichinomiya T, Cho S, Higashijima U, Matsumoto S, Maekawa T, Sumikawa K: High-dose fasudil preserves postconditioning against myocardial infarction under hyperglycemia in rats: role of mitochondrial KATP channels. Cardiovasc Diabetol. 2012, 11: 28-10.1186/1475-2840-11-28.
Matsumoto S, Cho S, Tosaka S, Higashijima U, Maekawa T, Hara T, Sumikawa K: Hyperglycemia raises the threshold of levosimendan- but not milrinone-induced postconditioning in rat hearts. Cardiovasc Diabetol. 2012, 11: 4-10.1186/1475-2840-11-4.
Zuanetti G, Latini R, Maggioni AP, Santoro L, Franzosi MG: Influence of diabetes on mortality in acute myocardial infarction: data from the GISSI-2 study. J Am Coll Cardiol. 1993, 22 (7): 1788-1794. 10.1016/0735-1097(93)90758-S.
Chyun D, Obata J, Kling J, Tocchi C: In-hospital mortality after acute myocardial infarction in patients with diabetes mellitus. Am J Crit Care. 2000, 9 (3): 168-179.
Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M: Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Eng J Med. 1998, 339 (4): 229-234. 10.1056/NEJM199807233390404.
Shah AM, Uno H, Kober L, Velazquez EJ, Maggioni AP, MacDonald MR, Petrie MC, McMurray JJ, Califf RM, Pfeffer MA, et al: The inter-relationship of diabetes and left ventricular systolic function on outcome after high-risk myocardial infarction. Eur J Heart Fail. 2010, 12 (11): 1229-1237. 10.1093/eurjhf/hfq179.
Tsang A, Hausenloy DJ, Mocanu MM, Carr RD, Yellon DM: Preconditioning the diabetic heart: the importance of Akt phosphorylation. Diabetes. 2005, 54 (8): 2360-2364. 10.2337/diabetes.54.8.2360.
Liu Y, Thornton JD, Cohen MV, Downey JM, Schaffer SW: Streptozotocin-induced non-insulin-dependent diabetes protects the heart from infarction. Circulation. 1993, 88 (3): 1273-1278. 10.1161/01.CIR.88.3.1273.
Kersten JR, Toller WG, Gross ER, Pagel PS, Warltier DC: Diabetes abolishes ischemic preconditioning: role of glucose, insulin, and osmolality. Am J Physiol Heart Circ Physiol. 2000, 278 (4): H1218-H1224.
Nieszner E, Posa I, Kocsis E, Pogatsa G, Preda I, Koltai MZ: Influence of diabetic state and that of different sulfonylureas on the size of myocardial infarction with and without ischemic preconditioning in rabbits. Exp Clin Endocrinol Diabetes. 2002, 110 (5): 212-218. 10.1055/s-2002-33069.
Kristiansen SB, Lofgren B, Stottrup NB, Khatir D, Nielsen-Kudsk JE, Nielsen TT, Botker HE, Flyvbjerg A: Ischaemic preconditioning does not protect the heart in obese and lean animal models of type 2 diabetes. Diabetologia. 2004, 47 (10): 1716-1721. 10.1007/s00125-004-1514-4.
Galagudza MM, Nekrasova MK, Syrenskii AV, Nifontov EM: Resistance of the myocardium to ischemia and the efficacy of ischemic preconditioning in experimental diabetes mellitus. Neurosci Behav Physiol. 2007, 37 (5): 489-493. 10.1007/s11055-007-0040-5.
Yadav HN, Singh M, Sharma PL: Involvement of GSK-3beta in attenuation of the cardioprotective effect of ischemic preconditioning in diabetic rat heart. Mol Cell Biochem. 2010, 343 (1–2): 75-81.
Kersten JR, Montgomery MW, Ghassemi T, Gross ER, Toller WG, Pagel PS, Warltier DC: Diabetes and hyperglycemia impair activation of mitochondrial K(ATP) channels. Am J Physiol Heart Circ Physiol. 2001, 280 (4): H1744-H1750.
Matsumoto S, Cho S, Tosaka S, Ureshino H, Maekawa T, Hara T, Sumikawa K: Pharmacological preconditioning in type 2 diabetic rat hearts: the roles of mitochondrial ATP-sensitive potassium channels and the phosphatidylinositol 3-kinase-Akt pathway. Cardiovasc Drugs Ther. 2009, 23 (4): 263-270. 10.1007/s10557-009-6184-5.
Gross ER, Hsu AK, Gross GJ: Diabetes abolishes morphine-induced cardioprotection via multiple pathways upstream of glycogen synthase kinase-3beta. Diabetes. 2007, 56 (1): 127-136. 10.2337/db06-0907.
Ghaboura N, Tamareille S, Ducluzeau PH, Grimaud L, Loufrani L, Croue A, Tourmen Y, Henrion D, Furber A, Prunier F: Diabetes mellitus abrogates erythropoietin-induced cardioprotection against ischemic-reperfusion injury by alteration of the RISK/GSK-3beta signaling. Basic Res Cardiol. 2011, 106 (1): 147-162. 10.1007/s00395-010-0130-3.
Drenger B, Ostrovsky IA, Barak M, Nechemia-Arbely Y, Ziv E, Axelrod JH: Diabetes blockade of sevoflurane postconditioning is not restored by insulin in the rat heart: phosphorylated signal transducer and activator of transcription 3- and phosphatidylinositol 3-kinase-mediated inhibition. Anesthesiology. 2011, 114 (6): 1364-1372. 10.1097/ALN.0b013e31820efafd.
Ueda Y, Kitakaze M, Komamura K, Minamino T, Asanuma H, Sato H, Kuzuya T, Takeda H, Hori M: Pravastatin restored the infarct size-limiting effect of ischemic preconditioning blunted by hypercholesterolemia in the rabbit model of myocardial infarction. J Am Coll Cardiol. 1999, 34 (7): 2120-2125. 10.1016/S0735-1097(99)00440-4.
Ungi I, Ungi T, Ruzsa Z, Nagy E, Zimmermann Z, Csont T, Ferdinandy P: Hypercholesterolemia attenuates the anti-ischemic effect of preconditioning during coronary angioplasty. Chest. 2005, 128 (3): 1623-1628. 10.1378/chest.128.3.1623.
Ikonomidis JS, Tumiati LC, Weisel RD, Mickle DA, Li RK: Preconditioning human ventricular cardiomyocytes with brief periods of simulated ischaemia. Cardiovasc Res. 1994, 28 (8): 1285-1291. 10.1093/cvr/28.8.1285.
Hassouna A, Loubani M, Matata BM, Fowler A, Standen NB, Galinanes M: Mitochondrial dysfunction as the cause of the failure to precondition the diabetic human myocardium. Cardiovasc Res. 2006, 69 (2): 450-458. 10.1016/j.cardiores.2005.11.004.
Kloner RA, Shook T, Przyklenk K, Davis VG, Junio L, Matthews RV, Burstein S, Gibson M, Poole WK, Cannon CP, et al: Previous angina alters in-hospital outcome in TIMI 4. A clinical correlate to preconditioning?. Circulation. 1995, 91 (1): 37-45. 10.1161/01.CIR.91.1.37.
Rezkalla SH, Kloner RA: Preconditioning and the human heart. Panminerva Med. 2005, 47 (2): 69-73.
Anzai T, Yoshikawa T, Asakura Y, Abe S, Akaishi M, Mitamura H, Handa S, Ogawa S: Preinfarction angina as a major predictor of left ventricular function and long-term prognosis after a first Q wave myocardial infarction. J Am Coll Cardiol. 1995, 26 (2): 319-327. 10.1016/0735-1097(95)80002-X.
Ishihara M, Sato H, Tateishi H, Kawagoe T, Shimatani Y, Kurisu S, Sakai K, Ueda K: Implications of prodromal angina pectoris in anterior wall acute myocardial infarction: acute angiographic findings and long-term prognosis. J Am Coll Cardiol. 1997, 30 (4): 970-975. 10.1016/S0735-1097(97)00238-6.
Kloner RA, Shook T, Antman EM, Cannon CP, Przyklenk K, Yoo K, McCabe CH, Braunwald E: Prospective temporal analysis of the onset of preinfarction angina versus outcome: an ancillary study in TIMI-9B. Circulation. 1998, 97 (11): 1042-1045. 10.1161/01.CIR.97.11.1042.
Ishihara M, Inoue I, Kawagoe T, Shimatani Y, Kurisu S, Nishioka K, Kouno Y, Umemura T, Nakamura S, Sato H: Diabetes mellitus prevents ischemic preconditioning in patients with a first acute anterior wall myocardial infarction. J Am Coll Cardiol. 2001, 38 (4): 1007-1011. 10.1016/S0735-1097(01)01477-2.
Lee TM, Chou TF: Impairment of myocardial protection in type 2 diabetic patients. J Clin Endocrinol Metab. 2003, 88 (2): 531-537. 10.1210/jc.2002-020904.
Ovunc K: Effects of glibenclamide, a K(ATP) channel blocker, on warm-up phenomenon in type II diabetic patients with chronic stable angina pectoris. Clin Cardiol. 2000, 23 (7): 535-539. 10.1002/clc.4960230713.
Sivaraman V, Hausenloy DJ, Wynne AM, Yellon DM: Preconditioning the diabetic human myocardium. J Cell Mol Med. 2010, 14 (6B): 1740-1746.
Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L'Huillier I, Aupetit JF, Bonnefoy E, Finet G, Andre-Fouet X, et al: Postconditioning the human heart. Circulation. 2005, 112 (14): 2143-2148. 10.1161/CIRCULATIONAHA.105.558122.
Freixa X, Bellera N, Ortiz-Perez JT, Jimenez M, Pare C, Bosch X, De Caralt TM, Betriu A, Masotti M: Ischaemic postconditioning revisited: lack of effects on infarct size following primary percutaneous coronary intervention. Eur Heart J. 2012, 33 (1): 103-112. 10.1093/eurheartj/ehr297.
Sorensson P, Saleh N, Bouvier F, Bohm F, Settergren M, Caidahl K, Tornvall P, Arheden H, Ryden L, Pernow J: Effect of postconditioning on infarct size in patients with ST elevation myocardial infarction. Heart (British Cardiac Society). 2010, 96 (21): 1710-1715. 10.1136/hrt.2010.199430.
Tarantini G, Favaretto E, Marra MP, Frigo AC, Napodano M, Cacciavillani L, Giovagnoni A, Renda P, De Biasio V, Plebani M, et al: Postconditioning during coronary angioplasty in acute myocardial infarction: the POST-AMI trial. Int J Cardiol. 2012, Epub ahead of print
Iliodromitis EK, Georgiadis M, Cohen MV, Downey JM, Bofilis E, Kremastinos DT: Protection from post-conditioning depends on the number of short ischemic insults in anesthetized pigs. Basic Res Cardiol. 2006, 101 (6): 502-507. 10.1007/s00395-006-0606-3.
Chiari PC, Bienengraeber MW, Pagel PS, Krolikowski JG, Kersten JR, Warltier DC: Isoflurane protects against myocardial infarction during early reperfusion by activation of phosphatidylinositol-3-kinase signal transduction: evidence for anesthetic-induced postconditioning in rabbits. Anesthesiology. 2005, 102 (1): 102-109. 10.1097/00000542-200501000-00018.
Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV: Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol. 2004, 44 (5): 1103-1110. 10.1016/j.jacc.2004.05.060.
Darling CE, Solari PB, Smith CS, Furman MI, Przyklenk K: 'Postconditioning' the human heart: multiple balloon inflations during primary angioplasty may confer cardioprotection. Basic Res Cardiol. 2007, 102 (3): 274-278. 10.1007/s00395-007-0643-6.
Wang G, Zhang S, Joggerst SJ, McPherson J, Zhao DX: Effects of the number and interval of balloon inflations during primary PCI on the extent of myocardial injury in patients with STEMI: does postconditioning exist in real-world practice?. J Invasive Cardiol. 2009, 21 (9): 451-455.
Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, Eisenberg PR, Bolli R, Casas AC, Molina-Viamonte V, et al: Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol. 1999, 34 (6): 1711-1720. 10.1016/S0735-1097(99)00418-0.
Kopecky SL, Aviles RJ, Bell MR, Lobl JK, Tipping D, Frommell G, Ramsey K, Holland AE, Midei M, Jain A, et al: A randomized, double-blinded, placebo-controlled, dose-ranging study measuring the effect of an adenosine agonist on infarct size reduction in patients undergoing primary percutaneous transluminal coronary angioplasty: the ADMIRE (AmP579 Delivery for Myocardial Infarction REduction) study. Am Heart J. 2003, 146 (1): 146-152. 10.1016/S0002-8703(03)00172-8.
Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW: A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). J Am Coll Cardiol. 2005, 45 (11): 1775-1780. 10.1016/j.jacc.2005.02.061.
Kloner RA, Forman MB, Gibbons RJ, Ross AM, Alexander RW, Stone GW: Impact of time to therapy and reperfusion modality on the efficacy of adenosine in acute myocardial infarction: the AMISTAD-2 trial. Eur Heart J. 2006, 27 (20): 2400-2405. 10.1093/eurheartj/ehl094.
Kitakaze M, Asakura M, Kim J, Shintani Y, Asanuma H, Hamasaki T, Seguchi O, Myoishi M, Minamino T, Ohara T, et al: Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. 2007, 370 (9597): 1483-1493. 10.1016/S0140-6736(07)61634-1.
Ozawa T, Toba K, Suzuki H, Kato K, Iso Y, Akutsu Y, Kobayashi Y, Takeyama Y, Kobayashi N, Yoshimura N, et al: Single-dose intravenous administration of recombinant human erythropoietin is a promising treatment for patients with acute myocardial infarction - randomized controlled pilot trial of EPO/AMI-1 study. Circ J. 2010, 74 (7): 1415-1423. 10.1253/circj.CJ-10-0109.
Voors AA, Belonje AM, Zijlstra F, Hillege HL, Anker SD, Slart RH, Tio RA, van 't Hof A, Jukema JW, Peels HO, et al: A single dose of erythropoietin in ST-elevation myocardial infarction. Eur Heart J. 2010, 31 (21): 2593-2600. 10.1093/eurheartj/ehq304.
Najjar SS, Rao SV, Melloni C, Raman SV, Povsic TJ, Melton L, Barsness GW, Prather K, Heitner JF, Kilaru R, et al: Intravenous erythropoietin in patients with ST-segment elevation myocardial infarction: REVEAL: a randomized controlled trial. JAMA. 2011, 305 (18): 1863-1872. 10.1001/jama.2011.592.
Ludman AJ, Yellon DM, Hasleton J, Ariti C, Babu GG, Boston-Griffiths E, Venugopal V, Walker M, Holdright D, Swanton H, et al: Effect of erythropoietin as an adjunct to primary percutaneous coronary intervention: a randomised controlled clinical trial. Heart (British Cardiac Society). 2011, 97 (19): 1560-1565. 10.1136/hrt.2011.223867.
Ferrario M, Arbustini E, Massa M, Rosti V, Marziliano N, Raineri C, Campanelli R, Bertoletti A, De Ferrari GM, Klersy C, et al: High-dose erythropoietin in patients with acute myocardial infarction: a pilot, randomised, placebo-controlled study. Int J Cardiol. 2011, 147 (1): 124-131. 10.1016/j.ijcard.2009.10.028.
Suh JW, Chung WY, Kim YS, Kim KI, Jeon EJ, Cho YS, Youn TJ, Chae IH, Kim CH, Choi DJ: The effect of intravenous administration of erythropoietin on the infarct size in primary percutaneous coronary intervention. Int J Cardiol. 2011, 149 (2): 216-220. 10.1016/j.ijcard.2010.02.002.
Jorgensen CH, Gislason GH, Andersson C, Ahlehoff O, Charlot M, Schramm TK, Vaag A, Abildstrom SZ, Torp-Pedersen C, Hansen PR: Effects of oral glucose-lowering drugs on long term outcomes in patients with diabetes mellitus following myocardial infarction not treated with emergent percutaneous coronary intervention–a retrospective nationwide cohort study. Cardiovasc Diabetol. 2010, 9: 54-10.1186/1475-2840-9-54.
Schramm TK, Gislason GH, Vaag A, Rasmussen JN, Folke F, Hansen ML, Fosbol EL, Kober L, Norgaard ML, Madsen M, et al: Mortality and cardiovascular risk associated with different insulin secretagogues compared with metformin in type 2 diabetes, with or without a previous myocardial infarction: a nationwide study. Eur Heart J. 2011, 32 (15): 1900-1908. 10.1093/eurheartj/ehr077.
Ye Y, Perez-Polo JR, Aguilar D, Birnbaum Y: The potential effects of anti-diabetic medications on myocardial ischemia-reperfusion injury. Basic Res Cardiol. 2011, 106 (6): 925-952. 10.1007/s00395-011-0216-6.
Klepzig H, Kober G, Matter C, Luus H, Schneider H, Boedeker KH, Kiowski W, Amann FW, Gruber D, Harris S, et al: Sulfonylureas and ischaemic preconditioning; a double-blind, placebo-controlled evaluation of glimepiride and glibenclamide. Eur Heart J. 1999, 20 (6): 439-446. 10.1053/euhj.1998.1242.
Mocanu MM, Maddock HL, Baxter GF, Lawrence CL, Standen NB, Yellon DM: Glimepiride, a novel sulfonylurea, does not abolish myocardial protection afforded by either ischemic preconditioning or diazoxide. Circulation. 2001, 103 (25): 3111-3116. 10.1161/01.CIR.103.25.3111.
Geisen K, Vegh A, Krause E, Papp JG: Cardiovascular effects of conventional sulfonylureas and glimepiride. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 1996, 28 (9): 496-507. 10.1055/s-2007-979841.
Nishida H, Sato T, Nomura M, Miyazaki M, Nakaya H: Glimepiride treatment upon reperfusion limits infarct size via the phosphatidylinositol 3-kinase/Akt pathway in rabbit hearts. J Pharmacol Sci. 2009, 109 (2): 251-256. 10.1254/jphs.08202FP.
Bhamra GS, Hausenloy DJ, Davidson SM, Carr RD, Paiva M, Wynne AM, Mocanu MM, Yellon DM: Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res Cardiol. 2008, 103 (3): 274-284. 10.1007/s00395-007-0691-y.
Kravchuk E, Grineva E, Bairamov A, Galagudza M, Vlasov T: The effect of metformin on the myocardial tolerance to ischemia-reperfusion injury in the rat model of diabetes mellitus type II. Exp Diabetes Res. 2011, 2011: 907496.
Wilding JP: PPAR agonists for the treatment of cardiovascular disease in patients with diabetes. Diabetes Obes Metab. 2012, Epub ahead of print
Chinda K, Chattipakorn S, Chattipakorn N: Cardioprotective effects of incretin during ischaemia-reperfusion. Diab Vasc Dis Res. 2012, Epub ahead of print
Yin M, Sillje HH, Meissner M, van Gilst WH, de Boer RA: Early and late effects of the DPP-4 inhibitor vildagliptin in a rat model of post-myocardial infarction heart failure. Cardiovasc Diabetol. 2011, 10: 85-10.1186/1475-2840-10-85.
Chinda K, Palee S, Surinkaew S, Phornphutkul M, Chattipakorn S, Chattipakorn N: Cardioprotective effect of dipeptidyl peptidase-4 inhibitor during ischemia-reperfusion injury. Int J Cardiol. 2012, Epub ahead of print
Huisamen B, Genis A, Marais E, Lochner A: Pre-treatment with a DPP-4 inhibitor is infarct sparing in hearts from obese, pre-diabetic rats. Cardiovasc Drugs Ther. 2011, 25 (1): 13-20. 10.1007/s10557-010-6271-7.
Ussher JR, Drucker DJ: Cardiovascular biology of the incretin system. Endocr Rev. 2012, 33 (2): 187-215. 10.1210/er.2011-1052.
Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, et al: Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Eng J Med. 2008, 359 (5): 473-481. 10.1056/NEJMoa071142.
Mewton N, Croisille P, Gahide G, Rioufol G, Bonnefoy E, Sanchez I, Cung TT, Sportouch C, Angoulvant D, Finet G, et al: Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. J Am Coll Cardiol. 2010, 55 (12): 1200-1205. 10.1016/j.jacc.2009.10.052.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
TaM has received research funding from Daiichi-Sankyo, Japan, Eisai, Japan, and Pfizer, Japan. TI and DS have no conflicts of interest or financial ties to disclose. TeM has received research funding from Takeda Pharmaceuticals, Japan, Novartis, Japan, Chugai Pharmaceutical, Japan, and Astellas Pharma, Japan.
Authors' contributions
TaM collected and arranged the data in sequence, drafted and correspond the manuscript. TI and DS contributed to collection and analysis of the data. TeM contributed to analysis and interpretation of data and revision. All authors have read and approved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Miki, T., Itoh, T., Sunaga, D. et al. Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovasc Diabetol 11, 67 (2012). https://doi.org/10.1186/1475-2840-11-67
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2840-11-67