
Abstract
The cardioprotection afforded by remote ischaemic conditioning (RIC) is mediated via a complex mechanism involving sensory afferent nerves, the vagus nerve, and release of a humoral blood-borne factor. However, it is unknown whether release of the protective factor depends on vagal activation or occurs independently. This study aimed to evaluate the co-dependence of the neural and humoral pathways of RIC, focussing on the vagus nerve and intrinsic cardiac ganglia. In the first study, anesthetised rats received bilateral cervical vagotomy or sham-surgery immediately prior to RIC (4 × 5 min limb ischemia–reperfusion) or sham-RIC. Venous blood plasma was dialysed across a 12–14 kDa membrane and dialysate perfused through a naïve-isolated rat heart prior to 35-min left anterior descending ischemia and 60-min reperfusion. In the second study, anesthetised rats received RIC (4 × 5-min limb ischemia–reperfusion) or control (sham-RIC). Dialysate was prepared and perfused through a naïve-isolated rat heart in the presence of the ganglionic blocker hexamethonium or muscarinic antagonist atropine, prior to ischemia–reperfusion as above. Dialysate collected from RIC-treated rats reduced infarct size in naïve rat hearts from 40.7 ± 6.3 to 23.7 ± 3.1 %, p < 0.05. Following bilateral cervical vagotomy, the protection of RIC dialysate was abrogated (42.2 ± 3.2 %, p < 0.05 vs RIC dialysate). In the second study, the administration of 50-μM hexamethonium (45.8 ± 2.5 %) or 100-nM atropine (36.5 ± 3.4 %) abrogated the dialysate-mediated protection. Release of a protective factor following RIC is dependent on prior activation of the vagus nerve. In addition, this factor appears to induce cardioprotection via recruitment of intrinsic cardiac ganglia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Remote ischemic conditioning describes the cardioprotective intervention, whereby brief periods of ischemia to an organ or tissue remote from the heart can protect the myocardium from a subsequent injurious ischemic insult [57]. This important discovery, further to that of classical ischemic conditioning [55], gave immediate potential for clinical translation. Indeed, since its inception in 1993, there have been many attempts to prove its efficacy in limiting injury associated with coronary artery bypass (CABG) surgery and acute myocardial infarction in patients, with varying levels of success [11, 14, 17, 30, 31, 53, 67, 71, 78, 80]. Perhaps a reason for this difficulty in translation is a significant gap in our understanding of the mechanism, in particular, how the protective message is communicated from the conditioned limb to the myocardium [33, 56]. This is the unique mechanistic trait of RIC, thus understanding how it occurs is of great importance, and we believe would aid more effective clinical translation.
The communication of RIC is classically thought to occur via one of two pathways. First, a humoral pathway whereby a factor is released in response to the conditioning stimulus which moves to the heart via the circulatory system and protects [20, 21, 46, 59, 66]. Second, a neural pathway whereby the conditioning stimulus induces sensory nerve firing which, via autonomic centres in the brainstem, leads to increased efferent autonomic tone to the heart, initiating cardioprotection [22, 26, 28, 47, 51]. These two pathways were originally thought of as mechanistically distinct; however, recent data have suggested that the two are interdependent. Indeed, release of the humoral mediator in response RIC is dependent on sensory innervation to the conditioned limb [37, 52, 60, 70]. The downstream target of sensory nerve activation leading to release of the humoral factor, however, is not clear. One possibility is the vagus nerve, which has been previously implicated in the RIC mechanism [22, 51]. Indeed, a recent study suggested vagal innervation to the gut was essential for the communication of RIC, perhaps, via release of a blood-borne factor [50]. However, although it suggests release of a factor is dependent on vagus nerve activation, these data presented does not absolutely prove this link as release of a humoral factor is not measured. This study investigates the hypothesis that the humoral mediator is released downstream of vagus nerve activation.
There exist within the heart intrinsic neural loops that are able to process sensory information from the myocardial milieu and modulate efferent autonomic output from intrinsic cardiac ganglia, without any necessary input from the central nervous system [3, 5–7]. These neural loops are at risk of destruction following myocardial infarction, and, indeed, remodel, such that the intrinsic cardiac nervous system (ICNS) no longer can function as normal [1, 29, 36, 58]. Vagus nerve stimulation has recently been demonstrated to ameliorate post-infarction remodelling of the ICNS [8]. Thus, given the putative importance of the vagus nerve in RIC, this study investigated the hypothesis that intrinsic cardiac ganglia are recruited by the humoral mediator of RIC as part of the transduction of protection within the myocardium.
Materials and methods
Materials
Hexamethonium (Sigma-Aldrich, Missouri, USA) was employed as a neuronal nicotinic acetylcholine receptor (nAChR) antagonist. Given it has affinity to the muscarinic M2 receptor above 100 μM [24], a dose of 50 µM was used for this study to aid specificity at nAChRs within cardiac ganglia. Atropine, a muscarinic acetylcholine receptor (mAChR) antagonist, was used at a dose of 100 nM. Although often used at micromolar doses in the literature, atropine has a high affinity for mAChRs (K d = 0.36 nM [72]). Thus, 100 nM was chosen, which has recently been demonstrated to effectively antagonise muscarinic agonism in isolated hearts [38].
Animals and ethical statement
All animals received humane care in accordance with the United Kingdom (Scientific Procedures) Act of 1986. Male Sprague–Dawley (SD) rats were bred at a central animal unit in University College London and were used at a weight of 250–300 g throughout the study.
In vivo procedure: donor rat
Male SD rats were anesthetised via an upper-left quadrant intraperitoneal injection of 20 % w/v sodium pentobarbitone (Animalcare, York, UK) at a dose of 0.05 ml/100 g + 0.05 ml. Once anesthesia was reached, confirmed via loss of pedal reflex, the animal was secured in a supine position on a heat mat. The rat was intubated via optic light trans-illumination of the trachea, using a modified 16G, 1.7 × 51 mm Abbocath-T intravenous cannula (B. Braun, PA, USA). This cannula was connected to either a PhysioSuite (Kent Scientific, CT, USA) or Small Animal Ventilator (Harvard Apparatus, Kent, UK), and ventilated with air supplemented with 0.5 l/min oxygen. Finally, core body temperature was maintained at 37.0 ± 0.5 °C via a rectal probe feeding back to a heat pad.
Model characterisation
Animals had a small cuff placed around their left hindlimb and were randomised to receive either (1) RIC protocol, consisting of 4 × 5-min cuff inflation to 200 mmHg with intermittent 5-min deflations, or, (2) sham-RIC protocol, consisting of a corresponding time period with no cuff inflation (see Fig. 1i). Following the procedure, a clamshell thoracotomy was performed and the heart was excised and perfused on a Langendorff apparatus, using the same method as described below. Following a 20-min period of stabilisation, hearts were subjected to a 35-min LAD ischemia and subsequent 60-min reperfusion [25] (Fig. 1ii). In a separate group of rats, blood was sampled via cardiac puncture following the protocol and prepared for dialysis as described below.

Design of experimental protocols: i displays in vivo procedures. Anaesthetised rats were subjected to either RIC (4 × 5 min hindlimb ischaemia/reperfusion) or sham protocols. Bilateral cervical vagotomy or sham-surgery immediately prior to either RIC (4 × 5-min limb ischaemia–reperfusion) or sham-RIC. ii At the end of this protocol, the heart was excised and perfused on a Langendorff rig before being subjected to a 35-min region ischaemia and 60-min reperfusion. iii Following the in vivo procedure, 9-ml blood was sampled and the plasma dialysed across a 12–14-kDa membrane. The dialysate was perfused through a naïve-isolated rat heart for 10 min prior to a regional ischaemia–reperfusion protocol, as described above. iv Isolated hearts were treated with either hexamethonium (50 μM) or atropine (100 nM) for 5 min prior to and the duration of dialysate perfusion/washout. Following each Langendorff experiment, infarct size (IS) was determined using TTC staining
Dialysate preparation
At the end of the protocol, ~9 ml of venous blood was harvested via right ventricular puncture, following clamshell thoracotomy, using a 1.5-in 19G Terumo needle (Egham, UK). Samples were immediately centrifuged at 1600g for 20 min at 21 °C to obtain plasma, followed by 10,000g for 30 min at 21 °C to obtain 4 ml of platelet-free plasma. This was dialysed across a 12–14-kDa membrane (Spectra/Por, Spectrum Laboratories, Inc., Rancho Dominguez, CA, USA) into 200 ml of modified Krebs-Henseleit buffer (KHB) (118-mM NaCl, 4.7-mM KCl, 1.22-mM MgSO4·7H2O, 1.21-mM KH2PO4, and 1.84-mM CaCl2·2H2O) for 24 h at 4 °C. Prior to perfusion through the naïve heart, the dialysate was supplemented with 25-mM NaHCO3 and 11-mM d-glucose, gassed with 95 % O2 and 5 % CO2 and warmed to 37.5 °C.
Naïve recipient hearts
Rats were anesthetised with an upper-left quadrant intraperitoneal injection of sodium pentobarbitone (60 mg/kg) (Animalcare, York, UK). Hearts were quickly excised via a clamshell thoracotomy and the aorta cannulated on a Langendorff apparatus and perfused with KHB (118-mM NaCl, 25-mM NaHCO3, 11-mM d-glucose, 4.7-mM KCl, 1.22-mM MgSO4·7H2O, 1.21-mM KH2PO4, and 1.84-mM CaCl2·2H2O (for detailed methods, see [9]). A fluid-filled latex balloon was inserted into the left ventricle to allow for measurement of functional parameters, including heart rate (HR) and left ventricular developed pressure (LVEDP). Coronary flow rate (CFR) was recorded throughout the protocol, and the temperature of the heart was maintained at 37.0 ± 0.5 °C. Finally, a 3–0 Mersilk suture (Ethicon, Edinburgh, UK) was inserted through the heart to surround the left anterior descending (LAD) coronary artery. Following a 20-min stabilisation, dialysate was perfused through the heart for 10 min with a subsequent 10-min washout before index ischemia. All hearts received a 35-min LAD ischemia, induced via reversible tightening of the suture, and 60-min reperfusion (Fig. 1iii). Although there is evidence that reperfusion can influence infarct size in mouse Langendorff [62], the use of 60-min reperfusion duration, as opposed to 120 min, in the rat Langendorff model does not influence the efficacy of triphenyl-tetrazolium (TTC) staining [25].
Study 1: bilateral cervical vagotomy and RIC in rats
Anesthetised SD rats were randomly assigned to one of the four groups: (1) Sham vagotomy + Control, the left and right vagus nerves were isolated at the mid-cervical level, but not severed, and the rat received a sham-RIC protocol; (2) Sham vagotomy + RIC, the same as group 1; however, the rat was subjected to RIC; (3) Vagotomy + Control, rats were subjected to bilateral cervical vagotomy and received a sham-RIC protocol; and (4) Vagotomy + RIC, rats were subjected to bilateral cervical vagotomy and received RIC (Fig. 1i). At the end of the protocol, blood was sampled via right ventricular puncture and dialysate prepared as described above. The dialysate was perfused through a naïve-isolated rat heart prior to IRI, also described above (Fig. 1iii).
Study 2: intrinsic cardiac nerves and RIC in rats
Isolated hearts were randomly assigned to one of the following six groups: (1) Control dialysate, hearts received dialysate prepared from an in vivo rat following sham-RIC; (2) RIC dialysate, hearts received dialysate prepared from an in vivo rat following in vivo RIC; (3) Control dialysate + Hexamethonium (50 μM); (4) RIC dialysate + Hexamethonium; (5) Control dialysate + Atropine (100 nM); and (6) RIC dialysate + Atropine (100 nM) (Fig. 1iv). In groups 3–6, drug perfusion was initiated 5 min prior to and for the duration of dialysate treatment. The choices of drug concentration were chosen carefully, as described above. All hearts were perfused with the dialysate for 10 min, with a subsequent 10-min washout period, prior to IRI, as described above.
Infarct size assessment
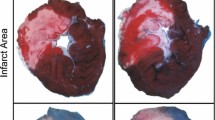
At the end of the reperfusion period, the LAD suture was re-tightened and 1 ml of 0.25 % Evans blue dye was perfused through the heart to delineate the area-at-risk of infarction. The hearts were then frozen at −20 °C before being sectioned into five-transverse slices and stained for viable tissue by immersion in 1 % triphenyl-tetrazolium chloride at 37 °C for 15 min. Following fixation in 10 % formalin for 24 h, the sections were digitally scanned to a computer for analysis. Analysis of infarct size (IS) as a proportion of area-at-risk (AAR) was calculated via planimetry using the ImageJ software (version 1.45, National Institutes of Health, USA). Infarct size was calculated a percentage of the area-at-risk (IS/AAR).
Bicinchoninic acid (BCA) assay to measure dialysate and plasma protein concentration
Plasma or dialysate samples were assayed for protein concentration using the BCA assay method, as described previously [68]. Briefly, 5 μl of each sample is added to a 96-well plate, followed by 195 μl of a 1:50 mix of copper II sulphate and bicinchoninic acid. This is allowed to incubate for 30 min at 37 °C before being subjected to colorimetric analysis at 470 nm.
Statistics
Data groups were first analysed for normality using the Kolmogorov–Smirnov test. Statistical differences between two groups were analysed using a student’s t test and more than two groups using a one-way analysis of variance (ANOVA) with Tukey’s multiple comparison post-test. Haemodynamic data in Figs. 4 and 5 were analysed using a two-way repeated measures ANOVA with Bonferroni’s post-test. All data are presented as mean ± standard error of the mean (SEM). Data groups were classed as significantly different with a p value less than 0.05. Notation of significance was as follows: * = p < 0.05, ** = p < 0.01, and *** = p < 0.001. Analysis was exclusively performed using GraphPad Prism version 5 for Windows (CA, USA).
Results
In vivo RIC induces significant cardioprotection
The RIC protocol of 4 × 5-min hindlimb ischemia–reperfusion, induced via a small blood-pressure cuff, was effective at inducing cardioprotection in vivo, as demonstrated by a significantly reduced infarct size in hearts subjected to RIC relative to sham procedure (sham IS/AAR = 61.7 ± 1.9 % vs RIC IS/AAR = 46.9 ± 4.6 %, p < 0.05) (Fig. 2d).

Characterisation of the rat dialysate model: a–c BCA protein assay was employed to measure protein concentration in the plasma following either RIC (4 × 5-min hindlimb ischaemia/reperfusion) or sham-surgery. No difference in plasma concentration was observed between pre- and post-dialysis groups, and no difference was observed following RIC relative to sham. Interestingly, a 400-fold reduction in protein concentration was observed within the dialysate when compared to plasma. d In vivo RIC or sham procedures preceded excision of the heart and perfusion on a Langendorff apparatus. These hearts were subjected to IRI, and those who had received RIC displayed reduced infarct size relative to control. e Dialysate was initially prepared using platelet-rich plasma. When perfused through a naïve-isolated heart, dialysate from both RIC and sham-treated rats gave a protected phenotype, although RIC dialysate still gave a significant additive protection. When platelets were removed from the plasma prior to dialysis (f), sham dialysate no longer gave a protective phenotype; however, RIC dialysate was able to significantly protect naïve hearts. Data were analysed via student’s t test, n = 6–8 per group, and expressed as mean ± SEM
Model characterisation: RIC dialysate is cardioprotective to a naïve-isolated rat heart
The dialysate transfer model has previously only been described in the rabbit [37, 61, 70] and mouse [34]. Whilst plasma transfer from pig to rat heart has been proven [66], it was important to characterise the dialysate model in the rat. We initially took platelet-rich plasma from sham- or RIC-treated rats, dialysed across a 12–14-kDa membrane into a 50-fold volume of buffer, and perfused this through naïve-isolated rat hearts prior to ischemia–reperfusion injury (IRI). The RIC dialysate provided a significant protection to the naïve-isolated heart (sham dialysate IS/AAR = 26.4 ± 1.0 % vs RIC dialysate IS/AAR = 16.7 ± 1.5 %, p < 0.01, n = 6–8 per group) (Fig. 2e). However, the infarct size in isolated hearts that were perfused with the sham dialysate was unexpectedly small in comparison to typical control infarct sizes. In contrast, when platelets were removed from the plasma by centrifugation, infarct sizes were more typical, and RIC remained fully effective (sham dialysate IS/AAR = 41.5 ± 1.7 % vs RIC dialysate IS/AAR = 25.2 ± 2.6 %, p < 0.001, n = 6–8 per group) (Fig. 2f). We, therefore, used platelet-free plasma in further studies.
The concentration of protein within the plasma and dialysate did not differ between sham and RIC groups (sham = 0.09 ± 0.02 μg/μl vs RIC = 0.11 ± 0.002 μg/μl, n = 6/group, p > 0.05). There were no differences observed in the plasma protein concentration between RIC and sham groups before or after dialysis. With the 50-fold dilution and 12–14-kDa dialysis cutoff, a 400-fold decrease in protein concentration was observed between plasma and dialysate.
Study 1: bilateral cervical vagotomy abolishes release of the humoral mediator following RIC
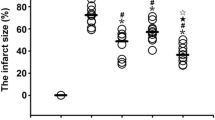
Sham surgical vagotomy did not influence the efficacy of RIC dialysate to protect a naïve, isolated rat heart (control dialysate from sham-surgery rat: IS/AAR = 40.7 ± 6.3 % vs RIC dialysate from sham-surgery rat IS/AAR = 23.7 ± 3.1, p < 0.05) (Fig. 3). When the vagus nerve was sectioned bilaterally at the cervical level, RIC dialysate no longer protected the naïve-isolated heart (control dialysate from vagotomised rat: IS/AAR = 31.4 ± 2.4 % vs RIC dialysate from vagotomised rat: IS/AAR = 42.2 ± 3.2 %, p < 0.05 vs sham-surgery RIC dialysate) (Fig. 3).

Bilateral cervical vagotomy abrogates dialysate-mediated protection of naïve-isolated rat hearts: the chart displays left ventricular infarct size as a proportion of the area-at-risk. Dialysate was prepared following in vivo vagotomy or sham-surgery matched with either RIC or sham protocols. This was perfused through a naïve-isolated heart prior to IRI. Sham vagotomy did not influence the ability of RIC dialysate to induce cardioprotection in the naïve heart. Bilateral cervical vagotomy, however, abrogated RIC dialysate protection in the naïve heart, suggesting that release of the blood-borne humoral mediator was inhibited. Data were analysed via one-way ANOVA with Tukey’s post-hoc test, and presented as mean ± SEM, with 6–8 animals per group
The hemodynamic data for the naïve-isolated hearts that received dialysate are shown in supplementary Fig. 5. Measurements were taken at the beginning of stabilisation, 5 min into the index ischemia and at the end of 60-min reperfusion. Those hearts treated with RIC dialysate did not demonstrate improved functional recovery as measured by coronary flow rate (CFR), left ventricular developed pressure (LVEDP), and heart rate (HR). In addition, dialysate prepared following bilateral cervical vagotomy did not affect the functional recovery when perfused through a naïve-isolated heart.
Study 2: the humoral RIC mediator exerts protection via intrinsic cardiac nerves
Figure 4 displays the infarct size chart and corresponding functional data from naïve-isolated rat hearts subjected to dialysate in the presence or absence of either the ganglionic blocker hexamethonium or the muscarinic antagonist atropine. Those hearts treated with RIC dialysate in the absence of either drug-induced powerful cardioprotection (sham dialysate IS/AAR = 40.1 ± 1.2 vs RIC dialysate IS/AAR = 27.6 ± 2.3, p < 0.05). In the presence of hexamethonium (50 μM), RIC dialysate was no longer able to protect naïve hearts (sham dialysate + Hex IS/AAR = 42.3 ± 4.3 % vs RIC dialysate + Hex IS/AAR = 45.8 ± 2.7 %, p > 0.05 vs sham dialysate). The muscarinic antagonist atropine (100 nM) also abrogated RIC dialysate-mediated cardioprotection (sham dialysate + atropine IS/AAR = 40.7 ± 4.8 % vs RIC dialysate + atropine IS/AAR = 36.5 ± 3.4 %, p > 0.05 vs sham dialysate) (Fig. 4).

Hexamethonium and atropine abrogate dialysate-mediated cardioprotection: isolated rat hearts were perfused with dialysate prepared following in vivo RIC or sham procedures. RIC dialysate significantly protected the naïve heart from IRI relative to sham. When the naïve heart was pre-treated with either the ganglionic antagonist hexamethonium (50 μM) or the muscarinic antagonist atropine (100 nM) abrogated this protection. Data expressed as mean ± SEM, n = 6–8 per group
The hemodynamic data again indicated that RIC dialysate did not significantly influence functional recovery of naïve hearts relative to control (Supp. Fig. 6). In addition, neither hexamethonium nor atropine affected functional recovery of the isolated hearts.
Discussion
Our study elucidates two novel aspects to the mechanism of RIC communication. First, release of the blood-borne humoral mediator is dependent on prior activation of the vagus nerve. Second, the humoral mediator exerts cardioprotection in the myocardium in part via the recruitment of intrinsic cardiac ganglia. Whilst the importance of the vagus nerve in RIC has been previously reported [22, 23, 50, 51], this study is the first to prove its involvement in the release of the humoral mediator. We have demonstrated that a plasma dialysate generated following in vivo RIC in rats is able to significantly protect a naïve-isolated rat heart from ischemia–reperfusion injury. Previous studies have prepared dialysate from an in vivo rabbit model [52, 60, 70] or, indeed, humans [34, 37, 64]. These used a 20-fold dilution gradient into the dialysate, which is 2.5-fold lower than the 50-fold dilution used in this study. This demonstrates both the conserved nature of the RIC mechanism across several mammalian species and the remarkable potency of cardioprotection offered by the factor(s). The interesting observation that platelet-rich plasma generates a cardioprotective dialysate is most likely a systematic effect. Platelets are thought to degranulate at low temperatures [54], and when activated are known to release several cardioprotective molecules that are smaller than 12–14 kDa, for example, stromal-derived factor 1α [13, 15, 16, 20, 49]. The plasma is dialysed at 4 °C, therefore, during this process, the platelets may degranulate and release these cardioprotective factors. However, this was not investigated in this study, and instead, platelet-free plasma was used for dialysis in the two subsequent experiments. Finally, recent evidence from our laboratory has suggested that the duration of reperfusion may influence the magnitude of protection induced by pre-conditioning [62], perhaps due to limitations of the TTC method of infarct analysis. The evidence in rat Langendorff studies, however, indicates that one can observe powerful cardioprotection at both 60 min [79] and 120 min [32] of reperfusion. Thus, the use of 60 min reperfusion can give an accurate account of infarct size within the confines of the TTC staining method.
Vagus nerve stimulation has been reported in the literature to offer significant cardioprotection from ischemia–reperfusion injury [22, 51, 65, 73]. Indeed, an important study by Mastitskaya et al. demonstrated that activation of a particular group of pre-ganglionic parasympathetic neurones, in the dorsal vagal motor nucleus (DVMN), was sufficient to induce powerful cardioprotection in vivo [51]. In a subsequent and very elegant experiment, the DVMN was genetically silenced using the allatostatin method [51, 76]. This process abrogated RIC-mediated cardioprotection, indicating that this group of pre-ganglionic parasympathetic neurones is fundamental for the communication of the protective message from the conditioned limb to the myocardium. Moreover, a recent study from the same group indicates vagal innervation to the stomach, and gut is responsible for RIC communication, suggesting the release of a blood-borne mediator following vagal recruitment [50]. The literature, however, is not fully in agreement with these data [23]. Donato and colleagues demonstrated that cervical but not sub-diaphragmatic vagotomy abrogated RIC, suggesting direct cardiac vagal innervation to be key for RIC communication. The absence of a sham sub-diaphragmatic vagotomy group, however, calls the result into question given the huge abdominal trauma associated with the abdominal surgery. In addition, release of a humoral cardioprotective mediator downstream of vagus nerve activation has not been demonstrated in the literature. The key result from our study, therefore, is that release of the humoral RIC mediator is dependent on prior activation of the vagus nerve. We further conclude that the humoral factor is released from a region of the body innervated by the vagus nerve below the cervical level. Whether this factor is released following non-cardiac vagal innervation, however, is not clear.
Release of the humoral mediator following RIC is dependent on an intact sensory innervation to the conditioned limb [37, 70]. In addition, two important studies demonstrated that direct femoral nerve stimulation, topical application of capsaicin or, indeed, transcutaneous electrical nerve stimulation generated a plasma dialysate that was able to protect a naïve heart from ischemia–reperfusion injury [52, 61]. These data suggest that the sensory afferent nerve is the sole means of communication from the conditioned limb, and that the factor is released downstream of nerve stimulation. Our study, perhaps, adds to the model by suggesting that the vagus nerve is the link between sensory nerve activation in the limb and release of the humoral mediator (Fig. 5).

Schematic of proposed mechanism of RIC communication (figure adapted from [56]): serial inflations and deflations of a cuff around the upper limb will activate sensory afferent nerves [70]. These, in turn, will convey their message to autonomic regions of the brainstem, leading to increased systemic efferent vagal tone. The vagus nerve will innervate an organ remote from the heart, which induces release of a dialysable cardioprotective factor less than 12–14 kDa in size. This factor will move to the heart via the blood and induce a protective phenotype within the myocardium, in part via the recruitment of intrinsic cardiac ganglia
Growing evidence suggests that cardiac neural control is hierarchical. Indeed, sensory information from the heart can be received by: (1) central nervous control from medullary autonomic centres in the brain, which provide information to the heart via autonomic efferent pre-ganglionic neurons; (2) intrathoracic extracardiac ganglia; (3) intrinsic cardiac ganglia [2–6, 35, 41, 48]. Intrinsic cardiac ganglia are able to process sensory information from the myocardium and directly activate efferent post-ganglionic nerve firing from intrinsic cardiac ganglia, thus neural control of the heart can occur without any extracardiac input [7, 41, 42]. The Langendorff perfused isolated heart is traditionally thought of as a denervated preparation; however, in light of recent studies in the literature described above, as well as the results from this paper, it appears that intrinsic neural loops remain intact in the isolated heart and continue to play an important role in its function and ability to withstand ischemia–reperfusion injury.
Transmission of a sensory message in a pre-ganglionic synapse within intrinsic cardiac ganglia is governed via the release of acetylcholine into the synaptic cleft, which will bind to and activate nicotinic acetylcholine receptors (nAChR), on the post-ganglionic nerve, causing a depolarisation and initiation of the nerve impulse [10, 19, 27]. Hexamethonium will antagonise the nAChR, thus preventing transmission of information at the ganglia. The observation, therefore, that hexamethonium abrogates dialysate-mediated protection suggests that the humoral factor recruits intrinsic cardiac ganglia as an essential process in the induction of cardioprotection. Muscarinic acetylcholine receptors (mAChR) are present primarily on the sarcolemma of cardiomyocytes [12]. They respond to acetylcholine released from parasympathetic post-ganglionic neurons, which innervate the ventricles [18, 39, 63, 74]. Indeed, their activation has been previously implicated in cardioprotection, with exogenous acetylcholine inducing powerful protection to isolated perfused rat hearts via an Akt-dependent mechanism [45]. Thus, the observation that atropine, a mAChR antagonist, abrogates dialysate-mediated cardioprotection suggests that the humoral factor induces increased intrinsic post-ganglionic parasympathetic nerve outflow. Previous evidence has demonstrated that hexamethonium and atropine abrogate in vivo RIC in rats [22, 26, 51], although the literature is not in full agreement [77]. However, it was not clear from these studies which ganglia are important for the transmission of the RIC protective message. In addition, evidence that the endothelium can be protected by RIC discounts the importance of intrinsic nerves in responding to the humoral mediator [43]. However, the protection offered to endothelium by RIC may be obtained via a different mechanism relative to the myocardium. Our data suggest that the intrinsic cardiac ganglia play an important role in this setting.
One interpretation of these data is that vagus nerve stimulation may be sufficient to evoke all of the protection conferred by RIC. In the literature, chronic vagal nerve stimulation failed to ameliorate cardiac remodelling or functional capacity in heart failure patients [81]. However, whether vagus nerve stimulation can protect the heart from acute myocardial infarction (AMI) in the clinic is unknown. Perhaps, the cuff inflation used currently to induce RIC could be replaced with non-invasive stimulation of the vagus nerve [69]. A recent clinical study demonstrated that the anesthetic propofol impedes the ability of RIC to protect the heart [44]. Propofol is known to be inhibitory to vagus nerve activity [75], thus suggesting the anesthetic prevents the communication of RIC at the level of the parasympathetic centres in the brainstem. Perhaps, RIC should be given, while the patient remains conscious, prior to administration of the anesthetic. Secondly, vagal tone is thought to depreciate with age [40]. Therefore, given the high average age of patients who suffer AMI, their diminished vagal tone may reduce the efficacy of RIC. Further study is required to elucidate the effect of age and anesthetics on RIC, both in terms of vagal tone and function of intrinsic cardiac ganglia. In addition, investigation into which branch of the vagus nerve is responsible for inducing release of the factor will help reveal the site of its release and improve the chance of discovering its identity.
Abbreviations
- AAR:
-
Area-at-risk
- AMI:
-
Acute myocardial infarction
- BCA:
-
Bicinchoninic acid
- CABG:
-
Coronary artery bypass surgery
- CFR:
-
Coronary flow rate
- DVMN:
-
Dorsal vagal motor nucleus
- IRI:
-
Ischemia–reperfusion injury
- IS:
-
Infarct size
- LAD:
-
Left anterior descending
- LVEDP:
-
Left ventricular end diastolic pressure
- mAChR:
-
Muscarinic acetylcholine receptor
- nAChR:
-
Nicotinic acetylcholine receptor
- RIC:
-
Remote ischemic conditioning
- SD:
-
Sprague–Dawley
References
Ajijola OA, Yagishita D, Reddy NK, Yamakawa K, Vaseghi M, Downs AM, Hoover DB, Ardell JL, Shivkumar K (2015) Remodeling of stellate ganglion neurons after spatially targeted myocardial infarction: neuropeptide and morphologic changes. Heart Rhythm. doi:10.1016/j.hrthm.2015.01.045
Ardell JL, Rajendran PS, Nier HA, KenKnight BH, Armour JA (2015) Central-peripheral neural network interactions evoked by vagus nerve stimulation: functional consequences on control of cardiac function. Am J Physiol Heart Circ Physiol 309:H1740–H1752. doi:10.1152/ajpheart.00557.2015
Armour JA (1991) Intrinsic cardiac neurons. J Cardiovasc Electrophysiol 2:331–341. doi:10.1111/j.1540-8167.1991.tb01330.x
Armour JA (2008) Potential clinical relevance of the “little brain” on the mammalian heart. Exp Physiol 93:165–176. doi:10.1113/expphysiol.2007.041178
Armour JA (2011) Physiology of the intrinsic cardiac nervous system. Heart Rhythm 8:739. doi:10.1016/j.hrthm.2011.01.033
Armour JA, Murphy DA, Yuan BX, Macdonald S, Hopkins DA (1997) Gross and microscopic anatomy of the human intrinsic cardiac nervous system. Anat Rec 247:289–298
Beaumont E, Salavatian S, Southerland EM, Vinet A, Jacquemet V, Armour JA, Ardell JL (2013) Network interactions within the canine intrinsic cardiac nervous system: implications for reflex control of regional cardiac function. J Physiol 591:4515–4533. doi:10.1113/jphysiol.2013.259382
Beaumont E, Southerland EM, Hardwick JC, Wright GL, Ryan S, Li Y, KenKnight BH, Armour JA, Ardell JL (2015) Vagus nerve stimulation mitigates intrinsic cardiac neuronal and adverse myocyte remodeling postmyocardial infarction. Am J Physiol Heart Circ Physiol 309:H1198–H1206. doi:10.1152/ajpheart.00393.2015
Bell RM, Mocanu MM, Yellon DM (2011) Retrograde heart perfusion: the Langendorff technique of isolated heart perfusion. J Mol Cell Cardiol 50:940–950. doi:10.1016/j.yjmcc.2011.02.018
Bibevski S, Zhou Y, McIntosh JM, Zigmond RE, Dunlap ME (2000) Functional nicotinic acetylcholine receptors that mediate ganglionic transmission in cardiac parasympathetic neurons. J Neurosci 20:5076–5082
Bøtker HE, Kharbanda R, Schmidt MR, Bøttcher M, Kaltoft AK, Terkelsen CJ, Munk K, Andersen NH, Hansen TM, Trautner S, Lassen JF, Christiansen EH, Krusell LR, Kristensen SD, Thuesen L, Nielsen SS, Rehling M, Sørensen HT, Redington AN, Nielsen TT (2010) Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet 375:727–734. doi:10.1016/S0140-6736(09)62001-8
Brodde O-E, Bruck H, Leineweber K, Seyfarth T (2001) Presence, distribution and physiological function of adrenergic and muscarinic receptor subtypes in the human heart. Basic Res Cardiol 96:528–538. doi:10.1007/s003950170003
Bromage DI, Davidson SM, Yellon DM (2014) Stromal derived factor 1 alpha: a chemokine that delivers a two-pronged defence of the myocardium. Pharmacol Ther 143(3):305–315. doi:10.1016/j.pharmthera.2014.03.009
Candilio L, Malik A, Ariti C, Barnard M, Di Salvo C, Lawrence D, Hayward M, Yap J, Roberts N, Sheikh A, Kolvekar S, Hausenloy DJ, Yellon DM (2015) Effect of remote ischaemic preconditioning on clinical outcomes in patients undergoing cardiac bypass surgery: a randomised controlled clinical trial. Heart 101:185–192. doi:10.1136/heartjnl-2014-306178
Chatterjee M, Gawaz M (2013) Platelet-derived CXCL12 (SDF-1α): basic mechanisms and clinical implications. J Thromb Haemost 11:1954–1967. doi:10.1111/jth.12404
Chatterjee M, Huang Z, Zhang W, Jiang L, Hultenby K, Zhu L, Hu H, Nilsson GP, Li N (2011) Distinct platelet packaging, release, and surface expression of proangiogenic and antiangiogenic factors on different platelet stimuli. Blood 117:3907–3911. doi:10.1182/blood-2010-12-327007
Cheung MMH, Kharbanda RK, Konstantinov IE, Shimizu M, Frndova H, Li J, Holtby HM, Cox PN, Smallhorn JF, Van Arsdell GS, Redington AN (2006) Randomized controlled trial of the effects of remote ischemic preconditioning on children undergoing cardiac surgery: first clinical application in humans. J Am Coll Cardiol 47:2277–2282. doi:10.1016/j.jacc.2006.01.066
Coote JH (2013) Myths and realities of the cardiac vagus. J Physiol 591:4073–4085. doi:10.1113/jphysiol.2013.257758
Dale H (1937) Transmission of nervous effects by acetylcholine: Harvey lecture, May 20, 1937. Bull N Y Acad Med 13:379–396
Davidson SM, Selvaraj P, He D, Boi-Doku C, Yellon RL, Vicencio JM, Yellon DM (2013) Remote ischaemic preconditioning involves signalling through the SDF-1alpha/CXCR4 signalling axis. Basic Res Cardiol 108:377
Dickson EW, Reinhardt CP, Renzi FP, Becker RC, Porcaro WA, Heard SO (1999) Ischemic preconditioning may be transferable via whole blood transfusion: preliminary evidence. J Thromb Thrombolysis 8:123–129
Donato M, Buchholz B, Rodriguez M, Perez V, Inserte J, Garcia-Dorado D, Gelpi RJ (2013) Role of the parasympathetic nervous system in cardioprotection by remote hindlimb ischaemic preconditioning. Exp Physiol 98:425–434
Donato M, Goyeneche MA, Garces M, Marchini T, Pérez V, Del Mauro J, Höcht C, Rodríguez M, Evelson P, Gelpi RJ (2016) Myocardial triggers involved in remote ischemic preconditioning activation. Exp Physiol. doi:10.1113/EP085535
Eglen RM, Michel AD, Cornett CM, Kunysz EA, Whiting RL (1989) The interaction of hexamethonium with muscarinic receptor subtypes in vitro. Br J Pharmacol 98:499–506
Ferrera R, Benhabbouche S, Bopassa JC, Li B, Ovize M (2009) One hour reperfusion is enough to assess function and infarct size with TTC staining in Langendorff rat model. Cardiovasc Drugs Ther 23:327–331. doi:10.1007/s10557-009-6176-5
Gho BC, Schoemaker RG, van den Doel MA, Duncker DJ, Verdouw PD (1996) Myocardial protection by brief ischemia in noncardiac tissue. Circulation 94:2193–2200
Gotti C, Clementi F (2004) Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol 74:363–396. doi:10.1016/j.pneurobio.2004.09.006
Gourine A, Gourine AV (2014) Neural mechanisms of cardioprotection. Physiology 29:133–140. doi:10.1152/physiol.00037.2013
Hardwick JC, Ryan SE, Beaumont E, Ardell JL, Southerland EM (2014) Dynamic remodeling of the guinea pig intrinsic cardiac plexus induced by chronic myocardial infarction. Auton Neurosci 181:4–12. doi:10.1016/j.autneu.2013.10.008
Hausenloy DJ, Candilio L, Evans R, Ariti C, Jenkins DP, Kolvekar S, Knight R, Kunst G, Laing C, Nicholas J, Pepper J, Robertson S, Xenou M, Clayton T, Yellon DM (2015) Remote ischemic preconditioning and outcomes of cardiac surgery. N Engl J Med 373:1408–1417. doi:10.1056/NEJMoa1413534
Hausenloy DJ, Mwamure PK, Venugopal V, Harris J, Barnard M, Grundy E, Ashley E, Vichare S, Di Salvo C, Kolvekar S, Hayward M, Keogh B, MacAllister RJ, Yellon DM (2007) Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: a randomised controlled trial. Lancet 370:575–579. doi:10.1016/S0140-6736(07)61296-3
Hausenloy DJ, Wynne AM, Yellon DM (2007) Ischemic preconditioning targets the reperfusion phase. Basic Res Cardiol 102:445–452. doi:10.1007/s00395-007-0656-1
Heusch G, Bøtker HE, Przyklenk K, Redington A, Yellon D (2015) Remote ischemic conditioning. J Am Coll Cardiol 65:177–195. doi:10.1016/j.jacc.2014.10.031
Hildebrandt HA, Kreienkamp V, Gent S, Kahlert P, Heusch G, Kleinbongard P (2016) Kinetics and signal activation properties of circulating factor(s) from healthy volunteers undergoing remote ischemic pre-conditioning. JACC Basic Transl Sci 1:3–13. doi:10.1016/j.jacbts.2016.01.007
Horackova M, Armour JA, Byczko Z (1999) Distribution of intrinsic cardiac neurons in whole-mount guinea pig atria identified by multiple neurochemical coding. A confocal microscope study. Cell Tissue Res 297:409–421
Janes RD, Johnstone DE, Armour JA (1987) Functional integrity of intrinsic cardiac nerves located over an acute transmural myocardial infarction. Can J Physiol Pharmacol 65:64–69
Jensen RV, Stottrup NB, Kristiansen SB, Botker HE (2012) Release of a humoral circulating cardioprotective factor by remote ischemic preconditioning is dependent on preserved neural pathways in diabetic patients. Basic Res Cardiol 107:285
Kalla M, Chotalia M, Coughlan C, Hao G, Crabtree MJ, Tomek J, Bub G, Paterson DJ, Herring N (2016) Protection against ventricular fibrillation via cholinergic receptor stimulation and the generation of nitric oxide. J Physiol. doi:10.1113/JP271588
Kawano H, Okada R, Yano K (2003) Histological study on the distribution of autonomic nerves in the human heart. Heart Vessels 18:32–39. doi:10.1007/s003800300005
Kelliher GJ, Conahan ST (1980) Changes in vagal activity and response to muscarinic receptor agonists with age. J Gerontol 35:842–849
Kember G, Armour JA, Zamir M (2011) Neural control of heart rate: the role of neuronal networking. J Theor Biol 277:41–47. doi:10.1016/j.jtbi.2011.02.013
Kember G, Armour JA, Zamir M (2013) Dynamic neural networking as a basis for plasticity in the control of heart rate. J Theor Biol 317:39–46. doi:10.1016/j.jtbi.2012.09.024
Kharbanda RK, Mortensen UM, White PA, Kristiansen SB, Schmidt MR, Hoschtitzky JA, Vogel M, Sorensen K, Redington AN, MacAllister R (2002) Transient limb ischemia induces remote ischemic preconditioning in vivo. Circulation 106:2881–2883
Kottenberg E, Thielmann M, Bergmann L, Heine T, Jakob H, Heusch G, Peters J (2012) Protection by remote ischemic preconditioning during coronary artery bypass graft surgery with isoflurane but not propofol—a clinical trial. Acta Anaesthesiol Scand 56:30–38. doi:10.1111/j.1399-6576.2011.02585.x
Krieg T, Qin Q, Philipp S, Alexeyev MF, Cohen MV, Downey JM (2004) Acetylcholine and bradykinin trigger preconditioning in the heart through a pathway that includes Akt and NOS. Am J Physiol Heart Circ Physiol 287:H2606–H2611. doi:10.1152/ajpheart.00600.2004
Li J, Rohailla S, Gelber N, Rutka J, Sabah N, Gladstone RA, Wei C, Hu P, Kharbanda RK, Redington AN (2014) MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res Cardiol 109:423
Lim SY, Yellon DM, Hausenloy DJ (2010) The neural and humoral pathways in remote limb ischemic preconditioning. Basic Res Cardiol 105:651–655. doi:10.1007/s00395-010-0099-y
Longpré J-P, Salavatian S, Beaumont E, Armour JA, Ardell JL, Jacquemet V (2014) Measure of synchrony in the activity of intrinsic cardiac neurons. Physiol Meas 35:549–566. doi:10.1088/0967-3334/35/4/549
Malik A, Bromage DI, He Z, Candilio L, Hamarneh A, Taferner S, Davidson SM, Yellon DM (2015) Exogenous SDF-1α protects human myocardium from hypoxia-reoxygenation injury via CXCR4. Cardiovasc Drugs Ther. doi:10.1007/s10557-015-6622-5
Mastitskaya S, Basalay M, Hosford PS, Ramage AG, Gourine A, Gourine AV (2016) Identifying the source of a humoral factor of remote (pre)conditioning cardioprotection. PLoS One 11:e0150108. doi:10.1371/journal.pone.0150108
Mastitskaya S, Marina N, Gourine A, Gilbey MP, Spyer KM, Teschemacher AG, Kasparov S, Trapp S, Ackland GL, Gourine AV (2012) Cardioprotection evoked by remote ischaemic preconditioning is critically dependent on the activity of vagal pre-ganglionic neurones. Cardiovasc Res 95:487–494
Merlocco AC, Redington KL, Disenhouse T, Strantzas SC, Gladstone R, Wei C, Tropak MB, Manlhiot C, Li J, Redington AN (2014) Transcutaneous electrical nerve stimulation as a novel method of remote preconditioning: in vitro validation in an animal model and first human observations. Basic Res Cardiol 109:406
Meybohm P, Bein B, Brosteanu O, Cremer J, Gruenewald M, Stoppe C, Coburn M, Schaelte G, Böning A, Niemann B, Roesner J, Kletzin F, Strouhal U, Reyher C, Laufenberg-Feldmann R, Ferner M, Brandes IF, Bauer M, Stehr SN, Kortgen A, Wittmann M, Baumgarten G, Meyer-Treschan T, Kienbaum P, Heringlake M, Schön J, Sander M, Treskatsch S, Smul T, Wolwender E, Schilling T, Fuernau G, Hasenclever D, Zacharowski K (2015) A multicenter trial of remote ischemic preconditioning for heart surgery. N Engl J Med 373:1397–1407. doi:10.1056/NEJMoa1413579
Murphy S, Gardner FH (1969) Effect of storage temperature on maintenance of platelet viability–deleterious effect of refrigerated storage. N Engl J Med 280:1094–1098. doi:10.1056/NEJM196905152802004
Murry CE, Jennings RB, Reimer KA (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74:1124–1136
Pickard JMJ, Bøtker HE, Crimi G, Davidson B, Davidson SM, Dutka D, Ferdinandy P, Ganske R, Garcia-Dorado D, Giricz Z, Gourine AV, Heusch G, Kharbanda R, Kleinbongard P, MacAllister R, McIntyre C, Meybohm P, Prunier F, Redington A, Robertson NJ, Suleiman MS, Vanezis A, Walsh S, Yellon DM, Hausenloy DJ (2015) Remote ischemic conditioning: from experimental observation to clinical application: report from the 8th Biennial Hatter Cardiovascular Institute workshop. Basic Res Cardiol 110:453. doi:10.1007/s00395-014-0453-6
Przyklenk K, Bauer B, Ovize M, Kloner RA, Whittaker P (1993) Regional ischemic “preconditioning” protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 87:893–899
Rajendran PS, Nakamura K, Ajijola OA, Vaseghi M, Armour JA, Ardell JL, Shivkumar K (2015) Myocardial infarction induces structural and functional remodeling of the intrinsic cardiac nervous system. J Physiol. doi:10.1113/JP271165
Rassaf T, Totzeck M, Hendgen-Cotta UB, Shiva S, Heusch G, Kelm M (2014) Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res 114(10):1601–1610. doi:10.1161/CIRCRESAHA.114.303822
Redington KL, Disenhouse T, Li J, Wei C, Dai X, Gladstone R, Manlhiot C, Redington AN (2013) Electroacupuncture reduces myocardial infarct size and improves post-ischemic recovery by invoking release of humoral, dialyzable, cardioprotective factors. J Physiol Sci 63:219–223
Redington KL, Disenhouse T, Strantzas SC, Gladstone R, Wei C, Tropak MB, Dai X, Manlhiot C, Li J, Redington AN (2012) Remote cardioprotection by direct peripheral nerve stimulation and topical capsaicin is mediated by circulating humoral factors. Basic Res Cardiol 107:241
Rossello X, Hall AR, Bell RM, Yellon DM (2015) Characterization of the Langendorff perfused isolated mouse heart model of global ischemia–reperfusion injury: impact of ischemia and reperfusion length on infarct size and LDH release. J Cardiovasc Pharmacol Ther 21:286–295. doi:10.1177/1074248415604462
Rysevaite K, Saburkina I, Pauziene N, Vaitkevicius R, Noujaim SF, Jalife J, Pauza DH (2011) Immunohistochemical characterization of the intrinsic cardiac neural plexus in whole-mount mouse heart preparations. Heart Rhythm 8:731–738. doi:10.1016/j.hrthm.2011.01.013
Shimizu M, Tropak M, Diaz RJ, Suto F, Surendra H, Kuzmin E, Li J, Gross G, Wilson GJ, Callahan J, Redington AN (2009) Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: evidence suggesting cross-species protection. Clin Sci (Lond) 117:191–200
Shinlapawittayatorn K, Chinda K, Palee S, Surinkaew S, Kumfu S, Kumphune S, Chattipakorn S, KenKnight BH, Chattipakorn N (2014) Vagus nerve stimulation initiated late during ischemia, but not reperfusion, exerts cardioprotection via amelioration of cardiac mitochondrial dysfunction. Heart Rhythm 11:2278–2287. doi:10.1016/j.hrthm.2014.08.001
Skyschally A, Gent S, Amanakis G, Schulte C, Kleinbongard P, Heusch G (2015) Across-species transfer of protection by remote ischemic preconditioning with species-specific myocardial signal transduction by reperfusion injury salvage kinase and survival activating factor enhancement pathways. Circ Res 117:279–288. doi:10.1161/CIRCRESAHA.117.306878
Sloth AD, Schmidt MR, Munk K, Kharbanda RK, Redington AN, Schmidt M, Pedersen L, Sorensen HT, Botker HE (2014) Improved long-term clinical outcomes in patients with ST-elevation myocardial infarction undergoing remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention. Eur Heart J 35:168–175
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85. doi:10.1016/0003-2697(85)90442-7
Stavrakis S, Humphrey MB, Scherlag BJ, Hu Y, Jackman WM, Nakagawa H, Lockwood D, Lazzara R, Po SS (2015) Low-level transcutaneous electrical vagus nerve stimulation suppresses atrial fibrillation. J Am Coll Cardiol 65:867–875. doi:10.1016/j.jacc.2014.12.026
Steensrud T, Li J, Dai X, Manlhiot C, Kharbanda RK, Tropak M, Redington A (2010) Pretreatment with the nitric oxide donor SNAP or nerve transection blocks humoral preconditioning by remote limb ischemia or intra-arterial adenosine. Am J Physiol Heart Circ Physiol 299:H1598–H1603
Thielmann M, Kottenberg E, Kleinbongard P, Wendt D, Gedik N, Pasa S, Price V, Tsagakis K, Neuhäuser M, Peters J, Jakob H, Heusch G (2013) Cardioprotective and prognostic effects of remote ischaemic preconditioning in patients undergoing coronary artery bypass surgery: a single-centre randomised, double-blind, controlled trial. Lancet 382:597–604. doi:10.1016/S0140-6736(13)61450-6
Thron CD, Waud DR (1968) The rate of action of atropine. J Pharmacol Exp Ther 160:91–105
Uitterdijk A, Yetgin T, te Lintel Hekkert M, Sneep S, Krabbendam-Peters I, van Beusekom HMM, Fischer TM, Cornelussen RN, Manintveld OC, Merkus D, Duncker DJ (2015) Vagal nerve stimulation started just prior to reperfusion limits infarct size and no-reflow. Basic Res Cardiol 110:508. doi:10.1007/s00395-015-0508-3
Ulphani JS, Cain JH, Inderyas F, Gordon D, Gikas PV, Shade G, Mayor D, Arora R, Kadish AH, Goldberger JJ (2010) Quantitative analysis of parasympathetic innervation of the porcine heart. Heart Rhythm 7:1113–1119. doi:10.1016/j.hrthm.2010.03.043
Wang X, Huang Z-G, Gold A, Bouairi E, Evans C, Andresen MC, Mendelowitz D (2004) Propofol modulates gamma-aminobutyric acid-mediated inhibitory neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Anesthesiology 100:1198–1205
Wehr M, Hostick U, Kyweriga M, Tan A, Weible AP, Wu H, Wu W, Callaway EM, Kentros C (2009) Transgenic silencing of neurons in the mammalian brain by expression of the allatostatin receptor (AlstR). J Neurophysiol 102:2554–2562. doi:10.1152/jn.00480.2009
Weinbrenner C, Nelles M, Herzog N, Sárváry L, Strasser RH (2002) Remote preconditioning by infrarenal occlusion of the aorta protects the heart from infarction: a newly identified non-neuronal but PKC-dependent pathway. Cardiovasc Res 55:590–601
White SK, Frohlich GM, Sado DM, Maestrini V, Fontana M, Treibel TA, Tehrani S, Flett AS, Meier P, Ariti C, Davies JR, Moon JC, Yellon DM, Hausenloy DJ (2015) Remote ischemic conditioning reduces myocardial infarct size and edema in patients with ST-segment elevation myocardial infarction. JACC Cardiovasc Interv 8:178–188. doi:10.1016/j.jcin.2014.05.015
Whittington HJ, Harding I, Stephenson CI, Bell R, Hausenloy DJ, Mocanu MM, Yellon DM (2013) Cardioprotection in the aging, diabetic heart: the loss of protective Akt signalling. Cardiovasc Res 99:694–704
Yellon DM, Ackbarkhan AK, Balgobin V, Bulluck H, Deelchand A, Dhuny MR, Domah N, Gaoneadry D, Jagessur RK, Joonas N, Kowlessur S, Lutchoo J, Nicholas JM, Pauvaday K, Shamloll O, Walker JM, Hausenloy DJ (2015) Remote ischemic conditioning reduces myocardial infarct size in STEMI patients treated by thrombolysis. J Am Coll Cardiol 65:2764–2765. doi:10.1016/j.jacc.2015.02.082
Zannad F, De Ferrari GM, Tuinenburg AE, Wright D, Brugada J, Butter C, Klein H, Stolen C, Meyer S, Stein KM, Ramuzat A, Schubert B, Daum D, Neuzil P, Botman C, Castel MA, D’Onofrio A, Solomon SD, Wold N, Ruble SB (2015) Chronic vagal stimulation for the treatment of low ejection fraction heart failure: results of the NEural Cardiac TherApy foR Heart Failure (NECTAR-HF) randomized controlled trial. Eur Heart J 36:425–433. doi:10.1093/eurheartj/ehu345
Acknowledgments
This work was funded by the British Heart Foundation (Grant No. FS12/70/30009)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pickard, J.M.J., Davidson, S.M., Hausenloy, D.J. et al. Co-dependence of the neural and humoral pathways in the mechanism of remote ischemic conditioning. Basic Res Cardiol 111, 50 (2016). https://doi.org/10.1007/s00395-016-0568-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-016-0568-z